 Hooi Hooi Ng
Hooi Hooi Ng Mariluz Soula
Mariluz Soula Bryan Rivas1
Bryan Rivas1 Kenneth J. Wilson
Kenneth J. Wilson Juan J. Marugan
Juan J. Marugan Alexander I. Agoulnik
Alexander I. Agoulnik
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 07 July 2021
Sec. Renal Physiology and Pathophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.650769
Diseases, such as diabetes and hypertension, often lead to chronic kidney failure. The peptide hormone relaxin has been shown to have therapeutic effects in various organs. In the present study, we tested the hypothesis that ML290, a small molecule agonist of the human relaxin receptor (RXFP1), is able to target the kidney to remodel the extracellular matrix and reduce apoptosis induced by unilateral ureteral obstruction (UUO). UUO was performed on the left kidney of humanized RXFP1 mice, where the right kidneys served as contralateral controls. Mice were randomly allocated to receive either vehicle or ML290 (30 mg/kg) via daily intraperitoneal injection, and kidneys were collected for apoptosis, RNA, and protein analyses. UUO significantly increased expression of pro-apoptotic markers in both vehicle- and ML290-treated mice when compared to their contralateral control kidneys. Specifically, Bax expression and Erk1/2 activity were upregulated, accompanied by an increase of TUNEL-positive cells in the UUO kidneys. Additionally, UUO induced marked increase in myofibroblast differentiation and aberrant remodeling on the extracellular matrix. ML290 suppressed these processes by promoting a reduction of pro-apoptotic, fibroblastic, and inflammatory markers in the UUO kidneys. Finally, the potent effects of ML290 to remodel the extracellular matrix were demonstrated by its ability to reduce collagen gene expression in the UUO kidneys. Our data indicate that daily administration of ML290 has renal protective effects in the UUO mouse model, specifically through its anti-apoptotic and extracellular matrix remodeling properties.
Chronic kidney failure is characterized by a progressive decline in kidney function, accompanied by a decrease in glomerular filtration rate. Primary causes of chronic kidney failure include diabetes and hypertension. The peptide hormone relaxin has potent anti-fibrotic and remodeling properties in various organs (Ng et al., 2019b), demonstrates clinical safety (Dschietzig et al., 2009; Dahlke et al., 2015), and may provide novel therapeutics for chronic kidney failure.
Long-term relaxin treatment modulates the renal hemodynamic changes to increase cardiac output and global arterial compliance, decrease systemic vascular resistance, with maintenance of the mean arterial pressure in conscious male and female normotensive and hypertensive rats (Debrah et al., 2005a). Similar hemodynamic changes are also observed in the angiotensin II-induced hypertensive model, but not in the spontaneously hypertensive and normotensive rats (Debrah et al., 2005b). Moreover, chronic infusion of relaxin in hypertensive rats attenuates renal inflammation, collagen, and lipid accumulation to improve overall renal function (Wang et al., 2017). The renoprotective effects of relaxin under hypertensive conditions are well reported in the literature, but the beneficial action of relaxin in the context of diabetes remains controversial. For example, relaxin fails to protect the kidneys from diabetes-mediated injuries in models of streptozotocin-induced endothelial nitric oxide synthase (NOS) knockout mice (Dschietzig et al., 2015) or the diabetic transgenic mRen-2 rats (Wong et al., 2013). In the Dahl salt-sensitive/resistant rats administered 8% NaCl diet, relaxin treatment reduces blood pressure, TGF-β level, and αSMA expression and increases neuronal NOS and endothelial NOS expression (Yoshida et al., 2012). Apart from the actions of relaxin treatment under diseased conditions, relaxin shows renoprotective effects in physiological aging as well. For instance, endogenous relaxin protects the male mice from developing kidney injury, as evidenced by a downregulation of creatinine and proteinuria, as well as a reduction in collagen deposition in the kidneys of wild-type mice when compared to relaxin-deficient mice (Samuel et al., 2004). Additionally, relaxin enhances glomerular filtration rate and effective renal plasma flow, while reducing effective renal vascular resistance and collagen deposition in the Munich-Wistar rats (Danielson et al., 2006). Recent clinical data demonstrate that relaxin increases creatinine clearance and reduces serum creatinine in acute (Teerlink et al., 2013) and chronic (Dschietzig et al., 2009) heart failure settings, respectively.
In the renal circulation, long-term relaxin treatment reduces renal vascular resistance and myogenic reactivity of small renal arteries and increases renal blood flow and glomerular filtration rate in conscious male and female intact or ovariectomized rats (Conrad, 1984). Notably, exogenous relaxin treatment in non-pregnant female and male rats recapitulates the hemodynamic profile associated with pregnancy (Danielson and Conrad, 2003; Danielson et al., 2006). These studies collectively show the important role of relaxin to maintain the renal hemodynamics during pregnancy. The endogenous role of relaxin is well-documented in the ovary (Anand-Ivell and Ivell, 2014), and indeed, women who had pregnancies occur through in vitro fertilization, egg donation, and embryo transfer due to ovarian insufficiency have impaired glomerular filtration rate, which would otherwise increase in normal pregnancy (Smith et al., 2006). This is due to the deficiency of ovarian function and corpus luteum in these women that leads to impaired relaxin production (Sherwood, 1994), indicating the vital role of relaxin to mediate gestational hyperfiltration.
Although relaxin has been shown to demonstrate beneficial actions in various models of kidney disease, it is worth noting that relaxin possesses the universal drawback of peptide-based therapies, such as its short half-life in vivo (Chen et al., 1993), which requires continuous infusion to achieve sustained dosage for the treatment of chronic diseases. Therefore, our discovery of a non-peptide small molecule agonist of the relaxin family peptide receptor 1 (RXFP1), ML290, as a result of a high-throughput screening and medicinal chemistry optimization campaign (Chen et al., 2013; Xiao et al., 2013) might serve as an alternative to the recombinant peptide ligands. ML290 is a specific agonist for human RXFP1 and does not activate the rodent receptor (Huang et al., 2015). In this study, we utilized a unique strain of mice generated in our laboratory with knock-in of the human RXFP1 (Kaftanovskaya et al., 2019) to test the therapeutic efficacies of ML290 in vivo. A plethora of studies reported the anti-fibrotic role of relaxin (Samuel et al., 2016), and we recently showed that ML290 replicates the protective functions of relaxin in a mouse model of liver fibrosis (Kaftanovskaya et al., 2019). On the other hand, the anti-apoptotic role of relaxin is inconsistent, especially in in vivo studies, and no studies to date have investigated the effects of ML290 in the context of apoptosis. Earlier work reported that relaxin treatment has no effect on apoptosis in renal fibroblasts (Heeg et al., 2005) or in an experimental model of renal fibrosis (Hewitson et al., 2010). Contrary to these studies, a recent study shows that relaxin attenuates apoptosis in several rodent models of kidney disease, including ischemia-reperfusion injury (Yoshida et al., 2013), aristolochic acid nephropathy (Yang et al., 2019), and cisplatin-induced nephrotoxicity models (Yoshida et al., 2014). Here, we sought to investigate whether ML290 can prevent apoptosis and aberrant matrix remodeling in the unilateral ureteral obstruction (UUO) mouse model.
This study used homozygous humanized RXFP1 (hRXFP1/hRXFP1; Kaftanovskaya et al., 2019) and Rxfp1-deficient male mice (Kaftanovskaya et al., 2015) on C57BL/6 background previously generated in our laboratory. All mice were bred and housed on a 12:12 h day/night cycle at a room temperature of 20 ± 2°C in the Florida International University Animal Care Facility. UUO procedures were performed on 6 weeks old mice under isoflurane anesthesia. Briefly, the ureter was ligated at two different positions to separate the kidney and bladder; one near the renal pelvis and the other one close to the bladder. Un-operated right kidneys were used as contralateral controls in this study. A day after the UUO surgery, mice were randomly allocated to receive either vehicle [3% DMSO, 10% 1-Methyl-2-pyrrolidinone (NMP), 17% Kolliphor® HS 15, 29% Poly (ethylene glycol) 400, and 41% phosphate-buffered saline] or ML290 (30 mg/kg; Wilson et al., 2018) via intraperitoneal injections for five consecutive days. Two days after the last injections, mice were anesthetized with isoflurane followed by cardiac puncture for blood collection. 20 μl of serum was used for osmolality measurement using the Advanced Micro Osmometer Model 3300 (Advanced Instruments, Norwood, MA). All experimental procedures were reviewed and approved by the FIU Institutional Animal Care and Use Committee under protocol AN16-003.
Total RNA was isolated from the kidneys by column purification using a Direct-zol RNA MiniPrep Plus kit (Zymo Research, Irvine, CA) following manufacturer’s protocol. First strand complementary DNA (cDNA) was synthesized using the Verso cDNA synthesis kit (Thermo Scientific, Waltham, MA) and 1 μg RNA in a final reaction volume of 20 μL. Target gene expression was assessed by quantitative PCR using –2ΔΔCt method with Gapdh as the housekeeping gene. Mouse or human (RXFP1)-specific PCR primers (Eurofins Scientific, Louisville, KY) were designed using the Universal Probe Library Web site (Roche, Indianapolis, IN; Table 1), and PCR reaction was determined by SYBR Green chemistry.

Table 1. Gene sequences of primers used for quantitative real-time PCR experiments.
Thirty μg of kidney protein lysates was subjected to SDS-PAGE and Western blot analysis using primary mouse/rabbit antibodies for phospho-p44/42 MAPK (pErk1/2; Thr202/Tyr204; 1:500, catalog no. 4370S, Cell Signaling, Danvers, MA), p44/42 MAPK (Erk1/2; 1:1,000, catalog no. 4695S, Cell Signaling), platelet-derived growth factor receptor-β (PDGFR-β; 1:300, catalog no. sc-374573, Santa Cruz Biotechnology, Dallas, TX), peroxisome proliferator-activated receptor gamma (PPARG; 1:250, catalog no. sc-7273, Santa Cruz Biotechnology), and alpha smooth muscle actin (αSMA; 1:500, catalog no. ab5694, Abcam, Cambridge, MA) overnight at 4°C. To normalize for the amount of protein, membranes were re-probed with a loading control antibody (β-tubulin, 1:1,000, catalog no. 05-661, Millipore Sigma, Burlington, MA). Protein expression was detected using the SuperSignal West Pico Chemiluminescent Substrate (catalog no. 34080, Thermo Scientific) after incubation with either anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody (Promega, Madison, WI). Original images of Western blots are shown in Supplementary Figures 1–8.
Paraformaldehyde-fixed paraffin-embedded kidneys were used for in situ apoptosis detection using the ApopTag Plus Peroxidase kit (Millipore Sigma) following manufacturer’s protocol. Non-specific binding of enzyme-conjugate was evaluated in the absence of terminal deoxynucleotidyl transferase (TdT) labeling. At least 10 non-overlapping images were analyzed for each animal using the Carl Zeiss Axio A1 microscope with an AxioCam MRc5 CCD camera (Oberkochen, Germany) at 20× magnification, and TUNEL-positive cells were counted using a hand tally counter in a blinded manner.
Paraformaldehyde-fixed paraffin-embedded kidneys were sectioned at 4.5 μm thickness and stained with 0.1% sirius red solution (Electron Microscopy Sciences, Hatfield, PA, United States; Ng et al., 2017). At least 10 non-overlapping bright field images at 20× magnification were analyzed for each animal using the Carl Zeiss Axio A1 microscope with an AxioCam MRc5 CCD camera (Oberkochen, Germany). Staining was quantified using ImageJ software (NIH, Bethesda, MD; Schneider et al., 2012) and presented as percentage of stained area within the analyzed image.
Fifty μl of undiluted kidney protein lysates was used for the detection of MMP2, MMP3, MMP8, MMP12, and proMMP9 by multiplexing technology (Eve Technologies, Calgary, AB). Protein concentration of each sample was calculated by fitting the fluorescence intensity values using a cubic spline regression applied against the standard curve fluorescence.
Two-way ANOVA with Tukey’s multiple comparison test assessed statistical differences in kidney weight, TUNEL staining, gene, and protein expression between groups. Un-paired student t-test assessed statistical differences in serum osmolality between vehicle- and ML290-treated mice. A level of p < 0.05 was considered statistically significant. All results are presented as mean ± SEM, where n represents the number of mice per group.
Unilateral ureteral obstruction induced a significant increase of apoptosis in the kidney from both vehicle- (p < 0.001) and ML290-treated (p = 0.01) mice (Figures 1A,B). Daily administration of ML290 decreased apoptosis in UUO kidney when compared to vehicle-treated mice, as evidenced by a significant (p = 0.004) reduction in the number of TUNEL-positive cells (Figures 1A,B). Interestingly, there was a significant (p < 0.001) increase in the expression of the pro-survival gene, Bcl2 in the UUO kidney of vehicle-treated mice when compared to their contralateral controls (Figure 1C). This effect was abolished in ML290-treated mice, with a concomitant increase (p = 0.01) in Bcl2 expression when compared to vehicle-treated mice in the contralateral control kidneys (Figure 1C). Consistent with our TUNEL staining, the pro-apoptotic gene, Bax, was significantly (p < 0.0001) upregulated in UUO kidneys from both vehicle- and ML290-treated mice when compared to their contralateral controls (Figure 1D). ML290 treatment significantly (p < 0.001) downregulated Bax expression in the UUO kidney when compared to vehicle-treated mice (Figure 1D). Importantly, Rxfp1−/− mice showed significant (p < 0.0001) increase in Bax expression after UUO surgery, but it was unaffected in the UUO kidneys of vehicle- and ML290-treated mice, indicating the specificity of the molecule in our humanized mice (Figure 1E). UUO surgery enhanced the pro-apoptotic activity of Erk1/2 in vehicle-treated mice, as evidenced by a significant (p < 0.001) increase in the ratio of phosphorylated Erk1/2 to total Erk1/2 protein (Figure 1F). These phenotypic changes induced by UUO were not apparent in the kidneys of ML290-treated mice (Figure 1F). There was a reduction (p = 0.05) in Erk1/2 activity in the UUO kidneys of ML290-treated mice when compared to the vehicle group, but it did not reach significance (Figure 1F). Despite the marked anti-apoptotic role of ML290 in vivo, the kidney weight of these mice was unaffected by either the UUO procedure or treatment after normalization to body weight. Daily intraperitoneal administration of ML290 did not affect serum osmolality (vehicle 388.1 ± 7.685 mOsm vs. ML290 381.3 ± 6.343 mOsm) in UUO mice when compared to vehicle-treated group.
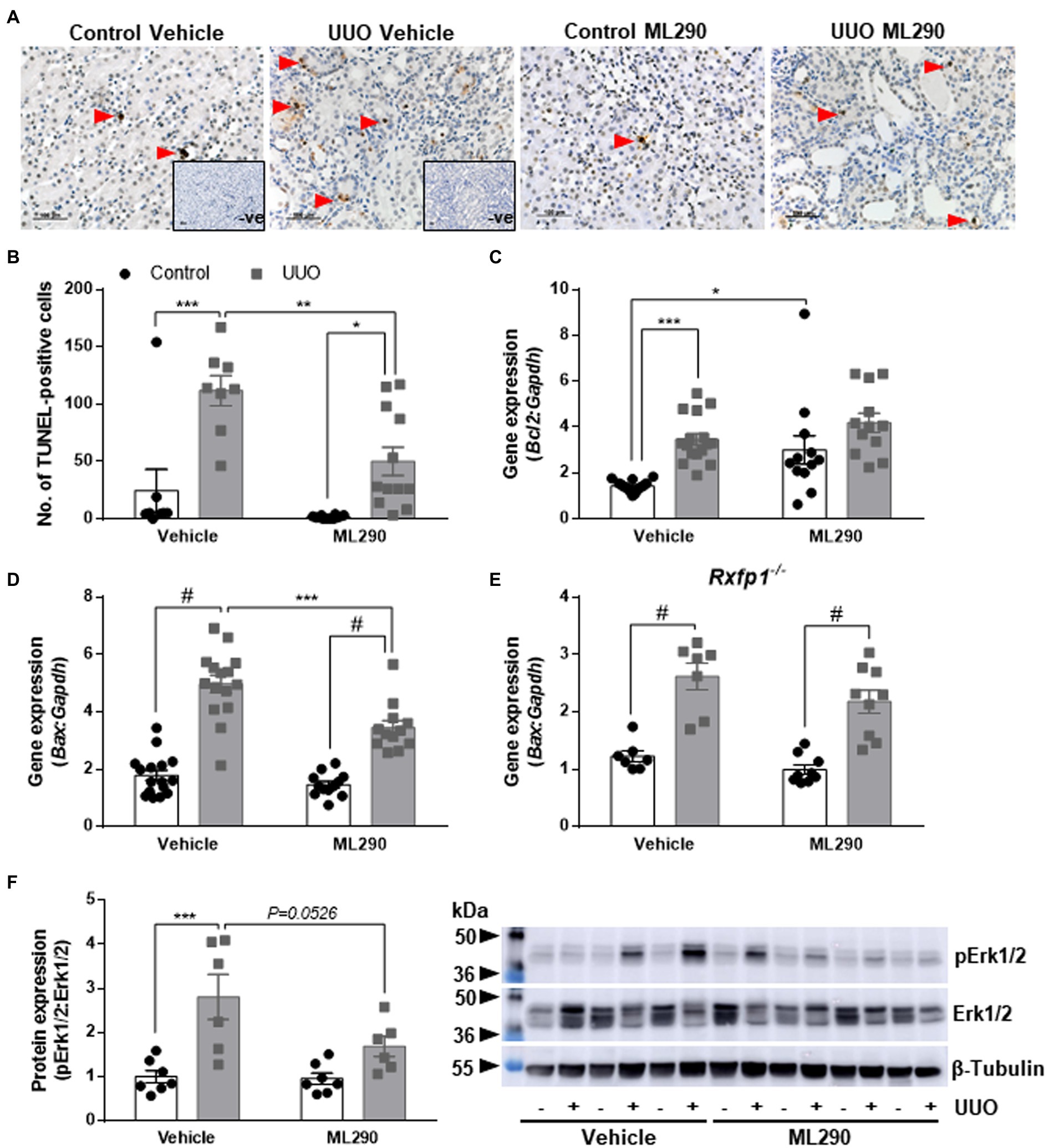
Figure 1. (A): Representative images of TUNEL staining. Red arrows indicate specific staining for apoptotic cells. There was no positive brown staining in negative control (without TdT enzyme; inset). Scale bar = 100 μm. (B): Quantitative analysis of TUNEL-positive cells in the control and unilateral ureteral obstruction (UUO) kidneys from vehicle- and ML290-treated humanized mice (n = 8–12 mice/group). (C–E): Bcl2 (C) and Bax gene expression in the control and UUO kidneys from vehicle- and ML290-treated humanized (D) (n = 12–16 mice/group) and Rxfp1-deficient (E) (n = 7–9 mice/group) mice. Gene expression is normalized to the housekeeping gene Gapdh and presented as fold change to control kidneys from vehicle-treated mice. (F): Densitometric ratio of phospho-Erk1/2 (44, 42 kDa) to total Erk1/2 (44, 42 kDa) protein expression in the control and UUO kidneys from vehicle- and ML290-treated humanized mice (n = 6–7 mice/group). β-tubulin (55 kDa) was used as the loading control. *p < 0.05, **p < 0.01, ***p < 0.001, and #p < 0.0001.
Unilateral ureteral obstruction significantly upregulated PDGFR-β (p < 0.0001; Figure 2A) and PPARG (p = 0.02; Figure 2B) protein expression in the kidneys of vehicle-treated mice when compared to their contralateral controls. This phenotype was abolished in ML290-treated mice, and there was a significant reduction in both the PDGFR-β (p < 0.0001; Figure 2A) and PPARG (p = 0.02; Figure 2B) expression in the UUO kidneys when compared to vehicle-treated mice. Similarly, Pparg gene expression was significantly (p < 0.0001) increased in the UUO kidneys of vehicle-treated, but not ML290-treated mice (Figure 2C). Consistent with PPARG protein expression in the UUO kidneys, ML290 significantly (p < 0.001) downregulated Pparg gene expression as well (Figure 2C). ML290-mediated actions on Pparg were not observed in the UUO kidneys of Rxfp1−/− mice, despite the significant increase of this nuclear transcription factor after UUO regardless of treatment (vehicle p = 0.002 and ML290 p < 0.0001; Figure 2D).
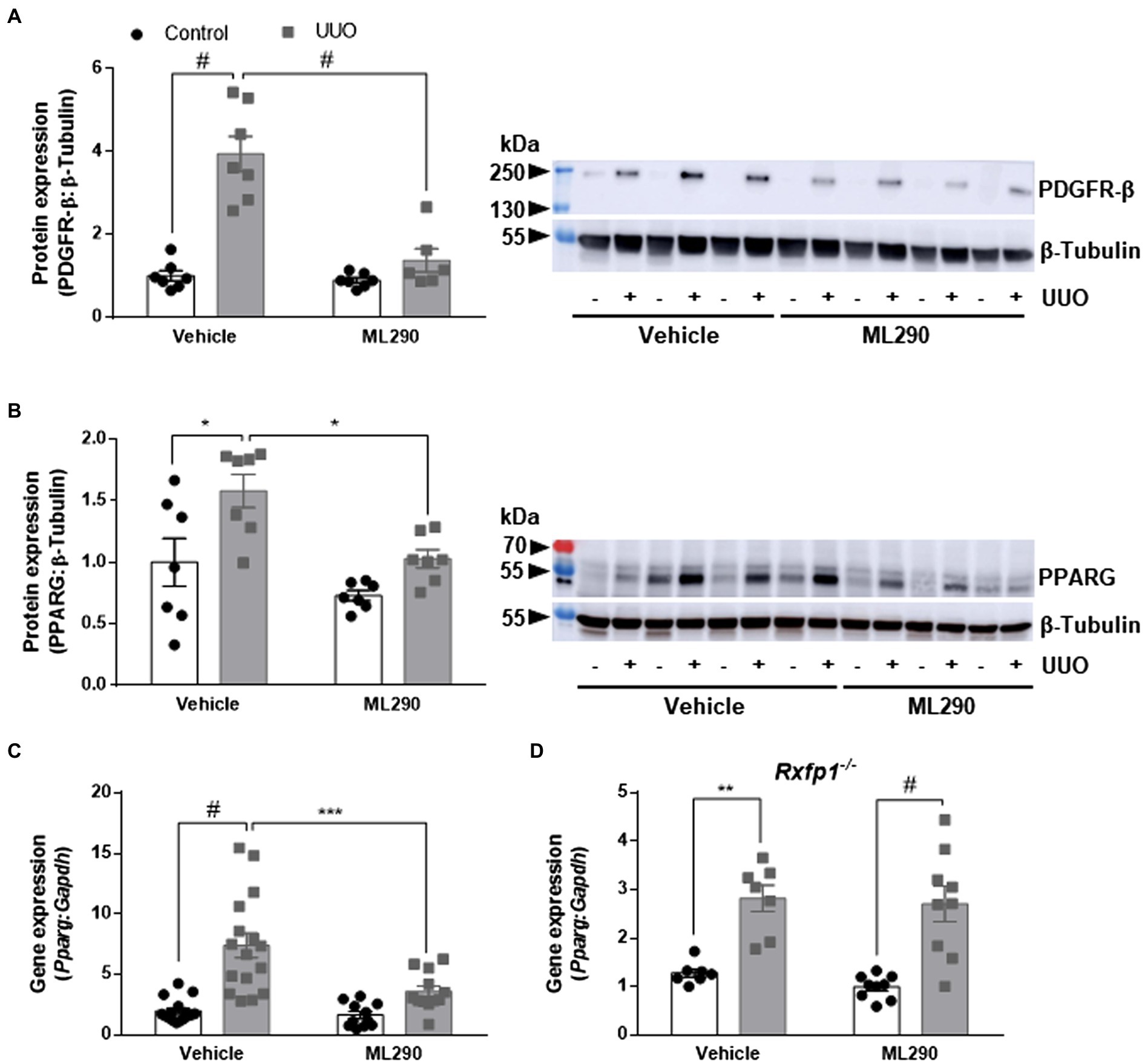
Figure 2. (A,B): Densitometry of PDGFR-β (A) (180–190 kDa) and PPARG (B) (54 kDa) protein expression in the control and UUO kidneys from vehicle- and ML290-treated humanized mice (n = 6–7 mice/group). β-tubulin (55 kDa) was used as the loading control. (C,D): Pparg gene expression in the control and UUO kidneys from vehicle- and ML290-treated humanized (C) (n = 12–16 mice/group) and Rxfp1-deficient (D) (n = 7–9 mice/group) mice. Gene expression is normalized to the housekeeping gene Gapdh and presented as fold change to control kidneys from vehicle-treated mice. *p < 0.05, **p < 0.01, ***p < 0.001, and #p < 0.0001.
UUO promoted inflammation in the kidneys of vehicle-treated but not ML290-treated mice, as evidenced by a significant (p < 0.0001) increase in Tnf gene expression when compared to their contralateral control kidney (Figure 3A). ML290 suppressed this inflammatory response by significantly (p = 0.04) reducing the expression of Tnf in the UUO kidney (Figure 3A). Inflammation in the kidney is often accompanied by macrophage infiltration. We demonstrated that UUO significantly increases the gene expression of a macrophage marker, Cd68 in both vehicle- (p < 0.0001) and ML290-treated (p < 0.001) mice (Figure 3B). Consistent with the reduction of Tnf in the UUO kidneys of ML290-treated mice, Cd68 expression was significantly (p = 0.01) downregulated when compared to vehicle-treated mice (Figure 3B).
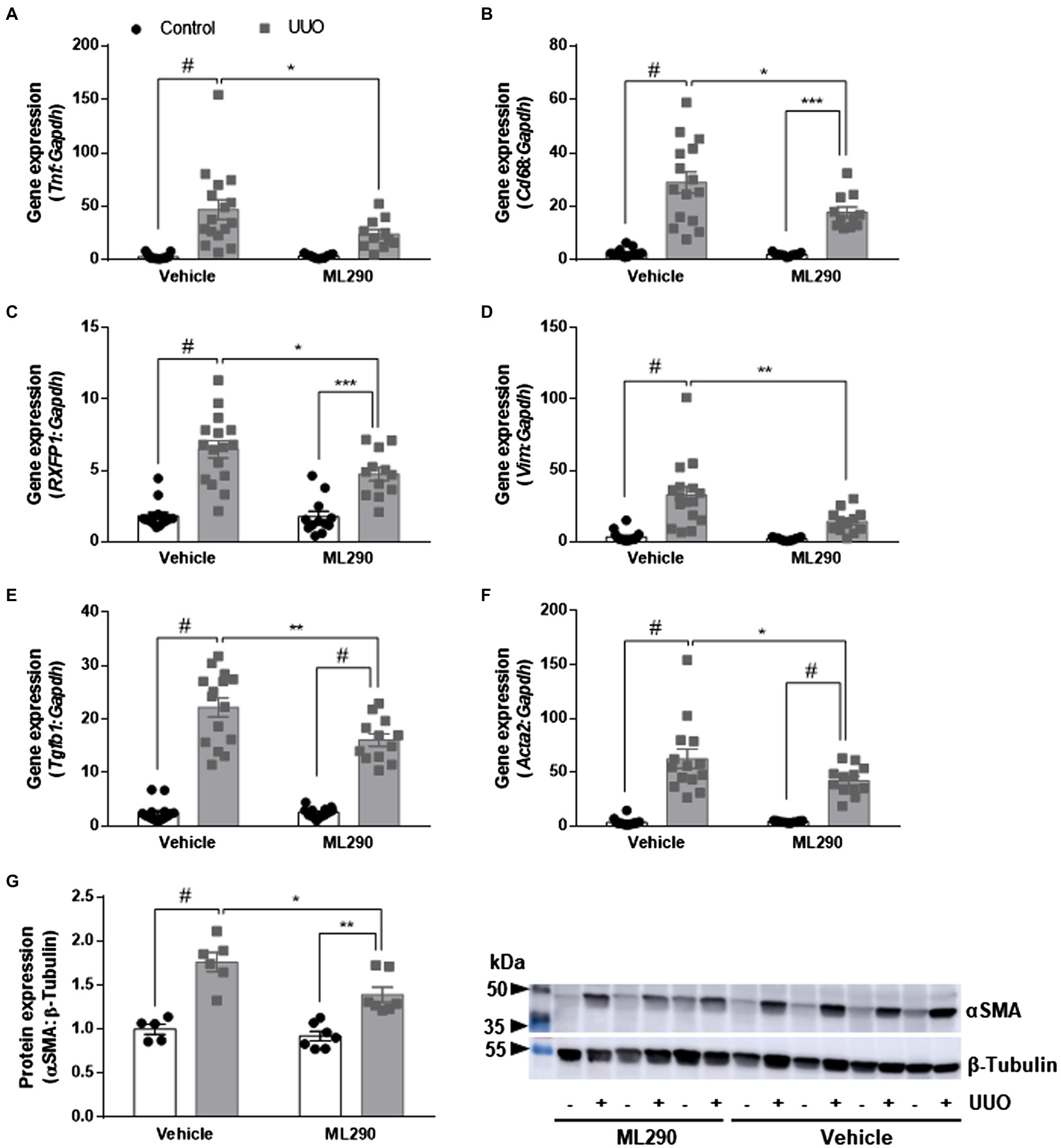
Figure 3. (A–F): Tnf (A), Cd68 (B), RXFP1 (C), Vim (D), Tgfb1 (E), and Acta2 (F) gene expression in the control and UUO kidneys from vehicle- and ML290-treated humanized mice (n = 12–16 mice/group). Gene expression is normalized to the housekeeping gene Gapdh and presented as fold change to control kidneys from vehicle-treated mice. (G): Densitometry of αSMA (42 kDa) protein expression in the control and UUO kidneys from vehicle- and ML290-treated humanized mice (n = 5–7 mice/group). β-tubulin (55 kDa) was used as the loading control. *p < 0.05, **p < 0.01, and #p < 0.0001.
Quantitative assessment of human RXFP1 by qPCR indicated significant increase of the receptor in UUO kidney of both vehicle- (p < 0.0001) and ML290-treated (p < 0.001) mice (Figure 3C). RXFP1 expression was significantly (p = 0.05) reduced in the UUO kidneys of ML290-treated mice compared to the vehicle group, an effect that may be associated with a reduction in fibroblasts activation (Figure 3C). Consistent with these findings, we found that UUO significantly (p < 0.0001) upregulated gene expression of several fibroblastic markers, such as Vim, Tgfb1, and Acta2 in vehicle-treated mice when compared to their contralateral control kidneys (Figures 3D–F). Notably, ML290 treatment for 5 days significantly reduced the expression of these fibroblastic markers (Vim p = 0.003, Tgfb1 p = 0.002, and Acta2 p = 0.04) in the UUO kidneys when compared to vehicle-treated mice (Figures 3D–F). Despite the profound action of ML290 to suppress fibroblast activation in the UUO kidney, Tgfb1 and Acta2 expression remained significantly (p < 0.0001) elevated when compared to their contralateral controls (Figures 3E,F). Protein analysis of αSMA was consistent with the expression at transcriptional level, revealing a significant upregulation of this protein induced by UUO in both vehicle- (p < 0.0001) and ML290-treated (p = 0.001) mice (Figure 3G). Collectively, we showed that ML290 treatment effectively prevented myofibroblast differentiation in the UUO kidneys, with significant (p = 0.02) inhibition on the αSMA production (Figure 3G).
Aberrant extracellular matrix accumulation was observed in the UUO kidneys of vehicle-treated mice when compared to their contralateral controls, as shown by a significant (p < 0.0001) increase in Col1a1, Col3a1, and Col4a2 gene expression (Figures 4A,C,D). In the UUO kidneys of ML290-treated mice, Col1a1 (p = 0.02) and Col4a2 (p < 0.0001) expression remained significantly elevated when compared to their contralateral controls (Figures 4A,D). There was, however, a significant reduction in the expression of these genes (Col1a1 p = 0.002, Col3a1 p = 0.02, and Col4a2 p = 0.02) in the UUO kidneys of ML290-treated mice when compared to the vehicle group (Figures 4A,C,D), suggesting the matrix remodeling actions of ML290 in vivo. To confirm the ML290-mediated inhibition on fibrogenesis was specific on the human RXFP1, we showed that Col1a1 expression was unaffected by ML290 treatment in the Rxfp1−/− mice, despite significant (p < 0.0001) increase of this gene in the UUO kidneys of vehicle- and ML290-treated mice (Figure 4B). Interestingly, we found that the interstitial collagen content was unaffected by ML290 treatment in the UUO kidneys when compared to vehicle-treated mice, as evidenced by similar level of sirius red staining in both the UUO groups (Supplementary Figure 9). In UUO kidneys, the aberrant accumulation of extracellular matrix was associated with a significant increase of Timp1 and Timp2 expression in the kidneys of both vehicle- (p < 0.0001) and ML290-treated (Timp1 p = 0.001 and Timp2 p < 0.0001) mice (Figures 4E,F). Interestingly, the extracellular matrix degrading gene, Mmp2 was also significantly upregulated in the UUO kidneys of vehicle- (p < 0.0001) and ML290-treated (p < 0.001) mice (Figure 4G). These findings suggest that the concomitant increase in Mmp2 may be a compensatory mechanism to overcome the impaired matrix turnover induced by UUO. In comparison to the UUO kidneys from vehicle-treated mice, ML290-treated mice had significantly lower Timp1 (p = 0.02), Timp2 (p = 0.04), and Mmp2 (p = 0.02) expression (Figures 4E–G), an effect that may be due to the already reduced collagen in their UUO kidneys. Despite the ability of ML290 to reduce Mmp2 expression at the transcriptional level, MMP2 protein expression was not significantly different from UUO kidneys of vehicle-treated mice (Figure 5A). UUO significantly (p < 0.0001) upregulated MMP2 level and significantly (p < 0.05) downregulated MMP3 level in the kidneys of both vehicle- and ML290-treated mice (Figures 5A,B). MMP8 (Figure 5C), MMP12 (Figure 5D), and proMMP9 (Figure 5E) levels were not affected by either UUO or ML290 treatment.
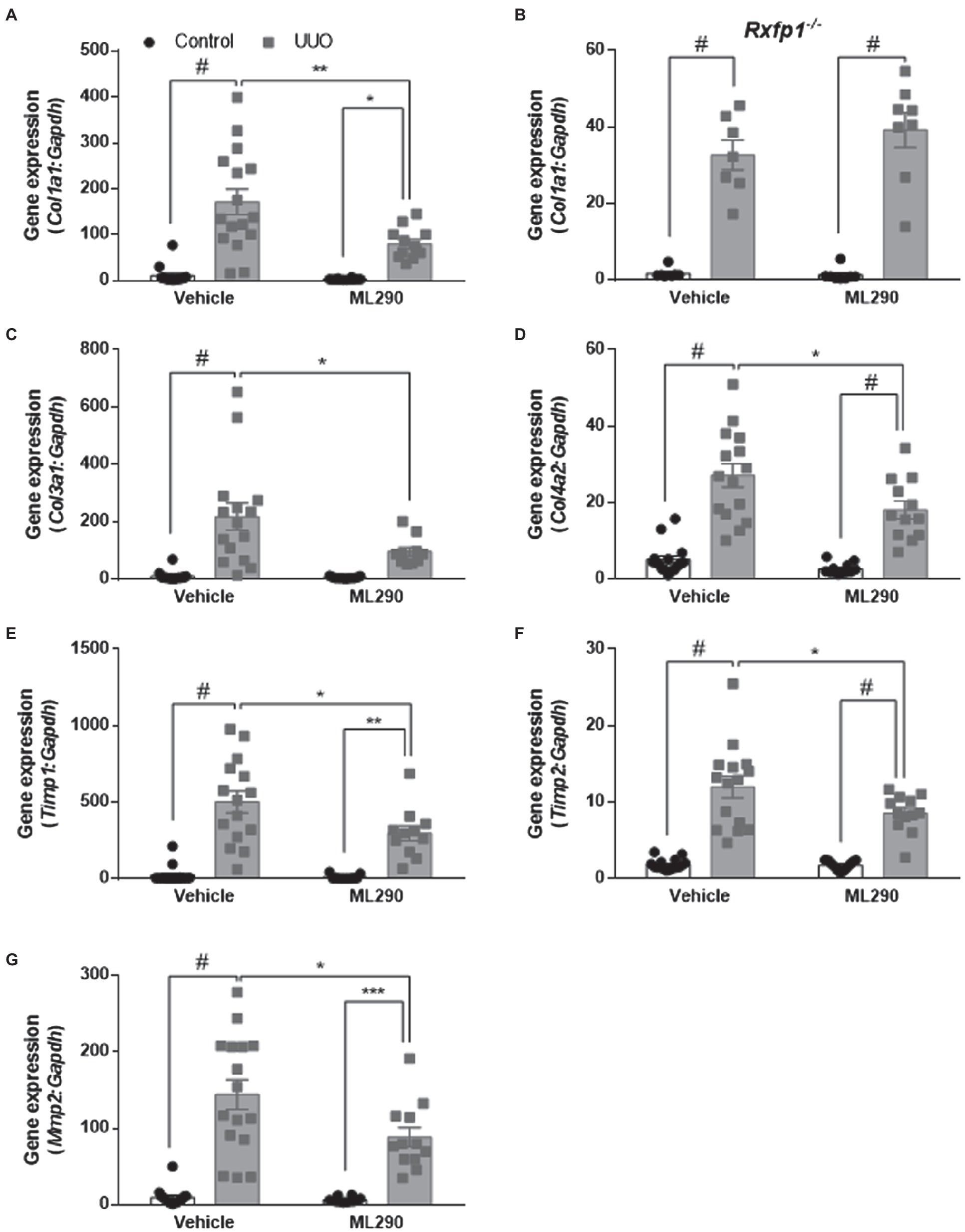
Figure 4. (A,C–G): Col1a1 (A), Col3a1 (C), Col4a2 (D), Timp1 (E), Timp2 (F), and Mmp2 (G) gene expression in the control and UUO kidneys from vehicle- and ML290-treated humanized mice (n = 12–16 mice/group). (B): Col1a1 gene expression in the control and UUO kidneys from vehicle- and ML290-treated Rxfp1-deficient mice (n = 7–8 mice/group). Gene expression is normalized to the housekeeping gene Gapdh and presented as fold change to control kidneys from vehicle-treated mice. *p < 0.05, **p < 0.01, ***p < 0.001, and #p < 0.0001.
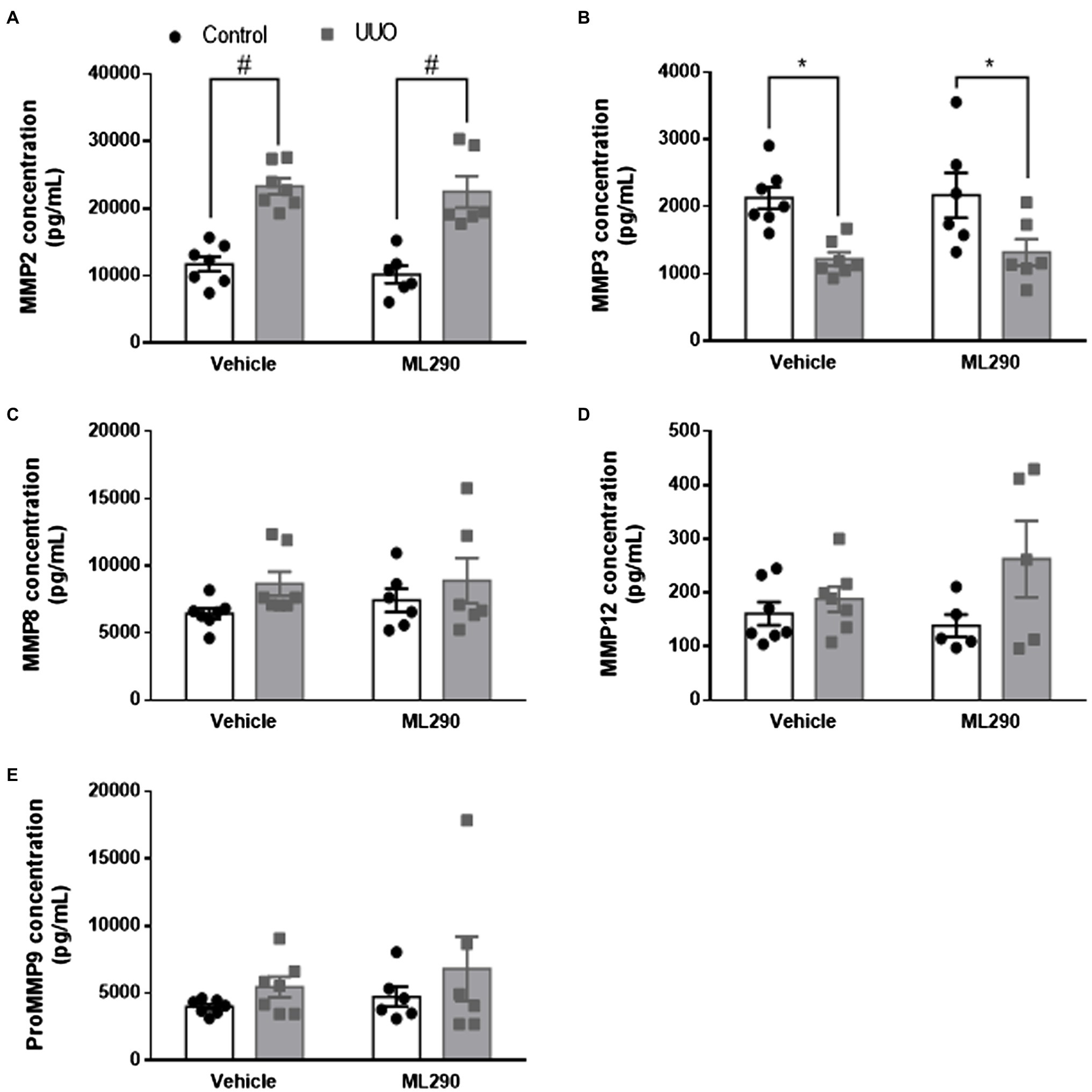
Figure 5. (A–E): MMP2 (A), MMP3 (B), MMP8 (C), MMP12 (D), and proMMP9 (E) concentration in the control and UUO kidneys from vehicle- and ML290-treated humanized mice (n = 5–7 mice/group). Protein concentration is expressed as pg/ml. *p < 0.05 and #p < 0.0001.
The aims of this study were to investigate the therapeutic potential of ML290 to reduce apoptosis in the kidney and to test the hypothesis that the non-peptide small molecule RXFP1 agonist ML290 suppresses aberrant extracellular matrix remodeling in the UUO mice. We demonstrated that daily administration of ML290 has potent anti-apoptotic actions in the UUO kidneys, primarily through the suppression of pro-apoptotic markers and promotion of pro-survival activity. Furthermore, ML290 impedes pericyte-myofibroblast differentiation in the UUO kidneys and inhibits fibrogenesis. The ML290-mediated effects were at least in part attributed to a reduction in inflammation and macrophage infiltration in the UUO kidneys.
Very little work to date has assessed the anti-apoptotic effects of relaxin in the kidneys. The majority of the research has focused on investigating relaxin’s anti-apoptotic actions in cardiomyocytes (Moore et al., 2007; Ng et al., 2017) and hepatocytes (Kageyama et al., 2018; Lee et al., 2019). Here, we showed that activation of the human RXFP1 by ML290 in humanized mice potently prevented UUO-induced apoptosis in the kidneys. These effects were evident by the detection of DNA breakage that occurs during apoptosis, and subsequent changes in the pro-apoptotic gene, Bax expression at transcriptional level. Conventionally, activation of Erk1/2 is related to cell survival, but several studies suggest that Erk1/2 could promote cell death under certain stimuli (Pearson et al., 2001). It is possible that the balance between the activation of pro-apoptotic or pro-survival pathways underscores the signals transmitted by Erk1/2 and determines whether a cell undergoes apoptosis. In fact, we found that the pro-survival gene, Bcl2 was simultaneously upregulated in the UUO kidneys, indicating a compensatory mechanism to overcome cell deaths that occurred in the UUO kidneys. Interestingly, Bcl2 expression was significantly increased in the contralateral control kidneys of ML290-treated mice when compared to their vehicle-treated counterparts. These data may suggest that ML290 promotes cell survival in the healthy kidneys to compensate for the reduced function of the left kidneys (UUO), since there was no apparent reduction in Bcl2 expression in the UUO kidneys of ML290-treated mice.
Furthermore, we demonstrated that UUO markedly upregulated expression of the receptor for PDGF-β only in vehicle-treated mice. This is consistent with our Bcl2 data as PDGF-β is one of the multiple major survival factors in several cell types of mesenchymal origin (Eitner et al., 2002). The PDGF-β receptor can be activated by various mediators, including TGF-β, fibroblast growth factor, and PDGF itself (Haberstroh et al., 1993; Floege and Johnson, 1995). Indeed, we found that Tgfb1 expression was increased in the UUO kidneys regardless of treatment, when there was significant activation of the PDGF-β receptor in these diseased kidneys. In line with these observations, we showed that PPAR-γ expression was significantly upregulated in the UUO kidneys of vehicle-treated mice, suggesting the enhanced differentiation of pericytes to myofibroblasts that ultimately contributes to the accumulation of pathological extracellular matrix in these kidneys. In addition to the aberrant remodeling of the extracellular matrix, overproduction of myofibroblasts in the kidneys also leads to inflammation and macrophage infiltration. These phenotypes were apparent in the UUO kidneys of vehicle-treated, but not ML290-treated mice. Notably, ML290 potently downregulated α-SMA and vimentin expression in the UUO kidneys to inhibit fibrogenesis in these diseased kidneys. Additionally, we found that human RXFP1 expression was increased in the UUO kidneys of both vehicle- and ML290-treated mice, an effect that may be in part due to fibroblasts activation (Fallowfield et al., 2014). Consistent with the α-SMA and vimentin data, hRXFP1 expression was reduced by ML290 treatment. Taken together, we demonstrated the target-specific actions of ML290 to regulate fibroblasts and pericyte-myofibroblast transition in this mouse model.
Relaxin is known to be a potent regulator of collagen turnover in the UUO mouse model (Hewitson et al., 2010; Huuskes et al., 2015). Here, we sought to determine whether the biased allosteric agonist of RXFP1 possesses similar action to the native ligand in this mouse model. ML290 treatment profoundly inhibited UUO-induced upregulation of collagen expression in the kidneys, an effect that may be attributed to its action on the tissue inhibitors of metalloproteinases (TIMP1 and TIMP2). We found that TIMPs expression was increased in the UUO kidneys to inhibit the degradation of extracellular matrix. Although expression of TIMPs remained significantly higher than the contralateral control kidneys in ML290-treated mice, these metalloproteinases were significantly downregulated in the diseased kidneys when compared to vehicle-treated mice. Interestingly, we found that MMP2 expression was reduced in the UUO kidneys after ML290 treatment, despite that it has been shown to activate MMP2 in the human cardiac fibroblasts (Kocan et al., 2017). It is possible that in the UUO kidneys where there was overproduction of collagen, MMP2 is activated to accelerate the degradation of matrix as a compensatory mechanism in these mice. Since there was lower level of collagen mRNA in the UUO kidneys of ML290-treated mice, activation of MMP2 was less pronounced. Surprisingly, we did not observe significant reduction in interstitial collagen content in the UUO kidneys of ML290-treated mice, despite the marked decrease in gene expression of several collagen markers (Col1a1, Col3a1, and Col4a2). This may be due to the short duration of our treatment in these UUO mice that was insufficient to induce morphological changes and collagen turnover in their kidneys.
Finally, it was important to demonstrate that the effects of ML290 resulted from activation of the human RXFP1, as this compound does not activate the rodent receptor (Huang et al., 2015; Ng et al., 2019a). We tested the gene expression of several markers that were markedly reduced by ML290 treatment, in the Rxfp1−/− mice. These genes were upregulated in the UUO kidneys regardless of vehicle or ML290 treatment, indicating that data obtained from the humanized mice resulted from activation of the human receptor.
Our proof-of-concept study revealed for the first time that ML290 prevented UUO-induced apoptosis and extracellular matrix remodeling in the kidneys of humanized mice. Detailed mechanistic studies should be carried out in the future to delineate the specific pathways activated by ML290 in more physiologically relevant pre-clinical animal models. Although ML290 is safe and tolerable in mice (Wilson et al., 2018; Kaftanovskaya et al., 2019), it is also important to evaluate the hemodynamics and functional outcomes of ML290 treatment in vivo, in particular those from diseased pre-clinical animal models. Future studies on the therapeutic efficacies of ML290 now warrant such enhanced translational value for future drug development.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Florida International University Institutional Animal Care and Use Committee under protocol AN16-003.
HN wrote the manuscript. HN, MS, BR, KW, JM, and AA designed the work or contributed to the acquisition, analysis, or interpretation of data for the work, critically revised the work for important intellectual content, agreed to be accountable for all aspects, including the accuracy or integrity of the work, and approved the final version of the work to be published. All authors contributed to the article and approved the submitted version.
This research was supported by the US National Institutes of Health (NIH) grant R01DK110167 (to AA). HN received the American Heart Association Postdoctoral Fellowship grant 19POST34380255. MS was supported by the NIH National Institute of General Medical Sciences (NIGMS) T34GM083688 fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fphys.2021.650769/full#supplementary-material
Anand-Ivell, R., and Ivell, R. (2014). Regulation of the reproductive cycle and early pregnancy by relaxin family peptides. Mol. Cell. Endocrinol. 382, 472–479. doi: 10.1016/j.mce.2013.08.010
Chen, S. A., Perlman, A. J., Spanski, N., Peterson, C. M., Sanders, S. W., Jaffe, R., et al. (1993). The pharmacokinetics of recombinant human relaxin in nonpregnant women after intravenous, intravaginal, and intracervical administration. Pharm. Res. 10, 834–838.
Chen, C. Z., Southall, N., Xiao, J., Marugan, J. J., Ferrer, M., Hu, X., et al. (2013). Identification of small-molecule agonists of human relaxin family receptor 1 (RXFP1) by using a homogenous cell-based cAMP assay. J. Biomol. Screen. 18, 670–677. doi: 10.1177/1087057112469406
Conrad, K. P. (1984). Renal hemodynamics during pregnancy in chronically catheterized, conscious rats. Kidney Int. 26, 24–29.
Dahlke, M., Ng, D., Yamaguchi, M., Machineni, S., Berger, S., Canadi, J., et al. (2015). Safety and tolerability of serelaxin, a recombinant human relaxin-2 in development for the treatment of acute heart failure, in healthy Japanese volunteers and a comparison of pharmacokinetics and pharmacodynamics in healthy Japanese and Caucasian populations. J. Clin. Pharmacol. 55, 415–422. doi: 10.1002/jcph.433
Danielson, L. A., and Conrad, K. P. (2003). Time course and dose response of relaxin-mediated renal vasodilation, hyperfiltration, and changes in plasma osmolality in conscious rats. J. Appl. Physiol. 95, 1509–1514. doi: 10.1152/japplphysiol.00545.2003
Danielson, L. A., Welford, A., and Harris, A. (2006). Relaxin improves renal function and histology in aging Munich Wistar rats. J. Am. Soc. Nephrol. 17, 1325–1333. doi: 10.1681/asn.2005121307
Debrah, D. O., Conrad, K. P., Danielson, L. A., and Shroff, S. G. (2005a). Effects of relaxin on systemic arterial hemodynamics and mechanical properties in conscious rats: sex dependency and dose response. J. Appl. Physiol. 98, 1013–1020. doi: 10.1152/japplphysiol.01083.2004
Debrah, D. O., Conrad, K. P., Jeyabalan, A., Danielson, L. A., and Shroff, S. G. (2005b). Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension 46, 745–750. doi: 10.1161/01.hyp.0000184230.52059.33
Dschietzig, T. B., Krause-Relle, K., Hennequin, M., von Websky, K., Rahnenfuhrer, J., Ruppert, J., et al. (2015). Relaxin-2 does not ameliorate nephropathy in an experimental model of Type-1 diabetes. Kidney Blood Press. Res. 40, 77–88. doi: 10.1159/000368484
Dschietzig, T., Teichman, S., Unemori, E., Wood, S., Boehmer, J., Richter, C., et al. (2009). Intravenous recombinant human relaxin in compensated heart failure: a safety, tolerability, and pharmacodynamic trial. J. Card. Fail. 15, 182–190. doi: 10.1016/j.cardfail.2009.01.008
Eitner, F., Ostendorf, T., Van Roeyen, C., Kitahara, M., Li, X., Aase, K., et al. (2002). Expression of a novel PDGF isoform, PDGF-C, in normal and diseased rat kidney. J. Am. Soc. Nephrol. 13, 910–917. doi: 10.1681/ASN.V134910
Fallowfield, J. A., Hayden, A. L., Snowdon, V. K., Aucott, R. L., Stutchfield, B. M., Mole, D. J., et al. (2014). Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology 59, 1492–1504. doi: 10.1002/hep.26627
Floege, J., and Johnson, R. J. (1995). Multiple roles for platelet-derived growth factor in renal disease. Miner. Electrolyte Metab. 21, 271–282.
Haberstroh, U., Zahner, G., Disser, M., Thaiss, F., Wolf, G., and Stahl, R. A. (1993). TGF-beta stimulates rat mesangial cell proliferation in culture: role of PDGF beta-receptor expression. Am. J. Phys. 264, F199–F205. doi: 10.1152/ajprenal.1993.264.2.F199
Heeg, M. H., Koziolek, M. J., Vasko, R., Schaefer, L., Sharma, K., Muller, G. A., et al. (2005). The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 68, 96–109. doi: 10.1111/j.1523-1755.2005.00384.x
Hewitson, T. D., Ho, W. Y., and Samuel, C. S. (2010). Antifibrotic properties of relaxin: in vivo mechanism of action in experimental renal tubulointerstitial fibrosis. Endocrinology 151, 4938–4948. doi: 10.1210/en.2010-0286
Huang, Z., Myhr, C., Bathgate, R. A., Ho, B. A., Bueno, A., Hu, X., et al. (2015). Activation of Relaxin family receptor 1 from different mammalian species by relaxin peptide and small-molecule agonist ML290. Front. Endocrinol. 6:128. doi: 10.3389/fendo.2015.00128
Huuskes, B. M., Wise, A. F., Cox, A. J., Lim, E. X., Payne, N. L., Kelly, D. J., et al. (2015). Combination therapy of mesenchymal stem cells and serelaxin effectively attenuates renal fibrosis in obstructive nephropathy. FASEB J. 29, 540–553. doi: 10.1096/fj.14-254789
Kaftanovskaya, E. M., Huang, Z., Lopez, C., Conrad, K., and Agoulnik, A. I. (2015). Conditional deletion of the relaxin receptor gene in cells of smooth muscle lineage affects lower reproductive tract in pregnant mice. Biol. Reprod. 92:91. doi: 10.1095/biolreprod.114.127209
Kaftanovskaya, E. M., Ng, H. H., Soula, M., Rivas, B., Myhr, C., Ho, B. A., et al. (2019). Therapeutic effects of a small molecule agonist of the relaxin receptor ML290 in liver fibrosis. FASEB J. 33, 12435–12446. doi: 10.1096/fj.201901046R
Kageyama, S., Nakamura, K., Fujii, T., Ke, B., Sosa, R. A., Reed, E. F., et al. (2018). Recombinant relaxin protects liver transplants from ischemia damage by hepatocyte glucocorticoid receptor: from bench-to-bedside. Hepatology 68, 258–273. doi: 10.1002/hep.29787
Kocan, M., Sarwar, M., Ang, S. Y., Xiao, J., Marugan, J. J., Hossain, M. A., et al. (2017). ML290 is a biased allosteric agonist at the relaxin receptor RXFP1. Sci. Rep. 7:2968. doi: 10.1038/s41598-017-02916-5
Lee, K. C., Hsieh, Y. C., Chan, C. C., Sun, H. J., Huang, Y. H., Hou, M. C., et al. (2019). Human relaxin-2 attenuates hepatic steatosis and fibrosis in mice with non-alcoholic fatty liver disease. Lab. Investig. 99, 1203–1216. doi: 10.1038/s41374-019-0240-y
Moore, X. L., Tan, S. L., Lo, C. Y., Fang, L., Su, Y. D., Gao, X. M., et al. (2007). Relaxin antagonizes hypertrophy and apoptosis in neonatal rat cardiomyocytes. Endocrinology 148, 1582–1589. doi: 10.1210/en.2006-1324
Ng, H. H., Esteban-Lopez, M., and Agoulnik, A. I. (2019a). Targeting the relaxin/insulin-like family peptide receptor 1 and 2 with small molecule compounds. Mol. Cell. Endocrinol. 487, 40–44. doi: 10.1016/j.mce.2018.12.013
Ng, H. H., Leo, C. H., Prakoso, D., Qin, C., Ritchie, R. H., and Parry, L. J. (2017). Serelaxin treatment reverses vascular dysfunction and left ventricular hypertrophy in a mouse model of type 1 diabetes. Sci. Rep. 7:39604. doi: 10.1038/srep39604
Ng, H. H., Shen, M., Samuel, C. S., Schlossmann, J., and Bennett, R. G. (2019b). Relaxin and extracellular matrix remodeling: mechanisms and signaling pathways. Mol. Cell. Endocrinol. 487, 59–65. doi: 10.1016/j.mce.2019.01.015
Pearson, G., Robinson, F., Beers Gibson, T., Xu, B. E., Karandikar, M., Berman, K., et al. (2001). Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22, 153–183. doi: 10.1210/edrv.22.2.0428
Samuel, C. S., Summers, R. J., and Hewitson, T. D. (2016). Antifibrotic actions of Serelaxin - new roles for an old player. Trends Pharmacol. Sci. 37, 485–497. doi: 10.1016/j.tips.2016.02.007
Samuel, C. S., Zhao, C., Bond, C. P., Hewitson, T. D., Amento, E. P., and Summers, R. J. (2004). Relaxin-1-deficient mice develop an age-related progression of renal fibrosis. Kidney Int. 65, 2054–2064. doi: 10.1111/j.1523-1755.2004.00628.x
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Smith, M. C., Murdoch, A. P., Danielson, L. A., Conrad, K. P., and Davison, J. M. (2006). Relaxin has a role in establishing a renal response in pregnancy. Fertil. Steril. 86, 253–255. doi: 10.1016/j.fertnstert.2005.11.070
Teerlink, J. R., Cotter, G., Davison, B. A., Felker, G. M., Filippatos, G., Greenberg, B. H., et al. (2013). Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet 381, 29–39. doi: 10.1016/s0140-6736(12)61855-8
Wang, D., Luo, Y., Myakala, K., Orlicky, D. J., Dobrinskikh, E., Wang, X., et al. (2017). Serelaxin improves cardiac and renal function in DOCA-salt hypertensive rats. Sci. Rep. 7:9793. doi: 10.1038/s41598-017-09470-0
Wilson, K. J., Xiao, J., Chen, C. Z., Huang, Z., Agoulnik, I. U., Ferrer, M., et al. (2018). Optimization of the first small-molecule relaxin/insulin-like family peptide receptor (RXFP1) agonists: activation results in an antifibrotic gene expression profile. Eur. J. Med. Chem. 156, 79–92. doi: 10.1016/j.ejmech.2018.06.008
Wong, S. E., Samuel, C. S., Kelly, D. J., Zhang, Y., Becker, G. J., and Hewitson, T. D. (2013). The anti-fibrotic hormone relaxin is not reno-protective, despite being active, in an experimental model of type 1 diabetes. Protein Pept. Lett. 20, 1029–1038. doi: 10.2174/0929866511320090009
Xiao, J., Huang, Z., Chen, C. Z., Agoulnik, I. U., Southall, N., Hu, X., et al. (2013). Identification and optimization of small-molecule agonists of the human relaxin hormone receptor RXFP1. Nat. Commun. 4:1953. doi: 10.1038/ncomms2953
Yang, X., Thorngren, D., Chen, Q., Wang, M., and Xie, X. (2019). Protective role of relaxin in a mouse model of aristolochic acid nephropathy. Biomed. Pharmacother. 115:108917. doi: 10.1016/j.biopha.2019.108917
Yoshida, T., Kumagai, H., Kohsaka, T., and Ikegaya, N. (2013). Relaxin protects against renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 305, F1169–F1176. doi: 10.1152/ajprenal.00654.2012
Yoshida, T., Kumagai, H., Kohsaka, T., and Ikegaya, N. (2014). Protective effects of relaxin against cisplatin-induced nephrotoxicity in rats. Nephron Exp. Nephrol. 128, 9–20. doi: 10.1159/000365852
Keywords: relaxin, relaxin family peptide receptor 1, apoptosis, extracellular matrix, unilateral ureteral obstruction
Citation: Ng HH, Soula M, Rivas B, Wilson KJ, Marugan JJ and Agoulnik AI (2021) Anti-apoptotic and Matrix Remodeling Actions of a Small Molecule Agonist of the Human Relaxin Receptor, ML290 in Mice With Unilateral Ureteral Obstruction. Front. Physiol. 12:650769. doi: 10.3389/fphys.2021.650769
Edited by:
Bellamkonda K. Kishore, University of Utah Health Care, United StatesReviewed by:
Mi Liu, Southern Medical University, ChinaCopyright © 2021 Ng, Soula, Rivas, Wilson, Marugan and Agoulnik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander I. Agoulnik, YWFnb3VsbmlAZml1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.