 Qianghua Xia
Qianghua Xia Xubo Huang
Xubo Huang Jieru Huang
Jieru Huang Yongfeng Zheng
Yongfeng Zheng Michael E. March
Michael E. March Jin Li
Jin Li Yongjie Wei
Yongjie Wei
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 25 March 2021
Sec. Striated Muscle Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.638983
This article is part of the Research Topic Autophagy and Mitophagy in Skeletal Muscle Health and Disease View all 6 articles
Skeletal muscle is the most abundant type of tissue in human body, being involved in diverse activities and maintaining a finely tuned metabolic balance. Autophagy, characterized by the autophagosome–lysosome system with the involvement of evolutionarily conserved autophagy-related genes, is an important catabolic process and plays an essential role in energy generation and consumption, as well as substance turnover processes in skeletal muscles. Autophagy in skeletal muscles is finely tuned under the tight regulation of diverse signaling pathways, and the autophagy pathway has cross-talk with other pathways to form feedback loops under physiological conditions and metabolic stress. Altered autophagy activity characterized by either increased formation of autophagosomes or inhibition of lysosome-autophagosome fusion can lead to pathological cascades, and mutations in autophagy genes and deregulation of autophagy pathways have been identified as one of the major causes for a variety of skeleton muscle disorders. The advancement of multi-omics techniques enables further understanding of the molecular and biochemical mechanisms underlying the role of autophagy in skeletal muscle disorders, which may yield novel therapeutic targets for these disorders.
Skeletal muscles, a type of highly organized and specialized tissue in vertebrates, make up about 40% of total body mass and play a central role in diverse activities, such as locomotion, macromolecule turnover and storage, energy metabolism, and oxygen consumption (Tortora and Anagnostakos, 1987; Frontera and Ochala, 2015). Under starvation conditions, skeletal muscles serve as a significant internal source of nutrients, energy, and cellular building blocks. Additionally, the different types of activities that skeletal muscles are involved in, especially prolonged or high-intensity exercises, generate reactive oxygen species (ROS), which results in the damage of macromolecules such as nucleic acids, proteins, lipids, and cellular components (Neel et al., 2013). Molecular signaling pathways and cellular processes in skeletal muscles are shaped to efficiently cope with such cellular damage as well as other types of injury.
The autophagy signaling pathway is essential for energy generation/consumption and macromolecule turnover processes in skeletal muscles. Abnormal autophagy in muscles results in cellular alterations such as mitochondrial damage, endoplasmic reticulum stress, impaired sarcomeric-protein turnover, and cell death (Bonaldo and Sandri, 2013), leading to the development of various types of skeletal muscle disease. The critical roles of autophagy in skeletal muscles have been gaining more attention over the past two decades. In this review, we discuss the physiological function of autophagy in maintaining cellular homeostasis of skeletal muscle and the role of autophagy in muscle disorders.
The term “autophagy,” which means “self-eating,” is a highly conserved process from yeast to plants and animals (Ohsumi, 2014; Levine and Kroemer, 2019). Genetic studies in yeast have identified essential Autophagy-related genes (ATG) (Tsukada and Ohsumi, 1993; Thumm et al., 1994; Harding et al., 1995; Titorenko et al., 1995; Yuan et al., 1997; Sakai et al., 1998; Mukaiyama et al., 2002; Parzych et al., 2018). A total of 42 ATG genes have been identified (Klionsky et al., 2016; Parzych et al., 2018; Delorme-Axford and Klionsky, 2019); they are highly conserved from yeast to human. Studies using reverse genetic tools have further illustrated the physiological and pathophysiological roles of autophagy in multiple cellular events in higher organisms, including human cells. The autophagy pathway comprises a series of highly organized sequential steps responsible for recruiting and degrading misfolded proteins and the recycling of the decomposition products.
A series of dynamic membrane events contribute to autophagosome formation, involving several steps of morphological change in the cell (Parzych and Klionsky, 2014). The sequential steps of autophagy involve the participation and interaction of the ATG proteins Figure 1. The ULK/Atg1 complex is responsible for the initiation of autophagosome formation. This complex consists of five members in yeast (Atg1, Atg13, Atg17, Atg29, and Atg31) and four members in mammals (ULK1/2, ATG13, FIP200/RB1CC1, and ATG101) (Papinski and Kraft, 2016). ULK1/2 (Atg1 in yeast) is the only core protein with serine/threonine kinase activity in the autophagy signaling pathway. Autophagy signaling is mediated by activation of the ULK/Atg1 complex prior to autophagosome assembly. The ULK/Atg1 complex acts as a bridge between the upstream nutrient or energy integrator mTOR and the downstream ATG proteins involved in autophagosome formation, phosphorylating a variety of downstream proteins. It is believed that downstream ATG proteins are not necessary for membrane recruitment of ULK/Atg1 complex at the initiation stage (Suzuki et al., 2007; Koyama-Honda et al., 2013).
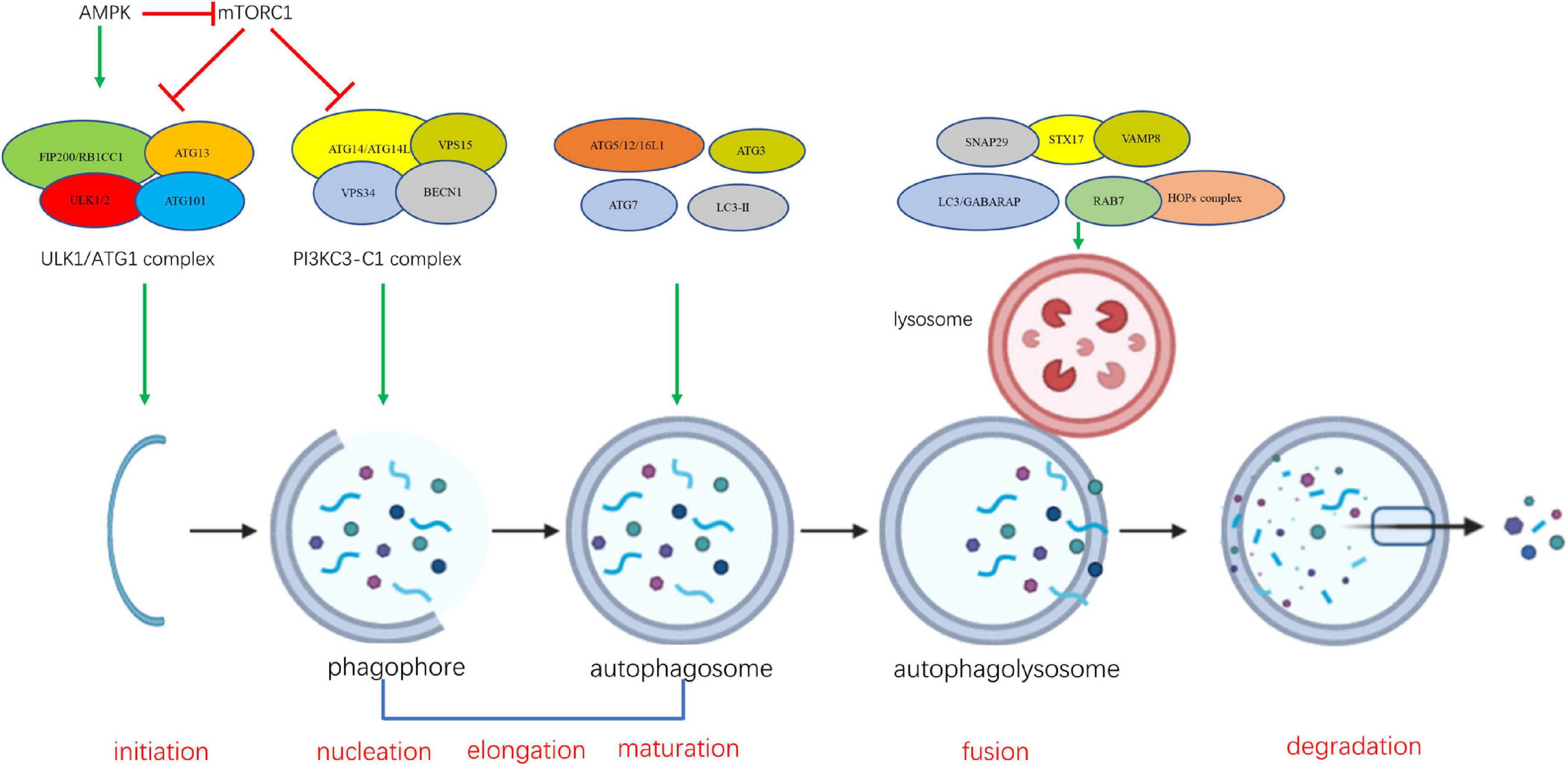
Figure 1. The sequential steps of autophagy involve the participation and interaction of the ATG proteins.
The formation of the class III phosphatidylinositol-3 kinase complexes I (PI3KC3-C1) is an essential event at the nucleation stage, which follows the formation of ULK/Atg1 complex (Kihara et al., 2001; Itakura et al., 2008). Vesicle nucleation leads to formation of the isolation phagophore (Levine and Kroemer, 2008). PI3KC3-C1 is composed of four components including the catalytic subunit PIK3C3 (also known as VPS34), BECLIN 1 (mammalian homolog of yeast Atg6), PIK3R4 (phosphoinositide-3-kinase regulatory subunit 4, also known as VSP15), and Atg14/ATG14L (Autophagy-Related Protein 14-Like Protein) (Kihara et al., 2001; Itakura et al., 2008; Matsunaga et al., 2009; Levine and Kroemer, 2019). During the initiation of autophagy, PI3KC3-C1 is activated and recruited to sites of phagophore nucleation on the endoplasmic reticulum (ER) and mitochondria to convert phosphatidylinositol (PI) into phosphatidylinositol 3-phosphate (PI3P) (Funderburk et al., 2010; Fan et al., 2011; Hamasaki et al., 2013). Although a number of studies have demonstrated that Atg14-containing complex PI3KC3-C1 is involved in the formation of autophagosome, several reports suggested that UVRAG-PI3KC3-C2 complex is critical for Vps34 function on endolysosomal and autophagolysosomal maturation (Itakura and Mizushima, 2009; Matsunaga et al., 2009; Nezis et al., 2014; Levine and Kroemer, 2019). PI3KC3-C2 shares PI3KC3, BECLIN1, and PIK3R4 with PI3KC3-C1, but contains a UVRAG (UV radiation resistance associated gene protein) instead of ATG14L (Itakura et al., 2008).
Vesicle nucleation is followed by the elongation and expansion of phagophore in the cytoplasm. The phagophore becomes a cup-shaped double membrane structure and begins to surround cytoplasmic material (Lamb et al., 2013). Two ubiquitin-like proteins, autophagy-related 12 (ATG12) and microtubule-associated protein 1 light chain 3 alpha/beta (MAP1LC3A/MAP1LC3B, LC3), a human homolog of yeast Atg8 (Ohsumi and Mizushima, 2004; Nakatogawa, 2013, 2014), play essential roles in the elongation and expansion of phagocytic membranes. ATG12 is conjugated to autophagy-related 5 (ATG5), mediated by E1 ubiquitin ligase-like conjugating enzyme autophagy-related 7 (ATG7) and E2 ubiquitin ligase like conjugating enzyme autophagy-related 10 (ATG10), and then the interacts with autophagy-related 16 (ATG16) non-covalently (Romanov et al., 2012; Walczak and Martens, 2013). LC3 is cleaved by the autophagy-related 4 (ATG4) cysteine peptidase at the C-terminal end to produce cytoplasmic LC3-I, which is also linked to phosphatidylthanolamine (PE) in a ubiquitin-like reaction that requires ATG7 and autophagy-related 3 (ATG3) to form LC3-phosphatidylethanolamine conjugate (LC3-II). LC3-II, in this lipid form, is integrated into the autophagosomal membrane and has been regarded as an autophagosomal marker (Fujita et al., 2008; Burman and Ktistakis, 2010).
Phagophore closure during the maturation stage results in sequestration of cytoplasmic component and formation of the autophagosome. As maturation proceeds, the autophagosome fuses with endosomes and vacuoles (in yeast and plant) or lysosomes (in metazoan cells), forming the autophagolysosome and leading to the degradation of the inner membrane and its contents. When the degradation is complete, the autophagolysosome becomes a residual body. The resulting breakdown products such as amino acids and fatty acids are sent back to the cytoplasm and are reused for cellular metabolism, providing an internal source of energy generation and building blocks for catabolism (Ohsumi, 2014; Sakakibara et al., 2015). The underlying mechanism by which autophagosome fusion is regulated is not clear. Recent studies suggest that Atg8 proteins may act as a master controller for the final fusion stages of autophagy (Nguyen et al., 2016; Yu and Melia, 2017; Kriegenburg et al., 2018). Several other proteins, such as SNAREs (syntaxin 17, SNAP29 and VAMP8), and tethering factors (HOPS complex, and the Rab GTPase, RAB7) are required for the fusion of the autophagosomal membrane with the lysosome (Itakura et al., 2012; Balderhaar and Ungermann, 2013; Schaaf et al., 2016; D’Agostino et al., 2017).
In skeletal muscles, autophagy is under the tight regulation of several signaling inputs and interacts with other signaling pathways Figure 2.
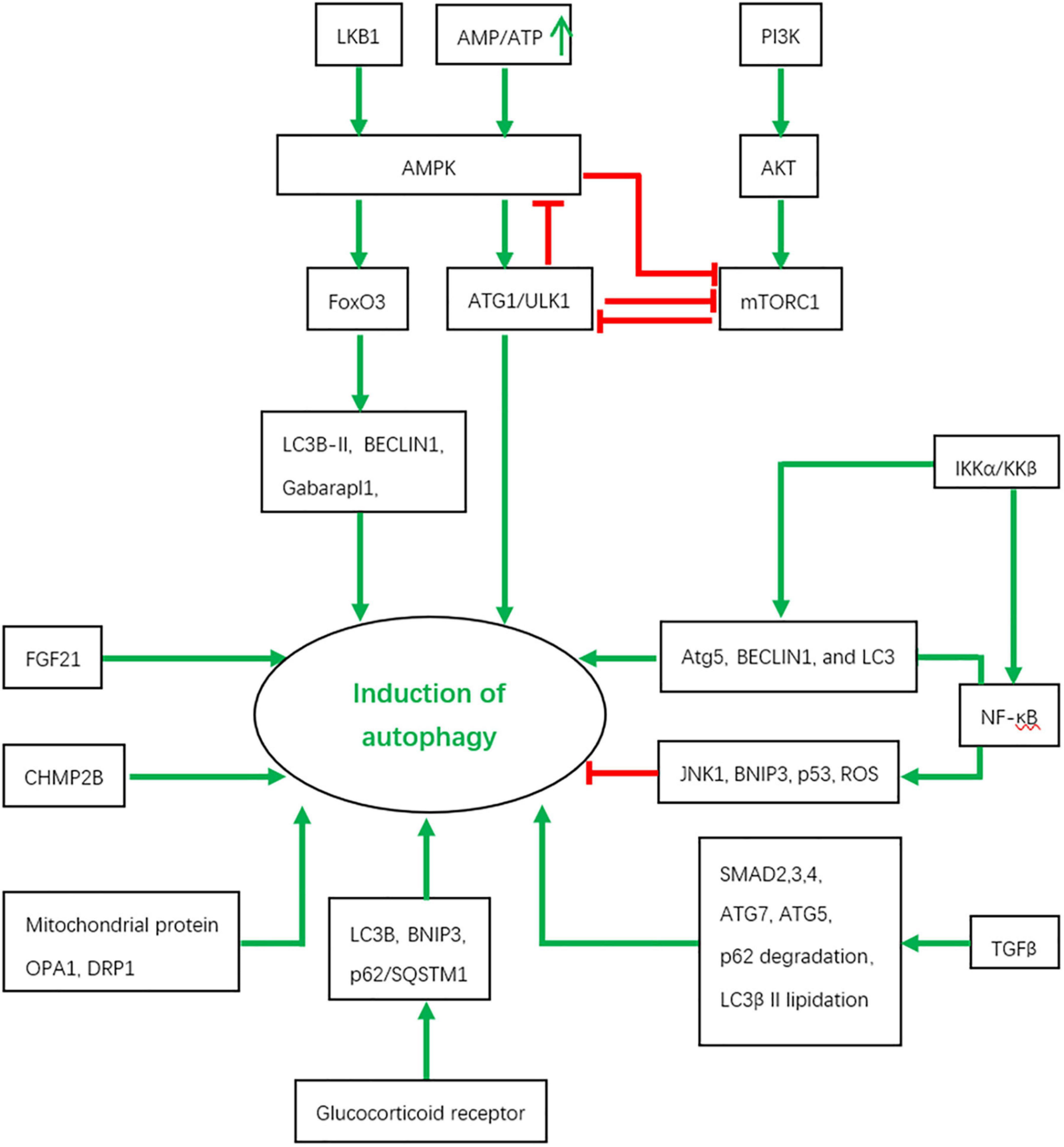
Figure 2. The regulation of autophagy by various signaling pathways and the interactions between them. Red color represents inhibition and green color indicates activation.
The AMPK and mammalian target of rapamycin complex 1 (mTORC1) signaling pathways, as central players in cell survival, proliferation and metabolism, have numerous interconnections with the core genes of the autophagy pathway.
AMPK acts as an energy sensor to monitor changes in the intracellular level of ATP, which is particularly critical in muscles where high rates of energy consumption occur. The growth of skeletal muscles depends on the balance between protein catabolism and anabolism. Atrophy occurs when the rate of muscle protein breakdown is higher than that of muscle protein synthesis. Activated by a rising AMP/ATP ratio, AMPK up-regulates ATP-producing catabolic pathways while suppresses ATP-consuming biosynthetic processes (Winder et al., 2000; Viollet et al., 2003; Wang et al., 2011). AMPK activity is also under the regulation of Liver kinase B1 (LKB1) and mTORC1 through a series of phosphorylation events in skeletal muscles, as revealed in cell culture and model organism studies (Bolster et al., 2002; Sakamoto et al., 2005; Koh et al., 2006; Williamson et al., 2006; Thomson et al., 2007, 2010; Ou et al., 2018).
AMPK modulates autophagy through at least two mechanisms in skeletal muscle. AMPK activation in mouse skeletal muscle results in a relocalization of transcription factor Forkhead box O3a (FoxO3a) to the nucleus where it induces the expression of autophagy-related genes including LC3B-II, Gabarapl1, and BECLIN1 through transcriptional regulation which we will discuss in more detail below, leading to autophagy (Sanchez et al., 2012). In addition, under nutrient starvation conditions, mammalian AMPK directly phosphorylates ULK1 at Ser 317 and Ser 777, promoting the initiation of autophagy which subsequently provides energy and nutrition (Kim et al., 2011; Ljubicic and Jasmin, 2013). AMPK activation releases ULK1 from the complex composed of AMPK, mTORC1, ULK1, FIP200, and Atg13, leading to autophagy activation (Hosokawa et al., 2009). These results suggest that a basal level of autophagy is required to degrade misfolding proteins and damaged organelles to maintain homeostasis under normal nutritional conditions, while autophagy is upregulated by AMPK activation to degrade proteins as a source of alternative nutrients and energy under stress responses such as starvation and exercise (Sanchez et al., 2012).
The mTOR signaling pathway is an evolutionarily conserved pathway which controls multiple cellular processes, including metabolism, protein synthesis, ribosome biogenesis, cell growth, differentiation, and autophagy (Wullschleger et al., 2006; Laplante and Sabatini, 2012). mTOR is classed into two distinct complexes, named mTORC1 and mTORC2 (Laplante and Sabatini, 2012). mTORC1 consists of raptor (regulatory associated protein of mTOR), mLST8 (the mammalian lethal with SEC13 protein 8), PRAS40 (the 40 kDa proline-rich Akt substrate), and DEPTOR (the DEP domain-containing mTOR-interacting protein), and is sensitive to rapamycin (Wullschleger et al., 2006). mTORC2 shares DEPTOR and mLST8 with mTORC1 and includes the distinct components rictor (rapamycin insensitive companion of mTOR) and mSIN1 (mammalian stress-activated map kinase-interacting protein 1) (Jacinto et al., 2004; Sarbassov et al., 2004).
Several studied have demonstrated that the mTORC1 signaling is essential for muscle function. Mice lacking muscle specific tuberous sclerosis complex (TSC) showed sustained activation of mTORC1 and unchanged levels of LC3I and LC3II under fed, basal, and starved conditions, suggesting constitutive and starvation-dependent autophagy is blocked. This impaired autophagy gives rise to a severe, late-onset myopathy. Rapamycin treatment restores autophagy and alleviates the myopathic phenotype of the mice. Although FoxO3 is activated, constitutive and starvation-induced autophagy is blocked by mTORC1-mediated inhibition of ULK1. mTORC1 phosphorylates ULK1 at several sites, such as Ser 757, which prevents interaction between ULK1 and AMPK (Kim et al., 2011; Bento et al., 2016). Paradoxically, abolishment of mTORC1 activity by deletion of raptor also induces autophagy, even though FoxO-dependent transcription of autophagy genes is reduced. These data demonstrate that mTORC1 is another upstream regulator for autophagy induction in skeletal muscle (Castets et al., 2013).
Other studies also show the feedback of ULK1 on AMPK and mTORC1. ULK1 can directly phosphorylate raptor and negatively regulate either mTORC1 activity or substrate binding (Alers et al., 2012). All three subunits of AMPK (AMPKα1, -β2, and -γ1) can serve as direct substrates of ULK1 and ULK2. Through such phosphorylation events, ULK1 confers negative regulation of AMPK kinase activity (Alers et al., 2012). Thus, in addition to being a regulatory target of AMPK and mTORC1, the autophagy pathway constitutes a negative regulatory feedback loop to both signaling pathways and maintains a finely balanced state of cellular homeostasis.
As previously mentioned, transcriptional regulation is a key component of autophagy regulation in skeletal muscle. A large number of studies have demonstrated that autophagy is under the control of multiple transcription factors such as Forkhead box O3 (FoxO3), nuclear factor κB (NF-κB), glucocorticoid receptors (GR), and SMAD. Autophagy genes are targets of these transcription factors under metabolic stress conditions.
FoxO3 functions as an activator of the transcription of autophagy genes ATG4, ATG8B, ATG12, LC3, BECLIN1, BNIP3, VPS34, ULK1, and ULK2 in skeletal muscle (Mammucari et al., 2007; Zhao et al., 2007; Sanchez et al., 2012; Di Malta et al., 2019). Importantly, FoxO3 plays a necessary and sufficient role in muscle atrophy through its effect on autophagy. Inhibition of Bnip3 largely blocks autophagy induced by FoxO3 (Sandri et al., 2004; Mammucari et al., 2007). A study using muscle-specific FoxO1,3,4–/– mice has identified that 29 of the 63 atrophy-related genes are controlled by FoxO under the fasting condition. The induction of autophagy-related genes like LC3, Gabarapl, Bnip3 etc. were abolished in the FoxO1,3,4–/– mice. Consistently, autophagy is severely impaired in these mice.
It is well known that the ubiquitin-mediated proteasome system (UPS) and autophagy/lysosome system are two major mechanisms for degradation of damaged or misfolded proteins. It has been found that both the autophagy-lysosome and the ubiquitin–proteasome system are under the control of FoxO proteins, contributing to skeletal muscle protein loss. FoxO family members regulate atrophy-related ubiquitin ligases atrogin1/MAFbx, MuRF1, TRIM63, MUSA1, SMART, UBC, USP14, and Ube4b, as well as other genes encoding proteasome subunits, which together are involved in muscle atrophy (Bodine et al., 2001; Gomes et al., 2001; Sandri et al., 2004; Milan et al., 2015). Genes functioning in other pathways connected to authophagy under stress conditions such as unfolded protein response were negatively affected in the FoxO 1,3,4–/– mice, too (Milan et al., 2015). These results strongly support the master regulating role of FoxO transcription factors in muscle atrophy.
Nuclear factor-κB (NF-κB) is an important molecule for multiple cellular responses. Hundreds of genes have been identified that are transcriptionally controlled by NF-κB (Zhang et al., 2017). Recent studies have associated NF-κB activation with the loss of skeletal muscle mass in different physiological and pathological conditions. It has been found that skeletal muscle-specific deletion of IKKβ, an upstream activator of NF-κB signaling, inhibited the expression of MuRF1 E3 ubiquitin ligase. Overexpression of the dominant negative mutant form of IκBα, an inhibitory protein of NF-κB also inhibited the degradation of proteins in muscle. These results suggest the important role of NF-κB activation in muscle-wasting (Bodine et al., 2001; Cai et al., 2004; Cao et al., 2005; Mourkioti et al., 2006).
NF-κB appears to have both activating and inhibitory effects on myogenesis or skeletal muscle formation. It can inhibit myogenic differentiation through transcriptional regulation of cyclin D1 and the transcription factor YinYang1 (YY1) which silences expression of myofibrillar genes (Guttridge et al., 1999; Mitin et al., 2001; Wang et al., 2007). Although NF-κB has been considered as a negative regulator of myogenesis, results from some reports support the role of NF-κB in pro-myogenesis. It has been found that insulin growth factor-II (IGF-II) stimulates NF-κB activation, which further induces the myogenic signaling pathway (Kaliman et al., 1999). These anti- or pro-myogenesis effects on muscles may be determined by a switch between the canonical and non-canonical NF-κB signaling pathway (Bakkar et al., 2008; Bakkar and Guttridge, 2010).
Many studies have described the relationship between NF-κB and autophagy. NF-κB signaling has been shown to be involved in autophagy in a context-dependent manner (Ben-Neriah and Karin, 2011; Hayden and Ghosh, 2012; Salminen et al., 2012; Verzella et al., 2016; Begalli et al., 2017; Bennett et al., 2018). One study has demonstrated that the proinflammatory cytokine TWEAK promotes skeletal muscle atrophy by activating the expression of several autophagy genes including BECLIN1, LC3B, and Atg5 via TRAF6-Mediated NF-κB activation (Dogra et al., 2007). Indeed, NF-κB activation is sufficient to induce the expression of genes involved in or related to the autophagy pathway such as BECLIN1 and the BAG3-HspB8 complex (Copetti et al., 2009; Nivon et al., 2012; Salminen et al., 2012; Rapino et al., 2015). Under starvation conditions, IKKα and IKKβ also stimulate the expression of Atg5, BECLIN1, and LC3 in a NF-κB-independent manner (Comb et al., 2011). On the other hand, NF-κB signaling also has inhibitory effects on autophagy under certain circumstances, which may be mediated by indirect mechanisms. NF-κB signaling may activate mTOR kinase, promote the expression of autophagy inhibitors such as Bfl-1/A1 (a Bcl-2 family member and a BECLIN1 binding partner) and block autophagy inducers such as JNK1, BNIP3, p53, and ROS (Edelstein et al., 2003; Djavaheri-Mergny et al., 2006; Papa et al., 2006; Balaburski et al., 2010; Kathania et al., 2011; Morgan and Liu, 2011; Sarkar et al., 2011; Salminen et al., 2012). These results indicate that the activation of NF-κB signaling in acute stress may induce autophagy while a delayed activation inhibits autophagy. This may represent a protective mechanism from cell death induced by autophagy (Salminen et al., 2012).
These studies demonstrate that NF-κB is a critical regulator of autophagy in skeletal muscle. Although the association of NF-κB with autophagy has been intensively investigated, the majority of these studies focused on cancer. Understanding of the role of NF-κB in muscle autophagy is still limited and future in-depth investigations are needed.
Glucocorticoid receptor (GR) signaling has been shown to be associated with protein synthesis and proteolysis in skeletal muscle. GR signaling is crucial for protein breakdown in muscle cells (McGrath and Goldspink, 1982; England and Price, 1995; Hasselgren, 1999; Long et al., 2008; Stahn and Buttgereit, 2008; Sun et al., 2008). During fasting, the expression of poly-ubiquitin mRNA is increased by administration of dexamethasone and the proteolysis pathway is activated in skeletal muscle. Muscle protein breakdown is decreased in adrenalectomized rats, which can be counteracted by glucocorticoid administration (Wing and Goldberg, 1993; Tiao et al., 1996; Mitch et al., 1999). These results suggest that GR signaling is required for protein breakdown in skeletal muscle. Glucocorticoids have been found to stimulate Foxo1 and 3 mRNA in muscle atrophy (Furuyama et al., 2003; Lecker et al., 2004) and activate the expression of UPS related proteins such as atrogin-1, Murf1, and Fbxo30 (Jagoe et al., 2002; Sandri et al., 2004; McLoughlin et al., 2009; Milan et al., 2015). The expression of the autophagy genes Map1lc3b and Bnip3 were induced in both control and glucocorticoid receptor knock-out (GRKO) mice under hypoxia and reduced food intake, but GRKO mice shows a blunted response with impaired expression of Foxo1. These data suggest that glucocorticoid receptor signaling may contribute to autophagy in the context of muscle atrophy through control of gene expression (de Theije et al., 2018). Furthermore, mTOR repressors REDD1 and KLF15 have been identified as direct target genes of the glucocorticoids. Expression of Redd1 induces autophagy and inhibits protein synthesis (Wang et al., 2006; Molitoris et al., 2011; Britto et al., 2014). KLF15 also activates the expression of atrogin-1 and MuRF1 genes and therefore induces skeletal muscle atrophy (Shimizu et al., 2011). One recent study has identified a new mechanism by which a selective NR3C1/glucocorticoid receptor modulator, Compound A (CpdA), has an off-target effect on autophagy. In the classic NR3C1/GR pathway, NR3C1 is recruited to the promoter of SQSTM1 and other NFE2L2-controlled genes following binding of the GR ligand dexamethasone. In contrast, the transcription factor NFE2L2 is recruited to the promoter of SQSTM1 by CpdA, suggesting a distinct activating mechanism of autophagy (Mylka et al., 2018). It should be noted that this mechanism has been identified only in mouse bone marrow-derived macrophages. Whether or not the mechanism can be extend to the skeletal muscles needed to be examined.
Transforming growth factor (TGF)-β signaling is one of the most important pathways in a variety of physiologic processes. Growth/differentiation factor-8 (GDF-8, myostatin), one of TGF-β family members, has been identified as a skeletal muscle-specific protein from early embryonic development till adulthood. Deletion of the GDF-8 gene results in a 3 fold increase in skeletal muscle mass, caused by hyperplasia or hypertrophy (McPherron et al., 1997). Multiple mutations in the GDF-8 gene have been identified in human and mammals, which lead to increased muscle mass (Grobet et al., 1997, 1998; Kambadur et al., 1997; McPherron and Lee, 1997; Szabo et al., 1998; Williams, 2004; Clop et al., 2006; Mosher et al., 2007). The binding of GDF8 to its receptors leads to the phosphorylation of the transcription factor SMAD2/3 and the recruitment of SMAD4 (Sartori et al., 2009). Activation of SMAD2/3 counteracts the inhibitory effect of JunB on FoxO3, contributing to atrophy (Sartori et al., 2009; Welle, 2009; Raffaello et al., 2010). SMAD3 expression also increases the promoter activity of the atrogin-1, MuRF1, and PGC1α, activates the PTEN 3′-UTR and FoxO response element reporters and inhibits the miR-29 promoter activity and mTOR in skeletal muscle leading to protein breakdown and skeletal muscle fiber atrophy (Goodman et al., 2013). These results suggest that GDF8 plays a critical role in muscle atrophy.
TGF-β1 treatment induces phosphorylation of SMAD2 and SMAD3,p62 degradation,LC3β II lipidation and the synthesis of collagen type Iα2 and fibronectin in primary human atrial myofibroblasts (Ghavami et al., 2015). On the other hand, autophagy may be negatively correlated with TGF-β signaling. One recent study has found that the prostaglandin-degrading enzyme, 15-PGDH, is elevated in muscles during aging, which may be responsible for age-related muscle atrophy. Inhibition of 15-PGDH by a small molecule or genetic depletion has beneficial effects on muscle rejuvenation, mediated by a series of events such as increased PGE2, restoration of mitochondrial function, decreased atrogene expression and TGF-β signaling, and increased autophagy (Palla et al., 2021). These results demonstrate the context-dependence of TGF-β-mediated effects in skeletal muscles.
Other regulatory molecules include mitochondrial proteins OPA1 (optic atrophy 1), DRP1 (Dynamin related protein 1), FGF21 (the fibroblast growth factor 21), and CHMP2B (the charged multivesicular body protein 2B). The mitochondrial dysfunction has been considered as one of the major characteristics of aging process and is associated with muscle loss. OPA1 has been well known for its roles in autophagy and crosstalk with Atg7 (Tezze et al., 2017; Larsson et al., 2019; Zaninello et al., 2020). Muscle-specific Drp1 deletion results in detrimental changes including mitochondrial dysfunction, autophagy impairment, muscle wasting and weakness, suggesting the critical role of mitochondrial dynamics in muscles (Favaro et al., 2019). FGF21 increases glucose uptake and fat utilization in adipocytes, resulting in enhanced mitochondrial oxidation and activation of AMPK. The skeletal muscle-specific deletion of Atg7 stimulates FGF21 expression in an ATG4-dependent manner (Kim et al., 2013). The mice lacking FGF21 showed a decrease in autophagy/lipophagy, which leads to accumulation of lipid and cardiac muscle cell disorganization (Ruperez et al., 2018). Moreover, the general autophagy flux is slightly reduced while the mitophagic flux is significantly decreased in FGF21 knockout mice under starved conditions, and Bnip3 is required for FGF21 induced muscle atrophy and mitophagy (Oost et al., 2019). Other studies showed that increased level of serum FGF21 is associated with multiple metabolic diseases such as muscle atrophy (Staiger et al., 2017; Tezze et al., 2017). Further studies are needed to clarify the physiological and pathological roles of FGF21 in muscle cells (Oost et al., 2019). CHMP2B is a subunit of endosomal sorting complex required for transport-III (ESCRT-III). CHMP2B has been shown to be involved in protein degradation pathways including autophagy and the endosome–lysosome pathway. siRNA-mediated depletion of ESCRT subunits or expression of the CHMP2B C-terminal truncated mutants impairs autophagic degradation, leading to the accumulation of autophagosomes (Filimonenko et al., 2007; Lee et al., 2007).
Skeletal muscle is often subjected to mechanical, heat, and oxidative stress, resulting in cell damage. Proteolysis is required for repair and regeneration in skeletal muscle. Both the ubiquitin/proteasome pathway and autophagy-mediated proteolysis contribute to protein turnover in skeletal muscles. Studies over the past two decades have revealed the connection between these two pathways in skeletal muscles. The transcription factor FoxO3 has been shown to stimulate the expression of many autophagy-related genes and the ubiquitin E3 ligases, atrogin-1/MAFbx (Sandri et al., 2004), as well as lysosomal proteolysis in muscle (Mammucari et al., 2007; Zhao et al., 2007; Bell et al., 2016). Autophagy inhibition results in muscle atrophy, loss-of-force production, myopathy phenotypes, and loss of muscle mass, which is similar to the phenotypes caused by disrupting the functions of atrogin-1 and MuRF1, two atrophy-related ubiquitin ligases, as well as deficiencies in genes involved in different catabolic pathways (Bodine et al., 2001; Baehr et al., 2011). Importantly, both Atrogin-1 and MuRF1 are stimulated in Atg7–/– muscles. Atg7 deletion also results in apoptosis in muscle cells. Muscle-specific Atg5–/– mice showed the same phenotypes as Atg7–/– mice (Raben et al., 2008). Thus the autophagy-lysosome pathway and ubiquitin-proteasome pathway function in parallel downstream of FoxO regulation and may be complementary to each other.
Thus AMPK, mTORC1, mTORC2 and other signaling activities interact with autophagy in the processes of controlling muscle growth, development, size control, atrophy, hypertrophy, and regeneration. The roles of these signalings in the transcriptional regulation of the autophagy pathway have been under intensive investigation in skeletal muscles under normal physiological and diseased pathological conditions. However, the complex and inextricably interwoven network of these signalings and how they are maintained at a finely balanced state remain fully elucidated. Furthermore, how perturbations to genes in these pathways lead to muscle disorders need to be further investigated by human genetics studies and functional studies using model organisms.
Accumulating evidence suggests the essential role of autophagy in maintaining cellular homeostasis. Autophagy provides energy and building blocks for metabolism and cellular renewal, controlling the level of amino acids, lipids, carbohydrates and nucleic acids during nutrient deprivation (Tsukada and Ohsumi, 1993; Otto et al., 2003; Scott et al., 2004; Kang et al., 2007). Also, autophagy plays an essential role in intracellular quality control, contributing to the degradation of damaged or aggregated proteins and basal protein turnover. Cells constitutively exhibit a basal autophagy level even under normal growth conditions and autophagy was further induced in response to metabolic stress (Mizushima, 2005). Studies have shown that the inhibition of autophagy results in accumulation of ubiquitinated protein aggregates and inclusion bodies in multiple cell types including muscle cells (Hara et al., 2006; Komatsu et al., 2006; Nakai et al., 2007; Ebato et al., 2008; Jung et al., 2008; Raben et al., 2008; Masiero et al., 2009), as well as abnormalities in mitochondria, peroxisomes, ER and Golgi (Komatsu et al., 2005; Jung et al., 2008). It has now become evident that autophagy involves a highly selective process for the removal of unwanted cellular components and damaged organelles in non-starved cells in addition to its non-selective bulk degradation mechanism (Reggiori et al., 2005; Kraft et al., 2009; Guimaraes et al., 2015).
Many studies have demonstrated the role of autophagy in muscle growth, atrophy, hypertrophy, regeneration and during exercise. In the Atg7 gene, the cysteine residue encoded by nucleotides in exon 14 is required for activation of substrates. Targeted deletion of exon 14 of Atg7 specifically in adult mice liver impaired autophagosome formation and inhibited the bulk degradation of proteins under fasting condition (Komatsu et al., 2005). In addition, muscle-specific Atg7 knockout mice showed abnormal mitochondria, disorganized sarcomere, reticulum distension, and aberrant concentric membranous structures (Komatsu et al., 2005; Masiero et al., 2009). They exhibited muscle phenotypes such as morphological properties of myopathy, muscle loss and degeneration under catabolic conditions (Masiero et al., 2009). ATG16L1 hypomorphic mice display decreased levels of autophagy, causing a significant reduction in the growth and generation of muscle fibers (Paolini et al., 2018). These data suggest a beneficial role of a basal level of autophagy in maintaining muscle mass and myofiber integrity under physiological conditions (Masiero et al., 2009). Stimulation of autophagy by caloric restriction and exercise may have beneficial effects on lifespan. Impairment of autophagy leads to muscle degeneration and wasting (Masiero et al., 2009; Carnio et al., 2014).
Physical activities have been shown to have an impact on muscle function, which is mediated by autophagy. A proper autophagic flux is required for the degradation of damaged cell organelles and proteins, which provides building blocks and energy during starvation and stress. Exercise and starvation may have the beneficial metabolic effects on human health through BCL2 mediated autophagy (He et al., 2012). On the other hand, excessive autophagy results in atrophy (Grumati et al., 2010). Therefore, an appropriate activation level of autophagy during exercise is critical for muscle homeostasis. Excessive autophagy associated with mutations in the LAMA2 gene resulted in massive muscle wasting. Laminin α2 chain deficiency in mice leads to the increased expression of expression of autophagy-related genes in muscle and the phenotypes of muscle fibrosis, atrophy, and apoptosis, suggesting the pathological role of excessive autophagy (Carmignac et al., 2011; Vainshtein et al., 2014). These findings provide the first evidence that autophagy flux is essential for normal physical activity and defective or excessive autophagy flux leads to muscular dystrophies. This further suggests that the activators of autophagy could serve as potential targets for the treatment of muscular dystrophies (Grumati et al., 2010, 2011; Grumati and Bonaldo, 2012).
Satellite cells are muscle stem cells, which usually reside in a mitotically quiescent state (in G0 phase) and are transcriptionally inactive (Schultz, 1978). The paired domain transcription factor Pax7 is the specific biomarker for all satellite cells (Seale et al., 2000). Once they receive stimuli such as hormones, nutrition, injury or disease, these cells will subsequently begin proliferating to replenish the stem cell pool or differentiating to generate new muscle fibers (Hansen-Smith et al., 1979; Chanoine et al., 1987; Brack and Rando, 2012; Yin et al., 2013). It has been shown that constitutive autophagy is required for satellite cells to maintain their stem cell fitness. Autophagy may provide an energy source for activation by degradation of substrates. SIRT1, a key nutrient sensor, is required for autophagy during satellite cell activation. Either a small molecular inhibitor of SIRT1 or genetic deletion could block autophagy in satellite cells (Tang and Rando, 2014). Failure of autophagy in aged satellite cells or genetically defective cells (such as those from Atg7 knockout mice) results in senescence, oxidative stress and mitochondrial dysfunction, and accumulation of organelles and proteins (Chakkalakal et al., 2012; Cosgrove et al., 2014; Sousa-Victor et al., 2014; Garcia-Prat et al., 2016). Reactivation of autophagy restores their stemness (Garcia-Prat et al., 2016). It has been shown that autophagy is activated in denervation atrophy (Schiaffino and Hanzlikova, 1972) and denervation leads to both a reduction in satellite cell numbers and progressive skeletal muscle atrophy (Schultz, 1978; Rodrigues Ade and Schmalbruch, 1995; Viguie et al., 1997). Thus autophagy is implicated in muscle regeneration. It determines the transition between the quiescence and senescence fate of muscle stem cells. On the other hand, it seems that satellite cells are not the reason for muscle fiber hypertrophy, although they are required for the de novo formation of new fibers and fiber regeneration. It is widely accepted that that muscle mass is increased primarily by hyperplasia but not the number of satellite cells (McCarthy et al., 2011; Fukada, 2018). Muscle fiber hypertrophy is functionally normal but regenerative process is significantly reduced in muscle depleted of satellite cells, suggesting that the presence of satellite cells does not determine muscle mass (Amthor et al., 2009; McCarthy et al., 2011; Sambasivan et al., 2011; Jackson et al., 2012; Bachman et al., 2018). In addition, activation of AKT results in an increase in hypertrophic fibers and enhances myofibrillar force without satellite cell proliferation (Blaauw et al., 2009). These findings demonstrate that atrophy may not be attributed to the decline in satellite cell number. Physical exercise and nutrition may be a better treatment for muscle weakness than a stem cell based therapeutical approach (Schwartz et al., 2016).
Autophagy plays a key role in different physiological and pathological processes in heart. Cardiac-specific deletion of Atg5 in mice results in hypertrophy, cardiac dilatation and contractile dysfunction accompanied by disorganized mitochondria, abnormal sarcomere structure and accumulation of misfolded proteins. The Atg5–/– mice develop cardiac dysfunction under stress conditions, demonstrating autophagy activity is required for cardiomyocyte homeostasis (Nakai et al., 2007). In contrast, overexpression of Atg5 stimulates autophagy and increases lifespan in mice (Klionsky et al., 2016). In addition, cardiac-specific Parkin deletion suppresses mitophagy and leads to a lethal cardiomyopathy in developing hearts (Gong et al., 2015).
Autophagy also contributes to ischemia and reperfusion. As discussed above, autophagy is induced by activation of AMPK signaling and inhibition of mTOR signaling. Autophagy was induced by AMPK activation and was suppressed by a dominant negative mutant of AMPK during ischemia, while autophagy was induced by BECLIN1 but not AMPK signaling during reperfusion, suggesting autophagy may play different roles in ischemia and reperfusion (Matsui et al., 2007). Further studies are needed to clarify the underlying mechanism of autophagy during ischemia and reperfusion injury. The roles of autophagy in heart failure vary depending on the context. Heart failure is characterized by cardiac hypertrophy, and can be induced by pressure overload through cardiac autophagy. Heterozygous deletion of BECLIN1, which leads to a reduction of cardiomyocyte autophagy, alleviates cardiac hypertrophy and dysfunction. In contrast, overexpression of BECLIN1 in cardiomyocytes worsens pathologic phenotypes (Zhu et al., 2007). However, phosphorylation of BECLIN1 induced by Mst1 impairs autophagy and induces cardiac dysfunction in heart failure. Mst1 gene deletion activates autophagy and diminishes cardiac remodeling and dysfunction in heart failure (Maejima et al., 2013). These results suggest that the beneficial or detrimental effect of autophagy on heart failure are determined by the specific conditions.
Loss of muscle mass has been found in a variety of diseases such as diabetes, AIDS, sepsis, cardiac disease, and chronic obstructive pulmonary disease (Jagoe and Engelen, 2003; Lecker et al., 2006). These diseases share the same cellular features of excessive protein breakdown which disrupts the balance between anabolism and catabolism, thereby leading to myofiber atrophy (Sandri, 2008). Microarray analyses also demonstrated the up-regulation of autophagy-related genes LC3 and GABARAPL1 in muscle wasting induced by different experimental conditions (Lecker et al., 2004). All the studies discussed above demonstrated that the autophagy level is required to be finely tuned and properly regulated to maintain the homeostasis of skeletal muscles during physiological and stressed conditions. Disruption of balanced autophagy leads to the pathogenesis of a variety of muscle disorders, which we discuss in more detail below.
Autophagic vacuoles have constantly been observed in skeletal myofibers in diseases like autophagic vacuolar myopathies (AVM) (Malicdan et al., 2008). Muscle disorders cause muscle weakness, muscle wasting and even paralysis, which severely affects patients’ mobility. Environmental factors such as bacterial infection, injury and other diseases like cancers can lead to muscle diseases. Genetic components can also contribute to the development of skeletal muscle disorders (Prior et al., 2007), including mutations in autophagy genes, which are summarized in Table 1.
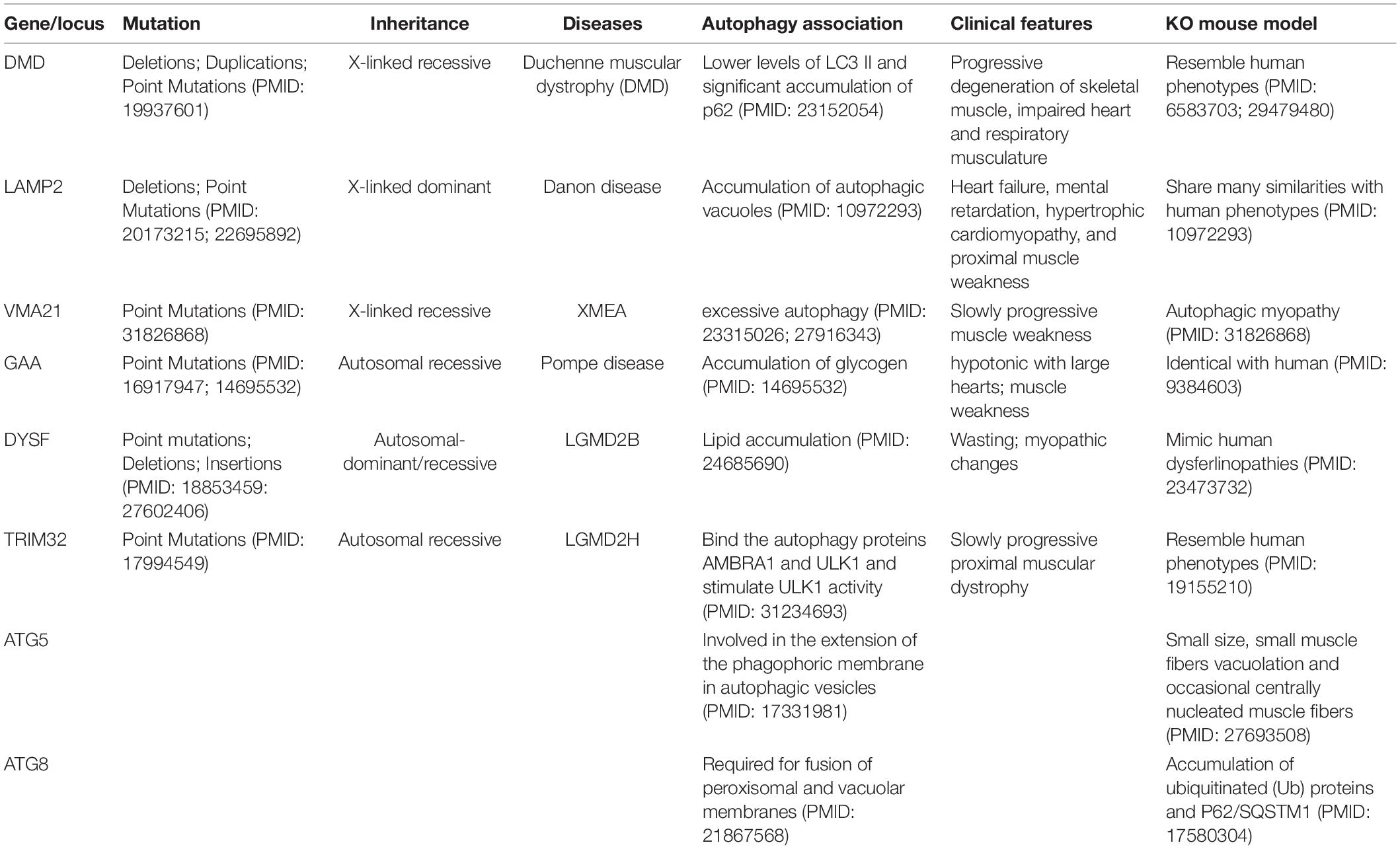
Table 1. The mutations in autophagy genes or deregulation of autophagy resulted in skeletal muscle disorders, and the mouse models with impaired autophagy pathway showed the phenotypes of abnormal skeletal muscles.
Duchenne Muscular Dystrophy (DMD) is the most common childhood form of muscular dystrophy. The prevalence is estimated to be 1 in every 3,500–5000 live male births (Emery, 1991; Romitti et al., 2015; Crisafulli et al., 2020). Because DMD is inherited in an X-linked pattern, it primarily affects males, with rare cases in females. Most children with DMD will need a wheelchair by their early teens. Later, heart problems develop into dilated cardiomyopathy with shortness of breath, an irregular heartbeat (arrhythmia), extreme tiredness (fatigue), and swelling of the legs and feet. These heart problems progressively get worse over time, and eventually become life-threatening in most patients (Ryder et al., 2017). DMD had been recognized as a metabolic dysfunction (Dreyfus et al., 1954; Hess, 1965; Di Mauro et al., 1967; Chi et al., 1987; Chinet et al., 1994), and in 1987 the mutations responsible for the disease were first identified in the dystrophin gene on the short arm of the X-chromosome (Koenig et al., 1987). The dystrophin gene is the largest known human gene containing 79 exons and spanning more than 2,200 kb, accounting for almost 0.1% of the entire human genome (Gao and McNally, 2015). The most common type of mutations in this gene are deletions of one or more exons, accounting for 60–70% of cases. The other mutations include small deletions, insertions, exonic duplications, splicing mutations, and point mutations (Flanigan et al., 2009; Tuffery-Giraud et al., 2009). These mutations may result in reading frame shift and subsequently produce truncated proteins with premature stop codons.
It has been shown that autophagy was impaired in DMD patients and mdx mouse models which closely mimic the human disease (De Palma et al., 2012). In mdx mice, mTOR was constitutively activated, leading to the down-regulation of LC3, Atg12, Bnip3 and Gabarapl1 at the molecular level. At the same time, a long-term low-protein diet treatment reactivated autophagy through inactivation of AKT (De Palma et al., 2012). Consistent with these observations in mdx mice, the expression and phosphorylation levels of AKT in the skeletal muscles and cardiac muscles of DMD patients were largely increased (Pichavant et al., 2011). In addition to the AKT signaling pathway, ROS generation by CYBB/NOX2 also led to autophagy deficiency in skeletal muscles of the mdx mice. Simvastatin treatment suppressed the generation of ROS and increased autophagy signaling. These findings suggest that autophagy may serve as a novel therapeutic target for DMD patients (De Palma et al., 2012; Whitehead et al., 2016).
It has also been found that P2RX7 (the purinergic receptor P2X, ligand-gated ion channel, 7) is stimulated in mdx mouse myoblasts and myofibers (Young et al., 2012). The large-pore formation of P2RX7 and HSP90 are required for the ATP-evoked autophagic death of dystrophic muscles (Young et al., 2015). Administration of Coomassie Brilliant Blue, the P2RX7 antagonist, leads to a reduction of degeneration-regeneration cycles in mdx mice, suggesting P2RX7 may act as a potential drug target for the treatment of the disease (Young et al., 2012; Bibee et al., 2014; Sinadinos et al., 2015).
Another molecule implicated in the pathogenesis of mdx mice is TNF receptor-associated factor 6 (TRAF6), which has a role in maintaining skeletal muscle mass. TRAF6 is upregulated in skeletal muscle of mdx mice. Inhibition of autophagy through the targeted deletion of TRAF6 in mdx mice appears to preserve skeletal muscle mass at the initial stage but exaggerates dystrophic phenotype at the late stage, suggesting the opposing effect of autophagy on skeletal muscles in mdx mice (Hindi et al., 2014).
Other approaches increasing autophagy or protein quality control have also been considered as potential therapeutic approaches for DMD. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc-1α) gene transfer results in an increase in Lc3 and Atg12 in mdx mouse muscles, indicating a beneficial effect of autophagy on dystrophic skeletal muscle (Hollinger et al., 2013). One recent study has demonstrated that the modulations of protein quality control mechanisms have been established in undifferentiated myoblasts derived from DMD patients, but misfolded/aggregated proteins are determined to take the path to autophagy rather than to proteasome. This change is caused by a switch from BAG1 to BAG3, NFκB activation, and up-regulation of BAG3/HSPB8 complexes. Restoration of the established mechanism of protein quality control may be a potential therapeutical target for DMD treatment (Wattin et al., 2018).
Collagen VI is a major extracellular matrix protein of skeletal muscle involved in cell adhesion and membrane stabilization. Collagen VI has been shown to be associated with of numerous physiological and pathological conditions. A number of mutations in Collagen VI have been identified in disorders of muscular dystrophy like Ullrich muscular dystrophies and Bethlem myopathies (Bonnemann, 2011). It has been suggested that reduced autophagocytic flux played a critical role in the pathogenesis of collagen VI deficiency (Grumati et al., 2010). Autophagy induction is impaired after physical exercises in collagen VI null mice, which has a detrimental effect on muscles (Grumati et al., 2011). Several molecular changes have been found in collagen VI null mice, such as reduced autophagosomes and LC3 lipidation, and impaired induction of BECLIN1 and Bnip3 (Grumati et al., 2010).
AVM is a group of rare genetic disorders that share common histopathological features on muscle biopsy with an excess of autophagic vacuoles and sarcolemmal characteristics (Munteanu et al., 2015). Mutations in genes related to autophagy have also been identified among patients with the spectrum of AVM (Table 1).
Danon disease, originally named as lysosomal glycogen storage disease with normal acid maltase, is the best-known AVM and is characterized by weakening of cardiomyopathy, weakening of myopathy, and neurological phenotypes like intellectual disability. Danon disease is caused by mutations in the gene encoding the Lysosome-associated membrane protein 2 (LAMP2), a membrane glycoprotein known to be related to autophagy. LAMP2 may play a role in controlling cell–cell or cell-extracellular matrix adhesion and maturation of autophagic vacuoles in addition to maintaining lysosomal structural integrity (Lippincott-Schwartz and Fambrough, 1986; Carlsson et al., 1988; Saitoh et al., 1992; Lichter-Konecki et al., 1999; Eskelinen, 2006). There are different splicing forms of LAMP2 in various tissues (Konecki et al., 1995), among which LAMP2A is the only isoform with positive amino acid residues at the carboxyl terminus tail responsible for substrate binding (Cuervo and Dice, 2000). A recent study has demonstrated that the Asn175 site at the linker region between N- and C-terminal subdomains of LAMP2 is critical for its role in autophagy. Loss of glycosylation at the Asn175 disrupts the interaction between Lamp2 and galectin-9 protein, which impairs endolysosome/lysosome function and cargo degradation (Sudhakar et al., 2020).
A variety of mutations in LAMP-2 have been reported in patients with Danon disease. Most of these mutations are nonsense or frameshift mutations, which result in truncated protein products (Brambatti et al., 2019; Gurka et al., 2020). Lack of the transmembrane and cytoplasmic domains at the C-terminal tail abolishes its function as a lysosomal membrane protein (D’Souza et al., 2014). Ample evidence indicates that deficiency of LAMP-2 causes mistargeting of certain lysosomal enzymes and impaired capacity for lysosomal degradation (Tanaka et al., 2000; Eskelinen et al., 2002), leading to disrupted phagosomal maturation, autophagosome-lysosome fusion, and accumulation of autophagosomes and resulting in myopathy and cardiomyopathy (Tanaka et al., 2000; Saftig et al., 2008). In contrast, overexpression of Lamp2 can alleviate autophagic flux blockade likely due to stimulation of cathepsin trafficking, which may improve cardiomyocyte resistance to lysosomal cell death (Cui et al., 2020).
X-linked myopathy with excessive autophagy (XMEA) is a rare disorder characterized by childhood onset of weakness and wasting, mainly in the proximal muscles of the lower extremities. Although the muscles including the anterior thigh, the ankle dorsiflexors, the hip girdles and the shoulder are affected, other organs, such as the heart appear to be normal in the majority of patients (Kalimo et al., 1988). Serum creatine kinase levels are dramatically elevated in the patients. The morphological abnormalities in muscle cells are easily observed with an optical microscope (Kalimo et al., 1988; Villanova et al., 1995; Minassian et al., 2002; Crockett et al., 2014).
XMEA is caused by mutations in the VMA21 gene at Xq28 encoding a chaperone protein for the lysosomal vacuolar ATPase (Ramachandran et al., 2013). Vacuolar ATPases are rotary proton pumps across the plasma membrane regulating the pH of intracellular organelles. VMA21 is required for the proper assembly of multiple proton pump subunits (Forgac, 2007). Loss of VMA21 disrupted the interaction between the major proteolipid subunit of V0 and another V0 subunit, Vph1p during assembly (Malkus et al., 2004). Loss of appropriate VMA21 activity results in the formation of autophagic vacuoles with sarcolemmal features (Crockett et al., 2014). Mechanistic study showed that reduced VMA21 level leads to increased lysosomal pH and decreased lysosomal degradative ability. Meanwhile, feedback upregulation of autophagosome formation and inhibition of the mTORC1 pathway results in accumulation of ineffective autolysosomes, cell vacuolation and tissue atrophy (Ramachandran et al., 2013). A recent study of in vitro cultured patient-derived myoblast cells revealed the possible mechanism by which VMA21 mutation triggers autophagy abnormity may contribute to XMEA development. This study demonstrated VMA21 mutation-associated autophagy defect leads to uncontrolled myoblast fusion and altered myoblast differentiation, which produced functionally inferior muscle cells. This observation also explains why muscles are the predominantly involved tissue in XMEA disorder (Fernandes et al., 2020).
Skeletal muscle of XMEA patients with VMA21 mutations is the main affected tissue, which may be due to the difference in V-ATPase demand between skeleton muscles and other organs (Ramachandran et al., 2013; Munteanu et al., 2015; Saraste et al., 2015). Additional study has identified a mutation (c.164-6T > G) resulting in much lower VMA21 expression and V-ATPase activities than those in classical XMEA; this patient does have comparatively mild cardiac hypertrophy (Munteanu et al., 2017). Another study revealed that the mutations c.10C > T, p.Arg18Gly and p.Asp63Gly in VMA21 were implicated in a congenital disorder of glycosylation with autophagic liver disease (Cannata Serio et al., 2020).
Pompe disease, also called type II glycogen storage disease, is a rare, autosomal recessive metabolic disorder characterized by α-glucosidase deficiency leading to accumulation of glycogen in the lysosomes (Dasouki et al., 2014). This disease has been classified into two subtypes according to the disease onset age. Infantile-onset Pompe disease is the severe subtype with which children develop symptoms under 12 months of age. This subtype is characterized by heart muscle malfunction or severe breathing problems, typically leading to death due to cardiac failure or respiratory abnormality within the first year of life without treatments. Late-onset Pompe disease is usually a mild form in which symptoms may begin at any time from late childhood to adulthood and progress more slowly than those in infantile-onset Pompe disease (Dasouki et al., 2014; Kohler et al., 2018).
Pompe disease is caused by the mutations in GAA gene encoding acid alpha-glucosidase which breaks down glycogen to glucose in the lysosome. The GAA gene is located on the long arm of chromosome 17 (17q25.2-q25.3) and contains 20 exons spanning 18.3 kb. Over 600 mutations in the GAA gene have been identified in patients with Pompe disease and missense mutations are the most common type (Pompe variant database1). Although most of the GAA mutations are rare, the variant rs386834236 (c.-32-13T > G) is common among Caucasian patients. The variant leads to the spicing out of the exon (Huie et al., 1994; Raben et al., 1996). The c.525delT and the c.2481 + 102_2646 + 31del mutations are overrepresented in the Dutch population, both of which lead to a reading frameshift with an early stop codon (Peruzzo et al., 2019). The severity of the disease depends on the degree of enzyme deficiency determined by the nature of the mutations in both alleles.
Dysfunction of acid alpha-glucosidase results in glycogen accumulation in the lysosomes, followed by lysosomal rupture in cardiac and skeletal muscles, leading to severe myofibril loss (Griffin, 1984; Thurberg et al., 2006). Autophagic accumulation in skeletal muscle was observed in Pompe patients (Fukuda et al., 2006) and further immunostaining with LAMP1 and LC3 in muscle fibers revealed extensive accumulation of autophagosomes, clustering of late endosomes and broken lysosomes (Raben et al., 2007; Raben et al., 2009). In a mouse model of Pompe disease, muscle-specific loss of GAA results in an autophagy defect due to the impaired autophagosomal–lysosomal fusion, and subsequently the phenotype of muscle atrophy (Murrow and Debnath, 2013). The enzyme acid alpha-glucosidase is produced as an inactive precursor which has to be glycosylated in the ER and phosphorylated in Golgi before maturation through proteolysis in the endosome/lysosomes (Moreland et al., 2005; Meena and Raben, 2020). Any mistake during this process would result in an acid alpha-glucosidase deficiency. The defective autophagic pathway in patients with Pompe Disease also leads to mitochondrial abnormalities detected in muscle biopsies. A number of therapeutic approaches have been developed to improve enzyme replacement for Pompe disease (Spampanato et al., 2013; Tian et al., 2019).
LGMD2B is an autosomal recessive disease characterized by proximal muscle weakness and wasting affecting shoulder girdles and pelvises with slow progression (Aoki, 1993). MMD1 is characterized by distal muscle weakness affecting the upper and lower limbs (Miyoshi et al., 1986). Both LGMD2B and MMD1 are caused by mutations in the gene dysferlin (DYSF) (Liu et al., 1998), which contains 55 exons, spanning a 230 kb genomic region located on chromosome 2p13.2. The protein product dysferlin is a type II transmembrane protein expressed mainly in muscle sarcolemma. It is involved in muscle contraction, calcium-mediated membrane fusion, and membrane regeneration. Dysferlin contains seven C2 domains (C2A to C2G) and two DysF domains, among which the C2A domain is responsible for Ca2+ and phospholipid binding.
Different types of DYSF mutations have been uncovered in LGMD2B and MMD patients, and the most frequently observed pathogenic variant is rs28937581 (c.2997 G > T; p.Trp999Cys) located in a DysF domain (Izumi et al., 2020). Crystal structures of the DysF domain indicate that mutations like p.Arg959Trp, p.Trp999Cys, and p.Arg1046His may disrupt an aromatic/arginine stack motif, leading to instability of the protein (Sula et al., 2014). DYSF mutations result in mRNA instability and degradation which further stimulates the autophagy and proteasome pathways (Barthelemy et al., 2011). Muscles from dysferlinopathy patients show elevated MuRF-1, LC3-II, p62/SQSTM1 and Bnip3 levels, and fiber atrophy phenotypically. Protein aggregates are found in these muscle fibers which is stained positive for p62. These observations suggest that altered proteasomal degradation of mutant dysferlin and autophagy level (Fanin et al., 2014). Insufficient membrane fusion and accumulation of vesicles have been observed in SJL/J mice carrying a splice-site mutation in the Dysf gene, implicating the role of dysferlin in autophagy process (Hino et al., 2009). Excessive mutant dysferlin may have a detrimental effect on muscle cells. Mutant dysferlin aggregates in the ER and induces autophagosome formation through eukaryotic translation initiation factor 2a (eIF2α) phosphorylation, therefore autophagy/lysosome is an important alternative to the ubiquitin proteasome system for the degradation of excess mutant dysferlin in ER-associated protein degradation (ERAD). Defects in the autophagy pathway lead to a more severe phenotype, exemplified by the increased aggregation of mutant dysferlin in the ER caused by Atg5 deficiency and dephosphorylation of eIF2α (Fujita et al., 2007). In myocytes developed from induced pluripotent stem cells from a patient carrying p.Trp999Cys mutation, nocodazole treatment increases dysferlin levels and improves membrane resealing, suggesting that dysferlin degradation may be a potential drug target for the treatment of dysferlinopathy (Kokubu et al., 2019).
LGMD2H is a relatively mild form of myopathies caused by mutations in the gene encoding Tripartite motif-containing protein 32 (TRIM32), a well-known E3 ubiquitin ligase (Shieh et al., 2011). A recent study showed that TRIM32 is required for autophagy induction in response to atrophic stimuli in vivo in mouse models. At the molecular level, TRIM32 interacts with the autophagy proteins AMBRA1 and ULK1, and thereby stimulates ULK1 activity through K63-linked ubiquitin chains. The pathogenic TRIM32 mutant p.Val591Met found in LGMD2H patients disrupts the interaction between TRIM32 and ULK1, therefore inhibiting autophagy induction, which leads to the exacerbated atrophy exhibited by an elevated level of ROS production and MuRF1 expression (Di Rienzo et al., 2019a, b). Another study showed that the disease-associated mutants p.Pro130Ser, p.Asp487Asn, p.Arg394His, and p.Val591Met inhibit autophagic degradation of p62/SQSTM1 in muscle cells (Overa et al., 2019). These studies link the stress of muscle inactivity caused by defects in ubiquitination to the impaired induction of autophagy machinery.
Sporadic inclusion body myositis (sIBM) is the most common form of acquired myopathy among adults aged over 50 years. Similar to Alzheimer’s disease, sIBM patients present the pathological feature of sporadic inclusion body myositis, which is characterized by abnormal accumulation of amyloid precursor protein (APP) and its proteolytic fragment, amyloid-β (Aβ). It has been shown that APP colocalizes with Atg8/LC3, and APP/beta-amyloid-containing autophagosomes are increased in muscle fibers of sIBM muscle biopsies, suggesting that the autophagy pathway is essential for the degradation of APP/beta-amyloid (Lunemann et al., 2007). sIBM tissues demonstrate damaged myofibres with obvious accumulation of p62/SQSTM1 and TDP-43 (Weihl et al., 2008; Salajegheh et al., 2009). Mechanistic study shows that the altered binding of the p62-ubiquitinated protein complex to LC3 in sIBM patients results in the early termination of autophagy at initiation stage, resulting in p62 protein aggregates (Nakano et al., 2017). Study of an animal model shows that resistance exercise may induce a hypertrophy signal, and alleviate autophagy and muscle atrophy, suggesting a preventive approach for sIBM (Jeong et al., 2017).
In summary, skeletal muscle is the most abundant tissue type in the human body, maintaining a finely tuned metabolism balance between catabolic and anabolic processes. Autophagy is an essential catabolic process responsible for the degradation of proteins and cellular organelles through the autophagosome-lysosome system with the involvement of evolutionally conserved ATG proteins. Autophagy plays a central role in maintaining cellular homeostasis through complex interactions with diverse signaling pathways under physiological conditions and metabolic stress in skeleton muscles. A basal level of autophagy is required for homeostasis in skeletal muscles due to the frequent turnover of protein and cytoplasmic components. Both deficient and excessive autophagy result in a pathological cascade and lead to muscular weakness and atrophy symptoms. Abnormal autophagy levels may also contribute to cell damage. Both increased formation of autophagosomes and inhibition of lysosome-autophagosome fusion cause myopathy. Mutations in autophagy genes and deregulation of autophagy pathways have been identified as one of the major causes of various muscle disorders.
Although advances have been made in understanding the role of autophagy in skeletal muscle disorders over the past two decades, much remains to be elucidated regarding the molecular mechanisms underlying abnormal autophagy activity in skeletal muscle disorders. Traditional experimental techniques employed in studies of other cell and tissue types can be utilized further to examine the contribution of autophagy to skeletal muscle disorders, especially to investigate whether the contribution is cell-type and tissue-type specific. Multi-omics techniques achieved astonishing advancement during the past 10 years and have been widely applied to the studies of complex human diseases. Future studies of skeletal muscle diseases can adopt these omics approaches. High-throughput sequencing at the DNA level can be applied to identify additional causal mutations, which may help to elucidate the underlying mutation landscape of skeletal muscle diseases. RNA sequencing of patient biopsies will generate altered expression profiling under different disease conditions compared to normal physiological conditions. For skeletal muscle diseases, proteomics studies to examine protein modification changes such as phosphorylation and ubiquitination levels, and metabolomics studies are particularly important. As we can see from the literature that we have reviewed, skeletal muscle disorders are clearly related to cellular metabolism. The change in autophagy and its interaction with other signaling pathways is reflected not only in gene expression but also in post-translational modification at the protein level. The application of multi-omics techniques will give a broader view of the influence of autophagy on skeletal muscle disorders and a deeper understanding of the contribution of autophagy to the pathogenesis of the diseases. Further understanding of the molecular and biochemical mechanisms underlying the role of autophagy in skeletal muscle disorders will help to develop new interventional and therapeutic strategies for the diseases.
YW and JL were responsible for conception and design of study. QX, XH, JH, YZ, and MM conducted literature search, summarization, and drafted the manuscript. YW and JL revised the manuscript. All authors have read and approved the manuscript.
This work was supported by the National Natural Science Foundation of China (81771769 and 32070718) and Guangzhou Key Medical Discipline Construction Project Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alers, S., Loffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 32, 2–11. doi: 10.1128/mcb.06159-11
Amthor, H., Otto, A., Vulin, A., Rochat, A., Dumonceaux, J., Garcia, L., et al. (2009). Muscle hypertrophy driven by myostatin blockade does not require stem/precursor-cell activity. Proc. Natl. Acad. Sci. U.S.A. 106, 7479–7484. doi: 10.1073/pnas.0811129106
Aoki, M. (1993). “Dysferlinopathy,” in GeneReviews, eds M. P. Adam, H. H. Ardinger, and R. A. Pagon (Seattle, WA:University of Washington).
Bachman, J. F., Klose, A., Liu, W., Paris, N. D., Blanc, R. S., Schmalz, M., et al. (2018). Prepubertal skeletal muscle growth requires Pax7-expressing satellite cell-derived myonuclear contribution. Development 145:dev167197. doi: 10.1242/dev.167197
Baehr, L. M., Furlow, J. D., and Bodine, S. C. (2011). Muscle sparing in muscle RING finger 1 null mice: response to synthetic glucocorticoids. J. Physiol. 589(Pt 19), 4759–4776. doi: 10.1113/jphysiol.2011.212845
Bakkar, N., and Guttridge, D. C. (2010). NF-kappaB signaling: a tale of two pathways in skeletal myogenesis. Physiol. Rev. 90, 495–511.
Bakkar, N., Wang, J., Ladner, K. J., Wang, H., Dahlman, J. M., Carathers, M., et al. (2008). IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J. Cell Biol. 180, 787–802. doi: 10.1083/jcb.200707179
Balaburski, G. M., Hontz, R. D., and Murphy, M. E. (2010). p53 and ARF: unexpected players in autophagy. Trends Cell Biol. 20, 363–369.
Balderhaar, H. J., and Ungermann, C. (2013). CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J. Cell Sci. 126(Pt 6), 1307–1316. doi: 10.1242/jcs.107805
Barthelemy, F., Wein, N., Krahn, M., Levy, N., and Bartoli, M. (2011). Translational research and therapeutic perspectives in dysferlinopathies. Mol. Med. 17, 875–882. doi: 10.2119/molmed.2011.00084
Begalli, F., Bennett, J., Capece, D., Verzella, D., D’Andrea, D., Tornatore, L., et al. (2017). Unlocking the NF-kappaB conundrum: embracing complexity to achieve specificity. Biomedicines 5:50. doi: 10.3390/biomedicines5030050
Bell, R. A., Al-Khalaf, M., and Megeney, L. A. (2016). The beneficial role of proteolysis in skeletal muscle growth and stress adaptation. Skelet. Muscle 6:16.
Ben-Neriah, Y., and Karin, M. (2011). Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 12, 715–723. doi: 10.1038/ni.2060
Bennett, J., Capece, D., Begalli, F., Verzella, D., D’Andrea, D., Tornatore, L., et al. (2018). NF-kappaB in the crosshairs: rethinking an old riddle. Int. J. Biochem. Cell Biol. 95, 108–112. doi: 10.1016/j.biocel.2017.12.020
Bento, C. F., Renna, M., Ghislat, G., Puri, C., Ashkenazi, A., Vicinanza, M., et al. (2016). Mammalian autophagy: how does it work?”. Annu. Rev. Biochem. 85, 685–713.
Bibee, K. P., Cheng, Y. J., Ching, J. K., Marsh, J. N., Li, A. J., Keeling, R. M., et al. (2014). Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASEB J. 28, 2047–2061. doi: 10.1096/fj.13-237388
Blaauw, B., Canato, M., Agatea, L., Toniolo, L., Mammucari, C., Masiero, E., et al. (2009). Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 23, 3896–3905. doi: 10.1096/fj.09-131870
Bodine, S. C., Latres, E., Baumhueter, S., Lai, V. K., Nunez, L., Clarke, B. A., et al. (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. doi: 10.1126/science.1065874
Bolster, D. R., Crozier, S. J., Kimball, S. R., and Jefferson, L. S. (2002). AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 277, 23977–23980. doi: 10.1074/jbc.c200171200
Bonaldo, P., and Sandri, M. (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 6, 25–39. doi: 10.1242/dmm.010389
Bonnemann, C. G. (2011). The collagen VI-related myopathies ullrich congenital muscular dystrophy and bethlem myopathy. Handb. Clin. Neurol. 101, 81–96.
Brack, A. S., and Rando, T. A. (2012). Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell 10, 504–514. doi: 10.1016/j.stem.2012.04.001
Brambatti, M., Caspi, O., Maolo, A., Koshi, E., Greenberg, B., Taylor, M. R. G., et al. (2019). Danon disease: gender differences in presentation and outcomes. Int. J. Cardiol. 286, 92–98. doi: 10.1016/j.ijcard.2019.01.020
Britto, F. A., Begue, G., Rossano, B., Docquier, A., Vernus, B., Sar, C., et al. (2014). REDD1 deletion prevents dexamethasone-induced skeletal muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 307, E983–E993.
Burman, C., and Ktistakis, N. T. (2010). Autophagosome formation in mammalian cells. Semin. Immunopathol. 32, 397–413. doi: 10.1007/s00281-010-0222-z
Cai, D., Frantz, J. D., Tawa, N. E. Jr., Melendez, P. A., Oh, B. C., Lidov, H. G., et al. (2004). IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119, 285–298. doi: 10.1016/j.cell.2004.09.027
Cannata Serio, M., Graham, L. A., Ashikov, A., Larsen, L. E., Raymond, K., Timal, S., et al. (2020). sMutations in the V-ATPase assembly factor VMA21 cause a congenital disorder of glycosylation with autophagic liver disease. Hepatology 72, 1968–1986. doi: 10.1002/hep.31218
Cao, P. R., Kim, H. J., and Lecker, S. H. (2005). Ubiquitin-protein ligases in muscle wasting. Int. J. Biochem. Cell Biol. 37, 2088–2097. doi: 10.1016/j.biocel.2004.11.010
Carlsson, S. R., Roth, J., Piller, F., and Fukuda, M. (1988). Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Major sialoglycoproteins carrying polylactosaminoglycan. J. Biol. Chem. 263, 18911–18919. doi: 10.1016/s0021-9258(18)37369-1
Carmignac, V., Svensson, M., Korner, Z., Elowsson, L., Matsumura, C., Gawlik, K. I., et al. (2011). Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 20, 4891–4902.
Carnio, S., Loverso, F., Baraibar, M. A., Longa, E., Khan, M. M., Maffei, M., et al. (2014). Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 8, 1509–1521. doi: 10.1016/j.celrep.2014.07.061
Castets, P., Lin, S., Rion, N., Di Fulvio, S., Romanino, K., Guridi, M., et al. (2013). Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 17, 731–744.
Chakkalakal, J. V., Jones, K. M., Basson, M. A., and Brack, A. S. (2012). The aged niche disrupts muscle stem cell quiescence. Nature 490, 355–360. doi: 10.1038/nature11438
Chanoine, C., d’Albis, A., Lenfant-Guyot, M., Janmot, C., and Gallien, C. L. (1987). Regulation by thyroid hormones of terminal differentiation in the skeletal dorsal muscle. IUrodelan, I., amphibians. Dev. Biol. 123, 33–42. doi: 10.1016/0012-1606(87)90424-6
Chi, M. M., Hintz, C. S., McKee, D., Felder, S., Grant, N., Kaiser, K. K., et al. (1987). Effect of Duchenne muscular dystrophy on enzymes of energy metabolism in individual muscle fibers. Metabolism 36, 761–767. doi: 10.1016/0026-0495(87)90113-2
Chinet, A. E., Even, P. C., and Decrouy, A. (1994). Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Experientia 50, 602–605. doi: 10.1007/bf01921731
Clop, A., Marcq, F., Takeda, H., Pirottin, D., Tordoir, X., Bibe, B., et al. (2006). A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 38, 813–818. doi: 10.1038/ng1810
Comb, W. C., Cogswell, P., Sitcheran, R., and Baldwin, A. S. (2011). IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene 30, 1727–1732. doi: 10.1038/onc.2010.553
Copetti, T., Bertoli, C., Dalla, E., Demarchi, F., and Schneider, C. (2009). p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell Biol. 29, 2594–2608. doi: 10.1128/mcb.01396-08
Cosgrove, B. D., Gilbert, P. M., Porpiglia, E., Mourkioti, F., Lee, S. P., Corbel, S. Y., et al. (2014). Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 20, 255–264. doi: 10.1038/nm.3464
Crisafulli, S., Sultana, J., Fontana, A., Salvo, F., Messina, S., and Trifiro, G. (2020). Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J. Rare Dis. 15:141.
Crockett, C. D., Ruggieri, A., Gujrati, M., Zallek, C. M., Ramachandran, N., Minassian, B. A., et al. (2014). Late adult-onset of X-linked myopathy with excessive autophagy. Muscle Nerve 50, 138–144. doi: 10.1002/mus.24197
Cuervo, A. M., and Dice, J. F. (2000). Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 113(Pt 24), 4441–4450.
Cui, L., Zhao, L. P., Ye, J. Y., Yang, L., Huang, Y., Jiang, X. P., et al. (2020). The lysosomal membrane protein Lamp2 alleviates lysosomal cell death by promoting autophagic flux in ischemic cardiomyocytes. Front. Cell Dev. Biol. 8:31. doi: 10.3389/fcell.2020.00031
D’Agostino, M., Risselada, H. J., Lurick, A., Ungermann, C., and Mayer, A. (2017). A tethering complex drives the terminal stage of SNARE-dependent membrane fusion. Nature 551, 634–638. doi: 10.1038/nature24469
Dasouki, M., Jawdat, O., Almadhoun, O., Pasnoor, M., McVey, A. L., Abuzinadah, A., et al. (2014). Pompe disease: literature review and case series. Neurol. Clin. 32, 751–776.
De Palma, C., Morisi, F., Cheli, S., Pambianco, S., Cappello, V., Vezzoli, M., et al. (2012). Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 3:e418.
de Theije, C. C., Schols, A., Lamers, W. H., Ceelen, J. J. M., van Gorp, R. H., Hermans, J. J. R., et al. (2018). Glucocorticoid receptor signaling impairs protein turnover regulation in hypoxia-induced muscle atrophy in male mice. Endocrinology 159, 519–534. doi: 10.1210/en.2017-00603
Delorme-Axford, E., and Klionsky, D. J. (2019). On the edge of degradation: autophagy regulation by RNA decay. Wiley Interdiscip. Rev. RNA 10:e1522. doi: 10.1002/wrna.1522
Di Malta, C., Cinque, L., and Settembre, C. (2019). Transcriptional regulation of autophagy: mechanisms and diseases. Front. Cell Dev. Biol. 7:114. doi: 10.3389/fcell.2019.00114
Di Mauro, S., Angelini, C., and Catani, C. (1967). Enzymes of the glycogen cycle and glycolysis in various human neuromuscular disorders. J. Neurol Neurosurg. Psychiatry 30, 411–415. doi: 10.1136/jnnp.30.5.411
Di Rienzo, M., Antonioli, M., Fusco, C., Liu, Y., Mari, M., Orhon, I., et al. (2019a). Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci. Adv. 5:eaau8857. doi: 10.1126/sciadv.aau8857
Di Rienzo, M., Piacentini, M., and Fimia, G. M. (2019b). A TRIM32-AMBRA1-ULK1 complex initiates the autophagy response in atrophic muscle cells. Autophagy 15, 1674–1676. doi: 10.1080/15548627.2019.1635385
Djavaheri-Mergny, M., Amelotti, M., Mathieu, J., Besancon, F., Bauvy, C., Souquere, S., et al. (2006). NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 281, 30373–30382. doi: 10.1074/jbc.m602097200
Dogra, C., Changotra, H., Wedhas, N., Qin, X., Wergedal, J. E., and Kumar, A. (2007). TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 21, 1857–1869. doi: 10.1096/fj.06-7537com
Dreyfus, J. C., Schapira, G., and Schapira, F. (1954). Biochemical study of muscle in progressive muscular dystrophy. J. Clin. Invest. 33, 794–797. doi: 10.1172/jci102950
D’Souza, R. S., Levandowski, C., Slavov, D., Graw, S. L., Allen, L. A., Adler, E., et al. (2014). Danon disease: clinical features, evaluation, and management. Circ. Heart Fail. 7, 843–849. doi: 10.1161/circheartfailure.114.001105
Ebato, C., Uchida, T., Arakawa, M., Komatsu, M., Ueno, T., Komiya, K., et al. (2008). Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 8, 325–332. doi: 10.1016/j.cmet.2008.08.009
Edelstein, L. C., Lagos, L., Simmons, M., Tirumalai, H., and Gelinas, C. (2003). NF-kappa B-dependent assembly of an enhanceosome-like complex on the promoter region of apoptosis inhibitor Bfl-1/A1. Mol. Cell Biol. 23, 2749–2761. doi: 10.1128/mcb.23.8.2749-2761.2003
Emery, A. E. (1991). Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul. Disord. 1, 19–29. doi: 10.1016/0960-8966(91)90039-u
England, B. K., and Price, S. R. (1995). Acidosis and glucocorticoids interact to provoke muscle protein and amino acid catabolism. Blood Purif. 13, 147–152. doi: 10.1159/000170197
Eskelinen, E. L. (2006). Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Aspects Med. 27, 495–502. doi: 10.1016/j.mam.2006.08.005
Eskelinen, E. L., Illert, A. L., Tanaka, Y., Schwarzmann, G., Blanz, J., Von Figura, K., et al. (2002). Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 13, 3355–3368.
Fan, W., Nassiri, A., and Zhong, Q. (2011). Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc. Natl. Acad. Sci. U.S.A. 108, 7769–7774. doi: 10.1073/pnas.1016472108
Fanin, M., Nascimbeni, A. C., and Angelini, C. (2014). Muscle atrophy, ubiquitin-proteasome, and autophagic pathways in dysferlinopathy. Muscle Nerve 50, 340–347. doi: 10.1002/mus.24167
Favaro, G., Romanello, V., Varanita, T., Morbidoni, V., Tezze, C., Albiero, M., et al. (2019). DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 10:2576.
Fernandes, S. A., Almeida, C. F., Souza, L. S., Lazar, M., Onofre-Oliveira, P., Yamamoto, G. L., et al. (2020). Altered in vitro muscle differentiation in X-linked myopathy with excessive autophagy. Dis. Model. Mech. 13:dmm041244. doi: 10.1242/dmm.041244
Filimonenko, M., Stuffers, S., Raiborg, C., Yamamoto, A., Malerod, L., Fisher, E. M., et al. (2007). Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 179, 485–500.
Flanigan, K. M., Dunn, D. M., von Niederhausern, A., Soltanzadeh, P., Gappmaier, E., Howard, M. T., et al. (2009). Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum. Mutat. 30, 1657–1666.
Forgac, M. (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929.
Frontera, W. R., and Ochala, J. (2015). Skeletal muscle: a brief review of structure and function. Calcif. Tissue Int. 96, 183–195.
Fujita, E., Kouroku, Y., Isoai, A., Kumagai, H., Misutani, A., Matsuda, C., et al. (2007). Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 16, 618–629. doi: 10.1093/hmg/ddm002
Fujita, N., Itoh, T., Omori, H., Fukuda, M., Noda, T., and Yoshimori, T. (2008). The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19, 2092–2100. doi: 10.1091/mbc.e07-12-1257
Fukada, S. I. (2018). The roles of muscle stem cells in muscle injury, atrophy and hypertrophy. J. Biochem. 163, 353–358. doi: 10.1093/jb/mvy019
Fukuda, T., Ahearn, M., Roberts, A., Mattaliano, R. J., Zaal, K., Ralston, E., et al. (2006). Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol. Ther. 14, 831–839. doi: 10.1016/j.ymthe.2006.08.009
Funderburk, S. F., Wang, Q. J., and Yue, Z. (2010). The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362. doi: 10.1016/j.tcb.2010.03.002
Furuyama, T., Kitayama, K., Yamashita, H., and Mori, N. (2003). Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 375(Pt 2), 365–371.
Gao, Q. Q., and McNally, E. M. (2015). The dystrophin complex: structure, function, and implications for therapy. Compr. Physiol. 5, 1223–1239. doi: 10.1002/cphy.c140048
Garcia-Prat, L., Martinez-Vicente, M., Perdiguero, E., Ortet, L., Rodriguez-Ubreva, J., Rebollo, E., et al. (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. doi: 10.1038/nature16187
Ghavami, S., Cunnington, R. H., Gupta, S., Yeganeh, B., Filomeno, K. L., Freed, D. H., et al. (2015). Autophagy is a regulator of TGF-beta1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. 6:e1696. doi: 10.1038/cddis.2015.36
Gomes, M. D., Lecker, S. H., Jagoe, R. T., Navon, A., and Goldberg, A. L. (2001). Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U.S.A. 98, 14440–14445. doi: 10.1073/pnas.251541198
Gong, G., Song, M., Csordas, G., Kelly, D. P., Matkovich, S. J., and Dorn, G. W. II (2015). Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350:aad2459.
Goodman, C. A., McNally, R. M., Hoffmann, F. M., and Hornberger, T. A. (2013). Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol. Endocrinol. 27, 1946–1957. doi: 10.1210/me.2013-1194
Griffin, J. L. (1984). Infantile acid maltase deficiency. Muscle, I., fiber destruction after lysosomal rupture. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 45, 23–36.
Grobet, L., Martin, L. J., Poncelet, D., Pirottin, D., Brouwers, B., Riquet, J., et al. (1997). A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat. Genet. 17, 71–74.
Grobet, L., Poncelet, D., Royo, L. J., Brouwers, B., Pirottin, D., Michaux, C., et al. (1998). Molecular definition of an allelic series of mutations disrupting the myostatin function and causing double-muscling in cattle. Mamm. Genome 9, 210–213.
Grumati, P., and Bonaldo, P. (2012). Autophagy in skeletal muscle homeostasis and in muscular dystrophies. Cells 1, 325–345. doi: 10.3390/cells1030325
Grumati, P., Coletto, L., Sabatelli, P., Cescon, M., Angelin, A., Bertaggia, E., et al. (2010). Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 16, 1313–1320. doi: 10.1038/nm.2247
Grumati, P., Coletto, L., Schiavinato, A., Castagnaro, S., Bertaggia, E., Sandri, M., et al. (2011). Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 7, 1415–1423. doi: 10.4161/auto.7.12.17877
Guimaraes, R. S., Delorme-Axford, E., Klionsky, D. J., and Reggiori, F. (2015). Assays for the biochemical and ultrastructural measurement of selective and nonselective types of autophagy in the yeast Saccharomyces cerevisiae. Methods 75, 141–150.
Gurka, J., Piherova, L., Majer, F., Chaloupka, A., Zakova, D., Pelak, O., et al. (2020). Danon disease is an underdiagnosed cause of advanced heart failure in young female patients: a LAMP2 flow cytometric study. ESC Heart Fail. 7, 2534–2543. doi: 10.1002/ehf2.12823
Guttridge, D. C., Albanese, C., Reuther, J. Y., Pestell, R. G., and Baldwin, A. S. Jr. (1999). NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell Biol. 19, 5785–5799.
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Hansen-Smith, F. M., Picou, D., and Golden, M. H. (1979). Muscle satellite cells in malnourished and nutritionally rehabilitated children. J. Neurol. Sci. 41, 207–221. doi: 10.1016/0022-510x(79)90040-6
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Harding, T. M., Morano, K. A., Scott, S. V., and Klionsky, D. J. (1995). Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 131, 591–602. doi: 10.1083/jcb.131.3.591
Hasselgren, P. O. (1999). Glucocorticoids and muscle catabolism. Curr. Opin. Clin. Nutr. Metab. Care 2, 201–205. doi: 10.1097/00075197-199905000-00002
Hayden, M. S., and Ghosh, S. (2012). NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234. doi: 10.1101/gad.183434.111
He, C., Bassik, M. C., Moresi, V., Sun, K., Wei, Y., Zou, Z., et al. (2012). Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515. doi: 10.1038/nature10758
Hess, J. W. (1965). Phosphorylase activity and glycogen, glucose-6-phosphate, and lactic acid content of human skeletal muscle in various myopathies. J. Lab. Clin. Med. 66, 452–463.
Hindi, S. M., Sato, S., Choi, Y., and Kumar, A. (2014). Distinct roles of TRAF6 at early and late stages of muscle pathology in the mdx model of Duchenne muscular dystrophy. Hum. Mol. Genet. 23, 1492–1505. doi: 10.1093/hmg/ddt536
Hino, M., Hamada, N., Tajika, Y., Funayama, T., Morimura, Y., Sakashita, T., et al. (2009). Insufficient membrane fusion in dysferlin-deficient muscle fibers after heavy-ion irradiation. Cell Struct. Funct. 34, 11–15. doi: 10.1247/csf.08033
Hollinger, K., Gardan-Salmon, D., Santana, C., Rice, D., Snella, E., and Selsby, J. T. (2013). Rescue of dystrophic skeletal muscle by PGC-1alpha involves restored expression of dystrophin-associated protein complex components and satellite cell signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R13–R23.
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991. doi: 10.1091/mbc.e08-12-1248
Huie, M. L., Chen, A. S., Tsujino, S., Shanske, S., DiMauro, S., Engel, A. G., et al. (1994). Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (-13T–>G) mutation in a majority of patients and a novel IVS10 (+1GT–>CT) mutation. Hum. Mol. Genet. 3, 2231–2236. doi: 10.1093/hmg/3.12.2231
Itakura, E., and Mizushima, N. (2009). Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy 5, 534–536. doi: 10.4161/auto.5.4.8062
Itakura, E., Kishi, C., Inoue, K., and Mizushima, N. (2008). Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 19, 5360–5372. doi: 10.1091/mbc.e08-01-0080
Itakura, E., Kishi-Itakura, C., and Mizushima, N. (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269. doi: 10.1016/j.cell.2012.11.001
Izumi, R., Takahashi, T., Suzuki, N., Niihori, T., Ono, H., Nakamura, N., et al. (2020). The genetic profile of dysferlinopathy in a cohort of 209 cases: genotype-phenotype relationship and a hotspot on the inner DysF domain. Hum. Mutat. 41, 1540–1554. doi: 10.1002/humu.24036
Jacinto, E., Loewith, R., Schmidt, A., Lin, S., Ruegg, M. A., Hall, A., et al. (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128. doi: 10.1038/ncb1183
Jackson, J. R., Mula, J., Kirby, T. J., Fry, C. S., Lee, J. D., Ubele, M. F., et al. (2012). Satellite cell depletion does not inhibit adult skeletal muscle regrowth following unloading-induced atrophy. Am. J. Physiol. Cell Physiol. 303, C854–C861.
Jagoe, R. T., and Engelen, M. P. (2003). Muscle wasting and changes in muscle protein metabolism in chronic obstructive pulmonary disease. Eur. Respir. J. Suppl. 46, 52s–63s.
Jagoe, R. T., Lecker, S. H., Gomes, M., and Goldberg, A. L. (2002). Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J. 16, 1697–1712. doi: 10.1096/fj.02-0312com
Jeong, J. H., Yang, D. S., Koo, J. H., Hwang, D. J., Cho, J. Y., and Kang, E. B. (2017). Effect of resistance exercise on muscle metabolism and autophagy in sIBM. Med. Sci. Sports Exerc. 49, 1562–1571. doi: 10.1249/mss.0000000000001286
Jung, H. S., Chung, K. W., Won Kim, J., Kim, J., Komatsu, M., Tanaka, K., et al. (2008). Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 8, 318–324. doi: 10.1016/j.cmet.2008.08.013
Kaliman, P., Canicio, J., Testar, X., Palacin, M., and Zorzano, A. (1999). Insulin-like growth factor-II, phosphatidylinositol 3-kinase, nuclear factor-kappaB and inducible nitric-oxide synthase define a common myogenic signaling pathway. J. Biol. Chem. 274, 17437–17444. doi: 10.1074/jbc.274.25.17437
Kalimo, H., Savontaus, M. L., Lang, H., Paljarvi, L., Sonninen, V., Dean, P. B., et al. (1988). X-linked myopathy with excessive autophagy: a new hereditary muscle disease. Ann. Neurol. 23, 258–265. doi: 10.1002/ana.410230308
Kambadur, R., Sharma, M., Smith, T. P., and Bass, J. J. (1997). Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 7, 910–916. doi: 10.1101/gr.7.9.910
Kang, C., You, Y. J., and Avery, L. (2007). Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 21, 2161–2171. doi: 10.1101/gad.1573107
Kathania, M., Raje, C. I., Raje, M., Dutta, R. K., and Majumdar, S. (2011). Bfl-1/A1 acts as a negative regulator of autophagy in mycobacteria infected macrophages. Int. J. Biochem. Cell Biol. 43, 573–585. doi: 10.1016/j.biocel.2010.12.014
Kihara, A., Noda, T., Ishihara, N., and Ohsumi, Y. (2001). Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 152, 519–530. doi: 10.1083/jcb.152.3.519
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, K. H., Jeong, Y. T., Oh, H., Kim, S. H., Cho, J. M., Kim, Y. N., et al. (2013). Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 19, 83–92. doi: 10.1038/nm.3014
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222.
Koenig, M., Hoffman, E. P., Bertelson, C. J., Monaco, A. P., Feener, C., and Kunkel, L. M. (1987). Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50, 509–517. doi: 10.1016/0092-8674(87)90504-6
Koh, H. J., Arnolds, D. E., Fujii, N., Tran, T. T., Rogers, M. J., Jessen, N., et al. (2006). Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol. Cell Biol. 26, 8217–8227. doi: 10.1128/mcb.00979-06
Kohler, L., Puertollano, R., and Raben, N. (2018). Pompe disease: from basic science to therapy. Neurotherapeutics 15, 928–942. doi: 10.1007/s13311-018-0655-y
Kokubu, Y., Nagino, T., Sasa, K., Oikawa, T., Miyake, K., Kume, A., et al. (2019). Phenotypic drug screening for dysferlinopathy using patient-derived induced pluripotent stem cells. Stem Cells Transl. Med. 8, 1017–1029. doi: 10.1002/sctm.18-0280
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Komatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., et al. (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434. doi: 10.1083/jcb.200412022
Konecki, D. S., Foetisch, K., Zimmer, K. P., Schlotter, M., and Lichter-Konecki, U. (1995). An alternatively spliced form of the human lysosome-associated membrane protein-2 gene is expressed in a tissue-specific manner. Biochem. Biophys. Res. Commun. 215, 757–767. doi: 10.1006/bbrc.1995.2528
Koyama-Honda, I., Itakura, E., Fujiwara, T. K., and Mizushima, N. (2013). Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 9, 1491–1499. doi: 10.4161/auto.25529
Kraft, C., Reggiori, F., and Peter, M. (2009). Selective types of autophagy in yeast. Biochim. Biophys. Acta 1793, 1404–1412. doi: 10.1016/j.bbamcr.2009.02.006
Kriegenburg, F., Ungermann, C., and Reggiori, F. (2018). Coordination of autophagosome-lysosome fusion by Atg8 family members. Curr. Biol. 28, R512–R518.
Lamb, C. A., Yoshimori, T., and Tooze, S. A. (2013). The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774. doi: 10.1038/nrm3696
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Larsson, L., Degens, H., Li, M., Salviati, L., Lee, Y. I., Thompson, W., et al. (2019). Sarcopenia: aging-related loss of muscle mass and function. Physiol. Rev. 99, 427–511.
Lecker, S. H., Goldberg, A. L., and Mitch, W. E. (2006). Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 17, 1807–1819. doi: 10.1681/asn.2006010083
Lecker, S. H., Jagoe, R. T., Gilbert, A., Gomes, M., Baracos, V., Bailey, J., et al. (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB. 18, 39–51. doi: 10.1096/fj.03-0610com
Lee, J. A., Beigneux, A., Ahmad, S. T., Young, S. G., and Gao, F. B. (2007). ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 17, 1561–1567. doi: 10.1016/j.cub.2007.07.029
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Levine, B., and Kroemer, G. (2019). biological functions of autophagy genes: a disease perspective. Cell 176, 11–42. doi: 10.1016/j.cell.2018.09.048
Lichter-Konecki, U., Moter, S. E., Krawisz, B. R., Schlotter, M., Hipke, C., and Konecki, D. S. (1999). Expression patterns of murine lysosome-associated membrane protein 2 (Lamp-2) transcripts during morphogenesis. Differentiation 65, 43–58. doi: 10.1046/j.1432-0436.1999.6510043.x
Lippincott-Schwartz, J., and Fambrough, D. M. (1986). Lysosomal membrane dynamics: structure and interorganellar movement of a major lysosomal membrane glycoprotein. J. Cell Biol. 102, 1593–1605. doi: 10.1083/jcb.102.5.1593
Liu, J., Aoki, M., Illa, I., Wu, C., Fardeau, M., Angelini, C., et al. (1998). Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 20, 31–36. doi: 10.1038/1682
Ljubicic, V., and Jasmin, B. J. (2013). AMP-activated protein kinase at the nexus of therapeutic skeletal muscle plasticity in Duchenne muscular dystrophy. Trends Mol. Med. 19, 614–624. doi: 10.1016/j.molmed.2013.07.002
Long, F., Wang, Y., Qi, H. H., Zhou, X., and Jin, X. Q. (2008). Rapid non-genomic effects of glucocorticoids on oxidative stress in a guinea pig model of asthma. Respirology 13, 227–232. doi: 10.1111/j.1440-1843.2008.01236.x
Lunemann, J. D., Schmidt, J., Schmid, D., Barthel, K., Wrede, A., Dalakas, M. C., et al. (2007). Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann. Neurol. 61, 476–483. doi: 10.1002/ana.21115
Maejima, Y., Kyoi, S., Zhai, P., Liu, T., Li, H., Ivessa, A., et al. (2013). Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 19, 1478–1488. doi: 10.1038/nm.3322
Malicdan, M. C., Noguchi, S., Nonaka, I., Saftig, P., and Nishino, I. (2008). Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromuscul. Disord. 18, 521–529. doi: 10.1016/j.nmd.2008.04.010
Malkus, P., Graham, L. A., Stevens, T. H., and Schekman, R. (2004). Role of Vma21p in assembly and transport of the yeast vacuolar ATPase. Mol. Biol. Cell 15, 5075–5091. doi: 10.1091/mbc.e04-06-0514
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. doi: 10.1016/j.cmet.2007.11.001
Masiero, E., Agatea, L., Mammucari, C., Blaauw, B., Loro, E., Komatsu, M., et al. (2009). Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515. doi: 10.1016/j.cmet.2009.10.008
Matsui, Y., Takagi, H., Qu, X., Abdellatif, M., Sakoda, H., Asano, T., et al. (2007). Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 100, 914–922. doi: 10.1161/01.res.0000261924.76669.36
Matsunaga, K., Saitoh, T., Tabata, K., Omori, H., Satoh, T., Kurotori, N., et al. (2009). Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396. doi: 10.1038/ncb1846
McCarthy, J. J., Mula, J., Miyazaki, M., Erfani, R., Garrison, K., Farooqui, A. B., et al. (2011). Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 138, 3657–3666. doi: 10.1242/dev.068858
McGrath, J. A., and Goldspink, D. F. (1982). Glucocorticoid action on protein synthesis and protein breakdown in isolated skeletal muscles. Biochem. J. 206, 641–645. doi: 10.1042/bj2060641
McLoughlin, T. J., Smith, S. M., DeLong, A. D., Wang, H., Unterman, T. G., and Esser, K. A. (2009). FoxO1 induces apoptosis in skeletal myotubes in a DNA-binding-dependent manner. Am. J. Physiol. Cell Physiol. 297, C548–C555.
McPherron, A. C., and Lee, S. J. (1997). Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. U.S.A. 94, 12457–12461. doi: 10.1073/pnas.94.23.12457
McPherron, A. C., Lawler, A. M., and Lee, S. J. (1997). Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387, 83–90. doi: 10.1038/387083a0
Meena, N. K., and Raben, N. (2020). Pompe disease: new developments in an old lysosomal storage disorder. Biomolecules 10:1339. doi: 10.3390/biom10091339
Milan, G., Romanello, V., Pescatore, F., Armani, A., Paik, J. H., Frasson, L., et al. (2015). Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 6:6670.
Minassian, B. A., Aiyar, R., Alic, S., Banwell, B., Villanova, M., Fardeau, M., et al. (2002). Narrowing in on the causative defect of an intriguing X-linked myopathy with excessive autophagy. Neurology 59, 596–601. doi: 10.1212/wnl.59.4.596
Mitch, W. E., Bailey, J. L., Wang, X., Jurkovitz, C., Newby, D., and Price, S. R. (1999). Evaluation of signals activating ubiquitin-proteasome proteolysis in a model of muscle wasting. Am. J. Physiol. 276, C1132–C1138.
Mitin, N., Kudla, A. J., Konieczny, S. F., and Taparowsky, E. J. (2001). Differential effects of Ras signaling through NFkappaB on skeletal myogenesis. Oncogene 20, 1276–1286. doi: 10.1038/sj.onc.1204223
Miyoshi, K., Kawai, H., Iwasa, M., Kusaka, K., and Nishino, H. (1986). Autosomal recessive distal muscular dystrophy as a new type of progressive muscular dystrophy. Seventeen cases in eight families including an autopsied case. Brain 109(Pt 1), 31–54. doi: 10.1093/brain/109.1.31
Mizushima, N. (2005). The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 12(Suppl. 2), 1535–1541. doi: 10.1038/sj.cdd.4401728
Molitoris, J. K., McColl, K. S., Swerdlow, S., Matsuyama, M., Lam, M., Finkel, T. H., et al. (2011). Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J. Biol. Chem. 286, 30181–30189. doi: 10.1074/jbc.m111.245423
Moreland, R. J., Jin, X., Zhang, X. K., Decker, R. W., Albee, K. L., Lee, K. L., et al. (2005). Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor. J. Biol. Chem. 280, 6780–6791. doi: 10.1074/jbc.m404008200
Morgan, M. J., and Liu, Z. G. (2011). Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 21, 103–115. doi: 10.1038/cr.2010.178
Mosher, D. S., Quignon, P., Bustamante, C. D., Sutter, N. B., Mellersh, C. S., Parker, H. G., et al. (2007). A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 3:e79. doi: 10.1371/journal.pgen.0030079
Mourkioti, F., Kratsios, P., Luedde, T., Song, Y. H., Delafontaine, P., Adami, R., et al. (2006). Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Invest. 116, 2945–2954. doi: 10.1172/jci28721
Mukaiyama, H., Oku, M., Baba, M., Samizo, T., Hammond, A. T., Glick, B. S., et al. (2002). Paz2 and 13 other PAZ gene products regulate vacuolar engulfment of peroxisomes during micropexophagy. Genes Cells 7, 75–90. doi: 10.1046/j.1356-9597.2001.00499.x
Munteanu, I., Kalimo, H., Saraste, A., Nishino, I., and Minassian, B. A. (2017). Cardiac autophagic vacuolation in severe X-linked myopathy with excessive autophagy. Neuromuscul. Disord. 27, 185–187. doi: 10.1016/j.nmd.2016.10.007
Munteanu, I., Ramachandran, N., Ruggieri, A., Awaya, T., Nishino, I., and Minassian, B. A. (2015). Congenital autophagic vacuolar myopathy is allelic to X-linked myopathy with excessive autophagy. Neurology 84, 1714–1716. doi: 10.1212/wnl.0000000000001499
Murrow, L., and Debnath, J. (2013). Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu. Rev. Pathol. 8, 105–137. doi: 10.1146/annurev-pathol-020712-163918
Mylka, V., Deckers, J., Ratman, D., De Cauwer, L., Thommis, J., De Rycke, R., et al. (2018). The autophagy receptor SQSTM1/p62 mediates anti-inflammatory actions of the selective NR3C1/glucocorticoid receptor modulator compound A (CpdA) in macrophages. Autophagy 14, 2049–2064. doi: 10.1080/15548627.2018.1495681
Nakai, A., Yamaguchi, O., Takeda, T., Higuchi, Y., Hikoso, S., Taniike, M., et al. (2007). The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624. doi: 10.1038/nm1574
Nakano, S., Oki, M., and Kusaka, H. (2017). The role of p62/SQSTM1 in sporadic inclusion body myositis. Neuromuscul. Disord. 27, 363–369. doi: 10.1016/j.nmd.2016.12.009
Nakatogawa, H. (2013). Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 55, 39–50. doi: 10.1042/bse0550039
Nakatogawa, H. (2014). [Roles and action mechanisms of two ubiquitin-like conjugation systems in autophagy]. Seikagaku 86, 441–451.
Neel, B. A., Lin, Y., and Pessin, J. E. (2013). Skeletal muscle autophagy: a new metabolic regulator. Trends Endocrinol. Metab. 24, 635–643. doi: 10.1016/j.tem.2013.09.004
Nezis, I. P., Vaccaro, M. I., Devenish, R. J., and Juhasz, G. (2014). Autophagy in development, cell differentiation, and homeodynamics: from molecular mechanisms to diseases and pathophysiology. Biomed. Res. Int. 2014:349623.
Nguyen, T. N., Padman, B. S., Usher, J., Oorschot, V., Ramm, G., and Lazarou, M. (2016). Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215, 857–874. doi: 10.1083/jcb.201607039
Nivon, M., Abou-Samra, M., Richet, E., Guyot, B., Arrigo, A. P., and Kretz-Remy, C. (2012). NF-kappaB regulates protein quality control after heat stress through modulation of the BAG3-HspB8 complex. J. Cell Sci. 125(Pt 5), 1141–1151. doi: 10.1242/jcs.091041
Ohsumi, Y. (2014). Historical landmarks of autophagy research. Cell Res. 24, 9–23. doi: 10.1038/cr.2013.169
Ohsumi, Y., and Mizushima, N. (2004). Two ubiquitin-like conjugation systems essential for autophagy. Semin. Cell Dev. Biol. 15, 231–236. doi: 10.1016/j.semcdb.2003.12.004
Oost, L. J., Kustermann, M., Armani, A., Blaauw, B., and Romanello, V. (2019). Fibroblast growth factor 21 controls mitophagy and muscle mass. J. Cachexia Sarcopenia Muscle 10, 630–642. doi: 10.1002/jcsm.12409
Otto, G. P., Wu, M. Y., Kazgan, N., Anderson, O. R., and Kessin, R. H. (2003). Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J. Biol. Chem. 278, 17636–17645. doi: 10.1074/jbc.m212467200
Ou, H., Liu, C., Feng, W., Xiao, X., Tang, S., and Mo, Z. (2018). Role of AMPK in atherosclerosis via autophagy regulation. Sci. China Life Sci. 61, 1212–1221. doi: 10.1007/s11427-017-9240-2
Overa, K. S., Garcia-Garcia, J., Bhujabal, Z., Jain, A., Overvatn, A., Larsen, K. B., et al. (2019). TRIM32, but not its muscular dystrophy-associated mutant, positively regulates and is targeted to autophagic degradation by p62/SQSTM1. J Cell Sci 132:jcs236596. doi: 10.1242/jcs.236596
Palla, A. R., Ravichandran, M., Wang, Y. X., Alexandrova, L., Yang, A. V., Kraft, P., et al. (2021). Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science 371:6528.
Paolini, A., Omairi, S., Mitchell, R., Vaughan, D., Matsakas, A., Vaiyapuri, S., et al. (2018). Attenuation of autophagy impacts on muscle fibre development, starvation induced stress and fibre regeneration following acute injury. Sci. Rep. 8:9062.
Papa, S., Bubici, C., Zazzeroni, F., Pham, C. G., Kuntzen, C., Knabb, J. R., et al. (2006). The NF-kappaB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ. 13, 712–729. doi: 10.1038/sj.cdd.4401865
Papinski, D., and Kraft, C. (2016). Regulation of autophagy by signaling through the Atg1/ULK1 complex. J. Mol. Biol. 428(9 Pt A), 1725–1741. doi: 10.1016/j.jmb.2016.03.030
Parzych, K. R., and Klionsky, D. J. (2014). An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal. 20, 460–473. doi: 10.1089/ars.2013.5371
Parzych, K. R., Ariosa, A., Mari, M., and Klionsky, D. J. (2018). A newly characterized vacuolar serine carboxypeptidase, Atg42/Ybr139w, is required for normal vacuole function and the terminal steps of autophagy in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 29, 1089–1099. doi: 10.1091/mbc.e17-08-0516
Peruzzo, P., Pavan, E., and Dardis, A. (2019). Molecular genetics of Pompe disease: a comprehensive overview. Ann. Transl. Med. 7:278. doi: 10.21037/atm.2019.04.13
Pichavant, C., Aartsma-Rus, A., Clemens, P. R., Davies, K. E., Dickson, G., Takeda, S., et al. (2011). Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol. Ther. 19, 830–840. doi: 10.1038/mt.2011.59
Prior, S. J., Roth, S. M., Wang, X., Kammerer, C., Miljkovic-Gacic, I., Bunker, C. H., et al. (2007). Genetic and environmental influences on skeletal muscle phenotypes as a function of age and sex in large, multigenerational families of African heritage. J. Appl. Physiol. 103, 1121–1127. doi: 10.1152/japplphysiol.00120.2007
Raben, N., Baum, R., Schreiner, C., Takikita, S., Mizushima, N., Ralston, E., et al. (2009). When more is less: excess and deficiency of autophagy coexist in skeletal muscle in Pompe disease. Autophagy 5, 111–113. doi: 10.4161/auto.5.1.7293
Raben, N., Hill, V., Shea, L., Takikita, S., Baum, R., Mizushima, N., et al. (2008). Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 17, 3897–3908. doi: 10.1093/hmg/ddn292
Raben, N., Nichols, R. C., Martiniuk, F., and Plotz, P. H. (1996). A model of mRNA splicing in adult lysosomal storage disease (glycogenosis type II). Hum. Mol. Genet. 5, 995–1000. doi: 10.1093/hmg/5.7.995
Raben, N., Takikita, S., Pittis, M. G., Bembi, B., Marie, S. K., Roberts, A., et al. (2007). Deconstructing Pompe disease by analyzing single muscle fibers: to see a world in a grain of sand. Autophagy 3, 546–552. doi: 10.4161/auto.4591
Raffaello, A., Milan, G., Masiero, E., Carnio, S., Lee, D., Lanfranchi, G., et al. (2010). JunB transcription factor maintains skeletal muscle mass and promotes hypertrophy. J. Cell Biol. 191, 101–113. doi: 10.1083/jcb.201001136
Ramachandran, N., Munteanu, I., Wang, P., Ruggieri, A., Rilstone, J. J., Israelian, N., et al. (2013). VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol. 125, 439–457. doi: 10.1007/s00401-012-1073-6
Rapino, F., Abhari, B. A., Jung, M., and Fulda, S. (2015). NIK is required for NF-kappaB-mediated induction of BAG3 upon inhibition of constitutive protein degradation pathways. Cell Death Dis. 6:e1692. doi: 10.1038/cddis.2014.584
Reggiori, F., Monastyrska, I., Shintani, T., and Klionsky, D. J. (2005). The actin cytoskeleton is required for selective types of autophagy, but not nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 16, 5843–5856. doi: 10.1091/mbc.e05-07-0629
Rodrigues Ade, C., and Schmalbruch, H. (1995). Satellite cells and myonuclei in long-term denervated rat muscles. Anat. Rec. 243, 430–437. doi: 10.1002/ar.1092430405
Romanov, J., Walczak, M., Ibiricu, I., Schuchner, S., Ogris, E., Kraft, C., et al. (2012). Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 31, 4304–4317. doi: 10.1038/emboj.2012.278
Romitti, P. A., Zhu, Y., Puzhankara, S., James, K. A., Nabukera, S. K., Zamba, G. K., et al. (2015). Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics 135, 513–521.
Ruperez, C., Lerin, C., Ferrer-Curriu, G., Cairo, M., Mas-Stachurska, A., Sitges, M., et al. (2018). Autophagic control of cardiac steatosis through FGF21 in obesity-associated cardiomyopathy. Int. J. Cardiol. 260, 163–170. doi: 10.1016/j.ijcard.2018.02.109
Ryder, S., Leadley, R. M., Armstrong, N., Westwood, M., de Kock, S., Butt, T., et al. (2017). The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J. Rare Dis. 12:79.
Saftig, P., Beertsen, W., and Eskelinen, E. L. (2008). LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy 4, 510–512. doi: 10.4161/auto.5724
Saitoh, O., Wang, W. C., Lotan, R., and Fukuda, M. (1992). Differential glycosylation and cell surface expression of lysosomal membrane glycoproteins in sublines of a human colon cancer exhibiting distinct metastatic potentials. J. Biol. Chem. 267, 5700–5711. doi: 10.1016/s0021-9258(18)42823-2
Sakai, Y., Koller, A., Rangell, L. K., Keller, G. A., and Subramani, S. (1998). Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J. Cell Biol. 141, 625–636. doi: 10.1083/jcb.141.3.625
Sakakibara, K., Eiyama, A., Suzuki, S. W., Sakoh-Nakatogawa, M., Okumura, N., Tani, M., et al. (2015). Phospholipid methylation controls Atg32-mediated mitophagy and Atg8 recycling. EMBO J. 34, 2703–2719. doi: 10.15252/embj.201591440
Sakamoto, K., McCarthy, A., Smith, D., Green, K. A., Grahame Hardie, D., Ashworth, A., et al. (2005). Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 24, 1810–1820. doi: 10.1038/sj.emboj.7600667
Salajegheh, M., Pinkus, J. L., Taylor, J. P., Amato, A. A., Nazareno, R., Baloh, R. H., et al. (2009). Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve 40, 19–31. doi: 10.1002/mus.21386
Salminen, A., Hyttinen, J. M., Kauppinen, A., and Kaarniranta, K. (2012). Context-dependent regulation of autophagy by IKK-NF-kappaB signaling: impact on the aging process. Int. J. Cell Biol. 2012:849541.
Sambasivan, R., Yao, R., Kissenpfennig, A., Van Wittenberghe, L., Paldi, A., Gayraud-Morel, B., et al. (2011). Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138, 3647–3656. doi: 10.1242/dev.067587
Sanchez, A. M., Csibi, A., Raibon, A., Cornille, K., Gay, S., Bernardi, H., et al. (2012). AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell Biochem. 113, 695–710. doi: 10.1002/jcb.23399
Sandri, M. (2008). Signaling in muscle atrophy and hypertrophy. Physiology 23, 160–170. doi: 10.1152/physiol.00041.2007
Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., et al. (2004). Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412. doi: 10.1016/s0092-8674(04)00400-3
Saraste, A., Koskenvuo, J. W., Airaksinen, J., Ramachandran, N., Munteanu, I., Udd, B., et al. (2015). No cardiomyopathy in X-linked myopathy with excessive autophagy. Neuromuscul. Disord. 25, 485–487.
Sarbassov, D. D., Ali, S. M., Kim, D. H., Guertin, D. A., Latek, R. R., Erdjument-Bromage, H., et al. (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302. doi: 10.1016/j.cub.2004.06.054
Sarkar, S., Korolchuk, V. I., Renna, M., Imarisio, S., Fleming, A., Williams, A., et al. (2011). Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 43, 19–32.
Sartori, R., Milan, G., Patron, M., Mammucari, C., Blaauw, B., Abraham, R., et al. (2009). Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 296, C1248–C1257.
Schaaf, M. B., Keulers, T. G., Vooijs, M. A., and Rouschop, K. M. (2016). LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 30, 3961–3978. doi: 10.1096/fj.201600698r
Schiaffino, S., and Hanzlikova, V. (1972). Studies on the effect of denervation in developing muscle. IThe, I., lysosomal system. J. Ultrastruct. Res. 39, 1–14.
Schultz, E. (1978). Changes in the satellite cells of growing muscle following denervation. Anat. Rec. 190, 299–311.
Schwartz, L. M., Brown, C., McLaughlin, K., Smith, W., and Bigelow, C. (2016). The myonuclear domain is not maintained in skeletal muscle during either atrophy or programmed cell death. Am. J. Physiol. Cell Physiol. 311, C607–C615.
Scott, R. C., Schuldiner, O., and Neufeld, T. P. (2004). Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 7, 167–178.
Seale, P., Sabourin, L. A., Girgis-Gabardo, A., Mansouri, A., Gruss, P., and Rudnicki, M. A. (2000). Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786.
Shieh, P. B., Kudryashova, E., and Spencer, M. J. (2011). Limb-girdle muscular dystrophy 2H and the role of TRIM32. Handb. Clin. Neurol. 101, 125–133.
Shimizu, N., Yoshikawa, N., Ito, N., Maruyama, T., Suzuki, Y., Takeda, S., et al. (2011). Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 13, 170–182. doi: 10.1016/j.cmet.2011.01.001
Sinadinos, A., Young, C. N., Al-Khalidi, R., Teti, A., Kalinski, P., Mohamad, S., et al. (2015). P2RX7 purinoceptor: a therapeutic target for ameliorating the symptoms of duchenne muscular dystrophy. PLoS Med. 12:e1001888. doi: 10.1371/journal.pmed.1001888
Sousa-Victor, P., Gutarra, S., Garcia-Prat, L., Rodriguez-Ubreva, J., Ortet, L., Ruiz-Bonilla, V., et al. (2014). Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321. doi: 10.1038/nature13013
Spampanato, C., Feeney, E., Li, L., Cardone, M., Lim, J. A., Annunziata, F., et al. (2013). Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 5, 691–706. doi: 10.1002/emmm.201202176
Stahn, C., and Buttgereit, F. (2008). Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 4, 525–533. doi: 10.1038/ncprheum0898
Staiger, H., Keuper, M., Berti, L., Hrabe, M., and de Angelis, and Haring, H. U. (2017). Fibroblast growth factor 21-metabolic role in mice and men. Endocr. Rev. 38, 468–488. doi: 10.1210/er.2017-00016
Sudhakar, J. N., Lu, H. H., Chiang, H. Y., Suen, C. S., Hwang, M. J., Wu, S. Y., et al. (2020). Lumenal Galectin-9-Lamp2 interaction regulates lysosome and autophagy to prevent pathogenesis in the intestine and pancreas. Nat. Commun. 11:4286.
Sula, A., Cole, A. R., Yeats, C., Orengo, C., and Keep, N. H. (2014). Crystal structures of the human Dysferlin inner DysF domain. BMC Struct. Biol. 14:3. doi: 10.1186/1472-6807-14-3
Sun, L., Trausch-Azar, J. S., Muglia, L. J., and Schwartz, A. L. (2008). Glucocorticoids differentially regulate degradation of MyoD and Id1 by N-terminal ubiquitination to promote muscle protein catabolism. Proc. Natl. Acad. Sci. U.S.A. 105, 3339–3344. doi: 10.1073/pnas.0800165105
Suzuki, K., Kubota, Y., Sekito, T., and Ohsumi, Y. (2007). Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12, 209–218. doi: 10.1111/j.1365-2443.2007.01050.x
Szabo, G., Dallmann, G., Muller, G., Patthy, L., Soller, M., and Varga, L. (1998). A deletion in the myostatin gene causes the compact (Cmpt) hypermuscular mutation in mice. Mamm. Genome 9, 671–672. doi: 10.1007/s003359900843
Tanaka, Y., Guhde, G., Suter, A., Eskelinen, E. L., Hartmann, D., Lullmann-Rauch, R., et al. (2000). Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906. doi: 10.1038/35022595
Tang, A. H., and Rando, T. A. (2014). Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 33, 2782–2797. doi: 10.15252/embj.201488278
Tezze, C., Romanello, V., Desbats, M. A., Fadini, G. P., Albiero, M., Favaro, G., et al. (2017). Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 25, 1374.e6–1389.e6.
Thomson, D. M., Hancock, C. R., Evanson, B. G., Kenney, S. G., Malan, B. B., Mongillo, A. D., et al. (2010). Skeletal muscle dysfunction in muscle-specific LKB1 knockout mice. J. Appl. Physiol. 108, 1775–1785. doi: 10.1152/japplphysiol.01293.2009
Thomson, D. M., Porter, B. B., Tall, J. H., Kim, H. J., Barrow, J. R., and Winder, W. W. (2007). Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am. J. Physiol. Endocrinol. Metab. 292, E196–E202.
Thumm, M., Egner, R., Koch, B., Schlumpberger, M., Straub, M., Veenhuis, M., et al. (1994). Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 349, 275–280. doi: 10.1016/0014-5793(94)00672-5
Thurberg, B. L., Lynch Maloney, C., Vaccaro, C., Afonso, K., Tsai, A. C., Bossen, E., et al. (2006). Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab. Invest. 86, 1208–1220. doi: 10.1038/labinvest.3700484
Tian, W., Ye, Z., Wang, S., Schulz, M. A., Van Coillie, J., Sun, L., et al. (2019). The glycosylation design space for recombinant lysosomal replacement enzymes produced in CHO cells. Nat. Commun. 10:1785.
Tiao, G., Fagan, J., Roegner, V., Lieberman, M., Wang, J. J., Fischer, J. E., et al. (1996). Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J. Clin. Invest. 97, 339–348. doi: 10.1172/jci118421
Titorenko, V. I., Keizer, I., Harder, W., and Veenhuis, M. (1995). Isolation and characterization of mutants impaired in the selective degradation of peroxisomes in the yeast Hansenula polymorpha. J. Bacteriol. 177, 357–363. doi: 10.1128/jb.177.2.357-363.1995
Tortora, G. J., and Anagnostakos, N. P. (1987). Principles of Anatomy and Physiology. New York, NY: Harper & Row.
Tsukada, M., and Ohsumi, Y. (1993). Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169–174. doi: 10.1016/0014-5793(93)80398-e
Tuffery-Giraud, S., Beroud, C., Leturcq, F., Yaou, R. B., Hamroun, D., Michel-Calemard, L., et al. (2009). Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum. Mutat. 30, 934–945. doi: 10.1002/humu.20976
Vainshtein, A., Grumati, P., Sandri, M., and Bonaldo, P. (2014). Skeletal muscle, autophagy, and physical activity: the menage a trois of metabolic regulation in health and disease. J. Mol. Med. 92, 127–137. doi: 10.1007/s00109-013-1096-z
Verzella, D., Fischietti, M., Capece, D., Vecchiotti, D., Del Vecchio, F., Cicciarelli, G., et al. (2016). Targeting the NF-kappaB pathway in prostate cancer: a promising therapeutic approach?”. Curr. Drug Targets 17, 311–320. doi: 10.2174/1389450116666150907100715
Viguie, C. A., Lu, D. X., Huang, S. K., Rengen, H., and Carlson, B. M. (1997). Quantitative study of the effects of long-term denervation on the extensor digitorum longus muscle of the rat. Anat. Rec. 248, 346–354. doi: 10.1002/(sici)1097-0185(199707)248:3<346::aid-ar7>3.0.co;2-n
Villanova, M., Louboutin, J. P., Chateau, D., Eymard, B., Sagniez, M., Tome, F. M., et al. (1995). X-linked vacuolated myopathy: complement membrane attack complex on surface membrane of injured muscle fibers. Ann. Neurol. 37, 637–645. doi: 10.1002/ana.410370514
Viollet, B., Andreelli, F., Jorgensen, S. B., Perrin, C., Flamez, D., Mu, J., et al. (2003). Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochem. Soc. Trans. 31(Pt 1), 216–219. doi: 10.1042/bst0310216
Walczak, M., and Martens, S. (2013). Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 9, 424–425. doi: 10.4161/auto.22931
Wang, H., Hertlein, E., Bakkar, N., Sun, H., Acharyya, S., Wang, J., et al. (2007). NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol. Cell Biol. 27, 4374–4387. doi: 10.1128/mcb.02020-06
Wang, H., Kubica, N., Ellisen, L. W., Jefferson, L. S., and Kimball, S. R. (2006). Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J. Biol. Chem. 281, 39128–39134. doi: 10.1074/jbc.m610023200
Wang, Y., Liang, Y., and Vanhoutte, P. M. (2011). SIRT1 and AMPK in regulating mammalian senescence: a critical review and a working model. FEBS Lett. 585, 986–994. doi: 10.1016/j.febslet.2010.11.047
Wattin, M., Gaweda, L., Muller, P., Baritaud, M., Scholtes, C., Lozano, C., et al. (2018). Modulation of protein quality control and proteasome to autophagy switch in immortalized myoblasts from duchenne muscular dystrophy patients. Int. J. Mol. Sci. 19:178. doi: 10.3390/ijms19010178
Weihl, C. C., Temiz, P., Miller, S. E., Watts, G., Smith, C., Forman, M., et al. (2008). TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 79, 1186–1189. doi: 10.1136/jnnp.2007.131334
Welle, S. L. (2009). Myostatin and muscle fiber size. Focus on “Smad2 and 3 transcription factors control muscle mass in adulthood” and “Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size”. Am. J. Physiol. Cell Physiol. 296, C1245–C1247.
Whitehead, N. P., Bible, K. L., Kim, M. J., Odom, G. L., Adams, M. E., and Froehner, S. C. (2016). Validation of ultrasonography for non-invasive assessment of diaphragm function in muscular dystrophy. J. Physiol. 594, 7215–7227. doi: 10.1113/jp272707
Williams, M. S. (2004). Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 351, 1030–1031; authorrely 1030–1031.
Williamson, D. L., Bolster, D. R., Kimball, S. R., and Jefferson, L. S. (2006). Time course changes in signaling pathways and protein synthesis in C2C12 myotubes following AMPK activation by AICAR. Am. J. Physiol. Endocrinol. Metab. 291, E80–E89.
Winder, W. W., Holmes, B. F., Rubink, D. S., Jensen, E. B., Chen, M., and Holloszy, J. O. (2000). Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. 88, 2219–2226. doi: 10.1152/jappl.2000.88.6.2219
Wing, S. S., and Goldberg, A. L. (1993). Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am. J. Physiol. 264(4 Pt 1), E668–E676.
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484.
Yin, H., Price, F., and Rudnicki, M. A. (2013). Satellite cells and the muscle stem cell niche. Physiol. Rev. 93, 23–67. doi: 10.1152/physrev.00043.2011
Young, C. N., Brutkowski, W., Lien, C. F., Arkle, S., Lochmuller, H., Zablocki, K., et al. (2012). P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: relationship to pathology and potential target for treatment. J. Cell Mol. Med. 16, 1026–1037. doi: 10.1111/j.1582-4934.2011.01397.x
Young, C. N., Sinadinos, A., Lefebvre, A., Chan, P., Arkle, S., Vaudry, D., et al. (2015). A novel mechanism of autophagic cell death in dystrophic muscle regulated by P2RX7 receptor large-pore formation and HSP90. Autophagy 11, 113–130. doi: 10.4161/15548627.2014.994402
Yu, S., and Melia, T. J. (2017). The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr. Opin. Cell Biol. 47, 92–98. doi: 10.1016/j.ceb.2017.03.010
Yuan, W., Tuttle, D. L., Shi, Y. J., Ralph, G. S., and Dunn, W. A. Jr. (1997). Glucose-induced microautophagy in Pichia pastoris requires the alpha-subunit of phosphofructokinase. J. Cell Sci. 110(Pt 16), 1935–1945.
Zaninello, M., Palikaras, K., Naon, D., Iwata, K., Herkenne, S., Quintana-Cabrera, R., et al. (2020). Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat. Commun. 11:4029.
Zhang, Q., Lenardo, M. J., and Baltimore, D. (2017). 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168, 37–57. doi: 10.1016/j.cell.2016.12.012
Zhao, J., Brault, J. J., Schild, A., Cao, P., Sandri, M., Schiaffino, S., et al. (2007). FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–483. doi: 10.1016/j.cmet.2007.11.004
Keywords: autophagy, AMPK, mTOR, muscle cell homeostasis, transcriptional regulation, skeletal muscle diseases
Citation: Xia Q, Huang X, Huang J, Zheng Y, March ME, Li J and Wei Y (2021) The Role of Autophagy in Skeletal Muscle Diseases. Front. Physiol. 12:638983. doi: 10.3389/fphys.2021.638983
Received: 08 December 2020; Accepted: 22 February 2021;
Published: 25 March 2021.
Edited by:
Marco Sandri, University of Padua, ItalyReviewed by:
Paolo Grumati, Telethon Institute of Genetics and Medicine (TIGEM), ItalyCopyright © 2021 Xia, Huang, Huang, Zheng, March, Li and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Li, amlubGlAZ3pobXUuZWR1LmNu; Yongjie Wei, d2VpeW9uZ2ppZUBnemhtdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.