 Valeria Rizzuto1,2,3
Valeria Rizzuto1,2,3 Tamara T. Koopmann4
Tamara T. Koopmann4 Adoración Blanco-Álvarez5Barbara Tazón-Vega5
Adoración Blanco-Álvarez5Barbara Tazón-Vega5 Amira Idrizovic1
Amira Idrizovic1 Cristina Díaz de Heredia6Rafael Del Orbe7Miriam Vara Pampliega7
Cristina Díaz de Heredia6Rafael Del Orbe7Miriam Vara Pampliega7 Pablo Velasco6
Pablo Velasco6 David Beneitez8
David Beneitez8 Gijs W. E. Santen4Quinten Waisfisz9Mariet Elting9Frans J. W. Smiers10Anne J. de Pagter10
Gijs W. E. Santen4Quinten Waisfisz9Mariet Elting9Frans J. W. Smiers10Anne J. de Pagter10 Jean-Louis H. Kerkhoffs11
Jean-Louis H. Kerkhoffs11 Cornelis L. Harteveld4
Cornelis L. Harteveld4 Maria del Mar Mañú-Pereira1*
Maria del Mar Mañú-Pereira1*- 1Translational Research in Child and Adolescent Cancer – Rare Anemia Disorders Research Laboratory, Vall d’Hebron Research Institute, ERN-EuroBloodNet Member, Barcelona, Spain
- 2Josep Carreras Leukaemia Research Institute, Badalona, Spain
- 3Department of Medicine, Universitat de Barcelona, Barcelona, Spain
- 4Department of Clinical Genetics, Leiden University Medical Center, ERN-EuroBloodNet Member, Leiden, Netherlands
- 5Hematologic Molecular Genetics Unit, Hematology Department, Hospital Universitari Vall d’Hebron, ERN-EuroBloodNet Member, Barcelona, Spain
- 6Oncohematologic Pediatrics Department, Hospital Universitari Vall d’Hebron, ERN-EuroBloodNet Member, Barcelona, Spain
- 7Hematology Department, Hospital Universitario Cruces, Barakaldo, Spain
- 8Red Blood Cell Disorders Unit, Hematology Department, Hospital Universitari Vall d’Hebron, ERN-EuroBloodNet Member, Barcelona, Spain
- 9Department of Clinical Genetics, VU Medical Center, Amsterdam, Netherlands
- 10Department of Pediatric Hematology, Leiden University Medical Center, Leiden, Netherlands
- 11Department of Hematology, HAGA City Hospital, The Hague, Netherlands
Unstable hemoglobinopathies (UHs) are rare anemia disorders (RADs) characterized by abnormal hemoglobin (Hb) variants with decreased stability. UHs are therefore easily precipitating, causing hemolysis and, in some cases, leading to dominant beta-thalassemia (dBTHAL). The clinical picture of UHs is highly heterogeneous, inheritance pattern is dominant, instead of recessive as in more prevalent major Hb syndromes, and may occur de novo. Most cases of UHs are not detected by conventional testing, therefore diagnosis requires a high index of suspicion of the treating physician. Here, we highlight the importance of next generation sequencing (NGS) methodologies for the diagnosis of patients with dBTHAL and other less severe UH variants. We present five unrelated clinical cases referred with chronic hemolytic anemia, three of them with severe blood transfusion dependent anemia. Targeted NGS analysis was performed in three cases while whole exome sequencing (WES) analysis was performed in two cases. Five different UH variants were identified correlating with patients’ clinical manifestations. Four variants were related to the beta-globin gene (Hb Bristol—Alesha, Hb Debrousse, Hb Zunyi, and the novel Hb Mokum) meanwhile one case was caused by a mutation in the alpha-globin gene leading to Hb Evans. Inclusion of alpha and beta-globin genes in routine NGS approaches for RADs has to be considered to improve diagnosis’ efficiency of RAD due to UHs. Reducing misdiagnoses and underdiagnoses of UH variants, especially of the severe forms leading to dBTHAL would also facilitate the early start of intensive or curative treatments for these patients.
Introduction
Beta-thalassemia major (BTHAL) is a well-known life-threatening condition characterized by severe transfusion-dependent anemia. BTHAL is an autosomal recessive disorder presenting with high frequencies in populations from the Mediterranean area. Currently, up to 257 genetic variants in the beta-globin gene (HBB) have been identified as BTHAL disease-causing, leading to a total or partial reduction of beta-globin chain synthesis. The clinical severity of BTHAL is related to the extent of imbalance between the alpha and non-alpha-globin chains, while clinical management consists of regular life-long red blood cell (RBC) transfusions and iron chelation therapy. At present, the only definitive cure is bone marrow transplant (Efremov, 2007; Galanello and Origa, 2010). Both BTHAL patients and carriers are usually easily diagnosed through routine laboratory tests. However, there is an ultra-rare condition overlapping BTHAL clinical manifestations known as dominant beta-thalassemia (dBTHAL), which is caused by the presence of certain unstable (UH) or hyper unstable (HUH) hemoglobinopathies.
UHs are a group of congenital disorders caused by mutations in globin genes leading to destabilization of hemoglobin (Hb) molecules as a consequence of (a) amino acid substitutions within the heme pocket, (b) disruption of secondary structure, (c) substitution in the hydrophobic interior of the subunit, (d) amino acid deletions, and (e) elongation of the subunit. Thus, altering any of the steps in globin processing, including subunit folding, heme interaction, dimerization, or tetramerization (Bunn and Forget, 1986). These abnormal Hb variants undergo rapid denaturation followed by precipitation, leading to the formation of Heinz bodies, which cause hemolysis of RBCs. Clinical manifestations may vary from asymptomatic to severely affected forms. Treatment is mainly symptomatic and based on transfusion requirements as for BTHAL (Steinberg et al., 2009; Thom et al., 2013).
UHs are dominantly inherited with a significant rate of de novo mutations. They generally do not separate from normal Hb using standard methods. Thus, diagnosis of dBTHAL can be challenging since it requires a high index of suspicion and the diagnosis may be delayed for years hampering the access to timely treatment interventions.
The study we present herein confirms the relevance of including globin genes in next generation sequencing (NGS) approaches for the diagnosis of rare anemia disorders (RADs), especially for cases with no family history in which the anemia is not easily explained.
Patients and Methods
Clinical Reports
Here we present five clinical cases diagnosed with UH after NGS analysis. Clinical data and laboratory findings are shown in Table 1.
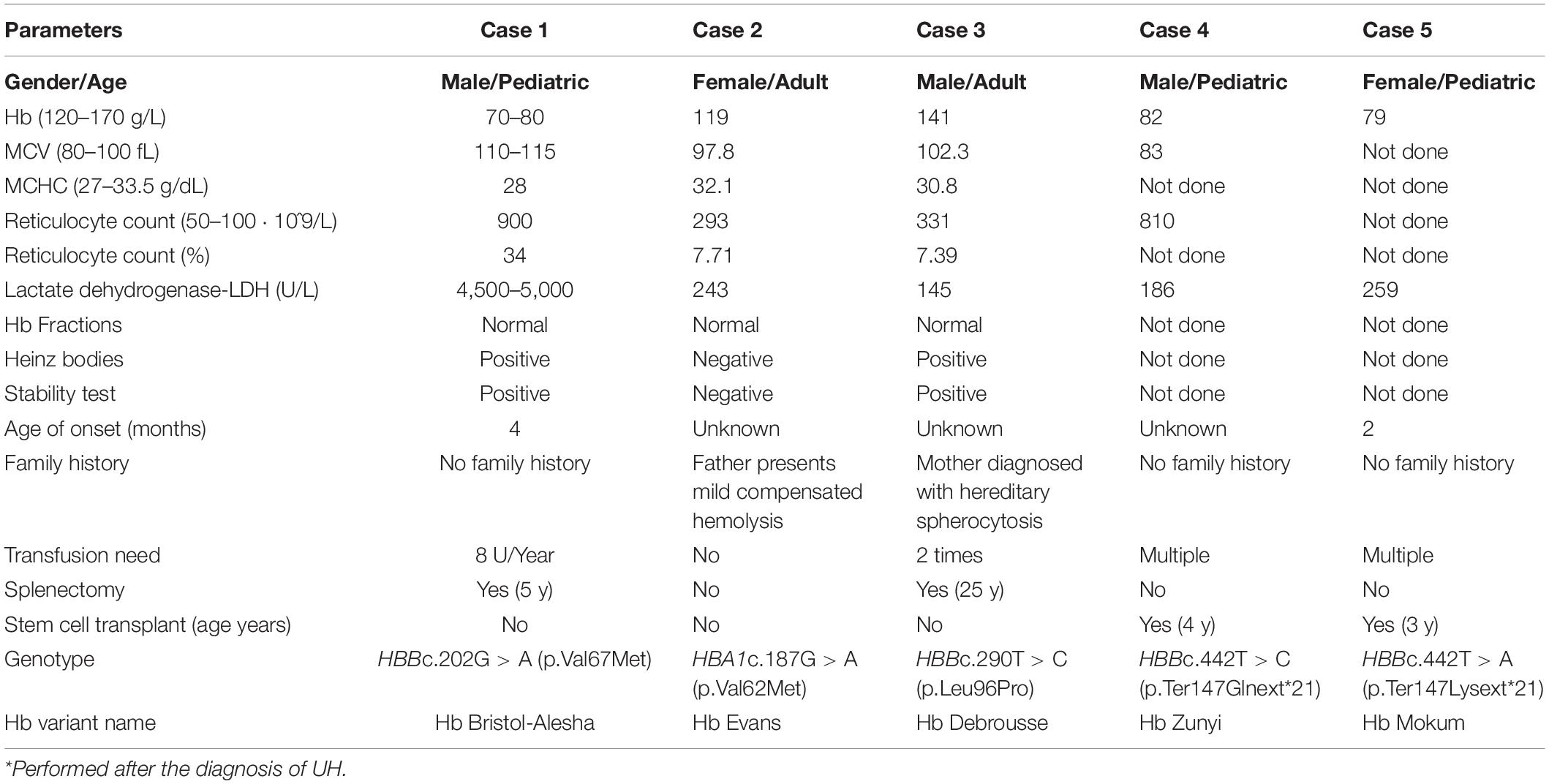
Table 1. Overview on clinical and genetic data of the five reported clinical cases.
The first case is a male pediatric patient referred with severe chronic blood dependent anemia since he was 4-month-old, asthenia, jaundice, and short stature. No family history of hemolytic anemia. Examination of blood smear revealed polychromasia, anisopoikilocytosis, basophil stippling, Cabot rings, schistocytes, and spherocytes. Separation and quantification of Hb fractions did not reveal any extraordinary peak and showed normal values for HbA2 and HbF. At 5-year-old he underwent splenectomy. After the surgery, Heinz bodies were present (Figures 1, 2) and isopropanol stability test, performed according to standard methodology, appeared positive (Figure 3). Family studies in both parents were strictly normal, including evaluation of Hb fractions. Enzyme activity assays, EMA-binding test, and osmotic gradient ectacytometry (LoRRca MaxSis) were performed to rule out hemolytic anemia due to RBC defects other than hemoglobinopathy. Results, although not strictly normal, did not reveal any RBC defect. However, they should be taken with caution since the patient was intensively transfused. Genetic analysis was performed on PKLR and G6PD genes failing to reveal any disease-causing mutation.
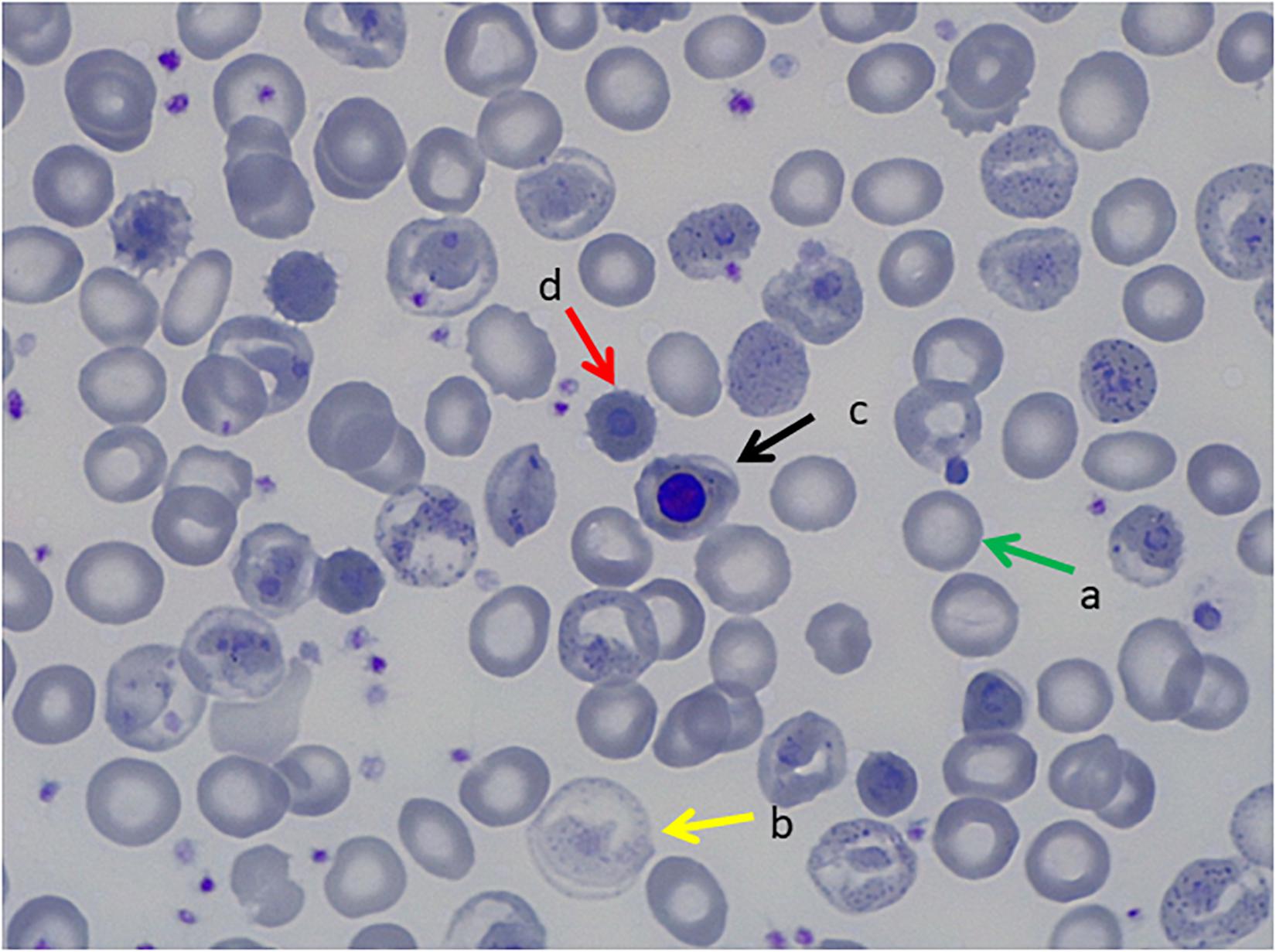
Figure 1. Peripheral Blood Smear, May Grunwald Giemsa Stain. (a) Transfused red blood cells, (b) non-transfused red blood cells with hemoglobinization abnormalities, (c) orthochromatic erythroblast, (d) erythrocitary inclusions that correspond to Heinz bodies.
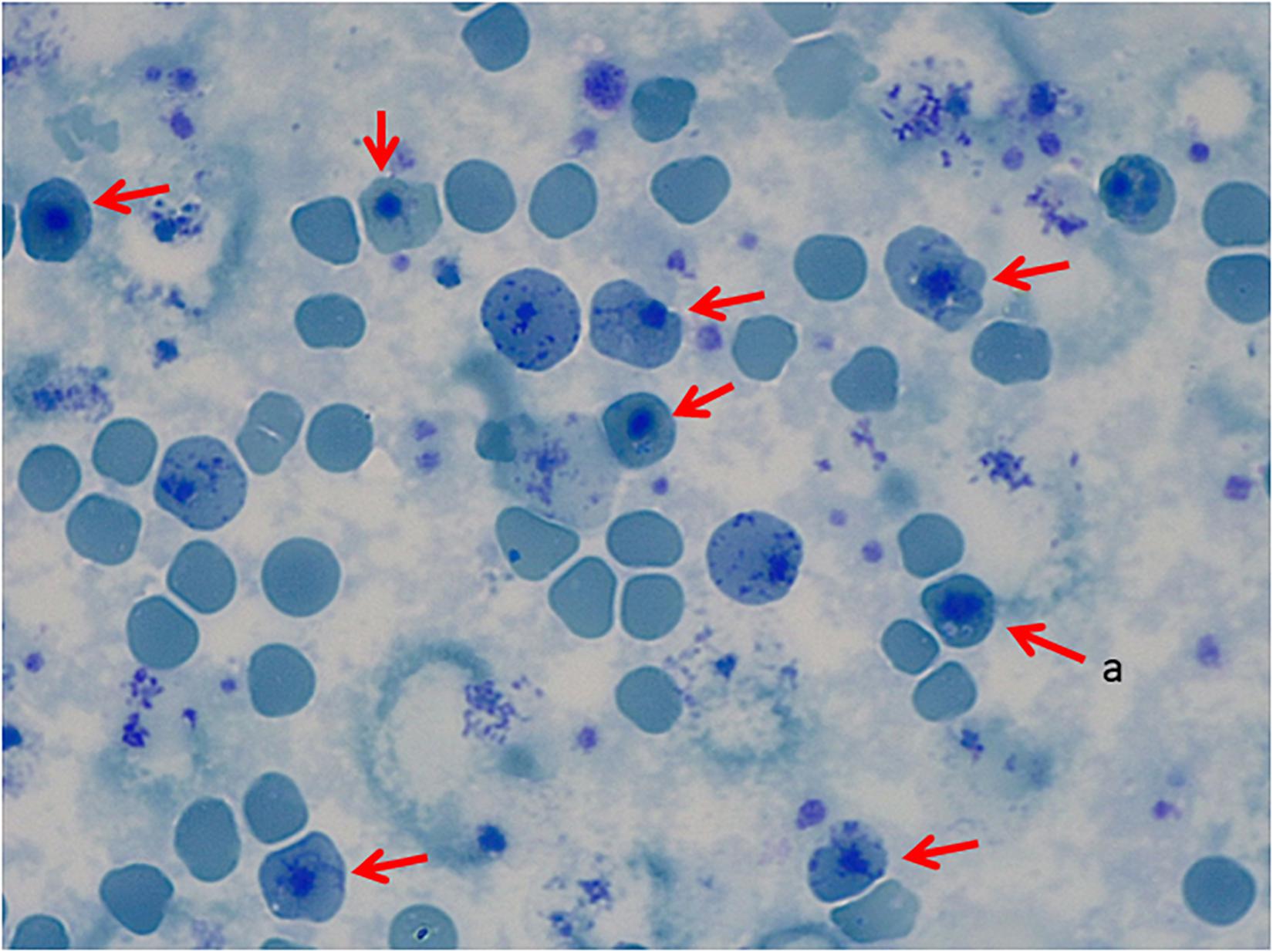
Figure 2. Peripheral Blood Smear, Brilliant Cresyl Blue Stain. (a) Heinz bodies.
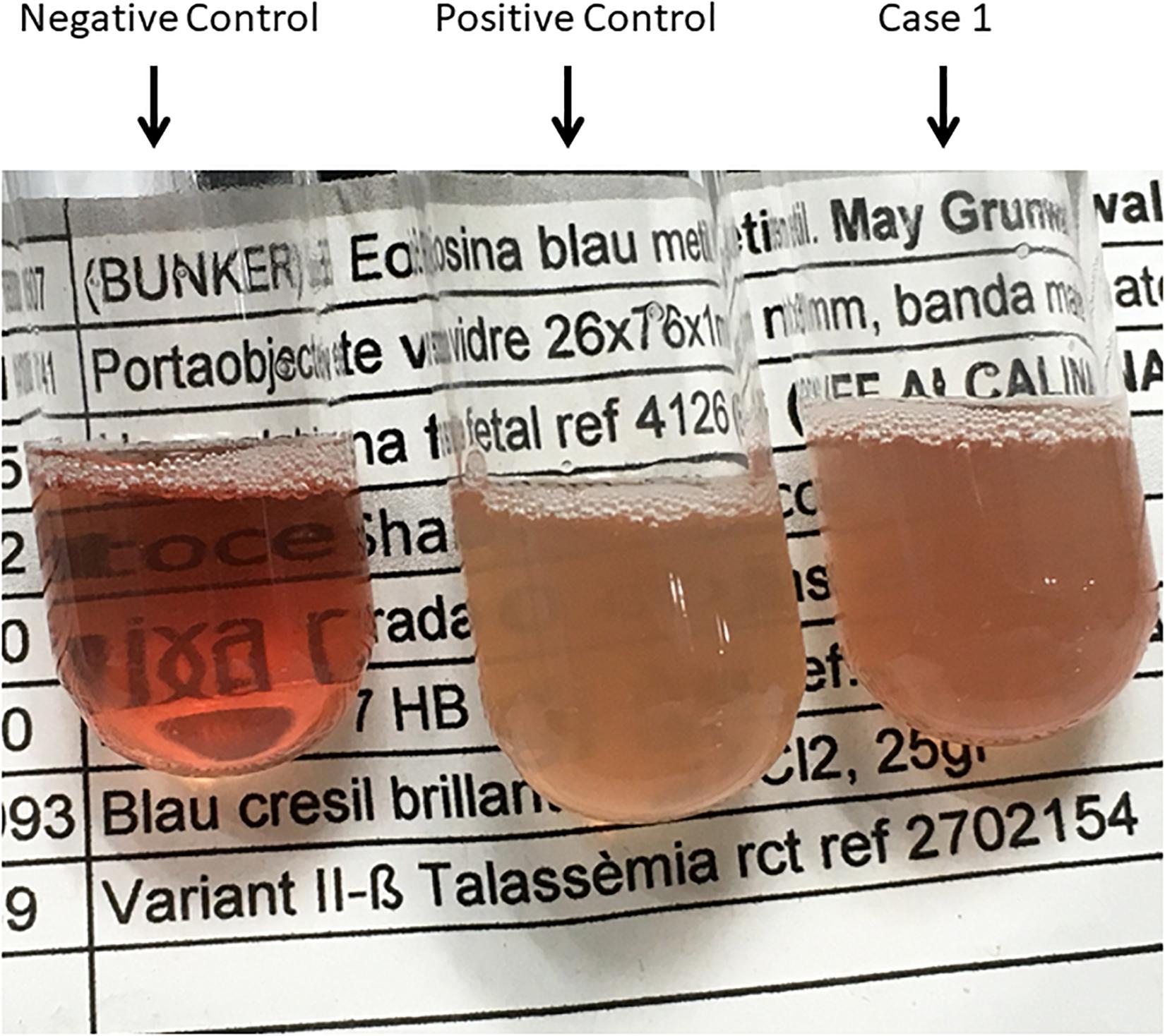
Figure 3. Isopropanol Test_01. Negative control (Hb AS), Positive control (Hb F), and Case 1.
The second case is a female adult patient with mild chronic compensated hemolysis referred for diagnosis when she was 20 years old. The father also presented with mild compensated hemolysis. No further examinations were performed before referral. Although the presence of extravascular hemolysis, the examination of blood smear was not informative. Separation and quantification of Hb fractions did not reveal any extra peaks and Heinz body and stability tests were normal. Further laboratory tests were performed to rule out hemolytic anemia due to RBC enzyme and membrane defects, including enzyme activity assays, EMA-binding test, and osmotic gradient ectacytometry (LoRRca MaxSis). All of them showed normal values.
The third case is a male adult patient. He presented with several episodes of hemolytic crises during childhood requiring blood transfusion on two occasions. He underwent splenectomy at the age of 25-year-old. The patient was diagnosed with hereditary spherocytosis (HS) following a previous HS diagnosis of his mother and the absence of abnormal Hb peaks by conventional electrophoresis.
Patients who underwent splenectomy neither clinically improved nor presented complications as pulmonary hypertension, thrombosis or increased hemolysis during 10-year follow-up.
The last two cases are two unrelated children who presented with macrocephaly and severe congenital anemia. The parents of both patients had no family history for abnormal Hb or thalassemia and had normal hematological features. Therefore, conventional testing for abnormal Hb was not performed. All siblings were unaffected.
The first of these two unrelated children is a male patient presenting with large head circumference and hepatosplenomegaly. Congenital dyserythropoietic anemia was suspected. However, no genetic analysis was performed for confirmation. He underwent successfully bone marrow transplant at the age of 4.
The second child is a female patient presenting with frontal bossing, macrocephaly, and severe anemia at the age of 2. Congenital dyserythropoietic anemia was suspected. Therefore, genetic analysis of CDAN1 and SEC23B genes was performed not revealing any disease-causing mutation. She underwent successfully bone marrow transplant when she was almost 3-year-old.
In all cases, RAD due to Hb variant was not suspected mainly due to the fact that parents did not present family history of RADs, except for case 3, RBC parameters were found to be normal and abnormal Hb fractions were absent when analyzed. Therefore, genetic testing was performed for genes associated with RADs other than globin genes, failing to show a conclusive diagnosis.
Genetics Analysis
Written informed consent was obtained from cases or legal guardian. Targeted NGS (t-NGS) analysis was performed in cases 1, 2, and 3 while whole exome sequencing (WES) analysis was performed in cases 4 and 5. For all the patients, genomic DNA was extracted from peripheral blood. For patients who underwent bone marrow transplant, DNA samples were previously stored.
The designed t-NGS panel covered 46 genes described as disease causing for RADs, including genes responsible for membrane disorders, enzyme defects, congenital dyserytrhopoietic anemia and the HBA1/HBA2 and HBB genes responsible for alpha and beta-globin chains, respectively. The full list of genes included is shown in Table 2. Exon and exon/intron boundaries were capture using a NimbleGen SeqCap EZ HyperCap (Roche) solution-based capture system followed by next generation sequencing on the MySeq (Illumina) with 150 bp paired-end reads. For the bioinformatics analysis, alignment to the hg38 genome was performed with BWA-MEM (Li H. 203 arXIV:1303.3997v2) and detection of changes with GATK1. Obtained variants were filtered and annotated based on variant effect, coverage (>30) and MAF (>0.05). Resulting variants were assessed for technique pitfalls through IGV. The nomenclature used was the recommended by HGVS2. Finally, disease-causing variants were prioritized based on inheritance pattern and VarSome3 for previous evidence as disease causing mutations or predictions score information. Variants were reported according to American College of Medical Genetics (ACMG) guidelines.
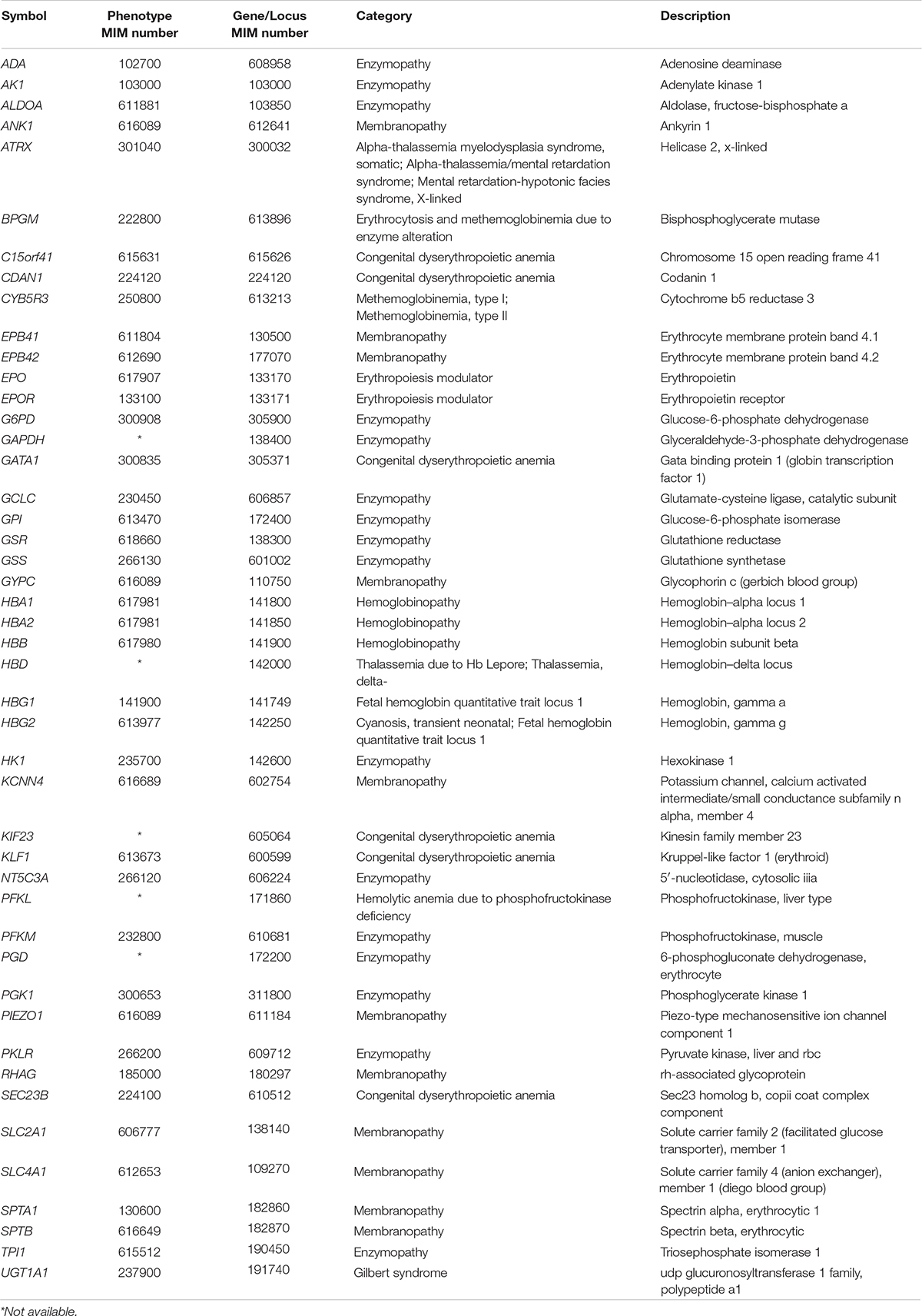
Table 2. List of genes included in the t-NGS approach.
For case 4, WES was performed in a trio approach (patient and both parents). Libraries were prepared using the Kapa HTP kit (Illumina, San Diego, CA, United States) and capture was performed using the SeqCap EZ Human Exome Library v3.0 (Roche NimbleGen Madison, WI, United States). Sequencing was done on an Illumina HiSeq2500 HTv4 (Illumina, San Diego, CA, United States) with paired-end 125-bp reads. Read alignment to hg19 and variant calling were done with a pipeline based on BWA-MEM0.7 and GATK 3.3.0. The median coverage of the captured target region was at least 98×. Variant annotation and prioritizing were done using Cartagenia Bench Lab NGS (Agilent Technologies). Variants located outside the exons and intron/exon boundaries and variants with a minor allele frequency (MAF) of >1% in control databases, including dbSNP1374, 1000 Genomes Project (phase 3)5, and Exome Variant Server (EVS), NHLBI Exome Sequencing Project National Heart, Lung, and Blood Institute GO Exome Sequencing Project (ESP6500 release)6 and in-house exome controls were excluded. Variants that fitted with a de novo or recessive mode of inheritance were further prioritized based on literature, predicted (deleterious) effects on protein function by e.g., truncating the protein, affecting splicing, amino acid change, and evolutionary conservation.
For case 5, WES was performed in a trio approach (patient and both parents). Exomes were captured using the Agilent SureSelectXT Human All Exon v5 (Agilent, Santa Clara, CA, United States) accompanied by Illumina paired-end sequencing on the HiSeq2000 (Illumina, San Diego, CA, United States). The in-house sequence analysis pipeline Modular GATK-Based Variant Calling Pipeline (MAGPIE) (LUMC Sequencing Analysis Support Core, LUMC) was used to call the SNVs/indels. LOVDplus (Leiden Genome Technology Center, LUMC, Leiden) was used for interpretation of variants.
Results
Genetic variants in globin genes responsible for UH or HUH were found in all five cases as shown in Table 1. All variants were confirmed by Sanger sequencing.
In case 1, variant HBB:c.202G > A (p.Val67Met) was found in exon 2 in the heterozygous state. This HBB variant is known as Hb Bristol-Alesha, a UH associated with moderate-severe hemolytic anemia. The variant was not found in the parents, suggesting a de novo variant in the patient.
In case 2, variant HBA1:c.187G > A (p.Val62Met) was found in exon 2 in the heterozygous state. This HBA1 variant is known as Hb Evans and is associated wild with mild hemolytic anemia and classified as UH. Parents were not sequenced.
In Case 3, variant HBB:c.290T > C (p.Leu96Pro) was found in in exon 2 in the heterozygous state. This HBB variant is known as Hb Debrousse and is described as a moderate UH. Parents were not sequenced. Nevertheless, antecedents of hemolytic anemia are present in the mother, suggesting a dominant inheritance pattern.
In cases 4 and 5, two missense stop-loss mutations at position 422 of the HBB gene were found. The first variant HBB:c.442T > C (p.Ter147Glnext∗21), found in case 4, is known as Hb Zunyi, while the second variant HBB:c.442T > A (p.Ter147Lysnext∗21), found in case 5, constitutes a novel variant which was called Hb Mokum. Both variants cause the loss of a stop codon and elongation of the translated beta-globin chain of 21 amino acids due to a new stop codon in the 3′ untranslated region (3′UTR) of the HBB gene. The variants were not found in the parents suggesting de novo variants in the patients.
According to the ACMG guidelines, all the variants were classified as pathogenic (Richards et al., 2015).
Discussion
We highlight the importance of including globin genes in the NGS analysis of RAD for enabling the diagnosis of UH. We present five clinical cases affected with RAD due to UH variants, four are related to the beta-globin gene (Hb Bristol—Alesha, Hb Debrousse, Hb Zunyi, and the novel Hb Mokum), meanwhile, one is related to the alpha-globin gene (Hb Evans). The use of NGS has been crucial for the final conclusive diagnosis.
The severity of RADs due to UHs depends on the mutation’s impact on protein stability and consequently on the degree of hemolysis and inefficient erythropoiesis. Patients’ RBCs typically display abnormal but unspecific morphology with microcytosis, hypochromia, moderate to severe anisopoikilocytosis, basophilic stippling, and inclusions that may become particularly prominent following splenectomy (Steinberg et al., 2009; Kent et al., 2014). UHs are commonly inherited in a dominant way or presented as de novo, although there are some examples of recessive inheritance leading to mild phenotypes. According to results obtained through the HbVar Query page (dated 14th January 2020), 1,534 Hb variants have been described so far due to mutations on either HBA1/HBA2 or the HBB genes. Up to 251 variants (16.4%) are classified as UH or HUH based on heat or isopropanol stability tests and/or low Hb abundancy (Giardine et al., 2007, 2014). It is worthy to highlight that all the HUH variants involving the HBB gene reported positive stability tests, meanwhile in most of the HUH involving the alpha-globin genes, hyper instability has been only deduced from low abundance. This must be cautiously taken since mutations in alpha-globin genes are lower expressed (<25%) than in beta-globin gene due to the existence of duplicated alpha-globin genes, HBA1 and HBA2, especially in mutations involving the HBA2 gene, as it encodes a 2-3–fold higher level of mRNA than HBA1 (Liebhaberts et al., 1986). Thus, the beta-globin gene is the first option to investigate for disease-causing mutations leading to RADs due to UHs/HUHs especially in cases with moderate to severe phenotypes.
Interestingly, Hb Bristol-Alesha is classified as a UH variant, not as a HUH as we expected based on the severity of the patient’s clinical picture. The change to methionine at position 67 of the beta-globin chain alters the hydrophobic heme pocket causing the instability of the protein (Kano et al., 2004). As described in previous clinical reports, at physical examination, splenomegaly and jaundice may be found. Iron overload and gallstones may develop due to the rapid turnover of RBCs.
Hb Debrousse, reported twice in literature, is a UH characterized by well-compensated chronic hemolytic anemia due to its high oxygen affinity. Hb Debrousse is caused by leucine to proline substitution at position 96 involving the hydrophobic environment of the proximal side of the heme. In the previously reported cases, Hb Debrousse discovery was possible after a Parvovirus B19 infection that caused a hemolytic crisis (Lacan et al., 1996). Indeed, since affected patients show a chronic well-compensated hemolytic anemia, the diagnosis of such a variant is unlikely until the globin genes are investigated. Such a study is usually performed only when some complications occur.
Hb Zunyi was recently reported for the first time as a de novo mutation in a Chinese child with severe anemia requiring blood transfusion, malnutrition, growth delay, splenomegaly and hepatomegaly (Su et al., 2019). In the study herein, we identified both Hb Zunyi and Hb Mokum as de novo mutations in the heterozygous state. Hb Zunyi and the novel Hb Mokum are stop-loss mutations at position 442 in HBB, resulting in an elongated beta-globin chain leading to HUHs. The extra amino acids in the elongated beta-globin chain (169 a.a.) are probably affecting its helical sequence, interfering with its tertiary structure and causing an unstable tetramer. Frameshift mutations in the HBB gene, resulting in the elongated beta-globin chain, have been described before but resulted in shorter beta-chains (max. 157 aa.) and milder phenotypes than the mutations described here (Su et al., 2019).
Finally, Hb Evans is classified as UH. It is consequence of a valine to methionine substitution at position 62 of the alpha2-globin chain encoding gene HBA2. Hb Evans has been reported in patients presenting with mild hemolytic anemia that was getting worse particularly in case of stress (Wilson et al., 1989).
The standard tests to detect abnormal anemias are High Precision Liquid Chromatography (HPLC) or conventional or capillary electrophoresis (CE). However, UHs/HUHs do not normally appear in the peak-patterns or appear as small peaks that may be mistaken for degradation products. In three of the five UH cases reported here, extra peaks were not detected. More confined methods are Heinz Bodies test or stability tests as isopropanol precipitation or heat stability tests, which are affordable screening techniques for UHs/HUHs variants. In the patient with Hb Bristol-Alesha, Heinz bodies were detected and heat stability test was positive only after splenectomy (Figures 1, 2), while in the other patients, Heinz bodies and heat stability test were not performed. Genetic analysis of globin genes should be performed for diagnosis confirmation. Inclusion of HBA1/HBA2 and HBB in NGS approaches will facilitate timely conclusive diagnosis. as a screening tool for hemolytic anemias will assist in reaching a definitive diagnosis sooner.
Furthermore, the occurrence of de novo mutations causing UHs/HUHs should also be considered in the analysis of genetic variants.
The usefulness of NGS in improving the diagnosis of RADs has already been demonstrated in several studies as well as its relevance in new gene discovery (Shang et al., 2017; Duez et al., 2018). In the case of overlapping phenotypes, which frustrate proper diagnosis, the use of NGS may be beneficial for ultra-rare RADs. In a recent publication, 36% of patients initially diagnosed with congenital dyserytrhopoietic anemia, received a final diagnosis of pyruvate kinase deficiency after NGS analysis (Russo et al., 2018). Nevertheless, in the majority of the t-NGS panels reported, globin genes are not included, since globin genes are quite short and molecular diagnosis of most common Hb disorders, such as sickle cell disease (SCD) and thalassemia syndromes, is well-established through Sanger sequencing and GAP-PCR/MLPA. Therefore, dBTHAL disorders due to UH/HUH may also benefit from NGS approaches for RADs by including globin genes, as presented herein.
Current literature on dBTHAL and UH/HUH variants is mainly composed of retrospective case reports, which makes evidenced-based management of this RAD unlikely. In addition, to benefit from the most adequate management it is necessary to achieve a diagnosis as early as possible. In conclusion, this study confirms the importance of NGS as a fundamental tool to early identify and treat UH/HUH in patients with RAD without an established diagnosis after standard methodologies.
Future challenges include a better understanding of disease characteristics and management, and consideration of bone marrow transplant as a curative option. Therefore, we encourage that these patients are referred to expert Units in referral centers for enabling basic and clinical research taking advantage of the already established European Reference Networks for rare hematological disorders, ERN-EuroBloodNet.
Data Availability Statement
The datasets generated for this study can be found in the online repositories. The names of the repository/repositories and accession number(s) can be found below: www.ithanet.eu and https://ithanet.eu/db/ithagenes?ithaID=3697.
Ethics Statement
Written informed consent was obtained from the cases/legal guardian for the publication of any potentially identifiable images or data included in this article.
Author Contributions
VR, MM-P, CLH, TK, and DB wrote the manuscript. All authors critically revised the manuscript.
Funding
This study was supported by funding from the authors’ institutions and the European Commission H2020-MSCA-ITN-2019, Grant Agreement N860436, “EVIDENCE.”
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was generated within the European Reference Network on Rare Hematological Diseases (ERN-EuroBloodNet, FPA 739541).
Footnotes
- ^ https://software.broadinstitute.org/gatk/
- ^ http://www.hgvs.org
- ^ https://varsome.com
- ^ http://www.ncbi.nlm.nih.gov/projects/SNP
- ^ http://www.internationalgenome.org/
- ^ http://evs.gs.washington.edu/EVS/
References
Bunn, H. F., and Forget, B. G. (1986). Hemoglobin: Molecular, Genetic and Clinical Aspects. Philadelphia, PA: W. B. Saunders Company, doi: 10.1016/0092-8674(87)90069-9
Duez, J., Carucci, M., Garcia-Barbazan, I., Corral, M., Perez, O., Luis Presa, J., et al. (2018). High-throughput microsphiltration to assess red blood cell deformability and screen for malaria transmission-blocking drugs. Nat. Protoc. 13, 1362–1376. doi: 10.1038/nprot.2018.035
Efremov, G. D. (2007). Dominantly inherited β-Thalassemia. Hemoglobin 31, 193–207. doi: 10.1080/03630260701290092
Galanello, R., and Origa, R. (2010). Beta-Thalassemia. Orphanet J. Rare Dis. 5:11. doi: 10.1186/1750-1172-5-11
Giardine, B., Borg, J., Viennas, E., Pavlidis, C., Moradkhani, K., Joly, P., et al. (2014). Updates of the hbvar database of human hemoglobin variants and Thalassemia Mutations. Nucleic Acids Res. 42, 1063–1069. doi: 10.1093/nar/gkt911
Giardine, B., van Baal, S., Kaimakis, P., Riemer, C., Miller, W., Samara, M., et al. (2007). HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum. Mutat. 28:206. doi: 10.1002/humu.9479
Kano, G., Morimoto, A., Hibi, S., Tokuda, C., Todo, S., and Sugimoto, T. (2004). Hb Bristol-Alesha presenting Thalassemia-Type hyperunstable hemoglobinopathy. Int. J. Hematol. 80, 410–415. doi: 10.1532/IJH97.04048
Kent, M. W., Oliveira, J. L., Hoyer, J. D., Swanson, K. C., Kluge, M. L., Dawson, D. B., et al. (2014). Hb grand junction (HBB: C.348-349delinsG; P.His117IlefsX42): a new hyperunstable hemoglobin variant. Hemoglobin 38, 8–12. doi: 10.3109/03630269.2013.853672
Lacan, P., Kister, J., Francina, A., Souillet, G., Galactéros, F., Delaunay, J., et al. (1996). Hemoglobin debrousse (B 96[FG3]Leu → Pro): a new unstable hemoglobin with twofold increased oxygen affinity. Am. J. Hematol. 51, 276–281. doi: 10.1002/(SICI)1096-8652(199604)51:4<276::AID-AJH5<3.0.CO;2-T
Liebhaberts, S. A., Cash, F. E., Ballad, S. K., and Human Gene Expression (1986). Human A-Globin gene expression. The dominant role of the Alpha 2-Locus in MRNA and protein synthesis. J. Biol. Chem. 261, 15327–15333.
Richards, S., Aziz, S., Bale, S., Bick, D., Das, S., Acmg Laboratory Quality Assurance Committee, et al. (2015). Standards and guidelines standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Russo, R., Manna, F., Gambale, A., Marra, R., Rosato, B. E., Caforio, P., et al. (2018). Multi-Gene panel testing improves diagnosis and management of patients with Hereditary Anemias. Am. J. Hematol. 93, 672–682. doi: 10.1002/ajh.25058
Shang, X., Peng, Z., Ye, Y., Asan, Zhang, X., Chen, Y., et al. (2017). Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine 23, 150–159. doi: 10.1016/j.ebiom.2017.08.015
Steinberg, M. H., Forget, B. G., Higgs, D. R., and Weatherall, D. J. (2009). Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management, Second Edition, Vol. 94. Cambridge: Cambridge University Press, i–iv. doi: 10.1017/CBO9780511596582
Su, Q., Chen, S., Wu, L., Tian, R., Yang, X., Huang, X., et al. (2019). Severe thalassemia caused by Hb Zunyi [B 147(HC3)Stop→Gln; HBB: C.442T>C)] on the β-Globin gene. Hemoglobin 43, 7–11. doi: 10.1080/03630269.2019.1582430
Thom, C. S., Dickson, C. F., Gell, D. A., and Weiss, M. J. (2013). Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harb. Perspect. Med. 3, 1–22. doi: 10.1101/cshperspect.a011858
Keywords: unstable hemoglobinopathies, dominant beta-thalassemia, next generation sequencing, whole exome sequencing, rare anemia disorders
Citation: Rizzuto V, Koopmann TT, Blanco-Álvarez A, Tazón-Vega B, Idrizovic A, Díaz de Heredia C, Del Orbe R, Pampliega MV, Velasco P, Beneitez D, Santen GWE, Waisfisz Q, Elting M, Smiers FJW, de Pagter AJ, Kerkhoffs J-LH, Harteveld CL and Mañú-Pereira MdM (2021) Usefulness of NGS for Diagnosis of Dominant Beta-Thalassemia and Unstable Hemoglobinopathies in Five Clinical Cases. Front. Physiol. 12:628236. doi: 10.3389/fphys.2021.628236
Received: 11 November 2020; Accepted: 13 January 2021;
Published: 05 February 2021.
Edited by:
Paola Bianchi, IRCCS Ca’ Granda Foundation Maggiore Policlinico Hospital, ItalyReviewed by:
James Hoyer, Mayo Clinic, United StatesTheodosia A. Kalfa, Cincinnati Children’s Hospital Medical Center, United States
Copyright © 2021 Rizzuto, Koopmann, Blanco-Álvarez, Tazón-Vega, Idrizovic, Díaz de Heredia, Del Orbe, Pampliega, Velasco, Beneitez, Santen, Waisfisz, Elting, Smiers, de Pagter, Kerkhoffs, Harteveld and Mañú-Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria del Mar Mañú-Pereira, bWFyLm1hbnVAdmhpci5vcmc=