 Hsin-Kuo Ko
Hsin-Kuo Ko An-Hsuan Lin3,4
An-Hsuan Lin3,4 Diahn-Warng Perng
Diahn-Warng Perng Tzong-Shyuan Lee
Tzong-Shyuan Lee Yu Ru Kou
Yu Ru Kou- 1Department of Chest Medicine, Taipei Veterans General Hospital, Taipei, Taiwan
- 2School of Medicine, National Yang-Ming University, Taipei, Taiwan
- 3Department of Physiology, School of Medicine, National Yang-Ming University, Taipei, Taiwan
- 4Department of Physiology, University of Kentucky, Lexington, KY, United States
- 5Graduate Institute and Department of Physiology, College of Medicine, National Taiwan University, Taipei, Taiwan
Toll-like receptor (TLR) 4 was originally thought to be the sole pattern recognition receptor for lipopolysaccharide (LPS). Transient receptor potential ankyrin 1 (TRPA1), a Ca2+-permeant channel, has been suggested as a non-TLR receptor membrane-bound sensor of LPS. We recently reported that TRPA1 is expressed in lung epithelial cells (LECs) and mediates lung inflammation induced by cigarette smoke. However, the role of TRPA1 in LPS-induced lung inflammation has not been conclusively defined, and its underlying cellular mechanisms remain unclear. In this study, our in vitro results showed that LPS sequentially produced a cascade of events, including the elevation of intracellular Ca2+, the activation of NADPH oxidase, increase in intracellular reactive oxygen species (ROS), the activation of mitogen-activated protein kinase (MAPK)/nuclear factor-kB (NF-κB) signaling, and the induction of IL-8. The increase in intracellular Ca2+ was inhibited by HC030031 (a TRPA1 antagonist) but was unaffected by TAK-242 (a TLR-4 inhibitor). The activation of NADPH oxidase was prevented by its inhibitor apocynin, EGTA (an extracellular Ca2+ chelator), and HC030031. The increase in intracellular ROS was attenuated by apocynin, N-acetyl-cysteine (NAC, a ROS scavenger), EGTA, and HC030031. The activation of the MAPK/NF-κB signaling was halted by NAC, EGTA, and HC030031. IL-8 induction was suppressed by HC030031 and TRPA1 siRNA, and further reduced by the combination of HC030031 and TAK-242. Our in vivo studies showed that trpa1–/– mice exhibited a reduced level of LPS-induced lung inflammation compared with wild-type mice as evidenced by the alleviations of increases in vascular permeability, inflammatory cell infiltration, inflammatory cytokine levels, oxidative stress, and MAPK signaling activation. Thus, in LECs, LPS may activate TRPA1 resulting in an increase in Ca2+ influx. The increased intracellular Ca2+ leads to NADPH oxidase activation, which causes an increase in intracellular ROS. The intracellular ROS activates the MAPK/NF-κB signaling resulting in IL-8 induction. This mechanism may possibly be at work to induce lung inflammation in mice.
Introduction
Sepsis is a systemic inflammatory state manifested by a complex interaction between infective microorganisms and host immune responses (Cecconi et al., 2018). Bacterial lipopolysaccharide (LPS), a constituent of the outer membrane of Gram-negative bacteria, is a potent inducer of pro-inflammatory activity (Neyen and Lemaitre, 2016). The lung epithelium is positioned at the interface with the environment and persistently exposed to airborne pathogens (Bals and Hiemstra, 2004). The recognition of pathogens by lung epithelial cells thus plays an important role in the lung inflammation induced by LPS (Diamond et al., 2000; Bals and Hiemstra, 2004). Toll-like receptor (TLR) 4 was originally thought to be the sole pattern recognition receptor for LPS (Neyen and Lemaitre, 2016), and its activation induces lung inflammation (Chen et al., 2019) or inflammatory responses in lung epithelial cells (Liu et al., 2018). Emerging evidence suggests that transient receptor potential ankyrin 1 (TRPA1) is a non-TLR membrane-bound sensor of LPS (Boonen et al., 2018a; Mazgaeen and Gurung, 2020). TRPA1 is a type of non-selective transmembrane cation channel that is involved in Ca2+ permeability (Fernandes et al., 2012). TRPA1 is predominately expressed in primary sensory neurons but also expressed in various non-neuronal cell types (Fernandes et al., 2012; Nassini et al., 2012; Tian et al., 2020), including lung epithelial cells (Nassini et al., 2012; Büch et al., 2013; Lin et al., 2015; Prandini et al., 2016). LPS can directly activate TRPA1 in mouse sensory neurons (Meseguer et al., 2014; Boonen et al., 2018b; Startek et al., 2018) and in human HEK293T cells transfected with TRPA1 (Soldano et al., 2016). However, studies on the functional importance of TRPA1 in LPS-induced consequences have reported equivocal findings. TRPA1 protects against LPS-induced pneumonitis (Hajna et al., 2020) and does not contribute to LPS-induced bladder inflammation (Kamei et al., 2018) in mice. However, TRPA1 mediates LPS-induced lung inflammation in mice (Liu et al., 2020) or inflammatory responses to LPS in vitro (Prandini et al., 2016; Yin et al., 2018). Thus, the role of TRPA1 in LPS-induced inflammation has not been conclusively defined.
LPS elevates the intracellular levels of NADPH oxidase-derived reactive oxygen species (ROS), which trigger inflammatory responses in lung epithelial cells (Cho et al., 2016; Dong and Yuan, 2018). NADPH oxidase is the primary enzyme system that generates ROS in mammalian cells (Bedard and Krause, 2007). LPS also induces lung inflammation via mitogen-activated protein kinase/nuclear factor-κB (MAPK/NF-κB) signaling (Cho et al., 2016; Jiang et al., 2016; Chen et al., 2019), which is sensitive to ROS (Mossman et al., 2006; Rahman and Adcock, 2006; Lin et al., 2017). We recently reported (Lin et al., 2015) that cigarette smoke can activate TRPA1 leading to an increase in Ca2+ influx in lung epithelial cells. The increased intracellular Ca2+ contributes to the activation of NADPH oxidase and results in increased intracellular ROS, which activates the MAPK/NF-κB signaling and leads to interleukin 8 (IL-8) induction (Lin et al., 2015). However, the underlying cellular mechanisms were not investigated in studies demonstrating the contribution of TRPA1 in LPS-induced inflammation (Prandini et al., 2016; Yin et al., 2018; Liu et al., 2020) and are yet to be determined.
The objectives of this study were to investigate the cellular mechanisms of the inflammatory role of TRPA1 in LPS-induced IL-8 responses using primary human bronchial epithelial cells (HBECs) with LPS exposure, and to determine the contribution of TRPA1 to LPS-induced lung inflammation using a murine model (Ueda et al., 2008).
Materials and Methods
Drugs and Reagents
Antibodies (Abs) and enzyme-linked immunosorbent assay (ELISA) kits for the detection of IL-8, IL-1β, IL-6, and macrophage inflammatory protein 2 (MIP-2) were purchased from R&D Systems (Minneapolis, MN, United States). Rabbit Ab against c-Jun N-terminal kinase (JNK) was obtained from Cell Signaling (Beverly, MA, United States). Mouse Ab against phospho-JNK was purchased from BD (San Jose, CA, United States). The Screen Quest Fluo-8 Medium Removal Calcium Assay Kit was obtained from AAT Bioquest (Sunnyvale, CA, United States). Membrane-permeable probes hydroethidine (HE) and 2’,7’-dichlorofluorescin diacetate were obtained from Molecular Probes (Eugene, OR, United States). The EnzyChrom NADP+/NADPH Assay Kit was purchased from BioAssay Systems (Hayward, CA, United States). Abs against extracellular signal-regulated kinase (ERK), phospho-ERK, and p65 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, United States). Escherichia coli LPS and mouse Abs against α-tubulin, HC030031, ethylene glycol tetraacetic acid (EGTA), N-acetyl-cysteine (NAC), apocynin, and TAK-242 were purchased from Sigma-Aldrich (St. Louis, MO, United States). Mouse Ab against TRPA1 was obtained from Calbiochem (San Diego, CA, United States). Mouse Ab against histone H1 was purchased from Millipore (Bedford, MA, United States). Mouse Ab against 4-hydroxynonenal (4-HNE) was purchased from Abcam (Cambridge, MA, United States). Scramble and TRPA1 small interfering RNAs (siRNAs) were purchased from Ambion (Austin, TX, United States). INTERFERin siRNA transfection reagent was obtained from Polyplus (New York, NY, United States). More information for LPS and antibodies used in this study are provided in Supplementary Table S1.
Cell Culture
HBECs (Cascade Biologics, Portland, OR, United States) were cultured in epithelial cell growth medium (medium 200; Cascade Biologics, United States) at 37°C in an incubator with 5% CO2 with a method described previously (Lin et al., 2015).
Assessment of Levels of Intracellular Ca2+
Intracellular Ca2+ levels were measured using Screen QuestTM Fluo-8 Medium Removal Calcium Assay Kit based on the instructions from the manufacturer.
Measurement of Levels of Intracellular ROS
Membrane-permeable HE was employed to assess the levels of ROS using the methods described previously (Liu et al., 2005); red fluorescent ethidium (ETH) is formed when HE is oxidized by ROS (Benov et al., 1998). HBECs were incubated in culture medium containing 10 μM HE at 37°C for 30 min. The cells were stimulated with LPS for the desired time and then washed and detached with trypsin/EDTA for the measurement of intracellular ROS levels. The fluorescence intensities of the culture medium and cells were analyzed using a multilabel counter (PerkinElmer, Waltham, MA, United States) at 530 nm excitation and 620 nm emission for ETH. Images of the cells were obtained using a Nikon TE2000-U florescence microscope (Tokyo, Japan).
Determination of NADPH Oxidase Activity
The activity of NADPH oxidase was examined using an EnzyChromTM NADP+/NADPH Assay Kit according to the manufacturer’s instructions. The assay kit measures the change in NADP+/NADPH ratio in the samples of cellular lysates to reflect the relative NADPH oxidase activity.
Transfection of siRNA in HBECs
HBECs were transfected with scramble or TRPA1 siRNA using INTERFERin siRNA transfection reagent for 24 h. The information of the TRPA1 siRNA was reported previously (Lin et al., 2015).
Mouse Model of LPS-Induced Lung Inflammation
All animal experiments were approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital (Approval Number: IACUC 2017-266). Briefly, 8 week old male trpa1–/– mice (Jackson Laboratory, Maine, United States) and wild-type C57BL/6J mice (National Laboratory Animal Center, Taipei, Taiwan) were randomly assigned to four study groups. Either E. coli LPS (20 mg/kg) or PBS (the vehicle) was administered via intraperitoneal injection (Ueda et al., 2008). The groups were PBS-wild-type, LPS-wild-type, PBS-trpa1–/–, and LPS-trpa1–/–. At the end of each experiment after 24 h of exposure to PBS or LPS, the mice were euthanized using CO2.
Bronchoalveolar Lavage Fluid (BALF) and Lung Tissues
The method of preparation of BALF in the right lung has been described previously (Lin et al., 2015). The BALF samples were centrifuged at 350 × g for 5 min at 4°C, and the supernatant of the first lavage fluid was stored at -80°C for the analysis of total protein using a Bio-Rad protein assay reagent (Bio-Rad Laboratories, Hercules, CA, United States). The cell pellets of the BALF samples were re-suspended in PBS for cell counting. Furthermore, the right lung was stored at -80°C for subsequent analysis.
Histological Assessments
The left lung was fixed with 4% paraformaldehyde and embedded in paraffin. The tissue blocks were cut into 8 μm sections. The sections were deparaffinized, rehydrated, stained with hematoxylin and eosin (H&E), and viewed under a microscope (Motic TYPE 102M, Xiamen, China). A pathologist who was blinded to the data performed the histological assessments based on a method described previously (Yu et al., 2012). The assessment was scored on a scale of 0 (normal) to 5 (maximal). The degree of leukocyte infiltration in peribronchiolar areas was judged by the number of infiltrated leucocytes. The sum of the score of cell infiltration and damage levels, including the thickening of the alveolar walls and epithelium yielded the lung inflammatory score.
Western Blot Analysis
Aliquots of cell lysates or tissue lysates were separated on 8–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transblotted onto ImmobilonTM-P membrane (Millipore). The blots were blocked and incubated with various primary antibodies and the appropriate secondary antibodies. Specific protein bands were detected using an enhanced chemiluminescence kit (PerkinElmer). Quantifications of band densities were performed using the ImageQuant 5.2 software (Healthcare Bio-Sciences, Philadelphia, PA, United States).
Measurement of 4-Hydroxynonenal (4-HNE) Level
The levels of 4-HNE-modified proteins in lung tissues were measured by Western blot analysis to serve as an index of oxidative stress (Connor et al., 2012).
Measurements of IL-8, IL-1β, IL-6, and MIP-2 Concentrations
The appropriate ELISA kits were employed to determine the concentrations of IL-8, IL-1β, and IL-6 in culture medium and the concentrations of MIP-2 in BALF and lung tissue.
Statistical Analysis
The results are presented as mean ± standard error of mean (SEM). Statistical evaluations involved one-way ANOVA followed by Dunnett’s test or Fisher’s least significant difference for multiple comparisons as appropriate. Differences were considered statistically significant at P < 0.05.
Results
LPS Increases the Production of Inflammatory Cytokines in HBECs
We first studied the effect of LPS on the production of IL-8 in HBECs. The exposure of HBECs to various concentrations (0–2 μg/ml) of LPS for 24 h resulted in a concentration-dependent increase in IL-8 levels in the cell medium (Figure 1A). Furthermore, the exposure of HBECs to 1 μg/ml LPS for up to 24 h increased the IL-8 levels in cell medium (Figure 1B) and increased the release of IL-1β and IL-6 from HBECs in a time-dependent manner (Figures 1C,D). Thus, 1 μg/ml LPS was selected as the standard challenge for subsequent studies.
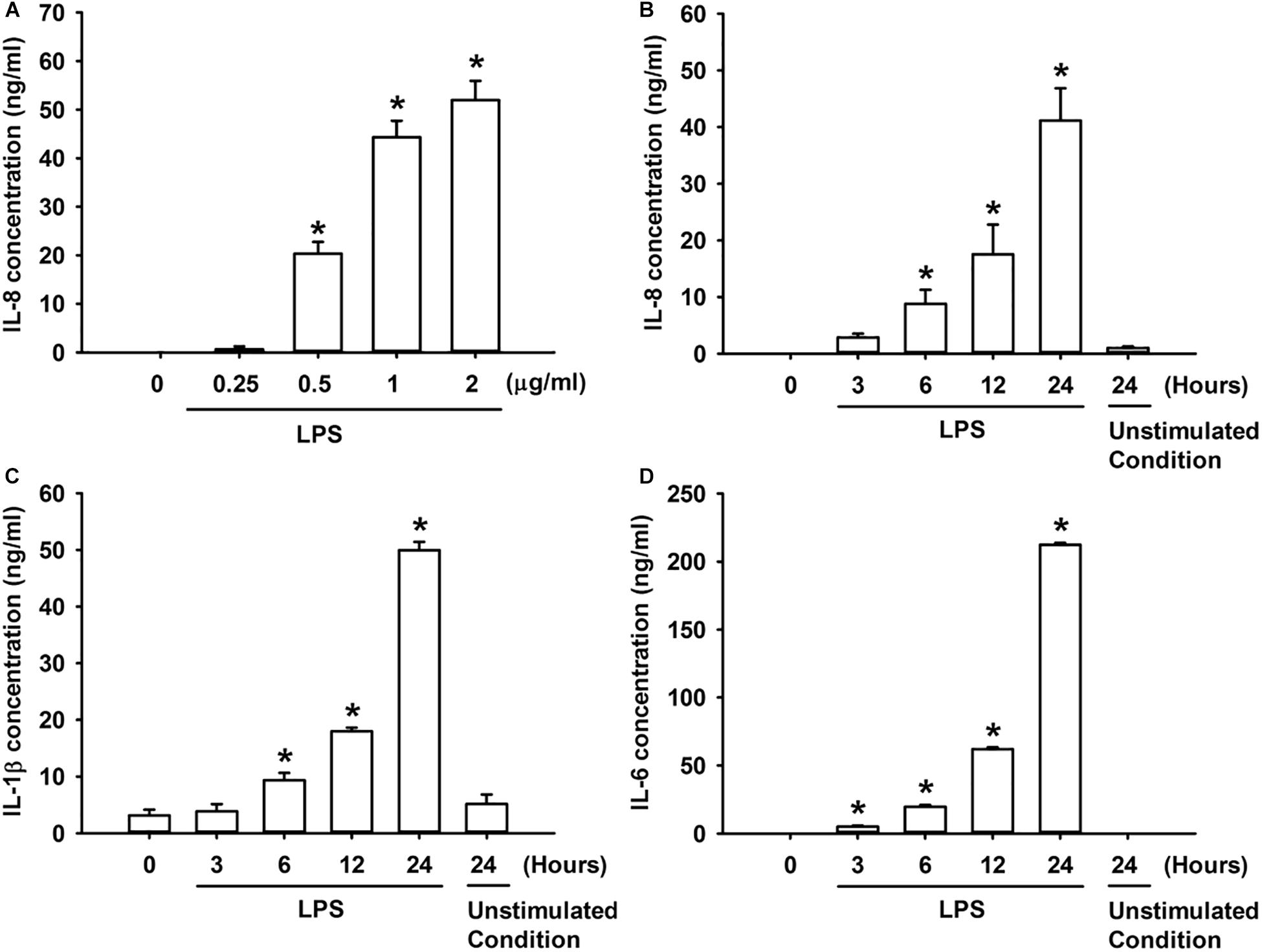
Figure 1. LPS increases the production of IL-8 (A,B), IL-1β (C), and IL-6 (D) in HBECs. (A) Cells were exposed to 0–2 μg/ml LPS for 24 h. (B–D) Cells were exposed to medium alone at time 0 for 24 h (unstimulated condition) or to 1 μg/ml LPS for the indicated times. Protein levels of IL-8 (A,B), IL-1β (C), and IL-6 (D) in the culture medium were determined by ELISA. Data in each group are mean ± SEM from four independent experiments. *P < 0.05 vs. time 0.
TRPA1 Mediates LPS-Induced IL-8 Expression in HBECs
We then explored the role of TRPA1 in the inflammatory responses of HBECs to LPS. The exposure of HBECs to LPS for 24 h increased the intracellular (Figure 2A) and extracellular expression of IL-8 (Figure 2B), which were largely or nearly prevented by pretreatment with the TRPA1 antagonist, HC030031 (9 μM); this dose was then adapted from our recent study (Lin et al., 2015). Additionally, pretreatment with TRPA1 siRNA (0, 25, and 50 nM) effectively reduced the expression of TRPA1 protein in HBECs (Figure 2C). Importantly, the induction of IL-8 by LPS was substantially attenuated by pretreatment with TRPA1 siRNA (50 nM), but was unaffected by pretreatment with scramble siRNA (Figure 2D).
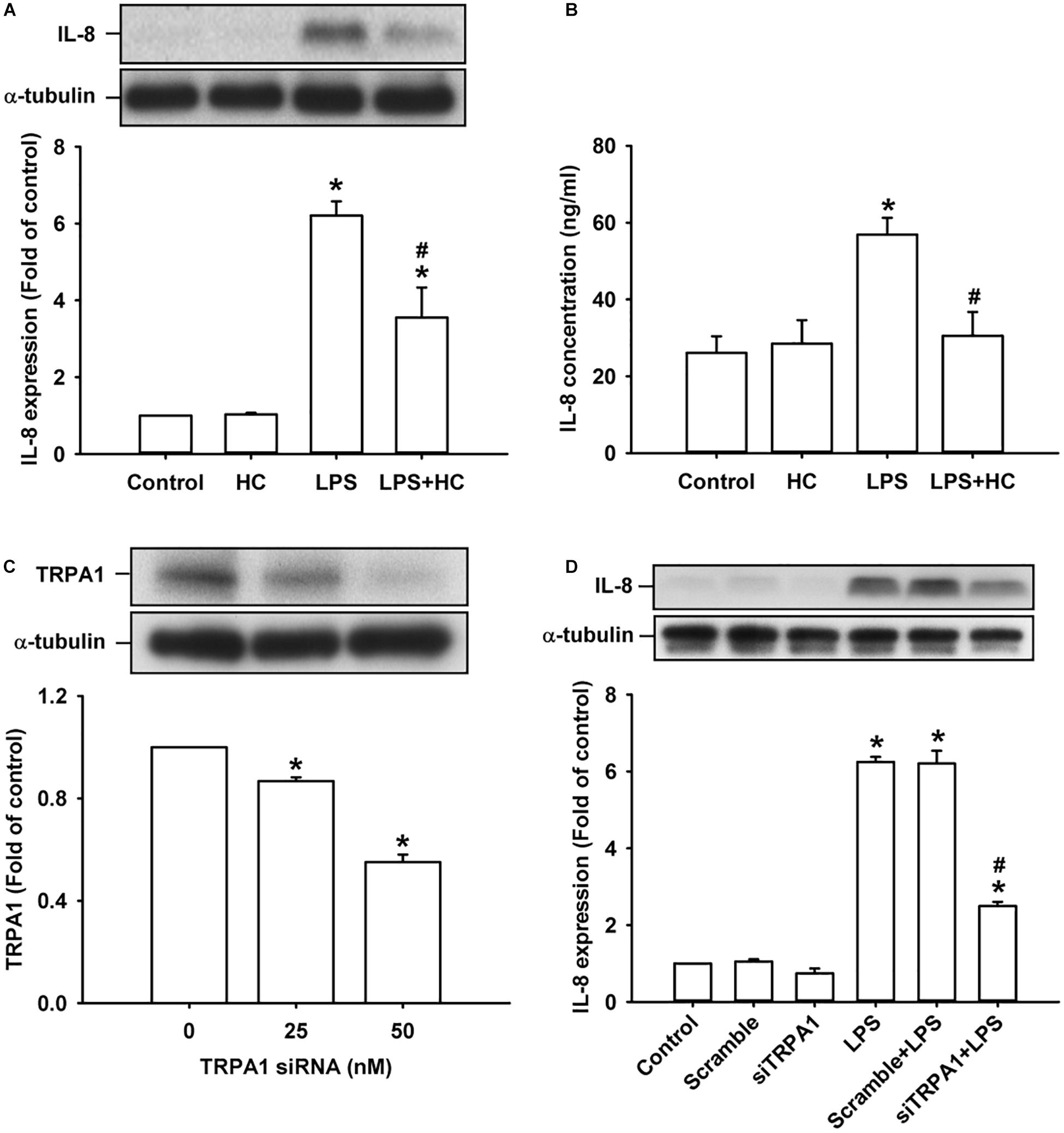
Figure 2. The LPS-induced IL-8 expression was alleviated by treatment with a TRPA1 antagonist or TRPA1 siRNA in HBECs. (A,B) Cells were exposed to medium alone or 1 μg/ml LPS for 24 h with or without pretreatment of HC030031 (HC, a TRPA1 antagonist, 9 μM). (C) Cells were treated with different concentrations of TRPA1 siRNA (0, 25, and 50 nM). (D) Cells were incubated with medium alone or 1 μg/ml LPS for 24 h with pretreatment of 50 nM siRNA (siTRPA1) or scramble siRNA. The protein levels in cell lysate (A,C,D) and cell medium (B) were measured using Western blot and ELISA, respectively. Data in each group are mean ± SEM from four independent experiments. *P < 0.05 vs. the control; #P < 0.05 vs. LPS alone.
TRPA1 Mediates LPS-Induced Increase in Intracellular ROS via the Ca2+-Dependent Activation of NADPH Oxidase in HBECs
We further studied the role of TRPA1 in evoking the early responses of HBECs to LPS. The exposure of HBECs to LPS immediately produced two peak increases in intracellular Ca2+ level at 30 s and 2 min after treatment initiation; this increase rapidly declined 3 min after treatment initiation (Figure 3A). The LPS-induced increase in intracellular Ca2+ level measured at 2 min was inhibited by pretreatment with TRPA1 antagonist, HC030031. The intracellular ROS level in HBEC was substantially increased 10 min after exposure to LPS and reached the peak 15 min after treatment initiation (Figure 3B). The LPS-induced increase in intracellular ROS was remarkably attenuated by pretreatment with EGTA (an extracellular Ca2+ chelator), apocynin (an inhibitor of NADPH oxidase), NAC (a ROS scavenger), or HC030031 (Figure 3C). Further analysis revealed that LPS considerably increased the activity of NADPH oxidase at 10 min after exposure, and this effect was markedly inhibited by pretreatment with EGTA, apocynin, or HC030031 (Figure 3D).
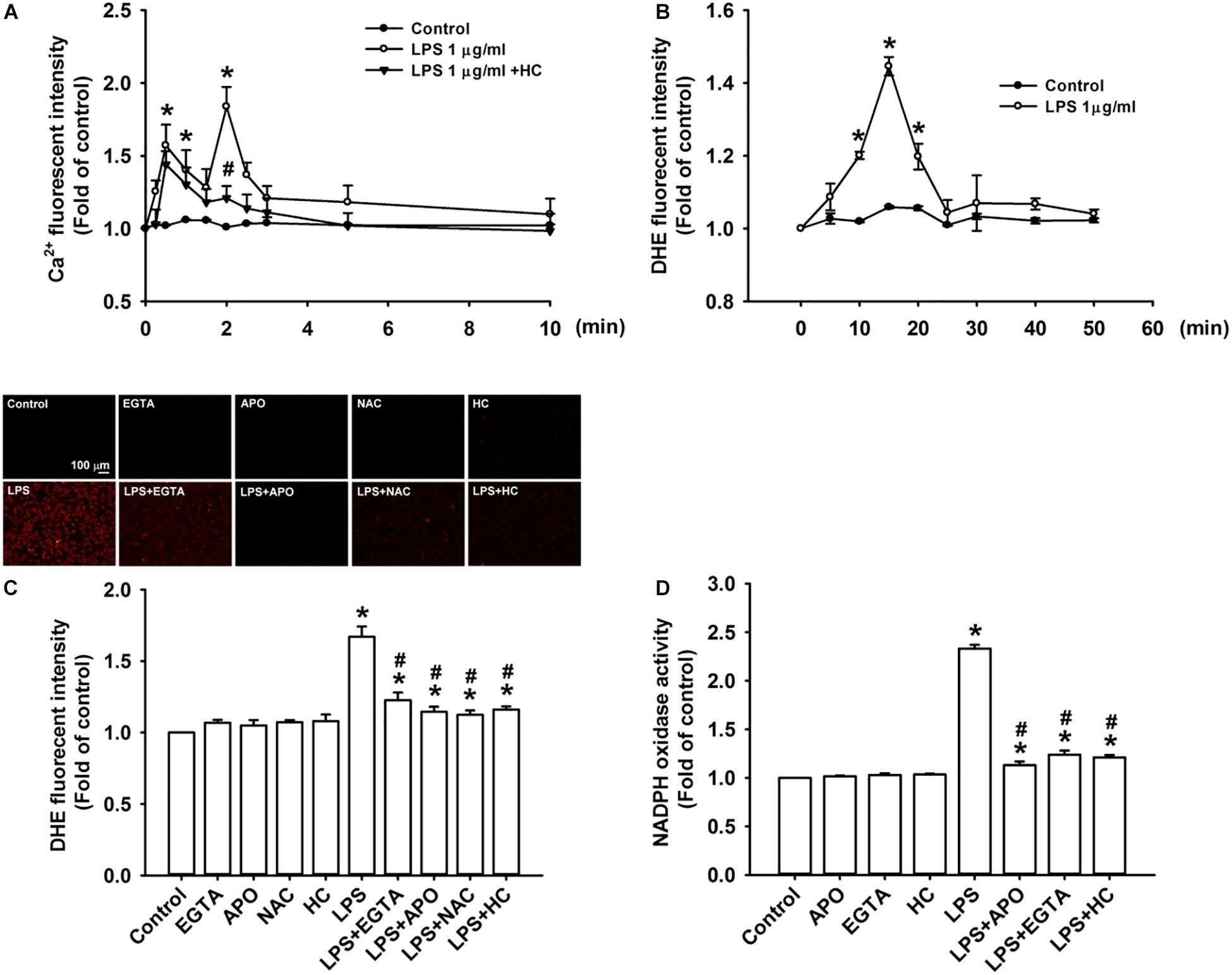
Figure 3. TRPA1 mediates LPS-induced increase in intracellular ROS via the Ca2+-dependent activation of NADPH oxidase in HBECs. (A) Cells were exposed to medium alone, 1 μg/ml LPS, or the combination of LPS and HC030031 (HC, 9 μM) for 0, 10, 30, 60, 90, and 120 s and 3, 5, and 10 min. (B) Cells were treated with or without LPS (1 μg/ml) for indicated times. (C) Cells were exposed to medium alone or 1 μg/ml LPS for 15 min after pretreatment with EGTA (500 μM), apocynin (APO, 150 μM), NAC (1 mM), or HC (9 μM). HC, EGTA, APO, and NAC are a TRPA1 antagonist, an extracellular Ca2+ chelator, an inhibitor of NADPH oxidase, and a ROS scavenger, respectively. (D) Cells were exposed to medium alone or 1 μg/ml LPS for 10 min after pretreatment with EGTA (500 μM), APO (150 μM), or HC (9 μM). Intracellular levels of Ca2+ (A) and ROS (B,C) and NADPH oxidase activity (D) were measured by Fluo-8, HE/DTH, and NADP+/NADPH fluorescent probe assays, respectively. Data in each group are mean ± SEM from four independent experiments. *P < 0.05 vs. the control; #P < 0.05 vs. LPS alone.
TRPA1 Mediates the LPS-Induced Activation of ERK/JNK/NF-κB Signaling in HBECs via the Functions of Intracellular Ca2+ and ROS
The activation of ERK, JNK, and NF-κB is known to be a signaling pathway that is central to the induction of IL-8 in HBECs (Lin et al., 2015, 2017). ERK and JNK are two MAPK subfamilies (Mossman et al., 2006). We found that the exposure of HBECs to LPS for 3 h increased the levels of phosphorylated ERK (Figure 4A) and phosphorylated JNK (Figure 4B). The incubation of HBECs with LPS for 9 h increased the expression of the p65 subunit of NF-κB in the nucleus (Figure 4C). This activation of MAPK/NF-κB signaling by LPS was greatly reduced by pretreatment with EGTA, NAC, or HC030031.
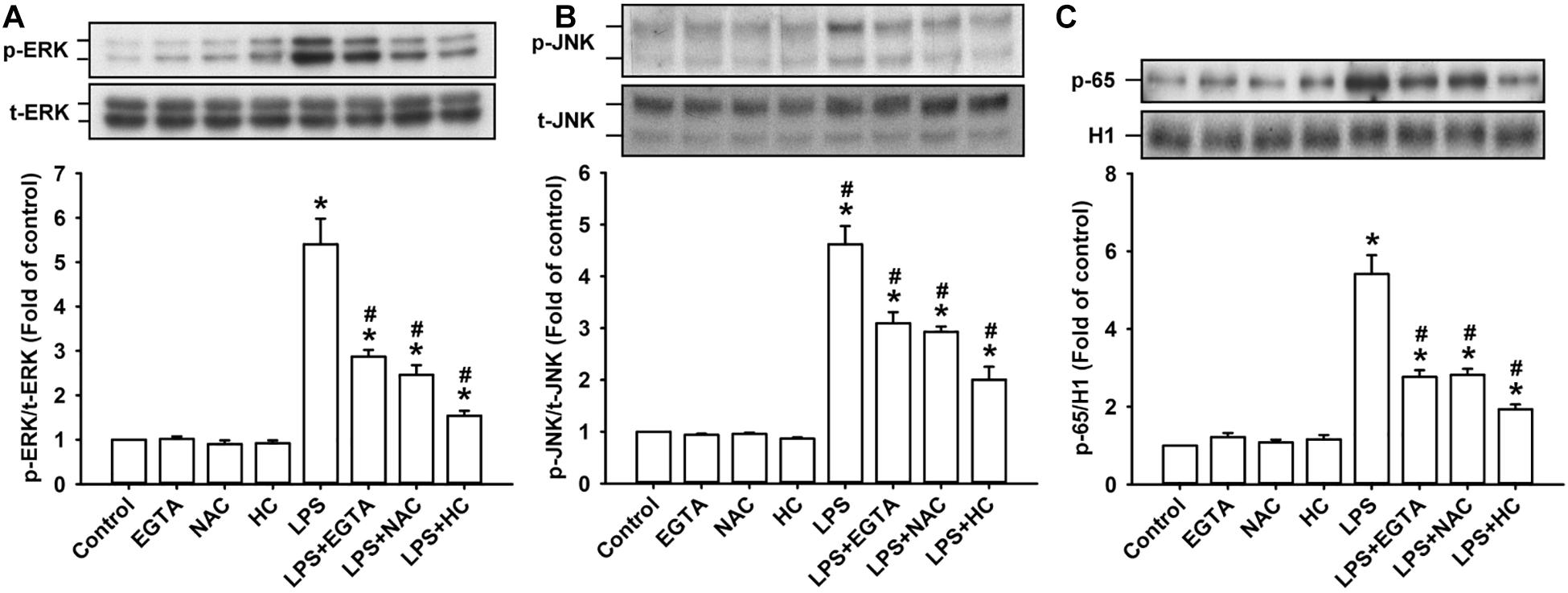
Figure 4. TRPA, intracellular Ca2+, and intracellular ROS contribute to the LPS-induced activation of the ERK/JNK/NF-κB signaling in HBECs. Cells were exposed to medium alone or 1 μg/ml LPS for 3 (A,B) and 9 h (C) and pretreated with EGTA (an extracellular Ca2+ chelator, 500 μM), NAC (ROS scavenger, 1 mM), or HC030031 (HC, a TRPA1 antagonist, 9 μM). Protein expression was analyzed by Western blot. The increased phosphorylation of kinases indicates the activation of ERK and JNK. The increased presence of the p65 subunit in the nucleus indicates the activation of NF-κB. Data in each group are mean ± SEM from four independent experiments. *P < 0.05 vs. the control; #P < 0.05 vs. LPS alone.
The Contribution of TRPA1 to LPS-Induced Responses in HBECs Is Independent of TLR4
The activation of TLR4 by LPS induces inflammatory responses in lung epithelial cells (Chen et al., 2019). We found that the exposure of HBECs to LPS for 2 min caused an increase in intracellular Ca2+ level, and this increase was unaffected by pretreatment with TAK-242 (a TLR4 inhibitor) (Matsunaga et al., 2011) as shown in Figure 5A. Also, the LPS-induced IL-8 production measured at 24 h after exposure in HBECs was substantially attenuated by pretreatment with HC030031 or TAK-242 alone (Figure 5B). Notably, the LPS-induced IL-8 production was further reduced by pretreatment with the combination of HC030031 and TAK-242 (Figure 5B).
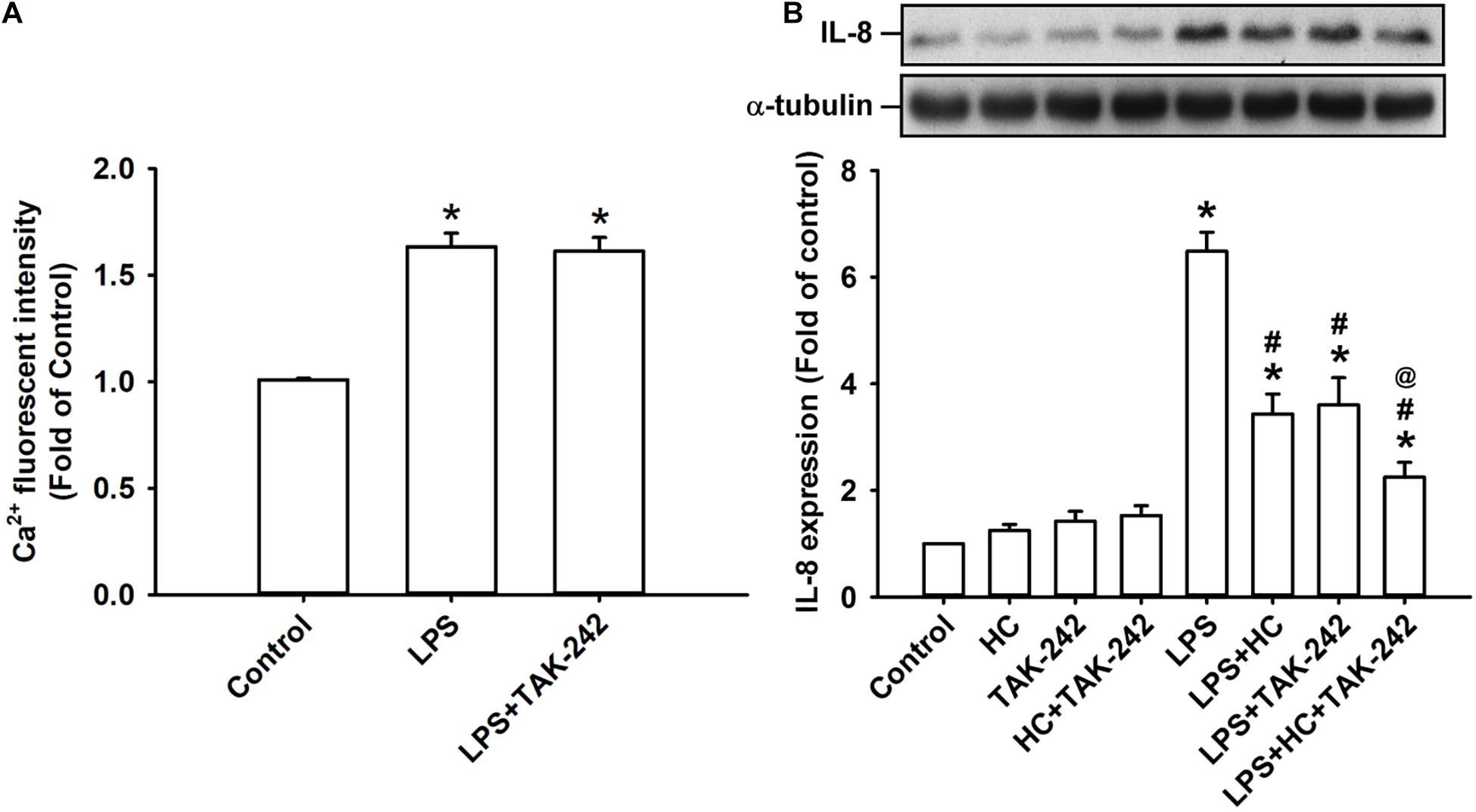
Figure 5. Both TRPA1 and TLR4 contribute to LPS-induced IL-8 expression. (A) Cells were exposed to medium alone, 1 μg/ml LPS for 24 h, or 1 μg/ml LPS with pretreatment with TAK-242 (a TLR4 antagonist, 1 μM). Intracellular levels of Ca2 + were measured by Fluo-8 fluorescent probe assays. (B) Cells were exposed to medium alone or 1 μg/ml LPS for 24 h and pretreated with HC030031 (HC, a TRPA1 antagonist, 9 mM), TAK-242 (a TLR4 antagonist, 1 μM), or the combination of HC and TAK-242. The protein levels in cell lysate were measured using Western blot. Data in each group are mean ± SEM from four independent experiments. *P < 0.05 versus the control; #P < 0.05 versus LPS alone; @P < 0.05 versus LPS + HC or LPS + TAK-242.
LPS-Induced Inflammation and Oxidative Stress in Lung Tissues Are Lessened in trpa1–/– Mice
The histological lung section of LPS-exposed wild-type mice displayed an extensive infiltration of inflammatory cells, thickening of the alveolar walls, and the presence of abnormal re-epithelialization (Figure 6A). These pathohistological changes were found to be lesser in the LPS-exposed trpa1–/– mice (Figure 6A). Comparisons of the group data of lung inflammatory scores confirmed the difference in the degree of lung inflammation between the LPS-exposed wild-type and trpa1–/– mice (Figure 6B). Furthermore, as compared to the PBS-exposed wild-type mice, LPS-exposed wild-type mice exhibited increases in total protein levels (Figure 7A), total cell counts (Figure 7B), differential cell counts (Figure 7C), and MIP-2 levels (Figure 7D) in BALF, and increases in MIP-2 levels (Figure 7E) in lung tissue samples. LPS-exposed wild-type mice also displayed increased levels of phosphorylated ERK (Figure 8A), phosphorylated JNK (Figure 8B), and 4-HNE (Figure 8C) in lung tissue samples. These indices of inflammation, the activation of MAPK signaling, and oxidative stress were remarkably lower in trpa1–/– mice exposed to LPS (Figures 6–8).
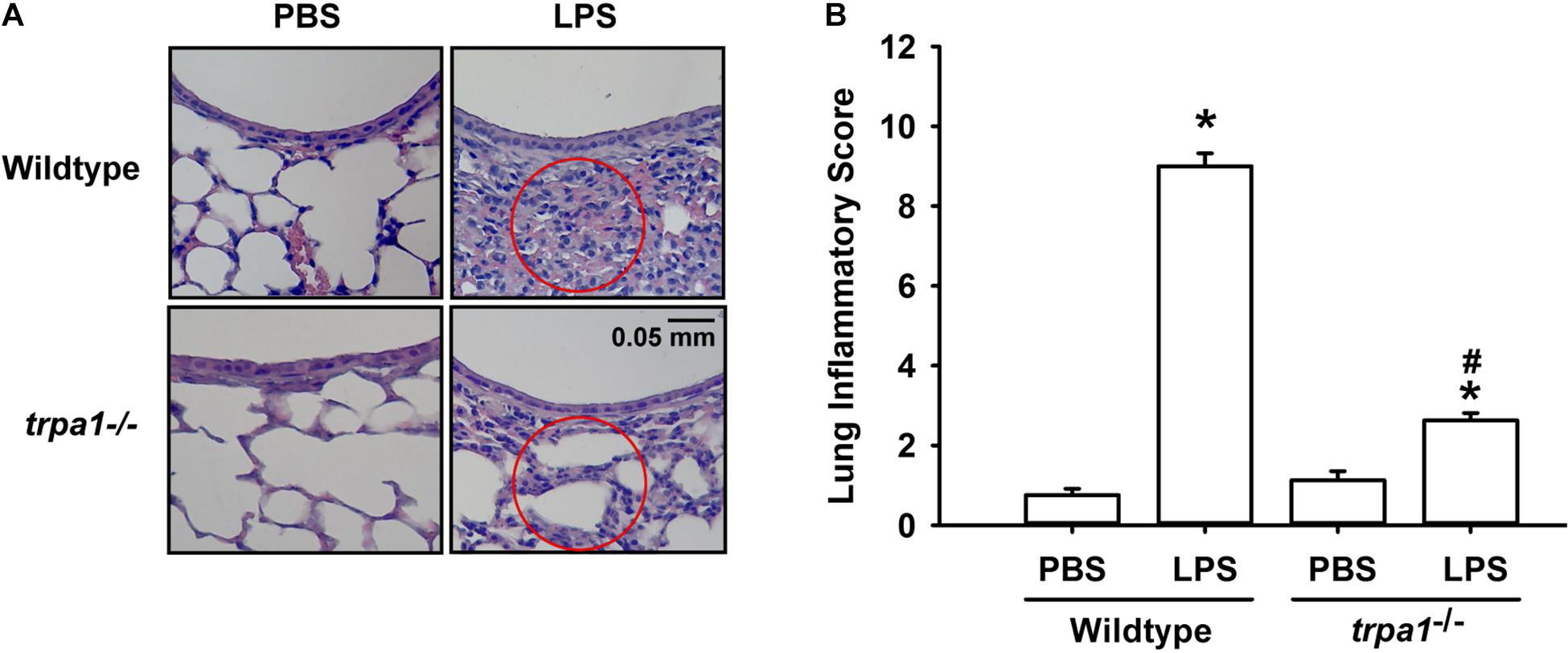
Figure 6. LPS-induced lung inflammation and lung inflammatory scores are reduced in trpa1– /– mice. Mice were treated with an intraperitoneal injection of E. coli LPS (20 mg/kg) or PBS (the vehicle). (A) Representative images of H&E-stained lung sections obtained from PBS- or LPS-exposed wild-type and trpa1– /– mice. Circles indicate the areas of inflammatory cell infiltration. (B) Lung inflammatory scores were calculated according to the sum of the levels of cell infiltration and damage levels assessed from lung sections. Data in each group are mean ± SEM from eight mice. *P < 0.05 vs. the PBS exposure group in both genotypes; #P < 0.05 vs. the LPS-exposed wild-type group.
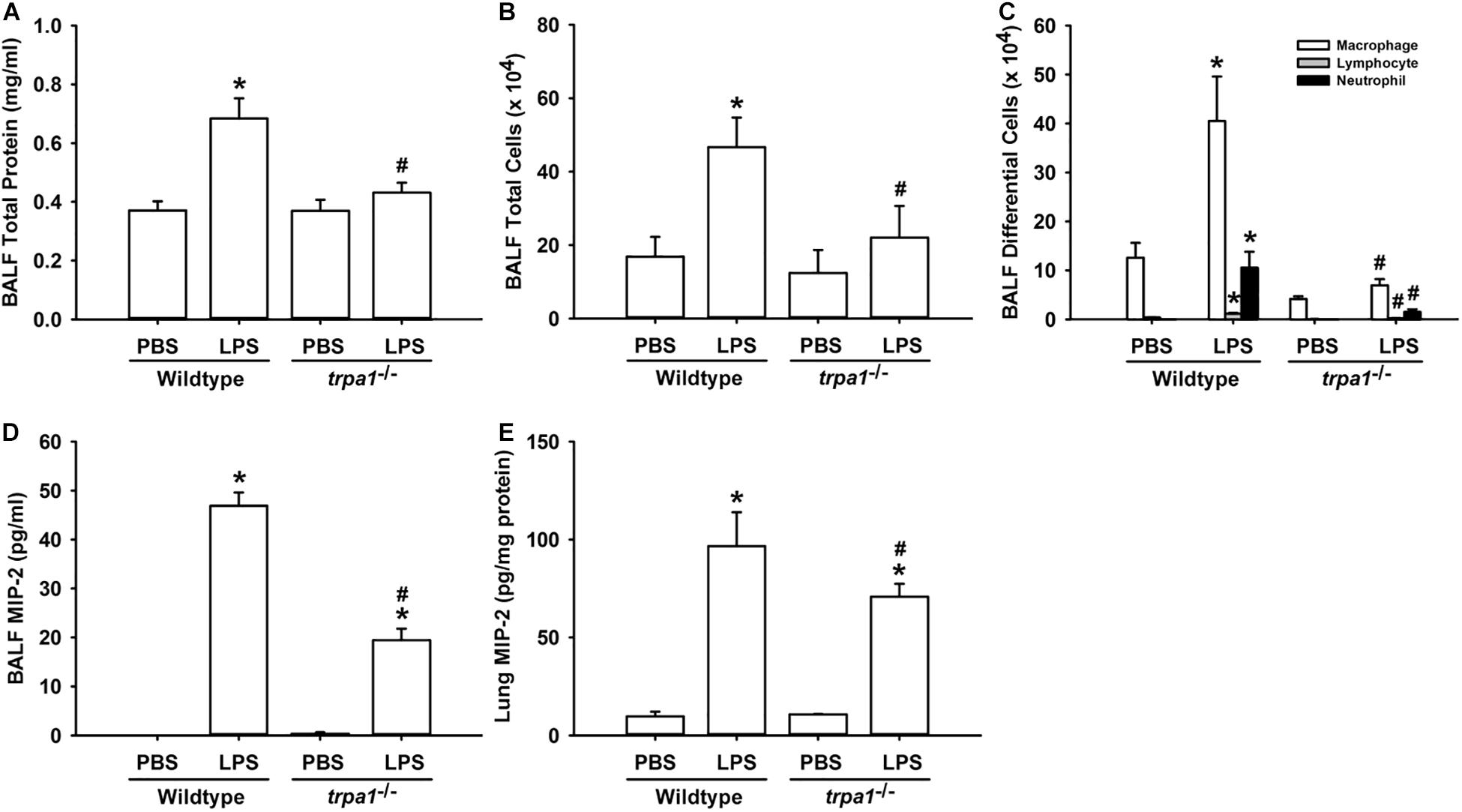
Figure 7. LPS-induced inflammatory responses in the lungs are alleviated in trpa1– /– mice. Mice were treated with an intraperitoneal injection of E. coli LPS (20 mg/kg) or PBS (the vehicle). Total protein contents (A), total cell count (B), and differential cell count (C) in the BALF at 24 h after PBS or LPS treatment as indications of lung inflammation. Levels of MIP-2 in BALF (D) and lung tissues (E) were analyzed by ELISA. Data in each group are mean ± SEM from eight mice. *P < 0.05 vs. the PBS-exposed group in both genotypes; #P < 0.05 vs. the LPS-exposed wild-type group.
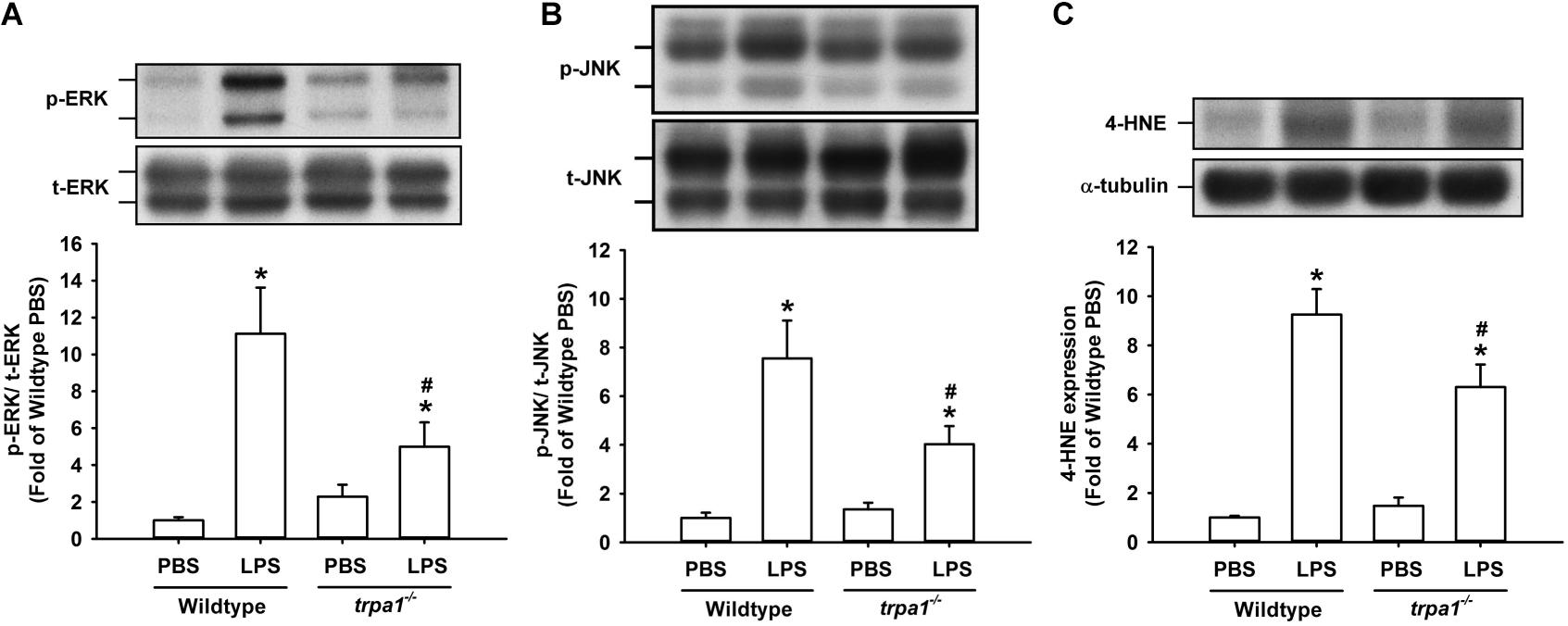
Figure 8. LPS-induced ERK/JNK activation and oxidative stress in lung tissues are reduced in trpa1– /– mice. Mice received an intraperitoneal injection of E. coli LPS (20 mg/kg) or PBS (the vehicle). (A,B) The increased phosphorylation of kinases indicates the activation of ERK and JNK. (C) LPS-induced increase in oxidative stress was indicated by increased 4-HNE expression. Protein expression was analyzed by Western blot. Data in each group are mean ± SEM from eight mice. *P < 0.05 vs. the PBS-exposed group in both genotypes; #P < 0.05 vs. the LPS-exposed wild-type group.
Discussion
Our in vitro studies showed that LPS increased the production of several inflammatory mediators, including IL-8, IL-1β, and IL-6, in HBECs (Figure 1). All these inflammatory mediators are important for the induction of LPS-induced lung inflammation (Tsuruta et al., 2007; Planagumà et al., 2015). We then chose IL-8 as the target response to study the contribution of TRPA1 and its underlying cellular mechanisms. The contribution of TRPA1 as evidenced by our finding is that TRPA1 antagonist or TRPA1 siRNA substantially reduced the LPS-induced IL-8 (Figure 2). Mechanistically, we demonstrated that LPS sequentially elevated Ca2+ level, increased ROS level, and activated MAPK/NF-κB signaling in HBECs. We identified the cascade of these cellular events using various experimental interventions. Initially, exposure to LPS activated the TRPA1 of HBECs resulting in an increase in Ca2+ influx (Figure 3). This increase in intracellular Ca2+ then promoted the NADPH oxidase activation, which led to the elevation of the intracellular ROS level (Figure 3). Finally, the increased ROS activated the MAPK/NF-κB signaling pathway (Figure 4), which resulted in the IL-8 induction. We further showed that the contribution of TRPA1 is at least in part independent of TRL4 using a TLR4 antagonist as the intervention (Figure 5). These observations suggest that the activation of lung epithelial TRPA1 by LPS contributes to the induction of IL-8 via a Ca2+-dependent signaling pathway in HBECs. This inflammatory signaling is similar to that reported by our recent study when cigarette smoke was used as the stimulus to activate lung epithelial TRPA1 (Lin et al., 2015).
NADPH oxidase-derived ROS (Cho et al., 2016; Dong and Yuan, 2018) and MAPK/NF-κB signaling (Cho et al., 2016; Jiang et al., 2016; Chen et al., 2019) are involved in LPS-induced lung inflammation. In this study, the LPS-induced increase in intracellular Ca2+ appears to be mediated though TRPA1. This increase in intracellular Ca2+ may serve as an important trigger to activate an inflammatory signaling for the induction of IL-8. We found that this Ca2+ signaling is essential for the activation of NADPH oxidase, which leads to an excess production of intracellular ROS. Indeed, intracellular Ca2+ has been shown to be an upstream signal for the NADPH oxidase activation during cellular stress (Trebak et al., 2010; Jiang et al., 2011). Our finding that intracellular ROS subsequently activated MAPK/NF-κB signaling is not surprising, because this pathway is ROS-sensitive (Mossman et al., 2006; Rahman and Adcock, 2006; Lin et al., 2017). We, however, cannot exclude the possibility that the TRPA1-mediated increase in intracellular Ca2+ also serves as a Ca2+ signal to activate downstream inflammatory signaling. For example, an increased intracellular Ca2+ can activate the MAPK/NF-κB pathway to induce IL-8 in lung epithelial cells (Carmona et al., 2010). Thus, lung epithelial TRPA1 may detect the presence of LPS and transduce this information into the transcriptional regulation of lung inflammation via a Ca2+-dependent signaling pathway. Our notion is in line with the current concept that TRPA1 is a non-TLR membrane-bound sensor of LPS (Boonen et al., 2018a; Mazgaeen and Gurung, 2020). This concept is supported by increasing evidence that LPS can directly activate TRPA1 (Meseguer et al., 2014; Soldano et al., 2016; Boonen et al., 2018b) via a mechanism that involves the mechanical perturbations of the plasma membrane (Boonen et al., 2018a; Startek et al., 2018; Mazgaeen and Gurung, 2020). In fact, TRPA1 is not the only type of TRP channel that can detect the presence of LPS (Boonen et al., 2018a; Mazgaeen and Gurung, 2020). For example, LPS also activates transient receptor potential vanilloid 4 (TRPV4) within seconds in lung epithelial cells and produce protective responses, including direct antimicrobial action and increase in airway clearance (Alpizar et al., 2017). In our study, we found that LPS produced two peak increases in intracellular Ca2+ level at 30 s and 2 min after exposure and the first peak increase was unaffected by pretreatment with TRPA1 antagonist (Figure 3A). Perhaps, the activation of other type(s) of TRP channels by LPS is responsible for the first peak increase in Ca2+. Collectively, the present study further characterized the LPS-triggered inflammatory signaling in lung epithelial cells and showed that the process depends on having a functional TRPA1 and an influx of Ca2+. This study provides additional evidence for the understanding of the early events involved in the induction of IL-8 by LPS in HBECs.
In this study, we further demonstrated that trpa1–/– mice displayed a lower level of LPS-induced lung inflammation compared with wild-type mice. This observation was based on our findings regarding alleviations of increases in vascular permeability, inflammatory cell infiltration, inflammatory cytokine levels, oxidative stress, and the activation of inflammatory MAPK signaling (Figures 6–8). In our previous study, we have shown that TRPA1 is expressed in the lung epithelium of wild-type mice as assessed by en face immunostaining (Lin et al., 2015). Other investigators have suggested that lung epithelium plays an important role in the initiation and progression of LPS-induced lung inflammation (Diamond et al., 2000; Bals and Hiemstra, 2004). Therefore, a lack of epithelial TRPA1 in the trpa1–/– mice may be at least in part responsible for the reduced lung inflammation observed in our trpa1–/– mice. TRPA1 is expressed in other types of lung cells, such as smooth muscle cells, fibroblasts, and macrophages (Nassini et al., 2012; Tian et al., 2020), all of which may participate in the development of LPS-induced lung inflammation. Accordingly, the lack of TRPA1 in these lung cells may also contribute to the reduction in lung inflammation that was observed in our trpa1–/– mice.
Previous findings regarding the role of TRPA1 in LPS-induced inflammation have been equivocal. By comparing the responses between wild-type and trpa1–/– mice, Hajna et al. (2020) demonstrated that TRPA1 protects against LPS-induced acute pneumonitis and hyperresponsiveness. Kamei et al. (2018), however, reported that TRPA1 plays no role, because TRPA1 knockout does not alter LPS-induced bladder inflammation in mice. Other studies showed that the pharmacological inhibition or gene silencing of TRPA1 alleviates LPS-induced lung inflammation in mice (Liu et al., 2020) or inflammatory responses in lung epithelial cells from patients with cystic fibrosis (Prandini et al., 2016) and in human osteoarthritic fibroblast-like synoviocytes (Yin et al., 2018), suggesting the inflammatory role of TRPA1. Our in vivo and in vitro studies provide strong evidence in favor of the contribution of TRPA1 to LPS-induced lung inflammation.
Our study had at least two limitations that warrant discussion. Firstly, instead of using a TRPA1 antagonist to treat wild-type mice, we used trpa1–/– mice as the model to delineate the contribution of TRPA1 to LPS-induced lung inflammation. Secondly, instead of intratracheal or intranasal installation LPS, our animals received intraperitoneal injection of LPS. Whether our findings can be confirmed by using a TRPA1 antagonist or intratracheal/intranasal installation of LPS requires further investigations.
In summary, our in vitro findings suggest that exposure to LPS activates lung epithelial TRPA1. This activation sequentially produces a cascade of related cellular events, including the elevation of Ca2+ level, increase in ROS level, the activation of MAPK/NF-κB signaling, and the induction of IL-8 in HBECs. Our in vivo findings indicate that TRPA1 knockout alleviates LPS-induced inflammation, oxidative stress, and the activation of MAPKs in the lungs of mice. Our findings offer novel information regarding the pathogenic mechanisms associated with LPS-induced lung inflammation and may be valuable for the development of potential therapies.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital (Approval Number: IACUC 2017-266).
Author Contributions
H-KK, A-HL, and D-WP conducted the studies, analyzed, and interpreted the data. H-KK and A-HL wrote the manuscript. YK and T-SL led the project, interpreted the data, and finished the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Ministry of Science and Technology (MOST 107-2320-B-010-027-MY3 and 108-2320-B-002-032-MY3), Taiwan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the KGSupport Academic Submission Services for assisting with language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.596314/full#supplementary-material
References
Alpizar, Y. A., Boonen, B., Sanchez, A., Jung, C., López-Requena, A., Naert, R., et al. (2017). TRPV4 activation triggers protective responses to bacterial lipopolysaccharides in airway epithelial cells. Nat. Commun. 8:1059. doi: 10.1038/s41467-017-01201-3
Bals, R., and Hiemstra, P. S. (2004). Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur. Respir. J. 23, 327–333. doi: 10.1183/09031936.03.00098803
Bedard, K., and Krause, K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Benov, L., Sztejnberg, L., and Fridovich, I. (1998). Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic. Biol. Med. 25, 826–831. doi: 10.1016/s0891-5849(98)00163-4
Boonen, B., Alpizar, Y. A., Meseguer, V. M., and Talavera, K. (2018a). TRP channels as sensors of bacterial endotoxins. Toxins 10:326. doi: 10.3390/toxins10080326
Boonen, B., Alpizar, Y. A., Sanchez, A., López-Requena, A., Voets, T., and Talavera, K. (2018b). Differential effects of lipopolysaccharide on mouse sensory TRP channels. Cell Calcium 73, 72–81. doi: 10.1016/j.ceca.2018.04.004
Büch, T. R., Schäfer, E. A., Demmel, M. T., Boekhoff, I., Thiermann, H., Gudermann, T., et al. (2013). Functional expression of the transient receptor potential channel TRPA1, a sensor for toxic lung inhalants, in pulmonary epithelial cells. Chem. Biol. Interact. 206, 462–471. doi: 10.1016/j.cbi.2013.08.012
Carmona, E. M., Lamont, J. D., Xue, A., Wylam, M., and Limper, A. H. (2010). Pneumocystis cell wall beta-glucan stimulates calcium-dependent signaling of IL-8 secretion by human airway epithelial cells. Respir. Res. 13 11:95. doi: 10.1186/1465-9921-11-95
Cecconi, M., Evans, L., Levy, M., and Rhodes, A. (2018). Sepsis and septic shock. Lancet 392, 75–87. doi: 10.1016/S0140-6736(18)30696-2
Chen, Y., Guo, S., Jiang, K., Wang, Y., Yang, M., and Guo, M. (2019). Glycitin alleviates lipopolysaccharide-induced acute lung injury via inhibiting NF-κB and MAPKs pathway activation in mice. Int. Immunopharmacol. 75:105749. doi: 10.1016/j.intimp.2019.105749
Cho, R. L., Yang, C. C., Lee, I. T., Lin, C. C., Chi, P. L., Hsiao, L. D., et al. (2016). Lipopolysaccharide induces ICAM-1 expression via a c-Src/NADPH oxidase/ROS-dependent NF-κB pathway in human pulmonary alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 310, L639–L657. doi: 10.1152/ajplung.00109.2014
Connor, A. J., Laskin, J. D., and Laskin, D. L. (2012). Ozone-induced lung injury and sterile inflammation. Role of toll-like receptor 4. Exp. Mol. Pathol. 92, 229–235. doi: 10.1016/j.yexmp.2012.01.004
Diamond, G., Legarda, D., and Ryan, L. K. (2000). The innate immune response of the respiratory epithelium. Immunol. Rev. 173, 27–38. doi: 10.1034/j.1600-065x.2000.917304.x
Dong, Z., and Yuan, Y. (2018). Accelerated inflammation and oxidative stress induced by LPS in acute lung injury: inhibition by ST1926. Int. J. Mol. Med. 41, 3405–3421. doi: 10.3892/ijmm.2018.3574
Fernandes, E. S., Fernandes, M. A., and Keeble, J. E. (2012). The functions of TRPA1 and TRPV1: moving away from sensory nerves. Br. J. Pharmacol. 166, 510–521. doi: 10.1111/j.1476-5381.2012.01851.x
Hajna, Z., Csekõ, K., Kemény, Á, Kereskai, L., Kiss, T., Perkecz, A., et al. (2020). Complex regulatory role of the TRPA1 receptor in acute and chronic airway inflammation mouse models. Int. J. Mol. Sci. 21:E4109. doi: 10.3390/ijms21114109
Jiang, F., Zhang, Y., and Dusting, G. J. (2011). NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 63, 218–242. doi: 10.1124/pr.110.002980
Jiang, Y., Zeng, Y., Huang, X., Qin, Y., Luo, W., Xiang, S., et al. (2016). Nur77 attenuates endothelin-1 expression via downregulation of NF-κB and p38 MAPK in A549 cells and in an ARDS rat model. Am. J. Physiol. Lung Cell. Mol. Physiol. 311, L1023–L1035. doi: 10.1152/ajplung.00043.2016
Kamei, J., Aizawa, N., Nakagawa, T., Kaneko, S., Kume, H., Homma, Y., et al. (2018). Attenuated lipopolysaccharide-induced inflammatory bladder hypersensitivity in mice deficient of transient receptor potential ankilin1. Sci. Rep. 8:15622. doi: 10.1038/s41598-018-33967-x
Lin, A. H., Liu, M. H., Ko, H. B., Perng, D. W., Lee, T. S., and Kou, Y. R. (2017). Inflammatory effects of menthol vs. non-menthol cigarette smoke extract on human lung epithelial cells: a double-hit on TRPM8 by reactive oxygen species and menthol. Front. Physiol. 8:263. doi: 10.3389/fphys.2017.00263
Lin, A. H., Liu, M. H., Ko, H. K., Perng, D. W., Lee, T. S., and Kou, Y. R. (2015). Lung epithelial TRPA1 transduces the extracellular ROS into transcriptional regulation of lung inflammation induced by cigarette smoke: the role of influxed Ca2 +. Mediators Inflamm. 2015:148367. doi: 10.1155/2015/148367
Liu, P. L., Chen, Y. L., Chen, Y. H., Lin, S. J., and Kou, Y. R. (2005). Wood smoke extract induces oxidative stress-mediated caspase-independent apoptosis in human lung endothelial cells: role of AIF and EndoG. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L739–L749. doi: 10.1152/ajplung.00099.2005
Liu, X., Yin, S., Chen, Y., Wu, Y., Zheng, W., Dong, H., et al. (2018). LPS-induced proinflammatory cytokine expression in human airway epithelial cells and macrophages via NF-κB, STAT3 or AP-1 activation. Mol. Med. Rep. 17, 5484–5491. doi: 10.3892/mmr.2018.8542
Liu, Z., Wang, P., Lu, S., Guo, R., Gao, W., Tong, H., et al. (2020). Liquiritin, a novel inhibitor of TRPV1 and TRPA1, protects against LPS-induced acute lung injury. Cell Calcium 88:102198. doi: 10.1016/j.ceca.2020.102198
Matsunaga, N., Tsuchimori, N., Matsumoto, T., and Ii, M. (2011). TAK-242 (resatorvid), a small-molecule inhibitor of toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 79, 34–41. doi: 10.1124/mol.110.068064
Mazgaeen, L., and Gurung, P. (2020). Recent advances in lipopolysaccharide recognition systems. Int. J. Mol. Sci. 21:379. doi: 10.3390/ijms21020379
Meseguer, V., Alpizar, Y. A., Luis, E., Tajada, S., Denlinger, B., Fajardo, O., et al. (2014). TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat. Commun. 5:3125. doi: 10.1038/ncomms4125
Mossman, B. T., Lounsbury, K. M., and Reddy, S. P. (2006). Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am. J. Respir. Cell. Mol. Biol. 34, 666–669. doi: 10.1165/rcmb.2006-0047SF
Nassini, R., Pedretti, P., Moretto, N., Fusi, C., Carnini, C., Facchinetti, F., et al. (2012). Transient receptor potential ankyrin 1 channel localized to non-neuronal airway cells promotes non-neurogenic inflammation. PLoS One 7:e42454. doi: 10.1371/journal.pone.0042454
Neyen, C., and Lemaitre, B. (2016). Sensing Gram-negative bacteria: a phylogenetic perspective. Curr. Opin. Immunol. 38, 8–17. doi: 10.1016/j.coi.2015.10.007
Planagumà, A., Domènech, T., Pont, M., Calama, E., García-González, V., and López, R. (2015). Combined anti CXC receptors 1 and 2 therapy is a promising anti-inflammatory treatment for respiratory diseases by reducing neutrophil migration and activation. Pulm. Pharmacol. Ther. 34, 37–45. doi: 10.1016/j.pupt.2015.08.002
Prandini, P., De Logu, F., Fusi, C., Provezza, L., Nassini, R., Montagner, G., et al. (2016). Transient receptor potential ankyrin 1 channels modulate inflammatory response in respiratory cells from patients with cystic fibrosis. Am. J. Respir. Cell. Mol. Biol. 55, 645–656. doi: 10.1165/rcmb.2016-0089OC
Rahman, I., and Adcock, I. M. (2006). Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 28, 219–242. doi: 10.1183/09031936.06.00053805
Soldano, A., Alpizar, Y. A., Boonen, B., Franco, L., López-Requena, A., Liu, G., et al. (2016). Gustatory-mediated avoidance of bacterial lipopolysaccharides via TRPA1 activation in Drosophila. Elife. 5:e13133. doi: 10.7554/eLife.13133
Startek, J. B., Talavera, K., Voets, T., and Alpizar, Y. A. (2018). Differential interactions of bacterial lipopolysaccharides with lipid membranes: implications for TRPA1-mediated chemosensation. Sci. Rep. 8:12010. doi: 10.1038/s41598-018-30534-2
Tian, C., Huang, R., Tang, F., Lin, Z., Cheng, N., Han, X., et al. (2020). Transient receptor potential ankyrin 1 contributes to lysophosphatidylcholine-induced intracellular calcium regulation and THP-1-derived macrophage activation. J. Membr. Biol. 253, 43–55. doi: 10.1007/s00232-019-00104-2
Trebak, M., Ginnan, R., Singer, H. A., and Jourd’heuil, D. (2010). Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid. Redox Signal. 12, 657–674. doi: 10.1089/ars.2009.2842
Tsuruta, Y., Park, Y. J., Siegal, G. P., Liu, G., and Abraham, E. (2007). Involvement of vitronectin in lipopolysaccaride-induced acute lung injury. J. Immunol. 179, 7079–7086. doi: 10.4049/jimmunol.179.10.7079
Ueda, J., Starr, M. E., Takahashi, H., Du, J., Chang, L. Y., Crapo, J. D., et al. (2008). Decreased pulmonary extracellular superoxide dismutase during systemic inflammation. Free Radic. Biol. Med. 45, 897–904. doi: 10.1016/j.freeradbiomed.2008.06.016
Yin, S., Wang, P., Xing, R., Zhao, L., Li, X., Zhang, L., et al. (2018). Transient receptor potential ankyrin 1 (TRPA1) mediates lipopolysaccharide (LPS)-induced inflammatory responses in primary human osteoarthritic fibroblast-like synoviocytes. Inflammation 41, 700–709. doi: 10.1007/s10753-017-0724-0
Keywords: lipopolysaccharide, TRPA1, calcium, reactive oxygen species, signaling pathway, lung epithelial cell, lung inflammation
Citation: Ko H-K, Lin A-H, Perng D-W, Lee T-S and Kou YR (2020) Lung Epithelial TRPA1 Mediates Lipopolysaccharide-Induced Lung Inflammation in Bronchial Epithelial Cells and Mice. Front. Physiol. 11:596314. doi: 10.3389/fphys.2020.596314
Received: 19 August 2020; Accepted: 30 October 2020;
Published: 17 November 2020.
Edited by:
John T. Fisher, Queen’s University, CanadaReviewed by:
Feng Li, Shanghai Jiao Tong University, ChinaChing Jung Lai, Tzu Chi University, Taiwan
Copyright © 2020 Ko, Lin, Perng, Lee and Kou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tzong-Shyuan Lee, bnR1dHNsZWVAbnR1LmVkdS50dw==; Yu Ru Kou, eXJrb3VAeW0uZWR1LnR3