
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 22 March 2021
Sec. Cardiac Electrophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.661429
This article is part of the Research Topic Inherited Arrhythmias of the Cardiac Sodium Channel Nav1.5 View all 8 articles
 Yang Zheng1,2
Yang Zheng1,2 Xiaoping Wan1
Xiaoping Wan1 Dandan Yang1
Dandan Yang1 Angelina Ramirez-Navarro1
Angelina Ramirez-Navarro1 Haiyan Liu1
Haiyan Liu1 Ji-Dong Fu1
Ji-Dong Fu1 Isabelle Deschênes1*
Isabelle Deschênes1*Nav1.5, encoded by the gene SCN5A, is the predominant voltage-gated sodium channel expressed in the heart. It initiates the cardiac action potential and thus is crucial for normal heart rhythm and function. Dysfunctions in Nav1.5 have been involved in multiple congenital or acquired cardiac pathological conditions such as Brugada syndrome (BrS), Long QT Syndrome Type 3, and heart failure (HF), all of which can lead to sudden cardiac death (SCD) – one of the leading causes of death worldwide. Our lab has previously reported that Nav1.5 forms dimer channels with coupled gating. We also found that Nav1.5 BrS mutants can exert a dominant-negative (DN) effect and impair the function of wildtype (WT) channels through coupled-gating with the WT. It was previously reported that reduction in cardiac sodium currents (INa), observed in HF, could be due to the increased expression of an SCN5A splice variant – E28D, which results in a truncated sodium channel (Nav1.5-G1642X). In this study, we hypothesized that this SCN5A splice variant leads to INa reduction in HF through biophysical coupling with the WT. We showed that Nav1.5-G1642X is a non-functional channel but can interact with the WT, resulting in a DN effect on the WT channel. We found that both WT and the truncated channel Nav1.5-G1642X traffic at the cell surface, suggesting biophysical coupling. Indeed, we found that the DN effect can be abolished by difopein, an inhibitor of the biophysical coupling. Interestingly, the sodium channel polymorphism H558R, which has beneficial effect in HF patients, could also block the DN effect. In summary, the HF-associated splice variant Nav1.5-G1642X suppresses sodium currents in heart failure patients through a mechanism involving coupled-gating with the wildtype sodium channel.
The voltage-gated cardiac sodium channel, known as Nav1.5 and encoded by the SCN5A gene, is the predominant voltage-gated sodium channel expressed in the heart. The channel initiates the cardiac action potential in myocytes by generating a rapid sodium influx. Dysfunction in Nav1.5 can lead to various cardiac arrhythmic diseases. Mutations in SCN5A have been linked to several inherited cardiac channelopathies including Long QT Syndrome Type 3 and Brugada syndrome (BrS) (Amin et al., 2010; Veerman et al., 2015). In addition to congenital disorders, reduction in sodium current, INa, was linked to human heart failure (HF) (Tomaselli and Zipes, 2004). However, the mechanisms leading to a reduction in INa are not fully understood. Pathological behaviors of the channel such as (1) reduction in sodium channel protein due to degradation or decrease in mRNA, (2) lower surface expression level and (3) abnormal kinetics such as changes in voltage-dependence of channel gating can all contribute to the decreased INa. Our lab previously demonstrated that cardiac sodium channels exist and gate as dimers and importantly, we showed that some Nav1.5 mutants exert dominant negative (DN) effect on the wildtype channel (WT) leading to a reduction in current through coupled-gating with the WT (Clatot et al., 2017). Notably, we were able to uncouple the channels by inhibiting the 14-3-3 accessory protein, resulting in removal of the DN effect (Clatot et al., 2017, 2018). We and the Lampert lab also showed a similar mechanism for the neuronal voltage-gated sodium channels Nav1.1, Nav1.2 and Nav1.7 (Clatot et al., 2017; Rühlmann et al., 2020). Therefore, uncoupling the Nav1.5 or other Navs from their pathogenic mutants could serve as a new strategy to develop clinical treatments for cardiac or neuronal diseases associated with Navs dysfunction.
There are also cardiac pathologies, such as HF, where decreases in INa are observed but without being linked to SCN5A mutations. It is very likely that the phenotypes can be associated with post-transcriptional and post-translational modifications of SCN5A. Alternative splicing or differential splicing is a post-transcriptional modulation of a gene that generates different combination of the translated regions, yielding different splice variants of the expressed gene. SCN5A splice variants have been studied for their implication in cardiac pathogenesis, and the C-terminal splice variants were recently shown to play a role in human cardiac pathology. Three C-terminal splice variants, E28B, E28C and E28D are associated with human HF, and were shown to activate unfolded protein response (Shang et al., 2007, 2008; Gao et al., 2013; Noyes et al., 2017). Among them, E28D is the most abundant one which gets significantly upregulated in human HF, and is also related with pulmonary arterial hypertension in clinical practice (Banerjee et al., 2020). SCN5A-E28D generates a non-functional truncated sodium channel Nav1.5-G1642X. This splice variant not only results in a nonfunctional channel but was also shown to reduce the WT current. In this study, we therefore aimed to investigate the dominant negative effect mechanisms produced by the E28D splice variant. Based on our earlier findings demonstrating dimerization and biophysical coupling of sodium channels, we hypothesized that this SCN5A splice variant leads to a reduction in INa through biophysical coupling with the WT. Our team previously found that the common Nav1.5-H558R can interact with multiple pathogenic SCN5A genetic mutations found in BrS or Long QT syndrome to rescue their pathogenic effect (Ye et al., 2003; Shinlapawittayatorn et al.,2011a,b). Interestingly, this Nav1.5-H558R polymorphism was reported to improve the survival of HF patients (Aleong et al., 2005). We therefore also explored if the presence of this polymorphism could influence the DN-effect produced by the splice variant.
The SCN5A splice variant G1642X, polymorphism H558R, and mutation S460A plasmids were created using the QuickChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instruction. The constructs were made on the pcDNA3.1 vector containing the N-terminus GFP-fused Nav1.5 (Clatot et al., 2012) (BD Biosciences, San Jose, CA, United States). The outcomes were verified by sequencing.
The plasmids of interest including the cardiac sodium channels and the 14-3-3 inhibitor difopein were transiently transfected into HEK 293 cells and human induced pluripotent stem cells derived cardiomyocytes (hiPSC-CMs). HEK 293 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin until they grew to about 60% confluence and were then used for transfection. During the transfection, the cells were maintained in DMEM without penicillin-streptomycin. The transfection was done with the reagent FUGENE® (Promega, Madison, WI, United States) according to the manufacturer’s instruction. The total amount of plasmid transfected into HEK293 cells was 0.6 μg for patch-clamp experiments; however, for the experiments where difopein was used, an additional 0.6 μg of difopein plasmids was transfected or 0.6 μg of empty pcDNA3.1 vector for the control group in order to balance the total amount of transfected DNA. For the biochemical experiments a total of 2.0 μg of the plasmids of interest were transfected into HEK293 cells. In the group where two plasmids of interest were co-expressed, i.e., WT and G1642X, 0.3 μg (for patch-clamp) or 1.0 μg (for biochemistry) of each DNA was transfected. In the group where only one sodium channel was expressed, 0.3 μg (for patch-clamp) or 1.0 μg (for biochemistry) of empty pcDNA3.1 vector was transfected in order to balance the total amount of transfected DNA. For hiPSC-CMs experiments, we used the commercially available iCells® (FujiFilm, Madison, WI, United States). The iCells® were cultured according to manufacturer’s instruction. We transfected 1.8 μg of Nav1.5-G1642X for the experimental group and 1.8 μg of YFP for the control group. The iCells® were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instruction.
Sodium currents were recorded at room temperature (21–23°C) by patch-clamp technique in whole-cell configuration, 24 h (HEK 293 cells) and 48 h (iCells®) after the transfection. The recordings were obtained using the Axopatch 200A amplifier (Molecular Devices, San Jose, CA, United States) and the Digidata 1440A digitizer (Molecular Devices, San Jose, CA, United States). The protocols were generated with the pCLAMP 14.2 software (Molecular Devices, San Jose, CA, United States). For HEK 293 cells the intracellular solution contained: NaCl 35 mM, CsF 105 mM, EGTA 10 mM, and Cs-HEPES 10 mM and the extracellular solution contained: NaCl 135 mM, KCl 4.5 mM, MgCl2 0.7 mM, CaCl2 2 mM, glucose 10 mM, and HEPES 10 mM. For the iPSC-CMs recording the intracellular solution contained: CsMes 130mM, TEACl 20 mM, MgCl2 1 mM, EGTA 10 mM, HEPES 10 mM, and MgATP 4 mM. The extracellular solution contained: NaCl 25 mM, CsCl 5.4 mM, MgCl2 1.8 mM, CaCl2-2H2O 1.8 mM, HEPES 10 mM, Glucose 10 mM and NMDG 105 mM. The pH of the iPSC-CM intracellular solution is adjusted to 7.2, and for the other solutions the pH was adjusted to 7.4. The current-voltage relationships of the sodium currents were recorded by holding the resting membrane potential at −120 mV and stepping from −80 mV to +60 mV in 10 mV interval (each step hold for 30 ms). The persistent or late sodium currents (INa–late) were recorded with a test pulse of 250 ms at −30 mV from a holding potential of −120 mV. Steady-state inactivation and recovery from inactivation were recorded using protocols as previously described (Poelzing et al., 2006). Briefly, the recovery from inactivation was recorded with a two-pulse protocol. Both the pre-pulse and the test-pulse duration are 30ms, stepping from −30 mV to −120 mV. The interval between the two pulses ranges from 1.8 ms to 70 ms. Currents of the recovery from inactivation were fit to the following equation:
The steady-state inactivation was studied with a 500 ms pre-pulse ranging from −140 mV to −30 mV, followed by a 30ms test pulse stepping from −120 mV to −30 mV. The currents for the steady-state inactivation were fit to a Boltzmann distribution using the following equation:
The fitting curves of steady-state inactivation and recovery from inactivation analysis were generated with Origin 10.1.1 software (OriginLab Corporation, Northampton, MA, United States). Throughout the whole-cell patch-clamp recordings, about 85% of the series resistance was compensated with a lag of about 10 μs. Leak subtraction protocol was used only for the persistent sodium current recordings.
HEK 293 cells were transfected with a total amount of 2.0 μg of DNA for the biochemical analysis. The cells were washed 48 h after transfection and collected for the surface biotinylation assays or co-immunoprecipitation (co-IP) experiments. Cell surface biotinylation assays were performed according to the manufacturer’s instruction and as previously described (Hoshi et al., 2014; Clatot et al., 2018). Briefly, the surface proteins were labeled at 4°C for 30 min with 0.25mg/ml Pierce Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific). The biotinylated proteins were then isolated through a Pierce NeutrAvidin Agarose resin column (Thermo Fisher Scientific). Co-IP experiments were performed with Dynabeads Protein G (Thermo Fisher Scientific) as previously described (Clatot et al., 2012, 2017). Briefly, the magnetic beads were washed twice with citrate-phosphate buffer (pH 5.4) and then incubated with 5 μg rabbit anti-GFP antibody (Invitrogen, Cat# A11122) for 2 h at room temperature. The incubated beads were then washed with citrate-phosphate buffer plus 0.1% Tween20 and incubated with precleared lysate sample (total amount of 400 mg protein) at 4°C overnight. Next, the sample was washed three times with lysis buffer before elution. The proteins were eluted with XT sample buffer (Bio-Rad) at 37°C for 1 h. Finally, the western blot experiments were performed to reveal the results of both biotinylation assays and co-IPs using the following primary antibodies: anti-Nav1.5 antibody 1:1000 (kind gift from Dr. Thomas Hund) (Hund et al., 2010), anti-HA antibody 1:1000 (Sigma, Cat# H6908), and anti-GFP antibody 1:2000 (Invitrogen, Cat# A11122).
Statistical analyses were performed using the standard statistical package in Origin 10.1.1 (OriginLab Corporation, Northampton, MA, United States). Student’s t-test was performed at a significance level p = 0.05 for single comparison after a normality test with the Shapiro-Wilk method for sample size 7–50. Two-side p values less than 0.05 were considered statistically significant. Results were presented as mean ± SEM.
The SCN5A-E28D splice variant results in a Nav1.5 truncated at the 1642th glycine residue therefore truncating the channel from the middle of the DIV S4 segment until the end of the C-terminus (Shang et al., 2007). It was previously shown that co-expression of this truncated splice variant with the WT channel leads to a reduction in WT currents (Shang et al., 2007). In order to study the mechanism by which Nav1.5-G1642X exerts this DN effect, we recorded sodium currents using whole-cell patch-clamping. We first confirmed that the splice variant is non-functional (n = 11) (Figures 1A,B). When Nav1.5-G1642X was co-expressed with the WT, the WT current was significantly (n = 47, p < 0.05) reduced by Nav1.5-G1642X although we transfected the same amount of WT DNA into each group, therefore suggesting a DN-effect (Figures 1A,B). For a DN effect, a reduction of 75% in current density would be expected when co-expressing the same level of WT and splice variant (Clatot et al., 2017), however, here the current was reduced by about 40% (Figures 1A,B). This could be explained by a partial degradation of the truncated Nav1.5-G1642X. We then quantified the expression level of the Nav1.5-WT and Nav1.5-G1642X, and found that about 40% of the truncated Nav1.5-G1642X was degraded (0.64 ± 0.11 arbitrary units) compared with the WT. Therefore, this explains why the current reduction was not of 75%. We also measured the persistent sodium current INa–late since an increased in persistent current is widely observed in HF. However, we found that Nav1.5-G1642X does not lead to an increase in the INa–late (n = 7) of the WT (Figure 1D). The C-terminus of Nav1.5 and the DIV S4 are known to modulate the channel inactivation (Cormier et al., 2002). Since we have previously shown that a channel with defective biophysical properties can modulate the gating properties of the WT channel (Shinlapawittayatorn et al., 2011b; Clatot et al., 2017), we questioned whether the kinetics of the WT channel would be influenced by the splice variant since it is lacking the DIV S4 and the C-terminus. However, we found that neither the conductance (n = 47, ns) (Figure 1C), the steady-state inactivation (n = 50, ns) (Figure 2A), nor the recovery from inactivation (n = 48, ns) (Figure 2B) of Nav1.5-WT were influenced by Nav1.5-G1642X.
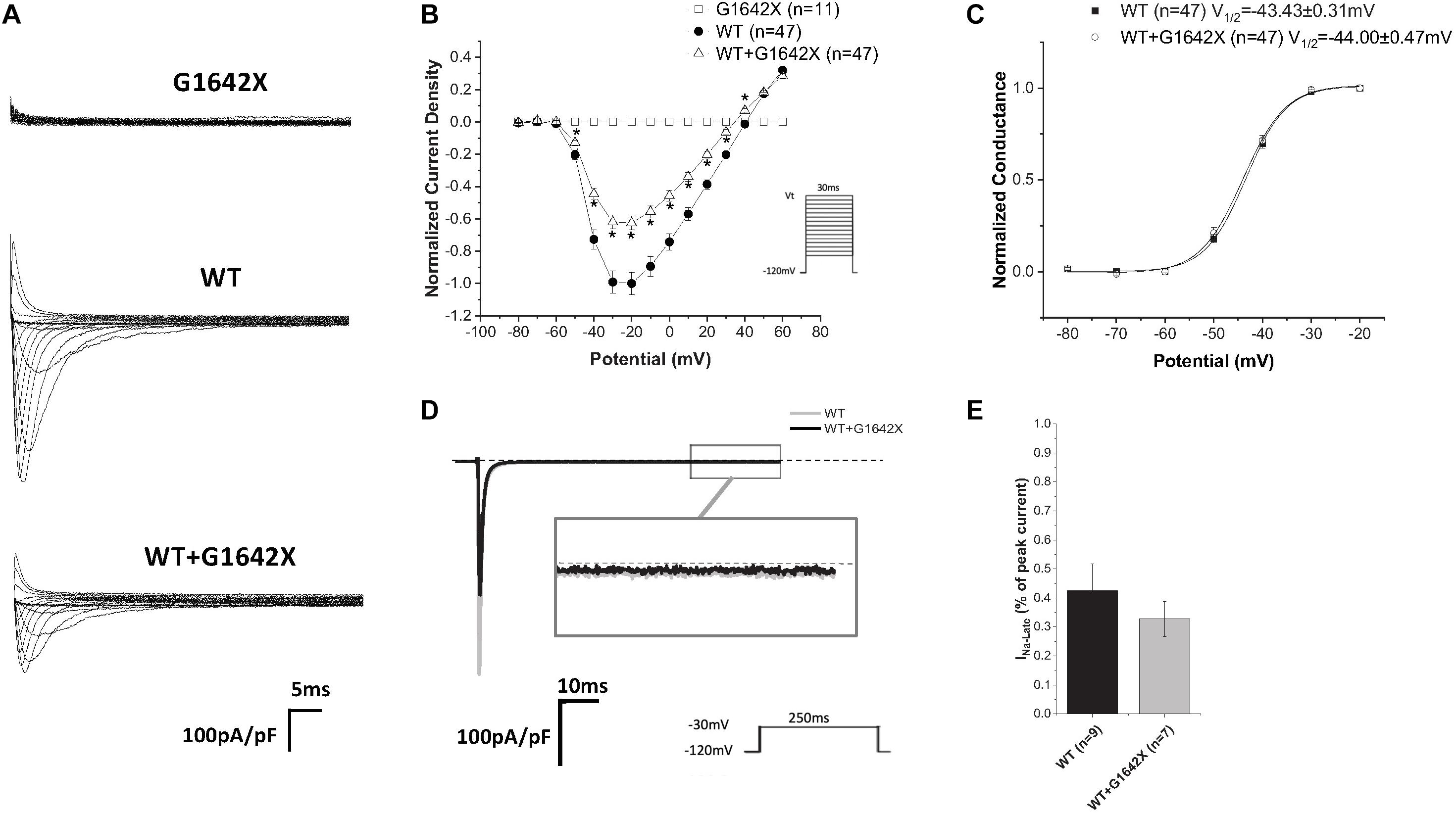
Figure 1. (A) Representative sodium current traces elicited using the indicated voltage protocol. (B) Current voltage relationships (I/V) curves. (C) Voltage-dependent conductance curves for Nav1.5-WT, Nav1.5-G1642X, and the co-expression of Nav1.5-WT and Nav1.5-G1642X recorded from HEK 293 cells. (D) Representative late sodium currents. (E) Quantification of the late sodium currents recorded from Nav1.5-WT and the co-expression of Nav1.5-WT with Nav1.5-G1642X. Voltage protocols used are included. *p < 0.05 compared to Nav1.5-WT.
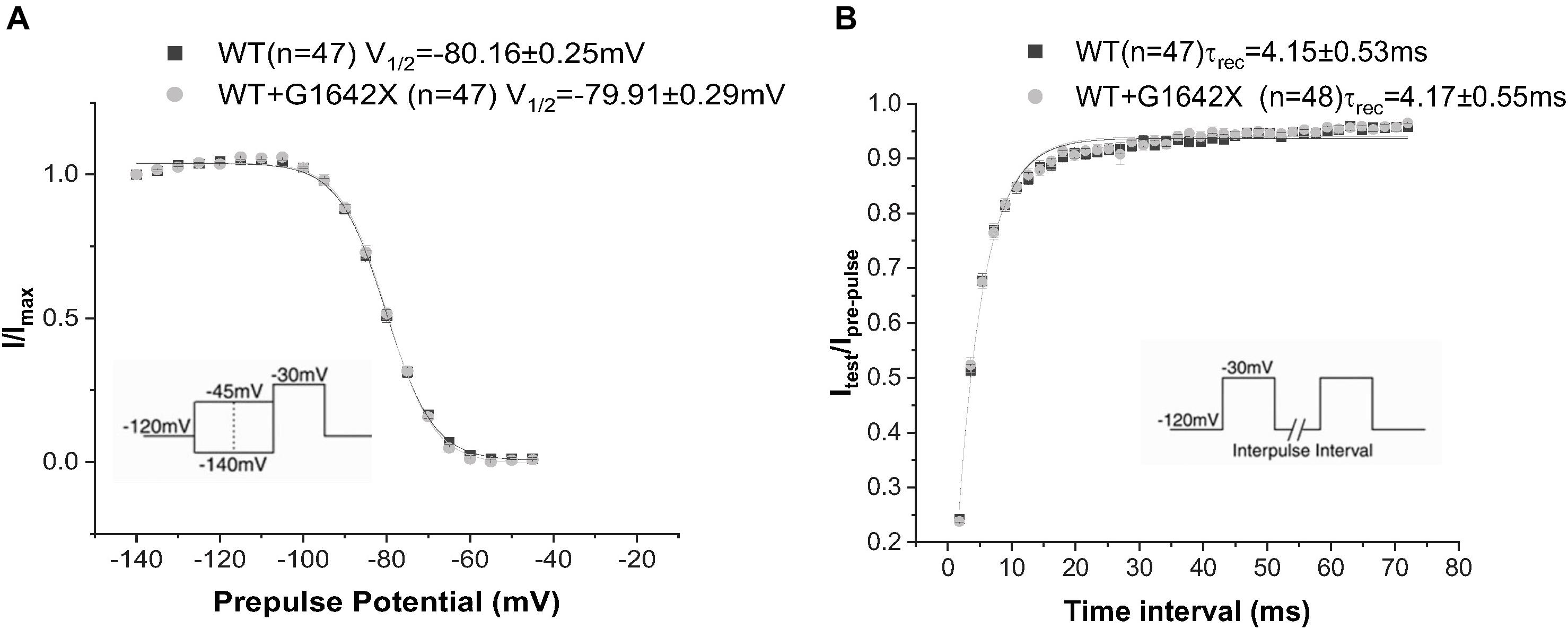
Figure 2. Recovery from inactivation (A) and steady-state inactivation (B) recorded from HEK 293 cells expressing Nav1.5-WT or Nav1.5-WT+Nav1.5-G1642X. The splice variant G1642X does not influence the recovery from inactivation and the steady-state inactivation of the Nav1.5-WT. Voltage protocols used are included.
We then investigated if this DN-effect of the splice variant could be reproduced in human cardiomyocytes, which better mimic what happens in patients. We transfected Nav1.5-G1642X into the commercially available iPSC-CMs iCells® and also observed a dramatic reduction (n = 7, p < 0.05) in endogenous INa when the cells were transfected with Nav1.5-G1642X compared to YFP transfected cells (Figures 3A,B).
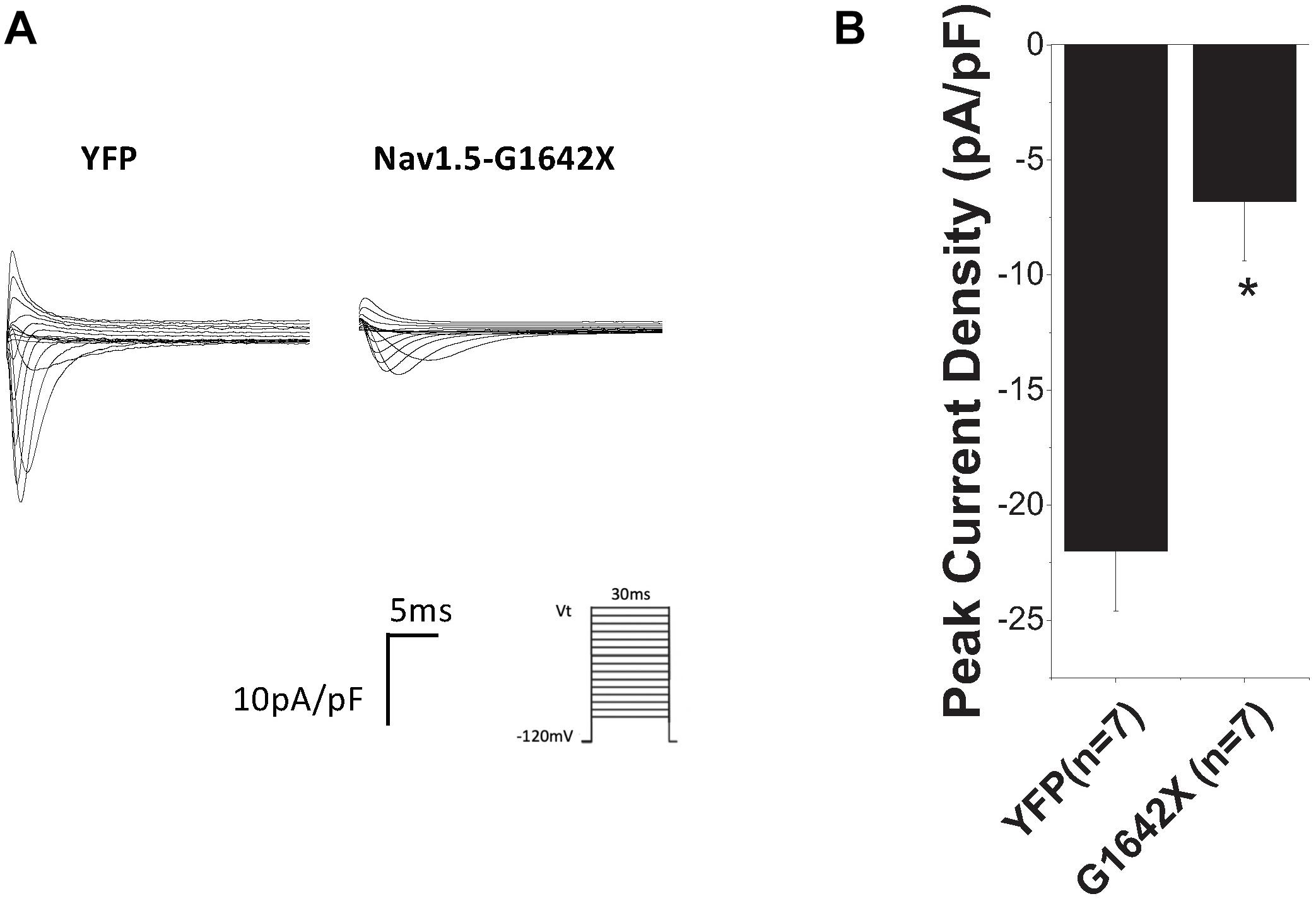
Figure 3. (A) Representative sodium current traces. (B) Peak current densities recorded from the commercially available iPSC-CM iCells®. The control group was cells transfected with YFP and currents recorded from these cells represent the endogenous sodium current present in these cells. This control current was then compared to currents recorded from iCells® transfected with Nav1.5-G1642X. Voltage protocol used is included. *p < 0.05 compared to YFP transfected cells.
Other than modulating the channel inactivation, the C-terminus of Nav1.5 can bind with multiple partner proteins which can regulate trafficking of the channel (Abriel, 2010). It is thus possible that since this splice variant is missing the whole C-terminus, that this could impair trafficking of Nav1.5-G1642X to the cell surface and withhold WT channels inside the cell through their interactions. To explore this possible mechanism of INa reduction, we measured the expression of the sodium channel at the cell surface through surface biotinylation assays. Similar to the electrophysiological studies, Nav1.5-G1642X was co-expressed with the WT to mimic the expression pattern from HF patients. In this experiment, we found that the splice variant Nav1.5-G1642X was able to traffic to the cell surface both when expressed alone and when co-expressed with WT. Importantly, the WT was also present at the surface therefore not explaining the DN-effect and reduction in INa observed (Figure 4). This demonstrates that Nav1.5-G1642X doesn’t affect Nav1.5 channel trafficking at the cell surface, suggesting that other mechanisms (e.g., biophysical coupling) might be taking place (Clatot et al., 2017, 2018).
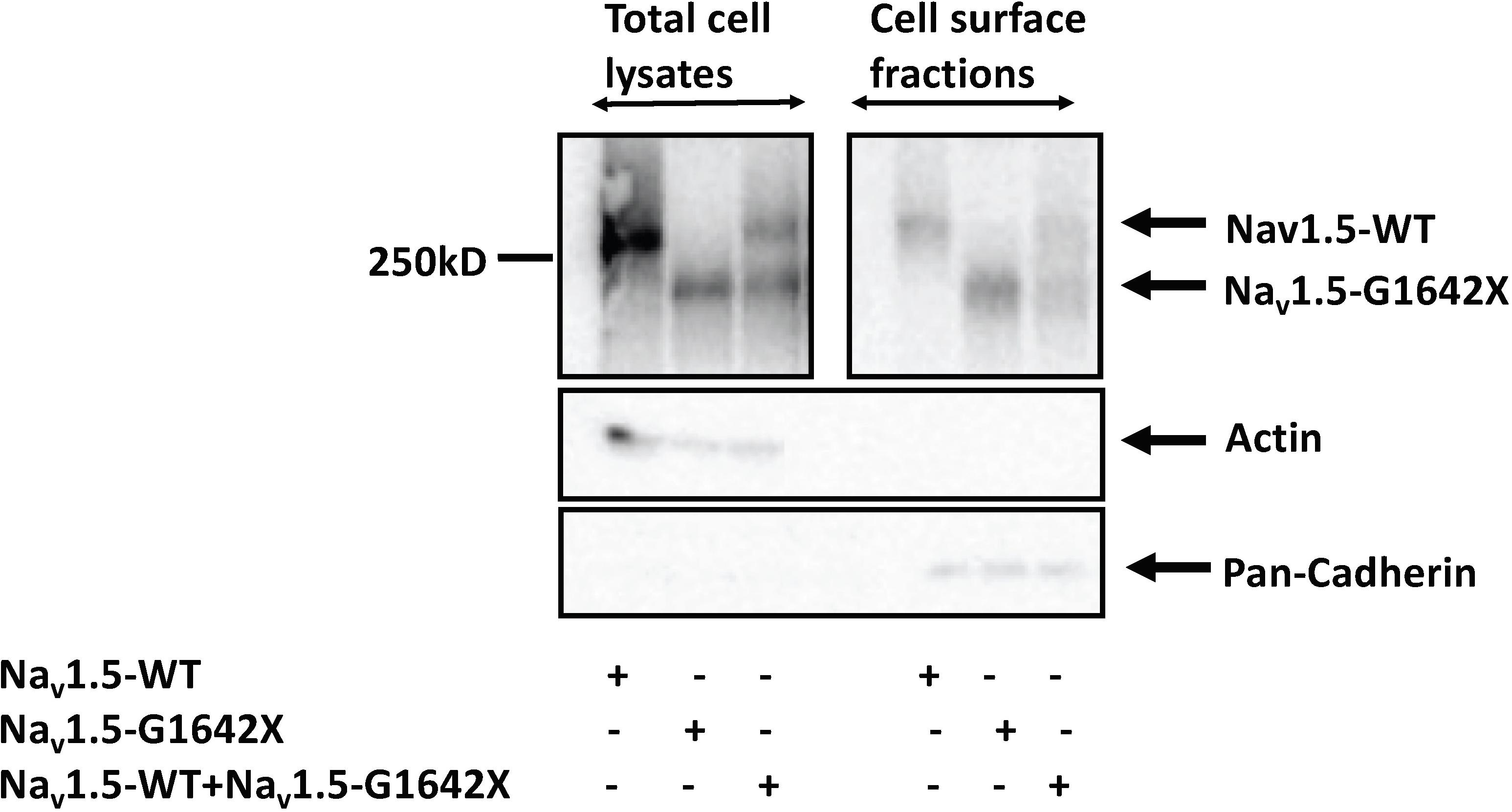
Figure 4. Surface Biotinylation assays showed that the Nav1.5-G1642X is expressed at the cell membrane. Nav1.5-WT is also present at the cell surface in presence of Nav1.5-G1642X. HEK 293 cells were transfected with either Nav1.5-WT, Nav1.5-G1642X, or Nav1.5-WT+Nav1.5-G1642X. Western blots were probed with a sodium channel antibody and both bands for Nav1.5-WT and Nav1.5-G1642X could be distinguished based on the smaller size of the truncated channel.
Our lab previously showed that Nav1.5 form dimers through interaction between DI-DII linker (amino acids 493–517), and this dimerization leads to coupled-gating. Similar results for the neuronal voltage-gated sodium channels Nav1.1, Nav1.2, and Nav1.7 were reported by us and other labs (Clatot et al., 2017; Rühlmann et al., 2020). We and others also demonstrated that the protein 14-3-3 is crucial for the regulation of the coupled-gating, and inhibition of 14-3-3 using the inhibitor difopein removed the coupled-gating of the Navs (Clatot et al., 2017, 2018; Rühlmann et al., 2020). In addition, we have shown that the modulation by 14-3-3 is dependent on phosphorylation of Nav1.5 Serine 460. Thus, other than direct 14-3-3 inhibition, the 14-3-3 activity and the Nav1.5 coupled-gating can be prohibited by mutating this Serine into an Alanine (S460A) (Clatot et al., 2017; Rühlmann et al., 2020). If Nav1.5-G1642X leads to a DN-effect through coupled-gating with the WT, we should be able to see the removal of the DN-effect by either using the 14-3-3 inhibitor difopein or with S460A. We first confirmed the interaction between Nav1.5-G1642X and the WT with co-IP experiments, and found that Nav1.5-WT (tagged with HA) was successfully pulled down by GFP antibody that targets the GFP-fused Nav1.5-G1642X (Figure 5). Next, we measured INa while inhibiting the biophysical coupling of the WT with co-expressed Nav1.5-G1642X by difopein administration (n = 29, ns) or by mutating Ser460 to alanine (n = 38, ns) in Nav1.5-G1642X. Importantly, both difopein and the S460A mutation removed the DN-effect of Nav1.5-G1642X, restoring current density to the WT alone level (Figures 6A,B). Although difopein did not change the current density, it shifted the voltage-dependent conductance curve toward a negative direction (n = 29, p < 0.05) (Figure 6C). These results demonstrate that the coupled-gating between the WT and Nav1.5-G1642X is essential to the DN-effect and the INa reduction.
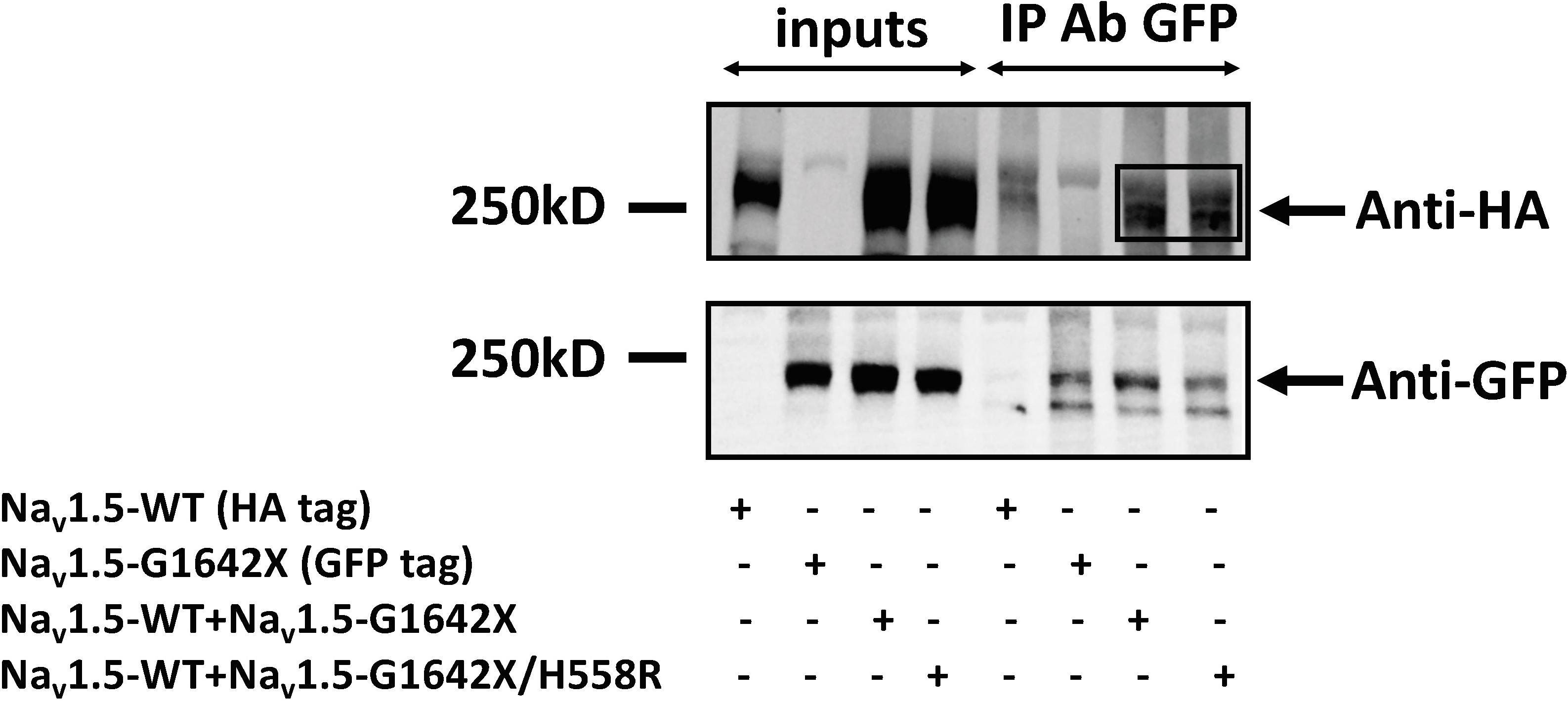
Figure 5. Co-Immunoprecipitation experiments for Nav1.5-WT-HA with Nav1.5-G1642X-GFP, and Nav1.5-WT-HA with Nav1.5-WT-G1642X/H558R-GFP. Complexes were coimmunoprecipitated using the anti-GFP antibody and revealed with both anti-GFP and anti-HA antibodies. Bands in the Anti-HA blot for the co-transfection conditions indicate interaction between Nav1.5-WT-HA and Nav1.5-G1642X-GFP, and between Nav1.5-WT-HA and Nav1.5-G1642X/H558R-GFP.
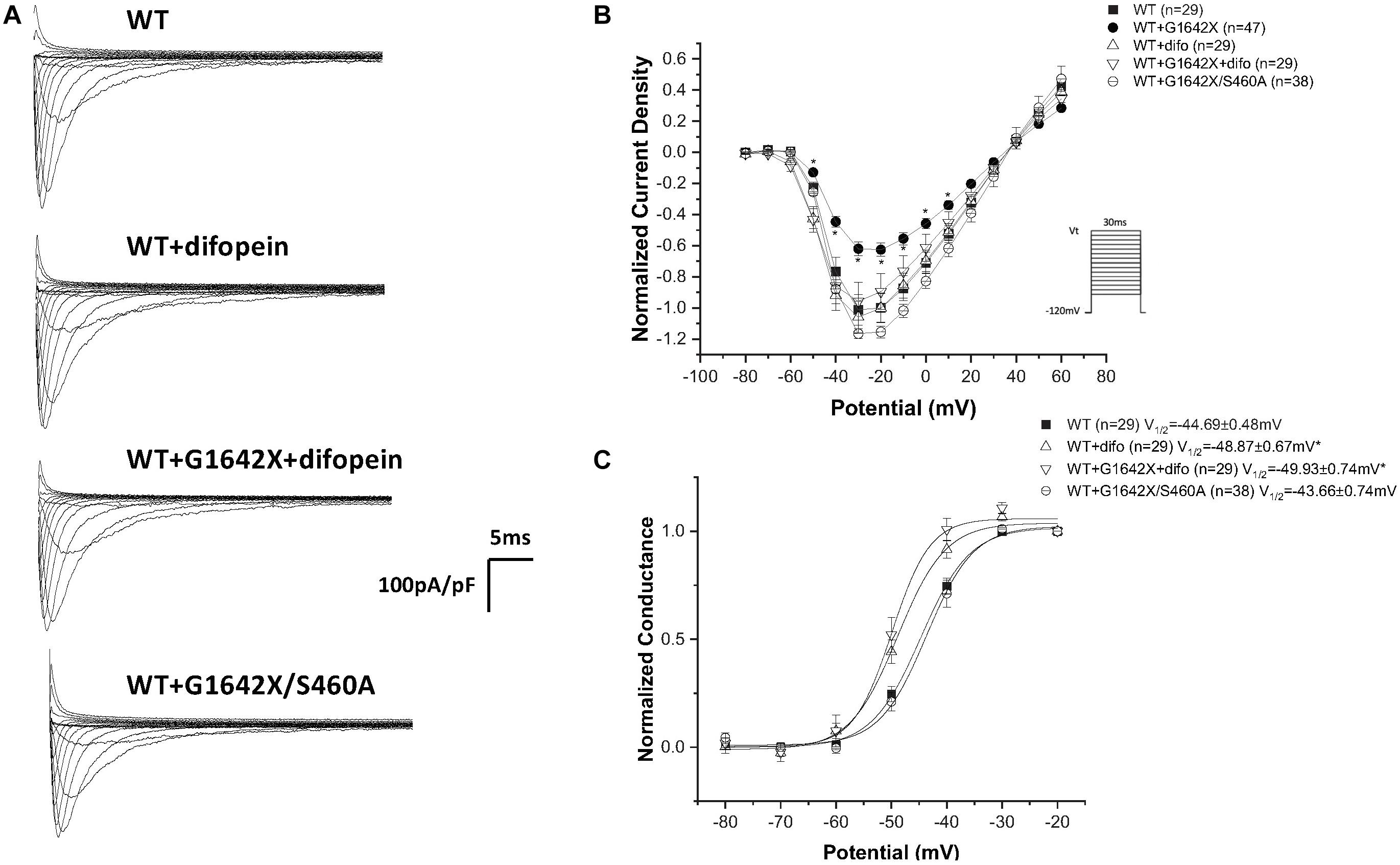
Figure 6. Inhibition of coupled-gating through modulation of 14-3-3. 14-3-3 was inhibited either through difopein (difo) or by mutating serine 460 to alanine (S460A) which removes Nav1.5 coupled-gating. (A) Representative sodium current traces. (B) Current Voltage Relationships (I/V) curves. (C) Voltage-dependent conductance curves for Nav1.5-WT, Nav1.5-G1642X, Nav1.5-WT+Nav1.5-G1642X, Nav1.5-WT+difo, Nav1.5-WT+Nav1.5-G1642X+difo, Nav1.5-WT+Nav1.5- G1642X/S460A recorded from HEK 293 cells. Voltage protocol used is included. *p < 0.05 compared to Nav1.5-WT. The Nav1.5-WT+Nav1.5-G1642X I/V curve shown in the right panel is reproduced from Figure 1 for easy comparison.
Nav1.5-H558R is a common polymorphism present in 30% of the population. It is the most frequent Nav1.5 polymorphism. This polymorphism is long known to be a genetic modifier of cardiac pathologies. Indeed, H558R can promote the functionality of mutated channels found in BrS, Long QT Syndrome and was also clinically observed to improve the survival of HF patients (Shinlapawittayatorn et al., 2011b; Aleong et al., 2005; Poelzing et al., 2006; Viswanathan et al., 2003; Marangoni et al., 2011; Matsumura et al., 2017). We therefore asked if the Nav1.5-H558R polymorphism could improve the survival of HF patients by also impairing the DN-effect caused by the HF splice variant Nav1.5-G1642X. We recorded INa for Nav1.5-H558R (n = 8, ns), and first tested the effect of Nav1.5-G1642X on Nav1.5-H558R by co-expressing the two channels together. We found that Nav1.5-G1642X did not produce a DN-effect on Nav1.5-H558R (n = 31, ns) (Figures 7A,C). We also inserted the H558R polymorphism on the G1642X construct (Nav1.5-G1642X/H558R) and co-expressed this construct with the WT Nav1.5. Interestingly, we found that Nav1.5-G1642X/H558R did not produce a DN-effect (n = 13, ns) (Figures 7A,B). This suggests that the presence of H558R disrupts the coupling. To confirm this, we also co-expressed difopein which we have shown can disrupt the coupling, with Nav1.5-G1642X and Nav1.5-H558R, and we did not observe any additional increase in the current density (n = 15, ns) (Figures 7A,B). This suggest that the coupling between the Nav1.5-H558R and the splice variant Nav1.5-G1642X is already disrupted. Difopein as in Figure 6C, also shifted the voltage-dependent conductance curve toward negative voltages (n = 15, p < 0.05) (Figure 7C). These results therefore suggest that the Nav1.5-H558R polymorphism removes this DN-effect by impairing the biophysical coupling between the splice variant and the WT resulting in restored INa. Finally, as observed with the WT, the splice variant did not change the steady-state inactivation (n = 13, ns) (Figure 8A) and the recovery from inactivation (n = 11, ns) (Figure 8B) of Nav1.5-H558R.

Figure 7. (A) Representative sodium current traces. (B) Current voltage relationships (I/V) curves. (C) Voltage-dependent conductance curves for Nav1.5-WT, Nav1.5-WT+Nav1.5-G1642X, Nav1.5-WT+Nav1.5-G1642X/H558R, Nav1.5-H558R, or Nav1.5-H558R+Nav1.5-G1642X, and Nav1.5-H558R+Nav1.5-G1642X+difopein recorded from HEK 293 cells. Voltage protocol used is included. *p < 0.05 compared to Nav1.5-WT. The splice variant does not exert DN effect on Nav1.5-H558R.
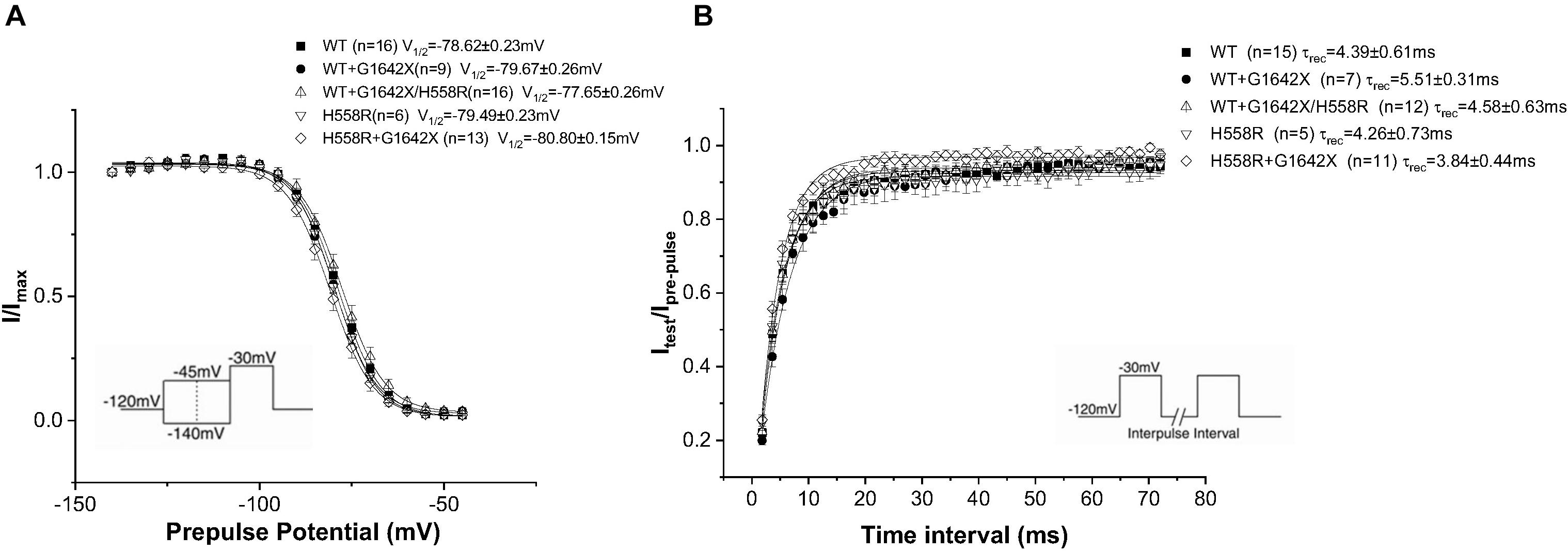
Figure 8. Recovery from inactivation (A) and steady-state inactivation (B) recorded from HEK 293 cells expressing either Nav1.5-WT, Nav1.5-WT+Nav1.5-G1642X, Nav1.5-WT+Nav1.5-G1642X/H558R, Nav1.5-H558R, or Nav1.5-H558R+Nav1.5-G1642X. The splice variant Nav1.5-G1642X does not influence the recovery from inactivation and the steady-state inactivation of the Nav1.5-H558R. Voltage protocols used are included.
Heart failure describes the state of the heart that fails to pump blood. It can cause over 250,000 annual deaths in the United States alone, and the number keeps growing with the aging population. Importantly more than half of patients with HF present with arrhythmias and SCD due to ion channel remodeling (Tomaselli and Zipes, 2004). Electrophysiological remodeling of Nav1.5 often occurs in HF resulting in reduction of the peak INa and an increase in the sustained late sodium current (INa–late). Reduction in INa is also commonly known to associate with BrS and conduction system diseases which can lead to HF and augment the risk of SCD (Valdivia et al., 2005; Remme and Bezzina, 2010; Mizusawa and Wilde, 2012). Splice variants in SCN5A naturally occur as a post-transcriptional regulation of Nav1.5 expression. To date, Nav1.5 splice variants have been found in the sequence of exon 6, exon 17, exon 18, exon 24 and exon 28. These splice variants can be either functional or nonfunctional and they can be developmentally regulated, i.e., exon 6 splice variant is also designated as the neonatal splice variant (Schroeter et al., 2010). The nonfunctional splice variant E28D associated with HF and other cardiac pathologies was shown to contribute to the reduction of INa (Gao et al., 2013; Noyes et al., 2017; Banerjee et al., 2020). In the present study, we found that although associated with HF, the SCN5A splice variant E28D (i.e., Nav1.5-G1642X) does not contribute to the increased INa–late observed in HF patients (Figures 1D,E). We therefore explored how Nav1.5-G1642X contributes to the reduction of INa seen in the failing heart. We demonstrated that the truncated channel Nav1.5-G1642X interacts with the WT Nav1.5 channel and exerts a dominant-negative effect, contributing to the decrease in INa seen in HF.
The mechanism behind INa reduction can vary depending on the genetic environment and the pathological condition. Previous studies reported that the reduction in cardiac sodium current can be induced by trafficking deficiency of the SCN5A mutants. For example, the mutant SCN5A-R282H identified in BrS patients was deficient in trafficking to the cell surface and therefore reduced the cardiac sodium currents (Poelzing et al., 2006). The trafficking efficiency can also be regulated by Nav1.5 partner proteins such as ankyrin-G and dystrophin therefore introducing other pathways to disturb the trafficking or surface expression of Nav1.5 (Abriel, 2010; Lang et al., 2018). In addition, abnormal post-translational modifications, such as phosphorylation and deglycosylation, were both shown to contribute to the decreased INa amplitude (Qu et al., 1996; Ufret-Vincenty et al., 2001). Moreover, our group reported that coupled-gating is one of the mechanisms leading to a decrease in INa. We found that the SCN5A-L325R BrS mutation exerted a DN-effect through coupled-gating with the SCN5A-WT. We revealed that the coupled-gating between the mutants and the WT could reduce the open probability and the coupling of the WT (Clatot et al., 2018). The decrease in sodium current observed in HF and the presence of HF-associated Nav1.5 splice variants agree with our previous studies with BrS mutants. Indeed, we show here that the splice variant Nav1.5-G1642X interacts with the WT (Figure 5), but has no influence on the channel trafficking, as both the truncated splice variant and the WT are trafficking at the cell surface (Figure 4). It demonstrated that much like BrS mutants, this truncated non-functional channel might exert a DN-effect through coupled-gating with the WT (Figures 1, 3), which would explain the significant reduction in sodium current seen in HF myocytes. When considering a mutant resulting in a DN effect for a dimeric channel, one would expect a reduction in sodium current of about 75% (Clatot et al., 2017). However, unlike what we observed with the BrS DN mutation L325R which leads to a ∼75% INa reduction (Clatot et al., 2017), the truncated Nav1.5-G1642X in the present study reduced the currents by about 40%. One potential explanation would be if there was a mild degradation of the truncated Nav1.5-G1642X channel, which is common for truncated proteins. We quantified the expression level of Nav1.5-WT and Nav1.5-G1642X, and found that about 40% of the truncated Nav1.5-G1642X was degraded compared with the WT (0.64 ± 0.11 arbitrary units). Therefore, this explains why the DN effect we observed here is not a 75% reduction in the current density. Nevertheless, consistent with our observations with BrS mutations, we demonstrated that the reduction in current observed here was also due to coupled gating between the two channels. In fact, inhibition of the coupled-gating by modulating 14-3-3 restored the reduced INa to its WT level (Figure 6).
Our group previously found that the α-subunits of Nav1.5 assemble and gate as dimers, and the protein 14-3-3 can regulate the coupled-gating of the Nav1.5 (Clatot et al., 2012, 2017, 2018). In our previous study, we also showed that the dimerization site for Nav1.5 is located on the DI-DII linker, which is known to be the ‘hotspot’ for Nav1.5 phosphorylation (Marionneau and Abriel, 2015). Since the 14-3-3 activity on its target proteins depends on a phosphoserine in a given putative binding motif (Dougherty and Morrison, 2004; Aitken, 2006; Allouis et al., 2006; Lau et al., 2007), mutating S460A on Nav1.5 can then modulate the activity between Nav1.5 and 14-3-3. Thus 14-3-3 inhibition through difopein and the S460A mutation could both remove Nav1.5 coupling. Our data suggest that although inhibiting 14-3-3 with difopein does not produce an obvious effect on the whole-cell current density, it shifts the voltage-dependent conductance curves toward negative voltages. We suspect that the shift is due to the regulation of 14-3-3 on Nav1.5 other than the coupling since we do not observe similar shifts through uncoupling with mutation S460A (Figure 6) or H558R (Figure 7). Additionally, we noticed that co-expression of the splice variant Nav1.5-G1642X/S460A did not produce a DN-effect on the WT (Figure 6). Importantly, we confirmed that Nav1.5-G1642X/S460A is also a non-functional channel (data not shown), which means that the S460A mutation does not lead to a gain-of-function effect which would explain the recovery of the functionality of Nav1.5-G1642X. It therefore appears that phosphorylation of S460 plays a critical role in the coupled-gating of Nav1.5 and could become an important target to inhibit DN-effects caused by BrS mutations or defective splice variants found in HF. The Nav1.5 serine 460 has been identified as a phosphorylated site by mass spectrometric experiments (Marionneau et al., 2012; Iqbal and Lemmens-Gruber, 2019). Therefore we speculate that phosphorylation of S460 is important for biophysical coupling and/or dimerization. Further investigations to study Serine 460, its phosphorylation and its role in the Nav1.5 coupling are currently underway in our lab. Our preliminary data do suggest an important role of the PKA phosphorylation in the Nav1.5 coupling since the PKA inhibitor KT5720 removed the DN effect caused by DN mutations (data not shown).
The single-nucleotide polymorphism H558R is the most common Nav1.5 polymorphism present in the population and has been demonstrated by multiple groups to modify the biophysical properties as well as the trafficking of pathogenic SCN5A mutations (Ye et al., 2003; Shinlapawittayatorn et al., 2011a; Makielski et al., 2003; Tan et al., 2005; Poelzing et al., 2006). One of our previous study even showed that using a fragment of only 20 amino acids of Nav1.5 containing H558R was able to rescue the trafficking of certain Nav1.5 mutants, indicating its definitive capacity in regulating Nav1.5 functioning (Shinlapawittayatorn et al., 2011b). Here, the H558R polymorphism was capable of rescuing the Nav1.5 loss-of-function resulting from the splice variant DN-effect. Considering that we are seeing similar restoration of currents in presence of H558R as we have seen with 14-3-3 inhibition, and considering the location of the H558R polymorphism is near the dimerization and 14-3-3 binding site, we speculate that a similar mechanism is involved. However, it is beyond the scope of this current study to elucidate this mechanism and future investigations will be focused on this rescuing effect.
Splice variants resulting in truncated channels could easily result in a non-produced or degraded protein which in itself would result in a reduction in current. Indeed, it has also been suggested that these HF-associated SCN5A splice variants could reduce the mRNA level of the WT SCN5A (Shang et al., 2007). However, the surface expression of the Nav1.5-G1642X (Figure 4) that we observed in the current study indicates that the truncated channel can be synthesized by the HEK 293 cells which is also consistent with the results in iPSC-CMs demonstrated by the Dudley group (Shang et al., 2008; Gao et al., 2013). This therefore supports that the mechanism we unveiled here showing a DN-effect between the splice variant and the WT due to biophysical coupling contributes, at least in part, to the INa reduction observed in HF patients. It is important to note that the ability to reproduce this DN-effect in human iPSC-CMs further support this mechanism in HF patients’ myocytes and explain the reduction in sodium current. Interestingly, we noticed that the reduction in INa in these cells (Figure 3) is even more significant than that of HEK 293 cells (Figure 1). One can speculate that in human iPSC-CMs, other splice variants such as the neonatal splice variant Nav1.5e may be more predominant due to the immature nature of the iPSC-CMs and might be more sensitive to this interaction. It is also possible that contrarily to HEK 293 cells where we control the level of expression of both the WT and the splice variant through the co-transfection of the same quantity of DNA, the ratio of the splice variant to the endogenous channel could be greater which would explain the larger reduction in current.
The Dudley group first demonstrated the upregulation of the splice variant E28D in heart failure which resulted in a decrease in sodium current (Shang et al., 2007). Here we demonstrated that this splice variant produces a DN-effect on the WT sodium channel through interaction with the channel and coupled-gating, further explaining how the splice variant can lead to a reduction in INa in HF patients. Future studies focusing on developing strategies to uncouple Nav1.5 without disturbing INa might be beneficial to all patients suffering from phenotypes related to SCN5A mutants or splice variants.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
YZ performed the experiments, collected the data, performed the analysis, and wrote the manuscript. XW, DY, and HL performed the experiments, collected the data, performed the analysis, and reviewed the manuscript. AR-N collected the data and reviewed the manuscript. J-DF conceived the study and reviewed the manuscript. ID conceived the study, and wrote and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by R01HL094450 and R01HL132520 to ID, R01HL139006 to ID and JD-F, and Heart Rhythm Society Fellowship to DY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abriel, H. (2010). Cardiac sodium channel Nav1.5 and interacting proteins: physiology and pathophysiology. J. Mol. Cell. Cardiol. 48, 2–11. doi: 10.1016/j.yjmcc.2009.08.025
Aitken, A. (2006). 14-3-3 proteins: a historic overview. Semin. Cancer Biol. 16, 162–172. doi: 10.1016/j.semcancer.2006.03.005
Aleong, R. G., Shah, A., Michalec, M., Bedi, M., McNamara, D., London, B., et al. (2005). The cardiac sodium channel H558R variant improves survival in heart failure. Heart Rhythm 2, S104–S105.
Allouis, M., Le Bouffant, F., Wilders, R., Péroz, D., Schott, J. J., Noireaud, J., et al. (2006). 14-3-3 is a regulator of the cardiac voltage-gated sodium channel Nav1.5. Circ. Res. 98, 1538–1546.
Amin, A. S., Asghari-Roodsari, A., and Tan, H. L. (2010). Cardiac sodium channelopathies. Pflugers Arch. Eur. J. Physiol. 460, 223–237.
Banerjee, D., Grammatopoulos, T. N., Palmisciano, A., Klinger, J. R., Krishnan, I., Whittenhall, M., et al. (2020). Alternative splicing of the cardiac sodium channel in pulmonary arterial hypertension. Chest 158, 735–738. doi: 10.1016/j.chest.2019.12.052
Clatot, J., Hoshi, M., Wan, X., Liu, H., Jain, A., Shinlapawittayatorn, K., et al. (2017). Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 8:2077.
Clatot, J., Zheng, Y., Girardeau, A., Liu, H., Laurita, K. R., Marionneau, C., et al. (2018). Mutant voltage-gated Na+ channels can exert a dominant negative effect through coupled gating. Am. J. Physiol. Heart Circ. Physiol. 315, H1250–H1257.
Clatot, J., Ziyadeh-Isleem, A., Maugenre, S., Denjoy, I., Liu, H., Dilanian, G., et al. (2012). Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Nav1.5 α-subunits. Cardiovasc. Res. 96, 53–63. doi: 10.1093/cvr/cvs211
Cormier, J. W., Rivolta, I., Tateyama, M., Yang, A. S., and Kass, R. S. (2002). Secondary structure of the human cardiac Na+ channel C terminus. Evidence for a role of helical structures in modulation of channel inactivation. J. Biol. Chem. 277, 9233–9241. doi: 10.1074/jbc.m110204200
Dougherty, M. K., and Morrison, D. K. (2004). Unlocking the code of 14-3-3. J. Cell Sci. 117, 1875–1884. doi: 10.1242/jcs.01171
Gao, G., Xie, A., Zhang, J., Herman, A. M., Jeong, E. M., Gu, L., et al. (2013). Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ. Arrhythm. Electrophysiol. 6, 1018–1024. doi: 10.1161/circep.113.000274
Hoshi, M., Du, X. X., Shinlapawittayatorn, K., Liu, H., Chai, S., Wan, X., et al. (2014). Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ. Cardiovasc. Genet. 7, 123–131. doi: 10.1161/circgenetics.113.000292
Hund, T. J., Koval, O. M., Li, J., Wright, P. J., Qian, L., Snyder, J. S., et al. (2010). A β IV-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519. doi: 10.1172/jci43621
Iqbal, S. M., and Lemmens-Gruber, R. (2019). Phosphorylation of cardiac voltage-gated sodium channel: potential players with multiple dimensions multiple kinases phosphorylate and regulate Na V 1.5 channel physi-ology and pathology. Cyclic AMP-dependent protein kinase (PKA), protein kinase C (PKC) and calcium. Acta Physiol. 225:13210.
Lang, D., Zhu, C., Zhou, H., Zhang, Y., Cai, Z., Wu, H., et al. (2018). Key role of the membrane trafficking of Nav1.5 channel protein in antidepressant-induced Brugada syndrome. Front. Physiol. 9:1230. doi: 10.3389/fphys.2018.01230
Lau, J. M. C., Jin, X., Ren, J., Avery, J., DeBosch, B. J., Treskov, I., et al. (2007). The 14-3-3 phosphoserine-binding protein is required for cardiomyocyte survival. Mol. Cell. Biol. 27, 1455–1466. doi: 10.1128/mcb.01369-06
Makielski, J. C., Ye, B., Valdivia, C. R., Pagel, M. D., Pu, J., Tester, D. J., et al. (2003). A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ. Res. 93, 821–828. doi: 10.1161/01.res.0000096652.14509.96
Marangoni, S., Di Resta, C., Rocchetti, M., Barile, L., Rizzetto, R., Summa, A., et al. (2011). A Brugada syndrome mutation (p.S216L) and its modulation by p.H558R polymorphism: standard and dynamic characterization. Cardiovasc. Res. 91, 606–616. doi: 10.1093/cvr/cvr142
Marionneau, C., and Abriel, H. (2015). Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J. Mol. Cell. Cardiol. 82, 36–47. doi: 10.1016/j.yjmcc.2015.02.013
Marionneau, C., Lichti, C. F., Lindenbaum, P., Charpentier, F., Nerbonne, J. M., Townsend, R. R., et al. (2012). Mass spectrometry-based identification of native cardiac Nav1.5 channel α subunit phosphorylation sites. J. Proteome Res. 11, 5994–6007. doi: 10.1021/pr300702c
Matsumura, H., Nakano, Y., Ochi, H., Onohara, Y., Sairaku, A., Tokuyama, T., et al. (2017). H558R, a common SCN5A polymorphism, modifies the clinical phenotype of Brugada syndrome by modulating DNA methylation of SCN5A promoters. J. Biomed. Sci. 24:91.
Mizusawa, Y., and Wilde, A. A. M. (2012). Brugada syndrome. Circ. Arrhythm. Electrophysiol. 5, 606–616.
Noyes, A. M., Zhou, A., Gao, G., Gu, L., Day, S., Andrew Wasserstrom, J., et al. (2017). Abnormal sodium channel mRNA splicing in hypertrophic cardiomyopathy. Int. J. Cardiol. 249, 282–286. doi: 10.1016/j.ijcard.2017.08.071
Poelzing, S., Forleo, C., Samodell, M., Dudash, L., Sorrentino, S., Anaclerio, M., et al. (2006). SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation 114, 368–376. doi: 10.1161/circulationaha.105.601294
Qu, Y., Rogers, J. C., Tanada, T. N., Catterall, W. A., and Scheuer, T. (1996). Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J. Gen. Physiol. 108, 75–379.
Remme, C. A., and Bezzina, C. R. (2010). REVIEW: sodium channel (Dys)function and cardiac arrhythmias. Cardiovasc. Ther. 28, 287–294. doi: 10.1111/j.1755-5922.2010.00210.x
Rühlmann, A. H., Körner, J., Hausmann, R., Bebrivenski, N., Neuhof, C., Detro-Dassen, S., et al. (2020). Uncoupling sodium channel dimers restores the phenotype of a pain-linked Nav1.7 channel mutation. Br. J. Pharmacol. 177, 4481–4496. doi: 10.1111/bph.15196
Schroeter, A., Walzik, S., Blechschmidt, S., Haufe, V., Benndorf, K., and Zimmer, T. (2010). Structure and function of splice variants of the cardiac voltage-gated sodium channel Nav1.5. J. Mol. Cell. Cardiol. 49, 16–24. doi: 10.1016/j.yjmcc.2010.04.004
Shang, L. L., Ge, G., and Dudley, S. C. (2008). Tail of the sodium channel. Channels (Austin) 2, 161–162. doi: 10.4161/chan.2.3.6189
Shang, L. L., Pfahnl, A. E., Sanyal, S., Jiao, Z., Allen, J., Banach, K., et al. (2007). Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ. Res. 101, 1146–1154. doi: 10.1161/circresaha.107.152918
Shinlapawittayatorn, K., Du, X. X., Liu, H., Ficker, E., Kaufman, E. S., and Deschênes, I. (2011a). A common SCN5A polymorphism modulates the biophysical defects of SCN5A mutations. Heart Rhythm 8, 455–462. doi: 10.1016/j.hrthm.2010.11.034
Shinlapawittayatorn, K., Dudash, L. A., Du, X. X., Heller, L., Poelzing, S., Ficker, E., et al. (2011b). A novel strategy using cardiac sodium channel polymorphic fragments to rescue trafficking-deficient SCN5A Mutations. Circ. Cardiovasc. Genet. 4, 500–509. doi: 10.1161/circgenetics.111.960633
Tan, B. H., Valdivia, C. R., Rok, B. A., Ye, B., Ruwaldt, K. M., Ruwaldt, K. M., et al. (2005). Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants. Heart Rhythm 2, 741–747. doi: 10.1016/j.hrthm.2005.04.021
Tomaselli, G. F., and Zipes, D. P. (2004). What causes sudden death in heart failure? Circ. Res. 95, 754–763. doi: 10.1161/01.res.0000145047.14691.db
Ufret-Vincenty, C. A., Baro, D. J., Lederer, W. J., Rockman, H. A., Quiñones, L. E., and Santana, L. F. (2001). Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J. Biol. Chem. 276, 28197–28203. doi: 10.1074/jbc.m102548200
Valdivia, C. R., Chu, W. W., Pu, J., Foell, J. D., Haworth, R. A., Wolff, M. R., et al. (2005). Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 38, 475–483. doi: 10.1016/j.yjmcc.2004.12.012
Veerman, C. C., Wilde, A. A. M., and Lodder, E. M. (2015). The cardiac sodium channel gene SCN5A and its gene product NaV1.5: role in physiology and pathophysiology. Gene 573, 177–187. doi: 10.1016/j.gene.2015.08.062
Viswanathan, P. C., Benson, D. W., and Balser, J. R. (2003). A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J. Clin. Invest. 111, 341–346. doi: 10.1172/jci200316879
Keywords: cardiac sodium channel, heart failure, arrhythima, electrophyiology, patch – clamp technique
Citation: Zheng Y, Wan X, Yang D, Ramirez-Navarro A, Liu H, Fu J-D and Deschênes I (2021) A Heart Failure-Associated SCN5A Splice Variant Leads to a Reduction in Sodium Current Through Coupled-Gating With the Wild-Type Channel. Front. Physiol. 12:661429. doi: 10.3389/fphys.2021.661429
Received: 30 January 2021; Accepted: 01 March 2021;
Published: 22 March 2021.
Edited by:
Mohamed-Yassine Amarouch, Sidi Mohamed Ben Abdellah University, MoroccoReviewed by:
Minoru Horie, Shiga University of Medical Science, JapanCopyright © 2021 Zheng, Wan, Yang, Ramirez-Navarro, Liu, Fu and Deschênes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Deschênes, SXNhYmVsbGUuZGVzY2hlbmVzQG9zdW1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.