
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 25 September 2020
Sec. Renal Physiology and Pathophysiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.586290
This article is part of the Research Topic Contributing Factors to Renal Dysfunction: Fetal Programming, Hormones and Epigenetic View all 12 articles
 Rogério Argeri1
Rogério Argeri1 Fernanda Thomazini1Débora Conte Kimura Lichtenecker1
Fernanda Thomazini1Débora Conte Kimura Lichtenecker1 Karina Thieme2
Karina Thieme2 Maria do Carmo Franco1†
Maria do Carmo Franco1† Guiomar Nascimento Gomes1*†
Guiomar Nascimento Gomes1*†The Barker hypothesis strongly supported the influence of fetal environment on the development of chronic diseases in later life. Multiple experimental and human studies have identified that the deleterious effect of fetal programming commonly leads to alterations in renal development. The interplay between environmental insults and fetal genome can induce epigenetic changes and lead to alterations in the expression of renal phenotype. In this review, we have explored the renal development and its functions, while focusing on the epigenetic findings and functional aspects of the renin-angiotensin system and its components.
The number of nephrons in humans is highly variable, ranging from 250,000 to 2 million per kidney (Bertram et al., 2011). There is an inverse correlation between the total number of nephrons and the risk of developing kidney disease and hypertension (Brenner et al., 1988; Keller et al., 2003). Nephrons are composed of specialized cells, such as epithelial, endothelial, and stromal cells. The epithelial cells originated from the intermediate mesoderm, which also originates two different progenitor cell populations: the ureteric bud (UB) and the metanephric mesenchyme (Little et al., 2007). Clusters of metanephric mesenchymal cells condense around each ureteric bud to form cap mesenchyme (CM). Then, follows a complex and highly coordinated process that depends on interactions between the UB (which will form the collecting duct system) and surrounding CM (which originates the intermediate progenitor cells that ultimately generate all cell types of the nephron; Costantini and Kopan, 2010; Little and McMahon, 2012; Nicolaou et al., 2015; Hurtado Del Pozo et al., 2018). Grobstein’s study was a pioneer in renal development. It showed that kidney development is a multi-stage process, which starts with a primary induction event from the UB, followed by mesenchymal-to-epithelial transition within the CM. The process ends with the completion of nephron patterning and elongation (Grobstein, 1956).
Renal disease is one of the common causes of mortality and morbidity worldwide (Luyckx et al., 2018). Interestingly, this disease can originate in early life (Hoy et al., 1999; Lackland et al., 2000; Eriksson et al., 2018). Barker proposed that insults during critical periods of fetal development can result in a growth deficit, characterized by low birth weight (LBW) and silent morpho-functional changes, that translate into kidney disease in the long term (Hales and Barker, 2001; Langley-Evans et al., 2003).
The human nephrogenesis requires an optimum balance and is completed by around 32–34 weeks of gestation. During this period, the kidneys can be influenced by insults in fetal environment. Evidence suggests that people with LBW have congenital deficit in the number of nephrons and are more susceptible to subsequent renal injury and functional decline in later life (Brenner et al., 1988; Schreuder et al., 2006; Eriksson et al., 2018). Reports that evaluated fetal kidney by ultrasound have supported these findings (Konje et al., 1996; Spencer et al., 2001). Interestingly, other studies have described a positive correlation between birth weight and number of nephrons (Feig et al., 2004; Hoy et al., 2005). Besides, an inverse correlation between the birth weight and glomerular size in the kidney, isolated from neonates, was observed (Mañalich et al., 2000). According to Brenner’s theory (Brenner et al., 1988), the reduced number of nephrons limits excretion of fluids and electrolytes, leading to volume expansion and development of hypertension; this alteration, in turn, damages the glomeruli, causing glomerulosclerosis and accelerating the loss of nephrons (Brenner et al., 1988; Hinchliffe et al., 1991; Schreuder et al., 2006). Conversely, the reduction in nephron number, by the reduction in renal mass or of congenital origin, is followed by compensatory renal growth that leads to hypertrophy of both glomeruli and tubules. This structural change, in addition to increasing glomerular filtration, results in the proximal and distal tubules growth favoring the reabsorption of the filtered volume. Thus, renal function is preserved; however, in long term may result in hypertension and kidney damage (Fong et al., 2014; McArdle et al., 2020).
The assessment of renal function is important to detect the extent and progression of renal diseases. Increasing evidence is available about the impact of fetal environment on the decline in renal function (Hoy et al., 1999; Giapros et al., 2007). An inverse correlation has been observed between birth weight and albumin-to-creatinine ratio in individuals from an Australian aboriginal community (Hoy et al., 1998). Other reports have observed a relationship between LBW with lower glomerular filtration rate (GFR) and albuminuria (Hoy et al., 1999, 2005; Keijzer-Veen et al., 2005). It has been described that LBW neonates, as a result of either prematurity or growth restriction, have increased levels of albuminuria (Aisa et al., 2016). Additionally, severe tubular injury, characterized by high levels of cathepsin B and N-acetyl-β-D-glucosaminidase (NAG) activity, was observed in these neonates (Aisa et al., 2016). In a twin study, Gielen et al. (2005) found that LBW population had lower creatinine clearance levels than the high birth weight population. Moderate renal disturbances were found in LBW children from 4 to 12 years of age (Monge et al., 1998). Another study has reported high levels of cystatin C, a marker of renal function, in LBW children (Franco et al., 2008b). Additionally, Barbati et al. (2016) have shown high levels of cystatin C in urine of LBW neonates. These authors observed an inverse correlation between cystatin C levels and renal volume (Barbati et al., 2016).
Several experimental studies were conducted to confirm the human data (Swanson and David, 2015). Studies demonstrated that restricted protein intake during pregnancy led to LBW, reduced nephron number, and glomerular enlargement in offspring (Merlet-Bénichou et al., 1994; Langley-Evans et al., 1999; Woods, 2000; Vehaskari et al., 2004; Mesquita et al., 2010). Additionally, protein restriction also increased: blood urea, urinary output, and urinary albumin excretion in resultant offspring (Nwagwu et al., 2000). It is believed that the renal changes occur in response to inadequate nutrition as adaptations to ensure survival (Langley-Evans, 2015). These adaptations would be adequate if in the postnatal period the nutritional conditions remained the same. However, if in the postnatal period the nutritional offer changes to a better nutritional standard, the occurred adaptations are no longer adequate and become harmful. This discrepancy between the phenotype developed and that suitable for a given environment is called “mismatch” (Gluckman et al., 2019).
Another important prenatal factor contributing to programming of the renal diseases is maternal diabetes mellitus (e.g., pre-existing type 1 or 2 and gestational diabetes). Although it is correlated with high, instead of LBW, is also associated with impairment of both GFR and renal plasma flow (Abi Khalil et al., 2010). The effect of maternal diabetes on fetal kidney volume is not well established (Hokke et al., 2019). In a recent study, instead of nephron number, the total renal and cortical volumes were assessed in newborns. Neonates of diabetic mothers, with inadequate glycemic control, had lower cortical and total renal volume, suggesting a lower nephron number. However, newborns of diabetic mothers, with strict glycemic control, had similar values as control newborns (Aisa et al., 2019). Thus, it is evident that variations in the maternal glycemic status can lead to different outcomes in the offspring’s kidneys (Aisa et al., 2019).
The studies about the impact of maternal diabetes on fetal development are of great importance since this metabolic disorder can promote several congenital anomalies (Steel et al., 1990). Some studies have demonstrated a high risk of congenital anomalies of the kidney and urinary tract (CAKUT) in children exposed to maternal diabetes (Shnorhavorian et al., 2011; Hsu et al., 2014). Dart et al. (2015) reported that pre-gestational diabetes increased the risk of CAKUT in infants by 67%. Maternal hyperglycemia is an important threat, as glucose crosses the placenta leading to fetal hyperinsulinemia (Higgins and Mc Auliffe, 2010). In turn, hyperinsulinemia stimulates leptin secretion, resulting in hyperleptinemia (Lepercq et al., 1998). Fetal exposure to non-physiological concentrations of these hormones is associated with hypothalamic dysfunction (Plagemann, 2004). Additionally, generation of reactive oxygen species can occur in response to increased glucose/insulin metabolism (Tozour et al., 2018). Therefore, maternal hyperglycemia can be threatening to the developing fetus by different mechanisms. Hence, it is crucial to maintain optimum blood glucose levels during pregnancy.
Experimental studies in female rats, in which diabetes mellitus was induced before the onset of pregnancy using streptozotocin (STZ), demonstrated that offspring developed glomerular hypertrophy and reduction of both GFR and urinary output (Rocha et al., 2005; Magaton et al., 2007). Additionally, renal vascular resistance and the thickness of interlobular arteries were increased in the offspring of diabetic mothers, suggesting vascular remodeling (Rocco et al., 2008); results recently confirmed by Dib et al. (2018). However, there are controversies regarding the nephron number in offspring of diabetic mothers (Rocha et al., 2005; Magaton et al., 2007; Rocco et al., 2008). Reduction in nephron number was observed in studies in which diabetes was induced by different protocols after confirmation of pregnancy (Amri et al., 1999; Tran et al., 2008; Hokke et al., 2013). However, in these studies, the fetus was exposed to the direct effects of STZ, in addition to hyperglycemia.
Many are the possible mechanisms involved in fetal programming, in Figure 1 are represented the most studied ones.
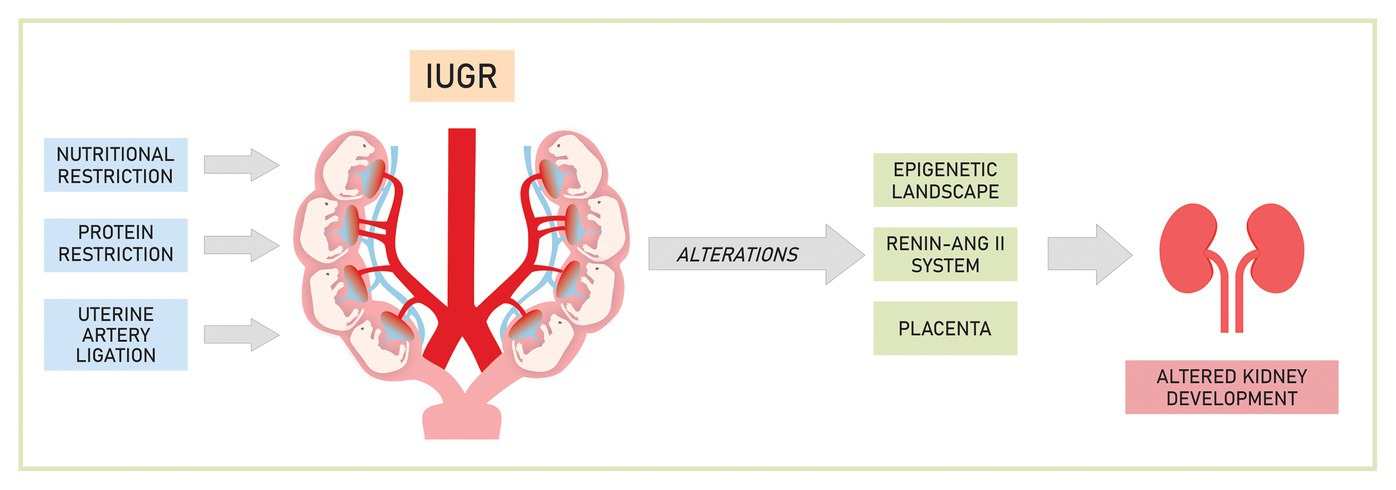
Figure 1. Mechanisms involved in the development of renal changes caused by experimental intrauterine growth restriction (IUGR).
The fetal environment can alter the epigenetic landscape, strongly impacting the reprogramming of gene expression (Dressler and Patel, 2015). Epigenetic modification refers to dynamic changes in chromatin that mediates interactions between environmental factors and the genome. These modifications alter chromatin structure, thereby modulating the accessibility of transcription factors and consequently determining the expression or repression of genes (Hurtado Del Pozo et al., 2018; Wanner et al., 2019). Some of the well-known epigenetic modifications are DNA methylation and post-translation modifications of histones (acetylation, methylation, phosphorylation, and ubiquitination; Hilliard and El-Dahr, 2016).
The role of DNA methylation in the nephron development and its function is not yet well defined. Recently, Wanner et al. (2019) reported that DNA methyltransferase 1 (Dnmt1), and not Dnmt3a/b, is the key regulator of prenatal renal programming, representing a fundamental link between nephron number and intrauterine environment. On the other hand, histone marks distinctively dictate nephrogenesis and affect the expression of several developmental genes, some of them involved in renal disease development (Harikumar and Meshorer, 2015). Studies have provided important information about spatiotemporal changes and dynamic functional states of the epigenome during nephrogenesis. These studies have demonstrated that promoter regions of specific genes in nephron progenitor cells are bivalent and carry both “active” (H3K4me3) and “repressive” (H3K9me3 and H3K27me3) histone marks, along with expression of their corresponding methyltransferases (Ash2l, G9a, Ezh2/Suz12, respectively; McLaughlin et al., 2013, 2014). This suggests that differentiation programs are silenced in these cells, however, can be activated in response to external factors that can be determined by maternal nutritional features during fetal life. Dnmt1 and H3K27me3 are critical in nephron progenitor cells (NPCs) self-renewal and differentiation (Huang et al., 2020). NPC express H3K4me3/K9me3/K27me3 but nascent nephrons retain H3K4me3 marks and downregulate H3K9me3/K27me3 (Hilliard and El-Dahr, 2016). Remarkably, alterations in H3K27me3 levels in differentiated cells have been linked to renal diseases (Majumder et al., 2018; Jia et al., 2019).
Gene-environment interactions during fetal development can persist in multiple generations. Some studies have reported that in utero exposure can affect fetal germ cells and F2 offspring, resulting in transgenerational programming. Epigenetic changes at birth can be associated with maternal nutrition and in the case of female fetuses they can be transmitted to the next generation, emphasizing that inheritance does not depend only on genetic factors but also involves epigenetic mechanisms (Gluckman et al., 2019). However, still little is known about these effects on kidney disease (Briffa et al., 2020).
It is well established that the renin-angiotensin system (RAS) plays an important role in normal morphological development of the kidney and renal function (Guron and Friberg, 2000). All the components of RAS are expressed in early gestation in both rats and human beings (McMillen and Robinson, 2005). In fact, the pharmacological blockade of RAS during renal development leads to altered renal structures (Machado et al., 2008; Madsen et al., 2010; Kett and Denton, 2011; de Almeida et al., 2017). Mutations and altered expressions in genes coding for RAS components are linked to congenital abnormalities of the kidney, confirming the importance of RAS in renal morphogenesis and metanephric organogenesis (Song et al., 2011). It was demonstrated that angiotensin II stimulates the growth and branching of the UB through its receptors (Yosypiv et al., 2008; Song et al., 2011). Also, angiotensin II stimulates the expression of Pax-2 (homeobox 2 gene), an anti-apoptotic factor, which is essential for renal development and repair (Hershkovitz et al., 2007). This process occurs through AT2 receptors (AT2r) and modulates nephrogenesis and renal development (Zandi-Nejad et al., 2006).
The expression of some RAS components could be sensitive to insult during fetal development contributing to later development of renal diseases. Evidence in the literature has demonstrated that fetal programming by several insults promote suppression of the fetal RAS, resulting in altered nephrogenesis (Woods et al., 2001; Dötsch et al., 2009; Mesquita et al., 2010; Moritz et al., 2010). Woods et al. (2001) showed reduction in messenger RNA (mRNA) expression and renin levels in newborns exposed to low protein diet in prenatal life. These authors suggest that such early alteration may be caused by lower intrarenal angiotensin II concentration during nephrogenesis and consequently impair the renal development.
It was observed that fewer ureteral branches sprouted out from the metanephros in the fetus exposed to low protein diet in utero (Mesquita et al., 2010). These changes occurred simultaneously with decreased expression of AT1 receptors (AT1r; on the 17th embryonic day), confirming maladjustment of RAS in the kidneys of these animals (Mesquita et al., 2010).
On the other hand, after the end of nephrogenesis, hyperactivity of RAS was observed in response to fetal programming. There is evidence that exposure to low protein diet, throughout gestation or during specific periods, results in an imbalance in the expression of AT1r and AT2r (Moritz et al., 2010). An increase in AT1r mRNA and protein and a decrease in AT2r gene expression were observed (McMullen et al., 2004; Vehaskari et al., 2004; McMullen and Langley-Evans, 2005). Moreover, it was reported high expression of both mRNA and proteins coding for Angiotensin II 1b receptor (AT1b-R) in adrenal glands isolated from adult offspring exposed to protein restriction during fetal life; and also, hypo-methylation in the promoter regions ofAT1b-R gene (Bogdarina et al., 2007). This epigenetic alteration could lead to higher transcriptional activity, thereby promoting higher expression of this receptor.
Considering maternal diabetes models, it has also been described that the expression of the mRNA coding for angiotensin 1–7 was significantly lower in the offspring of diabetic mothers (Magaton et al., 2007). Besides, increased expression of AT1r was observed in the kidneys of offspring from diabetic mothers, suggesting that changes in RAS might have contributed to the renal changes in this model (Yan et al., 2014).
Interestingly, the RAS blockade in early post-natal life seems to offset the negative effects of fetal programming on the kidneys (Manning and Vehaskari, 2005; Hsu et al., 2015; Watanabe et al., 2018). Early administration of aliskiren, a renin inhibitor, reduced angiotensinogen expression associated with increased renal AT2r and Mas protein in offspring exposed to nutrient restriction during gestation (Hsu et al., 2015). Also, post-weaning losartan (AT1r blocker) therapy completely stopped immune cell infiltration and intrarenal RAS activation in the kidneys isolated from adult offspring exposed to protein restriction during fetal life (Watanabe et al., 2018).
Additionally, altered expression of AT1r in areas of the brain involved in the regulation of blood pressure has been found in protein-restricted offspring (Pladys et al., 2004; de Lima et al., 2013). This change is probably associated with the autonomic changes found in these animals. Increased sympathetic tone seems to have an important role in the genesis of arterial hypertension in this experimental model, supporting this hypothesis are the high plasma concentration of catecholamines observed in protein-restricted offspring (Petry et al., 2000); and the fact that renal denervation prevented hypertension confirming the role of renal nerves activation in the offspring with restricted growth (Alexander et al., 2005; Custódio et al., 2017).
Regarding the changes in RAS in humans, Simonetti et al. (2008) observed higher salt sensitivity in LBW children, possibly as a result of increased aldosterone activity or alterations in AT1r expression or affinity. It has also been reported that circulating levels of angiotensin II and angiotensin-converting enzyme (ACE) activity were higher in healthy males with LBW (Franco et al., 2008a). Ajala et al. (2012) found that the presence of DD genotype in the ACE gene is associated with higher ACE activity in children with a history of LBW. Moreover, one study in LBW children has reported hypo-methylation in the promoter region of the ACE gene (Rangel et al., 2014). These authors also observed that LBW children have lower methylation levels along with higher ACE activity (Rangel et al., 2014).
The placenta, which forms the functional interface between the maternal and fetal circulations, is important for the normal development of the fetus. Usually, the placenta expresses the enzyme 11 beta-hydroxysteroid dehydrogenase type 2 (11β-HSD2) that inactivates maternal glucocorticoids, protecting the fetus from premature exposure to this hormone. Studies have shown that maternal nutritional restriction significantly reduces placental development and also the expression of 11β-HSD2, thus the fetus may be exposed to these hormones prematurely (Börzsönyi et al., 2012; Chapman et al., 2013).
In rats, premature exposure to corticosterone impacted kidney formation resulting in reduction of the nephron number, confirming the negative effects of early exposure to this hormone (Woods and Weeks, 2005; Singh et al., 2007).
Kidney disease is a concerning health problem in the modern world and can be considered as one of the common causes of morbidity and mortality. The fetal environment is an important period for the development of several adaptive mechanisms in various organ systems, leading to an increased risk of development of renal diseases in later life. Several reports from human and animal models suggest that the kidneys can be influenced by insults during fetal development in utero. The impairment in nephrogenesis leads to the process of glomerulosclerosis and loss of renal function in later life. Modifications in the epigenetic characteristics and RAS components are likely involved. Further studies are required to determine relationships between epigenetic alterations and RAS pathway abnormalities, and their ability to influence fetal programming of the kidney diseases.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This manuscript is a revision and for its elaboration no funding was received.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abi Khalil, C., Travert, F., Fetita, S., Rouzet, F., Porcher, R., Riveline, J. P., et al. (2010). Fetal exposure to maternal type 1 diabetes is associated with renal dysfunction at adult age. Diabetes 59, 2631–2636. doi: 10.2337/db10-0419
Aisa, M. C., Cappuccini, B., Barbati, A., Clerici, G., Torlone, E., Gerli, S., et al. (2019). Renal consequences of gestational diabetes mellitus in term neonates: a multidisciplinary approach to the DOHaD perspective in the prevention and early recognition of neonates of GDM mothers at risk of hypertension and chronic renal diseases in later life. J. Clin. Med. 8:429. doi: 10.3390/jcm8040429
Aisa, M. C., Cappuccini, B., Barbati, A., Orlacchio, A., Baglioni, M., and Di Renzo, G. C. (2016). Biochemical parameters of renal impairment/injury and surrogate markers of nephron number in intrauterine growth-restricted and preterm neonates at 30-40 days of postnatal corrected age. Pediatr. Nephrol. 31, 2277–2287. doi: 10.1007/s00467-016-3484-4
Ajala, A. R., Almeida, S. S., Rangel, M., Palomino, Z., Strufaldi, M. W., Puccini, R. F., et al. (2012). Association of ACE gene insertion/deletion polymorphism with birth weight, blood pressure levels, and ACE activity in healthy children. Am. J. Hypertens. 25, 827–832. doi: 10.1038/ajh.2012.50
Alexander, B. T., Hendon, A. E., Ferril, G., and Dwyer, T. M. (2005). Renal denervation abolishes hypertension in low-birth-weight offspring from pregnant rats with reduced uterine perfusion. Hypertension 45, 754–758. doi: 10.1161/01.HYP.0000153319.20340.2a
Amri, K., Freund, N., Vilar, J., Merlet-Bénichou, C., and Lelièvre-Pégorier, M. (1999). Adverse effects of hyperglycemia on kidney development in rats: in vivo and in vitro studies. Diabetes 48, 2240–2245. doi: 10.2337/diabetes.48.11.2240
Barbati, A., Cappuccini, B., Aisa, M. C., Grasselli, C., Zamarra, M., Bini, V., et al. (2016). Increased urinary cystatin-c levels correlate with reduced renal volumes in neonates with intrauterine growth restriction. Neonatology 109, 154–160. doi: 10.1159/000441273
Bertram, J. F., Douglas-Denton, R. N., Diouf, B., Hughson, M. D., and Hoy, W. E. (2011). Human nephron number: implications for health and disease. Pediatr. Nephrol. 26, 1529–1533. doi: 10.1007/s00467-011-1843-8
Bogdarina, I., Welham, S., King, P. J., Burns, S. P., and Clark, A. J. (2007). Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ. Res. 100, 520–526. doi: 10.1161/01.RES.0000258855.60637.58
Börzsönyi, B., Demendi, C., Pajor, A., Rigó, J. Jr., Marosi, K., Agota, A., et al. (2012). Gene expression patterns of the 11β-hydroxysteroid dehydrogenase 2 enzyme in human placenta from intrauterine growth restriction: the role of impaired feto-maternal glucocorticoid metabolism. Eur. J. Obstet. Gynecol. Reprod. Biol. 161, 12–17. doi: 10.1016/j.ejogrb.2011.12.013
Brenner, B. M., Garcia, D. L., and Anderson, S. (1988). Glomeruli and blood pressure. Less of one, more the other? Am. J. Hypertens. 1, 335–347. doi: 10.1093/ajh/1.4.335
Briffa, J. F., Wlodek, M. E., and Moritz, K. M. (2020). Transgenerational programming of nephron deficits and hypertension. Semin. Cell Dev. Biol. 103, 94–103. doi: 10.1016/j.semcdb.2018.05.025
Chapman, K., Holmes, M., and Seckl, J. (2013). 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 93, 1139–1206. doi: 10.1152/physrev.00020.2012
Costantini, F., and Kopan, R. (2010). Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev. Cell 18, 698–712. doi: 10.1016/j.devcel.2010.04.008
Custódio, A. H., de Lima, M. C., Vaccari, B., Boer, P. A., and Gontijo, J. A. R. (2017). Renal sodium handling and blood pressure changes in gestational protein-restricted offspring: role of renal nerves and ganglia neurokinin expression. PLoS One 12:e0179499. doi: 10.1371/journal.pone.0179499
Dart, A. B., Ruth, C. A., Sellers, E. A., Au, W., and Dean, H. J. (2015). Maternal diabetes mellitus and congenital anomalies of the kidney and urinary tract (CAKUT) in the child. Am. J. Kidney Dis. 65, 684–691. doi: 10.1053/j.ajkd.2014.11.017
de Almeida, L. F., Francescato, H. D. C., da Silva, C. G. A., Costa, R. S., and Coimbra, T. M. (2017). Calcitriol reduces kidney development disorders in rats provoked by losartan administration during lactation. Sci. Rep. 7:11472. doi: 10.1038/s41598-017-11815-8
de Lima, M. C., Scabora, J. E., Lopes, A., Mesquita, F. F., Torres, D., Boer, P. A., et al. (2013). Early changes of hypothalamic angiotensin II receptors expression in gestational protein-restricted offspring: effect on water intake, blood pressure and renal sodium handling. J. Renin-Angiotensin-Aldosterone Syst. 14, 271–282. doi: 10.1177/1470320312456328
Dib, A., Payen, C., Bourreau, J., Munier, M., Grimaud, L., Fajloun, Z., et al. (2018). In utero exposure to maternal diabetes is associated with early abnormal vascular structure in offspring. Front. Physiol. 9:350. doi: 10.3389/fphys.2018.00350
Dötsch, J., Plank, C., Amann, K., and Ingelfinger, J. (2009). The implications of fetal programming of glomerular number and renal function. J. Mol. Med. 87, 841–848. doi: 10.1007/s00109-009-0507-7
Dressler, G. R., and Patel, S. R. (2015). Epigenetics in kidney development and renal disease. Transl. Res. 165, 166–176. doi: 10.1016/j.trsl.2014.04.007
Eriksson, J. G., Salonen, M. K., Kajantie, E., and Osmond, C. (2018). Prenatal growth and CKD in older adults: longitudinal findings from the Helsinki birth cohort study, 1924-1944. Am. J. Kidney Dis. 71, 20–26. doi: 10.1053/j.ajkd.2017.06.030
Feig, D. I., Nakagawa, T., Karumanchi, S. A., Oliver, W. J., Kang, D. H., Finch, J., et al. (2004). Hypothesis: uric acid, nephron number, and the pathogenesis of essential hypertension. Kidney Int. 66, 281–287. doi: 10.1111/j.1523-1755.2004.00729.x
Fong, D., Denton, K. M., Moritz, K. M., Evans, R., and Singh, R. R. (2014). Compensatory responses to nephron deficiency: adaptive or maladaptive? Nephrology 19, 119–128. doi: 10.1111/nep.12198
Franco, M. C., Casarini, D. E., Carneiro-Ramos, M. S., Sawaya, A. L., Barreto-Chaves, M. L., and Sesso, R. (2008a). Circulating renin-angiotensin system and catecholamines in childhood: is there a role for birthweight? Clin. Sci. 114, 375–380. doi: 10.1042/cs20070284
Franco, M. C., Nishida, S. K., and Sesso, R. (2008b). GFR estimated from cystatin C versus creatinine in children born small for gestational age. Am. J. Kidney Dis. 51, 925–932. doi: 10.1053/j.ajkd.2008.02.305
Giapros, V., Papadimitriou, P., Challa, A., and Andronikou, S. (2007). The effect of intrauterine growth retardation on renal function in the first two months of life. Nephrol. Dial. Transplant. 22, 96–103. doi: 10.1093/ndt/gfl550
Gielen, M., Pinto-Sietsma, S. J., Zeegers, M. P., Loos, R. J., Fagard, R., de Leeuw, P. W., et al. (2005). Birth weight and creatinine clearance in young adult twins: influence of genetic, prenatal, and maternal factors. J. Am. Soc. Nephrol. 16, 2471–2476. doi: 10.1681/asn.2004030210
Gluckman, P. D., Hanson, M. A., and Low, F. M. (2019). Evolutionary and developmental mismatches are consequences of adaptive developmental plasticity in humans and have implications for later disease risk. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 374:20180109. doi: 10.1098/rstb.2018.0109
Grobstein, C. (1956). Trans-filter induction of tubules in mouse metanephrogenic mesenchyme. Exp. Cell Res. 10, 424–440. doi: 10.1016/0014-4827(56)90016-7
Guron, G., and Friberg, P. (2000). An intact renin-angiotensin system is a prerequisite for normal renal development. J. Hypertens. 18, 123–137. doi: 10.1097/00004872-200018020-00001
Hales, C. N., and Barker, D. J. (2001). The thrifty phenotype hypothesis. Br. Med. Bull. 60, 5–20. doi: 10.1093/bmb/60.1.5
Harikumar, A., and Meshorer, E. (2015). Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 16, 1609–1619. doi: 10.15252/embr.201541011
Hershkovitz, D., Burbea, Z., Skorecki, K., and Brenner, B. M. (2007). Fetal programming of adult kidney disease: cellular and molecular mechanisms. Clin. J. Am. Soc. Nephrol. 2, 334–342. doi: 10.2215/cjn.03291006
Higgins, M., and Mc Auliffe, F. (2010). A review of maternal and fetal growth factors in diabetic pregnancy. Curr. Diabetes Rev. 6, 116–125. doi: 10.2174/157339910790909431
Hilliard, S. A., and El-Dahr, S. S. (2016). Epigenetics mechanisms in renal development. Pediatr. Nephrol. 31, 1055–1060. doi: 10.1007/s00467-015-3228-x
Hinchliffe, S. A., Sargent, P. H., Howard, C. V., Chan, Y. F., and van Velzen, D. (1991). Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Lab. Investig. 64, 777–784.
Hokke, S. N., Armitage, J. A., Puelles, V. G., Short, K. M., Jones, L., Smyth, I. M., et al. (2013). Altered ureteric branching morphogenesis and nephron endowment in offspring of diabetic and insulin-treated pregnancy. PLoS One 8:e58243. doi: 10.1371/journal.pone.0058243
Hokke, S., de Zoysa, N., Carr, B. L., Abruzzo, V., Coombs, P. R., Allan, C. A., et al. (2019). Normal foetal kidney volume in offspring of women treated for gestational diabetes. Endocrinol. Diabetes Metab. 2:e00091. doi: 10.1002/edm2.91
Hoy, W. E., Hughson, M. D., Bertram, J. F., Douglas-Denton, R., and Amann, K. (2005). Nephron number, hypertension, renal disease, and renal failure. J. Am. Soc. Nephrol. 16, 2557–2564. doi: 10.1681/asn.2005020172
Hoy, W. E., Mathews, J. D., McCredie, D. A., Pugsley, D. J., Hayhurst, B. G., Rees, M., et al. (1998). The multidimensional nature of renal disease: rates and associations of albuminuria in an Australian aboriginal community. Kidney Int. 54, 1296–1304. doi: 10.1046/j.1523-1755.1998.00099.x
Hoy, W. E., Rees, M., Kile, E., Mathews, J. D., and Wang, Z. (1999). A new dimension to the Barker hypothesis: low birthweight and susceptibility to renal disease. Kidney Int. 56, 1072–1077. doi: 10.1046/j.1523-1755.1999.00633.x
Hsu, C. C., Jhang, H. R., Chang, W. T., Lin, C. H., Shin, S. J., Hwang, S. J., et al. (2014). Associations between dietary patterns and kidney function indicators in type 2 diabetes. Clin. Nutr. 33, 98–105. doi: 10.1016/j.clnu.2013.04.010
Hsu, C. N., Lee, C. T., Huang, L. T., and Tain, Y. L. (2015). Aliskiren in early postnatal life prevents hypertension and reduces asymmetric dimethylarginine in offspring exposed to maternal caloric restriction. J. Renin-Angiotensin-Aldosterone Syst. 16, 506–513. doi: 10.1177/1470320313514123
Huang, B., Liu, Z., Vonk, A., Zeng, Z., and Li, Z. (2020). Epigenetic regulation of kidney progenitor cells. Stem Cells Transl. Med. 9, 655–660. doi: 10.1002/sctm.19-0289
Hurtado Del Pozo, C., Garreta, E., Izpisúa Belmonte, J. C., and Montserrat, N. (2018). Modeling epigenetic modifications in renal development and disease withs organoids and genome editing. Dis. Model. Mech. 11:dmm035048. doi: 10.1242/dmm.035048
Jia, Y., Reddy, M. A., Das, S., Oh, H. J., Abdollahi, M., Yuan, H., et al. (2019). Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-β1-induced gene expression in mesangial cells and diabetic kidney. J. Biol. Chem. 294, 12695–12707. doi: 10.1074/jbc.RA119.007575
Keijzer-Veen, M. G., Schrevel, M., Finken, M. J., Dekker, F. W., Nauta, J., Hille, E. T., et al. (2005). Microalbuminuria and lower glomerular filtration rate at young adult age in subjects born very premature and after intrauterine growth retardation. J. Am. Soc. Nephrol. 16, 2762–2768. doi: 10.1681/asn.2004090783
Keller, G., Zimmer, G., Mall, G., Ritz, E., and Amann, K. (2003). Nephron number in patients with primary hypertension. N. Engl. J. Med. 348, 101–108. doi: 10.1056/NEJMoa020549
Kett, M. M., and Denton, K. M. (2011). Renal programming: cause for concern? Am. J. Phys. Regul. Integr. Comp. Phys. 300, R791–R803. doi: 10.1152/ajpregu.00791.2010
Konje, J. C., Bell, S. C., Morton, J. J., de Chazal, R., and Taylor, D. J. (1996). Human fetal kidney morphometry during gestation and the relationship between weight, kidney morphometry and plasma active renin concentration at birth. Clin. Sci. 91, 169–175. doi: 10.1042/cs0910169
Lackland, D. T., Bendall, H. E., Osmond, C., Egan, B. M., and Barker, D. J. (2000). Low birth weights contribute to high rates of early-onset chronic renal failure in the Southeastern United States. Arch. Intern. Med. 160, 1472–1476. doi: 10.1001/archinte.160.10.1472
Langley-Evans, S. C. (2015). Nutrition in early life and the programming of adult disease: a review. J. Hum. Nutr. Diet. 28(Suppl. 1), 1–14. doi: 10.1111/jhn.12212
Langley-Evans, S. C., Langley-Evans, A. J., and Marchand, M. C. (2003). Nutritional programming of blood pressure and renal morphology. Arch. Physiol. Biochem. 111, 8–16. doi: 10.1076/apab.111.1.8.15136
Langley-Evans, S. C., Welham, S. J., and Jackson, A. A. (1999). Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sci. 64, 965–974. doi: 10.1016/s0024-3205(99)00022-3
Lepercq, J., Cauzac, M., Lahlou, N., Timsit, J., Girard, J., Auwerx, J., et al. (1998). Overexpression of placental leptin in diabetic pregnancy: a critical role for insulin. Diabetes 47, 847–850. doi: 10.2337/diabetes.47.5.847
Little, M. H., Brennan, J., Georgas, K., Davies, J. A., Davidson, D. R., Baldock, R. A., et al. (2007). A high-resolution anatomical ontology of the developing murine genitourinary tract. Gene Expr. Patterns 7, 680–699. doi: 10.1016/j.modgep.2007.03.002
Little, M. H., and McMahon, A. P. (2012). Mammalian kidney development: principles, progress, and projections. Cold Spring Harb. Perspect. Biol. 4:a008300. doi: 10.1101/cshperspect.a008300
Luyckx, V. A., Tonelli, M., and Stanifer, J. W. (2018). The global burden of kidney disease and the sustainable development goals. Bull. World Health Organ. 96, 414–422D. doi: 10.2471/BLT.17.206441
Machado, F. G., Poppi, E. P., Fanelli, C., Malheiros, D. M., Zatz, R., and Fujihara, C. K. (2008). AT1 blockade during lactation as a model of chronic nephropathy: mechanisms of renal injury. Am. J. Physiol. Ren. Physiol. 294, F1345–F1353. doi: 10.1152/ajprenal.00020.2008
Madsen, K., Marcussen, N., Pedersen, M., Kjaersgaard, G., Facemire, C., Coffman, T. M., et al. (2010). Angiotensin II promotes development of the renal microcirculation through AT1 receptors. J. Am. Soc. Nephrol. 21, 448–459. doi: 10.1681/ASN.2009010045
Magaton, A., Gil, F. Z., Casarini, D. E., Cavanal Mde, F., and Gomes, G. N. (2007). Maternal diabetes mellitus-early consequences for the offspring. Pediatr. Nephrol. 22, 37–43. doi: 10.1007/s00467-006-0282-4
Majumder, S., Thieme, K., Batchu, S. N., Alghamdi, T. A., Bowskill, B. B., Kabir, M. G., et al. (2018). Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J. Clin. Invest. 128, 483–499. doi: 10.1172/JCI95946
Mañalich, R., Reyes, L., Herrera, M., Melendi, C., and Fundora, I. (2000). Relationship between weight at birth and the number and size of renal glomeruli in humans: a histomorphometric study. Kidney Int. 58, 770–773. doi: 10.1046/j.1523-1755.2000.00225.x
Manning, J., and Vehaskari, V. M. (2005). Postnatal modulation of prenatally programmed hypertension by dietary Na and ACE inhibition. Am. J. Phys. Regul. Integr. Comp. Phys. 288, R80–R84. doi: 10.1152/ajpregu.00309.2004
McArdle, Z., Schreuder, M. F., Moritz, K. M., Denton, K. M., and Singh, R. R. (2020). Physiology and pathophysiology of compensatory adaptations of a solitary functioning kidney. Front. Physiol. 11:725. doi: 10.3389/fphys.2020.00725
McLaughlin, N., Wang, F., Saifudeen, Z., and El-Dahr, S. S. (2014). In situ histone landscape of nephrogenesis. Epigenetics 9, 222–235. doi: 10.4161/epi.26793
McLaughlin, N., Yao, X., Li, Y., Saifudeen, Z., and El-Dahr, S. S. (2013). Histone signature of metanephric mesenchyme cell lines. Epigenetics 8, 970–978. doi: 10.4161/epi.25753
McMillen, I. C., and Robinson, J. S. (2005). Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol. Rev. 85, 571–633. doi: 10.1152/physrev.00053.2003
McMullen, S., Gardner, D. S., and Langley-Evans, S. C. (2004). Prenatal programming of angiotensin II type 2 receptor expression in the rat. Br. J. Nutr. 91, 133–140. doi: 10.1079/bjn20031029
McMullen, S., and Langley-Evans, S. C. (2005). Maternal low-protein diet in rat pregnancy programs blood pressure through sex-specific mechanisms. Am. J. Phys. Regul. Integr. Comp. Phys. 288, R85–R90. doi: 10.1152/ajpregu.00435.2004
Merlet-Bénichou, C., Gilbert, T., Muffat-Joly, M., Lelièvre-Pégorier, M., and Leroy, B. (1994). Intrauterine growth retardation leads to a permanent nephron deficit in the rat. Pediatr. Nephrol. 8, 175–180. doi: 10.1007/bf00865473
Mesquita, F. F., Gontijo, J. A., and Boer, P. A. (2010). Maternal undernutrition and the offspring kidney: from fetal to adult life. Braz. J. Med. Biol. Res. 43, 1010–1018. doi: 10.1590/s0100-879x2010007500113
Monge, M., García-Nieto, V. M., Domenech, E., Barac-Nieto, M., Muros, M., and Pérez-González, E. (1998). Study of renal metabolic disturbances related to renal lithiasis at school age in very-low-birth-weight children. Nephron 79, 269–273. doi: 10.1159/000045048
Moritz, K. M., Cuffe, J. S., Wilson, L. B., Dickinson, H., Wlodek, M. E., Simmons, D. G., et al. (2010). Review: sex specific programming: a critical role for the renal renin-angiotensin system. Placenta 31, S40–S46. doi: 10.1016/j.placenta.2010.01.006
Nicolaou, N., Renkema, K. Y., Bongers, E. M., Giles, R. H., and Knoers, N. V. (2015). Genetic, environmental, and epigenetic factors involved in CAKUT. Nat. Rev. Nephrol. 11, 720–731. doi: 10.1038/nrneph.2015.140
Nwagwu, M. O., Cook, A., and Langley-Evans, S. C. (2000). Evidence of progressive deterioration of renal function in rats exposed to a maternal low-protein diet in utero. Br. J. Nutr. 83, 79–85.
Petry, C. J., Dorling, M. W., Wang, C. L., Pawlak, D. B., and Ozanne, S. E. (2000). Catecholamine levels and receptor expression in low protein rat offspring. Diabet. Med. 17, 848–853. doi: 10.1046/j.1464-5491.2000.00392.x
Pladys, P., Lahaie, I., Cambonie, G., Thibault, G., Lê, N. L., Abran, D., et al. (2004). Role of brain and peripheral angiotensin II in hypertension and altered arterial baroreflex programmed during fetal life in rat. Pediatr. Res. 55, 1042–1049. doi: 10.1203/01.Pdr.0000127012.37315.36
Plagemann, A. (2004). ‘Fetal programming’ and ‘functional teratogenesis’: on epigenetic mechanisms and prevention of perinatally acquired lasting health risks. J. Perinat. Med. 32, 297–305. doi: 10.1515/jpm.2004.055
Rangel, M., dos Santos, J. C., Ortiz, P. H., Hirata, M., Jasiulionis, M. G., Araujo, R. C., et al. (2014). Modification of epigenetic patterns in low birth weight children: importance of hypomethylation of the ACE gene promoter. PLoS One 9:e106138. doi: 10.1371/journal.pone.0106138
Rocco, L., Gil, F. Z., da Fonseca Pletiskaitz, T. M., de Fátima Cavanal, M., and Gomes, G. N. (2008). Effect of sodium overload on renal function of offspring from diabetic mothers. Pediatr. Nephrol. 23, 2053–2060. doi: 10.1007/s00467-008-0884-0
Rocha, S. O., Gomes, G. N., Forti, A. L., do Carmo Pinho Franco, M., Fortes, Z. B., de Fátima Cavanal, M., et al. (2005). Long-term effects of maternal diabetes on vascular reactivity and renal function in rat male offspring. Pediatr. Res. 58, 1274–1279. doi: 10.1203/01.pdr.0000188698.58021.ff
Schreuder, M., Delemarre-van de Waal, H., and van Wijk, A. (2006). Consequences of intrauterine growth restriction for the kidney. Kidney Blood Press. Res. 29, 108–125. doi: 10.1159/000094538
Shnorhavorian, M., Bittner, R., Wright, J. L., and Schwartz, S. M. (2011). Maternal risk factors for congenital urinary anomalies: results of a population-based case-control study. Urology 78, 1156–1161. doi: 10.1016/j.urology.2011.04.022
Simonetti, G. D., Raio, L., Surbek, D., Nelle, M., Frey, F. J., and Mohaupt, M. G. (2008). Salt sensitivity of children with low birth weight. Hypertension 52, 625–630. doi: 10.1161/hypertensionaha.108.114983
Singh, R. R., Cullen-McEwen, L. A., Kett, M. M., Boon, W. M., Dowling, J., Bertram, J. F., et al. (2007). Prenatal corticosterone exposure results in altered AT1/AT2, nephron deficit and hypertension in the rat offspring. J. Physiol. 579, 503–513. doi: 10.1113/jphysiol.2006.125773
Song, R., Preston, G., and Yosypiv, I. V. (2011). Angiotensin II stimulates in vitro branching morphogenesis of the isolated ureteric bud. Mech. Dev. 128, 359–367. doi: 10.1016/j.mod.2011.07.002
Spencer, J., Wang, Z., and Hoy, W. (2001). Low birth weight and reduced renal volume in aboriginal children. Am. J. Kidney Dis. 37, 915–920. doi: 10.1016/s0272-6386(05)80006-x
Steel, J. M., Johnstone, F. D., Hepburn, D. A., and Smith, A. F. (1990). Can prepregnancy care of diabetic women reduce the risk of abnormal babies? BMJ 301, 1070–1074. doi: 10.1136/bmj.301.6760.1070
Swanson, A. M., and David, A. L. (2015). Animal models of fetal growth restriction: considerations for translational medicine. Placenta 36, 623–630. doi: 10.1016/j.placenta.2015.03.003
Tozour, J. N., Delahaye, F., Suzuki, M., Praiss, A., Zhao, Y., Cai, L., et al. (2018). Intrauterine hyperglycemia is associated with an impaired postnatal response to oxidative damage. Stem Cells Dev. 27, 683–691. doi: 10.1089/scd.2017.0232
Tran, S., Chen, Y. W., Chenier, I., Chan, J. S., Quaggin, S., Hébert, M. J., et al. (2008). Maternal diabetes modulates renal morphogenesis in offspring. J. Am. Soc. Nephrol. 19, 943–952. doi: 10.1681/ASN.2007080864
Vehaskari, V. M., Stewart, T., Lafont, D., Soyez, C., Seth, D., and Manning, J. (2004). Kidney angiotensin and angiotensin receptor expression in prenatally programmed hypertension. Am. J. Physiol. Ren. Physiol. 287, F262–F267. doi: 10.1152/ajprenal.00055.2004
Wanner, N., Vornweg, J., Combes, A., Wilson, S., Plappert, J., Rafflenbeul, G., et al. (2019). DNA methyltransferase 1 controls nephron progenitor cell renewal and differentiation. J. Am. Soc. Nephrol. 30, 63–78. doi: 10.1681/ASN.2018070736
Watanabe, I. K. M., Jara, Z. P., Volpini, R. A., Franco, M. D. C., Jung, F. F., and Casarini, D. E. (2018). Up-regulation of renal renin-angiotensin system and inflammatory mechanisms in the prenatal programming by low-protein diet: beneficial effect of the post-weaning losartan treatment. J. Dev. Orig. Health Dis. 9, 530–535. doi: 10.1017/s2040174418000296
Woods, L. L. (2000). Fetal origins of adult hypertension: a renal mechanism? Curr. Opin. Nephrol. Hypertens. 9, 419–425. doi: 10.1097/00041552-200007000-00014
Woods, L. L., Ingelfinger, J. R., Nyengaard, J. R., and Rasch, R. (2001). Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr. Res. 49, 460–467. doi: 10.1203/00006450-200104000-00005
Woods, L. L., and Weeks, D. A. (2005). Prenatal programming of adult blood pressure: role of maternal corticosteroids. Am. J. Phys. Regul. Integr. Comp. Phys. 289, R955–R962. doi: 10.1152/ajpregu.00455.2004
Yan, J., Li, X., Su, R., Zhang, K., and Yang, H. (2014). Long-term effects of maternal diabetes on blood pressure and renal function in rat male offspring. PLoS One 9:e88269. doi: 10.1371/journal.pone.0088269
Yosypiv, I. V., Boh, M. K., Spera, M. A., and El-Dahr, S. S. (2008). Downregulation of spry-1, an inhibitor of GDNF/ret, causes angiotensin II-induced ureteric bud branching. Kidney Int. 74, 1287–1293. doi: 10.1038/ki.2008.378
Keywords: renal function, programmed kidney disease, hypertension, nephron number, fetal environment
Citation: Argeri R, Thomazini F, Lichtenecker DCK, Thieme K, do Carmo Franco M and Gomes GN (2020) Programmed Adult Kidney Disease: Importance of Fetal Environment. Front. Physiol. 11:586290. doi: 10.3389/fphys.2020.586290
Edited by:
Adriana Castello Costa Girardi, University of São Paulo, BrazilReviewed by:
Michel Baum, University of Texas Southwestern Medical Center, United StatesCopyright © 2020 Argeri, Thomazini, Lichtenecker, Thieme, Franco and Gomes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guiomar Nascimento Gomes, Z3Vpb21hci5nb21lc0B1bmlmZXNwLmJy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.