 Emily M. Nakada
Emily M. Nakada Rui Sun
Rui Sun Utako Fujii
Utako Fujii James G. Martin
James G. Martin- 1Meakins-Christie Laboratories, Research Institute of the McGill University Health Centre (RI-MUHC), McGill University, Montreal, QC, Canada
- 2McGill University, Montreal, QC, Canada
The accumulation of unfolded/misfolded proteins in the endoplasmic reticulum (ER) causes ER stress and induces the unfolded protein response (UPR) and other mechanisms to restore ER homeostasis, including translational shutdown, increased targeting of mRNAs for degradation by the IRE1-dependent decay pathway, selective translation of proteins that contribute to the protein folding capacity of the ER, and activation of the ER-associated degradation machinery. When ER stress is excessive or prolonged and these mechanisms fail to restore proteostasis, the UPR triggers the cell to undergo apoptosis. This review also examines the overlooked role of post-translational modifications and their roles in protein processing and effects on ER stress and the UPR. Finally, these effects are examined in the context of lung structure, function, and disease.
Endoplasmic Reticulum Stress and the Unfolded Protein Response
Cells are normally in a state of proteostasis, whereby networks of signaling pathways work in concert to maintain the proper synthesis, folding, trafficking, and degradation of proteins. It is thought that a third of all proteins traffic through the endoplasmic reticulum (ER) for post-translational modifications (PTMs), folding, and trafficking (Huh et al., 2003). Under pathological or even physiological conditions, as well as in response to chronic stimuli, there is likely to be an accumulation of misfolded or unfolded proteins in the ER. This accumulation is referred to as ER stress and leads to the activation of the unfolded protein response (UPR) that inhibits de novo protein synthesis, while permitting the expression of protein-folding machinery and increasing degradation of unfolded proteins. If effective, the UPR attenuates ER stress and avoids cellular apoptosis (Hetz et al., 2015). Protein degradation or autophagy is an essential counterpart of protein synthesis and inhibition or a defect in autophagy leads to cell swelling. Autophagy is regulated by complex mechanisms which include pathways affecting cell metabolism, division, and autophagy, including the mevalonate pathway (Miettinen and Bjorklund, 2015). Further consideration of these pathways, however, is beyond the scope of this review.
The UPR Sensors
The UPR is a highly conserved response consisting of the three canonical receptors, protein kinase R-like ER kinase (PERK), inositol-requiring enzyme (IRE)1α, and activating transcription factor (ATF)6α, as well as the mediators that comprise each of their downstream signaling pathways (Hetz et al., 2015). Glucose-regulated protein 78 kDa (GRP78; binding immunoglobulin protein) binds all three receptors on the luminal surface of the ER membrane, where it acts as the master regulator of the UPR (Bertolotti et al., 2000; Shen et al., 2002). It simultaneously functions as a chaperone, directly aiding in the proper folding of unfolded proteins. Interestingly, in its role as a chaperone, GRP78 acts as the central regulator of the UPR. In response to ER stress, less GRP78 is bound to PERK, IRE1α, and ATF6α as it preferentially aids in the proper folding of proteins (Sundaram et al., 2018). GRP78 binds proteins with high promiscuity, recognizing and preferentially binding sequences containing hydrophobic amino acids that ordinarily would not be exposed in their properly folded state (Flynn et al., 1991). Thus, under conditions of high ER stress, GRP78 preferentially binds to unfolded proteins accumulating in the lumen of the ER, leaving PERK, IRE1α, and ATF6α unbound and in an active state. Consequently, all three receptors trigger mechanisms responsible for decreasing the unfolded protein load in an effort to return the cell to protein homeostasis. In contrast, under unstressed conditions, there are few unfolded proteins in the ER lumen, which consequently liberates GRP78 to bind the UPR receptors, which maintains them in their inactive state.
Inositol-requiring Enzyme 1α
Inositol-requiring enzyme1α, first discovered in 1993, is a type I transmembrane receptor consisting of both a protein kinase domain and an endoribonuclease domain that targets and splices out a sequence of 26 base pairs from the introns of mammalian X-box binding protein-1 (Xbp1) (Cox et al., 1993). This takes place following IRE1α disengagement from GRP78, its dimerization/oligomerization, autophosphorylation, conformational change, and activation of its ribonuclease (Figure 1). Spliced XBP1 (XBP1s), translated from Xbp1s mRNA, acts as a transcription factor that regulates the expression of ER stress-reducing chaperones and enzymes. Genes regulated by XBP1s are also involved in protein trafficking in and out of the ER and post-translational modifications (PTMs; Tirasophon et al., 1998; Tirosh et al., 2006; Oikawa et al., 2009). In contrast, unspliced XBP1 protein acts as a dominant-negative regulator of the UPR (Lee et al., 2003).
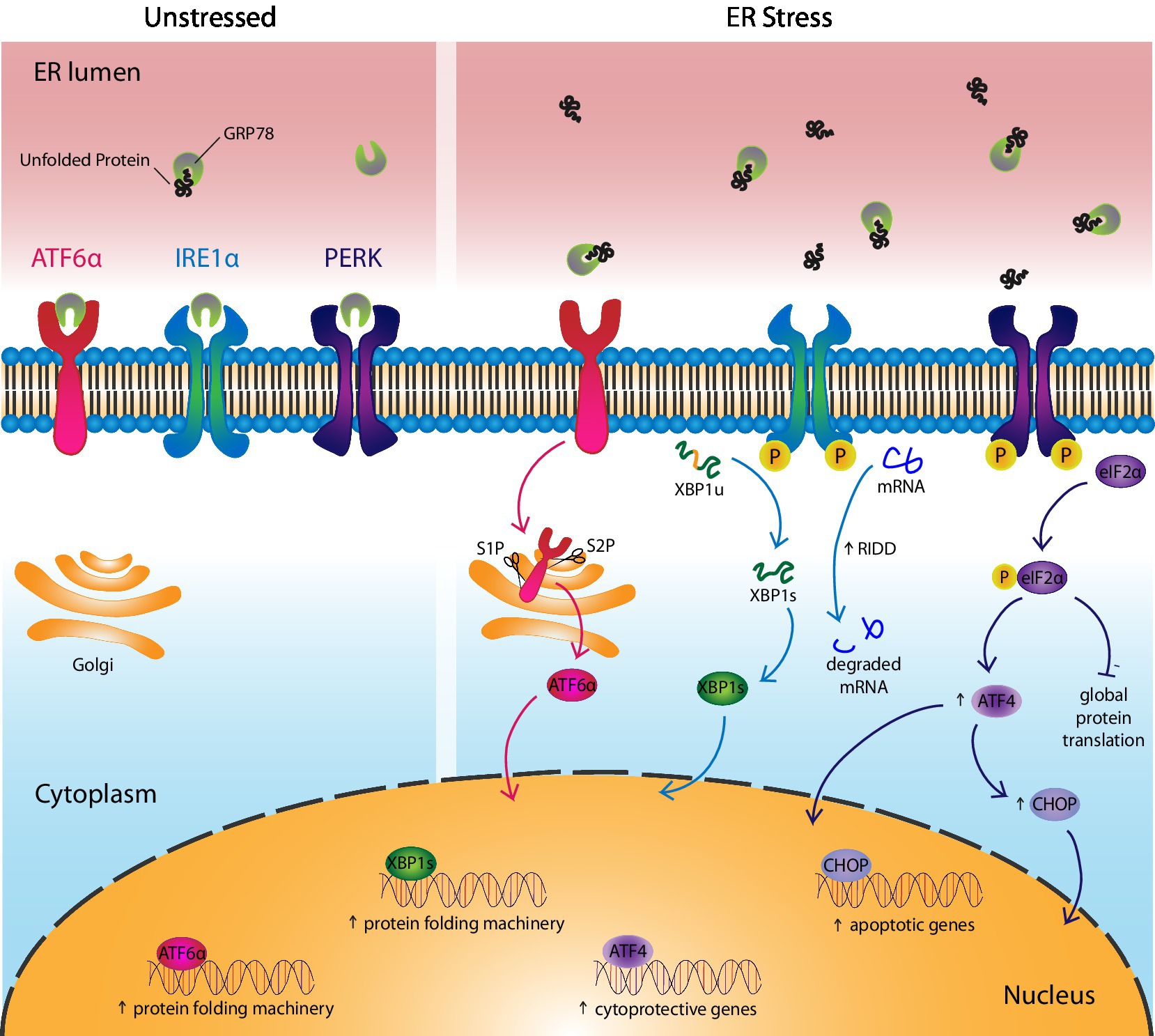
Figure 1. Cells respond to ER stress via UPR activation. The UPR consists of three canonical receptors, activating transcription factor 6α (ATF6α), inositol-requiring enzyme 1α (IRE1α), and protein kinase R-like ER kinase (PERK). (Left) Under unstressed conditions, GRP78, the UPR ligand and chaperone, binds the UPR receptors to maintain them in their inactive state. (Right) In response to ER stress, GRP78 leaves the UPR receptors and assists in the folding of unfolded proteins, thereby activating the receptors. More specifically, ATF6α translocates to the Golgi where it is cleaved by enzymes, site-1 protease and site-2 protease, before entering the nucleus as a transcription factor that upregulates genes involved in protein folding. IRE1α undergoes autophosphorylation and activates its endoribonuclease, which splices unspliced XBP1(u) mRNA into spliced XBP1(s). XBP1s protein acts as a transcription factor that upregulates genes involved in protein folding. IRE1α also reduces ER stress by splicing mRNAs into non-functional transcripts before they can be translated, a process called regulated IRE1-dependent decay. PERK autophosphorylates then phosphorylates eIF2α, which inhibits protein translation, with the exception of ATF4-regulated genes like CHOP. ATF4 upregulates cytoprotective genes and in the case of chronic ER stress, it induces apoptosis via CHOP.
There is also an XBP1-independent pathway downstream of the IRE1α sensor that regulates the UPR. In chronic ER stress, expression of XBP1 is reduced. The endoribonuclease activity of IRE1α switches from targeting Xbp1 for splicing to selectively degrading mRNAs that produce proteins that are notoriously difficult to fold accurately under ER stress conditions (Hollien and Weissman, 2006; Hollien et al., 2009; Hetz et al., 2011). This process is called the regulated IRE1-dependent decay (RIDD) pathway. This pathway alleviates ER stress by degrading mRNAs in the cytoplasm, before they can be translated into nascent proteins that aggravate ER stress. Finally, IRE1α plays an important role in the degradation of misfolded proteins via ER-associated degradation (ERAD), which is discussed in detail in the ERAD section of this review.
Activating Transcription Factor 6α
Unlike IRE1α, ATF6α is a type II transmembrane receptor first characterized as a UPR sensor in 1999 (Haze et al., 1999). ATF6α consists of a transmembrane domain that anchors it to the ER, a luminal domain that binds GRP78, and a basic leucine-zipper domain facing the cytoplasm (Hetz, 2012). The receptor is constitutively expressed as a 90 kDa protein that translocates to the Golgi after the release of GRP78, where it is activated following its cleavage into a 50 kDa product by site-1 protease (S1P) and site-2 protease (S2P; Figure 1; Ye et al., 2000; Shen et al., 2002). Activated ATF6α binds to ER stress-associated promoter sites in the nucleus to positively regulate ER stress-responsive genes like GRP78, GRP94, and calreticulin (CRT). It recognizes the ER stress response element with the sequence, CCAAT-N9-CCACG, in the promoter regions of these genes (Yoshida et al., 1998). Notably, ATF6β, which is also upregulated in response to ER stress, acts as a dominant negative regulator and may provide a mechanism through which the intensity and duration of ATF6α activation is regulated (Thuerauf et al., 2004). Although ATF6α is not essential for embryonic or post-natal development and despite considerable overlap in function with the other two UPR receptors, the importance of ATF6α appears central in orchestrating an effective chronic adaptive response to ER stress (Wu et al., 2007).
Protein Kinase R-like ER Kinase
Protein kinase R-like ER kinase is a type I transmembrane receptor that initiates a global reduction in protein synthesis, while selectively translating transcripts that extend the protein-folding capacity of the ER. PERK was first described in 1998 as a UPR sensor consisting of a luminal domain that binds GRP78, a transmembrane domain that traverses the ER membrane, and a cytoplasmic tail with protein kinase activity (Shi et al., 1998; Harding et al., 1999). Under ER stress conditions, PERK is released by GRP78, causing it to dimerize, autophosphorylate, and undergo a conformational change before phosphorylating eukaryotic initiation factor-2α (eIF2α; Figure 1). Phosphorylated (P)-eIF2α reduces protein translation by the competitive inhibition of eIF2β, a key component of an essential complex required in the initiation step of protein translation that allows transfer RNA binding to the AUG start codon (Gebauer and Hentze, 2004).
While P-eIF2α decreases global protein synthesis, it promotes the translation of select transcripts through alternative mechanisms like internal ribosomal entry sites or by bypassing inhibitory open reading frames (ORFs) upstream of target genes, as is the case with accessing the start codon of the Atf4 ORF (Harding et al., 2003; Ameri and Harris, 2008; Singleton and Harris, 2012). ATF4 regulates transcription of genes involved in cell metabolism, oxidative stress, and amino acid transport by binding C/ebp-Atf response element sequences of targeted genes (Kilberg et al., 2009). Many ATF4-regulated genes empower cells to respond to ER stress by increasing the protein folding capacity of the cell, including activating ATF6α by assisting in its synthesis and trafficking from the ER to the Golgi (Teske et al., 2011). However, under chronic ER stress conditions, the cell can undergo apoptosis through ATF4 upregulation of C/EBP Homologous Protein (CHOP) as part of the PERK-eIF2α-ATF4-CHOP axis. The details of this process are discussed in detail in the next section of the review.
Apoptosis
Although the cell responds to ER stress by increasing the protein-folding capacity of the cell, degrading misfolded/unfolded proteins, and decreasing de novo protein synthesis, the UPR can fall short of its ability to return the cell to proteostasis. Unalleviated ER stress-induced chronic UPR activation positively regulates CHOP expression to signal cellular apoptosis (Hu et al., 2018). CHOP, also known as growth arrest and DNA damage-inducible gene 153, is a transcription factor that is upregulated by the PERK-eIF2α-ATF4 axis, following ATF4-binding of the C/ebp-Atf response element sequence in its promoter. The IRE1α and ATF6α pathways of the UPR can also contribute to CHOP expression, but play secondary roles to that of PERK (Li et al., 2014).
C/EBP Homologous Protein consists of two functional domains, an N-terminal transcriptional activation domain and a C-terminal basic leucine zipper domain (Ubeda et al., 1996). CHOP functions by upregulating expression of pro-apoptotic and downregulating expression of anti-apoptotic members of the B cell lymphoma (BCL)2-family of proteins (Li et al., 2014). CHOP has been shown to downregulate expression of anti-apoptotic BCL2, BCL-XL, and myeloid cell leukemia sequence 1, while upregulating pro-apoptotic Bcl-2-like protein 11, which increases Bcl-2 homologous antagonist killer and Bcl-2-associated X (BAX) expression. BAX leads to the permeabilization of the mitochondrial membrane through the formation of permeability transition pores, allowing cytochrome c, second mitochondria-derived activator of caspases, apoptosis-inducing factor, and other apoptotic substrates to enter the cytoplasm where apoptosomes are formed. Activation of caspase-9, a component of the apoptosome, leads to the activation of execution caspases like caspases-3, -6, and -7, which cleave macromolecules and activate cytoplasmic endoribonucleases that proceed to cleave DNA, causing cell death (Siddiqui et al., 2015; Hu et al., 2018).
C/EBP Homologous Protein may also induce cell death through alternative mechanisms. Although controversial, studies show CHOP-induced death receptor 5 accumulation in the ER, in response to ER stress. This leads to caspase-8 activation, followed by the formation of the death-inducing signaling complex, which converges on activating the execution caspases to mediate apoptosis (Moon et al., 2013; Lu et al., 2014; Glab et al., 2017). CHOP-induced ER oxidoreductase (ERO)1α is another mechanism converging on the executing caspases that signal cell death. In a model of acute liver failure, ERO1α activity was shown to produce high levels of H2O2 in the process of oxidizing protein disulfide isomerases (PDIs) (Rao et al., 2015). PDIs are essential to the formation of disulfide bonds for proper protein folding and are therefore in high demand during ER stress. Thus, a potential consequence of accurately folding more proteins may be in elevating the production of H2O2, which could leak into the cytoplasm where it signals cell death through caspase-3.
ER-Associated Degradation
Because one-third of all proteins that traffic to the ER never exit the organelle as a functional protein, a mechanism of rapidly degrading terminally misfolded proteins in the ER is necessary. It is through ERAD that the proteins are retro-translocated to the cytoplasm where they are degraded. ERAD has long been considered a part of the UPR, as the expression of many of its mediators are regulated by UPR transcription factors; however, recent studies suggest that it is a separate protein quality control pathway that communicates closely with the UPR to maintain cellular proteostasis (Hwang and Qi, 2018). More specifically, IRE1α was recently determined to be an endogenous substrate of ERAD, making the ERAD machinery as much a regulator of, as it is regulated by, the UPR (Sun et al., 2015).
The ERAD machinery functions by first selecting appropriate substrates for degradation, followed by their retro-translocation to the cytosol, ubiquitinylation, and finally their proteasomal degradation (Figure 2). ERAD substrates are selected by OS9 or XTP3-B, lectin proteins that engage misfolded proteins and IRE1α, with the SEL1L-HRD1 ERAD complex (Fujimori et al., 2013; Sun et al., 2015). GRP78 competes with ERAD for these substrates, such that under basal conditions, GRP78-bound IRE1α is protected and stably expressed, whereas under ER stress, unbound IRE1α is more easily targeted for degradation. SEL1L is a cofactor for HRD1, a multi-spanning ubiquitin E3 ligase that forms a channel through the ER membrane through which ERAD substrates are transported to the cytoplasm. Substrate interaction with the SEL1L-HRD1 complex leads to its polyubiquitinylation, followed by proteasomal degradation (Sun et al., 2015; Wu and Rapoport, 2018; Li et al., 2020). In some instances, misfolded proteins that are too large, such as protein aggregates, are eliminated by ER-to-lysosome-associated degradation (Li et al., 2020). As the name suggests, these aggregates are engulfed by vesicles that are delivered to lysosomes where they are degraded.
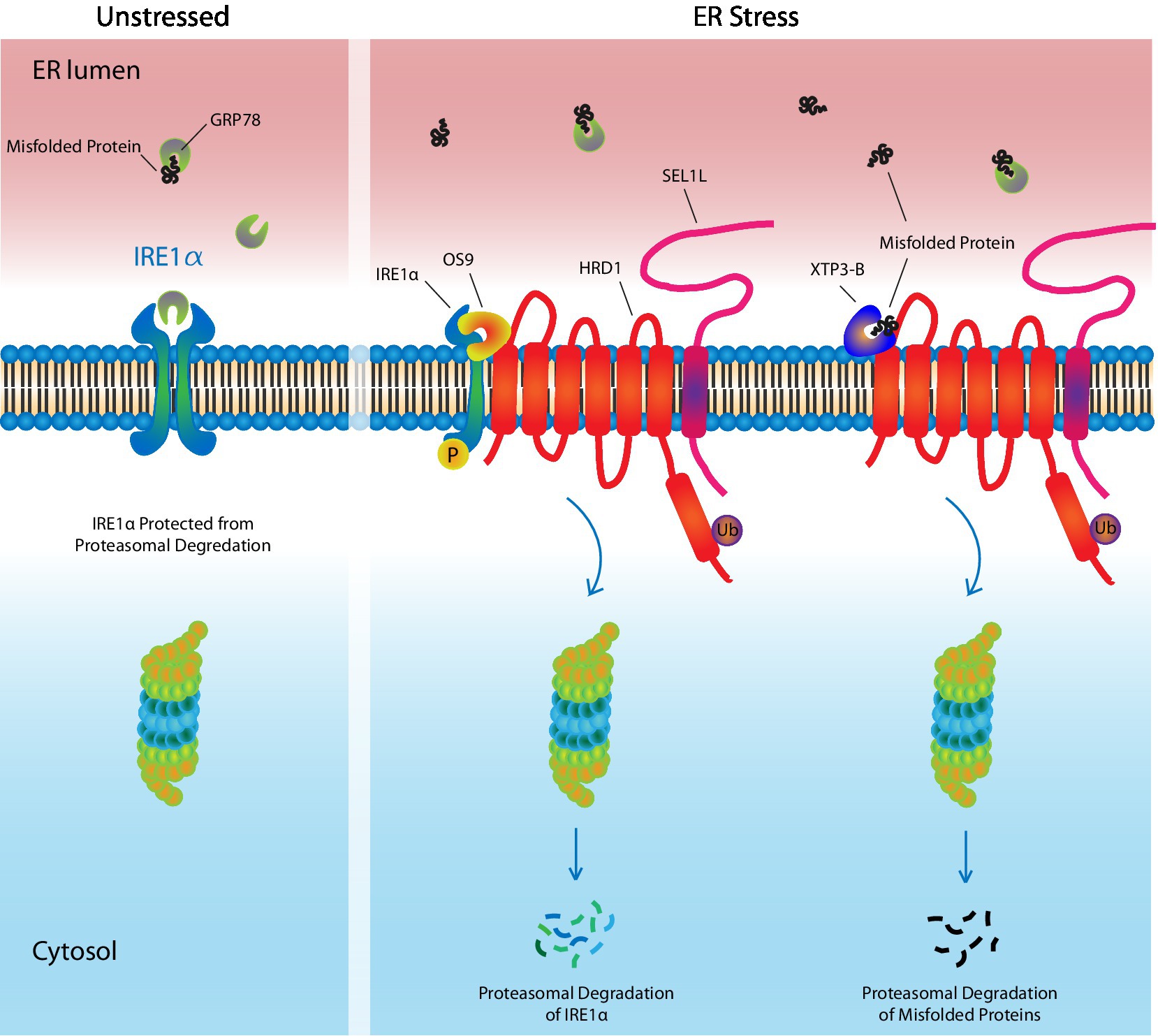
Figure 2. ER-Associated Degradation. Under unstressed conditions, the UPR receptor, IRE1α, is bound by the UPR ligand and chaperone, GRP78, which maintains it in an inactivate state, while also protecting it from targeted degradation by ERAD machinery. In response to ER stress, GRP78 leaves IRE1α and preferentially binds misfolded/unfolded proteins, which have accumulated in the ER lumen. This leaves IRE1α unprotected. OS9 and XTP3-B assist in targeting IRE1α, as well as misfolded proteins, to the HRD1/SEL1L complex where they undergo ubiquitinylation followed by proteasomal degradation, thereby reducing ER stress. Ub, ubiquitin.
Post-Translational Modifications
Approximately 5 amino acids are translated per second by each ribosome, with almost 2 min devoted to fully translating the average 438 residue protein in eukaryotic cells. In contrast, the average half time to properly fold proteins is between 30 and 60 min and takes an average of 1–2 h for proteins to be secreted (Braakman and Hebert, 2013; Sharma et al., 2019). So, while nascent proteins quickly enter the ER, the time-consuming course of protein-folding allows unfolded proteins to rapidly accumulate, in the absence of adequate protein-folding machinery. Protein folding can be a slow process because of the PTMs that proteins in the ER undergo, including signal peptide (SP) removal, N-linked glycosylation, disulfide bond (S–S) formation, palmitoylation, and proline hydroxylation (Ellgaard et al., 2016). In addition, many proteins, including most membrane glycoproteins and extracellular matrix proteins, undergo full or partial oligomerization in the ER before secretion (Hurtley and Helenius, 1989). This review will briefly summarize the three most common and well understood PTMs that occur in the ER, which are SP removal, N-linked glycosylation and S–S formation.
Signal Peptide Cleavage
The importance of the ER to the proper functioning of proteins cannot be overstated. The role of the ER begins with the SP, a short peptide sequence, generally within the first 25 amino acids translated by a ribosome that traffics novel proteins to specific organelles (Petersen et al., 2011). Although short in sequence, the SP consists of a hydrophobic N-terminal basic domain, a hydrophobic domain, and a polar domain containing the cleavage site (Hegde and Bernstein, 2006). The N-terminal and hydrophobic domains help position the peptide in a looped configuration during translocation to the ER. The cleavage-domain is oriented to face the lumen for immediate recognition and cleavage by the signal peptidase complex on the ER where translation continues (O’brien et al., 2014). The SP sequence can affect the efficiency of peptide cleavage, its maturation, and targeting, the last of which explains why some mature proteins can be directed to two distinct areas of the cell, such as CRT, which is co-localized to both the ER and the cytoplasm (Shaffer et al., 2005).
N-linked Glycosylation
Asparagine (N)-linked glycosylation is a highly conserved PTM with most secreted proteins from eukaryotic cells undergoing the alteration. In addition to its importance in protein folding, N-linked glycosylation is fundamental for molecular recognition, cell–cell communication, and protein stability (Braakman and Hebert, 2013; Mohanty et al., 2020). The enzymatic reaction involves the transfer of an oligosaccharide group from a donor substrate (lipid-linked oligosaccharide) to the acceptor substrate (asparagine residue) on newly synthesized proteins by the membrane-associated complex, oligosaccharyltransferase. Once transferred, N-linked oligosaccharides must be trimmed by glucosidases 1 and 2 to acquire a monoglucosylated glycan that can be recognized by the ER lectin molecules, calnexin (CNX) and CRT (Cherepanova et al., 2016). The lectin chaperones increase the efficiency of glycoprotein folding, prevent protein aggregation and premature exiting of the ER, and decrease misfolding by slowing down the kinetics of protein folding (Helenius, 1994; Price et al., 2012). The lectin chaperones recruit the oxidoreductase, PDI family A, member 3 (PDIA3; ERP57), and the peptidyl-prolyl isomerase, cyclophylin B, to assist in protein folding. Oligosaccharides on glycoproteins released by CNX and CRT may then be trimmed of a mannose residue by ER mannosidase I, before the glycoprotein is secreted or takes up permanent residence in the ER (Cherepanova et al., 2016). An error in N-linked glycosylation or excessive, sequential mannose trimming by ER degradation-enhancing α-mannosidases 1, 2 and 3, can lead to targeting of the misfolded glycoprotein for ERAD.
Disulfide Bond Formation
Oxidoreductases are enzymes that catalyze the transfer of electrons from one molecule, the donor/reductant, to another, the acceptor/oxidant. PDIs are thiol oxidoreductases that are essential in properly folding S–S-containing proteins. 29.5% of eukaryotic proteins are predicted to contain a S–S. While peptides of moderate length between 100 and 400 amino acids average less than 1 S–S, peptides less than 100 amino acids average a single bond, and large peptides with >400 amino acids average two bonds (Bosnjak et al., 2014). PDIs are involved in the formation, breakdown, and rearrangement of these bonds, meaning they oxidize, reduce, and isomerize S–Ss, respectively. During the formation of the disulfide bridges, PDIs oxidize thiol/sulfhydryl side chains (–SHs) on cysteine residues within and between peptide(s) to form intramolecular and intermolecular S–Ss, respectively (Figure 3; Ellgaard and Ruddock, 2005; Braakman and Hebert, 2013). These bonds often undergo isomerization before the protein achieves its final conformation. This involves an oxidized PDI that forms the initial bond, followed by the action of a reduced PDI that reduces the bond between the incorrect cysteine residues, before the now re-oxidized PDI can catalyze the new bond formation between the correct residues. These bonds help stabilize proteins in their correct tertiary and/or quaternary structures. To efficiently oxidize-SHs, PDIs require a highly oxidative environment like the ER lumen. In this environment, the oxidoreductase ERO1α can continuously re-oxidize PDIs (Appenzeller-Herzog, 2011). Recently, alternatives to ERO1α have been identified as PDI oxidants, including peroxiredoxin 4 and vitamin K epoxide reductase, but will not be discussed further (Wajih et al., 2007; Tavender et al., 2010).
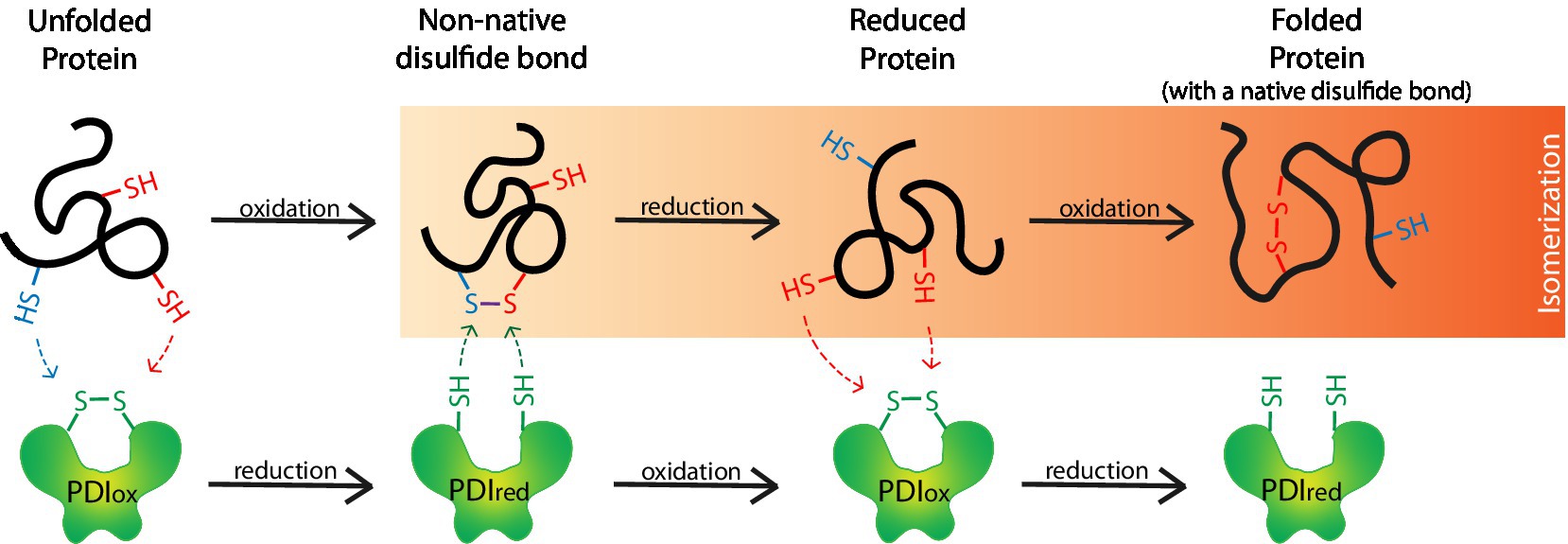
Figure 3. Protein disulfide isomerases (PDIs) form disulfide bridges that assist in the proper folding of proteins. PDIs (PDIox) oxidize thiol/sulfhydryl (–SH) side chains on unfolded proteins to form disulfide bonds (S–S) and are thereby reduced (PDIred). S–Ss often form between incorrect thiols (i.e., blue-SH with a red-SH) to form non-native S–Ss. When this occurs, the S–S undergoes isomerization whereby non-native S–Ss are reduced back to-SHs by a PDIred. A PDIox then oxidizes the correct-SHs (i.e., 2 red-SHs) on the reduced protein to form the correct native S–S and produce a properly folded protein.
More than 20 mammalian PDIs have been discovered that vary in their domains and activity, but all have at least one thioredoxin (Trx)-like domain. The number, location, redox potential, orientation, and electrostatic potential of their domains determine PDI function, including their ability to form, reduce and isomerize S–Ss, bind ERO1α and other substrates, retain proteins in the ER, traffic terminally misfolded proteins to the cytosol for proteasomal degradation, and whether they have chaperone activity (Okumura et al., 2015; Soares Moretti and Martins Laurindo, 2017). PDIA1, also simply referred to as PDI, was the first to be discovered and although ubiquitously expressed, is more highly expressed in secretory cells (Edman et al., 1985). It consists of four Trx-like domains (a, b, b' and a', starting from the N-terminus) in a “U” shape, with only the terminal ends having the catalytically active sequence Cys-X-X-Cys, and the b' domain binding substrate. PDIA1 in the oxidized state has a more open conformation compared to its reduced state, which could explain its ability to efficiently form disulfide bridges within and between a wide-range of substrates, bringing cysteine residues in close proximity to one another (Okumura et al., 2015). In contrast, a PDI like Erp27 is comprised of two non-catalytically active Trx-like domains, b and b', and is thought to bind and bring misfolded proteins to catalytically active PDIs like PDIA3 for S–S formation (Kober et al., 2013). Ultimately, PDIs are positively regulated by the UPR and contribute to the protein-folding machinery of the cell to attenuate ER stress.
Protein Processing in Lung Structure and Function
ER stress can occur under physiological conditions, including the G2/M phase of the cell cycle, in cells undergoing differentiation, and in secretory cells that continuously work on the maturation of proteins destined for secretion (Matsuzaki et al., 2015; Lee et al., 2019). However, acute and chronic ER stress, induced by endogenous and exogenous sources can challenge cells to return to proteostasis and may ultimately be detrimental to the proper functioning of cells, tissues, and organs. Tunicamycin (Tm), a chemical that induces ER stress by inhibiting N-linked glycosylation of proteins, has been administered to animals to study the effects of ER stress on the lungs. Tm was shown to worsen airway inflammation in an animal model of sepsis, enhance neutrophilic inflammation and airway hyperresponsiveness (AHR) in an ovalbumin-lipopolysaccharide model of asthma, and enhanced bleomycin-induced fibrosis (Lawson et al., 2011; Guo et al., 2017; Chen et al., 2020). Thus, augmenting ER stress in airway disease models in which ER stress is intrinsic to the disease, can worsen pathology.
Understanding the role of ER stress and the UPR can be difficult and is further complicated by the lack of methodology to quantify ER stress, considering the difficulty in producing a reliable reagent that can recognize all unfolded and misfolded proteins. Currently, the most reliable method measures ER dilation, typically by visualizing the expanded lumen of the ER by electron microscopy (Oslowski and Urano, 2011). Alternatively, mediators of the UPR, which are upregulated and/or activated in response to ER stress, are measured. However, because the UPR is a response to ER stress and not a direct measurement, it is important to correctly interpret the data. For example, an increase in the expression of GRP78 in the lungs of bleomycin-exposed mice would indicate an increase in ER stress. Deterioration of the disease in mice pre-treated with a siRNA targeting GRP78 could be due to either an increase or decrease in ER stress, following a decrease in chaperone activity provided by GRP78 or an increase in activation of the UPR with inadequate GRP78 to bind/inactivate the receptors, respectively. Thus, it is imperative that the role of ER stress and the UPR be interpreted alongside additional UPR mediators and readouts to discern whether a specific mediator of or the UPR in general plays a beneficial or harmful role in the pathogenesis of a disease.
Extracellular Matrix
Inhibition of the IRE1α pathway has been shown to improve TGFβ1-induced collagen and fibronectin production by fibroblasts from patients with idiopathic pulmonary fibrosis (IPF), cytokine-induced mucus production in human airway epithelial cells (AECs), and mucus production in the distal murine airway epithelia in murine models of fibrosis (Ghavami et al., 2018; Chen et al., 2019). GRP78 deficient mice showed greater airway remodeling, fibrosis, inflammation and mortality in one study, while CHOP deficient mice were protected from lung fibrosis in several murine models of fibrosis, including a bleomycin-induced model (Burman et al., 2018a; Borok et al., 2020). Thus, consistent with results from airway disease studies, GRP78 is likely to be protective, while CHOP expression may be damaging in IPF.
Idiopathic pulmonary fibrosis is a serious and often fatal interstitial lung disease characterized by fibrotic airway remodeling, progressive dyspnea, and respiratory failure (Burman et al., 2018b). Aberrant fibroblast, type II alveolar epithelial cell, and inflammatory cell activity are implicated in IPF progression. ER stress was first implicated in IPF with the discovery of mutations in surfactant protein C, a major protein secreted by type II alveolar epithelial cells, which can result in misfolding (Nogee et al., 2001). Since these cells are secretory in function, mutations in surfactant protein C can further elevate ER stress in these cells. The UPR markers GRP78, ERAD-enhancing α-mannosidase-like proteins, XBP1, CHOP, ATF4 and ATF6 have been detected at elevated levels in the lungs of IPF patients, especially in alveolar type II epithelial cells (Korfei et al., 2008; Lawson et al., 2008). These were accompanied by the increase in activation of pro-apoptotic pathways, specifically the cleavage of Bax and caspase-9. In addition, ER stress also promotes the epithelial to mesenchymal transition of alveolar type II epithelial cells, potentially contributing to the pool of pulmonary fibroblasts (PFs), culminating in the excessive deposition of extracurricular matrix (ECM; Tanjore et al., 2015; Kropski and Blackwell, 2018).
PFs are the primary cells responsible for the maintenance of healthy ECM in the parenchyma and disorders in their function can result in their differentiation into myofibroblasts, accompanied by the excessive production of ECM proteins and the stiffening and distortion of tissue as observed in interstitial lung diseases (Burman et al., 2018b). The elevated ER stress in PFs is associated with increased expression of GRP78 and all three of its receptors in PFs derived from IPF patients (Baek et al., 2012). TGFβ, the major growth factor that stimulates PF biosynthesis of ECM and differentiation into myofibroblasts, upregulates GRP78 and activates the IRE1α-XBP1 and ATF6α pathways in human PFs, which is in part due to oxidative stress (Baek et al., 2012; Ghavami et al., 2018). Inhibition of oxidative stress in cultured fibroblasts, using glutathione or N-acetyl cysteine, reduced TGFβ-induced GRP78, α-smooth muscle actin and type I collagen expression (Baek et al., 2012) Inhibition of ER stress with 4-phenylbutyric acid or GRP78 knock-down also reduced TGFβ-induced α-smooth muscle actin (αSMA) and type I collagen expression, while an IRE1 inhibitor alleviated TGFβ-induced myofibroblast differentiation and reduced their biosynthesis of collagen and fibronectin (Baek et al., 2012; Ghavami et al., 2018). In general, IRE1α activation drives myofibroblast differentiation by cleaving miR-150, a miRNA that suppresses αSMA expression (Heindryckx et al., 2016). In a bleomycin-induced murine model of fibrosis, an elevation in ER stress resulted in the activation of all three UPR-associated receptors in the whole lung and PFs, which was associated with PF proliferation and excessive collagen deposition (Baek et al., 2012; Hsu et al., 2017; Thamsen et al., 2019). ER stress inhibitors, tauroursodeoxycholic acid and 4-phenylbutyric acid inhibited PF proliferation through the reduced activation of the PI3K/AKT/mTOR pathway, subsequently ameliorating fibrosis and improving lung function (Hsu et al., 2017). Similarly, IRE1α-specific inhibition resulted in reduced lung collagen, hydroxyproline content and reversed bleomycin-induced fibrosis in mice (Thamsen et al., 2019).
Barrier Function
The primary role of AECs is to provide a physical barrier between the external environment and the inner milieu. This is accomplished through the mucociliary clearance (MCC) of inhaled microbes and small particles, the production and release of antimicrobial agents, and intercellular adherens and tight junctions (Ganesan et al., 2013). Adherens and tight junctions are located on the apicolateral membrane of epithelial cells and maintain contact with neighboring cells (Hartsock and Nelson, 2008). Tight junctions regulate the transport of ions and solutes in the intercellular space and consist of the transmembrane proteins, occludin and claudin, as well as scaffolding proteins zonula occludens-1, -2, and -3. The adherens junctions form, mature, and maintain cell-to-cell contact with adjacent cells and consist of the transmembrane proteins, E-cadherin and several intracellular proteins including p120-, α- and β-catenins.
In a murine model of allergic airways disease, the ER stress inhibitor, 4-phenylbutyric acid, prevented the allergen-induced loss of epithelial barrier function, represented by losses in zonula occludens-1 and E-cadherin expression, as well as reversing IL-25-induced loss of zonula occludens-1, E-cadherin, and trans-epithelial electrical resistance of 16HBE cells (Yuan et al., 2018). Although very little else is known about the role of ER stress and the UPR in maintaining the airway epithelial barrier, studies in other organ systems indicate that ER stress disrupts tight junctions between human retinal pigment epithelial cells and intestinal epithelial cells (Ma et al., 2016; Huang et al., 2019). Altogether, ER stress appears to decrease the protective physical barrier provided by tight and adherens junctions on epithelial cells and inhibiting allergen-induced loss of these junctions can be reversed using ER stress inhibitors.
Mucociliary Clearance
Mucociliary clearance is an innate defense mechanism of the lungs that is mediated by AECs and submucosal glands. The role of MCC is to trap, transport, and remove foreign particles, such as pollutants, pathogens, and allergens from the airways. Defects lead to bronchiectasis and other airway pathologies (Chalmers et al., 2018; Nikolic, 2018; Lucas et al., 2020). Cigarette smoke impairs mucociliary transport by inducing airway dehydration and increasing mucus viscosity, both in vitro and in vivo (Lin et al., 2020). ER stress has not yet been directly implicated. MCC function may be considered in three parts, namely, water and ion transport, mucin production, and ciliary beating. There is a variety of ion channels, including the cystic fibrosis (CF) transmembrane conductance regulator (CFTR), that allow AECs to control the trans-epithelial water flow and as such, controls the viscosity of the mucus and efficacy of the MCC (Hollenhorst et al., 2011). A deficiency or defect in CFTR expression on AECs can lead to severe respiratory impairment due to reduced Cl- secretion, resulting in increased Na+ reabsorption and excessive water absorption from fluid on the airway surface (Burch et al., 1995; Mall et al., 1998; Matsui et al., 1998). The increased viscosity and reduced MCC increase CF patient susceptibility to colonization by microbes like Staphylococcus aureus and Pseudomonas aeruginosa, resulting in chronic inflammation and airway damage (Cantin et al., 2015). Some proteins, like the CFTR, require the PTM, palmitoylation, before they can leave the ER. Preventing palmitoylation of the CFTR is known to inhibit the steady-state expression of both wild type and F508del mutant CFTR and reduce ion channel function on the surface of the cell. When corrected with specific protein acyl transferases regulating palmitoylation, CFTR expression is restored (Mcclure et al., 2014). Accumulating research also indicates that the IRE1α-XBP1 pathway of the UPR is more active in the lungs of CF patients, which promotes expression of pro-inflammatory cytokines like IL-8 (Ribeiro et al., 2005). Cultured human bronchial epithelial cells and alveolar macrophages from CF lungs express more XBP1s compared to controls and the inhibition of IRE1α decreases LPS-induced pro-inflammatory cytokine production in cultured CF cells.
The mucin component of the MCC traps pathogens and small particles. There are at least 21 genes encoding human mucins, 13 of which are expressed in AECs (Ma et al., 2018). Mucin maturation is achieved through an array of PTMs initiated in the ER where nascent mucin monomers undergo N-glycosylation before dimerizing and moving to the Golgi apparatus for O-glycosylation and higher-order multimerization (Ribeiro et al., 2005; Mcclure et al., 2014; Ma et al., 2018). Moreover IRE1β, acting through XBP1, is required for mucin production and its expression in bronchial epithelial cells is upregulated in the airways of asthmatics and CF patients (Martino et al., 2013). Expression of the ER molecular chaperone and member of the PDI family of ER proteins, Anterior Gradient (Agr)2, is increased with MUC5AC expression in the airway epithelium of Th2-high asthmatics and the lungs of mice with allergic airways disease (Schroeder et al., 2012). Immature MUC5AC can be found complexed with Agr2 and its expression reduced by 50% in allergen-challenged Agr2−/− mice. While its overall expression is reduced in Agr2−/− mice, more mucins accumulate at the level of the ER in these mice, where they contribute to ER stress. Tm-induced ER stress induces expression of MUC5AC and MUC5B through XBP1, ATF6α, and CHOP in human nasal epithelial cells (Kim et al., 2019).
Little is known about the contributions of ER stress and the UPR to ciliary beating. Ciliated cells are responsible for the propulsion of the mucus layer that covers the AECs. Proper ciliary function is absolutely required for effective MCC. In the apical region, there is an abundance of mitochondria that ensures ATP availability to sustain ciliary motion. Although its expression is not induced in response to ER stress, the PDI family member AGR3, is an ER resident protein that is uniquely expressed in ciliated cells of the airway epithelium where it regulates the ciliary beat frequency (Bonser et al., 2015). Agr3−/− mice have lower ciliary beat frequencies at baseline and in response to ATP. Finally, it is likely that ER stress would have adverse effects on the integrity of cilia, as their proteins require significant and various PTMs (Wloga et al., 2017).
Antioxidant Response
The lungs are exposed to thousands of liters of inhaled air each day that contain compounds with the potential to induce oxidative stress, defined as an imbalance of anti-and pro-oxidants. More specifically, oxidative stress can occur following exposure to exogenous oxidizing compounds like ozone, nitrous oxide, and chlorine, or endogenous sources like inflammatory cells that are recruited to the airways following exposure to particulates, allergens or microbes, but may also come from metabolically dysregulated cells or cells with a reduced anti-oxidant capacity (Knaapen et al., 2006; White and Martin, 2010; Kumarathasan et al., 2015). Cells respond to oxidative stress by means of an evolutionarily conserved antioxidant defense mechanism that is predominantly regulated by the transcription factor, nuclear factor erythroid 2-related factor (Nrf)2, and its endogenous inhibitor, Kelch-like ECH associated protein (Keap)1. Under normal conditions, Keap1 binds Nrf2 to sequester it in the cytoplasm and enhance its degradation. During oxidative stress, when electrophiles and reactive oxygen species (ROS) are present, Keap1 is dissociated from Nrf2, allowing Nrf2 to escape degradation and translocate to the nucleus where it positively regulates the expression of anti-oxidant genes like hemeoxygenase-1, the glutathione peroxidases, glutathione S-transferase, and NAD(P)H dehydrogenase quinone 1 by binding the antioxidant response elements in the promoter regions of these genes (Mcmahon et al., 2003; Holguin, 2013).
ER and oxidative stress are interconnected in their responses to physiological and pathological stressors. In fact, part of the anti-oxidant response and the UPR constitute pathways that comprise the integrated stress response (ISR), which hinges on the phosphorylation of eIF2α (Figure 4; Van’t Wout et al. 2014; Taniuchi et al., 2016). In eukaryotic cells, eIF2α is phosphorylated by four kinases, which are PERK, heme-regulated inhibitor (HRI) kinase, protein kinase R (PKR), and general control non-derepressible (GCN)2. While multiple stressors can activate the same kinase, a single stressor can also activate multiple kinases. In an in vitro study that knocked down expression of the four eIF2α kinases and re-introduced each kinase separately, the oxidative stress-inducers, H2O2 and carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone, phosphorylated eIF2α through HRI kinase and GCN2 (Taniuchi et al., 2016). Once activated, P-eIF2α inhibits global protein synthesis, thereby decreasing demand for oxidative folding, which reduces ROSs formed as a byproduct of the reaction and attenuates oxidative stress. P-eIF2α also reduces oxidative stress by upregulating ATF4, a mediator that has been shown to enhance Nrf2 recruitment to the anti-oxidant response element of oxidative stress-responsive genes like heme oxygenase-1 (He et al., 2001; Harding et al., 2003; Mimura et al., 2019). Finally, there is an eIF2α-independent mechanism through which the UPR regulates the antioxidant response. During ER stress, Nrf2 is directly phosphorylated by PERK, allowing its release from Keap1 so it can translocate to the nucleus where it upregulates expression of anti-oxidant genes (Cullinan et al., 2003; Cullinan and Diehl, 2004). Altogether, the UPR acts as a positive regulator of the antioxidant response via P-eIF2α inhibition of protein translation, ATF4 enhanced recruitment of Nrf2 to antioxidant response elements, and PERK phosphorylation of Nrf2.
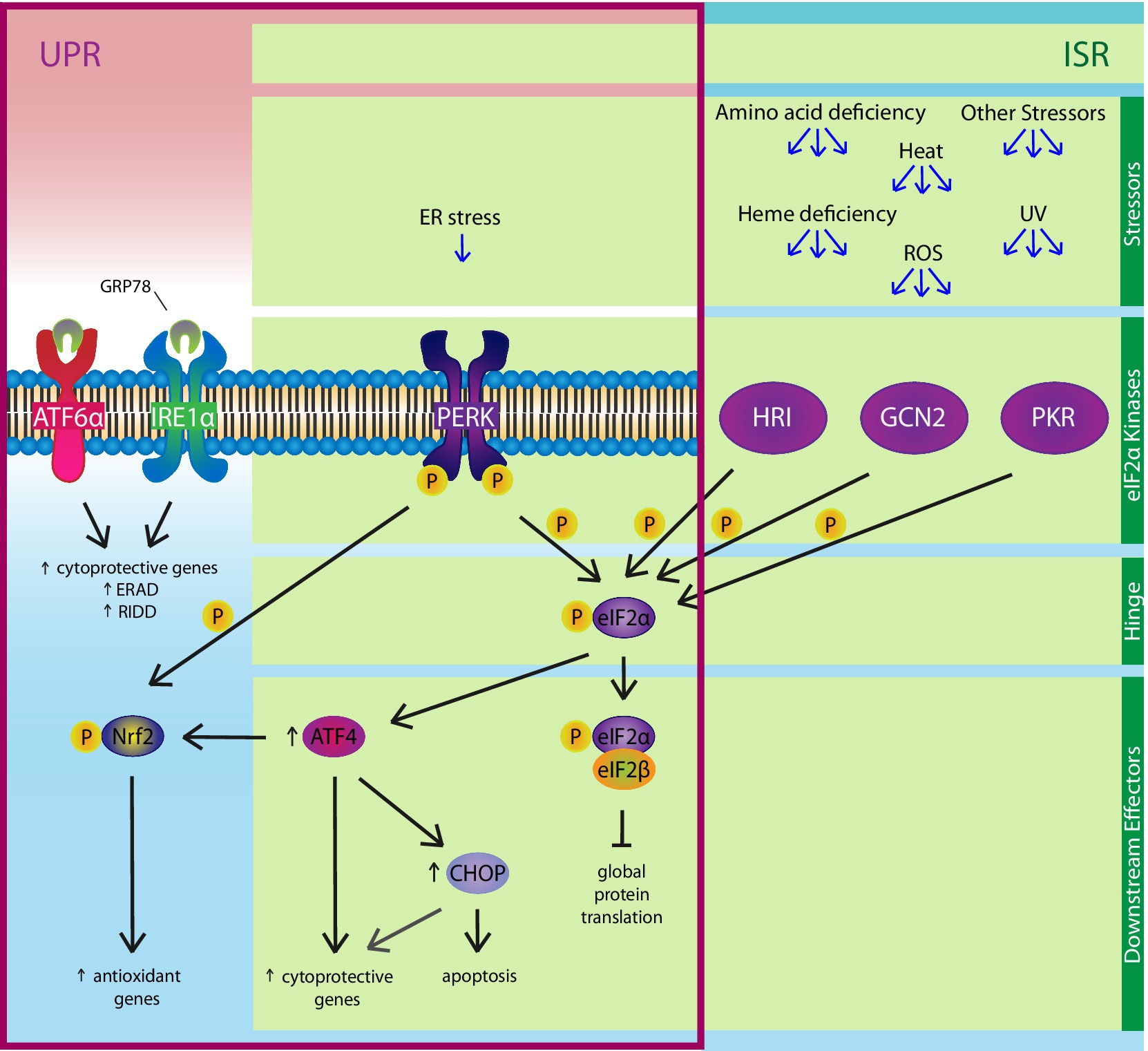
Figure 4. The Integrated Stress Response (ISR). The PERK pathway of the UPR is also a member of the ISR. Various stressors, including ER stress, amino acid deficiency, ultraviolet rays, heat, ROSs, and heme deficiency, can activate one or more of the four eIF2α kinases: PERK, HRI, GCN2, and PKR. The ISR hinges on eIF2α, which is phosphorylated by the four kinases. Phosphorylated eIF2α binds eIF2β, a key component of an essential complex involved in initiating protein translation, to inhibit global protein synthesis, except ATF4 and ATF4-regulated genes like CHOP. ATF4 positively regulates expression of cytoprotective genes, as well as upregulating CHOP, which can induce apoptosis under chronic ER stress conditions. Independent of the ISR, ER stress-induced activation of the PERK pathway can also enhance the anti-oxidant response by upregulating genes via the direct phosphorylation of nuclear factor erythroid 2-related factor (Nrf)2.
The redox potential of the cell and particularly that of the mitochondrion is tightly controlled and is essential for metabolism, cell growth and differentiation. When dysfunctional, oxidative stress may lead to mitophagy and autophagy. Cellular responses are initiated in the face of excessive oxidative stress in order to avoid these consequences and to promote cell survival (Dodson et al., 2013). Oxidative stress can interfere with the oxidative folding of unfolded proteins to induce ER stress. Correspondingly, dysregulated S–S formation in ER stress can allow ROSs to accumulate in the cell where they contribute to oxidative stress (Tavender and Bulleid, 2010). Most pathological airway studies on the topic focus on understanding the former and generally conclude that inhibiting oxidative stress (i.e., with antioxidants) leads to reduced ER stress, which decreases UPR activation and coincides with improved outcomes (Tagawa et al., 2008; Kenche et al., 2016; Wang et al., 2017a). However, consistent with topics covered in this review, we have focused on the latter, which is to examine the role of ER stress and the UPR on lung structure and function, in this case the antioxidant response in the lungs.
In a simple model of oxidative stress-induced airway injury, like hyperoxia, there is no concrete evidence of UPR activation that cannot also be attributed to the ISR, which shares the eIF2α-ATF4-CHOP axis (Figure 4). For example, in a murine model of hyperoxia-induced acute lung injury, CHOP expression increased, correlating with increased lung permeability and edema (Lozon et al., 2011). However, the expression of CHOP was confirmed to be downstream of the ISR eIF2α kinase, PKR, and not PERK. Interestingly, CHOP−/− mice were more sensitive to hyperoxia-induced acute lung injury than wild type mice and had a higher rate of mortality, indicating that CHOP expression is protective in this model. This could be the result of CHOP regulation of genes besides those related to apoptosis, which could be attributed to differences in the mechanism of CHOP activation, in this case by PKR (or HRI and GCN2) vs. PERK (Vij et al., 2008; Lozon et al., 2011; Yang et al., 2017). In other studies, hyperoxia attenuated the expression of UPR mediators GRP78 and PDIA3 (Gewandter et al., 2009; Xu et al., 2009). Both the overexpression and inhibition of GRP78 had no effect on ROS production or UPR activation, while overexpression and siRNA knockdown of PDIA3 increased and decreased hyperoxia-induced apoptosis of endothelial cells, respectively. Altogether, these studies indicate that ER stress and the UPR do not play significant roles in hyperoxia-induced airway injury, while activating the UPR in a model of disease without ER stress could aggravate rather than ameliorate oxidative stress-induced airway injury.
Expanding on our understanding of ER stress and the UPR in disease, we investigated their roles in complex models of oxidative stress-induced airway injury in which ER stress was also induced. In vivo and in vitro exposure to cigarette smoke extract is known to induce both stress responses (Lin et al., 2017b, 2019). Raising the protein folding capacity of lung cells by administering an ER stress inhibitor/chemical chaperone reduced cigarette smoke extract-induced airway remodeling and emphysema in the rat, which coincided with an augmentation in the antioxidant response (Lin et al., 2019). In a bleomycin-induced model of fibrosis, the adoptive transfer of mesenchymal stem cells reduced airway fibrosis and attenuated ER stress through PERK-Nrf2, but not the PERK-eIF2α-ATF4-CHOP pathway, suggesting that the ER stress-induced activation of the non-canonical PERK-Nrf2 pathway of the UPR may have a protective role in complex airway diseases (Ono et al., 2015; Lee et al., 2020). Similarly, activation of the PERK-Nrf2 pathway was suppressed in immortalized AECs, as well as blood cells and lung tissues from patients with CF and reversal of the pathway by salubrinal decreased inflammatory responses to flagellin and P. aeruginosa (Blohmke et al., 2012). Finally, the neutrophilic inflammation and edema that characterized lipopolysaccharide-induced acute lung injury were ameliorated through the PERK-Nrf2 pathway using the plant-derived alkaloid berberine (Liang et al., 2019). Thus, in contrast to hyperoxia-induced airway injury, disease outcomes could be improved by inhibiting ER stress or activating the PERK-Nrf2 pathway in complex airway diseases. Unfortunately, there are few other studies addressing the role of ER stress in airway diseases where the antioxidant response was is measured.
Bronchomotor Tone
Airway smooth muscles (ASMs) constrict in response to contractile agonists, which are the primary elements that increase bronchomotor tone and subsequently limit airflow (Martin et al., 2000). Pathological alterations in ASM characteristics have been extensively documented in airway inflammatory diseases, especially asthma and COPD (Bosken et al., 1990; Ozier et al., 2011). The increases in ASM mass observed in both diseases are likely the combined result of ASM cell (ASMC) hypertrophy and hyperplasia (Bosken et al., 1990; Ozier et al., 2011). These changes are proposed to contribute to overall elevated force generation and worsened airway narrowing (Lambert et al., 1993). The biological mechanisms mediating ASM remodeling are not fully elucidated and the precise role of ER stress is unknown.
It has been established that the phenotypes of smooth muscle cells in general display a dichotomy of either contractile or proliferative/secretory characteristics (Dekkers et al., 2012). Current evidence suggests that growth factors and inflammatory mediators in diseased airways promote the conversion of ASM to the proliferative phenotype and induce hyperplasia (Bentley and Hershenson, 2008). Pathways related to ER stress could dependently or independently participate in such processes, but there is as yet no direct evidence showing the relationship between ER stress and ASMC properties. However, research on other smooth muscles suggests that ER stress in general can act as a promoter of the proliferative smooth muscle phenotype. For example, fibroblast growth factor-2 upregulates ATF4 expression, which is directly responsible for inducing rat vascular smooth muscle proliferation (Malabanan et al., 2008). Platelet-derived growth factor also activates the IRE1α-XBP1 pathway of the UPR in vascular smooth muscle cells and drives proliferation via the downregulation of calponin h1 (Zeng et al., 2015). Vascular smooth muscle cells from mice with smooth muscle-specific knock-out of CHOP exhibit decreased proliferation induced by atherosclerotic lesions, due to the accumulation of the growth-inhibiting factor KLF4 (Zhou et al., 2015).
A recent study featuring the analysis of existing gene array data revealed an inverse relationship between the in vivo expression of ER stress markers and contractile proteins in human bladder smooth muscle and arterial smooth muscle (Zhu et al., 2020). The induction of ER stress in human bladder smooth muscle cells, using dithiothreitol or Tm, reduced contractile protein expression in an IRE1α-XBP1-dependent manner. Conversely, overexpression of myocardin, a master transcription factor of contractile genes and myocardin-related transcription factor suppressed the induction of ER stress. Though the proliferative properties of smooth muscle were not investigated, this study hinted that ER stress demonstrates a mutually antagonistic relationship with the contractile phenotype, potentially inducing pathological changes in smooth muscle. There is a potential that increased ER stress participates in the induction of the proliferative phenotype in ASMs through these processes, leading to remodeling and associated pathophysiology. Experimental evidence specifically conducted on ASM is required to confirm this hypothesis.
ASMs respond to a large variety of inflammatory mediators, which could induce ER stress. Notably, TNFα selectively activates the IRE1α-XBP1 pathway in cultured ASMCs but not the PERK or ATF6 pathways, which can be inhibited by the superoxide scavenger, tempol (Yap et al., 2020). IRE1α activation subsequently results in Mfn2 downregulation, a factor responsible for mitochondrial fusion and tethering to the ER, leading to an increase in mitochondrial fission (Delmotte and Sieck, 2019). Since mitochondria tethered to the ER absorb Ca2+ during its release from the ER and act as a buffering agent to control cytosolic [Ca2+], the authors also argue that the loss of Mfn2 due to IRE1α-XBP1 activation is responsible for TNFα-mediated mitochondrial dissociation from the ER (Delmotte et al., 2017). This corresponds to impaired Ca2+-buffering in the mitochondria and leads to increased cytosolic Ca2+ influx upon contractile agonist stimulation, subsequently contributing to the increased contractility of ASMCs (Delmotte and Sieck, 2019).
Tm-induced ER stress in murine ASMCs initiates synthesis of hyaluronan, an ECM protein observed in higher abundance within the asthmatic airway submucosa (Roche et al., 1989; Lauer et al., 2009). In this manner, ASMs secrete ECM proteins that directly contribute to the remodeling of the extracellular environment (Bourke et al., 2011). Conversely, ASMCs sense the ECM environment, which alters their contractile/proliferative phenotypes (Freyer et al., 2001). This may be accomplished through the increased infiltrative capacity of leukocytes as high hyaluronan content in the ECM has been shown to enhance leukocyte adhesion to ASM-derived matrix (Lauer et al., 2009). Mast cells and CD4+ T cells have been observed to infiltrate ASM bundles in higher numbers in asthmatics, where they potentially mediate some of the functional changes associated with asthma pathophysiology (Brightling et al., 2002; Ramos-Barbon et al., 2010). T cells can exert pro-proliferative effects on ASMCs via hyaluronan-specific binding (Al Heialy et al., 2013). Altogether, these data indicate that ER stress in ASMs play a role in ECM remodeling and the ECM can in turn enhance recruitment of leukocytes to ASMs where they induce ASMC proliferation.
Airway Inflammatory Response
The inflammatory response is a physiological response to injury. Inflammatory cells, including macrophages, eosinophils, neutrophils, and lymphocytes, are cells that migrate to the site of injury where they interact directly with the source of injury or infection and release mediators that coordinate the removal of harmful stimuli and initiate repair (Aghasafari et al., 2019). However, on occasion, the response does more damage than good, as is the case with some airway inflammatory diseases, such as COPD and asthma. The inflammatory profile of a disease can also vary based on the type of insult or injury, its duration, as well as genetic and epigenetic factors, health history, and condition of the host (Perez-Novo and Bachert, 2015; Wesolowska-Andersen and Seibold, 2015).
The immune response to injury almost always induces some degree of ER stress since among other considerations, inflammatory cytokines and chemokines rely heavily on the ER for their maturation; proliferating (immune) cells double their protein content before undergoing cell division; and de novo protein synthesis is essential for tissue repair and cell differentiation in response to injury (Iwakoshi et al., 2003b; Brunsing et al., 2008; Waldschmitt et al., 2014). Nevertheless, while ER stress is induced in airway inflammatory disease, less is known of the specific roles of the three canonical pathways of the UPR. Here, we address the role of the UPR in immune cell development, maturation, differentiation, and function. We also explore the profiles of UPR activation in the context of airway inflammatory disease and injury.
The highly conserved, IRE1α-XBP1 axis is the best studied of the three pathways of the UPR and is the most critical to the development, maturation, differentiation, survival, and function of most hematopoietic cells. A study looking at temporal changes in activity determined that the IRE1α-XBP1 pathway is active at early stages of T-lymphocyte development and differentiation, including CD4+CD8+ (double positive) thymic T cells, compared to mature T cells (Brunsing et al., 2008). IRE1α-XBP1 is also activated in CD8+ T cells, in response to bacterial and viral infections and the pathway plays an important role in terminal effector functions (Kamimura and Bevan, 2008). In CD4+ Th2 cells, the inhibition of IRE1α attenuates the secretion of interleukin (IL)-5, but not IL-4 (Poe et al., 2019). IL-5 is still produced, but is retained within the cell, indicating that IRE1α is specifically involved in the PTM and maturation of IL-5 that is required for its release. This pathway is also active at early stages of B-lymphocyte differentiation, including pro-B cells in the bone marrow and is less active in mature B cells (Brunsing et al., 2008). It is not essential for B cell cytokine production or survival, but is required for the terminal differentiation of plasma cells and the production and secretion of immunoglobulin M (Reimold et al., 2001; Iwakoshi et al., 2003a,b; Tirosh et al., 2005). The IRE1α-XBP1 pathway may be essential for early stage dendritic cell (DC) development, survival, and type-I interferon production in response to TLR9 agonists, but appear to be less important in committed CD11c-expressing DCs (Iwakoshi et al., 2007; Osorio et al., 2014). In granulocytes, XBP1 is required for eosinophil development, differentiation, and survival, in addition to the production of eosinophil granules (Bettigole et al., 2015). Although XBP1 is dispensable for neutrophil and basophil survival, an in vitro study using a human leukemia cell line shows that IRE1α activity is increased in differentiating neutrophils, while ATF6α and PERK activity are suppressed (Bettigole et al., 2015; Tanimura et al., 2018). Finally, an inhibitor of IRE1α kinase activity was shown to induce cell death in a mast cell leukemia cell line, indicating that this pathway may be important in mast cell survival (Mahameed et al., 2019). Altogether, IRE1α and its downstream mediators appear to be critical to the proper development, survival, and function of most, if not all, hematopoietic cells.
Apart from the IRE1α pathway, there is a significant gap in our understanding of the role of the UPR in inflammatory cell development and function. What is known is that differentiating macrophages have been shown to upregulate expression of the ER chaperones, GRP78 and GRP94, in addition to XBP1s (Dickhout et al., 2011). Macrophages may also rely on ER stress to differentiate into the M2 phenotype as the ER stress inhibitor, phenylbutyric acid, was shown to inhibit M2 differentiation (Oh et al., 2012). Although the precise arms of the UPR involved in regulating the M2 phenotype is unclear, there is evidence of both IRE1α and PERK activity. Similarly, the IRE1α and PERK pathways have been implicated in mast cell survival and DC production of IL-23 (Goodall et al., 2010; Marquez et al., 2017; Mahameed et al., 2019). GRP94-deficient B cells can survive, develop and even function properly (Randow and Seed, 2001). However, these cells produce significantly fewer antibodies following TLR activation and have defects in integrin formation (Melnick et al., 1992; Randow and Seed, 2001; Liu and Li, 2008; Wu et al., 2012; Pagetta et al., 2014). GRP78 is crucial for the assembly of immunoglobulin chains, binding the H and L domains, and it binds the TCR until assembly partners can come in to complete assembly (Haas and Wabl, 1983; Hendershot, 1990; Melnick et al., 1992; Vanhove et al., 2001). In hematopoietic stem cell progenitors, experiments in which the ER chaperone, CRT, was overexpressed or silenced indicated that CRT may be essential in the differentiation of erythroid cells and megakaryocytes (Salati et al., 2017). These studies indicate that the UPR and its mediators are important and even central to the maturation and function of many immune cells, which could make them ideal candidates for targeted therapy in complex diseases.
In previous sections, we addressed AECs and their importance in maintaining a physical barrier between the environment and the inner milieu and in MCC. However, AECs are also important participants in innate immune responses. These cells represent the first line of defense against harmful pathogens. Several chronic airway inflammatory diseases have been associated with increased epithelial proinflammatory cytokine production (Machen, 2006). There may also be evidence of ER stress; for example, airway infections activate XBP1 and increase Ca2+ stores to amplify Ca2+-dependent IL-8 secretion in vitro (Martino et al., 2009). Human epithelium in CF patients show higher IRE1α/XBP1 activation by ER stress and induces cytokine production (Hull-Ryde et al., 2021). ER stress boosts TLR-mediated IL-6 and IL-8 expression and secretion via PERK-and ATF6-mediated p38 and ERK activation in human primary bronchial epithelial cells (Mijosek et al., 2016). Additionally, house dust mite-induced ATF6 activation is associated with AEC death, hyperresponsiveness and subsequent airway fibrosis in mice (Hoffman et al., 2013). It also increases the production of IL-25, which increases CHOP and P-PERK expression and induces epithelial tight junction injury and cell apoptosis in human bronchial epithelial cells (Yuan et al., 2018). Cigarette-smoke increases the expression of CHOP, caspase-12 (an ER stress-induced mediator of apoptosis), and other markers of apoptosis in rat lungs. The nicotine component of cigarette smoke also increases the expression of CHOP, caspase-12, and apoptosis in human bronchial epithelial cells (Lin et al., 2017a). In infection, influenza A virus (IAV)-induced ER stress activates ATF6, but not CHOP. This activation of the ER stress response induces caspase12–dependent apoptosis of and TGFβ production by murine epithelial cells (Roberson et al., 2012). Deletion of Grp78 in alveolar type 2 cells in mice results in ER stress, apoptosis, senescence, and activation of TGFβ, with resulting lung fibrosis (Borok et al., 2020).
In inflammatory diseases of the airways, mechanisms that reduce ER stress and/or enhance UPR activation generally improve outcomes, including asthma. Asthma is a heterogeneous and complex disease in which the UPR is activated in response to the ER stress in the lungs (Pathinayake et al., 2018). Further enhancement of ER stress in an allergen-induced model of asthma by Tm administration increases airway cytokine production, inflammation, and AHR (Guo et al., 2017). In contrast, the attenuation of ER stress in murine models of asthma, via the administration of ER stress inhibitors like tauroursodeoxycholic acid, the epithelium-specific ablation of PDIA3, or the siRNA-targeted inhibition of PDIA3 and ATF6, attenuate allergen-induced ER stress, AHR, inflammation, and fibrosis (Hoffman et al., 2016; Siddesha et al., 2016; Nakada et al., 2019). In a genome-wide association study, the ORMDL3 (ORMDL sphingolipid biosynthesis regulator 3) gene was identified as having a strong association with asthma (Moffatt et al., 2007). This gene regulates ER stress by regulating Ca2+ signaling and increased expression leads to an attenuation of ER-mediated Ca2+ signaling and increases activation of the UPR, specifically activating the ATF6α arm (Cantero-Recasens et al., 2010; Miller et al., 2014). ORMDL3-deficient mice are protected in a murine model of asthma with reduced AHR, lung eosinophils, allergen-specific serum IgE, and IL-6 in response to the fungus, Alternaria alternata, while overexpression of ORMDL3 enhanced AHR in this model (Loser et al., 2017). Additionally, ORMDL3, which is predominantly expressed in AECs, is strongly associated with AHR, as well as airway remodeling, inflammation, and mucus hypersecretion, in other allergen-models of asthma (Miller et al., 2012, 2014; Oyeniran et al., 2015).
Several UPR-related mediators are upregulated in the lungs of tobacco smokers compared to non-smokers, including GRP78, CRT, and PDIA1 (Kelsen et al., 2008). Cigarettes are a major risk factor for the development of COPD, of which emphysema and chronic bronchitis are the most common features. An abnormal inflammatory response and ER stress are both characteristic of the disease with reports of increased expression of ERAD proteins, ubiquitinylated proteins, and accumulation of misfolded protein aggregates in the lungs of patients with severe emphysema (Min et al., 2011). Cigarette smoke affects the protein-folding capacity of the cell by damaging proteins involved in the proper folding of nascent proteins. For example, the lungs of mice exposed to cigarette smoke have 4–6 times higher oxidized and sulfenylated PDIA1 compared to control mice, but are completely deficient in the reduced form of PDIA1 (Kenche et al., 2013, 2016). A deficiency in the reduced form of PDIs adversely affects the isomerization of S–Ss as they require their reductase activity to break the initial bonds before rearrangement. Moreover, most PTMs to PDIs, including highly oxidized or sulfenylated PDIs, are associated with reduced enzymatic activity. Cysteine and tyrosine modifications to PDIA1 by cigarette smoke extract and some of its known pro-oxidative components like H2O2, peroxynitrite (a reactive nitrogen species), acrolein and hydroxyquinone (long lived free radicals) can alter PDIA1 structure and function, which effectively reduces overall protein folding, activates the UPR, and decreases chaperone, reductase, and isomerase activity (Kenche et al., 2016).
Strategies that reduce ER stress in the lungs of animals exposed to cigarettes have reduced airway inflammation, apoptosis, and remodeling (Lin et al., 2017b; Wang et al., 2017b; Lin et al., 2019). IRE1α, CHOP, and GRP78 that are upregulated in the lungs of cigarette smoke-exposed mice are reduced in mice treated with the ER stress inhibitor, 4-phenylbutyric acid (Wang et al., 2017b). Moreover, BALF chemokines and inflammatory cell numbers, including neutrophils, are significantly reduced in these mice. In another study, the overexpression of GRP78 and inhibition of the downstream, apoptosis-inducing UPR transcription factor, CHOP, significantly reduced cigarette smoke extract-induced apoptosis in an AEC line, further indications that enhancing the protein folding capacity of cells may be protective against cigarette smoke-induced airway injury and progression of COPD (Tagawa et al., 2008).
Patients with cystic fibrosis are prone to chronic airway infection and a corresponding and sustained inflammatory response that is detrimental to lung structure, health and function. The CF lung has significantly greater inflammation than the normal lung, which is associated with the activation of the IRE1α arm of the UPR (Lara-Reyna et al., 2019). IRE1α is upregulated in innate immune cells, mainly monocytes, neutrophils, and M1 macrophages. Activation of IRE1α leads to an increase in the metabolic activity of M1 macrophages from CF patients and its inhibition significantly reduces production of the inflammatory cytokines, IL-6 and TNFα. There is also an absence of PERK-eIF2α activity in the CF lung and in cultured AECs from ΔF508-CFTR patients, but activation of this pathway using an eIF2α agonist beneficially attenuated the robust inflammatory response to flagellin and P. aeruginosa (Nanua et al., 2006; Blohmke et al., 2012).
Viral Infection
The ER is central to the processing of proteins required to mount an effective host immune response to infection, but in many cases viruses like HIV, IAV, and hepatitis C virus hijack the host ER machinery to translate, fold and package structural proteins for virion production (Bagchi, 2020). The IAV structural glycoprotein, hemagglutinin (HA), coordinates viral fusion and entry into target cells, preferentially binding the α2,6- and α2,3- linked sialic acid receptors, which are expressed in abundance on the human airway epithelium (Couceiro et al., 1993; Shinya et al., 2006). During replication, HA interacts with ER chaperones like CNX and CRT for proper folding (Hebert et al., 1997). Additionally, PDIs are essential for the efficient oxidative folding of viral proteins, including PDIA3 on HA of IAV, PDIA1 on the E1 and E2 glycoproteins of hepatitis C virus, and PDIA3 on the F proteins of respiratory syncytial and Sendai viruses (Solda et al., 2006; Kim and Chang, 2018; Ozcelik et al., 2018; Piacentini et al., 2018). PDIA3 forms S–Ss between cysteine residues in HA and is significantly upregulated in mouse lungs following infection by various strains of IAV, as well as in human lung epithelial cells following infection with a pandemic, although not a seasonal strain of influenza (Chamberlain et al., 2019). Furthermore, epithelial-specific PDIA3 knockout mice have significantly lower viral burdens, less inflammation and better lung function. In addition, the ER stress inhibitors, tauroursodeoxycholic acid and 4-phenylbutyrate, are effective at, respectively, reducing the expression of viral proteins in human tracheobronchial epithelial cells and lowering the viral titer in the airways of mice infected with IAV (Hassan et al., 2012; Jung et al., 2019).
Infection by the respiratory virus, coronavirus (CoV) infectious bronchitis virus, activates the PERK pathway, but siRNA knockdown of the downstream mediator, CHOP, reduced apoptosis of infected cells and inhibited viral replication (Liao et al., 2013). Interestingly, primary bronchial epithelial cells from CF patients show less evidence of ER stress following rhinovirus infection than cells from healthy donors and therefore activate the UPR to a lesser degree (Schogler et al., 2019). In CF cells, the induction of ER stress with Tm or other chemical stressors is extremely effective at reducing rhinovirus replication and shedding. Altogether, these studies show a wide range of ER stress responses and patterns of UPR activation, but also highlight the therapeutic potential of targeting the UPR in viral infection.
In relation to ERAD, this IRE1α-XBP1-mediated pathway counters viral infections by degrading unfolded viral proteins, thereby limiting viral replication. This pathway has been shown to be activated in mouse embryonic fibroblasts and in a human alveolar epithelial cell line, in response to IAV infection (Frabutt et al., 2018; Jung et al., 2019). The HA glycoprotein alone is highly effective at activating IRE1α and the ERAD machinery (Frabutt et al., 2018). However, some viruses manipulate ERAD and secretory pathways to evade the host immune response by suppressing the expression of viral proteins on the cell surface where they can be recognized by immune cells, such as natural killer cells (Wilkinson et al., 2008). The human cytomegalovirus targets the major histocompatibility class I polypeptide-related sequence A (MICA), a stress-induced protein that is upregulated on the cell surface of virus-infected cells (Seidel et al., 2021). Specific receptors on natural killer cells then recognize this stress-induced ligand, allowing it to be targeted for elimination. During human cytomegalovirus infection, the signal peptide on the viral glycoprotein, US9, which has an unusually slow rate of cleavage, sustains its presence in the ER where it targets MICA for proteosomal degradation before it can be expressed on the surface of the cell.
Although GRP78 is largely localized to the ER, under ER stress conditions, a small fraction of the chaperone is translocated to the cell surface (Elfiky et al., 2020). Cell surface-GRP78 is upregulated in many cancer cells, including breast and prostate cancers and has become a target for cancer therapy (Tsai and Amy, 2018), In infection, cell surface-GRP78 can assist viral attachment and entry into the cell by binding pathogenic proteins, including the spike (S) protein on the outer envelope of viruses and coat proteins on fungi (Elfiky et al., 2020). Cell surface-GRP78 is expressed on several mammalian cells, including the human airway cell lines, A549, Beas2B, and Calu3 and is upregulated by a variety of viruses (Nain et al., 2017; Chu et al., 2018; Elfiky et al., 2020) The receptor-binding domain of the S protein of different members of the CoV family can interact with angiotensin-converting enzyme-2 (ACE2), dipeptidyl peptidase-4, and cell surface-GRP78, allowing the membranes of the virus and target cell to fuse (Chu et al., 2018; Allam et al., 2020). In Middle East Respiratory Syndrome (MERS)-CoV, cell suface-GRP78 does not independently allow nonpermissive cells to be infected by the virus, but facilitates entry of the virus into permissive cells in the presence of dipeptidyl peptidase-4 (Chu et al., 2018). In line with other CoVs, modeling studies predict cell surface-GRP78 binding to the receptor-binding domain of the S protein of Severe Acute Respiratory Syndrome (SARS)-CoV-2, the virus causing COVID-19 (Ibrahim et al., 2020). Moreover, the GRP78 binding site is predicted to overlap with the binding site of the ACE2 receptor, evidence that GRP78 may be a receptor directly utilized by SARS-CoV-2 to infect target cells (Aguiar et al., 2020). Serum GRP78 levels are also reported to be higher in COVID-19 positive patients compared to COVID-19 negative patients with pneumonia and healthy controls (Sabirli et al., 2021). Several candidate peptides and small molecules targeting the GRP78-binding site on the S protein of SARS-CoV-2 and the viral docking site on GRP78 have been identified, of which Satpdb18674 and epigallocatechin gallate are predicted to be the most effective (Allam et al., 2020). As of yet, no follow up studies have been performed to experimentally confirm the effectiveness of targeting the GRP78-S protein binding sites to inhibit SARS-CoV-2 infection and reduce viral load.
The spike protein of SARS-CoV-2 is synthesized in the ER of the infected cell where it undergoes protein modifications, including a predicted 22 N-and O-linked glycosylation sites on the S protein, before undergoing trimerization and further processing in the Golgi (Zhang et al., 2021). The receptor-binding motif and receptor-binding domain of the S protein of SARS-CoV-2 contain 1 and 3 S–Ss, respectively (Lan et al., 2020). They interact with ACE2 for cell entry and reducing S–Ss into thiols on the S protein and/or ACE2 are predicted to significantly impair binding and therefore infectivity (Hati and Bhattacharyya, 2020). Once again, the implications are that inhibitors of PDIs, PTMs, and ER stress have therapeutic potential as inhibitors of viral infection, including the COVID-19 causing SARS-CoV-2.
Summary
Upon the induction of ER stress, GRP78 dissociates from and activates the three key receptors that orchestrate the UPR, ATF6α, IRE1α and PERK. ATF6α is proteolytically cleaved to liberate its transcriptional factor domain, which in turn upregulate genes associated with the protein folding machinery. The endonuclease activity of IRE1α allows for its dual function of initiating RIDD, as well as activating XBP1 by alternative splicing of its mRNA, which promotes the expression of similar protein folding-associated genes. The receptor, along with unfolded proteins are degraded via ERAD, in an effort to reduce the source of ER stress. PERK phosphorylates eIF2α, which orchestrates the reduction in protein synthesis, promoting cytoprotective responses via ATF4-mediated transcriptional regulation and inducing apoptosis through the activation of CHOP in response to chronic ER stress. In addition, the ability of PERK to directly activate eIF2α and Nrf2 makes it a part of the ISR, which corresponds to signaling beyond the UPR, such as the activation of antioxidant responses, to restore cellular homeostasis. The PTM of peptides is also an important task of the ER, and dysregulation in the machinery involved in signal peptide cleavage, glycosylation and S–S formation will activate the UPR.
ER stress is associated with the pathogenesis of various lung diseases, and is associated with structural cell damage, dysfunction, and the inflammatory response. However, ER stress is not always pathological in that it plays an important role in immune cell development, cell division and other functions, when accompanied by a sufficient and appropriate UPR. Although it is not always clear whether ER stress is a cause or consequence of pathology, inhibiting ER stress and/or activating the UPR, in certain contexts, have some demonstrated therapeutic benefits. This is likely attributed to overlaps in stress responses and their pathways, including the pathways of and genes regulated by the three canonical UPR receptors, the ISR pathways hinging on eIF2α, and the many mediators that make up the protein-folding machinery in the ER like the chaperone and S–S-forming functions shared by many PDIs. Equally as important as the therapeutic potential of targeting the UPR is in its potential to induce cellular apoptosis. Thus, ER stress inhibitors, UPR and ISR activators, and other chemical modifiers affecting protein folding and degradation could in some cases augment rather than attenuate disease. It would be prudent to evaluate this approach as a therapy, on a case-to-case basis. Continued investigation into the roles of ER stress, the UPR and protein processing as they apply to the pathophysiology of pulmonary disease will provide us with a deeper understanding of how to navigate these complex diseases.
Author Contributions
EN, RS, and UF wrote the first draft of the manuscript. EN and JM contributed to the initial conception and layout of the review and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This review was supported by a Discovery grant NSERC 231926 from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Richard and Edith Strauss Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AEC, Airway epithelial cell; AGR, Anterior gradient; AHR, Airway hyperresponsiveness; ASM, Airway smooth muscle; ASMC, Airway smooth muscle cell; ATF4, Activating transcription factor 4; ATF6α, Activating transcription factor 6α; BCL2, B cell lymphoma 2; CF, Cystic fibrosis; CFTR, Cystic fibrosis transmembrane conductance regulator; CHOP, C/EBP Homologous Protein; CNX, Calnexin; CoV, Coronavirus; CRT, Calreticulin; DC, Dendritic cell; ECM, Extracellular matrix; eIF2α, Eukaryotic initiation factor-2α; ER, Endoplasmic reticulum; ERAD, ER-associated degradation machinery; ERO1α, ER oxidoreductase 1α; GCN2, General control nonderepressible 2; GRP78, Glucose-regulated protein 78 kDa; HA, Hemagglutinin; HIV, Human immunodeficiency virus; HRI, Heme-regulated inhibitor; IAV, Influenza A virus; IPF, Idiopathic pulmonary fibrosis; IRE1α, Inositol-requiring enzyme 1α; Keap1, Kelch-like ECH associated protein 1; MCC, Mucociliary clearance; MICA, Major histocompatibility class I polypeptide-related sequence A; Nrf2, Nuclear factor erythroid 2-related factor 2; ORF, Open reading frame; P, Phosphorylated; PDI, Protein disulfide isomerase; PDIA, Protein disulfide isomerase family A; PERK, Protein kinase R-like ER kinase; PKR, Protein Kinase R; PTM, Post translational modification; RIDD, Regulated IRE1-dependent decay; ROS, Reactive oxygen species; S, Spike; S1P, Site-1 protease; S2P, Site-2 protease; –SH, Thiol/sulfhydryl side chain; SP, Signal peptide; S–S, Disulfide bond; Tm, Tunicamycin; Trx, Thioredoxin; UPR, Unfolded protein response; XBP1, X-box binding protein-1.
References
Aghasafari, P., George, U., and Pidaparti, R. (2019). A review of inflammatory mechanism in airway diseases. Inflamm. Res. 68, 59–74. doi: 10.1007/s00011-018-1191-2
Aguiar, J. A., Tremblay, B. J., Mansfield, M. J., Woody, O., Lobb, B., Banerjee, A., et al. . (2020). Gene expression and in situ protein profiling of candidate SARS-CoV-2 receptors in human airway epithelial cells and lung tissue. Eur. Respir. J. 56:2001123. doi: 10.1183/13993003.01123-2020
Al Heialy, S., Risse, P. A., Zeroual, M. A., Roman, H. N., Tsuchiya, K., Siddiqui, S., et al. (2013). T cell-induced airway smooth muscle cell proliferation via the epidermal growth factor receptor. Am. J. Respir. Cell Mol. Biol. 49, 563–570. doi: 10.1165/rcmb.2012-0356OC
Allam, L., Ghrifi, F., Mohammed, H., El Hafidi, N., El Jaoudi, R., El Harti, J., et al. (2020). Targeting the GRP78-dependant SARS-CoV-2 cell entry by peptides and small molecules. Bioinform. Biol. Insights 14:1177932220965505. doi: 10.1177/1177932220965505
Ameri, K., and Harris, A. L. (2008). Activating transcription factor 4. Int. J. Biochem. Cell Biol. 40, 14–21. doi: 10.1016/j.biocel.2007.01.020
Appenzeller-Herzog, C. (2011). Glutathione-and non-glutathione-based oxidant control in the endoplasmic reticulum. J. Cell Sci. 124, 847–855. doi: 10.1242/jcs.080895
Baek, H. A., Kim, D. S., Park, H. S., Jang, K. Y., Kang, M. J., Lee, D. G., et al. (2012). Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 46, 731–739. doi: 10.1165/rcmb.2011-0121OC
Bagchi, P. (2020). Endoplasmic reticulum in viral infection. Int. Rev. Cell Mol. Biol. 350, 265–284. doi: 10.1016/bs.ircmb.2019.10.005
Bentley, J. K., and Hershenson, M. B. (2008). Airway smooth muscle growth in asthma: proliferation, hypertrophy, and migration. Proc. Am. Thorac. Soc. 5, 89–96. doi: 10.1513/pats.200705-063VS
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Bettigole, S. E., Lis, R., Adoro, S., Lee, A. H., Spencer, L. A., Weller, P. F., et al. (2015). The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat. Immunol. 16, 829–837. doi: 10.1038/ni.3225
Blohmke, C. J., Mayer, M. L., Tang, A. C., Hirschfeld, A. F., Fjell, C. D., Sze, M. A., et al. (2012). Atypical activation of the unfolded protein response in cystic fibrosis airway cells contributes to p38 MAPK-mediated innate immune responses. J. Immunol. 189, 5467–5475. doi: 10.4049/jimmunol.1103661
Bonser, L. R., Schroeder, B. W., Ostrin, L. A., Baumlin, N., Olson, J. L., Salathe, M., et al. (2015). The endoplasmic reticulum resident protein AGR3. Required for regulation of ciliary beat frequency in the airway. Am. J. Respir. Cell Mol. Biol. 53, 536–543. doi: 10.1165/rcmb.2014-0318OC
Borok, Z., Horie, M., Flodby, P., Wang, H., Liu, Y., Ganesh, S., et al. (2020). Grp78 loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 201, 198–211. doi: 10.1164/rccm.201902-0451OC
Bosken, C. H., Wiggs, B. R., Pare, P. D., and Hogg, J. C. (1990). Small airway dimensions in smokers with obstruction to airflow. Am. Rev. Respir. Dis. 142, 563–570. doi: 10.1164/ajrccm/142.3.563
Bosnjak, I., Bojovic, V., Segvic-Bubic, T., and Bielen, A. (2014). Occurrence of protein disulfide bonds in different domains of life: a comparison of proteins from the protein data Bank. Protein Eng. Des. Sel. 27, 65–72. doi: 10.1093/protein/gzt063
Bourke, J. E., Li, X., Foster, S. R., Wee, E., Dagher, H., Ziogas, J., et al. (2011). Collagen remodelling by airway smooth muscle is resistant to steroids and beta(2)-agonists. Eur. Respir. J. 37, 173–182.
Braakman, I., and Hebert, D. N. (2013). Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5:a013201. doi: 10.1101/cshperspect.a013201
Brightling, C. E., Bradding, P., Symon, F. A., Holgate, S. T., Wardlaw, A. J., and Pavord, I. D. (2002). Mast-cell infiltration of airway smooth muscle in asthma. N. Engl. J. Med. 346, 1699–1705. doi: 10.1056/NEJMoa012705
Brunsing, R., Omori, S. A., Weber, F., Bicknell, A., Friend, L., Rickert, R., et al. (2008). B-and T-cell development both involve activity of the unfolded protein response pathway. J. Biol. Chem. 283, 17954–17961. doi: 10.1074/jbc.M801395200
Burch, L. H., Talbot, C. R., Knowles, M. R., Canessa, C. M., Rossier, B. C., and Boucher, R. C. (1995). Relative expression of the human epithelial Na+ channel subunits in normal and cystic fibrosis airways. Am. J. Phys. 269, C511–C518. doi: 10.1152/ajpcell.1995.269.2.C511
Burman, A., Kropski, J. A., Calvi, C. L., Serezani, A. P., Pascoalino, B. D., Han, W., et al. (2018a). Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight 3:e99543. doi: 10.1172/jci.insight.99543
Burman, A., Tanjore, H., and Blackwell, T. S. (2018b). Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 68, 355–365. doi: 10.1016/j.matbio.2018.03.015
Cantero-Recasens, G., Fandos, C., Rubio-Moscardo, F., Valverde, M. A., and Vicente, R. (2010). The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Hum. Mol. Genet. 19, 111–121. doi: 10.1093/hmg/ddp471
Cantin, A. M., Hartl, D., Konstan, M. W., and Chmiel, J. F. (2015). Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J. Cyst. Fibros. 14, 419–430. doi: 10.1016/j.jcf.2015.03.003
Chalmers, J. D., Chang, A. B., Chotirmall, S. H., Dhar, R., and Mcshane, P. J. (2018). Bronchiectasis. Nat. Rev. Dis. Primers 4:45. doi: 10.1038/s41572-018-0042-3
Chamberlain, N., Korwin-Mihavics, B. R., Nakada, E. M., Bruno, S. R., Heppner, D. E., Chapman, D. G., et al. (2019). Lung epithelial protein disulfide isomerase A3 (PDIA3) plays an important role in influenza infection, inflammation, and airway mechanics. Redox Biol. 22:101129. doi: 10.1016/j.redox.2019.101129
Chen, G., Ribeiro, C. M. P., Sun, L., Okuda, K., Kato, T., Gilmore, R. C., et al. (2019). XBP1S regulates MUC5B in a promoter variant-dependent pathway in idiopathic pulmonary fibrosis airway epithelia. Am. J. Respir. Crit. Care Med. 200, 220–234. doi: 10.1164/rccm.201810-1972OC
Chen, H. G., Han, H. Z., Li, Y., Yu, Y. H., and Xie, K. L. (2020). Hydrogen alleviated organ injury and dysfunction in sepsis: The role of cross-talk between autophagy and endoplasmic reticulum stress: experimental research. Int. Immunopharmacol. 78:106049. doi: 10.1016/j.intimp.2019.106049
Cherepanova, N., Shrimal, S., and Gilmore, R. (2016). N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 41, 57–65. doi: 10.1016/j.ceb.2016.03.021
Chu, H., Chan, C. M., Zhang, X., Wang, Y., Yuan, S., Zhou, J., et al. (2018). Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 293, 11709–11726. doi: 10.1074/jbc.RA118.001897
Couceiro, J. N., Paulson, J. C., and Baum, L. G. (1993). Influenza virus strains selectively recognize sialyloligosaccharides on human respiratory epithelium; the role of the host cell in selection of hemagglutinin receptor specificity. Virus Res. 29, 155–165. doi: 10.1016/0168-1702(93)90056-S
Cox, J. S., Shamu, C. E., and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. doi: 10.1016/0092-8674(93)90648-A
Cullinan, S. B., and Diehl, J. A. (2004). PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 279, 20108–20117. doi: 10.1074/jbc.M314219200
Cullinan, S. B., Zhang, D., Hannink, M., Arvisais, E., Kaufman, R. J., and Diehl, J. A. (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23, 7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003
Dekkers, B. G., Bos, I. S., Zaagsma, J., and Meurs, H. (2012). Functional consequences of human airway smooth muscle phenotype plasticity. Br. J. Pharmacol. 166, 359–367. doi: 10.1111/j.1476-5381.2011.01773.x
Delmotte, P., and Sieck, G. C. (2019). Endoplasmic reticulum stress and mitochondrial function in airway smooth muscle. Front. Cell Dev. Biol. 7:374. doi: 10.3389/fcell.2019.00374
Delmotte, P., Zavaletta, V. A., Thompson, M. A., Prakash, Y. S., and Sieck, G. C. (2017). TNFalpha decreases mitochondrial movement in human airway smooth muscle. Am. J. Phys. Lung Cell. Mol. Phys. 313, L166–L176. doi: 10.1152/ajplung.00538.2016
Dickhout, J. G., Lhotak, S., Hilditch, B. A., Basseri, S., Colgan, S. M., Lynn, E. G., et al. (2011). Induction of the unfolded protein response after monocyte to macrophage differentiation augments cell survival in early atherosclerotic lesions. FASEB J. 25, 576–589. doi: 10.1096/fj.10-159319
Dodson, M., Darley-Usmar, V., and Zhang, J. (2013). Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic. Biol. Med. 63, 207–221. doi: 10.1016/j.freeradbiomed.2013.05.014
Edman, J. C., Ellis, L., Blacher, R. W., Roth, R. A., and Rutter, W. J. (1985). Sequence of protein disulphide isomerase and implications of its relationship to thioredoxin. Nature 317, 267–270. doi: 10.1038/317267a0
Elfiky, A. A., Baghdady, A. M., Ali, S. A., and Ahmed, M. I. (2020). GRP78 targeting: hitting two birds with a stone. Life Sci. 260:118317. doi: 10.1016/j.lfs.2020.118317
Ellgaard, L., Mccaul, N., Chatsisvili, A., and Braakman, I. (2016). Co-and post-translational protein folding in the ER. Traffic 17, 615–638. doi: 10.1111/tra.12392
Ellgaard, L., and Ruddock, L. W. (2005). The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32. doi: 10.1038/sj.embor.7400311
Flynn, G. C., Pohl, J., Flocco, M. T., and Rothman, J. E. (1991). Peptide-binding specificity of the molecular chaperone BiP. Nature 353, 726–730. doi: 10.1038/353726a0
Frabutt, D. A., Wang, B., Riaz, S., Schwartz, R. C., and Zheng, Y. H. (2018). Innate sensing of influenza A virus hemagglutinin glycoproteins by the host endoplasmic reticulum (ER) stress pathway triggers a potent antiviral response via ER-associated protein degradation. J. Virol. 92:e01690-17. doi: 10.1128/JVI.01690-17
Freyer, A. M., Johnson, S. R., and Hall, I. P. (2001). Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 25, 569–576. doi: 10.1165/ajrcmb.25.5.4605
Fujimori, T., Kamiya, Y., Nagata, K., Kato, K., and Hosokawa, N. (2013). Endoplasmic reticulum lectin XTP3-B inhibits endoplasmic reticulum-associated degradation of a misfolded alpha1-antitrypsin variant. FEBS J. 280, 1563–1575. doi: 10.1111/febs.12157
Ganesan, S., Comstock, A. T., and Sajjan, U. S. (2013). Barrier function of airway tract epithelium. Tissue Barriers 1:e24997. doi: 10.4161/tisb.24997
Gebauer, F., and Hentze, M. W. (2004). Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 5, 827–835. doi: 10.1038/nrm1488
Gewandter, J. S., Staversky, R. J., and O'reilly, M. A. (2009). Hyperoxia augments ER-stress-induced cell death independent of BiP loss. Free Radic. Biol. Med. 47, 1742–1752. doi: 10.1016/j.freeradbiomed.2009.09.022
Ghavami, S., Yeganeh, B., Zeki, A. A., Shojaei, S., Kenyon, N. J., Ott, S., et al. (2018). Autophagy and the unfolded protein response promote profibrotic effects of TGF-beta1 in human lung fibroblasts. Am. J. Phys. Lung Cell. Mol. Phys. 314, L493–L504. doi: 10.1152/ajplung.00372.2017
Glab, J. A., Doerflinger, M., Nedeva, C., Jose, I., Mbogo, G. W., Paton, J. C., et al. (2017). DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 24, 944–950. doi: 10.1038/cdd.2017.53
Goodall, J. C., Wu, C., Zhang, Y., Mcneill, L., Ellis, L., Saudek, V., et al. (2010). Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. U. S. A. 107, 17698–17703. doi: 10.1073/pnas.1011736107
Guo, Q., Li, H., Liu, J., Xu, L., Yang, L., Sun, Z., et al. (2017). Tunicamycin aggravates endoplasmic reticulum stress and airway inflammation via PERK-ATF4-CHOP signaling in a murine model of neutrophilic asthma. J. Asthma 54, 125–133. doi: 10.1080/02770903.2016.1205085
Haas, I. G., and Wabl, M. (1983). Immunoglobulin heavy chain binding protein. Nature 306, 387–389. doi: 10.1038/306387a0
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274.
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. doi: 10.1016/S1097-2765(03)00105-9
Hartsock, A., and Nelson, W. J. (2008). Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 1778, 660–669. doi: 10.1016/j.bbamem.2007.07.012
Hassan, I. H., Zhang, M. S., Powers, L. S., Shao, J. Q., Baltrusaitis, J., Rutkowski, D. T., et al. (2012). Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 287, 4679–4689. doi: 10.1074/jbc.M111.284695
Hati, S., and Bhattacharyya, S. (2020). Impact of thiol-disulfide balance on the binding of Covid-19 spike protein with angiotensin-converting enzyme 2 receptor. ACS Omega 5, 16292–16298. doi: 10.1021/acsomega.0c02125
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi: 10.1091/mbc.10.11.3787
He, C. H., Gong, P., Hu, B., Stewart, D., Choi, M. E., Choi, A. M., et al. (2001). Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 276, 20858–20865. doi: 10.1074/jbc.M101198200
Hebert, D. N., Zhang, J. X., Chen, W., Foellmer, B., and Helenius, A. (1997). The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J. Cell Biol. 139, 613–623. doi: 10.1083/jcb.139.3.613
Hegde, R. S., and Bernstein, H. D. (2006). The surprising complexity of signal sequences. Trends Biochem. Sci. 31, 563–571. doi: 10.1016/j.tibs.2006.08.004
Heindryckx, F., Binet, F., Ponticos, M., Rombouts, K., Lau, J., Kreuger, J., et al. (2016). Endoplasmic reticulum stress enhances fibrosis through IRE1alpha-mediated degradation of miR-150 and XBP-1 splicing. EMBO Mol. Med. 8, 729–744. doi: 10.15252/emmm.201505925
Helenius, A. (1994). How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol. Biol. Cell 5, 253–265. doi: 10.1091/mbc.5.3.253
Hendershot, L. M. (1990). Immunoglobulin heavy chain and binding protein complexes are dissociated in vivo by light chain addition. J. Cell Biol. 111, 829–837. doi: 10.1083/jcb.111.3.829
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. doi: 10.1038/ncb3184
Hetz, C., Martinon, F., Rodriguez, D., and Glimcher, L. H. (2011). The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 91, 1219–1243. doi: 10.1152/physrev.00001.2011
Hoffman, S. M., Chapman, D. G., Lahue, K. G., Cahoon, J. M., Rattu, G. K., Daphtary, N., et al. (2016). Protein disulfide isomerase-endoplasmic reticulum resident protein 57 regulates allergen-induced airways inflammation, fibrosis, and hyperresponsiveness. J. Allergy Clin. Immunol. 137, 822–832. doi: 10.1016/j.jaci.2015.08.018
Hoffman, S. M., Tully, J. E., Nolin, J. D., Lahue, K. G., Goldman, D. H., Daphtary, N., et al. (2013). Endoplasmic reticulum stress mediates house dust mite-induced airway epithelial apoptosis and fibrosis. Respir. Res. 14:141. doi: 10.1186/1465-9921-14-141
Holguin, F. (2013). Oxidative stress in airway diseases. Ann. Am. Thorac. Soc. 10(Suppl. 1), S150–S157. doi: 10.1513/AnnalsATS.201305-116AW
Hollenhorst, M. I., Richter, K., and Fronius, M. (2011). Ion transport by pulmonary epithelia. J. Biomed. Biotechnol. 2011:174306. doi: 10.1155/2011/174306
Hollien, J., Lin, J. H., Li, H., Stevens, N., Walter, P., and Weissman, J. S. (2009). Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323–331. doi: 10.1083/jcb.200903014
Hollien, J., and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107. doi: 10.1126/science.1129631
Hsu, H. S., Liu, C. C., Lin, J. H., Hsu, T. W., Hsu, J. W., Su, K., et al. (2017). Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 7:14272. doi: 10.1038/s41598-017-14612-5
Hu, H., Tian, M., Ding, C., and Yu, S. (2018). The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 9:3083. doi: 10.3389/fimmu.2018.03099
Huang, Y., Wang, Y., Feng, Y., Wang, P., He, X., Ren, H., et al. (2019). Role of endoplasmic reticulum stress-autophagy Axis in severe burn-induced intestinal tight junction barrier dysfunction in mice. Front. Physiol. 10:606. doi: 10.3389/fphys.2019.00606
Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S., et al. (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. doi: 10.1038/nature02026
Hull-Ryde, E. A., Minges, J. T., Martino, M. E. B., Kato, T., Norris-Drouin, J. L., and Ribeiro, C. M. P. (2021). IRE1alpha is a therapeutic target for cystic fibrosis airway inflammation. Int. J. Mol. Sci. 22:3063. doi: 10.3390/ijms22063063
Hurtley, S. M., and Helenius, A. (1989). Protein oligomerization in the endoplasmic reticulum. Annu. Rev. Cell Biol. 5, 277–307. doi: 10.1146/annurev.cb.05.110189.001425
Hwang, J., and Qi, L. (2018). Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 43, 593–605. doi: 10.1016/j.tibs.2018.06.005
Ibrahim, I. M., Abdelmalek, D. H., Elshahat, M. E., and Elfiky, A. A. (2020). COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 80, 554–562. doi: 10.1016/j.jinf.2020.02.026
Iwakoshi, N. N., Lee, A. H., and Glimcher, L. H. (2003a). The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 194, 29–38. doi: 10.1034/j.1600-065X.2003.00057.x
Iwakoshi, N. N., Lee, A. H., Vallabhajosyula, P., Otipoby, K. L., Rajewsky, K., and Glimcher, L. H. (2003b). Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4, 321–329. doi: 10.1038/ni907
Iwakoshi, N. N., Pypaert, M., and Glimcher, L. H. (2007). The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 204, 2267–2275. doi: 10.1084/jem.20070525
Jung, K. I., Ko, D. H., Shin, N., Pyo, C. W., and Choi, S. Y. (2019). Endoplasmic reticulum-associated degradation potentiates the infectivity of influenza A virus by regulating the host redox state. Free Radic. Biol. Med. 135, 293–305. doi: 10.1016/j.freeradbiomed.2019.03.021
Kamimura, D., and Bevan, M. J. (2008). Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J. Immunol. 181, 5433–5441. doi: 10.4049/jimmunol.181.8.5433
Kelsen, S. G., Duan, X., Ji, R., Perez, O., Liu, C., and Merali, S. (2008). Cigarette smoke induces an unfolded protein response in the human lung: a proteomic approach. Am. J. Respir. Cell Mol. Biol. 38, 541–550. doi: 10.1165/rcmb.2007-0221OC
Kenche, H., Baty, C. J., Vedagiri, K., Shapiro, S. D., and Blumental-Perry, A. (2013). Cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein disulfide isomerase. FASEB J. 27, 965–977. doi: 10.1096/fj.12-216234
Kenche, H., Ye, Z. W., Vedagiri, K., Richards, D. M., Gao, X. H., Tew, K. D., et al. (2016). Adverse outcomes associated with cigarette smoke radicals related to damage to protein-disulfide isomerase. J. Biol. Chem. 291, 4763–4778. doi: 10.1074/jbc.M115.712331
Kilberg, M. S., Shan, J., and Su, N. (2009). ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 20, 436–443. doi: 10.1016/j.tem.2009.05.008
Kim, M. H., Bae, C. H., Choi, Y. S., Na, H. G., Song, S. Y., and Kim, Y. D. (2019). Endoplasmic reticulum stress induces MUC5AC and MUC5B expression in human nasal airway epithelial cells. Clin. Exp. Otorhinolaryngol. 12, 181–189. doi: 10.21053/ceo.2018.00493
Kim, Y., and Chang, K. O. (2018). Protein disulfide isomerases as potential therapeutic targets for influenza A and B viruses. Virus Res. 247, 26–33. doi: 10.1016/j.virusres.2018.01.010
Knaapen, A. M., Gungor, N., Schins, R. P., Borm, P. J., and Van Schooten, F. J. (2006). Neutrophils and respiratory tract DNA damage and mutagenesis: a review. Mutagenesis 21, 225–236. doi: 10.1093/mutage/gel032
Kober, F. X., Koelmel, W., Kuper, J., Drechsler, J., Mais, C., Hermanns, H. M., et al. (2013). The crystal structure of the protein-disulfide isomerase family member ERp27 provides insights into its substrate binding capabilities. J. Biol. Chem. 288, 2029–2039. doi: 10.1074/jbc.M112.410522
Korfei, M., Ruppert, C., Mahavadi, P., Henneke, I., Markart, P., Koch, M., et al. (2008). Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 838–846. doi: 10.1164/rccm.200802-313OC
Kropski, J. A., and Blackwell, T. S. (2018). Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Invest. 128, 64–73. doi: 10.1172/JCI93560
Kumarathasan, P., Blais, E., Saravanamuthu, A., Bielecki, A., Mukherjee, B., Bjarnason, S., et al. (2015). Nitrative stress, oxidative stress and plasma endothelin levels after inhalation of particulate matter and ozone. Part. Fibre Toxicol. 12:28. doi: 10.1186/s12989-015-0103-7
Lambert, R. K., Wiggs, B. R., Kuwano, K., Hogg, J. C., and Pare, P. D. (1993). Functional significance of increased airway smooth muscle in asthma and COPD. J. Appl. Physiol. 74, 2771–2781. doi: 10.1152/jappl.1993.74.6.2771
Lan, J., Ge, J., Yu, J., Shan, S., Zhou, H., Fan, S., et al. (2020). Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220. doi: 10.1038/s41586-020-2180-5
Lara-Reyna, S., Scambler, T., Holbrook, J., Wong, C., Jarosz-Griffiths, H. H., Martinon, F., et al. (2019). Metabolic reprograming of cystic fibrosis macrophages via the IRE1alpha arm of the unfolded protein response results in exacerbated inflammation. Front. Immunol. 10:1789. doi: 10.3389/fimmu.2019.01789
Lauer, M. E., Mukhopadhyay, D., Fulop, C., De La Motte, C. A., Majors, A. K., and Hascall, V. C. (2009). Primary murine airway smooth muscle cells exposed to poly(I,C) or tunicamycin synthesize a leukocyte-adhesive hyaluronan matrix. J. Biol. Chem. 284, 5299–5312. doi: 10.1074/jbc.M807965200
Lawson, W. E., Cheng, D. S., Degryse, A. L., Tanjore, H., Polosukhin, V. V., Xu, X. C., et al. (2011). Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. U. S. A. 108, 10562–10567. doi: 10.1073/pnas.1107559108
Lawson, W. E., Crossno, P. F., Polosukhin, V. V., Roldan, J., Cheng, D. S., Lane, K. B., et al. (2008). Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am. J. Phys. Lung Cell. Mol. Phys. 294, L1119–L1126. doi: 10.1152/ajplung.00382.2007
Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003
Lee, D., Hokinson, D., Park, S., Elvira, R., Kusuma, F., Lee, J. M., et al. (2019). ER stress induces cell cycle arrest at the G2/M phase through eIF2alpha phosphorylation and GADD45alpha. Int. J. Mol. Sci. 20:6309. doi: 10.3390/ijms20246309
Lee, E. J., Cardenes, N., Alvarez, D., Sellares, J., Sembrat, J., Aranda, P., et al. (2020). Mesenchymal stem cells reduce ER stress via PERK-Nrf2 pathway in an aged mouse model. Respirology 25, 417–426. doi: 10.1111/resp.13646
Li, C., Xia, B., Wang, S., and Xu, J. (2020). Folded or degraded in endoplasmic reticulum. Adv. Exp. Med. Biol. 1248, 265–294. doi: 10.1007/978-981-15-3266-5_12
Li, Y., Guo, Y., Tang, J., Jiang, J., and Chen, Z. (2014). New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. Shanghai 46, 629–640. doi: 10.1093/abbs/gmu048
Liang, Y., Fan, C., Yan, X., Lu, X., Jiang, H., Di, S., et al. (2019). Berberine ameliorates lipopolysaccharide-induced acute lung injury via the PERK-mediated Nrf2/HO-1 signaling axis. Phytother. Res. 33, 130–148. doi: 10.1002/ptr.6206
Liao, Y., Fung, T. S., Huang, M., Fang, S. G., Zhong, Y., and Liu, D. X. (2013). Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J. Virol. 87, 8124–8134. doi: 10.1128/JVI.00626-13
Lin, F., Liao, C., Sun, Y., Zhang, J., Lu, W., Bai, Y., et al. (2017a). Hydrogen sulfide inhibits cigarette smoke-induced endoplasmic reticulum stress and apoptosis in bronchial epithelial cells. Front. Pharmacol. 8:675. doi: 10.3389/fphar.2017.00675
Lin, L., Hou, G., Han, D., Kang, J., and Wang, Q. (2019). Ursolic acid protected lung of rats From damage induced by cigarette smoke extract. Front. Pharmacol. 10:700. doi: 10.3389/fphar.2019.00700
Lin, L., Yin, Y., Hou, G., Han, D., Kang, J., and Wang, Q. (2017b). Ursolic acid attenuates cigarette smoke-induced emphysema in rats by regulating PERK and Nrf2 pathways. Pulm. Pharmacol. Ther. 44, 111–121. doi: 10.1016/j.pupt.2017.03.014
Lin, V. Y., Kaza, N., Birket, S. E., Kim, H., Edwards, L. J., Lafontaine, J., et al. (2020). Excess mucus viscosity and airway dehydration impact COPD airway clearance. Eur. Respir. J. 55:1900419 doi: 10.1183/13993003.00419-2019
Liu, B., and Li, Z. (2008). Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood 112, 1223–1230. doi: 10.1182/blood-2008-03-143107
Loser, S., Gregory, L. G., Zhang, Y., Schaefer, K., Walker, S. A., Buckley, J., et al. (2017). Pulmonary ORMDL3 is critical for induction of alternaria-induced allergic airways disease. J. Allergy Clin. Immunol. 139, 1496–1507. doi: 10.1016/j.jaci.2016.07.033
Lozon, T. I., Eastman, A. J., Matute-Bello, G., Chen, P., Hallstrand, T. S., and Altemeier, W. A. (2011). PKR-dependent CHOP induction limits hyperoxia-induced lung injury. Am. J. Phys. Lung Cell. Mol. Phys. 300, L422–L429. doi: 10.1152/ajplung.00166.2010
Lu, M., Lawrence, D. A., Marsters, S., Acosta-Alvear, D., Kimmig, P., Mendez, A. S., et al. (2014). Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345, 98–101. doi: 10.1126/science.1254312
Lucas, J. S., Davis, S. D., Omran, H., and Shoemark, A. (2020). Primary ciliary dyskinesia in the genomics age. Lancet Respir. Med. 8, 202–216. doi: 10.1016/S2213-2600(19)30374-1
Ma, J., Rubin, B. K., and Voynow, J. A. (2018). Mucins, mucus, and goblet cells. Chest 154, 169–176. doi: 10.1016/j.chest.2017.11.008
Ma, J. H., Wang, J. J., Li, J., Pfeffer, B. A., Zhong, Y., and Zhang, S. X. (2016). The role of IRE-XBP1 pathway in regulation of retinal pigment epithelium tight junctions. Invest. Ophthalmol. Vis. Sci. 57, 5244–5252. doi: 10.1167/iovs.16-19232
Machen, T. E. (2006). Innate immune response in CF airway epithelia: hyperinflammatory? Am. J. Physiol. Cell Physiol. 291, C218–C230. doi: 10.1152/ajpcell.00605.2005
Mahameed, M., Wilhelm, T., Darawshi, O., Obiedat, A., Tommy, W. S., Chintha, C., et al. (2019). The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 10:300. doi: 10.1038/s41419-019-1523-3
Malabanan, K. P., Kanellakis, P., Bobik, A., and Khachigian, L. M. (2008). Activation transcription factor-4 induced by fibroblast growth factor-2 regulates vascular endothelial growth factor-A transcription in vascular smooth muscle cells and mediates intimal thickening in rat arteries following balloon injury. Circ. Res. 103, 378–387. doi: 10.1161/CIRCRESAHA.107.168682
Mall, M., Bleich, M., Greger, R., Schreiber, R., and Kunzelmann, K. (1998). The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J. Clin. Invest. 102, 15–21. doi: 10.1172/JCI2729
Marquez, S., Fernandez, J. J., Teran-Cabanillas, E., Herrero, C., Alonso, S., Azogil, A., et al. (2017). Endoplasmic reticulum stress sensor IRE1alpha enhances IL-23 expression by human dendritic cells. Front. Immunol. 8:639. doi: 10.3389/fimmu.2017.00639
Martin, J. G., Duguet, A., and Eidelman, D. H. (2000). The contribution of airway smooth muscle to airway narrowing and airway hyperresponsiveness in disease. Eur. Respir. J. 16, 349–354. doi: 10.1034/j.1399-3003.2000.16b25.x
Martino, M. B., Jones, L., Brighton, B., Ehre, C., Abdulah, L., Davis, C. W., et al. (2013). The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol. 6, 639–654. doi: 10.1038/mi.2012.105
Martino, M. E., Olsen, J. C., Fulcher, N. B., Wolfgang, M. C., O'neal, W. K., and Ribeiro, C. M. (2009). Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. J. Biol. Chem. 284, 14904–14913. doi: 10.1074/jbc.M809180200
Matsui, H., Grubb, B. R., Tarran, R., Randell, S. H., Gatzy, J. T., Davis, C. W., et al. (1998). Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95, 1005–1015. doi: 10.1016/S0092-8674(00)81724-9
Matsuzaki, S., Hiratsuka, T., Taniguchi, M., Shingaki, K., Kubo, T., Kiya, K., et al. (2015). Physiological ER stress mediates the differentiation of fibroblasts. PLoS One 10:e0123578. doi: 10.1371/journal.pone.0123578
Mcclure, M. L., Wen, H., Fortenberry, J., Hong, J. S., and Sorscher, E. J. (2014). S-palmitoylation regulates biogenesis of core glycosylated wild-type and F508del CFTR in a post-ER compartment. Biochem. J. 459, 417–425. doi: 10.1042/BJ20131037
Mcmahon, M., Itoh, K., Yamamoto, M., and Hayes, J. D. (2003). Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 278, 21592–21600. doi: 10.1074/jbc.M300931200
Melnick, J., Aviel, S., and Argon, Y. (1992). The endoplasmic reticulum stress protein GRP94, in addition to BiP, associates with unassembled immunoglobulin chains. J. Biol. Chem. 267, 21303–21306. doi: 10.1016/S0021-9258(19)36608-6
Miettinen, T. P., and Bjorklund, M. (2015). Mevalonate pathway regulates cell size homeostasis and proteostasis through autophagy. Cell Rep. 13, 2610–2620. doi: 10.1016/j.celrep.2015.11.045
Mijosek, V., Lasitschka, F., Warth, A., Zabeck, H., Dalpke, A. H., and Weitnauer, M. (2016). Endoplasmic reticulum stress is a danger signal promoting innate inflammatory responses in bronchial epithelial cells. J. Innate Immun. 8, 464–478. doi: 10.1159/000447668
Miller, M., Rosenthal, P., Beppu, A., Mueller, J. L., Hoffman, H. M., Tam, A. B., et al. (2014). ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J. Immunol. 192, 3475–3487. doi: 10.4049/jimmunol.1303047
Miller, M., Tam, A. B., Cho, J. Y., Doherty, T. A., Pham, A., Khorram, N., et al. (2012). ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc. Natl. Acad. Sci. U. S. A. 109, 16648–16653. doi: 10.1073/pnas.1204151109
Mimura, J., Inose-Maruyama, A., Taniuchi, S., Kosaka, K., Yoshida, H., Yamazaki, H., et al. (2019). Concomitant Nrf2-and ATF4-activation by Carnosic acid cooperatively induces expression of cytoprotective genes. Int. J. Mol. Sci. 20:1706. doi: 10.3390/ijms20071706
Min, T., Bodas, M., Mazur, S., and Vij, N. (2011). Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. (Berl) 89, 577–593. doi: 10.1007/s00109-011-0732-8
Moffatt, M. F., Kabesch, M., Liang, L., Dixon, A. L., Strachan, D., Heath, S., et al. (2007). Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448, 470–473. doi: 10.1038/nature06014
Mohanty, S., Chaudhary, B. P., and Zoetewey, D. (2020). Structural insight into the mechanism of N-linked glycosylation by oligosaccharyltransferase. Biomol. Ther. 10:624. doi: 10.3390/biom10040624
Moon, D. O., Asami, Y., Long, H., Jang, J. H., Bae, E. Y., Kim, B. Y., et al. (2013). Verrucarin A sensitizes TRAIL-induced apoptosis via the upregulation of DR5 in an eIF2alpha/CHOP-dependent manner. Toxicol. In Vitro 27, 257–263. doi: 10.1016/j.tiv.2012.09.001
Nain, M., Mukherjee, S., Karmakar, S. P., Paton, A. W., Paton, J. C., Abdin, M. Z., et al. (2017). GRP78 is an important host factor for Japanese encephalitis virus entry and replication in mammalian cells. J. Virol. 91:e02274-16 doi: 10.1128/JVI.02274-16
Nakada, E. M., Bhakta, N. R., Korwin-Mihavics, B. R., Kumar, A., Chamberlain, N., Bruno, S. R., et al. (2019). Conjugated bile acids attenuate allergen-induced airway inflammation and hyperresponsiveness by inhibiting UPR transducers. JCI Insight 4:e98101 doi: 10.1172/jci.insight.98101
Nanua, S., Sajjan, U., Keshavjee, S., and Hershenson, M. B. (2006). Absence of typical unfolded protein response in primary cultured cystic fibrosis airway epithelial cells. Biochem. Biophys. Res. Commun. 343, 135–143. doi: 10.1016/j.bbrc.2006.02.137
Nikolic, A. (2018). Pathophysiology and genetics of bronchiectasis unrelated to cystic fibrosis. Lung 196, 383–392. doi: 10.1007/s00408-018-0121-y
Nogee, L. M., Dunbar, A. E. 3rd , Wert, S. E., Askin, F., Hamvas, A., and Whitsett, J. A. (2001). A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 344, 573–579. doi: 10.1056/NEJM200102223440805
O’brien, E. P., Ciryam, P., Vendruscolo, M., and Dobson, C. M. (2014). Understanding the influence of codon translation rates on cotranslational protein folding. Acc. Chem. Res. 47, 1536–1544. doi: 10.1021/ar5000117
Oh, J., Riek, A. E., Weng, S., Petty, M., Kim, D., Colonna, M., et al. (2012). Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J. Biol. Chem. 287, 11629–11641. doi: 10.1074/jbc.M111.338673
Oikawa, D., Kimata, Y., Kohno, K., and Iwawaki, T. (2009). Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 315, 2496–2504. doi: 10.1016/j.yexcr.2009.06.009
Okumura, M., Kadokura, H., and Inaba, K. (2015). Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic. Biol. Med. 83, 314–322. doi: 10.1016/j.freeradbiomed.2015.02.010
Ono, M., Ohkouchi, S., Kanehira, M., Tode, N., Kobayashi, M., Ebina, M., et al. (2015). Mesenchymal stem cells correct inappropriate epithelial-mesenchyme relation in pulmonary fibrosis using stanniocalcin-1. Mol. Ther. 23, 549–560. doi: 10.1038/mt.2014.217
Oslowski, C. M., and Urano, F. (2011). Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 490, 71–92. doi: 10.1016/B978-0-12-385114-7.00004-0
Osorio, F., Tavernier, S. J., Hoffmann, E., Saeys, Y., Martens, L., Vetters, J., et al. (2014). The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat. Immunol. 15, 248–257. doi: 10.1038/ni.2808
Oyeniran, C., Sturgill, J. L., Hait, N. C., Huang, W. C., Avni, D., Maceyka, M., et al. (2015). Aberrant ORM (yeast)-like protein isoform 3 (ORMDL3) expression dysregulates ceramide homeostasis in cells and ceramide exacerbates allergic asthma in mice. J. Allergy Clin. Immunol. 136, 1035–1046. doi: 10.1016/j.jaci.2015.02.031
Ozcelik, D., Seto, A., Rakic, B., Farzam, A., Supek, F., and Pezacki, J. P. (2018). Gene expression profiling of endoplasmic reticulum stress in hepatitis C virus-containing cells treated with an inhibitor of protein disulfide isomerases. ACS Omega 3, 17227–17235. doi: 10.1021/acsomega.8b02676
Ozier, A., Allard, B., Bara, I., Girodet, P. O., Trian, T., Marthan, R., et al. (2011). The pivotal role of airway smooth muscle in asthma pathophysiology. J. Allergy 2011:742710. doi: 10.1155/2011/742710
Pagetta, A., Tramentozzi, E., Tibaldi, E., Cendron, L., Zanotti, G., Brunati, A. M., et al. (2014). Structural insights into complexes of glucose-regulated Protein94 (Grp94) with human immunoglobulin G. relevance for Grp94-IgG complexes that form in vivo in pathological conditions. PLoS One 9:e86198. doi: 10.1371/journal.pone.0086198
Pathinayake, P. S., Hsu, A. C., Waters, D. W., Hansbro, P. M., Wood, L. G., and Wark, P. A. B. (2018). Understanding the unfolded protein response in the pathogenesis of asthma. Front. Immunol. 9:175. doi: 10.3389/fimmu.2018.00175
Perez-Novo, C. A., and Bachert, C. (2015). DNA methylation, bacteria and airway inflammation: latest insights. Curr. Opin. Allergy Clin. Immunol. 15, 27–32. doi: 10.1097/ACI.0000000000000130
Petersen, T. N., Brunak, S., Von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Piacentini, S., La Frazia, S., Riccio, A., Pedersen, J. Z., Topai, A., Nicolotti, O., et al. (2018). Nitazoxanide inhibits paramyxovirus replication by targeting the fusion protein folding: role of glycoprotein-specific thiol oxidoreductase ERp57. Sci. Rep. 8:10425. doi: 10.1038/s41598-018-28172-9
Poe, C., Youngblood, C., Hodge, K., and Kemp, K. (2019). Treatment of established TH2 cells with 4mu8c, an inhibitor of IRE1alpha, blocks IL-5 but not IL-4 secretion. BMC Immunol. 20:3. doi: 10.1186/s12865-018-0283-7
Price, J. L., Culyba, E. K., Chen, W., Murray, A. N., Hanson, S. R., Wong, C. H., et al. (2012). N-glycosylation of enhanced aromatic sequons to increase glycoprotein stability. Biopolymers 98, 195–211. doi: 10.1002/bip.22030
Ramos-Barbon, D., Fraga-Iriso, R., Brienza, N. S., Montero-Martinez, C., Verea-Hernando, H., Olivenstein, R., et al. (2010). T cells localize with proliferating smooth muscle alpha-actin+ cell compartments in asthma. Am. J. Respir. Crit. Care Med. 182, 317–324. doi: 10.1164/rccm.200905-0745OC
Randow, F., and Seed, B. (2001). Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat. Cell Biol. 3, 891–896. doi: 10.1038/ncb1001-891
Rao, J., Zhang, C., Wang, P., Lu, L., Qian, X., Qin, J., et al. (2015). C/EBP homologous protein (CHOP) contributes to hepatocyte death via the promotion of ERO1alpha signalling in acute liver failure. Biochem. J. 466, 369–378. doi: 10.1042/BJ20140412
Reimold, A. M., Iwakoshi, N. N., Manis, J., Vallabhajosyula, P., Szomolanyi-Tsuda, E., Gravallese, E. M., et al. (2001). Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300–307. doi: 10.1038/35085509
Ribeiro, C. M., Paradiso, A. M., Schwab, U., Perez-Vilar, J., Jones, L., O'neal, W., et al. (2005). Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J. Biol. Chem. 280, 17798–17806. doi: 10.1074/jbc.M410618200
Roberson, E. C., Tully, J. E., Guala, A. S., Reiss, J. N., Godburn, K. E., Pociask, D. A., et al. (2012). Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-beta release in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 46, 573–581. doi: 10.1165/rcmb.2010-0460OC
Roche, W. R., Beasley, R., Williams, J. H., and Holgate, S. T. (1989). Subepithelial fibrosis in the bronchi of asthmatics. Lancet 1, 520–524. doi: 10.1016/s0140-6736(89)90067-6
Sabirli, R., Koseler, A., Goren, T., Turkcuer, I., and Kurt, O. (2021). High GRP78 levels in Covid-19 infection: a case-control study. Life Sci. 265:118781. doi: 10.1016/j.lfs.2020.118781
Salati, S., Prudente, Z., Genovese, E., Pennucci, V., Rontauroli, S., Bartalucci, N., et al. (2017). Calreticulin affects hematopoietic stem/progenitor cell fate by impacting erythroid and megakaryocytic differentiation. Stem Cells Dev. 27, 225–236. doi: 10.1089/scd.2017.0137
Schogler, A., Caliaro, O., Brugger, M., Oliveira Esteves, B. I., Nita, I., Gazdhar, A., et al. (2019). Modulation of the unfolded protein response pathway as an antiviral approach in airway epithelial cells. Antivir. Res. 162, 44–50. doi: 10.1016/j.antiviral.2018.12.007
Schroeder, B. W., Verhaeghe, C., Park, S. W., Nguyenvu, L. T., Huang, X., Zhen, G., et al. (2012). AGR2 is induced in asthma and promotes allergen-induced mucin overproduction. Am. J. Respir. Cell Mol. Biol. 47, 178–185. doi: 10.1165/rcmb.2011-0421OC
Seidel, E., Dassa, L., Kahlon, S., Tirosh, B., Halenius, A., Seidel Malkinson, T., et al. (2021). A slowly cleaved viral signal peptide acts as a protein-integral immune evasion domain. Nat. Commun. 12:2061. doi: 10.1038/s41467-021-21983-x
Shaffer, K. L., Sharma, A., Snapp, E. L., and Hegde, R. S. (2005). Regulation of protein compartmentalization expands the diversity of protein function. Dev. Cell 9, 545–554. doi: 10.1016/j.devcel.2005.09.001
Sharma, A. K., Sormanni, P., Ahmed, N., Ciryam, P., Friedrich, U. A., Kramer, G., et al. (2019). A chemical kinetic basis for measuring translation initiation and elongation rates from ribosome profiling data. PLoS Comput. Biol. 15:e1007070. doi: 10.1371/journal.pcbi.1007070
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111. doi: 10.1016/S1534-5807(02)00203-4
Shi, Y., Vattem, K. M., Sood, R., An, J., Liang, J., Stramm, L., et al. (1998). Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18, 7499–7509. doi: 10.1128/MCB.18.12.7499
Shinya, K., Ebina, M., Yamada, S., Ono, M., Kasai, N., and Kawaoka, Y. (2006). Avian flu: influenza virus receptors in the human airway. Nature 440, 435–436. doi: 10.1038/440435a
Siddesha, J. M., Nakada, E. M., Mihavics, B. R., Hoffman, S. M., Rattu, G. K., Chamberlain, N., et al. (2016). Effect of a chemical chaperone, tauroursodeoxycholic acid, on HDM-induced allergic airway disease. Am. J. Phys. Lung Cell. Mol. Phys. 310, L1243–L1259. doi: 10.1152/ajplung.00396.2015
Siddiqui, W. A., Ahad, A., and Ahsan, H. (2015). The mystery of BCL2 family: bcl-2 proteins and apoptosis: an update. Arch. Toxicol. 89, 289–317. doi: 10.1007/s00204-014-1448-7
Singleton, D. C., and Harris, A. L. (2012). Targeting the ATF4 pathway in cancer therapy. Expert Opin. Ther. Targets 16, 1189–1202. doi: 10.1517/14728222.2012.728207
Soares Moretti, A. I., and Martins Laurindo, F. R. (2017). Protein disulfide isomerases: redox connections in and out of the endoplasmic reticulum. Arch. Biochem. Biophys. 617, 106–119. doi: 10.1016/j.abb.2016.11.007
Solda, T., Garbi, N., Hammerling, G. J., and Molinari, M. (2006). Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. J. Biol. Chem. 281, 6219–6226. doi: 10.1074/jbc.M513595200
Sun, S., Shi, G., Sha, H., Ji, Y., Han, X., Shu, X., et al. (2015). IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 17, 1546–1555. doi: 10.1038/ncb3266
Sundaram, A., Appathurai, S., Plumb, R., and Mariappan, M. (2018). Dynamic changes in complexes of IRE1alpha, PERK, and ATF6alpha during endoplasmic reticulum stress. Mol. Biol. Cell 29, 1376–1388. doi: 10.1091/mbc.E17-10-0594
Tagawa, Y., Hiramatsu, N., Kasai, A., Hayakawa, K., Okamura, M., Yao, J., et al. (2008). Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic. Biol. Med. 45, 50–59. doi: 10.1016/j.freeradbiomed.2008.03.003
Tanimura, A., Miyoshi, K., Horiguchi, T., Hagita, H., Fujisawa, K., and Noma, T. (2018). Mitochondrial activity and unfolded protein response are required for neutrophil differentiation. Cell. Physiol. Biochem. 47, 1936–1950. doi: 10.1159/000491464
Taniuchi, S., Miyake, M., Tsugawa, K., Oyadomari, M., and Oyadomari, S. (2016). Integrated stress response of vertebrates is regulated by four eIF2alpha kinases. Sci. Rep. 6:32886. doi: 10.1038/srep32886
Tanjore, H., Cheng, D. S., Degryse, A. L., Zoz, D. F., Abdolrasulnia, R., Lawson, W. E., et al. (2015). Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J. Biol. Chem. 290:3277. doi: 10.1074/jbc.A110.181164
Tavender, T. J., and Bulleid, N. J. (2010). Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J. Cell Sci. 123, 2672–2679. doi: 10.1242/jcs.067843
Tavender, T. J., Springate, J. J., and Bulleid, N. J. (2010). Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 29, 4185–4197. doi: 10.1038/emboj.2010.273
Teske, B. F., Wek, S. A., Bunpo, P., Cundiff, J. K., Mcclintick, J. N., Anthony, T. G., et al. (2011). The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 22, 4390–4405. doi: 10.1091/mbc.e11-06-0510
Thamsen, M., Ghosh, R., Auyeung, V. C., Brumwell, A., Chapman, H. A., Backes, B. J., et al. (2019). Small molecule inhibition of IRE1alpha kinase/RNase has anti-fibrotic effects in the lung. PLoS One 14:e0209824. doi: 10.1371/journal.pone.0209824
Thuerauf, D. J., Morrison, L., and Glembotski, C. C. (2004). Opposing roles for ATF6alpha and ATF6beta in endoplasmic reticulum stress response gene induction. J. Biol. Chem. 279, 21078–21084. doi: 10.1074/jbc.M400713200
Tirasophon, W., Welihinda, A. A., and Kaufman, R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. doi: 10.1101/gad.12.12.1812
Tirosh, B., Iwakoshi, N. N., Glimcher, L. H., and Ploegh, H. L. (2005). XBP-1 specifically promotes IgM synthesis and secretion, but is dispensable for degradation of glycoproteins in primary B cells. J. Exp. Med. 202, 505–516. doi: 10.1084/jem.20050575
Tirosh, B., Iwakoshi, N. N., Glimcher, L. H., and Ploegh, H. L. (2006). Rapid turnover of unspliced Xbp-1 as a factor that modulates the unfolded protein response. J. Biol. Chem. 281, 5852–5860. doi: 10.1074/jbc.M509061200
Tsai, Y.-L. L., and Amy, S. (2018). “Chapter 3-Cell surface GRP78: anchoring and translocation mechanisms and therapeutic potential in cancer” in Cell Surface GRP78, a New Paradigm in Signal Transduction Biology. ed. S. V. Pizzo (Academic Press, USA: Cambridge, Massachusetts).
Ubeda, M., Wang, X. Z., Zinszner, H., Wu, I., Habener, J. F., and Ron, D. (1996). Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol. Cell. Biol. 16, 1479–1489. doi: 10.1128/MCB.16.4.1479
Vanhove, M., Usherwood, Y. K., and Hendershot, L. M. (2001). Unassembled Ig heavy chains do not cycle from BiP in vivo but require light chains to trigger their release. Immunity 15, 105–114. doi: 10.1016/S1074-7613(01)00163-7
Van’t Wout, E. F., Hiemstra, P. S., and Marciniak, S. J. (2014). The integrated stress response in lung disease. Am. J. Respir. Cell Mol. Biol. 50, 1005–1009. doi: 10.1165/rcmb.2014-0019TR
Vij, N., Amoako, M. O., Mazur, S., and Zeitlin, P. L. (2008). CHOP transcription factor mediates IL-8 signaling in cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 38, 176–184. doi: 10.1165/rcmb.2007-0197OC
Wajih, N., Hutson, S. M., and Wallin, R. (2007). Disulfide-dependent protein folding is linked to operation of the vitamin K cycle in the endoplasmic reticulum. A protein disulfide isomerase-VKORC1 redox enzyme complex appears to be responsible for vitamin K1 2,3-epoxide reduction. J. Biol. Chem. 282, 2626–2635. doi: 10.1074/jbc.M608954200
Waldschmitt, N., Berger, E., Rath, E., Sartor, R. B., Weigmann, B., Heikenwalder, M., et al. (2014). C/EBP homologous protein inhibits tissue repair in response to gut injury and is inversely regulated with chronic inflammation. Mucosal Immunol. 7, 1452–1466. doi: 10.1038/mi.2014.34
Wang, Y., Gu, Y. H., Liu, M., Bai, Y., Liang, L. Y., and Wang, H. L. (2017a). TBHQ alleviated endoplasmic reticulum stress-apoptosis and oxidative stress by PERK-Nrf2 crosstalk in methamphetamine-induced chronic pulmonary toxicity. Oxidative Med. Cell. Longev. 2017:4310475. doi: 10.1155/2017/4310475
Wang, Y., Wu, Z. Z., and Wang, W. (2017b). Inhibition of endoplasmic reticulum stress alleviates cigarette smoke-induced airway inflammation and emphysema. Oncotarget 8, 77685–77695. doi: 10.18632/oncotarget.20768
Wesolowska-Andersen, A., and Seibold, M. A. (2015). Airway molecular endotypes of asthma: dissecting the heterogeneity. Curr. Opin. Allergy Clin. Immunol. 15, 163–168. doi: 10.1097/ACI.0000000000000148
White, C. W., and Martin, J. G. (2010). Chlorine gas inhalation: human clinical evidence of toxicity and experience in animal models. Proc. Am. Thorac. Soc. 7, 257–263. doi: 10.1513/pats.201001-008SM
Wilkinson, G. W., Tomasec, P., Stanton, R. J., Armstrong, M., Prod'homme, V., Aicheler, R., et al. (2008). Modulation of natural killer cells by human cytomegalovirus. J. Clin. Virol. 41, 206–212. doi: 10.1016/j.jcv.2007.10.027
Wloga, D., Joachimiak, E., Louka, P., and Gaertig, J. (2017). Posttranslational modifications of tubulin and cilia. Cold Spring Harb. Perspect. Biol. 9:a028159. doi: 10.1101/cshperspect.a028159
Wu, J., Rutkowski, D. T., Dubois, M., Swathirajan, J., Saunders, T., Wang, J., et al. (2007). ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364. doi: 10.1016/j.devcel.2007.07.005
Wu, S., Hong, F., Gewirth, D., Guo, B., Liu, B., and Li, Z. (2012). The molecular chaperone gp96/GRP94 interacts with toll-like receptors and integrins via its C-terminal hydrophobic domain. J. Biol. Chem. 287, 6735–6742. doi: 10.1074/jbc.M111.309526
Wu, X., and Rapoport, T. A. (2018). Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 53, 22–28. doi: 10.1016/j.ceb.2018.04.004
Xu, D., Perez, R. E., Rezaiekhaligh, M. H., Bourdi, M., and Truog, W. E. (2009). Knockdown of ERp57 increases BiP/GRP78 induction and protects against hyperoxia and tunicamycin-induced apoptosis. Am. J. Phys. Lung Cell. Mol. Phys. 297, L44–L51. doi: 10.1152/ajplung.90626.2008
Yang, Y., Liu, L., Naik, I., Braunstein, Z., Zhong, J., and Ren, B. (2017). Transcription factor C/EBP homologous protein in health and diseases. Front. Immunol. 8:1612. doi: 10.3389/fimmu.2017.01612
Yap, J., Chen, X., Delmotte, P., and Sieck, G. C. (2020). TNFalpha selectively activates the IRE1alpha/XBP1 endoplasmic reticulum stress pathway in human airway smooth muscle cells. Am. J. Phys. Lung Cell. Mol. Phys. 318, L483–L493. doi: 10.1152/ajplung.00212.2019
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Dave, U. P., Prywes, R., et al. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364. doi: 10.1016/S1097-2765(00)00133-7
Yoshida, H., Haze, K., Yanagi, H., Yura, T., and Mori, K. (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins: involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749. doi: 10.1074/jbc.273.50.33741
Yuan, X., Wang, J., Li, Y., He, X., Niu, B., Wu, D., et al. (2018). Allergy immunotherapy restores airway epithelial barrier dysfunction through suppressing IL-25-induced endoplasmic reticulum stress in asthma. Sci. Rep. 8:7950. doi: 10.1038/s41598-018-26221-x
Zeng, L., Li, Y., Yang, J., Wang, G., Margariti, A., Xiao, Q., et al. (2015). XBP 1-deficiency abrogates neointimal lesion of injured vessels via cross talk with the PDGF signaling. Arterioscler. Thromb. Vasc. Biol. 35, 2134–2144. doi: 10.1161/ATVBAHA.115.305420
Zhang, S., Go, E. P., Ding, H., Anang, S., Kappes, J. C., Desaire, H., et al. (2021). Analysis of glycosylation and disulfide bonding of wild-type SARS-CoV-2 spike glycoprotein. BioRXiv [Preprint]. doi: 10.1101/2021.04.01.438120
Zhou, A. X., Wang, X., Lin, C. S., Han, J., Yong, J., Nadolski, M. J., et al. (2015). C/EBP-homologous protein (CHOP) in vascular smooth muscle cells regulates their proliferation in aortic explants and atherosclerotic lesions. Circ. Res. 116, 1736–1743. doi: 10.1161/CIRCRESAHA.116.305602
Keywords: unfolded protein response, endoplasmic reticulum, integrated stress response, post-translational modifications, disulfide bonds, lung disease, lung function
Citation: Nakada EM, Sun R, Fujii U and Martin JG (2021) The Impact of Endoplasmic Reticulum-Associated Protein Modifications, Folding and Degradation on Lung Structure and Function. Front. Physiol. 12:665622. doi: 10.3389/fphys.2021.665622
Edited by:
Andrew John Halayko, University of Manitoba, CanadaReviewed by:
Amir A. Zeki, University of California, Davis, United StatesPawan Sharma, Thomas Jefferson University, United States
Copyright © 2021 Nakada, Sun, Fujii and Martin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James G. Martin, amFtZXMubWFydGluQG1jZ2lsbC5jYQ==