 Hannah E. Boycott
Hannah E. Boycott My-Nhan Nguyen
My-Nhan Nguyen Besarte Vrellaku
Besarte Vrellaku Katja Gehmlich
Katja Gehmlich Paul Robinson
Paul Robinson- 1Division of Cardiovascular Medicine, Radcliffe Department of Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom
- 2Division of Cardiovascular Medicine, Radcliffe Department of Medicine and British Heart Foundation Centre of Research Excellence Oxford, University of Oxford, Oxford, United Kingdom
- 3Institute of Cardiovascular Sciences, University of Birmingham, Birmingham, United Kingdom
The ability§ of the heart to adapt to changes in the mechanical environment is critical for normal cardiac physiology. The role of nitric oxide is increasingly recognized as a mediator of mechanical signaling. Produced in the heart by nitric oxide synthases, nitric oxide affects almost all mechano-transduction pathways within the cardiomyocyte, with roles mediating mechano-sensing, mechano-electric feedback (via modulation of ion channel activity), and calcium handling. As more precise experimental techniques for applying mechanical stresses to cells are developed, the role of these forces in cardiomyocyte function can be further understood. Furthermore, specific inhibitors of different nitric oxide synthase isoforms are now available to elucidate the role of these enzymes in mediating mechano-electrical signaling. Understanding of the links between nitric oxide production and mechano-electrical signaling is incomplete, particularly whether mechanically sensitive ion channels are regulated by nitric oxide, and how this affects the cardiac action potential. This is of particular relevance to conditions such as atrial fibrillation and heart failure, in which nitric oxide production is reduced. Dysfunction of the nitric oxide/mechano-electrical signaling pathways are likely to be a feature of cardiac pathology (e.g., atrial fibrillation, cardiomyopathy, and heart failure) and a better understanding of the importance of nitric oxide signaling and its links to mechanical regulation of heart function may advance our understanding of these conditions.
Introduction
The heart is a mechanically active organ. With every heartbeat cardiac cells are subjected to various mechanical forces, including stretch, shear, and strain (Waldman et al., 1985). Moreover, the mechanical forces experienced by the heart are not constant, varying with posture, exercise, stimulants (such as caffeine), and emotional state. The way that the heart responds to alterations in the mechanical environment has been the subject of investigation for decades. There is also huge medical relevance given that cardiac pathologies are often accompanied by a change in the mechanical environment where hypertrophy or increased fibrosis causes altered intrinsic tension in the heart chamber walls (Abrahams et al., 1987; Weber et al., 1988; Conrad et al., 1995; De Jong et al., 2011).
In this review we discuss the role of nitric oxide as a key modulator and of various mechanical forces in the cardiomyocyte. The production of NO is influenced by mechanical forces and NO is known to be a key mediator in mechano-chemo-transduction (MCT) during cardiac contraction. This provides therapeutic targets for treating mechanical stress-induced Ca2+ dysregulation, arrhythmias, and cardiomyopathy. Current understanding of the interaction between NO and MCT signaling pathways in the heart is incomplete, particularly whether mechanically ion-sensitive channels are regulated by NO, and how this affects the cardiac action potential.
Each defined mechanical force is governed by a defined set of protein interactors and intrinsic physiological processes:
1. Overall contractile force in cardiac muscle is generated principally by the actomyosin ATPase interactions of the sarcomeric thick and thin filaments, whilst the degree of acto-myosin interaction is controlled by Ca2+ binding to the regulatory troponin/tropomyosin complex on the actin filament.
2. The passive stiffness of the cardiomyocyte is generated by the giant protein titin along the intermediate filament of the sarcomere and extracellular collagen, where the amount and degree of cross linking within the collagen matrix can stiffen the myocardium in certain pro-fibrotic disease pathologies leading to diastolic dysfunction. Stiffness can also be influenced to a smaller extent by changes in the cytoskeletal tubulin network.
3. Myocardial stretch is a force on the cardiomyocyte in response to the hemodynamic load within the chambers of the heart. Molecular responses to increased stretch is termed stretch sensing and allows the heart to respond to stimulatory homeostasis of the sympathetic nervous system during periods of physiological arousal or compromise such as exercise or hemorrhage, to prevent circulatory congestion or failure.
4. Shear stress occurs as a result of fluid shear due to blood flow, and when laminar sheets of cardiomyocytes slide relative to each other when the heart contracts. This causes cardiomyocyte deformation and shear forces.
5. Preload/myocardial strain refers to the degree to which the myofilaments are initially stretched during ventricular filling. The sarcomere is able to quickly respond to alterations in preload due to the highly regular arrangement of its filaments to produce more contractile force and thereby increase the stroke volume of the heart in a process defined as the Frank-Starling mechanism (discussed in more detail below).
6. Afterload/myocardial stress is determined by the work the ventricular myocardium is required to perform in order to eject blood into circulation against the systemic vascular resistance. Changes in afterload affect the calcium handling and contractility, which may underlie the Anrep effect.
The intrinsic ability of the myocardium to adapt to increased stretch by increasing contractile force is embodied by the Frank-Starling mechanism (Frank, 1899; Starling, 1918; Sagawa et al., 1990), which links stroke volume and end diastolic pressure. The consequences of this are that increased diastolic filling (preload) leads to a stronger contraction and an increased stoke volume (Lakatta, 1987). The Frank Starling mechanism is underpinned by spatial rearrangement of sarcomeric filaments, bringing actin, and myosin filaments to closer proximity to increase the myosin duty ratio, when the sarcomere lengthens as the myocyte stretches, and increases calcium sensitivity of myofilaments. This leads to enhanced cross bridge formation, as well as stretch activated titin unfolding (Fukuda et al., 2009; Ait-Mou et al., 2016). The rapid response of the mechanism is accompanied by a slower, less pronounced increase in contractility known as the Anrep effect (von Anrep, 1912), where contractility is influenced by an increase in ventricular inotropy as a consequence of afterload-induced activity of tension dependent Na+/H+ exchangers (Cingolani et al., 2005). The Frank-Starling mechanism has been shown to be modulated by nitric oxide (NO) which affects the myofilament responsiveness to Ca2+ (Prendergast et al., 1997). In addition, the Anrep effect is modulated by NO; increased NO results in an increase in intracellular Ca2+ sparks (Petroff et al., 2001).
The role of the molecular gas NO in the cardiovascular system is well established, where it regulates a multitude of cellular processes. Endothelial cells synthesize and release NO which mediates diverse effects including vessel tone, hemostasis, blood pressure and vasculature remodeling (Naseem, 2005). The importance of NO for cardiomyocyte function is well recognized, with roles in ion channel regulation, Ca2+ homeostasis, contractility, energetics, cell growth, and survival (Shah and MacCarthy, 2000; Seddon et al., 2007).
Nitric oxide is synthesized by a family of enzymes known as nitric oxide synthases (NOS), which produce NO as they catalyze the conversion of L-arginine to L-citrulline. There are three isoforms of NOS (NOS1-3) each encoded by a specific gene. In the cardiovascular system “neuronal” NOS (nNOS or NOS1) and “endothelial” NOS (eNOS or NOS3) are expressed in cardiomyocytes (Feron et al., 1996); eNOS is expressed in the endothelium and fibroblasts. nNOS can also be found in the neurons which innervate the heart (Choate et al., 2008). The third isoform, “inducible” NOS (iNOS or NOS2) is produced in most cell types in response to stimuli such as hypoxia or cytokines [e.g., Tumor Necrosis Factor-α (TNFα)] (Shah, 2000).
Nitric oxide synthase-derived NO exerts its effects in a variety of ways. Firstly, NO is able to post-translationally modify target proteins primarily through the addition of a nitroso group, to the sulfhydryl side chain of cysteine, termed-S-nitrosylation (Kovacs and Lindermayr, 2013). Such modification results in an alteration in the target protein’s function (Gonzalez et al., 2009). The effects of direct NO modification of target proteins are limited by the relatively short diffusion distance of the molecule. Cellular carrier molecules such as S-nitrosoglutathione mediate longer range NO-signaling by acting as a carrier and a donor, transferring NO onto more distal targets (Such-Miquel et al., 2018). Exogenous application of glutathione exacerbates ventricular arrhythmias induced by stretch, which has been proposed to be a potential link between NO cycling and mechanical response (Such-Miquel et al., 2018). NO also activates the soluble guanylate cyclase (sGC)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG)-dependent phosphorylation pathway. Activation of this pathway results in the phosphorylation of target proteins, to inhibit the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK1/2/ERK1/2) pathway and activate c-Jun N-terminal kinase (JNK)1, 2, and 3 pathways, the ultimate result being cardio-protection and down regulation of genes involved in hypertrophy and regulation of genes involved in apoptosis (Pilz and Casteel, 2003). It has been speculated that the majority of nitrosylation is carried out by nNOS, whereas the cGMP pathway is primarily mediated by eNOS (Ziolo, 2008).
An imbalance in NO bioavailability has been implicated in the pathology of several cardiac pathologies and understanding of the processes underlying this dysregulation has been the subject of intense research. In pathological conditions NOS can become uncoupled leading to an increase in reactive oxygen species production rather than the physiological production of NO (reviewed in Roe and Ren, 2012). Uncoupling occurs when the tightly controlled flow of electrons through the NOS enzyme is redirected by the dissociation of the ferrous-dioxygen complex (Forstermann and Munzel, 2006), and has been linked to reduced levels of the cofactor tetrahydrobiopterin (BH4). Reduced levels of BH4 leading to NOS uncoupling are contributing factors in several pathologies, including atrial fibrillation, hypertension, ischemia/reperfusion injury and overload-induced heart failure (reviewed in Alkaitis and Crabtree, 2012). Increased superoxide production following NOS uncoupling increases the oxidative burden on the heart and affects cardiac performance (Singal et al., 1998).
Nitric oxide production is increased in response to various mechanical forces, a phenomenon which is of particular importance for cardiovascular function. For example, studies using NO sensitive dyes have shown that stretch of ventricular cardiomyocytes induces NO release (Petroff et al., 2001; Shim et al., 2017a). Furthermore, swelling of cardiomyocytes induced by hypotonic solution leads to an increase in NO production (Gonano et al., 2014). The response of cardiomyocytes to mechanical stimuli is of particular interest given the importance of the force-response phenomenon to heart function. For these reasons NO function in physiology and pathology has been the subject of intense investigation for several decades. Other redox molecules, particularly NADPH oxidase 2 and X-ROS signaling, are also altered in response to mechanical stimuli and have been reviewed elsewhere (Prosser et al., 2013; Ward et al., 2014). It has become clear from the literature over the last 10 years that NO appears to be the major modulator of mechano-transduction pathways in the heart via direct nitrosylation or activation of PKG. Here, we review these latest findings, with particular emphasis on three key areas: mechano-sensing, ion channel activity and calcium homeostasis.
Mechano-Sensing by the Cytoskeleton
In order to be transduced into a biological response, mechanical signals must be effectively sensed by the cell and transduced into meaningful signals. This process is mediated either by ion channels which can alter their activity in response to mechanical signals, or by the cytoskeleton which senses changes in mechanical load and alters cell function accordingly. The cytoskeleton is a network of tubules and filaments that aids in maintaining the structural and mechanical integrity of the cardiomyocyte (Sequeira et al., 2014). Importantly, the cytoskeleton is also involved in mechano-transduction in response to mechanical stress through its interaction with several membrane-associated proteins, which includes proteins associated with the integrin complex, dystrophin-glycoprotein complex (DGC), LIM domain proteins, and titin. Nitric oxide signaling has the ability to modulate cytoskeletal structure and function. For instance, the myofilament response to Ca2+ is reduced by high concentrations of NO via cGMP-dependent activation of PKG (Vila-Petroff et al., 1999) which can in turn regulate the expression of genes such as atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), β-Myosin and cardiac actin (Pilz and Casteel, 2003). The importance of NO signaling for cytoskeletal remodeling stems from post-translational modifications of various cytoskeletal and membrane-associated proteins, which influence cardiomyocyte contractility and/or relaxation.
Integrins and the Integrin Complex
The integrin complex is a group of cytoskeletal proteins essential for not only facilitating cellular adhesion, but also the sensing and integration of mechanical signals. Integrins are heterodimeric transmembrane receptors which associate the extracellular matrix with the actin cytoskeleton, whereby they transmit mechanical signals to the cytoskeleton (Wang et al., 1993). In cardiomyocytes, integrins contribute to preservation of cardiac function and have the ability to modulate the mechanical and electrical coupling in the heart (Valencik et al., 2006). The impact of NO signaling on cardiac integrins has been examined via the utilization of ligands containing the amino acid sequence of Arg-Gly-Asp (RGD) binding motif. Application of this peptide has been reported to modulate L-type Ca2+ currents (Wu et al., 1998) and affect Ca2+ release from the sarcoplasmic reticulum (Chan et al., 2001). In neonatal rat cardiomyocytes, pre-treatment with the NO donor SNAP enhanced Ca2+ release, whilst pre-treatment with the NOS inhibitor ng-nitroarginine methyl ester (L-NAME) abolished the release following integrin stimulation by RGD peptide (van der Wees et al., 2006), suggesting that stimulation of integrins promotes NO-mediated Ca2+ release from the sarcoplasmic reticulum. Further, this may be mediated by focal adhesion kinase (FAK) activation, as incubation of cardiomyocytes with the RGD peptide increased phosphorylation of FAKs, whilst incubation with SNAP induced phosphorylation of FAK (van der Wees et al., 2006). In addition to integrins modulating Ca2+ homeostasis via NO-signaling, it has been reported that the stimulatory effects of NO directly regulate integrin expression via cGMP signaling (Zhan et al., 2018).
Other components of the integrin complex are also important for regulating mechano-sensing and mechano-transduction. Talin and vinculin are intracellular cytoskeletal proteins that interact with both integrins and the actin cytoskeleton at focal adhesions. In response to external forces, talin, and vinculin alter their conformations to reinforce and stabilize the integrin-actin cytoskeleton connection (del Rio et al., 2009). The impact of NO signaling on talin and vinculin has been studied in skeletal muscle. Increased NO signaling in response to mechanical stimuli mediates skeletal talin and vinculin expression via a PKG-dependent manner, and prevents calpain-proteolytic degradation of talin (Koh and Tidball, 2000; Zhang et al., 2004), supporting a mechano-sensory role for NO, stabilizing the cytoskeleton via its interaction with talin and vinculin. In the heart, the loss of talin-1 and talin-2 is associated with disrupted expression of β1-integrin and associated proteins at the cardiomyocyte membrane, and promotes cardiomyocyte dysfunction leading to cardiomyopathy (Manso et al., 2017). Vinculin co-localizes at the costameres and intercalated discs of cardiomyocytes (Zemljic-Harpf et al., 2009) and its loss is associated with impaired cardiomyocyte organization (Zemljic-Harpf et al., 2007). These findings implicate the critical roles of talin and vinculin in integrin activation and maintenance of cardiomyocyte membrane stability. However, the impact of NO signaling on cardiac talin and vinculin remains to be investigated. The effect of NO on the integrin system are summarized in Figure 1.
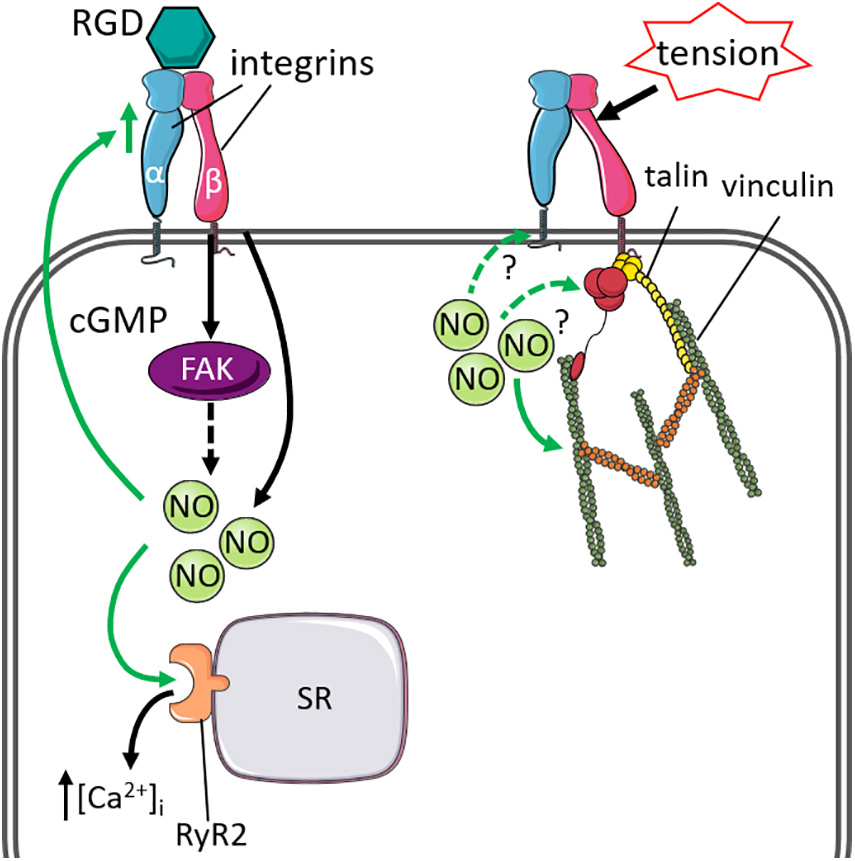
Figure 1. Cytoskeletal proteins linked to integrin that influence or are influenced by nitric oxide signaling and mechanical stimuli. Left: Stimulation of integrins by ligands containing the Arg-Gly-Asp motif (RGD) increased nitric oxide (NO)-mediated intracellular Ca2+ [(Ca2+)i] release from the sarcoplasmic reticulum (SR), via activation of ryanodine receptor 2 (RyR2). The stimulatory effects of NO can also regulate integrin expression via cGMP-signaling. Solid green lines indicate effect by NO. Right: Integrins form a complex with talin and vinculin to stabilize the actin-cytoskeleton under mechanical stress. The effect of cardiac NO signaling on the integrin-talin-vinculin complex remains to be investigated (green dashed lines).
Dystrophin and the Dystrophin Glycoprotein Complex
The DGC is a network of several membrane-associated proteins that provides structural integrity to the plasma membrane and facilitates transduction of mechanical forces from the sarcomere and intracellular cytoskeleton to the extracellular matrix during contraction (Lapidos et al., 2004). As an important component of this cytoskeletal complex, dystrophin is closely associated with nNOS (Figure 2), and loss of dystrophin has adverse consequences on the localization and regulation of nNOS (Brenman et al., 1995). In skeletal muscle, loss of dystrophin uncouples nNOS from the sarcolemma to the cytosol and depletes NO synthesis by > 70% (Brenman et al., 1995), and promotes skeletal muscle pathophysiology (Thomas et al., 2003; Percival et al., 2010; Froehner et al., 2015). In the heart, dystrophin is localized to the cardiomyocyte sarcolemmal membrane and T-tubules (Kaprielian et al., 2000; Garbincius and Michele, 2015), whilst nNOS is localized to the sarcoplasmic reticulum and sarcolemmal membrane (Xu et al., 1999; Oceandy et al., 2007; Carnicer et al., 2017). Loss of dystrophin at the sarcolemmal membrane thus displaces nNOS from the sarcolemmal membrane and contributes to altered nNOS regulation. This dysregulation also disrupts the Ca2+-cycling machinery in cardiomyocytes (Sears et al., 2003; Zhang et al., 2008), leading to impaired contractile function. Indeed, atrial dystrophin protein is repressed by atrial-specific upregulation of micro-RNA-31 (miR-31) in humans and goats with atrial fibrillation (Reilly et al., 2016). Reduced dystrophin protein disrupts the protein stability of nNOS and impairs NO synthesis, which promotes atrial electrical remodeling including shortening of action potential duration and increased repolarizing K+ currents (Reilly et al., 2016). These findings suggest important roles for dystrophin and NO signaling in cardiac electrophysiology and function. A study in dystrophin-deficient mdx mice previously reported associations between reduced nNOS activity or expression and cardiac ECG abnormalities (Bia et al., 1999). Similarly, transgenic overexpression of nNOS in aged mdx mice mitigated myocardial fibrosis and inflammation, cardiac ECG abnormalities and premature ventricular contractions (Wehling-Henricks et al., 2005). Enhancing NO signaling in response to impaired components of the DGC may be protective against mechanical stress. A study on dystrophin/utrophin-deficient cardiomyocytes used whole body periodic acceleration (a technique that induces pulsatile shear stress to the endothelium, by sinusoidal head to foot motion of the supine body) to increase endogenous expression of eNOS and nNOS (Adams et al., 2009). They showed that increased NO production ameliorated impaired cardiomyocyte contractile function, oxidative stress and elevated intracellular diastolic Ca2+ and Na+ concentrations that were induced by mechanical stress (Lopez et al., 2017).
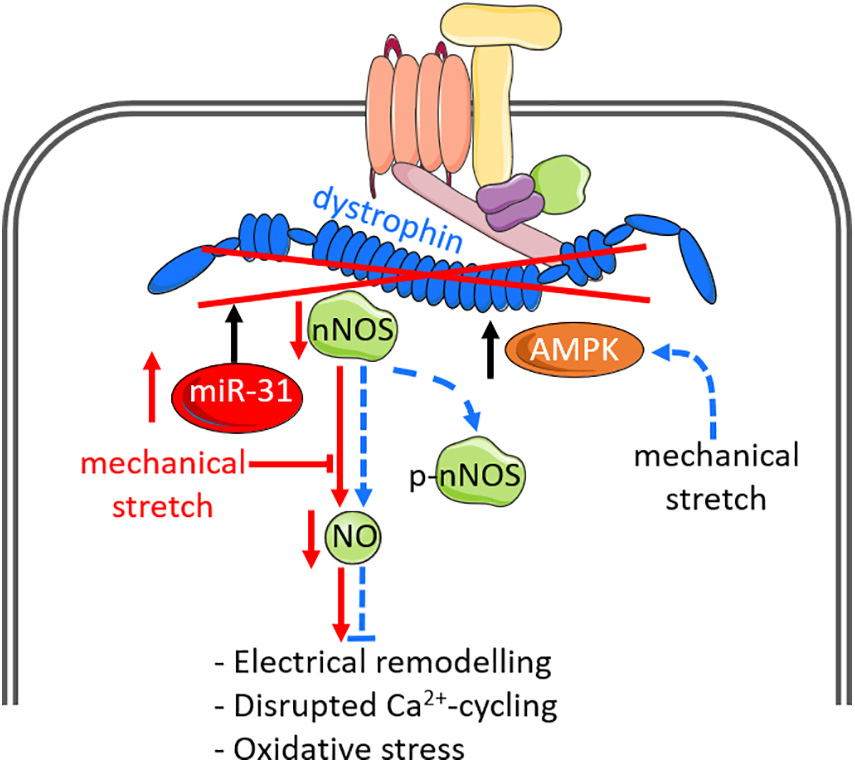
Figure 2. Nitric oxide signaling and mechanical stimuli affect dystro-sarcoglycan complex function and can modify disease pathology. Dystrophin is the largest component of the dystrophin-glycoprotein complex (DGC), and closely associates with neuronal nitric oxide synthase (nNOS). Loss of dystrophin, via upregulation of microRNA-31 or genetic mutations, leads to disassociation of nNOS from the DGC and impairs NO production. Loss of NO is associated with atrial electrical remodeling, disrupted Ca2+ cycling and oxidative stress. The DGC additionally acts as a mechano-sensor to regulate NO signaling. In normal cardiomyocytes in which dystrophin is intact (indicated by dashed blue lines), mechanical stretch enhances NO production via enhanced AMP-activated protein kinase (AMPK) activation and phosphorylation of nNOS-serine 1412 (p-nNOS), which increases nNOS activity. Such downstream effects by mechanical stretch are impaired in dystrophin-deficient cardiomyocytes. The impact of dystrophin-deficiency on NO production are indicated by solid red lines.
The DGC also functions as a mechano-sensor to directly regulate muscle NO signaling. In dystrophin-deficient mdx cardiomyocytes, mechanical stretch impairs cardiac NO production and is associated with impaired phosphorylation of nNOS-serine 1412 (Garbincius and Michele, 2015), a phosphorylated site known to enhance nNOS activity (Rameau et al., 2007), implicating the direct effects of dystrophin on mechanically activated NO signaling. Further investigations revealed that mechanical stretch promotes phosphorylation of nNOS by AMP-activated protein kinase (AMPK) activation, which is impaired in mdx cardiomyocytes, suggesting dystrophin is necessary for mechanically regulating AMPK and hence NO production (Garbincius and Michele, 2015). These findings suggest that the DGC and dystrophin not only serve as a passive scaffold for nNOS to bind on, but they also exert direct effects on NO production via AMPK activation and subsequent posttranslational modifications of nNOS.
LIM Domain Proteins
In addition to the integrin complex and DGC, there are other cytoskeletal proteins that exhibit mechanical sensing properties that are influenced by NO signaling. LIM domain proteins are a family of transcriptional and cytoskeletal proteins characterized by their LIM domains containing a double-zinc finger motif with protein-binding interfaces (Kadrmas and Beckerle, 2004). These proteins exhibit a diverse range of biological roles, including mechano-transduction (Smith et al., 2014). The LIM domain family proteins dynamically regulate the remodeling and repair of actin stress fibers, which reinforces the cytoskeleton in response to mechanical forces (Yoshigi et al., 2005; Smith et al., 2013, 2014). Two key LIM domain proteins that exhibit mechano-sensory roles and are influenced by NO signaling are Muscle LIM Protein (MLP) and Lipoma Preferred Partner (LPP). Whilst they are not structural components of the sarcomere or the cardiac cytoskeleton, both proteins transiently associate with these structures as part of their signaling function. Muscle Lim Protein (MLP) is a key nucleocytoplasmic shuttling protein located on the sarcomeric Z-disc that exhibits cardiac mechanical sensing properties (Knoll et al., 2002). The protein exhibits pleiotropic biological processes in the maintenance of cytoskeletal and sarcomere function including actin polymerization/depolymerization (Arber et al., 1997; Gupta et al., 2008; Papalouka et al., 2009). Deficiency of MLP is associated with misalignment of the Z-disc, loss of passive stretch sensing, and disruption of an interaction between MLP, telethonin, and titin, which forms a mechano-sensing complex at the Z-disc (Knoll et al., 2002). One of the roles of MLP is to inhibit protein kinase C-α (PKCα) at the Z disk, and loss of MLP can result in heart failure through removal of these inhibitory actions (Lange et al., 2016). Interestingly, NO exerts antihypertrophic effects by promoting transcriptional and translational downregulation of MLP via cGMP-dependent activation of PKG (Heineke et al., 2003), and inhibition of NO prevented upregulated MLP protein and sarcomere disarray (Hooper et al., 2013). Further, subcellular fractionation experiments revealed re-localization of MLP from the cytosol to the nucleus following inhibition of NO by L-NAME treatment (Hooper et al., 2013), in which nuclear MLP has been demonstrated to promote hypertrophic growth in response to mechanical stress (Boateng et al., 2009).
Lipoma-preferred partner (LPP) is another nucleocytoplasmic shuttling protein localized on focal adhesions and cell–cell junctions, and communicates between the nucleus and cytoskeleton (Petit et al., 2000). It has also been described as a cardiac mechano-sensitive protein highly expressed in cardiac fibroblasts and participates in the regulation of cellular adaptations in response to cardiac hypertrophy induced by hemodynamic overload (Hooper et al., 2012). LPP also associates with the actin cytoskeleton to inhibit actin polymerization (Hansen and Beckerle, 2006). Indeed, it is proposed that NO regulates LPP expression following mechanical stress.
Treatment of cardiac fibroblasts with L-NAME and subjected to 5% stretch rescued downregulated LPP protein levels induced by stretch (Hooper et al., 2012), suggesting that stretch-induced downregulation of LPP is dependent on NO production. In cardiomyocytes, mechanical stress may produce NO and promote subcellular localization of LPP that inhibits myocyte growth. Treatment of cardiomyocytes with L-NAME under control and with 10% stretch decreases nuclear and cytoskeletal expression of LPP protein levels (Hooper et al., 2013). Nuclear LPP represses of transcription factors related to cellular growth (Guo et al., 2006b), whilst cytoskeletal LPP limits the rate of actin polymerization by inhibiting vasodilator stimulator phosphoprotein activity (Bear et al., 2002). Thus, mechanically regulated NO may regulate cellular growth and actin polymerization in cardiomyocytes by altering the subcellular localization of LPP. Collectively, these findings highlight the significant impact NO production and NO-derived signaling exerts on the mechano-sensing properties of MLP and LPP.
Figure 3 summarizes the effect of NO and mechanical stress on MLP/LPP signaling.
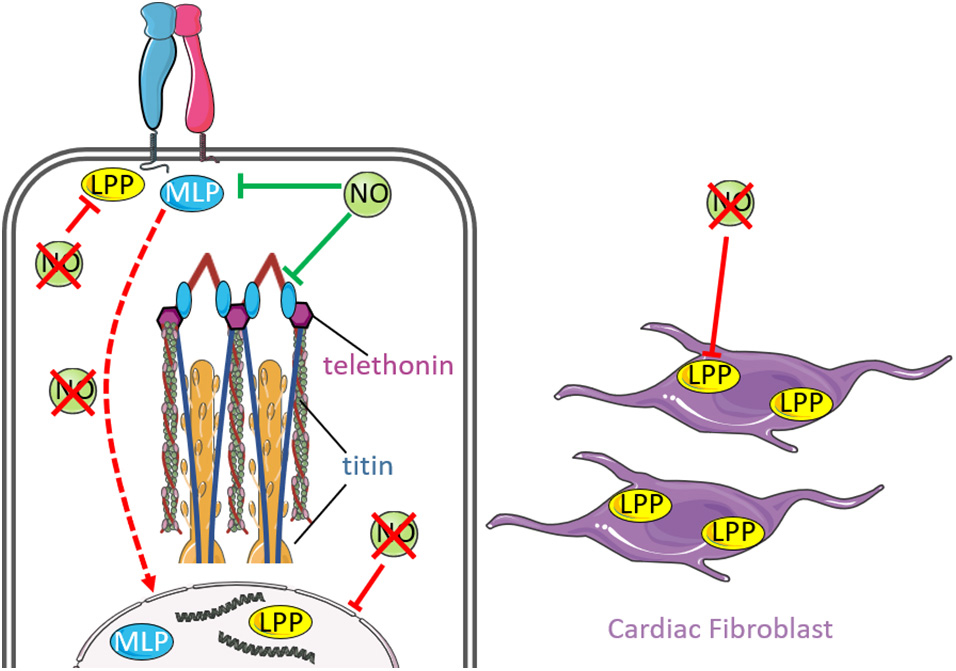
Figure 3. Nitric oxide signaling and mechanical stimuli affects signaling by MLP and LPP. Muscle LIM protein (MLP) is a nucleocytoplasmic protein that interacts with integrins at costameres and forms a mechano-sensing complex with titin and telethonin at the sarcomeric Z-disc (inset). Interestingly, enhanced NO downregulates MLP expression, which is associated with disruption of the MLP-titin-telethonin complex and misalignment of the Z-disc. Loss of NO promotes re-localization of MLP from the cytosol to the nucleus (red dashed arrow), where nuclear MLP has been demonstrated to promote hypertrophy in response to mechanical stress. Lipoma preferred partner (LPP) is another nucleo-cytoplsmic protein that is localized at focal adhesions, cell-cell junctions and communicates between the nucleus and cytoskeleton. The protein is highly expressed in cardiac fibroblasts and participates in cellular adaptations in response to mechanically induced cardiac hypertrophy. Nitric oxide has been proposed to regulates LPP expression following mechanical stress. Loss of NO is associated with reduced expression of nuclear and cytoskeletal LPP expression.
Titin
Titin is the largest protein in the sarcomere, forming the third filament system and is hence an important part of the cardiac cytoskeleton. It acts as a molecular “spring.” One titin molecule spans half a sarcomere from the Z-disc to the M-band (Linke, 2018). Whilst it is not actively involved in mechanical contractility, titin is the biggest contributor to elasticity and stretch activation (Linke, 2018). Titin is a mediator for mechano-sensory processes due to its ability to form multiple protein-protein interactions throughout the sarcomere (Kruger and Linke, 2009). The function of titin on passive tension is mediated by cyclic GMP-dependent PKG-mediated phosphorylation of several sites within the spring region of titin (Kruger et al., 2009; Leite-Moreira et al., 2018). PKG-mediated phosphorylation of titin at serine residue S469 alters the elasticity of N2B-unique sequence of titin, leading to reduced passive tension in human donor non-failing cardiac fibers (Kruger et al., 2009). Acute stretch of cardiomyocytes decreased passive tension, elevated PKG activity and phosphorylation of titin; the latter was dampened in acutely stretched cardiomyocytes that were pre-incubated with inhibition of PKG (Leite-Moreira et al., 2018). These observations further support a role of PKG in mediating phosphorylation of titin in response to stretch. Inhibition of NO, natriuretic peptide and NO scavenging in acutely stretched rabbit cardiomyocytes reduced passive tension decay that was similarly observed by inhibition of PKG only, suggesting that activation of cGMP-dependent PKG-mediated phosphorylation of titin may be partly mediated by the synergistic effects of NO and natriuretic peptide (Leite-Moreira et al., 2018). Other posttranslational modifications on titin, including S-glutathionylation (Alegre-Cebollada et al., 2014) and S-nitrosylation (Figueiredo-Freitas et al., 2015), also contribute to alterations in titin elasticity, supporting a role for NO signaling in regulating the elasticity of titin.
Ion Channel Modulation by Mechanical Stress
Contraction of the heart is initiated in response to the electrical depolarization of the tissue. Tissue depolarization occurs in the form of a wave of action potentials, originating in the sino-atrial node. However, the relationship between electrical signal and mechanical response is more nuanced than this simplistic view. The term “mechano-electrical coupling” has been coined to describe the dynamics of electrical and mechanical feedback in the context of cardiac function. Ion channels are well placed to integrate changes in the mechanical environment into an altered electrical response: sitting at the junction of cell membrane and signal transduction they are exquisitely placed to transform a mechanical signal into a biological response. Therefore, gating of ion channels in response to mechanical stimuli, results in changes to the ionic composition of the cell, which can act as a biological signal to drive responses to the change in mechanical environment. Numerous ion channels have been shown to be either directly gated by changes to the mechanical environment or mechanically sensitive (i.e., their function is modified by mechanical forces in an indirect manner through changes to the cytoskeleton or secondary messengers). The extensive number of cardiac ion channels regulated by mechanical stimuli has been excellently reviewed by Peyronnet and colleagues (Teng et al., 2014; Peyronnet et al., 2016). In the heart there are two main currents known to be activated in response to stretch. These are a stretch activated potassium current ISAC,K and the non-selective cation conductance Gns. ISAC,K is likely to be composed of a K2P channel such as TREK or TRAAK, a KATP channel, a BK channel (Reed et al., 2014) with contributions from the other many mechanically sensitive potassium channels (see Table 1). The molecular identity of Gns has not been categorically determined, but may be a TRP channel or Piezo 1 (reviewed in Reed et al., 2014; Peyronnet et al., 2016). Whilst there are many known mechanically sensitive ion channels which participate in the mechano-transduction process, many of these are classical voltage gated ion channels (which are generally considered to be gated by changes to the membrane potential) which are also able to be modulated by mechanical stimuli (Peyronnet et al., 2016). In order to be considered mechanically gated, three criteria have been proposed (Ridone et al., 2019). These are:
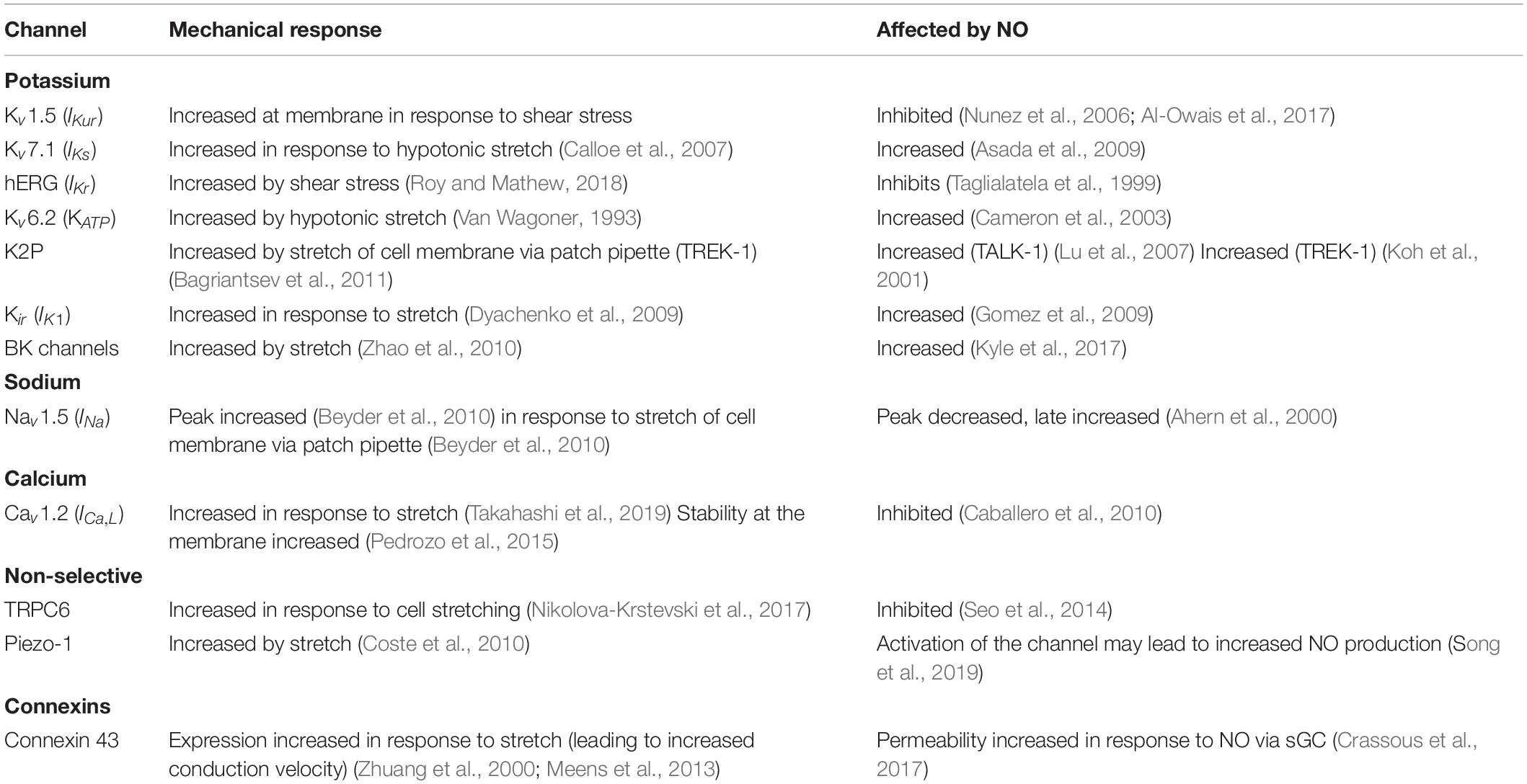
Table 1. Cardiac ion channels modulation by nitric oxide and mechanical stress.
1. Removal of the ion channel abolishes the mechano-sensory response of the cell.
2. The mechano-sensory response is disrupted if key structural residues are altered in the ion channel.
3. Over expression of the ion channel in a non-mechano-sensory cell renders it mechano-sensory.
In mammalian systems, the complexity and biological redundancy associated with mechano-transduction processes mean that fulfilment of all these criteria is difficult to prove. However, candidate ion channels which do meet these requirements are the Piezo 1 and 2 channels (see below).
Mechanically sensitive (rather than gated directly by mechanical stress) ion channels also pass current in response to changes in the mechanical environment. However, the mechanisms involved in their gating are diverse and may involve indirect processes. Some examples of the different ways in which mechanically sensitive ion channels gate are change in subcellular localization (Boycott et al., 2013) and change in stability of the channel at the membrane and changes to membrane tension (Honore, 2007; Pedrozo et al., 2015). There is also evidence that the cytoskeleton is a key regulator of ion channel mechano-sensitivity (Martinac, 2014), indeed the regulation of Kv1.5 by shear stress requires an intact microtubule network (Boycott et al., 2013).
The functional properties of some ion channels can be modified by NO, which can occur via the addition of a nitroso group to a thiol (Gonzalez et al., 2009), for example, on a cysteine residue. Such nitrosylation can have the effect of enhancing the channel activity or inhibiting it depending on the specific ion channel involved. In particular there are several channels which are both mechanically sensitive and modulated by NO, such as voltage dependent potassium channel-1.5 (Kv1.5) (Nunez et al., 2006), voltage dependent calcium channel-1.2 (Cav1.2) (Campbell et al., 1996) and voltage dependent sodium channel-1.5 (Nav1.5) (Ueda et al., 2008). NO also affects the transcriptional regulation of some ion channels via activation of the cGMP/PKG pathway. Certain ion channels, e.g., Kv1.5 can be modulated by NO through both direct nitrosylation and the PKG pathway (Nunez et al., 2006). This review will not evaluate the effects of NO on cardiac electrophysiology, as these have been excellently reviewed elsewhere (Tamargo et al., 2010), but will instead focus on the ion channels known to be affected by NO and mechanical stress, and the link between them.
There is some evidence that NO is released in response to mechanical stimuli (Petroff et al., 2001; Shim et al., 2017a), however, reports investigating the relationship between NO and mechano-electrical conduction (through modulation of ion channel activity) have been somewhat limited. At the level of the action potential, it has been reported that stretch of rat sino-atrial node cardiomyocytes prolongs the action potential duration (APD), and leads to fibrillation. The authors also found that the exogenous application of NO using the donor molecule S-Nitroso-N-acetylpenicillamine (SNAP) exacerbated the fibrillation, and prevented a return to control APD upon cessation of stretch. The authors conclude that increased NO during stretch is responsible for the opening of mechanically gated ion channels, and that in the presence of the NO donor this led to unresolvable fibrillation. The ionic currents underlying the findings were not described, and the role of endogenous NO in the response to stretch was not tested (Shim et al., 2017b). The major ionic currents known to be modulated by NO and mechanical stress and their potential contribution to the action potential is described in Figure 4.
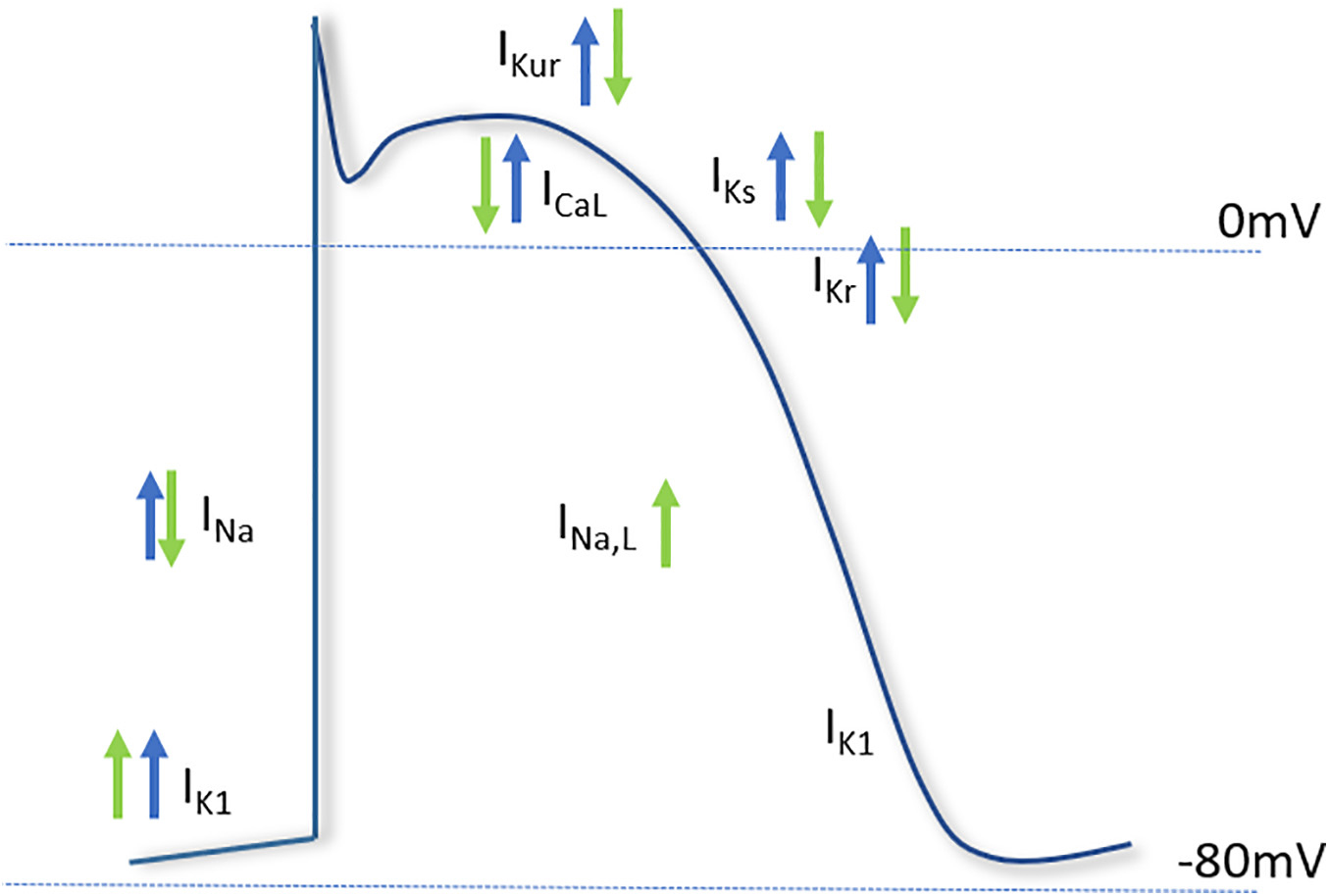
Figure 4. Major cardiac ion channels known to be affected by nitric oxide and mechanical stimuli. The effect of nitric oxide (green arrows) and mechanical stress (blue arrows) on the major currents of the cardiac action potential. The depolarizing sodium current (INa), responsible for the upstroke of the action potential, is increased by mechanical stress and decreased by NO. The late component of INa is increased by NO. The L-type calcium current (ICaL), involved in the plateau phase of the action potential, is inhibited by NO, but increased by mechanical stress. Of the repolarizing potassium currents, NO inhibits the ultrarapid delayed rectifier current (IKur), the rapid delayed rectifier (IKr) and slow delayed rectifier (IKs), whereas the inwardly rectifying potassium current (IK1) is increased by NO. Mechanical stress has been shown to activate the repolarizing currents IKur, IKs, IKr, and IK1.
The following section explores the evidence for regulation of specific cardiac ionic currents by mechanical stimuli, and the potential role for NO in this regulation.
The L-Type Calcium Channel
The L-type calcium current (LTCC) mediated by Cav1.2 is involved in action potential prolongation, maintaining phase two of the cardiac action potential. The influx of Ca2+ into cardiomyocytes following depolarization activates ryanodine receptors and leads to sarcoplasmic reticulum Ca2+ release and ultimately to cardiac contraction. As a result, the LTCC is critical for excitation-contraction coupling, linking the electrical signal of the action potential to the mechanical response of the cardiomyocyte manifested as contraction. A recent study demonstrated that the LTCC is involved in Ca2+ release in response to the stretch of a rat cardiomyocyte cell line (Takahashi et al., 2019). The channel is also affected by NO, which inhibits the channel. Ca2+ currents from mice lacking nNOS are larger and inactivate more slowly than wild type littermates, suggesting that NO tonically inhibits the channel (Caballero et al., 2010). Lack of nNOS is arrhythmogenic in mice, partially as a result of the removal of inhibition on the LTCC (Sears et al., 2003; Burger et al., 2009). Interestingly, nitrosylation of Cav1.2 is increased in rats in response to mechanical stress induced by isoproterenol treatment (Kentish et al., 2001; Sun et al., 2006). The nitrosylation is protective against ischemia-reperfusion injury, consistent with nitrosylation inhibiting the LTCC (Sun et al., 2006).
Sodium Current
The main depolarizing current in cardiomyocytes is mediated through the inward flow of Na+ ions, carried by the ion channel Nav1.5. The influx of Na+ into the cell initiates the depolarization necessary to reach the threshold for action potential generation. In cardiomyocytes, Nav1.5 forms a complex with nNOS and syntrophin at the sarcolemma (Ueda et al., 2008). Direct nitrosylation of Nav1.5 has been demonstrated in several studies and exerts a dual effect on the channel. Firstly, the peak current is decreased, secondly, the so-called late persistent current (which does not inactivate) is increased (Ahern et al., 2000). Nav1.5 is mechano-sensitive, with stretch increasing the peak current and accelerating inactivation (Peyronnet et al., 2016). In addition, Chorro et al. (2015) demonstrated that ranolazine (which selectively inhibits the late sodium current) application to Langendorff perfused rabbit hearts reduced stretch-induced ventricular fibrillation. The same group also showed that a more selective blocker of late inward sodium current (INa,L) (GS967) was similarly capable of preventing the stretch induced effects on cardiac refractoriness (Del Canto et al., 2018). Although the effect of NO in this process was not assessed it is conceivable that the stretch induced increase in late Na+ current could be mediated by NO.
TRPC6
Transient Receptor Potential C6 (TRPC6) channels are non-voltage gated cation channels. They are potentially involved in the atrial response to stretch, as endothelial TRPC6 changes its localization, expression and activity in response to stretch. This results in paracrine effects on cardiomyocytes which alters Ca2+ signaling (Nikolova-Krstevski et al., 2017). The authors suggest that the channel is directly modulated by stretch, as GsMTx-4 which disrupts TRPC6 interaction with the surrounding membrane lipids and can prevent the resultant effects of stretch on channel activity. Similarly, stretch of murine ventricular myocytes causes an increase in Gns, which may be a result of TRPC6 activation (Dyachenko et al., 2009). The stretch induced increase in Gns is dependent on the action of NO, generated by eNOS. The authors found that the effect of stretch also required superoxide, and conclude that peroxynitrite is the stretch dependent signaling molecule responsible for the increase in Gns. The TRPC6 channel can be inhibited by the PKG-pathway, and this regulation is necessary for the increase in Ca2+ seen upon stretch of myocytes with carbon-fiber rods (Seo et al., 2014).
Piezo-1
Discovered in 2010, Piezo-1 is a non-selective cation channel, which is directly gated by mechanical forces (Coste et al., 2010). Although the expression of Piezo-1 does not seem to be high in cardiomyocytes, there are intriguing hints that the protein may have a role to play in cardiomyocyte mechano-transduction. Piezo-1 is expressed in rodent myocardium, and is upregulated in response to heart failure and angiotensin II treatment (Liang et al., 2017). The protein is also expressed in human cardiac fibroblasts where it induces release of interleukin-6 (IL-6) (Blythe et al., 2019). In the endothelium, Piezo-1 mediates NO production in response to shear stress (Wang et al., 2016). The human cardiac cell line AC16 expresses Piezo-1 which is downregulated in response to prolonged stretching, and may modulate the phosphorylation (and hence activity) of eNOS (Wong et al., 2018). Interestingly, in drosophila neurons, Piezo-1 activation leads to an increase in intracellular Ca2+ which results in an increase in NO production and stimulation of the PKG pathway (Song et al., 2019). The extent to which Piezo-1 contributes to cardiac mechano-electrical signaling, and the involvement of NO remains to be explored. If Piezo-1 affects cardiac NO production it may be that the channel is important for the increase in NO production observed in response to mechanical stress, thus placing Piezo-1 at the start of the NO mediated mechanical signaling cascade.
Potassium Channels
Direct evidence linking potassium channels, NO and mechanical responsiveness is lacking at the present time. However, there are several channels which are both mechanically sensitive and modulated by NO, raising the possibility that a link may be present.
Interestingly, our group has shown that atrial cardiomyocytes taken from patients in chronic atrial fibrillation (a condition in which the mechanical environment is likely to be perturbed, in which nNOS is depleted, and the potassium currents IKur and IK1 are increased) leads to the electrical remodeling characteristic of the condition (Reilly et al., 2016). Further research is necessary to understand the role of NO in the mechanical regulation of cardiac K+ currents in health and disease. The use of specific NOS inhibitors to examine the effect of mechanical stress on K+ currents could be a useful tool to improve understanding of the role of NO in mediating the mechanical response of these channels.
Calcium Handling and Homeostasis
Precise regulation of intracellular Ca2+ levels are fundamental for the proper contractile response of cardiomyocytes. Within cardiac myocytes, Ca2+ signaling is tightly controlled in dedicated microdomains, myocyte mishandling of Ca2+ can cause both contractile dysfunction and arrhythmias in pathophysiological conditions (Schonleitner et al., 2017). During an action potential, Ca2+ enters the cell through the L-type Ca2+ channel, located in t-tubular membrane invaginations, during depolarization. The influx of a small amount of Ca2+ activates the underlying ryanodine receptors (RyR) located on the adjacent sarcoplasmic reticulum (SR) membrane. Electron microscopy and high resolution immunofluorescence images show that t-tubular L-type Ca2+ channels and RyR form regular and co-localized patterning within 10–12 nanometers in structures known as dyads (Page and Buecker, 1981; Koh et al., 2006). Ca2+ binding to the cytoplasmic extremity of the RyR channel pore triggers increased opening probability and concurrent release of Ca2+ from the SR via Ca2+ induced Ca2+ release (CICR) which amplifies the intracellular Ca2+ concentration 5–10-fold (Bers, 2002; del Rio et al., 2009; Schonleitner et al., 2017). Cytoplasmic Ca2+ influx from the SR is detected primarily by sarcomeric troponin C (TnC) on the actin filament, Ca2+ binding to the regulatory site II EF hand motif of TnC causes the release of the inhibitory peptide of troponin I from actin, and along with troponin T and tropomyosin activates actomyosin S1 ATPase dependent contraction of the cardiomyocyte (Tobacman, 1996; Bers, 2002). RyR located on the SR membrane mediate the release of Ca2+ from the SR when they are activated by the binding of Ca2+, meaning that RyR are central to Ca2+ amplification during CICR (Bers, 2002; Jian et al., 2014). For relaxation to occur, Ca2+ must be released by TnC in diastole and removed from the cytoplasm. This requires closure of the RyRs and Ca2+ to be pumped (1) back into the SR, by the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) and (2) out of the cell, largely by the sodium–calcium exchanger (NCX) and sarcolemmal Ca2+ ATPase (Eisner et al., 2017). The SERCA pump is regulated by the small transmembrane protein phospholamban (PLN) which acts as a molecular break and binds to SERCA when dephosphorylated, upon phosphorylation by either protein kinase A (PKA) or calcium-calmodulin Ca2+/calmodulin-dependent protein kinase II (CaMKII), PLN dissociates form SERCA and pentamerizes (Kranias and Hajjar, 2012). This process along with phosphorylation of key sarcomeric proteins and Ca2+ handling proteins such as troponin I, myosin regulatory light chain, RyR, and L-type Ca2+ channel forms the key processes that regulate the physiological fight or flight response in the heart in response to β-adrenergic stimulation (Bers, 2002; Najafi et al., 2016). The processes by which cardiomyocytes sense mechanical load and respond by changing Ca2+ dynamics have been termed mechano-chemo-transduction (MCT). Mechano-chemo-transduction pathways provide autoregulation of Ca2+ signaling and contractility in normal hearts, but may cause Ca2+ dysregulation in diseased hearts (Chen-Izu and Izu, 2017).
SR Ca2+ Release
Although Ca2+ release from the SR is usually triggered by opening of the L-type calcium channel, changes to RyR sensitivity may result in the localized spontaneous release of Ca2+ in a so-called Ca2+ spark. Under certain conditions, such as a high SR Ca2+ content, or increased RyR sensitization (such as channel phosphorylation by PKA or CAMKII), the Ca2+ spark can propagate to neighboring RyR clusters, and an arrhythmic Ca2+ wave is triggered (Schonleitner et al., 2017). Stretch-induced increases in Ca2+ spark frequency have been consistently observed in myocytes (Prosser et al., 2011), and are also found in response to shear stress and afterload (Jian et al., 2014). The existence of stretch induced Ca2+ sparks demonstrated that the Frank-Starling effect, previously thought to be Ca2+ independent, involves fast, local changes in Ca2+. These are believed to be a result of changes to the sensitivity of SR Ca2+ release channels (Schonleitner et al., 2017). Excessive mechanical loading and stress can cause a high rate of diastolic Ca2+ sparks, which are able to trigger spontaneous Ca2+ waves leading to arrhythmic events (Katra and Laurita, 2005; Jian et al., 2014).
RyR sensitivity can be affected by nitrosylation, which increases the activity of the channel (Wang et al., 2010; Johnson and Antoons, 2018). nNOS is localized between the SR and sarcolemma where it associates with various partner proteins, including SERCA and RyR. This proximity results in the modulation of RyR activity and SERCA by nNOS and increase in Ca2+ sparks and Ca2+ reuptake (Jian et al., 2014). nNOS increases RyR Ca2+ leak either directly via S-nitrosylation (Johnson and Antoons, 2018) or indirectly via CaMKII (Jian et al., 2014; Erickson et al., 2015). nNOS also facilitates SERCA Ca2+ reuptake (Vielma et al., 2016), which may compensate for increased SR Ca2+ leak (Carnicer et al., 2017). Redistribution of nNOS in the failing heart has been reported, and is thought to contribute to the pathology. This is potentially a consequence of the loss of micro domain structures seen in many pathological conditions (Schonleitner et al., 2017).
How the cell senses the mechanical stress stimulus and how the stimulus is transmitted to intracellular Ca2+ stores remain controversial. The study by Jian et al. (2014) demonstrated a relationship between mechanical load and NO production. Using a cell-in-gel system to recreate the forces experienced in the native cardiomyocyte, the authors showed that load mediated Ca2+ dynamics are reliant on NOS enzymes. The study also suggested differential effects for nNOS and eNOS in modulating different Ca2+ handling pathways. Both NOS isoforms are involved in elevating Ca2+ transients; however, afterload-induced spontaneous Ca2+ sparks are mediated by nNOS, but not eNOS. The explanation for the divergent effects of the NOS isoforms may be due to their different subcellular localizations (Jian et al., 2014).
In these settings, NOS produces NO by phosphorylation via the phosphoinositide 3-kinase/protein kinase B (PI3K-Akt) pathway (Petroff et al., 2001). nNOS is also centrally involved in stretch induced Ca2+ spark production, most likely via oxidation of CaMKII (Gutierrez et al., 2013; Jian et al., 2014) which increases RyR activity and SR Ca2+ load. Increased phosphorylation of RyR by CaMKII has been proposed to be a feature of hypertrophic cardiomyopathy genetic mutations (Robinson et al., 2018). Interestingly overexpression of iNOS specifically in cardiomyocytes of transgenic mice results in cardiac hypertrophy and sudden cardiac death (Mungrue et al., 2002), whilst energetic depletion as a result of pathogenic cardiomyopathy mutations results in increased the generation of reactive oxygen species by the mitochondria (Ashrafian et al., 2003; Sacchetto et al., 2019). The specific role of other NOS isoforms in this pathology is unknown.
CaMKII
Downstream of NOS signaling, CaMKII has been found to regulate Ca2+ homeostasis and cardiac function and also mediate the MCT effect on Ca2+ handling (Jian et al., 2014; Chen-Izu and Izu, 2017). CaMKII can phosphorylate several Ca2+-handling proteins including phospholamban, RyR, and the L-type Ca2+ channel, as well as myofilament proteins to stimulate Ca2+ sparks (Hidalgo et al., 2013; Jian et al., 2014). Recent reports have implied that NO can activate CaMKII in the heart (Erickson et al., 2011; Jian et al., 2014). Mechanical stress activates NOS which, through NO and ROS signaling, modulate CaMKII activity. These signaling pathways orchestrate the modulations of Ca2+ handling molecules to regulate Ca2+ dynamics in response to mechanical stress (Chen-Izu and Izu, 2017). Phosphorylation of the cardiac RyR is suggested to be important not only for normal cardiac excitation-contraction coupling (Marx et al., 2000; Wehrens et al., 2004) but also in exacerbating abnormalities in Ca2+ homeostasis that are seen in heart failure (Marx et al., 2000; Belevych et al., 2007) and cardiomyopathies (Robinson et al., 2018). It is known that CaMKII-dependent phosphorylation of RyR can give rise to an increase in open probability (Marx et al., 2000; Guo et al., 2006a). CaMKII contributes to afterload-induced Ca2+ sparks (Guo et al., 2006a) and the inhibition of both nNOS and CaMKII in cardiomyocytes eliminates the Ca2+ sparks, suggesting mechano-transduction activates nNOS and CaMKII (Jian et al., 2014). Mechano-chemo-transduction through NOS and CaMKII signaling pathways suggests possible therapeutic targets for treating mechanical stress-induced Ca2+ dysregulation, arrhythmias (Jian et al., 2014), and cardiomyopathy (Moolman et al., 1997; Alvarez et al., 1999). The effects of NO in mediating Ca2+ handling in the cardiomyocyte are shown in Figure 5.
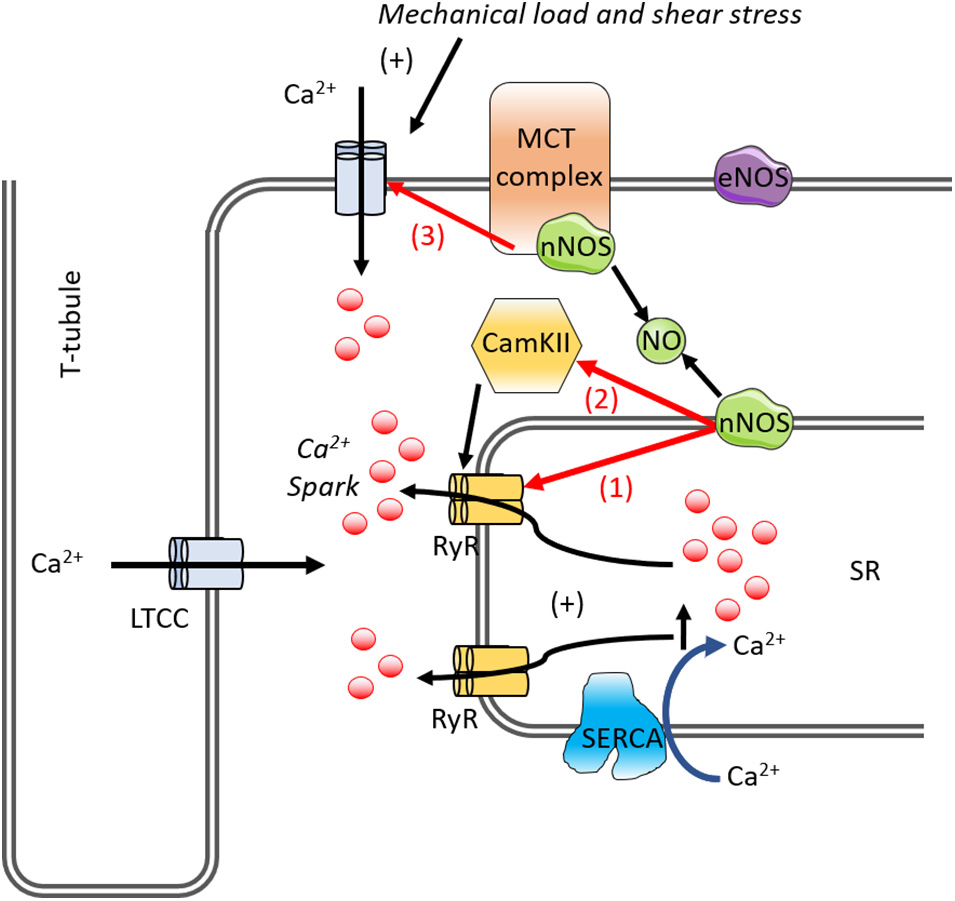
Figure 5. A diagram of the mechano-chemo transduction pathways in the cardiomyocyte. Mechano-transduction involves NO for RyR activation. In cardiomyocytes, mechanical load, and shear stress causes Ca2+ influx, Ca2+ binds to and activates the underlying RyR. This increases spontaneous Ca2+ spark via RyR. nNOS and CaMKII is activated promoting SR Ca2+ leak via RyR phosphorylation. nNOS increases RyR Ca2+ leak either (1) directly via S-nitrosylation or (2) indirectly via CaMKII. The (3) LTCC channel is affected by NO, inhibiting the channel. LTCC, L-type calcium current; NO, nitric oxide; nNOS, neuronal nitric oxide synthase; RyR, ryanodine receptor; SERCA, SR Ca2+-ATPase; SR, sarcoplasmic reticulum; CaMKII, Ca2+-calmodulin-dependent protein kinase II.
Discussion
The ability of the heart to respond appropriately to changes in the mechanical environment is important for proper cardiac function. Production of NO by nNOS and eNOS regulates every stage of mechano-electrical transduction; from initial sensing of mechanical signals through the integrin/cytoskeleton complexes, via control of the action potential by modulation of ion channel activity, through to mediating the calcium handling processes underpinning cardiac contraction.
Dysfunction of these processes in pathological conditions make understanding the complex role of NO vital in order to yield effective therapeutics based on NO replacement. It is clear that we still do not fully understand the way in which NO derived from mechanical signaling alters mechano-electrical signaling, and more work is needed, particularly in understanding how ionic currents are altered, and how this may result in arrhythmogenic activity. As technological advances occur it is becoming more feasible to investigate the contributions of different types of mechanical stresses to cardiac physiology. For example, acrylamide gels which allow approximation of native physiological and pathophysiological stiffness are used to investigate cardiomyocyte response to different surfaces (Pandey et al., 2018). A 3D cell-in-gel system allows the effect of preload to be investigated when the cardiomyocyte contracts again the gel (Shaw et al., 2013). Alternatively, myocytes can be adhered to stiff glass rods using MyotakTM to allow them to be stretched along the long axis (Prosser et al., 2011; Peyronnet et al., 2017). Similarly, force transducers and length controllers previously used to study chemically skinned cardiac trabeculae (Reiser et al., 1994; Simnett et al., 1994; Robinson et al., 2002), are now beginning to be used to study physiologically intact muscle strips in both cardiac and skeletal muscle (Tomalka et al., 2019). Magnetic tweezers can also be used to allow precise control of the strength and direction of force on myocytes (Chen et al., 2016). Finally, microdomain effects of stretch have been investigated using hydrojets from a micropipette to interrogate a highly specific area of the cell membrane, the jet causes indentation of the membrane in a similar way to atomic force microscopy, however, the area covered is more locally defined (Sanchez et al., 2008). These techniques have also been reviewed in greater detail (Schonleitner et al., 2017). The choice of technique used depends on the parameter to be measured, and attention should be paid to how the mechanical stress is affecting the cardiomyocyte (load free, stretch, shear etc.) reviewed in Chen-Izu and Izu (2017).
The effect on NO-regulated mechano-transduction/MCT in pathological conditions such as atrial fibrillation in which NO production is dramatically decreased (Reilly et al., 2016) remains to be fully investigated. There is some evidence, however, to suggest that changes to the mechanical environment of the cardiomyocyte are a key element of many disease pathologies. For example, mitral regurgitation causes shear stress in the atria and may thereby provide a mechanism for the development of atrial fibrillation (Grigioni et al., 2002) by affecting ion channel activity. Fibrosis in heart failure and cardiomyopathy can also stiffen the contractile mechanics of the heart (Fatkin and Graham, 2002), whist compromise in NO production is also seen in these disease states (Kelly et al., 1996; Kass et al., 2012). There are indications from recent literature that disease models have limited capacity to respond to increased challenge to the mechanical environment. For example, in atrial cardiomyocytes isolated from rats following the induction of atrial dilatation by trans-aortic constriction, patch clamping showed the ability of voltage gated potassium channels to respond to shear stress was reduced (Boycott et al., 2013). Similarly, subjecting cardiomyocytes from the mdx mouse lacking dystrophin to hypo-osmotic solutions to induce membrane stretch caused increased cytosolic Ca2+ only in the mdx cardiomyocytes (Leijendekker et al., 1996). Finally, experiments using cardiomyocytes plated on to substrates of increasing stiffness showed that obese diabetic rats progressing to heart failure had blunted Ca2+ handling with intrinsically stiffer cells due to increased myotubular networks (van Deel et al., 2017).
The extent to which NO can affect these processes and whether NO and stiffness changes work via independent biological signaling mechanisms remains to be investigated. The discovery of a synergistic link between the two would be of great clinical importance when trying to understand and prevent cardiac disease.
Author Contributions
HB conceived the manuscript, wrote the first draft, drew the first draft figures edited, and approved the final draft. M-NN and BV contributed to sections and figures to the first draft and edited subsequent drafts. PR edited all drafts, wrote discussion and drew figures. KG edited final draft. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Marie-Sklodowska-Curie (MCSA) individual fellowship (NO-STRESS—Nitric oxide regulation of repolarization in the heart: role of mechanical stress) to HB. British Heart Foundation (BHF) Programme Grant (Casadei) RG/16/12/32451 (For M-NN and BV), BHF Centre of Research Excellence Oxford Grant RE/13/1/30181 (HB, M-NN, BV, KG, and PR) and BHF Chair award (Watkins) CH/1992001/6764 (to PR). KG is supported by the British Heart Foundation (BHF) (FS/12/40/29712), the Oxford BHF Centre of Research Excellence (RE/13/1/30181), and the Wellcome Trust (201543/B/16/Z). The Institute of Cardiovascular Sciences, University of Birmingham, has received an Accelerator Award by the British Heart Foundation (AA/18/2/34218).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AKT, protein kinase B; AMPK, AMP-activated protein kinase; ANP, atrial natriuretic peptide; APD, action potential duration; BH4, Tetrahydrobiopterin; BNP, brain natriuretic peptide; CaMKII, calcium-calmodulin Ca2+/calmodulin-dependent protein kinase II; Cav1.2, voltage dependent calcium channel-1.2; CICR, Ca2+ induced Ca2+ release; DCG, dystrophin-glycoprotein complex; eNOS, endothelial nitric oxide synthase; ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; IL-6, Interleukin-6; INa,L, late inward sodium current; iNOS, inducible nitric oxide synthase; JNK, c-JunN-terminal kinase; Kv1.5, voltage dependent potassium channel-1.5; LIM domain, lin-11, islet-1, mec-3 domain; L-NAME, ng-nitroarginine methyl ester; LPP, lipoma-preferred partner; LTCC, L-type calcium current; MCT, mechano-chemo-transduction; MEK, mitogen-activated protein kinase kinase; mir31, micro-RNA-31; MLP, muscle LIM protein; Nav1.5, voltage dependent sodium channel-1.5; NCX, sodium–calcium exchanger; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NOS, nitric oxide synthase; PI3K, phosphoinositide 3-kinase; PKC-α, protein kinase C- α; PKG, protein kinase G; RyR, ryanodine receptor; SERCA, sarco-endoplasmic reticulum Ca2+ ATPase; sGC, guanylate cyclase; SNAP, S-nitroso -N-acetylpenicillamine; SR, sarcoplasmic reticulum; TNFα, tumor necrosis factor- α; TRPC6, transient receptor potential cation channel C6.
References
Abrahams, C., Janicki, J. S., and Weber, K. T. (1987). Myocardial hypertrophy in Macaca fascicularis, Structural remodeling of the collagen matrix. Lab Invest. 56, 676–683.
Adams, J. A., Wu, H., Bassuk, J. A., Arias, J., Uryash, A., and Kurlansky, P. (2009). Periodic acceleration (pGz) acutely increases endothelial and neuronal nitric oxide synthase expression in endomyocardium of normal swine. Peptides 30, 373–377. doi: 10.1016/j.peptides.2008.10.014
Ahern, G. P., Hsu, S. F., Klyachko, V. A., and Jackson, M. B. (2000). Induction of persistent sodium current by exogenous and endogenous nitric oxide. J. Biol. Chem. 275, 28810–28815. doi: 10.1074/jbc.m003090200
Ait-Mou, Y., Hsu, K., Farman, G. P., Kumar, M., Greaser, M. L., Irving, T. C., et al. (2016). Titin strain contributes to the Frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc. Natl. Acad. Sci. U.S.A. 113, 2306–2311. doi: 10.1073/pnas.1516732113
Alegre-Cebollada, J., Kosuri, P., Giganti, D., Eckels, E., Rivas-Pardo, J. A., Hamdani, N., et al. (2014). S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 156, 1235–1246. doi: 10.1016/j.cell.2014.01.056
Alkaitis, M. S., and Crabtree, M. J. (2012). Recoupling the cardiac nitric oxide synthases: tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail Rep. 9, 200–210. doi: 10.1007/s11897-012-0097-5
Al-Owais, M. M., Hettiarachchi, N. T., Boyle, J. P., Scragg, J. L., Elies, J., Dallas, M. L., et al. (2017). Multiple mechanisms mediating carbon monoxide inhibition of the voltage-gated K(+) channel Kv1.5. Cell Death Dis. 8:e3163. doi: 10.1038/cddis.2017.568
Alvarez, B. V., Perez, N. G., Ennis, I. L., Camilion De Hurtado, M. C., and Cingolani, H. E. (1999). Mechanisms underlying the increase in force and Ca(2+) transient that follow stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ. Res. 85, 716–722. doi: 10.1161/01.res.85.8.716
Arber, S., Hunter, J. J., Ross, J. Jr., Hongo, M., Sansig, G., Borg, J., et al. (1997). MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 88, 393–403. doi: 10.1016/s0092-8674(00)81878-4
Asada, K., Kurokawa, J., and Furukawa, T. (2009). Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel. J. Biol. Chem. 284, 6014–6020. doi: 10.1074/jbc.m807158200
Ashrafian, H., Redwood, C., Blair, E., and Watkins, H. (2003). Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet. 19, 263–268. doi: 10.1016/s0168-9525(03)00081-7
Bagriantsev, S. N., Peyronnet, R., Clark, K. A., Honore, E., and Minor, D. L. Jr. (2011). Multiple modalities converge on a common gate to control K2P channel function. EMBO J. 30, 3594–3606. doi: 10.1038/emboj.2011.230
Bear, J. E., Svitkina, T. M., Krause, M., Schafer, D. A., Loureiro, J. J., Strasser, G. A., et al. (2002). Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell 109, 509–521. doi: 10.1016/s0092-8674(02)00731-6
Belevych, A., Kubalova, Z., Terentyev, D., Hamlin, R. L., Carnes, C. A., and Gyorke, S. (2007). Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys. J. 93, 4083–4092. doi: 10.1529/biophysj.107.114546
Beyder, A., Rae, J. L., Bernard, C., Strege, P. R., Sachs, F., and Farrugia, G. (2010). Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J. Physiol. 588, 4969–4985. doi: 10.1113/jphysiol.2010.199034
Bia, B. L., Cassidy, P. J., Young, M. E., Rafael, J. A., Leighton, B., Davies, K. E., et al. (1999). Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. J. Mol. Cell Cardiol. 31, 1857–1862. doi: 10.1006/jmcc.1999.1018
Blythe, N. M., Muraki, K., Ludlow, M. J., Stylianidis, V., Gilbert, H. T. J., Evans, E. L., et al. (2019). Mechanically activated Piezo1 channels of cardiac fibroblasts stimulate p38 mitogen-activated protein kinase activity and interleukin-6 secretion. J. Biol. Chem. 294, 17395–17408. doi: 10.1074/jbc.ra119.009167
Boateng, S. Y., Senyo, S. E., Qi, L., Goldspink, P. H., and Russell, B. (2009). Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J. Mol. Cell. Cardiol. 47, 426–435. doi: 10.1016/j.yjmcc.2009.04.006
Boycott, H. E., Barbier, C. S., Eichel, C. A., Costa, K. D., Martins, R. P., Louault, F., et al. (2013). Shear stress triggers insertion of voltage-gated potassium channels from intracellular compartments in atrial myocytes. Proc. Natl. Acad. Sci. U.S.A. 110, E3955–E3964.
Brenman, J. E., Chao, D. S., Xia, H., Aldape, K., and Bredt, D. S. (1995). Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82, 743–752. doi: 10.1016/0092-8674(95)90471-9
Burger, D. E., Lu, X., Lei, M., Xiang, F. L., Hammoud, L., Jiang, M., et al. (2009). Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation 120, 1345–1354. doi: 10.1161/circulationaha.108.846402
Caballero, R., De La Fuente, M. G., Gomez, R., Barana, A., Amoros, I., Dolz-Gaiton, P., et al. (2010). In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J. Am. Coll. Cardiol. 55, 2346–2354. doi: 10.1016/j.jacc.2010.02.028
Calloe, K., Nielsen, M. S., Grunnet, M., Schmitt, N., and Jorgensen, N. K. (2007). KCNQ channels are involved in the regulatory volume decrease response in primary neonatal rat cardiomyocytes. Biochim. Biophys. Acta 1773, 764–773. doi: 10.1016/j.bbamcr.2007.02.008
Cameron, J. S., Hoffmann, K. E., Zia, C., Hemmett, H. M., Kronsteiner, A., and Lee, C. M. (2003). A role for nitric oxide in hypoxia-induced activation of cardiac KATP channels in goldfish (Carassius auratus). J. Exp. Biol. 206, 4057–4065. doi: 10.1242/jeb.00655
Campbell, D. L., Stamler, J. S., and Strauss, H. C. (1996). Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J. Gen. Physiol. 108, 277–293. doi: 10.1085/jgp.108.4.277
Carnicer, R., Suffredini, S., Liu, X., Reilly, S., Simon, J. N., Surdo, N. C., et al. (2017). The subcellular localisation of neuronal nitric oxide synthase determines the downstream effects of no on myocardial function. Cardiovasc. Res. 113, 321–331. doi: 10.1093/cvr/cvx002
Chan, W. L., Holstein-Rathlou, N. H., and Yip, K. P. (2001). Integrin mobilizes intracellular Ca(2+) in renal vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 280, C593–C603.
Chen, L., Maybeck, V., Offenhausser, A., and Krause, H. J. (2016). Implementation and application of a novel 2D magnetic twisting cytometry based on multi-pole electromagnet. Rev. Sci. Instrum 87:064301. doi: 10.1063/1.4954185
Chen-Izu, Y., and Izu, L. T. (2017). Mechano-chemo-transduction in cardiac myocytes. J. Physiol. 595, 3949–3958. doi: 10.1113/jp273101
Choate, J. K., Murphy, S. M., Feldman, R., and Anderson, C. R. (2008). Sympathetic control of heart rate in nNOS knockout mice. Am. J. Physiol. Heart Circ. Physiol. 294, H354–H361.
Chorro, F. J., Del Canto, I., Brines, L., Such-Miquel, L., Calvo, C., Soler, C., et al. (2015). Ranolazine attenuates the electrophysiological effects of myocardial stretch in Langendorff-Perfused Rabbit Hearts. Cardiovasc. Drugs Ther. 29, 231–241. doi: 10.1007/s10557-015-6587-4
Cingolani, H. E., Aiello, E. A., Perez, N. G., Ennis, I. L., and Camilion De Hurtado, M. C. (2005). “The Na(+)/H(+) Exchanger as the Main Protagonist following Myocardial Stretch: The Anrep Effect and Myocardial Hypertrophy,” in Mechanosensitivity in Cells and Tissues, eds A. Kamkin and I. Kiseleva (Moscow: Springer).
Conrad, C. H., Brooks, W. W., Hayes, J. A., Sen, S., Robinson, K. G., and Bing, O. H. (1995). Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation 91, 161–170. doi: 10.1161/01.cir.91.1.161
Coste, B., Mathur, J., Schmidt, M., Earley, T. J., Ranade, S., Petrus, M. J., et al. (2010). Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60. doi: 10.1126/science.1193270
Crassous, P. A., Shu, P., Huang, C., Gordan, R., Brouckaert, P., Lampe, P. D., et al. (2017). Newly identified NO-sensor guanylyl cyclase/connexin 43 association is involved in cardiac electrical function. J. Am. Heart Assoc. 6:e006397.
De Jong, A. M., Maass, A. H., Oberdorf-Maass, S. U., Van Veldhuisen, D. J., Van Gilst, W. H., and Van Gelder, I. C. (2011). Mechanisms of atrial structural changes caused by stretch occurring before and during early atrial fibrillation. Cardiovasc. Res. 89, 754–765. doi: 10.1093/cvr/cvq357
Del Canto, I., Santamaria, L., Genoves, P., Such-Miquel, L., Arias-Mutis, O., Zarzoso, M., et al. (2018). Effects of the Inhibition of Late Sodium Current by GS967 on Stretch-Induced Changes in Cardiac Electrophysiology. Cardiovasc. Drugs Ther. 32, 413–425. doi: 10.1007/s10557-018-6822-x
del Rio, A., Perez-Jimenez, R., Liu, R., Roca-Cusachs, P., Fernandez, J. M., and Sheetz, M. P. (2009). Stretching single talin rod molecules activates vinculin binding. Science 323, 638–641. doi: 10.1126/science.1162912
Dyachenko, V., Rueckschloss, U., and Isenberg, G. (2009). Modulation of cardiac mechanosensitive ion channels involves superoxide, nitric oxide and peroxynitrite. Cell Calcium 45, 55–64. doi: 10.1016/j.ceca.2008.06.002
Eisner, D. A., Caldwell, J. L., Kistamas, K., and Trafford, A. W. (2017). Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195. doi: 10.1016/b978-012436570-4/50007-8
Erickson, J. R., Nichols, C. B., Uchinoumi, H., Stein, M. L., Bossuyt, J., and Bers, D. M. (2015). S-Nitrosylation Induces Both Autonomous Activation and Inhibition of Calcium/Calmodulin-dependent Protein Kinase II delta. J. Biol. Chem. 290, 25646–25656. doi: 10.1074/jbc.m115.650234
Erickson, J. R., Patel, R., Ferguson, A., Bossuyt, J., and Bers, D. M. (2011). Fluorescence resonance energy transfer-based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ. Res. 109, 729–738. doi: 10.1161/circresaha.111.247148
Fatkin, D., and Graham, R. M. (2002). Molecular mechanisms of inherited cardiomyopathies. Physiol. Rev. 82, 945–980. doi: 10.1152/physrev.00012.2002
Feron, O., Belhassen, L., Kobzik, L., Smith, T. W., Kelly, R. A., and Michel, T. (1996). Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J. Biol. Chem. 271, 22810–22814. doi: 10.1074/jbc.271.37.22810
Figueiredo-Freitas, C., Dulce, R. A., Foster, M. W., Liang, J., Yamashita, A. M., Lima-Rosa, F. L., et al. (2015). S-Nitrosylation of Sarcomeric Proteins Depresses Myofilament Ca2+)Sensitivity in Intact Cardiomyocytes. Antioxid. Redox Signal. 23, 1017–1034. doi: 10.1089/ars.2015.6275
Forstermann, U., and Munzel, T. (2006). Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113, 1708–1714. doi: 10.1161/circulationaha.105.602532
Frank, O. (1899). Die Grundform des arteriellen pulses. Erste Abhandlung. Mathematische Analyse. Zeitschrift für Biologie. 37, 483–526.
Froehner, S. C., Reed, S. M., Anderson, K. N., Huang, P. L., and Percival, J. M. (2015). Loss of nNOS inhibits compensatory muscle hypertrophy and exacerbates inflammation and eccentric contraction-induced damage in mdx mice. Hum. Mol. Genet. 24, 492–505. doi: 10.1093/hmg/ddu469
Fukuda, N., Terui, T., Ohtsuki, I., Ishiwata, S., and Kurihara, S. (2009). Titin and troponin: central players in the frank-starling mechanism of the heart. Curr. Cardiol. Rev. 5, 119–124. doi: 10.2174/157340309788166714
Garbincius, J. F., and Michele, D. E. (2015). Dystrophin-glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 13663–13668. doi: 10.1073/pnas.1512991112
Gomez, R., Caballero, R., Barana, A., Amoros, I., Calvo, E., Lopez, J. A., et al. (2009). Nitric oxide increases cardiac IK1 by nitrosylation of cysteine 76 of Kir2.1 channels. Circ. Res. 105, 383–392. doi: 10.1161/circresaha.109.197558
Gonano, L. A., Morell, M., Burgos, J. I., Dulce, R. A., De Giusti, V. C., Aiello, E. A., et al. (2014). Hypotonic swelling promotes nitric oxide release in cardiac ventricular myocytes: impact on swelling-induced negative inotropic effect. Cardiovasc. Res. 104, 456–466. doi: 10.1093/cvr/cvu230
Gonzalez, D. R., Treuer, A., Sun, Q. A., Stamler, J. S., and Hare, J. M. (2009). S-Nitrosylation of cardiac ion channels. J. Cardiovasc. Pharmacol. 54, 188–195. doi: 10.1097/fjc.0b013e3181b72c9f
Grigioni, F., Avierinos, J. F., Ling, L. H., Scott, C. G., Bailey, K. R., Tajik, A. J., et al. (2002). Atrial fibrillation complicating the course of degenerative mitral regurgitation: determinants and long-term outcome. J. Am. Coll. Cardiol. 40, 84–92. doi: 10.1016/s0735-1097(02)01922-8
Guo, B., Sallis, R. E., Greenall, A., Petit, M. M., Jansen, E., Young, L., et al. (2006b). The LIM domain protein LPP is a coactivator for the ETS domain transcription factor PEA3. Mol. Cell Biol. 26, 4529–4538. doi: 10.1128/mcb.01667-05
Guo, T., Zhang, T., Mestril, R., and Bers, D. M. (2006a). Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 99, 398–406. doi: 10.1161/01.res.0000236756.06252.13
Gupta, M. P., Samant, S. A., Smith, S. H., and Shroff, S. G. (2008). HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J. Biol. Chem. 283, 10135–10146. doi: 10.1074/jbc.m710277200
Gutierrez, D. A., Fernandez-Tenorio, M., Ogrodnik, J., and Niggli, E. (2013). NO-dependent CaMKII activation during beta-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 100, 392–401. doi: 10.1093/cvr/cvt201
Hansen, M. D., and Beckerle, M. C. (2006). Opposing roles of zyxin/LPP ACTA repeats and the LIM domain region in cell-cell adhesion. J. Biol. Chem. 281, 16178–16188. doi: 10.1074/jbc.m512771200
Heineke, J., Kempf, T., Kraft, T., Hilfiker, A., Morawietz, H., Scheubel, R. J., et al. (2003). Downregulation of cytoskeletal muscle LIM protein by nitric oxide: impact on cardiac myocyte hypertrophy. Circulation 107, 1424–1432. doi: 10.1161/01.cir.0000055319.94801.fc
Hidalgo, C. G., Chung, C. S., Saripalli, C., Methawasin, M., Hutchinson, K. R., Tsaprailis, G., et al. (2013). The multifunctional Ca(2+)/calmodulin-dependent protein kinase II delta (CaMKIIdelta) phosphorylates cardiac titin’s spring elements. J. Mol. Cell Cardiol. 54, 90–97. doi: 10.1016/j.yjmcc.2012.11.012
Honore, E. (2007). The neuronal background K2P channels: focus on TREK1. Nat. Rev. Neurosci. 8, 251–261. doi: 10.1038/nrn2117
Hooper, C. L., Dash, P. R., and Boateng, S. Y. (2012). Lipoma preferred partner is a mechanosensitive protein regulated by nitric oxide in the heart. FEBS Open Bio 2, 135–144. doi: 10.1016/j.fob.2012.05.005
Hooper, C. L., Paudyal, A., Dash, P. R., and Boateng, S. Y. (2013). Modulation of stretch-induced myocyte remodeling and gene expression by nitric oxide: a novel role for lipoma preferred partner in myofibrillogenesis. Am. J. Physiol. Heart Circ. Physiol. 304, H1302–H1313. doi: 10.1016/j.fob.2012.05.005
Jian, Z., Han, H., Zhang, T., Puglisi, J., Izu, L. T., Shaw, J. A., et al. (2014). Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci. Signal. 7:ra27. doi: 10.1126/scisignal.2005046
Johnson, D. M., and Antoons, G. (2018). Arrhythmogenic Mechanisms in Heart Failure: Linking beta-Adrenergic Stimulation. Stretch, and Calcium. Front. Physiol. 9:1453. doi: 10.3389/fphys.2018.01453
Kadrmas, J. L., and Beckerle, M. C. (2004). The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 5, 920–931. doi: 10.1038/nrm1499
Kaprielian, R. R., Stevenson, S., Rothery, S. M., Cullen, M. J., and Severs, N. J. (2000). Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation 101, 2586–2594. doi: 10.1161/01.cir.101.22.2586
Kass, D. A., Carnicer, R., Crabtree, M. J., Sivakumaran, V., and Casadei, B. (2012). Nitric Oxide Synthases in Heart Failure. Antioxid. Redox Signal. 18, 1078-1099.
Katra, R. P., and Laurita, K. R. (2005). Cellular mechanism of calcium-mediated triggered activity in the heart. Circ. Res. 96, 535–542. doi: 10.1161/01.res.0000159387.00749.3c
Kelly, R. A., Balligand, J. L., and Smith, T. W. (1996). Nitric oxide and cardiac function. Circ. Res. 79, 363–380.
Kentish, J. C., Mccloskey, D. T., Layland, J., Palmer, S., Leiden, J. M., Martin, A. F., et al. (2001). Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ. Res. 88, 1059–1065. doi: 10.1161/hh1001.091640
Knoll, R., Hoshijima, M., Hoffman, H. M., Person, V., Lorenzen-Schmidt, I., Bang, M. L., et al. (2002). The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955. doi: 10.1016/s0092-8674(02)01226-6
Koh, S. D., Monaghan, K., Sergeant, G. P., Ro, S., Walker, R. L., Sanders, K. M., et al. (2001). TREK-1 regulation by nitric oxide and cGMP-dependent protein kinase. An essential role in smooth muscle inhibitory neurotransmission. J. Biol. Chem. 276, 44338–44346. doi: 10.1074/jbc.m108125200
Koh, T. J., and Tidball, J. G. (2000). Nitric oxide inhibits calpain-mediated proteolysis of talin in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 279, C806–C812.
Koh, X., Srinivasan, B., Ching, H. S., and Levchenko, A. (2006). A 3D Monte Carlo analysis of the role of dyadic space geometry in spark generation. Biophys. J. 90, 1999–2014. doi: 10.1529/biophysj.105.065466
Kovacs, I., and Lindermayr, C. (2013). Nitric oxide-based protein modification: formation and site-specificity of protein S-nitrosylation. Front. Plant Sci. 4:137. doi: 10.3389/fpls.2013.00137
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi: 10.1161/circresaha.111.259754
Kruger, M., Kotter, S., Grutzner, A., Lang, P., Andresen, C., Redfield, M. M., et al. (2009). Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 104, 87–94. doi: 10.1161/circresaha.108.184408
Kruger, M., and Linke, W. A. (2009). Titin-based mechanical signalling in normal and failing myocardium. J. Mol. Cell Cardiol. 46, 490–498. doi: 10.1016/j.yjmcc.2009.01.004
Kyle, B. D., Mishra, R. C., and Braun, A. P. (2017). The augmentation of BK channel activity by nitric oxide signaling in rat cerebral arteries involves co-localized regulatory elements. J. Cereb. Blood Flow Metab. 37, 3759–3773. doi: 10.1177/0271678x17691291
Lakatta, E. G. (1987). Starling’s law of the heart is explained by an intimate interaction of muscle length and myofilament calcium activation. J. Am. Coll. Cardiol. 10, 1157–1164. doi: 10.1016/s0735-1097(87)80361-3
Lange, S., Gehmlich, K., Lun, A. S., Blondelle, J., Hooper, C., Dalton, N. D., et al. (2016). MLP and CARP are linked to chronic PKCalpha signalling in dilated cardiomyopathy. Nat. Commun. 7:12120.
Lapidos, K. A., Kakkar, R., and Mcnally, E. M. (2004). The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res. 94, 1023–1031. doi: 10.1161/01.res.0000126574.61061.25
Leijendekker, W. J., Passaquin, A. C., Metzinger, L., and Ruegg, U. T. (1996). Regulation of cytosolic calcium in skeletal muscle cells of the mdx mouse under conditions of stress. Br. J. Pharmacol. 118, 611–616. doi: 10.1111/j.1476-5381.1996.tb15445.x
Leite-Moreira, A. M., Almeida-Coelho, J., Neves, J. S., Pires, A. L., Ferreira-Martins, J., Castro-Ferreira, R., et al. (2018). Stretch-induced compliance: a novel adaptive biological mechanism following acute cardiac load. Cardiovasc. Res. 114, 656–667. doi: 10.1093/cvr/cvy026
Liang, J., Huang, B., Yuan, G., Chen, Y., Liang, F., Zeng, H., et al. (2017). Stretch-activated channel Piezo1 is up-regulated in failure heart and cardiomyocyte stimulated by AngII. Am. J. Transl. Res. 9, 2945–2955.
Linke, W. A. (2018). Titin gene and protein functions in passive and active muscle. Annu. Rev. Physiol. 80, 389–411. doi: 10.1146/annurev-physiol-021317-121234
Lopez, J. R., Kolster, J., Zhang, R., and Adams, J. (2017). Increased constitutive nitric oxide production by whole body periodic acceleration ameliorates alterations in cardiomyocytes associated with utrophin/dystrophin deficiency. J. Mol. Cell Cardiol. 108, 149–157. doi: 10.1016/j.yjmcc.2017.06.004
Lu, Z., Gao, J., Zuckerman, J., Mathias, R. T., Gaudette, G., Krukenkamp, I., et al. (2007). Two-pore K+ channels. NO and metabolic inhibition. Biochem. Biophys. Res. Commun. 363, 194–196.
Manso, A. M., Okada, H., Sakamoto, F. M., Moreno, E., Monkley, S. J., Li, R., et al. (2017). Loss of mouse cardiomyocyte talin-1 and talin-2 leads to beta-1 integrin reduction, costameric instability, and dilated cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 114, E6250–E6259.
Martinac, B. (2014). The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity. Biochim. Biophys. Acta 1838, 682–691. doi: 10.1016/j.bbamem.2013.07.015
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/s0092-8674(00)80847-8
Meens, M. J., Pfenniger, A., Kwak, B. R., and Delmar, M. (2013). Regulation of cardiovascular connexins by mechanical forces and junctions. Cardiovasc. Res. 99, 304–314. doi: 10.1093/cvr/cvt095
Moolman, J. C., Corfield, V. A., Posen, B., Ngumbela, K., Seidman, C., Brink, P. A., et al. (1997). Sudden death due to troponin T mutations. J. Am. Coll. Cardiol. 29, 549–555. doi: 10.1016/s0735-1097(96)00530-x
Mungrue, I. N., Gros, R., You, X., Pirani, A., Azad, A., Csont, T., et al. (2002). Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J. Clin. Invest. 109, 735–743. doi: 10.1172/jci0213265
Najafi, A., Sequeira, V., Kuster, D. W., and Van Der Velden, J. (2016). beta-adrenergic receptor signalling and its functional consequences in the diseased heart. Eur. J. Clin. Invest. 46, 362–374. doi: 10.1111/eci.12598
Naseem, K. M. (2005). The role of nitric oxide in cardiovascular diseases. Mol. Aspects Med. 26, 33–65. doi: 10.1016/j.mam.2004.09.003
Nikolova-Krstevski, V., Wagner, S., Yu, Z. Y., Cox, C. D., Cvetkovska, J., Hill, A. P., et al. (2017). Endocardial TRPC-6 Channels Act as Atrial Mechanosensors and Load-Dependent Modulators of Endocardial/Myocardial Cross-Talk. JACC Basic Transl. Sci. 2, 575–590. doi: 10.1016/j.jacbts.2017.05.006
Nunez, L., Vaquero, M., Gomez, R., Caballero, R., Mateos-Caceres, P., Macaya, C., et al. (2006). Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc. Res. 72, 80–89. doi: 10.1016/j.cardiores.2006.06.021
Oceandy, D., Cartwright, E. J., Emerson, M., Prehar, S., Baudoin, F. M., Zi, M., et al. (2007). Neuronal nitric oxide synthase signaling in the heart is regulated by the sarcolemmal calcium pump 4b. Circulation 115, 483–492. doi: 10.1161/circulationaha.106.643791
Page, E., and Buecker, J. L. (1981). Development of dyadic junctional complexes between sarcoplasmic reticulum and plasmalemma in rabbit left ventricular myocardial cells. Morphometric analysis. Circ. Res. 48, 519–522. doi: 10.1161/01.res.48.4.519
Pandey, P., Hawkes, W., Hu, J., Megone, W. V., Gautrot, J., Anilkumar, N., et al. (2018). Cardiomyocytes Sense Matrix Rigidity through a Combination of Muscle and Non-muscle Myosin Contractions. Dev. Cell 45:661. doi: 10.1016/j.devcel.2018.05.016
Papalouka, V., Arvanitis, D. A., Vafiadaki, E., Mavroidis, M., Papadodima, S. A., Spiliopoulou, C. A., et al. (2009). Muscle LIM protein interacts with cofilin 2 and regulates F-actin dynamics in cardiac and skeletal muscle. Mol. Cell Biol. 29, 6046–6058. doi: 10.1128/mcb.00654-09
Pedrozo, Z., Criollo, A., Battiprolu, P. K., Morales, C. R., Contreras-Ferrat, A., Fernandez, C., et al. (2015). Polycystin-1 Is a cardiomyocyte mechanosensor that governs L-Type Ca2+ channel protein stability. Circulation 131, 2131–2142. doi: 10.1161/circulationaha.114.013537
Percival, J. M., Anderson, K. N., Huang, P., Adams, M. E., and Froehner, S. C. (2010). Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle. J. Clin. Invest. 120, 816–826. doi: 10.1172/jci40736
Petit, M. M., Fradelizi, J., Golsteyn, R. M., Ayoubi, T. A., Menichi, B., Louvard, D., et al. (2000). LPP, an actin cytoskeleton protein related to zyxin, harbors a nuclear export signal and transcriptional activation capacity. Mol. Biol. Cell 11, 117–129. doi: 10.1091/mbc.11.1.117
Petroff, M. G., Kim, S. H., Pepe, S., Dessy, C., Marban, E., Balligand, J. L., et al. (2001). Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat. Cell Biol. 3, 867–873. doi: 10.1038/ncb1001-867
Peyronnet, R., Bollensdorff, C., Capel, R. A., Rog-Zielinska, E. A., Woods, C. E., Charo, D. N., et al. (2017). Load-dependent effects of apelin on murine cardiomyocytes. Prog. Biophys. Mol. Biol. 130, 333–343. doi: 10.1016/j.pbiomolbio.2017.09.013
Peyronnet, R., Nerbonne, J. M., and Kohl, P. (2016). Cardiac Mechano-Gated Ion Channels and Arrhythmias. Circ. Res. 118, 311–329. doi: 10.1161/circresaha.115.305043
Pilz, R. B., and Casteel, D. E. (2003). Regulation of gene expression by cyclic GMP. Circ. Res. 93, 1034–1046. doi: 10.1161/01.res.0000103311.52853.48
Prendergast, B. D., Sagach, V. F., and Shah, A. M. (1997). Basal release of nitric oxide augments the Frank-Starling response in the isolated heart. Circulation 96, 1320–1329. doi: 10.1161/01.cir.96.4.1320
Prosser, B. L., Khairallah, R. J., Ziman, A. P., Ward, C. W., and Lederer, W. J. (2013). X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca(2)(+)]i. J. Mol. Cell Cardiol. 58, 172–181. doi: 10.1016/j.yjmcc.2012.11.011
Prosser, B. L., Ward, C. W., and Lederer, W. J. (2011). X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333, 1440–1445. doi: 10.1126/science.1202768
Rameau, G. A., Tukey, D. S., Garcin-Hosfield, E. D., Titcombe, R. F., Misra, C., Khatri, L., et al. (2007). Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J. Neurosci. 27, 3445–3455. doi: 10.1523/jneurosci.4799-06.2007
Reed, A., Kohl, P., and Peyronnet, R. (2014). Molecular candidates for cardiac stretch-activated ion channels. Glob. Cardiol. Sci. Pract. 2014, 9–25.
Reilly, S. N., Liu, X., Carnicer, R., Recalde, A., Muszkiewicz, A., Jayaram, R., et al. (2016). Up-regulation of miR-31 in human atrial fibrillation begets the arrhythmia by depleting dystrophin and neuronal nitric oxide synthase. Sci. Transl. Med. 8:340ra374.
Reiser, P. J., Westfall, M. V., Schiaffino, S., and Solaro, R. J. (1994). Tension production and thin-filament protein isoforms in developing rat myocardium. Am. J. Physiol. 267, H1589–H1596.
Ridone, P., Vassalli, M., and Martinac, B. (2019). Piezo1 mechanosensitive channels: what are they and why are they important. Biophys. Rev. 11, 795–805. doi: 10.1007/s12551-019-00584-5
Robinson, P., Liu, X., Sparrow, A., Patel, S., Zhang, Y. H., Casadei, B., et al. (2018). Hypertrophic cardiomyopathy mutations increase myofilament Ca(2+) buffering, alter intracellular Ca(2+) handling, and stimulate Ca(2+)-dependent signaling. J. Biol. Chem. 293, 10487–10499. doi: 10.1074/jbc.ra118.002081
Robinson, P., Mirza, M., Knott, A., Abdulrazzak, H., Willott, R., Marston, S., et al. (2002). Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J. Biol. Chem. 277, 40710–40716. doi: 10.1074/jbc.m203446200
Roe, N. D., and Ren, J. (2012). Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vascul. Pharmacol. 57, 168–172. doi: 10.1016/j.vph.2012.02.004
Roy, S., and Mathew, M. K. (2018). Fluid flow modulates electrical activity in cardiac hERG potassium channels. J. Biol. Chem. 293, 4289–4303. doi: 10.1074/jbc.ra117.000432
Sacchetto, C., Sequeira, V., Bertero, E., Dudek, J., Maack, C., and Calore, M. (2019). Metabolic alterations in inherited cardiomyopathies. J. Clin. Med. 8:2195. doi: 10.3390/jcm8122195
Sagawa, K., Lie, R. K., and Schaefer, J. (1990). Translation of Otto Frank’s paper “Die Grundform des Arteriellen Pulses”. J. Mol. Cell Cardiol. 22, 253–254. doi: 10.1016/0022-2828(90)91459-k
Sanchez, D., Johnson, N., Li, C., Novak, P., Rheinlaender, J., Zhang, Y., et al. (2008). Noncontact measurement of the local mechanical properties of living cells using pressure applied via a pipette. Biophys. J. 95, 3017–3027. doi: 10.1529/biophysj.108.129551
Schonleitner, P., Schotten, U., and Antoons, G. (2017). Mechanosensitivity of microdomain calcium signalling in the heart. Prog. Biophys. Mol. Biol. 130, 288–301. doi: 10.1016/j.pbiomolbio.2017.06.013
Sears, C. E., Bryant, S. M., Ashley, E. A., Lygate, C. A., Rakovic, S., Wallis, H. L., et al. (2003). Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ. Res. 92, e52–e59.
Seddon, M., Shah, A. M., and Casadei, B. (2007). Cardiomyocytes as effectors of nitric oxide signalling. Cardiovasc. Res. 75, 315–326. doi: 10.1016/j.cardiores.2007.04.031
Seo, K., Rainer, P. P., Lee, D. I., Hao, S., Bedja, D., Birnbaumer, L., et al. (2014). Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ. Res. 114, 823–832. doi: 10.1161/circresaha.114.302614
Sequeira, V., Nijenkamp, L. L., Regan, J. A., and Van Der Velden, J. (2014). The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim. Biophys. Acta 1838, 700–722. doi: 10.1016/j.bbamem.2013.07.011
Shah, A. M. (2000). Inducible nitric oxide synthase and cardiovascular disease. Cardiovasc. Res. 45, 148–155. doi: 10.1016/s0008-6363(99)00316-8
Shah, A. M., and MacCarthy, P. A. (2000). Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol. Ther. 86, 49–86. doi: 10.1016/s0163-7258(99)00072-8
Shaw, J., Izu, L., and Chen-Izu, Y. (2013). Mechanical analysis of single myocyte contraction in a 3-D elastic matrix. PLoS One 8:e75492. doi: 10.1371/journal.pone.0075492
Shim, A. L., Mitrokhin, V. M., Gorbacheva, L. R., Savinkova, I. G., Pustovit, K. B., Mladenov, M. I., et al. (2017a). Kinetics of Mechanical Stretch-Induced Nitric Oxide Production in Rat Ventricular Cardiac Myocytes. Bull. Exp. Biol. Med. 163, 583–585. doi: 10.1007/s10517-017-3853-4
Shim, A. L., Mitrokhin, V. M., Kazanski, V. E., Mladenov, M. I., and Kamkin, A. G. (2017b). Discrete stretch eliminates electrophysiological dose-dependent effects of nitric oxide donor SNAP in Rat Atrium. Bull. Exp. Biol. Med. 163, 705–709. doi: 10.1007/s10517-017-3885-9
Simnett, S. J., Lipscomb, S., Ashley, C. C., Potter, J. D., and Mulligan, I. P. (1994). The thiadiazinone EMD 57033 speeds the activation of skinned cardiac muscle produced by the photolysis of nitr-5. Pflug. Arch. 427, 550–552. doi: 10.1007/bf00374274
Singal, P. K., Khaper, N., Palace, V., and Kumar, D. (1998). The role of oxidative stress in the genesis of heart disease. Cardiovasc. Res. 40, 426–432. doi: 10.1016/s0008-6363(98)00244-2
Smith, M. A., Blankman, E., Deakin, N. O., Hoffman, L. M., Jensen, C. C., Turner, C. E., et al. (2013). LIM domains target actin regulators paxillin and zyxin to sites of stress fiber strain. PLoS One 8:e69378. doi: 10.1371/journal.pone.0069378
Smith, M. A., Hoffman, L. M., and Beckerle, M. C. (2014). LIM proteins in actin cytoskeleton mechanoresponse. Trends Cell Biol. 24, 575–583. doi: 10.1016/j.tcb.2014.04.009
ong, Y., Li, D., Farrelly, O., Miles, L., Li, F., Kim, S. E., et al. (2019). The mechanosensitive ion channel piezo inhibits axon regeneration. Neuron 102:e376.
Starling, E. H. (1918). The linacre lecture on the law of the heart given at cambridge, 1915. Nature 101, 43–43. doi: 10.1038/101043a0
Such-Miquel, L., Del Canto, I., Zarzoso, M., Brines, L., Soler, C., Parra, G., et al. (2018). Effects of S-Nitrosoglutathione on Electrophysiological Manifestations of Mechanoelectric Feedback. Cardiovasc. Toxicol. 18, 520–529. doi: 10.1007/s12012-018-9463-1
Sun, J., Picht, E., Ginsburg, K. S., Bers, D. M., Steenbergen, C., and Murphy, E. (2006). Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ. Res. 98, 403–411. doi: 10.1161/01.res.0000202707.79018.0a
Taglialatela, M., Pannaccione, A., Iossa, S., Castaldo, P., and Annunziato, L. (1999). Modulation of the K(+) channels encoded by the human ether-a-gogo-related gene-1 (hERG1) by nitric oxide. Mol. Pharmacol. 56, 1298–1308. doi: 10.1124/mol.56.6.1298
Takahashi, K., Hayashi, S., Miyajima, M., Omori, M., Wang, J., Kaihara, K., et al. (2019). L-type calcium channel modulates mechanosensitivity of the cardiomyocyte cell line H9c2. Cell Calcium 79, 68–74. doi: 10.1016/j.ceca.2019.02.008
Tamargo, J., Caballero, R., Gomez, R., and Delpon, E. (2010). Cardiac electrophysiological effects of nitric oxide. Cardiovasc. Res. 87, 593–600. doi: 10.1093/cvr/cvq214
Teng, J., Loukin, S., and Kung, C. (2014). Mechanosensitive ion channels in cardiovascular physiology. Exp. Clin. Cardiol. 20, 6550–6560.
Thomas, G. D., Shaul, P. W., Yuhanna, I. S., Froehner, S. C., and Adams, M. E. (2003). Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal Nitric oxide synthase. Circ. Res. 92, 554–560. doi: 10.1161/01.res.0000061570.83105.52
Tobacman, L. S. (1996). Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 58, 447–481. doi: 10.1146/annurev.ph.58.030196.002311
Tomalka, A., Rohrle, O., Han, J. C., Pham, T., Taberner, A. J., and Siebert, T. (2019). Extensive eccentric contractions in intact cardiac trabeculae: revealing compelling differences in contractile behaviour compared to skeletal muscles. Proc. Biol. Sci. 286:20190719. doi: 10.1098/rspb.2019.0719
Ueda, K., Valdivia, C., Medeiros-Domingo, A., Tester, D. J., Vatta, M., Farrugia, G., et al. (2008). Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. U.S.A. 105, 9355–9360. doi: 10.1073/pnas.0801294105
Valencik, M. L., Zhang, D., Punske, B., Hu, P., Mcdonald, J. A., and Litwin, S. E. (2006). Integrin activation in the heart: a link between electrical and contractile dysfunction? Circ. Res. 99, 1403–1410. doi: 10.1161/01.res.0000252291.88540.ac
van Deel, E. D., Najafi, A., Fontoura, D., Valent, E., Goebel, M., Kardux, K., et al. (2017). In vitro model to study the effects of matrix stiffening on Ca(2+) handling and myofilament function in isolated adult rat cardiomyocytes. J. Physiol. 595, 4597–4610. doi: 10.1113/jp274460
van der Wees, C. G., Bax, W. H., Van Der Valk, E. J., and Van Der Laarse, A. (2006). Integrin stimulation induces calcium signalling in rat cardiomyocytes by a NO-dependent mechanism. Pflug. Arch. 451, 588–595. doi: 10.1007/s00424-005-1402-x
Van Wagoner, D. R. (1993). Mechanosensitive gating of atrial ATP-sensitive potassium channels. Circ Res 72, 973–983. doi: 10.1161/01.res.72.5.973
Vielma, A. Z., Leon, L., Fernandez, I. C., Gonzalez, D. R., and Boric, M. P. (2016). Nitric oxide synthase 1 modulates basal and beta-adrenergic-stimulated contractility by rapid and reversible redox-dependent S-Nitrosylation of the Heart. PLoS One 11:e0160813. doi: 10.1371/journal.pone.0160813
Vila-Petroff, M. G., Younes, A., Egan, J., Lakatta, E. G., and Sollott, S. J. (1999). Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ. Res. 84, 1020–1031. doi: 10.1161/01.res.84.9.1020
von Anrep, G. (1912). On the part played by the suprarenals in the normal vascular reactions of the body. J. Physiol. 45, 307–317. doi: 10.1113/jphysiol.1912.sp001553
Waldman, L. K., Fung, Y. C., and Covell, J. W. (1985). Transmural myocardial deformation in the canine left ventricle. Normal in vivo three-dimensional finite strains. Circ. Res. 57, 152–163. doi: 10.1161/01.res.57.1.152
Wang, H., Viatchenko-Karpinski, S., Sun, J., Gyorke, I., Benkusky, N. A., Kohr, M. J., et al. (2010). Regulation of myocyte contraction via neuronal nitric oxide synthase: role of ryanodine receptor S-nitrosylation. J. Physiol. 588, 2905–2917. doi: 10.1113/jphysiol.2010.192617
Wang, N., Butler, J. P., and Ingber, D. E. (1993). Mechanotransduction across the cell surface and through the cytoskeleton. Science 260, 1124–1127. doi: 10.1126/science.7684161
Wang, S., Chennupati, R., Kaur, H., Iring, A., Wettschureck, N., and Offermanns, S. (2016). Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Invest. 126, 4527–4536. doi: 10.1172/jci87343
Ward, C. W., Prosser, B. L., and Lederer, W. J. (2014). Mechanical stretch-induced activation of ROS/RNS signaling in striated muscle. Antioxid Redox Signal. 20, 929–936. doi: 10.1089/ars.2013.5517
Weber, K. T., Janicki, J. S., Shroff, S. G., Pick, R., Chen, R. M., and Bashey, R. I. (1988). Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ. Res. 62, 757–765. doi: 10.1161/01.res.62.4.757
Wehling-Henricks, M., Jordan, M. C., Roos, K. P., Deng, B., and Tidball, J. G. (2005). Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet. 14, 1921–1933. doi: 10.1093/hmg/ddi197
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70.
Wong, T. Y., Juang, W. C., Tsai, C. T., Tseng, C. J., Lee, W. H., Chang, S. N., et al. (2018). Mechanical Stretching Simulates Cardiac Physiology and Pathology through Mechanosensor Piezo1. J. Clin. Med. 7:410. doi: 10.3390/jcm7110410
Wu, X., Mogford, J. E., Platts, S. H., Davis, G. E., Meininger, G. A., and Davis, M. J. (1998). Modulation of calcium current in arteriolar smooth muscle by alphav beta3 and alpha5 beta1 integrin ligands. J. Cell Biol. 143, 241–252. doi: 10.1083/jcb.143.1.241
Xu, K. Y., Huso, D. L., Dawson, T. M., Bredt, D. S., and Becker, L. C. (1999). Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 96, 657–662. doi: 10.1073/pnas.96.2.657
Yoshigi, M., Hoffman, L. M., Jensen, C. C., Yost, H. J., and Beckerle, M. C. (2005). Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J. Cell Biol. 171, 209–215. doi: 10.1083/jcb.200505018
Zemljic-Harpf, A., Manso, A. M., and Ross, R. S. (2009). Vinculin and talin: focus on the myocardium. J. Investig. Med. 57, 849–855. doi: 10.2310/jim.0b013e3181c5e074
Zemljic-Harpf, A. E., Miller, J. C., Henderson, S. A., Wright, A. T., Manso, A. M., Elsherif, L., et al. (2007). Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol. Cell Biol. 27, 7522–7537. doi: 10.1128/mcb.00728-07
Zhan, R., Wang, F., Wu, Y., Wang, Y., Qian, W., Liu, M., et al. (2018). Nitric oxide induces epidermal stem cell de-adhesion by targeting integrin beta1 and Talin via the cGMP signalling pathway. Nitric Oxide 78, 1–10. doi: 10.1016/j.niox.2018.04.001
Zhang, J. S., Kraus, W. E., and Truskey, G. A. (2004). Stretch-induced nitric oxide modulates mechanical properties of skeletal muscle cells. Am. J. Physiol. Cell Physiol. 287, C292–C299.
Zhang, Y. H., Zhang, M. H., Sears, C. E., Emanuel, K., Redwood, C., El-Armouche, A., et al. (2008). Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ. Res. 102, 242–249. doi: 10.1161/circresaha.107.164798
Zhao, H. C., Agula, H., Zhang, W., Wang, F., Sokabe, M., and Li, L. M. (2010). Membrane stretch and cytoplasmic Ca2+ independently modulate stretch-activated BK channel activity. J. Biomech. 43, 3015–3019. doi: 10.1016/j.jbiomech.2010.06.018
Zhuang, J., Yamada, K. A., Saffitz, J. E., and Kleber, A. G. (2000). Pulsatile stretch remodels cell-to-cell communication in cultured myocytes. Circ. Res. 87, 316–322. doi: 10.1161/01.res.87.4.316
Keywords: nitric oxide, nitric oxide synthase, cytoskeleton, cellular stress, mechano-electrical transduction, stretch, cardiomyocyte, calcium
Citation: Boycott HE, Nguyen M-N, Vrellaku B, Gehmlich K and Robinson P (2020) Nitric Oxide and Mechano-Electrical Transduction in Cardiomyocytes. Front. Physiol. 11:606740. doi: 10.3389/fphys.2020.606740
Received: 15 September 2020; Accepted: 23 November 2020;
Published: 15 December 2020.
Edited by:
T. Alexander Quinn, Dalhousie University, CanadaReviewed by:
Remi Peyronnet, University of Freiburg, GermanyYe Chen-Izu, University of California, Davis, United States
Copyright © 2020 Boycott, Nguyen, Vrellaku, Gehmlich and Robinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannah E. Boycott, aGFubmFoLmJveWNvdHRAY2FyZGlvdi5veC5hYy51aw==; aGFubmFoYm95Y290dEBob3RtYWlsLmNvbQ==; Paul Robinson, cGF1bHJAd2VsbC5veC5hYy51aw==