 Sumita Mishra
Sumita Mishra Brittany L. Dunkerly-Eyring
Brittany L. Dunkerly-Eyring Gizem Keceli
Gizem Keceli Mark J. Ranek
Mark J. Ranek- 1Division of Cardiology, Department of Medicine, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Department of Pharmacology and Molecular Sciences, Johns Hopkins University, Baltimore, MD, United States
Many forms of cardiac disease, including heart failure, present with inadequate protein quality control (PQC). Pathological conditions often involve impaired removal of terminally misfolded proteins. This results in the formation of large protein aggregates, which further reduce cellular viability and cardiac function. Cardiomyocytes have an intricately collaborative PQC system to minimize cellular proteotoxicity. Increased expression of chaperones or enhanced clearance of misfolded proteins either by the proteasome or lysosome has been demonstrated to attenuate disease pathogenesis, whereas reduced PQC exacerbates pathogenesis. Recent studies have revealed that phosphorylation of key proteins has a potent regulatory role, both promoting and hindering the PQC machinery. This review highlights the recent advances in phosphorylations regulating PQC, the impact in cardiac pathology, and the therapeutic opportunities presented by harnessing these modifications.
Introduction
Hearts are tasked with an immense challenge of maintaining protein homeostasis (proteostasis) in the face of disease conditions stemming from genetic mutations or environmental stressors (Willis and Patterson, 2013; Henning and Brundel, 2017). Pathological stressors can produce non-functional, misfolding-prone proteins. The failure to remove these proteins in a timely manner can compromise the integrity of intracellular proteins and impair the contractile apparatus and organelles; this results in decreased cell survival and function, culminating in the development of heart failure (Wang and Robbins, 2006; Wang et al., 2008). Indeed, many forms of heart disease present with the accumulation of ubiquitinated proteins and/or protein aggregates: hallmarks of inadequate or impaired PQC (Weekes et al., 2003; Day, 2013; Day et al., 2013). Therapeutic strategies to enhance PQC and induce cardioprotection are of great interest, however a better understanding of the molecular mechanisms regulating cardiomyocyte PQC is needed to facilitate the development of such a novel strategy.
Cardiomyocytes utilize elaborate intracellular PQC mechanisms to maintain proteostasis and counter disease progression (Figure 1). Briefly stated the first line of defense against impaired proteostasis are the molecular chaperones which bind to misfolded proteins, whereas degradation of proteins is carried out by the proteasome and lysosome (Wang et al., 2008; Ranek et al., 2018). Protein misfolding can expose a stretch of hydrophobic amino acids to the cytosol. These amino acids are recognized and bound by molecular chaperones to either promote protein re-folding, prevent misfolded proteins from aggregating, and/or facilitate the interaction between the misfolded protein and a ubiquitin ligase (Ranek et al., 2018). The UPS degrades proteins that have been targeted for degradation via a ubiquitin chain, canonically a lysine-48 linked chain (Wang et al., 2008). The UPS consists of the ubiquitination enzymes: an E1 ubiquitin activating enzyme, an E2 ubiquitin conjugating enzyme, and an E3 ubiquitin ligase; along with the barrel shaped structure known as the proteasome which contains the proteolytic activities (Wang et al., 2008, 2011). The lysosome degrades proteins, macromolecules, and whole organelles by processes encompassing autophagy, which involves the delivery of substrates to the lysosome for internalization and degradation (Zheng et al., 2011; Sciarretta et al., 2018b). Autophagy comes in many forms: microautophagy, macroautophagy, chaperone-mediated autophagy (CMA), CASA, and organelle-specific autophagies (e.g., mitophagy) (Henning and Brundel, 2017; Ghosh and Pattison, 2018; Sciarretta et al., 2018b).
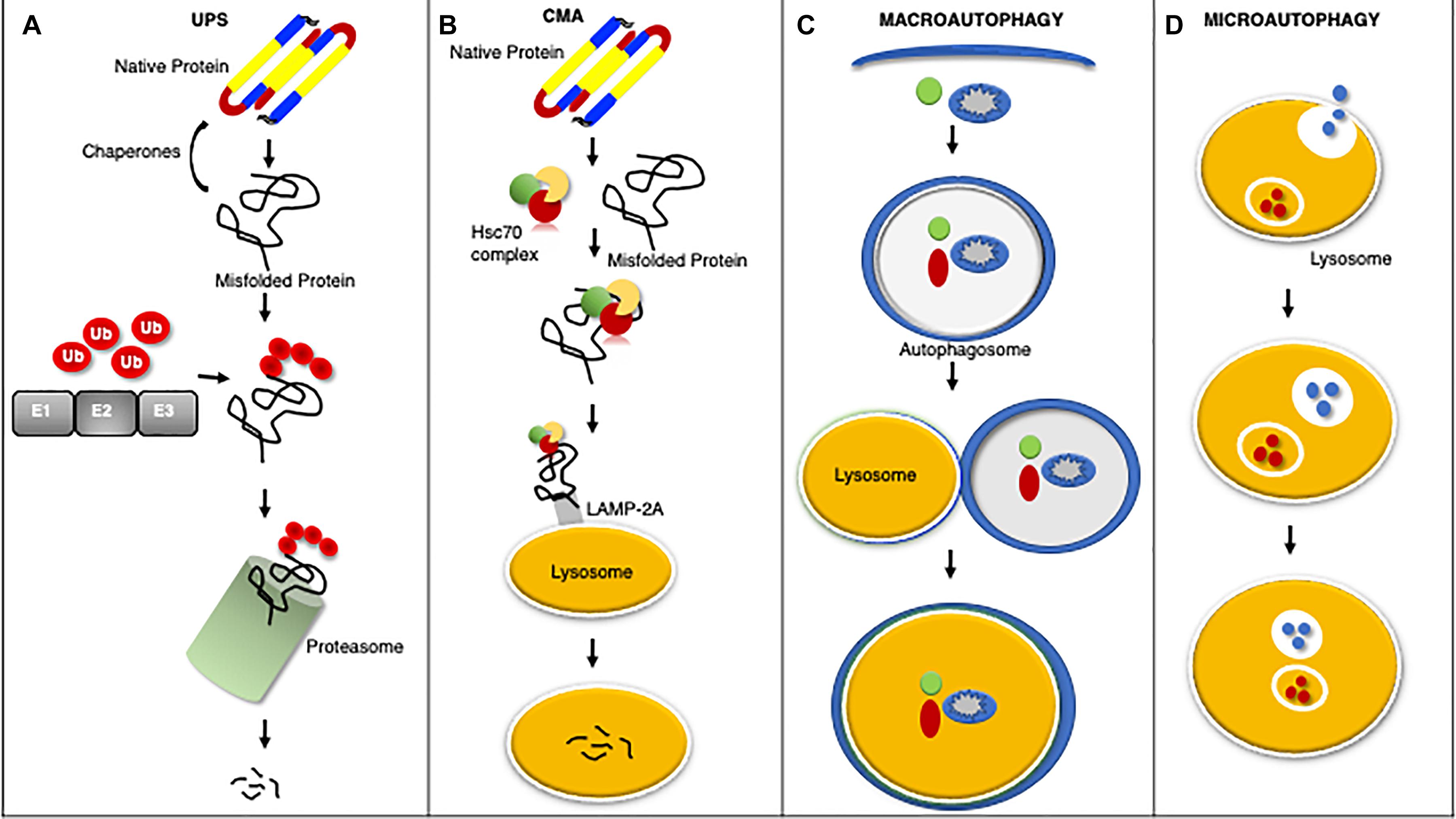
Figure 1. An overview of protein quality control systems. Misfolded proteins may regain native structure with the assistance of chaperones, however if proper re-folding cannot occur the misfolded protein will be catalyzed by the UPS through ubiquitination via a series of enzymatic reactions involving an ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), and ubiquitin ligase (E3) for degradation by the proteasome (A). Chaperone mediated autophagy (CMA) is a process by which the heat shock cognate 70 (HSC70) complex recognizes and binds select protein targets for internalization and degradation to the lysosome through the lysosome associated membrane protein 2A (LAMP2A) receptor (B). Macroautophagy is the bulk removal of proteins, protein aggregates, and organelles by first forming an autophagosome to surround the cargo followed by merger with the lysosome for degradation (C). Microautophagy is a process by which the lysosome invaginates to bring protein substrates into the lysosome for degradation (D).
A growing area of interest surrounds the role of PTMs in the regulation of PQC pathways. Various PTMs have been reported to promote or hinder cardiomyocyte PQC, including nitrosylation, oxidation, and ubiquitination among others (Wang et al., 2013). By doing so, PTMs have greatly expanded the specificity and capacity of protein degradation during cardiac disease. This review highlights the recent advances of arguably the most established PQC-regulating PTM, phosphorylation. Excellent reviews of the other PTMs have been described elsewhere (Christians and Benjamin, 2012; Scruggs et al., 2012; Wang et al., 2013; Penna et al., 2018; VerPlank and Goldberg, 2018). This review details the recent PQC phosphorylations that have been identified (Supplementary Table 1), especially those with influential roles in cardiac pathophysiology and some ubiquitous signaling pathways, and that can be clinically interrogated.
Impaired Protein Quality Control (PQC) in Cardiac Disease
Many forms of cardiac disease are characterized by an accumulation of ubiquitinated proteins and the presence of aberrant protein aggregation in the form of pre-amyloid oligomers (Ranek and Wang, 2009; Parry et al., 2015; Gilda and Gomes, 2017). Indeed, the majority of failing human hearts are characterized by impaired proteostasis, termed cardiac proteinopathies (Su and Wang, 2010; Zheng et al., 2011; Willis and Patterson, 2013). Studies conducted in mice revealed that inadequate cardiomyocyte PQC precedes and exacerbates cardiac pathogenesis (Figure 2) (Wang et al., 2011; Gilda and Gomes, 2017; Henning and Brundel, 2017). The Robbins lab overexpressed a misfolded protein surrogate with a polyglutamine expansion pre-amyloid oligomer (PQ83) in the heart, which impaired PQC leading to reduced cardiac function and ultimately failure (Pattison et al., 2008). Similar results were obtained in another mouse model expressing a mutated αB-crystallin (CryABR120G), a misfolded and aggregation prone protein, in cardiomyocytes alone (Wang et al., 2001, 2003). PQ83 and CryABR120G mice exemplify a pathological process termed proteotoxicity, which refers to the adverse effects induced by the presence of damaged or misfolded proteins (Sandri and Robbins, 2014). If not corrected, cardiac proteotoxicity will result in apoptosis/necrosis at the cellular level, decreased function at the organ level, and even premature death for the organism (Sandri and Robbins, 2014). Indeed, CryABR120G mice showed impaired PQC within 3 months of age and heart failure within 6 months of age (Wang et al., 2001, 2003; Kumarapeli and Wang, 2004). Stimulation of protein degradation via genetic upregulation of autophagy rescued the CryABR120G phenotype (Pattison et al., 2011). Interestingly exercise was also shown to attenuate pre-amyloid deposition, proteotoxicity, and heart failure progression (Maloyan et al., 2007). A recent study by the Wang lab utilized the CryABR120G as a model of proteinopathy that occurs with HFpEF, a devastating disease currently without a treatment (Sharma and Kass, 2014). CryABR120G mice were treated with a phosphodiesterase 1 inhibitor, which activated protein kinases A (PKA) and G (PKG), stimulated proteasome activities, and attenuated proteotoxic stress (Zhang et al., 2019). This study demonstrates the potential of targeting protein degradation as a heart failure treatment. Chronic inhibition of any one of the PQC machineries exacerbates cardiac pathogenesis following MI via closure of the left ventricular anterior descending artery or left ventricular PO via transaortic constriction (TAC), whereas the enhancement of cardiomyocyte PQC protects the myocardium facilitating increased proteostasis, cardiac function, and lifespan (Wang and Robbins, 2006; Wang et al., 2008, 2011; Su and Wang, 2010; Ranek et al., 2015).
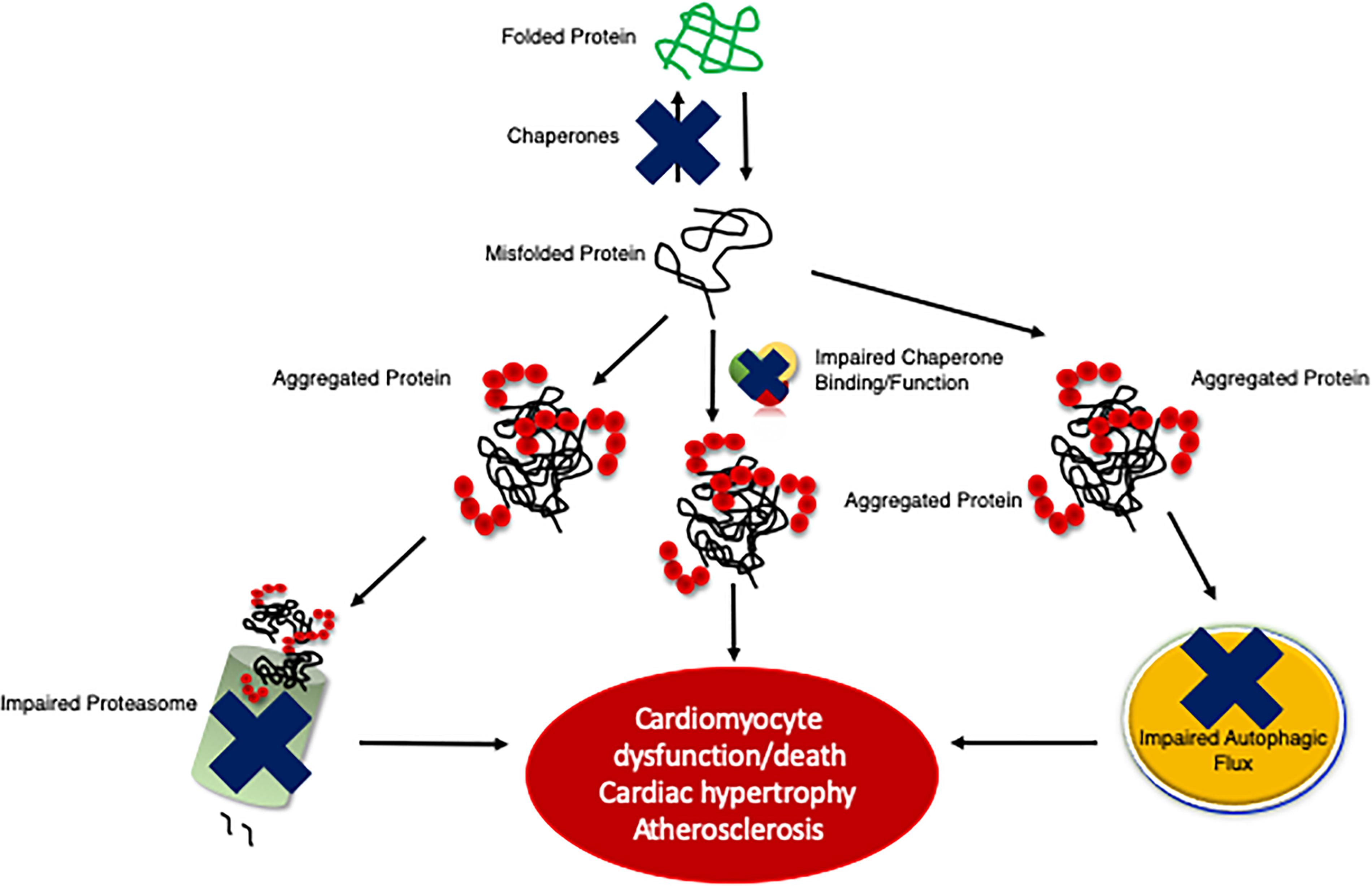
Figure 2. An illustration of the development of cardiac proteinopathy. Cardiomyocytes accumulate ubiquitinated and aggregated proteins as a result of impaired PQC mechanisms. The failure of cardiomyocyte chaperones activity to efficiently re-fold misfolded proteins results in misfolded proteins aggregating, which impairs the proteasome and overwhelms autophagy. Insufficient PQC exacerbates disease pathogeneses (e.g., cardiac hypertrophy and atherosclerosis).
Chaperones
The first line of defense against proteotoxicity are chaperones, which are a broad class of proteins that assist with refolding misfolded proteins, facilitating an interaction between misfolded proteins and a ubiquitination enzyme, and preventing proteins from aggregating together (Figure 1) (Willis and Patterson, 2010; Ranek et al., 2018). There are chaperones that are constitutively expressed (e.g., heat shock cognate 70, HSC70) and others, whose expression are induced by a cardiac stress [e.g., heat shock protein 70 (HSP70)] (Wu et al., 1985; Dworniczak and Mirault, 1987). Chaperones can protect cardiomyocytes against proteotoxicity and subsequent cell death during a stressful/pathological condition (Willis and Patterson, 2010; Tarone and Brancaccio, 2014; Penna et al., 2018). Indeed, overexpression of certain chaperones has protected hearts from cardiac disease (Willis and Patterson, 2010; Tarone and Brancaccio, 2014). Multiple studies have reported overexpression of HSP70 to be protective in mouse models of ischemia and ischemia-reperfusion injury (Marber et al., 1995; Plumier et al., 1995; Radford et al., 1996). Another study determined that HSP70 overexpression is protective in periods of brief ischemia where there is myocardial dysfunction but no infarct (Trost et al., 1998). In a pig model of ischemia-reperfusion, HSP90 transfection was protective and reduced the ischemic region (Kupatt et al., 2004). It was suggested that this protective effect was due to HSP90 mediated enhancement of NO formation (Depre et al., 2006). Injection of HSP22 into the swine heart was protective against MI in a NO-dependent manner (Chen et al., 2011), however, it seems that this is only effective acutely, as more recent work shows that chronic overexpression is deleterious (Morin et al., 2019). This discrepancy was due to increased ROS production and oxidative stress that occurs with chronic HSP22 expression that results in cardiac hypertrophy and shortened lifespan (Morin et al., 2019). Another study using a HSP22 overexpression transgenic mouse model reported cardioprotection with increased preconditioning to attenuate MI (Depre et al., 2006). Here, the authors noted a metabolic switch to favor glucose utilization and expression of anti-apoptotic factors over pro-apoptotic factors. These findings were supported by a recent study describing protection against doxorubicin induced cardiotoxicity in HSP22 transgenic mice (Lan et al., 2020).
One of the most recognized protective roles of cardiomyocyte chaperones is to maintain cardiac systole and diastole, through modulation of contractile proteins (Willis et al., 2009, 2010). Impaired cardiac function with increased ventricular stiffness are hallmarks of heart failure. Altered sarcomeric and calcium handling proteins contribute to diastolic left ventricular stiffness. Phospholamban regulates calcium uptake to the sarcoplasmic reticulum via SERCA for cardiomyocyte relaxation to occur. Qian et al. (2011) identified that HSP20 negatively regulates protein phosphatase 1 (PP1) activity via a direct interaction, favoring phosphorylation of phospholamban and increased cardiac function. The small heat shock protein HSPB7 modulates actin thin filament length by binding to monomeric actin and limiting its availability for polymerization (Wu et al., 2017). Zhu et al. (2009) applied single molecule force spectroscopy to determine the contour length and bending rigidity of the N2B-Us of titin and the effect of wild type and mutant R157H (harboring the dilated cardiomyopathy missense mutation), or R120G (desmin-related myopathy mutation) αB-crystallin (CryAB) on the molecular mechanics of the N2B-Us and its flanking Ig domains. CryAB functions as a chaperone that lowers the probability of Ig domain unfolding and the persistence length of the titin N2B-Us spring region. HSP27 or CryAB suppresses cardiomyocyte stiffness caused by stretch and low pH (Kotter et al., 2014). Collectively these studies demonstrate the strong cardioprotective potential of chaperones, however before this potential can be therapeutically leveraged, a greater understanding of the regulatory mechanisms involved in chaperone expression and function is needed (Willis and Patterson, 2010; Tarone and Brancaccio, 2014; Ranek et al., 2018).
Chaperones Regulated by Phosphorylation
Recent studies have identified protein kinases and phosphatases responsible for phosphorylation and dephosphorylation, respectively, of chaperones and the consequent effects on cardiac function (Figure 3). The role of phosphorylation of chaperones and co-chaperones varies depending on the site and even on the temporal regulation of the sites. Some phosphorylation events stabilize the chaperones and their interactions with their client proteins, while others are specific for their client proteins and will only function properly when phosphorylated at the appropriate sites.
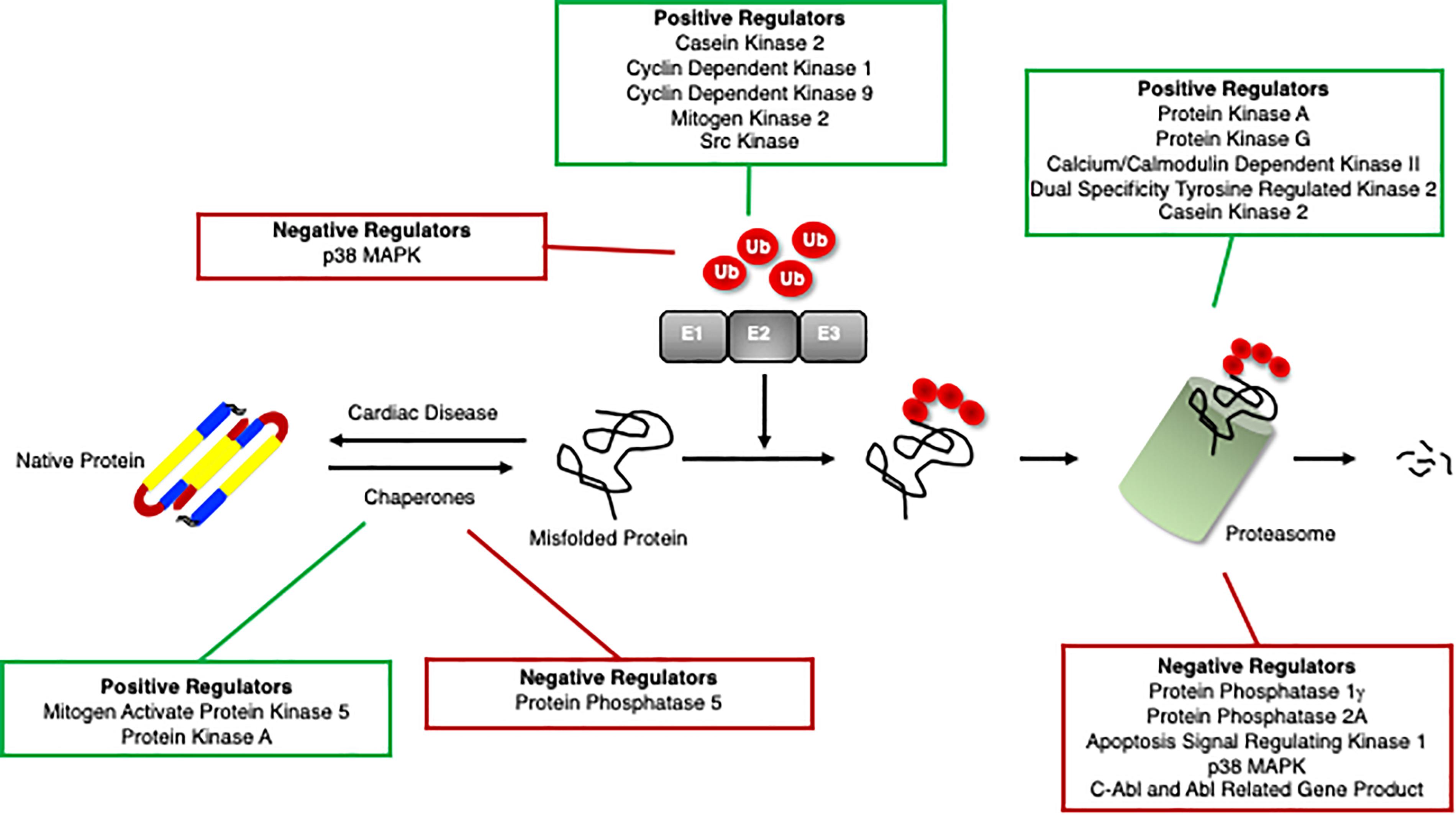
Figure 3. Positive and negative regulators of the ubiquitin proteasome system. Proteins that positively (green box) and negatively (red box) regulate cardiomyocyte chaperones, ubiquitination enzymes, and the proteasome.
The HSP70 family of chaperones support proper protein structure by unfolding and refolding misfolded proteins in an ATP-dependent manner (Sharma et al., 2010; Finka et al., 2015). Phosphorylation has a key role in regulating the function of HSP70s. HSC70 phosphorylation at threonine 38 controls cell cycle progression in a CDK-dependent manner by promoting G1 cyclin binding and subsequent degradation to promote G1/S transition in the cell cycle (Truman et al., 2012). While this study was done in yeast, this phosphorylation site is highly conserved and may play an important role in cardiovascular diseases which typically exhibit high expression of HSC70 (Boehm and Nabel, 2003). The vital role of HSP70s in redox homeostasis is precisely regulated by its phosphorylation at serine 631, which is required for superoxide dismutase 2 (SOD2) import and activation in mitochondria (Zemanovic et al., 2018). Once SOD2 restores the redox balance in mitochondria, HSP70 is dephosphorylated, and both HSP70 and SOD2 are ubiquitinated by CHIP and degraded (Zemanovic et al., 2018). Increased oxidative stress has been associated with the development and pathogenesis of cardiovascular disease. This precise regulation via phosphorylation is pivotal to both maintaining a homeostatic redox state and then binding to proteins damaged by oxidation. Regulatory phosphorylation can occur at the level of the chaperone as well as at the level of the co-chaperone. HSP40, a co-chaperone with HSP70, is phosphorylated by mitogen activated protein kinase 5 (MK5), resulting in enhanced ATP hydrolase function to increase the refolding capacity, thus PQC-maintaining ability, of HSP40/70 (Kostenko et al., 2014).
HSP90 proteins are chaperones with some similar, but different, functions to HSP70s in facilitating protein folding, and also directing proteins for degradation in order to avoid protein aggregation (Hohfeld et al., 2001). The decision-making process for whether to facilitate folding or to direct for degradation is mediated by binding to co-chaperones CHIP and HSP70-HSP90 organizing protein (HOP) (Muller et al., 2013). Phosphorylation is responsible for determining co-chaperone binding and macro-complex formation, such that phosphorylation in the C-termini of HSP70 and HSP90 prevents CHIP binding, but enhances HOP binding, which can assist with protein folding (Muller et al., 2013). Cells in a proliferative state have higher levels of phosphorylated HSP70 and HSP90 and those chaperones are preferentially bound to HOP (Muller et al., 2013). This has major implications in cancer due to the proliferative nature of those cells, but could also impact cardiovascular diseases like atherosclerosis, where vascular smooth muscle cell proliferation is problematic (Bennett et al., 2016). Bachman et al. (2018) demonstrated that the co-chaperone Cdc37 complexes with HSP90 and recruits client kinases, which phosphorylates HSP90 to provide a mechanism for highly specific tuning of the chaperone cycle depending on the client kinase that is recruited. Similarly, dephosphorylation of HSP90 by protein phosphatase 5 was shown to regulate the activity and interacting proteins of HSP90 (Haslbeck et al., 2015; Oberoi et al., 2016). These findings are of particular relevance to the heart as HSP90 is known to be involved in functioning of multiple steroid receptors (Pratt and Toft, 2003; Zhao et al., 2005) ARs play a role in multiple cardiovascular diseases including hypertension, stroke, and atherosclerosis, thus highlighting the therapeutic potential of modulating chaperones (Huang et al., 2016). HSP90 is bound to unliganded AR in the cytosol, however following phosphorylation of HSP90 at threonine 89 by PKA, the AR is released into the cytosol where it binds to HSP27 and migrates into the nucleus to modulates transcription via binding to androgen response elements (AREs) (Dagar et al., 2019).
The cardioprotection elicited by increased molecular chaperones in the heart is well-established (Willis and Patterson, 2010; Tarone and Brancaccio, 2014; Ranek et al., 2018), however, a better understanding of the regulation of chaperones via phosphorylation is desirable for the ability to better fine-tune chaperones’ functions. A mouse model that overexpressed a constitutively phosphorylated HSP20 at serine 16 (previously found to be PKA mediated), promoted fibrotic remodeling and heart failure (Gardner et al., 2019). This came as a surprise, as prior studies suggested that HSP20 phosphorylation at serine 16 was protective against beta-agonist-induced apoptosis in cardiomyocytes (Fan et al., 2004; Qian et al., 2009). A possible explanation for this disparity is the former study examining constitutive HSP20 phosphorylation at serine 16 was done in isolated cardiomyocytes, suggesting that acute phosphorylation of this site is protective, while the effects of chronic HSP20 activation are harmful. Gardner et al. (2019) utilized a ubiquitous knock-in mouse model and attributed this maladaptive response to a pro-fibrotic role of phosphorylated HSP20 that mediates IL-6 activation of cardiac fibroblasts. Another study identified HSP27 phosphorylation at serine 82 as being protective in biomechanically stressed mouse hearts (Collier et al., 2019). Here, they found that actin-binding protein filamin C (FLNC) and HSP27 are upregulated in mouse hearts subjected to left ventricular PO induced by TAC surgery (Collier et al., 2019). It was determined that these proteins interact, with phosphorylation of HSP27 facilitating their binding in a region of FLNC that is mechanosensing, while FLNC localizes to load-bearing sites (Collier et al., 2019). This suggests the mechanism chaperones are recruited in response to force destabilization is to maintain the integrity of the sarcomere and protect the myocardium. HSP27 phosphorylation has also been found to be upregulated in human platelets during ST-elevation post-MI, possibly offering a new measurable stress response for patients with MI (Kraemer et al., 2019).
Another chaperone with significant implications in cardiovascular disease is p97/valosin-containing protein (VCP)/Cdc48, an ATPase involved in a variety of functions to include ERAD, cell cycle progression, autophagy, and DNA repair (Parisi et al., 2018). This ATPase has been demonstrated to play a role in myofibrillar formation, both in limiting normal muscle growth but also contributing to muscle wasting diseases (Piccirillo and Goldberg, 2012). The function of p97 is regulated by phosphorylation at multiple sites (Baek et al., 2013). Phosphorylation of tyrosine 805 disrupts the interaction between p97 and PNGase, attenuating the ERAD pathway, thus PQC (Li et al., 2008). Additional studies found that tyrosine phosphorylation inhibits p97-ER membrane association and transitional ER assembly (Lavoie et al., 2000). Phosphorylation has also been shown to impact the ATPase activity of p97 as well as its association with ubiquitinated proteins (Klein et al., 2005; Mori-Konya et al., 2009). p97 has a role in the DNA damage response, as it is phosphorylated at serine 784 and accumulates at sites of DNA damage, possibly for DNA repair (Livingstone et al., 2005). Taken together, these studies demonstrate the regulatory role of phosphorylation and dephosphorylation on the function of chaperones, presenting many possibilities for targeting these modifications as a therapy.
Ubiquitination Enzymes
Proteins are targeted for degradation by the proteasome via ubiquitination, a sequential system involving the following enzymes: ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), ubiquitin ligase (E3), and possibly a ubiquitin elongation factor (E4) (Figure 1) (Hershko and Ciechanover, 1998; Goldberg, 2003; Zuo et al., 2020). In humans, there are only 2 E1s that have been identified, and as their name suggests, they are responsible for activating ubiquitin so that it can be conjugated to a protein. This process occurs in an ATP-dependent manner in which the C-terminus of ubiquitin is bound to an E1 cysteine via a thioester bond (Hershko and Ciechanover, 1998; Zuo et al., 2020). The E1 then transfers ubiquitin to one of approximately 40 E2s in humans through another thioester bond (Hershko and Ciechanover, 1998; Zuo et al., 2020). Finally, with an E3 the ubiquitin will be attached to a substrate protein. There are three families of E3s: HECT, RING and RBR, and these families differ slightly in the way that the ubiquitin is transferred to its substrate (Zheng and Shabek, 2017; George et al., 2018). HECT E3s will accept the ubiquitin first from the E2 to form a thioester bond, and will then transfer the ubiquitin to the substrate directly from the E3 (Zuo et al., 2020). RING E3s will bring together the ubiquitin-E2 complex and the substrate and simply mediate the transfer of ubiquitin from the E2 to the substrate (Zheng and Shabek, 2017). RBR ubiquitin ligases act as a hybrid protein of two domains, a canonical RING domain as well as a catalytic cysteine residue similar to the HECT domain (George et al., 2018). The specificity of the UPS is thought to lie within the ubiquitination step (Wang et al., 2008; Ranek et al., 2018). Given the importance of ubiquitination in PQC and degradation, it comes as no surprise that this process and selectivity is further regulated through phosphorylation of these ubiquitination enzymes (Figure 3).
Phosphorylations That Regulate Ubiquitination Enzymes
There is limited data into the sites phosphorylated and potential functional change of the two E1s in humans. Stephen et al. (1997) described multiple phosphorylation sites at the N-terminus of E1a. It was previously suggested that E1 could be phosphorylated by PKC, and that E1a serine 4 could be phosphorylated by Cdc2, a kinase that is key in cell cycle regulation (Kong and Chock, 1992; Nagai et al., 1995). In these studies, the function of these phosphorylation sites was not fully described. It was determined that while phosphorylation increases in various states of the cell cycle, the enzymatic activity of E1 was not changed (Stephen et al., 1996). Also, while E1a was found to localize to the nucleus, it was determined that phosphorylation was not required for this localization (Stephen et al., 1997). It is possible that this phosphorylation may increase the rate of nuclear targeting or possibly change the stability of interactions with E2 enzymes, but how phosphorylation may alter these roles is yet to be determined.
Functional alterations of E2 enzymes have been attributed to phosphorylation, albeit the knowledge about them in the heart is limited. A common theme of E2 enzymes and their regulation via phosphorylation is their involvement in the cell cycle. There is evidence of the cell cycle playing a role in cardiovascular diseases such as hypertrophy and atherosclerosis, so these mechanisms of regulation could have interesting therapeutic potential (Bicknell et al., 2003; Ahuja et al., 2007). In one such study of the involvement of an E2 in cell cycle regulation, Cdc34, in yeast was shown to be phosphorylated at serines 203, 222, and 231 in the acidic C-terminal tail domain by casein kinase 2 (CK2) (Sadowski et al., 2007). In an effort to understand the biological importance of these sites, they mutated them to alanine (phospho-null), or a glutamate or aspartate (phospho-mimetics). With the phospho-mutants, they observed differential kinetics through the cell cycle, from G1 through S into the G2/M phase (Sadowski et al., 2007). The authors were able to associate the cells with faster cell cycle kinetics with an increased rate of Sic1 degradation, and increased Skp, cullin, F-box containing complex (SCF)-mediated ubiquitination of Sic1 (Sadowski et al., 2007). Overall, this was a demonstration that phosphorylation can regulate the catalytic and cell cycle functions of Cdc34. In another study, Shchebet et al. (2012) demonstrated that the ubiquitin-conjugating enzyme E2 A (UBE2A) is directly phosphorylated by CDK9 at serine 120. Knocking down CDK9 reduced UBE2A phosphorylation, which then decreased monoubiquitination of histone H2B and PCNA, suggesting phosphorylation at serine 120 activates UBE2A (Shchebet et al., 2012). This finding is of particular interest to the heart, as a recent study identified monoubiquitination of H2B is a transcriptional regulator that controls expression of cilia genes, where mutations have been implicated in congenital heart disease (Robson et al., 2019). ER stress occurs in various cardiovascular diseases with impaired proteostasis (Ochoa et al., 2018). A study from 2017 identified ubiquitin-conjugating enzyme E2 J1 (UBE2J1) phosphorylation at serine 184 as being important for recovery of ER stress to maintain proteostasis (Elangovan et al., 2017). It was reported that UBE2J1 is phosphorylated at serine 184 by mitogen kinase 2 (MK2) as a mechanism to alleviate ER stress (Menon et al., 2013). Phosphorylated UBE2J1 has higher affinity for the E3 ligase, c-IAP1, and that cells expressing a phospho-null UBE2J1 cannot recover from ER stress (Elangovan et al., 2017). The authors also reported that phosphorylated UBE2J1 is degraded by the proteasome, potentially as a feedback loop (Elangovan et al., 2017).
An E3 ligase that is implicated in multiple diseases, including cardiovascular and neurodegenerative diseases, is Parkin, which is known to be regulated by phosphorylation (Wang et al., 2018). One study demonstrates phosphorylation negatively regulates Parkin’s protective function (Chen et al., 2018). Here, they use a mutant α-synuclein model that, when overexpressed, activates p38 MAPK, which then directly phosphorylates Parkin at serine 131. This disrupts PTEN-induced kinase 1 (PINK1)-Parkin mediated mitophagy and exacerbates the mitochondrial impairment induced by α-synuclein accumulation (Chen et al., 2018). A few studies have highlighted the importance of Parkin in the heart by performing knockout experiments. One such experiment knocked out Parkin in flies and observed accumulation of dysfunctional mitochondria and dilated cardiomyopathy (Bhandari et al., 2014). This phenotype could be rescued with cardiomyocyte-specific Parkin expression (Bhandari et al., 2014). Another study found that in response to MI the parkin knockout mice fared worse than WT mice in terms of survival and infarct size (Kubli et al., 2013). Collectively these studies suggest determining the kinases capable of phosphorylating Parkin in the heart and the role of this PTM during cardiac pathogenesis could provide a novel therapeutic avenue.
The neural precursor cell expressed developmentally down-regulated protein 4 (NEDD4) is a HECT domain ubiquitin ligase that is required for heart development, turnover of potassium and sodium channels, and maintenance of proteostasis (Fouladkou et al., 2010; Gilda and Gomes, 2017). FGFR1 activation results in tyrosine phosphorylation of four key sites (43, 365, 366, and 585) on NEDD4 (Persaud et al., 2014). Site-directed mutagenesis of these four tyrosines revealed that expression of a phospho-mimetic results in constitutive activation of NEDD4 ubiquitin ligase activity, while phospho-null NEDD4 decreased its ubiquitin ligase activity (Persaud et al., 2014). FGF signaling is known to play a pathophysiological role in the heart as evidenced by worsened cardiac hypertrophy in isoproterenol stimulated hearts of FGF2 transgenic mice (House et al., 2010; Itoh and Ohta, 2013). FGF2 KO mice were protected against cardiac hypertrophy and fibrosis following isoproterenol stimulation (Itoh and Ohta, 2013). FGF2 is mostly expressed in non-myocytes within the heart, suggesting cell-to-cell communication as the mode of cardiomyocyte regulation (Itoh and Ohta, 2013). Considering the requirement of NEDD4 in cardiac development, the presence of FGF in all cell types of the heart, and its role in heart disease, this signaling axis presents an interesting therapeutic target.
SUMOylation and neddylation are other PTMs that are analogous to ubiquitination, utilize a similar E1, E2, and E3 mechanism of conjugating either SUMO or NEDD8 to proteins, and are known to be involved in many different biological processes (Wilkinson and Henley, 2010; Zhou et al., 2019). There is evidence of the modifiers SUMO-1 and NEDD8 themselves being phosphorylated, but the functional implications are still unknown (Matic et al., 2008; Enchev et al., 2015). We are only beginning to understand the regulation of neddylation E1 (NEDD8-activating enzyme NAE) and E2s (UBE2M and UBE2F) by phosphorylation. Interestingly, the E3s that have been identified for neddylation can also function as ubiquitination E3s (Enchev et al., 2015). Furthermore, phosphorylation is responsible for converting an E3 from ubiquitin conjugating to NEDD8 conjugating (Batuello et al., 2015). Under growth factor stimulation, src kinase phosphorylates Mdm2 at tyrosines 281 and 302, recruits the NEDD8 E2 (Ubc12) and Mdm2, to neddylate p53 in human cancer cell lines (Batuello et al., 2015). This has interesting implications if this mechanism applies to other E3s. While phosphorylations have been identified on SUMOylation E1 and E2, the function of these modifications is not known (Tomanov et al., 2018). However, there are multiple studies that have found that phosphorylation modifies the function of SUMO E3s. One such study identified Ubc9 as being phosphorylated by CDK1 at serine 71 to enhance SUMOylation activity in vitro (Su et al., 2012). Another study in HEK 293T cells elucidates a feedback system whereby DNA-damage induced homeodomain interacting protein kinase 2 (HIPK2) phosphorylates proprotein convertase (Pc2) at threonine 495, which in turn controls Pc2 SUMOylation of HIPK2 in order to enhance its ability to mediate transcriptional repression (Roscic et al., 2006). Additional studies are needed to determine the potential role phosphorylation of the neddylation and SUMOylation proteins may have in cardiac disease pathogenesis.
The Proteasome
The UPS plays a key role in eukaryotic cells by degrading damaged, defective, and non-functional proteins (Figure 1) (Jentsch, 1992; Schwartz and Ciechanover, 1992; Ciechanover, 1994). This pathway represents non-lysosomal degradation of cellular proteins and plays a critical role in PQC (Goldberg, 2003; Divald and Powell, 2006). Proteins are destined to proteasome-mediated degradation by the formation of a ubiquitin chain on the target protein. Ubiquitinated proteins are fed through the proteasome core complex for degradation via an ATP driven process (Collins and Goldberg, 2017). The mammalian proteasome is a highly sophisticated multi subunit protease complex composed of the 20S catalytic core particle, within which proteins are degraded, and two 19S regulatory cap particles which locates and binds to ubiquitinated substrate (Budenholzer et al., 2017; Collins and Goldberg, 2017). The 20S particle is formed by two copies of 14 different subunits (α1–α7 and β1–β7) stacked in heptameric rings (Groll et al., 1997; Budenholzer et al., 2017). The active sites of the catalytic subunits (β1, β2, and β5) lines the central lumen of a chamber gated by an α subunit at its either end (Groll et al., 1997; Budenholzer et al., 2017). The proteasome activator 700 (PA700) or 19S cap has two distinct sub-components (Lasker et al., 2012). The base consists of six Rpt (1→6) subunits that have constitutive ATPase activity, plus two non-ATPase subunits, Rpn1 and Rpn2; and the lid structure contains 10 Rpn (3→12) subunits (Lasker et al., 2012; Budenholzer et al., 2017). The 19S subunit is responsible for binding and unfolding ubiquitinated proteins and feeding the protein substrate to the 20S proteasome for catalysis (Wang et al., 2008).
Several studies have shown a central role of the UPS in cardiovascular physiology and pathophysiology (Razeghi et al., 2006; Drews et al., 2010; Powell and Divald, 2010; Predmore et al., 2010; Zu et al., 2010; Li et al., 2011). Cardiac proteasomes exhibit distinct functional and subcellular distribution (Gomes et al., 2006, 2009; Drews et al., 2007; Powell et al., 2008; Dahlmann, 2016). A substantial number of studies have shown impaired proteasomal function contributes to heart disease (Su and Wang, 2010; Willis et al., 2010; Wang et al., 2011; Day, 2013), which is supported by findings of insufficient UPS activity in human end stage heart failure (Hein et al., 2003; Weekes et al., 2003; Predmore et al., 2010). Proteasome dysfunction has been reported in hypertrophic, ischemic, atrophic, desmin-related, and diabetic cardiomyopathies (Razeghi et al., 2006; Drews et al., 2010; Powell and Divald, 2010; Predmore et al., 2010; Zu et al., 2010; Li et al., 2011). Arguably the most well-studied cardiac proteinopathy mouse model is DRC caused by mutations in desmin, αB-crystallin (CryAB), and other related genes (Wang and Robbins, 2006). The DRC model features aberrant protein aggregates in myocytes, reduced cardiac function, and a shortened lifespan (Wang et al., 2001, 2003; Pattison et al., 2011). These hearts also have PFI, which was postulated to contribute further to the impaired proteostasis and the pathogenesis of DRC (Wang and Robbins, 2006; Su and Wang, 2010; Wang et al., 2011). Studies enhancing proteasome activity in DRC have supported this notion (Li et al., 2011; Ranek et al., 2013). PFI has also been observed in mouse hearts subjected to ischemia/reperfusion (I/R) injury and transaortic constriction (TAC)-induced cardiac PO (Wang et al., 2011). Expression of a catalytically inactive proteasome beta5 subunit negates the chymotrypsin-like activity, but not the caspase- or trypsin-like activities, and worsened cardiac PQC and function following I/R or TAC (Ranek et al., 2015).
Methods to enhance proteasome activities have been shown to be protective. Post-translational modifications are key regulatory mechanisms for proteasome function. More than 300 phosphorylation sites in proteasome subunits have been detected that can regulate protein stability, abundance, assembly, subcellular localization, substrate recognition and enzymatic activity of proteasome subunits (Benedict and Clawson, 1996; Satoh et al., 2001; Bose et al., 2004; Zong et al., 2006; Bingol et al., 2010; Guo et al., 2011, 2016; Djakovic et al., 2012; Sledz et al., 2013; Yuan et al., 2013; Li et al., 2015; Lokireddy et al., 2015; Wani et al., 2016). In humans, 20S cap and the 19S base subunits are more frequently phosphorylated than the 19S lid. Many kinases have been demonstrated to regulate proteasome activity in general and in heart tissues (Figure 3) (Powell and Divald, 2010; Sledz et al., 2013; Wang et al., 2013; Collins and Goldberg, 2017; VerPlank and Goldberg, 2018).
Phosphorylations Regulating the Proteasome
Protein Kinase A (PKA)
Protein kinase A was the first kinase identified to phosphorylate proteasome subunits and stimulate proteasome activities in vitro as well as in vivo (Zong et al., 2006, 2008; Asai et al., 2009; Gomes et al., 2009; Drews et al., 2010). A series of studies using bovine pituitaries, and later from murine hearts, identified PKA in a complex with endogenous 20S proteasomes (Pereira and Wilk, 1990; Zong et al., 2006; Drews et al., 2010). PKA phosphorylation of the proteasome has been detected on serine residues of the α 1-, α 2-, α 3-, β 2-, β 3-, and β7-subunits, and the threonine residues of the α 3-, β 3-, and β7-subunits of the 20S proteasome (Zong et al., 2006). Additionally studies demonstrated enhancement of proteasome activity via PKA stimulation. The addition of cAMP stimulates PKA activation and its interaction with the proteasome, which resulted in increased phosphorylation of 19S cap subunit Rpn6 at Ser14 (Lokireddy et al., 2015). Pathare and others have shown that raising cAMP levels increases the amount of double-capped 26S proteasomes, indicating increased stabilization of the proteasome complexes (Pathare et al., 2012; VerPlank et al., 2019). Rpn6 serine 14 phosphorylation has been detected in vivo in response to fasting, intense exercise, and hormonal cues which increases intracellular cAMP and PKA activity (VerPlank et al., 2019). PKA has also been reported to activate the proteasome via phosphorylation of the ATPase subunit Rpt6 (Zhang et al., 2007). Recombinant PKA directly phosphorylates Rpt6 at serine 120 in vitro, and expression of a Rpt6 phospho-silenced Ser120Ala mutation reduced the proteasome activity (Zhang et al., 2007). Phosphorylation of the 19S cap Rpt6 ATPase subunit initiates assembly of 26S proteasome, by stimulating the association of the 19S particle with the 20S proteasome (Satoh et al., 2001). Subsequent studies have reproduced the changes in proteasome activity by modulating PKA activity, however, they have failed to confirm phosphorylation of several proteasome subunit sites (VerPlank and Goldberg, 2018). There are various explanations for this discrepancy: a kinase may modify additional targets in an artificial setting, in vitro but not in vivo, only in specific cell types, and that kinases and phosphatases may co-purify with the proteasome which can add or remove phosphate groups during isolation (Guo et al., 2017; VerPlank and Goldberg, 2018). Nonetheless, these studies demonstrate PKA is a critical regulator of proteasome assembly and activity during physiological and pathological conditions.
Protein Phosphatases 1γ (PP1γ) and 2A (PP2A)
Dephosphorylation via protein phosphatases of the proteasome negatively regulates the activity of the proteasome. Protein phosphatase 1γ (PP1γ) was postulated to reverse the effect of PKA phosphorylation on 20S proteasome activity (Zhang et al., 2007). Protein phosphatase 2A (PP2A) has also been reported to interact with and dephosphorylate the native 20S subunits in cardiac proteasome (Zong et al., 2006). PP2A reduced serine phosphorylation on α1 and β7, and threonine phosphorylation on α1, were linked to suppressed proteasome activity. Echoing these results, pharmacological inhibition of PP2A by OA conferred increased proteasome activity (Gomes et al., 2006; Zong et al., 2006). These data demonstrate the regulatory role of these phosphatases, however whether they can be targeted during disease pathogenesis to enhance proteostasis remains to be determined.
Calcium/Calmodulin-Dependent Protein Kinase II (CaMKII)
A rise of intracellular calcium stimulates proteasome activity by way of calcium/calmodulin dependent protein kinase (CaMKII) phosphorylation of the proteasome (Djakovic et al., 2009). Purified CaMKIIα phosphorylated Rpt6 at serine 120 resulting in increased 26S proteasome activity in both neurons and HEK 293T cells (Djakovic et al., 2009). Phosphorylation of Rpt6 at serine 120 was found to be important for neuronal plasticity (Jarome et al., 2013). Activated CaMKIIα interacts with proteasome complex which is essential for proteasome redistribution to dendritic spines for degradation of polyubiquitinated proteins (Bingol et al., 2010; Djakovic et al., 2012; Hamilton et al., 2012). Interestingly in the presence of calcium, calmodulin binds to several proteasome subunits and proteasome-interacting proteins, which may alter 26S function allosterically without involvement of a kinase (Shen et al., 2005; Djakovic et al., 2009, 2012; Bingol et al., 2010). These findings are of particular relevance to the cardiac field as both intracellular calcium and activation of CaMKII are known to worsen cardiac disease pathogenesis (Anderson et al., 2011), although CaMKII regulation of the cardiac proteasomes and proteostasis in a diseased state is yet to be explored.
Dual-Specificity Tyrosine-Regulated Kinase 2 (DYRK2)
The proteasome regulates many cellular processes, including cell cycle progression where accumulating evidence suggests a pivotal role for proteasome phosphorylation. Guo and colleagues reported proteasomes purified from S phase of the cell cycle contained increased phosphorylated Rpt3 (of the 19S cap) at threonine 25 and remained high through G2 and M phases (Guo et al., 2016). Utilizing an unbiased screen of human kinases, they found that dual-specificity tyrosine-regulated kinase 2 (DYRK2) could catalyze Rpt3 phosphorylation at threonine 25 to promote cell proliferation (Guo et al., 2016). Prevention of this phosphorylation through expression of a phospho-null threonine to valine (Thr25Val) mutation in human breast cancer cells blocked the stimulation of the proteasomes by DYRK2, thus slowing the degradation of essential cell cycle regulatory factors like cyclin-dependent kinase inhibitor 1B (p27Kip1) and cyclin-dependent kinase inhibitor 1 (p21Cip1) (Guo et al., 2016). DYRK2 phosphorylation of Rpt3 threonine 25 increased substrate-stimulated ATP-hydrolysis, without changing basal ATPase activity, indicating that the modification promotes substrate translocation and degradation (Guo et al., 2016). The role of DYRK2 on the proteasome remains to be explored in striated muscle and as a potential therapeutic target for cardiac proteotoxicities.
Apoptosis Signal-Regulating Kinase 1 (ASK1)
Proteasome phosphorylation does not always result in increased activity. The ASK1 is a member of the mitogen-activated protein kinase kinase kinase (MKK) family and is activated by several cytotoxic stressors and apoptotic stimuli (Nishida et al., 2017). ASK1 negatively regulates the 26S proteasome under oxidative stress or apoptosis through phosphorylation of the 19S cap subunit Rpt5, however it remains unclear which sites of Rpt5 are modified by ASK1 (Um et al., 2010). Proteasome regulation by ASK1 could be an underlying mechanism of its role in cardiac pathogenesis. Deletion of ASK1 protected hearts from MI and PO as evidenced by decreased fibrotic remodeling and apoptosis with improved cardiac function (Yamaguchi et al., 2003). Similar results were obtained with pharmacological inhibition of ASK1 in a model of pulmonary arterial hypertension (Budas et al., 2018). Together these studies suggest the cardioprotective effects of ASK1 inhibition could be mediated by enhanced proteasome activities.
P38 Mitogen Activated Protein Kinase (MAPK)
The Rpn2 subunit of the 19S cap acts as a scaffold for other proteasome subunits and with other 19S subunits, regulates its gate opening of the 26S proteasome into the 20S core (Chen et al., 2016; Finley et al., 2016; Schweitzer et al., 2016). Phosphorylation of Rpn2 at threonine 273 by p38 MAPK suppresses proteasome function (Lee et al., 2010). It has been speculated that this phosphorylation of Rpn2 causes a conformational change, affecting the accessibility of substrates to the 20S core (Kors et al., 2019). Lee et al. (2010) reported purified 26S proteasomes from HeLa cells expressing activated p38 MAPK, had reduced proteolytic activities. The authors also showed that overexpression of an active mutant of p38 MAPK caused the accumulation of proteins that normally undergo rapid proteasomal degradation by ubiquitin-dependent and -independent pathways. p38 MAPK mediated phosphorylation of Rpn2 at threonine 273 is stimulated by sorbitol and NaCl-induced osmotic stress (Lee et al., 2010). Small molecule inhibitors of p38 MAPK and siRNA-mediated knockdown of its α-isoform stimulated proteasome peptidase activity and intracellular degradation of α-synuclein in neurons (Leestemaker et al., 2017). Of note, Tsai et al. (2015) has shown that another MAPK, ERK 2, could also phosphorylate the 19S cap subunit Rpn2 at threonine 273 in vitro. While p38 is known to be involved in that pathogenesis of cardiac disease (Yokota and Wang, 2016) its effect on the proteasome in the heart remains to be elucidated.
Casein Kinase II (CK2)
Casein kinase II (CK2) is a pivotal regulator of the proteasome and is dysregulated in heart failure. CK2 is co-purified with the 20S proteasome and has been demonstrated to phosphorylate the α7 subunit at serines 243 and 250, which is associated with stabilization of the 26S proteasome (Ludemann et al., 1993; Castano et al., 1996; Bose et al., 2004). The phosphorylation of serines is important for the interaction between the 19S cap proteins with the 20S regulatory proteins of proteasome complex (Bose et al., 2004). Interestingly, phosphorylation of the 20S proteasome α7 subunit at serine 250 is found to be significantly less in the tissues from end stage heart failure patients, which is consistent with the diminished activity of the UPS in these patients (Hein et al., 2003; Weekes et al., 2003; Predmore et al., 2010). However, CK2 expression was found to be higher by almost 70% (P = 0.019) in failing hearts (Day et al., 2013). The possible explanation could be due to by increased expression of PP1 and PP2A in myocardium of such patients which can dephosphorylate this site (Neumann et al., 1997; Zong et al., 2006; Hamdani et al., 2010), or increased oxidation and carbonylation of CK2 resulting in diminished activity of the kinase (Murtaza et al., 2008).
C-Abl and Abl-Related Gene Product (ARG)
As key regulators of cardiac growth and development, C-Abl and ARG (Abl-related gene product) are multi-functional tyrosine kinases, which also directly phosphorylate and regulate the proteasome (Liu et al., 2006; Qiu et al., 2010; Li et al., 2016). Phosphorylation of the α4-subunit at tyrosine 153 by c-Abl led to the inhibition of the 20S and 26S proteasome and decreased the degradation of ubiquitinated short-lived proteins in mouse and human cells (Liu et al., 2006; Li et al., 2015). Activation of c-Abl by H2O2 or γ-irradiation increased its interaction with the α4-subunit and inhibited proteasome function (Liu et al., 2006). Expression of a phospho-null α4 subunit tyrosine 153 mutant in HEK 293T cells resulted in downregulation of several cell cycle regulatory proteins and G1/S cell cycle arrest, indicating the prominent role of c-Abl/ARG in cell cycle control (Liu et al., 2006). In a separate study, it was shown that phosphorylation of α4 at a different site, tyrosine 106, by c-Abl/ARG regulated the turnover of the proteasome subunit itself (Li et al., 2015). These interesting studies could provide insight into the role of the proteasome in cardiac growth and development as well as its regulation.
Protein Kinase G (PKG)
Activation of PKG protects the myocardium against pressure-overload and ischemia-reperfusion (Takimoto et al., 2005; Nagayama et al., 2009; Zhang and Kass, 2011; Sasaki et al., 2014; Kokkonen and Kass, 2017; Ranek et al., 2019). There are also a plethora of therapeutic strategies capable of activating PKG in the heart (Dunkerly-Eyring and Kass, 2019). PKG activators have shown such immense promise in pre-clinical studies that many clinical trials were initiated with various PKG activation strategies to treat human heart failure (Dunkerly-Eyring and Kass, 2019; Pinilla-Vera et al., 2019; Oeing et al., 2020a). Stimulation of PKG via pharmacological activation of the muscarinic 2 receptor or inhibition of phosphodiesterase 5A, positively regulates cardiomyocyte proteasomal proteolytic activity (Ranek et al., 2013, 2014). Stimulation of PKG enhanced proteasome-mediated degradation of a misfolded protein substrate (a modified GFP harboring a degron sequence) and a bona fide misfolded protein CryABR120G (Ranek et al., 2013). The mechanism of action involved increased phosphorylation of the 20S subunit β5 and the 19S ATPase subunit Rpt6 upon PKG activation in cardiomyocytes (Ranek et al., 2013). The residues phosphorylated by PKG were not identified in this study. It is noteworthy that proteasome-mediated degradation of misfolded proteins was enhanced but not that of normal (properly folded) proteins with PKG stimulation, increasing the safety profile as a potential therapy (Ranek et al., 2013). Accordingly, PKG activators are safe and well-tolerated in human patients (Dunkerly-Eyring and Kass, 2019; Pinilla-Vera et al., 2019). A recent study that utilized human HFpEF myocardium biopsies found that in vitro treatment with an guanylyl cyclase 1 (GC-1) activator (to activate PKG) resulted in lower levels of inflammatory cytokines and oxidative stress (Kolijn et al., 2020). With oxidative stress being a known cause of protein damage and activation of the protein damage response, this reduction in oxidative stress can alleviate the burden on the PQC systems such as the proteasomal degradation pathway (Aiken et al., 2011). Taken together, the combination of promising pre-clinical studies, human clinical trials, and availability of pharmacological agents make activators of PKG an attractive therapeutic strategy for cardiac proteinopathies.
Autophagy
Autophagy is an intracellular self-degradative process responsible for the removal of misfolded proteins damaged organelles and by the lysosome. Cardiomyocytes utilize many forms of autophagy which differ by the method of degradation and the substrates targeted for degradation (Figure 1) (Sciarretta et al., 2018b). Macroautophagy begins with the formation of a double-membrane vesicle called phagophore, through a series of highly coordinated steps termed induction, nucleation and elongation, which engulf malformed proteins or whole organelles in the cytoplasm to form autophagosomes (Klionsky, 2005; Suzuki and Ohsumi, 2007; Levine and Kroemer, 2008). A fully formed autophagosome will then fuse with the acidic compartment of lysosome for the degradation of its cargo molecules (Sciarretta et al., 2018b). The non-selective lysosomal degradative process called microautophagy involves direct engulfment of cytoplasmic cargo at a boundary membrane by autophagic tubes, which mediate both invagination and vesicle scission into the lumen (Su et al., 2011). There are two types of autophagy in mammalian cells which involves degradation of cellular components via a chaperone. Chaperone-mediated autophagy (CMA) is a selective autophagic process mediated by a HSC70 chaperone complex, wherein the proteins for degradation are selected based on a consensus sequence of amino acid sequence present on the surface of protein (Kaushik and Cuervo, 2018; Juste and Cuervo, 2019). On the other hand, CASA is a highly selective autophagy of misfolded proteins following a chaperone-mediated formation of protein aggregates that are targeted to form autophagosomes (Ulbricht et al., 2015). The vast majority of studies into the role of autophagy in the heart and disease pathogenesis have investigated macroautophagy. This section describes our current knowledge into the roles and regulations of autophagy by phosphorylation in cardiac disease, primarily discussing macroautophagy due to the limited understanding of the regulations of other forms of autophagy.
Evidence of autophagic impairment in human heart disease was first reported in tissue samples from patients with dilated cardiomyopathy (Shimomura et al., 2001). Autophagy is repressed in failing hearts as evidenced by the accumulation of numerous autophagic vacuoles containing cytoplasmic material and organelles to be degraded within the degenerated cardiomyocytes, which is thought to contribute to pathological remodeling and heart failure (Shimomura et al., 2001). Reduced autophagy is also implicated in cardiovascular decline and aging (Eisenberg et al., 2016; Shirakabe et al., 2016). Autophagy protects the heart during ischemia and starvation by supplying substrates for maintaining cellular bioenergetics (Matsui et al., 2007; Hariharan et al., 2010; Zhai et al., 2011; Sciarretta et al., 2012a; Kubli et al., 2013). Activation of autophagy has protected the myocardium against pathological cardiac hypertrophy and proteotoxicity (Gao et al., 2006; Kuzman et al., 2007; Flynn et al., 2013; Dai et al., 2014). These seminal studies utilized a genetic overexpression of an autophagy protein to stimulate autophagy, while recent studies have identified critical PTMs that regulate autophagy. More than 300 PTMs have been characterized for various autophagic proteins that influence their structure and function (Xie et al., 2015; Botti-Millet et al., 2016). Here, we discuss the role of phosphorylation as a critical PTM and its functional relevance in fine-tuning the autophagic process (Figure 4).
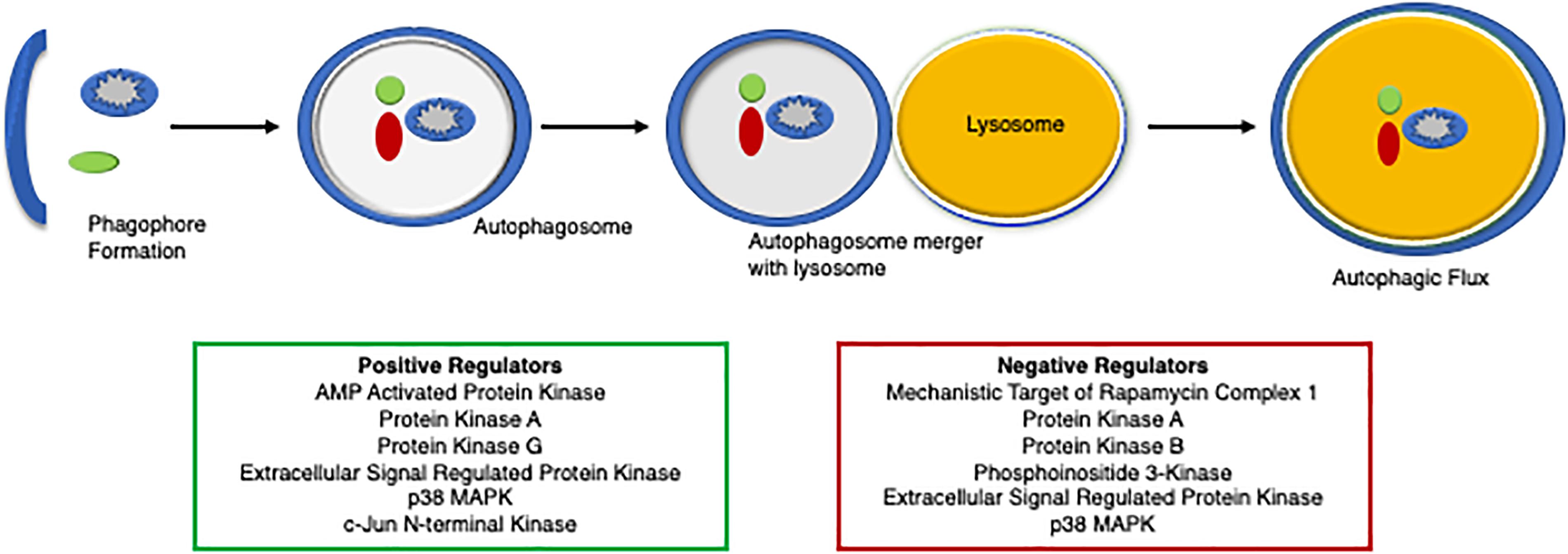
Figure 4. A diagram of the regulators of autophagy. An overview of protein degradation via autophagy and its regulators, both positively (green box) and negatively (red box).
Phosphorylations Regulating Autophagic Flux
Mechanistic Target of Rapamycin Complex 1 (mTORC1)
Arguably the most well-defined regulator of macroautophagy is the mechanistic (mammalian) target of rapamycin complex 1 (mTORC1), an atypical serine/threonine kinase that also regulates cellular metabolism and bioenergetics. Pro-growth signals and a nutrient-rich environment are associated with mTORC1 activation which negatively regulate macroautophagy. Cardiac PO, obesity, and metabolic syndrome are associated with hyperactivation of mTORC1 (Choi et al., 2012; Li et al., 2012; Ramos et al., 2012; Guo et al., 2013; Pires et al., 2017). Indeed, the hearts from pressure overloaded mice, ob/ob obese mice, mice with hypertrophic cardiomyopathies, and hearts from a swine model of metabolic syndrome exhibit mTORC1 activation, depressed autophagic flux, and cardiac functional impairment, all of which were rescued by inhibiting mTORC1 via rapamycin administration (Choi et al., 2012; Li et al., 2012; Ramos et al., 2012; Guo et al., 2013; Pires et al., 2017).
Macroautophagy is influenced by mTORC1 by various methods and pathways. Autophagosome formation is reduced through mTORC1 phosphorylation of autophagy related (Atg) 13, which decreases its affinity for Atg1 (also known as Unc-51 like autophagy activating protein kinase, Ulk1), resulting in the dissociation of the Atg13-Atg1-FIP200 complex (Papinski and Kraft, 2016). Direct phosphorylation of Atg1 (Ulk1) by mTORC1 is another strategy by which mTORC1 suppresses autophagosome formation (Sciarretta et al., 2012b, 2018a). Recently protein phosphatase 2A (PP2A) was identified as a phosphatase for Ulk1 at serine 637, which counteracts the autophagy inhibiting action of mTORC1 (Wong et al., 2015). The Ambra1 is a positive regulator of autophagy that is inhibited by mTORC1 phosphorylation at serine 52 (Nazio et al., 2013). The role of Ambra1 is not well-defined in cardiac disease, however it has been associated with the pathogenesis of Alzheimer’s Disease, aging, and tumor growth (Cianfanelli et al., 2015). Cardiomyocyte macroautophagy is tightly linked to the protein expression levels of Atg7 (Bhuiyan et al., 2013), which is negatively regulated by mTORC1 by reducing its expression level (Ramos et al., 2012). Overexpression of Atg7 enhances macroautophagy to protect the myocardium from cardiac proteinopathy, as evidenced by improved cardiac function and extended lifespan (Pattison et al., 2011). The TFEB is a master regulator, driving genes encoding autophagic proteins like ultraviolet radiation resistance associated gene (UVRAG), WD repeat domain phosphoinositide-interacting protein (WIPI), microtubule-associated protein 1 light chain 3 beta (MAPLC3B), sequestosome 1 (SQSTM1), vacuolar protein sorting-associated protein 11 (VPS11), VPS18, and Atg9B (Settembre et al., 2011). mTORC1 negatively regulates TFEB through phosphorylation at serines 142 and 211, which prevents the nuclear localization and transcriptional activity of TFEB and thereby decreasing macroautophagy (Napolitano et al., 2018). Buss et al. (2009) and others have shown that hyperactivated mTORC1 in the heart blunts autophagy and profoundly increases pathological cardiac remodeling in response to chronic ischemic injury, and can be reversed by direct mTORC1 inhibition with everolimus. Taneike et al. (2016) have shown that knocking out TSC2 (tuberin), an upstream negative regulator of mTORC1, results in constitutive stimulation of mTORC1 in the heart and subsequent cardiac chamber dilatation and dysfunction associated with an impairment of autophagic flux, which were reversed by pharmacological inhibition of mTORC1. GSK-3α knockout mice display premature death and age-related cardiac abnormalities, such as hypertrophy and sarcomere disruption, as a result of mTORC1 activation and autophagy inhibition (Zhou et al., 2013). Inactivation of Ras homolog enriched in brain (Rheb) (a positive mTORC1 regulator) protects cardiomyocytes during energy deprivation via depressing mTORC1 hyperactivation resulting in activation of autophagy, reduction of energy expenditure and attenuation of ER stress in high fat diet-induced metabolic syndrome (Sciarretta et al., 2012a). Similarly, long-term caloric restriction attenuates the LV diastolic dysfunction of the aged rat heart by reducing mTORC1 activity and enhancing autophagic flux (Shinmura et al., 2011). These studies demonstrate the regulatory capacity of mTORC1 on macroautophagy and cardioprotective effects that mTORC1 inhibition may have, however targeting mTORC1 as a treatment regimen is complex.
Considering mTORC1 is a critical regulator of many physiological processess, the role of mTORC1 and autophagy during cardiac development and pathogenesis is nuanced. Riehle et al. (2013) has shown that insulin/IGF-1 signaling through the insulin receptor substrate 1 proteins are vital to perinatal development of the heart. Reduced IRS signaling prevents the physiological activation of mTORC1 to suppress autophagy resulting in unrestrained autophagy in cardiomyocytes, which contributes to myocyte loss, heart failure, and premature death (Riehle et al., 2013). Knocking out raptor, the scaffolding protein of mTORC1, inhibits mTORC1 and leads to dilated cardiomyopathy (Shende et al., 2011). Furthermore, chronic mTORC1 inhibition with rapamycin is associated with immunosuppression (Thomson et al., 2009). Collectively these studies highlight the risk and reward that comes with broad mTORC1 inhibition. Inhibitors of mTORC1 that inhibit the pathological but not physiological roles of mTORC1 are of great interest.
AMP-Activated Protein Kinase (AMPK)
AMP-activated protein kinase is an intracellular metabolic energy sensor that functions to maintain homeostatic levels of ATP. Stimulation of AMPK results in inhibition of anabolic pathways consuming ATP and activation of catabolic pathways generating ATP. Cardiac insults such as myocardial ischemia, hypertrophy, and heart failure are associated with reduced intracellular ATP and subsequent activation of AMPK as a compensation to increase autophagy (Kubli and Gustafsson, 2014). Many studies have demonstrated that AMPK inhibits pathological cardiac hypertrophy by activating autophagy (Li et al., 2014). A study by Xie et al. (2011) suggested activation of AMPK and subsequent increase in cardiac autophagy reduced myocyte apoptosis in diabetic mouse hearts. AMPK activity was reduced in a mouse model of severe early-onset type-1 diabetes, which subsequently suppressed autophagy and increased cardiomyocyte apoptosis. Restoration of AMPK activity with metformin prevented the development of cardiomyopathy (Xie et al., 2011).
AMPK can stimulate autophagy through multiple mechanisms. AMPK activates TSC2 (an upstream negative of mTORC1) though phosphorylation at serine 1387 to inhibit mTORC1 (Inoki et al., 2003). AMPK also directly phosphorylates raptor (mTORC1 scaffold protein) at serines 722 and 792 to reduce mTORC1 activity by inducing 14-3-3 association (Gwinn et al., 2008). Atg1 (Ulk1) is positively and negatively regulated by phosphorylation. mTORC1-mediated phosphorylation of Ulk1 at serine 757 disrupts the Ulk1-AMPK interaction by inducing an unstructured and intrinsically disordered region in the protein (Kim J. et al., 2011; Shang et al., 2011; Khan and Kumar, 2012). Kim J. et al. (2011) have shown that under glucose starvation, AMPK promotes autophagy by phosphorylating Ulk1 at serines 317 and 777, thus activating Ulk1. Under conditions of mitochondrial stress, AMPK phosphorylates Ulk1 at serine 555 to enhance mitophagy by promoting translocation of Ulk1 to mitochondria. Tian et al. (2015) created a phosho-silenced Ulk1 by mutating serine 555, which blocked AMPK phosphorylation and subsequent mitophagy, demonstrating the necessity for this site to be targeted. AMPK phosphorylates beclin1 at serines 91 and 94 in response to glucose starvation, and serines 93 and 14 following exposure to ethanol (Kim et al., 2013; Hong-Brown et al., 2017). AMPK mediated phosphorylation of beclin1 at threonine 388 enhances the association of beclin1 with the VPS34-ATG14-VPS15 complex to stimulate autophagy and also reduces beclin1-Bcl2 complex formation (Zhang et al., 2016), which is known to inhibit autophagy and induce apoptosis. A beclin1T388A phospho-silenced mutant suppresses autophagy by inhibiting its interaction with ATG14 (Zhang et al., 2016). The role that phosphorylation of these sites on beclin1 has during cardiac disease remains to be explored but may hold potential as multiple studies demonstrated the important role of becin in the heart (Zhu and He, 2015; Maejima et al., 2016; Sun et al., 2018). AMPK is considered to be cardioprotective and is associated with enhancing cardiomyocyte PQC, thus represents a potential therapeutic target if the exact mechanisms of action can be well-defined during cardiac disease.
Protein Kinase A (PKA)
Autophagic flux has been reported to be negatively and positively regulated by PKA phosphorylation of various targets (Torres-Quiroz et al., 2015). In a nutrient-rich environment Atg1 is phosphorylated by PKA which keeps Atg1 largely cytosolic and dissociated from the pre-autophagosomal structure (PAS), which inhibits autophagosome formation to suppress autophagy. During starvation Atg1 is dephosphorylated and localized to the PAS to facilitate autophagy (He and Klionsky, 2009). Atg13 is an essential subunit of the Atg1 autophagy initiation complex, which following phosphorylation by PKA at serine 437 will translocate away from the PAS. Conversely, inactivation of PKA induces autophagy by reducing the inhibitory phosphorylations of Atg13 (serines 344, 437, and 581) and Atg1 (serines 508 and 515) allowing Atg13 localization at the PAS (Budovskaya et al., 2005; Stephan et al., 2009). PKA phosphorylation of microtubule-associated protein 1 light chain 3 (LC3) at serine 12 inhibits its lipidation, a critical step involved in the incorporation of LC3 into autophagosomes, resulting in inhibition of autophagy (Cherra et al., 2010). In a model of diabetic cardiomyopathy, Lin28a overexpression improved cardiac function and prevented apoptosis by activating PKA/RhoA/ROCK2-dependent signaling and by upregulating autophagy (Sun et al., 2016). Taken together, these studies suggest that, much like the positive and negative reports of stimulating autophagy in cardiac disease, PKA can also elicit protective or detrimental responses. These disparities are likely due to compartmentalization of activation within specific micro-domains. Further studies are needed to tease out the differential PKA regulation of autophagy.
Protein Kinase G (PKG)
The cardioprotective abilities of PKG have been demonstrated in the setting of various cardiac diseases including PO, MI, ischemia/reperfusion injury and is the subject of many clinical trials as a heart failure treatment (Kokkonen-Simon et al., 2018; Dunkerly-Eyring and Kass, 2019; Pinilla-Vera et al., 2019; Oeing et al., 2020b). ER stress during cardiac aging and heart failure was abrogated by activating PKG by way of sildenafil treatment (PDE5 inhibition) in isoproterenol-induced or TAC-induced hypertrophy, or swimming exercise (Gong et al., 2013; Chang et al., 2020). A new signaling paradigm was revealed in early 2019 whereby PKG phosphorylates TSC2 (aka tuberin) at serine 1365, which results in mTORC1 inhibition but only in the presence of a pathological stimulus (Ranek et al., 2019). Intriguingly, oxidation of PKG impairs the ability of PKG to target TSC2 S1365 in response to a pathological stimulus adding a layer of complexity to this pathway (Oeing et al., 2020b). Phosphorylation of TSC2 serine 1365 or expression of a knock in phosho-mimetic (S1365E) mouse had no effect on basal mTORC1 activity or macroautophagy, however during cardiac PO mTORC1 hyperactivation was blocked resulting in stimulated autophagic flux, reduced cardiomyocyte hypertrophy, and increased cardiac function and lifespan (Ranek et al., 2019). These findings are unique as inhibition of mTORC1, including phosphorylation regulations are associated with a total inhibition of mTORC1 activity, whereas modulation of TSC2 serine 1365 does not influence mTORC1 activity at baseline but has a potent suppression (phosphorylated or phospho-mimetic) or exacerbation (unphosphorylated or phospho-silenced) in the presence of a pathological hypertrophy stimulus (Ranek et al., 2019; Oeing et al., 2020b). In a recent study, activating PKG with an GC-1 activator has also been shown to reduce oxidative stress and enhance autophagy in H9c2 cardiomyocytes (Zhao et al., 2020). In this study, H9c2 cardiomyocytes were pretreated with an GC-1 activator, followed by doxorubicin, a known inducer of oxidative stress. These cells had an increase in autophagosome formation, indicating an increase in autophagic flux due to the activation of PKG. Collectively these findings make PKG an attractive therapeutic target.
Phosphoinositide 3-Kinase (PI3K) and Protein Kinase B (PKB)
Activation of the class I phosphoinositide 3-kinase (PI3K)-protein kinase B (Akt) pathway negatively regulates autophagy during cardiomyocyte stress (Mellor et al., 2013). Macroautophagy has been associated with attenuation and exacerbation of cardiac disease following myocardial ischemia-reperfusion (I/R) injury, which appears to be due to the timing and the method of autophagy activation (Sciarretta et al., 2012b, 2018b). Excessive autophagy is deleterious to cardiac function, and as Li X. et al. (2018) demonstrated, stimulation of the PI3K/Akt/mTOR pathway reduced myocardial I/R injury by suppressing excessive autophagy. Similarly, in a 12-weeks of high fructose diet model Mellor et al. (2011) found that PI3K-Akt pathway suppression resulted in a dramatic upregulation of autophagy. A recent study noted that in mice subjected to aortic banding to induce left ventricular PO, there was enhanced Akt activity with reductions in autophagic flux and cardiac function, whereas inhibiting Akt activation increased autophagic flux an cardiac function (Xu et al., 2019). A study out of the Ghigo lab in 2018 investigated the role of PI3Kγ on autophagy in anthracycline induced cardiotoxicity (Li M. et al., 2018). Wild type mice treated with doxycycline exhibited increased PI3Kγ activity, Akt activation, and Ulk1 phosphorylation which were associated with reduced autophagic flux, cell survival, and cardiac function (Li M. et al., 2018). PI3Kγ-inhibited mice had suppressed Akt activation and Ulk1 phosphorylation which coupled to increased autophagic flux, cell survival, and cardiac function (Li M. et al., 2018). In addition to increasing mTORC1 activity, activation of the PI3K/Akt pathway also negatively regulates autophagy by phosphorylating and inhibiting the Ulk1/2 complex in diabetic hearts (Mellor et al., 2013). Additionally, Akt inhibits autophagy through phosphorylation of forkhead transcription factor (FoxO) protein to impede transcription of autophagy related genes LC3, gamma-aminobutyric acid receptor-associated protein-like 1 (Gabarapl1), and Atg12 (Sengupta et al., 2009).
Extracellular Signal Regulated Protein Kinase (ERK)
Activation of the ERK regulates autophagy through maturation of the autophagic vacuoles (Corcelle et al., 2007). Treatment with trastuzumab (Herceptin), a monoclonal antibody commonly used as a therapy in HER2+ breast cancer, activated the ERK/mTORC1 pathway and inhibited autophagy in human primary cardiomyocytes resulting in increased production of ROS. Trastuzumab treatment interferes with cardiac HER2 signaling that leads to the phosphorylation of HER1-Y845/HER2-Y1248 and the activation of Erk. This in turn results in upregulation of the mTOR–Ulk1 pathway to mediate inhibition of autophagy in cardiomyocytes (Mohan et al., 2016). ERK is a negative regulator of TSC2, thereby activating mTORC1 which is known to suppress autophagy and accelerate cellular growth (Huang and Manning, 2008; Sciarretta et al., 2014, 2018a; Wang et al., 2019). Mitogen-activated protein kinase kinase (MEK) inhibition counteracted the protective effects of rapamycin on the induction of autophagy and attenuation of phenylephrine (PE)-induced cardiac hypertrophy, suggesting that ERK can also inhibit autophagy independent of mTORC1 (Gu et al., 2016). A highly selective inhibitor of MEK/ERK (U0126), reduced excessive autophagy, apoptosis, and infarct size in a model of cardiac I/R injury via the MEK/ERK/EGR-1 pathway (Wang et al., 2016). Studies in renal tubular epithelial cells and malignant human breast cells demonstrated that ERK positively regulates autophagy by facilitating the transition from LC3-I to LC3-II and increased production of beclin1 (Choi et al., 2010; Zeng et al., 2012). Further studies are needed to further determine the role of ERK regulation of autophagy in cardiac disease and identify the targets by which this occurs.
p38 MAPK
Similar to ERK, both activation and inhibition of autophagy have been reported with p38 MAPK stimulation (Webber and Tooze, 2010b). Autophagy-related (Atg) 5 is a crucial protein for autophagy to proceed. Atg5 is phosphorylated at threonine 75 by p38 MAPK leading to the inhibition of starvation-induced autophagy (Keil et al., 2013). The authors went on to create Atg5 phospho-silenced (threonine 75 to alanine, T75A) and phospho-mimetic (threonine 75 to glutamic acid, T75E) constructs, and these were used to transfect Atg5 deficient mouse embryonic fibroblasts (MEFs), thus the only Atg5 being expressed was the mutant constructs. Atg5 T75A MEFs contained autophagosomes in both the starved and serum fed conditions, suggesting enhanced basal autophagy, while the Atg5 T75E MEFs showed no autophagosome formation in either condition and had decreased autophagic flux (Keil et al., 2013). Another mechanism of inhibition is following a proinflammatory signal, whereupon p38α MAPK phosphorylates Atg1 (Ulk1) at serines 504 and 757 to disrupt its functional complex with Atg13 and reduce autophagy (He et al., 2018). Similarly, inhibition of p38 MAPK in myelogenous leukemic K562 cells increased beclin 1 expression and induction of autophagy (Colosetti et al., 2009). Alternatively, p38 MAPK was shown to phosphorylate the tumor suppressor protein p53 at serine 392, which enhanced its transcriptional activity causing increased expression of beclin1 resulting in stimulation of autophagy (Liu et al., 2009; Younce and Kolattukudy, 2010; Duan et al., 2011). Accordingly, in cultured neonatal rat cardiomyocytes exposed to 48 h of mechanical stretch and in mice following TAC, p38 MAPK inhibition caused a decrease in the autophagy marker LC3-II, suggesting a positive p38-autophagy relationship (Lin et al., 2014). Furthermore, Webber and Tooze provided evidence that the mAtg9–p38 MAPK interaction is important for autophagy and is regulated by the p38α activity (Webber and Tooze, 2010a). They identified p38 as a mAtg9 interacting protein and showed the interaction of mAtg9–p38 is required for starvation-induced mAtg9 trafficking and autophagosome formation, allowing the process of autophagy to proceed (Webber and Tooze, 2010a). The regulation of autophagy by p38 MAPK appears to be driven by the nature of the stimulus with potential sub-domain specificity.
c-Jun N-Terminal Kinase (JNK)
Increased autophagy has been reported with activation of c-Jun N-terminal Kinase (JNK), which has been attributed to a compensatory mechanism (Borsello et al., 2003; Jia et al., 2006; Li et al., 2006). JNK-dependent pathways are involved in the pathological mechanisms of myocardial hypertrophy and ischemia/reperfusion injury (He et al., 1999; Sun et al., 2012; Javadov et al., 2014). Other studies have shown that JNK activation is transient and varies depending on the severity and timing of oxidative stress during ischemia and reperfusion (Knight and Buxton, 1996; Laderoute and Webster, 1997; He et al., 1999; Fryer et al., 2001; Armstrong, 2004). JNK stimulates autophagy by phosphorylating anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) (Chen et al., 2008; Wei et al., 2008a; Geeraert et al., 2010; Nopparat et al., 2010), which is typically bound to beclin1 thereby inhibiting autophagy (Pattingre et al., 2005), however once phosphorylated will dissociate from beclin1, resulting in the induction of autophagy (Wei et al., 2008b; Mrakovcic and Frohlich, 2018). JNK phosphorylation of Bcl-2 is associated with cell survival (Pattingre et al., 2009). JNK has also been shown to induce expression of pro-autophagic proteins SQSTM1 (p62), Atg5 and Atg7 in response to oxidative stress and oncogenic transformations as a method to enhance autophagy (Byun et al., 2009; Wong et al., 2010; Kim M. J. et al., 2011). JNK can also induce the expression of beclin1 via phosphorylation of c-Jun (Li et al., 2009; Ren et al., 2010). In contrast to the above mentioned pro-autophagic role of JNK, Xu et al. (2011) have demonstrated that targeted deletion of JNK1, JNK2 and JNK3 in neurons increases autophagy. Furthermore, in a diabetic cardiomyopathy model and in Angiotensin II-treated hearts, induction of miR-221 stimulated the transcription of JNK/c-Jun which resulted in depressed autophagy and exacerbated cardiac hypertrophy (Qian et al., 2017). Collectively, these studies demonstrate that much like the other MAP kinases, the regulatory role of JNK is context and stimuli dependent.
Future Directions
Therapeutic strategies to enhance cardiac PQC are attractive, presenting a new direction for treatment of cardiac disease. Post-translational modifications, specifically phosphorylation and dephosphorylation, of key PQC proteins add selectivity and specificity to the protein degradation processes and provide a unique opportunity for pharmacological intervention. Indeed, some studies described here have demonstrated the cardioprotective potential of PQC enhancers. Here we have summarized the studies that investigated cardiac PQC (Supplementary Table 2). We also detailed other kinases that currently have not been linked to a protective pharmacological strategy, but may have immense potential. Phosphorylation of the proteins responsible for maintaining cardiac PQC during a pathological stimulus has added a new mechanism of regulation to this powerful system. With new drugs to target kinases and phosphatases, and advances in gene editing to create phospho-mimetic and -silenced proteins, the opportunities to manipulate these PQC systems are endless. Thus, elucidating the phosphorylations that regulate cardiac PQC is essential to advance our understanding and provide new therapeutic opportunities to interrogate cardiomyocyte PQC to treat cardiac disease.
Author Contributions
SM and BD-E wrote the manuscript in consultation with GK. MR conceived, designed, and directed the writing. All authors provided critical feedback, interpretation, and contributed to the final manuscript.
Funding
Supported by NIH – National Heart Lung and Blood Institute grant F31-HL143905 (BD-E), National Institute on Aging T32 training grant T32AG058527 (GK), American Heart Association 16SFRN28620000 (SM), NIH-NHLBI HL-135827 to David Kass, an American Heart Association Career Development Award 18CDA34110140 and Amyloidosis Foundation Donald C. Brockman Memorial Research Grant (MR).
Conflict of Interest
BD-E and MR co-inventors on a patent application (PCT: 448070145WO1) that was filed in July 2018 (provisional filed in June 2017). The patent relates to the use of TSC2 (S1365/S1364) modifications for immunological applications. MR is a co-founder and shareholder of Meta-T Cellular, a start-up company that aims to develop applications of this intellectual property for immune therapy.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.593585/full#supplementary-material
Abbreviations
Akt, protein kinase B; Ambra1, activating molecule in beclin1-regulated autophagy 1; AMPK, AMP-activated protein kinase; AR, androgen receptor; ARG, Abl-related gene product; ASK1, apoptosis signal-regulating kinase 1; Atg, autophagy-related gene; CamKII, calcium/calmodulin dependent protein kinase; CASA, chaperone-assisted autophagy; CDK, cyclin dependent kinase; CHIP, carboxyl terminus of HSC70 interacting protein; c-IAP 1, cellular inhibitor of apoptosis protein 1; CK2, casein kinase 2; CMA, chaperone-mediated autophagy; CryAB, α B-crystallin; DRC, desmin-related cardiomyopathy; DYRK2, dual-specificity tyrosine-regulated kinase 2; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; ERK, extracellular signal regulated protein kinase; FGF, fibroblast growth factor; FGFR1, fibroblast growth factor receptor 1; FLNC, filamin-c; Foxo, forkhead transcription factor; HECT, homologous to E6-AP carboxyl terminus; HFpEF, heart failure with a preserved ejection fraction; HOP, HSP70-HSP90 organizing protein; HSP, heat shock protein; I/R, ischemia/reperfusion; IRS, insulin receptor substrate 1; JNK, c-JunN-terminal Kinase; MDM2, murine double minute 2; MI, myocardial infarction; MK2, mitogen kinase 2; MKK, mitogen-activated protein kinase kinase; mTORC1, mechanistic target of rapamycin complex 1; NEDD4, neural precursor cell expressed developmentally down-regulated protein 4; OA, okadaic acid; PAS, pre-autophagosomal structure; PCNA, proliferating cell nuclear antigen; PFI, proteasome functional insufficiency; PI3K, phosphoinositide 3-kinase; PINK1, PTEN-induced kinase 1; PKA, protein kinase A; PKC, protein kinase C; PKG, protein kinase G; PO, pressure overload; PP1 γ, protein phosphatase 1 γ; PP2A, protein phosphatase 2A; PQ, expanded polyglutamine; PQC, protein quality control; PTM, post-translational modification; RBR, ring between ring; RING, really interesting new gene; SCF, skp, cullin, F-box containing complex; SOD, super oxide dismutase; TAC, transverse aortic constriction; TFEB, transcription factor EB; TSC2, tuberous sclerosis complex 2; UBE2A, ubiquitin-conjugating enzyme E2A; UBE2J1, ubiquitin-conjugating enzyme E2J1; Ulk1, Unc-51 like autophagy activating protein kinase; UPS, ubiquitin proteasome system.
References
Ahuja, P., Sdek, P., and MacLellan, W. R. (2007). Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 87, 521–544. doi: 10.1152/physrev.00032.2006
Aiken, C. T., Kaake, R. M., Wang, X., and Huang, L. (2011). Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteomics 10:R110006924. doi: 10.1074/mcp.R110.006924
Anderson, M. E., Brown, J. H., and Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell Cardiol. 51, 468–473. doi: 10.1016/j.yjmcc.2011.01.012
Armstrong, S. C. (2004). Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc. Res. 61, 427–436. doi: 10.1016/j.cardiores.2003.09.031
Asai, M., Tsukamoto, O., Minamino, T., Asanuma, H., Fujita, M., Asano, Y., et al. (2009). PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J. Mol. Cell Cardiol. 46, 452–462. doi: 10.1016/j.yjmcc.2008.11.001
Bachman, A. B., Keramisanou, D., Xu, W., Beebe, K., Moses, M. A., and Gelis, I. (2018). Phosphorylation induced cochaperone unfolding promotes kinase recruitment and client class-specific Hsp90 phosphorylation. Nat. Commun. 9:265. doi: 10.1038/s41467-017-02711-w
Baek, G. H., Cheng, H., Choe, V., Bao, X., Shao, J., Luo, S., et al. (2013). Cdc48: a swiss army knife of cell biology. J. Amino Acids 2013:183421. doi: 10.1155/2013/183421
Batuello, C. N., Hauck, P. M., Gendron, J. M., Lehman, J. A., and Mayo, L. D. (2015). Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proc. Natl. Acad. Sci. U.S.A. 112, 1749–1754. doi: 10.1073/pnas.1416656112
Benedict, C. M., and Clawson, G. A. (1996). Nuclear multicatalytic proteinase subunit RRC3 is important for growth regulation in hepatocytes. Biochemistry 35, 11612–11621. doi: 10.1021/bi960889p
Bennett, M. R., Sinha, S., and Owens, G. K. (2016). Vascular smooth muscle cells in atherosclerosis. Circ. Res. 118, 692–702. doi: 10.1161/CIRCRESAHA.115.306361
Bhandari, P., Song, M., Chen, Y., Burelle, Y., and Dorn, G. W. (2014). Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ. Res. 114, 257–265. doi: 10.1161/CIRCRESAHA.114.302734
Bhuiyan, M. S., Pattison, J. S., Osinska, H., James, J., Gulick, J., McLendon, P. M., et al. (2013). Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 123, 5284–5297. doi: 10.1172/JCI70877
Bicknell, K. A., Surry, E. L., and Brooks, G. (2003). Targeting the cell cycle machinery for the treatment of cardiovascular disease. J. Pharm. Pharmacol. 55, 571–591. doi: 10.1211/002235703765344487
Bingol, B., Wang, C. F., Arnott, D., Cheng, D., Peng, J., and Sheng, M. (2010). Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140, 567–578. doi: 10.1016/j.cell.2010.01.024
Boehm, M., and Nabel, E. G. (2003). The cell cycle and cardiovascular diseases. Prog. Cell Cycle Res. 5, 19–30.
Borsello, T., Croquelois, K., Hornung, J. P., and Clarke, P. G. (2003). N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur. J. Neurosci. 18, 473–485. doi: 10.1046/j.1460-9568.2003.02757.x
Bose, S., Stratford, F. L., Broadfoot, K. I., Mason, G. G., and Rivett, A. J. (2004). Phosphorylation of 20S proteasome alpha subunit C8 (alpha7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by gamma-interferon. Biochem. J. 378, 177–184. doi: 10.1042/bj20031122
Botti-Millet, J., Nascimbeni, A. C., Dupont, N., Morel, E., and Codogno, P. (2016). Fine-tuning autophagy: from transcriptional to posttranslational regulation. Am. J. Physiol. Cell Physiol. 311, C351–C362. doi: 10.1152/ajpcell.00129.2016
Budas, G. R., Boehm, M., Kojonazarov, B., Viswanathan, G., Tian, X., Veeroju, S., et al. (2018). ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 197, 373–385. doi: 10.1164/rccm.201703-0502OC
Budenholzer, L., Cheng, C. L., Li, Y., and Hochstrasser, M. (2017). Proteasome structure and assembly. J. Mol. Biol. 429, 3500–3524. doi: 10.1016/j.jmb.2017.05.027
Budovskaya, Y. V., Stephan, J. S., Deminoff, S. J., and Herman, P. K. (2005). An evolutionary proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 102, 13933–13938. doi: 10.1073/pnas.0501046102
Buss, S. J., Muenz, S., Riffel, J. H., Malekar, P., Hagenmueller, M., and Weiss, C. S. (2009). Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J. Am. Coll. Cardiol. 54, 2435–2446. doi: 10.1016/j.jacc.2009.08.031
Byun, J. Y., Yoon, C. H., An, S., Park, I. C., Kang, C. M., Kim, M. J., et al. (2009). The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis 30, 1880–1888. doi: 10.1093/carcin/bgp235
Castano, J. G., Mahillo, E., Arizti, P., and Arribas, J. (1996). Phosphorylation of C8 and C9 subunits of the multicatalytic proteinase by casein kinase II and identification of the C8 phosphorylation sites by direct mutagenesis. Biochemistry 35, 3782–3789. doi: 10.1021/bi952540s
Chang, P., Zhang, X., Zhang, M., Li, G., Hu, L., Zhao, H., et al. (2020). Swimming exercise inhibits myocardial ER stress in the hearts of aged mice by enhancing cGMPPKG signaling. Mol. Med. Rep. 21, 549–556. doi: 10.3892/mmr.2019.10864
Chen, J., Ren, Y., Gui, C., Zhao, M., Wu, X., Mao, K., et al. (2018). Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T alpha-synuclein model of Parkinson’s disease. Cell Death Dis. 9:700. doi: 10.1038/s41419-018-0722-7
Chen, J. L., Lin, H. H., Kim, K. J., Lin, A., Forman, H. J., and Ann, D. K. (2008). Novel roles for protein kinase Cdelta-dependent signaling pathways in acute hypoxic stress-induced autophagy. J. Biol. Chem. 283, 34432–34444. doi: 10.1074/jbc.M804239200
Chen, L., Lizano, P., Zhao, X., Sui, X., Dhar, S. K., Shen, Y. T., et al. (2011). Preemptive conditioning of the swine heart by H11 kinase/Hsp22 provides cardiac protection through inducible nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 300, H1303–H1310. doi: 10.1152/ajpheart.00979.2010
Chen, S., Wu, J., Lu, Y., Ma, Y. B., Lee, B. H., Yu, Z., et al. (2016). Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 113, 12991–12996. doi: 10.1073/pnas.1614614113
Cherra, S. J. III, Kulich, S. M., Uechi, G., Balasubramani, M., Mountzouris, J., Day, B. W., et al. (2010). Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 190, 533–539. doi: 10.1083/jcb.201002108
Choi, C. H., Jung, Y. K., and Oh, S. H. (2010). Autophagy induction by capsaicin in malignant human breast cells is modulated by p38 and extracellular signal-regulated mitogen-activated protein kinases and retards cell death by suppressing endoplasmic reticulum stress-mediated apoptosis. Mol. Pharmacol. 78, 114–125. doi: 10.1124/mol.110.063495
Choi, J. C., Wu, W., Muchir, A., Iwata, S., Homma, S., and Worman, H. J. (2012). Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J. Biol. Chem. 287, 40513–40524. doi: 10.1074/jbc.M112.404541
Christians, E. S., and Benjamin, I. J. (2012). Proteostasis and REDOX state in the heart. Am. J. Physiol. Heart Circ. Physiol. 302, H24–H37. doi: 10.1152/ajpheart.00903.2011
Cianfanelli, V., Nazio, F., and Cecconi, F. (2015). Connecting autophagy: AMBRA1 and its network of regulation. Mol. Cell Oncol. 2:e970059. doi: 10.4161/23723548.2014.970059
Ciechanover, A. (1994). The ubiquitin-proteasome proteolytic pathway. Cell 79, 13–21. doi: 10.1016/0092-8674(94)90396-4
Collier, M. P., Alderson, T. R., Nicholls, D., Gastall, H. Y., Allison, T. M., and Degiacomi, M. T. (2019). HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci. Adv. 5:eaav8421. doi: 10.1126/sciadv.aav8421
Collins, G. A., and Goldberg, A. L. (2017). The logic of the 26S proteasome. Cell 169, 792–806. doi: 10.1016/j.cell.2017.04.023
Colosetti, P., Puissant, A., Robert, G., Luciano, F., Jacquel, A., Gounon, P., et al. (2009). Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy 5, 1092–1098. doi: 10.4161/auto.5.8.9889
Corcelle, E., Djerbi, N., Mari, M., Nebout, M., Fiorini, C., Fenichel, P., et al. (2007). Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy 3, 57–59. doi: 10.4161/auto.3424
Dagar, M., Singh, J. P., Dagar, G., Tyagi, R. K., and Bagchi, G. (2019). Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J. Biol. Chem. 294, 8699–8710. doi: 10.1074/jbc.RA119.007420
Dahlmann, B. (2016). Mammalian proteasome subtypes: their diversity in structure and function. Arch. Biochem. Biophys. 591, 132–140. doi: 10.1016/j.abb.2015.12.012
Dai, D. F., Karunadharma, P. P., Chiao, Y. A., Basisty, N., Crispin, D., Hsieh, E. J., et al. (2014). Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13, 529–539. doi: 10.1111/acel.12203
Day, S. M. (2013). The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am. J. Physiol. Heart Circ. Physiol. 304, H1283–H1293. doi: 10.1152/ajpheart.00249.2012
Day, S. M., Divald, A., Wang, P., Davis, F., Bartolone, S., Jones, R., et al. (2013). Impaired assembly and post-translational regulation of 26S proteasome in human end-stage heart failure. Circ, Heart Fail. 6, 544–549. doi: 10.1161/CIRCHEARTFAILURE.112.000119
Depre, C., Wang, L., Sui, X., Qiu, H., Hong, C., Hedhli, N., et al. (2006). H11 kinase prevents myocardial infarction by preemptive preconditioning of the heart. Circ. Res. 98, 280–288. doi: 10.1161/01.RES.0000201284.45482.e8
Divald, A., and Powell, S. R. (2006). Proteasome mediates removal of proteins oxidized during myocardial ischemia. Free Radic. Biol. Med. 40, 156–164. doi: 10.1016/j.freeradbiomed.2005.09.022
Djakovic, S. N., Marquez-Lona, E. M., Jakawich, S. K., Wright, R., Chu, C., Sutton, M. A., et al. (2012). Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J. Neurosci. 32, 5126–5131. doi: 10.1523/JNEUROSCI.4427-11.2012
Djakovic, S. N., Schwarz, L. A., Barylko, B., DeMartino, G. N., and Patrick, G. N. (2009). Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 284, 26655–26665. doi: 10.1074/jbc.M109.021956
Drews, O., Tsukamoto, O., Liem, D., Streicher, J., Wang, Y., and Ping, P. (2010). Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ. Res. 107, 1094–1101. doi: 10.1161/CIRCRESAHA.110.222364
Drews, O., Wildgruber, R., Zong, C., Sukop, U., Nissum, M., Weber, G., et al. (2007). Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol. Cell. Proteomics 6, 2021–2031. doi: 10.1074/mcp.M700187-MCP200
Duan, W. J., Li, Q. S., Xia, M. Y., Tashiro, S., Onodera, S., and Ikejima, T. (2011). Silibinin activated p53 and induced autophagic death in human fibrosarcoma HT1080 cells via reactive oxygen species-p38 and c-Jun N-terminal kinase pathways. Biol. Pharm. Bull. 34, 47–53. doi: 10.1248/bpb.34.47
Dunkerly-Eyring, B., and Kass, D. A. (2019). Myocardial phosphodiesterases and their role in cGMP regulation. J. Cardiovasc. Pharmacol. 75, 483–493. doi: 10.1097/FJC.0000000000000773
Dworniczak, B., and Mirault, M. E. (1987). Structure and expression of a human gene coding for a 71 kd heat shock ‘cognate’ protein. Nucleic Acids Res. 15, 5181–5197. doi: 10.1093/nar/15.13.5181
Eisenberg, T., Abdellatif, M., Schroeder, S., Primessnig, U., Stekovic, S., Pendl, T., et al. (2016). Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438. doi: 10.1038/nm.4222
Elangovan, M., Chong, H. K., Park, J. H., Yeo, E. J., and Yoo, Y. J. (2017). The role of ubiquitin-conjugating enzyme Ube2j1 phosphorylation and its degradation by proteasome during endoplasmic stress recovery. J. Cell Commun. Signal. 11, 265–273. doi: 10.1007/s12079-017-0386-6
Enchev, R. I., Schulman, B. A., and Peter, M. (2015). Protein neddylation: beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 16, 30–44. doi: 10.1038/nrm3919
Fan, G. C., Chu, G., Mitton, B., Song, Q., Yuan, Q., and Kranias, E. G. (2004). Small heat-shock protein Hsp20 phosphorylation inhibits beta-agonist-induced cardiac apoptosis. Circ. Res. 94, 1474–1482. doi: 10.1161/01.RES.0000129179.66631.00
Finka, A., Sharma, S. K., and Goloubinoff, P. (2015). Multi-layered molecular mechanisms of polypeptide holding, unfolding and disaggregation by HSP70/HSP110 chaperones. Front. Mol. Biosci. 2:29. doi: 10.3389/fmolb.2015.00029
Finley, D., Chen, X., and Walters, K. J. (2016). Gates, channels, and switches: elements of the proteasome machine. Trends Biochem. Sci. 41, 77–93. doi: 10.1016/j.tibs.2015.10.009
Flynn, J. M., O’Leary, M. N., Zambataro, C. A., Academia, E. C., Presley, M. P., and Garrett, B. J. (2013). Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell 12, 851–862. doi: 10.1111/acel.12109
Fouladkou, F., Lu, C., Jiang, C., Zhou, L., She, Y., Walls, J. R., et al. (2010). The ubiquitin ligase Nedd4-1 is required for heart development and is a suppressor of thrombospondin-1. J. Biol. Chem. 285, 6770–6780. doi: 10.1074/jbc.M109.082347
Fryer, R. M., Patel, H. H., Hsu, A. K., and Gross, G. J. (2001). Stress-activated protein kinase phosphorylation during cardioprotection in the ischemic myocardium. Am. J. Physiol. Heart Circ. Physiol. 281, H1184–H1192. doi: 10.1152/ajpheart.2001.281.3.H1184
Gao, X. M., Wong, G., Wang, B., Kiriazis, H., Moore, X. L., Su, Y. D., et al. (2006). Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J. Hypertens. 24, 1663–1670. doi: 10.1097/01.hjh.0000239304.01496.83
Gardner, G. T., Travers, J. G., Qian, J., Liu, G. S., Haghighi, K., Robbins, N., et al. (2019). Phosphorylation of Hsp20 promotes fibrotic remodeling and heart failure. JACC Basic Transl. Sci. 4, 188–199. doi: 10.1016/j.jacbts.2018.11.007
Geeraert, C., Ratier, A., Pfisterer, S. G., Perdiz, D., Cantaloube, I., Rouault, A., et al. (2010). Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 285, 24184–24194. doi: 10.1074/jbc.M109.091553
George, A. J., Hoffiz, Y. C., Charles, A. J., Zhu, Y., and Mabb, A. M. (2018). A comprehensive atlas of E3 ubiquitin ligase mutations in neurological disorders. Front. Genet. 9:29. doi: 10.3389/fgene.2018.00029
Ghosh, R., and Pattison, J. S. (2018). Macroautophagy and chaperone-mediated autophagy in heart failure: the known and the unknown. Oxid. Med. Cell. Longev. 2018:8602041. doi: 10.1155/2018/8602041
Gilda, J. E., and Gomes, A. V. (2017). Proteasome dysfunction in cardiomyopathies. J. Physiol. 595, 4051–4071. doi: 10.1113/JP273607
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. doi: 10.1038/nature02263
Gomes, A. V., Young, G. W., Wang, Y., Zong, C., Eghbali, M., Drews, O., et al. (2009). Contrasting proteome biology and functional heterogeneity of the 20 S proteasome complexes in mammalian tissues. Mol. Cell. Proteom. 8, 302–315. doi: 10.1074/mcp.M800058-MCP200
Gomes, A. V., Zong, C., Edmondson, R. D., Li, X., Stefani, E., Zhang, J., et al. (2006). Mapping the murine cardiac 26S proteasome complexes. Circ. Res. 99, 362–371. doi: 10.1161/01.RES.0000237386.98506.f7
Gong, W., Duan, Q., Cai, Z., Chen, C., Ni, L., Yan, M., et al. (2013). Chronic inhibition of cGMP-specific phosphodiesterase 5 suppresses endoplasmic reticulum stress in heart failure. Br. J. Pharmacol. 170, 1396–1409. doi: 10.1111/bph.12346
Groll, M., Ditzel, L., Lowe, J., Stock, D., Bochtler, M., Bartunik, H. D., et al. (1997). Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386, 463–471. doi: 10.1038/386463a0
Gu, J., Hu, W., Song, Z. P., Chen, Y. G., Zhang, D. D., and Wang, C. Q. (2016). Rapamycin inhibits cardiac hypertrophy by promoting autophagy via the MEK/ERK/Beclin-1 pathway. Front. Physiol. 7:104. doi: 10.3389/fphys.2020.00104
Guo, R., Zhang, Y., Turdi, S., and Ren, J. (2013). Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim. Biophys. Acta 1832, 1136–1148. doi: 10.1016/j.bbadis.2013.03.013
Guo, X., Engel, J. L., Xiao, J., Tagliabracci, V. S., Wang, X., Huang, L., et al. (2011). UBLCP1 is a 26S proteasome phosphatase that regulates nuclear proteasome activity. Proc. Natl. Acad. Sci. U.S.A. 108, 18649–18654. doi: 10.1073/pnas.1113170108
Guo, X., Huang, X., and Chen, M. J. (2017). Reversible phosphorylation of the 26S proteasome. Protein Cell 8, 255–272. doi: 10.1007/s13238-017-0382-x
Guo, X., Wang, X., Wang, Z., Banerjee, S., Yang, J., Huang, L., et al. (2016). Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol. 18, 202–212. doi: 10.1038/ncb3289
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Hamdani, N., Borbely, A., Veenstra, S. P., Kooij, V., Vrydag, W., Zaremba, R., et al. (2010). More severe cellular phenotype in human idiopathic dilated cardiomyopathy compared to ischemic heart disease. J. Muscle Res. Cell Motil. 31, 289–301. doi: 10.1007/s10974-010-9231-8
Hamilton, A. M., Oh, W. C., Vega-Ramirez, H., Stein, I. S., Hell, J. W., Patrick, G. N., et al. (2012). Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 74, 1023–1030. doi: 10.1016/j.neuron.2012.04.031
Hariharan, N., Maejima, Y., Nakae, J., Paik, J., Depinho, R. A., and Sadoshima, J. (2010). Deacetylation of FoxO by sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 107, 1470–1482. doi: 10.1161/CIRCRESAHA.110.227371
Haslbeck, V., Eckl, J. M., Drazic, A., Rutz, D. A., Lorenz, O. R., Zimmermann, K., et al. (2015). The activity of protein phosphatase 5 towards native clients is modulated by the middle- and C-terminal domains of Hsp90. Sci. Rep. 5:17058. doi: 10.1038/srep17058
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi: 10.1146/annurev-genet-102808-114910
He, H., Li, H. L., Lin, A., and Gottlieb, R. A. (1999). Activation of the JNK pathway is important for cardiomyocyte death in response to simulated ischemia. Cell Death. Differ. 6, 987–991. doi: 10.1038/sj.cdd.4400572
He, Y., She, H., Zhang, T., Xu, H., Cheng, L., Yepes, M., et al. (2018). p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 217, 315–328. doi: 10.1083/jcb.201701049
Hein, S., Arnon, E., Kostin, S., Schonburg, M., Elsasser, A., Polyakova, V., et al. (2003). Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 107, 984–991. doi: 10.1161/01.CIR.0000051865.66123.B7
Henning, R. H., and Brundel, B. (2017). Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 14, 637–653. doi: 10.1038/nrcardio.2017.89
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hohfeld, J., Cyr, D. M., and Patterson, C. (2001). From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2, 885–890. doi: 10.1093/embo-reports/kve206
Hong-Brown, L. Q., Brown, C. R., Navaratnarajah, M., and Lang, C. H. (2017). FoxO1-AMPK-ULK1 regulates ethanol-induced autophagy in muscle by enhanced ATG14 association with the BECN1-PIK3C3 complex. Alcohol. Clin. Exp. Res. 41, 895–910. doi: 10.1111/acer.13377
House, S. L., House, B. E., Glascock, B., Kimball, T., Nusayr, E., Schultz, J. E., et al. (2010). Fibroblast growth factor 2 mediates isoproterenol-induced cardiac hypertrophy through activation of the extracellular regulated kinase. Mol. Cell. Pharmacol. 2, 143–154.
Huang, C. K., Lee, S. O., Chang, E., Pang, H., and Chang, C. (2016). Androgen receptor (AR) in cardiovascular diseases. J. Endocrinol. 229, R1–R16. doi: 10.1530/JOE-15-0518
Huang, J., and Manning, B. D. (2008). The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 412, 179–190. doi: 10.1042/BJ20080281
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/S0092-8674(03)00929-2
Itoh, N., and Ohta, H. (2013). Pathophysiological roles of FGF signaling in the heart. Front. Physiol. 4:247. doi: 10.3389/fphys.2013.00247
Jarome, T., Kwapis, J., Ruenzel, W., and Helmstetter, F. (2013). CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front. Behav. Neurosci. 7:115. doi: 10.3389/fnbeh.2013.00115
Javadov, S., Jang, S., and Agostini, B. (2014). Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives. Pharmacol. Ther. 144, 202–225. doi: 10.1016/j.pharmthera.2014.05.013
Jentsch, S. (1992). Ubiquitin-dependent protein degradation: a cellular perspective. Trends Cell. Biol. 2, 98–103. doi: 10.1016/0962-8924(92)90013-D
Jia, G., Cheng, G., Gangahar, D. M., and Agrawal, D. K. (2006). Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 84, 448–454. doi: 10.1111/j.1440-1711.2006.01454.x
Juste, Y. R., and Cuervo, A. M. (2019). Analysis of chaperone-mediated autophagy. Methods Mol. Biol. 1880, 703–727. doi: 10.1007/978-1-4939-8873-0_47
Kaushik, S., and Cuervo, A. M. (2018). The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381. doi: 10.1038/s41580-018-0001-6
Keil, E., Hocker, R., Schuster, M., Essmann, F., Ueffing, N., Hoffman, B., et al. (2013). Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death. Differ. 20, 321–332. doi: 10.1038/cdd.2012.129
Khan, S. H., and Kumar, R. (2012). Role of an intrinsically disordered conformation in AMPK-mediated phosphorylation of ULK1 and regulation of autophagy. Mol. Biosyst. 8, 91–96. doi: 10.1039/C1MB05265A
Kim, J., Kim, Y. C., Fang, C., Russell, R. C., Kim, J. H., Fan, W., et al. (2013). Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303. doi: 10.1016/j.cell.2012.12.016
Kim, J., Kundu, M., Viollet, B., and Guan, L. K. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, M. J., Woo, S. J., Yoon, C. H., Lee, J. S., An, S., Choi, Y. H., et al. (2011). Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 286, 12924–12932. doi: 10.1074/jbc.M110.138958
Klein, J. B., Barati, M. T., Wu, R., Gozal, D., and Sachleben, L. R. Jr. (2005). Akt-mediated valosin-containing protein 97 phosphorylation regulates its association with ubiquitinated proteins. J. Biol. Chem. 280, 31870–31881. doi: 10.1074/jbc.M501802200
Klionsky, D. J. (2005). The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 118, 7–18. doi: 10.1242/jcs.01620
Knight, R. J., and Buxton, D. B. (1996). Stimulation of c-Jun kinase and mitogen-activated protein kinase by ischemia and reperfusion in the perfused rat heart. Biochem. Biophys. Res. Commun. 218, 83–88. doi: 10.1006/bbrc.1996.0016
Kokkonen, K., and Kass, D. A. (2017). Nanodomain regulation of cardiac cyclic nucleotide signaling by phosphodiesterases. Annu. Rev. Pharmacol. Toxicol. 57, 455–479. doi: 10.1146/annurev-pharmtox-010716-104756
Kokkonen-Simon, K. M., Saberi, A., Nakamura, T., Ranek, M. J., Zhu, G., Bedja, D., et al. (2018). Marked disparity of microRNA modulation by cGMP-selective PDE5 versus PDE9 inhibitors in heart disease. JCI Insight. 3:e121739. doi: 10.1172/jci.insight.121739
Kolijn, D., Kovacs, A., Herwig, M., Lodi, M., Sieme, M., Alhaj, A., et al. (2020). Enhanced cardiomyocyte function in hypertensive rats with diastolic dysfunction and human heart failure patients after acute treatment with soluble guanylyl cyclase (sGC) Activator. Front. Physiol. 11:345. doi: 10.3389/fphys.2020.00345
Kong, S. K., and Chock, P. B. (1992). Protein ubiquitination is regulated by phosphorylation. An in vitro study. J. Biol. Chem. 267, 14189–14192.
Kors, S., Geijtenbeek, K., Reits, E., and Schipper-Krom, S. (2019). Regulation of proteasome activity by (Post-)transcriptional mechanisms. Front. Mol. Biosci. 6:48. doi: 10.3389/fmolb.2019.00048
Kostenko, S., Jensen, K. L., and Moens, U. (2014). Phosphorylation of heat shock protein 40 (Hsp40/DnaJB1) by mitogen-activated protein kinase-activated protein kinase 5 (MK5/PRAK). Int. J. Biochem. Cell Biol. 47, 29–37. doi: 10.1016/j.biocel.2013.11.004
Kotter, S., Unger, A., Hamdani, N., Lang, P., Vorgerd, M., Nagel-Steger, L., et al. (2014). Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J. Cell Biol. 204, 187–202. doi: 10.1083/jcb.201306077
Kraemer, B. F., Mannell, H., Lamkemeyer, T., Franz-Wachtel, M., and Lindemann, S. (2019). Heat-shock protein 27 (HSPB1) is upregulated and phosphorylated in human platelets during ST-elevation myocardial infarction. Int. J. Mol. Sci. 20:5968. doi: 10.3390/ijms20235968
Kubli, D. A., and Gustafsson, ÅB. (2014). Cardiomyocyte health: adapting to metabolic changes through autophagy. Trends Endocrinol. Metab. 25, 156–164. doi: 10.1016/j.tem.2013.11.004
Kubli, D. A., Zhang, X., Lee, Y., Hanna, R. A., Quinsay, M. N., Nguyen, C. K., et al. (2013). Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 288, 915–926. doi: 10.1074/jbc.M112.411363
Kumarapeli, A. R., and Wang, X. (2004). Genetic modification of the heart: chaperones and the cytoskeleton. J. Mol. Cell Cardiol. 37, 1097–1109. doi: 10.1016/j.yjmcc.2004.07.004
Kupatt, C., Dessy, C., Hinkel, R., Raake, P., Daneau, G., Bouzin, C., et al. (2004). Heat shock protein 90 transfection reduces ischemia-reperfusion-induced myocardial dysfunction via reciprocal endothelial NO synthase serine 1177 phosphorylation and threonine 495 dephosphorylation. Arterioscler. Thromb. Vasc. Biol. 24, 1435–1441. doi: 10.1161/01.ATV.0000134300.87476.d1
Kuzman, J. A., O’Connell, T. D., and Gerdes, A. M. (2007). Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology 148, 3477–3484. doi: 10.1210/en.2007-0099
Laderoute, K. R., and Webster, K. A. (1997). Hypoxia/reoxygenation stimulates Jun kinase activity through redox signaling in cardiac myocytes. Circ. Res. 80, 336–344. doi: 10.1161/01.RES.80.3.336
Lan, Y., Wang, Y., Huang, K., and Zeng, Q. (2020). Heat shock protein 22 attenuates doxorubicin-induced cardiotoxicity via regulating inflammation and apoptosis. Front. Pharmacol. 11:257. doi: 10.3389/fphys.2018.0257
Lasker, K., Forster, F., Bohn, S., Walzthoeni, T., Villa, E., Unverdorben, P., et al. (2012). Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. U.S.A. 109, 1380–1387. doi: 10.1073/pnas.1120559109
Lavoie, C., Chevet, E., Roy, L., Tonks, N. K., Fazel, A., Posner, B. I., et al. (2000). Tyrosine phosphorylation of p97 regulates transitional endoplasmic reticulum assembly in vitro. Proc. Natl. Acad. Sci. U.S.A. 97, 13637–13642. doi: 10.1073/pnas.240278097
Lee, S. H., Park, Y., Yoon, S. K., and Yoon, J. B. (2010). Osmotic stress inhibits proteasome by p38 MAPK-dependent phosphorylation. J. Biol. Chem. 285, 41280–41289. doi: 10.1074/jbc.M110.182188
Leestemaker, Y., de Jong, A., Witting, K. F., Penning, R., Schuurman, K., Rodenko, B., et al. (2017). Proteasome activation by small molecules. Cell Chem. Biol. 24, 725–736.e7. doi: 10.1016/j.chembiol.2017.05.010
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Li, C., Capan, E., Zhao, Y., Zhao, J., Stolz, D., Watkins, S. C., et al. (2006). Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 177, 5163–5168. doi: 10.4049/jimmunol.177.8.5163
Li, D., Dong, Q., Tao, Q., Gu, J., Cui, Y., Jiang, X., et al. (2015). c-Abl regulates proteasome abundance by controlling the ubiquitin-proteasomal degradation of PSMA7 subunit. Cell Rep. 10, 484–496. doi: 10.1016/j.celrep.2014.12.044
Li, D. D., Wang, L. L., Deng, R., Tang, J., Shen, Y., Guo, J. F., et al. (2009). The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 28, 886–898. doi: 10.1038/onc.2008.441
Li, G., Zhao, G., Schindelin, H., and Lennarz, W. J. (2008). Tyrosine phosphorylation of ATPase p97 regulates its activity during ERAD. Biochem. Biophys. Res. Commun. 375, 247–251. doi: 10.1016/j.bbrc.2008.08.018
Li, J., Horak, K. M., Su, H., Sanbe, A., Robbins, J., and Wang, X. (2011). Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J. Clin. Invest. 121, 3689–3700. doi: 10.1172/JCI45709
Li, J., Wilkinson, B., Clementel, V. A., Hou, J., O’Dell, T. J., and Coba, M. P. (2016). Long-term potentiation modulates synaptic phosphorylation networks and reshapes the structure of the postsynaptic interactome. Sci. Signal. 9:rs8. doi: 10.1126/scisignal.aaf6716
Li, M., Sala, V., Cimino, J., Cappello, P., Pianca, N., Margaria, J. P., et al. (2018). Phosphoinositide 3-kinase gamma inhibition protects from Anthracycline cardiotoxicity and reduces tumor growth. Circulation 138, 696–711. doi: 10.1161/CIRCULATIONAHA.117.030352
Li, X., Hu, X., Wang, J., Xu, W., Yi, C., Ma, R., et al. (2018). Inhibition of autophagy via activation of PI3K/Akt/mTOR pathway contributes to the protection of hesperidin against myocardial ischemia/reperfusion injury. Int. J. Mol. Med. 42, 1917–1924. doi: 10.3892/ijmm.2018.3794
Li, Y., Chen, C., Yao, F., Su, Q., Liu, D., Xue, R., et al. (2014). AMPK inhibits cardiac hypertrophy by promoting autophagy via mTORC1. Arch. Biochem. Biophys. 558, 79–86. doi: 10.1016/j.abb.2014.06.023
Li, Z. L., Woollard, J. R., Ebrahimi, B., Crane, J. A., Jordan, K. L., Lerman, A., et al. (2012). Transition from obesity to metabolic syndrome is associated with altered myocardial autophagy and apoptosis. Arterioscler. Thromb. Vasc. Biol. 32, 1132–1141. doi: 10.1161/ATVBAHA.111.244061
Lin, L., Tang, C., Xu, J., Ye, Y., Weng, L., Wei, W., et al. (2014). Mechanical stress triggers cardiomyocyte autophagy through angiotensin II type 1 receptor-mediated p38MAP kinase independently of angiotensin II. PLoS One 9:e89629. doi: 10.1371/journal.pone.089629
Liu, B., Cheng, Y., Zhang, B., Bian, H. J., and Bao, J. K. (2009). Polygonatum cyrtonema lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondria-mediated ROS-p38-p53 pathway. Cancer Lett. 275, 54–60. doi: 10.1016/j.canlet.2008.09.042
Liu, X., Huang, W., Li, C., Li, P., Yuan, J., Li, X., et al. (2006). Interaction between c-Abl and Arg tyrosine kinases and proteasome subunit PSMA7 regulates proteasome degradation. Mol. Cell. 22, 317–327. doi: 10.1016/j.molcel.2006.04.007
Livingstone, M., Ruan, H., Weiner, J., Clauser, K. R., Strack, P., Jin, S., et al. (2005). Valosin-containing protein phosphorylation at Ser784 in response to DNA damage. Cancer Res. 65, 7533–7540. doi: 10.1158/0008-5472.CAN-04-3729
Lokireddy, S., Kukushkin, N. V., and Goldberg, A. L. (2015). cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. U.S.A. 112, E7176–E7185. doi: 10.1073/pnas.1522332112
Ludemann, R., Lerea, K. M., and Etlinger, J. D. (1993). Copurification of casein kinase II with 20 S proteasomes and phosphorylation of a 30-kDa proteasome subunit. J. Biol. Chem. 268, 17413–17417.
Maejima, Y., Isobe, M., and Sadoshima, J. (2016). Regulation of autophagy by Beclin 1 in the heart. J. Mol. Cell Cardiol. 95, 19–25. doi: 10.1016/j.yjmcc.2015.10.032
Maloyan, A., Gulick, J., Glabe, C. G., Kayed, R., and Robbins, J. (2007). Exercise reverses preamyloid oligomer and prolongs survival in alphaB-crystallin-based desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 104, 5995–6000. doi: 10.1073/pnas.0609202104
Marber, M. S., Mestril, R., Chi, S. H., Sayen, M. R., Yellon, D. M., and Dillmann, W. H. (1995). Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J. Clin. Invest. 95, 1446–1456. doi: 10.1172/JCI117815
Matic, I., Macek, B., Hilger, M., Walther, T. C., and Mann, M. (2008). Phosphorylation of SUMO-1 occurs in vivo and is conserved through evolution. J. Proteome Res. 7, 4050–4057. doi: 10.1021/pr800368m
Matsui, Y., Takagi, H., Qu, X., Abdellatif, M., Sakoda, H., Asano, T., et al. (2007). Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 100, 914–922. doi: 10.1161/01.RES.0000261924.76669.36
Mellor, K. M., Bell, J. R., Young, M. J., Ritchie, R. H., and Delbridge, L. M. (2011). Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J. Mol. Cell Cardiol. 50, 1035–1043. doi: 10.1016/j.yjmcc.2011.03.002
Mellor, K. M., Reichelt, M. E., and Delbridge, L. M. (2013). Autophagic predisposition in the insulin resistant diabetic heart. Life Sci. 92, 616–620. doi: 10.1016/j.lfs.2012.03.042
Menon, M. B., Tiedje, C., Lafera, J., Ronkina, N., Konen, T., Kotlyarov, A., et al. (2013). Endoplasmic reticulum-associated ubiquitin-conjugating enzyme Ube2j1 is a novel substrate of MK2 (MAPKAP kinase-2) involved in MK2-mediated TNFalpha production. Biochem. J. 456, 163–172. doi: 10.1042/BJ20130755
Mohan, N., Shen, Y., Endo, Y., ElZarrad, M. K., and Wu, W. J. (2016). Trastuzumab, but not pertuzumab, dysregulates HER2 signaling to mediate inhibition of autophagy and increase in reactive oxygen species production in human cardiomyocytes. Mol. Cancer Ther. 15, 1321–1331. doi: 10.1158/1535-7163.MCT-15-0741
Mori-Konya, C., Kato, N., Maeda, R., Yasuda, K., Higashimae, N., Noguchi, M., et al. (2009). p97/valosin-containing protein (VCP) is highly modulated by phosphorylation and acetylation. Genes Cells 14, 483–497. doi: 10.1111/j.1365-2443.2009.01286.x
Morin, D., Long, R., Panel, M., Laure, L., Taranu, A., Gueguen, C., et al. (2019). Hsp22 overexpression induces myocardial hypertrophy, senescence and reduced life span through enhanced oxidative stress. Free Radic. Biol. Med. 137, 194–200. doi: 10.1016/j.freeradbiomed.2019.04.035
Mrakovcic, M., and Frohlich, L. F. (2018). p53-mediated molecular control of autophagy in tumor cells. Biomolecules 8:14. doi: 10.3390/biom8020014
Muller, P., Ruckova, E., Halada, P., Coates, P. J., Hrstka, R., Lane, D. P., et al. (2013). C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 32, 3101–3110. doi: 10.1038/onc.2012.314
Murtaza, I., Wang, H. X., Feng, X., Alenina, N., Bader, M., Prabhakar, B. S., et al. (2008). Down-regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J. Biol. Chem. 283, 5996–6004. doi: 10.1074/jbc.M706466200
Nagai, Y., Kaneda, S., Nomura, K., Yasuda, H., Seno, T., and Yamao, F. (1995). Ubiquitin-activating enzyme, E1, is phosphorylated in mammalian cells by the protein kinase Cdc2. J. Cell Sci. 108(Pt 6), 2145–2152.
Nagayama, T., Hsu, S., Zhang, M., Koitabashi, N., Bedja, D., Gabrielson, K. L., et al. (2009). Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J. Am. Coll. Cardiol. 53, 207–215. doi: 10.1016/j.jacc.2008.08.069
Napolitano, G., Esposito, A., Choi, H., Matarese, M., Benedetti, V., Monfregola, J., et al. (2018). mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 9:3312. doi: 10.1038/s41467-018-05862-6
Nazio, F., Strappazzon, F., Antonioli, M., Bielli, P., Cianfanelli, V., Bordi, M., et al. (2013). mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 15, 406–416. doi: 10.1038/ncb2708
Neumann, J., Eschenhagen, T., Jones, L. R., Linck, B., Schmitz, W., Scholz, H., et al. (1997). Increased expression of cardiac phosphatases in patients with end-stage heart failure. J. Mol. Cell Cardiol. 29, 265–272. doi: 10.1006/jmcc.1996.0271
Nishida, T., Hattori, K., and Watanabe, K. (2017). The regulatory and signaling mechanisms of the ASK family. Adv. Biol. Regul. 66, 2–22. doi: 10.1016/j.jbior.2017.05.004
Nopparat, C., Porter, J. E., Ebadi, M., and Govitrapong, P. (2010). The mechanism for the neuroprotective effect of melatonin against methamphetamine-induced autophagy. J. Pineal Res. 49, 382–389. doi: 10.1111/j.1600-079X.2010.00805.x
Oberoi, J., Dunn, D. M., Woodford, M. R., Mariotti, L., Schulman, J., Bourboulia, D., et al. (2016). Structural and functional basis of protein phosphatase 5 substrate specificity. Proc. Natl. Acad. Sci. U.S.A. 113, 9009–9014. doi: 10.1073/pnas.1603059113
Ochoa, C. D., Wu, R. F., and Terada, L. S. (2018). ROS signaling and ER stress in cardiovascular disease. Mol. Aspects Med. 63, 18–29. doi: 10.1016/j.mam.2018.03.002
Oeing, C. U., Mishra, S., Dunkerly-Eyring, B. L., and Ranek, M. J. (2020a). Targeting protein kinase G to treat cardiac proteotoxicity. Front. Physiol. 11:858. doi: 10.3389/fphys.2020.00858
Oeing, C. U., Nakamura, T., Pan, S., Mishra, S., Lin, B. L., Chen, A., et al. (2020b). PKG1alpha cysteine-42 redox state controls mTORC1 activation in pathological cardiac hypertrophy. Circ. Res. 127, 522–533. doi: 10.1161/CIRCRESAHA.119.315714
Papinski, D., and Kraft, C. (2016). Regulation of autophagy by signaling through the Atg1/ULK1 complex. J. Mol. Biol. 428, 1725–1741. doi: 10.1016/j.jmb.2016.03.030
Parisi, E., Yahya, G., Flores, A., and Aldea, M. (2018). Cdc48/p97 segregase is modulated by cyclin-dependent kinase to determine cyclin fate during G1 progression. EMBO J. 37:e98724. doi: 10.15252/embj.201798724
Parry, T. L., Melehani, J. H., Ranek, M. J., and Willis, M. S. (2015). Functional amyloid signaling via the inflammasome, necrosome, and signalosome: new therapeutic targets in heart failure. Front. Cardiovasc. Med. 2:25. doi: 10.3389/fphys.2018.0025
Pathare, G. R., Nagy, I., Bohn, S., Unverdorben, P., Hubert, A., Korner, R., et al. (2012). The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc. Natl. Acad. Sci. U.S.A. 109, 149–154. doi: 10.1073/pnas.1117648108
Pattingre, S., Bauvy, C., Carpentier, S., Levade, T., Levine, B., and Codogno, P. (2009). Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 284, 2719–2728. doi: 10.1074/jbc.M805920200
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Pattison, J. S., Osinska, H., and Robbins, J. (2011). Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ. Res. 109, 151–160. doi: 10.1161/CIRCRESAHA.110.237339
Pattison, J. S., Sanbe, A., Maloyan, A., Osinska, H., Klevitsky, R., and Robbins, J. (2008). Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation 117, 2743–2751. doi: 10.1161/CIRCULATIONAHA.107.750232
Penna, C., Sorge, M., Femmino, S., Pagliaro, P., and Brancaccio, M. (2018). Redox aspects of chaperones in cardiac function. Front. Physiol. 9:216. doi: 10.3389/fphys.2018.00216
Pereira, M. E., and Wilk, S. (1990). Phosphorylation of the multicatalytic proteinase complex from bovine pituitaries by a copurifying cAMP-dependent protein kinase. Arch. Biochem. Biophys. 283, 68–74. doi: 10.1016/0003-9861(90)90613-4
Persaud, A., Alberts, P., Mari, S., Tong, J., Murchie, R., Maspero, E., et al. (2014). Tyrosine phosphorylation of NEDD4 activates its ubiquitin ligase activity. Sci. Signal. 7:ra95. doi: 10.1126/scisignal.2005290
Piccirillo, R., and Goldberg, A. L. (2012). The p97/VCP ATPase is critical in muscle atrophy and the accelerated degradation of muscle proteins. EMBO J. 31, 3334–3350. doi: 10.1038/emboj.2012.178
Pinilla-Vera, M., Hahn, V. S., and Kass, D. A. (2019). Leveraging signaling pathways to treat heart failure with reduced ejection fraction. Circ. Res. 124, 1618–1632. doi: 10.1161/CIRCRESAHA.119.313682
Pires, K. M., Buffolo, M., Schaaf, C., David Symons, J., Cox, J., Abel, E. D., et al. (2017). Activation of IGF-1 receptors and Akt signaling by systemic hyperinsulinemia contributes to cardiac hypertrophy but does not regulate cardiac autophagy in obese diabetic mice. J. Mol. Cell Cardiol. 113, 39–50. doi: 10.1016/j.yjmcc.2017.10.001
Plumier, J. C., Ross, B. M., Currie, R. W., Angelidis, C. E., Kazlaris, H., Kollias, G., et al. (1995). Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J. Clin. Invest. 95, 1854–1860. doi: 10.1172/JCI117865
Powell, S. R., and Divald, A. (2010). The ubiquitin-proteasome system in myocardial ischaemia and preconditioning. Cardiovasc. Res. 85, 303–311. doi: 10.1093/cvr/cvp321
Powell, S. R., Samuel, S. M., Wang, P., Divald, A., Thirunavukkarasu, M., Koneru, S., et al. (2008). Upregulation of myocardial 11S-activated proteasome in experimental hyperglycemia. J. Mol. Cell Cardiol. 44, 618–621. doi: 10.1016/j.yjmcc.2007.12.009
Pratt, W. B., and Toft, D. O. (2003). Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. 228, 111–133. doi: 10.1177/153537020322800201
Predmore, J. M., Wang, P., Davis, F., Bartolone, S., Westfall, M. V., Dyke, D. B., et al. (2010). Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121, 997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557
Qian, J., Ren, X., Wang, X., Zhang, P., Jones, W. K., Molkentin, J. D., et al. (2009). Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ. Res. 105, 1223–1231. doi: 10.1161/CIRCRESAHA.109.200378
Qian, J., Vafiadaki, E., Florea, S. M., Singh, V. P., Song, W., Lam, C. K., et al. (2011). Small heat shock protein 20 interacts with protein phosphatase-1 and enhances sarcoplasmic reticulum calcium cycling. Circ. Res. 108, 1429–1438. doi: 10.1161/CIRCRESAHA.110.237644
Qian, L. B., Jiang, S. Z., Tang, X. Q., Zhang, J., Liang, Y. Q., Yu, H. T., et al. (2017). Exacerbation of diabetic cardiac hypertrophy in OVE26 mice by angiotensin II is associated with JNK/c-Jun/miR-221-mediated autophagy inhibition. Oncotarget 8, 106661–106671. doi: 10.18632/oncotarget.21302
Qiu, Z., Cang, Y., and Goff, S. P. (2010). c-Abl tyrosine kinase regulates cardiac growth and development. Proc. Natl. Acad. Sci. U.S.A. 107, 1136–1141. doi: 10.1073/pnas.0913131107
Radford, N. B., Fina, M., Benjamin, I. J., Moreadith, R. W., Graves, K. H., Zhao, P., et al. (1996). Cardioprotective effects of 70-kDa heat shock protein in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 93, 2339–2342. doi: 10.1073/pnas.93.6.2339
Ramos, F. J., Chen, S. C., Garelick, M. G., Dai, D. F., Liao, C. Y., Schreiber, K. H., et al. (2012). Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 4:144ra103. doi: 10.1126/scitranslmed.3003802
Ranek, M. J., Kost, C. K. Jr., Hu, C., Martin, D. S., and Wang, X. (2014). Muscarinic 2 receptors modulate cardiac proteasome function in a protein kinase G-dependent manner. J. Mol. Cell Cardiol. 69, 43–51. doi: 10.1016/j.yjmcc.2014.01.017
Ranek, M. J., Stachowski, M. J., Kirk, J. A., and Willis, M. S. (2018). The role of heat shock proteins and co-chaperones in heart failure. Philos. Trans. R. Soc. Lond. B Biol. Sci. 373:20160530. doi: 10.1098/rstb.2016.0530
Ranek, M. J., Terpstra, E. J., Li, J., Kass, D. A., and Wang, X. (2013). Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation 128, 365–376. doi: 10.1161/CIRCULATIONAHA.113.001971
Ranek, M. J., Vera, M. P., Oeing, C. U., Patel, C. H., Nakamura, T., Zhu, G., et al. (2019). PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 566, 264–269. doi: 10.1038/s41586-019-0895-y
Ranek, M. J., and Wang, X. (2009). Activation of the ubiquitin-proteasome system in doxorubicin cardiomyopathy. Curr. Hypertens. Rep. 11, 389–395. doi: 10.1007/s11906-009-0068-8
Ranek, M. J., Zheng, H., Huang, W., Kumarapeli, A. R., Li, J., Liu, J., et al. (2015). Genetically induced moderate inhibition of 20S proteasomes in cardiomyocytes facilitates heart failure in mice during systolic overload. J. Mol. Cell Cardiol. 85, 273–281. doi: 10.1016/j.yjmcc.2015.06.014
Razeghi, P., Baskin, K. K., Sharma, S., Young, M. E., Stepkowski, S., Essop, M. F., et al. (2006). Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem. Biophys. Res. Commun. 342, 361–364. doi: 10.1016/j.bbrc.2006.01.163
Ren, H., Fu, K., Mu, C., Li, B., Wang, D., and Wang, G. (2010). DJ-1, a cancer and Parkinson’s disease associated protein, regulates autophagy through JNK pathway in cancer cells. Cancer Lett. 297, 101–108. doi: 10.1016/j.canlet.2010.05.001
Riehle, C., Wende, A. R., Sena, S., Pires, K. M., Pereira, R. O., Zhu, Y., et al. (2013). Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J. Clin. Invest. 123, 5319–5333. doi: 10.1172/JCI71171
Robson, A., Makova, S. Z., Barish, S., Zaidi, S., Mehta, S., Drozd, J., et al. (2019). Histone H2B monoubiquitination regulates heart development via epigenetic control of cilia motility. Proc. Natl. Acad. Sci. U.S.A. 116, 14049–14054. doi: 10.1073/pnas.1808341116
Roscic, A., Moller, A., Calzado, M. A., Renner, F., Wimmer, V. C., Gresko, E., et al. (2006). Phosphorylation-dependent control of Pc2 SUMO E3 ligase activity by its substrate protein HIPK2. Mol. Cell. 24, 77–89. doi: 10.1016/j.molcel.2006.08.004
Sadowski, M., Mawson, A., Baker, R., and Sarcevic, B. (2007). Cdc34 C-terminal tail phosphorylation regulates Skp1/cullin/F-box (SCF)-mediated ubiquitination and cell cycle progression. Biochem. J. 405, 569–581. doi: 10.1042/BJ20061812
Sandri, M., and Robbins, J. (2014). Proteotoxicity: an underappreciated pathology in cardiac disease. J. Mol. Cell Cardiol. 71, 3–10. doi: 10.1016/j.yjmcc.2013.12.015
Sasaki, H., Nagayama, T., Blanton, R. M., Seo, K., Zhang, M., Zhu, G., et al. (2014). PDE5 inhibitor efficacy is estrogen dependent in female heart disease. J. Clin. Invest. 124, 2464–2471. doi: 10.1172/JCI70731
Satoh, K., Sasajima, H., Nyoumura, K. I., Yokosawa, H., and Sawada, H. (2001). Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 40, 314–319. doi: 10.1021/bi001815n
Schwartz, A. L., and Ciechanover, A. (1992). Ubiquitin-mediated protein modification and degradation. Am. J. Respir. Cell Mol. Biol. 7, 463–468. doi: 10.1165/ajrcmb/7.5.463
Schweitzer, A., Aufderheide, A., Rudack, T., Beck, F., Pfeifer, G., Plitzko, J. M., et al. (2016). Structure of the human 26S proteasome at a resolution of 3.9 A. Proc. Natl. Acad. Sci. U.S.A. 113, 7816–7821. doi: 10.1073/pnas.1608050113
Sciarretta, S., Forte, M., Frati, G., and Sadoshima, J. (2018a). New insights into the role of mTOR signaling in the cardiovascular system. Circ. Res. 122, 489–505. doi: 10.1161/CIRCRESAHA.117.311147
Sciarretta, S., Maejima, Y., Zablocki, D., and Sadoshima, J. (2018b). The Role of Autophagy in the Heart. Annu. Rev. Physiol. 80, 1–26. doi: 10.1146/annurev-physiol-021317-121427
Sciarretta, S., Volpe, M., and Sadoshima, J. (2014). Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 114, 549–564. doi: 10.1161/CIRCRESAHA.114.302022
Sciarretta, S., Zhai, P., Shao, D., Maejima, Y., Robbins, J., Volpe, M., et al. (2012a). Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation 125, 1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212
Sciarretta, S., Zhai, P., Volpe, M., and Sadoshima, J. (2012b). Pharmacological modulation of autophagy during cardiac stress. J. Cardiovasc. Pharmacol. 60, 235–241. doi: 10.1097/FJC.0b013e3182575f61
Scruggs, S. B., Zong, N. C., Wang, D., Stefani, E., and Ping, P. (2012). Post-translational modification of cardiac proteasomes: functional delineation enabled by proteomics. Am. J. Physiol. Heart Circ. Physiol. 303, H9–H18. doi: 10.1152/ajpheart.00189.2012
Sengupta, A., Molkentin, J. D., and Yutzey, K. E. (2009). FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 284, 28319–28331. doi: 10.1074/jbc.M109.024406
Settembre, C., Vetrini, F., Erdin, S., Erdin, S. U., Huynh, T., Medina, D., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. doi: 10.1126/science.1204592
Shang, L., Chen, S., Du, F., Li, S., Zhao, L., and Wang, X. (2011). Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U.S.A. 108, 4788–4793. doi: 10.1073/pnas.1100844108
Sharma, K., and Kass, D. A. (2014). Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ. Res. 115, 79–96. doi: 10.1161/CIRCRESAHA.115.302922
Sharma, S. K., De los Rios, P., Christen, P., Lustig, A., and Goloubinoff, P. (2010). The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat. Chem. Biol. 6, 914–920. doi: 10.1038/nchembio.455
Shchebet, A., Karpiuk, O., Kremmer, E., Eick, D., and Johnsen, S. A. (2012). Phosphorylation by cyclin-dependent kinase-9 controls ubiquitin-conjugating enzyme-2A function. Cell Cycle 11, 2122–2127. doi: 10.4161/cc.20548
Shen, X., Valencia, C. A., Szostak, J. W., Dong, B., and Liu, R. (2005). Scanning the human proteome for calmodulin-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 5969–5974. doi: 10.1073/pnas.0407928102
Shende, P., Plaisance, I., Morandi, C., Pellieux, C., Berthonneche, C., Zorzato, F., et al. (2011). Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 123, 1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066
Shimomura, H., Terasaki, F., Hayashi, T., Kitaura, Y., Isomura, T., and Suma, H. (2001). Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn. Circ. J. 65, 965–968. doi: 10.1253/jcj.65.965
Shinmura, K., Tamaki, K., Sano, M., Murata, M., Yamakawa, H., Ishida, H., et al. (2011). Impact of long-term caloric restriction on cardiac senescence: caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J. Mol. Cell Cardiol. 50, 117–127. doi: 10.1016/j.yjmcc.2010.10.018
Shirakabe, A., Ikeda, Y., Sciarretta, S., Zablocki, D. K., and Sadoshima, J. (2016). Aging and autophagy in the heart. Circ. Res. 118, 1563–1576. doi: 10.1161/CIRCRESAHA.116.307474
Sledz, P., Forster, F., and Baumeister, W. (2013). Allosteric effects in the regulation of 26S proteasome activities. J. Mol. Biol. 425, 1415–1423. doi: 10.1016/j.jmb.2013.01.036
Stephan, J. S., Yeh, Y. Y., Ramachandran, V., Deminoff, S. J., and Herman, P. K. (2009). The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc. Natl. Acad. Sci. U.S.A. 106, 17049–17054. doi: 10.1073/pnas.0903316106
Stephen, A. G., Trausch-Azar, J. S., Ciechanover, A., and Schwartz, A. L. (1996). The ubiquitin-activating enzyme E1 is phosphorylated and localized to the nucleus in a cell cycle-dependent manner. J. Biol. Chem. 271, 15608–15614. doi: 10.1074/jbc.271.26.15608
Stephen, A. G., Trausch-Azar, J. S., Handley-Gearhart, P. M., Ciechanover, A., and Schwartz, A. L. (1997). Identification of a region within the ubiquitin-activating enzyme required for nuclear targeting and phosphorylation. J. Biol. Chem. 272, 10895–10903. doi: 10.1074/jbc.272.16.10895
Su, H., Li, F., Ranek, M. J., Wei, N., and Wang, X. (2011). COP9 signalosome regulates autophagosome maturation. Circulation 124, 2117–2128. doi: 10.1161/CIRCULATIONAHA.111.048934
Su, H., and Wang, X. (2010). The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc. Res. 85, 253–262. doi: 10.1093/cvr/cvp287
Su, Y. F., Yang, T., Huang, H., Liu, L. F., and Hwang, J. (2012). Phosphorylation of Ubc9 by Cdk1 enhances SUMOylation activity. PLoS One 7:e34250. doi: 10.1371/journal.pone.0034250
Sun, L., Isaak, C. K., Zhou, Y., Petkau, J. C., Liu, Y., and Siow, Y. L. (2012). Salidroside and tyrosol from Rhodiola protect H9c2 cells from ischemia/reperfusion-induced apoptosis. Life Sci. 91, 151–158. doi: 10.1016/j.lfs.2012.06.026
Sun, S., Zhang, M., Lin, J., Hu, J., Zhang, R., Li, C., et al. (2016). Lin28a protects against diabetic cardiomyopathy via the PKA/ROCK2 pathway. Biochem. Biophys. Res. Commun. 469, 29–36. doi: 10.1016/j.bbrc.2015.11.065
Sun, Y., Yao, X., Zhang, Q. J., Zhu, M., Liu, Z. P., Ci, B., et al. (2018). Beclin-1-dependent autophagy protects the heart during sepsis. Circulation 138, 2247–2262. doi: 10.1161/CIRCULATIONAHA.117.032821
Suzuki, K., and Ohsumi, Y. (2007). Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 581, 2156–2161. doi: 10.1016/j.febslet.2007.01.096
Takimoto, E., Champion, H. C., Li, M., Belardi, D., Ren, S., Rodriguez, E. R., et al. (2005). Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 11, 214–222. doi: 10.1038/nm1175
Taneike, M., Nishida, K., Omiya, S., Zarrinpashneh, E., Misaka, T., Austin, R., et al. (2016). mTOR hyperactivation by ablation of tuberous sclerosis complex 2 in the mouse heart induces cardiac dysfunction with the increased number of small mitochondria mediated through the down-regulation of autophagy. PLoS One 11:e0152628. doi: 10.1371/journal.pone.0152628
Tarone, G., and Brancaccio, M. (2014). Keep your heart in shape: molecular chaperone networks for treating heart disease. Cardiovasc. Res. 102, 346–361. doi: 10.1093/cvr/cvu049
Thomson, A. W., Turnquist, H. R., and Raimondi, G. (2009). Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 9, 324–337. doi: 10.1038/nri2546
Tian, W., Li, W., Chen, Y., Yan, Z., Huang, X., Zhuang, H., et al. (2015). Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 589, 1847–1854. doi: 10.1016/j.febslet.2015.05.020
Tomanov, K., Nukarinen, E., Vicente, J., Mendiondo, G. M., Winter, N., and Nehlin, L. (2018). Sumoylation and phosphorylation: hidden and overt links. J. Exp. Bot. 69, 4583–4590. doi: 10.1093/jxb/ery167
Torres-Quiroz, F., Filteau, M., and Landry, C. R. (2015). Feedback regulation between autophagy and PKA. Autophagy 11, 1181–1183. doi: 10.1080/15548627.2015.1055440
Trost, S. U., Omens, J. H., Karlon, W. J., Meyer, M., Mestril, R., Covell, J. W., et al. (1998). Protection against myocardial dysfunction after a brief ischemic period in transgenic mice expressing inducible heat shock protein 70. J. Clin. Invest. 101, 855–862. doi: 10.1172/JCI265
Truman, A. W., Kristjansdottir, K., Wolfgeher, D., Hasin, N., Polier, S., Zhang, H., et al. (2012). CDK-dependent Hsp70 Phosphorylation controls G1 cyclin abundance and cell-cycle progression. Cell 151, 1308–1318. doi: 10.1016/j.cell.2012.10.051
Tsai, C. F., Wang, Y. T., Yen, H. Y., Tsou, C. C., Ku, W. C., Lin, P. Y., et al. (2015). Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 6:6622. doi: 10.1038/ncomms7622
Ulbricht, A., Gehlert, S., Leciejewski, B., Schiffer, T., Bloch, W., and Hohfeld, J. (2015). Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy 11, 538–546. doi: 10.1080/15548627.2015.1017186
Um, J. W., Im, E., Park, J., Oh, Y., Min, B., Lee, H. J., et al. (2010). ASK1 negatively regulates the 26 S proteasome. J. Biol. Chem. 285, 36434–36446. doi: 10.1074/jbc.M110.133777
VerPlank, J. S., and Goldberg, A. L. (2018). Exploring the regulation of proteasome function by subunit phosphorylation. Methods Mol. Biol. 1844, 309–319. doi: 10.1007/978-1-4939-8706-1_20
VerPlank, J. S., Lokireddy, S., Zhao, J., and Goldberg, A. L. (2019). 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 116, 4228–4237. doi: 10.1073/pnas.1809254116
Wang, A., Zhang, H., Liang, Z., Xu, K., Qiu, W., Tian, Y., et al. (2016). U0126 attenuates ischemia/reperfusion-induced apoptosis and autophagy in myocardium through MEK/ERK/EGR-1 pathway. Eur. J. Pharmacol 788, 280–285. doi: 10.1016/j.ejphar.2016.06.038
Wang, B., Nie, J., Wu, L., Hu, Y., Wen, Z., Dong, L., et al. (2018). AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ. Res. 122, 712–729. doi: 10.1161/CIRCRESAHA.117.312317
Wang, H., Liu, Y., Wang, D., Xu, Y., Dong, R., Yang, Y., et al. (2019). The upstream pathway of mTOR-mediated autophagy in liver diseases. Cells 8:1597. doi: 10.3390/cells8121597
Wang, X., Klevitsky, R., Huang, W., Glasford, J., Li, F., and Robbins, J. (2003). AlphaB-crystallin modulates protein aggregation of abnormal desmin. Circ. Res. 93, 998–1005. doi: 10.1161/01.RES.0000102401.77712.ED
Wang, X., Li, J., Zheng, H., Su, H., and Powell, S. R. (2011). Proteasome functional insufficiency in cardiac pathogenesis. Am. J. Physiol. Heart Circ. Physiol. 301, H2207–H2219. doi: 10.1152/ajpheart.00714.2011
Wang, X., Osinska, H., Klevitsky, R., Gerdes, A. M., Nieman, M., Lorenz, J., et al. (2001). Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ. Res. 89, 84–91. doi: 10.1161/hh1301.092688
Wang, X., Pattison, J. S., and Su, H. (2013). Posttranslational modification and quality control. Circ. Res. 112, 367–381. doi: 10.1161/CIRCRESAHA.112.268706
Wang, X., and Robbins, J. (2006). Heart failure and protein quality control. Circ. Res. 99, 1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2
Wang, X., Su, H., and Ranek, M. J. (2008). Protein quality control and degradation in cardiomyocytes. J. Mol. Cell Cardiol. 45, 11–27. doi: 10.1016/j.yjmcc.2008.03.025
Wani, P. S., Suppahia, A., Capalla, X., Ondracek, A., and Roelofs, J. (2016). Phosphorylation of the C-terminal tail of proteasome subunit alpha7 is required for binding of the proteasome quality control factor Ecm29. Sci. Rep. 6:27873. doi: 10.1038/srep27873
Webber, J. L., and Tooze, S. A. (2010a). Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 29, 27–40. doi: 10.1038/emboj.2009.321
Webber, J. L., and Tooze, S. A. (2010b). New insights into the function of Atg9. FEBS Lett. 584, 1319–1326. doi: 10.1016/j.febslet.2010.01.020
Weekes, J., Morrison, K., Mullen, A., Wait, R., Barton, P., and Dunn, M. J. (2003). Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 3, 208–216. doi: 10.1002/pmic.200390029
Wei, Y., Pattingre, S., Sinha, S., Bassik, M., and Levine, B. (2008a). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell. 30, 678–688. doi: 10.1016/j.molcel.2008.06.001
Wei, Y., Sinha, S., and Levine, B. (2008b). Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951. doi: 10.4161/auto.6788
Wilkinson, K. A., and Henley, J. M. (2010). Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145. doi: 10.1042/BJ20100158
Willis, M. S., and Patterson, C. (2010). Hold me tight: role of the heat shock protein family of chaperones in cardiac disease. Circulation 122, 1740–1751. doi: 10.1161/CIRCULATIONAHA.110.942250
Willis, M. S., and Patterson, C. (2013). Proteotoxicity and cardiac dysfunction–Alzheimer’s disease of the heart? N. Engl. J. Med. 368, 455–464. doi: 10.1056/NEJMra1106180
Willis, M. S., Schisler, J. C., Portbury, A. L., and Patterson, C. (2009). Build it up-Tear it down: protein quality control in the cardiac sarcomere. Cardiovasc. Res. 81, 439–448. doi: 10.1093/cvr/cvn289
Willis, M. S., Townley-Tilson, W. H., Kang, E. Y., Homeister, J. W., and Patterson, C. (2010). Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ. Res. 106, 463–478. doi: 10.1161/CIRCRESAHA.109.208801
Wong, C. H., Iskandar, K. B., Yadav, S. K., Hirpara, J. L., Loh, T., and Pervaiz, S. (2010). Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One 5:e9996. doi: 10.1371/journal.pone.09996
Wong, P. M., Feng, Y., Wang, J., Shi, R., and Jiang, X. (2015). Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 6:8048. doi: 10.1038/ncomms9048
Wu, B., Hunt, C., and Morimoto, R. (1985). Structure and expression of the human gene encoding major heat shock protein HSP70. Mol. Cell. Biol. 5, 330–341. doi: 10.1128/MCB.5.2.330
Wu, T., Mu, Y., Bogomolovas, J., Fang, X., Veevers, J., Nowak, R. B., et al. (2017). HSPB7 is indispensable for heart development by modulating actin filament assembly. Proc. Natl. Acad. Sci. U.S.A. 114, 11956–11961. doi: 10.1073/pnas.1713763114
Xie, Y., Kang, R., Sun, X., Zhong, M., Huang, J., Klionsky, D. J., et al. (2015). Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 11, 28–45. doi: 10.4161/15548627.2014.984267
Xie, Z., Lau, K., Eby, B., Lozano, P., He, C., Pennington, B., et al. (2011). Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes Metab. Res. Rev. 60, 1770–1778. doi: 10.2337/db10-0351
Xu, M., Wan, C. X., Huang, S. H., Wang, H. B., Fan, D., Wu, H. M., et al. (2019). Oridonin protects against cardiac hypertrophy by promoting P21-related autophagy. Cell Death Dis. 10:403. doi: 10.1038/s41419-019-1617-y
Xu, P., Das, M., Reilly, J., and Davis, R. J. (2011). JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 25, 310–322. doi: 10.1101/gad.1984311
Yamaguchi, O., Higuchi, Y., Hirotani, S., Kashiwase, K., Nakayama, H., Hikoso, S., et al. (2003). Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc. Natl. Acad. Sci. U.S.A. 100, 15883–15888. doi: 10.1073/pnas.2136717100
Yokota, T., and Wang, Y. (2016). p38 MAP kinases in the heart. Gene 575, 369–376. doi: 10.1016/j.gene.2015.09.030
Younce, C. W., and Kolattukudy, P. E. (2010). MCP-1 causes cardiomyoblast death via autophagy resulting from ER stress caused by oxidative stress generated by inducing a novel zinc-finger protein. MCPIP Biochem. J. 426, 43–53. doi: 10.1042/BJ20090976
Yuan, F., Ma, Y., You, P., Lin, W., Lu, H., Yu, Y., et al. (2013). A novel role of proteasomal beta1 subunit in tumorigenesis. Biosci. Rep. 33:e00050. doi: 10.1042/BSR20130013
Zemanovic, S., Ivanov, M. V., Ivanova, L. V., Bhatnagar, A., Michalkiewicz, T., Teng, R. J., et al. (2018). Dynamic phosphorylation of the C terminus of Hsp70 regulates the mitochondrial import of SOD2 and redox balance. Cell Rep 25, 2605–2616.e7. doi: 10.1016/j.celrep.2018.11.015
Zeng, Y., Yang, X., Wang, J., Fan, J., Kong, Q., and Yu, X. (2012). Aristolochic acid I induced autophagy extenuates cell apoptosis via ERK 1/2 pathway in renal tubular epithelial cells. PLoS One 7:e30312. doi: 10.1371/journal.pone.030312
Zhai, P., Sciarretta, S., Galeotti, J., Volpe, M., and Sadoshima, J. (2011). Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ. Res. 109, 502–511. doi: 10.1161/CIRCRESAHA.111.249532
Zhang, D., Wang, W., Sun, X., Xu, D., Wang, C., Zhang, Q., et al. (2016). AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy 12, 1447–1459. doi: 10.1080/15548627.2016.1185576
Zhang, F., Hu, Y., Huang, P., Toleman, C. A., Paterson, A. J., and Kudlow, J. E. (2007). Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J. Biol. Chem. 282, 22460–22471. doi: 10.1074/jbc.M702439200
Zhang, H., Pan, B., Wu, P., Parajuli, N., Rekhter, M. D., Goldberg, A. L., et al. (2019). PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci. Adv. 5:eaaw5870. doi: 10.1126/sciadv.aaw5870
Zhang, M., and Kass, D. A. (2011). Phosphodiesterases and cardiac cGMP: evolving roles and controversies. Trends Pharmacol. Sci. 32, 360–365. doi: 10.1016/j.tips.2011.02.019
Zhao, R., Davey, M., Hsu, Y. C., Kaplanek, P., Tong, A., Parsons, A. B., et al. (2005). Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 120, 715–727. doi: 10.1016/j.cell.2004.12.024
Zhao, X. X., Cho, H., Lee, S., Woo, J. S., Song, M. Y., Cheng, X. W., et al. (2020). BAY60-2770 attenuates doxorubicin-induced cardiotoxicity by decreased oxidative stress and enhanced autophagy. Chem. Biol. Interact. 328:109190. doi: 10.1016/j.cbi.2020.109190
Zheng, N., and Shabek, N. (2017). Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 86, 129–157. doi: 10.1146/annurev-biochem-060815-014922
Zheng, Q., Su, H., Ranek, M. J., and Wang, X. (2011). Autophagy and p62 in cardiac proteinopathy. Circ. Res. 109, 296–308. doi: 10.1161/CIRCRESAHA.111.244707
Zhou, J., Freeman, T. A., Ahmad, F., Shang, X., Mangano, E., Gao, E., et al. (2013). GSK-3α is a central regulator of age-related pathologies in mice. J. Clin. Invest. 123, 1821–1832. doi: 10.1172/JCI64398
Zhou, L., Jiang, Y., Luo, Q., Li, L., and Jia, L. (2019). Neddylation: a novel modulator of the tumor microenvironment. Mol. Cancer 18:77. doi: 10.1186/s12943-019-0979-1
Zhu, H., and He, L. (2015). Beclin 1 biology and its role in heart disease. Curr. Cardiol. Rev. 11, 229–237. doi: 10.2174/1573403X10666141106104606
Zhu, Y., Bogomolovas, J., Labeit, S., and Granzier, H. (2009). Single molecule force spectroscopy of the cardiac titin N2B element: effects of the molecular chaperone alphaB-crystallin with disease-causing mutations. J. Biol. Chem. 284, 13914–13923. doi: 10.1074/jbc.M809743200
Zong, C., Gomes, A. V., Drews, O., Li, X., Young, G. W., Berhane, B., et al. (2006). Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ. Res. 99, 372–380. doi: 10.1161/01.RES.0000237389.40000.02
Zong, C., Young, G. W., Wang, Y., Lu, H., Deng, N., Drews, O., et al. (2008). Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 8, 5025–5037. doi: 10.1002/pmic.200800387
Zu, L., Bedja, D., Fox-Talbot, K., Gabrielson, K. L., Van Kaer, L., Becker, L. C., et al. (2010). Evidence for a role of immunoproteasomes in regulating cardiac muscle mass in diabetic mice. J. Mol. Cell Cardiol. 49, 5–15. doi: 10.1016/j.yjmcc.2010.02.007
Keywords: phosphorylation, chaperones, proteasome, autophagy, ubiquitin enzymes, cardiac disease
Citation: Mishra S, Dunkerly-Eyring BL, Keceli G and Ranek MJ (2020) Phosphorylation Modifications Regulating Cardiac Protein Quality Control Mechanisms. Front. Physiol. 11:593585. doi: 10.3389/fphys.2020.593585
Received: 11 August 2020; Accepted: 28 September 2020;
Published: 12 November 2020.
Edited by:
Julian Stelzer, Case Western Reserve University, United StatesReviewed by:
Nazha Hamdani, Ruhr University Bochum, GermanyJohn Jeshurun Michael, Cornell University, United States
Copyright © 2020 Mishra, Dunkerly-Eyring, Keceli and Ranek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark J. Ranek, bXJhbmVrMUBqaG1pLmVkdQ==; bXJhbmVrMUBqaC5lZHU=
†These authors have contributed equally to this work