 Elisa Fermo1
Elisa Fermo1 Cristina Vercellati1
Cristina Vercellati1 Anna Paola Marcello1Ebru Yilmaz Keskin2
Anna Paola Marcello1Ebru Yilmaz Keskin2 Silverio Perrotta3
Silverio Perrotta3 Anna Zaninoni1
Anna Zaninoni1 Valentina Brancaleoni4
Valentina Brancaleoni4 Alberto Zanella1
Alberto Zanella1 Juri A. Giannotta1
Juri A. Giannotta1 Wilma Barcellini1
Wilma Barcellini1 Paola Bianchi1*
Paola Bianchi1*- 1UOS Fisiopatologia delle Anemie, UOC Ematologia, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 2Department of Pediatric Hematology and Oncology, Suleyman Demirel University, Isparta, Turkey
- 3Dipartimento della Donna, del Bambino e di Chirurgia Generale e Specialistica, Università degli Studi della Campania “Luigi Vanvitelli,” Naples, Italy
- 4UOC Medicina Generale, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
Congenital hemolytic anemias (CHAs) are heterogeneous and rare disorders caused by alterations in structure, membrane transport, metabolism, or red blood cell production. The pathophysiology of these diseases, in particular the rarest, is often poorly understood, and easy-to-apply tools for diagnosis, clinical management, and patient stratification are still lacking. We report the 3-years monocentric experience with a 43 genes targeted Next Generation Sequencing (t-NGS) panel in diagnosis of CHAs; 122 patients from 105 unrelated families were investigated and the results compared with conventional laboratory pathway. Patients were divided in two groups: 1) cases diagnosed with hematologic investigations to be confirmed at molecular level, and 2) patients with unexplained anemia after extensive hematologic investigation. The overall sensitivity of t-NGS was 74 and 35% for families of groups 1 and 2, respectively. Inside this cohort of patients we identified 26 new pathogenic variants confirmed by functional evidence. The implementation of laboratory work-up with t-NGS increased the number of diagnoses in cases with unexplained anemia; cytoskeleton defects are well detected by conventional tools, deserving t-NGS to atypical cases; the diagnosis of Gardos channelopathy, some enzyme deficiencies, familial siterosterolemia, X-linked defects in females and other rare and ultra-rare diseases definitely benefits of t-NGS approaches.
Introduction
Congenital hemolytic anemias (CHAs) comprise a group of very heterogeneous and rare disorders caused by alterations in structure, transport functions, metabolism, or defective production of red blood cells (RBCs). Since the pathophysiology of some rare forms is poorly understood, these disorders represent a group of diseases that still lack easy-to-apply tools for diagnosis, clinical management, and patient stratification.
The laboratory diagnostic pathway of CHAs was historically based on sequential steps using a panel of functional analyses that investigate the RBC membrane and metabolism. The first diagnostic step relies on hematological tests (complete blood count, red cell indices, hemolysis markers), peripheral blood smear examination, osmotic fragility tests and eosin-5-maleimide (EMA) binding test. The second includes biochemical tests such as quantitative assay of RBC enzymes activity, sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) for the diagnosis of membranopathies, and specialized investigations requiring instruments which are available only in reference Centers, as ektacytometry (King et al., 2015; Fermo et al., 2021). The molecular characterization of the affected genes by traditional Sanger sequencing was usually, up to some years ago, the last step allowing the definitive confirmation of the diagnosis. Despite this, even after extensive and complete investigation, the differential diagnosis of these disorders may be difficult, and some patients remain undiagnosed with a consequent negative impact on clinical follow up, risk of inappropriate therapeutic decisions and lack of access to new specific treatments.
The advent and recent progresses on next generation sequencing (NGS) technologies has radically changed the diagnostic approach to CHAs, often placing the genetic analysis as a first line screening tool; different NGS strategies have been developed in the last years including targeted panels and whole exome sequencing (WES), with a progressive reduction of costs that allowed their routinely use (Steinberg-Shemer and Tamary, 2020; Russo et al., 2020). Targeted-NGS panels have been developed and applied by several groups, being currently the preferred approach for the molecular diagnosis of CHAs; custom panels include different numbers of genes and have been reported to have a wide range of diagnostic efficacy (38–90%) depending on the number of genes included and on the characteristics of the patients studied (Agarwal et al., 2016; Del Orbe Barreto et al., 2016; Niss et al., 2016; Roy et al., 2016; Russo et al., 2018; Shefer Averbuch et al., 2018; Choi et al., 2019; Kedar et al., 2019a,b; Svidnicki et al., 2020). In this paper we report the 3 years monocentric experience in targeted-NGS and diagnosis of CHAs, and we compare the efficacy of this methodological approach with the conventional laboratory diagnosis.
Materials and Methods
Patients
Among the patients referred to our Institut (Pathophysiology of Anemias Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore of Milan–Italy) between 2017 and 2019 with a clinical suspect of CHAs we examined a cohort of 122 patients from 105 unrelated families (51% males and 49% females, median age 27 years, range 0–73 years). The inclusion criteria in the study were: a diagnostic indication for congenital hemoltic anemia; the specific request from clinicians to perform/complete diagnosis at molecular level; a specific scientific interest in cases with intra-family variability or atypical presentation; all cases with unexplained phenotype despite extensive investigations; signed informed consent to perform molecular investigations. Samples were collected from patients and controls during diagnostic procedures after obtaining informed consent and approval from the Institutional Human Research Committee. For patients under the age of 18, written informed consent was obtained from the parents. The procedures followed were in accordance with the Helsinki International ethical standards on human experimentation.
All patients but 18 (received after shipping from abroad) underwent extensive hematologic investigations, including: RBC morphology, complete blood count, hemolysis markers, screening for abnormal/unstable hemoglobins, direct antiglobulin test, osmotic fragility tests, EMA binding test (Bianchi et al., 2012), and RBC enzymes activities determination (hexokinase, glucosephosphate isomerase, phosphofructokinase, glyceraldehyde-P-dehydrogenase, phos phoglycerate kinase, pyruvate kinase, glucose-6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, adenylate kinase) (Beutler et al., 1977).
In the vast majority of cases we also performed Osmoscan analysis by LoRRca MaxSis (Laser-Assisted Optical Rotational Cell Analyzer, Mechatronics, NL) (Zaninoni et al., 2018, see Supplementary Materials for more details), and RBC membrane protein analysis by SDS–PAGE performed according to Laemmli (1970) and Fairbanks et al. (1971) with minor modifications (Mariani et al., 2008).
In the remaining 18 cases (15 families), when possible, a minimum panel of investigation was performed (i.e., EMA binding test, RBC enzyme assay, Osmoscan) and evaluated together with clinical and laboratory information received from the local centers.
In 60 patients from 51 families a definite diagnostic orientation was obtained after first and second level hematological investigations. In particular, 6 were suspected of RBC enzyme defects, 28 of structural membrane defects (hereditary spherocytosis, elliptocytosis, pyropoikilocytosis, HS/HE/HPP), 10 of stomatocytosis and 7 were classified as congenital dyserythropoietic anemia.
In the remaining 62 cases (from 54 families) the laboratory investigations did not allow a clear diagnosis.
NGS Sequencing and Analysis
Genomic DNA was extracted from peripheral blood, using standard manual methods and quantified by Nandrop One (Thermo Scientific, Italy). When available, relatives of affected cases were also enrolled to analyze allelic segregation and correctly assess the pathogenicity of each variant.
Using SureDesign software (Agilent) we created a NGS based panel containing 43 genes already described as disease causing for RBC membrane disorders (n = 14 genes), enzymopathies (n = 18), dyserythropoietic and sideroblastic anemias (n = 11) (Supplementary Table 1). For the probe design, coding regions, 5’UTR, 3’UTR, 50 bp flanking splice junctions were selected as regions of interest. Sequence length was set at 150 × 2 nucleotides and the average target coverage was 99.52%. The panel was validated on previously characterized patients affected by CHAs.
Libraries were obtained by HaloPlexHS Target Enrichment System Kit (Agilent) following the instruction’s manufacturer. Samples were pooled (average 22 samples) and loaded at 7 pM on MiSeq platform using a v2-300 cycle reagent kit (Illumina). Sequencing reads were aligned against reference genome (UCSC hg19) and variants were called and annotated using the SureCall software (Agilent Technologies).
Filter settings were as follows: variant call quality threshold > 100; minimum number of reads supporting variant allele > 10; mutant allele frequency ≥ 25%; minor allele frequency (MAF) < 1% of the population. Common pathogenic modifiers of hereditary spherocytosis known to be present above the 1% cutoff, e.g., SPTA1 LELY (Low Expression Lyon) allele (p.L1858V, found in association with the intronic variant c.6531-12 c > t), or the SPTA-Bughill allele (p.A970D) usually found in linkage with the LEPRA (Low Expression PRAgue) allele (SPTA c.4339-99 c > t) were also included.
Targeted filtering and annotation of protein-changing variants were performed using the wANNOVAR web tool1.
Variants were assessed by mutation prediction and conservation programs including SIFT2, polyphen-2 (Polymorphism Phenotyping v2)3 and MutationTaster4; pathogenicity was evaluated according to the guidelines of American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015) using the online tool VarSome5 (Kopanos et al., 2019).
Variants previously classified as pathogenic by databases such as ClinVar, HGMD, dbSNP, deleterious variants expected to produce truncated or abnormal protein, or splice site variants were considered as causatives. Variants of unknown significance (VUSs) were reported only if found in genes relevant to the primary indication for testing.
The mutations identified were confirmed by Sanger method (ABI PRISM 310 Genetic Analyzer, Applied Biosystems, Warrington, United Kingdom) using the Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Warrington, United Kingdom).
Nucleotide numbering reflects cDNA numbering with + 1 corresponding to the A of ATG translation initiation codon in the reference sequence, according to the nomenclature for the description of sequence variants of Human Genome Variation Society6. The initiation codon is codon 1.
According with ACMG guidelines, well-established in vitro functional studies were performed to assess pathogenicity of VUSs, in particular: spectrophotometric enzyme assay in the suspect of RBC enzyme defects, SDS-PAGE electrophoresis analysis of RBC membrane proteins in the suspect of membrane defects and CDAII, ektacytometry and patch-clamp analysis in case of novel missense variants in PIEZO1 and KCNN4 mutations.
Results
The 122 patients examined have been divided in two groups: (1) patients who reached a diagnosis after first and second level hematologic investigations, to be confirmed at molecular level; (2) patients with unexplained chronic hemolytic anemia after extensive hematologic investigations.
Patients belonging to group 1 (60 patients, 51 families) were selected among the entire cohort of patients with CHAs studied between 2017 and 2019 basing on complexity of clinical presentation, intra-family clinical variability or incomplete molecular characterization by Sanger (e.g., enzyme defects or CDA cases). All the available patients who didn’t reach a definitive diagnosis in the same period were investigated and included in group 2 (62 cases from 54 families, 39 of them studied in our Center, the other 15 received from abroad).
Group 1
We were able to identify pathogenic variants in 38/51 families belonging to Group 1, with an overall sensitivity of the t-NGS of 74% (Figure 1A and Table 1).
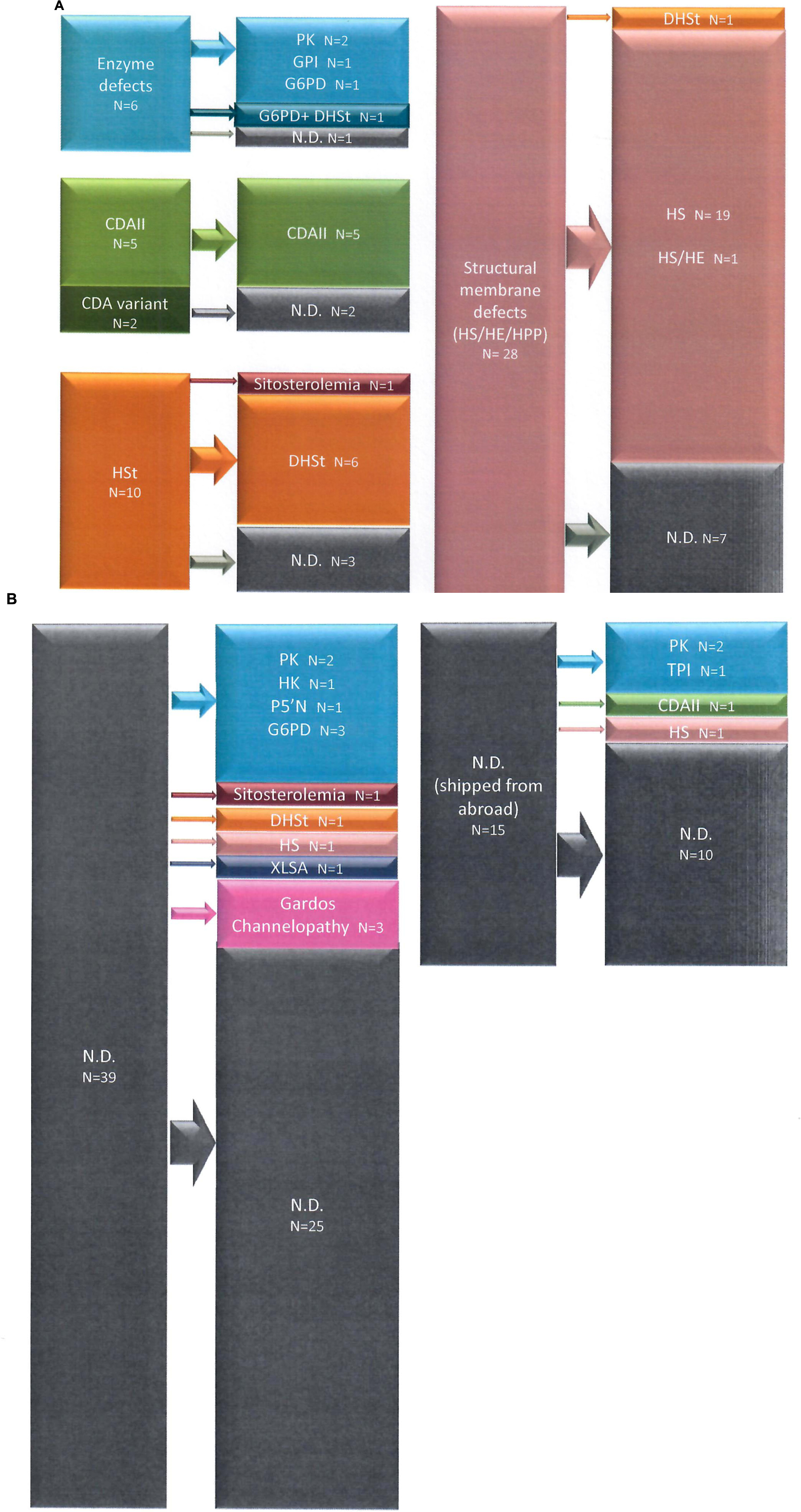
Figure 1. Diagnostic suspect after first and second level laboratory investigation and final diagnosis after t-NGS in Group 1 (A) and Group 2 (B) patients.
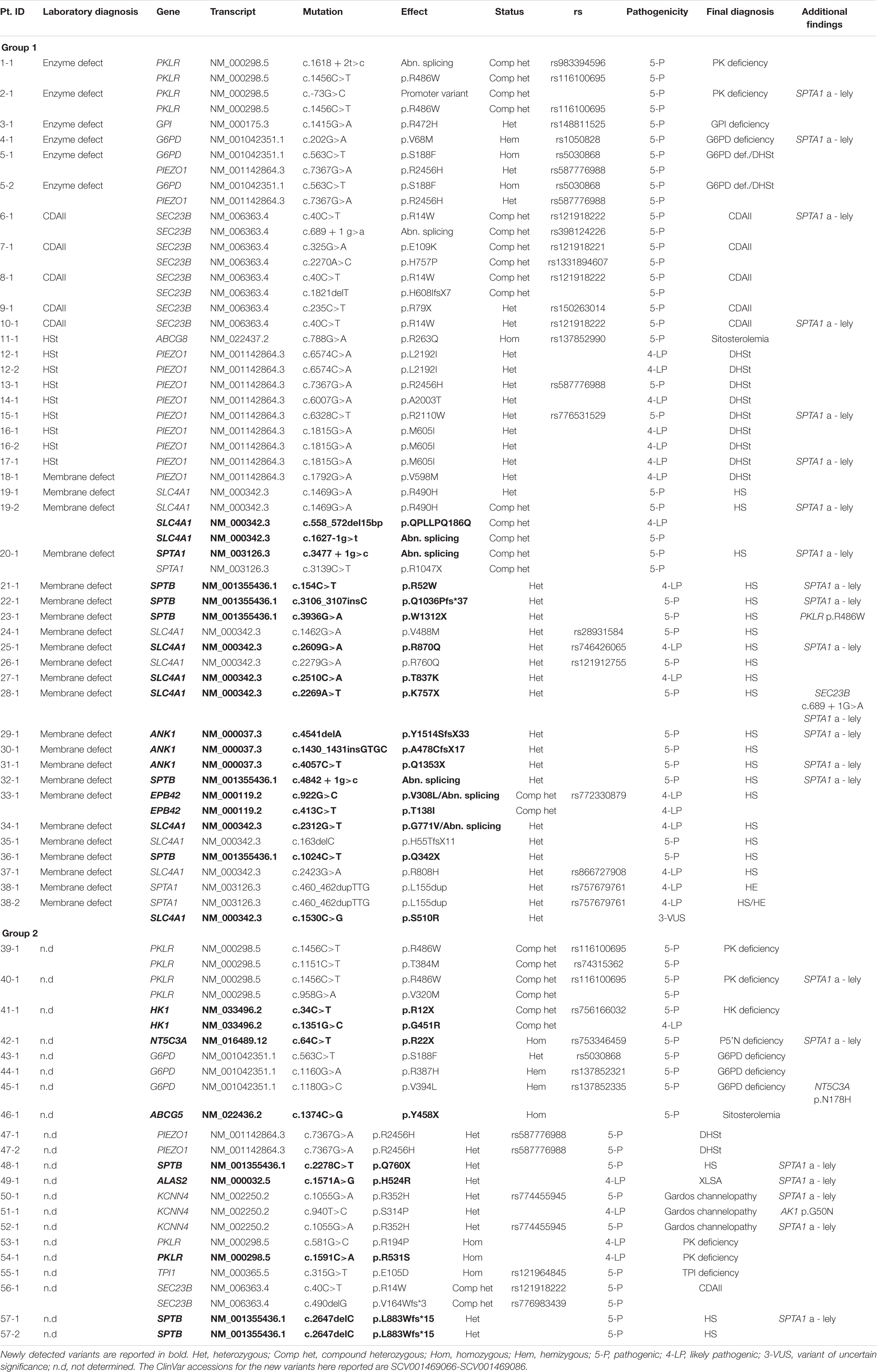
Table 1. Results of NGS analysis in 64 patients with congenital hemolytic anemia.
Enzyme Defects
Eight cases from six families had a biochemical diagnosis of enzyme defect; two were confirmed to have pyruvate kinase (PK) deficiency, three from two families glucose-6-phosphate dehydrogenase (G6PD) deficiency and one was found to have one mutation in GPI gene, in compound heterozygosity with the polymorphism c.489A > G (p.Gly163 = rs1801015), possibly associated to splicing alterations (Fermo et al., 2019); in two related cases no mutations in PKLR gene were found in spite of a slightly decreased PK activity detected by enzymatic assay.
Among these cases with enzyme defects, in four patients from three families a coinheritance of hereditary stomatocytosis was also suspected due to the presence of stomatocytes at peripheral blood smear and left-shifted Osmoscan curve; only in two of them (5-1 and 5-2) in addition to G6PD deficiency we found a mutation in PIEZO1 gene causing hereditary stomatocytosis.
Congenital Dyserythropoietic Anaemias (CDAs)
Seven cases had a suspect of congenital dyserythropoietic anemia; five were classified as CDAII on the basis of band 3 deglycosylation at SDS-PAGE analysis, and two had an atypical CDA variant with distinct morphological abnormalities at bone marrow examination. Three out of five CDAII cases displayed biallelic mutations in SEC23B gene, whereas in two cases (9-1 and 10-1) we were able to find only one pathogenic variant; in the two cases with the atypical CDA we didn’t find any pathological mutation in the genes associated to CDAs included in our panel.
Hereditary Stomatocytosis
Thirteen patients from 10 families had a suspect of hereditary stomatocytosis based on presence of stomatocytes at peripheral blood smear and/or abnormalities at ektacytometric curve; eight of them (from six families) were confirmed to have dehydrated stomatocytosis caused by PIEZO1 gene pathogenic variants. In one case previously diagnosed as overhydrated stomatocytosis (11-1) based on a clear right-shifted ektacytometric curve, we found mutations in ABCG8 gene responsible for sitosterolemia. In the remaining case no pathogenic variants in genes associated with hereditary stomatocytosis were identified.
Membrane Defects
We included in this group chronic hemolytic anemias due to altered RBC membrane structural organization, as HS, HE, HPP; 32 cases from 28 families have received such diagnosis based on first and second level laboratory investigations. In 23 cases from 21 families we found pathogenic variants. One patient with a previous diagnosis of HS was found to be affected by dehydrated stomatocytosis (DHSt); in the remaining cases, mutations in genes responsible for HS and HE were identified. In particular among them we studied 3 families with a complex phenotype: case 38-2 (mixed HS/HE phenotype) carried two variants in SPTA1 and SLC4A1, whereas the brother with typical HE (38-1) had only the SPTA1 variant. Case 20-1, presenting severe HPP with combined 68% spectrin and 56% ankyrin deficiency, had two different mutations in SPTA1 gene transmitted by the parents, one of them associated in cis with the alpha-LELY allele. Finally, case 19-2 (severe HS with 50% band3 deficiency) had 3 different variants in SLC4A1 gene, the father (19-1) presenting only one missense mutation, thus justifying intra-family clinical variability (Figure 2).
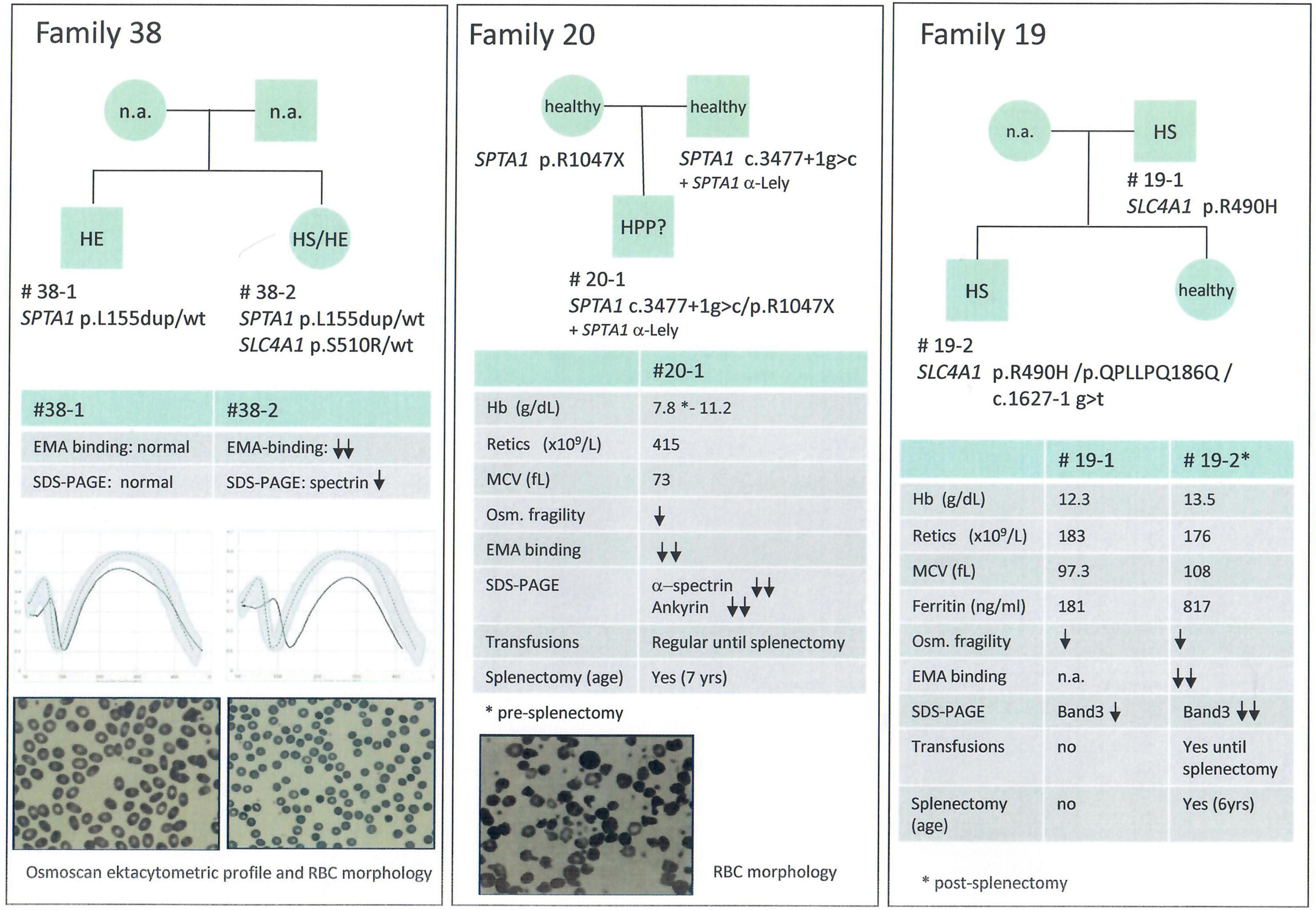
Figure 2. Families with erythrocyte membrane defects and complex phenoptypes and/or intrafamily variability.
Group 2
Inside this group we were able to identify pathogenic variants in 19/54 families (21/62 patients), with an overall sensitivity of the t-NGS of 35% (Figure 1B and Table 1).
Ten families were found to have pathogenic variants in RBC enzymes. In five of them (2 PK, 2 G6PD and 1 hexokinase (HK) deficiency, cases 39-1, 40-1, 43-1, 44-1, and 41-1) the enzyme activity was previously tested and found to be normal; in other four cases enzyme activity was not tested for sample unavailability. One case had known G6PD deficiency that was initially not considered during the laboratory screening based on chronic hemolysis. Triosephosphate isomerase (TPI) deficiency was diagnosed in a 7 months old child presenting at the time of the study only hemolytic anemia without neuromuscular manifestations.
Three unrelated cases were found to have pathogenic variants in KCNN4 gene associated with Gardos channelopathy, two of them carrying the common variant p.R352H and one a private variant p.S314P resulting in channel activation (Fermo et al., 2020).
Only one family with two affected siblings (47-1 and 47-2), addressed to our attention in the suspect of PK deficiency in absence of pathogenic variants in PKLR gene, carried a PIEZO1 pathogenic variant. Patient 46-1, who had 17% of stomatocytes at peripheral blood smear without any abnormality of the Osmoscan curve, was diagnosed with sitosterolemia due to ABCG5 mutation p.Y458X. Finally, NGS analysis allowed the diagnosis of X-linked sideroblastic anemia (XLSA) in a woman affected by unexplained macrocytic anemia since birth (case 49-1, ALAS2 mutation p.H524R). Human androgen receptor gene (HUMARA) analysis showed skewed X-Chromosome inactivation pattern; the mutation was transmitted by the asymptomatic mother.
New Variants Identified
Inside the analyzed cohort we identified 26 new variants, 20 of them associated with structural membrane defects (SLC4A1: p.QPLLPQ186Q, p.R870Q, p.T837K, p.K757X, p.G771V, p.S510R, c.1627-1g>t; SPTB: p.R52W, p.Q1036PfsX37, p.W1312X, p.Q342X, p.Q760X, p.L883WfsX15, c.4842+1g>c; ANK1: p.Y1514SfsX33, p.A478CfsX17, p.Q1353X; SPTA1: c.3477+1g>c; EPB42 p.V308L, p.T138I). The remaining new variants were identified in enzyme genes (PKLR: p.R531S; HK1: p.R12X, p.G451R; NT5C3A: p.R22X), and in genes associated with sitosterolemia (ABCG5: p.Y458X) and sideroblastic anemia (ALAS2: p.H524R). All new variants were classified as pathogenic or likely pathogenic by in silico analysis, according with transmission, clinical phenotype and functional laboratory evidences. Only one VUS variant in SLC4A1 gene was included and considered as pathogenic because clearly fitting with patients phenotype in a family with a suspected combined transmission of HS and HE (case 38-2) (Figure 2).
Discussion
The observed sensitivity of NGS in the two groups of patients analyzed is very different, varying from 74% (patients with a specific diagnostic orientation, Group 1) to 35% (hemolytic patients with no diagnosis after laboratory investigations, or not investigated due long shipping conditions, Group 2).
These data are in line with the available literature (Russo et al., 2018; Bianchi et al., 2020; Steinberg-Shemer and Tamary, 2020) and may explain the heterogeneous sensitivity that, independently from the number of genes analyzed, is often reported for these techniques. For example, the overall sensitivity was around 38.6% in a large series of 57 hemolytic patients with no definitive diagnosis (Roy et al., 2016) and 45.8% in a group of undiagnosed hemolytic patients studied with a combination of two-step t-NGS platforms covering about 100 genes (Russo et al., 2018). Different results are found when NGS is performed on well characterized patient subsets, for example in series of HS patients, where the reported sensitivity increased to 70–100% (Chonat et al., 2019; van Vuren et al., 2019; Xue et al., 2019; Vives-Corrons et al., 2021).
In this view, a sensitivity of 75%, as obtained in our Group 1, may be expected for t-NGS, considering some technical limitations of this technique (loss of variant calling, presence of pathogenic variants in not sequenced regions, copy number variations not always revealed), or to the presence of diseases-causing variants in new genes or genes not included in the panels. In such cases, results could be further improved by considering other technical approaches (WES or WGS) (Mansour-Hendili et al., 2020).
In a portion of cases reported in literature up to 45%, the genetic testing changed the initial diagnosis, in particular when transfusion dependence prevented laboratory investigations from being carried out, and the suspect was based only on clinical features (Russo et al., 2018). In our experience, when detailed clinical history is available together with fresh blood samples collected far from transfusions, first and second level laboratory diagnosis for CHAs has a good level of specificity, as demonstrated by the finding of only two patients who changed diagnosis after NGS analysis inside group 1: one patient previously diagnosed as HS had indeed a pathogenic variant in PIEZO1 gene, and one case with a long-lasting diagnosis of overhydrated stomatocytosis (done on morphological bases, with 18% of stomatocytes and right shift of ektacytometry curve) was indeed a case of familial sitosterolemia with homozygous mutation p.R263Q in ABCG8 gene (Berge et al., 2000). In both cases the molecular diagnosis changed the clinical management: in the first case the patient was warned about the thrombotic risk associated with splenectomy in hereditary stomatocytosis, and in the second case suggestions were given about dietary reduction of plant sterols consumption, to reduce cardiovascular risk that is associated with familial sitosterolemia (Tzavella et al., 2017). Patients affected by hereditary sitosterolemia may display an extremely variable clinical phenotype, in some cases restricted to abnormal hematological features, leading to misdiagnosis with hereditary stomatocytosis, immune thrombocytopenia (ITP), or Evans syndrome. The mechanisms of hematologic changes in these patients are so far unexplained, and a toxic effect of plant sterols on red blood cells has been hypothezied from in vitro experiments (Wang et al., 2012). The finding of two cases in our series, in line with increasing reports of the last years (Wang et al., 2014; Zheng et al., 2019), suggest that sitosterolemia may have an underestimated prevalence, and it should be taken in account in differential diagnosis of CHAs, in particular when stomatocytosis and concomitant macrothrombocytopenia are observed.
Combined defects of red cell membrane and/or metabolism are very rare, and the fact that carriership for a metabolic defect might modify the clinical expression of a membrane defect is still debated (van Zwieten et al., 2015; Fermo et al., 2017). However, the detection of cases with concomitant red cell defects has increased in the last years after the introduction of NGS analysis (Russo et al., 2018; Mansour-Hendili et al., 2020) suggesting that these conditions might not be so rare, also possibly due to protection toward malaria infection. In this series we found the association of G6PD deficiency and PIEZO1 stomatocytosis in two female patients (mother and daughter) with suspected hereditary stomatocytosis. Among families with membrane defects and complex intra-family phenotypes, we were able to identify a patient with mixed HS/HE who carried two variants in SPTA1 and SLC4A1 gene.
Notably, in addition to the disease-causing mutations, four patients also displayed heterozygous variants in other genes associated with hemolytic anemias (see Table 1). Although a synergistic effect of heterozygous variants has been hypothesized by some authors (van Zwieten et al., 2015; Pereira et al., 2016), in the presented cases the contribution of these variants to the phenotype remains to be determined.
A careful analysis of Group 2 put in light the limitations of laboratory work-up of CHAs. Only two among the undiagnosed families were found to have HS, suggesting that most of the HS patients can be correctly diagnosed during first and second level screening tools (Fermo et al., 2021). On the other hand, 10 out of the 19 families belonging to this group had an enzyme defect; in five of them enzymopathies were previously excluded because of normal enzyme activity, in the remaining cases the enzyme assays were not performed due to recent transfusions or unavailability of the appropriate sample. RBC enzyme defects are often detected by NGS in cases of unexplained anemia (Roy et al., 2016) or in patients misdiagnosed as CDAII (Shefer Averbuch et al., 2018; Russo et al., 2018), suggesting that this approach may greatly improve the diagnosis of these disorders.
In this series two PK deficient cases carrying known pathogenic variants (p.R486W, p.T384M, and p.V320M) repeatedly showed normal PK activity (14.7 and 18.8 IU/gHb, respectively, reference range 11.9–16.5), in absence of clear explanation (reticulocyte/WBC contamination excluded). Information on sensitivity of spectrophotometric enzyme assays is scanty, in particular for the rarer defects (Bianchi et al., 2019) but, as shown in the present cohort of patients, it will be possibly available in the next future through the systematic analysis of undiagnosed cases by NGS technologies.
Interestingly, we identified a triosephosphate isomerase deficiency in a 7 month child presenting at the time of the study only hemolytic anemia without neuromuscular manifestations. An early diagnosis allowed to inform and prepare the family and to offer bone marrow transplantation (BMT) as a therapeutic option before that neurological damage became evident (Conway et al., 2018); BMT was refused by the parents and the patient developed neurological signs at the age of 18 months. Early diagnosis in red cell disorders by NGS technologies opens interesting landscapes and perspectives in view of future therapeutic approaches for these diseases (Garcia-Gomez et al., 2016; Grace et al., 2019; Dessy-Rodriguez et al., 2020; Hrizo et al., 2021).
Among enzymopathies, we also identified 3 cases with undiagnosed G6PD deficiency, an heterozygous woman and two men with G6PD variants associated with chronic hemolysis, conditions in some cases underdiagnosed. A correct diagnosis enabled to give specific recommendation regarding contraindication for drugs associated with hemolytic anemia in G6PD -deficient patients (Luzzatto et al., 2020).
Differently from the PIEZO1 mutated cases, all the KCNN4 mutated patients belonged to group 2, confirming that molecular testing, and in particular NGS approaches, are the only diagnostic tools able to identify Gardos channelopathy, given the absence of a specific clinical or biological phenotype in this disorder; in fact, although ektacytometry, and some RBC and reticulocytes indices (Picard et al., 2019, 2021) may help to differentiate PIEZO1 from KCNN4 cases, no hematological parameter or specific laboratory test enable to differentiate Gardos channelopathy from other CHAs.
In conclusion, by considering the entire cohort here studied, the implementation of diagnostic work-up with t-NGS analysis increased the number of diagnoses of patients with unexplained anemia up to 35%. As reported in Figure 3, cytoskeleton defects (HS/HE) are well diagnosed by laboratory approaches, deserving NGS only to atypical cases; on the other hand, the diagnosis of Gardos channelopathy, some RBC enzyme defects, familial siterosterolemia, X-linked defects in females and other rare and ultra-rare conditions definitely benefits of NGS approaches. However, it should be always considered that a set of functional tests is required to validate the molecular results and assess pathogenicity of the new identified variants, in particular in highly polymorphic genes such as PIEZO1.
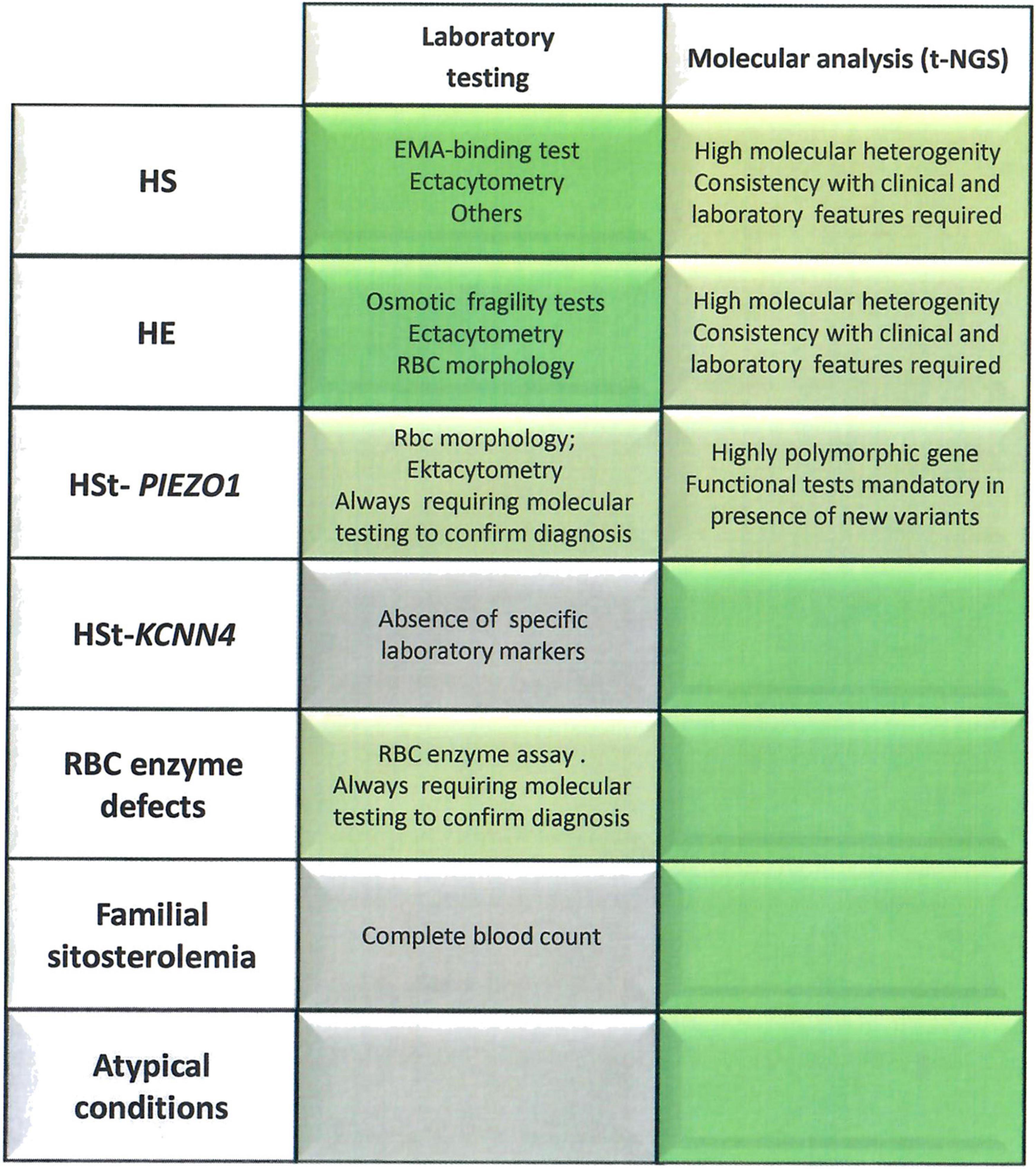
Figure 3. Advandages and limitations of laboratory approach vs. t-NGS technologies in congenital hemolytic anemias. The laboratory tests/parameters specifically useful for diagnosis of different diseases are reported. Brilliant green, exhaustive diagnostic approach; Green, requiring other approaches to confirm the diagnosis; gray, insufficient laboratory markers to reach the diagnosis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Table 1.
Ethics Statement
The studies involving human participants were reviewed and approved by Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
EF and PB designed the study and performed NGS analysis and molecular studies. CV, AnZ, and AM performed laboratory investigations. WB, AZ, SP, EK, and JG followed-up patients. EF and PB drafted the manuscript, all other authors revised and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by Fondazione IRCCS Ca’ Granda Policlinico Milano, Project number RC2020 175/05 and was generated in line with the European Reference Network on Rare Hematological Diseases (ERN-EuroBloodNet) FPA 739541.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.684569/full#supplementary-material
Supplementary Table 1 | Genes associated with CHAs investigated through a 43 genes-targeted Next Generation Sequencing panel.
Footnotes
- ^ http://wannovar.wglab.org/
- ^ http://sift.jcvi.org/
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://www.mutationtaster.org/
- ^ https://varsome.com/
- ^ www.hgvs.org/mutnomen
References
Agarwal, A. M., Nussenzveig, R. H., Reading, N. S., Patel, J. L., Sangle, N., Salama, M. E., et al. (2016). Clinical utility of next-generation sequencing in the diagnosis of hereditary haemolytic anaemias. Br. J. Haematol. 174, 806–814. doi: 10.1111/bjh.14131
Berge, K. E., Tian, H., Graf, G. A., Yu, L., Grishin, N. V., Schultz, J., et al. (2000). Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 290, 1771–1775.
Beutler, E., Blume, K. G., Kaplan, J. C., Lohr, G. W., Ramot, B., and Valentine, W. N. (1977). International Committee for Standardization in Haematology: recommended methods for red-cell enzyme analysis. Br. J. Haematol. 35, 331–340.
Bianchi, P., Fermo, E., Glader, B., Kanno, H., Agarwal, A., Barcellini, W., et al. (2019). Addressing the diagnostic gaps in pyruvate kinase deficiency: Consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am. J. Hematol. 94, 149–161. doi: 10.1002/ajh.25325
Bianchi, P., Fermo, E., Vercellati, C., Marcello, A. P., Porretti, L., Cortelezzi, A., et al. (2012). Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica 97, 516–523. doi: 10.3324/haematol.2011.052845
Bianchi, P., Vercellati, C., and Fermo, E. (2020). How will next generation sequencing (NGS) improve the diagnosis of congenital hemolytic anemia? Ann. Transl. Med. 8:268. doi: 10.21037/atm.2020.02.151
Choi, H. O., Choi, Q., Kim, J. A., Im, K. O., Park, S. N., Park, Y., et al. (2019). Molecular diagnosis of hereditary spherocytosis by multi-gene target sequencing in Korea: matching with osmotic fragility test and presence of spherocyte. Orphanet. J. Rare Dis. 14:114. doi: 10.1186/s13023-019-1070-0
Chonat, S., Risinger, M., Sakthivel, H., Niss, O., Rothman, J. A., Hsieh, L., et al. (2019). The Spectrum of SPTA1-Associated Hereditary Spherocytosis. Front. Physiol. 10:815. doi: 10.3389/fphys.2019.00815
Conway, A. J., Brown, F. C., Hortle, E. J., Burgio, G., Foote, S. J., Morton, C. J., et al. (2018). Bone marrow transplantation corrects haemolytic anaemia in a novel ENU mutagenesis mouse model of TPI deficiency. Dis. Model Mech. 11:dmm034678. doi: 10.1242/dmm.034678
Del Orbe Barreto, R., Arrizabalaga, B., De la Hoz, A. B., García-Orad, Á, Tejada, M. I, Garcia-Ruiz, J. C., et al. (2016). Detection of new pathogenic mutations in patients with congenital haemolytic anaemia using next-generation sequencing. Int. J. Lab. Hematol. 38, 629–338. doi: 10.1111/ijlh.12551
Dessy-Rodriguez, M., Fañanas-Baquero, S. A., Venturi, V., Payán-Pernía, S., Tornador, C., Hernandez, G., et al. (2020). Quintana Bustamante, O. Modelling Congenital Dyserythropoietic Anemia Type II through Gene Editing in Hematopoietic Stem and Progenitor Cells. Blood 136, (Suppl. 1):27.
Fairbanks, G., Steck, T. L., and Wallach, D. F. H. (1971). Electrophoretic analysis of the major polypeptides of the human erythrocyte membrane. Biochemistry 10, 2606–2617.
Fermo, E., Monedero-Alonso, D., Petkova-Kirova, P., Makhro, A., Pérès, L., Bouyer, G., et al. (2020). Gardos channelopathy: functional analysis of a novel KCNN4 variant. Blood Adv. 4, 6336–6341. doi: 10.1182/bloodadvances.2020003285
Fermo, E., Vercellati, C., and Bianchi, P. (2021). Screening tools for hereditary hemolytic anemia: new concepts and strategies. Expert Rev. Hematol. 1:11. doi: 10.1080/17474086.2021.1886919
Fermo, E., Vercellati, C., Marcello, A. P., Zaninoni, A., Aytac, S., Cetin, M., et al. (2019). Clinical and Molecular Spectrum of Glucose-6-Phosphate Isomerase Deficiency. Report of 12 New Cases. Front. Physiol. 10:467. doi: 10.3389/fphys.2019.00467
Fermo, E., Vercellati, C., Marcello, A. P., Zaninoni, A., van Wijk, R., Mirra, N., et al. (2017). Hereditary Xerocytosis due to Mutations in PIEZO1 Gene Associated with Heterozygous Pyruvate Kinase Deficiency and Beta-Thalassemia Trait in Two Unrelated Families. Case Rep. Hematol. 2017:2769570. doi: 10.1155/2017/2769570
Garcia-Gomez, M., Calabria, A., Garcia-Bravo, M., Benedicenti, F., Kosinski, P., López-Manzaneda, S., et al. (2016). Safe and Efficient Gene Therapy for Pyruvate Kinase Deficiency. Mol. Ther. 24, 1187–1198. doi: 10.1038/mt.2016.87
Grace, R. F., Rose, C., Layton, D. M., Galactéros, F., Barcellini, W., Morton, D. H., et al. (2019). Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency. N. Engl. J. Med. 381, 933–944. doi: 10.1056/NEJMoa1902678
Hrizo, S. L., Eicher, S. L., Myers, T. D., McGrath, I., Wodrich, A. P. K., Venkatesh, H., et al. (2021). Identification of protein quality control regulators using a Drosophila model of TPI deficiency. Neurobiol. Dis. 15:105299. doi: 10.1016/j.nbd.2021.105299
Kedar, P. S., Gupta, V., Dongerdiye, R., Chiddarwar, A., Warang, P. P., and Madkaikar, M. R. (2019a). Molecular diagnosis of unexplained haemolytic anaemia using targeted next-generation sequencing panel revealed (p.Ala337Thr) novel mutation in GPI gene in two Indian patients. J. Clin. Pathol. 72, 81–85. doi: 10.1136/jclinpath-2018-205420
Kedar, P. S., Harigae, H., Ito, E., Muramatsu, H., Kojima, S., Okuno, Y., et al. (2019b). Study of pathophysiology and molecular characterization of congenital anemia in India using targeted next-generation sequencing approach. Int. J. Hematol. 110, 618–626. doi: 10.1007/s12185-019-02716-9
King, M. J., Garçon, L., Hoyer, J. D., Iolascon, A., Picard, V., Stewart, G., et al. (2015). International Council for Standardization in Haematology. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int. J. Lab. Hematol. 37, 304–325. doi: 10.1111/ijlh.12335
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: the human genomic variant search engine. Bioinformatics 35, 1978–1980. doi: 10.1093/bioinformatics/bty897
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685.
Luzzatto, L., Ally, M., and Notaro, R. (2020). Glucose-6-phosphate dehydrogenase deficiency. Blood 136, 1225–1240. doi: 10.1182/blood.2019000944
Mansour-Hendili, L., Aissat, A., Badaoui, B., Sakka, M., Gameiro, C., Ortonne, V., et al. (2020). Exome sequencing for diagnosis of congenital hemolytic anemia. Orphanet. J. Rare Dis. 15:180. doi: 10.1186/s13023-020-01425-5
Mariani, M., Barcellini, W., Vercellati, C., Marcello, A. P., Fermo, E., Pedotti, P., et al. (2008). Clinical and hematologic features of 300 patients affected by hereditary spherocytosis grouped according to the type of the membrane protein defect. Haematologica 93, 1310–1317.
Niss, O., Chonat, S., Dagaonkar, N., Almansoori, M. O., Kerr, K., Rogers, Z. R., et al. (2016). Genotype-phenotype correlations in hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood Cells Mol. Dis. 61, 4–9. doi: 10.1016/j.bcmd.2016.07.003
Pereira, J., Bento, C., Maco, L., Gonzalez, A., Vagace, J., and Ribeiro, M. L. (2016). Congenital dyserythropoietic anemia associated to a GATA1 mutation aggravated by pyruvate kinase deficiency. Ann. Hematol. 95, 1551–1553. doi: 10.1007/s00277-016-2720-0
Picard, V., Guitton, C., Mansour-Hendili, L., Jondeau, B., Bendélac, L., Denguir, M., et al. (2021). Rapid Gardos Hereditary Xerocytosis Diagnosis in 8 Families Using Reticulocyte Indices. Front. Physiol. 11:602109. doi: 10.3389/fphys.2020.602109
Picard, V., Guitton, C., Thuret, I., Rose, C., Bendelac, L., Ghazal, K., et al. (2019). Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos channelopathy: a retrospective series of 126 patients. Haematologica 104, 1554–1564. doi: 10.3324/haematol.2018.205328
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Roy, N. B. A., Wilson, E. A., Henderson, S., Wray, K., Babbs, C., Okoli, S., et al. (2016). A novel 33-Gene targeted resequencing panel provides accurate, clinical-grade diagnosis and improves patient management for rare inherited anaemias. Br. J. Haematol. 175, 318–330. doi: 10.1111/bjh.14221
Russo, R., Andolfo, I., Manna, F., Gambale, A., Marra, R., Rosato, B. E., et al. (2018). Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am. J. Hematol. 93, 672–682. doi: 10.1002/ajh.25058
Russo, R., Marra, R., Rosato, B. E., Iolascon, A., and Andolfo, I. (2020). Genetics and Genomics Approaches for Diagnosis and Research Into Hereditary Anemias. Front. Physiol. 11:613559. doi: 10.3389/fphys.2020.613559
Shefer Averbuch, N., Steinberg-Shemer, O., Dgany, O., Krasnov, T., Noy-Lotan, S., Yacobovich, J., et al. (2018). Targeted next generation sequencing for the diagnosis of patients with rare congenital anemias. Eur. J. Haematol. 101, 297–304. doi: 10.1111/ejh.13097
Steinberg-Shemer, O., and Tamary, H. (2020). Impact of next-generation sequencing on the diagnosis and treatment of congenital anemias. Mol. Diagn Ther. 4, 397–407. doi: 10.1007/s40291-020-00478-3
Svidnicki, M. C. C. M., Zanetta, G. K., Congrains-Castillo, A., Costa, F. F., and Saad, S. T. O. (2020). Targeted next-generation sequencing identified novel mutations associated with hereditary anemias in Brazil. Ann. Hematol. 99, 955–962. doi: 10.1007/s00277-020-03986-8
Tzavella, E., Hatzimichael, E., Kostara, C., Bairaktari, E., Elisaf, M., and Tsimihodimos, V. (2017). Sitosterolemia: A multifaceted metabolic disorder with important clinical consequences. J. Clin. Lipidol. 11, 1095–1100. doi: 10.1016/j.jacl.2017.04.116
van Vuren, A., van der Zwaag, B., Huisjes, R., Lak, N., Bierings, M., Gerritsen, E., et al. (2019). The complexity of genotype-phenotype correlations in hereditary spherocytosis: a cohort of 95 patients. HemaSphere 3:4.
van Zwieten, R., van Oirschot, B. A., Veldthuis, M., Dobbe, J. G., Streekstra, G. J., van Solinge, W. W., et al. (2015). Partial pyruvate kinase deficiency aggravates the phenotypic expression of band 3 deficiency in a family with hereditary spherocytosis. Am. J. Hematol. 90, E35–E39. doi: 10.1002/ajh.23899
Vives-Corrons, J. L., Krishnevskaya, E., Rodriguez, I. H., and Ancochea, A. (2021). Characterization of hereditary red blood cell membranopathies using combined targeted next-generation sequencing and osmotic gradient ektacytometry. Int. J. Hematol. 113, 163–174. doi: 10.1007/s12185-020-03010-9
Wang, G., Cao, L., Wang, Z., Jiang, M., Sun, X., Bai, X., et al. (2012). Macrothrombocytopenia/stomatocytosis specially associated with phytosterolemia. Clin. Appl. Thromb. Hemost. 18, 582–587.
Wang, Z., Cao, L., Su, Y., Wang, G., Wang, R., Yu, Z., et al. (2014). Specific macrothrombocytopenia/hemolytic anemia associated with sitosterolemia. Am. J. Hematol. 89, 320–324. Google Scholar
Xue, J., He, Q., Xie, X., Su, A., and Cao, S. (2019). Clinical utility of targeted gene enrichment and sequencing technique in the diagnosis of adult hereditary spherocytosis. Ann. Transl. Med. 7:527.
Zaninoni, A., Fermo, E., Vercellati, C., Consonni, D., Marcello, A. P., et al. (2018). Use of Laser Assisted Optical Rotational Cell Analyzer (LoRRca MaxSis) in the Diagnosis of RBC Membrane Disorders, Enzyme Defects, and Congenital Dyserythropoietic Anemias: A Monocentric Study on 202 Patients. Front. Physiol. 9:451. doi: 10.3389/fphys.2018.00451
Keywords: congenital hemolytic anemia, targeted-NGS, pathogenic variants, red blood cells, differential diagnosis
Citation: Fermo E, Vercellati C, Marcello AP, Keskin EY, Perrotta S, Zaninoni A, Brancaleoni V, Zanella A, Giannotta JA, Barcellini W and Bianchi P (2021) Targeted Next Generation Sequencing and Diagnosis of Congenital Hemolytic Anemias: A Three Years Experience Monocentric Study. Front. Physiol. 12:684569. doi: 10.3389/fphys.2021.684569
Received: 23 March 2021; Accepted: 27 April 2021;
Published: 21 May 2021.
Edited by:
Angelo D’Alessandro, University of Colorado Denver, United StatesReviewed by:
Tiffany Thomas, Columbia University Irving Medical Center, United StatesMarianna H. Antonelou, National and Kapodistrian University of Athens, Greece
Copyright © 2021 Fermo, Vercellati, Marcello, Keskin, Perrotta, Zaninoni, Brancaleoni, Zanella, Giannotta, Barcellini and Bianchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Bianchi, cGFvbGEuYmlhbmNoaUBwb2xpY2xpbmljby5taS5pdA==