 Norliana Ghazali
Norliana Ghazali Normastura Abd Rahman
Normastura Abd Rahman Azlina Ahmad
Azlina Ahmad Sarina Sulong
Sarina Sulong Thirumulu Ponnuraj Kannan
Thirumulu Ponnuraj Kannan- 1School of Dental Sciences, Universiti Sains Malaysia, Kubang Kerian, Malaysia
- 2Human Genome Centre, School of Medical Sciences, Universiti Sains Malaysia, Kubang Kerian, Malaysia
Nonsyndromic cleft lip and or without cleft palate (NSCL/P) with the hypodontia is a common developmental abnormality in humans and animals. This study identified the genetic aberration involved in both NSCL/P and hypodontia pathogenesis. A cross-sectional study using genome-wide study copy number variation-targeted CytoScan 750K array carried out on salivary samples from 61 NSCL/P and 20 noncleft with and without hypodontia Malay subjects aged 7–13 years old. Copy number variations (CNVs) of SKI and fragile histidine triad (FHIT) were identified in NSCL/P and noncleft children using quantitative polymerase chain reaction (qPCR) as a validation analysis. Copy number calculated (CNC) for each gene determined with Applied Biosystems CopyCaller Software v2.0. The six significant CNVs included gains (12q14.3, 15q26.3, 1p36.32, and 1p36.33) and losses (3p14.2 and 4q13.2) in NSCL/P with hypodontia patients compared with the NSCL/P only. The genes located in these regions encoded LEMD3, IGF1R, TP73, SKI, FHIT, and UGT2β15. There were a significant gain and loss of both SKI and FHIT copy number in NSCL/P with hypodontia compared with the noncleft group (p < 0.05). The results supported that CNVs significantly furnish to the development of NSCL/P with hypodontia.
Introduction
Tooth agenesis (TA) is one of the most common developmental abnormalities in humans, described by the failure to develop one or more teeth (Ritwik and Patterson, 2018). TA is divided into three categories which are hypodontia, oligodontia, and anodontia (Klein et al., 2013). Hypodontia is the loss of one to five teeth, oligodontia is described as missing six or more teeth, and anodontia refers to the complete loss of tooth growth (Phan et al., 2016). Hypodontia is the most common dentofacial condition in humans (Al-Ani et al., 2017). Hypodontia can occur as an isolated condition without any other recognizable abnormality called nonsyndromic or associated with other structural deformities known as syndromic (Souza-Silva et al., 2018). The permanent dentition is more regularly affected than deciduous dentition (Abdulgani et al., 2017). A tooth development requires a complex process involving interaction between epithelial and mesenchymal signals, assisted by communication between signaling molecules and genetic pathways (Jussila and Thesleff, 2012). Various factors, such as those from wingless-related integration sites (Wnt), fibroblast growth factor (Fgf), bone morphogenic protein (BMP), and hedgehog (Hh) families, participate in the signaling mesenchymal interaction in tooth growth (Al-Ani et al., 2017). Alteration within one or more signaling pathways attributed to environmental influences (smoking, alcohol intake, chemotherapy, trauma, radiotherapy, and infection) or genetic factors could lead to abnormal tooth development (Rakhshan, 2015; Al-Ani et al., 2017).
The co-occurrence of CL/P (cleft lip with or without cleft palate) and hypodontia was reported frequently in human and animal models (Phan et al., 2016). Congenitally missing teeth or hypodontia is the most common dental anomalies found in CL/P patients (Rahman et al., 2004; Haque and Alam, 2015). Various studies revealed that MSX1 mutations led to the cleft palate (CP) and TA in humans (Kouskoura et al., 2011; Liang et al., 2012; Leslie and Marazita, 2013). Msx1-deficient mice revealed severe craniofacial anomalies, comprising the secondary palatal cleft and missing teeth (Nakatomi et al., 2010). The incidence of hypodontia was higher in more severe CL/P cases and presented as loss in maxillary lateral incisor tooth (Camporesi et al., 2010). The hypodontia among CL/P may significantly impact patient’s quality of life in terms of functional decrease, emotional distress, and social prosperity.
The most common orofacial cleft syndrome is the van der Woude syndrome (VWS), which found 2% of all syndromic CL/P, caused by mutation or deletion of the IRF6 gene in 68% of the cases (Kondo et al., 2002; de Lima et al., 2009). Letra et al. (2012) extensively investigated the role of IRF6 in the nonsyndromic orofacial cleft (OFCs) with TA situated outside the cleft region in a cohort of 134 Brazilian patients, thereby establishing a borderline-associated IRF6 marker (rs6588860) in the subgroup of the subjects exhibiting CP and TA. Nine genomic loci and 26 candidate genes were identified in the previous literature leading to the occurrence of those two congenital disabilities (Phan et al., 2016).
The variation in copy number has recently been identified as a significant cause of variation in the structural genome involving both duplications and sequence deletion (Redon et al., 2006). The previous study reported copy number variations (CNVs) in syndromic CL/P with hypodontia patients such as ectodermal dysplasia syndrome, Wolf Hirschhorn syndrome, and DiGeorge syndrome. The CNV in OFCs with TA found had associated with genomic loci such as in 1q21-q25, 1q32, 2q31.2-q32.2, 4p16.3, 8q24, and 16q22. However, the CNV and the related genes for nonsyndromic cleft lip with or without cleft palate (NSCL/P) with hypodontia remained with limited identification (Schinzel and Schmid, 1980; Wong et al., 1999). The causative genes and genomic loci of hypodontia among syndromic CL/P might also lead to the development in nonsyndromic CL/P. Therefore, a genome-wide association study (GWAS) was conducted to identify the contribution of CNV in the development of NSCL/P with hypodontia.
Materials and Methods
Study Population
A total of 81 individuals, including 61 NSCL/P cases and 20 noncleft aged 7–13, enrolled in the present comparative cross-sectional study. All the patient samples were recruited from one tertiary hospital in northern and eastern Malaysia between 2016 and 2018. The inclusion criteria for cases were NSCL/P children aged 7–13 years old, and those who had cleft palate only were excluded from this study. The subjects in the comparative group were noncleft children and those without any history of cleft. Noncleft children who have undergone orthodontic treatment were excluded from the study. The sample size for the prevalence of dental anomalies and genetic aberrations were calculated using the single proportion formula based on the prevalence of dental anomalies (Al-Kharboush et al., 2015) and CNV (Simioni et al., 2015). The precision was set at 2% giving a sample size of 42 and six subjects, respectively. However, this study recruited 61 NSCL/P patients and 100 subjects to determine the prevalence of dental anomalies. Dental panoramic tomography was taken for identification and confirmation of the number and morphology of the teeth. Exclusion criteria for the NSCL/P cases were those who had cleft palate only and patients with other syndromes such as ectodermal dysplasia or Axenfeld-Rieger syndromes. This study was approved by the Human Research Ethics Committee of Universiti Sains Malaysia (Reference number: USM/JPEPeM/140357). Both subjects and their guardians had become acquainted with the comprehensive research procedure and signed the informed consent before being enrolled in this study.
DNA Extraction
Saliva samples were collected from each patient and stored in sterile 50-ml conical tubes. Genomic DNA was extracted from saliva samples by following GeneAll Blood SV Mini Kit manual (General Biosystem, Seoul, South Korea), including lysis, binding, washing, and elution.
CytoScan 750K Array and Copy-Number Analysis
The Genome-Wide Human CytoScan 750K Array (Affymetrix, CA United States) was used to analyze genomic alterations according to the manufacturer’s protocol. A total of 250 ng of genomic DNA from NSCL/P with hypodontia and control samples (noncleft) were digested with the restriction enzyme Nsp1, then ligated to an adapter and PCR amplification using PCR on a single pair of primers that recognized the adapter sequence. The PCR products were analyzed by electrophoresis in 2% agarose using Tris-Borate-EDTA (TBE) to confirm the amplicon size between 150 and 2,000 bp in length. PCR products were combined from each sample and purified using magnetic beads (Agencourt AMPure, Beckman Coulter, Beverly, MA, United States). The purified PCR products were fragmented using DNase 1 and visualized on 4% TBE agarose gel to confirm that the fragment sizes ranged between 25 and 125 bp. The fragmented PCR products were subsequently end-labeled with biotin and hybridized to the array. The arrays were then washed, stained using GeneChips Fluidics Station 450 and Affymetrix GeneChip Command Console Software, version 1.2, followed by an Affymetrix Chromosome Analysis Suite version 3.1 (CHAS) Affymetrix United States. The data were normalized to baseline reference intensities using 270 HapMap samples and another 90 healthy noncleft in the software to calculate copy number. The Hidden Markov Model (HMM) available within the software package was used to determine the copy number states and their breakpoints. Based on HMM, the log-ratio thresholds were set at ≥0.58 and ≤−1, and used to categorize altered regions as copy number gains (amplification) and copy number losses (deletions). The alterations that only involved at least 25 conservative probes and more than 25 kbp in length were selected to avoid the false-positive CNVs.
Candidates CNV Analysis
This study compared overlapping regions in NSCL/P and noncleft children with or without hypodontia. Hypodontia-specific CNVs were considered potential candidate variants for hypodontia among subjects if they showed a statistically significant difference between patient CNV and CNV or overlapped genes. CNVs correlate with tooth’s craniofacial production and morphogenesis or are involved in a process suspected of affecting palatogenesis and odontogenesis.
Real-Time Quantitative PCR Validation
Two statistically significant genes were selected for validation by real-time quantitative PCR (qPCR) analysis on StepOnePlus Real-Time PCR system (Applied Biosystem, Forster City, CA, United States). The target information for the selected genes in the CNV region was entered into Assay Search Tool-Single Tube Taqman® Assay from Life Technologies website1 to obtain specific primer-probe pair. Two selected sequences were Homo sapiens SKI NCBI location Chr 2: 3,652,515-3,736,200, cytoband: 1p36.33 (Hs05780959_cn) and Fragile Histidine Triad Diadenosine Triphosphatase (FHIT) NCBI location Chr 3: 60555049–60579400, cytoband 3p14.2 (Hs03472126_cn). CNV performed using TaqMan Genotyping Master Mix for absolute quantitation of copy number using real-time qPCR. RNase P was used as endogenous control and NTC (reaction mixture without DNA template) as a negative control. qPCR was run with the following parameters: hold at 95°C for 10 min, followed by 95°C for 15 s and 60°C for 1 min for 40 cycles.
CNC for the gene is identified with Applied Biosystems CopyCaller Software v2.0. Several copies of the target sequence expected in the majority of samples were set at two. Samples were revised to attain optimum experimental conditions where samples are of high quality, copy number and reference assay have amplified, and sample replicates have parallel cycle threshold (CT) and difference of CT (∆CT) values. The number of copies of the target sequence in every test sample is regulated by relative quantitation (RQ) using the comparative CT (ΔΔCT) method. This method quantifies the Ct difference (ΔCt) between target and reference sequences, then compares the ΔCt values of test samples to a calibrator sample known to have two copies of the target sequence. Accepted copy number calls should have a confidence value >95% and Z-score <1.75 (Applied Biosystems CopyCaller Software v2.0).
Statistical Analysis
The analysis was carried out using SPSS, version 26.0. The frequency of the deletion or amplification of each CNV regions was compared between NSCL/P and noncleft with or without hypodontia using Fisher’s exact test to identify significant chromosome alterations. Chi-square tests were used to test the significant changes detected by CytoScan 750K Array. Statistical analysis for copy number data was performed using the Mann-Whitney test, and p < 0.05 was identified as statistically significant.
Results
Genome-Wide Assessment of CNVs in NSCL/P and Non-cleft With and Without Hypodontia
A total of 81 subjects were recruited in this study, including 61 NSCL/P patients and 20 noncleft subjects. In this NSCL/P, 31 (50.0%) were unilateral cleft lip and palate (UCLP), 13 (21%) were bilateral cleft lip and palate (BCLP), and 17 (27%) were cleft lip only (CL). LUCLP (34%) was the most common cleft type followed by BCLP (21.0%), RUCLP (16.0%), left cleft lip (14.0%), and right cleft lip (13.0%). The mean (SD) age for UCLP, BCLP, CL, and noncleft was 9.3 years (SD 1.8), 9.2 years (2.0), 9.1 years (2.0), and 5.5 years (3.5). Salivary samples from all subjects aged between 7 and 13 years old were subjected to genetic aberration assay using CytoScan 750K array. The subjects comprised four groups, such as NSCL/P with or without hypodontia and noncleft with or without hypodontia. All the samples exhibited chromosomal aberrations. The size of the detected CNVs varied from 25 kb to 2.5 Mb. Of the 721 genomic segments on the 81 samples, the analyses restricted to 558 genomic loci corresponding to the autosomes, including 196, showed amplification, and 362 showed deletion. The current study showed that the highest percentage of amplification occurred in chromosomes 1p, 2q, 12q, and 15q, and deletions observed for chromosomes 1q, 3p, 4q, 6p, and 7q among NSCL/P and noncleft with hypodontia. Identifying a candidate gene for NSCL/P with hypodontia in the CNVs selected regions by searching for copy number losses or copy number gains which involved the genes shared by multiple patients. The most recurrent amplified regions observed on chromosome 1p36.33, 1p36.32, 12q14.3, 14q32.33, and 15q26.3 loci and the most frequent deletion is found on chromosome 1q44, 3p14.2, 4q13.2, 6p25.3, and 7q34 among NSCL/P and noncleft with hypodontia.
Six of these unique candidate NSCL/P with hypodontia-specific CNVs (four gains and two losses) were significant, and these CNVs involved a total of eight genes. The six significant CNVs in NSCL/P with hypodontia included four gains (1p36.32, 1p36.33, 12q14.3, and 15q26.3) and two losses (3p14.2 and 4q13.2). The two significant CNVs among noncleft with hypodontia included one gain (1p36.32) and one copy number loss (4q13.2). The fisher exact analysis of the association between NSCL/P subjects in CNVs (gains and losses) are shown in Table 1. A total of six of these 334 CNVs were significantly different from NSCL/P only (p < 0.05). The significant CNVs included four gains (1p36.32, 1p36.33, 12q14.3, and 15q26.3) and two losses (3p14.2 and 4q13.2) and were found among NSCL/P with or without hypodontia. Besides, a total of 31 CNVs found in the noncleft with hypodontia did not overlap with the number of CNVs among noncleft-only children. Two of these were significantly different from a noncleft-only sample (p < 0.05) and are shown in Table 2. All the genomic loci that lay within the significant CNVs were employed to identify the phenotype like plausibly pathogenic sequence and genes of interest using Decipher database, OMIM, and Database of Genomic Variants. Table 3 shows several protein-coding genes identified from the overlapping on the significant copy number gains and losses. Two selected candidates’ genes were chosen for validation, SKI and FHIT, based on the literature (Vieira et al., 2007; Bliek et al., 2008) that the gene pathways were related to any tooth abnormalities, craniofacial deformities, and embryonic development.
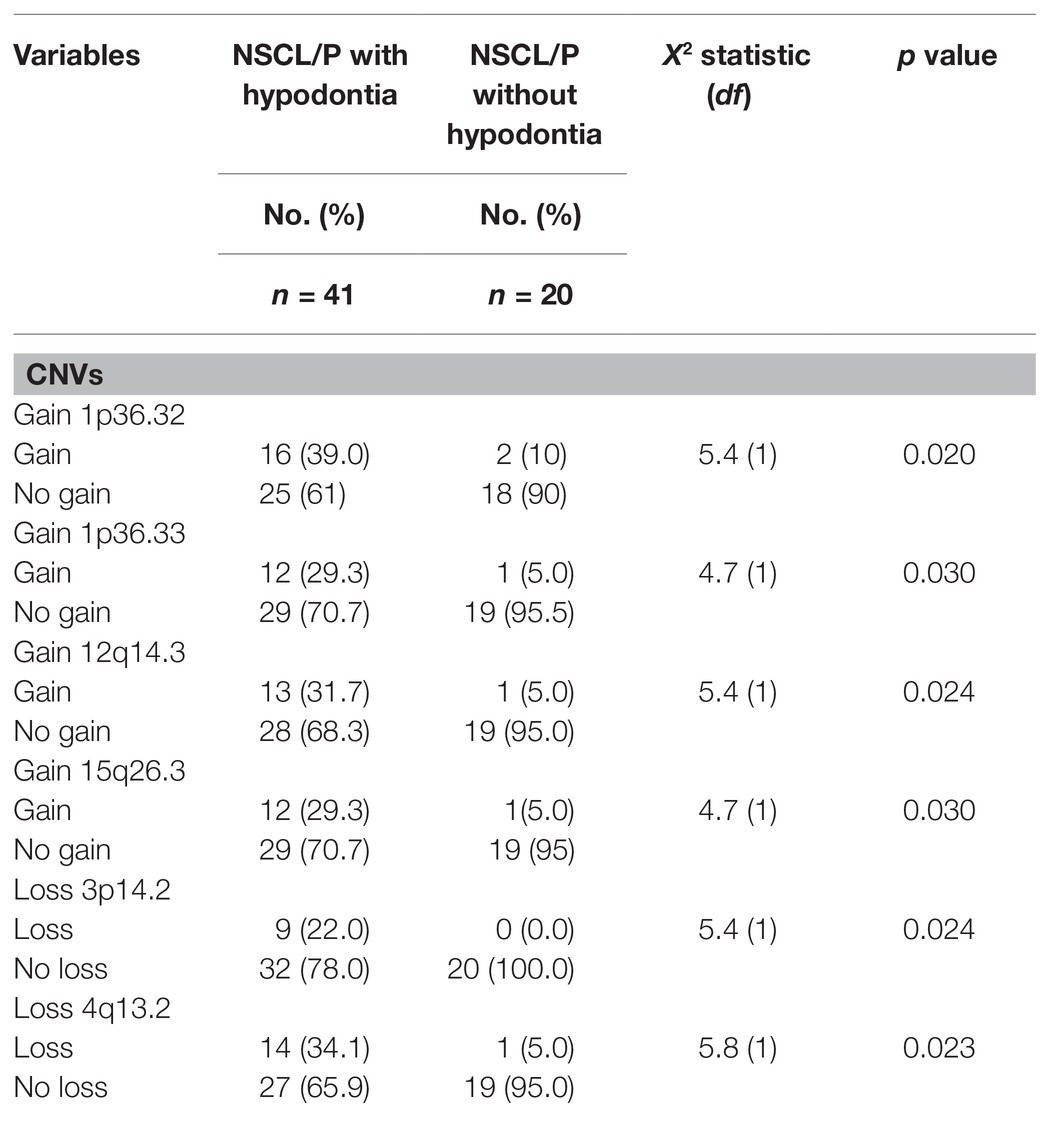
Table 1. Association between NSCL/P with or without hypodontia with copy number variations (CNVs).
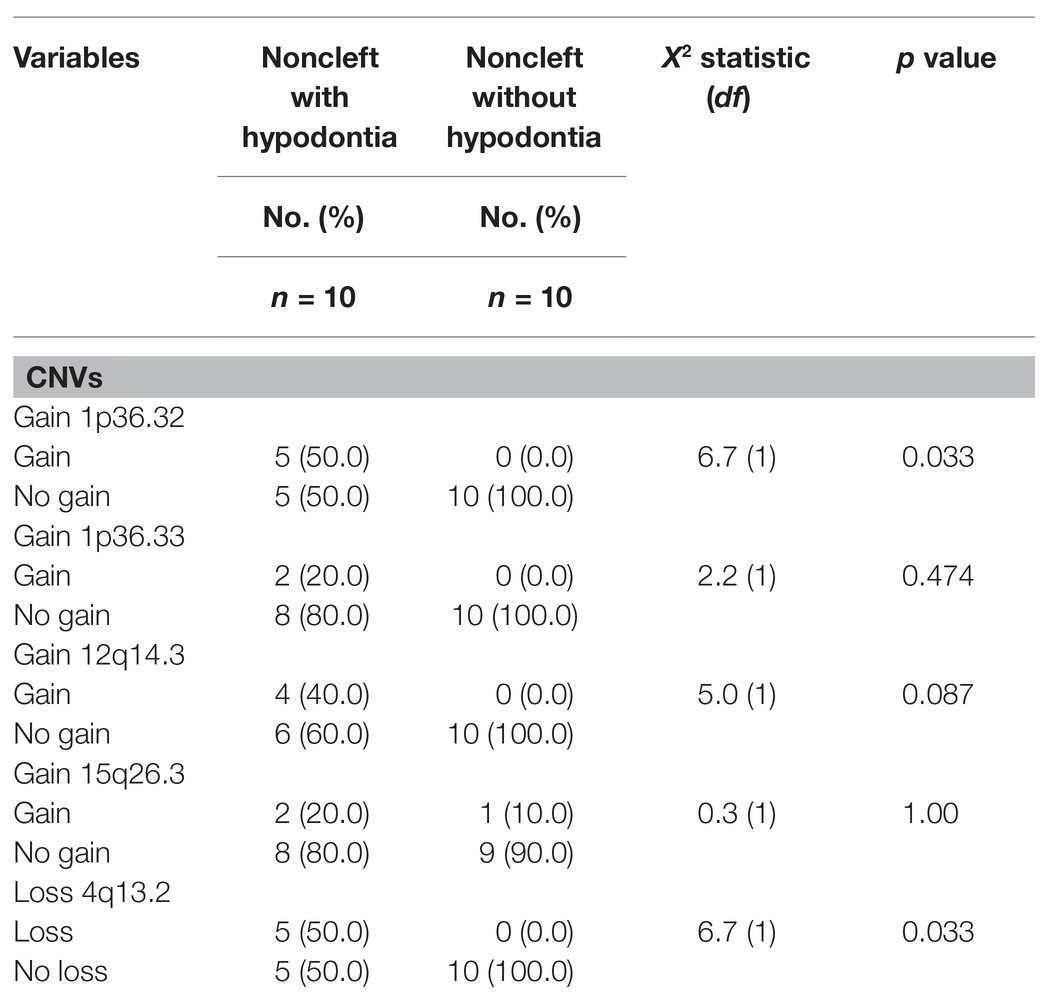
Table 2. Association between noncleft with or without hypodontia with copy number variations (CNVs).
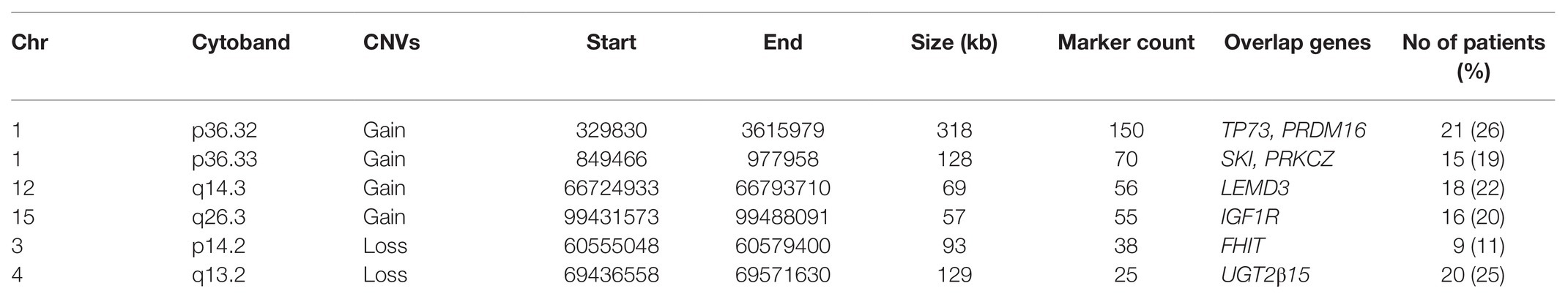
Table 3. Distribution of significant CNVs among NSCL/P and noncleft with and without hypodontia (n = 81).
Validation of CNVs by Real-Time qPCR
Validation of SKI and FHIT by CNV assay was performed as a continuous finding of copy number gain and copy number loss found by microarray-based copy number analysis 1p36.33 and 3p14.2 regions in NSCL/P with or without hypodontia. Microarray-based copy number analysis was performed with the Affymetrix CytoScan 750K array and visualized using the Affymetrix Chromosome Analysis Suite version 1.2.2. Figure 1 shows the 116-kb gain image at 1p36.33 (Chromosome 1: 2021784–2187344; GRCH37/hg19) present in a 10-year-old boy. The region overlaps with SKI and PRKCZ genes. Figure 2 shows image of the 26-kb gain at 3p14.2 (Chromosome 3:60,555,048-60,579,400; GRCH37/hg19) present in a 9-year-old girl. The region overlaps with the FHIT gene.
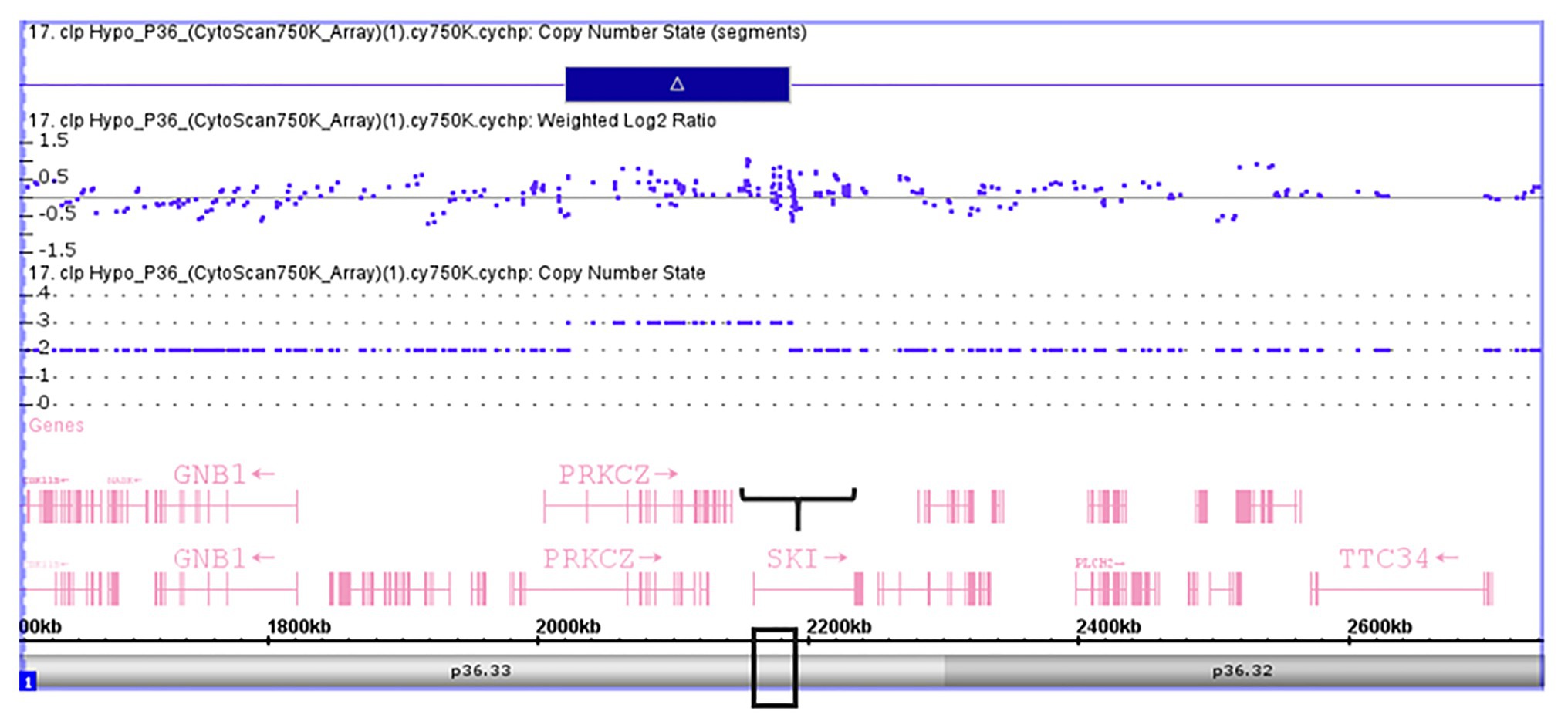
Figure 1. Microarray-based copy number analysis performed with the Affymetrix CytoScan 750K array and visualized using the Affymetrix Chromosome Analysis Suite version 1.2.2. Image of the 116 kb gain at 1p36.33 (Chromosome 1: 2021784-2187344, GRCH37/hg19) present in a boy 10 year-old. The region overlaps with SKI (black box).
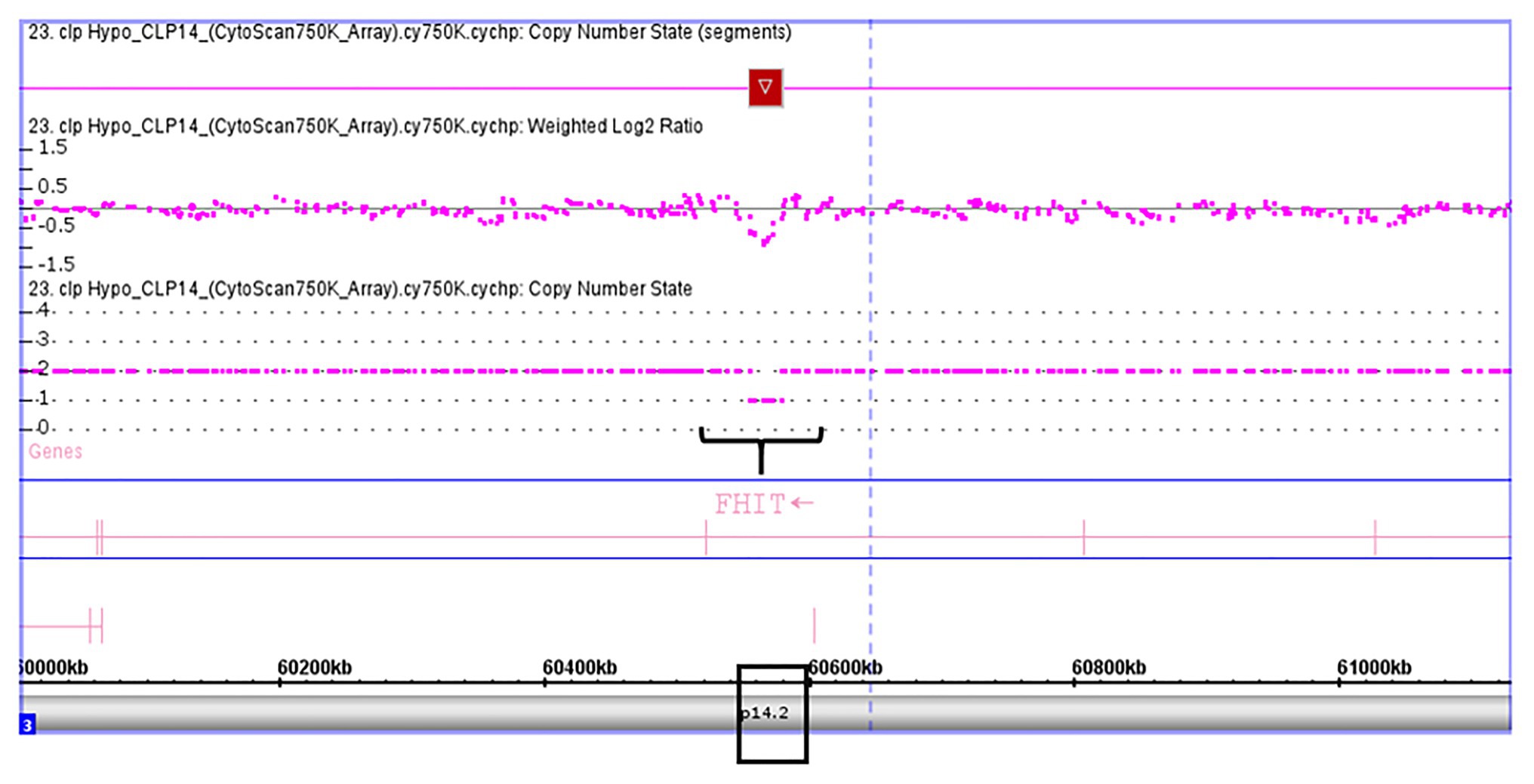
Figure 2. Microarray-based copy number analysis performed with the Affymetrix CytoScan 750K array and visualized using the Affymetrix Chromosome Analysis Suite version 1.2.2. Image of the 26 kb loss at 3p14.2 (Chromosome 3:60,555,048-60,579,400; GRCH37/hg19) present in a girl 9 year-old. The region overlaps with FHIT gene (black box).
Thirty subjects were recruited, including NSCL/P and noncleft hypodontia and normal control (noncleft hypodontia). The number calculated (CNC) for SKI and FHIT genes were determined. Figure 3 showed CNC of SKI gene in NSCL/P with hypodontia and noncleft with hypodontia compared with the normal control. From the findings, CNC for each individual in NSCL/P with hypodontia and noncleft with hypodontia showed CNC similar to the control group, 2.49 ≥ CNC ≥ 1.50 with one patient expressing more than 2.50 CNC. The CNC in the range of 2.49 ≥ CNC ≥ 1.50 may have a copy number predicted as two. From the 30 subjects, one of the NSCL/P with hypodontia patient showed a copy number predicted (CNP) at three, and the remaining 29 samples had CNP value equal to two.
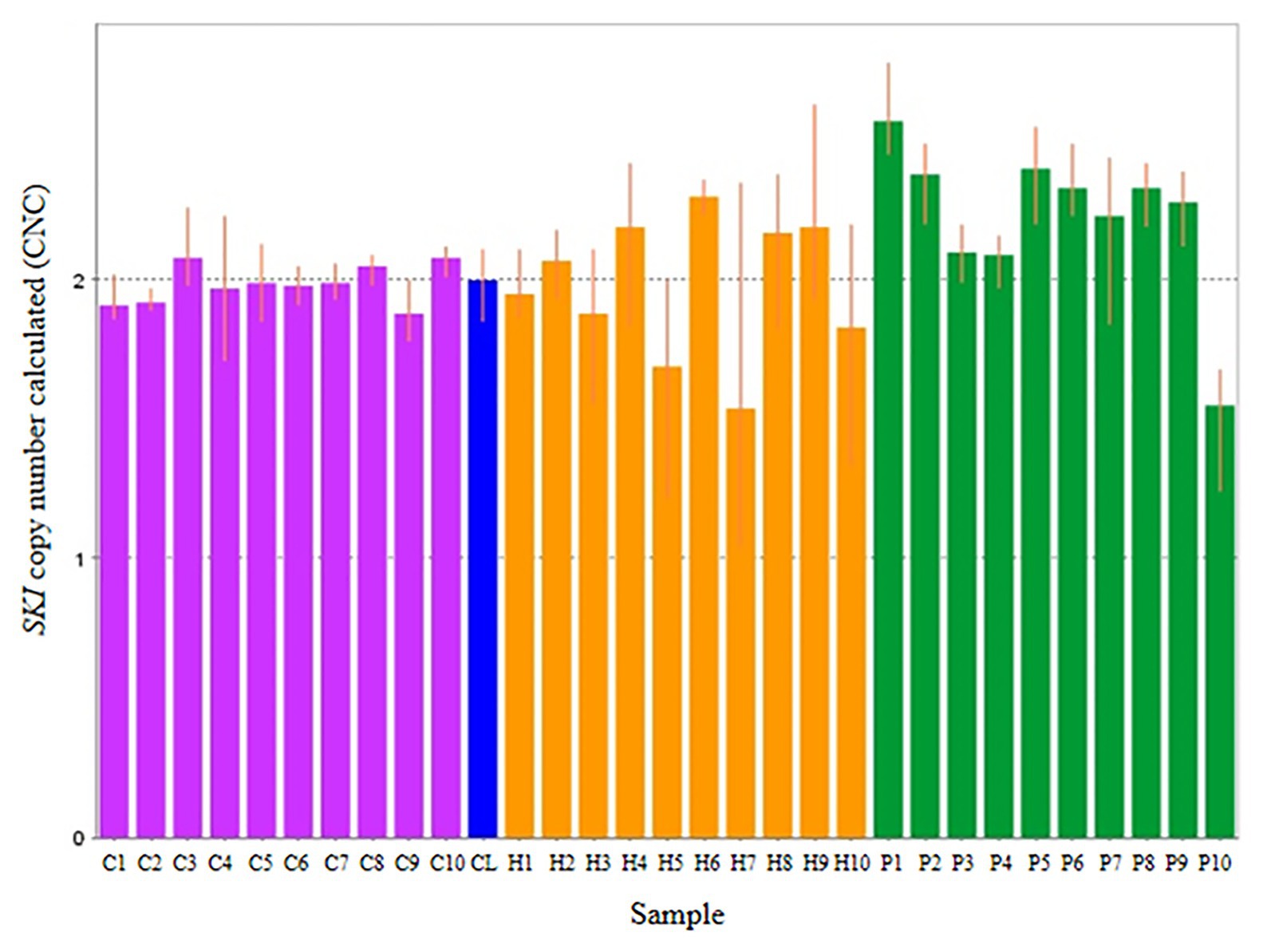
Figure 3. Representative of copy number calculated (CNC) of SKI in normal control (purple bars), non-cleft with hypodontia (yellow bars) NSCL/P with hypodontia (green bars) and calibrator (blue bar).
The scatter plot of the CNC data showed a distinct distribution of copy numbers between all the subjects (Figure 4). The straight line showed that the normal copy number fell at two. SKI copy number for NSCL/P with hypodontia was found scattered and exceeded two; meanwhile, most SKI copy numbers in normal controls were dispersed with less than copy number two. There was no significant difference in copy number of SKI (p = 0.9) in noncleft with hypodontia (1.98 ± 0.25) compared with the normal control group (1.98 ± 0.07), which means that both groups had normal copy number for SKI. However, there was a significant increase in SKI copy number in NSCL/P with hypodontia (2.2 ± 0.28, p = 0.02) compared with the normal control group (1.98 ± 0.07). These results revealed that SKI copy number gain was present among NSCL/P with hypodontia.
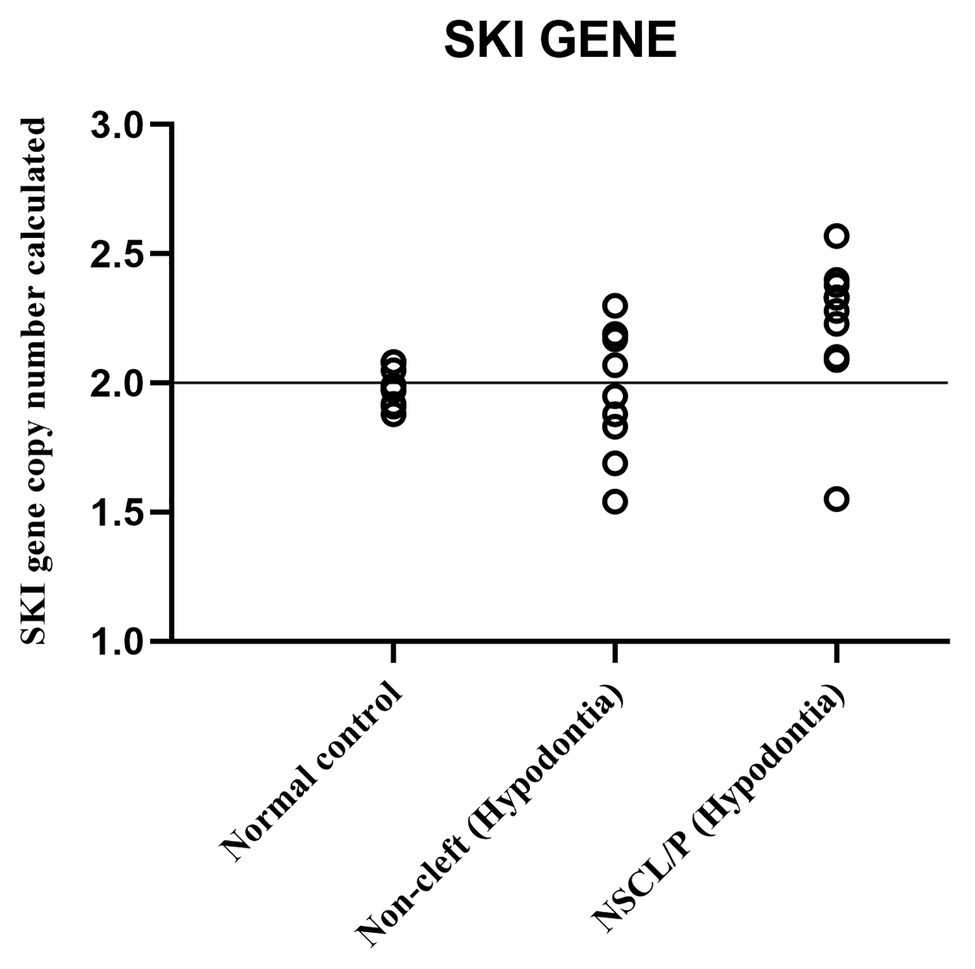
Figure 4. Scatter plot indicated copy number calculated value of SKI among NSCL/P with hypodontia, non-cleft with hypodontia and normal controls.
Copy number for FHIT was determined for NSCL/P with hypodontia as it showed copy number loss using Cytoscan 750K array. From the findings, the normal control, noncleft with hypodontia, and NSCL/P with hypodontia patients tested had a copy number of 2.49 ≥ CNC ≥ 1.50 except for two patients having CNC < 1.50 (Figure 5). Meanwhile, all the NSCL/P with hypodontia patients showed that CNC value did not exceed copy number two, including two samples with a copy number calculated as 1.55. However, scatter plot data had shown a distinct distribution of FHIT copy number between all the subjects (Figure 6). CNC among NSCL/P with hypodontia were lower than two compared with the normal control while noncleft with hypodontia had more copy number two. A significant decreased of FHIT copy number in NSCL/P with hypodontia (2.10 ± 0.231) compared with normal control (2.01 ± 0.069) was confirmed; p = 0.002. However, there was no significant difference in copy number of FHIT in noncleft with hypodontia (1.97 ± 0.09) compared with normal control.
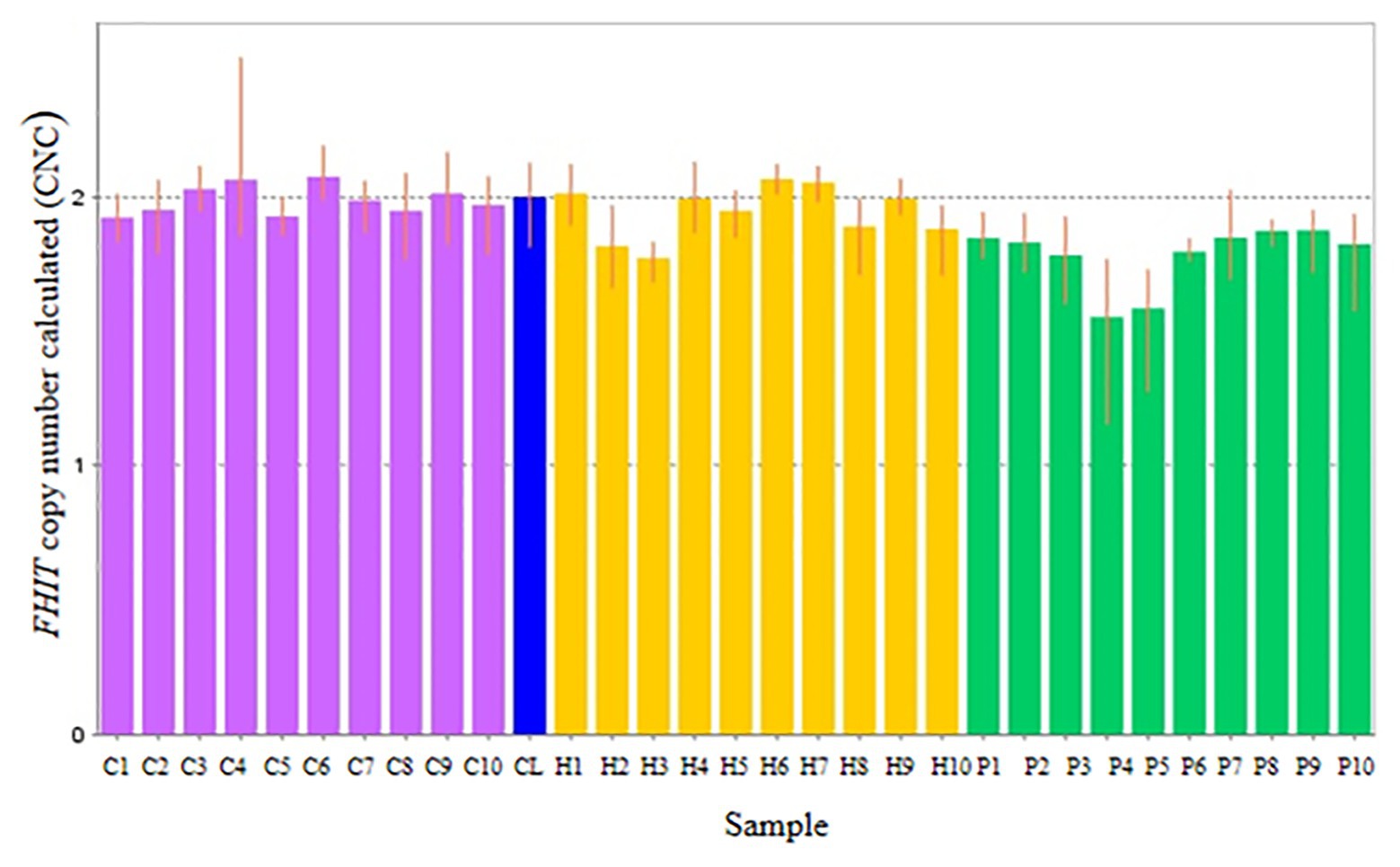
Figure 5. Representative of copy number calculated (CNC) of FHIT in normal control (purple bars), non-cleft with hypodontia (yellow bars) NSCL/P with hypodontia (green bars) and calibrator (blue bar).
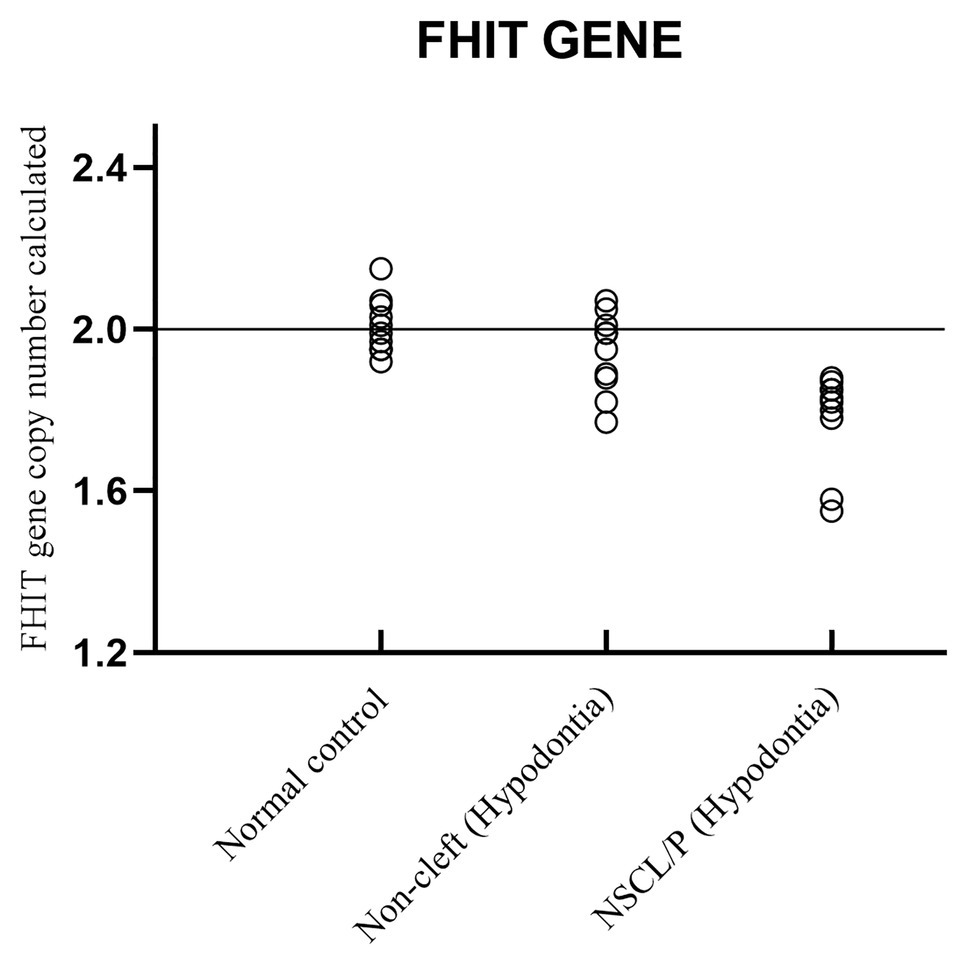
Figure 6. Scatter plot indicated copy number calculated value of FHIT among NSCL/P with hypodontia, non-cleft with hypodontia and normal control.
Discussion
Genome-Wide Assessment of CNVs in NSCL/P and Noncleft With and Without Hypodontia
In the recent study, 61 NSCL/P and 20 noncleft children were being recruited. A total of 558 genomic loci, including 196 amplification and 362 deletions, were detected among NSCL/P and noncleft with and without hypodontia. This result showed that the copy number loss is higher compared with the copy number gain. These results are highly consistent with those previously described in a systematic analysis study by Conte et al. (2016), with a total of 249 genomic deletions and 226 duplications from a cohort of 312 orofacial clefts reported in two publicly accessible databases of chromosome imbalance and phenotype in humans, DECIPHER and ECARUCA. This study supported the finding of CNVs with small and large chromosomal deletions among oral cleft patients and individual patients with multiple congenital disabilities (Phan et al., 2016). Our study identified a significant candidate CNVs on 1p36.32, 1p36.33, 3p14.2, 4q13.2, 12q14.3, and 15q26.3 among NSCL/P patients with hypodontia whereby study done by Yu et al. (2017) reported identified 14 novel loci and genetic heterogeneity among NSCL/P only. However, we consider these CNVs important candidates because several of the genes in these regions are functionally linked to normal embryonic and tooth development. Some genomic loci with deletion were reported of TA and syndromic orofacial clefts (OFCs; Phan et al., 2016). Van Der Woude syndrome characterized as cleft lip with or without cleft palate and hypodontia revealed 8 kb deletion or insertion in the region 1q32-q41 (Schutte et al., 2000). An ectodermal dysplasia-like syndrome patient showed a 26-Mb interstitial deletion involving the region 2q31.2-q33.2, with multiple phenotypes, including missing or abnormal teeth with cleft lip and palate (Rifai et al., 2010).
Therefore, quantitative PCR was performed to confirm the data array results. The genes SKI and FHIT were randomly selected from significant CNVs (1p36.33 and 3p14.2) regions to identify their association with NSCL/P and hypodontia.
Significant Copy Number Gain in SKI Among NSCL/P With Hypodontia
The present study showed a significantly increased copy number of the 1p36.33 region, encompassing the SKI gene. SKI encodes the nuclear proto-oncogene protein homolog of avian sarcoma viral (v-ski) oncogene. These findings are consistent with the previous research that showed SKI was the candidate gene for orofacial clefting (Berk et al., 1997); (Lu et al., 2005; Vieira et al., 2005); nevertheless, its role in tooth development remained unclear. The meta-analysis identified six new susceptible loci in European NSCL/P population including 1p36, 2p21, 3p11.1, 8q21.3, 13q31.1, and 15q22.2 (Ludwig et al., 2012). Most previous work investigated the single nucleotide polymorphism (SNP) and mutation of the SKI gene compared with the current study, which identified the copy number variation among the subjects. Duplication of the SKI gene has not yet been linked to orofacial clefting and tooth development directly. However, we found that SKI is involved in several signaling pathways important for the process, such as transforming factor-beta (TGF-β), BMP, and G-protein-coupled receptors (GPCR; Deheuninck and Luo, 2009; Parada and Chai, 2012).
SKI is an essential negative TGF-β signaling regulator, which interacts with mothers against decapentaplegic (SMADs) to suppress TGF-β signaling activity (Yang et al., 2015). SKI gene can block TGF-β signaling by interfering with the phosphorylation of SMAD2 and SMAD3 by activated TGF-β type 1 receptor (Buess et al., 2004). TGF-β signaling plays a crucial role in controlling the formation of palates in both palate epithelium and mesenchyme (Iwata et al., 2011). TGF-β is also critical for cell proliferation and differentiation in the dental pulp (Niwa et al., 2018). The finding may suggest that copy number gain of SKI may alter the building blocks of single proteins (amino acids) in the SKI protein. Many of the mutations modify the SKI protein region, which binds to SMAD proteins. It assumed that the changed SKI proteins could not be attached to SMAD proteins, which allow the unregulated prolongation of TGF-β signaling. Excessive TGF-β signaling affects gene activity regulation and is likely to interact with palatal and tooth growth. TGF-β3 is one of the main ligands of two serine/threonine kinase receptors, TGFβR1 and TFFβR2, investigated concerning syndromic cleft lip and palate (Loeys et al., 2005).
The SKI mutation was also associated with Shprintzen-Goldberg syndrome with distinctive facial features, hypotonia, and intellectual deficiency (Doyle et al., 2012; Schepers et al., 2015).
Significant Copy Number Loss in FHIT Among NSCL/P With Hypodontia
A lower 3p14.2 copy number containing a FHIT gene was identified among NSCL/P patients with hypodontia. FHIT gene is a prominent member of the histidine triad gene family and is considered a tumor suppressor gene. Deleting the FHIT gene has never been reported in disturbing the development of cleft lip and palate and the tooth. Thus, this would be the first finding that revealed FHIT dysregulation in patients affected with NSCL/P with hypodontia. The previous result has found a deletion on the similar locus of 3p14.2 in a patient of Waardenburg syndrome type 11A with cleft lip and palate (Lakhanpal et al., 2014). Another study found that the abnormal expression of FHIT may be associated with a variety of malignant tumors, such as lung cancer and breast cancer (Pekarsky et al., 2002; Ismail et al., 2011). The mechanism of FHIT in triggering NSCL/P and hypodontia formation was unknown. However, it was likely that loss of FHIT is involved in the related pathway and transcriptional activities including TGF-β, WNT signaling, β-activin transcription, and BMP (Pekarsky et al., 2002; Weiske et al., 2007 ; Prosseda et al., 2019). This finding supports the idea that TGF-β signaling and canonical Wnt/β-catenin pathways play an essential role in the development of tooth and palatogenesis (Tamura and Nemoto, 2016; Niwa et al., 2018; Reynolds et al., 2019). There are also various reports of genetic variations at the FHIT locus and loss of FHIT protein expression in preneoplasias, proposing a tumor-suppressive role for FHIT in the early stages of cancer progression (Waters et al., 2014; Malak et al., 2016; Kiss et al., 2017).
Weiske et al. (2007) reported that FHIT related with a lymphoid enhancer-binding factor β-catenin complex by directly binding to the β-catenin, a significant player in the canonical WNT pathway that is decontrolled in numerous form of human cancer. FHIT expresses the transcription of target genes such as cyclin D1, AXIN2, MMP-14, and survivin when binding to the β-catenin C-terminal domain (Weiske et al., 2007). AXIN2 likely plays a vital role in regulating beta-catenin stability in the WNT signaling pathway (Huraskin et al., 2016). AXIN2 also has independently associated with tooth agenesis and NSCL/P (Letra et al., 2009). The variations in WNT genes have been recognized in an individual with tooth agenesis (Dinckan et al., 2016). Therefore, the AXIN2 mutations could lead to an inefficient block of the WNT signaling pathway and altered the embryonic development of dental organs and predisposition to cancer (Otero et al., 2019). We hypothesized that reduced induction of FHIT might be triggering AXIN2 mutation and directly producing WNT dysregulation in the progression of lip and palate fusion and tooth development and finally leading to abnormal growth.
Validation through copy number analysis has strengthened results from the previous genome-wide association studies. This current study found a significant gain and loss of copy number of these genes, SKI and FHIT, respectively, in each of the selected NSCL/P with tooth-missing patients compared with normal groups. Both genes might have a significant contribution to the development of NSCL/P and hypodontia. Different gene copy numbers among individuals and populations reveal gene expression variations (De Smith et al., 2008). CNVs also overlap over 7,000 genes, many of which are essential in biological pathways. Many studies have reported that the number of gene copies has led to a cell modification of the transcription process and make a difference in the expression level (Stranger et al., 2007; Prestel et al., 2010). Hence, evidence for CNVs between genes and potential NSCL/P with hypodontia has been confirmed in this study. Significant copy number gain of SKI was observed on the subject members compared with the normal control, so we can conclude that SKI might give a minor contribution to the NSCL/P with hypodontia formation and could trigger the cleft occurrence. In addition, FHIT loss was reported to be associated with the NSCL/P with hypodontia development. This novel finding would help to shed light on the regulation of SKI and FHIT on the orofacial cleft and hypodontia formation. In clinical setting, early complications due to hypodontia can be part of prevention strategies and genetic counseling in a multidisciplinary clinic.
Conclusion
In conclusion, this study developed a GWAS study to explore the CNVs among NSCL/P patients and identify potential NSCL/P and hypodontia-related genes. We identified six significant CNVs with four genomic deletions and two duplication, including several genes, such as SKI, FHIT, TP73, LEMD3, IGF1R, and UGT2β15 genes. Our study enhances the reservoir of possible causative genes for NSCL/P and hypodontia genes for genetics studies. It provides a disease link to many of these genes known to contribute to several signaling pathways. Forthcoming human mutation analysis and animal model studies are required to confirm the role of the identified potential causative NSCL/P and hypodontia genes.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: (REPOSITORY: dbVar and ACCESSION ID: nstd202).
Ethics Statement
The studies involving human participants were reviewed and approved by Human Research Ethics Committee of Universiti Sains Malaysia (Reference number: USM/JPeM/140356). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
NG performed all the experiments, designed the methodology, and wrote the original draft. NA contributed to conceptualization, formal analysis, writing-reviewing, and editing the manuscripts. AA performed the data curation and visualization. SS contributed to software analysis and validation process. TPK supervised the research and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research received financial support from the Universiti Sains Malaysia Research University Grant (1001/PPSG/812128).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the voluntary patients and families for their full co-operation given to conduct this study.
Footnotes
References
Abdulgani, A., Watted, N., Azzaldeen, A., Mai, A., Borbély, P., and Abu-Hussein, M. (2017). Aetiological factors article in IOSR. J. Dent. Med. Sci. 16, 75–85. doi: 10.9790/0853-1601057585.
Al-Ani, A. H., Antoun, J. S., Thomson, W. M., Merriman, T. R., and Farella, M. (2017). Hypodontia: an update on its etiology, classification, and clinical management. Biomed. Res. Int. 2017:9378325. doi: 10.1155/2017/9378325
Al-Kharboush, G. H., Al-Balkhi, K. M., and Al-Moammar, K. (2015). The prevalence of specific dental anomalies in a group of Saudi cleft lip and palate patients. Saudi Dent. J. 27, 75–80. doi: 10.1016/j.sdentj.2014.11.007
Berk, M., Desai, S. Y., Heyman, H. C., and Colmenares, C. (1997). Mice lacking the ski proto-oncogene have defects in neurulation, craniofacial patterning, and skeletal muscle development. Genes Dev. 11, 2029–2039. doi: 10.1101/gad.11.16.2029
Bliek, B. J. B., Steegers-Theunissen, R. P. M., Blok, L. J., Santegoets, L. A. M., Lindemans, J., Oostra, B. A., et al. (2008). Genome-wide pathway analysis of folate-responsive genes to unravel the pathogenesis of orofacial clefting in man. Birth Defects Res. A Clin. Mol. Teratol. 82, 627–635. doi: 10.1002/bdra.20488
Buess, M., Terracciano, L., Reuter, J., Ballabeni, P., Boulay, J. -L., Laffer, U., et al. (2004). Amplification of SKI is a prognostic marker in early colorectal cancer. Neoplasia 6, 207–212. doi: 10.1593/neo.03442
Camporesi, M., Baccetti, T., Marinelli, A., Defraia, E., and Franchi, L. (2010). Maxillary dental anomalies in children with cleft lip and palate: a controlled study. Int. J. Paediatr. Dent. 20, 442–450. doi: 10.1111/j.1365-263X.2010.01063.x
Conte, F., Oti, M., Dixon, J., Carels, C. E. L., Rubini, M., and Zhou, H. (2016). Systematic analysis of copy number variants of a large cohort of orofacial cleft patients identifies candidate genes for orofacial clefts. Hum. Genet. 135, 41–59. doi: 10.1007/s00439-015-1606-x
Deheuninck, J., and Luo, K. (2009). Ski and SnoN, potent negative regulators of TGF-β signaling. Cell Res. 19, 47–57. doi: 10.1038/cr.2008.324
de Lima, R. L. L. F., Hoper, S. A., Ghassibe, M., Cooper, M. E., Rorick, N. K., Kondo, S., et al. (2009). Prevalence and nonrandom distribution of exonic mutations in interferon regulatory factor 6 in 307 families with Van der Woude syndrome and 37 families with popliteal pterygium syndrome. Genet. Med. 11, 241–247. doi: 10.1097/GIM.0b013e318197a49a
De Smith, A. J., Walters, R. G., Froguel, P., and Blakemore, A. I. (2008). Human genes involved in copy number variation: mechanisms of origin, functional effects and implications for disease. Cytogenet. Genome Res. 123, 17–26. doi: 10.1159/000184688
Dinckan, N., Uyguner, Z. O., Kayserili, H., and Letra, A. (2016). Association of AXIN2 gene polymorphisms with non-syndromic oligodontia in Turkish families. Dentistry 3000, 34–42. doi: 10.5195/D3000.2016.57
Doyle, A. J., Doyle, J. J., Bessling, S. L., Maragh, S., Lindsay, M. E., Schepers, D., et al. (2012). Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 44, 1249–1254. doi: 10.1038/ng.2421
Haque, S., and Alam, M. K. (2015). Common dental anomalies in cleft lip and palate patients. Malays. J. Med. Sci. 22, 55–60.
Huraskin, D., Eiber, N., Reichel, M., Zidek, L. M., Kravic, B., Bernkopf, D., et al. (2016). Wnt/β-catenin signaling via Axin 2 is required for myogenesis and, together with YAP/Taz and Tead1, active in IIa/IIx muscle fibers. Development 143, 3128–3142. doi: 10.1242/dev.139907
Ismail, H., Medhat, A. M., Karim, A. M., and Zakhary, N. I. (2011). Multiple patterns of FHIT gene homozygous deletion in Egyptian breast cancer patients. Int. J. Breast Cancer 2011:325947. doi: 10.4061/2011/325947
Iwata, J., Parada, C., and Chai, Y. (2011). The mechanism of TGF-β signaling during palate development. Oral Dis. 17, 733–744. doi: 10.1111/j.1601-0825.2011.01806.x
Jussila, M., and Thesleff, I. (2012). Signaling networks regulating tooth organogenesis and regeneration, and the specification of dental mesenchymal and epithelial cell lineages. Cold Spring Harb. Perspect. Biol. 4:a008425. doi: 10.1101/cshperspect.a008425
Kiss, D. L., Baez, W., Huebner, K., Bundschuh, R., and Schoenberg, D. R. (2017). Impact of FHIT loss on the translation of cancer-associated mRNAs. Mol. Cancer 16:179. doi: 10.1186/s12943-017-0749-x
Klein, O. D., Oberoi, S., Huysseune, A., Hovorakova, M., Peterka, M., and Peterkova, R. (2013). Developmental disorders of the dentition: an update. Am. J. Med. Genet. C Semin. Med. Genet. 163, 318–332. doi: 10.1002/ajmg.c.31382
Kondo, S., Schutte, B. C., Richardson, R. J., Bjork, B. C., Knight, A. S., Watanabe, Y., et al. (2002). Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 32, 285–289. doi: 10.1038/ng985
Kouskoura, T., Fragou, N., Alexiou, M., John, N., Sommer, L., Graf, D., et al. (2011). The genetic basis of craniofacial and dental abnormalities. Schweiz. Monatsschr. Zahnmed. 121, 636–646. doi: 10.5167/uzh-50358
Lakhanpal, M., Gupta, N., Rao, N. C., and Vashisth, S. (2014). Genetics of cleft lip and palate–is it still patchy. JSM Dent. 2, 1–4. Available at: http://www.jscimedcentral.com/Dentistry/dentistry-2-1030.pdf
Leslie, E. J., and Marazita, M. L. (2013). Genetics of cleft lip and cleft palate. Am. J. Med. Genet. C: Semin. Med. Genet. 163C, 246–258. doi: 10.1002/ajmg.c.31381
Letra, A., Fakhouri, W., Fonseca, R. F., Menezes, R., Kempa, I., Prasad, J. L., et al. (2012). Interaction between IRF6 and TGFA genes contribute to the risk of nonsyndromic cleft lip/palate. PLoS One 7:e45441. doi: 10.1371/journal.pone.0045441
Letra, A., Menezes, R., Granjeiro, J. M., and Vieira, A. R. (2009). AXIN2 and CDH1 polymorphisms, tooth agenesis, and oral clefts. Birth Defects Res. A Clin. Mol. Teratol. 85, 169–173. doi: 10.1002/bdra.20489
Liang, J., Zhu, L., Meng, L., Chen, D., and Bian, Z. (2012). Novel nonsense mutation in MSX 1 causes tooth agenesis with cleft lip in a Chinese family. Eur. J. Oral Sci. 120, 278–282. doi: 10.1111/j.1600-0722.2012.00965.x
Loeys, B. L., Chen, J., Neptune, E. R., Judge, D. P., Podowski, M., Holm, T., et al. (2005). A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 37, 275–281. doi: 10.1038/ng1511
Lu, W., Volcik, K., Zhu, H., Wen, S., Shaw, G. M., Lammer, E. J., et al. (2005). Genetic variation in the proto-oncogene SKI and risk for orofacial clefting. Mol. Genet. Metab. 86, 412–416. doi: 10.1016/j.ymgme.2005.06.003
Ludwig, K. U., Mangold, E., Herms, S., Nowak, S., Reutter, H., Paul, A., et al. (2012). Genome-wide meta-analyses of non-syndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 44, 968–971. doi: 10.1038/ng.2360
Malak, C. A. A., Elghanam, D. M., and Elbossaty, W. F. (2016). FHIT gene expression in acute lymphoblastic leukemia and its clinical significance. Asian Pac. J. Cancer Prev. 16, 8197–8201. doi: 10.7314/APJCP.2015.16.18.8197
Nakatomi, M., Wang, X. -P., Key, D., Lund, J. J., Turbe-Doan, A., Kist, R., et al. (2010). Genetic interactions between Pax9 and Msx1 regulate lip development and several stages of tooth morphogenesis. Dev. Biol. 340, 438–449. doi: 10.1016/j.ydbio.2010.01.031
Niwa, T., Yamakoshi, Y., Yamazaki, H., Karakida, T., Chiba, R., Hu, J. C. -C., et al. (2018). The dynamics of TGF-β in dental pulp, odontoblasts and dentin. Sci. Rep. 8, 1–14. doi: 10.1038/s41598-018-22823-7
Otero, L., Lacunza, E., Vasquez, V., Arbelaez, V., Cardier, F., and González, F. (2019). Variations in AXIN2 predict risk and prognosis of colorectal cancer. BDJ open 5, 1–6. doi: 10.1038/s41405-019-0022-z
Parada, C., and Chai, Y. (2012). “Roles of BMP signaling pathway in lip and palate development” in Frontiers of oral biology. ed. M. T. Cobourne (Basel, Switzerland: Karger Publishers), 60–70.
Pekarsky, Y., Palamarchuk, A., Huebner, K., and Croce, C. M. (2002). FHIT as tumor suppressor: mechanisms and therapeutic opportunities. Cancer Biol. Ther. 1, 232–236. doi: 10.4161/cbt.73
Phan, M., Conte, F., Khandelwal, K. D., Ockeloen, C. W., Bartzela, T., Kleefstra, T., et al. (2016). Tooth agenesis and orofacial clefting: genetic brothers in arms? Hum. Genet. 135, 1299–1327. doi: 10.1007/s00439-016-1733-z
Prestel, M., Feller, C., and Becker, P. B. (2010). Dosage compensation and the global re-balancing of aneuploid genomes. Genome Biol. 11:216. doi: 10.1186/gb-2010-11-8-216
Prosseda, S. D., Tian, X., Kuramoto, K., Boehm, M., Sudheendra, D., Miyagawa, K., et al. (2019). FHIT, a novel modifier gene in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 199, 83–98. doi: 10.1164/rccm.201712-2553OC
Rahman, N. A., Abdullah, N., Samsudin, A. R., Naing, L., and Sadiq, M. A. (2004). Dental anomalies and facial profile abnormality of the non-syndromic cleft lip and palate children in kelantan. Malays. J. Med. Sci. 11, 41–51.
Rakhshan, V. (2015). Congenitally missing teeth (hypodontia): a review of the literature concerning the etiology, prevalence, risk factors, patterns and treatment. Dent. Res. J. 12:1. doi: 10.4103/1735-3327.150286
Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D., et al. (2006). Global variation in copy number in the human genome. Nature 444, 444–454. doi: 10.1038/nature05329
Reynolds, K., Kumari, P., Rincon, L. S., Gu, R., Ji, Y., Kumar, S., et al. (2019). Wnt signaling in orofacial clefts: crosstalk, pathogenesis and models. Dis. Model. Mech. 12:dmm037051. doi: 10.1242/dmm.037051
Rifai, L., Port-Lis, M., Tabet, A. -C., Bailleul-Forestier, I., Benzacken, B., Drunat, S., et al. (2010). Ectodermal dysplasia-like syndrome with mental retardation due to contiguous gene deletion: further clinical and molecular delineation of del(2q32) syndrome. Am. J. Med. Genet. Part A 152, 111–117. doi: 10.1002/ajmg.a.33164
Ritwik, P., and Patterson, K. K. (2018). Diagnosis of tooth agenesis in childhood and risk for neoplasms in adulthood. Ochsner J. 18, 345–350. doi: 10.31486/toj.18.0060
Schepers, D., Doyle, A. J., Oswald, G., Sparks, E., Myers, L., Willems, P. J., et al. (2015). The SMAD-binding domain of SKI: a hotspot for de novo mutations causing Shprintzen–Goldberg syndrome. Eur. J. Hum. Genet. 23, 224–228. doi: 10.1038/ejhg.2014.61
Schinzel, A., and Schmid, W. (1980). Interstitial deletion of the long arm of chromosome 1, del (1)(q21-q25) in a profoundly retarded 8-year-old girl with multiple anomalies. Clin. Genet. 18, 305–313. doi: 10.1111/j.1399-0004.1980.tb00890.x
Schutte, B. C., Bjork, B. C., Coppage, K. B., Malik, M. I., Gregory, S. G., Scott, D. J., et al. (2000). A preliminary gene map for the Van der Woude syndrome critical region derived from 900 kb of genomic sequence at 1q32-q41. Genome Res. 10, 81–94.
Simioni, M., Araujo, T. K., Monlleo, I. L., Maurer-Morelli, C. V., and Gil-Da-Silva-Lopes, V. L. (2015). Investigation of genetic factors underlying typical orofacial clefts: mutational screening and copy number variation. J. Hum. Genet. 60, 17–25. doi: 10.1038/jhg.2014.96
Souza-Silva, B. N., Vieira, W. d. A., Bernardino, Í. d. M., Batista, M. J., Bittencourt, M. A. V., and Paranhos, L. R. (2018). Non-syndromic tooth agenesis patterns and their association with other dental anomalies: a retrospective study. Arch. Oral Biol. 96, 26–32. doi: 10.1016/j.archoralbio.2018.08.014
Stranger, B. E., Forrest, M. S., Dunning, M., Ingle, C. E., Beazley, C., Thorne, N., et al. (2007). Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853. doi: 10.1126/science.1136678
Tamura, M., and Nemoto, E. (2016). Role of the Wnt signaling molecules in the tooth. Jpn. Dent. Sci. Rev. 52, 75–83. doi: 10.1016/j.jdsr.2016.04.001
Vieira, A. R., Avila, J. R., Daack-Hirsch, S., Dragan, E., Félix, T. M., Rahimov, F., et al. (2005). Medical sequencing of candidate genes for non-syndromic cleft lip and palate. PLoS Genet. 1:e64. doi: 10.1371/journal.pgen.0010064
Vieira, A. R., Modesto, A., Meira, R., Barbosa, A. R. S., Lidral, A. C., and Murray, J. C. (2007). Interferon regulatory factor 6 (IRF6) and fibroblast growth factor receptor 1 (FGFR1) contribute to human tooth agenesis. Am. J. Med. Genet. A 143A, 538–545. doi: 10.1002/ajmg.a.31620
Waters, C. E., Saldivar, J. C., Hosseini, S. A., and Huebner, K. (2014). The FHIT gene product: tumor suppressor and genome "caretaker." Cell. Mol. Life Sci. 71, 4577–4587. doi: 10.1007/s00018-014-1722-0
Weiske, J., Albring, K. F., and Huber, O. (2007). The tumor suppressor Fhit acts as a repressor of β-catenin transcriptional activity. Proc. Natl. Acad. Sci. 104, 20344–20349. doi: 10.1073/pnas.0703664105
Wong, F. K., Karsten, A., Larson, O., Huggare, J., Hagberg, C., Larsson, C., et al. (1999). Clinical and genetic studies of Van der Woude syndrome in Sweden. Acta Odontol. Scand. 57, 72–76. doi: 10.1080/000163599428931
Yang, H., Zhan, L., Yang, T., Wang, L., Li, C., Zhao, J., et al. (2015). Ski prevents TGF-β-induced EMT and cell invasion by repressing SMAD-dependent signaling in non-small cell lung cancer. Oncol. Rep. 34, 87–94. doi: 10.3892/or.2015.3961
Keywords: cleft lip, cleft palate, hypodontia, DNA copy number variations, genome-wide association study
Citation: Ghazali N, Abd Rahman N, Ahmad A, Sulong S and Kannan TP (2021) Identification of Copy Number Variation Among Nonsyndromic Cleft Lip and or Without Cleft Palate With Hypodontia: A Genome-Wide Association Study. Front. Physiol. 12:637306. doi: 10.3389/fphys.2021.637306
Edited by:
Mohammad Khursheed Alam, Al Jouf University, Saudi ArabiaReviewed by:
Adith Venugopal, University of Puthisastra, CambodiaAnand Marya, University of Puthisastra, Cambodia
Siti Adibah Othman, University of Malaya, Malaysia
Copyright © 2021 Ghazali, Abd Rahman, Ahmad, Sulong and Kannan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Normastura Abd Rahman, bm9ybWFzdHVyYUB1c20ubXk=