 Ruomei Li
Ruomei Li Yidan Sun
Yidan Sun Zhengxi Chen
Zhengxi Chen Mengting Zheng
Mengting Zheng Yuhua Shan
Yuhua Shan Xiyu Ying
Xiyu Ying Mengjia Weng
Mengjia Weng Zhenqi Chen
Zhenqi Chen- 1Department of Orthodontics, Shanghai Key Laboratory of Stomatology, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 2Resident, Department of General Dentistry, Henry M. Goldman School of Dental Medicine, Boston University, Boston, MA, United States
Cleft palate, a common global congenital malformation, occurs due to disturbances in palatal growth, elevation, contact, and fusion during palatogenesis. The Fibroblast growth factor 9 (FGF9) mutation has been discovered in humans with cleft lip and palate. Fgf9 is expressed in both the epithelium and mesenchyme, with temporospatial diversity during palatogenesis. However, the specific role of Fgf9 in palatogenesis has not been extensively discussed. Herein, we used Ddx4-Cre mice to generate an Fgf9–/– mouse model (with an Fgf9 exon 2 deletion) that exhibited a craniofacial syndrome involving a cleft palate and deficient mandibular size with 100% penetrance. A smaller palatal shelf size, delayed palatal elevation, and contact failure were investigated to be the intrinsic causes for cleft palate. Hyaluronic acid accumulation in the extracellular matrix (ECM) sharply decreased, while the cell density correspondingly increased in Fgf9–/– mice. Additionally, significant decreases in cell proliferation were discovered in not only the palatal epithelium and mesenchyme but also among cells in Meckel’s cartilage and around the mandibular bone in Fgf9–/– mice. Serial sections of embryonic heads dissected at embryonic day 14.5 (E14.5) were subjected to craniofacial morphometric measurement. This highlighted the reduced oral volume owing to abnormal tongue size and descent, and insufficient mandibular size, which disturbed palatal elevation in Fgf9–/– mice. These results indicate that Fgf9 facilitates palatal growth and timely elevation by regulating cell proliferation and hyaluronic acid accumulation. Moreover, Fgf9 ensures that the palatal elevation process has adequate space by influencing tongue descent, tongue morphology, and mandibular growth.
Introduction
Cleft lip and/or palate is a common congenital birth defect affecting approximately 1 in 700 newborns (Dixon et al., 2011). The prevalence of cleft palate is 0.13–2.53‰ worldwide (Burg et al., 2016). Mice with cleft palate share similar developmental and genetic features with humans with cleft palate (Mittwoch, 2008). Secondary palate formation in both species is generally divided into three periods, comprising palatal growth and enlargement before embryonic day 13.5 (E13.5), elevation and contact before E14.5, and fusion before E15.5 (Li et al., 2019). Thus, effort has been made to explore the possible genetic factors responsible for cleft palate in mice, which represent the optimum model for identifying the underlying mechanism in humans (Juriloff and Harris, 2008; Burg et al., 2016; Li et al., 2019).
The 22 members of the fibroblast growth factor (FGF) family participate in multiple aspects of palate development. Mutations in these factors are involved in 3–5% of cases of non-syndromic cleft lip and palate in humans (Riley et al., 2007; Wu et al., 2015; Jin et al., 2018; Weng et al., 2018). The fibroblast growth factor 9 (FGF9) T > c mutation has been reported in a non-syndromic bilateral cleft lip and palate case (Riley and Murray, 2007). Moreover, cleft palate has been observed in Fgf9–/– mice with 40% penetrance, but the underlying mechanism has not been elaborated (Colvin et al., 2001).
Fgf9 is located on chromosome 14qC3, and it was initially recognized as a trophic factor for primary rat glial cells (Naruo et al., 1993). Moreover, it is also known as heparin-binding growth factor 9 and it has an affinity for fibroblast growth factor receptors 2 and 3 (FGFR2 and FGFR3) and heparin (Hecht et al., 1995; Harada et al., 2009). It was initially detected in the maternal uterine epithelium at E7.5 (Colvin et al., 1999). From E9.5 to E12.5, it is expressed in the ectoderm of the craniofacial region, with spatiotemporal variation (Colvin et al., 1999). It is then detected in the palatal epithelium at E13.5, and in both the epithelium and mesenchyme at E14.5 during palatal elevation (Iwata et al., 2012). Additionally, Fgf9 is involved in genetic cross talk with the Sox, Fgf, and Has families, all of which have unique and important functions in palatal growth and elevation (Lee and Saint–Jeannet, 2011; Loke et al., 2014; Chang et al., 2018; Wolk et al., 2020; Yonemitsu et al., 2020; Yue et al., 2020). Thus, it is rational to propose that Fgf9 plays an essential role during palatogenesis and possibly mediates palatal growth and elevation.
To verify our hypothesis and explore Fgf9’s possible functions, we generate an Fgf9 knockout murine model with 100% penetrance of cleft palate. Further, we provide evidence that Fgf9 mediates palatal growth and elevation basing on the examinations of histologic measurements, hyaluronic acid accumulation, cell proliferation, cell apoptosis analysis, etc.
Materials and Methods
Generation of Transgenic Mice
The strategy of generating an Fgf9 knockout allele is illustrated in Figure 1A. The recombinant vector of exon 2 (Fgf9–201, ENSMUST00000022545.13) of mouse Fgf9 (MGI: 104723) was constructed and transfected into SM–1 embryonic stem (ES) cells by electroporation. The positive ES cells were screened by G418 (350 Ag/mL) and ganciclovir (2 mmol/L) and then identified by Southern blotting. ES cells were microinjected into 102 blastocysts of C57BL/6J mice and then transferred into the uterus of pseudopregnant mice to obtain chimeric mice. Seven chimeric mice were identified and mated with Wildtype C57BL/6J mice. Four flox-NEO alleles (Fgf9FloxNeo) were selective from the F1 agouti offspring by PCR. Neo-free mice (Fgf9flox/+; Flp+) were generated by mating Fgf9FloxNeo with Flp mice. Finally, Fgf9flox/+ (Fgf9F/+) were generated by crossing Fgf9flox/+; Flp+ with Wildtype. Fgf9F/+ mice were sequenced to confirm correct targeted sites (forward primer CACTGGGCTCTAACTCTTC and reverse primer GACAATAATTTCCACCTCC) (Figure 1). Mice are housed in accredited animal facilities in specific pathogen-free conditions of Shanghai Ninth People’s Hospital (Shanghai, China).
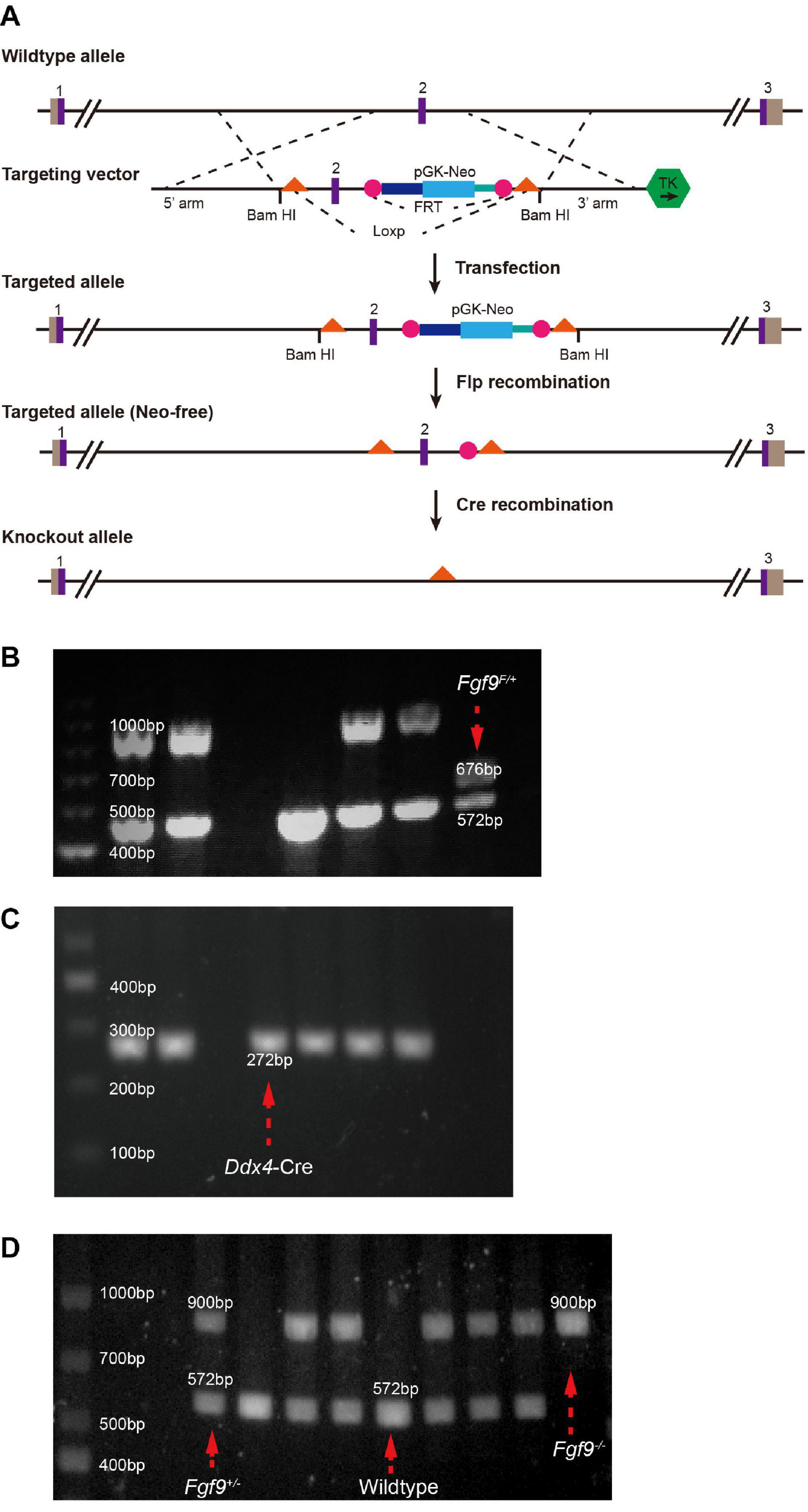
Figure 1. Generation of Fgf9 knockout allele. (A) Generations of Fgf9 allele lacking exon 2. Purple boxes, exons. Gray boxes, introns. Orange triangles, loxP sites. pGK-NEO, pGK-NEO cassette for positive selection. TK, thymidine kinase cassette included for negative selection. PCR-based genotyping of (B) Fgf9F/+ (F indicates a floxed allele), (C) Ddx4–Cre, (D) Fgf9+/–, Wildtype, and Fgf9–/– mice.
Ddx4-Cre mice purchased from Shanghai Model Organisms Center, Inc, Shanghai, China, were crossed with Fgf9F/+ to obtain Fgf9F/+; DDX4-Cre mice (Gallardo et al., 2007) (Figure 1C). Fgf9F/+; DDX4-Cre mice were crossed with Wildtype mice to generate Fgf9+/– mice. Moreover, male Fgf9+/– was crossed with female Fgf9+/– to generate Fgf9–/–, Fgf9+/–, and Wildtype embryos. Mice were genotyped by PCR (primer 1 ATTTGCTATGCACGGACAC, primer 2 CACTGGGCTCTAACTCTTC, and primer 3 GACAATAATTTCCACCTCC). Briefly, a 900-bp band and a 572-bp band represent Fgf9-null and Wildtype alleles, respectively (Figure 1D). The morning that the vaginal plug was identified was designated as embryonic day 0.5 (E0.5). All experiments were repeated on at least three littermates per genotype. The representative tissue of each genotype was chosen at random. Timed pregnant mice were euthanized using CO2, followed by thoracotomy to ensure death. The uterus with embryos was removed, and embryos were retrieved under a microscope. The tissues were further dissected and processed for analysis, as described below. The experience was undergone according to the protocols of the Animals’ Committee of Shanghai Ninth People’s Hospital (Shanghai, China).
Micro-CT Analysis and Skeletal Staining of Mouse Skulls
Skulls were isolated and fixed in 70% ethanol and scanned using SkyScan 1076 (Belgium) with a spatial resolution of 24 μm. Mice obtained at E18.5 were sacrificed, skinned, eviscerated, and fixed in 95% ethanol overnight and acetone overnight at room temperature before processing and staining with alcian blue and alizarin red, according to the protocols described by Ovchinnikov (2009).
Hematoxylin and Eosin (HE) Staining and Masson’s Trichrome (MASSON) Staining
Embryonic heads harvested from timed pregnant female mice were fixed in 4% paraformaldehyde (PFA) at 4°C. Samples were cut to a thickness of 5 μm. Moreover, sections through palatal shelves were selected from the anterior, middle, and posterior regions, with the middle region consisting of sections through the maxillary first molar tooth buds. HE staining was performed using standard procedure. For MASSON staining, sections were stained with Masson’s Trichrome Stain Kit (Solarbio, China). Briefly, the sections were treated sequentially with Bouin’s solution for 15 min, Weigert’s working hematoxylin for 10 min, Biebrich scarlet-acid fuchsin for 5 min, phosphotungstic/phosphomolybdic acid for 10 min, and aniline blue for 5 min.
In situ Hybridization
Embryos were collected at desired developmental stages and fixed in 4% paraformaldehyde overnight at 4°C. For section in situ hybridization, embryos were dehydrated through graded alcohols, embedded in paraffin, and sectioned at 7 μm. The sections through palatal shelves were selected from the anterior, middle, and posterior regions, with the middle region consisting of sections through the maxillary first molar tooth buds. The Fgf9 probe sequence was a generous gift from Dr. David M Ornitz (Colvin et al., 1999). Enhanced Sensitive ISH Detection Kit II (MK1032, Boster, China) was applied for staining. Sections were treated with proteinase K (in prewarmed 50 mM Tris for 10–20 min at 37°C) for antigen retrieval. Following the pre-hybridization step, the sections were incubated in a hybridization solution (2 μg/ml) at 37°C overnight. The slides were treated with a blocking buffer at 37°C for 30 min. Furthermore, biotinylated digoxin was applied at 37°C for 2 h.
Cell Proliferation Assay, Cell Apoptosis Assay, and Hyaluronic Acid-Binding Protein (HABP) Staining
Proliferation assays were performed by intraperitoneal injection of bromodeoxyuridine (BrdU) labeling reagent (100 μg/g body weight) into pregnant mice, which were sacrificed after 2 h, and embryos recovered and processed. Tissue sections from the comparable anterior, middle, and posterior palatal regions were stained with an antibody to BrdU (1:100, Abcam, United Kingdom). Sections were counterstained with DAPI solution for 7 min to visualize nuclei and were then rinsed in PBS. Two non-serial sections were counted for each region from three Wildtype, three Fgf9+/–, and three Fgf9–/– littermates at E13.5 and E14.5. Moreover, the mean values were recorded using ImageJ (MD, United States). Palatal shelves were divided into mesenchyme and epithelium for counting. BrdU-labeled cells were counted and calculated as the percentage of labeled cells among total nuclear-stained cells. P < 0.05 was considered as statistically significant (Student’s t-test). The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed using the Cell Apoptosis Detection Kit IV (CY3) (MK1016, Hubei, China) following the manufacturer’s instructions. Briefly, tissues were fixed in 4% paraformaldehyde and then dehydrated through an increasing graded ethanol series and processed for sectioning. Following rehydration steps, the sections were treated with Proteinase K (in 10 mM Tris–HCl, pH 8.0 for 10–15 min 37°C). The samples were incubated with TdT and DIG-d-UTP at 37°C for 2 h and a blocking buffer for 30 min at room temperature. Biotinylated anti-digoxin antibody (1:100) was applied for 30 min at 37°C. Sections were counterstained with DAPI solution for 7 min to visualize nuclei and were then rinsed in PBS. To evaluate the expression pattern of hyaluronic acid in the palate shelves at E13.5 and E14.5, the sections were stained with biotin-labeled HABP (1:200, 385911-50UG, Millipore, United States) and detected using Texas Red X-conjugated streptavidin (1:200, S6370, Thermo, United States). Images were taken using an Olympus fluorescence microscope.
Craniofacial Morphometric Measurements
The selected serial sections of embryonic heads from three different litters at E14.5 were applied for craniofacial morphometric measurements. The oral volume, tongue size, tongue height, tongue width, Intra-Meckel’s width, and oral height are evaluated.
Statistical Analysis
All quantitative data were presented as the mean ± SD as indicated by at least three independent experiments. IBM SPSS Statistics 25.0 (United States) was applied for analysis. P < 0.05 was considered as statistically significant (Student’s t-test).
Results
Fgf9–/– Embryos Exhibited Cleft Secondary Palate and Craniofacial Deformities
By dissecting pregnant Fgf9+/– mice that had mated with male Fgf9+/– mice, we harvested Fgf9–/– embryos which died shortly after birth with a significant cleft secondary palate (black star, 100% phenotype penetrance, n = 12, Figures 2A–A”). The inferior view of the Fgf9–/– palate showed a narrower palatal width, with palatal rugae (Figures 2A–A”). The top view of the Fgf9–/– mice showed a smaller head width (purple double-headed arrow) compared to their Wildtype and Fgf9+/– littermates (black double-headed arrow, Figures 2B–B”).
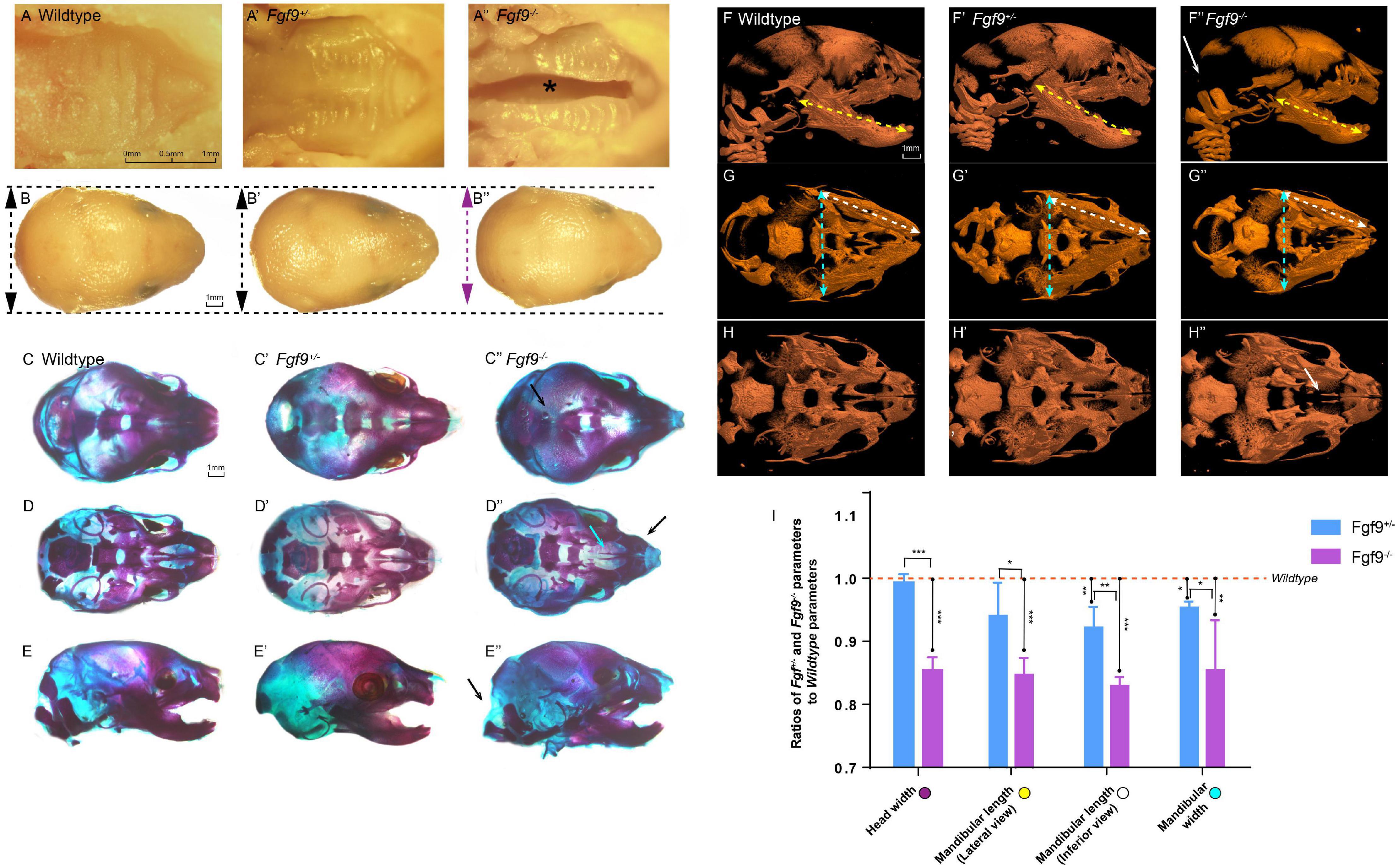
Figure 2. Craniofacial phenotypes of Wildtype, Fgf9+/–, and Fgf9–/– mice. Inferior views of (A) Wildtype, (A’) Fgf9+/–, and (A”) Fgf9–/– E18.5 embryos. “*” in (A”) indicates cleft secondary palate. Top views of (B) Wildtype, (B’) Fgf9+/–, and (B”) Fgf9–/– heads. Purple double-headed arrow in (B”) indicates narrow head width. Black double-headed arrows in (B,B’) indicate normal head widths. Top views of the skeletal staining of (C) Wildtype, (C’) Fgf9+/–, and (C”) Fgf9–/– heads. Black arrow in (C”) indicates the premature partial ossification. Inferior views of the skeletal staining of (D) Wildtype, (D’) Fgf9+/–, and (D”) Fgf9–/– heads. Blue arrow in (D”) indicates cleft secondary palate. Black arrow in (D”) indicates the abnormal circular tip of the premaxilla. Lateral views of the skeletal staining of (E) Wildtype, (E’) Fgf9+/–, and (E”) Fgf9–/– heads. Black arrow in (E”) indicates occipital deficiency. Lateral views of (F) Wildtype, (F’) Fgf9+/–, and (F”) Fgf9–/– embryos showing mandibular lengths (yellow double-headed arrows) on micro-CT scans. White arrows in (F”) indicate the occipital deficiency. Inferior views of (G) Wildtype, (G’) Fgf9+/–, and (G”) Fgf9–/– embryos showing mandibular widths (blue double-headed arrows) and mandibular lengths (white double-headed arrows) on micro-CT scans. Inferior views of (H) Wildtype, (H’) Fgf9+/–, and (H”) Fgf9–/– embryos. White arrow in (H”) indicates the cleft secondary palate. (I) Ratios of Fgf9+/– and Fgf9–/– parameters to Wildtype parameters. *P < 0.1, **P < 0.05, ***P < 0.001.
The skeletal staining and micro-computed tomography (CT) further showed the separation of the palatine and maxillary shelves, enabling a direct view of the vomer and presphenoid (Figures 2D”,H”). Premature parietal ossification can be seen in Figure 2C”. The abnormal circular tip of the premaxilla (black arrow) can be viewed distinctly based on the skeletal staining (Figure 2D”). Moreover, occipital deficiency was observed (Figures 2E”,F”). Several parameters, involving mandible length from lateral views (yellow double-headed arrows), mandibular length from inferior views (white double-headed arrows), and mandibular width from inferior views (green double-headed arrows), were analyzed based on micro-CT reconstruction (Figures 2F–H”). The ratios of the Fgf9+/– or Fgf9–/– parameters to the Wildtype parameters are displayed in Figure 2I. The head width (85.3% ± 1.2%), mandibular length (lateral view, 83.9% ± 2.7%; inferior view, 83.6% ± 1.8%), and mandibular width (85.4% ± 7.1%) were about 15% lower in the Fgf9–/– mice compared to the Wildtype mice. These results revealed a successfully established Fgf9 knockout mouse model exhibiting cleft secondary palate, smaller mandible size, occipital deficiency, premature parietal ossification, and malformed premaxilla. The 100% penetrance of cleft secondary palate provided an excellent foundation for exploring the function of Fgf9 during palatogenesis.
Fgf9–/– Embryos Displayed Defective Palatal Growth and Delayed Palatal Elevation
The histological analysis showed aberrant morphology, delayed palatal elevation, and fusion failure in the Fgf9–/– embryos (Figures 3–6).
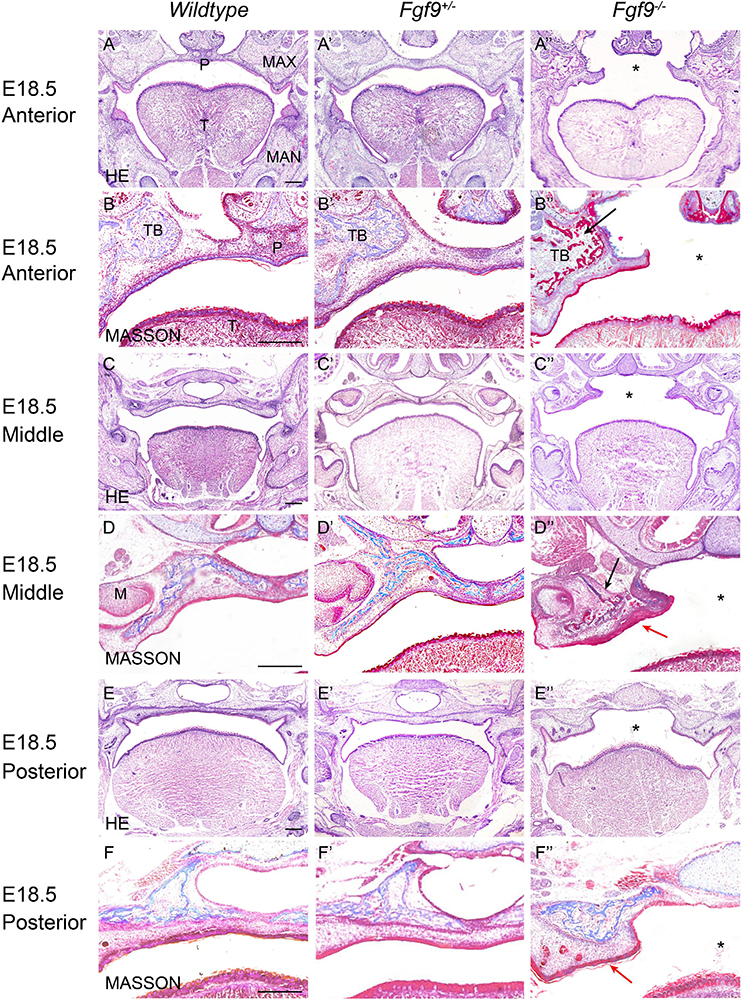
Figure 3. Aberrant morphology and fusion failure in Fgf9–/– embryos at E18.5. The HE staining of (A–A”) anterior, (C–C”) middle, and (E–E”) posterior palate in Wildtype, Fgf9+/– and Fgf9–/– embryos The magnified view showing MASSON staining of (B–B”) anterior, (D–D”) middle, and (F–F”) posterior palate in Wildtype, Fgf9+/–, and Fgf9–/– embryos. Black arrows indicate the premature trabecular bone development. Red arrows indicate the thickened cuticular layer. *, cleft secondary palate. P, palate. MAX, maxilla. MAN, mandible. T, tongue. TB, trabecular bone. M, molar. Scale bars = 200 μm.
At E18.5, Fgf9–/– embryos exhibited elevated and unfused palatal shelves with aberrant morphology (Figures 3A”,C”,E”). In the Wildtype embryos, trabecular bone was thick, from the alveolar bone to the middle of the palate. However, the Fgf9–/– trabecular bone was separated and thin, with premature palatal mineralization, especially in the anterior region (Figures 3B”,D”). Additionally, the cuticular layer of the oral epithelium of the unfused palate was thickened (Figures 3D”,F”).
To identify the causes of the abnormalities, the histological morphologies at crucial time points were evaluated (Figures 4–6). After the emergence of the palatal shelves from the maxillary prominence at E11.5, they grow and enlarge vertically on either side of the tongue (Li et al., 2017). At E12.5 and E13.5, while the vertical outgrowth is ongoing, Fgf9 mRNA primarily dominated the anterior and posterior epithelia, with a buccal–lingual gradient throughout the palate (Figures 4A–B’). No significant abnormalities were observed except for a relatively smaller size in the Fgf9–/– embryos (Figures 4C–G”).
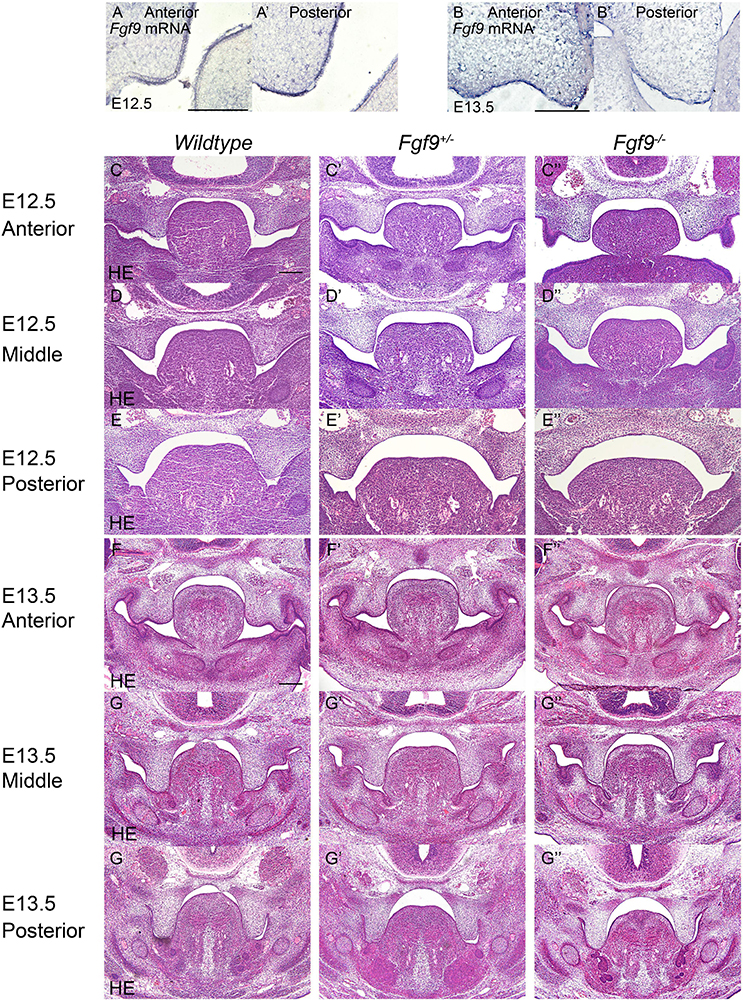
Figure 4. Fgf9 expression and phenotypes of Wildtype, Fgf9+/–, and Fgf9–/– embryos at E12.5 and E13.5. Location of Fgf9 mRNA at E12.5 in (A) anterior and (A’) posterior palatal shelves, and at E13.5 in (B) anterior and (B’) posterior palatal shelves. Fgf9–/– embryos exhibited no significant abnormalities at (C–E”) E12.5 and (D–G”) E13.5. Scale bars = 200 μm.
In Wildtype mice, the palate shelves had elevated to the horizontal position at E14.5, and Fgf9 mRNA occupied the medial edge epithelium and mesenchyme (where bone formations were being prepared), with dotted expression in the oral epithelium of the palate (Figures 5A,B). In some of the Fgf9+/– embryos, palatal elevation and contact were delayed, with an asynchronous elevation or without palatal shelf contact (Figures 5F–H). However, intriguingly, at the same time of E14.5, most of the Fgf9+/– embryos showed normal elevation without palatal shelf contact (Figure 5G). In other Fgf9+/– embryos, some shelves were in the process of elevation (Figure 5F), and in the remainder of the Fgf9+/– embryos, successful elevation and contact were observed (Figure 5H). In Wildtype palatogenesis, the asynchronous elevation and unfused palatal shelves are considered to be a normal and sequential phenomenon that occurs at around E14.5 (Yu and Ornitz, 2011). In contrast, a great number of the Fgf9+/– embryos exhibited delayed palatal elevation and shelf contact compared to their Wildtype littermates. Regarding the Fgf9–/– embryos, delayed elevation and aberrant short shelf were observed, probably resulting from impaired palatal shelf remodeling. Moreover, the disharmonious tongue–shelf relationship may act as an obstacle to shelf elevation and/or remodeling to reach the horizontal position (Figures 5C”,D”,E”).
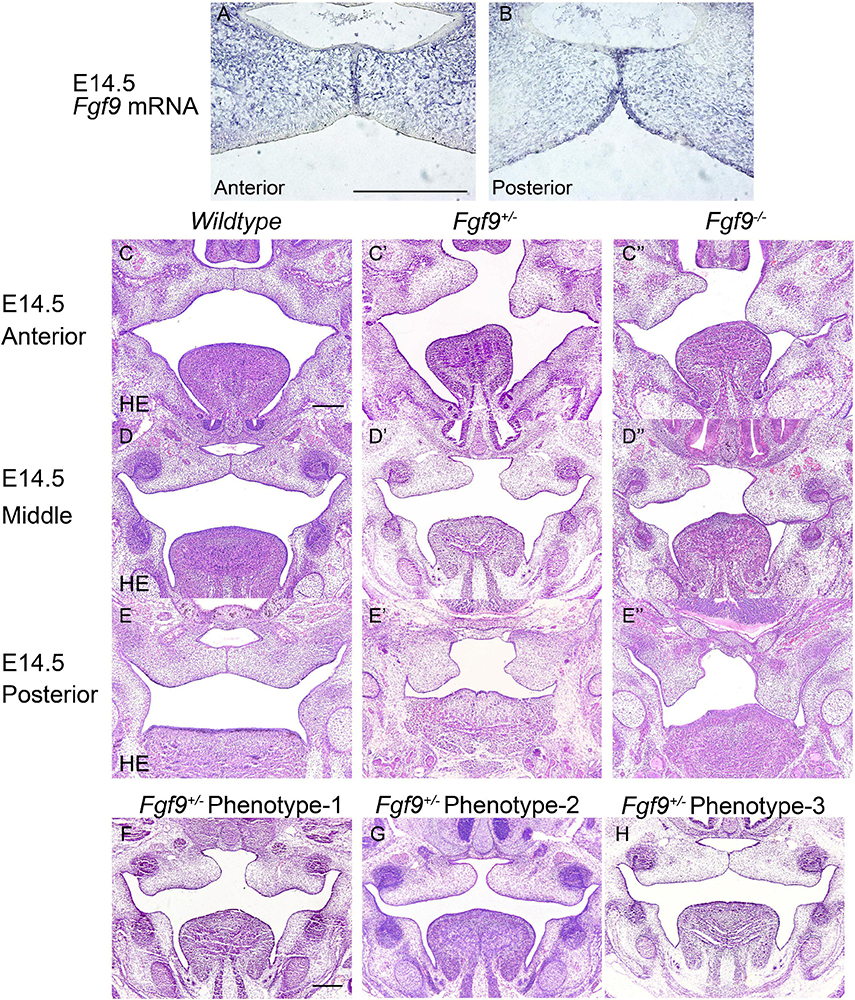
Figure 5. Fgf9 expression and phenotypes of Wildtype, Fgf9+/–, and Fgf9–/– embryos at E14.5. Location of (A) Fgf9 mRNA and (B) FGF9 protein in anterior and posterior regions at E14.5. HE staining of of (C–C”) anterior, (D–D”) middle, and (E–E”) posterior palate in Wildtype, Fgf9+/–, and Fgf9–/– embryos at E14.5. (F–H) HE staining of various types of delayed palatal elevation in Fgf9–/– embryos. Scale bars = 200 μm.
Based on the above evidence, we deduced that palatal shelves lacking Fgf9 were smaller during the vertical growth, and then palatal elevation, with aberrant shape, was delayed due to poor remodeling and the disharmonious tongue–shelf relationship.
Fgf9–/– Palatal Shelves Exhibited Aberrant Cell Proliferation and Increased Cell Density
To determine the immediate cause of the small palatal size in the Fgf9–/– embryos, we comprehensively assessed the palatal shelf width (L1), length (L2), 1/2 width (L3), and the mesenchymal cell density (Figures 6A–D’). The changes in these parameters in Wildtype and Fgf9–/– mice from E12.5 to E13.5 are shown in Figure 6E. The palatal shelves were smaller (Figures 6A–B’), while the mesenchymal cell density was higher in the Fgf9–/– embryos (Figures 6C–D’). From E12.5 to E13.5, when the shelves are actively growing and enlarging, the mesenchymal cell density decreased in both groups. Nevertheless, the cell density and the palatal shelf size of Fgf9–/– mice compared to Wildtype mice exhibited attenuated reduction and less growth, respectively (Figure 6E).
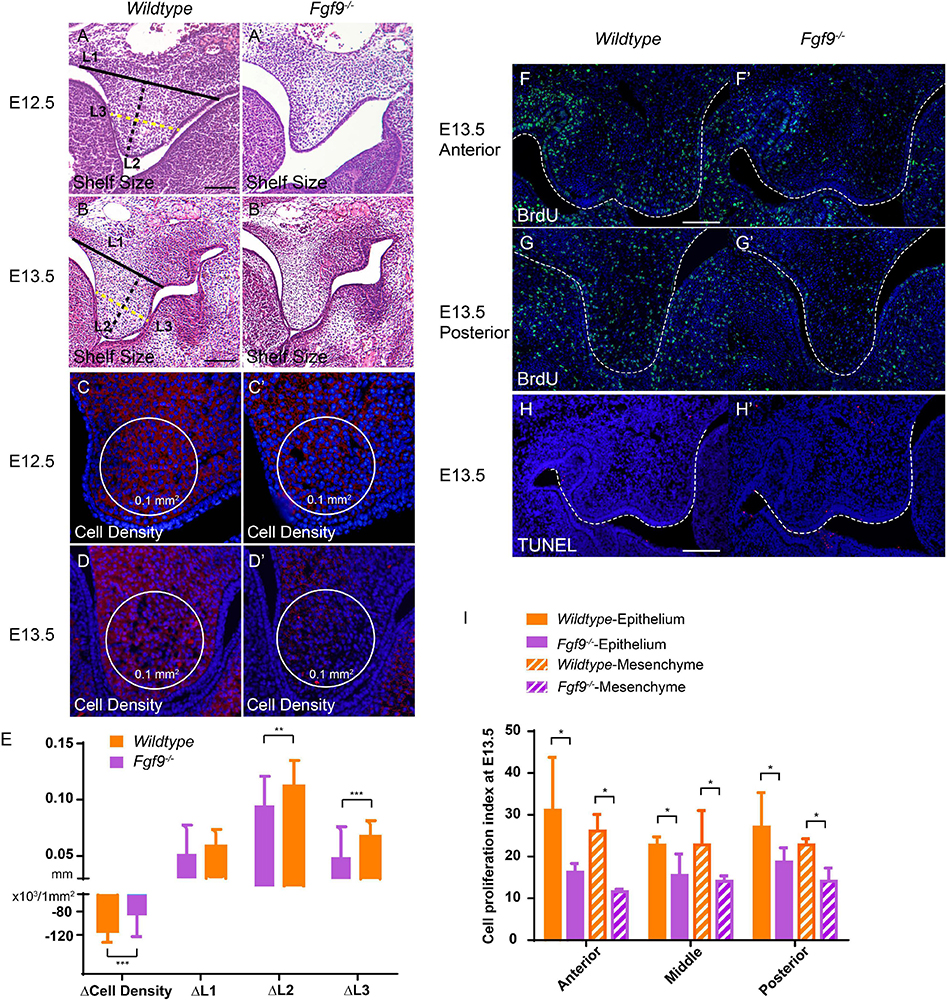
Figure 6. Fgf9–/– palatal shelves exhibit attenuated size increase and cell density decrease from E12.5 to E13.5. Selected serial sections of the anterior (n = 7), middle (n = 8), and posterior (n = 5) of the embryonic heads from three litters were measured. HE staining shows smaller palatal shelves in the Fgf9–/– embryos than the Wildtype littermates at (A,A’) E12.5 and (B,B’) E13.5. L1, L2, and L3 are shorter in the Fgf9–/– embryos. Cell density was higher in the Fgf9–/– embryos than the Wildtype littermates at (C,C’) E12.5 and (D,D’) E13.5. L1, L2, and L3 were shorter in the Fgf9–/– embryos. The cell density and palatal shelf size of Fgf9–/– embryos exhibited attenuated reduction and less growth, respectively (E). Coronal sections with BrdU immunofluorescence through (F,F’) anterior and (G,G’) posterior regions of Wildtype and Fgf9–/– embryos at E13.5. Coronal sections with TUNEL assays of (H) Wildtype and (H’) Fgf9–/– embryos at E13.5. (I) Quantification of cell proliferation index through anterior, middle, and posterior regions in the epithelium and mesenchyme of the Wildtype and Fgf9–/– embryos at E13.5. Full lines, black dotted lines, and yellow dotted lines indicate the width (L1), length (L2), and 1/2 width (L3) of palatal shelves, respectively. *P < 0.1, **P < 0.05, ***P < 0.001. Scale bars = 100 μm.
To detect the reasons for the above discoveries, we performed bromodeoxyuridine (BrdU) labeling and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays to examine cell proliferation and apoptosis. At E13.5, during the most active cell proliferation phase, many BrdU-labeled cells were located anteroposteriorly in the Wildtype palatal shelves. However, Fgf9–/– palate exhibited decreased BrdU-labeled cells in both the epithelium and mesenchyme (Figures 6F–G’,I). These reductions indicate sharp decreases in cell proliferation, giving rise to a smaller shelf size. The TUNEL assays showed a comparable apoptosis level in the Fgf9–/– and Wildtype mice (Figures 6H,H’). Together, these data show that the increased mesenchymal cell density in the Fgf9–/– mice owes to neither the impaired cell proliferation nor the abnormal apoptosis.
Abnormal HA Accumulation and Cell Proliferation Impaired Palatal Growth and Elevation in the Fgf9–/– Embryos
HA accumulation is believed to expand the ECM and generate osmotic pressure for palatal elevation (Goudy et al., 2010; Yonemitsu et al., 2020). To examine whether the increased cell density and abnormal palatal elevation resulted from insufficient HA content, we detected hyaluronic acid-binding protein (HABP) at E12.5 and E13.5.
At E12.5, Fgf9–/– mice comparing with Wildtype and Fgf9+/– mice exhibited reduced HA accumulation, especially in the anterior region, with a lingual-buccal gradient (Figures 7A–A”). At E13.5, HA accumulation was uniformly decreased in both Fgf9+/– and Fgf9–/– mice compared to Wildtype mice (Figures 7B–C”). Furthermore, the decrease was steep in the buccal area of the middle and posterior palatal shelves (Figures 7C–C”). The decrease in HA accumulation was more severe at E13.5 in both the Fgf9+/– and Fgf9–/– mice, matching the trend in cell density (Figure 7G). Thus, the increased cell density in the Fgf9–/– mice may be a result of impaired ECM expansion caused by decreased HA accumulation. Subsequently, the increased cell density, together with the impaired cell proliferation, is most likely to be the cause of the aberrant palatal elongation and expansion, leading to a smaller size. Moreover, the aberrant palatal shelves, along with the abnormal osmotic pressure owing to insufficient HA content, impairs palatal elevation. According to the above analysis, Fgf9 is required for HA accumulation to ensure tissue enlargement and osmotic pressure generation during palatal growth and elevation.
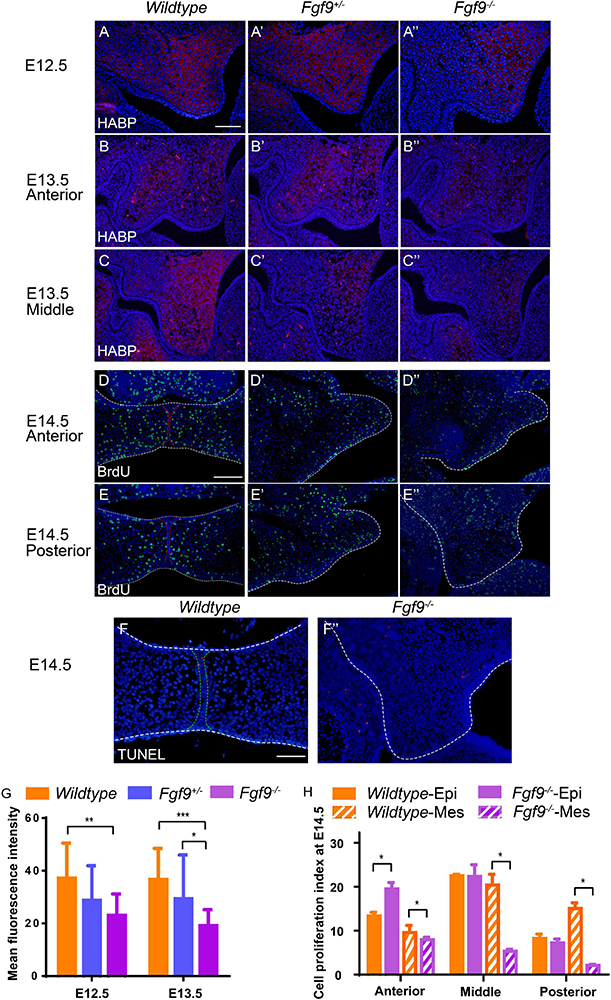
Figure 7. Fgf9–/– embryos have reduced HA accumulation and cell proliferation. Coronal sections with HABP staining of (A) Wildtype, (A’) Fgf9+/–, and (A”) Fgf9–/– embryos at E12.5. Coronal sections with HABP staining through (B–B”) anterior and (C–C”) middle regions of Wildtype, Fgf9+/–, and Fgf9–/– embryos at E13.5. Coronal sections with BrdU immunofluorescence through (D–D”) anterior and (E–E”) posterior regions of Wildtype, Fgf9+/–, and Fgf9–/– embryos at E14.5. Coronal sections with TUNEL assays of (F) Wildtype and (F”) Fgf9–/– embryos at E14.5. (G) Quantification of mean fluorescence intensity in Wildtype, Fgf9+/–, and Fgf9–/– embryos at E12.5 and E13.5. (H) Quantification of cell proliferation index through anterior, middle, and posterior regions in the epithelium and mesenchyme of the Wildtype and Fgf9–/– embryos at E14.5. HABP, hyaluronic acid binding protein. Epi, epithelium. Mes, mesenchyme. *P < 0.1, **P < 0.05, ***P < 0.001. Scale bars = 100 μm.
During and after palatal elevation, palatal shelves undergo remodeling and horizontal elongation. At E14.5, the mid-posterior regions of the Fgf9–/+ and Fgf9–/– palatal shelves were undergoing remodeling (Figures 7E’,E”). The cell proliferation sharply decreased in the Fgf9–/– palate, especially in the mesenchyme (Figures 7E–E”,H). In the anterior region in the Fgf9–/– mice, there were higher cell proliferation rates in shelves that had already elevated than in unelevated mid-posterior parts (Figures 7D”,E”). However, there was a lower cell proliferation rate compared to that in the Fgf9+/– mice, which shared a similar period of horizontal growth after elevation (Figures 7D–D”). The TUNEL assays showed a comparable apoptosis level in the Fgf9–/– and Wildtype mice at E14.5 (Figures 7F,F”).
These results indicate that in addition to the HA accumulation, decreased cell proliferation resulted in abnormal tissue remodeling and insufficient horizontal growth during and after elevation in the Fgf9–/– palatal shelves.
Inadequate Oral Volume Owing to Abnormal Tongue Shape and Descent, and Insufficient Mandibular Size in the Fgf9–/– Mice
To identify factors that may be involved in the aberrant palatal elevation in the Fgf9–/– embryos, craniofacial morphometric measurements, comprising oral volume, tongue size, tongue height, tongue width, intra-Meckel’s width (distance between the bilateral centers of Meckel’s cartilages), and oral height, were analyzed. The use of selected serial sections in the anterior (n = 7), middle (n = 8), and posterior (n = 5) parts of embryonic heads from three litters for morphometric measurements is illustrated in Figure 8A. Furthermore, the Fgf9+/– embryos that exhibited asynchronous elevation were investigated to get to the root of the abovementioned disharmonious tongue–shelf relationship.
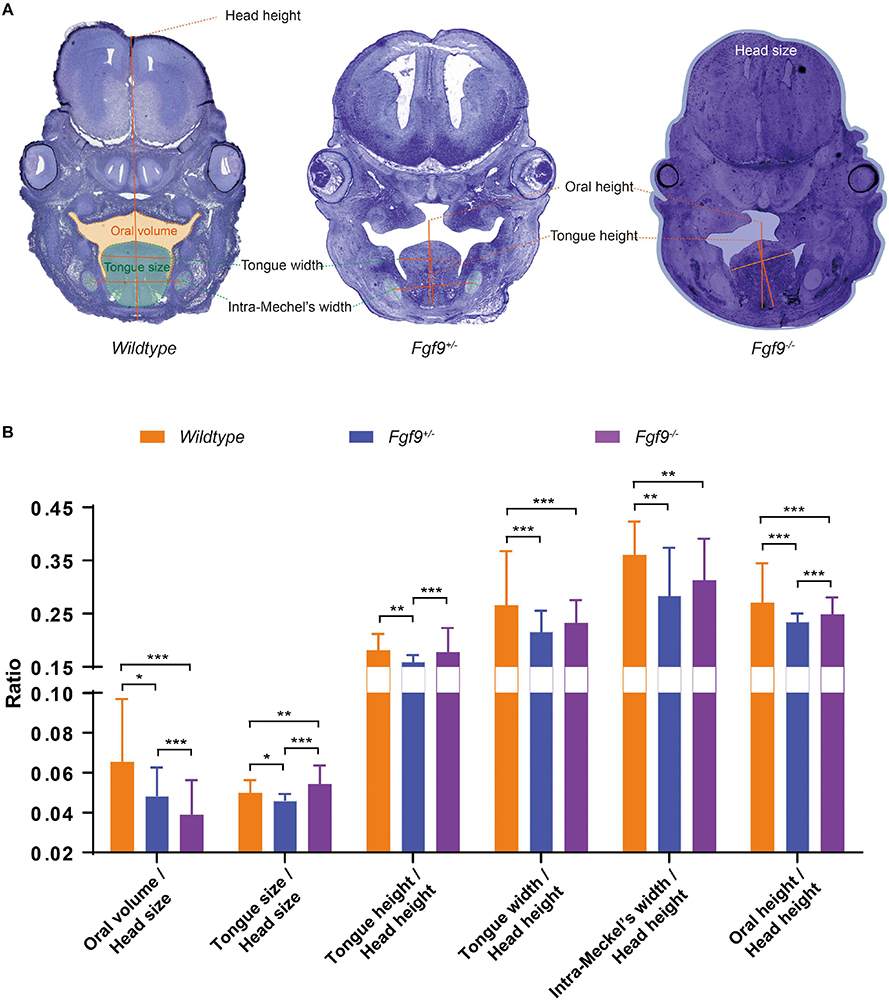
Figure 8. Craniofacial morphometric measurements among Wildtype, Fgf9+/–, and Fgf9–/– embryos during palatal elevation. (A) Oral volume, tongue size, tongue height, tongue width, intra-Meckel’s width (distance between the bilateral centers of Meckel’s cartilages), and oral height based on selected serial sections of embryonic heads from three litters. Orange, green, and blue regions indicate the oral volume, tongue size, and head size, respectively. (B) Ratios of craniofacial morphometric parameters. *P < 0.1, **P < 0.05, ***P < 0.001.
At E14.5, the palatal shelves had partially “flipped up” or undergone remodeling to reach the horizontal position in the Fgf9+/– and Fgf9–/– embryos (Figure 8A). At this time point, Fgf9–/– embryos had the smallest oral volume (oral volume/head size, Fgf9–/– = 0.039 ± 0.008 vs. Fgf9+/– = 0.052 ± 0.015 vs. Wildtype = 0.064 ± 0.023, P < 0.05) and the largest tongue size (tongue size/head size, Fgf9–/– = 0.055 ± 0.003 vs. Fgf9+/– = 0.046 ± 0.003 vs. Wildtype = 0.053 ± 0.005, P < 0.05, Figure 8B). The tongue position (tongue height/head height, Fgf9–/– = 0.174 ± 0.017 vs. Fgf9+/– = 0.160 ± 0.010, P < 0.05) and tongue width (tongue width/head height, Fgf9–/– = 0.230 ± 0.021 vs. Fgf9+/– = 0.220 + /− 0.023, P = 0.152) in the Fgf9+/– group was lower and shorter, respectively, than in the Fgf9–/– group. This indicates that timely tongue descent lay the foundation for later palatal elevation (Figure 8B).
The distance between the bilateral centers of Meckel’s cartilages (intra-Meckel’s width/head height) was used to measure the mandibular width, which was shorter in both Fgf9+/– and Fgf9–/– embryos compared to the Wildtype embryos (Fgf9–/– = 0.296 ± 0.058 vs. Fgf9+/– = 0.283 ± 0.099 vs. Wildtype = 0.33 ± 0.068, P < 0.05, Figure 8B). The oral cavity height (oral height/head height) indicates that the mandibular height was decreased in both Fgf9+/– and Fgf9–/– embryos (Fgf9–/ = 0.251 ± 0.020 vs. Fgf9+/– = 0.230 ± 0.021 vs. Wildtype = 0.292 ± 0.037, P < 0.05, Figure 8B). These observations might result from the aberrant mandibular development.
These results highlight that delayed tongue movement, insufficient mandibular size, and subsequently inadequate oral volume are impediments to palatal elevation in Fgf9–/– embryos.
Reduced Growth Potential in Meckel’s Cartilage and Intramembranous Ossification in the Fgf9–/– Mice
To investigate the possible reasons for insufficient mandibular growth in the Fgf9–/– mice, we examined Meckel’s cartilage development and intramembranous ossification.
E13.5, less than a day before palatal elevation in Wildtype mice, was explored. At this time point, Fgf9 mRNA was located in the perichondrium, periosteum, trabecular bone, and mesenchyme surrounding the trabecular bone and cartilage (Figure 9A). This indicates that Fgf9 may have a role during intramembranous ossification and Meckel’s cartilage development, which is required for appropriate mandibular growth.
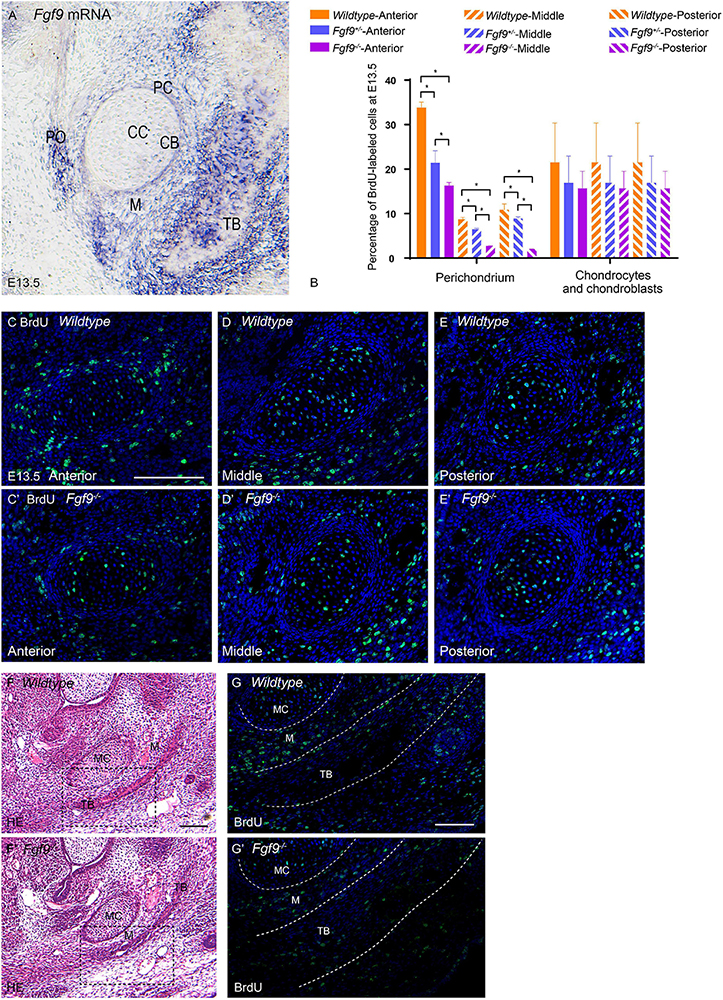
Figure 9. Growth potential of Meckel’s cartilage is impaired in Fgf9–/– embryos. (A) Fgf9 mRNA is located in the perichondrium, periosteum, trabecular bone, and mesenchyme surrounding bone and cartilage. (B) Quantification of BrdU-labeled cells in the perichondrium, chondrocytes, and chondroblasts in different sections of the palate. BrdU immunofluorescence of the anterior part of Meckel’s cartilage in (C) Wildtype and (C’) Fgf9–/– mice. BrdU immunofluorescence of the anterior part of Meckel’s cartilage in (D) Wildtype and (D’) Fgf9–/– mice. BrdU immunofluorescence of the anterior part of Meckel’s cartilage in (E) Wildtype and (E’) Fgf9–/– mice. HE staining of the intramembranous ossification region of (F) Wildtype and (F’) Fgf9–/– mice. The dotted-box regions in (F,F’) were examined with BrdU immunofluorescence, with magnified images in (G,G’), respectively. *P < 0.05. PC, perichondrium. CC, chondrocyte. CB, chondroblast. TB, trabecular bone. PO, periosteum. M, mesenchyme. MC, Meckel’s cartilage. Scale bars = 100 μm.
The BrdU labeling and TUNEL assays were used to detect cell proliferation and apoptosis, respectively, in Meckel’s cartilage and the surrounding mesenchymal and bone tissue (Figures 9B–E’). In Wildtype mice, the ratio of BrdU-labeled cells in the perichondrium was significantly higher in the anterior and posterior parts of the mandible (Figure 9B). This ratio underwent a reduction in the Fgf9–/– Meckel’s cartilage (Figure 9B).
In comparison to Wildtype mice, the Fgf9–/– trabecular bone at E13.5 was thinner (Figures 9F,F’). The mesenchyme around the Wildtype trabecular bone exhibited a high ratio of BrdU-labeled cells. However, the ratio was decreased in the same region of Fgf9–/– mice (Figures 9G,G’) and in the trabecular bone region of Fgf9–/– mice (Figures 9G,G’). There was no significant difference in the ratio of apoptotic cells among the three groups (data not shown).
The abovementioned results collectively indicate that the lack of Fgf9 expression diminished cell proliferation related to the perichondrium and mesenchyme around the trabecular bone, possibly leading to insufficient mandibular growth.
Discussion
Fgf9–/– Mouse Model Is Crucial for Revealing the Genetic Crosstalk in Palatogenesis
Multiple synostoses syndrome and craniosynostosis have been observed in FGF9 mutant mice and humans (Rodriguez–Zabala et al., 2017; Bird et al., 2020; Sentchordi–Montané et al., 2020). However, the syndrome of cleft palate, deficient mandibular size, premature partial ossification, and occipital deficiency in Fgf9 knockout mice generated in our study has not been reported in mice or humans.
Similar abnormalities of cleft palate and mandibular hypoplasia have been observed to be related to Sox family related mutations. Sox11 mutant mice exhibit cleft palate, cleft lip, and mandibular hypoplasia (Sock et al., 2004; Huang et al., 2016). Fgf9 was examined as the downstream target of Sox11. Moreover, Fgf9 mutant mice had comparatively milder abnormities compared to Sox11 mutant mice. In addition, similar phenotypes of premature craniofacial bone mineralization, cleft palate, and deficient mandibular size have been observed in Sox9 mutant mice (Bi et al., 2001; Lee and Saint–Jeannet, 2011). The palate of Sox9 + /− mice exhibited delayed palatal elevation that resembles that in Fgf9–/– mice (Mori–Akiyama et al., 2003). Fgf9 has been reported to be regulated by Sox9 in relation to sex determination (Loke et al., 2014). Thus, this tight link between Sox9 and Fgf9 may occur during palatogenesis.
Fgf9 cooperates closely with other FGF family members during palatogenesis. Fgf9-Fgfr2 regulate cell proliferation (Chang et al., 2018). Moreover, a complete loss of epithelial Fgfr2 leads to small palates and delayed elevation resembling that in Fgf9–/– mice (Rice et al., 2004). Furthermore, Fgf9 and Fgf18 have overlapping functions in skeletal development and they have been shown to interact in families with cases of cleft lip and palate (Wang et al., 2013; Hung et al., 2016). The gene–gene interaction test showed mesenchyme-specific Fgf18 knockout mice with a shortened mandible and cleft palate caused by impaired palatal elevation (Liu et al., 2002; Yue et al., 2020).
Conditional knockout of Has2, the gene encoding the hyaluronic acid synthase 2, results in small shelf size and failed palatal elevation, resembling the conditions in Fgf9–/– mice (Yonemitsu et al., 2020). Moreover, HA is a downstream target of Fgfs (Nikitovic et al., 2013; Bohrer et al., 2014; McCarthy et al., 2016; Virakul et al., 2016; Wolk et al., 2020). Many studies have shown that FGF family members share an intimate regulatory network with Has2, and Fgf9 may be a critical factor connecting the FGF family and Has2.
Therefore, the Fgf9–/– embryos obtained from pregnant Fgf9+/– mice that had mated with male Fgf9+/– mice, which had been established using Fgf9F/+ (F indicates a floxed allele) and Ddx4-Cre lines, provide an excellent model for studying these pathways and the genetic crosstalk. The Ddx4-Cre (also known as Vasa-Cre) mouse model, which is currently rarely utilized in research on palatogenesis, is promising for mediating global deletion. The Fgf9+/– mice generated from Ddx4-Cre recombination harbored “flox” deletion in their germ cells. This contributes to its efficiency and reliability regarding globally deleting a specific gene in the offspring comparing with using EIIa-Cre and Nestin-Cre (Gallardo et al., 2007). The major drawback is the lower fertility, especially in female Fgf9+/– mice, which makes obtaining adequate sample sizes more challenging. However, its efficiency and reliability make it an optimal choice for starting to explore a specific gene’s function in craniofacial development. Furthermore, global deletion lays a solid foundation for further investigation based on conditional knockouts.
The discussion below primarily focuses on how Fgf9 mediates palatogenesis based on our current findings and the findings of related studies.
Fgf9 Plays an Essential Role in the Epithelial–Mesenchymal Communication (EMC) That Underlies Palatal Growth and Elevation
As early as E12.5, Fgf9 is already found in the epithelium of palatal shelves. During vertical palatal growth, Fgf9–/– shelves failed to form regular morphology. Meanwhile, the mesenchymal cell density increased and the palatal elevation was delayed. After elevation, the bilateral malformed shelves failed to meet each other in the midline of the oral–nasal cavity. These successive abnormalities result from impaired cell proliferation and HA accumulation in the mesenchyme, which reveals that EMC is key during palatogenesis. This communication is based on the overlaying of the Fgf9 and Fgfr2 expression patterns in the epithelium of the developing palate and the resultant in modulation of palatal cell proliferation by Fgf9–Fgfr2 (Huang et al., 2016; Weng et al., 2018). Additionally, Fgfr2 combined with Fgf10, which is expressed in the palatal mesenchyme, along with Shh, plays an essential role in EMC (Colvin et al., 2001; Li et al., 2019). Studies have shown that Fgf9 participates in the Shh–Fgfr2–Fgf10 pathway during lung, cecal, and inner ear development (Colvin et al., 2001; Pirvola et al., 2004; Al Alam et al., 2012). Thus, future research should investigate the participation of Fgf9 in EMC underlying palatogenesis.
Fgf9-Induced Cell Proliferation and Hyaluronic Acid Accumulation Are of Comparable Importance in Palatal Vertical/Horizontal Growth and Elevation
For decades, there was a consensus that cell proliferation is the critical factor that induces palatal growth and that it is a requisite for palatal elevation (Li et al., 2019). However, in recent years continuous research has indicated the possible role of HA accumulation in ECM expansion; HA accumulation is no longer merely an internal force for palatal elevation but has emerged as a potential promoter of tissue expansion during palatal growth (Chiquet et al., 2016; Li et al., 2017; Lan et al., 2019; Yonemitsu et al., 2020). Combining recent related research with our results, we propose that impaired cell proliferation is not the only reason for aberrant palatal growth. If smaller palatal shelves occur as a result of reduced cell proliferation, the cell density in Fgf9–/– would be similar to that of the Wildtype, which was not the case. This conclusion is in line with the previous finding in lung development that Fgf9 regulates embryonic organ size by regulating mesenchymal expansion (Colvin et al., 2001). Moreover, the decrease in HA accumulation corresponds to the increase in cell density from E12.5 to E13.5. The evidence collectively indicates the unique and irreplaceable role of HA accumulation in palatal growth.
The aberrant palatal growth impairs subsequent palatal elevation in the Fgf9–/– embryos. At E14.5 in Fgf9–/– embryos, mesenchymal cell proliferation rates in the middle and posterior regions remained low, which impaired palatal remodeling. Further, the Fgf9–/– shelves that exhibited timely elevation had impaired cell proliferation compared to the Fgf9+/– shelves. This indicates the impairment after Fgf9–/– palatal elevation. Additionally, apoptosis was not observed in the distal portion of the palatal shelves which verified that palatal shelf remodeling does not involve apoptosis but a retraction or migration of cells instead.
Regarding HA accumulation during palatal elevation, it was decreased not only in the Fgf9–/– but also in the Fgf9+/– mice at E13.5. Intriguingly, there were varying degrees of delayed palatal in Fgf9+/– embryos, with some of the embryos showing similar palatal elevation to Fgf9–/– embryos, but without exhibiting the tongue-related obstacles to palatal elevation. Thus, it is beyond all doubt that decreased HA accumulation owing to partial ablation of Fgf9 can lead to delayed elevation. However, it is certainly not the sole reason for delayed elevation in the Fgf9–/– mice given the observation of tongue-related obstruction and, eventually, failed contact of the palatal shelves. In vitro organ culture without the tongue and mandible is required to confirm this potential conclusion.
Fgf9 Affects the Oral Volume Available for Palatal Elevation by Influencing Tongue Size and Descent
To further explore the factors involved in palatal elevation, the histomorphology of Wildtype, Fgf9+/–, and Fgf9–/– embryos was analyzed. The decrease in oral volume was a major cause of the disordered palatal elevation in Fgf9–/– model. Compared to the Wildtype mice, the tongue size and height in the Fgf9+/– mice decreased, making way for palatal shelf movement. However, a similar phenomenon did not appear in the Fgf9–/– mice; in fact, even a larger tongue size and height were observed compared to the Wildtype mice. The Fgf9–/– tongue might result from either abnormal tongue movement or intrinsic tongue malformation.
The Fgf9+/– mice underwent delayed palatal elevation. The shelves elevated in a wave-like manner from the posterior to the anterior palate, which is in line with the opinion of Professor Bush and Professor Jiang regarding Wildtype mice (Bush and Jiang, 2012). However, Fgf9–/– shelves asynchronously elevate from the anterior and middle of the palate with the posterior part unelevated, unlike in Fgf9+/– mice. The divergence might arise from abnormal tongue descent or tissue remodeling in the posterior palatal shelves.
Fgf9 Promotes Mandibular Enlargement by Enhancing Cell Proliferation in Meckel’s Cartilage and Intramembranous Ossification Site in Order to Provide Adequate Oral Volume
Besides the tongue descent that allows more space for palatal elevation, the mandible goes through a twisting motion to create space for unelevated shelves to find their way to the horizontal position (Yu and Ornitz, 2011). In Fgf9–/– mice, the smaller mandibular height decreases the oral volume and, along with the twisting motion, decreases the space for the anterior shelves to “flip-up” and for the mid-posterior shelves to undergo remodeling. Fgf9 is non-uniformly distributed and performs diverse functions in various developmental regions and at the various stages of endochondral and intramembranous ossification (Colvin et al., 1999; Govindarajan and Overbeek, 2006; Su et al., 2014). Fgf9 is located in the perichondrium, periosteum, trabecular bone, and related mesenchyme of the mandible, and it exhibits an identical expression pattern to that in developing limb cartilage (Hung et al., 2007). Cell proliferation is impaired in the perichondrium of Fgf9–/– Meckel’s cartilage. This might lead to aberrant cartilage growth potential (Yokohama–Tamaki et al., 2011), resulting in a smaller mandibular size. It is known that Meckel’s cartilage and condylar cartilage make different but equal contributions to mandibular development (Ramaesh and Bard, 2003; Yokohama–Tamaki et al., 2011). Future research is encouraged to focus on the mechanism of aberrant mandibular development in Fgf9–/– mice and whether condylar cartilage growth plays a role in palatal elevation.
In addition to the Meckel’s cartilage and condylar cartilage development, intramembranous ossification also contributes to the growth of the mandible (Parada and Chai, 2015). Fgf9 enhanced the mesenchymal cell proliferation rate at the initial site of intramembranous ossification. Moreover, Fgf9 even participated in the strengthening of the mandibular bone by enhancing the proliferation of the embedded cells. The separated and thin trabecular bone observed in the histological analysis of Fgf9–/– embryos provide further support for this. Nevertheless, further extensive research should be conducted on the functions of Fgf9 during mandibular formation.
Conclusion
In this study, we generated Fgf9 knockout mice with a cleft secondary palate and a small mandible. We dissected the mouse embryos and investigated the possible functions of Fgf9 during palatal development. We concluded that Fgf9 regulates cell proliferation, which involves the epithelial–mesenchymal communication. This facilitates vertical and horizontal palatal growth before elevation and contact, respectively. Additionally, by ensuring hyaluronic acid accumulation in the extracellular matrix space, Fgf9 is involved in palatal shelf expansion and the timely “flipped up” of the anterior shelves and the remodeling of the mid-posterior shelves. Moreover, Fgf9 benefits mandibular growth, which provides the elevating shelves with adequate space, by promoting Meckel’s cartilage growth and intramembranous ossification. Finally, Fgf9 regulates tongue size and descent, which allows more space for palatal elevation.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by the Animals Committee of Shanghai Ninth People’s Hospital.
Author Contributions
RL made substantial contribution to generation of the mouse model, data collection, analysis and interpretation, and drafting the manuscript. YS generated the mouse model. ZC made substantial contribution to the preparation of the experimental images. MZ made substantial contribution to the literature review. YS and XY made substantial contribution to the data collection and analysis. ZC and MW made substantial contribution to the concept and design of the study, data analysis, and critical revision for the important intellectual content. All authors read and approved the final manuscript.
Funding
This study was supported by grants from Clinical Research Program of 9th People’s Hospital, Shanghai Jiao Tong University School of Medicine No. JYLJ202004.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Al Alam, D., Sala, F. G., Baptista, S., Galzote, R., Danopoulos, S., Tiozzo, C., et al. (2012). FGF9–Pitx2–FGF10 signaling controls cecal formation in mice. Dev. Biol. 369, 340–348. doi: 10.1016/j.ydbio.2012.07.008
Bi, W., Huang, W., Whitworth, D. J., Deng, J. M., Zhang, Z., Behringer, R. R., et al. (2001). Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. U.S.A. 98, 6698–6703.
Bird, A. D., Croft, B. M., Harada, M., Tang, L., Zhao, L., Ming, Z., et al. (2020). Ovotesticular disorders of sex development in FGF9 mouse models of human synostosis syndromes. Hum. Mol. Genet. 29, 2148–2161. doi: 10.1093/hmg/ddaa100
Bohrer, L. R., Chuntova, P., Bade, L. K., Beadnell, T. C., Leon, R. P., Brady, N. J., et al. (2014). Activation of the FGFR–STAT3 pathway in breast cancer cells induces a hyaluronan–rich microenvironment that licenses tumor formation. Cancer Res. 74, 374–386. doi: 10.1158/0008-5472.CAN-13-2469
Burg, M. L., Chai, Y., Yao, C. A., Magee, W. III, and Figueiredo, J. C. (2016). Epidemiology, etiology, and treatment of isolated cleft palate. Front. Physiol. 7:67. doi: 10.3389/fphys.2016.00067
Bush, J. O., and Jiang, R. (2012). Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139, 231–243. doi: 10.1242/dev.067082
Chang, M. –M., Lai, M. –S., Hong, S. –Y., Pan, B. –S., Huang, H., Yang, S. –H., et al. (2018). FGF9/FGFR2 increase cell proliferation by activating ERK1/2, Rb/E2F1, and cell cycle pathways in mouse Leydig tumor cells. Cancer Sci. 109, 3503–3518. doi: 10.1111/cas.13793
Chiquet, M., Blumer, S., Angelini, M., Mitsiadis, T. A., and Katsaros, C. (2016). Mesenchymal remodeling during palatal shelf elevation revealed by extracellular matrix and F–Actin expression patterns. Front. Physiol. 7:392. doi: 10.3389/fphys.2016.00392
Colvin, J. S., Feldman, B., Nadeau, J. H., Goldfarb, M., and Ornitz, D. M. (1999). Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev. Dyn. 216, 72–88. doi: 10.1002/(SICI)1097-0177(199909)216:1<72::AID-DVDY9<3.0.CO;2-9
Colvin, J. S., White, A. C., Pratt, S. J., and Ornitz, D. M. (2001). Lung hypoplasia and neonatal death in Fgf9–null mice identify this gene as an essential regulator of lung mesenchyme. Development 128, 2095–2106.
Dixon, M. J., Marazita, M. L., Beaty, T. H., and Murray, J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167–178. doi: 10.1038/nrg2933
Gallardo, T., Shirley, L., John, G. B., and Castrillon, D. H. (2007). Generation of a germ cell–specific mouse transgenic Cre line, Vasa–Cre. Genesis 45, 413–417. doi: 10.1002/dvg.20310
Goudy, S., Law, A., Sanchez, G., Baldwin, H. S., and Brown, C. (2010). Tbx1 is necessary for palatal elongation and elevation. Mech. Dev. 127, 292–300. doi: 10.1016/j.mod.2010.03.001
Govindarajan, V., and Overbeek, P. A. (2006). FGF9 can induce endochondral ossification in cranial mesenchyme. BMC Dev. Biol. 6:7. doi: 10.1186/1471-213X-6-7
Harada, M., Murakami, H., Okawa, A., Okimoto, N., Hiraoka, S., Nakahara, T., et al. (2009). FGF9 monomer–dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat. Genet. 41, 289–298. doi: 10.1038/ng.316
Hecht, D., Zimmerman, N., Bedford, M., Avivi, A., and Yayon, A. (1995). Identification of fibroblast growth factor 9 (FGF9) as a high affinity, heparin dependent ligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. Growth Fact. 12, 223–233. doi: 10.3109/08977199509036882
Huang, H., Yang, X., Bao, M., Cao, H., Miao, X., Zhang, X., et al. (2016). Ablation of the Sox11 Gene results in clefting of the secondary palate resembling the pierre robin sequence. J. Biol. Chem. 291, 7107–7118. doi: 10.1074/jbc.M115.690875
Hung, I. H., Schoenwolf, G. C., Lewandoski, M., and Ornitz, D. M. (2016). A combined series of Fgf9 and Fgf18 mutant alleles identifies unique and redundant roles in skeletal development. Dev. Biol. 411, 72–84. doi: 10.1016/j.ydbio.2016.01.008
Hung, I. H., Yu, K., Lavine, K. J., and Ornitz, D. M. (2007). FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev. Biol. 307, 300–313. doi: 10.1016/j.ydbio.2007.04.048
Iwata, J. I., Tung, L., Urata, M., Hacia, J. G., Pelikan, R., Suzuki, A., et al. (2012). Fibroblast growth factor 9 (FGF9)–pituitary homeobox 2 (PITX2) pathway mediates transforming growth factor β (TGFβ) signaling to regulate cell proliferation in palatal mesenchyme during mouse palatogenesis. J. Biol. Chem. 287, 2353–2363. doi: 10.1074/jbc.M111.280974
Jin, J. Z., Lei, Z., Lan, Z. J., Mukhopadhyay, P., and Ding, J. (2018). Inactivation of Fgfr2 gene in mouse secondary palate mesenchymal cells leads to cleft palate. Reprod. Toxicol. 77, 137–142. doi: 10.1016/j.reprotox.2018.03.004
Juriloff, D. M., and Harris, M. J. (2008). Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. A Clin. Mol. Teratol. 82, 63–77. doi: 10.1002/bdra.20430
Lan, Y., Qin, C., and Jiang, R. (2019). Requirement of hyaluronan Synthase–2 in craniofacial and palate development. J. Dent. Res. 98, 1367–1375. doi: 10.1177/0022034519872478
Lee, Y. –H., and Saint–Jeannet, J. –P. (2011). Sox9 function in craniofacial development and disease. Genesis 49, 200–208. doi: 10.1002/dvg.20717
Li, C., Lan, Y., and Jiang, R. (2017). Molecular and cellular mechanisms of palate development. J. Dent. Res. 96, 1184–1191. doi: 10.1177/0022034517703580
Li, R., Chen, Z., Yu, Q., Weng, M., and Chen, Z. (2019). The function and regulatory network of Pax9 gene in palate development. J. Dent. Res. 98, 277–287. doi: 10.1177/0022034518811861
Liu, Z., Xu, J., Colvin, J. S., and Ornitz, D. M. (2002). Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 16, 859–869. doi: 10.1101/gad.965602
Loke, J., Pearlman, A., Radi, O., Zuffardi, O., Giussani, U., Pallotta, R., et al. (2014). Mutations in MAP3K1 tilt the balance from SOX9/FGF9 to WNT/β–catenin signaling. Hum. Mol. Genet. 23, 1073–1083. doi: 10.1093/hmg/ddt502
McCarthy, N., Sidik, A., Bertrand, J. Y., and Eberhart, J. K. (2016). An Fgf–Shh signaling hierarchy regulates early specification of the zebrafish skull. Dev. Biol. 415, 261–277. doi: 10.1016/j.ydbio.2016.04.005
Mittwoch, U. (2008). Different gene expressions on the left and the right: a genotype/phenotype mismatch in need of attention. Ann. Hum. Genet. 72(Pt 1), 2–9. doi: 10.1111/j.1469-1809.2007.00402.x
Mori–Akiyama, Y., Akiyama, H., Rowitch, D. H., and de Crombrugghe, B. (2003). Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. U.S.A. 100, 9360–9365. doi: 10.1073/pnas.1631288100
Naruo, K., Seko, C., Kuroshima, K., Matsutani, E., Sasada, R., Kondo, T., et al. (1993). Novel secretory heparin–binding factors from human glioma cells (glia–activating factors) involved in glial cell growth. Purification and biological properties. J. Biol. Chem. 268, 2857–2864.
Nikitovic, D., Kouvidi, K., Karamanos, N. K., and Tzanakakis, G. N. (2013). The roles of hyaluronan/RHAMM/CD44 and their respective interactions along the insidious pathways of fibrosarcoma progression. Biomed. Res. Int. 2013:929531. doi: 10.1155/2013/929531
Ovchinnikov, D. (2009). Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harb. Protoc. 2009:db.rot5170. doi: 10.1101/pdb.prot5170
Parada, C., and Chai, Y. (2015). Mandible and tongue development. Curr. Top. Dev. Biol. 115, 31–58. doi: 10.1016/bs.ctdb.2015.07.023
Pirvola, U., Zhang, X., Mantela, J., Ornitz, D. M., and Ylikoski, J. (2004). Fgf9 signaling regulates inner ear morphogenesis through epithelial–mesenchymal interactions. Dev. Biol. 273, 350–360. doi: 10.1016/j.ydbio.2004.06.010
Ramaesh, T., and Bard, J. B. (2003). The growth and morphogenesis of the early mouse mandible: a quantitative analysis. J. Anat. 203, 213–222. doi: 10.1046/j.1469-7580.2003.00210.x
Rice, R., Spencer–Dene, B., Connor, E. C., Gritli–Linde, A., McMahon, A. P., Dickson, C., et al. (2004). Disruption of Fgf10/Fgfr2b–coordinated epithelial–mesenchymal interactions causes cleft palate. J. Clin. Invest. 113, 1692–1700. doi: 10.1172/jci20384
Riley, B. M., Mansilla, M. A., Ma, J., Daack–Hirsch, S., Maher, B. S., Raffensperger, L. M., et al. (2007). Impaired FGF signaling contributes to cleft lip and palate. Proc. Natl. Acad. Sci. U.S.A. 104, 4512–4517. doi: 10.1073/pnas.0607956104
Riley, B. M., and Murray, J. C. (2007). Sequence evaluation of FGF and FGFR gene conserved non–coding elements in non–syndromic cleft lip and palate cases. Am. J. Med. Genet. A 143a, 3228–3234. doi: 10.1002/ajmg.a.31965
Rodriguez–Zabala, M., Aza–Carmona, M., Rivera–Pedroza, C. I., Belinchón, A., Guerrero–Zapata, I., Barraza–García, J., et al. (2017). FGF9 mutation causes craniosynostosis along with multiple synostoses. Hum. Mutat. 38, 1471–1476. doi: 10.1002/humu.23292
Sentchordi–Montané, L., Diaz–Gonzalez, F., Cátedra–Vallés, E. V., and Heath, K. E. (2020). Identification of the third FGF9 variant in a girl with multiple synostosis – comparison of the genotype:phenotype of FGF9 variants in humans and mice. Clin. Genet. 99, 309–312. doi: 10.1111/cge.13876
Sock, E., Rettig, S. D., Enderich, J., Bösl, M. R., Tamm, E. R., and Wegner, M. (2004). Gene targeting reveals a widespread role for the high–mobility–group transcription factor Sox11 in tissue remodeling. Mol. Cell Biol. 24, 6635–6644. doi: 10.1128/mcb.24.15.6635-6644.2004
Su, N., Jin, M., and Chen, L. (2014). Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res. 2:14003. doi: 10.1038/boneres.2014.3
Virakul, S., Heutz, J. W., Dalm, V. A., Peeters, R. P., Paridaens, D., van den Bosch, W. A., et al. (2016). Basic FGF and PDGF–BB synergistically stimulate hyaluronan and IL–6 production by orbital fibroblasts. Mol. Cell Endocrinol. 433, 94–104. doi: 10.1016/j.mce.2016.05.023
Wang, H., Zhang, T., Wu, T., Hetmanski, J. B., Ruczinski, I., Schwender, H., et al. (2013). The FGF and FGFR Gene family and risk of cleft lip with or without cleft palate. Cleft Palate Craniofac. J. 50, 96–103. doi: 10.1597/11-132
Weng, M., Chen, Z., Xiao, Q., Li, R., and Chen, Z. (2018). A review of FGF signaling in palate development. Biomed. Pharmacother. 103, 240–247. doi: 10.1016/j.biopha.2018.04.026
Wolk, A., Hatipoglu, D., Cutler, A., Ali, M., Bell, L., Qi, J. H., et al. (2020). Role of FGF and hyaluronan in choroidal neovascularization in sorsby fundus dystrophy. Cells 9:608. doi: 10.3390/cells9030608
Wu, W., Gu, S., Sun, C., He, W., Xie, X., Li, X., et al. (2015). Altered FGF signaling pathways impair cell proliferation and elevation of palate shelves. PLoS One 10:e0136951. doi: 10.1371/journal.pone.0136951
Yokohama–Tamaki, T., Maeda, T., Tanaka, T. S., and Shibata, S. (2011). Functional analysis of CTRP3/cartducin in Meckel’s cartilage and developing condylar cartilage in the fetal mouse mandible. J. Anat. 218, 517–533. doi: 10.1111/j.1469-7580.2011.01354.x
Yonemitsu, M. A., Lin, T. –Y., and Yu, K. (2020). Hyaluronic acid is required for palatal shelf movement and its interaction with the tongue during palatal shelf elevation. Dev. Biol. 457, 57–68. doi: 10.1016/j.ydbio.2019.09.004
Yu, K., and Ornitz, D. M. (2011). Histomorphological study of palatal shelf elevation during murine secondary palate formation. Dev. Dyn. 240, 1737–1744. doi: 10.1002/dvdy.22670
Keywords: secondary palate development, fibroblast growth factor 9, hyaluronic acid, mandibular growth, tongue movement
Citation: Li R, Sun Y, Chen Z, Zheng M, Shan Y, Ying X, Weng M and Chen Z (2021) The Fibroblast Growth Factor 9 (Fgf9) Participates in Palatogenesis by Promoting Palatal Growth and Elevation. Front. Physiol. 12:653040. doi: 10.3389/fphys.2021.653040
Received: 13 January 2021; Accepted: 11 March 2021;
Published: 20 April 2021.
Edited by:
Charles Evans Wood, University of Florida, United StatesReviewed by:
Han Sung Jung, Yonsei University, South KoreaRichard C. Pelikan, Oklahoma Medical Research Foundation, United States
Copyright © 2021 Li, Sun, Chen, Zheng, Shan, Ying, Weng and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mengjia Weng, dzRtNGozQDEyNi5jb20=; Zhenqi Chen, b3J0aG9jaGVuQHllYWgubmV0