 Esteban R. Quezada
Esteban R. Quezada Alexis Díaz-Vegas
Alexis Díaz-Vegas Enrique Jaimovich
Enrique Jaimovich Mariana Casas
Mariana Casas- Center for Exercise, Metabolism, and Cancer, Physiology and Biophysics Program, Biomedical Sciences Institute (ICBM), Faculty of Medicine, University of Chile, Santiago, Chile
The slow calcium transient triggered by low-frequency electrical stimulation (ES) in adult muscle fibers and regulated by the extracellular ATP/IP3/IP3R pathway has been related to muscle plasticity. A regulation of muscular tropism associated with the MCU has also been described. However, the role of transient cytosolic calcium signals and signaling pathways related to muscle plasticity over the regulation of gene expression of the MCU complex (MCU, MICU1, MICU2, and EMRE) in adult skeletal muscle is completely unknown. In the present work, we show that 270 0.3-ms-long pulses at 20-Hz ES (and not at 90 Hz) transiently decreased the mRNA levels of the MCU complex in mice flexor digitorum brevis isolated muscle fibers. Importantly, when ATP released after 20-Hz ES is hydrolyzed by the enzyme apyrase, the repressor effect of 20 Hz on mRNA levels of the MCU complex is lost. Accordingly, the exposure of muscle fibers to 30 μM exogenous ATP produces the same effect as 20-Hz ES. Moreover, the use of apyrase in resting conditions (without ES) increased mRNA levels of MCU, pointing out the importance of extracellular ATP concentration over MCU mRNA levels. The use of xestospongin B (inhibitor of IP3 receptors) also prevented the decrease of mRNA levels of MCU, MICU1, MICU2, and EMRE mediated by a low-frequency ES. Our results show that the MCU complex can be regulated by electrical stimuli in a frequency-dependent manner. The changes observed in mRNA levels may be related to changes in the mitochondria, associated with the phenotypic transition from a fast- to a slow-type muscle, according to the described effect of this stimulation frequency on muscle phenotype. The decrease in mRNA levels of the MCU complex by exogenous ATP and the increase in MCU levels when basal ATP is reduced with the enzyme apyrase indicate that extracellular ATP may be a regulator of the MCU complex. Moreover, our results suggest that this regulation is part of the axes linking low-frequency stimulation with ATP/IP3/IP3R.
Introduction
Skeletal muscle can modify its phenotype to adapt to different external stimuli, such as disuse (Goldspink et al., 1986; Kneppers et al., 2019), hypoxia (Baresic et al., 2014; Nguyen et al., 2016), physical exercise (Yan et al., 2012; Joseph et al., 2016), among others (Gundersen, 2011), in a process known as muscle plasticity. According to this, different muscles of the body differ in their phenotype depending on the function they perform, expressing different isoforms of contractile proteins and metabolic enzymes. Accordingly, muscles (and, in a more general manner, motor units) have been broadly classified into three groups: slow-fatigue resistance, fast-fatigue resistance, and fast-fatigable. The model of muscle phenotype adaptation to different types of exercise is intimately linked to patterned electrical stimulation (ES) from the motoneurons innervating them. The first evidence involving frequency of ES in triggering molecular events associated with muscle plasticity was originated on in vivo models of cross-innervation, where a transition from fast to slow phenotype was observed in fast muscles innervated with α-motoneurons belonging to slow motor units presenting a low-frequency, long-lasting firing pattern (Eccles et al., 1962; Salmons and Vrbova, 1969). We have demonstrated that the inositol triphosphate receptor (IP3R) mediates the frequency-dependent induced changes (excitation–transcription coupling process) in gene expression involved in muscle plasticity of adult skeletal muscle. In particular, our results show that IP3R (and associated calcium signals) has a role in the activation of transcriptional programs associated with a slow phenotype that are activated at low frequencies of stimulation (Jorquera et al., 2013), partially emulating muscle changes induced by aerobic training (Klitgaard et al., 1990; Casas et al., 2010; Jorquera et al., 2013).
Excitation–transcription coupling is a process linking patterned depolarization of the muscle fibers with the activation of specific signaling pathways downstream. After a train of electrical stimuli, the process is triggered by the activation of the voltage-dependent L-type calcium channel Cav1.1 or dihydropyridine receptor (DHPR) (Jaimovich et al., 2000; Araya et al., 2003). At low frequencies of stimulation, DHPR activates the release of adenosine triphosphate (ATP), from the inside of the muscle fiber to the extracellular medium, through type 1 pannexin channels (Panx1). Extracellular ATP and its metabolites can thus act in an autocrine and paracrine manner, activating purinergic receptors, which, in turn, activate phosphatidylinositol 3-kinase (PI3K) and downstream pathway that favors the production of the second messenger 1,4,5 triphosphate (IP3) (Araya et al., 2003; Buvinic et al., 2009; Jorquera et al., 2013). Subsequently, IP3 binds to the membrane receptor of the sarcoplasmic reticulum (SR) and the nuclear envelope, causing Ca2+ release from the SR and the consequent increase in the cytosolic and nuclear Ca2+ concentration, which modulates the activity of several transcription factors to foster transcription (Carrasco et al., 2003).
During muscle contraction, the energy requirements of the muscle fiber are increased several times compared to rest (Weibel and Hoppeler, 2005). In muscle cells, mitochondria are the main source of ATP, and its function can be stimulated by various molecules, such as adenosine diphosphate (ADP), adenosine monophosphate (AMP), and Ca2+ (Lazarowski et al., 2003; Yi et al., 2011). In the mitochondrial matrix, different enzymes, such as isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, have Ca2+ as a co-factor (Cortassa et al., 2003). Consequently, increases in the intramitochondrial concentration of Ca2+ increase the activity of these enzymes, increasing the speed of the Krebs cycle as well as the production of reduced compounds (NADH and FADH2) that feed the electron transport chain and ATP synthesis (Diaz-Vegas et al., 2019). Therefore, Ca2+ entry into the mitochondria is key to maintain the balance between the metabolic requirements and the synthesis of ATP in the skeletal muscle (Baughman et al., 2011; Tarasov et al., 2012; Diaz-Vegas et al., 2019).
The mitochondrial calcium uniporter (MCU) is a highly selective Ca2+ channel located in the inner mitochondrial membrane. MCU mediates an electrogenic Ca2+ influx from the intermembrane space to the mitochondrial matrix (Wan et al., 1989; O’Donnell et al., 1998; Carafoli, 2010). Furthermore, MCU is associated with different regulatory proteins that modulate their affinity for Ca2+ (Fan et al., 2020). MCU and its regulatory proteins are collectively known as the MCU complex, where MCU is the protein forming the pore of the channel (Chaudhuri et al., 2013; Shanmughapriya et al., 2015). The main components of the MCU complex expressed in adult skeletal muscle are MCU, essential MCU regulator (EMRE), mitochondrial Ca2+ uptake 1 (MICU1), and mitochondrial Ca2+ uptake 2 (MICU2) (Murgia and Rizzuto, 2015). Among these, the MICU1/MCU ratio appears to be particularly important for the regulation of mitochondrial Ca2+ uptake (Paillard et al., 2017). Increases in cytosolic Ca2+ levels generate an increase in mitochondrial Ca2+ through MCU. This process takes place after depolarization of the muscle fiber, where a high increase in intracellular Ca2+ concentration occurs. The consequent rise in mitochondrial Ca2+ depends on the activation of ryanodine receptor 1 (RyR1) (a fast component of Ca2+ release) as well as IP3R (a slow component of calcium release) (Diaz-Vegas et al., 2018).
There is evidence pointing to a possible role of MCU in muscle plasticity. Indeed, the silencing of MCU has been suggested to induce muscular atrophy, and its overexpression generates muscular hypertrophy in murine models (Mammucari et al., 2015), although these results are somehow controversial (Kwong et al., 2018). Also, 9 weeks of strength training or high-frequency EE (60 Hz) in humans induces hypertrophy and increases MCU protein levels in skeletal muscle (Zampieri et al., 2016). Interestingly, microarray analysis of muscles overexpressing MCU or underexpressing MCU showed differential changes in expression of genes related to sarcomere organization, calcium regulation, differentiation, and development. Notably, when MCU was overexpressed, an increase in expression of genes related to Ca2+ homeostasis was observed (Chemello et al., 2015).
Recently, the role of Ca2+ in the control of expression of MCU has been studied using the Ca2+ ionophore ionomycin to increase intracellular calcium concentration (Shanmughapriya et al., 2015). The results showed that CREB binds to the MCU promoter and alterations in cytosolic Ca2+ levels induced changes in MCU levels. Interestingly, these results suggest the existence of a crosstalk between cytosolic Ca2+ levels and the control of mitochondrial Ca2+ buffering capacity mechanisms (Shanmughapriya et al., 2015). Studies using more physiological stimuli are thus needed to further explore this mechanism. Furthermore, in hippocampal and cortical neurons, a reduction of MCU levels after increases in cytosolic Ca2+ through activation of NMDA receptor has been described (Qiu et al., 2013).
These works reveal the importance of understanding the role that transient changes in cytosolic Ca2+ levels (induced by a physiological stimulus) play in the regulation of gene expression of MCU, MICU1.1, MICU2, and EMRE in a tissue such as skeletal muscle where Ca2+ is a key factor. Such regulation could modulate the Ca2+ buffering efficiency of mitochondria, generating a physiological control loop of intracellular Ca2+ signals. In the case of adult skeletal muscle, a phenotypic change is observed under low-frequency ES, with modifications, among others, in the expression of oxidative related metabolic enzymes and an increase in mitochondria content (Hood et al., 1989; Putman et al., 2004; Petrie et al., 2015). The possible changes in mitochondrial proteins related to its Ca2+ buffering capacity is a subject that remains poorly studied until now.
In this work, we show a decrease in mRNA levels of MCU, MICU1, MICU2, and EMRE specifically after low-frequency ES. Moreover, the changes observed appear in line with an asymmetric distribution of some of these proteins between fast and slow phenotype muscle fibers.
Results
20-Hz ES Produces a Decrease in mRNA Levels of the MCU Complex
We have previously demonstrated that ES of muscle fibers with 270 pulses, 0.3 ms long, at 20 Hz, induces changes in mRNA levels related to a slow-to-fast phenotypic transition, whereas the same amount of pulses at 90 Hz induces the inverse effect (Jorquera et al., 2013). We evaluated the effect of ES on mRNA levels of the MCU complex finding a significant decrease in MCU mRNA levels 30 min (1.00 ± 0.06 vs 0.81 ± 0.05 at 30 min) and 1 h (1.002 ± 0.061 vs 0.669 ± 0.216 at 1 h) after 20-Hz ES of isolated fdb muscle fibers (Figure 1A). High-frequency, 90-Hz ES did not produce changes in MCU mRNA levels (Figure 1B). Moreover, a decrease in the mRNA levels of MICU1 (1.03 ± 0.23; vs 0.761 ± 0.091 at 20 Hz), MICU2 (1.01 ± 0.16; vs 0.83 ± 0.07 at 20 Hz), and EMRE (1.01 ± 0.17 vs 0.73 ± 0.15 at 20 Hz) was also observed 1 h after 20-Hz ES, whereas 90-Hz ES did not generate any changes in mRNA levels of these genes (Figures 1C–E).
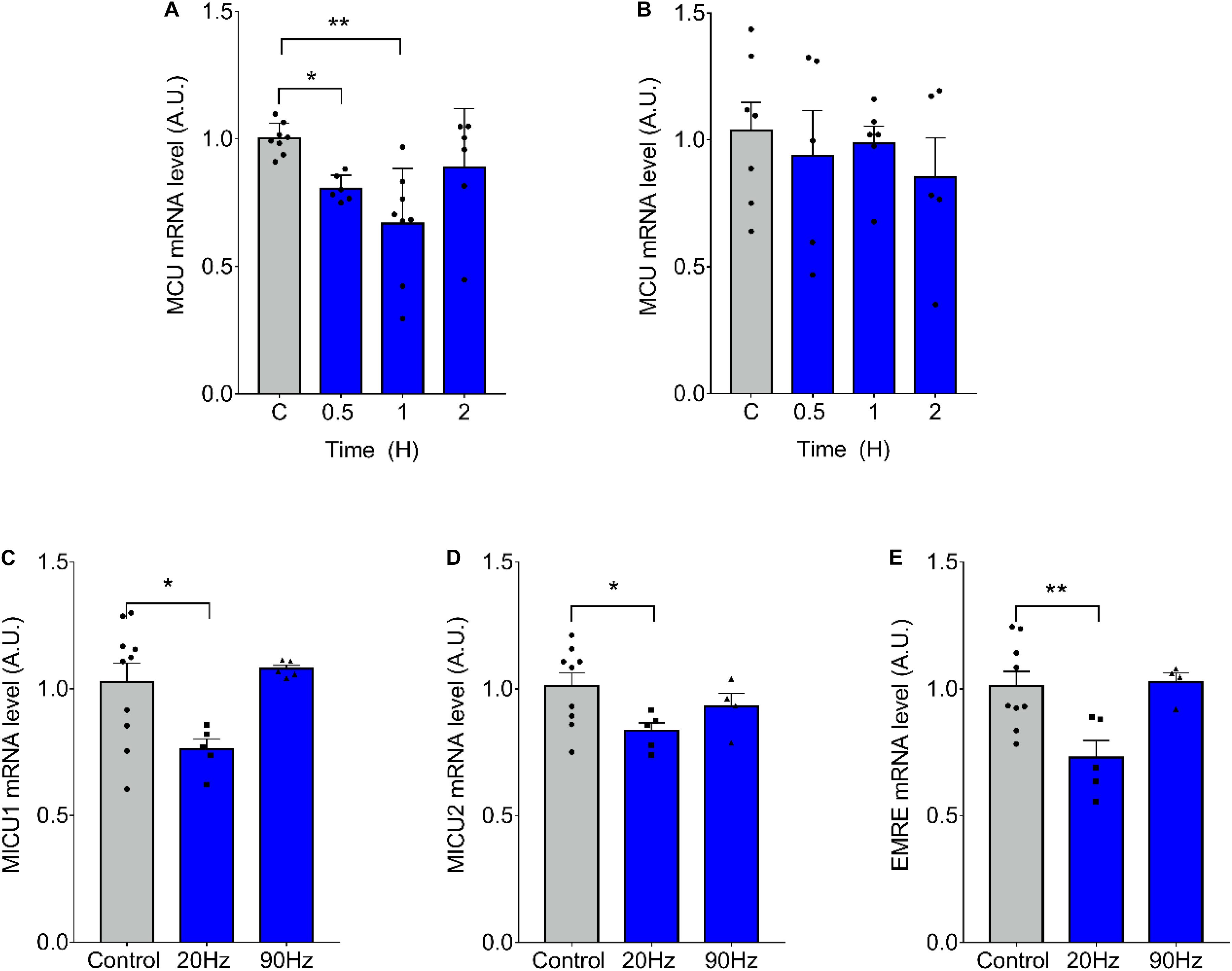
Figure 1. Low-frequency electrical stimulation (ES) transiently reduces mRNA levels of MCU complex in skeletal muscle fibers. Muscle fibers isolated from the Flexor Digitorium Brevis (fdb) muscle were stimulated with 270 pulses, 0.3 ms each (A) at 20 Hz (n = 6–8 per condition) or (B) 90 Hz (n = 5–7 per condition). mRNA levels of MCU decreased 1h post-ES (A); while high-frequency stimulation produced no change (B). The low frequency stimulation also decreased mRNA levels of MICU1 (n = 5–10 per condition) (C), MICU2 (n = 4–9 per condition) (D) and EMRE (n = 4–9 per condition) (E) 1 h post ES. For (A) using the Kruskal–Wallis test with post hoc Dunn’s was applied. Values are presented as mean ± SEM. 18S was used as normalizer. *p < 0.05; **p < 0.01; ***p < 0.001.
Changes in mRNA Levels of the MCU Complex Are Dependent on Extracellular ATP and IP3R
To evaluate the downstream signaling after low-frequency ES, responsible for the observed changes in mRNA levels of MCU complex genes, we searched to determine the role of extracellular ATP. We have demonstrated that 20-Hz ES induces a release of ATP to the extracellular milieu trough Pannexin-1 channels (Jorquera et al., 2013). The ATP released acts over purinergic receptors to activate signaling cascades that activate, among others, the production of IP3 and the release of Ca2+ through IP3R, inducing changes in transcriptional activity of several genes (Casas et al., 2010). Figure 2 shows changes in MCU mRNA level in fdb muscle fibers pre-incubated with 2 U/ml of the ecto-nucleotidase apyrase to reduce extracellular ATP levels. The decrease observed in mRNA levels of MCU, MICU1, MICU2, and EMRE after a 20-Hz ES was absent when fibers were pre-incubated with apyrase (Figures 2A–E), when compared to control fibers. A significant increase in MCU mRNA levels (C = 1.02 ± 0.14; Apy = 1.32 ± 0.19) was also observed after incubation with apyrase (Figure 2A) in basal conditions (without ES). This effect of apyrase was not observed in MICU1, MICU2, and EMRE mRNA levels.
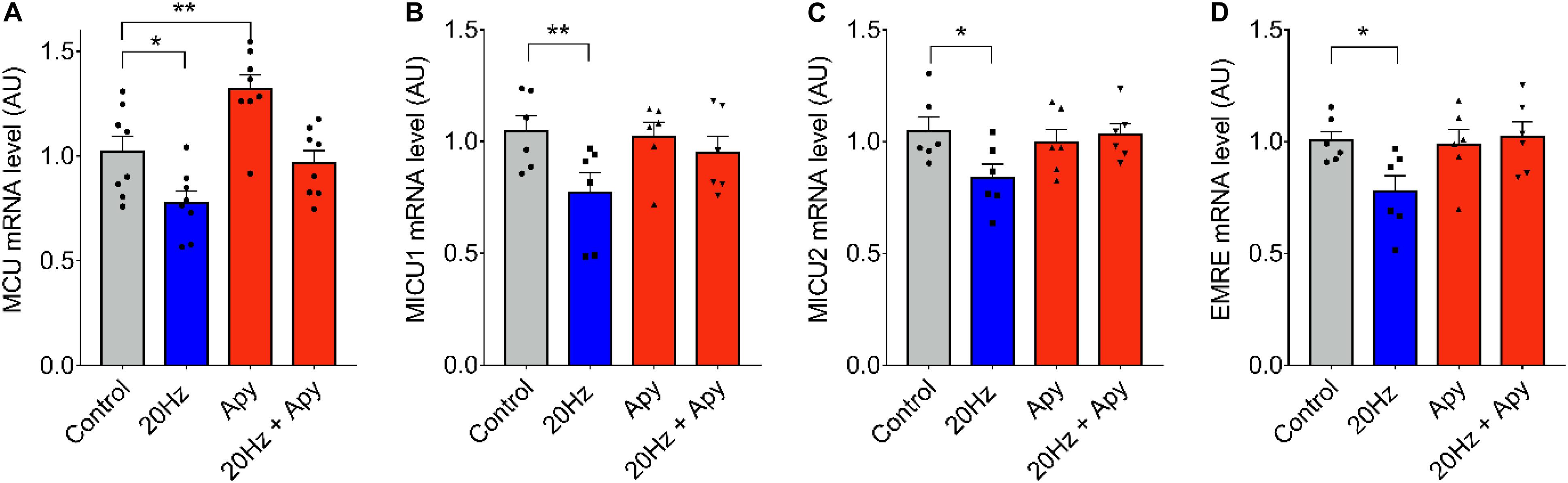
Figure 2. Low-frequency ES-dependent reduction in mRNA levels of MCU complex is dependent on extracellular ATP. Muscle fibers isolated from the fdb muscle were pre-incubated 30 min with apyrase (2 U/ml) before ES (as in Figure 1). The mRNA was extracted 1 h after the ES. MCU mRNA levels increased with pre-incubation with apyrase in the absence of ES (A). Preincubation with apyrase prevented the low frequency-dependent reduction in mRNA levels of MCU (n = 8) (A), MICU1 (n = 6) (B), MICU2 (n = 6) (C), and EMRE (n = 6) (D). Values are presented as mean ± SEM. 18s was used as normalizer. *p < 0.05; **p < 0.01; ***p < 0.001.
To evaluate if ATP alone (in absence of ES) can induce the observed changes in mRNA levels after 20-Hz ES, we stimulated fdb muscle fibers with 30 μM of exogenous ATP and measured mRNA levels of the MCU complex. We observed that ATP exposure resulted in a significant decrease (C = 1.02 ± 0.20; 0.5 h = 0.74 ± 0.20) in mRNA levels of MCU after 30 min (Figure 3A). The same was observed at 30 min for mRNA levels of MICU1 (Control = 1.01 ± 0.17; 30 μM = 0.78 ± 0.17), MICU2 (Control = 1.02 ± 0.15; 30 μM = 0.79 ± 0.25), and EMRE (Control = 1.01 ± 0.13; 30 μM = 0.74 ± 0.15) after (Figures 3B–D). Similar effects were produced by extracellular ATP in an ex vivo model of complete fdb muscle (Supplementary Figure 1).
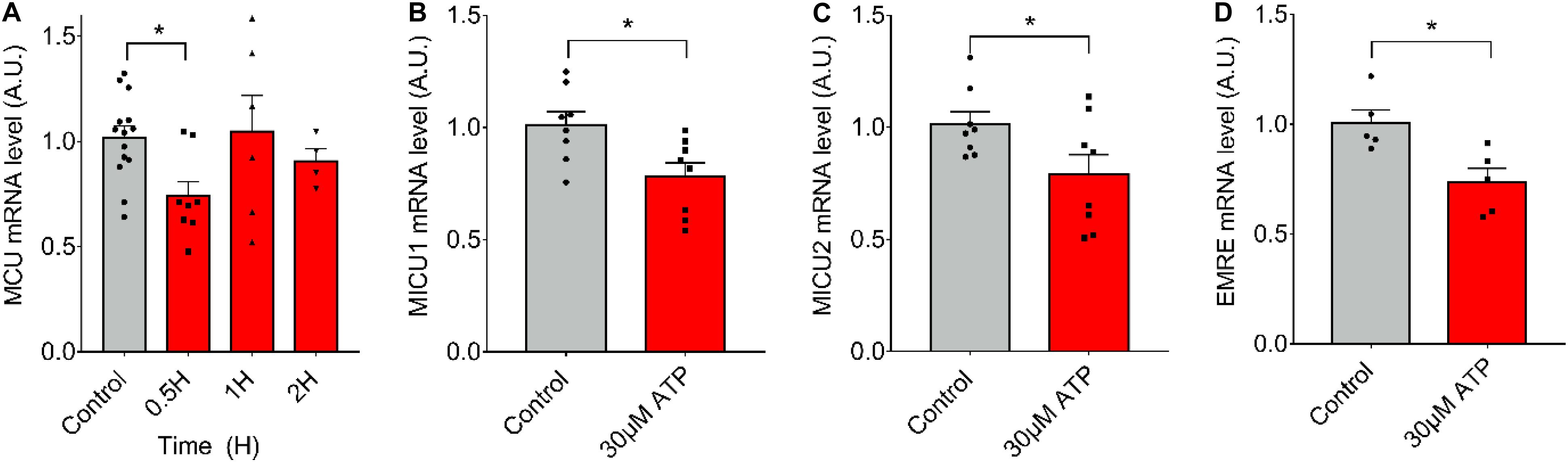
Figure 3. Extracellular ATP is sufficient to reduce mRNA levels of the MCU complex in skeletal muscle fibers. Muscle fibers fdb muscle and stimulated with exogenous ATP (30 μM). A decrease in mRNA levels of all the MCU complex components after 30 min of stimulation is observed. mRNA levels are shown normalized by mRNA level of control fibers. mRNA levels for MCU (n = 4–14) (A), MICU1 (n = 8) (B), MICU2 (n = 8) (C), and EMRE (n = 5) (D) were measured. Values are presented as mean ± SEM. 18S was used as normalizer. *p < 0.05; **p < 0.01; ***p < 0.001.
As it was previously mentioned, signaling downstream ATP release and purinergic receptors activation can be mediated by the production of IP3 and Ca2+ released through IP3R. To test the role of IP3R, we blocked this intracellular Ca2+ channel using xestospongin B. No changes in MCU, MICU1, 0.MICU2, and EMRE mRNA levels after 20 Hz ES were observed when fibers were pre-incubated with 10 μM of xestospongin B (Figures 4A–D), suggesting a role of Ca2+ released through IP3R in the regulation of mRNA levels of the MCU complex.
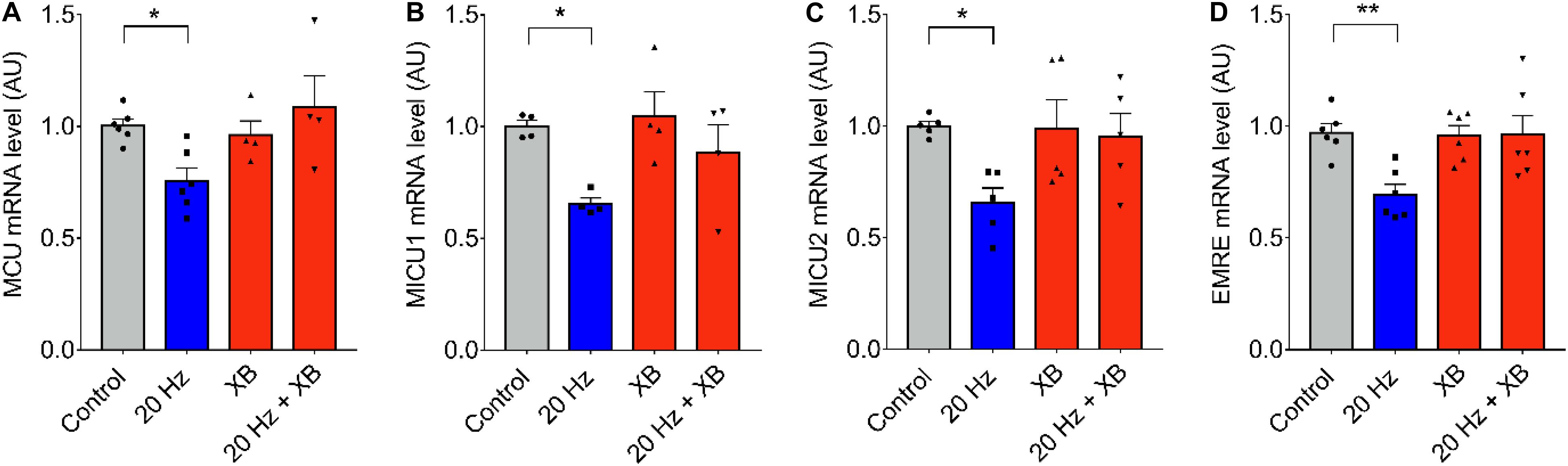
Figure 4. IP3R is involved in low-frequency ES-dependent reduction of mRNA levels of MCU complex. Muscle fibers fdb muscle were pretreated with the IP3R blocker, XB (10 μM) by 30 min before being electrically stimulated (as in Figure 1). The mRNA was extracted 1 h after electrical stimulation. Preincubation with XB prevented the low frequency-dependent reduction in mRNA levels of MCU (n = 6) (A), MICU1 (n = 5) (B), MICU2 (n = 5) (C), and EMRE (n = 6) (D). For (C) using the Kruskal–Wallis test with post hoc Dunn’s was applied. Values are presented as mean ± SEM. 18S was used as normalizer. *p < 0.05; **p < 0.01; ***p < 0.001.
Mitochondria From Fast Muscles Have a Higher Content of MCU Complex Proteins
We found a decrease in mRNA levels of the MCU complex after low-frequency ES. Considering that low-frequency ES is appropriate for slow-type motor units and that this type of ES can trigger transcriptional changes related to muscle fast-to-slow phenotype transitions, we hypothesized that proteins of the MCU complex could be differentially expressed in slow compared with fast phenotype adult muscles. We evaluated the levels of MCU and MICU1 proteins in a fast (fdb) and a slow (soleus) muscle by Western blot. We found a smaller amount of MCU (fdb = 1.00 ± 0.18; sol = 0.47 ± 0.13) (Figure 5A) and MICU1 (fdb = 1.00 ± 0.32; sol = 0.55 ± 0.24) (Figure 6A) in soleus muscle compared with fdb muscles. The protein content was normalized by TOM20, indicating that the relative amount of the MCU complex is lower in mitochondria from soleus muscle compared to fdb. This difference is not significant when normalized by total proteins (Figures 5C, 6C). This is probably due to the intrinsic difference in mitochondria content between these muscles, a difference that is compensated by the normalization of MCU protein content with a marker of mitochondria such as TOM20. MCU, MICU1, and TOM20 membranes are shown (Supplementary Figures 2, 3).
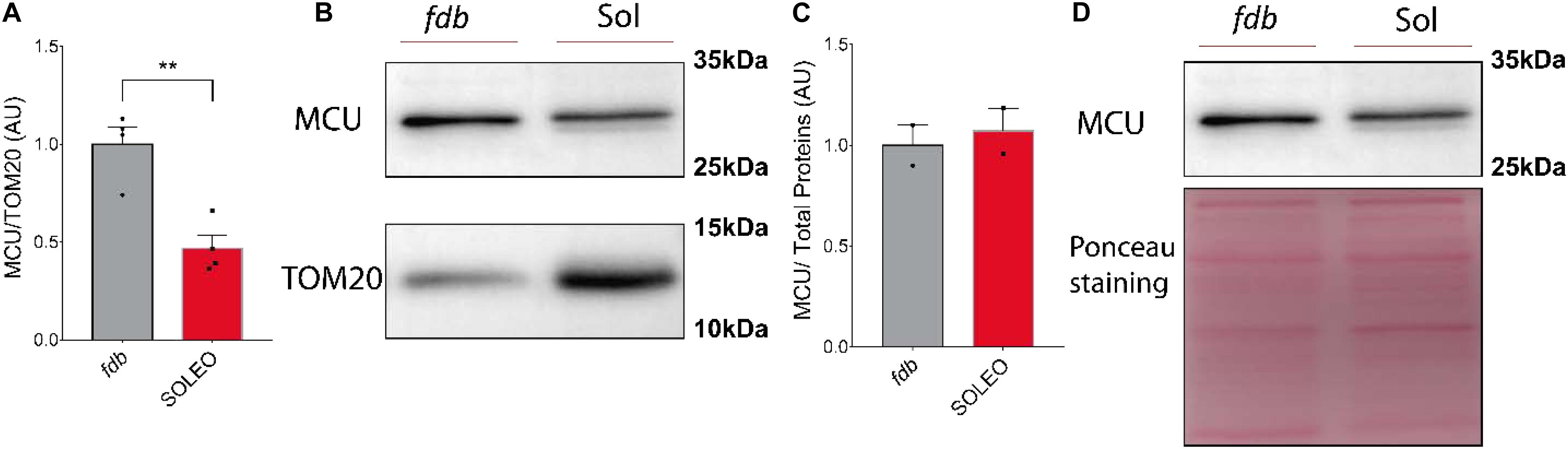
Figure 5. Mitochondria from Flexor Digitorium Brevis (fdb) muscle presented a higher content of MCU protein than soleus (sol) muscle. Representative Western Blot and quantification show the levels of the MCU protein in homogenates of fdb and Sol normalized by mitochondrial content using TOM20 as a normalizing protein (n = 4) (A,B). When MCU content was normalized by total protein content, no differences between these two muscles were observed (n = 2) (C,D). Values are presented as mean ± S.E.M. Values are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
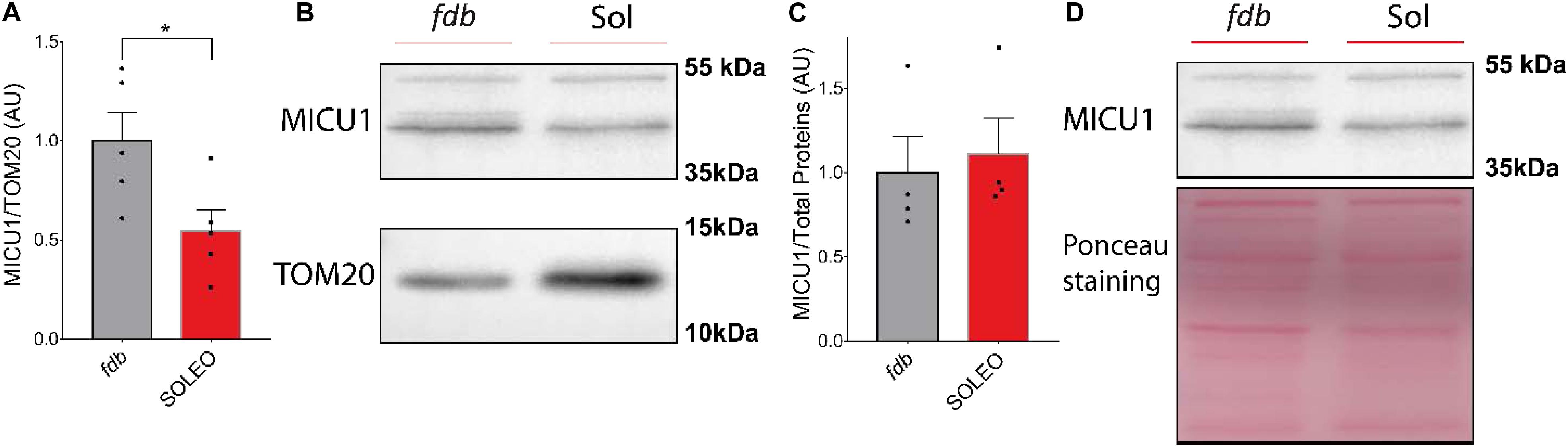
Figure 6. Mitochondria from Flexor Digitorium Brevis (fdb) muscle presented a higher content of MICU1 protein than mitochondria from soleus (sol) muscle. Representative Western Blot and quantification show the levels of the MICU1 protein in homogenates of fdb and sol, normalized by a mitochondrial marker (n = 5) (A,B) and total protein (n = 4) (C,D). For (C) using the Mann-Whitney test was applied. Values are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
We have demonstrated that low-frequency ES results in a decrease in the mRNA levels of MCU, MICU1, MICU2, and EMRE, while high-frequency ES does not generate modifications. Our laboratory has described a fine-tuned mechanism that relates the decoding of the frequency of stimulation by Cav1.1, ATP release, IP3R activation, and transcription changes related to fast-to-slow muscle phenotype transition (Jorquera et al., 2013). Therefore, if there is a differential calcium management between different types of muscle fibers (Carroll et al., 1997), it is to be expected that genes that regulate mitochondrial calcium uptake could be regulated by different stimulation frequencies. In the present work, we showed that mRNA levels of MCU, MICU1, MICU2, and EMRE are also regulated in a frequency-dependent manner, being affected only by low-frequency ES. We have pooled the data from all controls and 20 Hz from the different experiments (Supplementary Figure 4). The observed changes suggest a new process related to the phenotypic transition of a fast to slow muscle fiber, in this case, related to mitochondrial proteins other than those related to oxidative metabolism, classically described before in the plasticity process from fast to slow muscle phenotype transition.
Our results showed that in fibers stimulated at 20 Hz after pre-incubation with apyrase, mRNA of the MCU complex did not decrease (as it does when they are only ES at 20 Hz), showing no significant statistical difference between control and 20 Hz + Apy conditions, indicating that the effect of 20-Hz ES in the decrease of MCU complex mRNAs is lost when fibers are pre-incubated with apyrase. The same is observed when fibers are pre-incubated with xestospongin B (IP3R blocker). No statistical difference was found between mRNA levels of the MCU complex from fibers electrically stimulated at 20 Hz compared to those electrically stimulated at 20 Hz and pre-incubated with apyrase or xestospongin B. However, these results, together with those showing an increase in mRNA levels of the MCU complex after stimulation with exogenous ATP, although not conclusive, are consistent with the idea that the transcriptional effects observed on MCU, MICU1, MICU2, and EMRE after low-frequency ES would be mediated by slow calcium transients activated by the extracellular ATP/IP3 production/IP3R signaling pathway (Buvinic et al., 2009; Jorquera et al., 2013). The role of mitochondrial Ca2+ uptake over cytosolic Ca2+ signals after muscle fiber depolarization has been shown previously to play a role in excitation–contraction coupling (Yi et al., 2011). Moreover, there is also evidence that protein levels of MCU are susceptible to change after exercise and to ES in humans (Zampieri et al., 2016). Interestingly, it has been postulated that protective synaptic activity is related to a decrease in MCU protein levels, which is lost in the presence of a CaM kinase inhibitor, suggesting a role of cytoplasmic Ca2+ in MCU regulation (Qiu et al., 2013). On the other hand, the reduction in mRNA levels of the MCU complex is observed at shorter times (30 min compared to 60 min for ES) when fibers are stimulated with exogenous ATP; this phenomenon could be related to the final concentration of ATP reached in the T-tubules and their affinity for purinergic receptors (Lazarowski et al., 2003) or the activation of these receptors without the process involving depolarization sensing by Cav1.1 and triggering of ATP release through pannexin-1 channels.
The results showing an increase in mRNA levels of MCU in the presence of apyrase, in the absence of further stimulus, suggest that basal levels of ATP present in the extracellular medium would be inhibiting or repressing the transcription of this gene.
Our data provide new information on the relative amount of MCU and MICU1 in muscle of different phenotypes, showing the existence of higher protein levels of MCU and MICU1 per mitochondria in fdb than those belonging to the soleus. Therefore, the decrease in mRNA of the MCU complex after low-frequency ES of isolated fibers of the fdb muscle would favor a lower level of the MCU and MICU1 per mitochondrion, such as that present in a slow muscle. This could be considered an early metabolic response to the phenotypic shift from fast to slow muscle fiber (Loucif et al., 2020). The gradual increase in the number of mitochondria, together with a decrease in MCU complex content in response to a low-frequency stimulus, will allow adapting mitochondrial Ca2+ homeostasis to finally reach that of a slow muscle. Besides, the higher levels of TOM20 in slow muscle compared to fast muscle are consistent with that described with other mitochondrial proteins, such as ATP synthase and succinate dehydrogenase (Schiaffino and Reggiani, 2011; Khodabukus and Baar, 2015). Moreover, the kinetics of Ca2+ entry to the mitochondria have also been reported to be different between different fiber types (Sembrowich et al., 1985; Picard et al., 2008). Interestingly, it has been described that MCU overexpression causes neuronal death (Granatiero et al., 2019). On the other hand, stimulation of cortical and hippocampal neurons results in a decrease in mRNA and protein levels of MCU (Qiu et al., 2013), which have been associated with a protective effect preventing mitochondrial Ca2+ overload, thus preventing cytochrome C from triggering cell death. Likewise, it has been observed that the decrease in MCU in a model of cells from colon cancer results in a resistance to apoptosis (Marchi et al., 2013; Nemani et al., 2018). It appears then that a fine regulation of the MCU protein complex is needed to balance protection and cell death after different stimuli. In this sense, it has been proposed that the concentration of mitochondrial Ca2+ necessary to exceed the threshold to trigger the opening of the mitochondrial permeability transition pore (mPTP) is lower in a slow fiber compared to a fast fiber (Picard et al., 2008, 2012). Therefore, since the muscle contraction of a slow phenotype fiber results in regular and prolonged elevations in cytosolic (and possibly mitochondrial) (Ca2+), we can hypothesize that low-frequency ES appropriate for slow-type muscles could induce a decrease in the MCU complex to regulate the entry of Ca2+ to the mitochondria and to prevent mPTP opening and thus protect the muscle fiber from triggering cell death. Even if muscle contraction in slow phenotype muscle relies strongly on mitochondrial production of ATP, which depends on mitochondrial Ca2+, the lower protein abundance of MCU and MICU1 per mitochondria in the soleus muscle could be compensated in the first place by the higher mitochondrial content in slow phenotype muscle and also by phenotypic adaptations of the slow muscle, such as an increase in sensitivity to Ca2+ by the enzymes isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, maintaining the metabolism according to energy requirements (McCormack and Denton, 1989; Hurst et al., 2017). Not only has the protein level been described as a regulator of mitochondrial calcium uptake (Qiu et al., 2013), the stoichiometry of MCU/MICU1 (Paillard et al., 2017), mitochondrial endoplasmic reticulum interaction (Ainbinder et al., 2015), calcium release from the IP3R (Diaz-Vegas et al., 2018), and mitochondrial membrane potential (Diaz-Vegas et al., 2018) have also been described. Also, it has been described that an adaptive process of a cell is supported by a previous metabolic change, such as the case of metabolic changes associated with the activation of T-cell (Loucif et al., 2020). Exercise increases transcription factors of mitochondrial biogenesis and muscle hypertrophy in humans (Ruas et al., 2012). Deletion of PGC-1α (regulator of mitochondrial biogenesis) decreases the number and size of the mitochondria and the mass of the soleus and affects muscle performance (Leone et al., 2005). Thus, the changes observed at the level of the MCU complex in fdb could signify a prior process for the regulation of cytoplasmic Ca2+ signals mediated by the mitochondria, changing the regulation of genes related to contractility and favoring muscle plasticity. Therefore, future research is required to evaluate changes induced by ES in protein expression of the MCU complex and the consequences of a reduction of the MCU complex in the different functions of a skeletal muscle fiber, in the muscle plasticity process.
Materials and Methods
Animals
This study was carried out following the guidelines of the Bioethics Committee of the Faculty Medicine, University of Chile (FONDECYT #1151293). Eight- to 10-week-old male C57/BL6J mice were obtained from the Central Animal Facility of the Faculty of Medicine, University of Chile. Mice were kept in a room with controlled temperature in a light–dark cycle of 12 h and fed ad libitum.
Adult FBD Fiber Isolation
The isolated adult muscle fibers were obtained from the fdb muscle by enzymatic digestion of the whole muscle with 450–500 units/ml of collagenase type II (Worthington) for 90 min, followed by mechanical dissociation with Pasteur pipettes of different diameters as described previously (Casas et al., 2010). Fibers were plated in ECM (Sigma)-covered 35-mm plates in culture medium [Dulbecco’s modified Eagle’s medium (DMEM), 10% horse serum, and 1% penicillin/streptomycin]. Fibers were used for analysis 20 h after seeding.
Electrical Stimulation
The skeletal muscle fibers were electrically stimulated by field stimulation with a device consisting of parallel platinum wires with alternate polarity, as described previously (Casas et al., 2010). The protocol consisted of 270 pulses, 0.3 ms each at 20 Hz or 90 Hz (Jorquera et al., 2013).
mRNA Isolation, cDNA, and Real-Time qPCR
Total mRNA was isolated from the muscle complete of the fdb by using TRIzol® reagent (Invitrogen) according to the manufacturer’s protocol. The same extraction protocol was used to obtain total RNA from isolated fibers after ES. The cDNA was obtained by reverse transcription reaction of 1 μg of total RNA using random primer and polyDT primers. Real-time qPCR was performed according to the recommendations of EvaGreen® qPCR Mix Plus (ROX) using the following primers: MCU-fw: 5′-GTGCGCCTGTTTGTAACTCA-3′ and MCU-rv: 5′-CAAGACTCGCTAAGCCCTTT-3′, MICU1.1-fw: 5′-CTTTG ATGGAAAGGAGTTCTGGC-3′ and MICU1.1-rv: 5′-CCTCCA TGTCTACCTCTCCGT-3′, MICU2-fw: 5′-TGGAGCACGACG GAGAGTAT-3′ and MICU2-rv: 5′-GCCAGCTTCTTGACCA GTGT-3′, EMRE-fw: 5′-AACTTCGCTGCTCTGCTTGA-3′ and EMRE-rv: 5′-TGAGGCTGAGGGCTTTCCTT-3′, 18 s. The design of primers was performed using the AmplifX program and validated by Primer-BLAST.
Western Blot
The fdb and soleus samples were homogenized with an electric homogenizer (Fluko, Shanghai, China) in a lysis buffer containing 20 mM Tris–HCl (pH 7.5), 1% Triton X-100, 2 mM EDTA, 20 mM NaF, 1 mM Na2P2O7, 10% glycerol, 150 mM NaCl, 10 mM Na3VO4, 1 mM PMSF, and protease inhibitors (CompleteTM, Roche Applied Science). Proteins were separated using SDS-PAGE and transferred to PVDF membranes. The following antibodies and their dilutions were used: MCU (1:2,000; HPA016480, Sigma), MICU1 (1:2,000; HPA037480, Sigma), and TOM20 (1:10,000; ab186735, ABCAM). The protein bands in the blots were visualized using a WESTAR Supernova detection kit (Cyanagen, Bologna, Italy), super-resolution, and, further, ChemiDocTM MP System (Bio-Rad, United States). The intensity of the bands was determined with ImageJ densitometry analysis.
Statistical analysis
The results were expressed as mean ± standard error (±SEM). For the difference between data groups, the two-tailed paired t test was used. For comparison of more than two groups, the one-tailed one-way ANOVA was used followed by Dunnett’s multiple comparisons test. In cases where the data could not pass Levene’s equal variance test and Shapiro–Wilk test, Mann–Whitney test, and Kruskal–Wallis test with post hoc Dunn’s, a non-parametric test, as indicated in the figure legend, was applied. The level of significance was set at p < 0.05. All statistical analyses were performed in GraphPad Prism 7 and SPSS25.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Comité Institucional de Cuidado y Uso de Animales (CICUA).
Author Contributions
EQ performed experiments, analyzed results, and contributed to write the manuscript. AD-V analyzed results and contributed to design experiments. EJ planned experiments, discussed results, and contributed to wrote the manuscript. MC design the project, planned experiments, analyzed and discussed results, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Financed by Fondecyt 1151293, VID-Enl09, Líneas de apoyo a la investigación financiadas por ICBM 2020.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.601313/full#supplementary-material
References
Ainbinder, A., Boncompagni, S., Protasi, F., and Dirksen, R. T. (2015). Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calc. 57, 14–24. doi: 10.1016/j.ceca.2014.11.002
Araya, R., Liberona, J. L., Cardenas, J. C., Riveros, N., Estrada, M., Powell, J. A., et al. (2003). Dihydropyridine receptors as voltage sensors for a depolarization-evoked, IP3R-mediated, slow calcium signal in skeletal muscle cells. J. Gen. Physiol. 121, 3–16. doi: 10.1085/jgp.20028671
Baresic, M., Salatino, S., Kupr, B., van Nimwegen, E., and Handschin, C. (2014). Transcriptional network analysis in muscle reveals AP-1 as a partner of PGC-1alpha in the regulation of the hypoxic gene program. Mol. Cell. Biol. 34, 2996–3012. doi: 10.1128/mcb.01710-13
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Buvinic, S., Almarza, G., Bustamante, M., Casas, M., Lopez, J., Riquelme, M., et al. (2009). ATP released by electrical stimuli elicits calcium transients and gene expression in skeletal muscle. J. Biol. Chem. 284, 34490–34505. doi: 10.1074/jbc.m109.057315
Carafoli, E. (2010). The fateful encounter of mitochondria with calcium: how did it happen? Biochim. Biophys. Acta 1797, 595–606. doi: 10.1016/j.bbabio.2010.03.024
Carrasco, M. A., Riveros, N., Rios, J., Muller, M., Torres, F., Pineda, J., et al. (2003). Depolarization-induced slow calcium transients activate early genes in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 284, C1438–C1447.
Carroll, S. L., Klein, M. G., and Schneider, M. F. (1997). Decay of calcium transients after electrical stimulation in rat fast- and slow-twitch skeletal muscle fibres. J. Physiol. 501(Pt 3), 573–588. doi: 10.1111/j.1469-7793.1997.573bm.x
Casas, M., Figueroa, R., Jorquera, G., Escobar, M., Molgo, J., and Jaimovich, E. (2010). IP(3)-dependent, post-tetanic calcium transients induced by electrostimulation of adult skeletal muscle fibers. J. Gen. Physiol. 136, 455–467. doi: 10.1085/jgp.200910397
Chaudhuri, D., Sancak, Y., Mootha, V. K., and Clapham, D. E. (2013). MCU encodes the pore conducting mitochondrial calcium currents. eLife 2:e00704.
Chemello, F., Mammucari, C., Gherardi, G., Rizzuto, R., Lanfranchi, G., and Cagnin, S. (2015). Gene expression changes of single skeletal muscle fibers in response to modulation of the mitochondrial calcium uniporter (MCU). Genom. Data 5, 64–67. doi: 10.1016/j.gdata.2015.05.023
Cortassa, S., Aon, M. A., Marban, E., Winslow, R. L., and O’Rourke, B. (2003). An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 84, 2734–2755. doi: 10.1016/s0006-3495(03)75079-6
Diaz-Vegas, A., Eisner, V., and Jaimovich, E. (2019). Skeletal muscle excitation-metabolism coupling. Arch. Biochem. Biophys. 664, 89–94.
Diaz-Vegas, A. R., Cordova, A., Valladares, D., Llanos, P., Hidalgo, C., Gherardi, G., et al. (2018). Mitochondrial calcium increase induced by RyR1 and IP3R channel activation after membrane depolarization regulates skeletal muscle metabolism. Front. Physiol. 9:791. doi: 10.3389/fphys.2018.00791
Eccles, J. C., Eccles, R. M., and Kozak, W. (1962). Further investigations on the influence of motoneurones on the speed of muscle contraction. J. Physiol. 163, 324–339. doi: 10.1113/jphysiol.1962.sp006978
Fan, M., Zhang, J., Tsai, C.-W., Orlando, B. J., Rodriguez, M., Xu, Y., et al. (2020). Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 582, 129–133. doi: 10.1038/s41586-020-2309-6
Goldspink, D. F., Morton, A. J., Loughna, P., and Goldspink, G. (1986). The effect of hypokinesia and hypodynamia on protein turnover and the growth of four skeletal muscles of the rat. Pfluger. Archiv. 407, 333–340. doi: 10.1007/bf00585311
Granatiero, V., Pacifici, M., Raffaello, A., De Stefani, D., and Rizzuto, R. (2019). Overexpression of mitochondrial calcium uniporter causes neuronal death. Oxid. Med. Cell Longev. 2019:1681254.
Gundersen, K. (2011). Excitation-transcription coupling in skeletal muscle: the molecular pathways of exercise. Biol. Rev. Cambr. Philos. Soc. 86, 564–600. doi: 10.1111/j.1469-185x.2010.00161.x
Hood, D. A., Zak, R., and Pette, D. (1989). Chronic stimulation of rat skeletal muscle induces coordinate increases in mitochondrial and nuclear mRNAs of cytochrome-c-oxidase subunits. Eur. J. Biochem. 179, 275–280. doi: 10.1111/j.1432-1033.1989.tb14551.x
Hurst, S., Hoek, J., and Sheu, S. S. (2017). Mitochondrial Ca(2+) and regulation of the permeability transition pore. J. Bioenerget. Biomembr. 49, 27–47.
Jaimovich, E., Reyes, R., Liberona, J. L., and Powell, J. A. (2000). IP(3) receptors, IP(3) transients, and nucleus-associated Ca(2+) signals in cultured skeletal muscle. Am. J. Physiol. Cell Physiol. 278, C998–C1010.
Jorquera, G., Altamirano, F., Contreras-Ferrat, A., Almarza, G., Buvinic, S., Jacquemond, V., et al. (2013). Cav1.1 controls frequency-dependent events regulating adult skeletal muscle plasticity. J. Cell Sci. 126(Pt 5), 1189–1198. doi: 10.1242/jcs.116855
Joseph, A. M., Adhihetty, P. J., and Leeuwenburgh, C. (2016). Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J. Physiol. 594, 5105–5123. doi: 10.1113/jp270659
Khodabukus, A., and Baar, K. (2015). Contractile and metabolic properties of engineered skeletal muscle derived from slow and fast phenotype mouse muscle. J. Cell. Physiol. 230, 1750–1757. doi: 10.1002/jcp.24848
Klitgaard, H., Mantoni, M., Schiaffino, S., Ausoni, S., Gorza, L., Laurent-Winter, C., et al. (1990). Function, morphology and protein expression of ageing skeletal muscle: a cross-sectional study of elderly men with different training backgrounds. Acta Physiol. Scand. 140, 41–54. doi: 10.1111/j.1748-1716.1990.tb08974.x
Kneppers, A., Leermakers, P., Pansters, N., Backx, E., Gosker, H., van Loon, L., et al. (2019). Coordinated regulation of skeletal muscle mass and metabolic plasticity during recovery from disuse. FASEB J. 33, 1288–1298. doi: 10.1096/fj.201701403rrr
Kwong, J. Q., Huo, J., Bround, M. J., Boyer, J. G., Schwanekamp, J. A., Ghazal, N., et al. (2018). The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight. 3:e121689.
Lazarowski, E. R., Boucher, R. C., and Harden, T. K. (2003). Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol. Pharmacol. 64, 785–795. doi: 10.1124/mol.64.4.785
Leone, T. C., Lehman, J. J., Finck, B. N., Schaeffer, P. J., Wende, A. R., Boudina, S., et al. (2005). PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3:e101. doi: 10.1371/journal.pbio.0030101
Loucif, H., Dagenais-Lussier, X., Beji, C., Telittchenko, R., Routy, J. P., and van Grevenynghe, J. (2020). Plasticity in T-cell mitochondrial metabolism: a necessary peacekeeper during the troubled times of persistent HIV-1 infection. Cytok. Growth Fact. Rev. 55, 26–36. doi: 10.1016/j.cytogfr.2020.02.004
Mammucari, C., Gherardi, G., Zamparo, I., Raffaello, A., Boncompagni, S., Chemello, F., et al. (2015). The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 10, 1269–1279. doi: 10.1016/j.celrep.2015.01.056
Marchi, S., Lupini, L., Patergnani, S., Rimessi, A., Missiroli, S., Bonora, M., et al. (2013). Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 23, 58–63. doi: 10.1016/j.cub.2012.11.026
McCormack, J. G., and Denton, R. M. (1989). The role of Ca2+ ions in the regulation of intramitochondrial metabolism and energy production in rat heart. Mol. Cell. Biochem. 89, 121–125.
Murgia, M., and Rizzuto, R. (2015). Molecular diversity and pleiotropic role of the mitochondrial calcium uniporter. Cell Calc. 58, 11–17. doi: 10.1016/j.ceca.2014.11.001
Nemani, N., Shanmughapriya, S., and Madesh, M. (2018). Molecular regulation of MCU: Implications in physiology and disease. Cell Calc. 74, 86–93. doi: 10.1016/j.ceca.2018.06.006
Nguyen, D. D., Kim, G., and Pae, E. K. (2016). Modulation of muscle fiber compositions in response to hypoxia via pyruvate dehydrogenase kinase-1. Front. Physiol. 7:e604. doi: 10.3389/fphys.2016.00604
O’Donnell, J. M., Doumen, C., LaNoue, K. F., White, L. T., Yu, X., Alpert, N. M., et al. (1998). Dehydrogenase regulation of metabolite oxidation and efflux from mitochondria in intact hearts. Am. J. Physiol. 274(2 Pt 2), H467–H476.
Paillard, M., Csordas, G., Szanda, G., Golenar, T., Debattisti, V., Bartok, A., et al. (2017). Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 18, 2291–2300. doi: 10.1016/j.celrep.2017.02.032
Petrie, M., Suneja, M., and Shields, R. K. (2015). Low-frequency stimulation regulates metabolic gene expression in paralyzed muscle. J. Appl. Physiol. 118, 723–731. doi: 10.1152/japplphysiol.00628.2014
Picard, M., Csukly, K., Robillard, M. E., Godin, R., Ascah, A., Bourcier-Lucas, C., et al. (2008). Resistance to Ca2+-induced opening of the permeability transition pore differs in mitochondria from glycolytic and oxidative muscles. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R659–R668.
Picard, M., Hepple, R. T., and Burelle, Y. (2012). Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am. J. Physiol. Cell Physiol. 302, C629–C641.
Putman, C. T., Dixon, W. T., Pearcey, J. A., Maclean, I. M., Jendral, M. J., Kiricsi, M., et al. (2004). Chronic low-frequency stimulation upregulates uncoupling protein-3 in transforming rat fast-twitch skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R1419–R1426.
Qiu, J., Tan, Y. W., Hagenston, A. M., Martel, M. A., Kneisel, N., Skehel, P. A., et al. (2013). Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 4:2034.
Ruas, J. L., White, J. P., Rao, R. R., Kleiner, S., Brannan, K. T., Harrison, B. C., et al. (2012). A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151, 1319–1331. doi: 10.1016/j.cell.2012.10.050
Salmons, S., and Vrbova, G. (1969). The influence of activity on some contractile characteristics of mammalian fast and slow muscles. J. Physiol. 201, 535–549. doi: 10.1113/jphysiol.1969.sp008771
Schiaffino, S., and Reggiani, C. (2011). Fiber types in mammalian skeletal muscles. Physiol. Rev. 91, 1447–1531. doi: 10.1152/physrev.00031.2010
Sembrowich, W. L., Quintinskie, J. J., and Li, G. (1985). Calcium uptake in mitochondria from different skeletal muscle types. J. Appl. Physiol. 59, 137–141. doi: 10.1152/jappl.1985.59.1.137
Shanmughapriya, S., Rajan, S., Hoffman, N. E., Zhang, X., Guo, S., Kolesar, J. E., et al. (2015). Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci. Signal. 8:ra23. doi: 10.1126/scisignal.2005673
Tarasov, A. I., Semplici, F., Ravier, M. A., Bellomo, E. A., Pullen, T. J., Gilon, P., et al. (2012). The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PLoS One 7:e39722. doi: 10.1371/journal.pone.0039722
Wan, B., LaNoue, K. F., Cheung, J. Y., and Scaduto, R. C. Jr. (1989). Regulation of citric acid cycle by calcium. J. Biol. Chem. 264, 13430–13439.
Weibel, E. R., and Hoppeler, H. (2005). Exercise-induced maximal metabolic rate scales with muscle aerobic capacity. J. Exper. Biol. 208(Pt 9), 1635–1644. doi: 10.1242/jeb.01548
Yan, Z., Lira, V. A., and Greene, N. P. (2012). Exercise training-induced regulation of mitochondrial quality. Exerc. Sport Sci. Rev. 40, 159–164. doi: 10.1097/jes.0b013e3182575599
Yi, J., Ma, C., Li, Y., Weisleder, N., Rios, E., Ma, J., et al. (2011). Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J. Biol. Chem. 286, 32436–32443. doi: 10.1074/jbc.m110.217711
Keywords: mitochondria, calcium handling, muscle plasticity, ATP release, IP3R
Citation: Quezada ER, Díaz-Vegas A, Jaimovich E and Casas M (2021) Changes in Gene Expression of the MCU Complex Are Induced by Electrical Stimulation in Adult Skeletal Muscle. Front. Physiol. 11:601313. doi: 10.3389/fphys.2020.601313
Received: 31 August 2020; Accepted: 27 November 2020;
Published: 26 January 2021.
Edited by:
Bruno Bastide, Lille University of Science and Technology, FranceReviewed by:
Kevin Foskett, University of Pennsylvania, United StatesRoland Malli, Medical University of Graz, Austria
Copyright © 2021 Quezada, Díaz-Vegas, Jaimovich and Casas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariana Casas, bWNhc2FzQG1lZC51Y2hpbGUuY2w=