 Kathy Pham
Kathy Pham Keval Parikh
Keval Parikh Erica C. Heinrich
Erica C. Heinrich
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 26 May 2021
Sec. Environmental, Aviation and Space Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.676782
This article is part of the Research TopicTime Domains of Hypoxia Adaptation: Evolutionary Insights and Applications, Volume IIView all 12 articles
The key regulators of the transcriptional response to hypoxia and inflammation (hypoxia inducible factor, HIF, and nuclear factor-kappa B, NF-κB, respectively) are evolutionarily conserved and share significant crosstalk. Tissues often experience hypoxia and inflammation concurrently at the site of infection or injury due to fluid retention and immune cell recruitment that ultimately reduces the rate of oxygen delivery to tissues. Inflammation can induce activity of HIF-pathway genes, and hypoxia may modulate inflammatory signaling. While it is clear that these molecular pathways function in concert, the physiological consequences of hypoxia-induced inflammation and how hypoxia modulates inflammatory signaling and immune function are not well established. In this review, we summarize known mechanisms of HIF and NF-κB crosstalk and highlight the physiological consequences that can arise from maladaptive hypoxia-induced inflammation. Finally, we discuss what can be learned about adaptive regulation of inflammation under chronic hypoxia by examining adaptive and maladaptive inflammatory phenotypes observed in human populations at high altitude. We aim to provide insight into the time domains of hypoxia-induced inflammation and highlight the importance of hypoxia-induced inflammatory sensitization in immune function, pathologies, and environmental adaptation.
Inflammation plays a key role in the physiological response to hypoxic stress. Tissues experience hypoxia during injury, infection, hypoperfusion, ischemia, or hypoxemia secondary to sleep apnea, pulmonary disease, anemia, high-altitude exposure, or other causes (Celli et al., 2004; McNicholas, 2009; Brill and Wedzicha, 2014; Hirota, 2015; Couzin-Frankel, 2020; Tobin et al., 2020; Xie et al., 2020). Cellular hypoxia can trigger the expression of several inflammatory mediators which signal tissue damage and initiate survival responses. However, while hypoxia-induced inflammation may serve a protective role by initiating an immune response and promoting tissue healing, it can also contribute to several pathologies, particularly in the context of chronic hypoxia. In this review, we summarize the known crosstalk between the transcriptional responses to hypoxia and inflammation and highlight the physiological consequences that occur as a result of maladaptive hypoxia-induced inflammation. Finally, we review what we know about adaptive regulation of inflammation under chronic hypoxia by investigating inflammatory phenotypes in human populations adapted to high altitude.
The transcriptional response to hypoxia is controlled by the hypoxia-inducible factor (HIF) signaling cascade (Semenza, 2009). HIF is a heterodimer protein composed of an oxygen-sensitive alpha subunit and constitutively expressed beta subunit (Biddlestone et al., 2015). The three HIF isoforms (HIF-1, HIF-2, and HIF-3) have some overlapping roles but also demonstrate distinct functions in different cell types (Carroll and Ashcroft, 2006; Dengler et al., 2014; Watts and Walmsley, 2019). Under normoxic conditions, HIF-α is hydroxylated by oxygen-dependent prolyl hydroxylases (PHDs 1–3 in humans; Figure 1). Upon hydroxylation, HIF-α is ubiquitinated by the von Hippel-Lindau tumor suppressor protein (pVHL) and degraded by proteasomes. Since PHD requires oxygen as a co-substrate, its activity decreases under hypoxic conditions, allowing HIF-α to dimerize with HIF-β and translocate to the nucleus. The HIF complex can then bind to hypoxia response elements (5'-RCGTG-3') in gene promoters to regulate expression of at least 100 genes to coordinate increased oxygen supply to hypoxic tissue (Kaelin and Ratcliffe, 2008; Semenza, 2009). HIF pathway activity is associated with activation of genes involved in metabolic adaptation, such as phosphoglycerate kinase (PGK) and lactate dehydrogenase A (LDHA), vascularization via vascular endothelial growth factor (VEGF), as well as red blood cell production via erythropoietin (EPO), and several other genes involved in improving oxygen delivery and use efficiency (Dengler et al., 2014; Villafuerte et al., 2014).
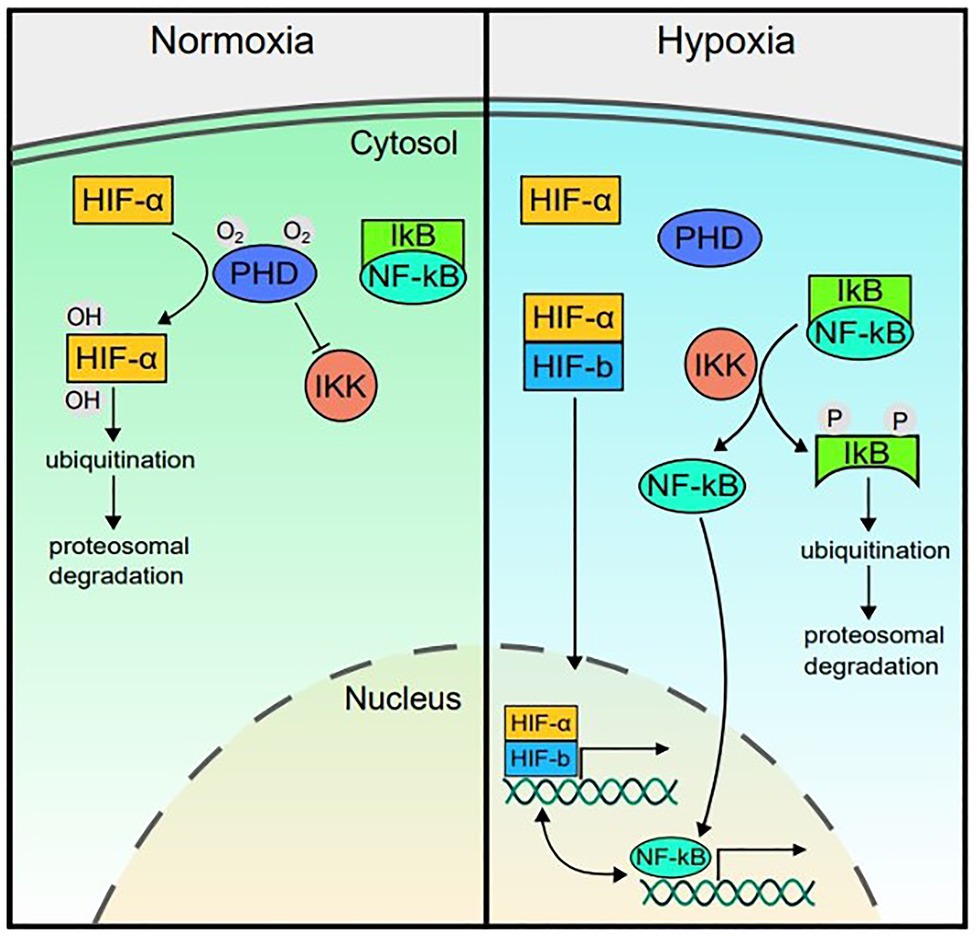
Figure 1. Hypoxia inducible factor (HIF)-nuclear factor (NF)-kappa B (NF-κB) crosstalk. In normoxic conditions, prolyl hydroxylases (PHDs) hydroxylate HIF-α and the IKKβ subunit of the IκB kinase (IKK) complex, marking them for degradation and thereby reducing transcriptional activity of HIF and repressing (but not completely blocking) NF-kB activity. In hypoxia, PHD activity decreases since it utilizes oxygen as a cofactor. Therefore, HIF-α is stabilized and can dimerize with the constitutively active HIF-β subunit. The complex translocates to the nucleus to upregulate expression of genes involved in the hypoxia response. In hypoxia, with reduced PHD activity, the rate of IKK degradation of IκB increases, releasing repression of NF-κB and allowing it to translocate to the nucleus at higher rates and upregulate inflammatory gene expression. PHD, prolyl hydroxylase; HIF, hypoxia-inducible factor; NF-κB, Nuclear factor kappa B; IKK, IκB kinase complex (composed of IKK-α, IKK-β, and IKK-γ subunits); and IκB, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor.
While the HIF pathway primarily responds to hypoxia, HIF expression is also increased in response to non-hypoxic stimuli, including bacterial lipopolysaccharide (LPS), tumor necrosis factor-α (TNF-α), reactive oxygen species, hepatocyte growth factor, and interleukin (IL)-18 via crosstalk with the nuclear factor-κB (NF-κB) pathway (Figueroa et al., 2002; Zhou et al., 2003; Frede et al., 2006; Taylor, 2008). The NF-κB transcription factor is a master regulator of inflammation. NF-κB is kept localized in the cytoplasm by inhibitory IκB proteins (Lawrence, 2009; Oeckinghaus and Ghosh, 2009), which thereby inhibit DNA binding by NF-κB (Beg and Jr, 1993; Mitchell et al., 2016). In response to inflammatory stimuli and microbial products, the IκB kinase (IKK) complex phosphorylates IκB, leading to IκB ubiquitination and proteasomal degradation (Israël, 2010). With the degradation of IκB, NF-κB can translocate to the nucleus and upregulate key downstream inflammatory pathways (Hayden and Ghosh, 2004; Perkins, 2006; Mitchell et al., 2016; Figure 1).
The NF-κB pathway can also upregulate HIF-1α (BelAiba et al., 2007). NF-κB subunits bind to the NF-κB binding element within the HIF-1α gene promoter region and induce HIF-1α mRNA expression (Van Uden et al., 2008). Several studies support this NF-κB-dependent HIF-1α expression. For example, in cell culture models (HEK293 cells and pulmonary artery smooth muscle cells); NF-κB transfection resulted in increased HIF-1α mRNA and protein expression. Additionally, when these cells were co-transfected with a mutated dominant negative IκB (which cannot be phosphorylated by IKK) to reduce NF-κB translocation to the nucleus, HIF-1α mRNA and protein expression decreased (BelAiba et al., 2007; Bonello et al., 2007; Görlach and Bonello, 2008).
In addition to this direct link, the HIF and NF-κB pathways also share common regulators. Like HIF-α, the IKK complex responsible for regulating NF-κB activity is also a target of PHD and therefore its NF-κB regulatory activity is oxygen dependent. In normoxic conditions, PHD hydroxylates IKKβ, therefore repressing NF-κB nuclear translocation and transcriptional activity. When PHD is rendered inactive in hypoxic conditions, the IKK complex can proceed to remove IκB from NF-κB, increasing its rate of translocation to the nucleus and upregulating inflammatory gene expression (Figure 1; Cummins et al., 2006; Taylor, 2008).
At short timescales, and at the tissue level, inflammatory signaling in response to hypoxia is an adaptive mechanism which evolved to promote cell survival during infection, injury, or oxygen limitation (Walmsley et al., 2014). However, chronic and/or systemic hypoxia can produce maladaptive inflammation which can contribute to disease development. For example, in a clinical context, the crosstalk between hypoxia and inflammation may contribute to several inflammation-mediated metabolic and cardiovascular comorbidities that accompany hypoxia-promoted diseases such as chronic obstructive pulmonary disease or obstructive sleep apnea (Tasali and Ip, 2008; Quercioli et al., 2010; Cavaillès et al., 2013). This can also be investigated in the context of high-altitude exposure, where inflammatory signaling pathways and immune function must respond and adapt to acute, chronic, or lifelong hypoxemia. Here, we will examine our current understanding of how the interaction between hypoxia and inflammation contributes to high-altitude illness and what we might learn about the adaptive regulation of hypoxia-induced inflammation from high-altitude native populations (Figure 2).

Figure 2. Predicted contributions of inflammation to high-altitude illnesses, erythrocytosis, and immune function. Hypoxic conditions lead to increases in pro-inflammatory mediators, which may play a role in the development of high-altitude illnesses [(Acute Mountain Sickness (AMS), high-altitude pulmonary hypertension (HAPH), high-altitude pulmonary edema (HAPE), and high-altitude cerebral edema (HACE)] and erythrocytosis, or modify immune function. (1) Julian et al. (2011); (2) Liu et al. (2017); (3) Wang et al. (2018); (4) Varatharaj and Galea (2017); (5) Wilkins et al. (2015); (6) Brito et al. (2020); (7) Mishra et al. (2016); (8) Song et al. (2016); (9) Zhou et al. (2017); (10) Jackson et al. (2010); (11) Liao et al. (2018); (12) Bennett et al. (2019); (13) Imagawa et al. (1990); (14) Haase (2013); (15) Kvamme et al. (2013); (16) Baze et al. (2011); (17) Facco et al. (2005); and (18) Feuerecker et al. (2019).
High-altitude illnesses are a common risk for sojourners to high altitude (>2,500 m elevation). Within the first week of exposure, sojourners often present with Acute Mountain Sickness (AMS), which is characterized by headache, nausea, fatigue, and gastrointestinal issues (Roach et al., 2018). AMS typically resolves with acclimatization (Gallagher and Hackett, 2004). However, in serious cases, sojourners may develop severe and potentially fatal illnesses, such as high-altitude pulmonary edema (HAPE), high-altitude pulmonary hypertension (HAPH), or high-altitude cerebral edema (HACE; Gallagher and Hackett, 2004; Mehta et al., 2008; Luks et al., 2017). The incidence and severity of HAPH, HAPE and HACE vary depending on multiple factors, such as ascent time, altitude, and timely recognition and treatment (Gallagher and Hackett, 2004). Despite several decades of examining the physiology of AMS, HAPH, HAPE, and HACE, some questions remain regarding the pathophysiology of these conditions and the extent to which inflammation contributes to their onset and progression.
AMS development results from a complex network of physiological responses to hypoxia (i.e., inflammation, vasogenic edema, and acidosis) as well as anatomical factors (i.e., insufficient cerebrospinal fluid production, varied cerebral venous blood flow; West, 2004; Luks et al., 2017). While there is substantial research on potential contributors to AMS (reviewed in Imray et al., 2010; Luks et al., 2017), the exact biological pathways and molecular mechanisms behind AMS development remain unclear.
Recent studies support a potential role of inflammation in AMS. There is a general consensus that pro-inflammatory cytokines and other inflammatory markers (most notably C-Reactive protein (CRP), IL-1β, and IL-6) are increased in individuals acutely exposed to hypoxia or high altitude (Hartmann et al., 2000; Song et al., 2016; Lundeberg et al., 2018; Wang et al., 2018; Malacrida et al., 2019; Kammerer et al., 2020). In some cases, inflammatory mediator expression appears to differ across individuals who develop AMS and those who do not. For example, individuals who develop AMS have been reported to show decreased plasma levels of IL-10 and increased IL-1β, IL-6, and TNF-α compared to non-AMS controls (Liu et al., 2017; Wang et al., 2018). Furthermore, the association between plasma IL-6 and AMS score has been identified by multiple independent groups (Boos et al., 2016; Wang et al., 2018).
While these studies indicate that some circulating pro-inflammatory markers may be associated with AMS development, it is not yet clear what specific role they may play. It is possible that hypoxia initiates the release of inflammatory and angiogenic mediators which disrupt the blood-brain barrier and promote vasogenic edema. A complementary hypothesis is that protection from AMS may be driven by a robust anti-inflammatory response which protects against potential increased blood-brain barrier permeability caused by acute systemic inflammation (Varatharaj and Galea, 2017). This is supported by data from Julian et al. (2011), who found higher levels of anti-inflammatory marker (IL-1RA, HSP70, and adrenomedullin) expression in AMS-resistant compared to AMS-susceptible individuals (Julian et al., 2011).
Finally, both steroids and nonsteroidal anti-inflammatory drugs (NSAIDS) equally reduce AMS incidence despite their different modes of action (Dumont et al., 2000; Gertsch et al., 2012; Zheng et al., 2014). This suggests that both COX-2 mediated inflammation as well as analgesic mechanisms that mediate nociception contribute to AMS symptomology (Hartmann et al., 2000; Song et al., 2016; Kanaan et al., 2017). Indeed, inflammatory mediators released during tissue injury can activate nociceptors and can contribute to pain hypersensitivity (Kidd and Urban, 2001). However, Lundeberg et al. (2018) note no change in AMS symptoms in individuals receiving Ibuprofen at normal recommended doses of 400mg three times a day (Lundeberg et al., 2018), perhaps, due to the lower dosage compared to other studies which did observe a reduction in AMS symptoms with a higher Ibuprofen dose (600 mg three times a day; Gertsch et al., 2010, 2012; Lipman et al., 2012).
HACE is a severe and potentially fatal complication that can occur in individuals who travel above 2000 m. HACE is accompanied by symptoms including headache, ataxia, declines in cognitive function, and can lead to seizures (Hackett, 1999). As with AMS, HACE is most common with rapid ascent. HACE is frequently preceded by some AMS symptoms, and both illnesses are influenced by cerebral hemodynamics. As a result, HACE is sometimes considered a more severe form of AMS, however, additional distinct pathophysiological mechanisms contribute to HACE development (Hackett and Roach, 2004; Brugniaux et al., 2007; Li et al., 2018). HACE can develop spontaneously at very high altitudes in acclimatized individuals (Clarke, 1988). HACE appears to occur as a result of hypoxia-mediated cerebral vasodilation and a subsequent impairment of the autoregulation of cerebral blood flow, loss of blood-brain barrier integrity, and a rise in intracranial pressure (Hackett, 2000). Ultimately, cytotoxic and vasogenic edema leads to microvascular disruption and microbleeds (Hackett et al., 2019).
While some aspects of HACE pathophysiology remain uncertain, the underlying mechanisms producing HACE are likely similar to those of AMS and inflammation may play a role in its development (Bailey et al., 2009; Song et al., 2016; Zhou et al., 2017). In both mouse and rat models, pro-inflammatory cytokines are significantly increased in the brain cortex after hypoxia exposure. Furthermore, when pre-treated with bacterial LPS to produce a systemic inflammatory response, subsequent hypoxia exposure results in cerebral edema (Song et al., 2016; Zhou et al., 2017). It is proposed that pre-existing inflammation increases aquaporin 4 (AQP4) activity in astrocytes via toll-like receptor 4 (TLR4), mitogen-activated protein kinase (MAPK), and NF-κB signaling, thereby increasing astrocyte permeability. Together, these data indicate that when challenged with a combination of hypoxia and inflammation, the combination of increased pro-inflammatory cytokines and increased blood vessel permeability produces vasogenic edema (Song et al., 2016; Zhou et al., 2017).
Although this data provide intriguing support for a role of hypoxia-induced inflammation and HIF-NF-kB crosstalk in HACE onset, it remains unclear if inflammation is a proximal cause of AMS or HACE. Nonetheless, inflammation likely plays a contributing role in susceptibility and progression of these illnesses. Further work is required to reach a unified explanation for HACE development.
HAPH occurs due to general hypoxic pulmonary vasoconstriction (HPV). Under typical conditions, local HPV aids in redistributing blood flow away from lung regions with poor ventilation and improves pulmonary gas exchange. However, at high altitude, global reductions in alveolar oxygen pressure can lead to HPV throughout the lung and increase mean pulmonary artery pressures (Swenson, 2013). HAPH is estimated to impact up to 10% of high-altitude residents (León-Velarde et al., 2005). The clinical presentation of HAPH includes dyspnea, general fatigue, exercise intolerance, and in severe cases, chest pain and mental alterations, and ultimately cor pulmonale.
While acute HAPH is primarily driven by increased vasomotor tone, chronic hypoxic stress and persistent pulmonary hypertension can produce vascular remodeling and exacerbate pulmonary artery pressures (Groves et al., 1987; Wilkins et al., 2015). The hallmark of vascular remodeling in chronic hypoxia is increased vessel muscularization (Arias-Stella and Saldana, 1963; Hislop and Reid, 1976). It is predicted that inflammation plays an important role in this remodeling process. Chronic hypoxia produces a proinflammatory microenvironment in pulmonary artery walls (Burke et al., 2009). Resident fibroblasts, immune cells, and progenitor cells in the vascular adventitia respond to this local cellular stress by releasing additional inflammatory mediators and growth factors which impact vascular wall cell phenotypes and contribute to increased muscularization (Stenmark et al., 2013). This inflammatory microenvironment also promotes recruitment, retention, and differentiation of additional inflammatory cells (Burke et al., 2009).
HAPE occurs most commonly during rapid ascent. This is due to exaggerated HPV which causes acute pulmonary hypertension, increased capillary permeability, and alveolar fluid buildup (Talbot et al., 2005; Dunham-Snary et al., 2017; Brito et al., 2020; Swenson, 2020; Sydykov et al., 2021). HAPE onset is primarily due to this non-inflammatory hydrostatic pulmonary edema (Swenson et al., 2002). However, inflammation may play a downstream role in HAPE pathology. Several studies report increased inflammatory mediators and chemokines in bronchoalveolar fluid in later stages of the illness including leukotriene B4 and complement fragments (C5a; Schoene et al., 1986, 1988), plasma endothelin-1 (ET-1; Droma et al., 1996), lactate dehydrogenase, IL-1β, IL-6, IL-8, TNF-α, and IL-1RA (Kubo et al., 1996, 1998). Several of these studies also report increased leukocyte counts in bronchoalveolar lavage fluid. This late-stage inflammation could exacerbate pulmonary vascular leakage by causing endothelial damage and increasing capillary permeability (Mishra et al., 2016).
Some studies have also suggested that inflammation contributes to HAPE susceptibility. Individuals with a history of HAPE are more susceptible to developing HAPE again upon re-entry to high altitude (Lakshminarayan and Pierson, 1975; Bärtsch, 1999; Gallagher and Hackett, 2004). This increased susceptibility is attributed to higher baseline inflammation (Mishra et al., 2016). Furthermore, one report suggests that HAPE-susceptible individuals also demonstrate reduced lung function compared to HAPE-resistant individuals (Gupta et al., 2017), which would limit the adaptive compensatory ventilatory response to hypoxia. The reduced lung function in these individuals is further correlated with increased plasma CRP (Shaaban et al., 2006; Hancox et al., 2007). This association between chronic inflammation and poor lung function is hypothesized to contribute to HAPE-susceptibility, although it does not explain the direct cause of onset. Additionally, pre-existing pulmonary vascular remodeling, driven partially by inflammation, may exacerbate pulmonary pressures and blood flow distribution and thereby increase HAPE susceptibility (Wilkins et al., 2015).
Erythropoiesis is tightly regulated to maintain homeostatic balance between red blood cell production and degradation. This balance is crucial for optimal oxygen delivery to tissues. An inadequate number of erythrocytes lead to tissue hypoxia, while high erythrocyte concentrations can increase blood viscosity, impair blood flow, and increase risk of thrombosis (Keohane et al., 2013). This is particularly relevant in Chronic Mountain Sickness (CMS), a clinical syndrome commonly presented in high-altitude natives and life-long residents, which frequently coincides with excessive erythrocytosis (Hb ≥ 19 g/dL for women, Hb ≥ 21 g/dL for men) in some high altitude groups (León-Velarde et al., 2005; Oberholzer et al., 2020). The HIF pathway plays a key role in regulating erythropoiesis. HIF activation results in increased transcription of the EPO gene in the kidney and liver, and EPO serum concentration can increase up to several 100-fold in response (Imagawa et al., 1990; Haase, 2013). Additionally, HIF activity plays a role in regulating iron uptake from the gut and mobilizing erythroid progenitor cells in the bone marrow (Nemeth, 2008).
Chronic inflammation typically leads to anemia through several mechanisms: prioritizing myeloid cell production, sequestering iron, and increasing erythroid turnover rate. Inflammation impacts crucial sites of erythrocyte production to redistribute resources toward myeloid cell production and therefore reduces lymphoid and erythroid output. For example, expression of pro-inflammatory IL-1β, interferon gamma (IFNγ), and IL-6 skew multipotential hematopoietic progenitors toward myeloid cell development (Tie et al., 2019). This is an adaptive response since myeloid cells are needed to fight infection; however, this comes at the cost of bone marrow erythropoiesis. Additionally, while iron is an essential component of hemoglobin synthesis, it is also an essential micronutrient to pathogens. During inflammation, increased IL-6 induces hepcidin production, interrupting iron absorption from the gut and blocking iron release from macrophages, leading to hypoferremia (Nemeth et al., 2004; Nemeth, 2008; Zaritsky et al., 2009; Weiss et al., 2019). By decreasing iron availability, the immune system limits pathogen proliferation; however, a consequence of iron sequestration is that this also limits iron availability for red blood cell production.
When steady state erythropoiesis becomes insufficient, or red blood cells are broken down at high rates, a compensatory extramedullary mechanism called stress erythropoiesis is initiated to prevent lethal anemia (Paulson et al., 2011). Stress erythrocytosis produces a burst of new erythrocytes to maintain red cell concentrations until bone marrow erythropoiesis recovers. The mechanism of stress erythropoiesis has been studied extensively in mice, where it occurs in the spleen and liver in response to anemia, hypoxia, or sterile inflammation. Studies in mice revealed that sterile inflammation produced by phenylhydrazine injections lead to decreased erythroid progenitor cells in the bone marrow and increased erythroid progenitor cells in the spleen. It was discovered that bone marrow erythroid progenitor cells and splenic erythroid progenitor cells respond to different factors, indicating that splenic progenitors are distinct from bone marrow progenitors. This splenic stress erythropoiesis response depends on TLR signaling molecules Myd88 and TRIF (Jackson et al., 2010). Furthermore, pro-inflammatory cytokines TNF-α and IL-1β promote the expansion of splenic erythroid progenitors (Liao et al., 2018; Bennett et al., 2019). It is currently unknown if similar mechanisms are responsible for stress erythropoiesis in humans (Kim et al., 2015; Mairbäurl, 2018). Future studies may investigate biomarkers of stress erythropoiesis signaling to determine if this protective mechanism is active during high-altitude exposure or other forms of hypoxic stress.
Immune cells are exposed to hypoxia when they are recruited to sites of inflammation. In physiological immunological niches (bone marrow, placenta, gastrointestinal tract mucosal surfaces, and lymph nodes), the maintenance of sustained and moderate physiological hypoxia is an adaptive mechanism to regulate metabolic pathways and immune homeostasis. However, in pathological immunological niches (tumors and chronically inflamed and ischemic tissue), severe and unregulated hypoxia can lead to maladaptive inflammation and disease development (Taylor and Colgan, 2017). In either case, the immune cell response to hypoxia hinges upon the ability to mount a transcriptional response. Therefore, HIF activity is essential to immune cell survival and function.
Several studies demonstrate the significance of HIF signaling in immune function. HIF-1α deletion in myeloid cells (granulocytes and monocytes/macrophages) impairs their mobility, aggregation, antibacterial activity, and survival (Cramer et al., 2003; Walmsley et al., 2005). On an organismal level, HIF-1α deletion in macrophages can reduce mortality in LPS-induced sepsis in mice (Peyssonnaux et al., 2007). HIF-2α can also directly regulate pro-inflammatory cytokine expression in myeloid cells (Imtiyaz et al., 2010). In addition to HIF’s essential role in immune cell function, NF-kB is also critical to cell survival in hypoxia. Walmsley et al. (2005) demonstrated that the transcription of the NF-κB p65 subunit is regulated by hypoxia. Neutrophils cultured in hypoxia and treated with NF-κB inhibitors had reduced survival rates, suggesting that activation of the NF-κB pathway promotes neutrophil survival in hypoxia (Walmsley et al., 2005).
Despite this important interplay between hypoxia and inflammation in immune function, we know little about how inflammatory signaling and immune function adapt to chronic sustained or chronic intermittent hypoxia. Studies in animal models demonstrate potential blunting or sensitization of inflammatory responses to infection as a result of chronic hypoxia exposure. A study in salmon exposed to chronic hypoxia for 58 days found blunted expression of pro-inflammatory genes in macrophages and the head kidney in response to a viral inflammatory stimulus (Kvamme et al., 2013). Alternatively, in a mouse model, 36 days of hypoxia exposure lead to an enhanced immunological response to LPS illustrated by higher antibody titers and higher TNF-α expression, indicating that hypoxia may stimulate innate and adaptive immune responses (Baze et al., 2011).
Among studies in humans, chronic hypoxia also appears to modulate immune function and inflammatory signaling. A study of women exposed to high altitude (5,050 m) for 21 days showed increased white blood cells, reductions in CD3+ and CD4+ T-cells, an increase in natural killer cells, and a decrease in IFNγ expression by circulating T-cells (Facco et al., 2005). Another recent study in humans exposed to high altitude (3,232 m) for up to 11 months shows that cytokine expression in response to inflammatory stimuli were also higher than sea-level values (Feuerecker et al., 2019). The possibility that chronic hypoxia exposure in humans leads to immune sensitization and hypersensitivity to inflammatory stimuli warrants future study. Since several critical and chronic illnesses are associated with hypoxemia (e.g., sepsis, acute respiratory distress syndrome, chronic obstructive pulmonary disease, and sleep apnea), it is possible that inflammatory dysregulation in these conditions may be caused in part by this concurrent hypoxic stress.
High altitude environments are physiologically stressful due to low atmospheric pressure and oxygen limitation. Despite this, humans have survived in these environments for thousands of years and different high-altitude native populations exhibit distinct physiological adaptations that may be associated with genetic variants. In this section, we discuss what can be learned from high-altitude acclimatized and adapted populations about how inflammatory pathways respond to chronic hypoxia.
Several studies have identified genes under evolutionary selection in high-altitude native populations. HIF pathway genes, particularly EGLN1 and EPAS1, are consistently highlighted across studies. Additionally, several genes associated with inflammation also show signals of selection but have received less attention (Foll et al., 2014). The inflammatory pathway genes IL6, IL1A, IL1B, NOS1, NOS2, and TNF show signals of evolutionary selection in both Andean and Tibetan high-altitude native populations (Beall, 2007; Bigham and Lee, 2014; Moore, 2017). NOS1 and NOS3 have also been shown to be under positive selection in the Sherpa population (Droma et al., 2006; Zhang et al., 2017). Additional inflammation-related genes have been reported to be under selection in individual groups, including HLA-DQB1, PPARA, and TGFBR3 in Tibetans (Yang et al., 2017), PPARA in Sherpas (Horscroft et al., 2017), BRINP3, DUOX2, and CLC in Andeans (Crawford et al., 2017; Jacovas et al., 2018), and AIMP1 in Ethiopians (Scheinfeldt et al., 2012). Table 1 summarizes inflammation-related genes found to be under evolutionary selection in high altitude human populations.
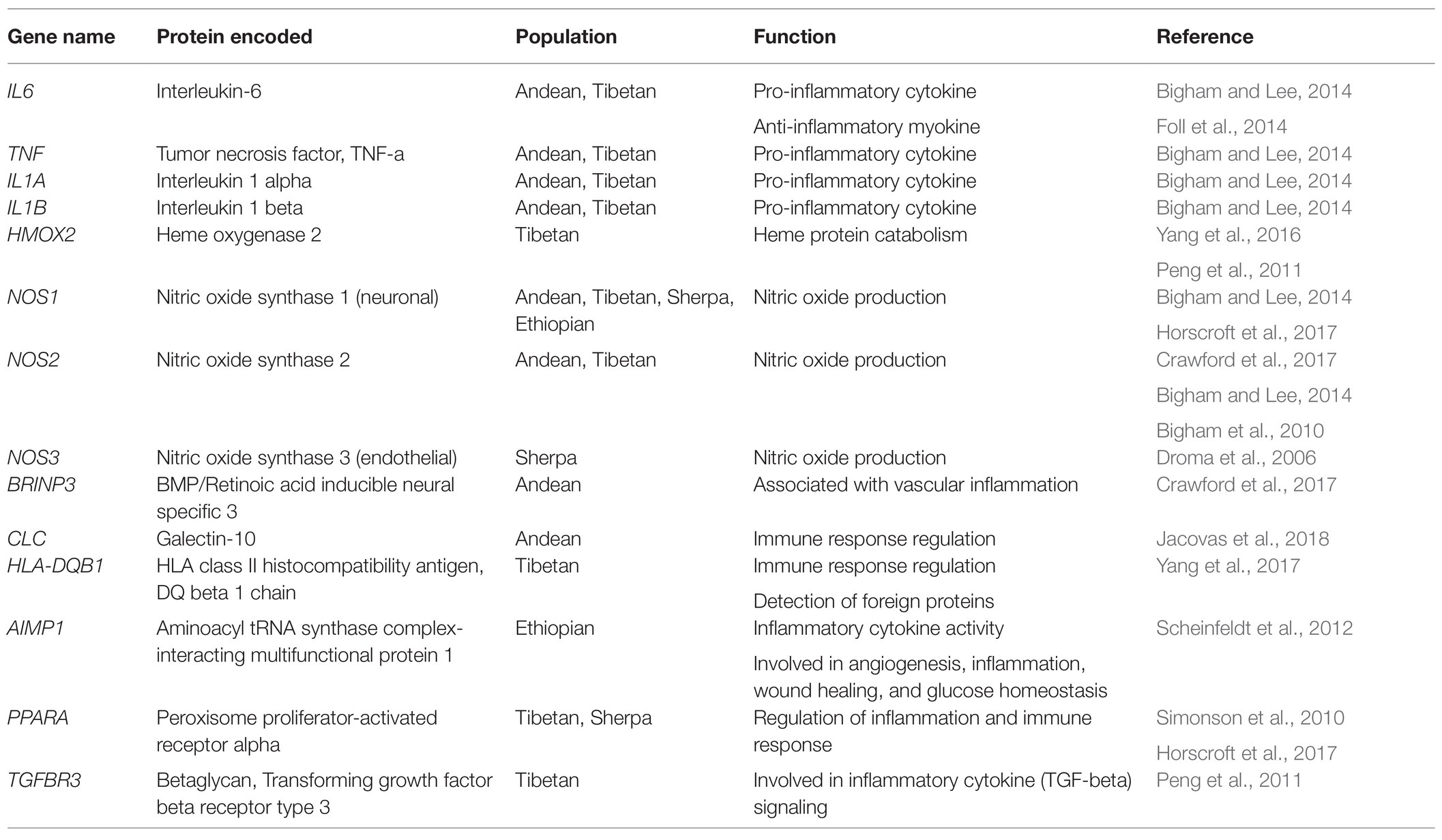
Table 1. Inflammation-related genes identified as top candidates showing signals of evolutionary selection in high-altitude human populations.
IL6 is particularly interesting given that its expression has been associated with AMS development. IL-6 is also noted to increase hematopoietic stem cell proliferation under hypoxic conditions, and its effects on red cell production are synergistic with other pro-inflammatory cytokines, including IL-1α and TNF-α (Faquin et al., 1992). Together, this data suggest that IL-6 plays a role in adaptation to chronic hypoxia. It is currently unknown what the precise adaptive IL6 variants are, but it is possible that blunting IL-6 expression in response to chronic hypoxia or chronic intermittent hypoxia may provide an advantage by preventing hypoxia-induced chronic low-grade systemic inflammation. Another important inflammatory gene under selection in both Tibetan and Andean groups is TNF (Moore, 2017), a multifunctional pro-inflammatory cytokine. TNF-α levels have also been found to be elevated in individuals traveling acutely to high altitude (Lundeberg et al., 2018). Like IL6, the adaptive TNF variant is unknown but modulation of TNF expression in response to chronic hypoxia may be important for preventing chronic inflammation at high altitude.
In addition to these genetic changes, epigenetic mechanisms also play a critical role in regulating expression of inflammatory genes and likely influence adaptation of inflammatory pathways to chronic hypoxia (Bayarsaihan, 2011). Epigenetic changes alter gene expression without altering the underlying genetic code. These changes include DNA methylation, histone modifications, and microRNA expression. These epigenetic changes play key roles in environmental adaptation during development and exposure in adulthood (Fernández et al., 2014). There are several HIF-dependent and independent mechanisms by which hypoxia significantly impacts epigenetic modifications (Kim and Park, 2020). For example, HIF induces expression of several histone methyltransferases and demethylases (Batie et al., 2019). Additionally, hydroxylation of histone methyl transferases as well as activity of histone demethylases, are oxygen dependent. DNA methylation patterns are also altered by hypoxia, in part due to the impact on ten-eleven translocation (TET) enzyme expression and activity (Thienpont et al., 2016). The HIF consensus binding site also contains a CpG dinucleotide, indicating that expression of HIF-pathway genes may be dependent on DNA methylation (Wenger et al., 2005; Chen et al., 2020; D’Anna et al., 2020). Given that immune cell phenotypes, including inflammatory macrophage phenotypes, can be regulated by epigenetic modifications, hypoxia has the potential to significantly alter immune cell function via its impact on epigenetics (Davis and Gallagher, 2019). These changes may occur within or across generations of exposure. Future work should investigate if high-altitude acclimatized or adapted groups develop particular patterns of DNA methylation or histone modification, which protect against chronic hypoxia-induced inflammation. Over generations, mutations at loci containing CpG sites could also assist in blunting inflammatory gene expression in the face of chronic hypoxic stress.
Nitric oxide (NO) is a natural vasodilator that plays a crucial role in regulating vasodilation in vascular smooth muscle. Exhaled NO is also used as marker of airway inflammation (Kharitonov and Barnes, 2001; Birrell et al., 2006). The vasodilatory function of NO protects against pulmonary hypertension at high altitude (Feelisch, 2018). Individuals exposed acutely to high altitude tend to show reduced gas phase NO, which typically returns to baseline levels after a couple days then exceeds baseline levels by 5 days. However, gas phase NO has been demonstrated to remain low in HAPE-sensitive individuals, perhaps, indicating that deficits in pulmonary epithelial NO synthesis contribute to exaggerated pulmonary vasoconstriction and edema (Duplain et al., 2000). Tibetan and Bolivian high-altitude adapted populations also show elevated exhaled NO compared to sea-level residents (Beall et al., 2001, 2012; Erzurum et al., 2007). This phenotype correlates with higher pulmonary blood flow and protection from HAPH. However, He et al. (2017) demonstrate that enhanced NO production may not be unique to high-altitude native populations since individuals of Han Chinese ancestry living at high altitude (3,660–3,700 m) demonstrated an even higher average NO metabolite production than Tibetans at the same altitude (He et al., 2017). Another study reports that the native Sherpa population has a lower level of circulating NO in serum and no differences in circulating NO metabolites in comparison to low-altitude samples (Droma et al., 2006; Horscroft et al., 2017). Interestingly, this finding contrasts the high exhaled NO among Tibetans. This discrepancy in NO substrate bioavailability in Tibetans vs. Sherpas also suggests that these two high-altitude populations may have some distinct adaptation in the nitric oxide pathway (Zhang et al., 2017).
Elevated NO may also be an important adaptation of the immune system to chronic hypoxia-induced inflammation. Under normal physiological conditions, NO plays an anti-inflammatory role and inhibits platelet aggregation and rolling, adherence, and transmigration of leukocytes (Grisham et al., 1998; Coleman, 2001). During inflammatory reactions, the production of the inducible form of nitric oxide synthase in many immune cells, including monocytes, macrophages, and neutrophils leads to very large increases in NO by up to 1,000-fold (Cook and Cattell, 1996; Sharma et al., 2007). NO can then become oxidized to reactive nitrogen oxide species which nitrosate thiol groups in glutathione. This inhibits the activity of many proteins including mitochondrial enzymes and transcription factors. As a result, elevated NO production can impair pathogen function (Sethi and Dikshit, 2000; Bogdan, 2001; Coleman, 2001). At high concentrations, NO also stabilizes HIF by inhibiting HIF PHDs (Taylor and Moncada, 2010). Therefore, NO production during periods of inflammation permit increased expression of HIF-pathway genes as a protective mechanism since tissue inflammation often leads to cellular hypoxia. However, Lüneburg et al. (2016) demonstrated in a rat model that while there is an increase in endothelial nitric oxide synthase (eNOS; NOS3) in chronic hypoxia and chronic intermittent hypoxia conditions, NO bioavailability may be impaired due to increases in asymmetric dimethylarginine (ADMA), a competitive nitric oxide synthase inhibitor that regulates NO production, and hypoxia-induced increase in oxidative stress (Lüneburg et al., 2016). ADMA displaces L-arginine, a NOS substrate, and therefore an increase in ADMA concentrations may result in lower NO substrate bioavailability (Brito et al., 2007). Oxidative stress may lead to superoxide radical production, which may mediate NO degradation and attenuate NO bioavailability (Siques et al., 2014).
Since both NOS1 (nitric oxide synthase 1, neuronal) and NOS2 (nitric oxide synthase 2, inducible) are under selection in multiple high-altitude groups, it is clear that adjusting NO production is a key adaptive phenotype for life at high altitude. While the precise adaptive mechanism is unknown, it likely involves protection against pulmonary hypertension and/or modulation of hypoxia-induced inflammation.
Carbon monoxide (CO) is emerging as a potential therapeutic target for inflammatory modulation due to its anti-inflammatory, anti-apoptotic, and anti-proliferative effects (Knauert et al., 2013; Ryter and Choi, 2016). CO is also used as a clinical marker for inflammation and red blood cell turnover since endogenous CO is only produced through the heme oxygenase (HO) pathway (Maines, 1997). HO is responsible for catabolizing free heme proteins into Fe2+, CO, and biliverdin. These end products provide cytoprotective benefits and protect cells from programmed cell death in response to pro-inflammatory agonists (Gozzelino et al., 2010). The HO-CO pathway may be a key regulator of hypoxia-induced inflammation since the gene encoding heme oxygenase-1 (HMOX1) is upregulated in response to HIF-pathway activation and HO-1 can inhibit NF-κB (Seldon et al., 2007).
High altitude exposure has been shown to increased exhaled CO levels, and carboxyhemoglobin levels are positively associated with hematocrit in Andean high-altitude residents (Tift et al., 2020). This is suspected to be caused by increased red blood cell turnover, a persistent stress erythropoiesis response, or a unique HO/CO pathway mechanism. Interestingly, the gene encoding the constitutively expressed heme oxygenase-2 (HMOX2) is under evolutionary selection in Tibetan high-altitude native groups (Yang et al., 2016). The adaptive HMOX2 variants in Tibetans are predicted to play a critical role in regulating red blood cell turnover and potentially contribute to the low hemoglobin concentrations in this group by increasing heme oxygenase activity. Due to the key role that the HO-CO pathway plays in linking the molecular responses to hypoxia and inflammation, as well as evidence that HO is a top candidate for evolutionary selection in high-altitude populations, future research should continue to investigate potential therapeutic uses of exogenous CO and HO pathway activation for modulating inflammatory responses, especially in hypoxia-promoted pathologies.
HIF-NF-κB crosstalk plays an essential role in the transcriptional response to both hypoxia and inflammation. However, additional research is necessary to understand the physiological implications of these pathway interactions at an organismal level. Several studies have demonstrated that select inflammatory mediators are upregulated at high altitude, or in the presence of acute hypoxia, even in the absence of infection. Since these studies have focused on candidate inflammatory markers, large scale transcriptomic or proteomic studies would provide a better understanding of how inflammatory networks are shifted during chronic hypoxia. Further, our understanding of the time domains of hypoxia-induced inflammation and the impact on immune function may also provide insight into pathology of high-altitude illnesses and highlight the importance of hypoxia-induced inflammatory sensitization in critical and chronic illnesses including chronic obstructive pulmonary disorder, sepsis, and COVID-19. This work may identify novel therapeutic targets for mitigating excessive inflammation in patients with concomitant hypoxemia and systemic inflammation.
KaP drafted versions of the manuscript with input and revisions from KeP and EH. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Arias-Stella, J., and Saldana, M. (1963). The terminal portion of the pulmonary arterial tree in people native to high altitudes. Circulation 28, 915–925. doi: 10.1161/01.CIR.28.5.915
Bailey, D. M., Bärtsch, P., Knauth, M., and Baumgartner, R. W. (2009). Emerging concepts in acute mountain sickness and high-altitude cerebral edema: From the molecular to the morphological. Cell. Mol. Life Sci. 66, 3583–3594. doi: 10.1007/s00018-009-0145-9
Batie, M., Frost, J., Frost, M., Wilson, J. W., Schofield, P., and Rocha, S. (2019). Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 363, 1222–1226. doi: 10.1126/science.aau5870
Bayarsaihan, D. (2011). Epigenetic mechanisms in inflammation. J. Dent. Res. 90, 9–17. doi: 10.1177/0022034510378683
Baze, M. M., Hunter, K., and Hayes, J. P. (2011). Chronic hypoxia stimulates an enhanced response to immune challenge without evidence of an energetic tradeoff. J. Exp. Biol. 214, 3255–3268. doi: 10.1242/jeb.054544
Beall, C. (2007). Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc. Natl. Acad. Sci. U. S. A. 104, 8655–8660. doi: 10.1073/pnas.0701985104
Beall, C. M., Laskowski, D., and Erzurum, S. C. (2012). Nitric oxide in adaptation to altitude. Free Radic. Biol. Med. 52, 1123–1134. doi: 10.1016/j.freeradbiomed.2011.12.028
Beall, C. M., Laskowski, D., Strohl, K. P., Soria, R., Villena, M., Vargas, E., et al. (2001). Pulmonary nitric oxide in mountain dwellers. Nature 414, 411–412. doi: 10.1038/35106641
Beg, A. A., and Jr, A. S. B. (1993). The IKB proteins: multifunctional regulators of Rel/NF-KB transcription factors. Genes Dev. 7, 2064–2070. doi: 10.1101/gad.7.11.2064
BelAiba, R. S., Bonello, S., Zähringer, C., Schmidt, S., Hess, J., Kietzmann, T., et al. (2007). Hypoxia up-regulates hypoxia-inducible factor-1α transcription by involving phosphatidylinositol 3-kinase and nuclear factor κB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 18, 4691–4697. doi: 10.1091/mbc.E07-04-0391
Bennett, L. F., Liao, C., Quickel, M. D., Yeoh, B. S., Vijay-Kumar, M., Hankey-Giblin, P., et al. (2019). Inflammation induces stress erythropoiesis through heme-dependent activation of SPI-C. Sci. Signal. 12, 1–14. doi: 10.1126/scisignal.aap7336
Biddlestone, J., Bandarra, D., and Rocha, S. (2015). The role of hypoxia in inflammatory disease (review). Int. J. Mol. Med. 35, 859–869. doi: 10.3892/ijmm.2015.2079
Bigham, A., Bauchet, M., Pinto, D., Mao, X., Akey, J. M., Mei, R., et al. (2010). Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 6:e1001116. doi: 10.1371/journal.pgen.1001116
Bigham, A. W., and Lee, F. S. (2014). Human high-altitude adaptation: forward genetics meets the HIF pathway. Genes Dev. 28, 2189–2204. doi: 10.1101/gad.250167.114
Birrell, M. A., McCluskie, K., Hardaker, E., Knowles, R., and Belvisi, M. G. (2006). Utility of exhaled nitric oxide as a noninvasive biomarker of lung inflammation in a disease model. Eur. Respir. J. 28, 1236–1244. doi: 10.1183/09031936.00048506
Bogdan, C. (2001). Nitric oxide and the immune response. Nat. Immunol. 2, 907–916. doi: 10.1038/ni1001-907
Bonello, S., Zähringer, C., BelAiba, R. S., Djordjevic, T., Hess, J., Michiels, C., et al. (2007). Reactive oxygen species activate the HIF-1α promoter via a functional NFκB site. Arterioscler. Thromb. Vasc. Biol. 27, 755–761. doi: 10.1161/01.ATV.0000258979.92828.bc
Boos, C. J., Woods, D. R., Varias, A., Biscocho, S., Heseltine, P., and Mellor, A. J. (2016). High altitude and acute mountain sickness and changes in circulating endothelin-1, interleukin-6, and interleukin-17a. High Alt. Med. Biol. 17, 25–31. doi: 10.1089/ham.2015.0098
Brill, S. E., and Wedzicha, J. A. (2014). Oxygen therapy in acute exacerbations of chronic obstructive pulmonary disease. Int. J. COPD 9, 1241–1252. doi: 10.2147/COPD.S41476
Brito, J., Siqués, P., León-Velarde, F., De La Cruz, J. J., López, V., and Herruzo, R. (2007). Chronic intermittent hypoxia at high altitude exposure for over 12 years: assessment of hematological, cardiovascular, and renal effects. High Alt. Med. Biol. 8, 236–244. doi: 10.1089/ham.2007.8310
Brito, J., Siques, P., and Pena, E. (2020). Long-term chronic intermittent hypoxia: a particular form of chronic high-altitude pulmonary hypertension. Pulm. Circ. 10, 5–12. doi: 10.1177/2045894020934625
Brugniaux, J. V., Hodges, A. N. H., Hanly, P. J., and Poulin, M. J. (2007). Cerebrovascular responses to altitude. Respir. Physiol. Neurobiol. 158, 212–223. doi: 10.1016/j.resp.2007.04.008
Burke, D. L., Frid, M. G., Kunrath, C. L., Karoor, V., Anwar, A., Wagner, B. D., et al. (2009). Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, 238–250. doi: 10.1152/ajplung.90591.2008
Carroll, V. A., and Ashcroft, M. (2006). Role of hypoxia-inducible factor (HIF)-1α versus HIF-2α in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res. 66, 6264–6270. doi: 10.1158/0008-5472.CAN-05-2519
Cavaillès, A., Brinchault-Rabin, G., Dixmier, A., Goupil, F., Gut-Gobert, C., Marchand-Adam, S., et al. (2013). Comorbidities of COPD. Eur. Respir. Rev. 22, 454–475. doi: 10.1183/09059180.00008612
Celli, B. R., MacNee, W., Agusti, A., Anzueto, A., Berg, B., Buist, A. S., et al. (2004). Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur. Respir. J. 23, 932–946. doi: 10.1183/09031936.04.00014304
Chen, S., Yang, J., Wei, Y., and Wei, X. (2020). Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell. Mol. Immunol. 17, 36–49. doi: 10.1038/s41423-019-0315-0
Clarke, C. (1988). High altitude cerebral oedema. Int. J. Sports Med. 9, 170–174. doi: 10.1055/s-2007-1024999
Coleman, J. W. (2001). Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 1, 1397–1406. doi: 10.1016/S1567-5769(01)00086-8
Cook, H. T., and Cattell, V. (1996). Role of nitric oxide in immune-mediated diseases. Clin. Sci. 91, 375–384. doi: 10.1042/cs0910375
Couzin-Frankel, J. (2020). The mystery of the pandemic’s ‘happy hypoxia.’. Science 368, 455–456. doi: 10.1126/science.368.6490.455
Cramer, T., Yamanishi, Y., Clausen, B. E., Förster, I., Pawlinski, R., Mackman, N., et al. (2003). HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112, 645–657. doi: 10.1016/S0092-8674(03)00154-5
Crawford, J. E., Amaru, R., Song, J., Julian, C. G., Racimo, F., Cheng, J. Y., et al. (2017). Natural selection on genes related to cardiovascular health in high-altitude adapted andeans. Am. J. Hum. Genet. 101, 752–767. doi: 10.1016/j.ajhg.2017.09.023
Cummins, E. P., Berra, E., Comerford, K. M., Ginouves, A., Fitzgerald, K. T., Seeballuck, F., et al. (2006). Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc. Natl. Acad. Sci. U. S. A. 103, 18154–18159. doi: 10.1073/pnas.0602235103
D’Anna, F., Van Dyck, L., Xiong, J., Zhao, H., Berrens, R. V., Qian, J., et al. (2020). DNA methylation repels binding of hypoxia-inducible transcription factors to maintain tumor immunotolerance. Genome Biol. 21:182. doi: 10.1186/s13059-020-02087-z
Davis, F. M., and Gallagher, K. A. (2019). Epigenetic mechanisms in monocytes/macrophages regulate inflammation in cardiometabolic and vascular disease. Arterioscler. Thromb. Vasc. Biol. 39, 623–634. doi: 10.1161/ATVBAHA.118.312135
Dengler, V. L., Galbraith, M. D., and Espinosa, J. M. (2014). Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 49, 1–15. doi: 10.3109/10409238.2013.838205
Droma, Y., Hanaoka, M., Basnyat, B., Arjyal, A., Neupane, P., Pandit, A., et al. (2006). Genetic contribution of the endothelial nitric oxide synthase gene to high altitude adaptation in Sherpas. High Alt. Med. Biol. 7, 209–220. doi: 10.1089/ham.2006.7.209
Droma, Y., Hayano, T., Takabayashi, Y., Koizumi, T., Kubo, K., Kobayashi, T., et al. (1996). Endothelin-1 and interleukin-8 in high altitude pulmonary oedema. Eur. Respir. J. 9, 1947–1949. doi: 10.1183/09031936.96.09091947
Dumont, L., Mardirosoff, C., and Tramèr, M. R. (2000). Efficacy and harm of pharmacological prevention of acute mountain sickness: quantitative systematic review. Br. Med. J. 321, 267–272. doi: 10.1136/bmj.321.7256.267
Dunham-Snary, K. J., Wu, D., Sykes, E. A., Thakrar, A., Parlow, L. R. G., Mewburn, J. D., et al. (2017). Hypoxic pulmonary vasoconstriction: from molecular mechanisms to medicine. Chest 151, 181–192. doi: 10.1016/j.chest.2016.09.001
Duplain, H., Sartori, C., Lepori, M., Egli, M., Allemann, Y., Nicod, P., et al. (2000). Exhaled nitric oxide in high-altitude pulmonary edema: role in the regulation of pulmonary vascular tone and evidence for a role against inflammation. Am. J. Respir. Crit. Care Med. 162, 221–224. doi: 10.1164/ajrccm.162.1.9908039
Erzurum, S. C., Ghosh, S., Janocha, A. J., Xu, W., Bauer, S., Bryan, N. S., et al. (2007). Higher blood flow and circulating NO products offset high-altitude hypoxia among Tibetans. Proc. Natl. Acad. Sci. U. S. A. 104, 17593–17598. doi: 10.1073/pnas.0707462104
Facco, M., Zilli, C., Siviero, M., Ermolao, A., Travain, G., Baesso, I., et al. (2005). Modulation of immune response by the acute and chronic exposure to high altitude. Med. Sci. Sports Exerc. 37, 768–774. doi: 10.1249/01.MSS.0000162688.54089.CE
Faquin, W. C., Schneider, T. J., and Goldberg, M. A. (1992). Effect of inflammatory cytokines on hypoxia-induced erythropoietin production. Blood 79, 1987–1994.
Feelisch, M. (2018). Enhanced nitric oxide production is a universal response to hypoxic stress. Natl. Sci. Rev. 5, 532–533. doi: 10.1093/nsr
Fernández, A. F., Toraño, E. G., Urdinguio, R. G., Lana, A. G., Fernández, I. A., and Fraga, M. F. (2014). The epigenetic basis of adaptation and responses to environmental change: perspective on human reproduction. Adv. Exp. Med. Biol. 753, 97–117. doi: 10.1007/978-1-4939-0820-2_6
Feuerecker, M., Crucian, B. E., Quintens, R., Buchheim, J. I., Salam, A. P., Rybka, A., et al. (2019). Immune sensitization during 1 year in the Antarctic high-altitude concordia environment. Allergy Eur. J. Allergy Clin. Immunol. 74, 64–77. doi: 10.1111/all.13545
Figueroa, Y. G., Chan, A. K., Ibrahim, R., Tang, Y., Burow, M. E., Alam, J., et al. (2002). NF-κB plays a key role in hypoxia-inducible factor-1-regulated erythropoietin gene expression. Exp. Hematol. 30, 1419–1427. doi: 10.1016/S0301-472X(02)00934-7
Foll, M., Gaggiotti, O. E., Daub, J. T., Vatsiou, A., and Excoffier, L. (2014). Widespread signals of convergent adaptation to high altitude in Asia and America. Am. J. Hum. Genet. 95, 394–407. doi: 10.1016/j.ajhg.2014.09.002
Frede, S., Stockmann, C., Freitag, P., and Fandrey, J. (2006). Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-κB. Biochem. J. 396, 517–527. doi: 10.1042/BJ20051839
Gallagher, S. A., and Hackett, P. H. (2004). High-altitude illness. Emerg. Med. Clin. North Am. 22, 329–355. doi: 10.1016/j.emc.2004.02.001
Gertsch, J. H., Corbett, B., Holck, P. S., Mulcahy, A., Watts, M., Stillwagon, N. T., et al. (2012). Altitude sickness in climbers and efficacy of NSAIDs trial (ASCENT): randomized, controlled trial of ibuprofen versus placebo for prevention of altitude illness. Wilderness Environ. Med. 23, 307–315. doi: 10.1016/j.wem.2012.08.001
Gertsch, J. H., Lipman, G. S., Holck, P. S., Merritt, A., Mulcahy, A., Fisher, R. S., et al. (2010). Prospective, double-blind, randomized, placebo-controlled comparison of acetazolamide versus ibuprofen for prophylaxis against high altitude headache: the headache evaluation at altitude trial (HEAT). Wilderness Environ. Med. 21, 236–243. doi: 10.1016/j.wem.2010.06.009
Görlach, A., and Bonello, S. (2008). The cross-talk between NF-kappaB and HIF-1: further evidence for a significant liaison. Biochem. J. 412, 17–19. doi: 10.1042/bj20080920
Gozzelino, R., Jeney, V., and Soares, M. P. (2010). Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50, 323–354. doi: 10.1146/annurev.pharmtox.010909.105600
Grisham, M. B., Granger, D. N., and Lefer, D. J. (1998). Modulation of leukocyte-endothelial interactions by reactive metabolites of oxygen and nitrogen: relevance to ischemic heart disease. Free Radic. Biol. Med. 25, 404–433. doi: 10.1016/S0891-5849(98)00094-X
Groves, B. M., Reeves, J. T., Sutton, J. R., Wagner, P. D., Cymerman, A., Malconian, M. K., et al. (1987). Operation Everest II: elevated high-altitude pulmonary resistance unresponsive to oxygen. J. Appl. Physiol. 63, 521–530. doi: 10.1152/jappl.1987.63.2.521
Gupta, R. K., Soree, P., Desiraju, K., Agrawal, A., and Singh, S. B. (2017). Subclinical pulmonary dysfunction contributes to high altitude pulmonary edema susceptibility in healthy non-mountaineers. Sci. Rep. 7:14892. doi: 10.1038/s41598-017-14947-z
Haase, V. H. (2013). Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 27, 41–53. doi: 10.1016/j.blre.2012.12.003
Hackett, P. H. (1999). The cerebral etiology of high-altitude cerebral edema and acute mountain sickness. Wilderness Environ. Med. 10, 97–109. doi: 10.1580/1080-6032(1999)010[0097:TCEOHA]2.3.CO;2
Hackett, P. H. (2000). High altitude cerebral edema and acute mountain sickness: a pathophysiology update. Adv. Exp. Med. Biol. 474, 23–45. doi: 10.1007/978-1-4615-4711-2_2
Hackett, P. H., and Roach, R. C. (2004). High altitude cerebral edema. High Alt. Med. Biol. 5, 136–146. doi: 10.1089/1527029041352054
Hackett, P. H., Yarnell, P. R., Weiland, D. A., and Reynard, K. B. (2019). Acute and evolving MRI of high-altitude cerebral edema: microbleeds, edema, and pathophysiology. Am. J. Neuroradiol. 40, 464–469. doi: 10.3174/ajnr.A5897
Hancox, R. J., Poulton, R., Greene, J. M., Filsell, S., McLachlan, C. R., Rasmussen, F., et al. (2007). Systemic inflammation and lung function in young adults. Thorax 62, 1064–1068. doi: 10.1136/thx.2006.076877
Hartmann, G., Tschöp, M., Fischer, R., Bidlingmaier, C., Riepl, R., Tschöp, K., et al. (2000). High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and c-reactive protein. Cytokine 12, 246–252. doi: 10.1006/cyto.1999.0533
Hayden, M. S., and Ghosh, S. (2004). Signaling to NF-κB. Genes Dev. 18, 2195–2224. doi: 10.1101/gad.1228704
He, Y., Qi, X., Ouzhuluobu, Liu, S., Li, J., Zhang, H., et al. (2017). Blunted nitric oxide regulation in Tibetans under high-altitude hypoxia. Natl. Sci. Rev. 6, 1–6. doi: 10.1093/nsr/nwy037
Hirota, K. (2015). Involvement of hypoxia-inducible factors in the dysregulation of oxygen homeostasis in sepsis. Cardiovasc. Haematol. Disord. Targets 15, 29–40. doi: 10.2174/1871529X15666150108115553
Hislop, A., and Reid, L. (1976). New findings in pulmonary arteries of rats with hypoxia induced pulmonary hypertension. Br. J. Exp. Pathol. 57, 542–554.
Horscroft, J. A., Kotwica, A. O., Laner, V., West, J. A., Hennis, P. J., Levett, D. Z. H., et al. (2017). Metabolic basis to sherpa altitude adaptation. Proc. Natl. Acad. Sci. U. S. A. 114, 6382–6387. doi: 10.1073/pnas.1700527114
Imagawa, S., Goldberg, M. A., and Bunn, H. F. (1990). Regulation of the erythropoietin gene. Adv. Exp. Med. Biol. 94, 75–85. doi: 10.1182/blood.v94.6.1864
Imray, C., Wright, A., Subudhi, A., and Roach, R. (2010). Acute mountain sickness: pathophysiology, prevention, and treatment. Prog. Cardiovasc. Dis. 52, 467–484. doi: 10.1016/j.pcad.2010.02.003
Imtiyaz, H. Z., Williams, E. P., Hickey, M. M., Patel, S. A., Durham, A. C., Yuan, L.-J., et al. (2010). Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Invest. 120, 2699–2714. doi: 10.1172/JCI39506
Israël, A. (2010). The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2:a000158. doi: 10.1101/cshperspect.a000158
Jackson, A., Nanton, M. R., O’Donnell, H., Akue, A. D., and McSorley, S. J. (2010). Innate immune activation during salmonella infection initiates extramedullary erythropoiesis and splenomegaly. J. Immunol. 185, 6198–6204. doi: 10.4049/jimmunol.1001198
Jacovas, V. C., Couto-Silva, C. M., Nunes, K., Lemes, R. B., de Oliveira, M. Z., Salzano, F. M., et al. (2018). Selection scan reveals three new loci related to high altitude adaptation in native Andeans. Sci. Rep. 8:12733. doi: 10.1038/s41598-018-31100-6
Julian, C. G., Subudhi, A. W., Wilson, M. J., Dimmen, A. C., Pecha, T., and Roach, R. C. (2011). Acute mountain sickness, inflammation, and permeability: new insights from a blood biomarker study. J. Appl. Physiol. 111, 392–399. doi: 10.1152/japplphysiol.00391.2011
Kaelin, W. G., and Ratcliffe, P. J. (2008). Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402. doi: 10.1016/j.molcel.2008.04.009
Kammerer, T., Faihs, V., Hulde, N., Stangl, M., Brettner, F., Rehm, M., et al. (2020). Hypoxic-inflammatory responses under acute hypoxia: In vitro experiments and prospective observational expedition trial. Int. J. Mol. Sci. 21:1034. doi: 10.3390/ijms21031034
Kanaan, N. C., Peterson, A. L., Pun, M., Holck, P. S., Starling, J., Basyal, B., et al. (2017). Prophylactic acetaminophen or ibuprofen result in equivalent acute mountain sickness incidence at high altitude: a prospective randomized trial. Wilderness Environ. Med. 28, 72–78. doi: 10.1016/j.wem.2016.12.011
Keohane, C., McMullin, M. F., and Harrison, C. (2013). The diagnosis and management of erythrocytosis. BMJ 347:f6667. doi: 10.1136/bmj.f6667
Kharitonov, S. A., and Barnes, P. J. (2001). Exhaled markers of inflammation. Curr. Opin. Allergy Clin. Immunol. 1, 217–224. doi: 10.1097/00130832-200106000-00004
Kidd, B. L., and Urban, L. A. (2001). Mechanisms of inflammatory pain. Br. J. Anaesth. 87, 3–11. doi: 10.1093/bja/87.1.3
Kim, T. S., Hanak, M., Trampont, P. C., and Braciale, T. J. (2015). Stress-associated erythropoiesis initiation is regulated by type 1 conventional dendritic cells. J. Clin. Invest. 125, 3965–3980. doi: 10.1172/JCI81919
Kim, I., and Park, J. W. (2020). Hypoxia-driven epigenetic regulation in cancer progression: a focus on histone methylation and its modifying enzymes. Cancer Lett. 489, 41–49. doi: 10.1016/j.canlet.2020.05.025
Knauert, M., Vangala, S., Haslip, M., Lee, P. J., and Ryter, S. W. (2013). Therapeutic applications of carbon monoxide. Oxidative Med. Cell. Longev. 2013:360815. doi: 10.1155/2013/360815
Kubo, K., Hanaoka, M., Hayano, T., Miyahara, T., Hachiya, T., Hayasaka, M., et al. (1998). Inflammatory cytokines in BAL fluid and pulmonary hemodynamics in high-altitude pulmonary edema. Respir. Physiol. 111, 301–310. doi: 10.1016/S0034-5687(98)00006-1
Kubo, K., Hanaoka, M., Yamaguchi, S., Hayano, T., Hayasaka, M., Koizumi, T., et al. (1996). Cytokines in bronchoalveolar lavage fluid in patients with high altitude pulmonary oedema at moderate altitude in Japan. Thorax 51, 739–742. doi: 10.1136/thx.51.7.739
Kvamme, B. O., Gadan, K., Finne-Fridell, F., Niklasson, L., Sundh, H., Sundell, K., et al. (2013). Modulation of innate immune responses in Atlantic salmon by chronic hypoxia-induced stress. Fish Shellfish Immunol. 34, 55–65. doi: 10.1016/j.fsi.2012.10.006
Lakshminarayan, S., and Pierson, D. J. (1975). Recurrent high altitude pulmonary edema with blunted chemosensitivity. Am. Rev. Respir. Dis. 111, 869–872. doi: 10.1164/arrd.1975.111.6.869
Lawrence, T. (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1:a001651. doi: 10.1101/cshperspect.a001651
León-Velarde, F., Maggiorini, M., Reeves, J. T., Aldashev, A., Asmus, I., Bernardi, L., et al. (2005). Consensus statement on chronic and subacute high altitude diseases. High Alt. Med. Biol. 6, 147–157. doi: 10.1089/ham.2005.6.147
Li, Y., Zhang, Y., and Zhang, Y. (2018). Research advances in pathogenesis and prophylactic measures of acute high altitude illness. Respir. Med. 145, 145–152. doi: 10.1016/j.rmed.2018.11.004
Liao, C., Sandeep Prabhu, K., and Paulson, R. F. (2018). Monocyte-derived macrophages expand the murine stress erythropoietic niche during the recovery from anemia. Blood 132, 2580–2593. doi: 10.1182/blood-2018-06-856831
Lipman, G. S., Kanaan, N. C., Holck, P. S., Constance, B. B., and Gertsch, J. H. (2012). Ibuprofen prevents altitude illness: a randomized controlled trial for prevention of altitude illness with nonsteroidal anti-inflammatories. Ann. Emerg. Med. 59, 484–490. doi: 10.1016/j.annemergmed.2012.01.019
Liu, B., Chen, J., Zhang, L., Gao, Y., Cui, J., Zhang, E., et al. (2017). IL-10 dysregulation in acute mountain sickness revealed by transcriptome analysis. Front. Immunol. 8:628. doi: 10.3389/fimmu.2017.00628
Luks, A. M., Swenson, E. R., and Bärtsch, P. (2017). Acute high-altitude sickness. Eur. Respir. Rev. 26:160096. doi: 10.1183/16000617.0096-2016
Lundeberg, J., Feiner, J. R., Schober, A., Sall, J. W., Eilers, H., and Bickler, P. E. (2018). Increased cytokines at high altitude: lack of effect of ibuprofen on acute mountain sickness, physiological variables, or cytokine levels. High Alt. Med. Biol. 19, 249–258. doi: 10.1089/ham.2017.0144
Lüneburg, N., Siques, P., Brito, J., Arriaza, K., Pena, E., Klose, H., et al. (2016). Long-term chronic intermittent hypobaric hypoxia in rats causes an imbalance in the asymmetric dimethylarginine/nitric oxide pathway and ros activity: a possible synergistic mechanism for altitude pulmonary hypertension? Pulm. Med. 2016:6578578. doi: 10.1155/2016/6578578
Maines, M. D. (1997). The heme oxygenase system: a regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 37, 517–554. doi: 10.1146/annurev.pharmtox.37.1.517
Mairbäurl, H. (2018). Neocytolysis: how to get rid of the extra erythrocytes formed by stress erythropoiesis upon descent from high altitude. Front. Physiol. 9:345. doi: 10.3389/fphys.2018.00345
Malacrida, S., Giannella, A., Ceolotto, G., Reggiani, C., Vezzoli, A., Mrakic-Sposta, S., et al. (2019). Transcription factors regulation in human peripheral white blood cells during hypobaric hypoxia exposure: an in-vivo experimental study. Sci. Rep. 9:9901. doi: 10.1038/s41598-019-46391-6
McNicholas, W. T. (2009). Chronic obstructive pulmonary disease and obstructive sleep apnea: overlaps in pathophysiology, systemic inflammation, and cardiovascular disease. Am. J. Respir. Crit. Care Med. 180, 692–700. doi: 10.1164/rccm.200903-0347PP
Mehta, S. R., Chawla, A., and Kashyap, A. S. (2008). Acute mountain sickness, high altitude cerebral oedema, high altitude pulmonary oedema: the current concepts. Med. J. Armed Forces India 64, 149–153. doi: 10.1016/S0377-1237(08)80062-7
Mishra, K. P., Sharma, N., Soree, P., Gupta, R. K., Ganju, L., and Singh, S. B. (2016). Hypoxia-induced inflammatory chemokines in subjects with a history of high-altitude pulmonary edema. Indian J. Clin. Biochem. 31, 81–86. doi: 10.1007/s12291-015-0491-3
Mitchell, S., Vargas, J., and Hoffmann, A. (2016). Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 8, 227–241. doi: 10.1002/wsbm.1331
Moore, L. G. (2017). Human genetic adaptation to high altitudes: current status and future prospects. Quat. Int. 461, 4–13. doi: 10.1016/j.quaint.2016.09.045
Nemeth, E. (2008). Iron regulation and erythropoiesis. Curr. Opin. Hematol. 15, 169–175. doi: 10.1097/MOH.0b013e3282f73335
Nemeth, E., Rivera, S., Gabayan, V., Keller, C., Taudorf, S., Pedersen, B. K., et al. (2004). IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest. 113, 1271–1276. doi: 10.1172/JCI200420945
Oberholzer, L., Lundby, C., Stauffer, E., Ulliel-Roche, M., Hancco, I., Pichon, A., et al. (2020). Reevaluation of excessive erythrocytosis in diagnosing chronic mountain sickness in men from the world’s highest city. Blood 136, 1884–1888. doi: 10.1182/blood.2019004508
Oeckinghaus, A., and Ghosh, S. (2009). The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1:a000034. doi: 10.1101/cshperspect.a000034
Paulson, R. F., Shi, L., and Wu, D. C. (2011). Stress erythropoiesis: new signals and new stress progenitor cells. Curr. Opin. Hematol. 18, 139–145. doi: 10.1097/MOH.0b013e32834521c8
Peng, Y., Yang, Z., Zhang, H., Cui, C., Qi, X., Luo, X., et al. (2011). Genetic variations in tibetan populations and high-altitude adaptation at the Himalayas. Mol. Biol. Evol. 28, 1075–1081. doi: 10.1093/molbev/msq290
Perkins, N. D. (2006). Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 25, 6717–6730. doi: 10.1038/sj.onc.1209937
Peyssonnaux, C., Cejudo-Martin, P., Doedens, A., Zinkernagel, A. S., Johnson, R. S., and Nizet, V. (2007). Cutting edge: essential role of hypoxia inducible factor-1α in development of lipopolysaccharide-induced sepsis. J. Immunol. 178, 7516–7519. doi: 10.4049/jimmunol.178.12.7516
Quercioli, A., Mach, F., and Montecucco, F. (2010). Inflammation accelerates atherosclerotic processes in obstructive sleep apnea syndrome (OSAS). Sleep Breath. 14, 261–269. doi: 10.1007/s11325-010-0338-3
Roach, R. C., Hackett, P. H., Oelz, O., Bärtsch, P., Luks, A. M., MacInnis, M. J., et al. (2018). The 2018 Lake Louise acute mountain sickness score. High Alt. Med. Biol. 19, 4–6. doi: 10.1089/ham.2017.0164
Ryter, S. W., and Choi, A. M. K. (2016). Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 167, 7–34. doi: 10.1016/j.trsl.2015.06.011
Scheinfeldt, L. B., Soi, S., Thompson, S., Ranciaro, A., Woldemeskel, D., Beggs, W., et al. (2012). Genetic adaptation to high altitude in the Ethiopian highlands. Genome Biol. 13:R1. doi: 10.1186/gb-2012-13-1-r1
Schoene, R. B., Hackett, P. H., Henderson, W. R., Sage, E. H., Chow, M., Roach, R. C., et al. (1986). High-altitude pulmonary edema: characteristics of lung lavage fluid. JAMA J. Am. Med. Assoc. 256, 63–69. doi: 10.1001/jama.1986.03380010067027
Schoene, R. B., Swenson, E. R., Pizzo, C. J., Hackett, P. H., Roach, R. C., Mills, W. J., et al. (1988). The lung at high altitude: Bronchoalveolar lavage in acute mountain sickness and pulmonary edema. J. Appl. Physiol. 64, 2605–2613. doi: 10.1152/jappl.1988.64.6.2605
Seldon, M. P., Silva, G., Pejanovic, N., Larsen, R., Gregoire, I. P., Filipe, J., et al. (2007). Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of nf-κb rela phosphorylation at serine 276. J. Immunol. 179, 7840–7851. doi: 10.4049/jimmunol.179.11.7840
Semenza, G. L. (2009). Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 24, 97–106. doi: 10.1152/physiol.00045.2008
Sethi, S., and Dikshit, M. (2000). Modulation of polymorphonuclear leukocytes function by nitric oxide. Thromb. Res. 100, 223–247. doi: 10.1016/S0049-3848(00)00320-0
Shaaban, R., Kony, S., Driss, F., Leynaert, B., Soussan, D., Pin, I., et al. (2006). Change in C-reactive protein levels and FEV1 decline: a longitudinal population-based study. Respir. Med. 100, 2112–2120. doi: 10.1016/j.rmed.2006.03.027
Sharma, J. N., Al-Omran, A., and Parvathy, S. S. (2007). Role of nitric oxide in inflammatory diseases. Inflammopharmacology 15, 252–259. doi: 10.1007/s10787-007-0013-x
Simonson, T. S., Yang, Y., Huff, C. D., Yun, H., Qin, G., Witherspoon, D. J., et al. (2010). Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75. doi: 10.1126/science.1189406
Siques, P., López De Pablo, Á. L., Brito, J., Arribas, S. M., Flores, K., Arriaza, K., et al. (2014). Nitric oxide and superoxide anion balance in rats exposed to chronic and long term intermittent hypoxia. Biomed. Res. Int. 2014:610474. doi: 10.1155/2014/610474
Song, T. T., Bi, Y. H., Gao, Y. Q., Huang, R., Hao, K., Xu, G., et al. (2016). Systemic pro-inflammatory response facilitates the development of cerebral edema during short hypoxia. J. Neuroinflammation 13:63. doi: 10.1186/s12974-016-0528-4
Stenmark, K. R., Yeager, M. E., El Kasmi, K. C., Nozik-Grayck, E., Gerasimovskaya, E. V., Li, M., et al. (2013). The adventitia: essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 75, 23–47. doi: 10.1146/annurev-physiol-030212-183802
Swenson, E. R. (2013). Hypoxic pulmonary vasoconstriction. High Alt. Med. Biol. 14, 101–110. doi: 10.1089/ham.2013.1010
Swenson, E. R. (2020). Early hours in the development of high-altitude pulmonary edema: time course and mechanisms. J. Appl. Physiol. 128, 1539–1546. doi: 10.1152/japplphysiol.00824.2019
Swenson, E. R., Maggiorini, M., Mongovin, S., Simon, R., Gibbs, J., Greve, I., et al. (2002). Pathogenesis of high-altitude pulmonary edema: inflammation is not an etiologic factor. J. Am. Med. Assoc. 287, 2228–2235. doi: 10.1001/jama.287.17.2228
Sydykov, A., Mamazhakypov, A., Maripov, A., Kosanovic, D., Weissmann, N., Ghofrani, H. A., et al. (2021). Pulmonary hypertension in acute and chronic high altitude maladaptation disorders. Int. J. Environ. Res. Public Health 18:1692. doi: 10.3390/ijerph18041692
Talbot, N. P., Balanos, G. M., Dorrington, K. L., and Robbins, P. A. (2005). Two temporal components within the human pulmonary vascular response to ∼2 h of isocapnic hypoxia. J. Appl. Physiol. 98, 1125–1139. doi: 10.1152/japplphysiol.00903.2004
Tasali, E., and Ip, M. S. M. (2008). Obstructive sleep apnea and metabolic syndrome: alterations in glucose metabolism and inflammation. Proc. Am. Thorac. Soc. 5, 207–217. doi: 10.1513/pats.200708-139MG
Taylor, C. T. (2008). Interdependent roles for hypoxia inducible factor and nuclear factor-κB in hypoxic inflammation. J. Physiol. 586, 4055–4059. doi: 10.1113/jphysiol.2008.157669
Taylor, C. T., and Colgan, S. P. (2017). Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 17, 774–785. doi: 10.1038/nri.2017.103
Taylor, C. T., and Moncada, S. (2010). Nitric oxide, cytochrome c oxidase, and the cellular response to hypoxia. Arterioscler. Thromb. Vasc. Biol. 30, 643–647. doi: 10.1161/ATVBAHA.108.181628
Thienpont, B., Steinbacher, J., Zhao, H., D’Anna, F., Kuchnio, A., Ploumakis, A., et al. (2016). Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537, 63–68. doi: 10.1038/nature19081
Tie, R., Li, H., Cai, S., Liang, Z., Shan, W., Wang, B., et al. (2019). Interleukin-6 signaling regulates hematopoietic stem cell emergence. Exp. Mol. Med. 51, 1–12. doi: 10.1038/s12276-019-0320-5
Tift, M. S., Alves de Souza, R. W., Weber, J., Heinrich, E. C., Villafuerte, F. C., Malhotra, A., et al. (2020). Adaptive potential of the heme oxygenase/carbon monoxide pathway during hypoxia. Front. Physiol. 11:886. doi: 10.3389/fphys.2020.00886
Tobin, M. J., Laghi, F., and Jubran, A. (2020). Why COVID-19 silent hypoxemia is baffling to physicians. Am. J. Respir. Crit. Care Med. 202, 356–360. doi: 10.1164/rccm.202006-2157CP
Van Uden, P., Kenneth, N. S., and Rocha, S. (2008). Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem. J. 412, 477–484. doi: 10.1042/BJ20080476
Varatharaj, A., and Galea, I. (2017). The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 60, 1–12. doi: 10.1016/j.bbi.2016.03.010
Villafuerte, F. C., Macarlupú, J. L., Anza-Ramírez, C., Corrales-Melgar, D., Vizcardo-Galindo, G., Corante, N., et al. (2014). Decreased plasma soluble erythropoietin receptor in high-altitude excessive erythrocytosis and Chronic Mountain sickness. J. Appl. Physiol. 117, 1356–1362. doi: 10.1152/japplphysiol.00619.2014
Walmsley, S., Harris, A., Thompson, A. A. R., and Whyte, M. K. B. (2014). HIF-mediated innate immune responses: cell signaling and therapeutic implications. Hypoxia 2, 47–58. doi: 10.2147/hp.s50269
Walmsley, S. R., Print, C., Farahi, N., Peyssonnaux, C., Johnson, R. S., Cramer, T., et al. (2005). Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J. Exp. Med. 201, 105–115. doi: 10.1084/jem.20040624
Wang, C., Jiang, H., Duan, J., Chen, J., Wang, Q., Liu, X., et al. (2018). Exploration of acute phase proteins and inflammatory cytokines in early stage diagnosis of acute mountain sickness. High Alt. Med. Biol. 19, 170–177. doi: 10.1089/ham.2017.0126
Watts, E. R., and Walmsley, S. R. (2019). Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol. Med. 25, 33–46. doi: 10.1016/j.molmed.2018.10.006
Weiss, G., Ganz, T., and Goodnough, L. T. (2019). Anemia of inflammation. Blood 133, 40–50. doi: 10.1182/blood-2018-06-856500
Wenger, R. H., Stiehl, D. P., and Camenisch, G. (2005). Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005:re12. doi: 10.1126/stke.3062005re12
West, J. B. (2004). The physiologic basis of high-altitude diseases. Ann. Intern. Med. 141:789. doi: 10.7326/0003-4819-141-10-200411160-00010
Wilkins, M. R., Ghofrani, H. A., Weissmann, N., Aldashev, A., and Zhao, L. (2015). Pathophysiology and treatment of high-altitude pulmonary vascular disease. Circulation 131, 582–590. doi: 10.1161/CIRCULATIONAHA.114.006977
Xie, J., Covassin, N., Fan, Z., Singh, P., Gao, W., Li, G., et al. (2020). Association between hypoxemia and mortality in patients with COVID-19. Mayo Clin. Proc. 95, 1138–1147. doi: 10.1016/j.mayocp.2020.04.006
Yang, J., Jin, Z. B., Chen, J., Huang, X. F., Li, X. M., Liang, Y. B., et al. (2017). Genetic signatures of high-altitude adaptation in Tibetans. Proc. Natl. Acad. Sci. U. S. A. 114, 4189–4194. doi: 10.1073/pnas.1617042114
Yang, D., Peng, Y., Ouzhuluobu, B., Cui, C., Bianba, Wang, L., et al. (2016). HMOX2 functions as a modifier gene for high-altitude adaptation in Tibetans. Hum. Mutat. 37, 216–223. doi: 10.1002/humu.22935
Zaritsky, J., Young, B., Wang, H. J., Westerman, M., Olbina, G., Nemeth, E., et al. (2009). Hepcidin—A potential novel biomarker for iron status in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 4, 1051–1056. doi: 10.2215/CJN.05931108
Zhang, C., Lu, Y., Feng, Q., Wang, X., Lou, H., Liu, J., et al. (2017). Differentiated demographic histories and local adaptations between Sherpas and Tibetans. Genome Biol. 18:115. doi: 10.1186/s13059-017-1242-y
Zheng, C. R., Chen, G. Z., Yu, J., Qin, J., Song, P., Bian, S. Z., et al. (2014). Inhaled budesonide and oral dexamethasone prevent acute mountain sickness. Am. J. Med. 127, 1001–1009.e2. doi: 10.1016/j.amjmed.2014.04.012
Zhou, Y., Huang, X., Zhao, T., Qiao, M., Zhao, X., Zhao, M., et al. (2017). Hypoxia augments LPS-induced inflammation and triggers high altitude cerebral edema in mice. Brain Behav. Immun. 64, 266–275. doi: 10.1016/j.bbi.2017.04.013
Keywords: hypoxia, inflammation, high altitude, hypoxia inducible factor, nuclear factor-κB
Citation: Pham K, Parikh K and Heinrich EC (2021) Hypoxia and Inflammation: Insights From High-Altitude Physiology. Front. Physiol. 12:676782. doi: 10.3389/fphys.2021.676782
Edited by:
Francisco C. Villafuerte, Universidad Peruana Cayetano Heredia, PeruReviewed by:
Patricia Siques, Arturo Prat University, ChileCopyright © 2021 Pham, Parikh and Heinrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erica C. Heinrich, ZXJpY2EuaGVpbnJpY2hAbWVkc2NoLnVjci5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.