 Thomas SitharaKonstantinos Drosatos*
Thomas SitharaKonstantinos Drosatos*- Center for Translational Medicine, Lewis Katz School of Medicine at Temple University, Philadelphia, PA, United States
Aging is a process that can be accompanied by molecular and cellular alterations that compromise cardiac function. Although other metabolic disorders with increased prevalence in aged populations, such as diabetes mellitus, dyslipidemia, and hypertension, are associated with cardiovascular complications; aging-related cardiomyopathy has some unique features. Healthy hearts oxidize fatty acids, glucose, lactate, ketone bodies, and amino acids for producing energy. Under physiological conditions, cardiac mitochondria use fatty acids and carbohydrate mainly to generate ATP, 70% of which is derived from fatty acid oxidation (FAO). However, relative contribution of nutrients in ATP synthesis is altered in the aging heart with glucose oxidation increasing at the expense of FAO. Cardiac aging is also associated with impairment of mitochondrial abundance and function, resulting in accumulation of reactive oxygen species (ROS) and activation of oxidant signaling that eventually leads to further mitochondrial damage and aggravation of cardiac function. This review summarizes the main components of pathophysiology of cardiac aging, which pertain to cardiac metabolism, mitochondrial function, and systemic metabolic changes that affect cardiac function.
Introduction
Cardiac aging is an intrinsic process, accompanied by structural and functional changes of the heart, which increase vulnerability to stress and cardiovascular-related mortality and morbidity. The most common features of cardiac aging include progressive degeneration of myocardium, impaired cardiac conduction system, aortic valvular stenosis and mitral valvular insufficiency, increased wall thickening, and stiffening of the arteries. Aging-related cardiovascular complications also include hypertension, atrial fibrillation, and atherosclerosis.
Aged myocardium presents with left ventricular hypertrophy, increased left ventricular end-diastolic pressure, lower fractional shortening, diastolic dysfunction, fibrosis, cardiomyocyte apoptosis, oxidative stress, inflammation, and lower exercise capacity (Dai et al., 2012). Age-related cardiac complications that lead to heart failure with preserved ejection fraction (HFpEF) have higher prevalence in women than men (Merz and Cheng, 2016). This difference has been attributed to differential dependence of cardiac function on estrogen signaling, which attenuates in post-menopausal women (Mahmoodzadeh et al., 2006).
Many studies have explored the association of cardiac complications with alterations in metabolic pathways (Lopaschuk, 2017). As the world population grows older and cardiovascular complications become more frequent, the involvement of alterations in cardiac metabolism in cardiac aging pathophysiology is warranted. The objective of this article pertains to better understanding of metabolic amendments that occur during cardiac aging including intermediary metabolism and mitochondrial function and may unravel novel factors that account for pathophysiology in aged hearts.
Cardiac Metabolism and Function in the Healthy Heart
A healthy heart pumps about 7,000 litters of blood per day, which requires a significant amount of ATP that is acquired through oxidation of fatty acids, glucose, amino acids, ketone bodies, and lactate. Fatty acid oxidation (FAO) is the primary source for cardiac ATP synthesis (Lopaschuk et al., 2010). Depending on cardiac work load, hormonal status, fasting vs. feeding state and availability of energy substrates, and the ratio of energy supply from different substrates is adjusted accordingly (Ramos-Roman et al., 2012; Viscarra and Ortiz, 2013; Karwi et al., 2018).
Lipoprotein- or adipose tissue-derived free fatty acids are transported into the heart via the transmembrane fatty acid transporter, cluster of differentiation (CD36). For the uptake of lipids from triglyceride-rich lipoprotein, lipoprotein lipase-mediated lipolysis precedes, while whole lipoprotein uptake is also possible (Coburn et al., 2000; Bharadwaj et al., 2010; Son et al., 2018). Fatty acids are then converted to fatty acyl-CoA by acyl-CoA synthase and fatty acylcarnitine by carnitine palmitoyltransferase I (CPT-I)β (Tomec and Hoppel, 1975), which is transported into the mitochondrial matrix, where it is converted back to long-chain acyl-CoA by carnitine palmitoyltransferase II (CPT-II; Hoppel et al., 1998). β-oxidation processes fatty acyl-CoAs to acetyl-CoA yielding NADH and FADH2. Further acetyl-CoA oxidation within the mitochondrial matrix by the tricarboxylic acid cycle (TCA cycle or Krebs cycle) produces three NADHs and one FADH2 and GTP. The NADH and FADH2 are further oxidized by electron transport chain, which leads to ATP generation (Ma and Li, 2015).
Glucose is obtained by cardiomycoytes either from circulation or from hydrolysis of intracellular glycogen stores. Glucose serves as the main cardiomyocyte energy production fuel in the fetal hearts. Glucose uptake by cardiomyocytes occurs through insulin-dependent glucose transporters (GLUT4) or via the insulin-independent GLUT1. On entering the cell, glucose is phosphorylated by hexokinase to glucose-6-phosphate (G-6-P), which is used for glycolysis, glycogen synthesis, the hexosamine biosynthesis pathway, or the pentose phosphate pathway. Pyruvate produced during glycolysis is transferred via mitochondrial pyruvate carrier (MPC) to mitochondrial matrix, where it is converted to acetyl-CoA by pyruvate dehydrogenase (PDH), and enters the TCA cycle (Ma and Li, 2015). PDH complex (PDC) activity is inhibited by PDH kinases (PDKs) that respond to allosteric modifiers derived from glycolysis and FAO. Ketone bodies are produced in the liver during starvation and fasting. There are three types of ketone bodies that the heart uses: acetone, acetoacetate, and β-hydroxybutyrate (βOHB; Cotter et al., 2013). βOHB-dehydrogenase oxidizes βOHB to acetoacetate in mitochondria. Acetoacetate is then converted to acetoacetyl-CoA by the rate limiting enzyme succinyl-CoA:3-oxoacid-CoA-transferase (SCOT). Finally, acetoacetyl-CoA is converted into acetyl-CoA by acetyl-CoA acetyltransferase (Williams et al., 1971) and enters the TCA cycle and electron transport chain for generating ATP (Figure 1).

Figure 1. Metabolic pathways in normal heart.
Alterations in Lipid Metabolism During Cardiac Aging
Considering the heavy reliance of cardiac ATP synthesis on FAO, it is not surprising that either deprivation or oversupply of fatty acids can be detrimental (Noh et al., 2006). An imbalance between fatty acid uptake and oxidation in aged hearts leads to accumulation of medium-chain acyl-carnitines (Koves et al., 2008) and long-chain fatty acids that are incorporated in triglycerides, phospholipids, and other lipid subspecies, such as diacylglycerols and ceramides. Higher diacylglycerols and ceramides content has been associated with cardiac lipotoxicity, which has been observed in the elderly (Drosatos and Schulze, 2013), as well as in mouse models of cardiac aging along with lower cardiac FAO and higher serum non-esterified fatty acids (Kates et al., 2003; Rodriguez-Calvo et al., 2007; Wang et al., 2015).
Higher cardiac lipid content in aging may be sustained by increased expression of cardiac CD36, which mediates fatty acid transport (Koonen et al., 2007). In accordance, aged CD36-deficient mice have lower levels of intramyocardial lipids, higher mitochondria-derived ATP synthesis, and improved cardiac function compared with aged wild-type mice. While CD36 upregulation and fatty acid uptake seem to be detrimental for cardiac aging, no difference was detected in fasting-induced cardiac lipoprotein lipase activity of aged rats compared with young animals (Bergo et al., 1997). This indicates continuous release of lipoprotein-derived fatty acids in aged hearts, which in combination with higher CD36 expression, explains the increased cardiac lipid content in aging. However, neither inhibition nor activation of lipoprotein lipase offers significant therapeutic potential. Nevertheless, both deletion (Noh et al., 2006) and constitutive activation of lipoprotein lipase (Yagyu et al., 2003) result in cardiomyopathy, due to energetic failure and increased ceramide content (Park et al., 2008), Protein Kinase C signaling activation, and β-adrenergic receptor desensitization (Drosatos et al., 2011).
In humans, age-dependent decline in myocardial fatty acid utilization and ATP production has been observed (Kates et al., 2003). This finding correlates with lower expression of cardiac peroxisome proliferator-activated receptors (PPAR)α (Iemitsu et al., 2002), which is a central transcriptional regulator of proteins that are involved in cardiac energy metabolism (Pol et al., 2015). However, activation of PPARα does not seem to be a desirable intervention, due to similarities of constitutive cardiomyocyte-specific expression of PPARα with cardiomyopathy in diabetes (Finck et al., 2002). On the other hand, cardiomyocyte PPARγ constitutive expression is also toxic for the heart (Son et al., 2007), unless PPARα is inhibited (Son et al., 2010). Thus, as aged hearts have lower PPARα expression (Iemitsu et al., 2002); activation of PPARγ emerges as a potential therapeutic direction that warrants further investigation in models of cardiac aging. Thus the combination of increased lipid uptake and reduced lipid oxidation during cardiac aging may account for the observed cardiac lipotoxicity and associated complications.
Carbohydrate Oxidative Metabolism in Aged Heart
Aged hearts have higher glycolysis and glucose catabolism at the expense of FAO (Lakatta and Yin, 1982). It is also noted that during aging, PDC phosphorylation by PDK is lower, which favors mitochondrial pyruvate oxidation (Moreau et al., 2004). Another study has also reported lower PDK4 expression in aged hearts compared with young hearts (Hyyti et al., 2010). The downregulation of PDK4 may be accounted for by the suppression of the expression of cardiac PPARα that has been reported in aging-associated cardiomyopathy (Iemitsu et al., 2002; Pol et al., 2015). Insulin resistance constitutes another layer of control of glucose utilization that pertains to glucose uptake by the tissues. In elderly, insulin resistance is reckoned as an independent risk factor for coronary heart disease (Butler et al., 2006). It is observed that IGF-1 deficiency enhances resistance of cardiomyocytes against aging (Li et al., 2008). Also, suppression of phosphoinositide 3-kinase (PI3K) weakens senescence and inflammatory markers expression and decreases in accumulation of the aging pigment and lipofuscin (Inuzuka et al., 2009). Thus, shift from FAO to glucose oxidation may constitute a compensatory action of cardiomyocytes aiming to balance the energetic deficiency caused by suppression of FAO. However, the increased glucose utilization in aged hearts and the alleviating effects of insulin signaling inhibition suggest that increased glucose utilization contributes in aging-related cardiomyopathy and may constitute a therapeutic target.
Oxidation of Ketone Bodies in Aged Heart
Recent studies have suggested ketone bodies as an efficient alternative fuel for failing hearts (Takahara et al., 2021), which overall energy production without interfering with fatty acid or glucose catabolism (Ho et al., 2019). In light of the association of increased glucose catabolism with cardiac aging, ketone bodies oxidation emerges as a potential resource that may help alleviate aging-related cardiovascular complication. Adaptation from glycolysis to ketone oxidation is delayed in aged rats, which take longer time than young animals to produce high levels of circulating ketone bodies either when fed on ketogenic diet or following fasting (Okuda et al., 1987; Hernandez et al., 2018).
Ketone body production is increased by approximately 2-fold between 3 and 30 months of age in mice (Robinson and Williamson, 1980). Cardiac SCOT, which is a mitochondrial enzyme involved in the extrahepatic metabolism of ketone bodies, shows increased activity in aged rats due to higher nitro-hydroxyl modification of tryptophan 372 that is proximal to the active site (Rebrin et al., 2007). The beneficial metabolic effect of ketone bodies may rely on their antioxidant properties that pertain to the increased ratio between reduced and oxidized glutathione (Veech et al., 2001; Cahill and Veech, 2003) or their stimulatory effect on succinate oxidation (Balietti et al., 2009). The anti-oxidant effect of ketone bodies, which has been demonstrated by various studies (Mallet and Sun, 2003; Squires et al., 2003), may account for their protective effect in aged hearts as oxidative stress is one of the main manifestations of cardiac aging. The anti-oxidant effect of ketone bodies may also account for the improvements in mitochondrial repair mechanisms (Thai et al., 2019) or in mitophagy flux (Thai et al., 2019) in the aged hearts.
Nutritional interventions with cyclic ketogenic diet in old mice improve heart rate, fractional shortening, left aortic valve pressure gradient, and ventricular mass (Newman et al., 2017). At the same time, ketone bodies incur anti-oxidative and anti-inflammatory effects and counteract age-related cardiac remodeling (Shimazu et al., 2013; Puchalska and Crawford, 2017). Despite the beneficial metabolic effects of ketone bodies, it has been reported that they compete with fatty acids and inhibit their oxidation in diabetic hearts (Hasselbaink et al., 2003; Stanley et al., 2003), which results in lipid accumulation. Studies with isolated cardiomyocytes showed that chronic exposure to βOHB induces insulin resistance and suppresses glucose uptake and oxidation (Pelletier et al., 2007). Taking into account the association of glucose oxidation with cardiotoxicity in aging, ketone bodies hold promise for therapeutic interventions that aim to alleviate cardiomyopathy in the elderly.
Mitochondrial Function in Cardiac Aging
Due to the high-energy demands of the heart, cardiomyocytes are rich in mitochondria, which account for ~35% of total cellular volume. Therefore, the heart is highly vulnerable to mitochondrial anomalies. There are two types of cardiac mitochondria with varying activities: the subsarcolemmal mitochondria (SSM) and the interfibrillar mitochondria (IFM) that are located between the myofibrils (Palmer et al., 1977). Aging primarily decreases content and function of IFM, associated with lower activity of mitochondrial respiratory chain complexes III and IV, impaired oxidative phosphorylation, and increased production of reactive oxygen species (ROS) that leads to injury (Fannin et al., 1999; Judge et al., 2005). Studies in humans and animals have reported that aged hearts have larger mitochondria with disrupted structure and inner membrane cristae (Sachs et al., 1977; Dai and Rabinovitch, 2009). Other studies have also reported lower content and activity of cytochrome oxidase in aged human hearts. This event has deleterious effect on cardiac energy production and exerts cytotoxicity (Fujioka et al., 2011).
Cardiac mitochondria of the elderly are more sensitive to lipid peroxidation and dysfunction when exposed to exogenous iron and H2O2 (Sawada and Carlson, 1987; Cusack et al., 1991). Aged mice have significantly lower expression of myocardial mitochondrial deacetylase sirtuin (SIRT)3, which counteracts oxidative stress. In young animals, nuclear factor erythroid-2 related factor (NRF-2), a highly conserved redox-sensitive transcription factor, is activated by ROS production and increases the expression of several antioxidant genes. However, in aging, ROS-dependent activation of NRF-2 is defective, which aggravates the deleterious effects of ROS (Warabi et al., 2007; Ungvari et al., 2011). Manganese superoxide dismutase, which transforms toxic superoxide to hydrogen peroxide and diatomic oxygen, thus preventing accumulation of mitochondrial ROS, is found to be reduced in aged hearts (Li et al., 2018). Along these lines, supplementation of polyunsaturated fatty acid-containing diet with the anti-oxidant coenzyme Q10 in 24-month old rats increased catalase activity, reduced H2O2 generation, improved cytochrome oxidase activity in cardiac mitochondria, and extended life span (Ochoa et al., 2005; Quiles et al., 2010).
In addition, mitochondrial protein carbonylation is increased with aging, which further aggravates mitochondrial oxidative damage (Dai et al., 2012). Mitochondrial DNA (mtDNA), which lacks protective histones, is particularly vulnerable to oxidative damage leading to point mutations and deletions, inflammation, apoptosis, telomere shortening, necrosis, and immunological dysfunction (Larsson, 2010; Steenman and Lande, 2017).
Besides oxidative stress, mitochondrial biogenesis is also compromised during cardiac aging. PGC-1α, which stimulates mitochondrial biogenesis, serves as a transcriptional coactivator of nuclear receptors, like PPARs, Estrogen-related receptor alpha (ERRα), nuclear factor erythroid-2 related factor (NRF)-1 and NRF-2 (Sanchis-Gomar et al., 2014; Dorn et al., 2015), and regulator of cardiac fatty acid utilization (Arany et al., 2005; Rowe et al., 2010). PGC-1α protein levels are lower in aged mice, compared to young mice (Conley et al., 2007; Vina et al., 2009) similar to what is observed in humans with heart failure (Sihag et al., 2009). PGC-1α knockout in young mice mimics age-related impairments in mitochondrial gene expression. On the other hand, overt activation of PGC-1α accelerates cardiac aging and reduces life span of old wild type mice because of mitochondrial damage and ROS accumulation (Zhu et al., 2019), indicating a delicate balance between PGC1α activation and inhibition for cardiac homeostasis. Indeed, moderate PGC-1α activation inhibits age-related cardiac remodeling changes, such as apoptosis and collagen accumulation, and increases the expression of genes involved in myocardial contractility, metabolism, biogenesis, dynamics, and calcium handling (Whitehead et al., 2018). In accordance with the genetic interventions, mild activators of SIRT1 and PGC-1α, such as resveratrol, incur beneficial effects in mitochondria of senescent HL-1 cardiomyocytes (Ren et al., 2017).
Mitochondrial function is regulated by cardiolipin, which is a main phospholipid of the inner mitochondrial membrane. Cardiolipin preserves the morphology and architecture of the mitochondrial membrane and regulates the activity of mitochondrial enzymes and proteins (Paradies et al., 2019). Some studies have attributed mitochondrial complications during aging to lower cardiolipin content that alters fluidity of the inner mitochondrial membrane. During oxidative stress, cardiolipin undergoes oxidation that disrupts the electron transport chain, leading to mitochondrial dysfunction, cytochrome c release, and cellular apoptosis (Paradies et al., 1998; Ott et al., 2002; Chicco and Sparagna, 2007).
Mitochondrial quality in cardiomyocytes is maintained through a balance between mitophagy and biogenesis. The mitochondrial defects that are observed during cardiac aging are further aggravated by impaired fission, fusion, autophagy, and lysosomal degradation. To correct flaws in mitochondrial function, there is a quality control process that includes biogenesis of new mitochondria and degradation or recycling of the old ones. Accumulation of dysfunctional mitochondria due to oxidative stress, mtDNA mutations, lowering of mitochondrial membrane potential, and defective ATP synthesis stimulate cellular apoptosis and premature cardiac aging. The frequency of mtDNA mutations in older mice is 1,000-fold higher than that for nuclear genes. Accordingly, human cardiac mtDNA deletions begin at the age of 40 years and accumulate gradually with age (Corral-Debrinski et al., 1992; Khaidakov et al., 2003).
Thus, reductions in mitochondrial number along with accumulation of defective mitochondria that constitute a source of ROS are major contributors of the pathophysiology of cardiac aging.
Autophagy in Cardiac Aging
Autophagy is a highly conserved homeostatic mechanism in cells. It includes micro-autophagy, macro-autophagy, and chaperone-mediated autophagy and is regulated by a series of autophagy related genes (ATG). It is involved in degradation and recycling of cellular components, removal of misfolded proteins and damaged organelles, and internal “recycling” of nutrients (Rubinsztein et al., 2011). In cells with low proliferative capacity, like cardiomyocytes, accumulation of lethal proteins, and dysfunctional organelles is a critical event that impairs cellular homeostasis. As age advances the rate of autophagy and mitophagy decrease in heart as well as in other tissues (Hoshino et al., 2013; Xu et al., 2016). The decline in autophagy has a significant impact on the aging-associated accumulation of defective mitochondria and consequent ROS generation in cariomyocytes leading to mitochondrial dysfunction, cardiac aging, and myocardial injury (Mammucari and Rizzuto, 2010; Rezzani et al., 2012). Accordingly, autophagy is a vital mechanism for maintenance of homeostasis and adaptation to stress that the heart undergoes throughout the aging process (Levine and Kroemer, 2008). Lower autophagy that occurs during cardiac aging, results in the accumulation of protein aggregates, oxidized and damaged proteins, and lipofuscin (Dai et al., 2010). Lipofuscin is rich in iron and therefore, it causes lysosomal rupture, oxidative stress, and further mitochondrial damage (Brunk and Terman, 2002). Mouse models of diet-induced obesity with impaired autophagy have compromised myocardial geometry and function (Xu et al., 2013), which indicates that autophagy also alleviates cardiac lipotoxicity.
Protein carbonylation and ubiquitination in aged hearts along with reduced protein turnover rates (Basisty et al., 2018) compromise cardiac proteasome function that impairs protein quality control and leads to accumulation of damaged and misfolded proteins. It is also noted alteration in the expression of heat shock proteins (HSP), which function as molecular chaperones facilitating protein folding and targeting improperly folded proteins to degradative pathways are reduced in aged cardiomyocytes (Bodyak et al., 2002). Higher levels of ROS in aged cardiomyocytes stimulate formation of lipofuscin aggregates, consisting of lipids and aggregated liposomal proteins in lysosomes and which distort autophagy by preventing the fusion of autophagosomes with lysosomes leads to accumulation of aberrant proteins and dysfunctional mitochondria, less effective stress response, and functional loss in cardiomyocytes (Taneike et al., 2010).
Stimulation of autophagy improves cardiac function and prolongs the lifespan in many organisms (Miyamoto, 2019). Selective elimination of damaged mitochondria by autophagy (mitophagy) is an essential component of this mechanism. Rapamycin, which inhibits mTOR and induces autophagy, improves cardiac function and extends life span in C57BL/6 mice, which is also accompanied by increased mitochondrial biogenesis and fatty acid catabolism (Dai et al., 2014; Zhang et al., 2014; Quarles et al., 2020). Another intervention that activated autophagy, which rely on overexpression of ATG5, extended life span in mice. Accordingly, ATG5 deficiency in mouse hearts induces age-associated cardiomyopathy (Taneike et al., 2010; Pyo et al., 2013).
Beclin 1 regulates the initial stage of autophagy in mammalian cells by leading to the formation of a double-membrane structure that engulfs cytoplasmic material to form autophagosomes. Bcl-2 is inhibiting activity of beclin 1 by directly binding to it (Qu et al., 2003; Pattingre et al., 2005). Disruption of the Beclin1-Bcl2 complex stimulates autophagy and inhibits age-induced cardiac fibrosis, hypertrophy, apoptotic cell death, and delays cardiac aging (Fernandez et al., 2018).
Parkin is an E3 ligase that mediates selective identification and removal of damaged mitochondria by autophagy. Parkin-deficient mice accumulate unusual mitochondria in the heart as they age, while parkin transgenic mice display increased mitophagy and are resistant to cardiac aging (Hoshino et al., 2013; Kubli et al., 2013).
The mitochondrial membrane depolarization-dependent PTEN-induced putative kinase (PINK)1/Parkin pathway is involved in tagging defective mitochondria, which is vital for maintaining cardiac function during aging (Nguyen et al., 2016). Autophagy is controlled by various metabolism-related pathways, including the IGF-1/Akt pathway (Ock et al., 2016), the mammalian target of rapamycin (mTORC1) pathway (Harrison et al., 2009), the AMP-activated protein kinase (AMPK) pathway (Kim et al., 2011), Forkhead box O (FoxO), the transcription factor EB (TFEB) pathway (Fullgrabe et al., 2016), ROS (Shirakabe et al., 2016), and sirtuins (Alcendor et al., 2007).
Thus, aging-related cardiomyopathy is associated with lower autophagic flux. Hence, autophagy is a promising therapeutic target for cardiac aging.
Metabolic Deficiency and Inflammation in Cardiac Aging
A close link between cellular oxidative stress and inflammation exists and contributes in the cardiac aging process (De La Fuente, 2002). Inflammation is stimulated by accumulation of senescent cardiomyocytes that leads to senescence-associated secretory phenotype (SASP). GDF15, Tgfb2, and Edn3 are nontypical SASP that are secreted by cardiomyocytes. Chronic stimulation of the immune system attenuates immune response, which is typical for inflammation-associated aging (“inflammaging”; Douziech et al., 2002) that is controlled by NF-κB (Salminen et al., 2008a). Inhibition of aging-related NF-κB activation by the longevity gene, SIRT1 delays aging (Salminen et al., 2008b; Cevenini et al., 2013). Circulating levels of activin, a senescence-associated secreted protein has been correlated with age-related heart failure (Li et al., 2020). Thus, defects in the metabolic machinery of aged cardiomyocytes exert pro-inflammatory effects that compromise cardiac function further.
Epilogue
Heart is an organ with high energetic demands that are achieved through oxidation of various substrates. Utilization of fatty acids, which is the major substrate for energy production, is reduced during cardiac aging at the expense of higher carbohydrate oxidation. This leads to accumulation of toxic lipids in the heart. These events are associated with mitochondrial dysfunction and eventual accumulation of ROS that further aggravate cardiac dysfunction. The higher inflammatory status and impaired autophagy compromise reparative processes that could further worsen aging-related cardiomyopathy (Figure 2).
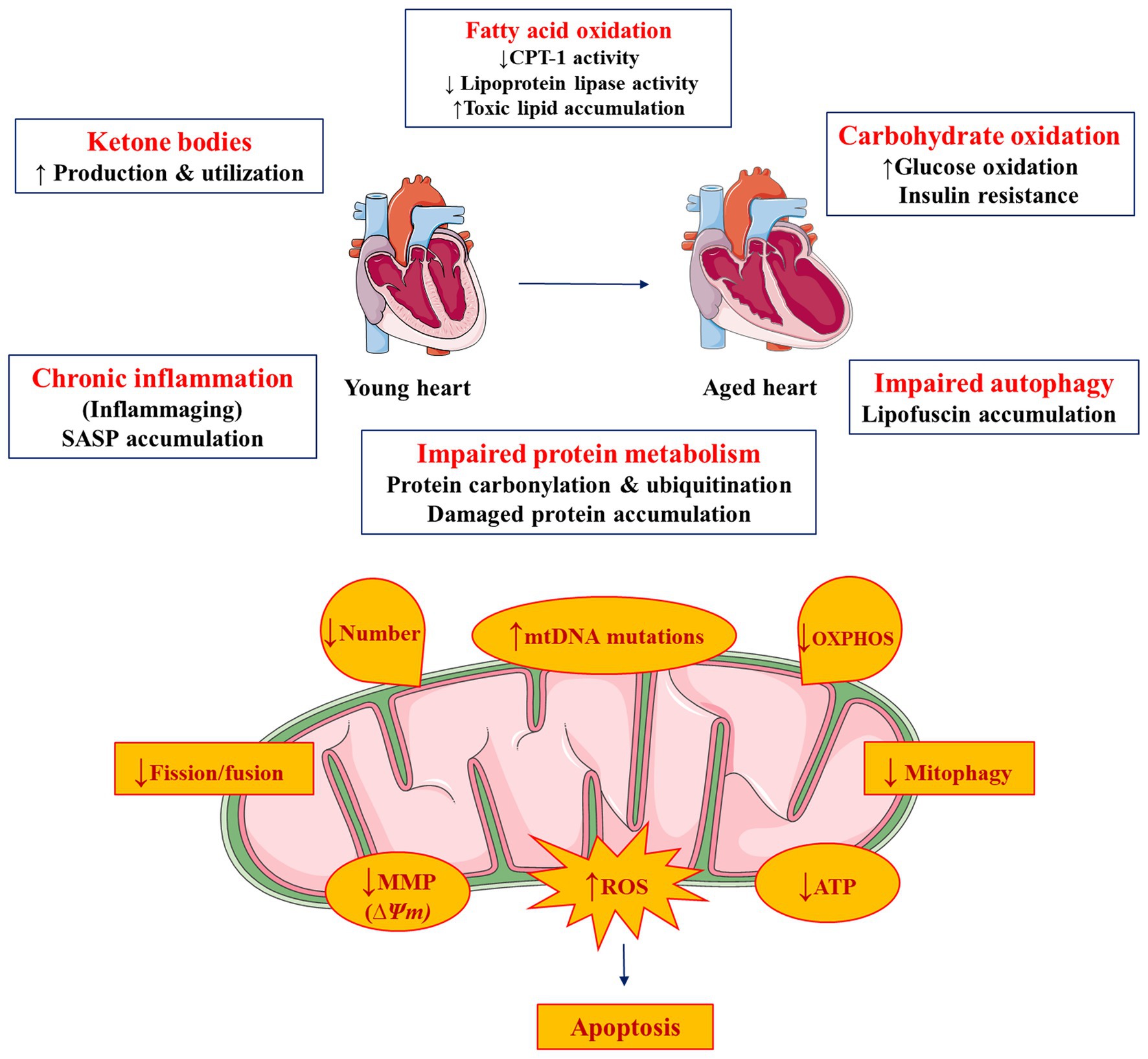
Figure 2. Metabolic and mitochondrial complications in cardiac aging. Figure was produced using Servier medical art (http://www.servier.com).
The variety of metabolic pathways that are affected during cardiac aging suggests potential novel therapeutic interventions. Particularly, interventions that aim to negate aging-induced defects in mitochondria and intermediate metabolism are warranted. Based on clinical observations and preclinical interventions, treatments that will stimulate oxidation of fatty acids and ketone bodies, while they will simultaneously limit glucose catabolism, maintain normal autophagic flux, and limit mitochondrial ROS accumulation hold significant therapeutic potential. Thus, additional studies aiming to elucidate metabolic changes that occur during the progression of cardiac aging are warranted, in order to identify novel therapeutic avenues that may eventually translate to treatments of aging-related cardiovascular complications in humans.
Author Contributions
TS reviewed the literature and wrote the manuscript. KD reviewed the literature and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Heart, Lung and Blood Institute (HL130218 and HL151924) and the National Institute for General Medical Sciences (GM135399) of the National Institutes of Health (KD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alcendor, R. R., Gao, S., Zhai, P., Zablocki, D., Holle, E., Yu, X., et al. (2007). Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100, 1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a
Arany, Z., He, H., Lin, J., Hoyer, K., Handschin, C., Toka, O., et al. (2005). Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 1, 259–271. doi: 10.1016/j.cmet.2005.03.002
Balietti, M., Fattoretti, P., Giorgetti, B., Casoli, T., Di Stefano, G., Solazzi, M., et al. (2009). A ketogenic diet increases succinic dehydrogenase activity in aging cardiomyocytes. Ann. N. Y. Acad. Sci. 1171, 377–384. doi: 10.1111/j.1749-6632.2009.04704.x
Basisty, N., Meyer, J. G., and Schilling, B. (2018). Protein turnover in aging and longevity. Proteomics 18:e1700108. doi: 10.1002/pmic.201700108
Bergo, M., Olivecrona, G., and Olivecrona, T. (1997). Regulation of adipose tissue lipoprotein lipase in young and old rats. Int. J. Obes. Relat. Metab. Disord. 21, 980–986. doi: 10.1038/sj.ijo.0800506
Bharadwaj, K. G., Hiyama, Y., Hu, Y., Huggins, L. A., Ramakrishnan, R., Abumrad, N. A., et al. (2010). Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J. Biol. Chem. 285, 37976–37986. doi: 10.1074/jbc.M110.174458
Bodyak, N., Kang, P. M., Hiromura, M., Sulijoadikusumo, I., Horikoshi, N., Khrapko, K., et al. (2002). Gene expression profiling of the aging mouse cardiac myocytes. Nucleic Acids Res. 30, 3788–3794. doi: 10.1093/nar/gkf497
Brunk, U. T., and Terman, A. (2002). The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 269, 1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x
Butler, J., Rodondi, N., Zhu, Y., Figaro, K., Fazio, S., Vaughan, D. E., et al. (2006). Metabolic syndrome and the risk of cardiovascular disease in older adults. J. Am. Coll. Cardiol. 47, 1595–1602. doi: 10.1016/j.jacc.2005.12.046
Cahill, G. F. Jr., and Veech, R. L. (2003). Ketoacids? Good medicine? Trans. Am. Clin. Climatol. Assoc. 114, 149–161 discussion 162-143.
Cevenini, E., Monti, D., and Franceschi, C. (2013). Inflamm-ageing. Curr. Opin. Clin. Nutr. Metab. Care 16, 14–20. doi: 10.1097/MCO.0b013e32835ada13
Chicco, A. J., and Sparagna, G. C. (2007). Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Phys. Cell Phys. 292, C33–C44. doi: 10.1152/ajpcell.00243.2006
Coburn, C. T., Knapp, F. F. Jr., Febbraio, M., Beets, A. L., Silverstein, R. L., and Abumrad, N. A. (2000). Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 275, 32523–32529. doi: 10.1074/jbc.M003826200
Conley, K. E., Marcinek, D. J., and Villarin, J. (2007). Mitochondrial dysfunction and age. Curr. Opin. Clin. Nutr. Metab. Care 10, 688–692. doi: 10.1097/MCO.0b013e3282f0dbfb
Corral-Debrinski, M., Shoffner, J. M., Lott, M. T., and Wallace, D. C. (1992). Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat. Res. 275, 169–180. doi: 10.1016/0921-8734(92)90021-g
Cotter, D. G., Schugar, R. C., and Crawford, P. A. (2013). Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 304, H1060–H1076. doi: 10.1152/ajpheart.00646.2012
Cusack, B. J., Mushlin, P. S., Andrejuk, T., Voulelis, L. D., and Olson, R. D. (1991). Aging alters the force-frequency relationship and toxicity of oxidative stress in rabbit heart. Life Sci. 48, 1769–1777. doi: 10.1016/0024-3205(91)90215-W
Dai, D. F., Chen, T., Johnson, S. C., Szeto, H., and Rabinovitch, P. S. (2012). Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 16, 1492–1526. doi: 10.1089/ars.2011.4179
Dai, D. F., Chen, T., Wanagat, J., Laflamme, M., Marcinek, D. J., Emond, M. J., et al. (2010). Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 9, 536–544. doi: 10.1111/j.1474-9726.2010.00581.x
Dai, D. F., Karunadharma, P. P., Chiao, Y. A., Basisty, N., Crispin, D., Hsieh, E. J., et al. (2014). Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13, 529–539. doi: 10.1111/acel.12203
Dai, D. F., and Rabinovitch, P. S. (2009). Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc. Med. 19, 213–220. doi: 10.1016/j.tcm.2009.12.004
De La Fuente, M. (2002). Effects of antioxidants on immune system ageing. Eur. J. Clin. Nutr. 56(Suppl. 3), S5–S8. doi: 10.1038/sj.ejcn.1601476
Dorn, G. W. 2nd, Vega, R. B., and Kelly, D. P. (2015). Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 29, 1981–1991. doi: 10.1101/gad.269894.115
Douziech, N., Seres, I., Larbi, A., Szikszay, E., Roy, P. M., Arcand, M., et al. (2002). Modulation of human lymphocyte proliferative response with aging. Exp. Gerontol. 37, 369–387. doi: 10.1016/S0531-5565(01)00204-2
Drosatos, K., Bharadwaj, K. G., Lymperopoulos, A., Ikeda, S., Khan, R., Hu, Y., et al. (2011). Cardiomyocyte lipids impair beta-adrenergic receptor function via PKC activation. Am. J. Physiol. Endocrinol. Metab. 300, E489–E499. doi: 10.1152/ajpendo.00569.2010
Drosatos, K., and Schulze, P. C. (2013). Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 10, 109–121. doi: 10.1007/s11897-013-0133-0
Fannin, S. W., Lesnefsky, E. J., Slabe, T. J., Hassan, M. O., and Hoppel, C. L. (1999). Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch. Biochem. Biophys. 372, 399–407. doi: 10.1006/abbi.1999.1508
Fernandez, A. F., Sebti, S., Wei, Y., Zou, Z., Shi, M., Mcmillan, K. L., et al. (2018). Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140. doi: 10.1038/s41586-018-0162-7
Finck, B. N., Lehman, J. J., Leone, T. C., Welch, M. J., Bennett, M. J., Kovacs, A., et al. (2002). The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Invest. 109, 121–130. doi: 10.1172/JCI0214080
Fujioka, H., Moghaddas, S., Murdock, D. G., Lesnefsky, E. J., Tandler, B., and Hoppel, C. L. (2011). Decreased cytochrome c oxidase subunit VIIa in aged rat heart mitochondria: immunocytochemistry. Anat. Rec. 294, 1825–1833. doi: 10.1002/ar.21486
Fullgrabe, J., Ghislat, G., Cho, D. H., and Rubinsztein, D. C. (2016). Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 129, 3059–3066. doi: 10.1242/jcs.188920
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. doi: 10.1038/nature08221
Hasselbaink, D. M., Glatz, J. F., Luiken, J. J., Roemen, T. H., and Van Der Vusse, G. J. (2003). Ketone bodies disturb fatty acid handling in isolated cardiomyocytes derived from control and diabetic rats. Biochem. J. 371, 753–760. doi: 10.1042/bj20021617
Hernandez, A. R., Hernandez, C. M., Campos, K. T., Truckenbrod, L. M., Sakarya, Y., Mcquail, J. A., et al. (2018). The antiepileptic ketogenic diet alters hippocampal transporter levels and reduces adiposity in aged rats. J. Gerontol. A Biol. Sci. Med. Sci. 73, 450–458. doi: 10.1093/gerona/glx193
Ho, K. L., Zhang, L., Wagg, C., Al Batran, R., Gopal, K., Levasseur, J., et al. (2019). Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 115, 1606–1616. doi: 10.1093/cvr/cvz045
Hoppel, C. L., Kerner, J., Turkaly, P., Turkaly, J., and Tandler, B. (1998). The malonyl-CoA-sensitive form of carnitine palmitoyltransferase is not localized exclusively in the outer membrane of rat liver mitochondria. J. Biol. Chem. 273, 23495–23503. doi: 10.1074/jbc.273.36.23495
Hoshino, A., Mita, Y., Okawa, Y., Ariyoshi, M., Iwai-Kanai, E., Ueyama, T., et al. (2013). Cytosolic p53 inhibits parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 4:2308. doi: 10.1038/ncomms3308
Hyyti, O. M., Ledee, D., Ning, X. H., Ge, M., and Portman, M. A. (2010). Aging impairs myocardial fatty acid and ketone oxidation and modifies cardiac functional and metabolic responses to insulin in mice. Am. J. Physiol. Heart Circ. Physiol. 299, H868–H875. doi: 10.1152/ajpheart.00931.2009
Iemitsu, M., Miyauchi, T., Maeda, S., Tanabe, T., Takanashi, M., Irukayama-Tomobe, Y., et al. (2002). Aging-induced decrease in the PPAR-alpha level in hearts is improved by exercise training. Am. J. Physiol. Heart Circ. Physiol. 283, H1750–H1760. doi: 10.1152/ajpheart.01051.2001
Inuzuka, Y., Okuda, J., Kawashima, T., Kato, T., Niizuma, S., Tamaki, Y., et al. (2009). Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation 120, 1695–1703. doi: 10.1161/CIRCULATIONAHA.109.871137
Judge, S., Jang, Y. M., Smith, A., Hagen, T., and Leeuwenburgh, C. (2005). Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 19, 419–421. doi: 10.1096/fj.04-2622fje
Karwi, Q. G., Uddin, G. M., Ho, K. L., and Lopaschuk, G. D. (2018). Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 5:68. doi: 10.3389/fcvm.2018.00068
Kates, A. M., Herrero, P., Dence, C., Soto, P., Srinivasan, M., Delano, D. G., et al. (2003). Impact of aging on substrate metabolism by the human heart. J. Am. Coll. Cardiol. 41, 293–299. doi: 10.1016/s0735-1097(02)02714-6
Khaidakov, M., Heflich, R. H., Manjanatha, M. G., Myers, M. B., and Aidoo, A. (2003). Accumulation of point mutations in mitochondrial DNA of aging mice. Mutat. Res. 526, 1–7. doi: 10.1016/s0027-5107(03)00010-1
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Koonen, D. P., Febbraio, M., Bonnet, S., Nagendran, J., Young, M. E., Michelakis, E. D., et al. (2007). CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation 116, 2139–2147. doi: 10.1161/CIRCULATIONAHA.107.712901
Koves, T. R., Ussher, J. R., Noland, R. C., Slentz, D., Mosedale, M., Ilkayeva, O., et al. (2008). Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56. doi: 10.1016/j.cmet.2007.10.013
Kubli, D. A., Quinsay, M. N., and Gustafsson, A. B. (2013). Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun. Integr. Biol. 6:e24511. doi: 10.4161/cib.24511
Lakatta, E. G., and Yin, F. C. (1982). Myocardial aging: functional alterations and related cellular mechanisms. Am. J. Phys. 242, H927–H941. doi: 10.1152/ajpheart.1982.242.6.H927
Larsson, N. G. (2010). Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 79, 683–706. doi: 10.1146/annurev-biochem-060408-093701
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Li, Q., Ceylan-Isik, A. F., Li, J., and Ren, J. (2008). Deficiency of insulin-like growth factor 1 reduces sensitivity to aging-associated cardiomyocyte dysfunction. Rejuvenation Res. 11, 725–733. doi: 10.1089/rej.2008.0717
Li, H., Hastings, M. H., Rhee, J., Trager, L. E., Roh, J. D., and Rosenzweig, A. (2020). Targeting age-related pathways in heart failure. Circ. Res. 126, 533–551. doi: 10.1161/CIRCRESAHA.119.315889
Li, Y., Ma, Y., Song, L., Yu, L., Zhang, L., Zhang, Y., et al. (2018). SIRT3 deficiency exacerbates p53/Parkin mediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int. J. Mol. Med. 41, 3517–3526. doi: 10.3892/ijmm.2018.3555
Lopaschuk, G. D. (2017). Metabolic modulators in heart disease: past, present, and future. Can. J. Cardiol. 33, 838–849. doi: 10.1016/j.cjca.2016.12.013
Lopaschuk, G. D., Ussher, J. R., Folmes, C. D., Jaswal, J. S., and Stanley, W. C. (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258. doi: 10.1152/physrev.00015.2009
Ma, Y., and Li, J. (2015). Metabolic shifts during aging and pathology. Compr. Physiol. 5, 667–686. doi: 10.1002/cphy.c140041
Mahmoodzadeh, S., Eder, S., Nordmeyer, J., Ehler, E., Huber, O., Martus, P., et al. (2006). Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. 20, 926–934. doi: 10.1096/fj.05-5148com
Mallet, R. T., and Sun, J. (2003). Antioxidant properties of myocardial fuels. Mol. Cell. Biochem. 253, 103–111. doi: 10.1023/A:1026009519783
Mammucari, C., and Rizzuto, R. (2010). Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 131, 536–543. doi: 10.1016/j.mad.2010.07.003
Merz, A. A., and Cheng, S. (2016). Sex differences in cardiovascular ageing. Heart 102, 825–831. doi: 10.1136/heartjnl-2015-308769
Miyamoto, S. (2019). Autophagy and cardiac aging. Cell Death Differ. 26, 653–664. doi: 10.1038/s41418-019-0286-9
Moreau, R., Heath, S. H., Doneanu, C. E., Harris, R. A., and Hagen, T. M. (2004). Age-related compensatory activation of pyruvate dehydrogenase complex in rat heart. Biochem. Biophys. Res. Commun. 325, 48–58. doi: 10.1016/j.bbrc.2004.10.011
Newman, J. C., Covarrubias, A. J., Zhao, M., Yu, X., Gut, P., Ng, C. P., et al. (2017). Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 26, 547.e548–557.e548. doi: 10.1016/j.cmet.2017.08.004
Nguyen, T. N., Padman, B. S., and Lazarou, M. (2016). Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol. 26, 733–744. doi: 10.1016/j.tcb.2016.05.008
Noh, H. L., Okajima, K., Molkentin, J. D., Homma, S., and Goldberg, I. J. (2006). Acute lipoprotein lipase deletion in adult mice leads to dyslipidemia and cardiac dysfunction. Am. J. Physiol. Endocrinol. Metab. 291, E755–E760. doi: 10.1152/ajpendo.00111.2006
Ochoa, J. J., Quiles, J. L., Huertas, J. R., and Mataix, J. (2005). Coenzyme Q10 protects from aging-related oxidative stress and improves mitochondrial function in heart of rats fed a polyunsaturated fatty acid (PUFA)-rich diet. J. Gerontol. A Biol. Sci. Med. Sci. 60, 970–975. doi: 10.1093/gerona/60.8.970
Ock, S., Lee, W. S., Ahn, J., Kim, H. M., Kang, H., Kim, H. S., et al. (2016). Deletion of IGF-1 receptors in cardiomyocytes attenuates cardiac aging in male mice. Endocrinology 157, 336–345. doi: 10.1210/en.2015-1709
Okuda, Y., Kawai, K., and Yamashita, K. (1987). Age-related change in ketone body metabolism: diminished glucagon effect on ketogenesis in adult rats. Endocrinology 120, 2152–2157. doi: 10.1210/endo-120-5-2152
Ott, M., Robertson, J. D., Gogvadze, V., Zhivotovsky, B., and Orrenius, S. (2002). Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. U. S. A. 99, 1259–1263. doi: 10.1073/pnas.241655498
Palmer, J. W., Tandler, B., and Hoppel, C. L. (1977). Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 252, 8731–8739. doi: 10.1016/S0021-9258(19)75283-1
Paradies, G., Paradies, V., Ruggiero, F. M., and Petrosillo, G. (2019). Role of cardiolipin in mitochondrial function and dynamics in health and disease: molecular and pharmacological aspects. Cell 8:728. doi: 10.3390/cells8070728
Paradies, G., Ruggiero, F. M., Petrosillo, G., and Quagliariello, E. (1998). Peroxidative damage to cardiac mitochondria: cytochrome oxidase and cardiolipin alterations. FEBS Lett. 424, 155–158. doi: 10.1016/S0014-5793(98)00161-6
Park, T. S., Hu, Y., Noh, H. L., Drosatos, K., Okajima, K., Buchanan, J., et al. (2008). Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res. 49, 2101–2112. doi: 10.1194/jlr.M800147-JLR200
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Pelletier, A., Tardif, A., Gingras, M. H., Chiasson, J. L., and Coderre, L. (2007). Chronic exposure to ketone bodies impairs glucose uptake in adult cardiomyocytes in response to insulin but not vanadate: the role of PI3-K. Mol. Cell. Biochem. 296, 97–108. doi: 10.1007/s11010-006-9303-7
Pol, C. J., Lieu, M., and Drosatos, K. (2015). PPARs: protectors or opponents of myocardial function? PPAR Res. 2015:835985. doi: 10.1155/2015/835985
Puchalska, P., and Crawford, P. A. (2017). Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25, 262–284. doi: 10.1016/j.cmet.2016.12.022
Pyo, J. O., Yoo, S. M., Ahn, H. H., Nah, J., Hong, S. H., Kam, T. I., et al. (2013). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4:2300. doi: 10.1038/ncomms3300
Qu, X., Yu, J., Bhagat, G., Furuya, N., Hibshoosh, H., Troxel, A., et al. (2003). Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 112, 1809–1820. doi: 10.1172/JCI20039
Quarles, E., Basisty, N., Chiao, Y. A., Merrihew, G., Gu, H., Sweetwyne, M. T., et al. (2020). Rapamycin persistently improves cardiac function in aged, male and female mice, even following cessation of treatment. Aging Cell 19:e13086. doi: 10.1111/acel.13086
Quiles, J. L., Pamplona, R., Ramirez-Tortosa, M. C., Naudi, A., Portero-Otin, M., Araujo-Nepomuceno, E., et al. (2010). Coenzyme Q addition to an n-6 PUFA-rich diet resembles benefits on age-related mitochondrial DNA deletion and oxidative stress of a MUFA-rich diet in rat heart. Mech. Ageing Dev. 131, 38–47. doi: 10.1016/j.mad.2009.11.004
Ramos-Roman, M. A., Sweetman, L., Valdez, M. J., and Parks, E. J. (2012). Postprandial changes in plasma acylcarnitine concentrations as markers of fatty acid flux in overweight and obesity. Metabolism 61, 202–212. doi: 10.1016/j.metabol.2011.06.008
Rebrin, I., Bregere, C., Kamzalov, S., Gallaher, T. K., and Sohal, R. S. (2007). Nitration of tryptophan 372 in succinyl-CoA:3-ketoacid CoA transferase during aging in rat heart mitochondria. Biochemistry 46, 10130–10144. doi: 10.1021/bi7001482
Ren, X., Chen, L., Xie, J., Zhang, Z., Dong, G., Liang, J., et al. (2017). Resveratrol ameliorates mitochondrial elongation via Drp1/parkin/PINK1 signaling in senescent-Like Cardiomyocytes. Oxidative Med. Cell. Longev. 2017:4175353. doi: 10.1155/2017/4175353
Rezzani, R., Stacchiotti, A., and Rodella, L. F. (2012). Morphological and biochemical studies on aging and autophagy. Ageing Res. Rev. 11, 10–31. doi: 10.1016/j.arr.2011.09.001
Robinson, A. M., and Williamson, D. H. (1980). Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 60, 143–187. doi: 10.1152/physrev.1980.60.1.143
Rodriguez-Calvo, R., Serrano, L., Barroso, E., Coll, T., Palomer, X., Camins, A., et al. (2007). Peroxisome proliferator-activated receptor alpha down-regulation is associated with enhanced ceramide levels in age-associated cardiac hypertrophy. J. Gerontol. A Biol. Sci. Med. Sci. 62, 1326–1336. doi: 10.1093/gerona/62.12.1326
Rowe, G. C., Jiang, A., and Arany, Z. (2010). PGC-1 coactivators in cardiac development and disease. Circ. Res. 107, 825–838. doi: 10.1161/CIRCRESAHA.110.223818
Rubinsztein, D. C., Marino, G., and Kroemer, G. (2011). Autophagy and aging. Cell 146, 682–695. doi: 10.1016/j.cell.2011.07.030
Sachs, H. G., Colgan, J. A., and Lazarus, M. L. (1977). Ultrastructure of the aging myocardium: a morphometric approach. Am. J. Anat. 150, 63–71. doi: 10.1002/aja.1001500105
Salminen, A., Huuskonen, J., Ojala, J., Kauppinen, A., Kaarniranta, K., and Suuronen, T. (2008a). Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 7, 83–105. doi: 10.1016/j.arr.2007.09.002
Salminen, A., Kauppinen, A., Suuronen, T., and Kaarniranta, K. (2008b). SIRT1 longevity factor suppresses NF-kappaB-driven immune responses: regulation of aging via NF-kappaB acetylation? BioEssays 30, 939–942. doi: 10.1002/bies.20799
Sanchis-Gomar, F., Garcia-Gimenez, J. L., Gomez-Cabrera, M. C., and Pallardo, F. V. (2014). Mitochondrial biogenesis in health and disease. molecular and therapeutic approaches. Curr. Pharm. Des. 20, 5619–5633. doi: 10.2174/1381612820666140306095106
Sawada, M., and Carlson, J. C. (1987). Changes in superoxide radical and lipid peroxide formation in the brain, heart and liver during the lifetime of the rat. Mech. Ageing Dev. 41, 125–137. doi: 10.1016/0047-6374(87)90057-1
Shimazu, T., Hirschey, M. D., Newman, J., He, W., Shirakawa, K., Le Moan, N., et al. (2013). Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. doi: 10.1126/science.1227166
Shirakabe, A., Ikeda, Y., Sciarretta, S., Zablocki, D. K., and Sadoshima, J. (2016). Aging and autophagy in the heart. Circ. Res. 118, 1563–1576. doi: 10.1161/CIRCRESAHA.116.307474
Sihag, S., Cresci, S., Li, A. Y., Sucharov, C. C., and Lehman, J. J. (2009). PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 46, 201–212. doi: 10.1016/j.yjmcc.2008.10.025
Son, N. H., Basu, D., Samovski, D., Pietka, T. A., Peche, V. S., Willecke, F., et al. (2018). Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Invest. 128, 4329–4342. doi: 10.1172/JCI99315
Son, N. H., Park, T. S., Yamashita, H., Yokoyama, M., Huggins, L. A., Okajima, K., et al. (2007). Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J. Clin. Invest. 117, 2791–2801. doi: 10.1172/JCI30335
Son, N. H., Yu, S., Tuinei, J., Arai, K., Hamai, H., Homma, S., et al. (2010). PPARgamma-induced cardiolipotoxicity in mice is ameliorated by PPARalpha deficiency despite increases in fatty acid oxidation. J. Clin. Invest. 120, 3443–3454. doi: 10.1172/JCI40905
Squires, J. E., Sun, J., Caffrey, J. L., Yoshishige, D., and Mallet, R. T. (2003). Acetoacetate augments beta-adrenergic inotropism of stunned myocardium by an antioxidant mechanism. Am. J. Physiol. Heart Circ. Physiol. 284, H1340–H1347. doi: 10.1152/ajpheart.00473.2002
Stanley, W. C., Meadows, S. R., Kivilo, K. M., Roth, B. A., and Lopaschuk, G. D. (2003). Beta-hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am. J. Physiol. Heart Circ. Physiol. 285, H1626–H1631. doi: 10.1152/ajpheart.00332.2003
Steenman, M., and Lande, G. (2017). Cardiac aging and heart disease in humans. Biophys. Rev. 9, 131–137. doi: 10.1007/s12551-017-0255-9
Takahara, S., Soni, S., Maayah, Z. H., Ferdaoussi, M., and Dyck, J. R. B. (2021). Ketone therapy for heart failure: current evidence for clinical use. Cardiovasc. Res. cvab068. doi: 10.1093/cvr/cvab068 [Ebup ahead of print]
Taneike, M., Yamaguchi, O., Nakai, A., Hikoso, S., Takeda, T., Mizote, I., et al. (2010). Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6, 600–606. doi: 10.4161/auto.6.5.11947
Thai, P. N., Seidlmayer, L. K., Miller, C., Ferrero, M., Dorn, G. W. II, Schaefer, S., et al. (2019). Mitochondrial quality control in aging and heart failure: influence of ketone bodies and mitofusin-stabilizing peptides. Front. Physiol. 10:382. doi: 10.3389/fphys.2019.00382
Tomec, R. J., and Hoppel, C. L. (1975). Carnitine palmitoyltransferase in bovine fetal heart mitochondria. Arch. Biochem. Biophys. 170, 716–723. doi: 10.1016/0003-9861(75)90169-1
Ungvari, Z., Bailey-Downs, L., Sosnowska, D., Gautam, T., Koncz, P., Losonczy, G., et al. (2011). Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 301, H363–H372. doi: 10.1152/ajpheart.01134.2010
Veech, R. L., Chance, B., Kashiwaya, Y., Lardy, H. A., and Cahill, G. F. Jr. (2001). Ketone bodies, potential therapeutic uses. IUBMB Life 51, 241–247. doi: 10.1080/152165401753311780
Vina, J., Gomez-Cabrera, M. C., Borras, C., Froio, T., Sanchis-Gomar, F., Martinez-Bello, V. E., et al. (2009). Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 61, 1369–1374. doi: 10.1016/j.addr.2009.06.006
Viscarra, J. A., and Ortiz, R. M. (2013). Cellular mechanisms regulating fuel metabolism in mammals: role of adipose tissue and lipids during prolonged food deprivation. Metabolism 62, 889–897. doi: 10.1016/j.metabol.2012.12.014
Wang, X., West, J. A., Murray, A. J., and Griffin, J. L. (2015). Comprehensive metabolic profiling of age-related mitochondrial dysfunction in the high-fat-fed ob/ob mouse heart. J. Proteome Res. 14, 2849–2862. doi: 10.1021/acs.jproteome.5b00128
Warabi, E., Takabe, W., Minami, T., Inoue, K., Itoh, K., Yamamoto, M., et al. (2007). Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 42, 260–269. doi: 10.1016/j.freeradbiomed.2006.10.043
Whitehead, N., Gill, J. F., Brink, M., and Handschin, C. (2018). Moderate modulation of cardiac PGC-1alpha expression partially affects age-associated transcriptional remodeling of the heart. Front. Physiol. 9:242. doi: 10.3389/fphys.2018.00242
Williams, D. L., Spray, G. H., Hems, R., and Williamson, D. H. (1971). Metabolic effects of propionate in normal and vitamin B 12-deficient rats. Biochem. J. 124, 501–507. doi: 10.1042/bj1240501
Xu, X., Hua, Y., Nair, S., Zhang, Y., and Ren, J. (2013). Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J. Mol. Cell Biol. 5, 61–63. doi: 10.1093/jmcb/mjs055
Xu, X., Pang, J., Chen, Y., Bucala, R., Zhang, Y., and Ren, J. (2016). Macrophage migration inhibitory factor (MIF) deficiency exacerbates aging-induced cardiac remodeling and dysfunction despite improved inflammation: role of autophagy regulation. Sci. Rep. 6:22488. doi: 10.1038/srep22488
Yagyu, H., Chen, G., Yokoyama, M., Hirata, K., Augustus, A., Kako, Y., et al. (2003). Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J. Clin. Invest. 111, 419–426. doi: 10.1172/JCI16751
Zhang, Y., Bokov, A., Gelfond, J., Soto, V., Ikeno, Y., Hubbard, G., et al. (2014). Rapamycin extends life and health in C57BL/6 mice. J. Gerontol. A Biol. Sci. Med. Sci. 69, 119–130. doi: 10.1093/gerona/glt056
Keywords: cardiac aging, fatty acid oxidation, carbohydrate metabolism, ketone bodies, autophagy, mitochondria
Citation: Sithara T and Drosatos K (2021) Metabolic Complications in Cardiac Aging. Front. Physiol. 12:669497. doi: 10.3389/fphys.2021.669497
Edited by:
Adam R. Wende, University of Alabama at Birmingham, United StatesReviewed by:
Manoja Kumar Brahma, Université libre de Bruxelles, BelgiumJunco Shibayama Warren, The University of Utah, United States
John Reyes Ussher, University of Alberta, Canada
Copyright © 2021 Sithara and Drosatos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Konstantinos Drosatos, drosatos@temple.edu