 Laura Caracuel1,2Esther Sastre1,2María Callejo1Raquel Rodrigues-Díez2,3,4Ana B. García-Redondo1,2,4Isabel Prieto2,5Carlos Nieto6Mercedes Salaices2,3,4Ma Ángeles Aller7Jaime Arias7
Laura Caracuel1,2Esther Sastre1,2María Callejo1Raquel Rodrigues-Díez2,3,4Ana B. García-Redondo1,2,4Isabel Prieto2,5Carlos Nieto6Mercedes Salaices2,3,4Ma Ángeles Aller7Jaime Arias7 Javier Blanco-Rivero1,2,4*
Javier Blanco-Rivero1,2,4*- 1Departamento de Fisiología, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain
- 2Instituto de Investigación Hospital Universitario La Paz, Madrid, Spain
- 3Departamento de Farmacología y Terapéutica, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain
- 4Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares, Madrid, Spain
- 5Departamento de Cirugía General y Digestiva, Hospital Universitario la Paz, Madrid, Spain
- 6Departamento de Cirugía Cardiaca, Hospital Universitario la Paz, Madrid, Spain
- 7Cátedra de Cirugía, Facultad de Medicina, Universidad Complutense de Madrid, Madrid, Spain
The acute-on-chronic liver failure (ACLF) is a syndrome characterized by liver decompensation, hepatic encephalopathy (HE) and high mortality. We aimed to determine the mechanisms implicated in the development of HE-associated cerebral vasculopathy in a microsurgical liver cholestasis (MHC) model of ACLF. Microsurgical liver cholestasis was induced by ligating and extracting the common bile duct and four bile ducts. Sham-operated and MHC rats were maintained for eight postoperative weeks Bradykinin-induced vasodilation was greater in middle cerebral arteries from MHC rats. Both Nω-Nitro-L-arginine methyl ester and indomethacin diminished bradykinin-induced vasodilation largely in arteries from MHC rats. Nitrite and prostaglandin (PG) F1α releases were increased, whereas thromboxane (TX) B2 was not modified in arteries from MHC. Expressions of endothelial nitric oxide synthase (eNOS), inducible NOS, and cyclooxygenase (COX) 2 were augmented, and neuronal NOS (nNOS), COX-1, PGI2 synthase, and TXA2S were unmodified. Phosphorylation was augmented for eNOS and unmodified for nNOS. Altogether, these endothelial alterations might collaborate to increase brain blood flow in HE.
Introduction
Liver cholestasis is a well-known clinical syndrome that may be induced by multiple pathologies (Rodríguez-Garay, 2003). Independently of its origin, liver cholestasis is clinically characterized by jaundice, discolored urine, pale stools, pruritus, spleen enlargement, liver cirrhosis, hepatorenal syndrome, and portal hypertension, the latter due to an increase in portal vein resistance (Boyer, 2007). Alterations in endothelium-derived vasoactive factors are determinant in the circulatory disturbances observed in liver cholestasis, leading to the development of portal hypertension and to a splanchnic and systemic vasodilation (Pateron et al., 2000; Iwakiri and Groszmann, 2007). Different experimental models of liver cholestasis have shown a decompensation after six postoperative weeks, together with hepatic encephalopathy (HE) and ascites, leading to an acute-on-chronic liver failure (ACLF; García-Moreno et al., 2005; Gilsanz et al., 2017). This decompensation can aggravate these cardiovascular disturbances, causing hypotension, decreased effective blood volume, and increased cardiac output (Møller et al., 2001; Sastre et al., 2016b; Caracuel et al., 2019), eventually leading to patient death.
Hepatic encephalopathy is a complex neuropsychiatric syndrome present in around 30% of the patients with ACLF (Lizardi-Cervera et al., 2003). Although insufficient clearance of toxins from blood, hyperammonemia, increased oxidative stress and enhanced inflammatory pathways are pivotal in the pathophysiology of HE, abnormalities in cerebral blood flow are also implicated in the development of this syndrome (Shawcross et al., 2004; Shawcross and Jalan, 2005). Both increases and decreases in cerebral vasculature resistance have been reported in HE, depending on the seriousness of the pathology, and can lead to either a decrease or an increase in cerebral blood flow, respectively (Guevara et al., 1998; Hollingsworth et al., 2010; Sunil et al., 2012; Dam et al., 2013; Zheng et al., 2013). In fact, an increased cerebral blood flow has been reported to correlate with raised intracranial pressure in patients with ACLF (Kerbert et al., 2017).
Several factors modulate physiological regulation of cerebral blood flow, including neural, metabolic and endothelial factors. Focusing on the last, endothelium exerts a profound influence on blood flow in cerebral arteries by releasing nitric oxide (NO) and prostanoids, among other factors (Andresen et al., 2006; Peterson et al., 2011), thus regulating the tone of underlying smooth muscle. Under inflammatory conditions, the role of vascular endothelium can be modified in cerebral vessels (Hernanz et al., 2004; Maccarrone et al., 2017), leading to increases in NO and prostanoids release.
Despite the reports regarding modifications in cerebral blood flow in HE, to the best of our knowledge the studies concerning possible endothelial modifications in cerebral vasculature in ACLF are scarce. Thus, the objective of this study was to determine the mechanisms implicated in the development of cerebral vasculopathy in microsurgical liver cholestasis (MHC), a model of ACLF, which develops HE (García-Moreno et al., 2005; Gilsanz et al., 2017). Specifically, we analyzed whether MHC modifies the endothelial function in rat middle cerebral artery (MCA), and the possible alterations of the endothelial vasoactive factors in cerebral vasculature.
Materials and Methods
Ethical Statements
All experimental procedures were approved by the Ethical Committee of the Universidad Autónoma de Madrid, and the Comunidad de Madrid (PROEX 322/16), are in compliance with NIH guidelines, with the ARRIVE guidelines (Animal Research: Reporting in Vivo Experiments) for how to REPORT animal experiments, and follow the European Parliament Directive 2010/63/EU guidelines.
Animals
Thirty-four male Wistar rats (Initial weight: 337.9 ± 6.91 g) were purchased from Harlan Ibérica SL, Barcelona, Spain and housed in the Animal Facility of the Universidad Autónoma de Madrid (Registration number EX-021U). Rats were held in groups of 2 in appropriate cages in controlled environmental conditions (20–24°C, 55% relative humidity and 12 h light-dark cycle). All rats had access to drinking water and specific rat chow ad libitum.
Animals were randomly divided into two groups: Sham-operated (SO; n = 17), in which the common bile duct was dissected; and MHC (n = 17), in which the extrahepatic biliary tract was resected (García-Moreno et al., 2005; Sastre et al., 2016b; Gilsanz et al., 2017; Caracuel et al., 2019). Surgery was performed under aseptic but not sterile conditions. In summary, rats were anesthetized with Ketamine hydrochloride (100 mg/kg) and xylazine (12 mg/kg) i.m. In the SO group, the bile duct and its lobular branches were dissected. In the MHC group, the extrahepatic bile tract was resected using a binocular operatory microscope (Zeiss, OPMI 1-FR). First, the common bile duct was ligated (silk 4/0) and sectioned close to the beginning of its intrapancreatic portion. Dissection and sectioning between ligatures of all biliary branches that drain the hepatic lobes is possible using a binocular operatory microscope (Zeiss, OPMI 1-FR). The dissection and excision of the bile ducts from the four liver lobes of the rat was done without injuring either the portal and/or, most especially, the arterial vascularization of these lobes. The abdomen was closed in two layers by continuous running sutures using an absorbable suture (3/0 polyglycolic acid) and silk (3/0). Buprenorphine s.c. (0.05 mg/kg/8 h) was administered postoperatively for analgaesia the first 24 h after the surgery.
Systolic Blood Pressure
Systolic blood pressure (SBP) was measured using the tail-cuff method 8 weeks after the surgery was performed (Xavier et al., 2010; Sastre et al., 2016b; Caracuel et al., 2019).
Portal Vein Pressure Measurement
After an overnight fasting, splenic pulp pressure, an indirect measurement of portal pressure (PP) was measured in four animals from each group, by inserting a fluid filled 20-gauge needle into the splenic parenchyma (Sastre et al., 2016b; Caracuel et al., 2019). The needle was joined to a PE-50 tube, and then connected to a pressure recorder (PowerLab 200 ML 201) and to a transducer (Sensonor SN-844) with a Chart V 4.0 computer program (ADI Instruments), that was calibrated before each experiment. The pressure reading was considered satisfactory when a stable recording was produced and respiratory variations were observed. Previous studies have demonstrated the excellent correlation between splenic pulp pressure and PP (Kravetz et al., 1986). These animals were discarded for the following experiments.
Animal Euthanasia and Sample Collection
After an overnight fasting, animals were sacrificed by CO2 inhalation followed by decapitation. Ascitic liquid was carefully extracted to measure its volume. Liver and spleen were extracted, rinsed in saline solution and weighed. Kidneys were extracted, rinsed in saline buffer, and frozen in liquid nitrogen and stored at −70°C, until use. Brain was removed and placed in cold Krebs–Henseleit solution (KHS, in mmol/L: NaCl 115; CaCl2 2.5; KCl 4.6; KH2PO4 1.2; MgSO4⋅7H2O 1.2; NaHCO3 25; glucose 11.1, Na2 EDTA 0.03) at 4°C. Cerebral arteries were dissected under a stereoscopic magnifying glass (Euromex Holland, Barcelona).
Gene Expression Studies
Total RNA was isolated from 1/4 rat renal samples with TriPure Isolation Reagent (Sigma). cDNA was synthesized using the NZY First-Strand cDNA Synthesis Kit (Nzytech) using 2 μg of total RNA, following the manufacturer’s instructions. Real Time PCR was performed in 7500 Fast ABI System (Life Technologies Inc.) to detect gene expression of Rat NGAL (FW: 5′-GAGCGATTCGTCAGCTTTGC-3′; Rv:5′ATTGGTCGGTGGGAACAGAG-3′) and Rat KIM-1 (FW:5′-AAGCCGAGCAAACATTAGTGC-3′; RV:5′-TGAGCTAGAATTCAGCCACACA-3′). mRNA copy numbers were calculated for each sample by the instrument software using Ct value (“arithmetic fit point analysis for the lightcycler”). Results are expressed in copy numbers, calculated relative to control rat, after normalization with rat β2 microglobulin (Fw: 5′ ACCGTGATCTTTCTGGTGCTTG-3′; Rv: 5′ TAGCAGTTG AGGAAGTTGGGCT-3′).
Vascular Reactivity Experiments
For vascular reactivity experiments, we used MCA segments of 2 mm in length (internal diameter: SO: 231.4 ± 1.8 μm, (n = 6); MHC: 233.8 ± 1.4 μm (n = 4); P > 0.05) were mounted in a wire myograph for measurement of isometric tension according to a described method (Hernanz et al., 2004). Segment contractility was tested by an initial exposure to a high-KCl solution (120 mmol/L KCl-KHS), observing a similar vasoconstriction in MCA segments from both SO and MHC animals (in mN/mm: SO: 2.216 ± 0.65; MHC: 2.194 ± 0.38; P > 0.05).
Endothelium integrity was determined by the ability of 1 μmol/L bradykinin (BK) to relax segments precontracted with 10 μmol/L 5-hydroxytryptamine (5HT, serotonin). Afterward, concentration-response curves for BK (0.1 nmol/L to 10 μmol/L) were performed in 5HT-precontracted MCA segments from SO and MHC rats. The effects of the non-selective NO synthase inhibitor Nω -nitro-L-arginine methyl ester (L-NAME, 0.1 mmol/L) or the non-specific cyclooxygenase (COX) inhibitor indomethacin (10 μmol/L) on the concentration-response curves for BK were investigated. The inhibitors were added 30 min before the concentration-response curve was performed. The inhibitors did not alter the arterial basal tone.
The vasodilator response to the NO donor, diethylamine NON-Oate, (DEA-NO, 0.1 nmol/L–0.1 mmol/L) was determined in 5HT-precontracted MCA segments from both experimental groups.
NADPH Oxidase Activity
The specific superoxide anion production generated by NADPH oxidase activity was determined as previously described (Llévenes et al., 2018). For that purpose, frozen cerebral vasculature was homogenized in an ice-cold buffer containing 20 mmol/L KH2PO4, 1 mmol/L EGTA and 150 mmol/L sucrose. The reaction was started by the addition of a lucigenin (5 μmol/L)/NADPH (100 μmol/L) mixture to the tissue homogenate. Chemiluminescence was determined every 2.4 s for 5 min in a plate luminometer (AutoLumat LB 953, Berthold, Germany). Buffer blank was subtracted from each reading. Luminescence was normalized by protein concentration, and data were expressed as chemiluminiscence units/μg protein.
Vasoactive Factor Production
The production of NO, TXA2 and PGI2 were measured using the commercial kits Nitric Oxide Assay Kit (Abcam Laboratories), Thromboxane (TX) B2 ELISA kit (Cayman Chemical) and 6-keto Prostaglandin (PG) F1α ELISA Kit (Cayman Chemical), respectively. For sample collecting, the cerebral vasculature was pre-incubated for 30 min in 2 mL of KHS at 37°C, continuously gassed with a 95% O2–5% CO2 mixture (stabilization period). This was followed by two washout periods of 10 min in a bath of 0.2 mL of KHS. Then, the medium was collected to measure basal release. Afterward, arteries were subjected to 10 μmol/L 5-HT for 2 min, and then a BK concentration curve (0.1 nmol/L to 10 μmol/L) was applied at 1 min intervals. Afterward, the medium was collected to measure the BK-induced vasoactive factor release. For NO production measurement, some samples were collected in presence of 5 μmol/L L-NPA (specific neuronal nitric oxide synthase – nNOS-inhibitor), 1 μmol 1400W (specific inducible NOS – iNOS-inhibitor) or 0.1 mmol/L L-NAME. The inhibitors were added 30 min before the concentration-response curve to BK was performed. Samples were immediately frozen in liquid nitrogen and conserved at −70°C until the assays were performed. The different assays were carried out according to the manufacturers’ instructions.
Western Blot Analysis
For Western blot analysis, the cerebral vasculature were homogenized in a buffer composed of 1 mmol/L sodium vanadate (a protease inhibitor), 1% SDS, and 0.01 mol/L pH 7.4 Tris–HCl. Protein content was determined using a DCTM Protein Assay (Bio-Rad Laboratories, Hercules, CA, United States). Homogenates containing 30 μg protein were electrophoretically separated on a 7.5% (iNOS, eNOS, PS1177–eNOS, nNOS, and PS1417 nNOS), or 10% (COX-1, COX-2, TXA2 synthase, and PGI2 synthase) SDS–polyacrylamide gel (SDS–PAGE), and then transferred to polyvinyl difluoride membranes (Bio Rad Immun-Blot w) overnight at 4°C, 230 mA, using a Bio-Rad Mini Protean Tetra system (Bio-Rad Laboratories, Hercules, CA, United States) containing 25 mmol/L Tris, 190 mmol/L glycine, 20% methanol, and 0.05% SDS. PageRuler Prestained Protein Ladder (Thermo Scientific) was used as molecular mass markers. The membranes were blocked for 3 h at room temperature in a Tris-buffered-saline solution (100 mmol/L, 0.9% w/v NaCl, 0.1% SDS) with 5% bovine serum albumin before being incubated overnight at 4°C with mouse monoclonal anti-eNOS (1:1000, Transduction Laboratories), rabbit polyclonal anti-PS1177 eNOS (1:1000, Abcam laboratories), mouse monoclonal anti-nNOS (1:1000, Transduction Laboratories), rabbit polyclonal anti-PS1417 nNOS (1:1000, Abcam laboratories); mouse monoclonal anti-iNOS (1:1000, Transduction Laboratories); rabbit monoclonal anti-COX-1 (1:1000 dilution, Abcam laboratories); rabbit polyclonal anti-COX-2 (1:2000 dilution, Cayman Chemical); rabbit polyclonal anti-TXA2 synthase (1:500 dilution, Cayman Chemical); or rabbit polyclonal anti-PGI2 synthase (1:500 dilution, Cayman Chemical). After washing, the membrane was incubated with a 1:2000 dilution of the appropriate secondary antibody (anti-mouse or anti-rabbit immunoglobulin G antibody) conjugated to horseradish peroxidase (GE Healthcare, Little Chalfont, United Kingdom). The membrane was thoroughly washed and the immunocomplexes were detected using an enhanced horseradish peroxidase/luminol chemiluminescence system (ECL Plus, GE Healthcare, Little Chalfont, United Kingdom). Finally, the images were developed and quantified using the Quantity One software (v. 4.6.6, Biorad, Spain). The same membranes were used to determine β-actin expression (1:50000 dilution, Sigma-Aldrich, Spain) and the content of the latter was used to correct protein expression.
Drugs and Solutions
Drugs used were 5-hydroxytryptamine creatinine sulfate (serotonin), bradykinin, sodium vanadate, L-NAME (Nω -nitro-l-arginine methyl ester), indomethacin, SDS, Trizma-base and bovine serum albumin (Sigma-Aldrich; Spain). Stock solutions (10 mmol/L) were made in bidistilled water and kept at −20°C. Appropriate dilutions were made in KHS on the day of the experiment.
Data Analysis
Graph representation and statistical analysis were performed using GraphPad Prism 8.0 software (CA, United States). The relaxations induced by BK were expressed as a percentage of the initial contraction elicited by 5HT (in mN: SO: 1.18 + 0.15; MHC: 1.29 + 0.19, P > 0.05). Non-lineal regressions were performed. Areas under the curve (AUC) were calculated from the individual concentration-response plots. Results were expressed as mean ± standard error of the mean (SEM). In vascular reactivity experiments, statistical analysis was performed by means of two-way analysis of variance (ANOVA). For differences of AUC (dAUC), vasoactive substance release and Western blot experiments, the ROUT method was used to identify and remove outliers. Moreover, a Shapiro–Wilk test was applied to confirm the normality of the population data, followed by a Student’s t-test statistical analysis. P < 0.05 was considered significant.
Results
Animal Evolution
All MHC animals showed jaundice and choluria. Paraesophageal, splenorenal and pararectal collateral vessels developed in MHC animals (Data not shown). Final body weight and body weight gain were lower in MHC animals. Systolic blood pressure was lower, and PP was enhanced in the MHC group. Moreover, both spleen and liver hypertrophy, and extravasation of ascitic fluid were present in MHC animals (Table 1). We also found that both neutrophil gelatinase-associated lipocalin (NGAL), and Kidney injury molecule 1 (KIM-1), renal damage markers, were enhanced after MHC (Table 2). In addition, a jaundice-derived color and an inflammatory phenotype were found in rats submitted to MHC (Figure 1). Altogether, these results confirm that MHC is an appropriate experimental model for studying hepatic and extrahepatic complications of this pathology.

Table 1. Effect of microsurgical liver cholestasis (MHC) on body weight (BW), body weight gain (BWG), systolic blood pressure (SBP), portal pressure (PP), liver weight (LW), spleen weight (SW) and ascitic liquid extravasation in Wistar rats.

Table 2. NGAL and KIM-1 levels in kidneys from Sham-Operated (SO) and microsurgical liver cholestasis (MHC) rats.
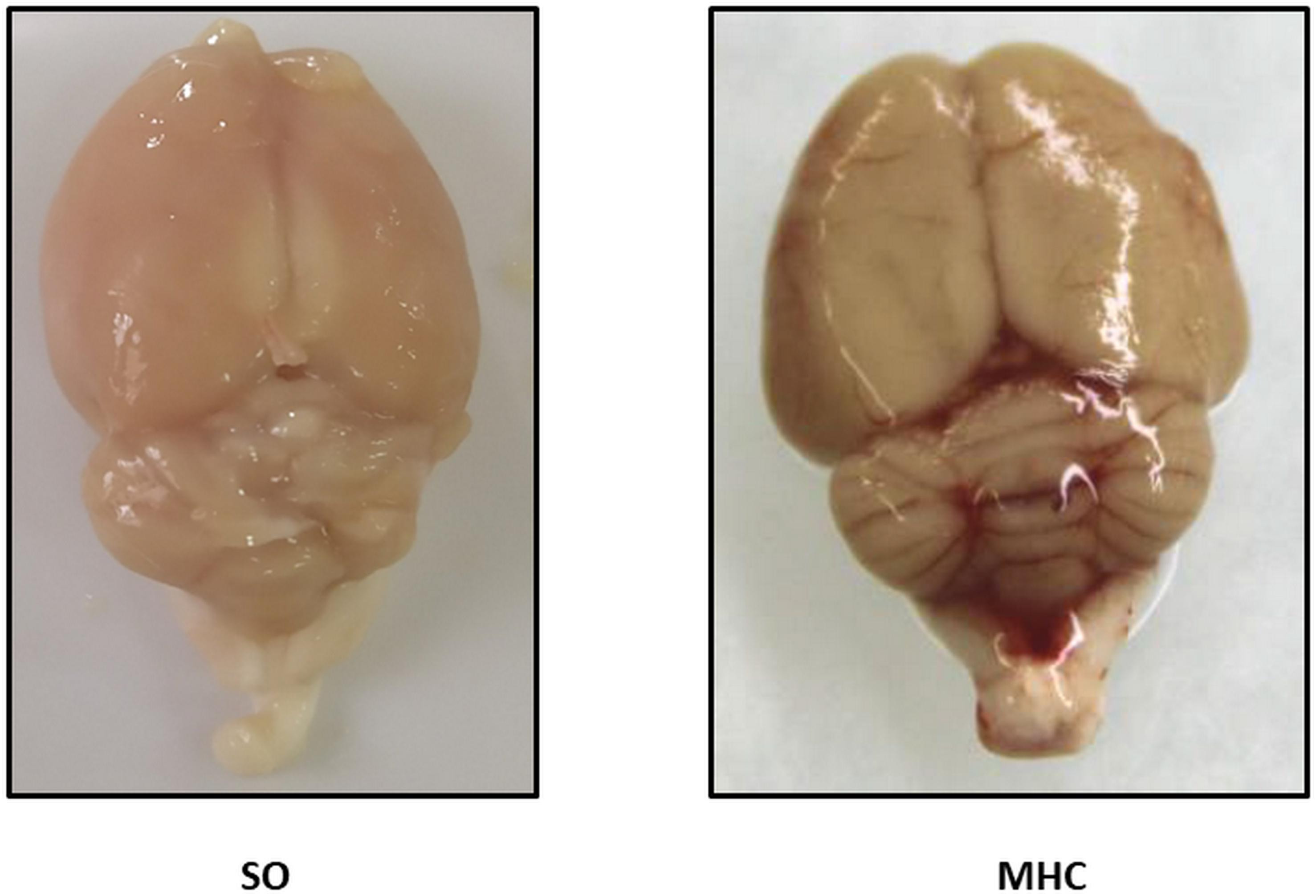
Figure 1. Representative pictures showing the brains from Sham-Operated (SO) and rats submitted to microsurgical liver cholestasis (MHC).
Bradykinin-Induced Vasodilation
Large cerebral arteries, like MCA, strongly contribute to total cerebrovascular resistance (Faraci and Heistad, 1990, 1998). For that reason, we analyzed whether MHC could modify the endothelium dependent BK-induced response in MCA. We observed that BK produced a concentration-dependent vasodilator response in MCA from both groups, which was greater in MHC animals (Figure 2).
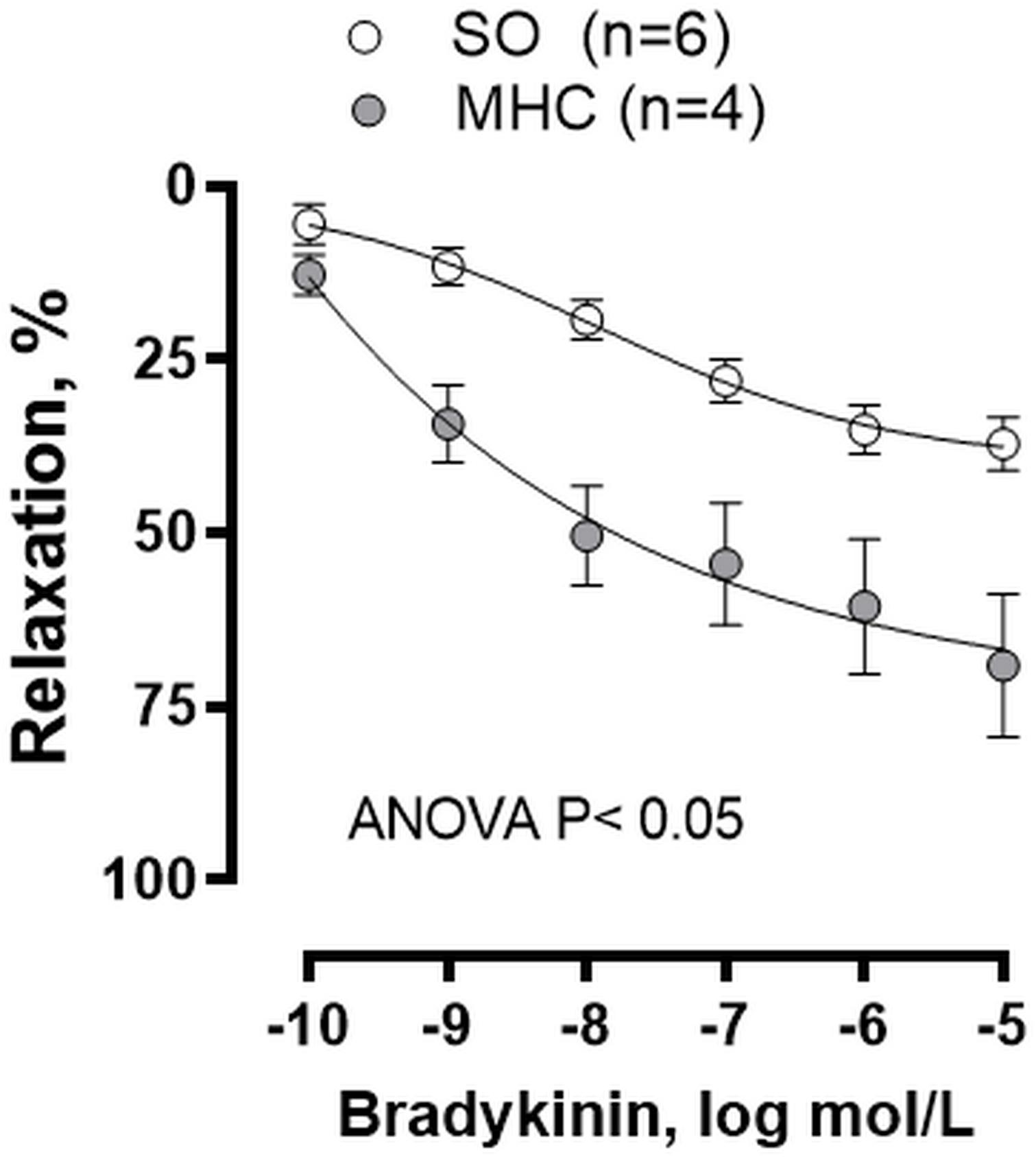
Figure 2. Effect of microsurgical liver cholestasis (MHC) on the concentration-dependent relaxation to bradykinin in rat middle cerebral arteries. Sham-Operated (SO): n = 6; MHC: n = 4. Results (mean ± SEM) were expressed as a percentage of the initial contraction elicited by 5HT.
Role of Endothelium-Derived NO on the Vasodilator Response to BK
One of the pivotal endothelial vasoactive factors modified by inflammation is NO, which exerts a vasodilator effect in cerebral vessels (Faraci and Heistad, 1998; Andresen et al., 2006). To determine a different role of NO in SO and MHC animals, we preincubated MCA segments with the non-selective NO synthase inhibitor L-NAME. This drug diminished BK-induced vasodilation in MCA segments from both groups, being this decrease greater in arteries from rats submitted to MHCcerebral vasculature from MHC (Figures 3A,B, and dAUC). It is also remarkable that the vasodilator role to NO donor DEA-NO was similar in MCA segments from both experimental groups (Figure 3C). This result rules out possible differences in smooth muscle sensitivity to NO in our experimental conditions.
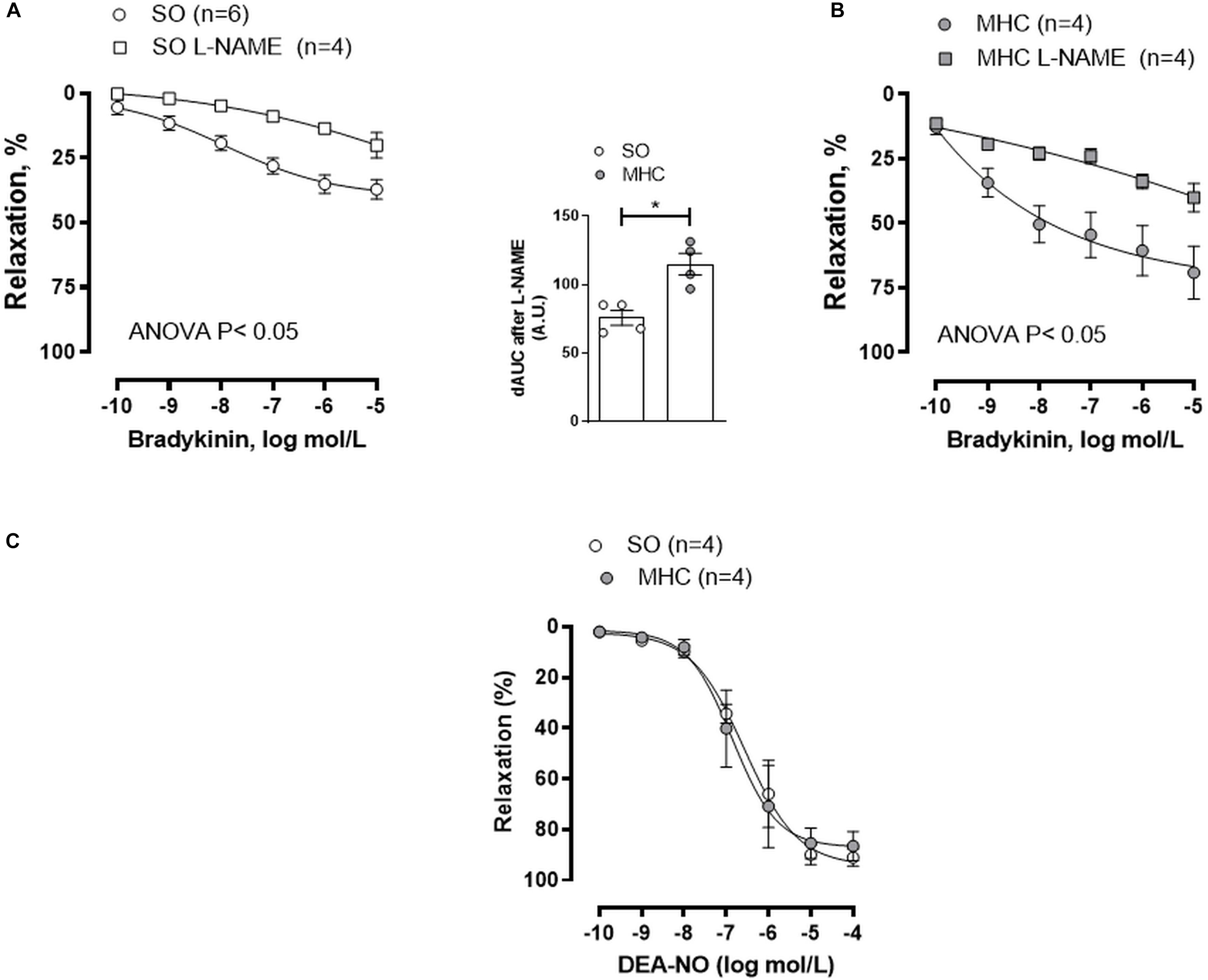
Figure 3. Effect of preincubation with NOS inhibitor L-NAME on the concentration-dependent relaxation to bradykinin in rat middle cerebral arteries from Sham-Operated (SO), (A); (n = 4–6 segments from different animals) and microsurgical liver cholestasic (MHC), (B); (n = 4 segments from different animals) rats. Results (mean ± SEM) were expressed as a percentage of the initial contraction elicited by 5HT. The attached graph shows the differences of area under the curve (dAUC) to bradykinin for segments in the absence or presence of 0.1 mmol/L L-NAME. dAUC values are expressed as a percentage of the difference of the corresponding AUC for the segments in the absence of L-NAME. (C) Vasodilator response to NO donor DEA-NO in arteries from SO and MHC rats (n = 4 segments from different animals in each group). Results (mean ± SEM) were expressed as a percentage of the initial contraction elicited by 5HT.
To analyze whether this differential role of NO was due to NO release, we analyzed the release of the stable NO metabolite nitrite, finding that BK-induced nitrite release was greater in the cerebral vasculature from MHC animals (Figure 4A).
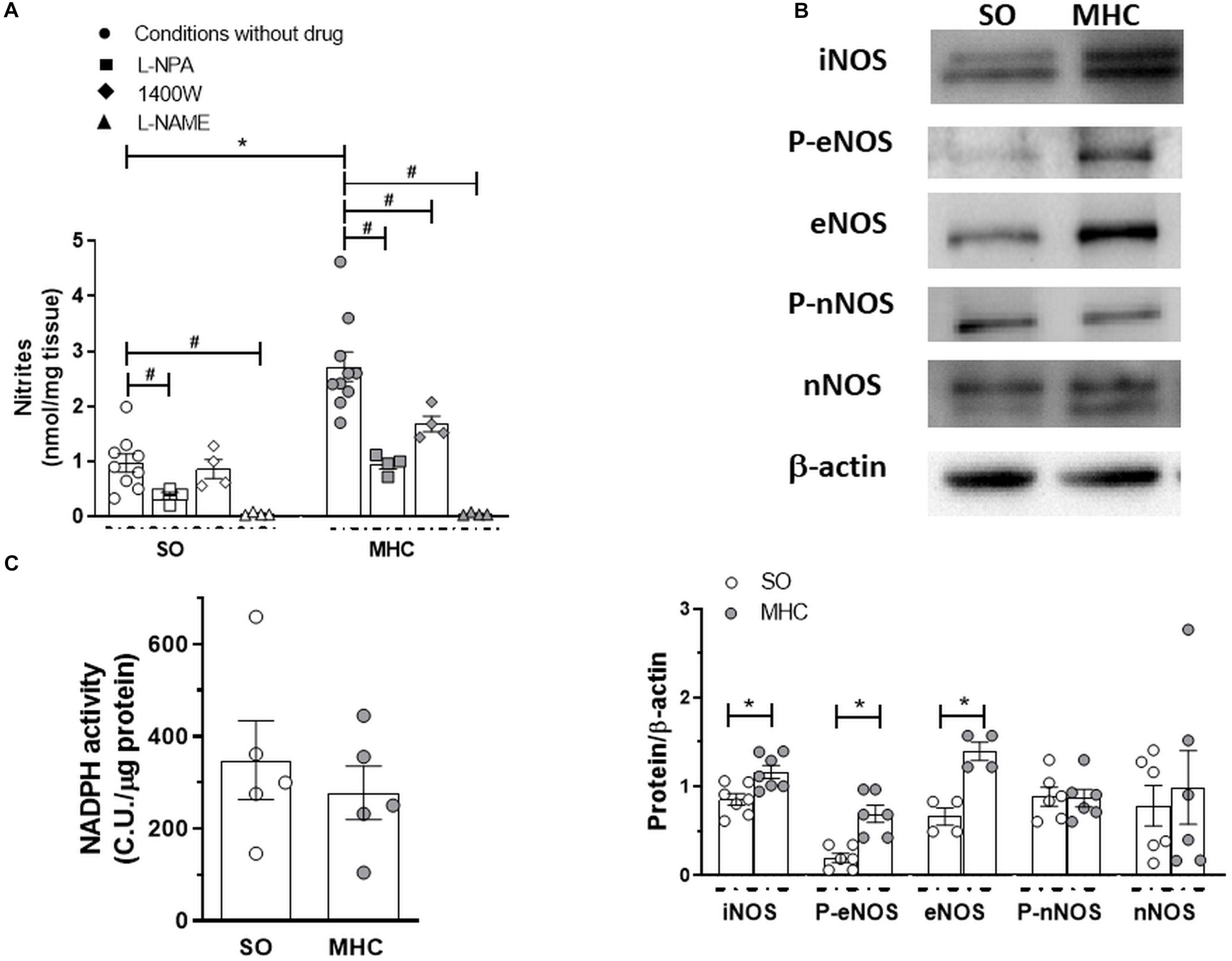
Figure 4. (A) Bradykinin-induced nitrite release in cerebral vasculature from Sham-Operated (SO; n = 4-9 animals) and microsurgical liver cholestasic (MHC; n = 4–10 animals) rats. Effect of preincubation with specific nNOS inhibitor L-NPA, specific iNOS inhibitor 1400W, or non-specific NOS inhibitor L-NAME. Data (Mean ± SEM) were expressed as nmol nitrites/mg tissue. *P < 0.05 SO vs MHC (Student t-test). #P < 0.05 conditions without drug vs. conditiosmn with drug (Student t-test). (B) Western blot analysis for and inducible nitric oxide synthase (iNOS), total and phosphorylated endothelial nitric oxide synthase (eNOS) in the Ser 1177 residue (P-eNOS), and total and phosphorylated neuronal nitric oxide synthase (nNOS) in the Ser 1417 residue (P-nNOS), in cerebral arteries from Sham-Operated (SO) and microsurgical liver cholestasis (MHC) rats. The blots are representative of 4–7 segments from each group. Lower panels show densitometry analysis for the expression of each protein. Results (mean ± SEM) were expressed as the relation between the signal obtained for the protein analyzed and the signal obtained for β-actin. *P < 0.05 SO vs MHC (Student t test). (C) NADPH activity in cerebral vessels from SO and MHC rats (n = 5 different animals from each group). Results (mean ± SEM) were expressed as chemiluminiscence units (CU.)/μg protein.
Multiple enzymes are implicated in the NO synthesis. When analyzing the influence of different NOS inhibitors in nitrite release, we found that the specific nNOS inhibitor L-NPA diminished BK-induced nitrite release in a similar extent in cerebral vasculature from both experimental groups (in percentage of inhibition: SO: 61.3 + 6.5; MHC: 65.1 + 3.1; P > 0.05). In addition, the specific iNOS inhibitor 1400W significantly decreased BK-induced nitrite release only in arteries from MHC rats, while the non-specific NOS inhibitor L-NAME abolished this release in the cerebral vasculature from both SO and MHC rats (Figure 4A). We also observed an increase in iNOS expression in arteries from MHC rats. Moreover, both eNOS expression and phosphorylation in Ser 1177 were enhanced in MHC cerebral arteries, while neither nNOS expression nor Ser1417 phosphorylation were modified by MHC (Figure 4B).
Aside from NO release, it is also important to remark that NO function also depends on its bioavailability. Liver pathologies enhance oxidative stress in different tissues, including vascular tissue (Xavier et al., 2010), thereby reducing NO function. The fact that NADPH oxidase activity, the main producer of superoxide anion, was similar in cerebral vasculature from both experimental rules (Figure 4C), allowed us to rule out possible alterations in NO bioavailability.
Role of COX-Derived Prostanoids on the Vasodilator Response to BK
Modifications of other vasoactive factors could also be involved in the pathogenesis of arterial vasodilation in MHC (Theodorakis et al., 2003). To analyze the possible participation of prostanoids in the enhanced BK-induced vasodilation observed in MHC rats, we preincubated MCA with the unspecific COX inhibitor indomethacin. We observed that BK-induced vasodilation was not modified by indomethacin in MCA from SO animals, while it was diminished in arteries from MHC rats (Figures 5A,B).
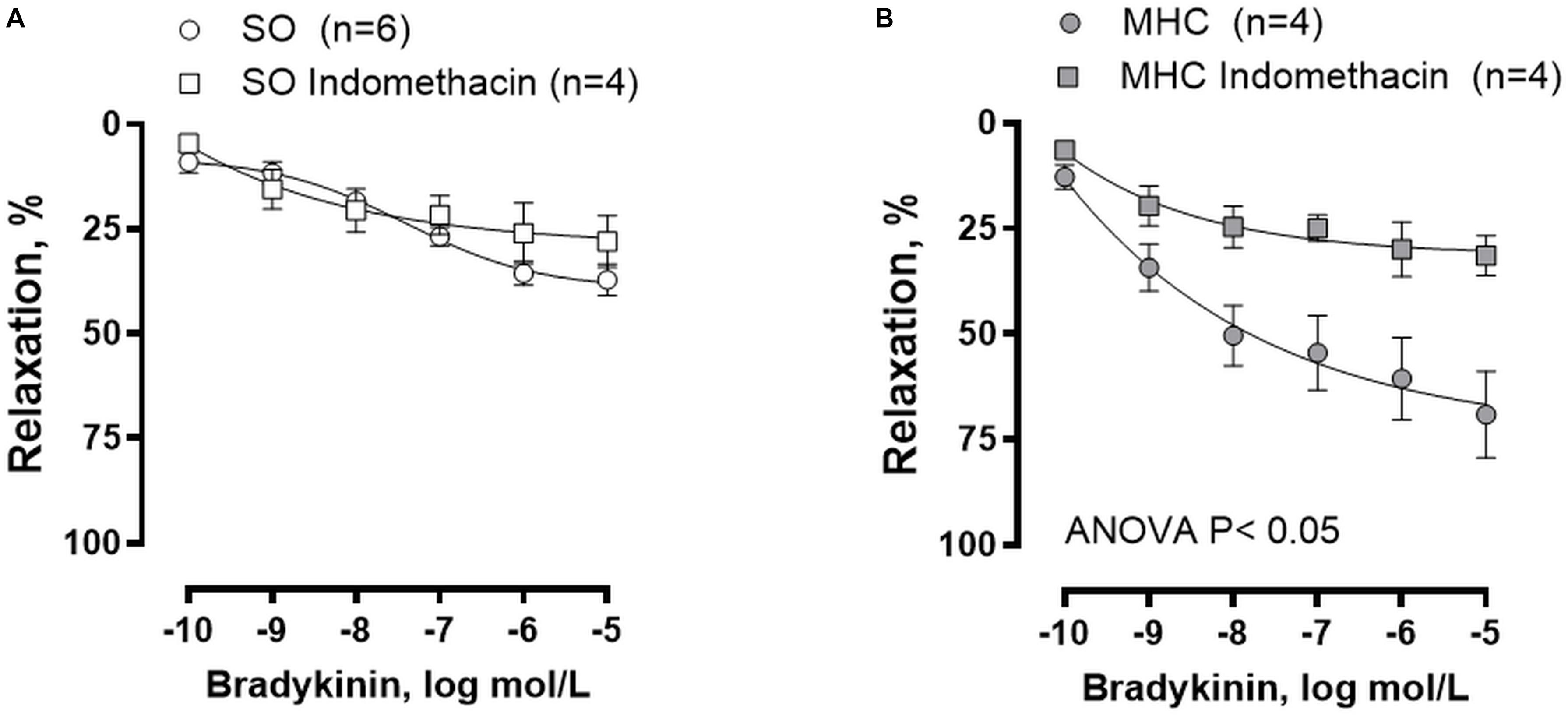
Figure 5. Effect of preincubation with COX inhibitor indomethacin on the concentration-dependent relaxation to bradykinin in rat middle cerebral arteries from Sham-Operated (SO), (A); (n = 4–6 segments from different animals) and microsurgical liver cholestasic (MHC), (B); (n = 4 segments from different animals) rats. Results (mean ± SEM) were expressed as a percentage of the initial contraction elicited by 5HT.
An overexpression of the enzymes implicated in prostanoid synthesis has been reported in different vascular beds in rats with liver pathologies (Xavier et al., 2010). When analyzing COX-1, we observed a similar expression in cerebral arteries from both experimental groups, while the expression of COX-2 was greater in cerebral arteries from MHC animals (Figure 6A).
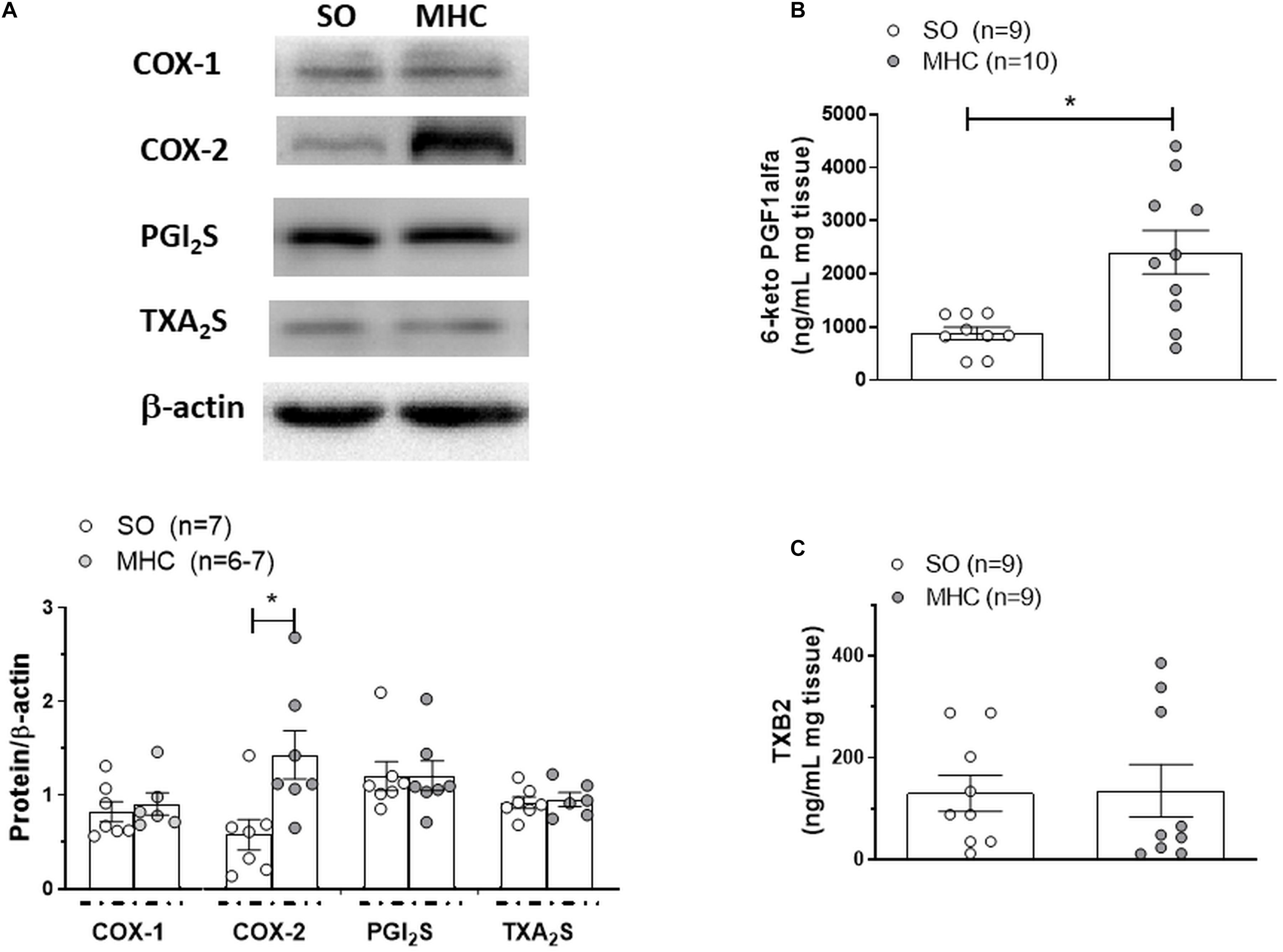
Figure 6. (A) Western blot analysis for cyclooxygenase (COX) 1 and 2, PGI2 synthase (PGI2S) and TXA2 synthase (TXA2S) in cerebral arteries from Sham-Operated (SO) and microsurgical liver cholestasis (MHC) rats. The blots are representative of 6–7 segments from each group. Lower panels show densitometry analysis for the expression of each protein. Results (mean ±SEM) were expressed as the relation between the signal obtained for the protein analyzed and the signal obtained for β-actin. *P < 0.05 SO vs MHC (Student t-test). Effect of microsurgical liver cholestasis (MHC) on cerebral artery 6-Keto PGF1α release (B); (SO: n = 9 animals; MHC: n = 10 animals) and TXB2 release (C); (n = 10 animals from each experimental group) Results (mean ±tometry analysis for the expression of each protein. Results (mean SEM) were expressed as pg prostanoid/mL mg tissue. *P < 0.05 SO vs MHC (Student t-test).
Among the prostanoids implicated in the regulation of vascular tone, both vasodilator PGI2 and vasoconstrictor TXA2 have a relevant role in cerebral vessels (Andresen et al., 2006; Peterson et al., 2011; Maccarrone et al., 2017). When analyzing the expressions of PGI2 synthase and TXA2 synthase, the enzymes implicated in their synthesis, we observed no differences among groups (Figure 6A). Moreover, when analyzing 6-keto PGF1α and TXB2, the respective stable metabolites of PGI2 and TXA2, we observed that MCH enhanced BK-induced 6-keto PGF1α release but did not modify BK-induced TXB2 release (Figures 6B,C).
Discussion
The present study analyses the alterations in the cerebrovascular function in rats submitted to MHC, a model of ACLF, which develops HE. The results obtained show an enhanced endothelium-dependent BK-induced vasodilation in MHC rats, which is due to increases in the vasodilator factors NO and PGI2 release.
Extrahepatic cholestasis is the most common model to study the vascular complications of obstructive liver cholestasis. Rats with MHC develop ACLF symptoms such as hepatomegaly, PH, enlarged spleen, collateral portosystemic circulation, and ascites, as reported earlier (Aller et al., 2012; Sastre et al., 2016b; Caracuel et al., 2019). It is also remarkable that we previously described alterations in albumin, total protein, bilirubin, and transaminases after this experimental procedure (Gilsanz et al., 2017; Caracuel et al., 2019). We can also highlight that these animals developed hepatorenal syndrome, since both the renal damage markers NGAL and KIM-1, were enhanced in kidneys from MHC rats. In is also interesting to remark the jaundice-derived color and the inflammatory phenotype that we observed in brains from rats submitted to MHC. In addition, a previous study from our group showed alterations in different brain structures, such as the hippocampus, several weeks after this surgery was performed (García-Moreno et al., 2002). These alterations were accentuated after 8 weeks of the surgery (Braissant et al., 2019), consequently developing behavioral changes including both cognitive and motor impairment, which worsen together with the severity of the pathology (García-Moreno et al., 2002; Giménez-Garzó et al., 2015; Braissant et al., 2019). Altogether, these data allow us to confirm that MHC is an appropriate experimental model for studying hepatic and extrahepatic complications, including HE alterations, developed in ACLF.
Microsurgical liver cholestasis is characterized by the development of systemic vascular complications, which can be the origin of the HE associated to ACLF (Jalan and Williams, 2002; Wright et al., 2014). Moreover, previous studies have reported that a systemic proinflammatory state can activate microglia and consequently produce a neuroinflammatory situation, therefore worsening the neuropsychiatric symptoms of HE (Butterworth, 2013; Rama Rao et al., 2014; Macías-Rodríguez et al., 2015). This proinflammatory state can modify the release of endothelial vasoactive factors in several vascular beds, including cerebral vessels (Faraci and Heistad, 1998; Andresen et al., 2006), consequently altering cerebral blood flow and vascular resistance, and contributing to the development of the brain abnormalities observed in this pathology. Large cerebral arteries, like MCA, strongly contribute to total cerebrovascular resistance, being the main determinants of local microvascular pressure (Faraci and Heistad, 1990, 1998). For that reason, we analyzed whether MHC could modify the endothelium dependent BK-induced response in MCA. We observed an augmented BK vasodilation in vessels from MHC rats, as has been reported in other vascular beds (Xavier et al., 2010; Hollenberg and Waldman, 2016; Caracuel et al., 2019). As we commented in the introduction section, alterations in the balance of vasodilator and vasoconstrictor agents are frequent in liver pathologies, leading to blood flow alterations in multiple vascular beds (Xavier et al., 2010; Bolognesi et al., 2014; Bosch et al., 2015; Sastre et al., 2016b; Caracuel et al., 2019). One of the pivotal endothelial vasoactive factors modified by inflammation is NO, which exerts a vasodilator effect in cerebral vessels both in vivo and ex vivo by activating soluble guanylate cyclase and/or producing smooth muscle hyperpolarization through potassium channel opening (Faraci and Heistad, 1998; Andresen et al., 2006). A dysregulation of NO production is a common denominator of most of the symptoms accompanying liver pathologies. The role of NO is reported to vary in different vascular beds. Thus, a decrease in NO levels has been described in portal vein, increasing the resistance in this vascular bed. Conversely, increases in NO have also been reported in systemic vasculature and plasma, where they participate in the development of hyperdynamic circulation in splanchnic and systemic circulation (Bolognesi et al., 2014; Bosch et al., 2015; Hollenberg and Waldman, 2016). We observed that, similar to earlier reports in different splanchnic and systemic vascular beds (Xavier et al., 2010; Sastre et al., 2016b; Caracuel et al., 2019), BK-induced nitrite (the stable NO metabolite) levels were significantly increased in cerebral arteries from MHC rats compared to SO animals. This result correlates with the fact that preincubation with unspecific NOS inhibitor L-NAME diminished BK-induced relaxation to a greater extent in MCA from MHC rats, hence confirming a major functional role for NO in this experimental group. In addition, the fact that the vasodilator role to NO donor DEA-NO was similar in MCA segments from both experimental groups allowed us to rule out possible differences in both smooth muscle sensitivity to NO and in the NO signaling pathway in MCA due to MHC.
NO can be synthetized through the action of both constitutive eNOS and nNOS, and inducible iNOS. The increase in iNOS expression in splanchnic vasculature suggests involvement by iNOS-derived NO in the development of hyperdynamic circulation in different liver pathologies (Bhimani et al., 2003; Ferguson et al., 2006; Xavier et al., 2010). Given the fact that iNOS plays a relevant role in NO synthesis in brain under inflammatory conditions (Hernanz et al., 2004; Rama Rao et al., 2014), we analyzed possible differences in iNOS expression under our experimental conditions, finding an increase in this enzyme expression in cerebral arteries from MHC animals. Consequently, the increase in iNOS expression in MHC animals would explain the augmented NO release observed in this experimental group. The fact that the specific iNOS inhibitor 1400W reduced nitrite release only in cerebral vessels from MHC rats corroborates this hypothesis, thereby confirming the relevant role of iNOS in the vascular alterations in MHC.
We must also take into account that both constitutive isoforms eNOS and nNOS are also present in vascular endothelium of cerebral arteries (Andresen et al., 2006; Iwakiri and Groszmann, 2007). An increase in eNOS expression was described in the splanchnic and systemic vascular beds in several liver pathology models (Wright et al., 2012; Lin et al., 2014), and eNOS-derived NO has been described to be high in cerebral vessels under inflammatory status (Ando et al., 2004; Maccarrone et al., 2017). Thus, we cannot rule out possible alterations in eNOS expression/activation in cerebral arteries from MHC animals. An increase in eNOS expression was found in cerebral vessels from MHC rats. Additionally, we observed an augmented eNOS phosphorylation at Ser1177, indicating enhanced eNOS activation, as we previously described in mesenteric vascular bed (Xavier et al., 2010; Caracuel et al., 2019).
Regarding nNOS-derived NO, it is implicated in the increased vasodilation observed in splanchnic and systemic vasculature in liver pathologies, including ACLF (Xu et al., 2000; Sastre et al., 2016b), while several reports have described that nNOS is present in cultured endothelial cells from different vascular beds, exerting an anti-inflammatory role (Chakrabarti et al., 2012). We found no modifications in either nNOS expression or phosphorylation on Ser1417. Moreover, the specific inhibition of nNOS diminished NO release in a similar extent in both experimental groups. These results contrast with these studies previously mentioned. The different tissues and experimental model could explain this discrepancy. Altogether, these results show that the observed increase in NO release in cerebral arteries from MHC rats could be due to augmented eNOS and iNOS activity.
Multiple studies have shown that MHC induced a systemic increase of oxidative stress. Specially, enhanced pro-oxidative biomarkers and diminished antioxidant mechanisms have been reported in brain from MHC rats (Ommati et al., 2020). In blood vessels, superoxide anions can modulate the role of NO, diminishing its bioavailability. Although there are several sources of reactive oxygen species, the enzyme NADPH oxidase is the main producer of the excessive superoxide anions in vascular tissue (Miller et al., 2010; Sastre et al., 2016a; Llévenes et al., 2018). When analyzing the activity of this enzyme, we found no differences between our experimental groups. This result might contrast with the fact that liver pathologies enhance vascular oxidative stress in other vascular beds (Xavier et al., 2010), while we also found no differences in superoxide anions in superior mesenteric artery from MHC rats (unpublished results from our group). It is interesting to remark that an enhanced systemic oxidative stress systemic does not necessary correlate with local alterations in superoxide anions (Torres et al., 2019; Llévenes et al., 2020). In addition, we cannot rule out the participation of other sources of vascular superoxide anions. Since the functional analysis of NO-induced vasodilation showed no differences between SO and MHC rats, we can infer that the oxidative stress participation in cerebral vasculature might not change due to MHC.
Aside from excess NO generation in the splanchnic circulation, data from eNOS and iNOS knockout mice suggests that modifications of vasoactive factors other than NO could be involved in the pathogenesis of arterial vasodilation in liver pathologies (Theodorakis et al., 2003). Endothelial prostanoids, synthetized through COX activation, participate in the regulation of vascular tone in healthy situations, depending on the vascular bed analyzed, but their production can be modified under certain inflammatory pathological situations (Brian et al., 2001; Hernanz et al., 2004; Mollace et al., 2005; Blanco-Rivero et al., 2007; Peterson et al., 2011; Wiggers et al., 2016). In fact, multiple studies have already focused on the role of COX-derived vasodilator (PGI2) and vasoconstrictor (TXA2) prostanoids in the vascular disturbances observed in liver pathologies (Xavier et al., 2010; Macías-Rodríguez et al., 2015; Caracuel et al., 2019). Therefore, we aimed to determine the possible differential influence of prostanoids on the BK-induced vasodilation in cerebral vessels from SO and MHC rats. For that purpose, we incubated MCA segments with the non-specific COX inhibitor indomethacin, observing that this drug exerted no influence in vessels from SO animals, as previously described (Wiggers et al., 2016), while it diminished BK-induced vasodilation in arteries from MHC animals. Previously, an overexpression of COX-2 was described in splanchnic vasculature in liver pathologies (Xavier et al., 2010). What is more, alterations in constitutive COX-1 expression have also been reported in several tissues, including endothelium and brain tissue (Zheng et al., 2013; Kerbert et al., 2017). Therefore, we aimed to investigate possible alterations in the expression of both COX isoforms. We found a greater COX-2 expression in cerebral arteries from MHC rats, while COX-1 was not modified in our experimental conditions. Therefore, the increase in COX-2 expression we observed in cerebral arteries suggests the presence of alterations in prostanoid release in this vascular bed.
One of the main vasodilator prostanoid present in cerebral arteries is PGI (Andresen et al., 2006; Xavier et al., 2010; Peterson et al., 2011; Maccarrone et al., 2017). We observed an increase in BK-induced 6-keto PGF1 α (the stable PGI2 metabolite) release in cerebral arteries from MHC animals, similarly to that reported in aorta and mesenteric resistance arteries in diverse liver pathologies (González-Correa et al., 1996; Blanco-Rivero et al., 2009; Xavier et al., 2010). Since no differences in PGI2 synthase expression were observed, we could attribute the increased PGI2 release in MHC cerebral arteries to the augmented COX-2 expression, but we cannot exclude a possible enhancement in PGI2S activity.
Aside from vasodilator PGI2, COX-derived vasoconstrictor TXA2 also has a relevant role in the regulation of vascular tone in cerebral arteries (Hou et al., 2000; Andresen et al., 2006; Peterson et al., 2011). In fact, inter-relations between both prostanoids have been described (Cheng et al., 2002; Mollace et al., 2005). Modifications in TXA2 participation have been described in cerebral vasculature in different pathologies (Andresen et al., 2006). Regarding liver pathologies, both increases and decreases in TXA2 participation have been described in portal vein and splanchnic vasculature, respectively (Iwakiri and Groszmann, 2007; Gatta et al., 2008; Xavier et al., 2010), but, to the best of our knowledge, no reports regarding possible modifications in this vasoconstrictor factor have been observed in cerebral vessels. When measuring the stable TXA2 metabolite, TXB2, we found no differences between its release in cerebral arteries from SO and MHC rats, agreeing with the observation that TXA2 synthase expression was similar in both experimental groups. Altogether, these data show augmented PGI2 release in cerebral arteries from MHC rats, mainly due to the increase in COX-2 expression, while no modification in TXA2 release was observed.
An extravasation of ascitic fluid to the abdominal cavity is found in this MHC model after a six-week evolution of the pathology, being this symptom characteristic of ACLF. Regarding cerebral vasculature, the vasculopathy that we observed in the present study could be implicated in the development of HE related to an ACLF, characterized by a brain edema and hypoxia situation, which might consequently lead to CNS ischemia (Faraci and Heistad, 1990; Theodorakis et al., 2003; Sawhney et al., 2016). In fact, after a longer evolution of MHC (8 weeks or more), the animals die after going into a coma. Therefore, it would be interesting in the future to determine the pathogenic influence of vasoactive mediators in the development of the endothelial permeability that causes the HE related to ACLF.
In conclusion, we observed an enhanced BK-induced vasodilation observed in MCA from MHC rats, due to increased NO and PGI2. This augmented vasodilation might collaborate to increase brain blood flow in HE, and consequently be implicated in the brain alterations observed in ACLF.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by All experimental procedures were approved by the Ethical Committee of the Universidad Autónoma de Madrid, and the Comunidad de Madrid.
Author Contributions
LC, ES, MC, RR-D, and AG-R performed the experiments and statistical analyses and the systolic blood pressure measurements. IP and CN performed some experimental procedures, the surgical techniques, and the portal pressure measurements. IP, MA, and JA collaborated in the discussion of the results and the writing of the manuscript. MS and JB-R performed some experiments and statistical analyses, discussed the results, and wrote this manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Ministerio de Economía y Competitividad (SAF2016-80305-P), CiberCV (Grant number: CB16/11/00286), the European Regional Development Grant (FEDER) (Comunidad de Madrid, grant number B2017/BMD-3676), and R + D projects for young researchers, Universidad Autónoma de Madrid-Comunidad de Madrid (SI1-PJI-2019-00321). RR-D received a fellowship from Juan de la Cierva Program (IJCI-2017-31399).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aller, M. A., Arias, N., Prieto, I., Agudo, S., Gilsanz, G., Lorente, L., et al. (2012). A half century (1961-2011) of applying microsurgery to experimental liver research. World J. Hepatol. 4, 199–208. doi: 10.4254/wjh.v4.i7.199
Ando, H., Zhou, J., Macova, M., Imboden, H., and Saavedra, J. M. (2004). Angiotensin II AT1 receptor blockade reverses pathological hypertrophy and inflammation in brain microvessels of spontaneously hypertensive rats. Stroke 35, 1726–1731. doi: 10.1161/01.str.0000129788.26346.18
Andresen, J., Shafi, N. I., and Bryan, R. M. Jr. (2006). Endothelial influences on cerebrovascular tone. J. Appl. Physiol. (1985). 100, 318–327. doi: 10.1152/japplphysiol.00937.2005
Bhimani, E. K., Serracino-Inglott, F., Sarela, A. I., Batten, J. J., and Mathie, R. T. (2003). Hepatic and mesenteric nitric oxide synthase expression in a rat model of CCl(4)-induced cirrhosis. J. Surg. Res. 113, 172–178. doi: 10.1016/s0022-4804(03)00163-x
Blanco-Rivero, J., Aller, M. A., Arias, J., Ferrer, M., and Balfagón, G. (2009). Long-term portal hypertension increases the vasodilator response to acetylcholine in rat aorta: role of prostaglandin I2. Clin. Sci. (Lond). 117, 365–374. doi: 10.1042/cs20080499
Blanco-Rivero, J., Márquez-Rodas, I., Xavier, F. E., Aras-López, R., Arroyo-Villa, I., Ferrer, M., et al. (2007). Long-term fenofibrate treatment impairs endothelium-dependent dilation to acetylcholine by altering the cyclooxygenase pathway. Cardiovasc. Res. 75, 398–407. doi: 10.1016/j.cardiores.2007.03.006
Bolognesi, M., Di Pascoli, M., Vererdo, A., and Gatta, A. (2014). Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis. World J. Gastroenterol. 20, 2555–2563. doi: 10.3748/wjg.v20.i10.2555
Bosch, J., Groszmann, R. J., and Shah, V. H. (2015). Evolution in the understanding of the pathophysiological basis of portal hypertension: how changes in paradigm are leading to successful new treatments. J. Hepatol. 62, S121–S130.
Boyer, J. L. (2007). New perspectives for the treatment of cholestasis: lessons from basic science applied clinically. J. Hepatol. 46, 365–371. doi: 10.1016/j.jhep.2006.12.001
Braissant, O., Rackayová, V., Pierzchala, K., Grosse, J., McLin, V. A., and Cudalbu, C. (2019). Longitudinal neurometabolic changes in the hippocampus of a rat model of chronic hepatic encephalopathy. J. Hepatol. 71, 505–515. doi: 10.1016/j.jhep.2019.05.022
Brian, J. E., Faraci, F. M., and Moore, S. A. (2001). COX-2-dependent delayed dilatation of cerebral arterioles in response to bradykinin. Am. J. Physio. Heart Circ. Physiol. 280, H2023–H2029.
Butterworth, R. F. (2013). The liver-brain axis in liver failure: neuroinflammation and encephalopathy. Nat. Rev. Gastroenterol. Hepatol. 10, 522–528. doi: 10.1038/nrgastro.2013.99
Caracuel, L., Sastre, E., Llévenes, P., Prieto, I., Funes, T., Aller, M. A., et al. (2019). Acute-on-chronic liver disease enhances phenylephrine-induced endothelial nitric oxide release in rat mesenteric resistance arteries through enhanced PKA, PI3K/AKT and cGMP signalling pathways. Sci. Rep. 9:6993.
Chakrabarti, S., Jiang, Y., and Davidge, S. T. (2012). Neuronal nitric oxide synthase regulates endothelial inflammation. J. Leukoc. Biol. 91, 947–956. doi: 10.1189/jlb.1011513
Cheng, Y., Austin, S. C., Rocca, B., Koller, B. H., Coffman, T. M., Grosser, T., et al. (2002). Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 296, 539–541. doi: 10.1126/science.1068711
Dam, G., Keiding, S., Munk, O. L., Ott, P., Vilstrup, H., Bak, L. K., et al. (2013). Hepatic encephalopathy is associated with decreased cerebral oxygen metabolism and blood flow, not increased ammonia uptake. Hepatology 57, 258–265. doi: 10.1002/hep.25995
Faraci, F. M., and Heistad, D. D. (1990). Regulation of large cerebral arteries and cerebral microvascular pressure. Circ. Res. 66, 8–17. doi: 10.1161/01.res.66.1.8
Faraci, F. M., and Heistad, D. D. (1998). Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol. Rev. 78, 53–97. doi: 10.1152/physrev.1998.78.1.53
Ferguson, J. W., Dover, A. R., Chia, S., Cruden, N. L. M., Hayes, P. C., and Newby, D. E. (2006). Inducible nitric oxide synthase activity contributes to the regulation of peripheral vascular tone in patients with cirrhosis and ascites. Gut 55, 542–546. doi: 10.1136/gut.2005.076562
García-Moreno, L. M., Aller, M. A., Conejo, N. M., Gómez, M. A., Martín, F. R., Arias, J., et al. (2002). Brain Ag-NOR activity in cholestatic rats with hepatic encephalopathy. Hepatol. Res. 24:275. doi: 10.1016/s1386-6346(02)00132-8
García-Moreno, L. M., Conejo, N. M., González-Pardo, H., Aller, M. A., Nava, M. P., Arias, J., et al. (2005). Evaluation of two experimental models of hepatic encephalopathy in rats. Braz. J. Med. Biol. Res. 38, 127–132. doi: 10.1590/s0100-879x2005000100019
Gatta, A., Bolognesi, M., and Merkel, C. (2008). Vasoactive factors and hemodynamic mechanisms in the pathophysiology of portal hypertension in cirrhosis. Mol. Aspects Med. 29, 119–129. doi: 10.1016/j.mam.2007.09.006
Gilsanz, C., Aller, M. A., Fuentes-Julian, S., Prieto, I., Bázquez-Martínez, A., Argudo, S., et al. (2017). Adipose-derived mesenchymal stem cells slow disease progression of acute-on-chronic liver failure. Biomed. Pharmacother. 91, 776–787. doi: 10.1016/j.biopha.2017.04.117
Giménez-Garzó, C., Salhi, D., Urios, A., Ruíz-Sauri, A., Carda, C., Montoliu, C., et al. (2015). Rats with mild bile duct ligation show hepatic encephalopathy with cognitive and motor impairment in the absence of cirrhosis: effects of alcohol ingestion. Neurochem. Res. 40, 230–240. doi: 10.1007/s11064-014-1330-2
González-Correa, J. A., De La Cruz, J. P., Lucena, I., and Sánchez de la Cuesta, F. (1996). Effect of cyclosporin A on platelet aggregation and thromboxane/prostacyclin balance in a model of extrahepatic cholestasis in the rat. Thromb. Res 81, 367–381. doi: 10.1016/0049-3848(96)00008-4
Guevara, M., Bru, C., Ginès, P., Fernández-Esparrach, G., Sort, P., Bataller, R., et al. (1998). Increased cerebrovascular resistance in cirrhotic patients with ascites. Hepatology 28, 39–44. doi: 10.1002/hep.510280107
Hernanz, R., Briones, A. M., Alonso, M. J., Vila, E., and Salaices, M. (2004). Hypertension alters role of iNOS, COX-2, and oxidative stress in bradykinin relaxation impairment after LPS in rat cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 287, H225–H234.
Hollenberg, S. M., and Waldman, B. (2016). The circulatory system in liver disease. Crit. Care Clin. 32, 331–342. doi: 10.1016/j.ccc.2016.02.004
Hollingsworth, K. G., Jones, D. E., Taylor, R., Frith, J., Blamire, A. M., and Newton, J. L. (2010). Impaired cerebral autoregulation in primary biliary cirrhosis: implications for the pathogenesis of cognitive decline. Liver Int. 30, 878–885. doi: 10.1111/j.1478-3231.2010.02259.x
Hou, X., Gobeil, F. Jr., Peri, K., Speranza, G., Marrache, A. M., Lachapelle, P., et al. (2000). Augmented vasoconstriction and thromboxane formation by 15-F2t-isoprostane (8-Iso-Prostaglandin F2α) in immature pig periventricular brain microvessels. Stroke 31, 516–525. doi: 10.1161/01.str.31.2.516
Iwakiri, Y., and Groszmann, R. J. (2007). Vascular endothelial dysfunction in cirrhosis. J. Hepatol. 46, 927–934. doi: 10.1016/j.jhep.2007.02.006
Jalan, R., and Williams, R. (2002). Acute-on-chronic liver failure: pathophysiological basis of therapeutic options. Blood Purif. 20, 252–261. doi: 10.1159/000047017
Kerbert, A. J. C., Verspaget, H. W., Navarro, ÀA., Jalan, R., Solà, E., Benten, D., et al. (2017). Copeptin in acute decompensation of liver cirrhosis: relationship with acute-on-chronic liver failure and short-term survival. Crit. Care 21, 321.
Kravetz, D., Sikuler, E., and Groszmann, R. J. (1986). Splanchnic and systemic hemodynamics in portal hypertensive rats during hemorrhage and blood volume restitution. Gastroenterology 90(5 Pt 1), 1232–1240. doi: 10.1016/0016-5085(86)90390-2
Lin, L. H., Jin, J., Nashelsky, M. B., and Talman, W. T. (2014). Acid-sensing ion channel 1 and nitric oxide synthase are in adjacent layers in the wall of rat and human cerebral arteries. J. Chem. Neuroanat. 6, 161–168. doi: 10.1016/j.jchemneu.2014.10.002
Lizardi-Cervera, J., Almeda, P., Guevara, L., and Uribe, M. (2003). Hepatic encephalopathy: a review. Ann. Hepatol. 2, 122–130.
Llévenes, P., Balfagón, G., and Blanco-Rivero, J. (2018). Thyroid hormones affect nitrergic innervation function in rat mesenteric artery: role of the PI3K/AKT pathway. Vascul. Pharmacol. 108, 36–45. doi: 10.1016/j.vph.2018.05.001
Llévenes, P., Rodrigues-Díez, R., Cros-Brunsó, L., Prieto, M. I., Casaní, L., Balfagón, G., et al. (2020). Beneficial effect of a multistrain synbiotic prodefen® plus on the systemic and vascular alterations associated with metabolic syndrome in rats: the role of the neuronal nitric oxide synthase and protein kinase A. Nutrients 12:117. doi: 10.3390/nu12010117
Maccarrone, M., Ulivi, L., Giannini, N., Montano, V., Ghiadoni, L., Bruno, R. M., et al. (2017). Endothelium and oxidative stress: the pandor’s box of cerebral (and Non-Only) small vessel disease? Curr. Mol. Med. 17, 169–180.
Macías-Rodríguez, R. U., Duarte-Rojo, A., Cantú-Brito, C., Sauerbruch, T., Ruiz-Margáin, A., Trebicka, J., et al. (2015). Cerebral haemodynamics in cirrhotic patients with hepatic encephalopathy. Liver Int. 35, 344–352. doi: 10.1111/liv.12557
Miller, A. A., Budzyn, K., and Sobey, C. G. (2010). Vascular dysfunction in cerebrovascular disease: mechanisms and therapeutic intervention. Clin. Sci. (Lond) 119, 1–17. doi: 10.1042/cs20090649
Mollace, V., Muscoli, C., Masini, E., Cuzzocrea, S., and Salvemini, D. (2005). Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol. Rev. 57, 217–252. doi: 10.1124/pr.57.2.1
Møller, S., Bendtsen, F., and Henriksen, J. H. (2001). Vasoactive substances in the circulatory dysfunction of cirrhosis. Scand. J. Clin. Lab. Invest. 61, 421–429. doi: 10.1080/00365510152567059
Ommati, M. M., Amjadinia, A., Mousavi, K., Azarpira, N., Jamshidzadeh, A., and Heidari, R. (2020). N-acetyl cysteine treatment mitigates biomarkers of oxidative stress in different tissues of bile duct ligated rats. Stress 22, 1–16. doi: 10.1080/10253890.2020.1777970
Pateron, D., Tazi, K. A., Sogni, P., Heller, J., Chagneau, C., Poirel, O., et al. (2000). Role of aortic nitric oxide synthase 3 (eNOS) in the systemic vasodilation of portal hypertension. Gastroenterology 119, 196–200. doi: 10.1053/gast.2000.8554
Peterson, E. C., Wang, Z., and Britz, G. (2011). Regulation of cerebral blood flow. Int. J. Vasc. Med. 2011:823525.
Rama Rao, K. V., Jayakumar, A. R., and Norenberg, M. D. (2014). Brain edema in acute liver failure: mechanisms and concepts. Metab. Brain Dis. 29, 927–936. doi: 10.1007/s11011-014-9502-y
Rodríguez-Garay, E. A. (2003). Cholestasis: human disease and experimental animal models. Ann. Hepatol. 2, 150–158. doi: 10.1016/s1665-2681(19)32126-x
Sastre, E., Caracuel, L., Blanco-Rivero, J., Xavier, F., and Balfagón, G. (2016a). Biphasic effect of diabetes on neuronal nitric oxide release in rat mesenteric arteries. PLoS One 11:e0156793. doi: 10.1371/journal.pone.0156793
Sastre, E., Caracuel, L., Prieto, I., Llévenes, P., Aller, M. A., Arias, J., et al. (2016b). Decompensated liver cirrhosis and neural regulation of mesenteric vascular tone in rats: role of sympathetic, nitrergic and sensory innervations. Sci. Rep. 6:31076.
Sawhney, R., Holland-Fischer, P., Rosselli, M., Mookerjee, R. P., Agarwal, B., and Jalan, R. (2016). Role of ammonia, inflammation and cerebral oxygenation in brain dysfunction of acute on chronic liver failure patients. Liver Transpl. 22, 732–742. doi: 10.1002/lt.24443
Shawcross, D., Davies, N., Williams, R., and Jalan, R. (2004). Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J. Hepatol. 40, 247–254. doi: 10.1016/j.jhep.2003.10.016
Shawcross, D., and Jalan, R. (2005). The pathophysiologic basis of hepatic encephalopathy: central role for ammonia and inflammation. Cell Mol. Life Sci. 62, 2295–2304. doi: 10.1007/s00018-005-5089-0
Sunil, H. V., Mittal, B. R., Kurmi, R., Chawla, R. J., and Dhiman, R. K. (2012). Brain perfusion single photon emission computed tomography abnormalities in patients with minimal hepatic encephalopathy. J. Clin. Exp. Hepatol. 2, 116–121. doi: 10.1016/s0973-6883(12)60099-1
Theodorakis, N. G., Wang, Y. N., Skill, N. J., Metz, M. A., Cahill, P. A., Redmond, E. M., et al. (2003). The role of nitric oxide synthase isoforms in extrahepatic portal hypertension: studies in gene-knockout mice. Gastroenterology 124, 1500–1508. doi: 10.1016/s0016-5085(03)00280-4
Torres, S., Fabersani, E., Marquez, A., and Gauffin-Cano, P. (2019). Adipose tissue inflammation and metabolic syndrome. The proactive role of probiotics. Eur. J. Nutr. 58, 27–43. doi: 10.1007/s00394-018-1790-2
Wiggers, G. A., Furieri, L. B., Briones, A. M., Avendaño, M. S., Peçanha, F. M., Vassallo, D. D., et al. (2016). Cerebrovascular endothelial dysfunction induced by mercury exposure at low concentrations. Neurotoxicology 53, 282–289. doi: 10.1016/j.neuro.2016.02.010
Wright, G., Sharifi, Y., Jover-Cobos, M., and Jalan, R. (2014). The brain in acute on chronic liver failure. Metab. Brain Dis. 29, 965–973.
Wright, G., Vairappan, B., Stadlbauer, V., Mookerjee, R. P., Davies, N. A., and Jalan, R. (2012). Reduction in hyperammonaemia by ornithine phenylacetate prevents lipopolysaccharide- induced brain edema and coma in cirrhotic rats. Liver. Int. 32, 410–419.
Xavier, F. E., Blanco-Rivero, J., Sastre, E., Badimón, L., and Balfagón, G. (2010). Simultaneous inhibition of TXA(2) and PGI(2) synthesis increases NO release in mesenteric resistance arteries from cirrhotic rats. Clin. Sci. (Lond). 119, 283–292. doi: 10.1042/cs20090536
Xu, L., Carter, E. P., Ohara, M., Martin, P. Y., Rogachev, B., Morris, K., et al. (2000). Neuronal nitric oxide synthase and systemic vasodilation in rats with cirrhosis. Am. J. Physiol. Renal. Physiol. 279, F1110–F1115.
Zheng, G., Zhang, L. J., Zhong, J., Wang, Z., Qi, R., Shi, D., et al. (2013). Cerebral blood flow measured by arterial-spin labeling MRI: a useful biomarker for characterization of minimal hepatic encephalopathy in patients with cirrhosis. Eur. J. Radiol. 82, 1981–1988. doi: 10.1016/j.ejrad.2013.06.002
Keywords: acute-on-chronic liver failure, hepatic encephalopathy, cerebral vasculature, bradykinin, nitric oxide, prostaglandin I2
Citation: Caracuel L, Sastre E, Callejo M, Rodrigues-Díez R, García-Redondo AB, Prieto I, Nieto C, Salaices M, Aller MÁ, Arias J and Blanco-Rivero J (2020) Hepatic Encephalopathy-Associated Cerebral Vasculopathy in Acute-on-Chronic Liver Failure: Alterations on Endothelial Factor Release and Influence on Cerebrovascular Function. Front. Physiol. 11:593371. doi: 10.3389/fphys.2020.593371
Received: 10 August 2020; Accepted: 23 October 2020;
Published: 20 November 2020.
Edited by:
Carlos R. Tirapelli, University of São Paulo, BrazilReviewed by:
Eliana Hiromi Akamine, University of São Paulo, BrazilCristina Antoniali, São Paulo State University, Brazil
Copyright © 2020 Caracuel, Sastre, Callejo, Rodrigues-Díez, García-Redondo, Prieto, Nieto, Salaices, Aller, Arias and Blanco-Rivero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier Blanco-Rivero, amF2aWVyLmJsYW5jb0B1YW0uZXM=