
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 13 January 2021
Sec. Mitochondrial Research
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.615038
 Jonathan M. Memme1,2†
Jonathan M. Memme1,2† David A. Hood1,2*†
David A. Hood1,2*†Mitochondrial dysfunction is common to many organ system disorders, including skeletal muscle. Aging muscle and diseases of muscle are often accompanied by defective mitochondrial ATP production. This manuscript will focus on the pre-clinical evidence supporting the use of regular exercise to improve defective mitochondrial metabolism and function in skeletal muscle, through the stimulation of mitochondrial turnover. Examples from aging muscle, muscle-specific mutations and cancer cachexia will be discussed. We will also examine the effects of exercise on the important mitochondrial regulators PGC-1α, and Parkin, and summarize the effects of exercise to reverse mitochondrial dysfunction (e.g., ROS production, apoptotic susceptibility, cardiolipin synthesis) in muscle pathology. This paper will illustrate the breadth and benefits of exercise to serve as “mitochondrial medicine” with age and disease.
Mitochondria are the principal organelles tasked with the provision of cellular energy in the form of ATP, which is supplied in equilibrium with the energy demands placed on the cell. Changes within the cellular environment also provoke the mitochondrial network to regulate other important functions, such as initiating mitochondrial-nuclear retrograde signals, maintaining redox and Ca2+ homeostasis, as well as determining cell fate (Hood et al., 2019). The complex and multifaceted influence of mitochondria on cellular functions requires that the organelle network exist in a constant state of flux that allows for precise remodeling in order to match morphology and function of mitochondria to these imposing demands (Eisner et al., 2018; Guan et al., 2019). An intriguing aspect of mitochondrial biology is the tissue specificity that these organelles exhibit (Fernández-Vizarra et al., 2011). Skeletal muscle in particular comprises a large volume of total body mass (∼40%) and is highly metabolic with varying concentrations of mitochondria that provide ATP to the working muscle for locomotion, posture, and the generation of fine motor skills. Additionally, skeletal muscle displays distinct mitochondrial characteristics that are dependent on the location within the cell, and the metabolic requirements within those cellular compartments. Thus, given the relative proportion of body mass and the metabolic nature of skeletal muscle, the status of the mitochondrial pool within this tissue has considerable ramifications for systemic health.
Within the skeletal muscle myofiber, mitochondria exist in two distinct locales, which correspond with unique conformations and roles (Kayar et al., 1988; Cogswell et al., 1993; Ferreira et al., 2010). Subsarcolemmal (SS) mitochondria appear to be essential to provide energy for nuclear gene transcription and transport across the membrane (Kirkwood et al., 1986; Ogata and Yamasaki, 1997). Intermyofibrillar (IMF) mitochondria, localized along the sarcomeric proteins of the myofibrils and exhibit a more elongated appearance forming a reticular network throughout muscle fibers in close proximity to the transverse tubules and the sarcoplasmic reticulum (Kirkwood et al., 1986; Ogata and Yamasaki, 1997; Vincent et al., 2019). As such, these organelles supply the ATP used by myosin ATPases to achieve muscle contraction, and also play an important role in Ca2+ signaling (Boncompagni et al., 2009). Glancy et al. (2015) have further classified mitochondrial subpopulations based on their morphologies and proximity to various cellular structures. These uniquely situated mitochondrial structures comprise a network of interconnected organelles and serve to provide a pathway for energy distribution throughout the cell in the form of proton-motive force (PMF) (Glancy et al., 2017). Thus, the extent and location of the mitochondrial network is determined by the interplay of various ATP demand pathways, and regulated by mitochondrial quality control (MQC) machinery, which help to maintain the viability and quality of the organelle pool.
One of the most remarkable features of skeletal muscle mitochondria is the dynamic plasticity that they exhibit. For instance, prolonged aerobic exercise training is capable of eliciting improvements in mitochondrial content, mass, and overall function, whereas chronic muscle disuse produces the opposite effect (Tryon et al., 2014; Memme et al., 2019). Mitochondrial synthesis (biogenesis) is a result of increases in the expression of nuclear and mtDNA genes encoding mitochondrial proteins, while fusion proteins such as Mfn1/2 and Opa1/2 promote the merger of adjacent organelles, thus collectively expanding the mitochondrial network while improving efficiency (Iqbal et al., 2013; Mishra and Chan, 2016). Conversely, mitochondrial segments that malfunction are selectively removed, first through their segregation by fission machinery proteins such as Drp1, Fis1 and Mff in order to be cleared by the lysosome through mitophagy (Iqbal et al., 2013; Mishra and Chan, 2016). The balance of these opposing processes is imperative to mitochondrial quality and function, and any loss of equilibrium in these processes can contribute to mitochondrial impairments that are detrimental to tissue health. In fact, conditions such as advancing age, mitochondrial mutations, and diseases such as cancer are associated with deficiencies in the MQC machinery, leading to organelle dysfunction and pathogenesis (Figure 1). Conversely, exercise is perhaps the most potent stimulus for the activation of both mitochondrial biogenesis and mitophagy, and the consequent preservation and improvement of metabolic health within skeletal muscle. Therefore, it is important to understand how these processes are regulated at the molecular level.
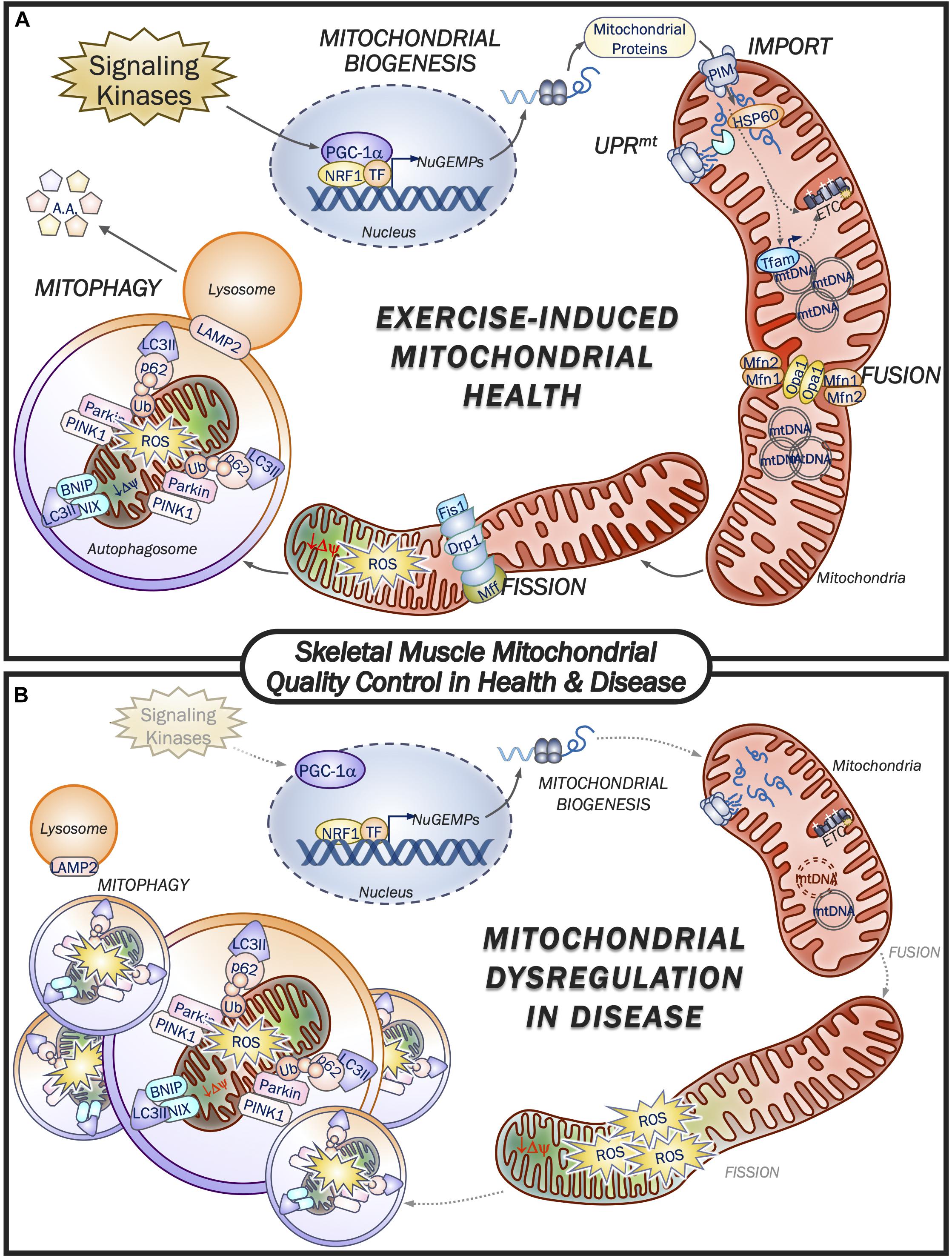
Figure 1. Skeletal muscle mitochondrial quality control in health and disease. (A) Exercise is a potent promotor of mitochondrial health by eliciting increases in mitochondrial quality control machinery. Upstream signaling kinases respond to muscle contractile activity leading to activation of PGC-1α and augmented transcription of nuclear genes encoding mitochondrial proteins (NuGEMPs). Newly translated mitochondrial-targeted proteins are then transported into the mitochondrion via the protein import machinery (PIM) and are subsequently met by resident chaperones and proteases to facilitate their incorporation within the organelle. The UPRmt is responsible for regulating the expression of mitochondrial chaperones and proteases in the presence of a stress stimulus such as exercise. As mitochondrial volume within the myofiber expands, fusion machinery adjoins neighboring organelles’ inner and outer membranes to facilitate transfer of metabolites, mtDNA, etc. Dysfunctional mitochondrial segments that lose their membrane potential and emit excessive ROS are cleaved from the reticulum by the fission proteins, thereby allowing mitophagy machinery to selectively encapsulate and degrade these non-functioning organelles for delivery to the lysosome. Exercise activates both the biogenesis and mitophagy pathways of MQC to promote the health and viability of the mitochondrial pool within muscle. (B) Mitochondrial dysfunction is a hallmark feature of disease, including cancer, aging and associated mitochondrial disorders. In general, activation of signaling kinases is impaired and reduces the drive for mitochondrial biogenesis from the nucleus, while mtDNA copy number can be reduced or have a high proportion of mutated mtDNA, which can serve to blunt mitochondrial expansion. Reductions in fission and fusion machinery impair mitochondrial morphology and shift the fission:fusion ratio in favor of network fragmentation and organelle ROS production. Further exacerbating the mitochondrial derangements is the impaired mitochondrial removal via reduced mitophagic clearance by the lysosome, thus contributing to the poor morphology and function of the mitochondrial network. As described in the text, exercise can reverse many of the pathways that lead to mitochondrial dysfunction, thereby restoring muscle health.
Mitochondrial biogenesis is responsible for the increased synthesis of mitochondrial proteins that are derived from both nuclear and mitochondrial genomes. While there are approximately 1,200 proteins that are localized within mitochondria, mtDNA contributes a very small, but nonetheless important fraction, including 13 ETC subunit proteins synthesized by the organelle’s distinct protein synthesis machinery (Anderson et al., 1981). Signaling toward mitochondrial biogenesis is first initiated by kinases such as AMPK, p38 MAPK, CaMK, and PKA, which become activated upon the sensing of energetic and redox imbalances, increases in intracellular Ca2+ levels, or adrenergic signaling, respectively (Winder et al., 2000; Puigserver et al., 2001; Handschin et al., 2003; Ojuka et al., 2003; Irrcher et al., 2009; Ristow et al., 2009). These signaling kinases converge on the master regulator of mitochondrial biogenesis, PGC-1α, along with related factors such as PGC-1β and PRC, which bind various transcription factors on the promotor regions of a multitude of nuclear genes encoding mitochondrial proteins (NuGEMPs), thus increasing their expression (Hood, 2001; Handschin and Spiegelman, 2006; Scarpulla, 2011; Scarpulla et al., 2012). Perhaps the most notable PGC-1α-transcription factor interaction is through Nuclear Respiratory Factor-1 (NRF1) to promote the expression of mitochondrial transcription factor A (Tfam), which in turn mediates mtDNA transcription, thereby connecting the expression of the mitochondrial and nuclear genomes (Gordon et al., 2001; Scarpulla, 2011; Figure 1). While PGC-1α is considered to be the master regulator of mitochondrial biogenesis, it may not always be necessary to achieving mitochondrial adaptations following exercise, since training-induced increases in mitochondrial content (Leick et al., 2008) and function (Adhihetty et al., 2009; Ballmann et al., 2016) can be achieved in PGC-1 knockout animals. Indeed, additional regulators contributing to mitochondrial regulation include SirT1 (Menzies et al., 2013), which is capable of activating PGC-1α through its deacetylase function, as well as p53, another well-established transcription factor that translocates to both the nucleus and mitochondria to regulate the expression of mtDNA- and nDNA-derived transcripts (Heyne et al., 2004; Park et al., 2009; Saleem et al., 2009; Saleem and Hood, 2013; Beyfuss et al., 2018). Moreover, the regulatory regions of nuclear genes encoding mitochondrial proteins are heterogeneous and therefore, the regulation of organelle synthesis requires multiple regulators of transcription (Lenka et al., 1998; Hood, 2001). A number of transcription factors associated with exercise-induced mitochondrial biogenesis have been established, such as CREB, the estrogen related receptors (ERRα,β,γ), c-myc, specificity protein-1 (Sp-1), upstream stimulatory factor-1 (USF-1), as well as nuclear respiratory factors 1 and 2 (NRF1 and NRF2) (see Islam et al., 2020 for review; Hood et al., 2011; Knudsen et al., 2020). Each transcription factor when bound to target gene promoter sequences can upregulate the expression of mitochondrial proteins.
Equally important to the synthesis of new mitochondrial proteins is the removal of mitochondria that begin to emit excessive ROS, or that have lost their membrane potential, therefore becoming dysfunctional (Ljubicic et al., 2010; Lira et al., 2013; Kim et al., 2018). Mitophagy is the mitochondrial-specific form of autophagy whereby a double membrane phagophore engulfs damaged organelles to form an autophagosome, that is subsequently broken down by the lysosome and recycled (Wei et al., 2015). Although several mitophagy pathways have been elucidated, the most well-established pathway is mediated by the interaction of PINK1 and the E3-ubiquitin ligase, Parkin. Under basal conditions, PINK1 is imported into the mitochondria and degraded, however, as the mitochondrial membrane potential dissipates, PINK1 import is impaired. As a result PINK1 stabilizes on the OM and recruits Parkin, leading to the ubiquitination of OM proteins (Geisler et al., 2010). The nucleation of the phagophore membrane requires maturation of LC3-I into its lipidated form, LC3-II and is initiated by upstream activation of the Beclin1 complex. Various autophagy (ATG) proteins such as ATG7 conjugate LC3 with phosphatidylethanolamine, thus allowing for its incorporation within the membrane structure (Tanida et al., 2004). The adapter protein p62/SQSTM1 simultaneously binds to ubiquitin bound to the tagged mitochondrion along with LC3 embedded in the phagophore membrane, thus forming the autophagosome. This structure then traverses microtubule tracks to the lysosome where it can fuse, and the contents are degraded (Geisler et al., 2010; Vainshtein and Hood, 2016; Figure 1). The transcription factors TFEB and TFE3 are two critical regulators of genes involved in lysosomal biogenesis as well as autophagosomal machinery (Settembre and Ballabio, 2011; Mansueto et al., 2017). TFEB and TFE3 have been shown to be activated via common signals (i.e., AMPK, Ca2+) that are known to promote mitochondrial biogenesis, and they may also play a role in initiating mitochondrial biogenesis directly, or in concert with PGC-1α (Kim et al., 2014; Vainshtein and Hood, 2016; Mansueto et al., 2017; Erlich et al., 2018).
The UPRmt monitors the mitochondrial environment and appropriately mounts a response in order to either match an imposed demand, or to mitigate damage to the organelle network. In this way, the UPRmt provides an intermediate between synthesis and degradation of mitochondria. Recent work has identified the transcription factor ATF4, along with its downstream targets ATF5 and CHOP, as key regulators of the UPRmt, which are induced by disruptions in mitochondrial proteostasis and increasing levels of ROS, such as with exercise when protein import is accelerated (Memme et al., 2016; Mesbah Moosavi and Hood, 2017; Oliveira and Hood, 2018). Together, the activation of these transcription factors provides a mitochondria-to-nucleus retrograde signal, as they translocate to the nucleus to promote the upregulation of mitochondrial chaperones and proteases to help nascent polypeptides either achieve their mature configuration, or degrade terminally misfolded proteins, respectively (Granata et al., 2017; Melber and Haynes, 2018). The proteolytic cleavage of misfolded proteins generates peptide fragments that can directly suppress organellular import, contributing to PINK1 stabilization on the outer membrane and the subsequent induction of mitophagy. Furthermore, evidence suggests that ATF4 is intricately linked to TFEB/TFE3 signaling in the presence of cellular stress (Martina et al., 2016). Thus, the UPRmt matches the health status of the organelle to the appropriate signaling response and is upregulated in the early stages of an exercise stimulus, preceding improvements in mitochondrial content and function (Memme et al., 2016; Mesbah Moosavi and Hood, 2017). However, while an increasing volume of research suggests the importance of the UPRmt in the maintenance of mitochondrial health, the influence of this process in disease, and the activation of the pathway by exercise remains to be fully understood.
The natural process of aging is associated with progressive reductions in the strength, function and size of muscle, the defining features of a condition known as sarcopenia (Rosenberg, 2011). Dysfunctional mitochondria are considered to play a primary role in the development of sarcopenia as they are important players in regulating many of the aforementioned processes that become dysregulated (Heber et al., 1996; Rapizzi et al., 2002; Kujoth et al., 2005; Schaap et al., 2006).
Mitochondrial decline is characteristic of aged muscle, and when viewed under the microscope, the organelle network maintains a distinctly fragmented appearance with aberrantly enlarged organelles, yet considerably thinner SS and IMF regions as compared to young muscle (Short et al., 2005; Iqbal et al., 2013; Leduc-Gaudet et al., 2015). The observed mitochondrial fragmentation can be accounted for by the reduced expression of both fission and fusion proteins, resulting in a net increase in the fission:fusion ratio (Ibebunjo et al., 2013; Iqbal et al., 2013). Diminished content is explained by reductions in both the expression of mitochondrial genes and corresponding proteins, as well as increased rates of mitophagy flux (Baraibar et al., 2013; Liu et al., 2013; Carter et al., 2015, 2018a; Chen et al., 2018). Therefore, synthesis of mitochondrial enzymes is reduced, while concurrently, mitochondrial proteins are being degraded at a faster rate. PGC-1α is likewise reduced at the transcript level in older muscle, while mtDNA mutations become more prevalent with advancing age (Wanagat et al., 2001; Bua et al., 2006; Conley et al., 2007; Chabi et al., 2008; Koltai et al., 2012; Carter et al., 2018b). The consequences of these reductions have functional implications since mitochondrial respiratory function is commonly found to be reduced, and augmented uncoupling of O2 consumption to the synthesis of ATP is observed (Boffoli et al., 1994; Rooyackers et al., 1996; Marcinek et al., 2005). Furthermore, as strength is correlated to muscle fiber cross sectional area, the contribution of mitochondrial derangements to muscle atrophy is considerable. Decreases in PGC-1α allow FoxO3a, a pro-atrophy gene that is suppressed by PGC-1α, to become elevated, while it has likewise been observed that atrophy is most prevalent in fibers harboring > 80% mtDNA defects (Wanagat et al., 2001; Bua et al., 2006; Sandri et al., 2006). Additionally, insufficient clearance of maladaptive organelles in aged tissue leads to overproduction of ROS, activation of apoptosis and subsequent DNA fragmentation, ultimately promoting regional atrophy along the myofiber (Bua et al., 2002; Chabi et al., 2008; Gouspillou et al., 2014).
The question of whether the skeletal muscle decline with aging is a byproduct of increased sedentarism or intrinsic to the aging process remains to be unequivocally determined experimentally. However, the merits of adopting a physical activity regimen is unquestionable, as continuous exercise can promote favorable organelle health, and mitigate some of the associated declines in muscle quality seen with age. Indeed, endurance training can ameliorate the decreased expression observed in many of the affected proteins that are diminished in aged muscle, such as those involved in energy metabolism and generation of ATP (Lanza et al., 2008). While life-long participation in high-level physical activity is our best means of promoting and preserving mitochondrial health in aged muscle, adoption of an exercise routine later in life is nonetheless capable of achieving levels of mitochondrial content and function that are comparable to those found in young muscle (Short et al., 2003; Lanza et al., 2008; Joseph et al., 2012; Carter et al., 2015). However, the sensitivity of aged muscle to the exercise stimulus appears to be impaired, as the activation of the important signaling kinases is blunted, as is the exercise-induced activation of mitochondrial clearance via mitophagy (Ljubicic et al., 2009; Chen et al., 2018). While basal mitophagy flux is elevated with age, it is unclear whether this is intended to be protective, or contributes to the mitochondrial defect since dysfunction still predominates aged muscle, as evidenced by increased ROS emission (O’Leary et al., 2013; Carter et al., 2018a; Chen et al., 2018). Nevertheless, exercise can mitigate the excessive accumulation of ROS and attenuate activation of atrophy-inducing apoptosis (Ljubicic et al., 2009; Di Meo et al., 2019). Exercise also reverses the age-induced reductions in PGC-1αtranscription, restoring it to levels observed in younger cohorts (Carter et al., 2018b). In addition to endurance exercise, resistance type training also promotes muscle health by promoting muscle hypertrophy while also recruiting the fusion of satellite cells that do not harbor mtDNA mutations, despite the reduced stimulus for mitochondrial biogenesis that this training modality offers (Taivassalo, 1999; Kowald and Kirkwood, 2014; Carter et al., 2015). Thus, continuous participation in regular exercise training, whether adopted throughout life, or much later, is an established and non-pharmacological approach to maintaining mitochondrial health, leading to improved skeletal muscle function and overall quality of life.
As mitochondria are integral organelles in the maintenance of many cellular processes, the pathogenesis of various diseases can be traced to mitochondrial irregularities that may be inherent or acquired via lifestyle factors. More than 300 disease-associated mtDNA mutations have been identified, which can be attributed to either genetic inheritance, or the vulnerability of mtDNA and its exposure to high levels of ROS in the mitochondrial matrix (Li et al., 2019). However, the investigation of mitochondrial disorders (MD) is complex, and often present with varied pathological phenotypes that manifest in a range of tissues including brain, liver, eye, heart, and skeletal muscle (Alston et al., 2017).
Mutations to either mitochondrial or nuclear-derived genes can produce mitochondrial defects that most often present within skeletal muscle, either as the lone affected tissue or as a predominant feature of a multisystem disorder, thus making it the tissue of choice for the study of MD (Taylor et al., 2004; McFarland et al., 2010). Skeletal muscle can be routinely sampled via the muscle biopsy technique, which allows full characterization of the functional and morphological impairments to mitochondria (Phadke, 2017). Mutations of mtDNA commonly affect mitochondrial tRNAs and protein encoding regions, and are associated with mtDNA instability leading to derangements in copy number and quality (Russell and Turnbull, 2014; Rusecka et al., 2018). The broad range of mutations can impact various aspects of mitochondrial quality control, not limited to an effect on ETC function or impaired ATP production. Clinical detection of MD can be made histologically via the presence of ragged red fibers (RRFs), COX-negative fibers, as well as SDH deficiency (Phadke, 2017). Among the numerous MDs, common diseases include MELAS, Kearns-Sayre syndrome, Leigh syndrome, MERRF, and rhabdomyolysis, which present with weakness, fatigue and severe myopathy among a spectrum of other symptoms (Taylor and Turnbull, 2005; Alston et al., 2017; Phadke, 2017; Rusecka et al., 2018; Yan et al., 2019). Additionally, derangements in nuclear-transcribed mitochondrial genes can contribute to the etiology of various other conditions (Li et al., 2019; Sharma and Sampath, 2019). For instance, mutations in the Tafazzin gene promotes skeletal muscle and cardiac myopathy associated with Barth Syndrome due to impaired maturation of the inner membrane phospholipid cardiolipin that is required for membrane stability (Schlame and Ren, 2006). Likewise, defects in the mitophagy protein parkin prevents the clearance of dysfunctional mitochondria, contributing to the development of Parkinson’s disease (Dawson and Dawson, 2010). Evidently, the multifaceted regulation of mitochondrial content, morphology and function presents numerous opportunities for mutation and the development of various diseases.
The reliance of mitochondria on nuclear-encoded genes that are essential for mtDNA transcription makes the development of targeted therapies difficult. However, exercise is a proven stimulus for the improvement of mitochondrial content and function within skeletal muscle, and despite a reduced exercise capacity in patients with MD, evidence suggests that adhering to a continuous exercise regimen may serve as the most complete therapeutic intervention for the restoration of mitochondrial function. Preclinical studies have shown that PGC-1α may provide a useful therapeutic target as its activation may alleviate some of the observed myopathy and mitochondrial respiratory defects that are present (Srivastava et al., 2009; Fiuza-Luces et al., 2019). As such, the ability of endurance exercise to activate multiple signaling kinases that converge on PGC-1α suggests that it is a potent stimulus. Studies have illustrated that exercise is a viable intervention strategy that is effective in increasing citrate synthase activity, ETC complexes and overall patient aerobic fitness (Taivassalo et al., 1999, 2001, 2006; Jeppesen et al., 2006, 2009; Porcelli et al., 2016; Fiuza-Luces et al., 2018). Similar to the benefits described in aged muscle, strength training may also aid in promoting muscle mass and strength in MD patients, while reducing mtDNA heteroplasmy as a result of satellite cell recruitment, without any observable negative secondary effects (Kraemer et al., 2002; Murphy et al., 2008; Groennebaek and Vissing, 2017). Moreover, the combination of resistance with endurance training promoted significant improvements in the quality and function of skeletal and respiratory muscle in MD patients (Fiuza-Luces et al., 2018). Studies employing the mtDNA mutator mice, which harbor mutations in mitochondrial polymerase γ, have also suggested a potential role for exercise-induced p53 localization to the mitochondria to aid in the restoration of organellular morphological and functional defects (Safdar et al., 2016). Interestingly, there is evidence to suggest that endurance training promotes the restoration of mitochondrial defects associated with mtDNA mutations in the brains of patients with neurogenerative diseases such as Alzheimer’s (Picard et al., 2016). Thus, exercise elicits multiple signaling cascades that improve mitochondrial health, and does so across a range of affected tissues, which is particularly important given the multifaceted nature of MD.
While mitochondrial dysfunction is a hallmark feature of cancer tumorigenesis, in skeletal muscle, mitochondrial dysfunction contributes to the progressive muscle wasting associated with cancer cachexia. Approximately 80% of late stage cancer patients experience cachexia, which impacts morbidity and contributes to 20–40% of all cancer mortalities (Melstrom et al., 2007; Tisdale, 2009; Prado et al., 2013). The consensus definition of cachexia describes three stages of the condition: (1) Precachexia, involving weight loss < 5% along with signs of anorexia and impaired glucose tolerance; (2) cachexia, diagnosed when weight loss exceeds 2–5%; and (3) refractory cachexia, referring to variable levels of muscle wasting whereby the cancer is highly pro-catabolic with poor prognosis (Fearon et al., 2011). Thus, treatments are most effective when administered early, however, clinical management remains a major challenge as patient population, stage, type of tumor, and chemotherapies may contribute to the progression of cachexia.
The prevailing belief is that the progression of cachexia is the result of tumor-induced immune cell activation leading to systemic inflammation and the release of potent cytokines TNFα, IFNγ, IL-1, and IL-6, which promote changes in metabolism, energy wasting, cancer-induced anorexia, and skeletal muscle protein catabolism (Dwarkasing et al., 2016; Van Norren et al., 2017). The progressive nature of cachexia is based on the convergence of imbalanced proteostasis, myofiber degeneration and structural remodeling, as well as altered mitochondrial function (Van Der Ende et al., 2018). Overactivation of the ubiquitin proteasome system (UPS) and the autophagy-lysosome system contribute to advanced protein turnover, while impaired mTOR signaling contributes to impaired synthesis of new proteins (Llovera et al., 1995; Lorite et al., 1998; Gordon et al., 2013; Sandri, 2016). A consistent feature of cancer cachexia is the vast remodeling of the mitochondrial network and the array of changes in the expression level of regulatory genes and proteins involved in mitochondrial biogenesis (Cornwell et al., 2014; Judge et al., 2014; Shum et al., 2015). Most studies indicate that PGC-1α protein is significantly reduced with cancer cachexia, while genome-wide transcriptome datasets show that PGC-1β gene expression is similarly diminished (Fontes-Oliveira et al., 2013; Puppa et al., 2014; Sheng et al., 2017; Van Der Ende et al., 2018). Activators of PGC-1α SirT1 and MEF2C, are also found to decrease in cachectic muscle, as well as downstream targets, ERRα and Tfam, altogether indicating impaired drive for organelle synthesis (White et al., 2011; Shum et al., 2012; Zhuang et al., 2016; Chen et al., 2017; Van Der Ende et al., 2018). Indeed, it appears that the suppression of PGC-1α is a key event in the progression of cancer cachexia and is directly related to elevations in circulating IL-6 (Baltgalvis et al., 2008; White et al., 2012). Additionally, morphology proteins, Drp1, along with Opa1 and Mfn2 are reduced in cancer- and chemotherapy-induced cachexia, thus leading to increased organelle toxicity, and reduced organelle efficiency (Barreto et al., 2016; Van Der Ende et al., 2018). Microscopically, mitochondria in cachectic tissue appear swollen and are often contained in vesicle-like structures indicative of their incomplete clearance by the mitophagy machinery (Fontes-Oliveira et al., 2013; Tzika et al., 2013; Pin et al., 2015). Coincidentally, increases in autophagosomal markers such as p62, BNIP3, and the LC3-II:LC3-I ratio, along with the appearance of undegraded mitochondria further suggest impairments in the function of the lysosomes and their ability to fuse with, and digest the fully formed mitochondria-containing autophagosomes (Pin et al., 2015; Brown et al., 2017; Segatto et al., 2017).
Although more work is needed in this area, an increasing volume of evidence suggests that impairments in mitochondrial function play a causal role in the muscle wasting observed in cancer. In particular, over a 4 weeks time course following C26 tumor cell inoculation, one study found that changes in mitochondrial content and function preceded the corresponding atrophy, while another reported that mitochondrial dysfunction directly influenced amino acid metabolism, and diminished protein synthesis (Brown et al., 2017; Kunzke et al., 2020). Interestingly, a study in which Drp1 was selectively knocked out within rodent muscle demonstrated the robust myopathy, fiber degeneration/regeneration, and mitochondrial impairments that are characteristic of cachexia (Favaro et al., 2019). Notably, the corresponding mitochondrial dysfunction in these animals lead to the activation of ATF4, consequently inducing release of the myokine FGF21, which largely accounted for the diminished body mass (Favaro et al., 2019). These reports highlight a primary role for mitochondria in the pathogenesis of cancer cachexia and suggest the importance of targeted therapies that improve the mitochondrial condition, thus helping to suppress muscle wasting.
It is clear that mitochondria are highly relevant to not only tumorigenesis but also the progression of cancer cachexia. Therefore, it is reasonable to presume that regular exercise, with its well known effect on improving mitochondrial content and function, would provide a potent and viable therapeutic option for cancer patients. Multiple studies in both rodent and human tissue have reported that continuous exercise training in cachectic patients is capable of improving muscle mass and strength (Deuster et al., 1985; Salomão et al., 2010; Penna et al., 2011). Interestingly, both endurance and resistance type training modalities are sufficient to elicit these responses (Norton et al., 1979; Jaweed et al., 1983; Otis et al., 2007; Schmidt et al., 2015; Ballarò et al., 2019a; Sato et al., 2019). While considerable work is still required to dissect the molecular mechanisms at play, the current literature suggest that the signaling events associated with exercise reverse the pro-cachectic signals that promote skeletal muscle decline and frailty. Treadmill training has been also been shown to similarly augment mTOR signaling and augment mitochondrial quality control in the presence of systemic IL-6 overexpression, a major contributor to the activation of cachexia in cancer patients (White et al., 2012, 2013). Along with activation of PGC-1α, exercise also signals improvements in mitochondrial quality by mediating the expression of mitophagy proteins such as BNIP3, PINK1, and Parkin, along with fission/fusion machinery, and reduces ROS via augmented redox homeostasis (Pigna et al., 2016; Ballarò et al., 2019a,b). Moreover, the exercise stimulus is capable of enhancing oxidative capacity in tumor bearing mice, and aerobic fitness was found to be directly correlated to survival, suggesting the importance of mitochondrial health in tumor-bearing subjects (Hardee et al., 2016; Pigna et al., 2016). In addition, patients diagnosed with p53-mutation cancers, such as soft-tissue sarcomas, or cancers of the colon, lung, breast, liver, brain, and hemopoietic tissues, are likely able to improve muscle health through exercise training. p53 maintains normal mitochondrial content and function in muscle, but when mutated, leads to reduced mitochondrial function and causes cancer. Studies initially by Saleem et al. (2009), and subsequently by Beyfuss et al. (2018) found that muscle mitochondrial content and function can still adapt to exercise in the absence of p53, and muscle health benefits are still possible. However, it should also be noted that in animals studies involving late stage cachexia, exercise had no beneficial effect, and in fact reduced their survival. Thus, it appears that exercise is most effective as an early intervention strategy to either prevent or to slow down cachexia progression. The optimal exercise parameters, such as type, duration, intensity and frequency, to properly prescribe as part of training program to patients remains to be determined. However, it is clear that exercise provides a multifactorial treatment option that can be employed early in a patient’s diagnosis to improve strength, mobility, energy metabolism and overall quality of life.
As mitochondrial quality control is highly dynamic and relies on the convergence of multiple processes to regulate the function of the organelle network, the development of therapeutic interventions that adequately address all aspects of mitochondrial remodeling remains a challenge to identify. Moreover, the metabolic nature of skeletal muscle, and the relative proportion of body mass that it comprises, make muscle a unique tissue to study in the context of various diseases. Exercise performed on a regular basis in either continuous or interval training formats remains our one true treatment modality capable of augmenting mitochondrial health in muscle, by coordinately inducing both synthesis and recycling of mitochondrial proteins, and therefore serving as mitochondrial medicine for muscle. Indeed, this signaling toward mitochondrial turnover begins with the very first bout of exercise, which triggers the onset of biogenesis as well as mitophagy (Vainshtein and Hood, 2016). Ultimately, long-term training-induced adaptations can also be used to identify specific factors that can potentially be targeted pharmaceutically as adjuncts to promote mitochondrial health in the presence of an exercise stimulus, or perhaps independently for those incapable of adopting a training program. However, before this can be accomplished, there are still many aspects of mitochondrial signaling associated with exercise that remain elusive. For instance, while the balance of synthesis and degradation is well understood, other key regulators of mitochondrial remodeling require further analyses, such as the UPRmt, as well as TFEB and TFE3 as master regulators of lysosomal biogenesis and autophagy proteins. The interplay of these processes and their regulators with exercise warrants considerable research effort to establish a clear understanding of the mechanistic features that drive the sustenance of a high quality mitochondrial pool, and the consequent maintenance of muscle health with age, cancer and mitochondrial disease.
JM and DH contributed to writing, editing and reviewing this manuscript, and accompanying figure. Both authors contributed to the article and approved the submitted version.
This work was supported by a grant from the Canadian Institutes for Health Research (CIHR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; ATF4, activating transcription factor 4; ATF5, activating transcription factor 5; ATP, adenosine triphosphate; BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; C26, colon-26 adenocarcinoma tumor cells; CaMK, Ca2+/calmodulin-dependent protein kinase; CHOP, CCAAT/enhancer-binding protein (C/EBP) homologous protein; COX, cytochrome c oxidase; Drp1, dynamin related protein 1; eIF2 α, eukaryotic initiation factor 2 α; ERR α, estrogen related receptor α; ETC, electron transport chain; FGF21, fibroblast growth factor 21; Fis1, mitochondrial fission protein 1; FoxO3a, forkhead box O3a; IFN γ, interferon γ; IL-1, interleukin 1; IL-6, interleukin 6; IM, inner mitochondrial membrane; IMF, intramyofibrillar; LC3-I/II, microtubule-associated proteins 1A/1B light chain 3; MD, mitochondrial disorder; MEF2C, myocyte enhancer factor 2C; MELAS, mitochondrial myopathy, encephalomyopathy, lactic acidosis and stroke-like episodes; Mff, mitochondrial fission factor; Mfn1/2, mitofusin 1&2; MQC, mitochondrial quality control; mtDNA, mitochondrial DNA; mTOR, mammalian target of rapamycin; NIX, BNIP3-like protein; NRF-1, nuclear respiratory factor 1; OM, outer mitochondrial membrane; Opa1/2, optic atrophy 1&2; p38 MAPK, p38 mitogen activated protein kinase; p53, tumor protein 53; p62, ubiquitin-binding protein p62 aka sequestosome 1; PIM, protein import machinery; PINK1, PTEN-induced kinase 1; PKA, protein kinase A; PRC, PGC-related co-activator; ROS, reactive oxygen species; PGC-1 α/β, PPAR γ coactivator, 1 α &1 β; RRF, ragged red fibers; SDH, succinate dehydrogenase; Sirt1, sirtuin 1; SS, subsarcolemmal; Tfam, mitochondrial transcription factor A; TFE3, transcription factor binding to IGHM enhancer 3; TFEB, transcription factor EB; TNF α, tumor necrosis factor α; UPRmt, mitochondrial unfolded protein response; UPS, ubiquitin proteosome system; VDAC, voltage dependent anion channel.
Adhihetty, P. J., Uguccioni, G., Leick, L., Hidalgo, J., Pilegaard, H., and Hood, D. A. (2009). The role of PGC-1 on mitochondrial function and apoptotic susceptibility in muscle. AJP Cell Physiol. 297, C217–C225. doi: 10.1152/ajpcell.00070.2009
Alston, C. L., Rocha, M. C., Lax, N. Z., Turnbull, D. M., and Taylor, R. W. (2017). The genetics and pathology of mitochondrial disease. J. Pathol. 241, 236–250. doi: 10.1002/path.4809
Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H., Coulson, A. R., Drouin, J., et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. doi: 10.1038/290457a0
Ballarò, R., Beltrà, M., De Lucia, S., Pin, F., Ranjbar, K., Hulmi, J. J., et al. (2019a). Moderate exercise in mice improves cancer plus chemotherapy−induced muscle wasting and mitochondrial alterations. FASEB J. 33, 5482–5494. doi: 10.1096/fj.201801862R
Ballarò, R., Penna, F., Pin, F., Gómez-Cabrera, M. C., Viña, J., and Costelli, P. (2019b). Moderate exercise improves experimental cancer cachexia by modulating the redox homeostasis. Cancers (Basel) 11, 1–20. doi: 10.3390/cancers11030285
Ballmann, C., Tang, Y., Bush, Z., Glenn, X., and Rowe, C. (2016). Adult expression of PGC-1 and-1 in skeletal muscle is not required for endurance exercise-induced enhancement of exercise capacity. Am. J. Physiol. Endocrinol. Metab. 311, 928–938. doi: 10.1152/ajpendo.00209.2016.-Exercise
Baltgalvis, K. A., Berger, F. G., Pena, M. M. O., Davis, J. M., Muga, S. J., and Carson, J. A. (2008). Interleukin-6 and cachexia in ApcMin/+ mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R393–R401. doi: 10.1152/ajpregu.00716.2007
Baraibar, M. A., Gueugneau, M., Duguez, S., Butler-Browne, G., Bechet, D., and Friguet, B. (2013). Expression and modification proteomics during skeletal muscle ageing. Biogerontology 14, 339–352. doi: 10.1007/s10522-013-9426-7
Barreto, R., Mandili, G., Witzmann, F. A., Novelli, F., Zimmers, T. A., and Bonetto, A. (2016). Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front. Physiol. 7:472. doi: 10.3389/fphys.2016.00472
Beyfuss, K., Erlich, A. T., Triolo, M., and Hood, D. A. (2018). The role of p53 in determining mitochondrial adaptations to endurance training in skeletal muscle. Sci. Rep. 8, 14710. doi: 10.1038/s41598-018-32887-0
Boffoli, D., Scacco, S. C., Vergari, R., Solarino, G., Santacroce, G., and Papa, S. (1994). Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1226, 73–82. doi: 10.1016/0925-4439(94)90061-2
Boncompagni, S., Rossi, A. E., Micaroni, M., Beznoussenko, G. V., Polishchuk, R. S., Dirksen, R. T., et al. (2009). Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell 20, 1058–1067. doi: 10.1091/mbc.E08
Brown, J. L., Rosa-Caldwell, M. E., Lee, D. E., Blackwell, T. A., Brown, L. A., Perry, R. A., et al. (2017). Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia. Sarcopenia Muscle 8, 926–938. doi: 10.1002/jcsm.12232
Bua, E. A., Johnson, J., Herbst, A., Delong, B., McKenzie, D., Salamat, S., et al. (2006). Mitochondrial DNA–deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 79, 469–480. doi: 10.1086/507132
Bua, E. A., McKiernan, S. H., Wanagat, J., McKenzie, D., and Aiken, J. M. (2002). Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J. Appl. Physiol. 92, 2617–2624. doi: 10.1152/japplphysiol.01102.2001
Carter, H. N., Chen, C. C. W., and Hood, D. A. (2015). Mitochondria, muscle health, and exercise with advancing age. Physiology 30, 208–223. doi: 10.1152/physiol.00039.2014
Carter, H. N., Kim, Y., Erlich, A. T., Zarrin-khat, D., and Hood, D. A. (2018a). Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 596, 3567–3584. doi: 10.1113/JP275998
Carter, H. N., Pauly, M., Tryon, L. D., and Hood, D. A. (2018b). Effect of contractile activity on PGC-1α transcription in young and aged skeletal muscle. J. Appl. Physiol. 124, 1605–1615. doi: 10.1152/japplphysiol.01110.2017
Chabi, B., Ljubicic, V., Menzies, K. J., Huang, J. H., Saleem, A., and Hood, D. A. (2008). Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 7, 2–12. doi: 10.1111/j.1474-9726.2007.00347.x
Chen, C., Ju, R., Zhu, L., Li, J., Chen, W., Zhang, D., et al. (2017). Carboxyamidotriazole alleviates muscle atrophy in tumor-bearing mice by inhibiting NF-κB and activating SIRT1. Naunyn. Schmiedebergs. Arch. Pharmacol. 390, 423–433. doi: 10.1007/s00210-017-1345-8
Chen, C. C. W., Erlich, A. T., Crilly, M. J., and Hood, D. A. (2018). Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. 315, E404–E415. doi: 10.1152/ajpendo.00391.2017
Cogswell, A. M., Stevens, R. J., and Hood, D. A. (1993). Properties of skeletal muscle mitochondria from subsarcolemmal and intermyofibrillar isolated regions. Am. J. Physiol. Cell Physiol. Physiol. 264, C383–C389. doi: 10.1152/ajpcell.1993.264.2.C383
Conley, K. E., Marcinek, D. J., and Villarin, J. (2007). Mitochondrial dysfunction and age. Curr. Opin. Clin. Nutr. Metab. Care 10, 688–692. doi: 10.1097/MCO.0b013e3282f0dbfb
Cornwell, E. W., Mirbod, A., Wu, C. L., Kandarian, S. C., and Jackman, R. W. (2014). C26 cancer-induced muscle wasting Is IKKb-dependent and NF-kappaB-independent. PLoS One 9:e87776. doi: 10.1371/journal.pone.0087776
Dawson, T. M., and Dawson, V. L. (2010). The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 25(Suppl. 1) S32–S39. doi: 10.1002/mds.22798
Deuster, P. A., Morrison, S. D., and Ahrens, R. A. (1985). Endurance exercise modifies cachexia of tumor growth in rats. Med. Sci. Sports Exerc. 17, 385–392. doi: 10.1249/00005768-198506000-00015
Di Meo, S., Napolitano, G., and Venditti, P. (2019). Mediators of physical activity protection against ROS-linked skeletal muscle damage. Int. J. Mol. Sci. 20, 3024. doi: 10.3390/IJMS20123024
Dwarkasing, J. T., Witkamp, R. F., Boekschoten, M. V., Ter Laak, M. C., Heins, M. S., and van Norren, K. (2016). Increased hypothalamic serotonin turnover in inflammation-induced anorexia. BMC Neurosci. 17:26. doi: 10.1186/s12868-016-0260-0
Eisner, V., Picard, M., and Hajnóczky, G. (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 20, 755–765. doi: 10.1038/s41556-018-0133-0
Erlich, A. T., Brownlee, D. M., Beyfuss, K., and Hood, D. A. (2018). Exercise induces TFEB expression and activity in skeletal muscle in a pgc-1α-dependent manner. Am. J. Physiol. Cell Physiol. 314, C62–C72. doi: 10.1152/ajpcell.00162.2017
Favaro, G., Romanello, V., Varanita, T., Andrea Desbats, M., Morbidoni, V., Tezze, C., et al. (2019). DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 10, 2576. doi: 10.1038/s41467-019-10226-9
Fearon, K., Strasser, F., Anker, S. D., Bosaeus, I., Bruera, E., Fainsinger, R. L., et al. (2011). Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495. doi: 10.1016/S1470-2045(10)70218-7
Fernández-Vizarra, E., Enríquez, J. A., Pérez-Martos, A., Montoya, J., and Fernández-Silva, P. (2011). Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 11, 207–213. doi: 10.1016/j.mito.2010.09.011
Ferreira, R., Vitorino, R., Alves, R. M. P., Appell, H. J., Powers, S. K., Duarte, J. A., et al. (2010). Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 10, 3142–3154. doi: 10.1002/pmic.201000173
Fiuza-Luces, C., Díez-Bermejo, J., Fernández-De, L. A., Torre, M., Rodríguez-Romo, G., Sanz-Ayán, P., et al. (2018). Health benefits of an innovative exercise program for mitochondrial disorders. Med. Sci. Sports Exerc. 50, 1142–1151. doi: 10.1249/MSS.0000000000001546
Fiuza-Luces, C., Valenzuela, P. L., Laine-Menéndez, S., Fernández-de la Torre, M., Bermejo-Gómez, V., Rufián-Vázquez, L., et al. (2019). Physical exercise and mitochondrial disease: insights from a mouse model. Front. Neurol. 10:790. doi: 10.3389/fneur.2019.00790
Fontes-Oliveira, C. C., Busquets, S., Toledo, M., Penna, F., Aylwin, M. P., Sirisi, S., et al. (2013). Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim. Biophys. Acta 1830, 2770–2778. doi: 10.1016/j.bbagen.2012.11.009
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Glancy, B., Hartnell, L. M., Combs, C. A., Fenmou, A., Sun, J., Murphy, E., et al. (2017). Power grid protection of the muscle mitochondrial reticulum. Cell Rep. 19, 487–496. doi: 10.1016/j.celrep.2017.03.063
Glancy, B., Hartnell, L. M., Malide, D., Yu, Z. X., Combs, C. A., Connelly, P. S., et al. (2015). Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523, 617–620. doi: 10.1038/nature14614
Gordon, B. S., Kelleher, A. R., and Kimball, S. R. (2013). Regulation of muscle protein synthesis and the effects of catabolic states. Int. J. Biochem. Cell Biol. 45, 2147–2157. doi: 10.1016/j.biocel.2013.05.039
Gordon, J. W., Rungi, A. A., Inagaki, H., and Hood, D. A. (2001). Effects of contractile activity on mitochondrial transcription factor A expression in skeletal muscle. J. Appl. Physiol. 90, 389–396. doi: 10.1152/jappl.2001.90.1.389
Gouspillou, G., Sgarioto, N., Kapchinsky, S., Purves-Smith, F., Norris, B., Pion, C. H., et al. (2014). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 28, 1621–1633. doi: 10.1096/fj.13-242750
Granata, C., Oliveira, R. S. F., Little, J. P., Renner, K., and Bishop, D. J. (2017). Sprint-interval but not continuous exercise increases PGC-1α protein content and p53 phosphorylation in nuclear fractions of human skeletal muscle. Sci. Rep. 7, 44227. doi: 10.1038/srep44227
Groennebaek, T., and Vissing, K. (2017). Impact of resistance training on skeletal muscle mitochondrial biogenesis, content, and function. Front. Physiol. 8:713. doi: 10.3389/fphys.2017.00713
Guan, Y., Drake, J. C., and Yan, Z. (2019). Exercise-induced mitophagy in skeletal muscle and heart. Exerc. Sport Sci. Rev. 47, 151–156. doi: 10.1249/JES.0000000000000192
Handschin, C., Rhee, J., Lin, J., Tarr, P. T., and Spiegelman, B. M. (2003). An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc. Natl. Acad. Sci. U.S.A. 100, 7111–7116. doi: 10.1073/pnas.1232352100
Handschin, C., and Spiegelman, B. M. (2006). Peroxisome proliferator-activated receptor γ Coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 27, 728–735. doi: 10.1210/er.2006-0037
Hardee, J. P., Mangum, J. E., Gao, S., Sato, S., Hetzler, K. L., Puppa, M. J., et al. (2016). Eccentric contraction-induced myofiber growth in tumor-bearing mice. J. Appl. Physiol. 120, 29–37. doi: 10.1152/japplphysiol.00416.2015
Heber, D., Ingles, S., Ashley, J. M., Maxwell, M. H., Lyons, R. F., and Elashoff, R. M. (1996). Clinical detection of sarcopenic obesity by bioelectrical impedance analysis. Am. J. Clin. Nutr. 64, 472S–477S. doi: 10.1093/ajcn/64.3.472S
Heyne, K., Mannebach, S., Wuertz, E., Knaup, K. X., Mahyar-Roemer, M., and Roemer, K. (2004). Identification of a putative p53 binding sequence within the human mitochondrial genome. FEBS Lett. 578, 198–202. doi: 10.1016/j.febslet.2004.10.099
Hood, D. A. (2001). Invited review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. 90, 1137–1157. doi: 10.1152/jappl.2001.90.3.1137
Hood, D. A., Memme, J. M., Oliveira, A. N., and Triolo, M. (2019). Maintenance of skeletal muscle mitochondria in health. Exer. Aging. Annu. Rev. Physiol. 81, 19–41. doi: 10.1146/annurev-physiol-020518-114310
Hood, D. A., Uguccioni, G., Vainshtein, A., and D’souza, D. (2011). Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle: implications for health and disease. Comp. Physiol. 1, 1119–1134. doi: 10.1002/cphy.c100074
Ibebunjo, C., Chick, J. M., Kendall, T., Eash, J. K., Li, C., Zhang, Y., et al. (2013). Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 33, 194–212. doi: 10.1128/MCB.01036-12
Iqbal, S., Ostojic, O., Singh, K., Joseph, A.-M., and Hood, D. A. (2013). Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 48, 963–970. doi: 10.1002/mus.23838
Irrcher, I., Ljubicic, V., and Hood, D. A. (2009). Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 296, C116–C123. doi: 10.1152/ajpcell.00267.2007
Islam, H., Hood, D. A., and Gurd, B. J. (2020). Looking beyond PGC-1α: emerging regulators of exercise-induced skeletal muscle mitochondrial biogenesis and their activation by dietary compounds. Appl. Physiol. Nutr. Metab. 45, 11–23. doi: 10.1139/apnm-2019-0069
Jaweed, M. M., Herbison, G. J., Miller, E. E., and Ditunno, J. F. (1983). Compensatory hypertrophy of the soleus in tumor-bearing rats. J. Neurol. Sci. 61, 171–179. doi: 10.1016/0022-510x(83)90003-5
Jeppesen, T. D., Dunø, M., Schwartz, M., Krag, T., Rafiq, J., Wibrand, F., et al. (2009). Short- and long-term effects of endurance training in patients with mitochondrial myopathy. Eur. J. Neurol. 16, 1336–1339. doi: 10.1111/j.1468-1331.2009.02660.x
Jeppesen, T. D., Schwartz, M., Olsen, D. B., Wibrand, F., Krag, T., Dunø, M., et al. (2006). Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 129, 3402–3412. doi: 10.1093/brain/awl149
Joseph, A. M., Adhihetty, P. J., Buford, T. W., Wohlgemuth, S. E., Lees, H. A., Nguyen, L. M.-D., et al. (2012). The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 11, 801–809. doi: 10.1111/j.1474-9726.2012.00844.x
Judge, S. M., Wu, C.-L., Beharry, A. W., Roberts, B. M., Ferreira, L. F., Kandarian, S. C., et al. (2014). Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 14:997. doi: 10.1186/1471-2407-14-997
Kayar, S. R., Hoppeler, H., Mermod, L., and Weibel, E. R. (1988). Mitochondrial size and shape in equine skeletal muscle: a three-dimensional reconstruction study. Anat. Rec. 222, 333–339. doi: 10.1002/ar.1092220405
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2014). AMPK and mTOR regualte autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, Y., Triolo, M., Erlich, A. T., and Hood, D. A. (2018). Regulation of autophagic and mitophagic flux during chronic contractile activity-induced muscle adaptations. Pflügers Arch. 471, 431–440. doi: 10.1007/s00424-018-2225-x
Kirkwood, S. P., Munn, E. A., and Brooks, G. A. (1986). Mitochondrial reticulum in limb skeletal muscle. Am. J. Physiol. 251, C395–C402. doi: 10.1152/ajpcell.1986.251.3.C395
Knudsen, N. H., Stanya, K. J., Hyde, A. L., Chalom, M. M., Alexander, R. K., Liou, Y.-H., et al. (2020). Interleukin-13 drives metabolic conditioning of muscle to endurance exercise. Science 368, eaat3987. doi: 10.1126/science.aat3987
Koltai, E., Hart, N., Taylor, A. W., Goto, S., Ngo, J. K., Davies, K. J. A., et al. (2012). Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Integr. Comp. Physiol. 303, R127–R134. doi: 10.1152/ajpregu.00337.2011
Kowald, A., and Kirkwood, T. B. L. (2014). Transcription could be the key to the selection advantage of mitochondrial deletion mutants in aging. Proc. Natl. Acad. Sci. U.S.A. 111, 2972–2977. doi: 10.1073/pnas.1314970111
Kraemer, W. J., Ratamess, N. A., and French, D. N. (2002). Resistance training for health and performance. Curr. Sports Med. Rep. 1, 165–171. doi: 10.1249/00149619-200206000-00007
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484. doi: 10.1126/science.1112125
Kunzke, T., Buck, A., Prade, V. M., Feuchtinger, A., Prokopchuk, O., Martignoni, M. E., et al. (2020). Derangements of amino acids in cachectic skeletal muscle are caused by mitochondrial dysfunction. J. Cachexia. Sarcopenia Muscle 11, 226–240. doi: 10.1002/jcsm.12498
Lanza, I. R., Short, D. K., Short, K. R., Raghavakaimal, S., Basu, R., Joyner, M. J., et al. (2008). Endurance exercise as a countermeasure for aging. Diabetes 57, 2933–2942. doi: 10.2337/db08-0349.I.R.L
Leduc-Gaudet, J.-P., Picard, M., Pelletier, F. S.-J., Sgarioto, N., Auger, M.-J., Vallée, J., et al. (2015). Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923–17937. doi: 10.18632/oncotarget.4235
Leick, L., Wojtaszewski, J. F. P., Johansen, S. T., Kiilerich, K., Comes, G., Hellsten, Y., et al. (2008). PGC-1alpha is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 294, E463–E474. doi: 10.1152/ajpendo.00666.2007
Lenka, N., Vijayasarathy, C., Mullick, J., and Avadhani, N. G. (1998). Structural organization and transcription regulation of nuclear genes encoding the mammalian Cytochrome c oxidase complex. Prog. Nucleic Acid Res. Mol. Biol. 61, 309–344. doi: 10.1016/S0079-6603(08)60830-2
Li, H., Slone, J., Fei, L., and Huang, T. (2019). Mitochondrial DNA variants and common diseases: a mathematical model for the diversity of age-related mtDNA mutations. Cells 8, 608. doi: 10.3390/cells8060608
Lira, V. A., Okutsu, M., Zhang, M., Greene, N. P., Laker, R. C., Breen, D. S., et al. (2013). Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 27, 4184–4193. doi: 10.1096/fj.13-228486
Liu, D., Sartor, M. A., Nader, G. A., Pistilli, E. E., Tanton, L., Lilly, C., et al. (2013). Microarray analysis reveals novel features of the muscle aging process in men and women. J. Gerontol. Ser. A 68, 1035–1044. doi: 10.1093/gerona/glt015
Ljubicic, V., Joseph, A.-M., Adhihetty, P. J., Huang, J. H., Saleem, A., Uguccioni, G., et al. (2009). Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging (Albany. NY). 1, 818–830. doi: 10.18632/aging.100083
Ljubicic, V., Menzies, K. J., and Hood, D. A. (2010). Mitochondrial dysfunction is associated with a pro-apoptotic cellular environment in senescent cardiac muscle. Mech. Ageing Dev. 131, 79–88. doi: 10.1016/j.mad.2009.12.004
Llovera, M., Garcia-Martinez, C., Agell, N., Lopez-Soriano, F. J., and Argiles, J. M. (1995). Muscle wasting associated with cancer cachexia is linked to an important activation of the atp-dependent ubiquitin-mediated proteolysis. Int. J. Cancer 61, 138–141. doi: 10.1002/ijc.2910610123
Lorite, M., Thompson, M., Drake, J., Carling, G., and Tisdale, M. (1998). Mechanism of muscle protein degradation induced by a cancer cachectic factor. Br. J. Cancer 78, 850–856. doi: 10.1038/bjc.1998.592
Mansueto, G., Armani, A., Viscomi, C., D’Orsi, L., De Cegli, R., Polishchuk, E. V., et al. (2017). Transcription factor EB controls metabolic flexibility during exercise. Cell Metab. 25, 182–196. doi: 10.1016/j.cmet.2016.11.003
Marcinek, D. J., Schenkman, K. A., Ciesielski, W. A., Lee, D., and Conley, K. E. (2005). Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J. Physiol. 569, 467–473. doi: 10.1113/jphysiol.2005.097782
Martina, J. A., Diab, H. I., Brady, O. A., and Puertollano, R. (2016). TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 35, 479–495. doi: 10.15252/embj.201593428
McFarland, R., Taylor, R. W., and Turnbull, D. M. (2010). A neurological perspective on mitochondrial disease. Lancet Neurol. 9, 829–840. doi: 10.1016/S1474-4422(10)70116-2
Melber, A., and Haynes, C. M. (2018). UPR mt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 28, 281–295. doi: 10.1038/cr.2018.16
Melstrom, L. G., Melstrom, K. A., Ding, X. Z., and Adrian, T. E. (2007). Mechanisms of skeletal muscle degradation and its therapy in cancer cachexia. Histol. Histopathol. 22, 805–814. doi: 10.14670/HH-22.805
Memme, J. M., Erlich, A. T., Phukan, G., and Hood, D. A. (2019). Exercise and mitochondrial health. J. Physiol. doi: 10.1113/JP278853 [Epub ahead of print],
Memme, J. M., Oliveira, A. N., and Hood, D. A. (2016). Chronology of UPR; activation in skeletal muscle adaptations to chronic contractile activity. Am. J. Physiol. Cell Physiol. 310, C1024–C1036. doi: 10.1152/ajpcell.00009.2016
Menzies, K. J., Singh, K., Saleem, A., and Hood, D. A. (2013). Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J. Biol. Chem. 288, 6968–6979. doi: 10.1074/jbc.M112.431155
Mesbah Moosavi, Z. S., and Hood, D. A. (2017). The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 312, C583–C594. doi: 10.1152/ajpcell.00320.2016
Mishra, P., and Chan, D. C. (2016). Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387. doi: 10.1083/jcb.201511036
Murphy, J. L., Blakely, E. L., Schaefer, A. M., He, L., Wyrick, P., Haller, R. G., et al. (2008). Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 131, 2832–2840. doi: 10.1093/brain/awn252
Norton, J. A., Lowry, S. F., and Brennan, M. F. (1979). Effect of work-induced hypertrophy on skeletal muscle of tumor- and nontumor-bearing rats. J. Appl. Physiol. 46, 654–657. doi: 10.1152/jappl.1979.46.4.654
Ogata, T., and Yamasaki, Y. (1997). Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat. Rec. 248, 214–223. doi: 10.1002/(SICI)1097-0185(199706)248:2<214::AID-AR8<3.0.CO;2-S
Ojuka, E. O., Jones, T. E., Han, D.-H., Chen, M., and Holloszy, J. O. (2003). Raising Ca2+ in L6 myotubes mimics effects of exercise on mitochondrial biogenesis in muscle. FASEB J. 17, 675–681. doi: 10.1096/fj.02-0951com
O’Leary, M. F. N., Vainshtein, A., Iqbal, S., Ostojic, O., and Hood, D. A. (2013). Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 304, C422–C430. doi: 10.1152/ajpcell.00240.2012
Oliveira, A. N., and Hood, D. A. (2018). Effect of Tim23 knockdown in vivo on mitochondrial protein import and retrograde signaling to the UPRmt in muscle. Am. J. Physiol. Cell Physiol. 315, C516–C526.
Otis, J. S., Lees, S. J., and Williams, J. H. (2007). Functional overload attenuates plantaris atrophy in tumor-bearing rats. BMC Cancer 7:146. doi: 10.1186/1471-2407-7-146
Park, J.-Y., Wang, P.-Y., Matsumoto, T., Sung, H. J., Ma, W., Choi, J. W., et al. (2009). p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ. Res. 105, 705–712. doi: 10.1161/CIRCRESAHA.109.205310
Penna, F., Busquets, S., Pin, F., Toledo, M., Baccino, F. M., López-Soriano, F. J., et al. (2011). Combined approach to counteract experimental cancer cachexia: eicosapentaenoic acid and training exercise. J. Cachexia. Sarcopenia Muscle 2, 95–104. doi: 10.1007/s13539-011-0028-4
Phadke, R. (2017). Myopathology of adult and paediatric mitochondrial diseases. J. Clin. Med. 6, 64. doi: 10.3390/jcm6070064
Picard, M., Wallace, D. C., and Burelle, Y. (2016). The rise of mitochondria in medicine. Mitochondrion 30, 105–116. doi: 10.1016/j.mito.2016.07.003
Pigna, E., Berardi, E., Aulino, P., Rizzuto, E., Zampieri, S., Carraro, U., et al. (2016). aerobic exercise and pharmacological treatments counteract cachexia by modulating autophagy in colon cancer. Sci. Rep. 6, 1–14. doi: 10.1038/srep26991
Pin, F., Busquets, S., Toledo, M., Camperi, A., Lopez-Soriano, F. J., Costelli, P., et al. (2015). Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 6, 43202–43215. doi: 10.18632/oncotarget.6439
Porcelli, S., Marzorati, M., Morandi, L., and Grassi, B. (2016). Home-based aerobic exercise training improves skeletal muscle oxidative metabolism in patients with metabolic myopathies. J. Appl. Physiol. 121, 699–708. doi: 10.1152/japplphysiol.00885.2015
Prado, C. M., Sawyer, M. B., Ghosh, S., Lieffers, J. R., Esfandiari, N., Antoun, S., et al. (2013). Central tenet of cancer cachexia therapy: do patients with advanced cancer have exploitable anabolic potential? Am. J. Clin. Nutr. 98, 1012–1019. doi: 10.3945/ajcn.113.060228
Puigserver, P., Rhee, J., Lin, J., Wu, Z., Yoon, J. C., Zhang, C. Y., et al. (2001). Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ Coactivator-1. Mol. Cell 8, 971–982. doi: 10.1016/S1097-2765(01)00390-2
Puppa, M. J., Murphy, E. A., Fayad, R., Hand, G. A., and Carson, J. A. (2014). Cachectic skeletal muscle response to a novel bout of low-frequency stimulation. J. Appl. Physiol. 116, 1078–1087. doi: 10.1152/japplphysiol.01270.2013
Rapizzi, E., Pinton, P., Szabadkai, G., Wieckowski, M. R., Vandecasteele, G., Baird, G., et al. (2002). Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624. doi: 10.1083/jcb.200205091
Ristow, M., Zarse, K., Oberbach, A., Klöting, N., Birringer, M., Kiehntopf, M., et al. (2009). Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8665–8670. doi: 10.1073/pnas.0903485106
Rooyackers, O. E., Adey, D. B., Ades, P. A., and Nair, K. S. (1996). Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 93, 15364–15369. doi: 10.1073/pnas.93.26.15364
Rosenberg, I. H. (2011). Sarcopenia: origins and clinical relevance. Clin. Geriatr. Med. 27, 337–339. doi: 10.1016/j.cger.2011.03.003
Rusecka, J., Kaliszewska, M., Bartnik, E., and Tonska, K. (2018). Nuclear genes involved in mitochondrial diseases caused by instability of mitochondrial DNA Joanna. J. Appl. Genomics 59, 43–57. doi: 10.1371/journal.pone.0069534
Russell, O., and Turnbull, D. (2014). Mitochondrial DNA disease-molecular insights and potential routes to a cure. Exp. Cell Res. 325, 38–43. doi: 10.1016/j.yexcr.2014.03.012
Safdar, A., Khrapko, K., Flynn, J. M., Saleem, A., De Lisio, M., Johnston, A. P. W., et al. (2016). Exercise-induced mitochondrial p53 repairs mtDNA mutations in mutator mice. Skelet. Muscle 6, 7. doi: 10.1186/s13395-016-0075-9
Saleem, A., Adhihetty, P. J., and Hood, D. A. (2009). Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genomics 37, 58–66. doi: 10.1152/physiolgenomics.90346.2008
Saleem, A., and Hood, D. A. (2013). Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-Tfam-mitochondrial DNA complex in skeletal muscle. J. Physiol. 591, 3625–3636. doi: 10.1113/jphysiol.2013.252791
Salomão, E. M., Toneto, A. T., Silva, G. O., and Gomes-Marcondes, M. C. C. (2010). Physical exercise and a leucine-rich diet modulate the muscle protein metabolism in Walker tumor-bearing rats. Nutr. Cancer 62, 1095–1104. doi: 10.1080/01635581.2010.492082
Sandri, M. (2016). Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 54, 11–19. doi: 10.1016/j.semcdb.2015.11.002
Sandri, M., Lin, J., Handschin, C., Yang, W., Arany, Z. P., Lecker, S. H., et al. (2006). PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. U.S.A. 103, 16260–16265. doi: 10.1073/pnas.0607795103
Sato, S., Gao, S., Puppa, M. J., Kostek, M. C., Wilson, L. B., and Carson, J. A. (2019). High-frequency stimulation on skeletal muscle maintenance in female cachectic mice. Med. Sci. Sports Exerc. 51, 1828–1837. doi: 10.1249/MSS.0000000000001991
Scarpulla, R. C. (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278. doi: 10.1016/j.bbamcr.2010.09.019
Scarpulla, R. C., Vega, R. B., and Kelly, D. P. (2012). Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 23, 459–466. doi: 10.1016/j.tem.2012.06.006
Schaap, L. A., Pluijm, S. M. F., Deeg, D. J. H., and Visser, M. (2006). Inflammatory markers and loss of muscle mass (Sarcopenia) and strength. Am. J. Med. 119, 526.e9–526.e17. doi: 10.1016/J.AMJMED.2005.10.049
Schlame, M., and Ren, M. (2006). Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455. doi: 10.1016/j.febslet.2006.07.022
Schmidt, M. E., Wiskemann, J., Armbrust, P., Schneeweiss, A., Ulrich, C. M., and Steindorf, K. (2015). Effects of resistance exercise on fatigue and quality of life in breast cancer patients undergoing adjuvant chemotherapy: a randomized controlled trial. Int. J. Cancer 137, 471–480. doi: 10.1002/ijc.29383
Segatto, M., Fittipaldi, R., Pin, F., Sartori, R., Dae Ko, K., Zare, H., et al. (2017). Epigenetic targeting of bromodomain protein BRD4 counteracts cancer cachexia and prolongs survival. Nat. Commun. 8, 1707. doi: 10.1038/s41467-017-01645-7
Settembre, C., and Ballabio, A. (2011). TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy 7, 1379–1381. doi: 10.4161/auto.7.11.17166
Sharma, P., and Sampath, H. (2019). Mitochondrial DNA Integrity: Role in Health and Disease. Cells 8, 100. doi: 10.3390/cells8020100
Sheng, Q., Xiao, X., Prasadan, K., Chen, C., Ming, Y., Fusco, J., et al. (2017). Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci. Rep. 7, 16348. doi: 10.1038/s41598-017-16485-0
Short, K. R., Bigelow, M. L., Kahl, J., Singh, R., Coenen-Schimke, J., Raghavakaimal, S., et al. (2005). Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. U.S.A. 102, 5618–5623. doi: 10.1073/pnas.0501559102
Short, K. R., Vittone, J. L., Bigelow, M. L., Proctor, D. N., Rizza, R. A., Coenen-Schimke, J. M., et al. (2003). Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 52, 1888–1896. doi: 10.2337/diabetes.52.8.1888
Shum, A. M. Y., Fung, D. C. Y., Corley, S. M., McGill, M. C., Bentley, N. L., Tan, T. C., et al. (2015). Cardiac and skeletal muscles show molecularly distinct responses to cancer cachexia. Physiol. Genomics 47, 588–599. doi: 10.1152/physiolgenomics.00128.2014
Shum, A. M. Y., Mahendradatta, T., Taylor, R. J., Painter, A. B., Moore, M. M., Tsoli, M., et al. (2012). Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging (Albany NY). 4, 133–143. doi: 10.18632/aging.100436
Srivastava, S., Diaz, F., Iommarini, L., Aure, K., Lombes, A., and Moraes, C. T. (2009). PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum. Mol. Genet. 18, 1805–1812. doi: 10.1093/hmg/ddp093
Taivassalo, T. (1999). Gene shifting: a novel therapy for mitochondrial myopathy. Hum. Mol. Genet. 8, 1047–1052. doi: 10.1093/hmg/8.6.1047
Taivassalo, T., De Stefano, N., Chen, J., Karpati, G., Arnold, D. L., and Argov, Z. (1999). Short-term aerobic training response in chronic myopathies. Muscle Nerve 22, 1239–1243. doi: 10.1002/(sici)1097-4598(199909)22:9<1239::aid-mus11<3.0.co;2-w
Taivassalo, T., Gardner, J. L., Taylor, R. W., Schaefer, A. M., Newman, J., Barron, M. J., et al. (2006). Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain 129, 3391–3401. doi: 10.1093/brain/awl282
Taivassalo, T., Shoubridge, E. A., Chen, J., Kennaway, N. G., DiMauro, S., Arnold, D. L., et al. (2001). Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann. Neurol. 50, 133–141. doi: 10.1002/ana.1050
Tanida, I., Ueno, T., and Kominami, E. (2004). LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518. doi: 10.1016/j.biocel.2004.05.009
Taylor, R. W., Schaefer, A. M., Barron, M. J., Mcfarland, R., and Turnbull, D. M. (2004). The diagnosis of mitochondrial muscle disease. Neuromuscul. Disord. 14, 237–245. doi: 10.1016/j.nmd.2003.12.004
Taylor, R. W., and Turnbull, D. M. (2005). Mitochondrial DNA Mutations in Human Disease. Nat. Rev. Genet. 6, 389–402. doi: 10.1038/nrg1606
Tisdale, M. J. (2009). Mechanisms of cancer cachexia. Physiol. Rev. 89, 381–410. doi: 10.1152/physrev.00016.2008
Tryon, L. D., Vainshtein, A., Memme, J. M., Crilly, M. J., and Hood, D. A. (2014). Recent advances in mitochondrial turnover during chronic muscle disuse. Integr. Med. Res. 3, 161–171. doi: 10.1016/j.imr.2014.09.001
Tzika, A. A., Fontes-Oliveira, C. C., Shestov, A. A., Constantinou, C., Psychogios, N., Righi, V., et al. (2013). Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 43, 886–894. doi: 10.3892/ijo.2013.1998
Vainshtein, A., and Hood, D. A. (2016). The regulation of autophagy during exercise in skeletal muscle. J. Appl. Physiol. 120, 664–673. doi: 10.1152/japplphysiol.00550.2015
Van Der Ende, M., Grefte, S., Plas, R., Meijerink, J., Witkamp, R. F., Keijer, J., et al. (2018). Mitochondrial dynamics in cancer-induced cachexia. BBA Rev. Cancer 1870, 137–150. doi: 10.1016/j.bbcan.2018.07.008
Van Norren, K., Dwarkasing, J. T., and Witkamp, R. F. (2017). The role of hypothalamic inflammation, the hypothalamic-pituitary-Adrenal axis and serotonin in the cancer anorexia-cachexia syndrome. Curr. Opin. Clin. Nutr. Metab. Care 20, 396–401. doi: 10.1097/MCO.0000000000000401
Vincent, A. E., White, K., Davey, T., Philips, J., Ogden, R. T., Lawess, C., et al. (2019). Quantitative 3D mapping of the human skeletal muscle mitochondrial network. Cell Rep. 26, 996–1009.e4. doi: 10.1016/j.celrep.2019.01.010
Wanagat, J., Cao, Z., Pathare, P., and Aiken, J. M. (2001). Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 15, 322–332. doi: 10.1096/fj.00-0320com
Wei, H., Liu, L., and Chen, Q. (2015). Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta - Mol. Cell Res. 1853, 2784–2790. doi: 10.1016/j.bbamcr.2015.03.013
White, J. P., Baltgalvis, K. A., Puppa, M. J., Sato, S., Baynes, J. W., and Carson, J. A. (2011). Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am. J. Physiol. Integr. Comp. Physiol. 300, R201–R211. doi: 10.1152/ajpregu.00300.2010
White, J. P., Puppa, M. J., Gao, S., Sato, S., Welle, S. L., and Carson, J. A. (2013). Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am. J. Physiol. Endocrinol. Metab. 304, E1042–E1052. doi: 10.1152/ajpendo.00410.2012
White, J. P., Puppa, M. J., Sato, S., Gao, S., Price, R. L., Baynes, J. W., et al. (2012). IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2, 14. doi: 10.1186/2044-5040-2-14
Winder, W. W., Holmes, B. F., Rubink, D. S., Jensen, E. B., Chen, M., and Holloszy, J. O. (2000). Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. 88, 2219–2226. doi: 10.1152/jappl.2000.88.6.2219
Yan, C., Duanmu, X., Zeng, L., Liu, B., and Song, Z. (2019). Mitochondrial DNA: distribution, mutations, and elimination. Cells 8, 379. doi: 10.3390/cells8040379
Keywords: mitochondrial quality control, skeletal muscle, aging, cancer, exercise as medicine
Citation: Memme JM and Hood DA (2021) Molecular Basis for the Therapeutic Effects of Exercise on Mitochondrial Defects. Front. Physiol. 11:615038. doi: 10.3389/fphys.2020.615038
Received: 26 October 2020; Accepted: 16 December 2020;
Published: 13 January 2021.
Edited by:
Ildikò Szabò, University of Padua, ItalyReviewed by:
Zsolt Radak, Semmelweis University, HungaryCopyright © 2021 Memme and Hood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Hood, ZGhvb2RAeW9ya3UuY2E=
†ORCID: Jonathan M. Memme, orcid.org/0000-0002-6192-4122; David A. Hood, orcid.org/0000-0002-9553-152X
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.