 Caroline Barbé
Caroline Barbé Audrey Loumaye1
Audrey Loumaye1 Olli Ritvos
Olli Ritvos Jean-Paul Thissen
Jean-Paul Thissen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 17 June 2021
Sec. Striated Muscle Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.677746
This article is part of the Research Topic Modulators of Skeletal Muscle Hypertrophy: Mechanisms to Lifestyle Strategies View all 8 articles
Skeletal muscle, the most abundant tissue in the body, plays vital roles in locomotion and metabolism. Understanding the cellular processes that govern regulation of muscle mass and function represents an essential step in the development of therapeutic strategies for muscular disorders. Myostatin, a member of the TGF-β family, has been identified as a negative regulator of muscle development. Indeed, its inhibition induces an extensive skeletal muscle hypertrophy requiring the activation of Smad 1/5/8 and the Insulin/IGF-I signaling pathway, but whether other molecular mechanisms are involved in this process remains to be determined. Using transcriptomic data from various Myostatin inhibition models, we identified Pak1 as a potential mediator of Myostatin action on skeletal muscle mass. Our results show that muscle PAK1 levels are systematically increased in response to Myostatin inhibition, parallel to skeletal muscle mass, regardless of the Myostatin inhibition model. Using Pak1 knockout mice, we investigated the role of Pak1 in the skeletal muscle hypertrophy induced by different approaches of Myostatin inhibition. Our findings show that Pak1 deletion does not impede the skeletal muscle hypertrophy magnitude in response to Myostatin inhibition. Therefore, Pak1 is permissive for the skeletal muscle mass increase caused by Myostatin inhibition.
Myostatin (MSTN), a member of the TGF-β family primarily expressed in skeletal muscle, is a negative regulator of skeletal muscle mass, as shown by the marked increase in skeletal muscle mass following MSTN inhibition or gene deletion (Mcpherron et al., 1997; Grobet et al., 2003). Several approaches may be used to inhibit MSTN. Among these, the physiological MSTN binding protein, Follistatin (FS), and the soluble Activin IIB receptor (sActRIIB), have been shown to be promising therapeutic strategies for reversing muscle wasting (Busquets et al., 2012) and dystrophies (Rodino-Klapac et al., 2013). Previous works from us and others have demonstrated that muscle fiber hypertrophy induced by MSTN inhibition results from increased protein synthesis and requires the activation of Smad 1/5/8 (Winbanks et al., 2013) and the Insulin/IGF-I pathway (Kalista et al., 2012; Winbanks et al., 2012; Barbé et al., 2015). However, the molecular mechanisms responsible for the muscle phenotype induced by MSTN inhibition remain not fully understood.
The p21-activated kinase 1 (PAK1) is a 68-kDa Serine/Threonine kinase of the PAK1–6 family, which plays a role in many cellular processes as evidenced by its more than 40 substrates identified to date. PAK proteins are divided into two groups, with PAK1–3 belonging to group 1 and PAK 4–6 belonging to group 2 (Bokoch, 2003). From Group 1 PAKs, only PAK1 and PAK2 have been detected in skeletal muscle (Tunduguru et al., 2014; Joseph et al., 2017). While Pak1 or Pak2 deletion does not affect muscle mass, mice lacking both Pak1 and Pak2 display reduced muscle mass indicating a potential redundancy of these two proteins (Joseph et al., 2017). Recent evidences have highlighted the role of Pak1 in muscle mass regulation (Martinelli et al., 2016; Joseph et al., 2017; Cerquone Perpetuini et al., 2018), but its implication in muscle hypertrophy caused by MSTN inhibition has never been explored to date. In a first step, we identified Pak1 as the only gene consistently upregulated in skeletal muscle following various MSTN inhibition models. Given the anti-atrophic role of Pak1 in skeletal muscle (Cerquone Perpetuini et al., 2018) and its upregulation in response to MSTN inhibition, we hypothesized that increased Pak1 could mediate the muscle mass increase induced by MSTN inhibition. Then, we explored the role of Pak1 in both the magnitude and the onset of skeletal muscle hypertrophy following MSTN inhibition. Our findings show that Pak1 deletion did not affect the magnitude of muscle hypertrophy caused by MSTN inhibition.
All the mouse experiments were performed with the approval of the Committee for Ethical Practices in Animal Experiments of the Catholic University of Louvain (Brussels, Belgium). Male mice were housed under standardized conditions of light (12:12 h light–dark cycle) and temperature (22 ± 2°C) with free access to standard chow pellets and water. Animals were euthanized by decapitation after CO2 administration and tibialis anterior (TA), gastrocnemius (GC), soleus (SOL), and extensor digitorum longus (EDL) muscles were collected for further analysis. For a complete overview of the different animal models including the age at the start of the experiment, see Table 1.

Table 1. Overview of the different animal models.
Different animal models of MSTN inhibition were used in this work: FVB MSTN knockout (KO) mice and their control wild-type (WT) littermates (Grobet et al., 2003); C57Bl/6 transgenic (mTrFS) mice and their control WT littermates (Lee and Mcpherron, 2001); C57Bl/6 mice transfected with human FS gene (pM1-hFS288) (Barbé et al., 2015); and C57Bl/6 mice treated by intraperitoneal (ip) injections with sActRIIB (Hulmi et al., 2013). The sActRIIB is a soluble ligand-binding domain of Activin receptor type IIB fused to Fc domain IgG that causes MSTN and Activin A inhibition (Hoogaars et al., 2012).
FVB mice transfected with murine MSTN gene (pcDNA-MSTN) (Durieux et al., 2007) and FVB mice subcutaneously injected with dexamethasone (DEXA, Aacidexam®; Organon BioSciences) (Gueugneau et al., 2018) were used.
To investigate the role of Pak1 in muscle hypertrophy induced by MSTN inhibition, C57Bl/6 Pak1 KO mice and their control WT littermates were used (Allen et al., 2009).
Total RNA was isolated from the frozen TA and GC muscles using TRIzol reagent® as described by the manufacturer. Recovery was 1 μg/mg of muscle. Reverse transcription and real-time quantitative PCR were done as previously described (Dehoux et al., 2004). Primers were tested in order to avoid primers dimers, self-priming formation or unspecific amplifications and Glyceraldehyde-3-Phosphate Dehydrogenase (Gapdh) was used as reporter gene. Primers were designed to have standardized optimal PCR conditions. Accession numbers for the sequences and primers used were: Pak1: NM_011035.2 (TCCGCCAGATGCTTTGACCCG-AATGG CCACCTCCTGCCCTGT), Pak2: NM_177326.3 (AGCACCGG AGGAAAAGATCC-TGCCAGTGAACTCTCCCGTA), and Gapdh: AF106860 (TGCACCACCAACTGCTTA-GGATGCA GGGATGATGTTC). We confirmed that the expression values of GAPDH normalized to RNA were not affected by MSTN inhibition in our different animal models. This was confirmed at the protein level, showing that the abundance of GAPDH protein was not affected by FS overexpression, Mstn deletion, sActRIIB treatment, DEXA treatment and Pak1 deletion, making GAPDH a valuable control.
Frozen GC muscles, previously pestled in liquid nitrogen, were homogenized (100 mg/ml) with Ultraturrax (IKA Labortechnik) in ice-cold pH 7.0 buffer containing 20 mM Tris, 270 mM sucrose, 5 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 50 mM β-glycerophosphate, 5 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM DTT (1,4-dithiothreitol), 1% (v/v) Triton X-100, and 10% protease inhibitor cocktail (Roche Applied Science). Homogenates were centrifuged at 10,000 × g for 10 min at 4°C and supernatants were immediately stored at −80°C. Equal amounts of proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. Membranes were incubated overnight at 4°C in TBST containing 1% bovine serum albumin with the following primary antibodies: anti-phospho-Smad3 Ser423/425 (Cell signaling #9520) and anti-Smad3 (Cell signaling #9523) at 1:250 dilution; anti-phospho-AKT Ser473 (Cell signaling #9271), anti-AKT (Cell signaling #9272), anti-phospho-4E-BP1 Ser65 (Cell signaling #9451), anti-4E-BP1 (Cell signaling #9452), anti-phospho-P70S6K Thr389 (Cell signaling #9205), anti-phospho-FOXO3a Ser253 (Cell signaling #9466), anti-FOXO3a (Cell signaling #2497), and anti-PAK2 (Cell signaling #2615) at 1:500 dilution; anti-S6 (Cell signaling #2217) and anti-PAK1 (Cell signaling #2602) at 1:1,000 dilution; anti-phospho-S6 (Cell signaling #4858) at 1:2,000 dilution. Then membranes were incubated with a goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (Cell Signaling #7074) at 1:2,000 dilution for 1 h and developed using Enhanced Chemiluminescence (ECL) Western blotting Detection System Plus (GE Healthcare). Developed films were scanned and signal quantification was determined using Image J software as described before (Gueugneau et al., 2018). Ponceau Red staining was used as a loading control (Moritz, 2017).
The pcDNA-MSTN plasmid coding for the murine MSTN was kindly provided by D. Freyssenet (Saint Étienne, France) (Durieux et al., 2007). The pM1-hFS288 c-myc plasmid coding for the human FS containing 288 amino acids was constructed as previously described (Gilson et al., 2009; Kalista et al., 2012). Empty plasmids (pcDNA and pM1) were used as control plasmids. Plasmids were amplified in Escherichia coli top 10 F’ (Invitrogen) and purified with an EndoFree plasmid giga kit (QIAGEN). Plasmids were stored at −80°C. The day before injection, 30 μg of plasmid were lyophilized and resuspended in 30 μl 0.9% NaCl solution.
Each animal was anesthetized with a mixture of 100 mg/kg ketamine (Anesketin®; Pfizer) and 15 mg/kg xylazine hydrochloride (Rompun®; Bayer) administered by ip injection. Thirty microliters of plasmid solution (1 μg/μl) were injected into each TA muscle using a Hamilton syringe with a 30-gauge needle (pcDNA or pM1 in the right TA and pcDNA-MSTN or pM1-hFS288 in the left TA), and the muscles were then electroporated using the electroporation conditions described by Bloquel et al. (2004) (8 pulses of 200 V/cm and 20 ms per pulse at 2 Hz). The mice were euthanized 14 or 17 days after electroporation of pcDNA-MSTN or pM1-hFS288, respectively.
Tibialis anterior muscles were dissected and immediately fixed with buffered formalin for 48 h and embedded in paraffin for histological analysis. Serial sections (5 μm thick) were cut and mounted on glass slides (Superfrost Plus; Menzel-Glaser). Muscle sections were deparaffinized and blocked in PBS-BSA (5%) containing normal goat serum (4%) for 30 min at RT. To evaluate the effect of FS on muscle fibers cross-sectional area (CSA), FS-c-myc was detected by immunohistochemistry with a rabbit polyclonal anti-c-myc (1:800; Bethyl Laboratories) for 1 h at RT. Primary antibody was detected by applying for 30 min at RT a biotinylated second antibody, which was a goat anti-rabbit conjugated to peroxidase-labeled polymer (Dako). Peroxidase activity was revealed with DAB substrate (Chemicon International), which produces a brown stain. The sections were counterstained with Mayer’s hematoxylin, rinsed, and mounted in Faramount (Dako). Muscle fiber CSA were measured with a microscope Axio-Star (Carl Zeiss) coupled to a Zeiss Axiocam digital camera MRc and to image analyzer software (Axiovision software version 4.7; Carl Zeiss). All the positive muscle fibers in the FS-transfected TA muscles were measured (18 ± 3 in Pak1 KO mice vs. 69 ± 4 in WT mice). Two hundred negative fibers, randomly chosen in the contralateral TA transfected with insertless plasmid pM1, were measured and considered as controls.
Maximal muscle force was determined by a grip test of both forelimb and combined fore- and hindlimb muscles. Limb strength was recorded using a grid connected to a sensor (Bioseb - Panlab). The mice were gently laid on the top of the grid so that their front paws (forelimb test) or both their four paws (fore and hindlimb test) can grip the grid. Then mice were pulled back steadily until the grip was released down the complete length of the grid. Two series of three tests were performed at an interval of 15 min. Results are presented as the highest value of force recorded, related to body weight. Endurance tolerance was determined by an incremental treadmill exercise test. The test was started at low velocity (5 m/min) and increments of 1 m/min) every minute were applied until 18 m/min for a maximum running time of 60 min.
Results are presented as means ± standard error of the mean (SEM) or, when applicable, means ± SEM with individual data points shown. Statistical analyses were performed using unpaired t-test to compare muscles from two different experimental conditions. Interactions between FS electroporation or sActRIIB treatment and Pak1 deletion were assessed using a two-way ANOVA followed by Bonferroni post-tests. Fiber CSA distribution statistical analysis was performed using χ2 Pearson test. Statistical significance was set at P-value < 0.05.
To decipher the molecular mechanisms behind the muscle phenotype induced by MSTN inhibition, we sought to compare eight published transcriptomic analyses encompassing various models of MSTN inhibition: three models of MSTN gene deletion, whose two prenatal (Steelman et al., 2006; Rahimov et al., 2011) and one postnatal (Welle et al., 2009); two models of postnatal MSTN inhibition with either acute (sActRIIB 1x) or chronic (sActRIIB 4x) sActRIIB treatment (Rahimov et al., 2011); three models of FS-induced MSTN inhibition, whose two postnatal with either acute (72 h post-transfection) or chronic (7–14 days post-transfection) FS electroporation (Davey et al., 2016) and one prenatal with muscle–specific transgenic mTrFS mice (Barbé et al., 2017). Prior to the comparison, the microarray data were uploaded for chip matching and official gene symbols. Only differentially expressed genes with P-value < 0.05 were integrated in the comparative analysis, except for data published by Welle et al. (2009), for which a P-value < 0.01 was applied. With a 1.5-fold cut-off, 135 genes were commonly regulated in at least one of each MSTN inhibition model, namely MSTN deletion, sActRIIB treatment, and FS overexpression. Differential expression of 20 genes (8 upregulated and 12 downregulated) was shared between at least 6 from the 8 transcriptomes compared (Table 2).
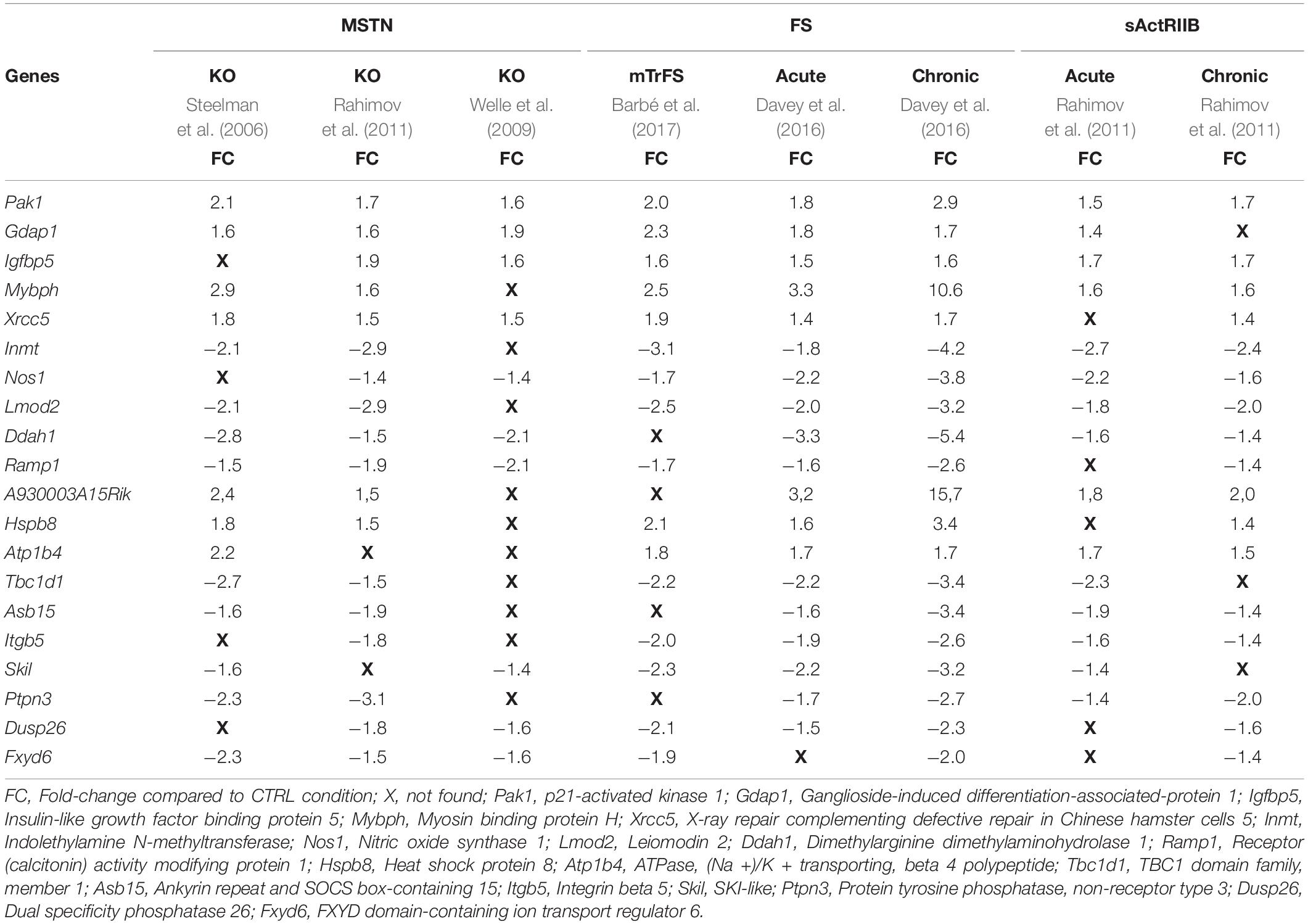
Table 2. Commonly regulated genes following MSTN inhibition.
Then, we investigated the biologic processes coordinated by these 20 differentially expressed genes according to information extracted from Database for Annotation, Visualization and Integrated Discovery (DAVID)1 and NCBI2. Consistent with the molecular mechanisms responsible for the muscle hypertrophy induced by MSTN inhibition, several genes were associated to Insulin/IGF-I and Smad signaling pathways (Table 3). Among the three genes associated to Insulin/IGF-I signaling (Pak1, Igfbp5, and Tbc1d1), Pak1 was the only gene consistently upregulated in all MSTN inhibition models compared in Table 2. Given the role of Pak1 in muscle mass regulation (Martinelli et al., 2016; Joseph et al., 2017; Cerquone Perpetuini et al., 2018), we investigated its action in the muscle mass increase induced by MSTN inhibition.
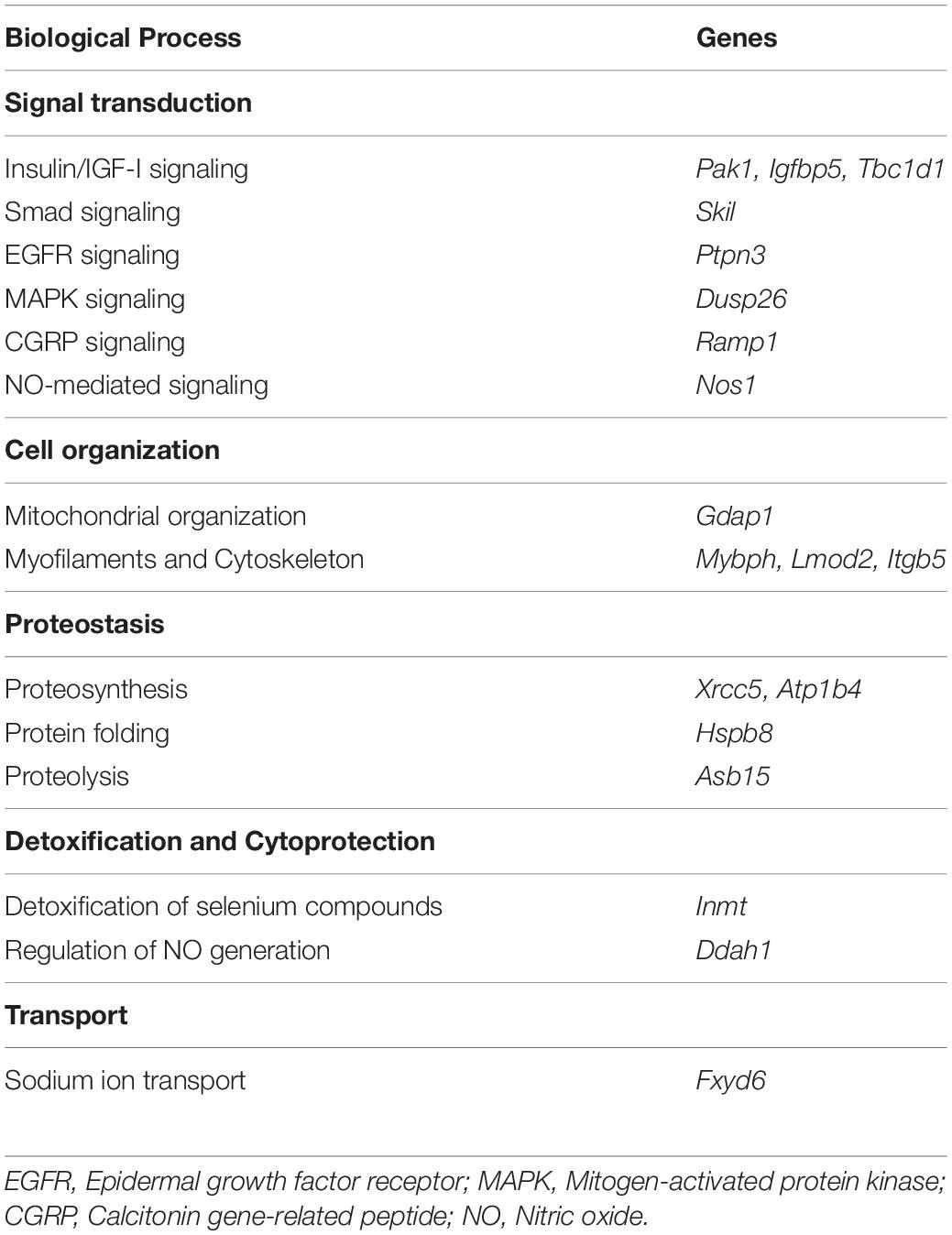
Table 3. Biological processes regulated by the commonly regulated genes following MSTN inhibition.
As illustrated in Figures 1A,B, Pak1 was the only gene consistently upregulated in all MSTN inhibition models. To confirm the validity of these results, the muscle mRNA levels of Pak1 were assessed by RT-qPCR in several models of MSTN inhibition. As expected, muscle Pak1 expression was systematically increased in skeletal muscles from mTrFS (+66%, P < 0.01), MSTN KO (+82%, P < 0.01) and 4 times sActRIIB-injected mice (+52%, P < 0.01) compared to WT or CTRL mice, as was muscle mass (mTrFS: +77%, P < 0.001; MSTN KO: +53%, P < 0.001; and sActRIIB: +25%, P < 0.001) (Figure 1C). In contrast, muscle Pak1 expression was decreased in atrophied muscles following MSTN transfection (Pak1 mRNA: −18%, P < 0.01; muscle mass: −9%, P < 0.05), compared to CTRL pcDNA-transfected muscles (Figure 1D). Moreover, mice treated with DEXA, a well-known atrophic agent acting in part through the induction of MSTN (Schakman et al., 2013), also showed a similar decrease in Pak1 mRNA and muscle mass (Pak1 mRNA: −46%, P < 0.05; muscle mass: −13%, P < 0.01), compared to their control littermates (Figure 1D). Finally, as shown in Figures 1E,F, overexpression of FS and sActRIIB treatment increased not only Pak1 mRNA but also its protein expression in skeletal muscle (mTrFS: 8.2 ± 1.3-fold vs. WT, P < 0.001; sActRIIB: 2.0 ± 0.2-fold vs. CTRL, P < 0.01). Altogether, these results confirm that muscle PAK1 levels are consistently upregulated in response to MSTN inhibition, suggesting PAK1 could contribute to driving the hypertrophic muscle phenotype induced by MSTN inhibition.
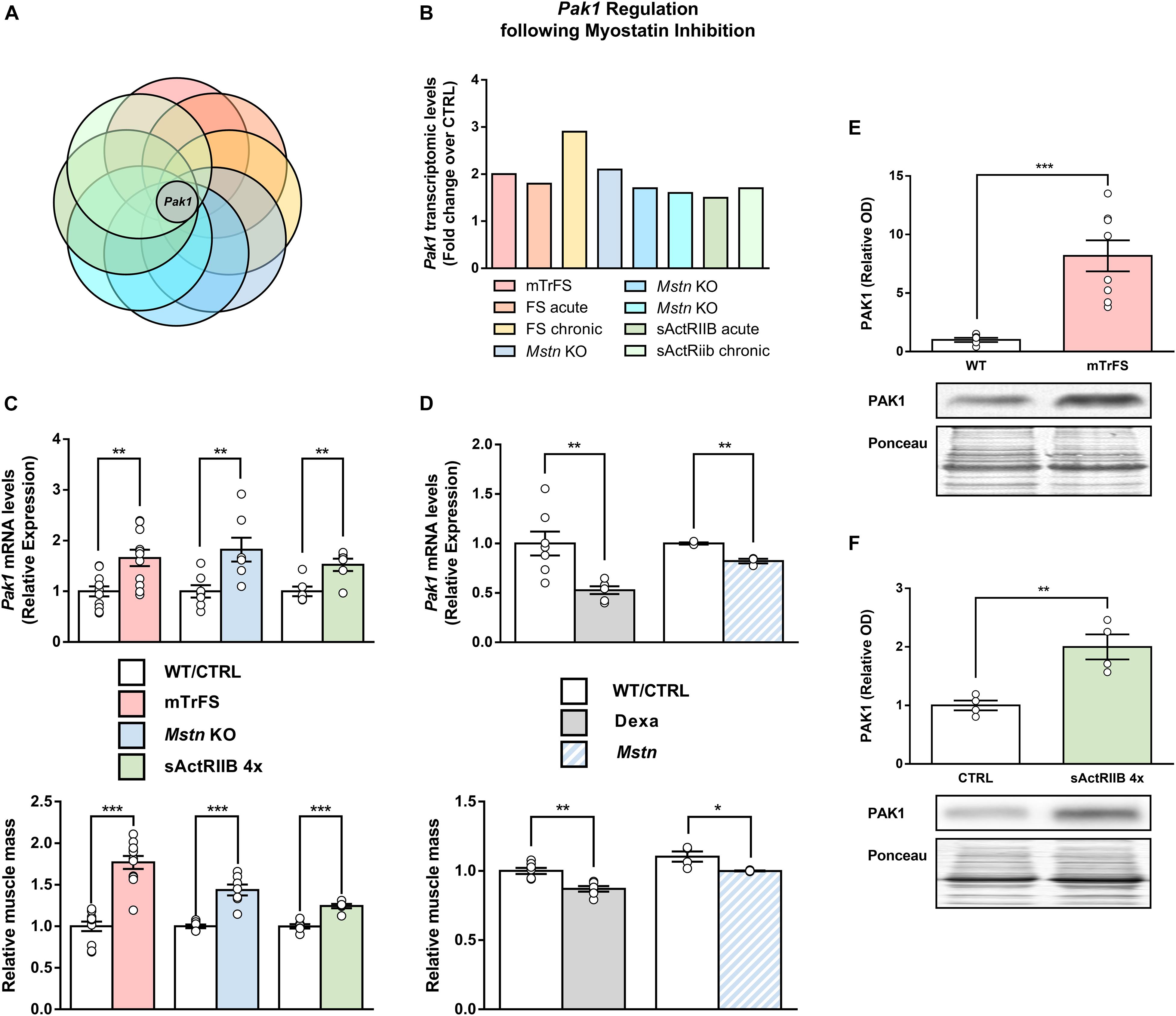
Figure 1. Global comparative transcriptomic analysis of skeletal muscles from mouse models of MSTN inhibition identified Pak1 as a MSTN-responsive gene. (A) Venn diagram comparing the differentially expressed genes into muscles from eight different models of pre- and postnatal MSTN inhibition with Pak1 as the only gene commonly regulated between all the transcriptomes compared. (B) Pak1 mRNA fold-changes for each muscle transcriptome studied. (C) Muscle Pak1 mRNA levels and muscle weight from TA muscles of mTrFS vs. WT mice (n = 11/group); from TA muscles of four times sActRIIB-injected vs. PBS-injected mice (n = 6/group) and from GC muscles of MSTN KO vs. WT mice (n = 7/group). (D) Muscle Pak1 mRNA levels and muscle weight from GC muscles of DEXA-treated vs. saline-treated mice (n = 7/group) and from MSTN-transfected vs. CTRL pcDNA-transfected TA muscles (n = 3/group). Representative western blot image and densitometric analysis of PAK1 protein levels in GC muscles from (E) mTrFS vs. WT C57Bl/6 mice (n = 8/group) and from (F) sActRIIB-injected vs. PBS-injected mice (n = 4/group). Results are expressed as means ± SEM and statistical analysis was performed using unpaired t-test (*P < 0.05; **P < 0.01, and ***P < 0.001 vs. CTRL/WT).
To investigate the role of PAK1 in mediating the MSTN inhibition effect on muscle mass and function, we investigated the consequences of Pak1 deletion on muscle homeostasis. The absence of Pak1 in skeletal muscle from Pak1 KO mice was confirmed by western blot (Figure 2A). There was no difference in body weight nor muscle mass between Pak1 KO and WT mice (Figures 2B,C). Two different functional tests were used to investigate the global muscular force (grip test) and the resistance to fatigue (treadmill running test) which were unchanged under Pak1 deletion (Figures 2D–F).
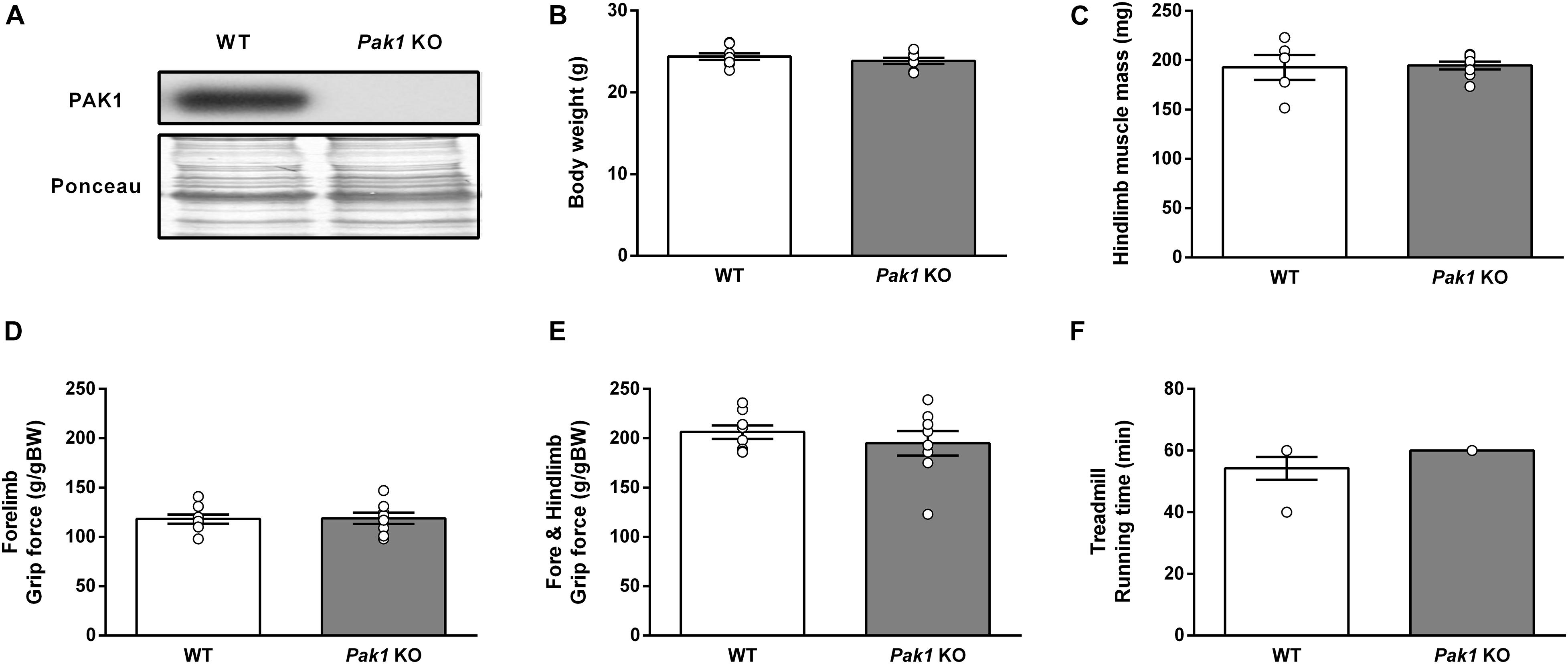
Figure 2. Deletion of Pak1 does not alter muscle mass and function. (A) Representative western blot image of muscle PAK1 protein levels in GC muscles from Pak1 KO vs. WT mice. (B) Body weight, and (C) hindlimb muscle mass (including TA, GC, SOL, and EDL muscles) from Pak1 KO vs. WT mice (n = 5–8/group). (D,E) Muscle strength from Pak1 KO vs. WT mice (n = 7–8/group). Mice were lowered on a grid connected to a sensor to measure the muscle force of their forelimbs or of both their forelimb and hindlimb and data were then expressed in gram force relative to body weight (g/gBW). (F) Endurance tolerance from Pak1 KO vs. WT mice (n = 7–8/group). Mice were submitted to an incremental treadmill running exercise (from 5 m/min with added 1 m/min every min until maximum 18 m/min) for a maximum running time of 60 min. Results are expressed as means ± SEM and statistical analysis was performed using unpaired t-test to compare results from Pak1 KO and WT mice.
Given the role of Pak1 in muscle mass regulation (Joseph et al., 2017; Cerquone Perpetuini et al., 2018) and its increased muscle levels in response to MSTN inhibition, we investigated the effect of Pak1 deletion on muscle hypertrophy induced by FS electroporation. For this purpose, we overexpressed FS by DNA electrotransfer in TA muscle from Pak1 KO mice. Surprisingly, the absence of Pak1 did not affect the FS-induced skeletal muscle hypertrophy. Indeed, we observed a similar increase in muscle fiber CSA from Pak1 KO (+96%, 3.443 ± 306 vs. 1.831 ± 194 μm2, P < 0.01) and WT mice (+85%, 4.121 ± 452 vs. 2.218 ± 101 μm2, P < 0.01) in response to FS overexpression (Figures 3A,C). These results were supported by the analysis of fiber size distribution showing a similar shift to a greater proportion of large fibers among the FS-transfected than among CTRL fibers in the two groups of mice (P < 0.001, Figure 3B). These results demonstrate that FS retains its hypertrophic effect toward skeletal muscle despite the absence of Pak1.
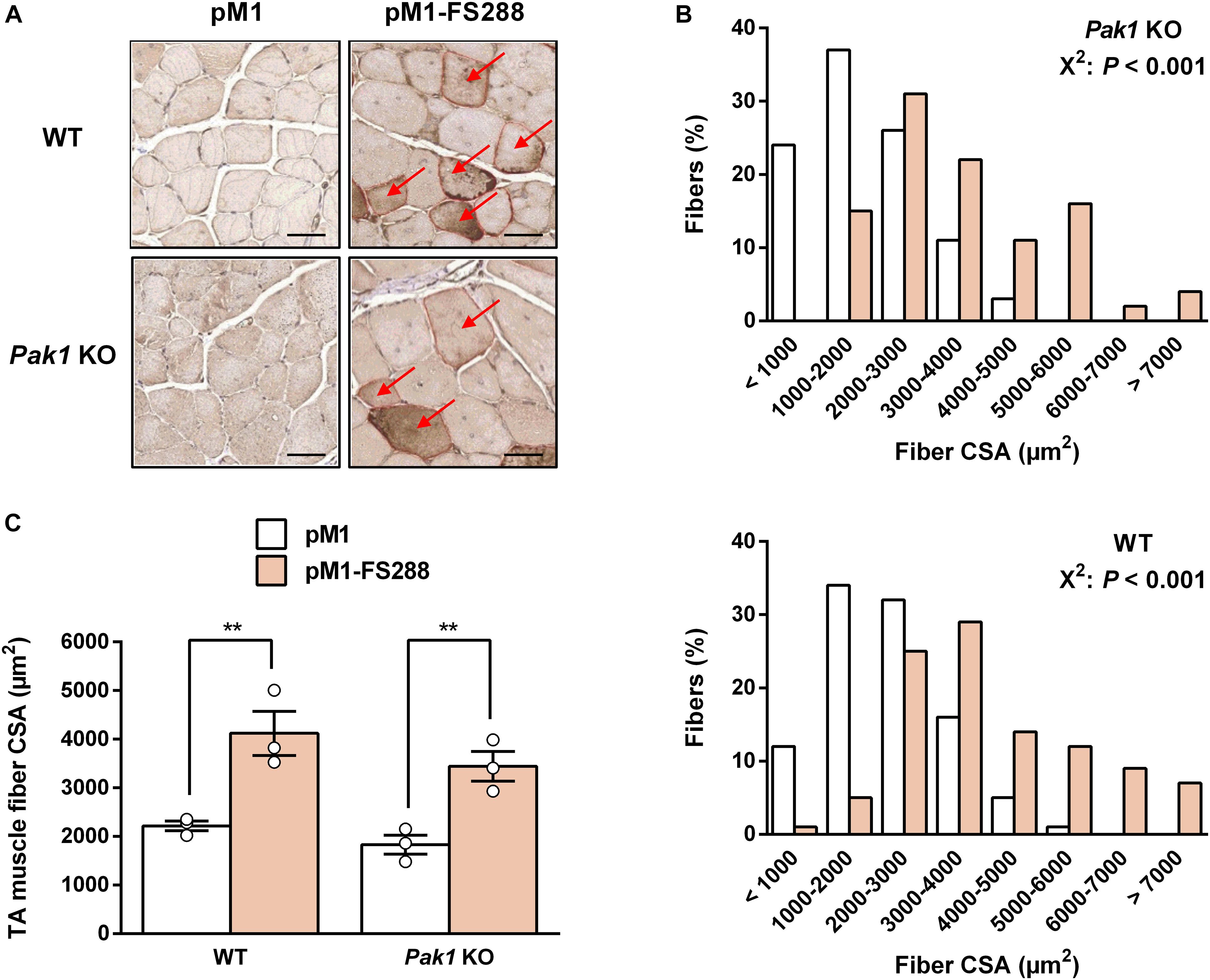
Figure 3. Follistatin induces skeletal muscle hypertrophy despite the absence of Pak1. (A) Representative FS-c-myc immunochemistry image from TA muscle of Pak1 KO and WT littermate mice. Red arrows indicate C-myc positive fibers. Scale bar = 50 μm. (B) Muscle fiber size distribution from pM1-FS288-transfected vs. pM1-transfected TA muscles of Pak1 KO (up panel) and WT (bottom panel) mice (n = 3/group). (C) Muscle fiber CSA assessed 17 days after electroporation of pM1-hFS288 (orange columns) or pM1 (white columns) in TA muscles from Pak1 KO vs. WT littermate mice (n = 3/group). Results are expressed as mean ± SEM. Statistical analysis was performed using two-way ANOVA test and Bonferroni post-tests (FS effect: **P < 0.01). Fiber CSA distribution statistical analysis was performed using χ2 Pearson test (P < 0.001).
Although Pak1 deletion does not change the extent of muscle hypertrophy induced by FS overexpression, we could not rule out that Pak1 does not regulate the onset of skeletal muscle hypertrophy. Indeed, the transcriptomic data from acute models of MSTN inhibition (sActRIIB 1x and FS acute; Figure 1B) show an acute upregulation of Pak1 suggesting that its increase may not result from muscle mass increase but could be involved in its onset. Previous results demonstrated that 4 injections of sActRIIB induces a maximal hypertrophy (Hoogaars et al., 2012; Hulmi et al., 2013). However, we observed that muscle Pak1 mRNA was already upregulated after one (+64%, P < 0.01) or 2 (+49%, P < 0.05) injections of sActRIIB compared to PBS-injected mice (Figure 4A), while muscle hypertrophy only appears after 4 injections (Figures 4C,D). For this reason, we investigated the effect of Pak1 deletion at the start and the end of muscle hypertrophy process induced by sActRIIB treatment, namely after 2 and 4 injections, respectively.
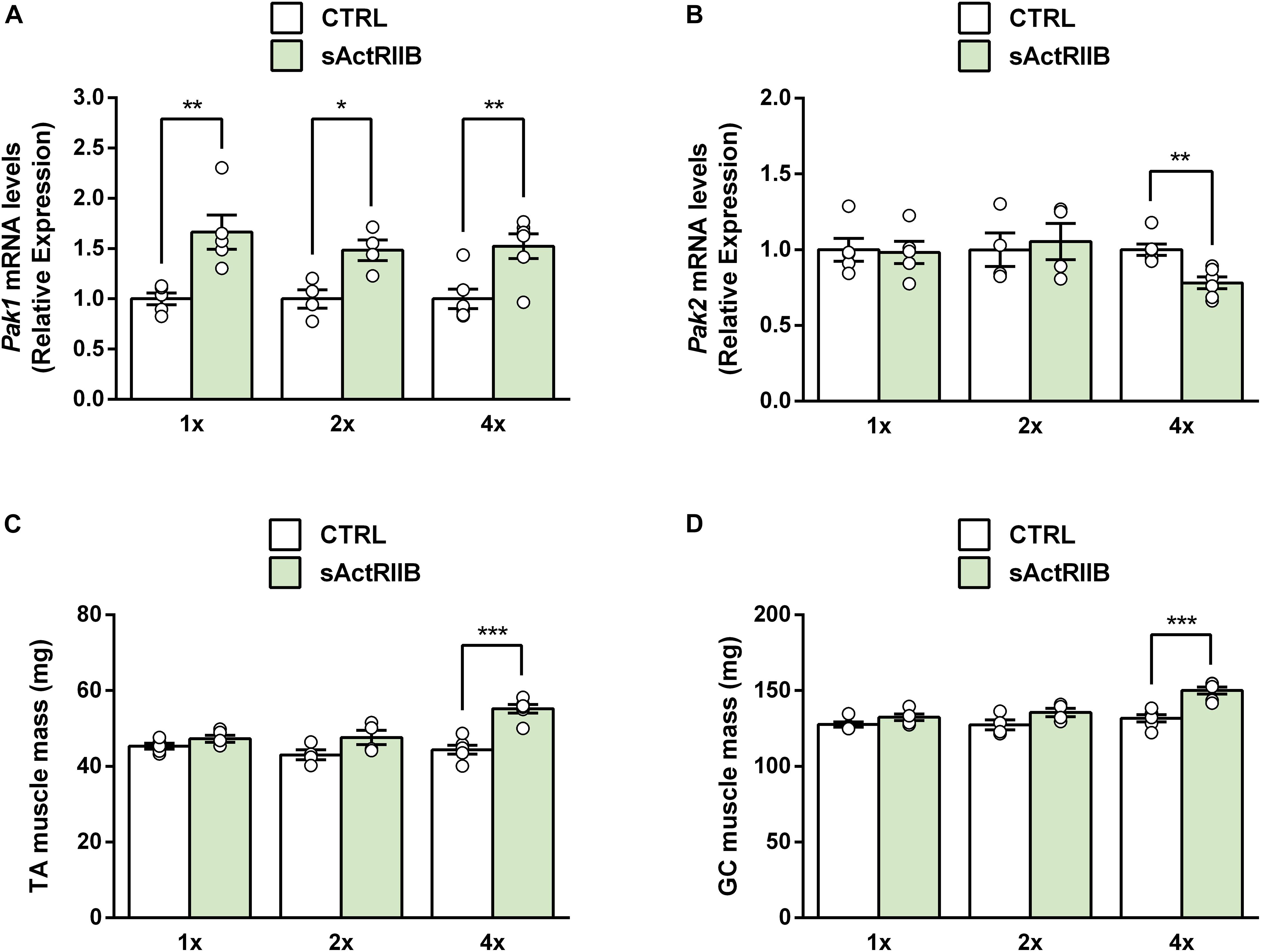
Figure 4. Muscle Pak1 and Pak2 mRNA levels in response to skeletal muscle hypertrophy induced by MSTN inhibition. (A) TA muscle Pak1 and (B) Pak2 mRNA levels, (C) TA and (D) GC muscle weight after one, 2 or 4 injections of sActRIIB (green columns) or PBS (white columns) in WT mice (n = 4–6/group). Results are expressed as means ± SEM. Statistical analysis was performed using unpaired t-test to compare results from sActRIIB-treated and PBS-treated mice (*P < 0.05; **P < 0.01, and ***P < 0.001 vs. CTRL).
In response to 2 injections of sActRIIB, while skeletal muscle hypertrophy was only starting in WT mice (TA muscle: +11%, 48 ± 2 vs. 43 ± 1 mg, ns; GC muscle: +6%, 136 ± 3 vs. 127 ± 3 mg, ns), we observed a significant increase in muscle weight from Pak1 KO (TA muscle: +33%, 49 ± 2 vs. 37 ± 3 mg, P < 0.05; GC muscle: +29%, 153 ± 11 vs. 118 ± 10 mg, P < 0.05) (Figures 5A,B). However, in response to 4 injections of sActRIIB, the maximal magnitude of skeletal muscle hypertrophy was similar in Pak1 KO (TA muscle: +26%, 51 ± 2 vs. 40 ± 1 mg, P < 0.001; GC muscle: +21%, 157 ± 8 vs. 130 ± 3 mg, P < 0.01) and WT mice (TA muscle: +25%, 56 ± 1 vs. 44 ± 2 mg, P < 0.001; GC muscle: +23%, 173 ± 4 vs. 141 ± 3 mg, P < 0.01) (Figures 5D,E). These results confirm that Pak1 deletion does not impede the skeletal muscle hypertrophy, regardless of the MSTN inhibition model.
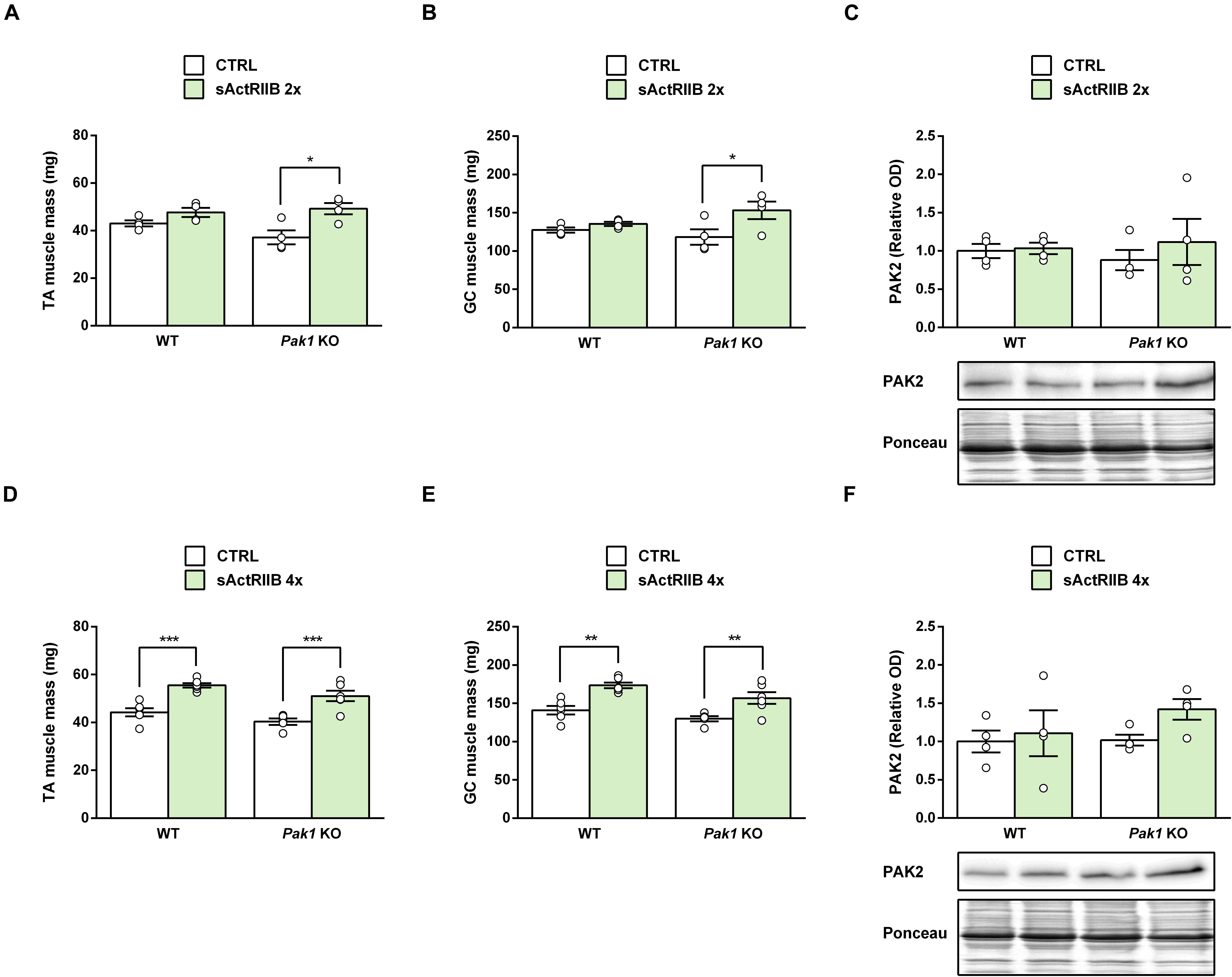
Figure 5. sActRIIB induces skeletal muscle hypertrophy despite the absence of Pak1. (A,D) TA and (B,E) GC muscle weight and (C,F) TA muscle PAK2 protein levels after 2 injections (n = 4/group) or 4 injections (n = 4–6/group) of sActRIIB (green columns) or PBS (white columns) in Pak1 KO vs. WT mice. Results are expressed as means ± SEM. Statistical analysis was performed using two-way ANOVA test and Bonferroni post-tests (sActRIIB effect: *P < 0.05; **P < 0.01, and ***P < 0.001).
Because PAK1 and PAK2 seem to be redundant, we hypothesized that Pak1 deletion could be compensated by Pak2 upregulation. However, we did not observed any significant change in TA muscle PAK2 protein levels following Pak1 deletion (Figures 5C,F). In response to sActRIIB, we observed that TA muscle Pak2 mRNA was downregulated only after 4 injections of sActRIIB (−23%, P < 0.01) compared to PBS-injected mice (Figure 4B), once the skeletal muscle mass increased (Figures 4C,D). However, we did not observe any significant change in muscle PAK2 protein levels of Pak1 KO and WT mice in response to 2 or 4 sActRIIB-injections (Figures 5C,F).
To gain new insights on the molecular mechanisms responsible for the observed changes in muscles from Pak1 KO mice after 2 sActRIIB-injections, we investigated proteins involved in muscle growth. In line with previous data (Hulmi et al., 2013), the phosphorylation state of AKT at Ser473 was unchanged (Figures 6A,B). In contrast, we observed an increased phosphorylation of S6 ribosomal protein at Ser235/236 (Pak1 KO mice: 2.3 ± 0.6-fold; WT mice: 2.2 ± 0.4-fold; P < 0.01, n = 4/group; Figures 6A,C), indicating activated mTORC1 signaling. Interestingly, phosphorylation of 4E-BP1 at Ser65 and P70S6K at Thr389 seemed to be more increased by sActRIIB in Pak1 KO (P-4E-BP1: 1.6 ± 0.3-fold, P < 0.05; P-P70S6K: 2.7 ± 0.6-fold, P = 0.14; n = 4/group) than in WT mice (P-4E-BP1: 1.2 ± 0.1-fold, ns; P-P70S6K: 1.6 ± 0.5-fold, ns; n = 4/group) (Figures 6A,D,E). Although the small number of samples creates low statistical power, our results suggest that Pak1 deletion could increase the rate of translation initiation through activating phosphorylation of P70S6K and inactivating phosphorylation of 4E-BP1. Since Smad3 and FBXO32 emerged as the downstream effectors in the anti-hypertrophic action of Pak1 in the heart (Liu et al., 2011; Tsui et al., 2015), we investigated these signaling proteins in skeletal muscle from Pak1 KO mice. However, we did not observe any change in FBXO32 protein expression, nor in P-FOXO3a or P-Smad3 levels in Pak1 KO muscles (Figure 7).
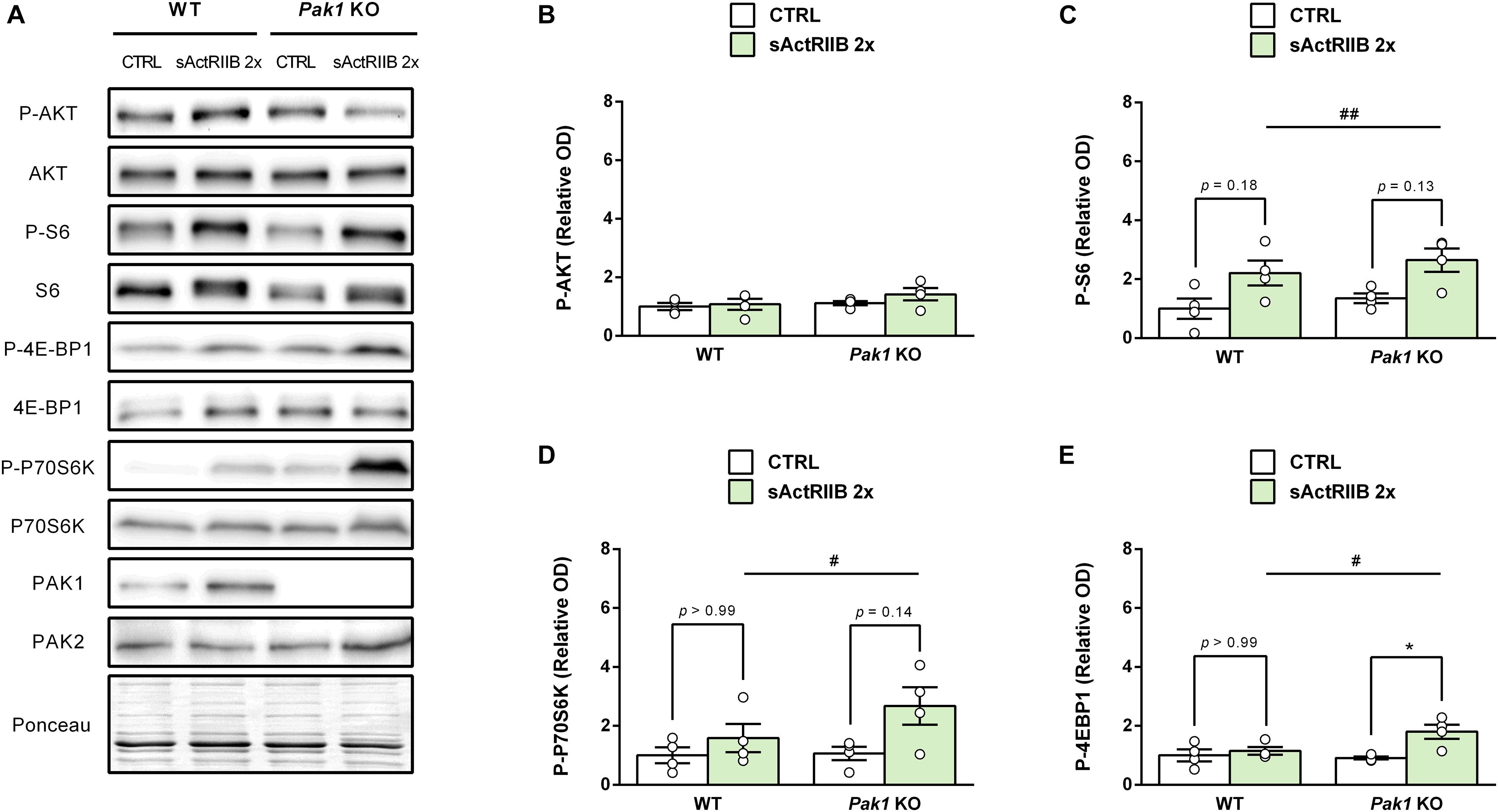
Figure 6. Pak1 deletion tends to increase the sActRIIB-induced 4E-BP1 inactivating phosphorylation. (A) Representative western blot images and densitometric analysis of (B) P-AKT, (C) P-S6, (D) P-P70S6K, and (E) P-4E-BP1 levels in GC muscles from Pak1 KO and WT mice after 2 injections of sActRIIB (green columns) or PBS (white columns) (n = 4/group). The results are expressed as mean ± SEM. Statistical analysis was performed using two-way ANOVA tests (sActRIIB effect: #P < 0.05 and ##P < 0.01) and Bonferroni post-tests (sActRIIB effect: *P < 0.05).
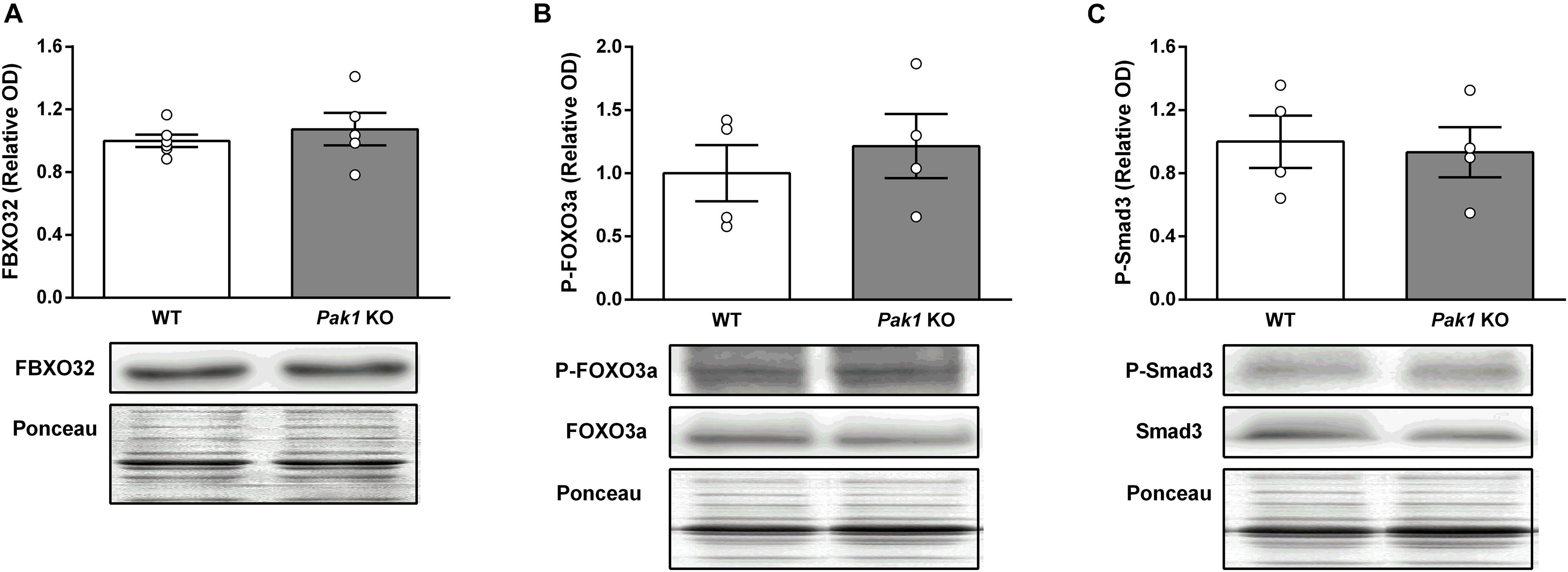
Figure 7. Pak1 deletion does not increase Smad3 phosphorylation nor FBXO32 protein expression in skeletal muscle. (A) GC muscle FBXO32 protein levels, (B) P-FOXO3a, and (C) P-Smad3 levels in GC muscles from Pak1 KO and WT mice (n = 4–6/group). The results are expressed as mean ± SEM. Statistical analysis was performed using unpaired t-test to compare results from Pak1 KO and WT mice.
By comparing changes in muscle transcriptome associated with muscle hypertrophy induced by various MSTN inhibition approaches, we identified Pak1 as the only gene systematically upregulated regardless the MSTN inhibition model considered. This in silico observation was confirmed by RT-qPCR analysis: muscle Pak1 mRNA levels were increased in parallel to skeletal muscle hypertrophy in mTrFS, MSTN KO, and sActRIIB-treated mice. At the opposite, we observed a decreased expression of muscle Pak1 mRNA levels in atrophied muscles following MSTN transfection and DEXA treatment. Using Pak1 KO mice, we investigated the effect of Pak1 deletion on muscle hypertrophy induced by FS electroporation and sActRIIB treatment. Although we observed a slight acceleration in the sActRIIB-induced muscle hypertrophy, our results show that FS and sActRIIB retain their anabolic effect toward skeletal muscle even in the absence of Pak1. Taken together, our results demonstrate that Pak1 is dispensable for the muscle mass increase caused by MSTN inhibition.
Several studies examined the transcriptomic changes caused by MSTN deletion during pre- (Steelman et al., 2006; Chelh et al., 2009; Rahimov et al., 2011) and post-development (Welle et al., 2009) or in response to pre- (Barbé et al., 2017) and postnatal MSTN inhibition (Rahimov et al., 2011; Miao et al., 2015; Davey et al., 2016). Due to the extremely large number of data resulting from these high-throughput experimental techniques, interpretation of these analysis is made difficult. Although we penned only one of these transcriptomic analyses, our work combines in a unique way all the results obtained from various models of MSTN inhibition and offers a short list of 20 genes (8 upregulated and 12 downregulated) whose mRNA levels appear commonly regulated in most (75%) MSTN inhibition models compared. In this study, we highlighted Pak1 as a likely mediator of the skeletal muscle hypertrophy induced by MSTN inhibition based on its consistent induction in all MSTN inhibition models. The fact that Pak1 is also induced by the Insulin/IGF-I pathway (Tunduguru et al., 2014), which contributes to the hypertrophy induced by the inhibition of MSTN (Kalista et al., 2012; Barbé et al., 2015), supports this hypothesis. The role of the 19 other regulated genes remains to be investigated.
p21-activated kinase 1 is the best characterized member of the PAK kinases and has been involved in actin cytoskeleton remodeling (Sells et al., 1997). In recent years, a role for Pak1 in muscle mass regulation has emerged, notwithstanding some inconsistencies. Pak1 has been first suggested to contribute to the regulation of cardiac muscle hypertrophy. Indeed, while some authors found that Pak1 is essential for AKT activity in cardiomyocytes (Mao et al., 2008), others reported an anti-hypertrophic action of Pak1 in the heart (Liu et al., 2011). Regarding the skeletal muscle, Joseph et al. (2017) demonstrated that muscle-specific combined Pak1 and Pak2 deletion resulted in reduced skeletal muscle mass. Moreover, a decreased expression of Pak1 was observed in cancer-associated cachectic muscles from Yoshida hepatoma-bearing rats (Martinelli et al., 2016) and colon adenocarcinoma C26-bearing mice (Cerquone Perpetuini et al., 2018). Most of all, Pak1 overexpression prevented the muscle atrophy observed in cachectic muscles suggesting that Pak1 might have an anti-atrophic action in skeletal muscle (Cerquone Perpetuini et al., 2018). Reasoning that PAK1 has a central role in skeletal muscle mass regulation and its muscle levels increase in response to MSTN inhibition, we hypothesized that PAK1 could mediate the skeletal muscle hypertrophy induced by MSTN inhibition. Although this assumption was never been investigated, it was suggested that the anti-hypertrophic action of PAK1 in cardiac muscle implicates Smad3 (Tsui et al., 2015), the main downstream effector of the atrophic effect of MSTN (Sartori et al., 2009). Moreover, it has been evidenced that FS overexpression amplifies insulin-stimulated phosphorylation of PAK1 at its activation site Thr423 (Han et al., 2019), while our previous data have demonstrated the role of Insulin signaling in the FS-induced skeletal muscle hypertrophy (Kalista et al., 2012; Barbé et al., 2015). For these reasons, we investigated the effect of Pak1 deletion on skeletal muscle hypertrophy induced by MSTN inhibition.
In the present work, we show that Pak1 deletion does not alter skeletal muscle mass, just as ablation of Pak1 has no effect on muscle fiber size (Joseph et al., 2017), suggesting that, in basal conditions, changes in Pak1 expression levels are not sufficient to affect muscle mass. In contrast, in pathological conditions, deletion of Pak1 is sufficient to amplify cardiac muscle hypertrophy in response to pressure overload (Liu et al., 2011), while Pak1 overexpression mitigates muscle atrophy caused by cancer cachexia (Cerquone Perpetuini et al., 2018). In addition, we demonstrate that Pak1 deletion does not affect skeletal muscle function. The absence of consequences of Pak1 deletion on skeletal muscle mass and function is striking, giving the many roles of this protein in particular on the actin cytoskeleton. Since we used a developmental KO model, compensation for the lack of Pak1 is still possible. In particular, Pak1 and Pak2 have been shown to be redundant regulators of postnatal myogenesis (Joseph et al., 2017). Therefore, we may not exclude that Pak2 compensates for the lack of Pak1. However, the absence of changes in TA muscle PAK2 protein levels observed in Pak1 KO mice does not support this hypothesis.
An interesting finding in the current study is that Pak1 is induced prior to the muscle mass increase suggesting that increased Pak1 levels do not result from muscle hypertrophy but from MSTN inhibition itself and may modulate the rate of the anabolic process. Indeed, our results showed that 2 sActRIIB injections were sufficient to induce muscle hypertrophy in Pak1 KO mice, while 4 injections are needed to increase muscle mass at the same extent in WT mice. These results suggest a negative role for Pak1 in the rate of muscle hypertrophy process. Although we did not measure the muscle fiber CSA, the sActRIIB is well known to increase muscle fiber CSA in parallel to muscle mass, without affecting the number of muscle fibers (Cadena et al., 2010; Pistilli et al., 2010; Hulmi et al., 2013; Relizani et al., 2014). After 2 sActRIIB injections, we observed a slight increase in phosphorylated 4E-BP1 and P70SK in Pak1 KO mice suggesting that Pak1 could curb muscle hypertrophy through inhibiting these protein targets. Supporting this hypothesis, Smad3, and FBXO32 has emerged as the downstream effectors in the anti-hypertrophic action of Pak1 in the heart (Liu et al., 2011; Tsui et al., 2015). However, we did not observe any change in Smad3 phosphorylation nor FBXO32 protein expression in skeletal muscle from Pak1 KO mice. If these results are due to genetic compensation for the lack of Pak1 or if Pak1 does not signal through Smad3 and FBXO32 in skeletal muscle remains unknown. Therefore, although these findings may seem appealing, more research is definitely needed before defending this thesis, given that our sample size was low and that muscle fiber CSA was not closely assessed in this experiment.
The major discovery in the current study is that Pak1 is dispensable for the skeletal muscle hypertrophy induced by MSTN inhibition. Due to the plateau effect occurring with the sActRIIB injected four times at 10 mg/kg (Hoogaars et al., 2012; Hulmi et al., 2013), we cannot exclude Pak1 deletion would not affect the magnitude of muscle hypertrophy induced by the sActRIIB. However, the similar results observed in response to FS overexpression suggest that muscle hypertrophy induced by MSTN inhibition does not rely on Pak1. Moreover, the dispensable nature of Pak1 for the muscle hypertrophy caused by MSTN inhibition is in line with the recent study of Møller et al. (2020) who have demonstrated that muscle-specific genetic ablation of Pak2, but not the whole-body Pak1 knockout, impairs the insulin-induced muscle glucose uptake, the second main effect of MSTN inhibition toward muscle (Akpan et al., 2009; Guo et al., 2009; Morissette et al., 2009; Cleasby et al., 2014). In our work, we observed that, in response to sActRIIB, the muscle Pak2 mRNA levels were decreased only once the muscle mass increased. Knowing that combined deletion of Pak1 and Pak2 reduces muscle mass (Joseph et al., 2017), the decreased Pak2 levels might result from muscle hypertrophy and acts as a brake for the anabolic signal. Alternatively, given the role of Pak2 in the insulin-stimulated glucose uptake in skeletal muscle, we might assume that changes observed in muscle Pak2 mRNA levels would curb the increased insulin sensitivity induced by the sActRIIB as a negative feedback. However, we did not observe any significant change in PAK2 protein levels from Pak1 KO and WT mice treated with sActRIIB.
To the best of our knowledge, we are the first to have investigated the role of Pak1 in the skeletal muscle hypertrophy induced by MSTN inhibition. The strength of our observations stems in part from the use of different MSTN inhibition models. Moreover, the sActRIIB seems to have clinical relevance as therapeutic strategy for reversing muscle wasting, such as sarcopenia and cachexia, as well as muscular dystrophies (Saitoh et al., 2017). In this regard, defining the key mediators of MSTN actions on skeletal muscle mass is essential to prevent and to remedy the potential adverse effects of investigational drugs. In this regard, our work demonstrates that, Pak1 is permissive for the anabolic action induced by MSTN inhibition. Further studies will need to investigate more deeply the role of group I PAKs in the skeletal muscle hypertrophy induced by MSTN inhibitors.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Committee for Ethical Practices in Animal Experiments of the Catholic University of Louvain (Brussels, Belgium).
CB, AL, OR, and J-PT conceived and planned the experiments. CB, AL, and PL carried out the experiments. CB, AL, PL, and J-PT analyzed and interpreted the data. CB wrote the manuscript with support from J-PT. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
This study was funded by grants from Fonds de la Recherche Scientifique Médicale (FRSM-FNRS, Belgium). Fonds spéciaux de Recherche (FSR) (Université catholique de Louvain, Belgium) and Bourses du Patrimoine (Faculté de Médecine, Université catholique de Louvain, Belgium). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. SJ Lee (Johns Hopkins University School of Medicine, United States), Pr. EA Richter (University of Copenhagen, Denmark), and Pr. D Freyssenet (University of Saint-Etienne, France) for the genetically modified mTrFS, the Pak1 KO mice and the pcDNA-MSTN plasmid, respectively. Thanks to L Poreba, C Ploquin, A Essaghir, and O Schakman who also contributed to some parts of this work.
Akpan, I., Goncalves, M. D., Dhir, R., Yin, X., Pistilli, E. E., Bogdanovich, S., et al. (2009). The effects of a soluble activin type IIB receptor on obesity and insulin sensitivity. Int. J. Obes. 33, 1265–1273. doi: 10.1038/ijo.2009.162
Allen, J. D., Jaffer, Z. M., Park, S. J., Burgin, S., Hofmann, C., Sells, M. A., et al. (2009). p21-activated kinase regulates mast cell degranulation via effects on calcium mobilization and cytoskeletal dynamics. Blood 113, 2695–2705. doi: 10.1182/blood-2008-06-160861
Barbé, C., Bray, F., Gueugneau, M., Devassine, S., Lause, P., Tokarski, C., et al. (2017). Comparative Proteomic and Transcriptomic Analysis of Follistatin-Induced Skeletal Muscle Hypertrophy. J. Proteome Res. 16, 3477–3490. doi: 10.1021/acs.jproteome.7b00069
Barbé, C., Kalista, S., Loumaye, A., Ritvos, O., Lause, P., Ferracin, B., et al. (2015). Role of IGF-I in follistatin-induced skeletal muscle hypertrophy. Am. J. Physiol. Endocrinol. Metab. 309, E557–E567.
Bloquel, C., Fabre, E., Bureau, M. F., and Scherman, D. (2004). Plasmid DNA electrotransfer for intracellular and secreted proteins expression: new methodological developments and applications. J. Gene Med. 6(Suppl. 1), S11–S23.
Bokoch, G. M. (2003). Biology of the p21-activated kinases. Annu. Rev. Biochem. 72, 743–781. doi: 10.1146/annurev.biochem.72.121801.161742
Busquets, S., Toledo, M., Orpi, M., Massa, D., Porta, M., Capdevila, E., et al. (2012). Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J. Cachexia Sarcopenia Muscle 3, 37–43. doi: 10.1007/s13539-011-0049-z
Cadena, S. M., Tomkinson, K. N., Monnell, T. E., Spaits, M. S., Kumar, R., Underwood, K. W., et al. (2010). Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J. Appl. Physiol. 109, 635–642. doi: 10.1152/japplphysiol.00866.2009
Cerquone Perpetuini, A., Re Cecconi, A. D., Chiappa, M., Martinelli, G. B., Fuoco, C., Desiderio, G., et al. (2018). Group I Paks support muscle regeneration and counteract cancer-associated muscle atrophy. J. Cachexia Sarcopenia Muscle 9, 727–746. doi: 10.1002/jcsm.12303
Chelh, I., Meunier, B., Picard, B., Reecy, M. J., Chevalier, C., Hocquette, J. F., et al. (2009). Molecular profiles of Quadriceps muscle in myostatin-null mice reveal PI3K and apoptotic pathways as myostatin targets. BMC Genomics 10:196. doi: 10.1186/1471-2164-10-196
Cleasby, M. E., Jarmin, S., Eilers, W., Elashry, M., Andersen, D. K., Dickson, G., et al. (2014). Local overexpression of the myostatin propeptide increases glucose transporter expression and enhances skeletal muscle glucose disposal. Am. J. Physiol. Endocrinol. Metab. 306, E814–E823.
Davey, J. R., Watt, K. I., Parker, B. L., Chaudhuri, R., Ryall, J. G., Cunningham, L., et al. (2016). Integrated expression analysis of muscle hypertrophy identifies Asb2 as a negative regulator of muscle mass. JCI Insight 1:e85477.
Dehoux, M., Van Beneden, R., Pasko, N., Lause, P., Verniers, J., Underwood, L., et al. (2004). Role of the insulin-like growth factor I decline in the induction of atrogin-1/MAFbx during fasting and diabetes. Endocrinology 145, 4806–4812. doi: 10.1210/en.2004-0406
Durieux, A. C., Amirouche, A., Banzet, S., Koulmann, N., Bonnefoy, R., Pasdeloup, M., et al. (2007). Ectopic expression of myostatin induces atrophy of adult skeletal muscle by decreasing muscle gene expression. Endocrinology 148, 3140–3147. doi: 10.1210/en.2006-1500
Gilson, H., Schakman, O., Kalista, S., Lause, P., Tsuchida, K., and Thissen, J. P. (2009). Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am. J. Physiol. Endocrinol. Metab. 297, E157–E164.
Grobet, L., Pirottin, D., Farnir, F., Poncelet, D., Royo, L. J., Brouwers, B., et al. (2003). Modulating skeletal muscle mass by postnatal, muscle-specific inactivation of the myostatin gene. Genesis 35, 227–238. doi: 10.1002/gene.10188
Gueugneau, M., D’hose, D., Barbe, C., De Barsy, M., Lause, P., Maiter, D., et al. (2018). Increased Serpina3n release into circulation during glucocorticoid-mediated muscle atrophy. J. Cachexia Sarcopenia Muscle 9, 929–946. doi: 10.1002/jcsm.12315
Guo, T., Jou, W., Chanturiya, T., Portas, J., Gavrilova, O., and Mcpherron, A. C. (2009). Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS One 4:e4937. doi: 10.1371/journal.pone.0004937
Han, X., Møller, L. L. V., De Groote, E., Bojsen-Møller, K. N., Davey, J., Henríquez-Olguin, C., et al. (2019). Mechanisms involved in follistatin-induced hypertrophy and increased insulin action in skeletal muscle. J. Cachexia Sarcopenia Muscle 10, 1241–1257. doi: 10.1002/jcsm.12474
Hoogaars, W. M., Mouisel, E., Pasternack, A., Hulmi, J. J., Relizani, K., Schuelke, M., et al. (2012). Combined effect of AAV-U7-induced dystrophin exon skipping and soluble activin Type IIB receptor in mdx mice. Hum. Gene Ther. 23, 1269–1279. doi: 10.1089/hum.2012.056
Hulmi, J. J., Oliveira, B. M., Silvennoinen, M., Hoogaars, W. M., Ma, H., Pierre, P., et al. (2013). Muscle protein synthesis, mTORC1/MAPK/Hippo signaling, and capillary density are altered by blocking of myostatin and activins. Am. J. Physiol. Endocrinol. Metab. 304, E41–E50.
Joseph, G. A., Lu, M., Radu, M., Lee, J. K., Burden, S. J., Chernoff, J., et al. (2017). Group I Paks Promote Skeletal Myoblast Differentiation In Vivo and In Vitro. Mol. Cell Biol. 37, e222–e216.
Kalista, S., Schakman, O., Gilson, H., Lause, P., Demeulder, B., Bertrand, L., et al. (2012). The type 1 insulin-like growth factor receptor (IGF-IR) pathway is mandatory for the follistatin-induced skeletal muscle hypertrophy. Endocrinology 153, 241–253. doi: 10.1210/en.2011-1687
Lee, S. J., and Mcpherron, A. C. (2001). Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. U S A. 98, 9306–9311. doi: 10.1073/pnas.151270098
Liu, W., Zi, M., Naumann, R., Ulm, S., Jin, J., Taglieri, D. M., et al. (2011). Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation 124, 2702–2715. doi: 10.1161/circulationaha.111.048785
Mao, K., Kobayashi, S., Jaffer, Z. M., Huang, Y., Volden, P., Chernoff, J., et al. (2008). Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. J. Mol. Cell Cardiol. 44, 429–434. doi: 10.1016/j.yjmcc.2007.10.016
Martinelli, G. B., Olivari, D., Re Cecconi, A. D., Talamini, L., Ottoboni, L., Lecker, S. H., et al. (2016). Activation of the SDF1/CXCR4 pathway retards muscle atrophy during cancer cachexia. Oncogene 35, 6212–6222. doi: 10.1038/onc.2016.153
Mcpherron, A. C., Lawler, A. M., and Lee, S. J. (1997). Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387, 83–90. doi: 10.1038/387083a0
Miao, Y., Yang, J., Xu, Z., Jing, L., Zhao, S., and Li, X. (2015). RNA sequencing identifies upregulated kyphoscoliosis peptidase and phosphatidic acid signaling pathways in muscle hypertrophy generated by transgenic expression of myostatin propeptide. Int. J. Mol. Sci. 16, 7976–7994. doi: 10.3390/ijms16047976
Møller, L. L. V., Jaurji, M., Kjøbsted, R., Joseph, G. A., Madsen, A. B., Knudsen, J. R., et al. (2020). Insulin-stimulated glucose uptake partly relies on p21-activated kinase (PAK)2, but not PAK1, in mouse skeletal muscle. J. Physiol. 598, 5351–5377. doi: 10.1113/jp280294
Morissette, M. R., Cook, S. A., Buranasombati, C., Rosenberg, M. A., and Rosenzweig, A. (2009). Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am. J. Physiol. Cell Physiol. 297, C1124–C1132.
Moritz, C. P. (2017). Tubulin or Not Tubulin: Heading Toward Total Protein Staining as Loading Control in Western Blots. Proteomics 17:201600189.
Pistilli, E. E., Bogdanovich, S., Mosqueira, M., Lachey, J., Seehra, J., and Khurana, T. S. (2010). Pretreatment with a soluble activin type IIB receptor/Fc fusion protein improves hypoxia-induced muscle dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R96–R103.
Rahimov, F., King, O. D., Warsing, L. C., Powell, R. E., Emerson, C. P. Jr., Kunkel, L. M., et al. (2011). Gene expression profiling of skeletal muscles treated with a soluble activin type IIB receptor. Physiol. Genomics 43, 398–407. doi: 10.1152/physiolgenomics.00223.2010
Relizani, K., Mouisel, E., Giannesini, B., Hourdé, C., Patel, K., Morales Gonzalez, S., et al. (2014). Blockade of ActRIIB signaling triggers muscle fatigability and metabolic myopathy. Mol. Ther. 22, 1423–1433. doi: 10.1038/mt.2014.90
Rodino-Klapac, L. R., Janssen, P. M., Shontz, K. M., Canan, B., Montgomery, C. L., Griffin, D., et al. (2013). Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Hum. Mol. Genet. 22, 4929–4937. doi: 10.1093/hmg/ddt342
Saitoh, M., Ishida, J., Ebner, N., Anker, S. D., Springer, J., and Von Haehling, S. (2017). Myostatin inhibitors as pharmacological treatment for muscle wasting and muscular dystrophy. JCSM Clin. Rep. 2, 1–10.
Sartori, R., Milan, G., Patron, M., Mammucari, C., Blaauw, B., Abraham, R., et al. (2009). Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 296, C1248–C1257.
Schakman, O., Kalista, S., Barbé, C., Loumaye, A., and Thissen, J. P. (2013). Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 45, 2163–2172. doi: 10.1016/j.biocel.2013.05.036
Sells, M. A., Knaus, U. G., Bagrodia, S., Ambrose, D. M., Bokoch, G. M., and Chernoff, J. (1997). Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7, 202–210. doi: 10.1016/s0960-9822(97)70091-5
Steelman, C. A., Recknor, J. C., Nettleton, D., and Reecy, J. M. (2006). Transcriptional profiling of myostatin-knockout mice implicates Wnt signaling in postnatal skeletal muscle growth and hypertrophy. FASEB J. 20, 580–582. doi: 10.1096/fj.05-5125fje
Tsui, H., Zi, M., Wang, S., Chowdhury, S. K., Prehar, S., Liang, Q., et al. (2015). Smad3 Couples Pak1 With the Antihypertrophic Pathway Through the E3 Ubiquitin Ligase, Fbxo32. Hypertension 66, 1176–1183. doi: 10.1161/hypertensionaha.115.06068
Tunduguru, R., Chiu, T. T., Ramalingam, L., Elmendorf, J. S., Klip, A., and Thurmond, D. C. (2014). Signaling of the p21-activated kinase (PAK1) coordinates insulin-stimulated actin remodeling and glucose uptake in skeletal muscle cells. Biochem. Pharmacol. 92, 380–388. doi: 10.1016/j.bcp.2014.08.033
Welle, S., Burgess, K., and Mehta, S. (2009). Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. Am. J. Physiol. Endocrinol. Metab. 296, E567–E572.
Winbanks, C. E., Chen, J. L., Qian, H., Liu, Y., Bernardo, B. C., Beyer, C., et al. (2013). The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. J. Cell Biol. 203, 345–357. doi: 10.1083/jcb.201211134
Keywords: myostatin, follistatin, sActRIIB, PAK1, skeletal muscle hypertrophy
Citation: Barbé C, Loumaye A, Lause P, Ritvos O and Thissen J-P (2021) p21-Activated Kinase 1 Is Permissive for the Skeletal Muscle Hypertrophy Induced by Myostatin Inhibition. Front. Physiol. 12:677746. doi: 10.3389/fphys.2021.677746
Received: 08 March 2021; Accepted: 26 May 2021;
Published: 17 June 2021.
Edited by:
Vandre Casagrande Figueiredo, University of Kentucky, United StatesReviewed by:
Craig Andrew Goodman, The University of Melbourne, AustraliaCopyright © 2021 Barbé, Loumaye, Lause, Ritvos and Thissen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Barbé, Y2Fyb2xpbmUuYmFyYmVAdWNhLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.