
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 23 December 2020
Sec. Vascular Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.602930
This article is part of the Research TopicCell Communication in Vascular BiologyView all 16 articles
 Kjestine Schmidt1,2
Kjestine Schmidt1,2 Cor de Wit1,2*
Cor de Wit1,2*The endothelium controls vascular tone adopting blood flow to tissue needs. It releases chemical mediators [e.g., nitric oxide (NO), prostaglandins (PG)] and exerts appreciable dilation through smooth muscle hyperpolarization, thus termed endothelium-dependent hyperpolarization (EDH). Initially, EDH was attributed to release of a factor, but later it was suggested that smooth muscle hyperpolarization might be derived from radial spread of an initial endothelial hyperpolarization through heterocellular channels coupling these vascular cells. The channels are indeed present and formed by connexins that enrich in gap junctions (GJ). In vitro data suggest that myoendothelial coupling underlies EDH-type dilations as evidenced by blocking experiments as well as simultaneous, merely identical membrane potential changes in endothelial and smooth muscle cells (SMCs), which is indicative of coupling through ohmic resistors. However, connexin-deficient animals do not display any attenuation of EDH-type dilations in vivo, and endothelial and SMCs exhibit distinct and barely superimposable membrane potential changes exerted by different means in vivo. Even if studied in the exact same artery EDH-type dilation exhibits distinct features in vitro and in vivo: in isometrically mounted vessels, it is rather weak and depends on myoendothelial coupling through connexin40 (Cx40), whereas in vivo as well as in vitro under isobaric conditions it is powerful and independent of myoendothelial coupling through Cx40. It is concluded that EDH-type dilations are distinct and a significant dependence on myoendothelial coupling in vitro does not reflect the situation under physiologic conditions in vivo. Myoendothelial coupling may act as a backup mechanism that is uncovered in the absence of the powerful EDH-type response and possibly reflects a situation in a pathophysiologic environment.
The endothelium contributes importantly to the regulation of vascular tone and thereby controls organ perfusion. Endothelial cells (ECs) communicate in different languages (or signaling pathways) to smooth muscle cells (SMCs), one of which is the release of chemical substances or mediators that diffuse through the narrow intercellular space before reaching membrane or intracellular proteins as receptors. The outstanding molecules acting as chemical mediators in this pathway of intercellular communication are PG [cyclooxygenase (COX) products] and the gaseous transmitter nitric oxide (NO) which is produced by endothelial NO-synthase and easily diffuses through plasma membranes to reach its target, the intracellularly localized soluble guanylyl cyclase (sGC; Qian and Fulton, 2013; Luo et al., 2016; Nava and Llorens, 2019). Apart from these molecules, other chemical entities have been suggested to be released from ECs and relax smooth muscle, among them K+ ions, lipophilic compounds (e.g., epoxyeicosanoids, EETs), proteins (C-type natriuretic peptide), radicals [hydrogen peroxide (H2O2)], and other gaseous molecules [CO, hydrogen sulfide (H2S); Figure 1; Feletou and Vanhoutte, 2009; Garland et al., 2011; Feletou et al., 2012; Ellinsworth et al., 2014, 2016; Feletou, 2016; Freed and Gutterman, 2017; Garland and Dora, 2017].
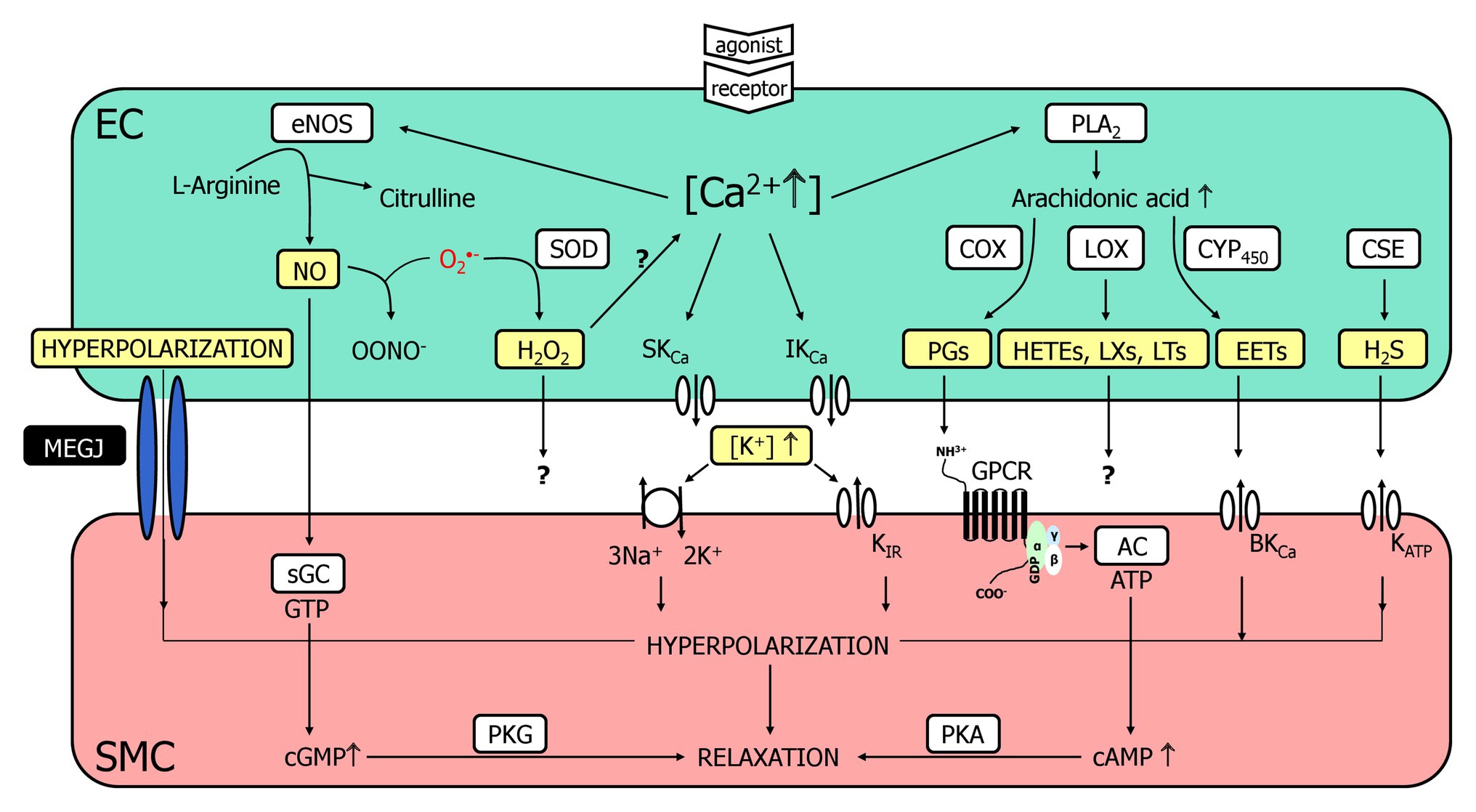
Figure 1. A multitude of endothelial molecules and mechanisms (depicted in yellow boxes) relax vascular smooth muscle. The well-accepted chemical mediators such as nitric oxide (NO) and prostaglandins (PG) are shown, and a variety of substances that have been suggested to underly the endothelium-dependent hyperpolarization (EDH)-type dilation. Importantly, a completely distinct pathway, namely radial electrotonic spread of endothelial hyperpolarizations through myoendothelial coupling was also proposed to initiate the EDH-type dilation. However, the evidence was mostly acquired in vitro, whereas data obtained in vivo argue against this hypothesis. This justifies reconsideration of other molecules as outlined. For further details, see text. AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; COX, cyclooxygenase; CSE, cystathionine-lyase; CYP450, cytochrome P450 epoxygenase; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; EETs, epoxyeicosatrienoic acids; GPCR, G-protein coupled receptor; HETEs, hydroxyeicosatrienoic acids; H2O2, hydrogen peroxide; H2S, hydrogen sulfide; KCa, Ca2+-activated K+-channel with small (SKCa), intermediate (IKCa) or big conductance (BKCa); KATP, ATP-sensitive K+-channels; KIR, inwardly rectifying K+-channel; LOX, lipoxygenase; LTs, leukotrienes; LXs, lipoxines; MEGJ, myoendothelial gap junctions; NO, nitric oxide; OONO−, peroxynitrite anion; O2−, superoxide anion; PGs, prostaglandins; PKA, protein kinase A; PKG, protein kinase G; PLA2, phospholipase A2; sGC, soluble guanylyl cyclase; SMC, smooth muscle cell; SOD; superoxide dismutase.
However, this is a “sticky” business as it is difficult to imagine that several of these compounds are easily and quickly transferable. The smooth muscle relaxes by the opening of K+-channels, which induce relaxation likely through closure of voltage-gated Ca2+-channels representing a rather speedy signaling pathway. Since the dilaton is depending on the presence of ECs and characterized by smooth muscle hyperpolarization, this obscure mediator has been termed endothelium-derived hyperpolarizing factor (EDHF) assuming that also this mechanism communicates in the same language, i.e., release of a mediator as for the classical autacoids, NO, and PG. However, this view was challenged and a direct electrotonic way of communication was suggested. Charge is transferred from ECs to smooth muscle through intercellular channels [myoendothelial gap junctions (GJs), MEGJs] driven by an initial endothelial hyperpolarization that spreads radially into the vessel wall (Figure 1; de Wit and Griffith, 2010; Freed and Gutterman, 2017; Garland and Dora, 2017).
We describe in this short review the structural requirements and illuminate the experimental evidence obtained in vivo that argues against or in favor of the hypothesis that EDHF indeed is an EDH-type dilation rather than being a freely diffusible molecule. Other important functions of myoendothelial coupling, including the transfer of calcium or intracellular messengers, such as inositol-1,4,5-triphosphate (IP3) that modulate vascular responses (Looft-Wilson et al., 2017; Biwer et al., 2018; Wei et al., 2018; Wilson et al., 2019) are reviewed in detail elsewhere (Billaud et al., 2014; Straub et al., 2014; Pogoda and Kameritsch, 2019; Pohl, 2020) and are not addressed in detail in this manuscript. This aspect is mainly examined in vitro and due to the lack of experimental data, it is currently unknown if such a signaling pathway is of importance in vivo.
Gap junctions are composed of connexin proteins and six of these molecules assemble to form a hexameric hemichannel. Two of them from adjacent cells dock together and interlink the cytoplasms of these cells through a channel that is tightly sealed against the extracellular environment. The intercellular channels either connect equal (homocellular) or different cell types (heterocellular, e.g., myoendothelial coupling) and allow transfer of small molecules (< 1 kDa) and ions. The latter equals current flow along an electrochemical potential difference and, ultimately, permits the transfer of membrane potential changes between interconnected cells. Hundreds of such channels are located at GJ where adjoining plasma membranes are only 2 nm apart (Saez et al., 2003; Nielsen et al., 2012; Grosely and Sorgen, 2013). GJ can be visualized by electron microscopy and appears as an electron-dense pentalaminar area that can be easily identified between adjacent ECs because of their abundance and the large size of the plaques. They are found to a lesser extent between SMCs, which is possibly due to the smaller plaque size and the limited number of GJ (Sandow et al., 2012). Alternatively, the presence of GJ may be implicated from the demonstration of connexin protein expression by immunostaining. A tremendous amount of publications detected connexins in vascular cells (ECs and SMCs) but this imaging technique does not allow to distinguish homocellular GJ from heterocellular GJ (de Wit, 2004; Figueroa et al., 2004). Moreover, connexin staining is indicated in many cases but the demonstration of a proper functional localization, i.e., inside the plasma membrane, is lacking.
The family of connexin genes is comprised of ~20 members (in humans 21, in mice 20), and the proteins are named according to their theoretical molecular mass; for example, connexin40 (Cx40) has a predicted molecular mass of about 40 kDa (Sohl and Willecke, 2004). In the cardiovascular system, Cx40, Cx37, Cx43, and Cx45 are expressed (Haefliger et al., 2004; Morel, 2014). Their expression pattern is not specific for ECS or SMCs, but a subtype preponderance was found: Cx40 and Cx37 prevail in the endothelium throughout the vascular tree and reportedly Cx43 is also expressed specifically in larger vessels (Gustafsson et al., 2003; Sandow et al., 2003; Rummery and Hill, 2004; de Wit et al., 2006; Haddock et al., 2006; Hakim et al., 2008; Sorensen and Holstein-Rathlou, 2012; Pohl, 2020). Cx43 and Cx45 are the dominant subtypes expressed in SMCs (Hill et al., 2002; Wolfle et al., 2007; Schmidt et al., 2012), but Cx37 was also reported for smooth muscle (Sandow et al., 2003; Alonso et al., 2010a), whereas Cx40 is only found in a very few instances in these cells. Interestingly, Cx40 and Cx37 are also found abundantly in renin producing cells and serve important functions in renin secretion (Wagner and Kurtz, 2013).
Having the expression pattern in mind, MEGJ may be composed of Cx40, Cx37, and Cx43 provided from the endothelial side and Cx43, Cx45, and Cx37 from smooth muscle (Sandow et al., 2009, 2012). This important information is experimentally difficult to obtain in vessels, however, in an in vitro co-culture system, myoendothelial junctions were composed of Cx40 and Cx43 provided by both cell types. This occured despite the expression of Cx37 in endothelial and SMCs in this artificial system but this connexin was excluded from these junctions (Isakson and Duling, 2005). Experimental data from intact vessels are scarce: in rat basilar artery using serial section and immunoelectron microscopy, Haddock et al. (2006) demonstrated myoendothelial junctions in which Cx40 and Cx37 were located, others confirmed the presence of Cx37 and Cx40 in different vessels (Mather et al., 2005; Isakson et al., 2008), and also Cx43 was identified at MEGJ (Straub et al., 2010; Pogoda et al., 2014).
Recently, Cx37 was shown to be enriched specifically at sites where endothelial and SMCs come into contact (the internal elastic lamina) in murine small skeletal muscle arteries. Functionally, the authors observed in a culture system, a modulation of Cx37 permeability by NO specifically for this connexin (compared to Cx40 and Cx43) and conclude from these findings that NO exerts a specific modulatory effect on myoendothelial junctions due to the fact that Cx37 is the main Cx at this strategic location, and NO preferentially affects its function (Pogoda et al., 2014). The inhibitory effect of NO on Ca2+ permeability of MEGJ was elicited through enhanced phosphorylation of Cx37 at position 332 (Y332) by inhibition of the protein tyrosine phosphatase SHP-2. This effect prevented in isolated vessels the Ca2+ drainage from the endothelium into the smooth muscle and enhanced thereby Ca2+ levels in ECs promoting vasodilation induced by vasoactive agonists (Pogoda et al., 2017). Taken together, these data are somewhat contradictory (Cx37) and can be only regarded as an initial step on the bumpy road with many experimental obstacles to uncover the subtypes of Cx being localized at the MEGJ.
Direct injection of electrical current into ECs in isolated arterioles evoked endothelial hyperpolarizations that were conducted electrotonically (i.e., passively) to subjacent SMCs and, conversely, action potentials originating in the arterial media were rapidly conducted to the endothelium (Emerson and Segal, 2000, 2001; Yamamoto et al., 2001). The signal amplitude was reduced in either direction by about 15%, without change in form, suggesting that myoendothelial junctions behave as ohmic resistors that mediate the heterocellular spread of membrane potential changes without rectification (Yamamoto et al., 2001). Circumferential and subsequent longitudinal spread of hyperpolarization was also demonstrated in conduit vessels (strips of rabbit iliac and porcine coronary artery), from which the endothelium had been partly removed, following hyperpolarization of the residual endothelium (Griffith et al., 2002; Selemidis and Cocks, 2007). A direct heterocellular communication invoking larger molecules was visualized using dyes that diffused from initially loaded ECs into SMCs in the media via GJs (Griffith et al., 2002; Kansui et al., 2008; Saliez et al., 2008; Billaud et al., 2009).
If such molecules are able to diffuse through GJ it opens the possibility that non-electrotonic mechanisms contribute to vascular tone regulation, e.g., diffusion of IP3 and/or Ca2+ ions in either direction (Isakson et al., 2007; Isakson, 2008). For example, the transfer of Ca2+ from activated smooth muscle to endothelium has been implicated in a feedback mechanism that evokes an endothelial dilation, thereby counterbalancing the constriction initiated in the smooth muscle by e.g., phenylephrine (Dora et al., 1997; Yashiro and Duling, 2000; Kerr et al., 2012; Biwer et al., 2018; Wei et al., 2018; Wilson et al., 2019). This feedback is tightly controlled (Straub et al., 2012; Biwer et al., 2018) and, interestingly, the artificial modulation of heterocellular contact in larger arteries may alter their vascular function (Shu et al., 2019). The hypothesis that myoendothelial junctions serve as conduits for charge transfer and represent mechanistically the EDH-type dilation instead of an EDHF that traverses the extracellular space has been reviewed excellently in more detail elsewhere (Griffith, 2004; Figueroa and Duling, 2009; de Wit and Griffith, 2010; Garland et al., 2011; Ellinsworth et al., 2014; Freed and Gutterman, 2017; Garland and Dora, 2017).
Proof of concept additionally requires blockage of the gap junctional pathway and the abrogation of EDH-type responses. However, GJs are difficult to block efficiently and comcomitantly avoid nonspecific effects. Application of short interfering peptides (so-called connexin-mimetic peptides) is a useful strategy that provides even certain specificity for different connexin subtypes depending on the amino acid sequences of the peptide. In fact, numerous studies have shown that they are able to suppress EDHF-type smooth muscle hyperpolarizations and dilations in vitro (Griffith, 2004; de Wit and Griffith, 2010). In some vessels, connexin-mimetic peptides targeted against Cx37 and/or Cx40 attenuated endothelium-dependent smooth muscle hyperpolarization (Chaytor et al., 2005), whereas in other arteries, peptide combinations to target Cx37, Cx40, and Cx43 collectively were required to block of EDH-type responses (Chaytor et al., 2001; Matchkov et al., 2006).
Aiming at the protein structure with a blocking antibody provides a distinct, very specific approach to verify the functional necessity of connexins. Mather et al. (2005) loaded ECs with antibodies targeted against the cytoplasmic tail of Cx40 and observed an abrogation of EDH-type responses in rat mesenteric small arteries. This occurred without affecting increases in endothelial calcium and, importantly, antibodies directed against Cx37 and Cx43 were inactive suggesting that Cx40 is a key player in the response (Mather et al., 2005). The endothelium opposes myogenic constrictions through NO release (de Wit et al., 1998) and EDH-type dilations possibly through Ca2+ transfer from smooth muscle to endothelium as outlined above. The latter opposing mechanism was abrogated in mice carrying a mutant Cx40 protein, which also impaired chemical coupling (Chaston et al., 2013).
Taken together, there is compelling evidence that myoendothelial coupling through connexins provides a functional pathway that allows electrotonic spread of membrane potential changes (and signaling molecules). Blockade of this pathway attenuates or even abrogates EDH-type dilations in vitro. Specifically, Cx40 (and Cx37) seem to be invoked and appear to be a required brick of the intercellular signaling pathway.
Collecting evidence in vivo is more cumbersome; however, an efficient strategy is the use of mice deficient for connexins. We published the first study on vascular connexin function by using Cx40-deficient mice in which we demonstrated a crucial role for Cx40 to conduct endothelium-dependent dilations longitudinally along the vessel wall, thereby coordinating vascular cellular behavior (de Wit et al., 2000; Wolfle et al., 2007) that was shortly later confirmed by Figueroa et al. (2003). However, Cx40 is not required for radial electrotonic signal transmission because acetylcholine-induced EDH-type dilations were nearly preserved in skeletal muscle arterioles studied by intravital microscopy in anaesthestized Cx40-deficent mice. Later, we re-examined these responses in more detail and demonstrated a very minor (if any) attenuation of EDH-type dilations upon acetylcholine (Milkau et al., 2010).
In awake Cx40-deficient mice, intraarterial acetylcholine reduced blood pressure to the same absolute level as in wildtype controls, despite their initially elevated arterial pressure (de Wit et al., 2003). This hypotensive response can be attributed to the EDHF phenomenon because it is unaffected by NO-synthase inhibition and is preserved in mice with targeted disruption of the NO-cyclic guanosine monophosphate (cGMP) pathway (Koeppen et al., 2004). Furthermore, resistance changes in the renal circulation in response to acetylcholine were comparable in wildtype and Cx40-deficient mice (Just et al., 2009).
Collectively, these results argue against the hypothesis that Cx40 is universally required for EDH-type signaling in vivo. These differences may relate indeed due to in vivo vs. in vitro vascular behavior or caused by different vessel sizes. Arteries studied in vitro are usually larger (> 100 μm) than arterioles accessible for intravital microscopy examined in vivo (20–50 μm). The latter arterioles are those that provide the vascular resistance, and their behavior is revealed by pressure drop or resistance measurements in intact organs or animals.
To exclude effects related to vessel size differences, we performed measurements on a small murine artery (gracilis artery) that exhibits a diameter (~130 μm) allowing in vitro as well as in vivo examination because the artery is located at a spot that is accessible for intravital microscopy. This small artery exhibits in vitro an acetylcholine-induced dilation that is mediated to a considerable part by an EDH-type mechanism (~60%, the remaining portion is mediated by NO and PG). Thus, the EDH-type dilation is larger than in a conducting artery (femoral artery), which relies mostly on NO but it is smaller than in arterioles. Arteriolar responses in the microcirculation in vivo are nearly exclusively mediated by EDH-type dilation (Si et al., 2006; Brahler et al., 2009; Wolfle et al., 2009).
In any case, acetylcholine-induced dilations were attenuated in gracilis arteries harvested from Cx40-deficient mice compared to wildtype mice if studied isometrically in vitro using wire myography. Blockade of NO-synthase and COX nearly abrogated the dilation in Cx40-deficient arteries but uncovered EDH-type dilations in wildtype mice. This demonstrates that EDH-type dilations require Cx40 in this setting and suggest that myoendothelial coupling requiring Cx40 supports this pathway (Figure 2, left; Boettcher and de Wit, 2011). However, if the exact same vessel was examined at isobaric conditions (pressure myograph), the dilation upon acetylcholine was firstly not attenuated by NO-synthase and COX inhibition in wildtype and secondly fully preserved in Cx40-deficient vessels. This demonstrates that EDH-type dilations, firstly, are more powerful at these experimental conditions (isobaric study) and, secondly, are fully preserved in the absence of Cx40 and, thus, most likely independent of myoendothelial coupling. The exact same results were found if this vessel was studied in the anesthetized mouse in vivo (Figure 2, left panel; Boettcher and de Wit, 2011).
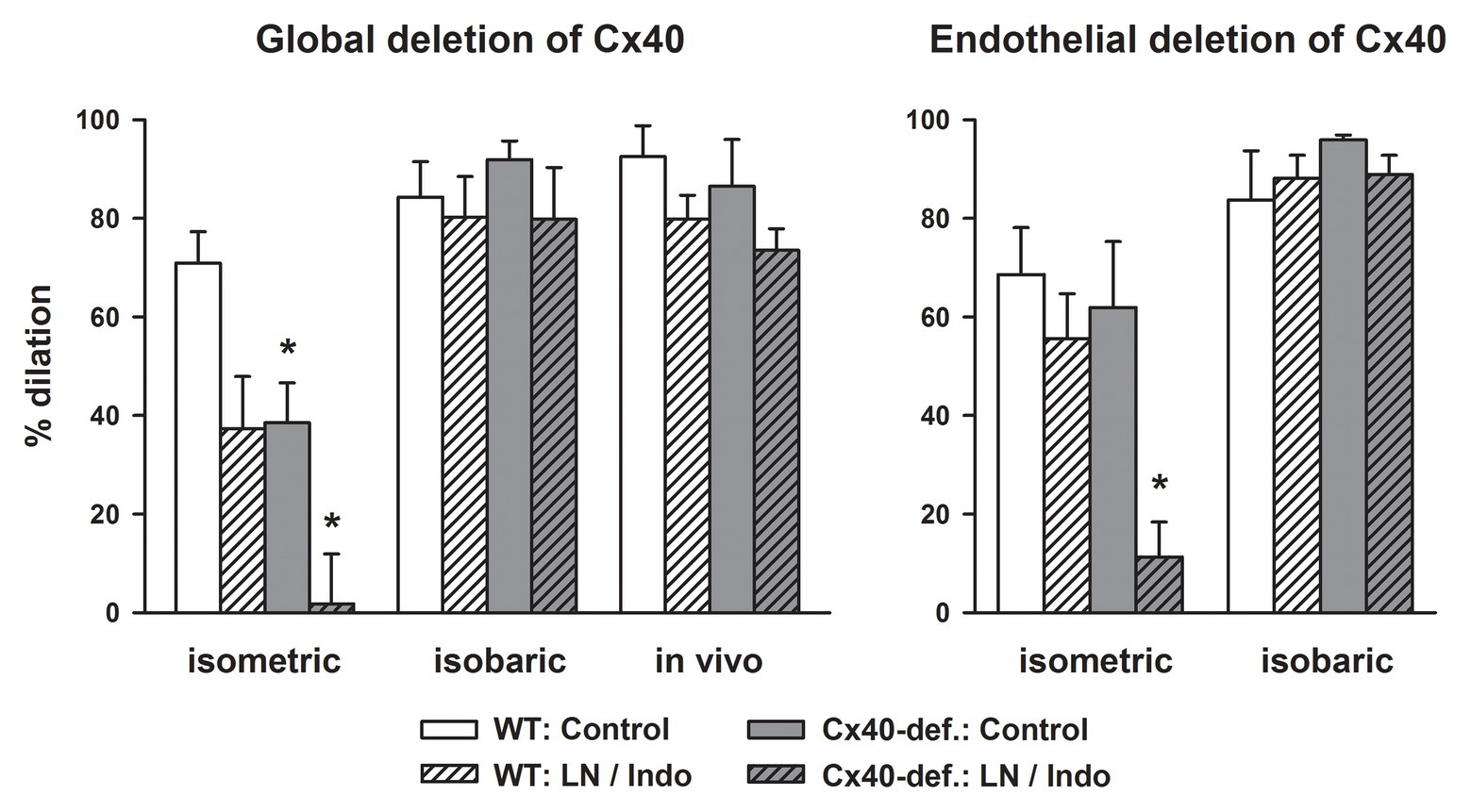
Figure 2. EDH-type dilations are abrogated in gracilis arteries in the absence of connexin40 (Cx40) only if studied in vitro in an isometric setting. Acetylcholine (3 μM) induced dilations in gracilis artery studied in vitro either in a wire- (isometric) or a pressure-myograph (isobaric) or in vivo in anesthetized mice. Cx40 was either deleted globally (left) or in an endothelial-specific manner (right) and compared to respective wildtype mice. Under isometric conditions, blockade of NO-synthase and COX reduced the response in wildtype and abrogated it in Cx40-deficient mice demonstrating a dependence of EDH-type dilations on Cx40 and supposedly on myoendothelial coupling in this setting. In marked contrast, EDH-type dilations were completely preserved in Cx40-deficient mice under isobaric conditions and in vivo. Thus, under these conditions, EDH-type dilations are independent of the presence of Cx40. Interestingly, EDH was more powerful under these experimental conditions and induced near full dilation in all genotypes. * indicates p < 0.05 vs. wildtype; LN/Indo, L-nitro-arginine (300 μM) and indometacin (3 μM) to block NO-synthase and COX; Cx40-def, Cx40-deficient mice either global or endothelial-cell specific deletion using Cre recombinase driven by TIE2 in mice carrying homozygously a Cx40 floxed gene. The full data set is published (Boettcher and de Wit, 2011).
In line with this, an attenuation of the EDH-type dilation in Cx40-deficient vessels was also observed in renal arteries (interlobar vessels) that were studied isometrically in vitro using wire myography. In addition to myoendothelial coupling, endothelial K+ release contributed to the EDH-type dilation in these vessels since a blockade of inwardly rectifying K+-channel (KIR) and the sodium pump reduced the responses in both, wildtype and Cx40-deficient mice (Brasen et al., 2018). Although the responses were not studied in vivo in this study, acetylcholine infusion decreased vascular resistance in the renal circulation measured in vivo to a comparable degree in wildtype and Cx40-deficient mice (Just et al., 2009) indicating again intact EDH-type dilations in Cx40-deficient mice in vivo.
Since mice globally deficient for Cx40 are hypertensive due to an abrogated feedback of pressure on renin release (Wagner et al., 2007, 2010; Schweda et al., 2009), hypertension might be a causative factor in impairing the EDH-type dilation through modifying myoendothelial junctions by an alteration of connexin expression (Figueroa et al., 2006; Meens et al., 2013). Therefore, we examined arteries from mice with EC-specific deletion of Cx40, which are normotensive (Wagner et al., 2010; Jobs et al., 2012). However, arteries exhibited similar characteristics: The acetylcholine-induced dilation in isometrically mounted vessels was abrogated after the blockade of NO synthase and COX in the absence of endothelial Cx40 but not in wildtype vessels. This confirmed an EDH-type dilation in wildtype vessels that is not found in the absence of endothelial Cx40 and indicates that this part of the acetylcholine-response is dependent on myoendothelial coupling in this setting.
In marked contrast, a fully intact EDH-type dilation upon acetylcholine was found in vessels from endothelial-specific Cx40-deficient mice if mounted isobarically (Figure 2, right; Boettcher and de Wit, 2011). As before, the EDH-type dilation in the isobaric setting was more powerful in both genotypes, and an attenuation of the response after NO synthase and COX inhibition was neither observed in wildtype vessels nor in vessels deficient for endothelial Cx40 (Figure 2, right). This suggests that this EDH-type dilation, which was only observed in the isobaric setting or in vivo, is independent of Cx40. Furthermore, this powerful response is even capable to replace the dilatory function of other endothelial mediators (mainly NO). Importantly, EC-specific Cx40-deficient mice are not hypertensive (Wagner et al., 2010; Jobs et al., 2012) excluding that hypertension modulates connexin expression and that such an alteration had caused the defects observed in globally Cx40-deficient mice.
The divergent results observed in isobaric or isometric vessel study are striking. A key experimental difference is the change in wall tension during constriction and dilation. In isometric conditions, wall tension decreases during dilations and rises during constriction whereas the opposite is true for isobaric (and in vivo) conditions. These different experimental setups have also shown to modulate the sensitivity of rat mesenteric arteries for vasoconstrictors (Buus et al., 1994). This may be related to changes in membrane potential because a small initial depolarization did offset the differences in agonist sensitivity (Dunn et al., 1994). Modulatory effects of experimental conditions were also demonstrated on EDH-type dilations: Lowering the extracellular Ca2+ concentration shifted the type of endothelial KCa channel initiating the EDH response from KCa2.3 to KCa3.1. This also affected the link to the vessel relaxation, which shifted from myoendothelial coupling to the activation of the Na+/K+ ATPase, thus defining distinct EDHF pathways depending on experimental conditions (Dora et al., 2008).
Since ECs express both connexins, Cx40 and Cx37, in abundant amounts, the question arises why Cx37 is not able to replace the function of Cx40. Contrary to an anticipated compensatory upregulation in Cx40-deficient vessels, Cx37 expression is actually downregulated in the aortic endothelium (Simon and McWhorter, 2003), and Cx37 cannot be detected by immunostaining in arterioles (de Wit, 2010; Jobs et al., 2012). Recent work identified spare Cx37 expression in skeletal muscle arterioles and, in addition, demonstrated the presence of myoendothelial junctions in Cx40-deficient mice (Howitt et al., 2013). Interestingly, Cx40 and Cx37 are located tightly together suggesting physical interaction (Alonso et al., 2010b). Thus, Cx40 seems to be required to locate Cx37 appropriately in the endothelial plasma membrane, and a potentially remaining Cx37 expression is functionally insufficient. Conversely, deficiency of Cx37 did not result in noticeable defects of vascular regulation (Figueroa and Duling, 2008) or renin secretion (Wagner et al., 2009) suggesting that the role of Cx37 can be replaced by Cx40.
Acetylcholine activates muscarinic G-protein coupled receptors (GPCRs) that initiate a Ca2+-dependent signaling pathway ultimately leading to endothelial hyperpolarization by means of the activation of endothelial Ca2+-dependent K+-channels (KCa3.1, KCa2.3; Brahler et al., 2009). This last element in the cascade can be activated pharmacologically by compounds, such as SKA-31 (Sankaranarayanan et al., 2009). In fact, SKA-31 exhibits a higher sensitivity toward KCa3.1 and produces arteriolar dilation in vivo specifically via activation of this endothelial ion channel and the ensuing endothelial hyperpolarization supposedly occurs without increases of endothelial Ca2+. This even more “pure” EDH-type dilation is likewise unimpeded in Cx40-deficient arterioles examined in the microcirculation in vivo (Radtke et al., 2013). Similarly, the arterial pressure decreases upon systemic application of SKA-31 were not attenuated in either global or endothelial-specific Cx40-deficient mice (Radtke et al., 2013). This verifies that endothelial hyperpolarization through activation of KCa3.1 initiates an EDH-type dilation which is independent of Cx40 in vivo and thus likely independent of myoendothelial junctions.
Taken together, a strong powerful EDH-type dilation exists in vivo that acts independent of Cx40. We suggest that we need to reconsider the “sticky” business, which may not be so sticky at all in search for this mediator (Figure 1). Importantly, this powerful mechanism is lost in vitro in certain experimental settings (isometric setup). At such conditions, myoendothelial coupling may take over and provide a remaining, less powerful EDH-type dilation that is, in fact, dependent on Cx40.
Having provided evidence that lack of Cx40 (and Cx37) leaves EDH-type dilations fully intact in vivo it remains to be considered if myoendothelial coupling exists in vivo at all. It may well be that heterocellular coupling is turned off during physiologic vascular control and is only activated during special (pathophysiologic?) conditions. In contrast to the findings with isolated arteries and arterioles described above, a number of studies have failed to demonstrate tight electric myoendothelial coupling in arterioles in vivo (de Wit et al., 2008). For example, electrophysiological measurements in vivo demonstrated differences in the resting membrane potential of endothelial and SMCs in the order of 10 mV in murine skeletal muscle arterioles (Siegl et al., 2005). Additionally, acetylcholine-induced hyperpolarization of SMCs was selectively blocked by a specific K+-channel blocker without modifying endothelial hyperpolarization (Siegl et al., 2005). Conversely, adenosine dilated arterioles by activating smooth muscle KATP channels but did not hyperpolarize the endothelium (de Wit, 2010); whereas in isolated conduit arteries, KATP mediated hyperpolarization is transmitted to the endothelium via MEGJ (Murai et al., 1999). Also in hamster arterioles in vivo, smooth muscle depolarizations did not affect the membrane potential of ECs: hyperpolarizations initiated by acetylcholine differed in amplitude and form between endothelial and SMCs (Welsh and Segal, 1998). Again, endothelial and smooth muscle hyperpolarization exhibited differential sensitivity to inhibitors (Welsh and Segal, 2000). Finally, selective destruction of endothelial or SMCs along the conduction pathway suggests that hyperpolarization is not freely transferred between the two cell types (Budel et al., 2003).
These experimental findings do not provide evidence for functional myoendothelial coupling that aligns the membrane potential of endothelial and SMCs in vivo. The data rather suggest that the membrane potential of these distinct cell populations is regulated independently reflecting their specific functions. Interestingly, connexins at the myoendothelial junctions are subject to functional regulation (Straub et al., 2010; Westcott and Segal, 2013), which may provide an explanation for the discrepant in vivo findings. Although significant charge transfer through MEGJ was not convincingly demonstrated in vivo, significant contributions of myoendothelial coupling to vascular function may well be of importance, for example the transfer of intracellular messengers (Looft-Wilson et al., 2017; Biwer et al., 2018; Wei et al., 2018; Wilson et al., 2019; Pohl, 2020).
Many in vitro studies demonstrate an important role for myoendothelial coupling by transferring charge from endothelial to SMCs to elicit EDH-type dilations. However, in vivo a distinct, more powerful mechanism is responsible for such dilations that easily overcome the lack of myoendothelial coupling. The evidence for efficient charge transfer through myoendothelial coupling in vivo is scarce. This is not related to a lack of data, in fact, many laboratories have attempted to tackle this important question, but failed to find solid evidence documenting myoendothelial coupling. In fact, these efforts have accumulated strong indications for the opposite, i.e., endothelial and SMCs “do their own thing” with respect to membrane potential. However, under certain experimental conditions, myoendothelial coupling supports EDH-type dilation, but this EDH-type response is rather weak compared to the EDH-mechanism acting in vivo.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alonso, F., Boittin, F. X., Beny, J. L., and Haefliger, J. A. (2010b). Loss of connexin40 is associated with decreased endothelium-dependent relaxations and eNOS levels in the mouse aorta. Am. J. Physiol. Heart Circ. Physiol. 299, H1365–H1373. doi: 10.1152/ajpheart.00029.2010
Alonso, F., Krattinger, N., Mazzolai, L., Simon, A., Waeber, G., Meda, P., et al. (2010a). An angiotensin II- and NF-kappaB-dependent mechanism increases connexin 43 in murine arteries targeted by renin-dependent hypertension. Cardiovasc. Res. 87, 166–176. doi: 10.1093/cvr/cvq031
Billaud, M., Lohman, A. W., Johnstone, S. R., Biwer, L. A., Mutchler, S., and Isakson, B. E. (2014). Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol. Rev. 66, 513–569. doi: 10.1124/pr.112.007351
Billaud, M., Marthan, R., Savineau, J. P., and Guibert, C. (2009). Vascular smooth muscle modulates endothelial control of vasoreactivity via reactive oxygen species production through myoendothelial communications. PLoS One 4:e6432. doi: 10.1371/journal.pone.0006432
Biwer, L. A., Good, M. E., Hong, K., Patel, R. K., Agrawal, N., Looft-Wilson, R., et al. (2018). Non-endoplasmic reticulum-based calr (calreticulin) can coordinate heterocellular calcium signaling and vascular function. Arterioscler. Thromb. Vasc. Biol. 38, 120–130. doi: 10.1161/ATVBAHA.117.309886
Boettcher, M., and de Wit, C. (2011). Distinct endothelium-derived hyperpolarizing factors emerge in vitro and in vivo and are mediated in part via connexin 40-dependent myoendothelial coupling. Hypertension 57, 802–808. doi: 10.1161/HYPERTENSIONAHA.110.165894
Brahler, S., Kaistha, A., Schmidt, V. J., Wolfle, S. E., Busch, C., Kaistha, B. P., et al. (2009). Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119, 2323–2332. doi: 10.1161/CIRCULATIONAHA.108.846634
Brasen, J. C., de Wit, C., and Sorensen, C. M. (2018). Myoendothelial coupling through Cx40 contributes to EDH-induced vasodilation in murine renal arteries: evidence from experiments and modelling. Acta Physiol. 222:e12906. doi: 10.1111/apha.12906
Budel, S., Bartlett, I. S., and Segal, S. S. (2003). Homocellular conduction along endothelium and smooth muscle of arterioles in hamster cheek pouch: unmasking an NO wave. Circ. Res. 93, 61–68. doi: 10.1161/01.RES.0000080318.81205.FD
Buus, N. H., VanBavel, E., and Mulvany, M. J. (1994). Differences in sensitivity of rat mesenteric small arteries to agonists when studied as ring preparations or as cannulated preparations. Br. J. Pharmacol. 112, 579–587. doi: 10.1111/j.1476-5381.1994.tb13114.x
Chaston, D. J., Baillie, B. K., Grayson, T. H., Courjaret, R. J., Heisler, J. M., Lau, K. A., et al. (2013). Polymorphism in endothelial connexin40 enhances sensitivity to intraluminal pressure and increases arterial stiffness. Arterioscler. Thromb. Vasc. Biol. 33, 962–970. doi: 10.1161/ATVBAHA.112.300957
Chaytor, A. T., Bakker, L. M., Edwards, D. H., and Griffith, T. M. (2005). Connexin-mimetic peptides dissociate electrotonic EDHF-type signalling via myoendothelial and smooth muscle gap junctions in the rabbit iliac artery. Br. J. Pharmacol. 144, 108–114. doi: 10.1038/sj.bjp.0706046
Chaytor, A. T., Martin, P. E., Edwards, D. H., and Griffith, T. M. (2001). Gap junctional communication underpins EDHF-type relaxations evoked by ACh in the rat hepatic artery. Am. J. Physiol. Heart Circ. Physiol. 280, H2441–H2450. doi: 10.1152/ajpheart.2001.280.6.H2441
de Wit, C. (2004). Connexins pave the way for vascular communication. News Physiol. Sci. 19, 148–153. doi: 10.1152/nips.01520.2004
de Wit, C. (2010). Different pathways with distinct properties conduct dilations in the microcirculation in vivo. Cardiovasc. Res. 85, 604–613. doi: 10.1093/cvr/cvp340
de Wit, C., Boettcher, M., and Schmidt, V. J. (2008). Signaling across myoendothelial gap junctions—fact or fiction? Cell Commun. Adhes. 15, 231–245. doi: 10.1080/15419060802440260
de Wit, C., and Griffith, T. M. (2010). Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers Arch. 459, 897–914. doi: 10.1007/s00424-010-0830-4
de Wit, C., Jahrbeck, B., Schafer, C., Bolz, S. S., and Pohl, U. (1998). Nitric oxide opposes myogenic pressure responses predominantly in large arterioles in vivo. Hypertension 31, 787–794. doi: 10.1161/01.HYP.31.3.787
de Wit, C., Roos, F., Bolz, S. S., Kirchhoff, S., Kruger, O., Willecke, K., et al. (2000). Impaired conduction of vasodilation along arterioles in connexin40 deficient mice. Circ. Res. 86, 649–655. doi: 10.1161/01.RES.86.6.649
de Wit, C., Roos, F., Bolz, S. S., and Pohl, U. (2003). Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol. Genomics 13, 169–177. doi: 10.1152/physiolgenomics.00169.2002
de Wit, C., Wolfle, S. E., and Hopfl, B. (2006). Connexin-dependent communication within the vascular wall: contribution to the control of arteriolar diameter. Adv. Cardiol. 42, 268–283. doi: 10.1159/000092575
Dora, K. A., Doyle, M. P., and Duling, B. R. (1997). Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc. Natl. Acad. Sci. U. S. A. 94, 6529–6534. doi: 10.1073/pnas.94.12.6529
Dora, K. A., Gallagher, N. T., McNeish, A., and Garland, C. J. (2008). Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ. Res. 102, 1247–1255. doi: 10.1161/CIRCRESAHA.108.172379
Dunn, W. R., Wellman, G. C., and Bevan, J. A. (1994). Enhanced resistance artery sensitivity to agonists under isobaric compared with isometric conditions. Am. J. Phys. 266, H147–H155. doi: 10.1152/ajpheart.1994.266.1.H147
Ellinsworth, D. C., Earley, S., Murphy, T. V., and Sandow, S. L. (2014). Endothelial control of vasodilation: integration of myoendothelial microdomain signalling and modulation by epoxyeicosatrienoic acids. Pflugers Arch. 466, 389–405. doi: 10.1007/s00424-013-1303-3
Ellinsworth, D. C., Sandow, S. L., Shukla, N., Liu, Y., Jeremy, J. Y., and Gutterman, D. D. (2016). Endothelium-derived hyperpolarization and coronary vasodilation: diverse and integrated roles of epoxyeicosatrienoic acids, hydrogen peroxide, and gap junctions. Microcirculation 23, 15–32. doi: 10.1111/micc.12255
Emerson, G. G., and Segal, S. S. (2000). Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries—role in vasomotor control. Circ. Res. 87, 474–479. doi: 10.1161/01.RES.87.6.474
Emerson, G. G., and Segal, S. S. (2001). Electrical activation of endothelium evokes vasodilation and hyperpolarization along hamster feed arteries. Am. J. Physiol. Heart Circ. Physiol. 280, H160–H167. doi: 10.1152/ajpheart.2001.280.1.H160
Feletou, M. (2016). Endothelium-dependent hyperpolarization and endothelial dysfunction. J. Cardiovasc. Pharmacol. 67, 373–387. doi: 10.1097/FJC.0000000000000346
Feletou, M., Kohler, R., and Vanhoutte, P. M. (2012). Nitric oxide: orchestrator of endothelium-dependent responses. Ann. Med. 44, 694–716. doi: 10.3109/07853890.2011.585658
Feletou, M., and Vanhoutte, P. M. (2009). EDHF: an update. Clin. Sci. 117, 139–155. doi: 10.1042/CS20090096
Figueroa, X. F., and Duling, B. R. (2008). Dissection of two Cx37-independent conducted vasodilator mechanisms by deletion of Cx40: electrotonic versus regenerative conduction. Am. J. Physiol. Heart Circ. Physiol. 295, H2001–H2007. doi: 10.1152/ajpheart.00063.2008
Figueroa, X. F., and Duling, B. R. (2009). Gap junctions in the control of vascular function. Antioxid. Redox Signal. 11, 251–266. doi: 10.1089/ars.2008.2117
Figueroa, X. F., Isakson, B. E., and Duling, B. R. (2004). Connexins: gaps in our knowledge of vascular function. Physiology 19, 277–284. doi: 10.1152/physiol.00008.2004
Figueroa, X. F., Isakson, B. E., and Duling, B. R. (2006). Vascular gap junctions in hypertension. Hypertension 48, 804–811. doi: 10.1161/01.HYP.0000242483.03361.da
Figueroa, X. F., Paul, D. L., Simon, A. M., Goodenough, D. A., Day, K. H., Damon, D. N., et al. (2003). Central role of connexin40 in the propagation of electrically activated vasodilation in mouse cremasteric arterioles in vivo. Circ. Res. 92, 793–800. doi: 10.1161/01.RES.0000065918.90271.9A
Freed, J. K., and Gutterman, D. D. (2017). Communication is key: mechanisms of intercellular signaling in vasodilation. J. Cardiovasc. Pharmacol. 69, 264–272. doi: 10.1097/FJC.0000000000000463
Garland, C. J., and Dora, K. A. (2017). EDH: endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol. 219, 152–161. doi: 10.1111/apha.12649
Garland, C. J., Hiley, C. R., and Dora, K. A. (2011). EDHF: spreading the influence of the endothelium. Br. J. Pharmacol. 164, 839–852. doi: 10.1111/j.1476-5381.2010.01148.x
Griffith, T. M. (2004). Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br. J. Pharmacol. 141, 881–903. doi: 10.1038/sj.bjp.0705698
Griffith, T. M., Chaytor, A. T., Taylor, H. J., Giddings, B. D., and Edwards, D. H. (2002). cAMP facilitates EDHF-type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions. Proc. Natl. Acad. Sci. U.S.A. 99, 6392–6397. doi: 10.1073/pnas.092089799
Grosely, R., and Sorgen, P. L. (2013). A history of gap junction structure: hexagonal arrays to atomic resolution. Cell Commun. Adhes. 20, 11–20. doi: 10.3109/15419061.2013.775256
Gustafsson, F., Mikkelsen, H. B., Arensbak, B., Thuneberg, L., Neve, S., Jensen, L. J., et al. (2003). Expression of connexin 37, 40 and 43 in rat mesenteric arterioles and resistance arteries. Histochem. Cell Biol. 119, 139–148. doi: 10.1007/s00418-002-0493-0
Haddock, R. E., Grayson, T. H., Brackenbury, T. D., Meaney, K. R., Neylon, C. B., Sandow, S. L., et al. (2006). Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40. Am. J. Physiol. Heart Circ. Physiol. 291, H2047–H2056. doi: 10.1152/ajpheart.00484.2006
Haefliger, J. A., Nicod, P., and Meda, P. (2004). Contribution of connexins to the function of the vascular wall. Cardiovasc. Res. 62, 345–356. doi: 10.1016/j.cardiores.2003.11.015
Hakim, C. H., Jackson, W. F., and Segal, S. S. (2008). Connexin isoform expression in smooth muscle cells and endothelial cells of hamster cheek pouch arterioles and retractor feed arteries. Microcirculation 15, 503–514. doi: 10.1080/10739680801982808
Hill, C. E., Rummery, N., Hickey, H., and Sandow, S. L. (2002). Heterogeneity in the distribution of vascular gap junctions and connexins: implications for function. Clin. Exp. Pharmacol. Physiol. 29, 620–625. doi: 10.1046/j.1440-1681.2002.03699.x
Howitt, L., Chaston, D. J., Sandow, S. L., Matthaei, K. I., Edwards, F. R., and Hill, C. E. (2013). Spreading vasodilatation in the murine microcirculation: attenuation by oxidative stress-induced change in electromechanical coupling. J. Physiol. 591, 2157–2173. doi: 10.1113/jphysiol.2013.250928
Isakson, B. E. (2008). Localized expression of an ins(1,4,5)P3 receptor at the myoendothelial junction selectively regulates heterocellular Ca2+ communication. J. Cell Sci. 121, 3664–3673. doi: 10.1242/jcs.037481
Isakson, B. E., Best, A. K., and Duling, B. R. (2008). Incidence of protein on actin bridges between endothelium and smooth muscle in arterioles demonstrates heterogeneous connexin expression and phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 294, H2898–H2904. doi: 10.1152/ajpheart.91488.2007
Isakson, B. E., and Duling, B. R. (2005). Heterocellular contact at the myoendothelial junction influences gap junction organization. Circ. Res. 97, 44–51. doi: 10.1161/01.RES.0000173461.36221.2e
Isakson, B. E., Ramos, S. I., and Duling, B. R. (2007). Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ. Res. 100, 246–254. doi: 10.1161/01.RES.0000257744.23795.93
Jobs, A., Schmidt, K., Schmidt, V. J., Lubkemeier, I., van Veen, T. A. B., Kurtz, A., et al. (2012). Defective Cx40 maintains Cx37 expression but intact Cx40 is crucial for conducted dilations irrespective of hypertension. Hypertension 60, 1422–1429. doi: 10.1161/HYPERTENSIONAHA.112.201194
Just, A., Kurtz, L., de Wit, C., Wagner, C., Kurtz, A., and Arendshorst, W. J. (2009). Connexin 40 mediates the tubuloglomerular feedback contribution to renal blood flow autoregulation. J. Am. Soc. Nephrol. 20, 1577–1585. doi: 10.1681/ASN.2008090943
Kansui, Y., Garland, C. J., and Dora, K. A. (2008). Enhanced spontaneous Ca2+ events in endothelial cells reflect signalling through myoendothelial gap junctions in pressurized mesenteric arteries. Cell Calcium 44, 135–146. doi: 10.1016/j.ceca.2007.11.012
Kerr, P. M., Tam, R., Ondrusova, K., Mittal, R., Narang, D., Tran, C. H. T., et al. (2012). Endothelial feedback and the myoendothelial projection. Microcirculation 19, 416–422. doi: 10.1111/j.1549-8719.2012.00187.x
Koeppen, M., Feil, R., Siegl, D., Feil, S., Hofmann, F., Pohl, U., et al. (2004). cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension 44, 952–955. doi: 10.1161/01.HYP.0000147661.80059.ca
Looft-Wilson, R. C., Goodell, C. R., Mutch, C. A., Mutchler, S. M., Miller, K. L., and Guraya, M. (2017). Increased myoendothelial feedback is associated with increased connexin37 and IK1 channel expression in mesenteric arteries of diet-induced hyperhomocysteinemic mice. Microcirculation 24:e12398. doi: 10.1111/micc.12398
Luo, W., Liu, B., and Zhou, Y. (2016). The endothelial cyclooxygenase pathway: insights from mouse arteries. Eur. J. Pharmacol. 780, 148–158. doi: 10.1016/j.ejphar.2016.03.043
Matchkov, V. V., Rahman, A., Bakker, L. M., Griffith, T. M., Nilsson, H., and Aalkjaer, C. (2006). Analysis of effects of connexin-mimetic peptides in rat mesenteric small arteries. Am. J. Physiol. Heart Circ. Physiol. 291, H357–H367. doi: 10.1152/ajpheart.00681.2005
Mather, S., Dora, K. A., Sandow, S. L., Winter, P., and Garland, C. J. (2005). Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ. Res. 97, 399–407. doi: 10.1161/01.RES.0000178008.46759.d0
Meens, M. J., Pfenniger, A., Kwak, B. R., and Delmar, M. (2013). Regulation of cardiovascular connexins by mechanical forces and junctions. Cardiovasc. Res. 99, 304–314. doi: 10.1093/cvr/cvt095
Milkau, M., Kohler, R., and de Wit, C. (2010). Crucial importance of the endothelial K+ channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J. 24, 3572–3579. doi: 10.1096/fj.10-158956
Morel, S. (2014). Multiple roles of connexins in atherosclerosis- and restenosis-induced vascular remodelling. J. Vasc. Res. 51, 149–161. doi: 10.1159/000362122
Murai, T., Muraki, K., Imaizumi, Y., and Watanabe, M. (1999). Levcromakalim causes indirect endothelial hyperpolarization via a myo-endothelial pathway. Br. J. Pharmacol. 128, 1491–1496. doi: 10.1038/sj.bjp.0702956
Nava, E., and Llorens, S. (2019). The local regulation of vascular function: from an inside-outside to an outside-inside model. Front. Physiol. 10:729. doi: 10.3389/fphys.2019.00729
Nielsen, M. S., Axelsen, L. N., Sorgen, P. L., Verma, V., Delmar, M., and Holstein-Rathlou, N. H. (2012). Gap junctions. Compr. Physiol. 2, 1981–2035. doi: 10.1002/cphy.c110051
Pogoda, K., Fuller, M., Pohl, U., and Kameritsch, P. (2014). NO, via its target Cx37, modulates calcium signal propagation selectively at myoendothelial gap junctions. Cell Commun. Signal. 12:33. doi: 10.1186/1478-811X-12-33
Pogoda, K., and Kameritsch, P. (2019). Molecular regulation of myoendothelial gap junctions. Curr. Opin. Pharmacol. 45, 16–22. doi: 10.1016/j.coph.2019.03.006
Pogoda, K., Mannell, H., Blodow, S., Schneider, H., Schubert, K. M., Qiu, J., et al. (2017). NO augments endothelial reactivity by reducing myoendothelial calcium signal spreading: a novel role for Cx37 (Connexin 37) and the protein tyrosine phosphatase SHP-2. Arterioscler. Thromb. Vasc. Biol. 37, 2280–2290. doi: 10.1161/ATVBAHA.117.309913
Pohl, U. (2020). Connexins: key players in the control of vascular plasticity and function. Physiol. Rev. 100, 525–572. doi: 10.1152/physrev.00010.2019
Qian, J., and Fulton, D. (2013). Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 4:347. doi: 10.3389/fphys.2013.00347
Radtke, J., Schmidt, K., Wulff, H., Kohler, R., and de Wit, C. (2013). Activation of KCa3.1 by SKA-31 induces arteriolar dilation and lowers blood pressure in normo- and hypertensive connexin40-deficient mice. Br. J. Pharmacol. 170, 293–303. doi: 10.1111/bph.12267
Rummery, N. M., and Hill, C. E. (2004). Vascular gap junctions and implications for hypertension. Clin. Exp. Pharmacol. Physiol. 31, 659–667. doi: 10.1111/j.1440-1681.2004.04071.x
Saez, J. C., Berthoud, V. M., Branes, M. C., Martinez, A. D., and Beyer, E. C. (2003). Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359–1400. doi: 10.1152/physrev.00007.2003
Saliez, J., Bouzin, C., Rath, G., Ghisdal, P., Desjardins, F., Rezzani, R., et al. (2008). Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation 117, 1065–1074. doi: 10.1161/CIRCULATIONAHA.107.731679
Sandow, S. L., Gzik, D. J., and Lee, R. M. K. W. (2009). Arterial internal elastic lamina holes: relationship to function? J. Anat. 214, 258–266. doi: 10.1111/j.1469-7580.2008.01020.x
Sandow, S. L., Looft-Wilson, R., Doran, B., Grayson, T. H., Segal, S. S., and Hill, C. E. (2003). Expression of homocellular and heterocellular gap junctions in hamster arterioles and feed arteries. Cardiovasc. Res. 60, 643–653. doi: 10.1016/j.cardiores.2003.09.017
Sandow, S. L., Senadheera, S., Bertrand, P. P., Murphy, T. V., and Tare, M. (2012). Myoendothelial contacts, gap junctions, and microdomains: anatomical links to function? Microcirculation 19, 403–415. doi: 10.1111/j.1549-8719.2011.00146.x
Sankaranarayanan, A., Raman, G., Busch, C., Schultz, T., Zimin, P. I., Hoyer, J., et al. (2009). Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol. Pharmacol. 75, 281–295. doi: 10.1124/mol.108.051425
Schmidt, V. J., Jobs, A., von Maltzahn, J., Worsdorfer, P., Willecke, K., and de Wit, C. (2012). Connexin45 is expressed in vascular smooth muscle but its function remains elusive. PLoS One 7:e42287. doi: 10.1371/journal.pone.0042287
Schweda, F., Kurtz, L., de Wit, C., Janssen-Bienhold, U., Kurtz, A., and Wagner, C. (2009). Substitution of connexin40 with connexin45 prevents hyperreninemia and attenuates hypertension. Kidney Int. 75, 482–489. doi: 10.1038/ki.2008.637
Selemidis, S., and Cocks, T. (2007). Smooth muscle mediates circumferential conduction of hyperpolarization and relaxation to focal endothelial cell activation in large coronary arteries. Naunyn Schmiedebergs Arch. Pharmacol. 375, 85–94. doi: 10.1007/s00210-007-0149-7
Shu, X., Ruddiman, C. A., Keller, T. C. S., Keller, A. S., Yang, Y., Good, M. E., et al. (2019). Heterocellular contact can dictate arterial function. Circ. Res. 124, 1473–1481. doi: 10.1161/CIRCRESAHA.118.313926
Si, H., Heyken, W. T., Wolfle, S. E., Tysiac, M., Schubert, R., Grgic, I., et al. (2006). Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+ activated K+ channel. Circ. Res. 99, 537–544. doi: 10.1161/01.RES.0000238377.08219.0c
Siegl, D., Koeppen, M., Wolfle, S. E., Pohl, U., and de Wit, C. (2005). Myoendothelial coupling is not prominent in arterioles within the mouse cremaster microcirculation in vivo. Circ. Res. 97, 781–788. doi: 10.1161/01.RES.0000186193.22438.6c
Simon, A. M., and McWhorter, A. R. (2003). Decreased intercellular dye-transfer and downregulation of non-ablated connexins in aortic endothelium deficient in connexin37 or connexin40. J. Cell Sci. 116, 2223–2236. doi: 10.1242/jcs.00429
Sohl, G., and Willecke, K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228–232. doi: 10.1016/j.cardiores.2003.11.013
Sorensen, C. M., and Holstein-Rathlou, N. H. (2012). Cell-cell communication in the kidney microcirculation. Microcirculation 19, 451–460. doi: 10.1111/j.1549-8719.2011.00149.x
Straub, A. C., Johnstone, S. R., Heberlein, K. R., Rizzo, M. J., Best, A. K., Boitano, S., et al. (2010). Site-specific connexin phosphorylation is associated with reduced heterocellular communication between smooth muscle and endothelium. J. Vasc. Res. 47, 277–286. doi: 10.1159/000265562
Straub, A. C., Lohman, A. W., Billaud, M., Johnstone, S. R., Dwyer, S. T., Lee, M. Y., et al. (2012). Endothelial cell expression of haemoglobin alpha regulates nitric oxide signalling. Nature 491, 473–477. doi: 10.1038/nature11626
Straub, A. C., Zeigler, A. C., and Isakson, B. E. (2014). The myoendothelial junction: connections that deliver the message. Physiology 29, 242–249. doi: 10.1152/physiol.00042.2013
Wagner, C., de Wit, C., Kurtz, L., Grunberger, C., Kurtz, A., and Schweda, F. (2007). Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ. Res. 100, 556–563. doi: 10.1161/01.RES.0000258856.19922.45
Wagner, C., Jobs, A., Schweda, F., Kurtz, L., Kurt, B., Lopez, M. L. S., et al. (2010). Selective deletion of connexin 40 in renin-producing cells impairs renal baroreceptor function and is associated with arterial hypertension. Kidney Int. 78, 762–768. doi: 10.1038/ki.2010.257
Wagner, C., and Kurtz, A. (2013). Distribution and functional relevance of connexins in renin-producing cells. Pflugers Arch. 465, 71–77. doi: 10.1007/s00424-012-1134-7
Wagner, C., Kurtz, L., Schweda, F., Simon, A. M., and Kurtz, A. (2009). Connexin 37 is dispensable for the control of the renin system and for positioning of renin-producing cells in the kidney. Pflugers Arch. 459, 151–158. doi: 10.1007/s00424-009-0707-6
Wei, R., Lunn, S. E., Tam, R., Gust, S. L., Classen, B., Kerr, P. M., et al. (2018). Vasoconstrictor stimulus determines the functional contribution of myoendothelial feedback to mesenteric arterial tone. J. Physiol. 596, 1181–1197. doi: 10.1113/JP274797
Welsh, D. G., and Segal, S. S. (1998). Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am. J. Phys. 274, H178–H186. doi: 10.1152/ajpheart.1998.274.1.H178
Welsh, D. G., and Segal, S. S. (2000). Role of EDHF in conduction of vasodilation along hamster cheek pouch arterioles in vivo. Am. J. Physiol. Heart Circ. Physiol. 278, H1832–H1839. doi: 10.1152/ajpheart.2000.278.6.H1832
Westcott, E. B., and Segal, S. S. (2013). Perivascular innervation: a multiplicity of roles in vasomotor control and myoendothelial signaling. Microcirculation 20, 217–238. doi: 10.1111/micc.12035
Wilson, C., Zhang, X., Buckley, C., Heathcote, H. R., Lee, M. D., and McCarron, J. G. (2019). Increased vascular contractility in hypertension results from impaired endothelial calcium signaling. Hypertension 74, 1200–1214. doi: 10.1161/HYPERTENSIONAHA.119.13791
Wolfle, S. E., Schmidt, V. J., Hoepfl, B., Gebert, A., Alcolea, S., Gros, D., et al. (2007). Connexin45 cannot replace the function of connexin40 in conducting endothelium-dependent dilations along arterioles. Circ. Res. 101, 1292–1299. doi: 10.1161/CIRCRESAHA.107.163279
Wolfle, S. E., Schmidt, V. J., Hoyer, J., Kohler, R., and de Wit, C. (2009). Prominent role of KCa3.1 in endothelium-derived hyperpolarizing factor-type dilations and conducted responses in the microcirculation in vivo. Cardiovasc. Res. 82, 476–483. doi: 10.1093/cvr/cvp060
Yamamoto, Y., Klemm, M. F., Edwards, F. R., and Suzuki, H. (2001). Intercellular electrical communication among smooth muscle and endothelial cells in Guinea-pig mesenteric arterioles. J. Physiol. 535, 181–195. doi: 10.1111/j.1469-7793.2001.00181.x
Keywords: connexins, gap junctions, endothelium-dependent hyperpolarization, myoendothelial coupling, microcirculation
Citation: Schmidt K and de Wit C (2020) Endothelium-Derived Hyperpolarizing Factor and Myoendothelial Coupling: The in vivo Perspective. Front. Physiol. 11:602930. doi: 10.3389/fphys.2020.602930
Edited by:
Zsolt Bagi, Augusta University, United StatesReviewed by:
Daniel Henrion, INSERM U1083 Physiopathologie Mitochondriale et Cardiovasculaire, FranceCopyright © 2020 Schmidt and de Wit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cor de Wit, ZGV3aXRAdW5pLWx1ZWJlY2suZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.