 Mi Rong Lee1
Mi Rong Lee1 Doo-Hyung Lee
Doo-Hyung Lee Jae Su Kim
Jae Su Kim
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 19 May 2021
Sec. Invertebrate Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.643389
This article is part of the Research TopicEntomopathogenic Fungi for the Control of Arthropod PestsView all 12 articles
The longhorned tick, Haemaphysalis longicornis (Acari: Ixodidae), is a hard tick and a vector for severe fever with thrombocytopenia syndrome (SFTS) virus. The number of patients infected with SFTS is rapidly increasing. Recently, the invertebrate pathogen Metarhizium anisopliae JEF-290 was reported to be useful to control the tick as an alternative to chemical acaricides, which are not easily applicable in human living areas where the tick is widely spread. In this study, we analyzed how the tick and the fungal pathogen interact at the transcriptional level. Field-collected tick nymphs were treated with JEF-290 conidia at 1 × 108 conidia/ml. In the early stage of infection with 2.5% mortality, the infected ticks were subjected to RNA sequencing, and non-infected ticks and fungal masses served as controls. Fungus and tick genes were mostly up-regulated at the early stage of infection. In the gene set enrichment analysis of the infecting fungus, catabolic processes that included lipids, phospholipids, and detoxification processes, the response to oxidative stress, and toxic substances were significantly up-regulated. In this fungal up-regulation, various lipase, antioxidant enzyme, and hydrolase genes were highly transcribed. The gene set enrichment analysis of the infected tick showed that many peptide synthesis processes including translation, peptide metabolism, ribonucleotide metabolism, and energy production processes that included ATP generation and ADP metabolism were significantly up-regulated. Structurally, mitochondria and ribosome subunit genes in ticks were highly transcribed to upregulate these processes. Together these results indicate that JEF-290 initiates process that infects the tick while the tick actively defends against the fungal attack. This work provides background to improve our understanding of the early stage of fungal infection in longhorned tick.
Haemaphysalis longicornis (Acari: Ixodidae), the longhorned tick is an important vector for human disease, and known to transmit severe fever with thrombocytopenia syndrome virus (SFTSV), Rickettsia japonica, and Coxiella burnetii (Mahara, 1997). SFTSV, which belongs to the Phlebovirus genus in the family Bunyaviridae, was first reported in China in 2009, and then spread throughout Asia with reports in North America also (Denic et al., 2011; McMullan et al., 2012). SFTSV causes vomiting, diarrhea, high fever, and thrombocytopenia, and the mortality ranges from 2% to as high as 30% (Gai et al., 2012). Previous studies confirmed that infection occurs person to person through blood contact or bodily secretions from infected patients (Liu et al., 2012). The longhorned tick is also an important pest for livestock such as deer, sheep, and cattle. In New Zealand and Australia, the tick can reduce cattle production by 25% (Heath, 2016). This tick also mediates cattle diseases from Theileria species, which are threats to the livestock industry (Hammer et al., 2015).
The longhorned tick is a three-host ixodid tick, which takes blood meals on three different hosts at each developmental stage until engorged. The engorged adult females lay more than 2,000 eggs in the late spring and early summer. The eggs hatch into six-legged larvae in late summer and early autumn. The ticks overwinter at the nymphal stage, and become active in the following spring. The ticks suck blood for 5–7 days, and they can survive for many month without blood meal at all stages (Heath, 2016). The longhorned tick consists of a diploid and triploid population which are bisexual and obligatory parthenogenetic, respectively, and an aneuploidy population is capable of both bisexual and parthenogenetic reproduction which make longhorned tick a very interesting case of tick cytogenetics and reproduction (Oliver, 1977).
Several management tools are available to control tick (Lee et al., 2015), but most of them are not environmentally sound or effective. Most of the commonly used pesticides against ticks include pyrethroid and carbamate. Pyrethroid insecticides have high insecticidal activity but have a negative impact on the environment. In addition, mite resistance to pyrethroid-based acaricides has been recently reported (Hernandez et al., 2018). Carbamate insecticides are also highly toxic to beneficial insects, such as bees, so their use is limited. ticks inhabit parks, mountains, and lakesides, and it is difficult to spray acaricides near areas of human habitation. For this reason, various methods using biological control agents have recently been studied. Cymbopogon citratus essential oil has a relatively high mortality rate in longhorned ticks (Agwunobi et al., 2020). Another report showed that the antlion Euroleon coreaus could become a new predator for the ticks (Nwanade et al., 2020). However, control methods using predators and plant extracts have disadvantages as they are difficult to apply in large areas and rarely show high control activity (George et al., 2004; Quadros et al., 2020).
Entomopathogenic fungi may be an alternative strategy to control ticks. This fungal group is a facultative pathogen to arthropods, causes pathogenicity specifically to insects, mites and ticks, and has been used to control various pests (Charnley and Collins, 2007; Fernandes et al., 2012). Although studies on entomopathogenic fungi–mediated control of tick are still lacking, the possible effects of entomopathogenic fungi on other ticks have been shown. Dermacentor variabilis, Ixodes scapularis, and Rhipicephalus sanguineus showed relatively high susceptibility when Beauveria bassiana or Metarhizium anisopliae was applied to target ticks (Kirkland et al., 2004). In addition, entomopathogenic fungi have high virulence against engorged females and tick eggs (Perinotto et al., 2012). Recently, studies have shown that selected B. bassiana strains show high virulence against longhorned tick, and confirmed the characteristics of the strains (Zhendong et al., 2019). However, the biological control of longhorned tick using entomopathogenic fungi is unknown.
A previous study of transcriptome analysis of small planthopper, Laodephax striatellus has provided the information about changes of gene expression pattern upon chemical pesticide-treatment, and demonstrated that suppression of the upregulated genes synergistically improved the insecticidal effect of the pesticide (Fang et al., 2020). In considering the use of microbials on targeting pests, it is also important to understand how the two organisms respond to each other at transcriptional level because it will provide information of pathogenicity and immunity related genes which might help study of putative virulence-modulating genes. Many studies have used transcriptome sequencing of ticks to determine their defense mechanisms when parasitize their hosts (Charrier et al., 2018; Araujo et al., 2019). In addition, tick-borne diseases have been studied at the transcriptional level. In a study in Australia, RNA sequencing was conducted to study the diseases caused by various ticks (Harvey et al., 2019). In Europe, various studies have been conducted to identify diseases that ticks can carry (Vayssier-Taussat et al., 2013; Pettersson et al., 2017). However, little research has been done on the defense mechanisms in ticks during fungal infection and the fungal mechanisms during tick infection.
In this study, the entire genome of JEF-290 was sequenced by PacBio sequencing technology, application of M. anisopliae JEF-290 for the control of longhorned tick was evaluated, and the interaction between tick and fungal pathogen at the early stage of infection was investigated by using transcriptome analyses. To compare the gene expression level of JEF-290 and longhorned tick under infection, differently expressed genes (DEGs), gene ontology (GO), and gene set enrichment analyses were conducted, and the results were analyzed to identify potentially important tick genes for defense and fungal genes for infection. This study will help clarify and modulate how the longhorned tick and JEF-290 respond to each other at the onset of fungal infection.
M. anisopliae JEF-290 was obtained from the Insect Microbiology and Biotechnology Laboratory (IMBL), Jeonbuk National University, South Korea. The fungal isolate was grown on quarter strength Sabouraud dextrose agar (1/4SDA; Difco, United States) in the dark at 25 ± 1°C and stored in 20% (v/v) glycerol at −80°C. This isolate has high acaricidal activity against the longhorned tick (Lee et al., 2019).
For whole genome sequencing of M. anisopliae JEF-290 at Macrogen1 (Macrogen Inc., Seoul, South Korea), genomic DNA and RNA were extracted from 7-day-old fungal mycelia. DNA quantity was assessed by using Victor 3 fluorometry (Perkin-Elmer) with Pico-green® fluorescent nucleic acid stain (Thermo Fisher Scientific). To assess DNA quality, gel electrophoresis was performed. The concentration of genomic DNA was measured using a Nano Drop spectrophotometer (Thermo Fisher Scientific) and a Qubit fluorometer (Life Technology). For PacBio RS sequencing, 8 μg of input genomic DNA was used for 20 kb library preparation. For gDNA with a size range less than 17 Kb, the Agilent 2100 Bioanalyzer system (Agilent Technologies, Palo Alto, Cambridge, United States) was used to determine the actual size distribution. Genomic DNA was sheared with g-TUBE (Covaris Inc., Woburn, MA, United States) and purified using AMPure PB magnetic beads (Beckman Coulter Inc., Brea, CA, United States). The gDNA concentration was measured using both a Nano Drop spectrophotometer and a Qubit fluorometer, and approximately 200 ng μl–1 of gDNA was run on a field-inversion gel. A total of 10 μl of library was prepared using the PacBio DNA Template Prep Kit 1.0 (for 3∼10 Kb). SMRT bell templates were annealed using the PacBio DNA/Polymerase Binding Kit P6. The PacBio DNA Sequencing Kit 4.0 and eight SMRT cells were used for sequencing. Subsequent steps were based on the PacBio Sample Net-Shared Protocol2. The genome sequences of entomopathogenic isolates such as M. anisopliae, M. rileyi, M. acridum, and M. robertsii were subjected to orthologous analysis. Orthologous and paralogous gene clustering analysis was performed using OrthoMCL (v.2.0.3). Data were analyzed in blastp (v2.2.25 +) (E-value 1e-5), and sequences with less than 10 amino acids or stop codon ratios exceeding 20% were excluded.
Wild populations of tick nymphs were collected several times from a grassland field near a rural community in Seongnam City, Korea using a carbon dioxide trapping method (Miles, 1968) from April until July in 2017 and 2018. Dry ice (2.5 kg) was placed into traps (36 × 40 cm) and traps were left in the grass field for 7 days. The collected ticks were identified based on a tick handling manual and training handbook (Yamaguti et al., 1971). Because the collected ticks consisted of approximately 95 percent of longhorned tick and less than five percent of H. flava, the larvae which are difficult to identify morphologically were removed from the collected colony, and only the nymphs were subjected to identification to collect ticks.
Seventy microliter of conidial suspension of M. anisopliae JEF-290 (1 × 108 conidia/ml) was spread on a 1/4SDA plate, and cultured at 27°C for 14 days to harvest conidia by vortexing from a sporulated agar disc (5 mm diameter) in 1.0 ml of 0.03% siloxane solution for 60 sec. The concentration of newly harvested conidia was adjusted to 1 × 108 conidia/ml. Approximately 180 nymphs were dipped into the conidial suspension for approximately 30 sec, placed on a filter paper-layered petri dish, and incubated at 27°C. The ticks were collected when the tick mortality rate reached 3.0%.
Longhorned tick RNA was extracted from the infected 180 nymphs, and same number of non-infected nymphs. On the nitrocellulose membrane liner covering 1/4SDA culture petri-dishes, JEF-290 was inoculated as described above. Five days after inoculation, actively proliferating mycelia and conidia were harvested for RNA extraction as controls. Total RNAs of these samples were extracted with TRIzol reagent (Invitrogen Life Technologies, CA, United States) following the manufacturer’s instructions. RNA purity and integrity were quantified by an ASP-2680 spectrophotometer (ACTGene, Piscataway, NJ, United States) and an Agilent 2100 Bionalyzer system.
Libraries of infected tick, non-infected tick and non-infecting fungus were made using the Truseq RNA kit (Illumina, San Diego, CA, United States) following the manufacturer’s protocol at Macrogen.
The first step was to purify mRNA containing poly-A using poly-T oligo-attached magnetic beads and fragment the samples into small pieces at elevated temperature with divalent cations. The cleaved RNA fragments were reverse-transcribed into first strand cDNA with random primers.
Ampure XP beads were used in the second strand reaction mix to generate double-stranded cDNA by removing the RNA template and synthesizing the replacement strand. The 3′ overhang was removed, and polymerase was used to fill the 5′ overhang using an End Repair (ERP) mix. The adapter was then ligated to the fragment by attaching the “A” nucleotide to the 3′ end of the blunt fragment and giving a core reaction single “T” nucleotide at the 3′ end of the adapter. Multiple indexing adapters were ligated to the ends of the double-stranded cDNA and then enriched by PCR to create DNA library templates. In each isolate, infecting and non-infecting samples were sequenced in parallel using an Illumina HiSeq 2000 sequencer with a read length of 101 bp. The read quality was verified by fastQC v.0.11.8 (Andrews, 2010) and the quality was filtered to remove low quality sequences with a Phred score of 30 or less using NGS QC Toolkit v.2.3.3 (Patel and Jain, 2012). For efficient and robust de novo reconstruction of transcriptomes, Trinity (ver 2.8.3) was used3. TransDecoder (version 5.5.0) was used to identify candidate coding regions within the transcription sequence. Contigs with more than 90% of sequence identities were clustered using cd-hit-est v.4.8.1 (Fu et al., 2012) to remove isoforms, and the in silico cDNA library was constructed.
To quantify transcript abundances, Kallisto (ver 0.45.0) was used to build an index form from the fasta form of target sequences and non-infecting and infecting libraries were compared. Transcripts per million (TPM) of non-infecting and infecting samples was calculated. Raw signals were normalized using a log2-based transformation. Fold-change statistical tests were performed and log2|FC| 2 was defined as statistically significant differential expression. Contigs were blasted using the Blast2Go program with a local blast. The statistical significance threshold was 1.0E-10 and the number of blast hits was set to one. GO analysis of up- and down-regulated contigs was performed using InterPro (online) in the Blast2Go program. The public EMBL-EBI database was used to scan sequences against InterPro’s signatures. Up- and down-regulated contigs were annotated at GO level 2. In addition, using the Blast2Go program, the tick contigs were mapped to the local immune database that based on ImmunoDB4 was prepared by downloading immune genes of Drosophila melanogaster from NCBI to analyze the up-regulation of immune-related contigs in the infected tick.
Six and five randomly selected genes of tick and M. anisopliae JEF-290 were validated using qRT-PCR, respectively. The RNA samples from infected tick and non-infected tick, and non-infecting JEF-290 were subjected to reverse transcription (RT) using an AccuPower® RT PreMix (Bioneer, Daejeon, South Korea) with oligo (dT) 15 primer (Promega, MI, United States). The qRT-PCR primers were designed at SnapDragon5. qRT-PCR was performed using Thunderbird® SYBR® qPCR mix (QPS-201, TOYOBO, Japan) in a 96-well Bio-Rad CFX96 Real-Time PCR System (Bio-Rad, United States). PCR conditions were as follows: denaturation for 1 min at 95°C, and then 40 cycles of 15 s at 95°C, 1 min at 60°C followed by melting, increased 0.5°C per 5 s started from 65 to 95°C. The total RNA without reverse transcription was used as a negative control, and different actin genes from tick and JEF-290 were independently used as an internal control to normalize relative expression level. In this validation, other housekeeping genes were tried but they worked well, so actin genes were finally used. All experiments were performed in triplicate. △Ct (threshold cycle) was calculated as (Ct value of up-regulated genes)–(Ct value of actin) and subjected to the calculation of fold change value (2−ΔΔCt).
DEG contigs of infecting M. anisopliae JEF-290 and infected longhorned tick with more than twofold change difference were subjected to GO enrichment analysis to identify the significantly involved functional gene groups in infection and defense. Blastn was performed with an E-value of 1.0E-10 and non-blasted and non-GO ID contigs were removed. As a reference, Ixodes scapularis and Beauveria bassiana were used. Functional genes set enrichment was performed using the g:Profiler web server6. The related p-value was corrected for multiple testing using the Benjamini-Hochberg False Discovery Rate (FDR) procedure with a threshold of 0.05.
As a result of whole genome sequencing of M. anisopliae JEF-290, a total of 11,868,389,082 bases were identified with N50 of 26,851 bp (Supplementary Table 1). The number of reads was 746,811 and the mean subread length was 15.9 Kb. A total of 18 contigs were assembled and the whole genome size was analyzed to be 42.85 Mb with 13,634 coding genes, which was larger than other Metarhizium species (Table 1). Comparison of the orthologs of the six different genus of entomopathogenic fungi revealed that all strains shared 5,320 genes, and JEF-290, M. robertstii, M. riley, and M. acridum shared 698 genes (Supplementary Figure 1).
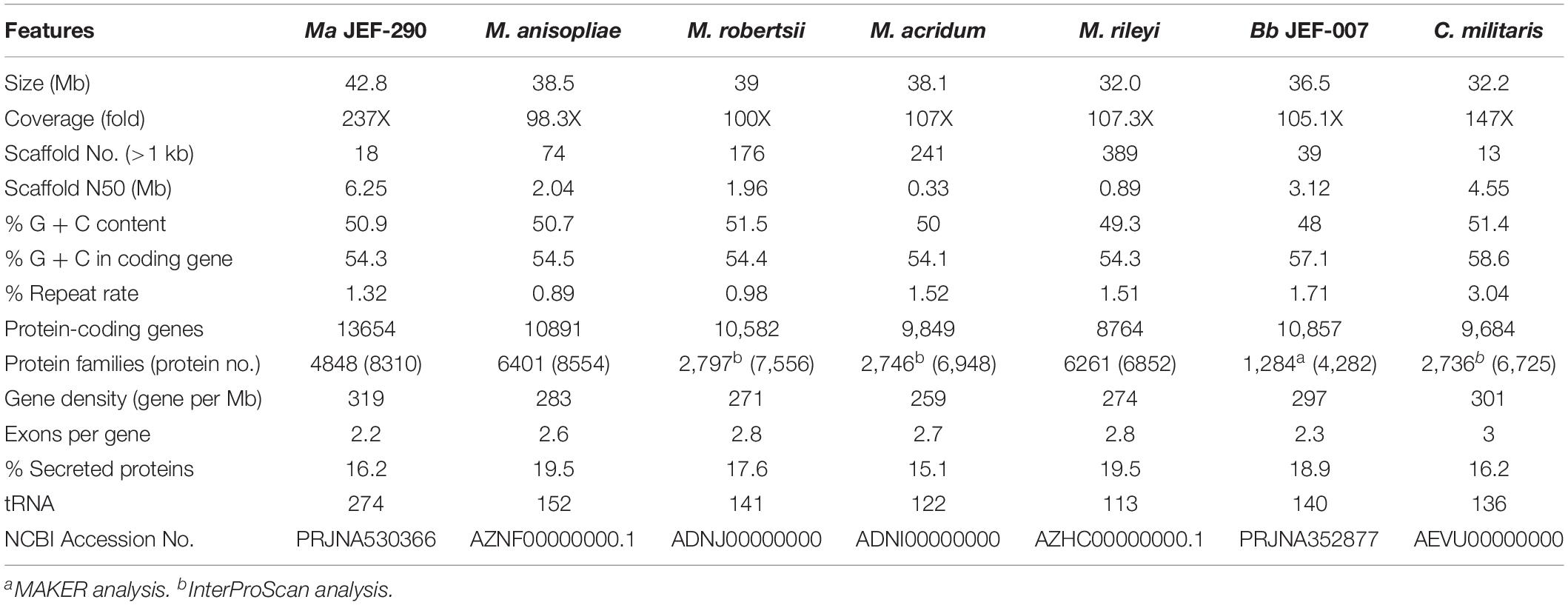
Table 1. Genome features of M. anisopliae JEF-290 and other entomopathogenic fungi.
Under laboratory conditions, application of M. anisopliae JEF-290 to nymphs resulted in high control efficacy and mycosis (Figure 1). The survival rate of nymphs in the non-treated control was 90.0% at 15 days post-application, but JEF-290 treatment resulted in approximately 66.7% mortality at 15 days and >90% of mortality at 30 days [F(2, 143) = 241.9, p < 0.001]. Fungal mycosis was observed on the cadavers of infected nymphs.
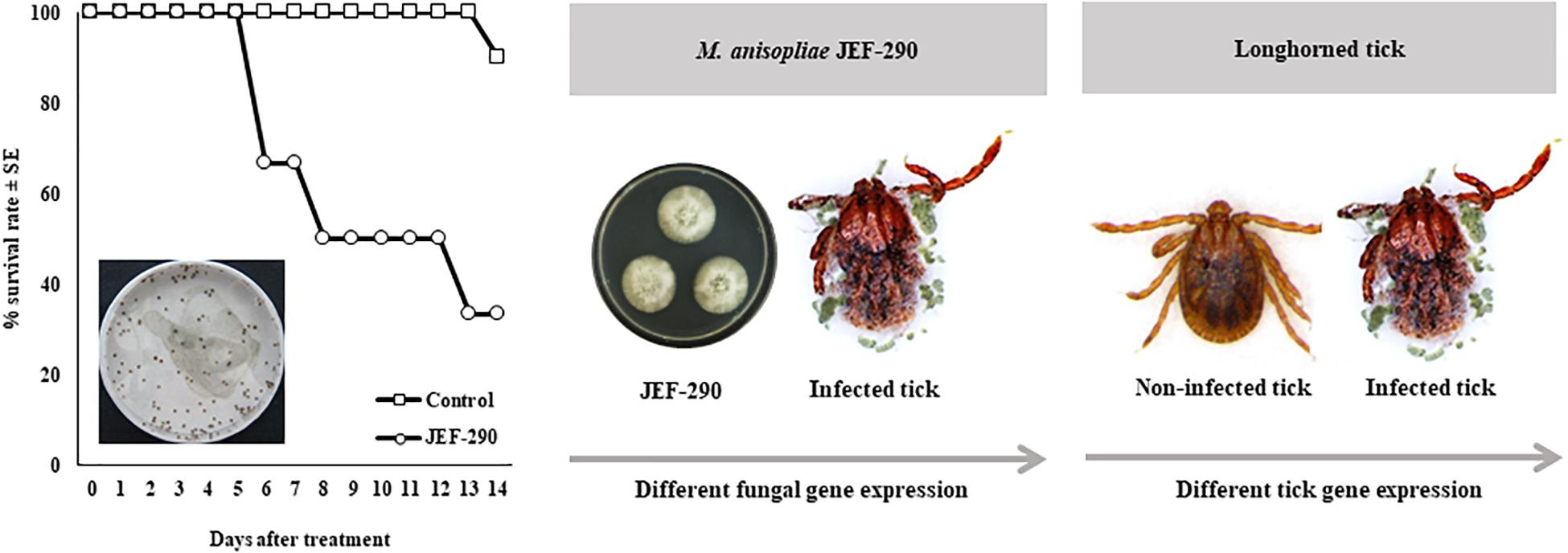
Figure 1. Virulence of M. anisopliae JEF-290 against nymphs of longhorn ticks in laboratory conditions.
From analyzing the RNA sequences, 8.767 Gb (longhorned tick), 8.549 Gb (JEF-290) and 11.224 Gb (infected tick) of raw sequences were identified (Supplementary Table 2). Filtering and de novo assembly were performed based on the raw data above to obtain a sequence of 27.2, 16.5, and 28.9 Mb, respectively. The assembled contigs of longhorned tick, JEF-290 and infected tick were clustered with more than 90% identity using the CD-hit-est program, and the numbers of clustered contigs were 33,099 (longhorned tick), 15,044 (JEF-290), and 36,292 (infected tick), respectively. The N50 values of the samples were 1101 b (longhorned tick), 1554 b (JEF-290), and 1056 b (infected tick). In the DEG analysis, relatively large numbers of tick and fungal genes were up-regulated when the tick was infected by the fungus (Figure 2). Among the tick genes, 1216 genes were up-regulated and 165 genes were down-regulated (|FC| > 2). Among the fungal genes, 137 genes were up-regulated and 11 genes were down-regulated (|FC| > 2) (Supplementary Table 3). qRT-PCR was performed on the randomly selected genes (Supplementary Table 4), and the results confirmed that gene expression levels determined by qRT-PCR and RNA-seq analysis results were similar to each other although limited numbers of internal control genes were used (Supplementary Figure 2).
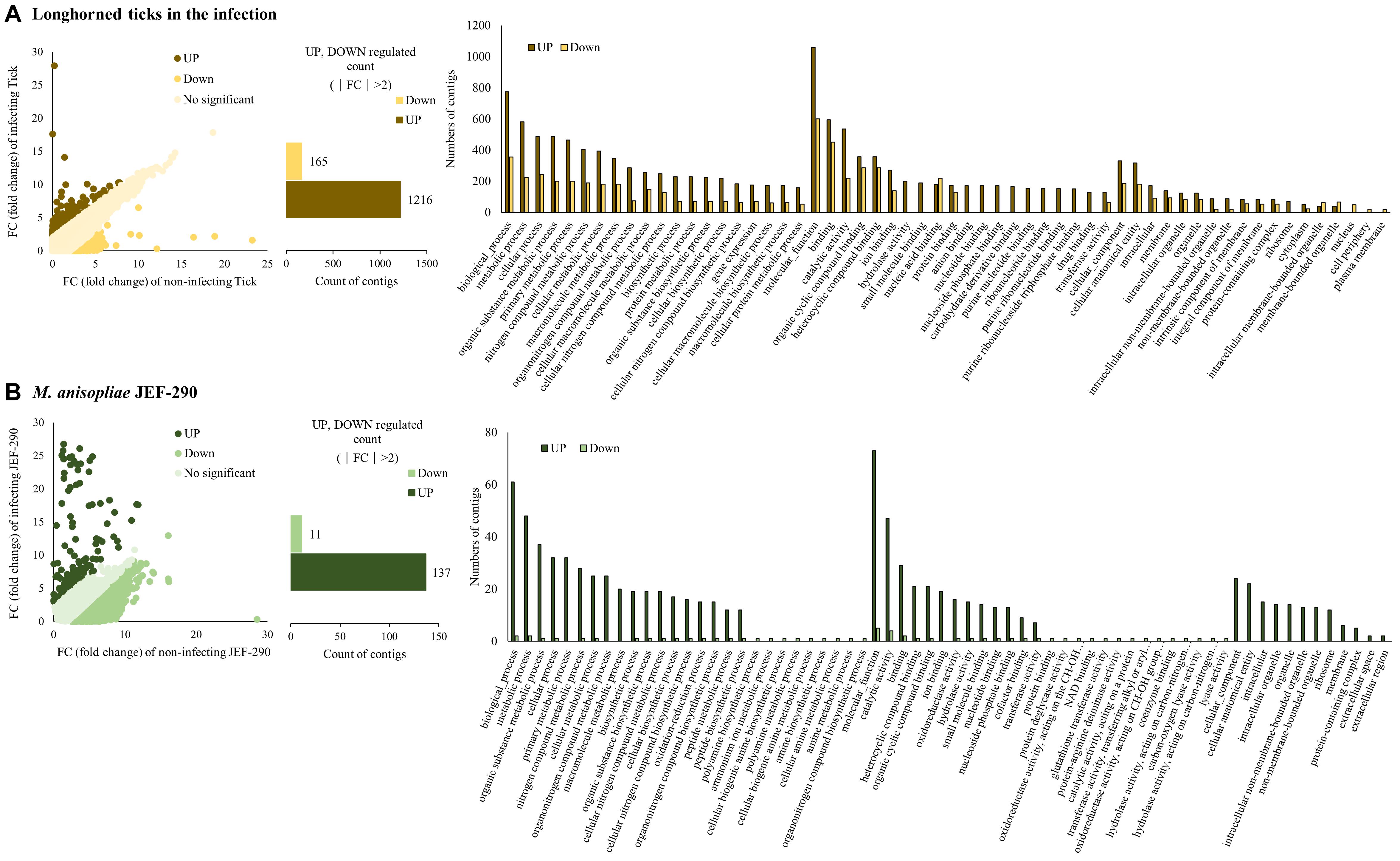
Figure 2. Differentially expressed genes (DEG) of (A) longhorned tick and (B) M. anisopliae JEF-290 at early stage of infection. Longhorned ticks were exposed to 14 days old fungus cultured plates for 14 days. As a non-infecting control, 5 days cultured fungus was used for RNA extraction. In each isolate, the numbers of up- and down-regulated contigs (|fold change| > 2) were analyzed and GO analyses of DEGs were conducted using Blast2Go program.
In the GO analysis of the infected tick, fungal GO terms were almost up-regulated, but GO terms of longhorned tick were mostly up-regulated with partial down-regulation (Figure 2). In the GO of longhorned tick genes, 1,381 DEGs were classified in the following three GO terms: biological process, cellular components, and molecular function (Figure 2A). Many longhorned tick contigs belonging to biological process and molecular function showed a trend that more of these contigs were up-regulated while the contigs belonging cellular component showed relatively less changes of gene expression.
The up-regulated longhorned tick GO terms (FC > 2) included 1,216 genes that included biological process (54%), molecular function (36%), and cellular component (11%), and these GO terms were mainly metabolism, catalytic activity, and bindings. The contigs annotated as being involved in metabolic process (GO:0008152), cellular process (GO:0009987), and organic substance metabolic process (GO:0071704), primary metabolic process (GO:0044238), catalytic activity (GO:0003824), hydrolase activity (GO:0016787), small molecule binding (GO:0036094), anion binding (GO:0043168), and nucleotide binding (GO:0000166) were upregulated noteworthily. The down-regulated longhorned tick GO terms (FC < 2) included 165 genes that included biological process (59%), molecular function (29%), and cellular component (11%). The contigs annotated as being involved in organic cyclic compound binding (GO:0097159), heteroclyclic compound binding (GO:1901363), nucleic acid binding (GO:0003676), and protein binding (GO:0005515) were down-regulated noteworthily.
In the GO of M. anisopliae JEF-290, 295 DEGs were categorized as three GO types: biological process, cellular component, and molecular function (Figure 2B). The up-regulated fungal GO terms (FC > 2) included 137 genes that included biological process (53%), molecular function (36%) and cellular component (12%). The up-regulated GO terms were mainly metabolic process and binding. The contigs annotated as being involved in organic substance metabolic process (GO:0071704), nitrogen compound metabolic process (GO:0006807), macromolecule metabolic process (GO:0043170), cellular metabolic process (GO:0044237), biosynthetic process (GO:0009058), oxidoreductase activity (GO:0016491), hydrolase activity (GO:0016787), transferase activity (GO:0016740), cellular anatomical entity (GO:0110165), and intracellular organelle (GO:0043229) were up-regulated noteworthily. The down-regulated fungal GO terms (FC < 2) included 11 genes that included biological process (43%) and molecular function (57%).
Mapping of the longhorned tick contigs to a local immune database from ImmunoDB revealed that three serpin genes were up-regulated at the early stage of fungal infection (Table 2). Other immune-related genes did not show any significant up- and down-regulations. From the mapping, two uncharacterized genes (Infect_tick_13855 and Infect_tick_25741) and three serpin genes (Infect_tick_1165, Infect_tick_1329 and Infect_tick_13414) were upregulated when the tick was infected by M. anisopliae JEF-290. Other immune-related genes were mapped to the database, but most of the genes were not strongly affected by the fungal infection (|FC| < 1): D. melanogaster cactus, pelle and caspar genes, and peroxidasin, thioredoxin peroxidase, and other serpin genes. Serpin 28DC, 55B, and 43Ab genes were upregulated, but the other mapped serpin genes (serpin 27A, 28F, 28B, 42Dc, 42De, 43Aa, 43Ad, 77Ba, and 100A) were not significantly changed.
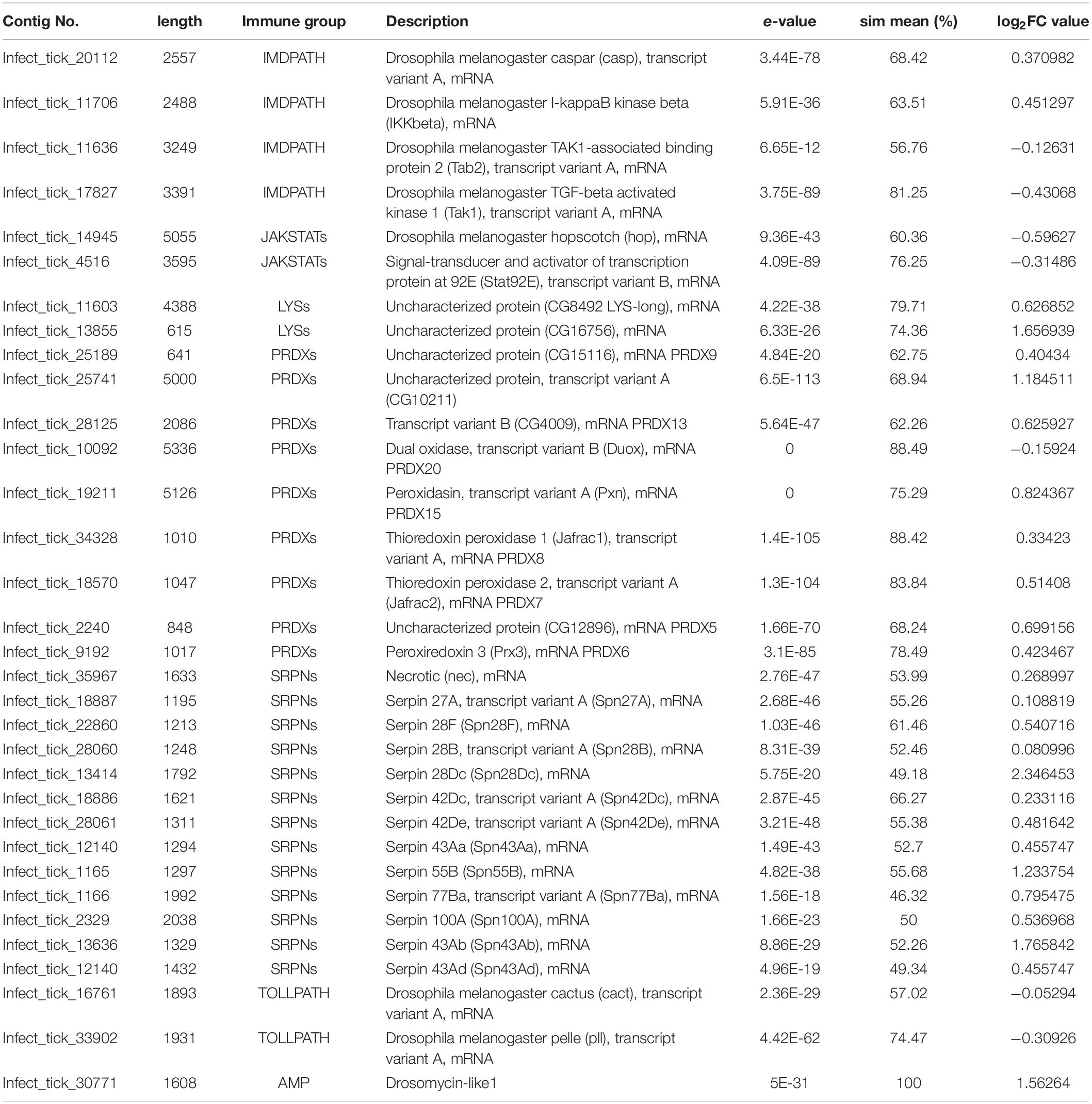
Table 2. Immune-related genes of infected longhorned tick at early stage of infection.
Gene set enrichment analysis of the DEGs showed that the GO terms related to peptide synthesis processes and cellular energy production of longhorned tick were over-represented while those of catabolic processes were over-represented in M. anisopliae JEF-290 was working on catabolic processes to degrade metabolites (Figure 3). In the infected tick, seven tick pathways and 30 tick GO terms were significantly involved in the fungal infection (Figure 3A). The major up-regulated GO terms were peptide biosynthetic process (GO:0043043), translation (GO:0006412), amide biosynthetic processes (GO:0043604), and peptide metabolic processes (GO:0006518). Up-regulated longhorned tick genes were related to the following pathways: 18 genes were related to carbon metabolism (KEGG:01200), nine genes to glycolysis/gluconeogenesis (KEGG:00010), 14 genes to ribosomes (KEGG:03010), nine genes to biosynthesis of amino acids (KEGG:01230), seven genes to pyruvate metabolism (KEGG:00620), six genes to the citrate cycle (KEGG:00020), and senven genes to glyoxylate/dicarboxylate metabolism (KEGG:00630). In the enriched pathways of infected tick, the RNA-binding translation regulator IRP (FC = 6.0) and enolase (FC = 5.) were highly up-regulated in the TCA cycle and glycolysis pathways, respectively (Figure 4). The infected tick also significantly upregulated peptide production and energy production pathways. The 26 GO terms in JEF-290 were significantly involved in the fungal infection (Figure 3B). The major up-regulated GO terms were lipid catabolic processes, phospholipid catabolic processes, cellular oxidant detoxification, cellular detoxification, and response to oxidative stress. JEF-290 significantly up-regulated lipid degradation and detoxification against the host defense (Figure 3B).
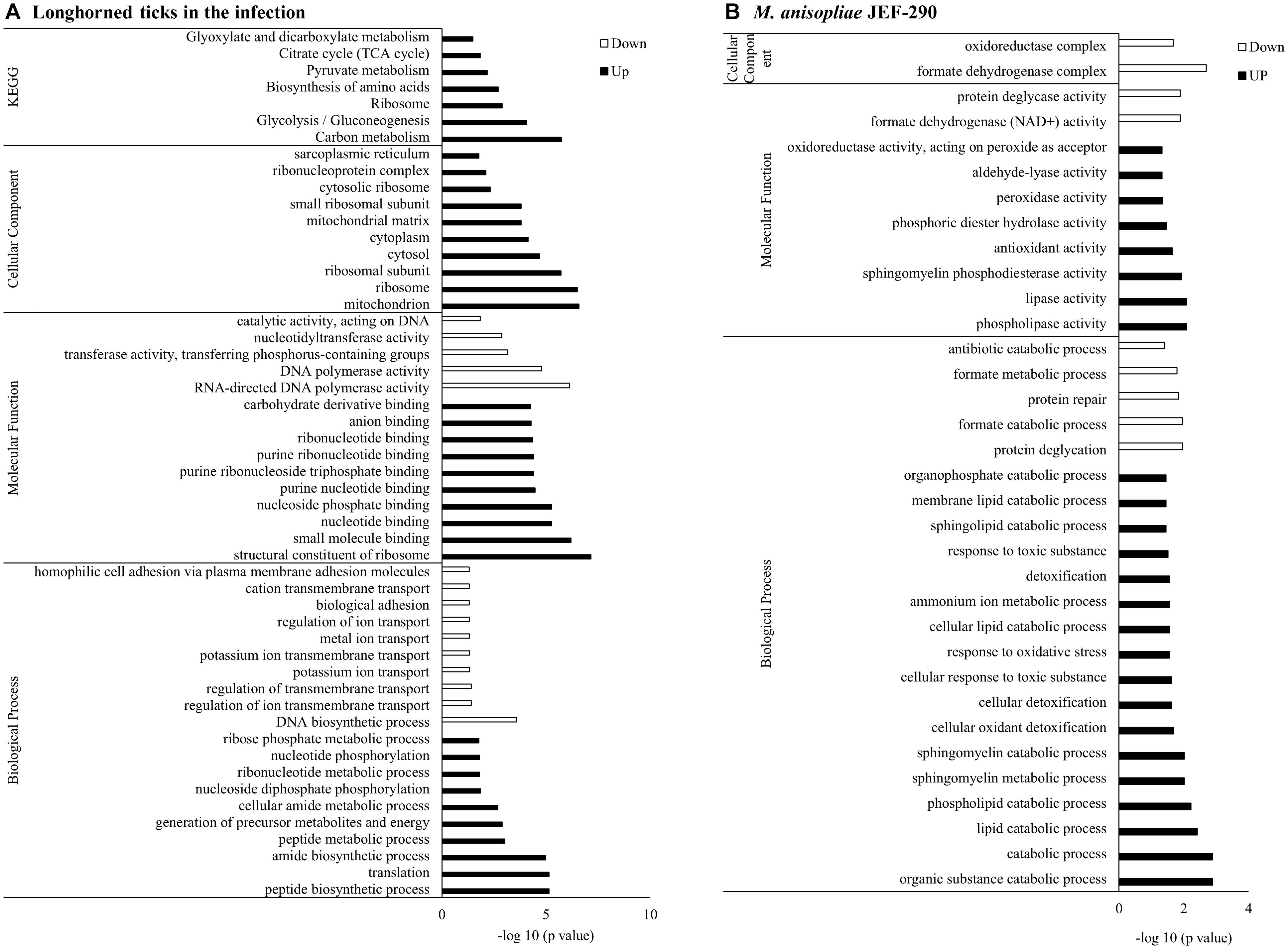
Figure 3. GO enrichment of longhorned tick (A) and M. anisopliae JEF-290 (B) DEGs at early stage of infection. Longhorned tick in the infection and M. anisopliae JEF-290 that showed more than ± 2 fold change difference were subjected to GO enrichment analysis using g:Profiler to investigate significant functional differences when infecting ticks and M. anisopliae JEF-290. The M. anisopliae does not provide enough gene IDs for enrichment analysis, so alternatively Beauveria bassiana with much larger gene IDs was used as a reference and the p-value was corrected for multiple testing using the FDR procedure with a threshold of 0.05.
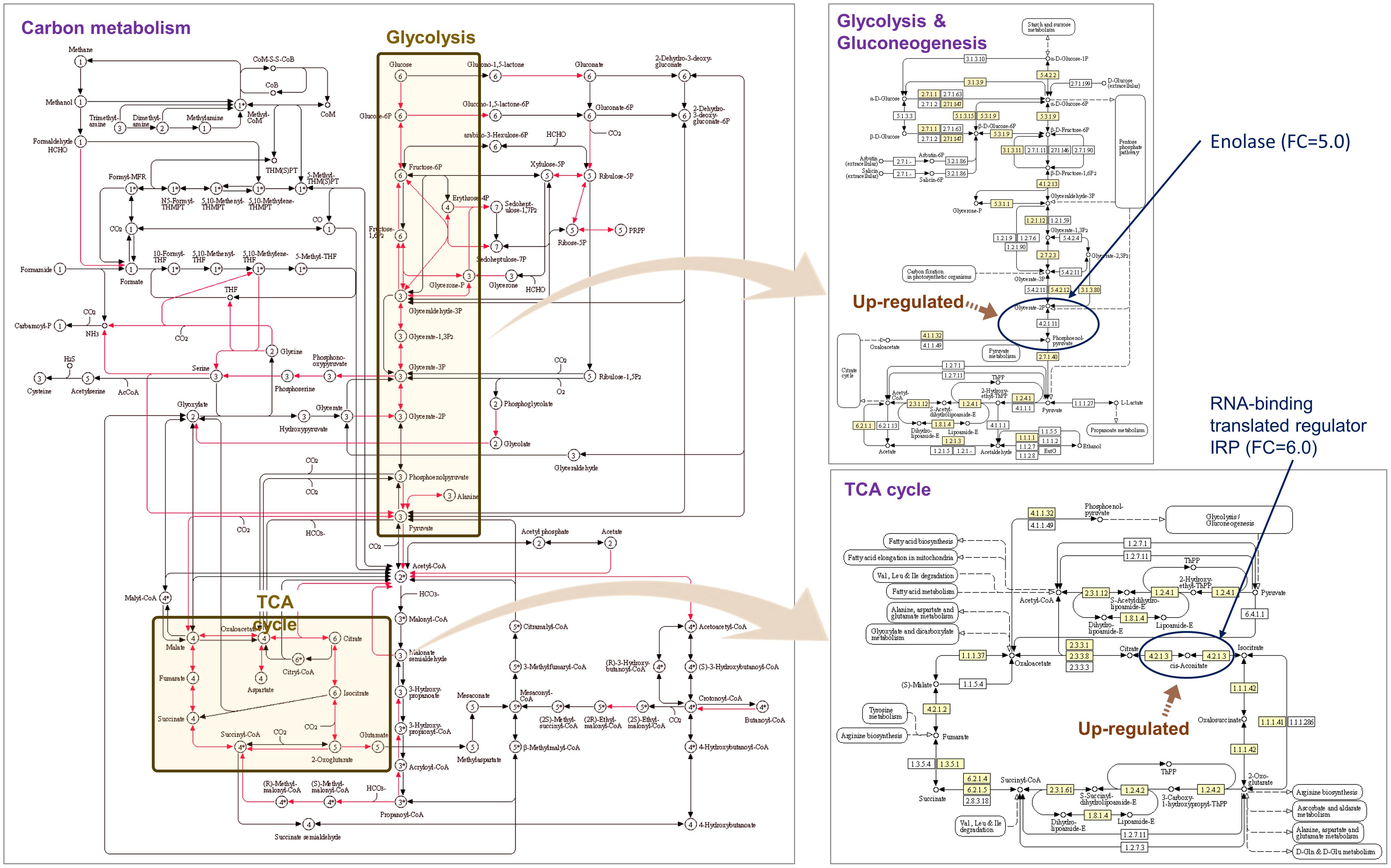
Figure 4. Enriched pathways of infected longhorned tick. KEGG pathway were summarized. Yellow box parts of the KEGG pathway can be involved with Glycolysis and Gluconeogenesis and TCA cycle (figure from KEGG database).
In this RNA-sequencing study, the whole genome of M. anisopliae was very useful for fungal gene characterization. The genome size of M. anisopliae JEF-290 is 42.8 Mb, which is larger than those of other Metarhizium species (32∼39 Mb). JEF-290 was found to contain 99 specific genes those were not found in the other analyzed species. Many JEF-290 genes were shared with other Metarhizium species and other genus of entomopathogenic fungi, however, this isolate has some unique genes which were found to be involved in a variety of fungal life activities. Alpha-beta hydrolase is a structure-related enzyme with various catalytic functions (Holmquist, 2000). In general, the alpha-beta hydrolase enzyme is involved in the breakdown of cellular metabolites (Hotelier et al., 2004). The unique ankyrin repeat protein is involved in protein-protein interactions. It exists in most organisms and is involved in cell cycle control, transcriptional regulation, cell signaling, development, differentiation, apoptosis, cellular scaffolding, and plant defense (Li et al., 2006). The ubiquitin-protein ligase in JEF-290 is actively involved in the protein conversion mechanisms that play an important role in the regulation of various cellular functions. Ubiquitin-protein ligase is also involved in activating toll-like receptors (Chuang and Ulevitch, 2004). The ribonuclease H-like (RNHL) superfamily group is involved in nucleic acid-related biological processes including replication, homologous recombination, DNA repair, transposition, and RNA interference (Majorek et al., 2014). Transposase-like proteins are generally known as mobile genetic units and have the potential to be mobilized by stress.
In the early infection stage, JEF-290 possibly initiates infection steps while the longhorned tick recognizes the fungal infection, and is attempting to defend against infection. However, once the infected hosts died, the fungus becomes saprophytic. Comparative transcriptome analysis of the early stage of fungal infection may provide more informative interactions between the tick and entomopathogenic fungus, including fungal initiation of infection and host response to the fungal infection. On the other hand, it would not be possible to observe much of an interactive response of ticks against fungal infection in the late infection stage showing more than 90% of host mortality, therefore RNA-sequencing would not provide meaningful information of the interaction.
In the analysis of longhorned tick and JEF-290 transcripts, many up-regulated contigs were found in both pathogen and host at the early infection stage. Our results showed that the longhorned tick recognizes JEF-290 infection, and increases the expression of defense and immune related contigs. However, the contigs which are supposed to transcribe very short mRNAs, such as antimicrobial peptide genes were not be able to be analyzed properly because the standard sequencing library preparation protocol does not target such short mRNAs. Therefore the library preparation protocol need to be modified for further study of low molecules. Additionally, cloning and sequencing of the transcripts is indispensable for reliable validation of Illumina sequencing results and further functional study using qPCR.
From the mapping of longhorned tick contigs to the immunoDB, three serpin genes were identified as up-regulated. Serpin is a well-known family of serine protease inhibitors that are typically around 45 kDa. Serpin regulates the arthropod immune reaction through the inhibition of serine protease reactions that initiate the melanization cascade and antimicrobial peptide production (Reichhart et al., 2011; Meekins et al., 2016), therefore the insects, ticks, and mites have serpin genes in their genomes (Mulenga et al., 2009; Rodriguez-Valle et al., 2015; Rider et al., 2015). Forty five serpin genes were found in the blacklegged tick Ixodes scapularis, 22 in the cattle tick Rhipicephalus microplus, and only 10 in the scabies mite Sarcoptes scabiei. Some immune-responsive serpins function at the early stage of microbial infection, and are highly up-regulated in the response to microbial infection or physical injury (Meekins et al., 2016). Several studies on arthropod serpins have been reported, but still an interactive response between arthropods and entomopathogenic fungi did not fully explain the up- and down-regulation of serpin gene-expression.
In the gene set enrichment analysis of longhorned tick, pathways related to protein synthesis and energy production such as translation, peptide metabolism, ribonucleotide metabolism, energy production process including ATP generation, and ADP metabolism were up-regulated. These results suggest that the longhorned tick actively secreted protective substances against the invasion of JEF-290. Of the several pathways obtained from the enrichment analysis of longhorned tick, the RNA-binding translational regulator IRP gene was significantly involved in the management of fungal challenge in most of the significant pathways. When the longhorned tick was infected by the fungus, this gene was highly up-regulated (FC = 6.0). RNA binding proteins (RBP) regulate the translation of mRNA by interacting with 5’ and 3’ untranslated regions (UTR) of mRNA (Harvey et al., 2018). RBP is involved in the assembly of mRNA to the ribosomes and regulates protein synthesis or alternatively suppresses translation (Moore and Lindern, 2018). Among the RBPs, iron regulatory protein (IRP) binds to RNA stem-loops of UTR to manipulate mRNA translation (Zhang et al., 2002) and maintains homeostasis of iron concentrations in mammalians including in response to blood-sucking ticks (Anderson et al., 2012). IRP regulates not only iron homeostasis-related mRNAs, but also other mRNAs such as mitochondrial acotinase (ACO2) mRNA, which has an iron responsive element (IRE) and encodes a protein involved in the tricarboxylic acid cycle (TCA) catalyzing citrate to isocitrate (Wang and Pantopoulos, 2011). Acotinase has a 4Fe-4S iron-sulfur cluster as an IRE that directly interacts with the IRP. We speculate that when longhorned tick is infected by JEF-290, IRP possibly binds to the IRE of acotinase mRNA of tick for gene regulation, however, clear evidence remains to be demonstrated.
In the gene set enrichment analysis of JEF-290, fungal pathways such as catabolic processes including lipid, phospholipid, sphingomycelin, and sphingolipid pathways, and detoxification processes including response to oxidative stress and toxic substances were to be up-regulated. We speculate that JEF-290 easily degrades insect cuticles during penetration of the host insect body. In addition, degrading host defense substances possibly makes the fungal invasion much easier. In entomopathogenic fungi, the cytochrome P450 (CYP450) gene encodes a protein that is involved in the degradation of lipid and wax layers of arthropods (Shin et al., 2020). In RNA-sequencing of western flower thrips infected with B. bassiana ERL836 or JEF-007, two different CYP genes (CYP539B1 and CYP655C1) were identified in the infecting fungal isolates (Kim et al., 2020). CYP52 and CYP53 degrade insect lipid layers and assimilate cuticular hydrocarbons (Huarte-Bonnet et al., 2018). In fungal detoxification, most substrate-degrading fungi contain a detoxification system, named the xenomic network, which includes the cytochrome P450 (CYP450) family (Phase I) and glutathione transferase (GST) family (Phase II) (Schrand et al., 2010; Meux et al., 2011). These proteins are involved in the modification of exogenous compounds through oxidative reaction and conjugation, respectively. As discussed above, entomopathogenic fungi produce various CYPs, and they are involved in both fatty acid degradation and fungal detoxification against the arthropod immune response perhaps to work as a cuticle penetrator and defender against arthropods. More studies need to be conducted to explore this potential mechanism.
In summary, genes from the longhorned tick and JEF-290 fungus were mostly up-regulated at the early stage of fungal infection. Our results indicated that JEF-290 initiates infection of the longhorned tick by degrading host cuticles with a detoxification tool while the longhorned tick was actively defending against the fungal attack by producing a large amount of energy by inducing catabolism processes (Figure 5). These findings may provide a strong background to understand the early stage of fungal infection of the longhorned tick. Comparative transcriptome analysis needs to be combined with genetic variation study for better understanding of the tick and fungus interaction, and searching for virulence-related marker genes of both pathogen and host.
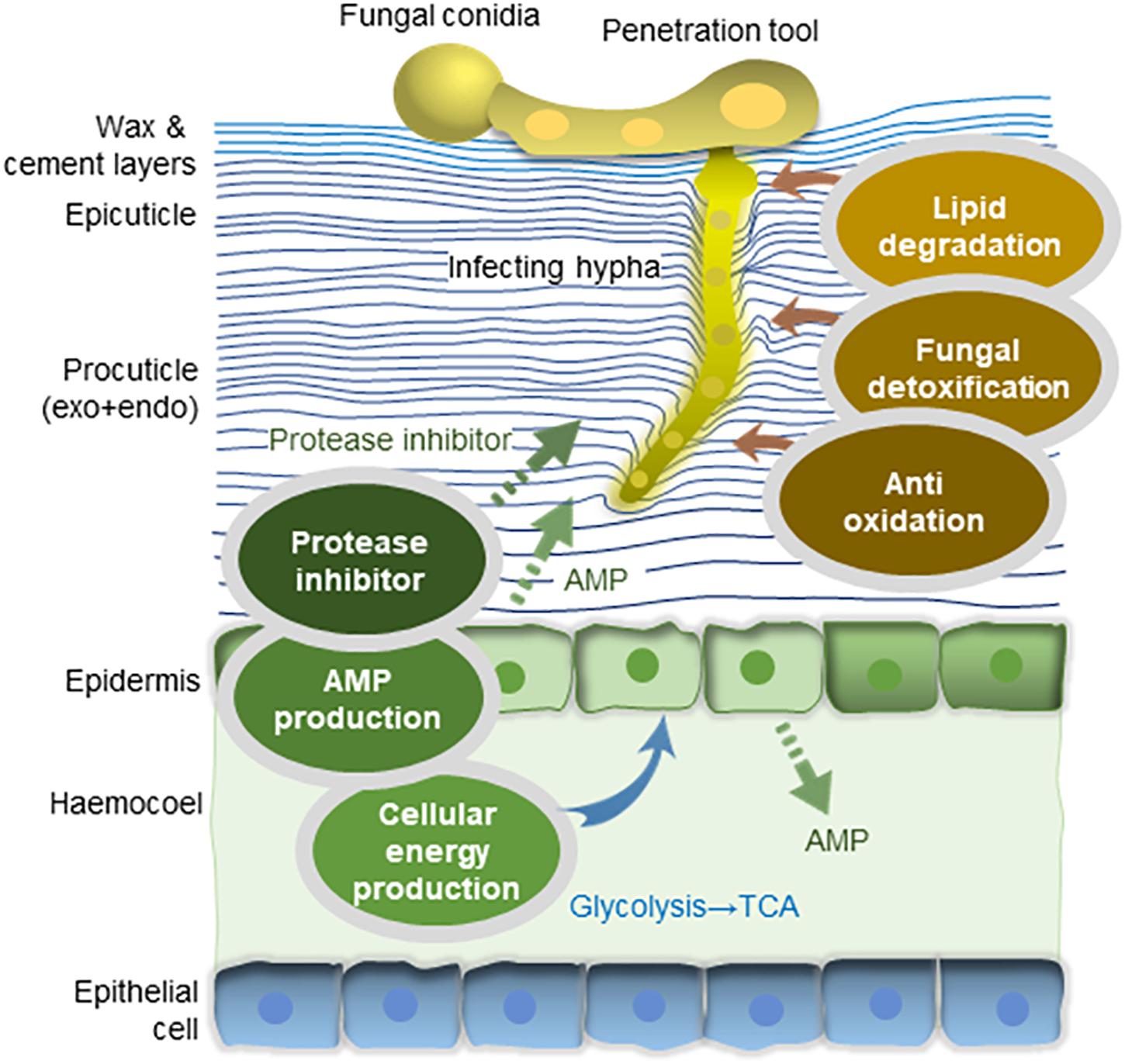
Figure 5. Interaction of M. anisopliae JEF-290 and longhorned tick at early stage of fungal infection. Genes from the longhorned tick and JEF-290 fungus were mostly up-regulated at the early stage of fungal infection. Our results indicate that JEF-290 initiates infection of the longhorned tick by degrading host cuticles with a detoxification tool, while the longhorned tick was actively defending against the fungal attack by producing a large amount of energy by inducing catabolism processes (original illustration).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
ML designed this work and analyzed RNA-sequencing raw data. JK and WK analyzed the RNA-sequencing data. ML and SP extracted DNA and RNA from the samples. D-HL collected longhorned tick from fields and identified. JK designed the whole experiments and wrote the manuscript. All authors approved the submission of this manuscript.
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant No. 2020R1I1A3057522) and by funds (HD17A0031) from Research of Korea Centers for Disease Control and Prevention.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We appreciate many students at Gacheon University for supporting tick collections in field areas and Bruce L. Parker and Cheryl Frank Sullivan (University of Vermont, United States) for comments about biological control of ticks.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.643389/full#supplementary-material
Supplementary Figure 1 | Reciprocal analysis of M. anisopliae JEF-290 and other entomopathogenic fungi.
Supplementary Figure 2 | Validation of RNA-sequencing data using qRT-PCR.
Supplementary Table 1 | Raw data and assembly of M. anisopliae JEF-290 contigs for whole genome prediction. The sequencing of whole genomes using Pac Bio RSII technology with error correction.
Supplementary Table 2 | Summary of the longhorned tick, M. anisopliae JEF-290 and infected longhorned tick in silico cDNA library.
Supplementary Table 3 | TPM values and fold changes of JEF-290 and longhorned tick contigs.
Supplementary Table 4 | Primers used in qRT-PCR for validation of RNA-sequencing.
Agwunobi, D. O., Pei, T., Yang, J., Wang, X., Lv, L., Shen, R., et al. (2020). Expression profiles of glutathione S-transferases genes in semi-engorged Haemaphysalis longicornis (Acari: Ixodidae) exposed to Cymbopogon citratus essential oil. Syst. Appl. Acarol. 25, 918–930. doi: 10.11158/saa.25.5.12
Anderson, C. P., Shen, M., Eisenstein, R. S., and Leibold, E. A. (2012). Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 1823, 1468–1483. doi: 10.1016/j.bbamcr.2012.05.010
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Araujo, R. N., Silva, N. C., Mendes-Sousa, A., Paim, R., Costa, G. C., Dias, L. R., et al. (2019). RNA-seq analysis of the salivary glands and midgut of the Argasid tick Ornithodoros rostratus. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-42899-z
Charnley, A. K., and Collins, S. A. (2007). 10 Entomopathogenic Fungi and Their Role in Pest Control. Environ. Microb. Relationsh. 4:159. doi: 10.1007/978-3-540-71840-6_10
Charrier, N. P., Couton, M., Voordouw, M. J., Rais, O., Durand-Hermouet, A., Hervet, C., et al. (2018). Whole body transcriptomes and new insights into the biology of the tick Ixodes ricinus. Parasit. Vectors 11:364. doi: 10.1186/s13071-018-2932-3
Chuang, T. H., and Ulevitch, R. J. (2004). Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat. Immunol. 5, 495–502. doi: 10.1038/ni1066
Denic, S., Janbeih, J., Nair, S., Conca, W., Tariq, W. U., and Al-Salam, S. (2011). Acute thrombocytopenia, leucopenia, and multiorgan dysfunction: the first case of SFTS Bunyavirus outside China? Case Rep. Infect. Dis. 2011:204056. doi: 10.1155/2011/204056
Fang, Y., Park, M. G., Choi, J. Y., Park, D. H., Wang, M., Kim, H. J., et al. (2020). Insecticidal and synergistic activity of dsRNAs targeting buprofezin−specific genes against the small brown planthopper, Laodelphax striatellus. Arch. Insect Biochem. Physiol. 105:e21739. doi: 10.1002/arch.21739
Fernandes, ÉK., Bittencourt, V. R., and Roberts, D. W. (2012). Perspectives on the potential of entomopathogenic fungi in biological control of ticks. Exp. Parasitol. 130, 300–305. doi: 10.1016/j.exppara.2011.11.004
Fu, L., Niu, B., Zhu, Z., and Wu S. Li, W. (2012). CD-HIT: accelerated for clustering the next generation sequencing data. Bioinformatics 28:3150. doi: 10.1093/bioinformatics/bts565
Gai, Z. T., Zhang, Y., Liang, M. F., Jin, C., Zhang, S., Zhu, C. B., et al. (2012). Clinical progress and risk factors for death in severe fever with thrombocytopenia syndrome patients. J. Infect. Dis. 206, 1095–1102. doi: 10.1093/infdis/jis472
George, J. E., Pound, J. M., and Davey, R. B. (2004). Chemical control of ticks on cattle and the resistance of these parasites to acaricides. Parasitology 129, S353–S366. doi: 10.1017/S0031182003004682
Hammer, J. F., Emery, D., Bogema, D. R., and Jenkins, C. (2015). Detection of Theileria orientalis genotypes in Haemaphysalis longicornis ticks from southern Australia. Parasit. Vectors 8:229. doi: 10.1186/s13071-015-0839-9
Harvey, E., Rose, K., Eden, J. S., Lo, N., Abeyasuriya, T., Shi, M., et al. (2019). Extensive diversity of RNA viruses in Australian ticks. J. Virol. 93:18. doi: 10.1128/JVI.01358-18
Harvey, R. F., Smith, T. S., Mulroney, T., Queiroz, R. M. L., Pizzinga, M., Dezi, V., et al. (2018). Trans-acting translational regulatory RNA binding proteins. Wiley Interdiscip. Rev. RNA 9:e1465. doi: 10.1002/wrna.1465
Heath, A. (2016). Biology, ecology and distribution of the tick, Haemaphysalis longicornis Neumann (Acari: Ixodidae) in New Zealand. N Z. Vet. J. 64, 10–20. doi: 10.1080/00480169.2015.1035769
Hernandez, E. P., Kusakisako, K., Talactac, M. R., Galay, R. L., Hatta, T., Fujisaki, K., et al. (2018). Glutathione S-transferases play a role in the detoxification of flumethrin and chlorpyrifos in Haemaphysalis longicornis. Parasit. Vectors 11, 1–14. doi: 10.1186/s13071-018-3044-9
Holmquist, M. (2000). Alpha/beta-hydrolase fold enzymes structures, functions and mechanisms. Curr. Protein Pept. Sci. 1, 209–235. doi: 10.2174/1389203003381405
Hotelier, T., Renault, L., Cousin, X., Negre, V., Marchot, P., and Chatonnet, A. (2004). ESTHER, the database of the α/β−hydrolase fold superfamily of proteins. Nucleic Acids Res. 32, 145–147. doi: 10.1093/nar/gkh141
Huarte-Bonnet, C., Kumar, S., Saparrat, M. C., Girotti, J. R., Santana, M., Hallsworth, J. E., et al. (2018). Insights into hydrocarbon assimilation by Eurotialean and Hypocrealean Fungi: roles for CYP52 and CYP53 clans of cytochrome P450 genes. Appl. Biochem. Biotechnol. 184, 1047–1060. doi: 10.1007/s12010-017-2608-z
Kim, S., Kim, J. C., Lee, S. J., Lee, M. R., Park, S. E., Li, D., et al. (2020). Beauveria bassiana ERL836 and JEF-007 with similar virulence show different gene expression when interacting with cuticles of western flower thrips, Frankniella occidentalis. BMC Genomics 21:1–12. doi: 10.1186/s12864-020-07253-y
Kirkland, B. H., Westwood, G. S., and Keyhani, N. O. (2004). Pathogenicity of entomopathogenic fungi Beauveria bassiana and Metarhizium anisopliae to Ixodidae tick species Dermacentor variabilis, Rhipicephalus sanguineus, and Ixodes scapularis. J. Med. Entomol. 41, 705–711. doi: 10.1603/0022-2585-41.4.705
Lee, D. W., Chang, K. S., Kim, M. J., Ahn, Y. J., Jo, H. C., and Kim, S. I. (2015). Acaricidal activity of commercialized insecticides against Haemaphysalis longicornis (Acari: Ixodidae) nymphs. J. Asia Pacific Entomol. 18, 715–718. doi: 10.1016/j.aspen.2015.09.004
Lee, M. R., Li, D., Lee, S. J., Kim, J. C., Kim, S., Park, S. E., et al. (2019). Use of Metarhizum aniopliae s.l. to control soil-dwelling longhorned tick, Haemaphysalis longicornis. J. Invertebr. Pathol. 166:107230. doi: 10.1016/j.jip.2019.107230
Li, J., Mahajan, A., and Tsai, M. D. (2006). Ankyrin repeat: a unique motif mediating protein- protein interactions. Biochemistry 45, 15168–15178. doi: 10.1021/bi062188q
Liu, Y., Li, Q., Hu, W., Wu, J., Wang, Y., Mei, L., et al. (2012). Person-to-person transmission of severe fever with thrombocytopenia syndrome virus. Vector Borne Zoonotic Dis. 12, 156–160. doi: 10.1089/vbz.2011.0758
Mahara, F. (1997). Japanese spotted fever: report of 31 cases and review of the literature. Emerg. Infect. Dis. 3, 105–111. doi: 10.3201/eid0302.970203
Majorek, K. A., Dunin-Horkawicz, S., Steczkiewicz, K., Muszewska, A., Nowotny, M., Ginalski, K., et al. (2014). The RNase H-like superfamily: new members, comparative structural analysis and evolutionary classification. Nucleic Acids Res. 42, 4160–4179. doi: 10.1093/nar/gkt1414
McMullan, L. K., Folk, S. M., Kelly, A. J., MacNeil, A., Goldsmith, C. S., Metcalfe, M. G., et al. (2012). A new phlebovirus associated with severe febrile illness in Missouri. N. Engl. J. Med. 367, 834–841. doi: 10.1056/NEJMoa1203378
Meekins, D. A., Kanost, M. R., and Michel, K. (2016). Serpins in arthropod biology. Semin. Cell Dev. Biol. 62, 105–119. doi: 10.1016/j.semcdb.2016.09.001
Meux, E., Prosper, P., Ngadin, A., Didierjean, C., Morel, M., Dumarçay, S., et al. (2011). Glutathione Transferases of Phanerochaete chrysosporium S-glutathionyl-p-hydroquinone reductase belongs to a new structural class. J. Biol. Chem. 286, 9162–9173. doi: 10.1074/jbc.M110.194548
Miles, V. I. (1968). A carbon dioxide bait trap for collecting ticks and fleas from animal burrows. J. Med. Entomol. 5, 491–495. doi: 10.1093/jmedent/5.4.491
Moore, K., and Lindern, M. V. (2018). RNA Binding Proteins and Regulation of mRNA Translation in Erythropoiesis. Front. Physiol. 9:910. doi: 10.3389/fphys.2018.00910
Mulenga, A., Khumthong, R., and Chalaire, K. C. (2009). Ixodes scapularis tick serine proteinase inhibitor (serpin) gene family; annotation and transcriptional analysis. BMC Genomics 10:217. doi: 10.1186/1471-2164-10-217
Nwanade, C. F., Yang, X., Bao, R., Yu, Z., and Liu, J. (2020). A case of rapid predation of the tick Haemaphysalis longicornis (Acari: Ixodidae) by antlion Euroleon coreaus. Int. J. Acarol. 46, 1–2. doi: 10.1080/01647954.2020.1765868
Oliver, J. H. Jr. (1977). Cytogenetics of mites and ticks. Annu. Rev. Entomol. 22, 407–429. doi: 10.1146/annurev.en.22.010177.002203
Patel, R. K., and Jain, M. (2012). NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Perinotto, W. M. S., Angelo, I. C., Golo, P. S., Quinelato, S., Camargo, M. G., Sá, F. A., et al. (2012). Susceptibility of different populations of ticks to entomopathogenic fungi. Exp. Parasitol. 130, 257–260. doi: 10.1016/j.exppara.2011.12.003
Pettersson, J. H. O., Shi, M., Bohlin, J., Eldholm, V., Brynildsrud, O. B., Paulsen, K. M., et al. (2017). Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 7, 1–7. doi: 10.1038/s41598-017-11439-y
Quadros, D. G., Johnson, T. L., Whitney, T. R., Oliver, J. D., and Oliva Chávez, A. S. (2020). Plant-Derived Natural Compounds for Tick Pest Control in Livestock and Wildlife: Pragmatism or Utopia? Insects 11:490. doi: 10.3390/insects11080490
Reichhart, J. M., Gubb, D., and Leclerc, V. (2011). The Drosophila serpins: multiple functions in immunity and morphogenesis. Methods Enzymol. 499, 205–225. doi: 10.1016/B978-0-12-386471-0.00011-0
Rider, S. D. Jr., Morgan, M. S., and Arlian, L. G. (2015). Draft genome of the scabies mite. Parasit. Vectors 8, 1–14. doi: 10.1186/s13071-015-1198-2
Rodriguez-Valle, M., Xu, T., Kurscheid, S., and Lew-Tabor, A. E. (2015). Rhipicephalus microplus serine protease inhibitor family: annotation, expression and functional characterisation assessment. Parasit. Vectors 8, 1–9. doi: 10.1186/s13071-014-0605-4
Schrand, A. M., Rahman, M. F., Hussain, S. M., Schlager, J. J., Smith, D. A., and Syed, A. F. (2010). Metal−based nanoparticles and their toxicity assessment. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2, 544–568. doi: 10.1002/wnan.103
Shin, T. Y., Lee, M. R., Park, S. E., Lee, S. J., Kim, W. J., and Kim, J. S. (2020). Pathogenesis−related genes of entomopathogenic fungi. Arch. Insect Biochem. Physiol. 105:e21747. doi: 10.1002/arch.21747
Vayssier-Taussat, M., Moutailler, S., Michelet, L., Devillers, E., Bonnet, S., Cheval, J., et al. (2013). Next generation sequencing uncovers unexpected bacterial pathogens in ticks in western Europe. PLoS One 8:e81439. doi: 10.1371/journal.pone.0081439
Wang, J., and Pantopoulos, K. (2011). Regulation of cellular iron metabolism. Biochem. J. 434, 365–381. doi: 10.1042/BJ20101825
Yamaguti, N., Tipton, V. J., Keegan, H. L., and Toshioka, S. (1971). Ticks of Japan, Korea, and the Ryukyu islands. Biol. Ser. 15, 8–1971.
Zhang, D., Dimopoulos, G., Wolf, A., Minana, B., Kafatos, F. C., and Winzerling, J. J. (2002). Cloning and molecular characterization of two mosquito iron regulatory proteins. Insect. Biochem. Mol. Biol. 32, 579–589. doi: 10.1016/s0965-1748(01)00138-2
Keywords: Metarhizium anisopliae, Haemaphysalis longicornis, transcription, catabolic process, peptides synthesis
Citation: Lee MR, Kim JC, Park SE, Lee SJ, Kim WJ, Lee D-H and Kim JS (2021) Interactive Gene Expression Between Metarhizium anisopliae JEF-290 and Longhorned Tick Haemaphysalis longicornis at Early Stage of Infection. Front. Physiol. 12:643389. doi: 10.3389/fphys.2021.643389
Received: 18 December 2020; Accepted: 22 April 2021;
Published: 19 May 2021.
Edited by:
Isabele da Costa Angelo, Universidade Federal Rural do Rio de Janeiro, BrazilReviewed by:
Krishnendu Mukherjee, University Hospital Münster, GermanyCopyright © 2021 Lee, Kim, Park, Lee, Kim, Lee and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jae Su Kim, anNraW0xMEBqYm51LmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.