 Angela Della Sala
Angela Della Sala Giulia Prono2
Giulia Prono2 Emilio Hirsch
Emilio Hirsch Alessandra Ghigo
Alessandra Ghigo- 1Molecular Biotechnology Center, Department of Molecular Biotechnology and Health Sciences, University of Turin, Turin, Italy
- 2Kither Biotech S.r.l, Turin, Italy
Cystic fibrosis transmembrane conductance regulator (CFTR) is an anion channel expressed on the apical membrane of epithelial cells, where it plays a pivotal role in chloride transport and overall tissue homeostasis. CFTR constitutes a unique member of the ATP-binding cassette transporter superfamily, due to its distinctive cytosolic regulatory (R) domain carrying multiple phosphorylation sites that allow the tight regulation of channel activity and gating. Mutations in the CFTR gene cause cystic fibrosis, the most common lethal autosomal genetic disease in the Caucasian population. In recent years, major efforts have led to the development of CFTR modulators, small molecules targeting the underlying genetic defect of CF and ultimately rescuing the function of the mutant channel. Recent evidence has highlighted that this class of drugs could also impact on the phosphorylation of the R domain of the channel by protein kinase A (PKA), a key regulatory mechanism that is altered in various CFTR mutants. Therefore, the aim of this review is to summarize the current knowledge on the regulation of the CFTR by PKA-mediated phosphorylation and to provide insights into the different factors that modulate this essential CFTR modification. Finally, the discussion will focus on the impact of CF mutations on PKA-mediated CFTR regulation, as well as on how small molecule CFTR regulators and PKA interact to rescue dysfunctional channels.
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-activated chloride and bicarbonate channel expressed at the apical surface of secretory epithelia, including the airways, sweat glands, gastrointestinal tract, and other tissues (Riordan et al., 1989; Rommens et al., 1989; Riordan, 2008). The CFTR has a critical role in transepithelial ion and fluid secretion and homeostasis, and mutations in this gene have been implied in the pathogenesis of cystic fibrosis (CF; Li and Naren, 2005). CF, the most common life-shortening rare disease among Caucasians, is an autosomal recessive genetic disease affecting around 32,000 individuals in Europe and about 85,000 individuals worldwide (Zolin et al., 2020). The absence of a functional CFTR leads to a decrease in chloride ion secretion that, together with the consequent alteration of water homeostasis, results in the accumulation of dehydrated mucus, recurrent bacterial infection, and ultimately organ failure. Although CFTR mutations cause a multiorgan disease, respiratory failure is the major cause of morbidity and mortality for CF patients (Ratjen et al., 2015). Although CF nowadays remains an incurable disease, the identification of the defective CFTR gene in 1989 (Riordan et al., 1989; Rommens et al., 1989) has prompted significant advances in the development of molecular therapies aimed at addressing the underlying cellular defect (De Boeck and Amaral, 2016; Veit et al., 2016).
The CFTR gene, expressed on the long arm of chromosome 7 (Kerem et al., 1989), encodes for a unique member of the large protein superfamily of ATP-binding cassette (ABC) transporters (Klein et al., 1999), which carries a cytosolic regulatory domain that is actively phosphorylated (Collins, 1992; Serohijos et al., 2008). Whereas most of the ABC transporters are active pumps using ATP as the energy source for the transport of small molecules, CFTR is an ATP-gated ion channel where ATP hydrolysis controls channel opening (Dean et al., 2001). Cystic fibrosis transmembrane conductance regulator, like other ABC transporters, is composed of two nucleotide-binding domains (NBDs), involved in channel regulation through ATP binding and hydrolysis, and two transmembrane domains (TMDs), containing six helices that span the plasma membrane to form the ion channel pore (Riordan et al., 1989; Callebaut et al., 2018). In the CFTR, however, the NDB1 domain is linked to the NDB2 one by an additional structural region, a distinctive cytoplasmic regulatory (R) domain with many charged residues and multiple phosphorylation sites that allow the tight regulation of channel activity and gating.
Importantly, the gating of the channel is strictly coupled to the phosphorylation of the R domain by protein kinase A (PKA; Egan et al., 1992; Vergani et al., 2005a,b) and protein kinase C (Chappe et al., 2003; Seavilleklein et al., 2008). PKA-dependent phosphorylation triggers large conformational changes that remove the R region from its position and allow NBDs dimerization to occur. Consequently, ATP binds to the CFTR leading to the opening and activation of the channel, while ATP hydrolysis closes it (Gunderson and Kopito, 1995; Figure 1). In addition to regulating the gating of the channel, PKA-mediated phosphorylation is involved in the regulation of multiple processes, such as CFTR trafficking and stability at the plasma membrane (Chin et al., 2017a).
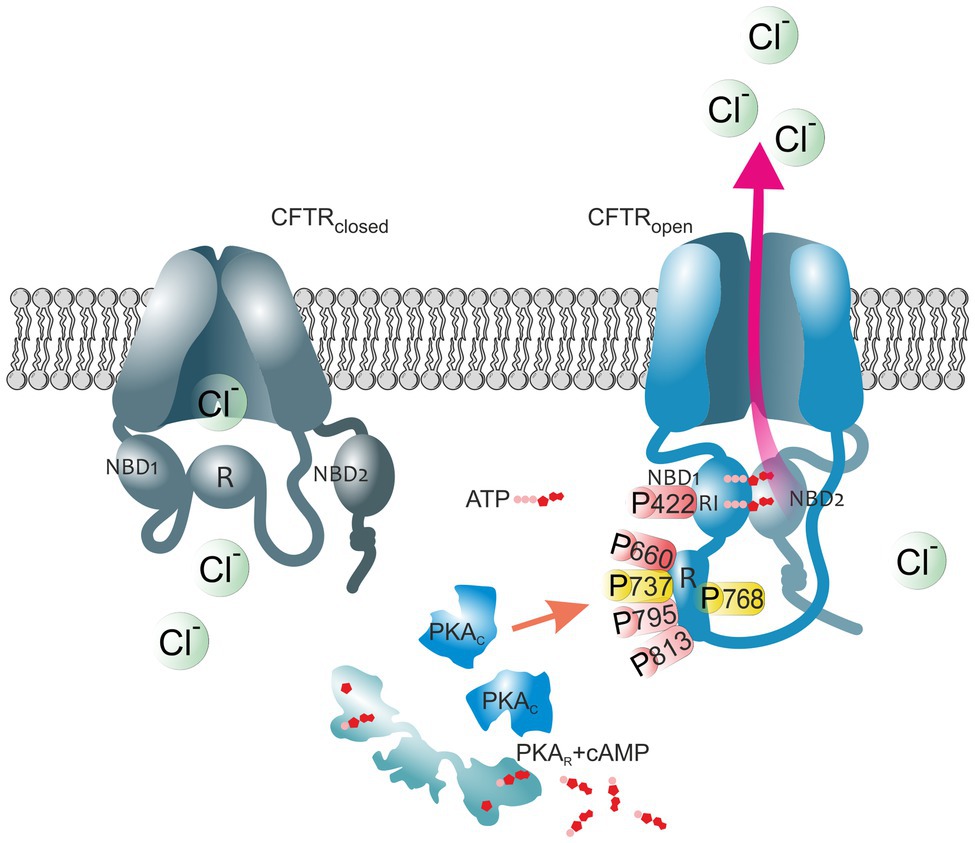
Figure 1. The impact of PKA-mediated phosphorylation on CFTR channel. The cystic fibrosis transmembrane conductance regulator (CFTR) structure consists of two membrane spanning domains, two nucleotide-binding domains (NBD1 and NBD2), and the unique cytoplasmic regulatory (R) domain. Among these, the latter represents a critical site of channel regulation due to its enrichment in protein kinase A (PKA) consensus motifs, with multiple serines and threonines as phosphorylation targets. Moreover, there is an additional phosphorylation site in the regulatory insertion (RI) segment of NBD1. In the closed CFTR state (left channel), the NBD1 interacts with the R domain creating steric hindrances which prevent it from dimerization with NBD2. Upon PKA-dependent phosphorylation (right channel), large conformational changes occur that decrease this interaction leading to the release of the R region from its inhibitory position and allow NBDs dimerization, ultimately ATP binding and CFTR-mediated chloride secretion. Thus, the open probability of the channel is dependent on the access of PKA to the main phospho-sites (S422, S660, S737, S768, S795, and S813). Phospho-sites critical for CFTR channel activation are shown in red. Phosphoserines S737 and S768 are shown in yellow since they have been shown to either activate or inhibit CFTR gating. The role of the major PKA consensus sites in the regulation of CFTR function is detailed in Table 1.
This review will discuss the role of PKA in CFTR activity regulation, and how this physiological mechanism of channel activation is disrupted by CF mutations. Furthermore, in light of the recent advances in the development of CFTR modulators, we will discuss the impact of these novel molecules on the PKA-mediated regulation of mutant channels.
CFTR Gating Requires PKA-Mediated Phosphorylation
Although the molecular mechanisms underlying CFTR activation have been studied for years, several questions remain unanswered. This section discusses how channel gating strictly depends on the intrinsic structure of the CFTR protein and several posttranslational modifications, primarily phosphorylation.
One of the major challenges to the understanding of the regulation by the R region of the opening and the gating of CFTR has been the resolution of the CFTR structure. A crucial breakthrough has been represented by the construction of medium-high-resolution models of the 3D structures of the full-length CFTR, first from zebrafish (Zhang and Chen, 2016) and then from humans (Liu et al., 2017). These molecular structures of the CFTR, determined using cryo-electron microscopy, provide insights into a dephosphorylated, ATP-free conformation, which represents a closed and inactive state of the channel (Zhang and Chen, 2016; Liu et al., 2017). Of note, Liu and colleagues with the resolution of the human CFTR structure reveal a previously unresolved helix belonging to the R region docked in an inward-facing conformation between the two halves of CFTR, where it acts as a steric block precluding channel opening (Liu et al., 2017). On the other hand, activation of the CFTR channel is strictly coupled to the formation of a closed NBD dimer, and thereby to the initial conformational changes driven by the PKA-dependent phosphorylation of the R domain (Liu et al., 2017; Mihalyi et al., 2020).
Thus, the open probability of the channel is dependent on the access of PKA to the multiple consensus sites in the R domain (Liu et al., 2017), including 18 potential phosphorylation sites (12 serines and 8 threonines) on a 200 residues long domain. Specifically, in the fully phosphorylated protein, eight phosphoserines (residue positions 660, 700, 712, 737, 753, 768, 795, and 813) and partial phosphorylation of serine at position 670 (Baker et al., 2007) have been detected by mass spectrometry (Townsend et al., 1996; Neville et al., 1997) and NMR (Baker et al., 2007). Recently, Lucaks and colleagues implemented a novel CFTR affinity enrichment method, followed by an advanced mass spectrometry technique, to establish the phospho-occupancy of CFTR upon PKA phosphorylation (Schnur et al., 2019). Among the previously described 15 PKA consensus sites, they identified 10 PKA phospho-sites in the R and NBD1 domain of CFTR (serine 422, 660, 670, 686, 700, 712, 737, 753, 768, and 795; Schnur et al., 2019). Table 1 provides a comprehensive description of the major phospho-sites and their effect on CFTR channel activity regulation. The role of each phosphoserine in the modulation of CFTR activity is quite complex and multiple experiments have been attempted to address this problem by site-direct mutagenesis, where the putative phosphorylation sites in the R region were substituted with alanines (Kanelis et al., 2010; Marasini et al., 2012, 2013). Of note, among such studies, the replacement of serine at position 700, 795, and 813 revealed a decrease in channel open probability (Rich et al., 1993; Vais et al., 2004; Chen, 2020), whereas mutations of serine 737 and 768 increased the activity, thus implying a phospho-dependent inhibitory effect of these residues on the CFTR channel function (Wilkinson et al., 1997; Kongsuphol et al., 2009). Accordingly, phosphorylation of these inhibitory sites by AMPK inhibits CFTR-mediated chloride secretion by maintaining the channel in a closed state (Kongsuphol et al., 2009). These findings appear to be in apparent contrast with the results of the work by Riordan and colleagues aimed at investigating whether individual PKA phospho-sites act cooperatively or distinctly in the regulation of channel activity. This study investigated the impact of the reintroduction of S737 and S768 in a CFTR variant insensitive to PKA, namely, the 15SA mutant, in which the 15 PKA consensus sites were replaced by alanines (Hegedus et al., 2009). Reintroduction of S737 or S768 or both restored a significant level of channel activation by PKA, thus indicating that phosphorylation of these sites stimulates CFTR channel activity. Furthermore, a mutant CFTR in which all phosphorylation sites have been removed, completely eliminates the PKA-dependent regulation of the channel activity (Xie et al., 2002). In line with this finding, a CFTR mutant containing six or more serine-to-aspartate substitutions mimicking the effect of phosphorylation results in channel opening even in the absence of PKA (Rich et al., 1993).

Table 1. PKA phospho-sites in CFTR and their role in channel activity regulation.
Overall, these experiments highlight that the effects of PKA-dependent phosphorylation of the R domain are correlated to conformational changes triggered by the negative charges of the phosphate group introduced on phosphoserines (Zhang et al., 2017, 2018; Schnur et al., 2019).
As a confirmation, circular dichroism, X-ray scattering, and NMR experiments showed a reduced density of the phosphorylated structure, suggesting that upon phosphorylation the R domain becomes entirely disordered and less compact (Baker et al., 2007; Bozoky et al., 2013). Further evidence of the structure of phosphorylated CFTR, in its ATP-bound conformation, comes from cryo-electron microscopy (Zhang et al., 2017, 2018). These images, comparing the conformation in a phosphorylated and ATP-bound state (Zhang and Chen, 2016; Liu et al., 2017) with the dephosphorylated and ATP-free conformation (Zhang et al., 2017, 2018), define the structural changes of the two human and zebrafish CFTR orthologs. These structures further reveal the clearly distinct position of the R domain after phosphorylation. Upon PKA binding, phosphorylation promotes the release of the R domain from its inhibitory position, causing NBDs dimerization and flipping of the TMDs into an outward-facing conformation, ultimately leading to channel opening and activation of its ATPase function (Sorum et al., 2015; Figure 1).
On the other hand, how phosphorylation triggers the ATPase activity of CFTR has remained unclear. Whereas PKA-mediated phosphorylation of the R domain is absolutely required (Chang et al., 1993; Winter and Welsh, 1997; Seibert et al., 1999), whether ATP binding is essential for CFTR gating is still controversial. In the following paragraph, we will describe how phosphorylated CFTR channels are gated through ATP-dependent and independent mechanisms.
Effects of PKA-Mediated Phosphorylation on ATP Binding
The widely accepted model of CFTR activation establishes that phosphorylated CFTR channels are gated through an ATP-dependent mechanism that is based on conformational changes (Sorum et al., 2015). In these models, the ATP molecule powers the gating cycle by inducing the opening through NBD1:NBD2 dimerization as well as the closing through its hydrolysis (Higgins and Linton, 2004). While PKA phosphorylation promotes the release of the R domain from its position, the binding of ATP triggers the dimerization of the NBDs that, in turn, leads to channel opening and activation (Vergani et al., 2003, 2005b; Csanady et al., 2010). Therefore, the phosphorylation of the R domain increases the rate of channel opening by stimulating conformational changes that enhance the affinity for ATP (Winter and Welsh, 1997).
Following this obligatory modification, the ATP generally bound only to NBD1 is released, and it binds to the two sites at the NBD1:NBD2 interface (Aleksandrov et al., 2018). Recent studies by Csana and colleagues support a model wherein ATP binding to both NBDs enhances their dimerization and consequently channel opening (Mihalyi et al., 2016). Instead, the ATPase activity at NBD2 subsequently promotes destabilization of the NBD dimer, leading to channel closure (Mihalyi et al., 2016). Additionally, Lewis and colleagues have found that in the CFTR closed state, the NBDs dimer is prevented, due to the inhibitory interaction of the R domain with the regulatory insertion (RI) domain of NDB1 (Lewis et al., 2004). Although the majority of the canonical phosphorylation sites are located within the R domain of CFTR, an additional residue (S422) in the RI region plays a key regulatory function (Lewis et al., 2004). In particular, S422 of the RI domain interacts with S660 in the R domain, creating steric hindrances on NBD1, which prevent it from dimerization with NBD2 (Hudson et al., 2012; Dawson et al., 2013). Upon phosphorylation by PKA of these two serines, the alpha-helical content of both the RI and the R regions decreases (Hudson et al., 2012; Dawson et al., 2013). Therefore, phosphorylation by PKA weakens the interaction between the R domain and NBD1, ultimately allowing the formation of the NBD1:NBD2 dimer (Aleksandrov et al., 2018).
Different from the wild-type CFTR channel, where the gating is dependent on ATP binding, in gating mutants where ATP binding is impaired, the introduction of a second mutation, like the K978C or the NBD2 deletion, restores the open conformation of the channel, but PKA-mediated phosphorylation of the R domain is still required for full channel activation (Eckford et al., 2012). These examples of ATP-independent CFTR channel appear to be regulated by phosphorylation, via allosteric interactions between the R domain and the NBD1 region (Eckford et al., 2012). Nonetheless, further studies are required to understand the molecular basis for such ATP-independent gating and CFTR regulation.
Overall, phosphorylation represents an additional level of control of CFTR activity besides ATP binding and hydrolysis. Given that the ATP concentrations within a cell are high enough for constant activation of the channel, phosphorylation is essential for the prevention of futile cycling of the channel between the open and closed conformations (Higgins and Linton, 2004).
Effects of CF-Causing Mutations on PKA-Mediated Phosphorylation of CFTR
Considering the essential role of PKA-dependent phosphorylation of the intrinsically disordered R domain in modulating CFTR gating (Hallows et al., 2003; Billet et al., 2015, 2016), it is plausible that CF-causing mutations may disturb this mechanism of channel regulation. The most common CFTR mutation, consisting of the deletion of phenylalanine (F508del) located at the interface between NBD1 and the intracellular loop 4, interferes with the correct R domain folding and assembly (Riordan, 2005; Hwang et al., 2018). As a consequence, the aberrant channel is targeted to early ER-associated degradation (Farinha and Amaral, 2005). Moreover, the resulting thermal instability of this mutant form further contributes to its removal from the plasma membrane (Okiyoneda et al., 2010), which has been shown to contain only 2% of mature F508del-CFTR compared to the normal wild-type (WT) channel amount (Van Goor et al., 2011). Importantly, Wang and colleagues demonstrated that the phosphorylation rate of F508del-CFTR by PKA is significantly lower than that of WT-CFTR, suggesting that the abnormal protein does not constitute a good substrate for the kinase (Wang et al., 2000). The same results were obtained from the analysis of the other two disease-causing CFTR mutations, R170G and A1067T, both occurring in coupling helices that allow the correct channel activation by phosphorylation (Chin et al., 2017b).
Furthermore, a relevant study by Pasyk et al. exploited mass spectrometry to quantitatively assess PKA-mediated phosphorylation of serine 660 of F508del-CFTR at the ER and reported a drastic reduction of phosphorylation upon forskolin stimulation in comparison with the WT form (Pasyk et al., 2009). Interestingly, COPI-mediated retrograde trafficking from the Golgi to the ER, which prevents misfolded F508del-CFTR from successfully reaching the apical cell membrane (Rennolds et al., 2008; Okiyoneda et al., 2010), has been linked to phosphorylation-dependent interactions between the channel and 14-3-3 proteins (Liang et al., 2012). In particular, selective binding of 14-3-3 protein isoforms to specific PKA-phosphorylated sites within the R region decreases this retrograde retrieval, ultimately resulting in augmented CFTR biogenesis. Unfortunately, whether the impaired phosphorylation caused by the deletion of F508 negatively impacts this protein-protein interaction has yet to be elucidated.
Another relevant process that contributes to the maintenance of channel density at the plasma membrane is the endosomal trafficking of the CFTR, which ensures the internalization and recycling of abundant or misfolded proteins (Moore et al., 2007). Notably, Holleran and collaborators revealed that cell surface F508del-CFTR is targeted to lysosomes and displays defective PKA-regulated exocytosis, consisting of significantly slower rates of translocation to the plasma membrane from the Rab11+/EHD1+ endosomal recycling compartment upon PKA challenge (Holleran et al., 2013). Therefore, these data suggest that the abnormal phosphorylation of the mutant CFTR by PKA could, at least partly, account for the defective peripheral trafficking of the channel. Moreover, it has been shown that the excision of the RI polypeptide from the NBD1 of F508del protein, which contains the additional PKA consensus site S422, may be beneficial for the life cycle of the mutant channel (Aleksandrov et al., 2010). Specifically, removal of this region was found to promote the escape of F508del-CFTR from the ER quality control machinery and thus the increased apical surface stability. In addition, recent evidence confirmed that the amount of phosphorylated S422, which represents a minor PKA target site, is decreased in the aberrant F508del protein (Pankow et al., 2019). However, further analyses are required to clarify whether the presence of the RI hampers the F508del-CFTR stability independently of its phosphorylation state and to evaluate the biological consequence of its reduced phosphorylation in the mutant CFTR.
Another important CFTR genetic defect that could potentially interfere with PKA-mediated phosphorylation of the channel is the G551D mutation, a class III mutation with a worldwide frequency of ∼4% (CFF patient registry, 2019). The disease-causing substitution occurs in the NDB1 domain and strongly impairs the ATP-dependent channel gating, without hindering channel trafficking to the plasma membrane (Gregory et al., 1991; Bompadre et al., 2007). Interestingly, Chang and colleagues demonstrated that the R domain of this mutant form undergoes normal phosphorylation despite its lack of ATP binding (Chang et al., 1993). In contrast, a very recent study showed that G551D-CFTR exhibits defective phosphorylation-dependent activation as a result of a decreased sensitivity to PKA-mediated phosphorylation. Stimulation of mutant channels with high doses of PKA induced a remarkable increase in their activity, thus suggesting that the intrinsic phosphorylation defect of G551D-CFTR might be one of the major causes of low basal functionality. Moreover, phosphorylation of the serine residue 737 of G551D occurred at a lesser extent compared to WT-CFTR, highlighting a possible causative factor of the slower activation rate of the aberrant protein (Wang et al., 2020).
Taken together, abnormal PKA-mediated phosphorylation underlies multiple molecular defects observed in mutant CFTR channels and represents a promising therapeutic target for the treatment of CF. Therefore, the following paragraph will focus on how currently available CFTR modulators impact on the phosphorylation of the channel by PKA.
Effects of CFTR Modulators on PKA-Dependent Regulation of CFTR
In recent years, major advances have been made in the field of precision therapy against the underlying cause of CF. A variety of small molecules, designed to target specific channel defects and collectively known as CFTR modulators, have been developed to improve or even restore the expression, activity, and stability of defective CFTR variants (Lopes-Pacheco, 2016). Of outmost relevance are CFTR correctors, which rescue the misprocessing of mutant CFTRs, and potentiators, that are intended to restore the defective cAMP-dependent chloride channel activity at the cell surface (Lopes-Pacheco, 2016). Nonetheless, how these therapeutic agents act on the PKA-mediated phosphorylation of CFTR mutants is still largely unaddressed.
Importantly, there are some paradigmatic exceptions of CFTR modulating drugs whose relationship with PKA-dependent phosphorylation has been investigated and will be now discussed. The potentiator VX-770 (ivacaftor) is approved both as a single therapy for G551D mutants and as a combination with either the corrector VX-809 (lumacaftor; Connett, 2019) or VX-445 and VX-661 for F508del carriers (Bear, 2020). Importantly, ivacaftor was found to enhance channel activity in an ATP-independent manner (Eckford et al., 2012). However, non-phosphorylated CFTR does not exhibit any significant ion current increase upon VX-770 stimulation, suggesting that its potentiating action is strictly dependent on the phosphorylation state of the channel (Eckford et al., 2012). Conversely, Jih and collaborators demonstrated that this potentiator can increase the activity of a CFTR lacking the R domain, thus arguing with the previous hypotheses of a phosphorylation-dependent mechanism (Jih and Hwang, 2013). Recently, a study by Uliyakina et al. revealed that the absence of the RI domain strongly emphasizes VX-809-mediated rescue of F508del-CFTR but negatively impacts the channel currents stimulated by VX-770 (Uliyakina et al., 2020). Therefore, these two modulators display a contradictory behavior in the absence of the unique region containing the PKA phosphorylation site, S422, but future analyses are needed to dissect the role of RI in mutant CFTR. Additionally, another molecule, VRT-532, which had been formerly identified as a potentiator (Van Goor et al., 2006), showed to significantly amplify the activity of G551D-CFTR mutants. Despite its direct interaction with the aberrant channel (Pasyk et al., 2009), VRT-532 did not induce an increase in CFTR phosphorylation, suggesting that its mechanism of action occurs at stages that are downstream of the PKA kinase activity (Pyle et al., 2011).
Conversely, correctors based on 3-(2-benzyloxy-phenyl)-5-chloromethyl-isoxazoles, like UCCF-152, were found to stimulate potent and rapid phosphorylation of the R domain of WT, temperature-rescued F508del-CFTR and G551D-CFTR, while also increasing iodide currents, leading to their classification as CFTR activators (Pyle et al., 2011). The specific residues phosphorylated upon the interaction with UCCF-152 have not been yet identified, but this isoxazole neither raises cellular cAMP levels nor directly activates PKA, suggesting a possible enhancement of the propensity of the R domain to be phosphorylated by releasing steric hindrances (Sammelson et al., 2003). To date, no additional analyses have been performed to further characterize UCCF-152 as a candidate CF-treating agent. The impact of CF-causing mutations on PKA phosphorylation of CFTR, and the effect of CFTR modulators on the activity of mutant channels is summarized in Table 2.
Table 2. Effects of CFTR modulators on PKA-dependent regulation of CFTR mutants.
Overall, our current knowledge of how CFTR modulators interfere or promote PKA-dependent phosphorylation of the channel remains scarce, and future efforts are needed to allow a better understanding of their impact on this essential molecular modification of the channel.
Conclusion
In conclusion, PKA-dependent phosphorylation plays a key role in multiple steps during the life cycle of CFTR. While interactions resulting in phosphorylation at the CFTR R domain regulate channel opening and activity, other phosphorylation events at the C and N terminal ends of CFTR modulate channel stability and trafficking at the PM. Importantly, CFTR mutations leading to CF impair different steps of CFTR biogenesis that are regulated by these phosphorylation events. Targeting PKA-mediated phosphorylation thus represents a promising strategy to rescue the activity of different CF-causing CFTR variants. Nonetheless, how correctors and potentiators, including their highly effective combinations, like the recently approved Trikafta, impact on the PKA-mediated phosphorylation of CFTR still needs to be thoroughly investigated. Future studies in this direction might help to maximize therapeutic efficacy, ultimately normalizing the life expectancy of CF patients.
Author Contributions
ADS designed the overall layout and wrote the bulk of the manuscript. GP was involved in the writing of the manuscript and referencing. EH reviewed the manuscript and produced the figure. AG was involved in the editing and critical revision of the manuscript. AG and EH acquired the funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Italian Cystic Fibrosis Research Foundation (FFC#8/2018 to EH), Compagnia di Sanpaolo (CSTO161109 to EH), Cariplo Foundation (#2018-0498 to EH), Roche Foundation (Bando Roche per la Ricerca 2019 to AG), and Telethon Foundation (GGP20079 to AG).
Conflict of Interest
AG and EH are founders and board members of Kither Biotech, a company focused on the development of PI3K inhibitors for airway diseases not in conflict with statements in this article. The other co-authors declare no conflict of interest.
References
Aleksandrov, L. A., Fay, J. F., and Riordan, J. R. (2018). R-domain phosphorylation by protein kinase A stimulates dissociation of unhydrolyzed ATP from the first nucleotide-binding site of the cystic fibrosis transmembrane conductance regulator. Biochemistry 57, 5073–5075. doi: 10.1021/acs.biochem.8b00646
Aleksandrov, A. A., Kota, P., Aleksandrov, L. A., He, L., Jensen, T., Cui, L., et al. (2010). Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J. Mol. Biol. 401, 194–210. doi: 10.1016/j.jmb.2010.06.019
Baker, J. M., Hudson, R. P., Kanelis, V., Choy, W. Y., Thibodeau, P. H., Thomas, P. J., et al. (2007). CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat. Struct. Mol. Biol. 14, 738–745. doi: 10.1038/nsmb1278
Bear, C. E. (2020). A therapy for most with cystic fibrosis. Cell 180:211. doi: 10.1016/j.cell.2019.12.032
Billet, A., Jia, Y., Jensen, T. J., Hou, Y. X., Chang, X. B., Riordan, J. R., et al. (2016). Potential sites of CFTR activation by tyrosine kinases. Channels 10, 247–251. doi: 10.1080/19336950.2015.1126010
Billet, A., Jia, Y., Jensen, T., Riordan, J. R., and Hanrahan, J. W. (2015). Regulation of the cystic fibrosis transmembrane conductance regulator anion channel by tyrosine phosphorylation. FASEB J. 29, 3945–3953. doi: 10.1096/fj.15-273151
Bompadre, S. G., Sohma, Y., Li, M., and Hwang, T. C. (2007). G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J. Gen. Physiol. 129, 285–298. doi: 10.1085/jgp.200609667
Bozoky, Z., Krzeminski, M., Muhandiram, R., Birtley, J. R., Al-Zahrani, A., Thomas, P. J., et al. (2013). Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactions. Proc. Natl. Acad. Sci. U. S. A. 110, E4427–E4436. doi: 10.1073/pnas.1315104110
Callebaut, I., Chong, P. A., and Forman-Kay, J. D. (2018). CFTR structure. J. Cyst. Fibros. 17, S5–S8. doi: 10.1016/j.jcf.2017.08.008
Chang, X. B., Tabcharani, J. A., Hou, Y. X., Jensen, T. J., Kartner, N., Alon, N., et al. (1993). Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J. Biol. Chem. 268, 11304–11311. doi: 10.1016/S0021-9258(18)82125-1
Chappe, V., Hinkson, D. A., Zhu, T., Chang, X. B., Riordan, J. R., and Hanrahan, J. W. (2003). Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J. Physiol. 548, 39–52. doi: 10.1113/jphysiol.2002.035790
Chen, J. H. (2020). Protein kinase A phosphorylation potentiates cystic fibrosis transmembrane conductance regulator gating by relieving autoinhibition on the stimulatory C terminus of the regulatory domain. J. Biol. Chem. 295, 4577–4590. doi: 10.1074/jbc.RA119.008427
Chin, S., Hung, M., and Bear, C. E. (2017a). Current insights into the role of PKA phosphorylation in CFTR channel activity and the pharmacological rescue of cystic fibrosis disease-causing mutants. Cell. Mol. Life Sci. 74, 57–66. doi: 10.1007/s00018-016-2388-6
Chin, S., Yang, D., Miles, A. J., Eckford, P. D. W., Molinski, S., Wallace, B. A., et al. (2017b). Attenuation of phosphorylation-dependent activation of cystic fibrosis transmembrane conductance regulator (CFTR) by disease-causing mutations at the transmission Interface. J. Biol. Chem. 292, 1988–1999. doi: 10.1074/jbc.M116.762633
Collins, F. S. (1992). Cystic fibrosis: molecular biology and therapeutic implications. Science 256, 774–779. doi: 10.1126/science.1375392
Connett, G. J. (2019). Lumacaftor-ivacaftor in the treatment of cystic fibrosis: design, development and place in therapy. Drug Des. Devel. Ther. 13, 2405–2412. doi: 10.2147/DDDT.S153719
Csanady, L., Vergani, P., and Gadsby, D. C. (2010). Strict coupling between CFTR's catalytic cycle and gating of its cl- ion pore revealed by distributions of open channel burst durations. Proc. Natl. Acad. Sci. U. S. A. 107, 1241–1246. doi: 10.1073/pnas.0911061107
Dawson, J. E., Farber, P. J., and Forman-Kay, J. D. (2013). Allosteric coupling between the intracellular coupling helix 4 and regulatory sites of the first nucleotide-binding domain of CFTR. PLoS One 8:e74347. doi: 10.1371/journal.pone.0074347
Dean, M., Rzhetsky, A., and Allikmets, R. (2001). The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166. doi: 10.1101/gr.GR-1649R
De Boeck, K., and Amaral, M. D. (2016). Progress in therapies for cystic fibrosis. Lancet Respir. Med. 4, 662–674. doi: 10.1016/S2213-2600(16)00023-0
Eckford, P. D., Li, C., Ramjeesingh, M., and Bear, C. E. (2012). Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 287, 36639–36649. doi: 10.1074/jbc.M112.393637
Egan, M., Flotte, T., Afione, S., Solow, R., Zeitlin, P. L., Carter, B. J., et al. (1992). Defective regulation of outwardly rectifying cl- channels by protein kinase A corrected by insertion of CFTR. Nature 358, 581–584. doi: 10.1038/358581a0
Farinha, C. M., and Amaral, M. D. (2005). Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 25, 5242–5252. doi: 10.1128/MCB.25.12.5242-5252.2005
Gregory, R. J., Rich, D. P., Cheng, S. H., Souza, D. W., Paul, S., Manavalan, P., et al. (1991). Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol. Cell. Biol. 11, 3886–3893. doi: 10.1128/MCB.11.8.3886
Gunderson, K. L., and Kopito, R. R. (1995). Conformational states of CFTR associated with channel gating: the role ATP binding and hydrolysis. Cell 82, 231–239. doi: 10.1016/0092-8674(95)90310-0
Hallows, K. R., Mccane, J. E., Kemp, B. E., Witters, L. A., and Foskett, J. K. (2003). Regulation of channel gating by AMP-activated protein kinase modulates cystic fibrosis transmembrane conductance regulator activity in lung submucosal cells. J. Biol. Chem. 278, 998–1004. doi: 10.1074/jbc.M210621200
Hegedus, T., Aleksandrov, A., Mengos, A., Cui, L., Jensen, T. J., and Riordan, J. R. (2009). Role of individual R domain phosphorylation sites in CFTR regulation by protein kinase A. Biochim. Biophys. Acta 1788, 1341–1349. doi: 10.1016/j.bbamem.2009.03.015
Higgins, C. F., and Linton, K. J. (2004). The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11, 918–926. doi: 10.1038/nsmb836
Holleran, J. P., Zeng, J., Frizzell, R. A., and Watkins, S. C. (2013). Regulated recycling of mutant CFTR is partially restored by pharmacological treatment. J. Cell Sci. 126, 2692–2703. doi: 10.1242/jcs.120196
Hudson, R. P., Chong, P. A., Protasevich, I., Vernon, R., Noy, E., Bihler, H., et al. (2012). Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 287, 28480–28494. doi: 10.1074/jbc.M112.371138
Hwang, T. C., Yeh, J. T., Zhang, J., Yu, Y. C., Yeh, H. I., and Destefano, S. (2018). Structural mechanisms of CFTR function and dysfunction. J. Gen. Physiol. 150, 539–570. doi: 10.1085/jgp.201711946
Jih, K. Y., and Hwang, T. C. (2013). Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc. Natl. Acad. Sci. U. S. A. 110, 4404–4409. doi: 10.1073/pnas.1215982110
Kanelis, V., Hudson, R. P., Thibodeau, P. H., Thomas, P. J., and Forman-Kay, J. D. (2010). NMR evidence for differential phosphorylation-dependent interactions in WT and DeltaF508 CFTR. EMBO J. 29, 263–277. doi: 10.1038/emboj.2009.329
Kerem, B., Rommens, J. M., Buchanan, J. A., Markiewicz, D., Cox, T. K., Chakravarti, A., et al. (1989). Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073–1080. doi: 10.1126/science.2570460
Klein, I., Sarkadi, B., and Varadi, A. (1999). An inventory of the human ABC proteins. Biochim. Biophys. Acta 1461, 237–262. doi: 10.1016/s0005-2736(99)00161-3
Kongsuphol, P., Cassidy, D., Hieke, B., Treharne, K. J., Schreiber, R., Mehta, A., et al. (2009). Mechanistic insight into control of CFTR by AMPK. J. Biol. Chem. 284, 5645–5653. doi: 10.1074/jbc.M806780200
Lewis, H. A., Buchanan, S. G., Burley, S. K., Conners, K., Dickey, M., Dorwart, M., et al. (2004). Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23, 282–293. doi: 10.1038/sj.emboj.7600040
Li, C., and Naren, A. P. (2005). Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol. Ther. 108, 208–223. doi: 10.1016/j.pharmthera.2005.04.004
Liang, X., Da Paula, A. C., Bozoky, Z., Zhang, H., Bertrand, C. A., Peters, K. W., et al. (2012). Phosphorylation-dependent 14-3-3 protein interactions regulate CFTR biogenesis. Mol. Biol. Cell 23, 996–1009. doi: 10.1091/mbc.E11-08-0662
Liu, F., Zhang, Z., Csanady, L., Gadsby, D. C., and Chen, J. (2017). Molecular structure of the human CFTR ion channel. Cell 169, 85.e8–95.e8. doi: 10.1016/j.cell.2017.02.024
Lopes-Pacheco, M. (2016). CFTR modulators: shedding light on precision medicine for cystic fibrosis. Front. Pharmacol. 7:275. doi: 10.3389/fphar.2016.00275
Marasini, C., Galeno, L., and Moran, O. (2012). Thermodynamic study of the native and phosphorylated regulatory domain of the CFTR. Biochem. Biophys. Res. Commun. 423, 549–552. doi: 10.1016/j.bbrc.2012.05.165
Marasini, C., Galeno, L., and Moran, O. (2013). A SAXS-based ensemble model of the native and phosphorylated regulatory domain of the CFTR. Cell. Mol. Life Sci. 70, 923–933. doi: 10.1007/s00018-012-1172-5
Mihalyi, C., Iordanov, I., Torocsik, B., and Csanady, L. (2020). Simple binding of protein kinase A prior to phosphorylation allows CFTR anion channels to be opened by nucleotides. Proc. Natl. Acad. Sci. U. S. A. 117, 21740–21746. doi: 10.1073/pnas.2007910117
Mihalyi, C., Torocsik, B., and Csanady, L. (2016). Obligate coupling of CFTR pore opening to tight nucleotide-binding domain dimerization. elife 5:e18164. doi: 10.7554/eLife.18164
Moore, C. A., Milano, S. K., and Benovic, J. L. (2007). Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 69, 451–482. doi: 10.1146/annurev.physiol.69.022405.154712
Neville, D. C., Rozanas, C. R., Price, E. M., Gruis, D. B., Verkman, A. S., and Townsend, R. R. (1997). Evidence for phosphorylation of serine 753 in CFTR using a novel metal-ion affinity resin and matrix-assisted laser desorption mass spectrometry. Protein Sci. 6, 2436–2445. doi: 10.1002/pro.5560061117
Okiyoneda, T., Barriere, H., Bagdany, M., Rabeh, W. M., Du, K., Hohfeld, J., et al. (2010). Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329, 805–810. doi: 10.1126/science.1191542
Pankow, S., Bamberger, C., and Yates, J. R. 3rd. (2019). A posttranslational modification code for CFTR maturation is altered in cystic fibrosis. Sci. Signal. 12:eaan7984. doi: 10.1126/scisignal.aan7984
Pasyk, S., Li, C., Ramjeesingh, M., and Bear, C. E. (2009). Direct interaction of a small-molecule modulator with G551D-CFTR, a cystic fibrosis-causing mutation associated with severe disease. Biochem. J. 418, 185–190. doi: 10.1042/BJ20081424
Pyle, L. C., Ehrhardt, A., Mitchell, L. H., Fan, L., Ren, A., Naren, A. P., et al. (2011). Regulatory domain phosphorylation to distinguish the mechanistic basis underlying acute CFTR modulators. Am. J. Phys. Lung Cell. Mol. Phys. 301, L587–L597. doi: 10.1152/ajplung.00465.2010
Ratjen, F., Bell, S. C., Rowe, S. M., Goss, C. H., Quittner, A. L., and Bush, A. (2015). Cystic fibrosis. Nat. Rev. Dis. Primers. 1:15010. doi: 10.1038/nrdp.2015.10
Rennolds, J., Tower, C., Musgrove, L., Fan, L., Maloney, K., Clancy, J. P., et al. (2008). Cystic fibrosis transmembrane conductance regulator trafficking is mediated by the COPI coat in epithelial cells. J. Biol. Chem. 283, 833–839. doi: 10.1074/jbc.M706504200
Rich, D. P., Berger, H. A., Cheng, S. H., Travis, S. M., Saxena, M., Smith, A. E., et al. (1993). Regulation of the cystic fibrosis transmembrane conductance regulator Cl- channel by negative charge in the R domain. J. Biol. Chem. 268, 20259–20267. doi: 10.1016/S0021-9258(20)80723-6
Riordan, J. R. (2005). Assembly of functional CFTR chloride channels. Annu. Rev. Physiol. 67, 701–718. doi: 10.1146/annurev.physiol.67.032003.154107
Riordan, J. R. (2008). CFTR function and prospects for therapy. Annu. Rev. Biochem. 77, 701–726. doi: 10.1146/annurev.biochem.75.103004.142532
Riordan, J. R., Rommens, J. M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., et al. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073. doi: 10.1126/science.2475911
Rommens, J. M., Iannuzzi, M. C., Kerem, B., Drumm, M. L., Melmer, G., Dean, M., et al. (1989). Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245, 1059–1065. doi: 10.1126/science.2772657
Sammelson, R. E., Ma, T., Galietta, L. J., Verkman, A. S., and Kurth, M. J. (2003). 3-(2-Benzyloxyphenyl) isoxazoles and isoxazolines: synthesis and evaluation as CFTR activators. Bioorg. Med. Chem. Lett. 13, 2509–2512. doi: 10.1016/S0960-894X(03)00482-7
Schnur, A., Premchandar, A., Bagdany, M., and Lukacs, G. L. (2019). Phosphorylation-dependent modulation of CFTR macromolecular signalling complex activity by cigarette smoke condensate in airway epithelia. Sci. Rep. 9:12706. doi: 10.1038/s41598-019-48971-y
Seavilleklein, G., Amer, N., Evagelidis, A., Chappe, F., Irvine, T., Hanrahan, J. W., et al. (2008). PKC phosphorylation modulates PKA-dependent binding of the R domain to other domains of CFTR. Am. J. Phys. Cell Phys. 295, C1366–C1375. doi: 10.1152/ajpcell.00034.2008
Seibert, F. S., Chang, X. B., Aleksandrov, A. A., Clarke, D. M., Hanrahan, J. W., and Riordan, J. R. (1999). Influence of phosphorylation by protein kinase A on CFTR at the cell surface and endoplasmic reticulum. Biochim. Biophys. Acta 1461, 275–283. doi: 10.1016/s0005-2736(99)00163-7
Serohijos, A. W., Hegedus, T., Aleksandrov, A. A., He, L., Cui, L., Dokholyan, N. V., et al. (2008). Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U. S. A. 105, 3256–3261. doi: 10.1073/pnas.0800254105
Sorum, B., Czege, D., and Csanady, L. (2015). Timing of CFTR pore opening and structure of its transition state. Cell 163, 724–733. doi: 10.1016/j.cell.2015.09.052
Townsend, R. R., Lipniunas, P. H., Tulk, B. M., and Verkman, A. S. (1996). Identification of protein kinase A phosphorylation sites on NBD1 and R domains of CFTR using electrospray mass spectrometry with selective phosphate ion monitoring. Protein Sci. 5, 1865–1873. doi: 10.1002/pro.5560050912
Uliyakina, I., Botelho, H. M., Da Paula, A. C., Afonso, S., Lobo, M. J., Felicio, V., et al. (2020). Full rescue of F508del-CFTR processing and function by CFTR modulators can be achieved by removal of two regulatory regions. Int. J. Mol. Sci. 21:4524. doi: 10.3390/ijms21124524
Vais, H., Zhang, R., and Reenstra, W. W. (2004). Dibasic phosphorylation sites in the R domain of CFTR have stimulatory and inhibitory effects on channel activation. Am. J. Phys. Cell Phys. 287, C737–C745. doi: 10.1152/ajpcell.00504.2003
Van Goor, F., Hadida, S., Grootenhuis, P. D., Burton, B., Stack, J. H., Straley, K. S., et al. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U. S. A. 108, 18843–18848. doi: 10.1073/pnas.1105787108
Van Goor, F., Straley, K. S., Cao, D., Gonzalez, J., Hadida, S., Hazlewood, A., et al. (2006). Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Phys. Lung Cell. Mol. Phys. 290, L1117–L1130. doi: 10.1152/ajplung.00169.2005
Veit, G., Avramescu, R. G., Chiang, A. N., Houck, S. A., Cai, Z., Peters, K. W., et al. (2016). From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 27, 424–433. doi: 10.1091/mbc.E14-04-0935
Vergani, P., Basso, C., Mense, M., Nairn, A. C., and Gadsby, D. C. (2005a). Control of the CFTR channel's gates. Biochem. Soc. Trans. 33, 1003–1007. doi: 10.1042/BST20051003
Vergani, P., Lockless, S. W., Nairn, A. C., and Gadsby, D. C. (2005b). CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880. doi: 10.1038/nature03313
Vergani, P., Nairn, A. C., and Gadsby, D. C. (2003). On the mechanism of MgATP-dependent gating of CFTR Cl- channels. J. Gen. Physiol. 121, 17–36. doi: 10.1085/jgp.20028673
Wang, W., Fu, L., Liu, Z., Wen, H., Rab, A., Hong, J. S., et al. (2020). G551D mutation impairs PKA-dependent activation of CFTR channel that can be restored by novel GOF mutations. Am. J. Phys. Lung Cell. Mol. Phys. 319, L770–L785. doi: 10.1152/ajplung.00262.2019
Wang, F., Zeltwanger, S., Hu, S., and Hwang, T. C. (2000). Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J. Physiol. 524, 637–648. doi: 10.1111/j.1469-7793.2000.00637.x
Wilkinson, D. J., Strong, T. V., Mansoura, M. K., Wood, D. L., Smith, S. S., Collins, F. S., et al. (1997). CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am. J. Phys. 273, L127–L133. doi: 10.1152/ajplung.1997.273.1.L127
Winter, M. C., and Welsh, M. J. (1997). Stimulation of CFTR activity by its phosphorylated R domain. Nature 389, 294–296. doi: 10.1038/38514
Xie, J., Adams, L. M., Zhao, J., Gerken, T. A., Davis, P. B., and Ma, J. (2002). A short segment of the R domain of cystic fibrosis transmembrane conductance regulator contains channel stimulatory and inhibitory activities that are separable by sequence modification. J. Biol. Chem. 277, 23019–23027. doi: 10.1074/jbc.M201661200
Zhang, Z., and Chen, J. (2016). Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell 167, 1586–1597. doi: 10.1016/j.cell.2016.11.014
Zhang, Z., Liu, F., and Chen, J. (2017). Conformational changes of CFTR upon phosphorylation and ATP binding. Cell 170, 483–491. doi: 10.1016/j.cell.2017.06.041
Zhang, Z., Liu, F., and Chen, J. (2018). Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. U. S. A. 115, 12757–12762. doi: 10.1073/pnas.1815287115
Keywords: cystic fibrosis transmembrane conductance regulator, protein kinase A, phosphorylation, cystic fibrosis, VX770, VX809, F508del-CFTR mutation
Citation: Della Sala A, Prono G, Hirsch E and Ghigo A (2021) Role of Protein Kinase A-Mediated Phosphorylation in CFTR Channel Activity Regulation. Front. Physiol. 12:690247. doi: 10.3389/fphys.2021.690247
Edited by:
Fabio Mammano, University of Padua, ItalyReviewed by:
Steven S. An, Rutgers Institute for Translational Medicine and Science, United StatesJohn Cuppoletti, University of Cincinnati, United States
Copyright © 2021 Della Sala, Prono, Hirsch and Ghigo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Ghigo, YWxlc3NhbmRyYS5naGlnb0B1bml0by5pdA==; Emilio Hirsch, ZW1pbGlvLmhpcnNjaEB1bml0by5pdA==
†These authors have contributed equally to this work and share senior authorship