
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 20 October 2020
Sec. Membrane Physiology and Membrane Biophysics
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.585907
This article is part of the Research TopicEmerging Roles of Monovalent Ion Transporters in Intracellular SignalingView all 7 articles
 Adrián Rafael Murillo-de-Ozores1,2
Adrián Rafael Murillo-de-Ozores1,2 María Chávez-Canales3
María Chávez-Canales3 Paola de los Heros4
Paola de los Heros4 Gerardo Gamba1,5
Gerardo Gamba1,5 María Castañeda-Bueno1*
María Castañeda-Bueno1*The role of Cl– as an intracellular signaling ion has been increasingly recognized in recent years. One of the currently best described roles of Cl– in signaling is the modulation of the With-No-Lysine (K) (WNK) – STE20-Proline Alanine rich Kinase (SPAK)/Oxidative Stress Responsive Kinase 1 (OSR1) – Cation-Coupled Cl– Cotransporters (CCCs) cascade. Binding of a Cl– anion to the active site of WNK kinases directly modulates their activity, promoting their inhibition. WNK activation due to Cl– release from the binding site leads to phosphorylation and activation of SPAK/OSR1, which in turn phosphorylate the CCCs. Phosphorylation by WNKs-SPAK/OSR1 of the Na+-driven CCCs (mediating ions influx) promote their activation, whereas that of the K+-driven CCCs (mediating ions efflux) promote their inhibition. This results in net Cl– influx and feedback inhibition of WNK kinases. A wide variety of alterations to this pathway have been recognized as the cause of several human diseases, with manifestations in different systems. The understanding of WNK kinases as Cl– sensitive proteins has allowed us to better understand the mechanistic details of regulatory processes involved in diverse physiological phenomena that are reviewed here. These include cell volume regulation, potassium sensing and intracellular signaling in the renal distal convoluted tubule, and regulation of the neuronal response to the neurotransmitter GABA.
The chloride (Cl–) anion is an important component of all known living beings, where it plays several roles in homeostatic and rheostatic processes in all types of cells. In humans, extracellular Cl– concentration is maintained relatively constant, between 100 and 116 mmol/L, due to tight regulation by the kidneys and intestine (Boulpaep and Boron, 2016). It is notable that there is interspecies variability of plasma Cl– concentration. For instance, normal levels in rats and mice are in the range of 90–132 mmol/L and 106–131 mmol/L, respectively (Lea et al., 2018).
Intracellular Cl– concentration ([Cl–]i) varies wildly among different cell types within an organism. For example, it has been reported that most adult neurons have relatively low [Cl–]i (5–15 mmol/L) (Kakazu et al., 1999; Yamada et al., 2004; Glykys et al., 2014), and [Cl–]i of renal epithelial cells such as the ones of the distal convoluted tubule (DCT) has been estimated to be between 10 and 20 mmol/L (Beck et al., 1988; Boettger et al., 2002; Weinstein, 2005; Terker et al., 2015b). Conversely, olfactory sensory neurons (Reuter et al., 1998) and some cells from secretory epithelia, such as pancreatic (O’Doherty and Stark, 1983) and salivary acinar cells (Foskett, 1990), have a [Cl–]i as high as 60–65 mmol/L. Additionally, [Cl–]i can be dynamically modulated by different stimuli, such as cholinergic agonists (Foskett, 1990), cAMP levels (Xie and Schafer, 2004), lectin-stimulation (Lai et al., 2003), and extracellular potassium concentration ([K+]e) (Terker et al., 2015b). These reports exemplify the wide variation of [Cl–]i, which is important for the role that this anion plays in the physiology of specific cell types.
While some of the most studied roles for Cl– in physiology are related to cell volume regulation (Hoffmann et al., 2009), establishment of resting membrane potential (Funabashi et al., 2010; Hutter, 2017), and acid-base balance (Seifter and Chang, 2016), it is now becoming clear that this anion is involved in intracellular signaling pathways involved in the regulation of a wider variety of cellular processes, such as gene expression, cell proliferation, apoptosis, among others (reviewed in Valdivieso and Santa-Coloma, 2019; Wilson and Mongin, 2019; Lüscher et al., 2020). For instance, published evidence suggests that [Cl–]i can modulate the activity of different kinases, such as the MAPKs p38, JNK and ERK (Ohsawa et al., 2010; Wu et al., 2016), as well as SGK1 (Zhang et al., 2018), although it is still unclear whether direct effects of Cl– ions on the kinases themselves are responsible. However, the With-No-lysine (K) (WNK) family of kinases is one example where the direct Cl– binding to the enzyme’s active site (Piala et al., 2014) that modulates kinase activity (Bazua-Valenti et al., 2015) has been thoroughly studied (detailed in sections below). These observations support the novel proposed role of [Cl–]i as a second messenger (Valdivieso and Santa-Coloma, 2019; Wilson and Mongin, 2019; Lüscher et al., 2020), responsible for modulating the activity of several proteins.
Transmembrane transport proteins determine the Cl– permeability of each cell type. Ion channels that facilitate large Cl– fluxes across cell membranes include the CLC family (reviewed extensively in Jentsch and Pusch, 2018), the cystic fibrosis transmembrane conductance regulator (CFTR) channel (Csanády et al., 2019), the volume-regulated anion channel (VRAC) channel (Osei-Owusu et al., 2018), and Ca2+-activated Cl– channels (CaCCs) such as anoctamins (Pedemonte and Galietta, 2014). The direction of Cl– flux through ion channels is solely determined by the electrochemical gradient of this ion across the membrane. However, secondary active transporters can set the [Cl–]i at levels that diverge from the electrochemical equilibrium by coupling Na+ influx or K+ efflux to the movement of Cl–. Transporters with this type of activity are all members of the SLC12 family of solute carriers described below.
The SLC12 family of solute carriers is comprised by the electroneutral cation-coupled Cl– cotransporters (CCCs). Seven members of this family are arranged in two branches, depending on their ability to use Na+ as one of the transported cations coupled to Cl–. The Na+-dependent branch includes the Na+-K+-2Cl– cotransporters, known as NKCC1 and NKCC2, and the Na+-Cl– cotransporter, NCC (Gamba, 2005). NKCC1 is expressed in many epithelial and non-epithelial cells (Delpire et al., 1994). Within epithelial cells it is expressed in the basolateral membrane, except in the choroid plexus of the brain, where it is expressed apically (Delpire et al., 1994; Wu et al., 1998). NKCC2 is exclusively expressed in the apical membrane of the thick ascending limb of Henle’s loop in the kidney (Gamba et al., 1994) and NCC is present in the apical membrane of the distal convoluted tubule in the kidney and in osteoblasts in bone (Gamba et al., 1994; Dvorak et al., 2007). Identity degree at the amino acid level among these transporters is between 50 and 60%. The Na+-independent branch is composed of four K+-Cl– cotransporters, known as KCC1 to KCC4. Of these four, KCC2 is exclusively expressed in neurons, while the other three KCCs are present in many cells throughout the body. Identity degree among KCCs is about 60% and between the Na+- dependent and independent branches is around 25% (Gamba, 2005; Arroyo et al., 2013).
The CCCs are secondary active transporters whose activity is driven by the Na+ and K+ gradients generated by the Na+-K+-ATPase. The Na+-driven transporters move ions from the extracellular space into the cytoplasm, while the K+-driven transporters (of the Na+-independent branch) mediate ion extrusion from the cells. In non-epithelial cells, the sustained activity of the Na+-K+-ATPase maintains a low Na+ and high K+ intracellular concentration, respectively. Thus, it is considered that the net effect of the activity of CCCs is the modulation of the [Cl–]i. Because of this, the expression of Na+-driven and K+-driven members of the SLC12 family in the same cell constitutes a system for the dynamic modulation of [Cl–]i. This, for example, is particularly relevant in neurons, where, as explained below in detail, the type and magnitude of the response to neurotransmitters that activate Cl– channels in the postsynaptic membrane depends on the electrochemical Cl– gradient (Kahle et al., 2008). Regulation of [Cl–]i also plays a relevant role for the regulation of cell volume (de los Heros et al., 2018). In epithelial cells, the CCCs works in conjunction with other apical and/or basolateral channels and transporters to carry out transepithelial ion transport (Gamba, 2005). Thus, the major physiological roles of the CCCs are modulation of [Cl–]i, cell volume regulation, and transepithelial ion transport. For this reason, the CCCs are implicated in many organs’ and systems’ physiological processes (Gamba, 2005).
A variety of human and animal diseases and phenotypes in knockout mice models have been helpful in revealing the many roles of CCCs in physiology (Delpire and Mount, 2002) (Table 1). Inactivating mutations in the renal cotransporters NKCC2 and NCC are the cause of the Bartter syndrome type I (Simon et al., 1996a) and Gitelman disease (Simon et al., 1996b), respectively. NKCC2 constitutes the main apical entryway for Na+ and Cl– in the thick ascending limb of Henle’s loop and NCC plays a similar role in the downstream adjacent nephron segment known as the distal convoluted tubule. Decreased Na+ reabsorption in these segments is not only associated with volume depletion and low blood pressure, but also with hypokalemic metabolic alkalosis due to increased K+ secretion in the aldosterone sensitive distal nephron that is stimulated by the increased distal Na+ delivery (Gamba, 2005). The phenotype of both, type I Bartter syndrome and Gitelman syndrome patients, is exclusively the consequence of the lack of activity of these transporters in the nephron, suggesting that indeed their expression is very restricted to the kidney and, if expressed elsewhere, like NCC in bone, its role in other tissues is not essential. Regarding NKCC1, knockout mice were generated and studied before human mutations in this gene were found. These mice have a wide variety of phenotypic alterations, such as small size, inner ear dysfunction, male infertility, altered pain perception, defects in intestinal transit, decreased saliva production, and low blood pressure, among others (Gagnon and Delpire, 2013). Delpire et al. (2016) described a human patient with respiratory weakness, endocrine and pancreatic abnormalities, and multi-organ failure. Genetic analysis revealed a heterozygous 11-bp deletion in exon 22 of SLC12A2 (encoding NKCC1) (Delpire et al., 2016) that causes a frameshift resulting in a truncated protein lacking the last 187 amino acid residues of the C-terminus. This mutation causes the mislocalization of the cotransporter to the apical membrane in epithelial cells (Koumangoye et al., 2018). The same mutation has been shown to cause a similar, although milder, phenotype in mice (Koumangoye et al., 2020). Macnamara et al. (2019) reported a novel syndrome, named Kilquist syndrome, in a patient harboring a large homozygous deletion in SLC12A2, from intron 1 through exon 7. Such mutation leads to aberrant splicing between exons 1 and 8, introducing a frameshift that would produce a truncated protein. Molecular analysis showed lower mRNA levels and absence of the NKCC1 protein in the patient’s fibroblasts. Phenotypic features similar to the ones observed in NKCC1 knockout (−/−) mice were reported, including global developmental delay, bilateral sensorineural hearing loss, gastrointestinal abnormalities, and xerostomia (Macnamara et al., 2019). In addition, a missense variant that increases the activity of NKCC1 was described to be associated with schizophrenia (Merner et al., 2016).
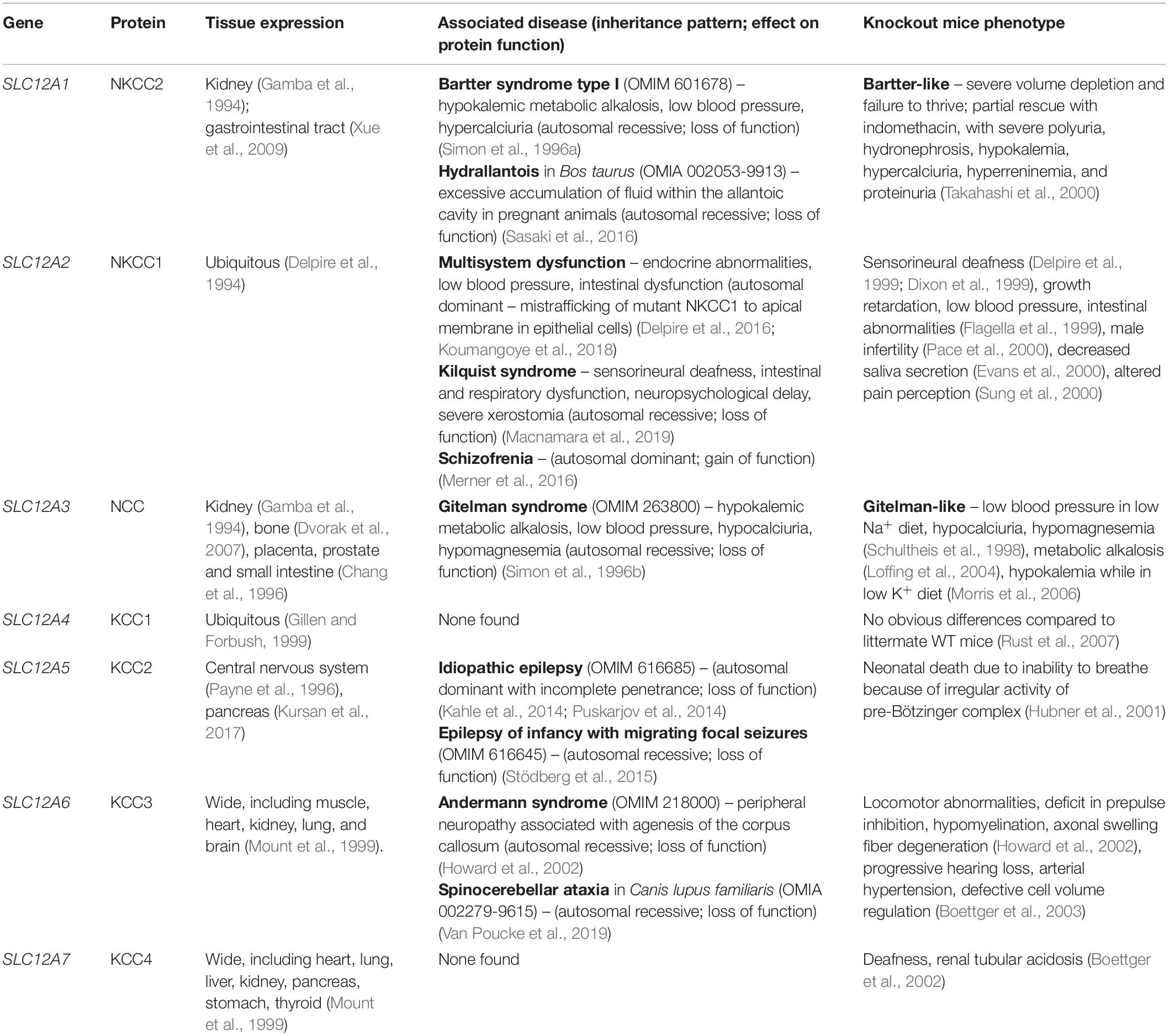
Table 1. SLC12 cotransporters: associated genetic diseases and phenotype of knockout models.
Mutations in KCC2 have been implicated in a variety of epileptic syndromes (Kahle et al., 2014; Puskarjov et al., 2014) and mutations in KCC3 are the cause of a very complex inherited neurological disease known as Andermann’s syndrome in which patients exhibit absence of the corpus callosum in the brain, together with a variety of neurodegenerative and psychiatric manifestations (Howard et al., 2002). No pathogenic mutations in KCC4 have been described in humans. However, KCC4–/– mice display deafness and renal tubular acidosis, suggesting that this transporter plays an important role in inner ear and kidney physiology (Boettger et al., 2002). Finally, global, constitutive disruption of KCC1 in mice has no phenotypic consequences, suggesting that the absence of KCC1 can be compensated by other K+-Cl– cotransporters (Rust et al., 2007).
The Na+-driven and K+-driven cotransporters are regulated in opposite ways. They are all regulated by a kinase cascade in which the With No lysine (K) kinases (WNKs) phosphorylate and regulate the STE20-Proline Alanine rich Kinase (SPAK), and the Oxidative Stress Responsive Kinase 1 (OSR1). SPAK and OSR1 in turn phosphorylate the CCCs (Richardson and Alessi, 2008; Gagnon and Delpire, 2012; Alessi et al., 2014). Phosphorylation of the Na+-driven cotransporters, which occur in a cluster of serine-threonine residues located in the intracellular N-terminal domain, results in upregulation of cotransporter activity. In contrast, phosphorylation of K+-driven transporters that occurs in threonine residues of the C-terminal cytoplasmic domain results in downregulation of cotransporter activity. Thus, activation of this kinase cascade is able to simultaneously activate the Na+-(K+)-Cl– influx and prevent the K+-Cl– efflux, increasing [Cl–]i and intracellular osmolarity, while inactivation of the phosphorylating cascade and/or activation of dephosphorylating pathways result in opposite effects.
Stimuli such as the decrease in [Cl–]i or the decrease in cell volume (cell shrinkage) result in activation of the WNK-SPAK/OSR1 phosphorylation pathway, increasing the activity of the Na+-(K+)-Cl– cotransporters and inhibiting the K+-Cl– cotransporters (Gagnon et al., 2006; Zagórska et al., 2007; Arroyo et al., 2013; Piala et al., 2014; Bazua-Valenti et al., 2015; de los Heros et al., 2018). This results in the increase of [Cl–]i or in the net influx of ions that contribute to the regulatory volume increase response. In contrast, an increase in [Cl–]i or cell volume (cell swelling) inhibits the phosphorylating pathway and activates protein phosphatases, thus resulting in cotransporters dephosphorylation, and the consequent decrease in [Cl–]i or the net efflux of ions that contribute to the regulatory volume decrease response. Thus, a negative feedback loop integrated by the monovalent cation-Cl– cotransporters of the CCC family, the WNK-SPAK/OSR1 kinase cascade, and protein phosphatases serve to regulate the [Cl–]i and/or the cell volume, which in turn modulate the activity of the cotransporters via the kinases and phosphatases. This feedback loop has implications in several physiological processes that are discussed in this work.
The WNK family of Ser/Thr kinases is comprised of four members in mammals: WNK1, WNK2, WNK3, and WNK4. Unlike most kinases, WNKs have their conserved catalytic Lys residue involved in ATP binding located in subdomain I, instead of in subdomain II, a characteristic that earned them their name (Xu et al., 2000) (Figure 1). Structurally, WNK kinases are composed by a relatively small regulatory N-terminal domain, followed by a highly conserved kinase domain (divided in the 12 subdomains characteristic of Ser/Thr kinases), and a large C-terminal domain with regulatory functions conferred by a variety of domains and binding sites for different interacting proteins (McCormick and Ellison, 2011; Gagnon and Delpire, 2012) (Figure 2). WNK1, WNK3, and WNK4 are expressed in a wide variety of tissues, such as heart, brain, lung, liver, muscle, kidney, testis, and colon, among others (Xu et al., 2000; Holden et al., 2004; Kahle et al., 2004; Vitari et al., 2005; Murillo-de-Ozores et al., 2018), while WNK2 is only expressed in brain, heart, and colon (Veríssimo and Jordan, 2001) (Table 2).
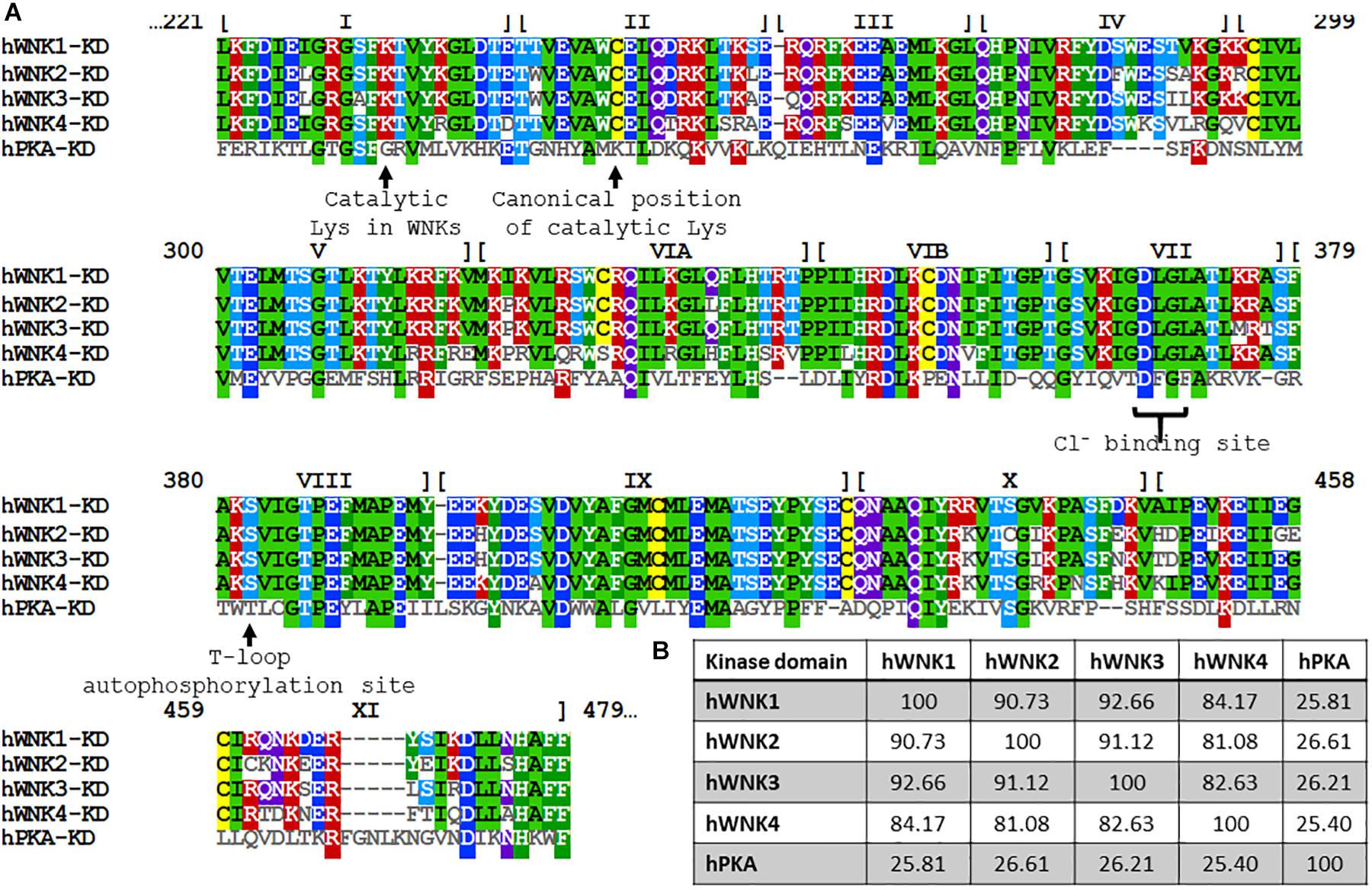
Figure 1. Alignment of the primary structure of kinase domains of human WNKs and the PKA. (A) Amino acid sequence alignment of the kinase domains of human WNK1 (UniProt accession number: Q9H4A3), WNK2 (Q9Y3S1), WNK3 (Q9BYP7), WNK4 (Q96J92) and PKA (P17612). Numbers at the top represent the residue numbers of WNK1. The roman numerals indicate the subdomains within the kinase domain. PKA was included for reference and comparison of functional residues, such as the positioning of catalytic Lys and critical residues involved in Cl– binding in WNKs. (B) Percentage identity among kinase domains is significantly higher than identity among full-length proteins (see Figure 2). Notably, WNK4’s kinase domain is the most divergent one. Alignment and percentage identity matrix were generated in Clustal Omega (EMBL-EBI).
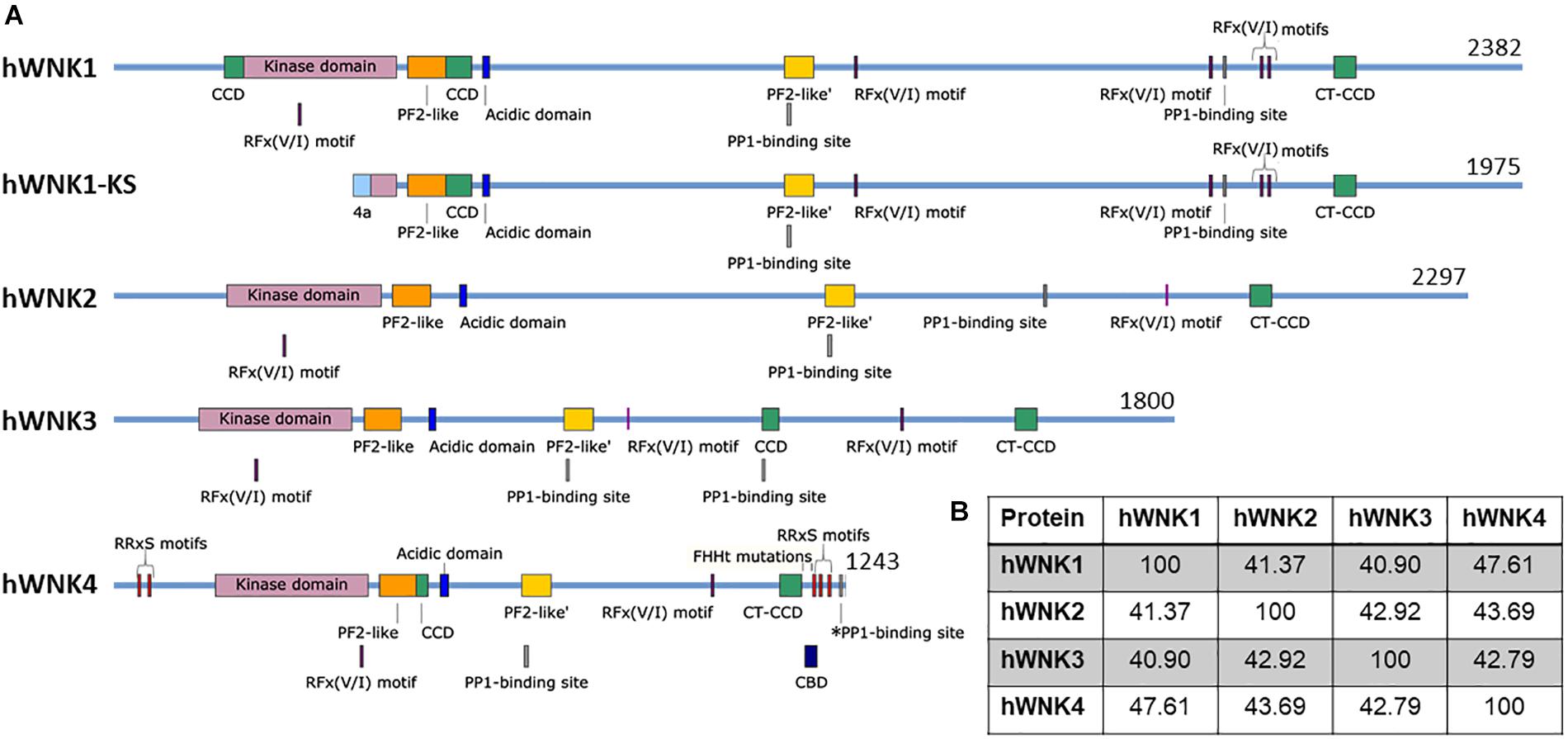
Figure 2. Schematic representation of human WNK kinases. (A) Four genes encoding WNK kinases exist in humans. An alternative promoter gives rise to the kinase-deficient kidney-specific (KS) isoform of WNK1 (Delaloy et al., 2003). Several confirmed and putative domains and binding sites are indicated, such as the kinase domain [Pfam (EMBL-EBI)] (El-Gebali et al., 2019), the PF2-like [Pfam (EMBL-EBI)] and PF2-like’ domains [similar to PF2 (PASK/Fray 2) domains in SPAK and OSR1] (Gagnon and Delpire, 2012), the acidic domain (responsible for interaction with KLHL3 and therefore, WNK degradation) (Ohta et al., 2013), RFx(V/I) motifs which mediate interaction with SPAK/OSR1 (Villa et al., 2007), predicted PP1-binding sites (RVxF motifs) (ELM, Kumar et al., 2020) and a confirmed PP1-binding site in WNK4 (*PP1) (Murillo-de-Ozores et al., 2018), the coiled-coil domains (CCD) (predicted by PCOILS, Gruber et al., 2006), including the C-terminal CCD (CT-CCD) mediating WNK-WNK interaction (Thastrup et al., 2012), the CaM-binding domain (CBD, studied in WNK4 but conserved in other WNKs) (Na et al., 2012), and the RRxS motifs that are phosphorylated by PKC/PKA and/or SGK1 in WNK4 (Rozansky et al., 2009; Na et al., 2012; Castañeda-Bueno et al., 2017). All reported FHHt mutations in WNK4 are located in the acidic domain, with the exception of K1169E (Zhang et al., 2011) and R1185C (Wilson et al., 2001) which are located in its C-terminus, while WNK1 FHHt mutations are intronic deletions that affect gene expression. It is noteworthy that alternative splicing is responsible for producing several isoforms of WNK3 and WNK1 (Holden et al., 2004; Vidal-Petiot et al., 2012), while proteolytic processing produces C-terminally-truncated WNK4 proteins (Murillo-de-Ozores et al., 2018). Finally, it is important to emphasize that some of these sites are based on prediction and experimental evidence will be necessary to assess their particular role. Figures were made in SnapGene software (from Insightful Science; available at snapgene.com). (B) Percent identity among WNK kinases shows the highest similarity between WNK1 and WNK4. Percentage identity matrix was generated in Clustal Omega (EMBL-EBI) (Sievers et al., 2011).

Table 2. Components of the WNK-SPAK/OSR1 pathway: associated genetic diseases, and phenotype of knockout models.
The first evidence linking WNK kinases to the CCCs was the discovery of mutations in the genes WNK1 and WNK4 in Familial Hyperkalemic Hypertension (FHHt), also known as Pseudohypoaldosteronism type 2 (PHAII), or Gordon syndrome (Wilson et al., 2001). FHHt is mainly driven by overactivation of NCC in the distal nephron (Lalioti et al., 2006; Grimm et al., 2017). Further studies showed that WNKs activate N(K)CCs and inhibit KCCs by phosphorylating and activating the downstream kinases SPAK and OSR1 (Moriguchi et al., 2005; Vitari et al., 2005; Alessi et al., 2014).
SPAK and OSR1 are two highly similar Ser/Thr kinases of the Ste20 family, that display wide tissue expression (extensively reviewed by Gagnon and Delpire, 2013) (Table 2). Initially, these kinases were shown to bind the CCCs by a yeast two-hybrid screen (Piechotta et al., 2002) and it was later shown that they are responsible for direct phosphorylation of the CCCs (Dowd and Forbush, 2003; de los Heros et al., 2014). SPAK and OSR1 contain a domain called PF2 in their C-terminal region (also called CCT), that mediates binding with RFx(V/I) motifs in WNK kinases (Villa et al., 2007) and in CCCs (Piechotta et al., 2002). Accordingly, mice with a mutation in SPAK PF2 domain (L502A) display lower SPAK and CCCs phosphorylation levels (Zhang J. et al., 2015). Interestingly, it has been proposed that two regions with a similar tertiary structure exist in WNK kinases themselves (PF2-like and PF2-like’) (Gagnon and Delpire, 2013). Mutation of PF2-like’ in WNK4 prevents SPAK phosphorylation (Murillo-de-Ozores et al., 2018), and although the role of these regions is currently unknown, they might play a role in WNK binding with each other and/or with the CCCs (Moon et al., 2013).
The role of WNK kinases as Cl– sensing proteins was suggested since their initial characterization. It was shown that the reduction in [Cl–]i induced kinase autophosphorylation and activation (Moriguchi et al., 2005). Increased activation and phosphorylation of NKCC1, NKCC2, and NCC by lowering [Cl–]i also suggested that WNK kinases were likely modulated by [Cl–]i (Breitwieser et al., 1990; Lytle and Forbush, 1996; Pacheco-Alvarez et al., 2006; Ponce-Coria et al., 2008).
Conclusive evidence that WNK kinases are Cl–−sensitive proteins came from X-ray crystallography studies performed by Piala et al. (2014). Crystallographic structure of the kinase domain of rat WNK1 showed the presence of a Cl– anion bound directly to the kinase, specifically to the backbone amides of Gly370 and Leu371, located in the N-terminus of the activation loop, with additional hydrophobic interactions with Phe283, Leu299, Leu369, and Leu371 (a Cl– binding pocket structurally similar to the one observed in ClC transporters). Direct Cl– binding stabilizes a WNK1 inactive conformation, while decreased [Cl–] or mutation of the Cl– binding site, by substituting Leu369 for a Phe (L369F), resulted in increased autophosphorylation and activation of WNK1 kinase. Interestingly, as the Cl– binding site is located in the catalytic site of the kinase, it might overlap with the canonical positioning of the catalytic Lys in other kinases. Thus, the unique placement of this Lys in WNKs in subdomain I might permit Cl– binding. Cl–-sensitivity of WNK kinases seems to be a conserved regulatory mechanism, as it has been shown that Cl– also inhibits Drosophila melanogaster WNK (DmWNK) autophosphorylation (Sun et al., 2018).
Reports of WNK4 effect over NCC activity were initially discordant, because evidence showed inhibitory modulation in vitro (Wilson et al., 2003; Yang et al., 2003), while in vivo evidence pointed to WNK4 as an activator of NCC (Castaneda-Bueno et al., 2012). Later, it was described that this discrepancy could be explained by WNK4 regulation by [Cl–]i. Bazua-Valenti et al. (2015) showed that while WNK4 coexpression does not upregulate NCC in basal conditions in Xenopus laevis oocytes, decreasing [Cl–]i promotes WNK4’s activating phosphorylation and WNK4-mediated NCC activation. Mutation of the WNK4 Cl–-binding site (L322F in human WNK4) turned it into a constitutively active kinase that upregulated NCC activity, even without Cl– depletion. These series of experiments not only helped to understand the different effects of WNK4 over NCC function, but also confirmed WNK kinases as key Cl– sensing proteins that regulate the activity of the CCCs.
Analysis of WNK1, WNK3, and WNK4 autophosphorylation upon Cl– depletion in X. laevis oocytes (Bazua-Valenti et al., 2015), as well as in vitro kinase assays (Terker et al., 2015a) have suggested different Cl– sensitivities for these three kinases. While WNK1 and WNK4 autophosphorylation was increased by incubating oocytes in a hypotonic low Cl– media, WNK3 phosphorylation was not affected by this maneuver as it was already phosphorylated in basal conditions (Bazua-Valenti et al., 2015). In vitro kinase assays, incubating the recombinant kinase domains of WNK1, WNK3, or WNK4 with their substrate SPAK in buffers with different [Cl–], showed that WNK4 was inhibited in a lower range of [Cl–]s than WNK1 or WNK3 (Terker et al., 2015a). Coexpression of NCC with Cl–-insensitive mutants of WNK1 (L369F/L371F) and WNK4 (L322F/L324F) dramatically increased NCC phosphorylation when compared to pNCC levels in the presence of their wild type (WT) counterparts. However, WNK3 L295F/L297F did not affect NCC differently from WT WNK3 as this kinase is already active even in cells with high [Cl–]i (∼70 mM in HEK cells and ∼55 mM in oocytes) (Bazua-Valenti et al., 2015; Terker et al., 2015a; Pacheco-Alvarez et al., 2020). These studies suggested that WNK3 activity is independent of [Cl–]i. Thus, although WNK3 can modify [Cl–]i through the regulation of the CCCs, [Cl–]i is not the main regulator of WNK3 activity, as we discuss below.
It is worth mentioning that there are some differences in the specific values of [Cl–] that inhibit WNK activity (Piala et al., 2014; Terker et al., 2015a; Sun et al., 2018). Discrepancies could be due to the use of different substrates’ phosphorylation as a surrogate for WNK activity (such as myelin basic protein, OSR1, SPAK, or WNK itself). Additionally, in vitro assays have been performed using only the kinase domain of WNK kinases, while it is possible that the N- and C-terminal regions could affect Cl– sensitivity. However, in vitro experiments have served as proof of concept to demonstrate that the kinase activity of WNKs can be directly modulated by Cl– binding. This phenomenon has more recently been corroborated in vivo in flies expressing a Cl– insensitive DmWNK (L421F). DmWNK regulates K+-flux in the fly’s Malpighian tubule and the DmWNK-L421F is more active than its WT counterpart (Sun et al., 2018). Moreover, Chen and collaborators generated mice with a Cl– insensitive WNK4 (L319F/L321F). These mice display an altered renal phenotype, reminiscent of FHHt, as shall be explained in a later section, showing that WNK4 is indeed a physiological Cl– sensitive protein (Chen et al., 2019).
These recent findings related to WNKs regulation by [Cl–]i are helping to elucidate how the WNK-SPAK/OSR1-CCCs signaling pathway is involved in diverse physiological processes such as regulation of cell volume (Pacheco-Alvarez et al., 2020), potassium sensing and signaling in the renal distal convoluted tubule (Terker et al., 2015b), and differential neuronal response to GABA (Alessi et al., 2014). These processes will be discussed in the following sections.
Excluding bacterial and plant cells, all other cells are challenged constantly by changes in their volume due to differences in osmotic pressures between the intracellular and the extracellular milieu. Cell volume changes are proportional to the osmotic challenge they face. Water diffuses from the least concentrated solution to the more concentrated one, to equilibrate the osmotic pressure, causing cells to swell or shrink as osmotic balance is restored. However, basal cell volume must be promptly restored to minimize disruption of cellular functions and organization (Hoffmann et al., 2009).
When cells are exposed to hypotonic conditions, the resulting cell swelling triggers the regulatory volume decrease (RVD) response (Figure 3). The early phase of RVD involves activation of ion transporters that mediate K+ and Cl– efflux. Water molecules will follow these ions until cell volume is restored. On the contrary, when cells are exposed to hypertonic conditions, cell shrinkage triggers the regulatory volume increase (RVI) response, which involves intracellular solute accumulation causing the osmotic influx of water and the recovery to normal cell volume. Na+, K+, and Cl– ions influx occurs in the early phase of this response (de los Heros et al., 2018; Delpire and Gagnon, 2018). Transport systems on the membrane are activated within seconds of volume deviation. Transport proteins involved in RVD include the K+-Cl– cotransporters (KCCs), the volume-regulated Cl– channel VRAC, as well as K+ channels. For RVI, the main molecular players involved are the Na+-H+ exchanger and the Na+-K+-2Cl– cotransporter (NKCC1) (Koivusalo et al., 2009). In this section we will focus on the mechanisms for CCCs’ activation and deactivation during cell volume regulation.
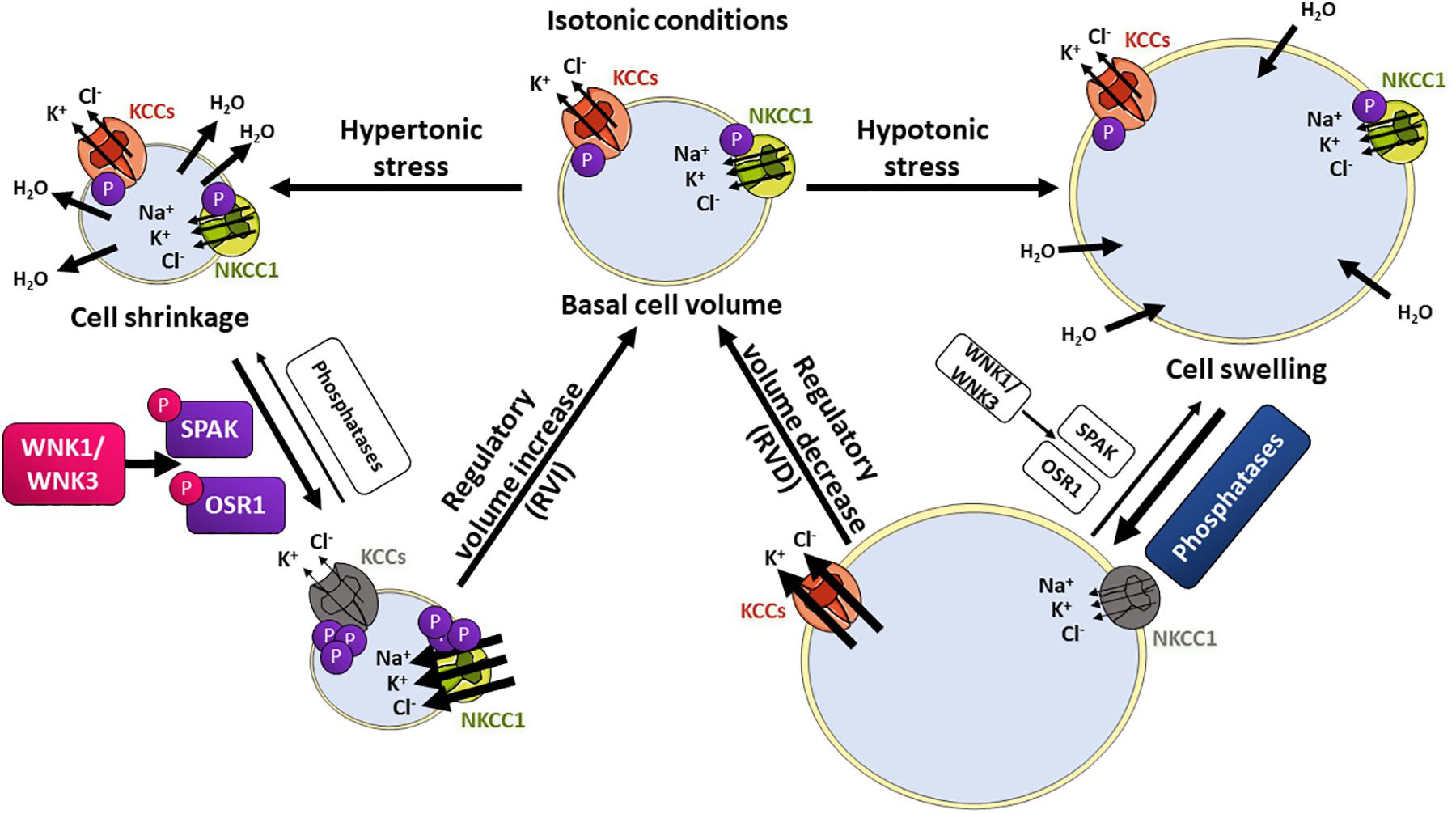
Figure 3. The WNK/SPAK/CCC signaling pathway in cell volume homeostasis. Changes in extracellular osmolarity induce cell shrinkage or swelling. Hypertonic stress with higher extracellular solute concentrations will induce water loss and cell shrinkage. This activates the regulatory volume increase (RVI) response, in which influx of Na+, K+, and Cl– ions is stimulated to restore normal cell volume. On the contrary, lower extracellular solute concentrations (hypotonic stress) will induce water molecules entry and swelling, activating the regulatory volume decrease (RVD) response, in which K+ and Cl– efflux is stimulated, followed by water loss and cell volume restoration. Transport proteins involved in RVI include the NKCC1 (green) cotransporter that is phosphorylated and activated by the SPAK and OSR1 kinases, which in turn are activated by phosphorylation mediated by the volume-sensitive kinases WNK1 and WNK3. The KCC cotransporters (orange) are inhibited by WNK-SPAK/OSR1-mediated phosphorylation. During RVD, KCCs and NKCC1 are dephosphorylated by phosphatases, which results in net K+ and Cl– efflux.
As mentioned above, a key signaling event for modulation of CCCs in response to cell volume changes is the phosphorylation or dephosphorylation of specific residues in their N- and C-terminal tails. For instance, during cell swelling, dephosphorylation of conserved C-terminal threonine residues in KCCs induces their activation, while dephosphorylation of conserved N-terminal residues in NKCC1 inhibits its activity (Lytle and Forbush, 1992; Darman and Forbush, 2002; Rinehart et al., 2009). This results in increased K+-Cl– efflux and decreased Na+ influx, promoting water to follow ion movement. In contrast, during cell shrinkage, increasing phosphorylation of the above-mentioned sites, results in NKCC1 activation and KCCs inhibition, and thus, increased Na+, K+, and Cl– influx, with the consequent influx of water that follows. This reciprocal regulation has led to propose the existence of a volume-sensing enzyme cascade that regulates both NKCCs and KCCs in opposite ways. The identity of the sensors and transducers have been studied and examined, and the WNK-SPAK/OSR1 signaling cascade has emerged as the cascade involved in such regulation, with protein phosphatases playing also an essential regulatory role (de Los Heros et al., 2006; Zagórska et al., 2007; Pacheco-Alvarez et al., 2020).
Different groups have shown that WNK1 activity is stimulated by hypertonic stress. For instance, Xu et al. (2000) and Zagórska et al. (2007) showed that WNK1 immunoprecipitated from HEK293 cells grown in hypertonic media displayed higher levels of autophosphorylation and higher ability to phosphorylate OSR1, respectively. Hypertonic stress promoted WNK1 autophosphorylation at S382, the T-loop’s residue, an event that promotes kinase activation (Xu et al., 2002; Zagórska et al., 2007). Hypertonic stress has also been shown to promote activation of SPAK and OSR1 in cellular models (Chen et al., 2004; Anselmo et al., 2006; Zagórska et al., 2007). Such activation correlates with kinases’ phosphorylation at activating sites targeted by WNK kinases (T-loop and S-motif sites). Additionally, siRNA-mediated WNK1 depletion in HeLa cells has been shown to partially prevent sorbitol-induced phosphorylation and activation of SPAK and OSR1 (Zagórska et al., 2007), suggesting that WNK1 activation in these cells is at least partially responsible for SPAK/OSR1 activation.
Regarding the mechanisms implicated in the modulation of WNK1 activity by hyperosmotic stress, it is interesting to note that Zagórska et al. (2007) observed that the truncated 1-667 WNK1 protein retained the ability of becoming activated by this stimulus, suggesting that the domain or domains involved in kinase activation are located within this region. In a more recent work, Naguro et al. (2012) showed that the MAP3K apoptosis signal-regulated kinase 3 (ASK3) is an osmotic stress-sensitive protein that can bind WNK1 and regulate its T-loop’s phosphorylation. More specifically, they showed that ASK3 becomes phosphorylated and activated when stimulated by hypotonic stress and inhibited by hypertonic stress. They also showed that, when activated by hypotonicity, ASK3 inhibits WNK1 autophosphorylation, because in ASK3 depleted cells, but not in control cells, an increase in WNK1-mediated OSR1 phosphorylation was observed when the cells were subjected to hypotonic stimulation. Given that hypotonic stress promotes [Cl–]i reduction, for example, via activation of the VRAC Cl– channel, an appealing hypothesis is that ASK3 may be important for preventing Cl– depletion-induced WNK1 activation under this condition.
As it was discussed previously, among the WNK family, WNK1 and WNK4 are sensitive to [Cl–]i, while WNK3 constitutes the non-Cl–-sensitive member of the family and it might function as a cell volume-sensitive kinase. Indeed, WNK3 has shown unique biochemical and functional regulatory properties over the CCCs that have led to propose this protein as possible candidate for the volume sensor kinase. Initial characterization of WNK3 in 2005 performed in Xenopus laevis oocytes showed that WNK3 is a potent activator of NKCC1, NKCC2, and NCC, and an inhibitor of KCCs. In contrast, a catalytically inactive version of WNK3 (WNK3-KD) had opposite effects, most likely due to dominant negative effects on the endogenous WNK-SPAK/OSR1 cascade (Kahle et al., 2005; Rinehart et al., 2005; de Los Heros et al., 2006). No other WNK kinase has been shown to possess the ability to regulate all N(K)CCs and KCCs in such manner. It was also shown that WNK3 regulation of KCCs function, depend on phosphatase activity. By using phosphatase inhibitors, protein phosphatase 1 and 2B were proposed to be involved in the signaling pathway.
Experiments performed in HEK293 cells showed that WNK3 has a direct effect on the RVD and RVI volume compensatory mechanisms in mammalian cells. Cruz-Rangel et al. (2012) observed that cells that overexpress WNK3, achieved through stable transfection, showed less efficient RVD, which correlated with lower KCC activity and decreased Cl– efflux. In contrast, cells overexpressing WNK3-KD showed more efficient RVD, as well and higher KCC activity and Cl– efflux. Later on, in 2016, Zhang and coworkers used a combination of screening methods (kinome wide siRNA-screens, kinase inhibitor screen, and a kinase trapping- Orbitrap mass spectrometry screen) that allowed them to identify the major endogenous WNK kinase responsible for stimulating basal KCC3 C-terminal phosphorylation (T991 and T1048) in HEK293 cells (Zhang et al., 2016). WNK3 was identified in all three screening methods, suggesting that WNK3 basal activity is high in these cells. Subsequent targeted knockout experiments in the same model showed that not only WNK3 knockout, but also WNK1 knockout decreased KCC3 and NKCC1 phosphorylation in this model. They showed that HEK293 cells expressing KCC3 and transiently transfected with wild type WNK3 swelled under hypotonic stimulation, while expression of WNK3-KD prevented hypotonic swelling. This effect of WNK3-KD overexpression was reversed in the presence of the KCCs inhibitor furosemide, revealing that increased KCC activity was responsible for the more efficient RVD response in these cells. Finally, treatment of cells expressing wild type WNK3 with the SPAK/OSR1 inhibitor STOCK1S-50699, also prevented cells from swelling under hypotonic stress, demonstrating that inhibition of the WNK3-SPAK/OSR1 pathway promoted RVD. These experiments established the WNK3-SPAK/OSR1 complex as an integral component of the Cl–/volume sensitive kinase system regulating the CCCs.
More recently, using the X. laevis oocytes heterologous expression system we have shown that WNK3 activity towards NCC is not affected neither by changes in [Cl–]i, nor by eliminating the putative Cl– binding sites on the kinase (Pacheco-Alvarez et al., 2020). Instead, the activating phosphorylation of WNK3’s T-loop, is modulated by changes in extracellular osmolarity (increased by hypertonicity and decreased by hypotonicity), whereas phosphorylation of WNK4 in the homologous residue is not affected by such stimuli (Figure 4). In contrast, WNK4 T-loop phosphorylation is stimulated by [Cl–]i depletion. This supports the hypothesis that, at least toward the CCCs, while WNK4 and WNK1 are the Cl– sensitive kinases, WNK3 is instead involved in volume-sensing. Key questions that remain open are whether this kinase can act as the actual cellular osmosensor or whether it is regulated instead by another protein with such activity. Also, whether WNK3 activity modulation by cell volume could also affect other players in the RVI or RVD response remains to be determined.
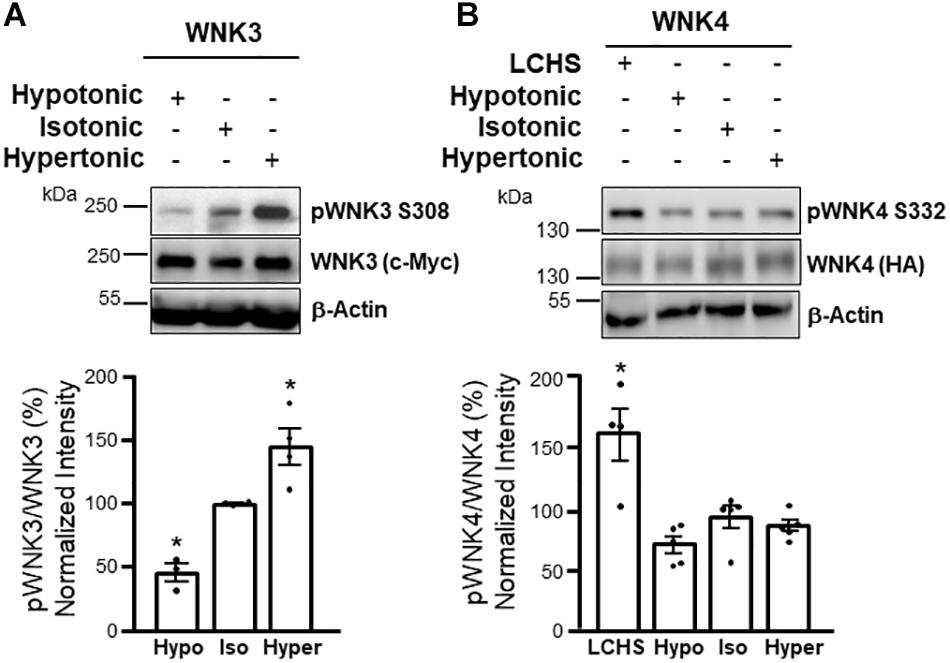
Figure 4. WNK3 phosphorylation is modulated by cell volume changes, while WNK4 phosphorylation is not altered by these stimuli, but responds to [Cl–]i depletion. Western blot assays (upper panels) and corresponding results from densitometric analysis (lower panels) show that T-loop autophosphorylation of WNK3 (S308 in human WNK3) is decreased by incubation of oocytes in hypotonic media, while it is increased by incubation in hypertonic media (A). In contrast, WNK4 T-loop autophosphorylation (S332 in mouse WNK4) is not affected by these maneuvers, but it is increased by low Cl– hypotonic stress (LCHS) that promotes [Cl–]i depletion (B). These results suggest that WNK3 activity primarily responds to changes in cell volume, while WNK4 is mainly regulated by [Cl–]i. *p < 0.05 vs control. Modified from Pacheco-Alvarez et al. (2020) with permission.
In vivo model experiments analyzed the role of the WNK3-SPAK/OSR1-CCC complex in cerebral edema, a condition where volume homeostasis of brain cells is disrupted. It is worth mentioning that increased glial cell volume is the major contributor to cerebral edema, given that in the mammalian brain glia outnumber neurons and also because glia, unlike neurons, express aquaporins that render them more vulnerable to osmotic stress (Kahle et al., 2015). NKCC1 and KCC3 are highly expressed in astrocytes where they participate in cell volume regulation (Pearson et al., 2001; Su et al., 2002).
Middle cerebral artery occlusion (MCAO) was performed to produce brain ischemia, a maneuver that induces cerebral edema, in wild type (WT), WNK3–/–, and SPAK–/– mice (Begum et al., 2015; Zhang et al., 2016; Zhao et al., 2017). In comparison to WT mice, WNK3–/– and SPAK–/– mice showed reduced cerebral edema and infarct volume after MCAO, as well as less demyelination and faster neurobehavioral recovery. Phosphorylation levels of KCC3 and NKCC1 in brain homogenates of WNK3–/– mice after brain ischemia were decreased compared to those in wild type mice (Begum et al., 2015; Zhang et al., 2016). It was suggested that these effects on CCCs phosphorylation could account for the decreased cerebral edema and other improved outcomes. Supporting the key role of SPAK in the regulation of NKCC1, KCC2, and KCC3 activity in brain tissue, it was shown that SPAK-CCT domain knock-in mice (SPAKL502A/L502A), in which SPAK is unable to bind CCCs, have lower levels of phosphorylated NKCC1, KCC2, and KCC3 in brain homogenates. Co-immunoprecipitation experiments of KCC3 with SPAK performed with brain lysates of these mice confirmed that the KCC3-SPAK interaction is disrupted. Therefore, these results place the WNK3-SPAK complex as a “volume sensor-transducer” in mammalian brain that regulates CCC activity to achieve volume homeostasis.
The fine tuning of urinary Na+ and K+ excretion takes place within the mammalian distal nephron of which the distal convoluted tubule (DCT) is the very first segment. The DCT actively participates in Na+, Ca2+, and Mg2+ reabsorption, and thus, its activity has an impact on blood pressure, Ca2+ and Mg2+ homeostasis. NCC constitutes the apical entry pathway for Na+ and Cl– to DCT cells and its activity is the rate-limiting step for NaCl reabsorption in this segment. In addition, even though no net K+ reabsorption or secretion occurs in the DCT, NCC activity has an important impact on renal K+ handling, and thus, renal K+ excretion (Subramanya and Ellison, 2014; Bazúa-Valenti et al., 2016). This is evidenced by the phenotype displayed by patients with Gitelman’s syndrome, a genetic disease caused by inactivating mutations in the SLC12A3 gene that encodes NCC (Simon et al., 1996b). Patients present with hypotension, hypocalciuria, and hypomagnesemia, but also, one of the most notable features is renal K+ loss and hypokalemia. NCC activity affects renal K+ handling by indirectly affecting the activity of the secretory K+ apparatus of the aldosterone sensitive distal nephron (ASDN) comprised by the connecting tubule and the cortical collecting duct. In these segments aldosterone drives K+ secretion by stimulating the concerted action of apical epithelial Na+ channels (ENaC) and Renal Outer Medullary K+ channels (ROMK). Electrogenic Na+ reabsorption via ENaC generates a lumen-negative transepithelial potential that drives K+ secretion via ROMK. Big K+ (BK) channels are also important contributors to K+ secretion under certain conditions (Meneton et al., 2004; Murillo-de-Ozores et al., 2019). The mechanisms explaining NCC’s impact on K+ secretion by the ASDN are currently controversial. It was initially thought that, by affecting distal Na+ delivery, NCC activity could impact on ENaC’s activity, and thus K+ secretion. However, some recent works have failed to confirm this mechanism, and instead, recent data point out to a more complex interaction that involves remodeling of distal tubule segments (Hunter et al., 2014; Grimm et al., 2017).
Whichever the mechanism, the importance that NCC plays on modulation of renal K+ excretion is evidenced by the fact that physiological mechanisms exist to modulate NCC activity in response to changes in dietary K+ intake. NCC activity, assessed by measuring activating phosphorylation (Pacheco-Alvarez et al., 2006), increases in mouse models subjected to low K+ diets and decreases in mouse models subjected to high K+ diets (Vallon et al., 2009; Sorensen et al., 2013; Castaneda-Bueno et al., 2014; Terker et al., 2015a). When this mechanism is broken, alterations in K+ homeostasis occur, like it is observed in Gitelman’s syndrome, caused by loss of function of NCC, or in Familial Hyperkalemic Hypertension (FHHt), which appears to be mainly caused by overactivation of NCC (Wilson et al., 2001; Lalioti et al., 2006). As the disease name indicates, FHHt patients present hypertension with hyperkalemia, as well as hyperchloremic metabolic acidosis. FHHt-causative mutations do not occur in the SLC12A3 gene, but in genes encoding proteins that participate in the regulation of NCC activity. These genes include, as previously discussed, two that encode the WNK kinases WNK1 and WNK4 (Wilson et al., 2001), and two more (CUL3 and KLHL3) that encode components of a protein complex with ubiquitin ligase activity that regulate WNK1 and WNK4 ubiquitylation and degradation (Boyden et al., 2012; Louis-Dit-Picard et al., 2012; Ohta et al., 2013; Shibata et al., 2013; Wakabayashi et al., 2013).
The regulation of [Cl–]i in DCT cells is a key part of the signaling mechanism that mediates regulation of NCC in response to the subtle changes in extracellular K+ concentration ([K+]e) resulting, for example, from variations in dietary K+ content. As explained below in detail, expression of a specific subset of monovalent ion channels in the basolateral membrane of these cells allows translating changes of extracellular K+ levels into changes in membrane potential that in turn drive Cl– fluxes that alter [Cl–]i (Terker et al., 2015b). Such fluctuations in [Cl–]i are sensed by the Cl– sensitive WNK4, the master regulator of the signaling cascade involved in the regulation of NCC (Figure 5). In this section we describe the molecular players involved in this relatively novel signaling mechanism, as well as an overview of the in vitro and in vivo evidence that has been key for the description of this pathway.
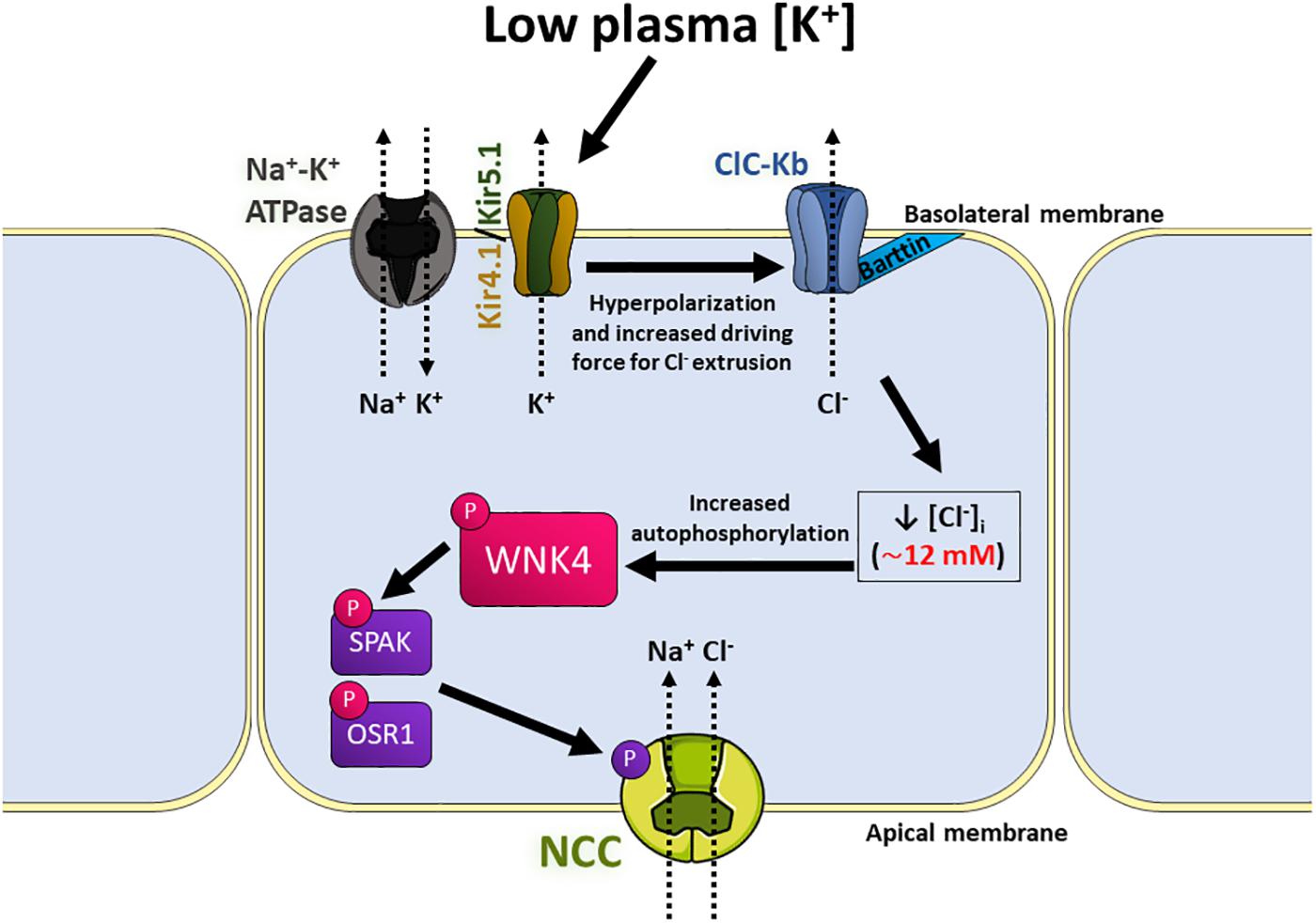
Figure 5. Model of the signaling pathway that activates NCC in response to low extracellular [K+] in the distal convoluted tubule (DCT). Low K+ intake can subtly decrease plasma [K+]. This upregulates the basolateral K+ conductance mediated by Kir4.1/5.1 heterotetramers located in the basolateral membrane of DCT cells, which causes hyperpolarization of membrane potential and increases the driving force for Cl– efflux through the ClC-Kb channels, also located in the basolateral membrane by its association with its β-subunit, Barttin. This process lowers [Cl–]i, and therefore allows dissociation of a Cl– anion from the kinase domain of WNK4, leading to kinase activation and autophosphorylation. Activated WNK4 then phosphorylates the kinases SPAK and OSR1, who phosphorylate and activate NCC, increasing NaCl reabsorption by these cells.
A decrease in NCC phosphorylation (pNCC) levels can be observed within 15 minutes after an acute K+ oral load. This effect parallels the rise in plasma K+ levels and precedes the rise in plasma aldosterone and activation of ENaC. Interestingly, the rapid natriuresis and kaliuresis induced by the K+ load seem to be dependent on the inhibition of NCC, because they are not observed in NCC–/– mice (Sorensen et al., 2013). Decreased pNCC levels are also observed in mice in which hyperkalemia is induced pharmacologically by treatment with amiloride (Terker et al., 2015b) or in genetic mouse models with hyperkalemia, like in mice with specific deletion of ENaC subunits in the nephron (Perrier et al., 2016; Boscardin et al., 2017, 2018). Conversely, in rodent models with hypokalemia, e.g., in hyperaldosteronism models, NCC activation is observed (Kim et al., 1998; Terker et al., 2016). Finally, it has been shown by Terker et al. (2015a) that, across physiological levels of [K+]e, a linear correlation is observed between pNCC levels and [K+]e, and that subtle changes occur in [K+]e in response to modest changes in dietary K+ content, that are responsible for variations in pNCC observed.
Terker et al. (2015b) based on experiments performed in HEK293 cells, proposed that changes in [K+]e modulate pNCC by inducing changes in the [Cl–]i, that in turn affect the activity of the WNK-SPAK/OSR1 pathway. The physiological relevance of this model was supported by ex vivo experiments performed by Penton et al. (2016) in which isolated mice kidneys were perfused or mouse kidney slices were incubated with solutions containing variable [K+]. They confirmed that the [K+]e has a direct effect on pNCC levels in the DCT. Moreover, such effects were not observed when changes in [Cl–]i were prevented. Finally, they showed that whereas the activation of NCC on low [K+] is completely Cl–-dependent, Cl–-independent mechanisms also exist for the high [K+]-induced dephosphorylation of NCC.
Recent works support the idea that K+ channels formed by Kir4.1/5.1 heterotetramers are key for the direct sensing of extracellular [K+] by DCT cells. Inwardly rectifying K+ (Kir) channels are formed by homo- or hetero-tetramers, where each subunit is composed of two transmembrane regions with amino- and carboxyl-terminal regions located in the cytoplasm. These channels are expressed in a wide variety of cell types and are responsible for different functions (extensively reviewed by Hibino et al. (2010), one of which is the maintenance of resting membrane potential (Su and Wang, 2016).
In mouse microdissected DCT tubules, basolateral inwardly rectifying K+ channels identified as Kir4.1/5.1 heterotetramers were characterized by patch-clamp experiments (Lourdel et al., 2002; Zhang et al., 2014), and the expression of these channels was confirmed by RT-PCR (Lourdel et al., 2002). Immunostaining assays have also shown basolateral localization of Kir4.1 (Bockenhauer et al., 2009; Zhang et al., 2014; Cuevas et al., 2017) and Kir5.1 (Tucker et al., 2000; Zhang C. et al., 2015) in the DCT, and proteomic data from microdissected tubules from rats shows high levels of Kir4.1 and Kir5.1 in the DCT (Limbutara et al., 2020). Coincidentally, in vivo interaction between Kir4.1 and Kir5.1 was first found in kidney samples (Tanemoto et al., 2000).
The activity of these channels is key for DCT function. In humans, loss of function mutations in the gene KCNJ10 (encoding Kir4.1) are the cause of EAST/SESAME (epilepsy, ataxia, sensorineural deafness, and renal tubulopathy/seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance) syndrome (Bockenhauer et al., 2009; Scholl et al., 2009), a complex disease characterized, among other manifestations, by hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria, a phenotype reminiscent of Gitelman syndrome. Accordingly, Kir4.1 global knockout mice have reduced levels of NCC (Zhang et al., 2014). As these mice display early lethality, mice with reduced Kir4.1 expression in the kidney (Malik et al., 2018) and kidney-specific-knockout mice (Cuevas et al., 2017) have also been generated and they also display decreased levels of expression and activity of NCC. DCT atrophy is also observed (Saritas et al., 2018).
Evidence supporting the role of the Kir4.1/5.1 heterotetramer in the establishment of membrane potential of DCT cells and its modulation by changes in [K+]e include the following. Genetic disruption of Kcnj10 in mice abolishes the basolateral K+ conductance of DCT cells and promotes depolarization (Zhang et al., 2014; Cuevas et al., 2017). NCC regulation by changes in dietary K+ content is completely blunted in kidney-specific-Kir4.1–/– mice (Cuevas et al., 2017; Wang et al., 2018). Additionally, the activity of Kir4.1/Kir5.1 channels in the DCT has been shown to be modulated by [K+]e, as hyperpolarization and higher basolateral K+ conductance is observed in the DCTs of mice on low K+ diet. In contrast, high K+ intake decreases basolateral K+ currents and depolarizes DCT cells. These phenomena are not observed in cells from kidney-specific Kir4.1–/– mice (Wang et al., 2018). All these findings together have led to the proposal of naming Kir4.1 as the ‘potassium sensor’ of the kidney.
Kir4.1 absence is not compensated by Kir5.1, as this latter subunit alone does not seem to form functional channels on the cell membrane (Pessia et al., 1996; Tanemoto et al., 2005). However, Kir5.1 does play an important role in establishing the sensitivity to [K+]e of DCT cells. The phenotype of Kir5.1–/– mice differs from that of Kir4.1–/– mice. Mice lacking Kir5.1 have higher NCC activity, measured as thiazide-sensitive natriuresis (Paulais et al., 2011), and total and phosphorylated NCC levels (Wu et al., 2019). The DCT cells of these mice display higher basolateral K+ conductance and hyperpolarization compared to DCT cells of WT mice. Regulation of DCT basolateral K+ conductance and levels of pNCC and NCC in response to changes in dietary K+ content was impaired in Kir5.1–/– mice (Wu et al., 2019).
Kir4.1/5.1 heterotetramers have different properties to Kir4.1 homotetramers (Pessia et al., 1996), such as increased intracellular pH sensitivity, as demonstrated by in vitro (Tanemoto et al., 2000; Tucker et al., 2000) and ex vivo experiments (Paulais et al., 2011). It has been suggested that decreased sensibility of Kir4.1 homotetramers to inhibition by H+ ions may explain the higher DCT basolateral K+ conductance of DCT cells on Kir5.1–/– mice (Paulais et al., 2011).
The ClC family comprises nine genes that encode four plasmalemmal Cl– channels (ClC-1, -2, -Ka and -Kb) and five intracellular Cl–-H+ antiporters (ClC-3 to -7) (reviewed extensively by Jentsch and Pusch, 2018). ClC proteins assemble into homodimers, where each subunit mediates ion movement across the membrane.
ClC-Ka and ClC-Kb channels display different characteristics to the rest of the family, as they lack the ‘gating glutamate’ present in other family members and therefore, their voltage dependence is nearly ohmic (Waldegger and Jentsch, 2000). This allows them to mediate transmembrane movement of Cl– over a wide range of membrane voltages, permitting the constant transepithelial transport of Cl– (Jentsch and Pusch, 2018). Additionally, both ClC-Ka and ClC-kB require the presence of the β-subunit Barttin (Estévez et al., 2001), which acts like a chaperone that promotes localization of these channels in the plasma membrane (Waldegger et al., 2002).
In the kidney, ClC-Ka and -Kb channels play a prominent role. Both, human ClC-Ka (named ClC-K1 in mouse and rat) and ClC-Kb (ClC-K2 in mouse and rat) were initially identified and cloned from kidney (Kieferle et al., 1994), although their expression (as well as Barttin’s) is also observed in epithelial cells of the inner ear (Estévez et al., 2001). While the main site of expression of ClC-Ka in the kidney is the thin ascending limb of Henle’s loop, where it plays a role in urine concentration (Matsumura et al., 1999), ClC-Kb is primarily expressed in the basolateral membrane of the thick ascending limb of Henle’s loop (TAL), DCT, and α-intercalated cells of the ASDN, as shown by RT-PCR (Vitzthum et al., 2002), immunostaining (Hennings et al., 2017), and proteomics (Limbutara et al., 2020).
Loss of function mutations in CLCNKB (which encodes ClC-Kb) and BSND (encoding Barttin) are the cause of Bartter syndrome type III (Simon et al., 1997) and type IV (Birkenhager et al., 2001), respectively (see SLC12 section). These types of Bartter often share characteristics with Gitelman’s syndrome, such as normo- or hypocalciuria and blunted response to thiazides (Konrad et al., 2000; Seyberth and Schlingmann, 2011; Cruz and Castro, 2013). This suggests that ClC-Kb and Barttin activities are not only relevant for NKCC2-mediated salt reabsorption in the TAL, but also for NCC-mediated salt reabsorption in the DCT.
Patch-clamp assays performed in microdissected tubules from ClC-K2–/– mice have shown that ClC-K2 constitutes the main basolateral Cl– conductance of TAL and DCT cells. Accordingly, ClC-K2–/– mice have a Bartter type III-like phenotype with severe renal salt and potassium wasting. Furosemide-sensitive, as well as thiazide-sensitive NaCl transport are completely abolished (Grill et al., 2016; Hennings et al., 2017). Decreased levels of total and phosphorylated NCC are also observed (Hennings et al., 2017). While global Barttin–/– mice die a few days after birth because of severe dehydration (Rickheit et al., 2008), hypomorphic mice for Barttin (with low expression levels of a mutated Barttin) are able to thrive and recapitulate a phenotype similar to Bartter syndrome type IV (Nomura et al., 2011). Interestingly, in baseline conditions these mice have similar levels of NCC expression and phosphorylation to WT mice, despite being hypokalemic and hypovolemic, which suggests impaired physiological response of the DCT (Nomura et al., 2018).
As explained in the previous section, changes in [K+]e regulate the membrane potential of the DCT, thanks to the basolateral expression of Kir4.1/5.1 channels. This affects the driving force for Cl– movement through ClC-Kb channels, as suggested by the reduced basolateral Cl– conductance observed in Kir4.1–/– mice (Zhang et al., 2014). Therefore, low [K+]e promotes hyperpolarization, which increases Cl– efflux and decreases [Cl–]i, whereas, increased [K+]e promotes depolarization, lowers Cl– efflux, and increases [Cl–]i (Terker et al., 2015b; Murthy and O’Shaughnessy, 2019). As explained previously in detail, [Cl–]i is an important regulator of the WNK4-SPAK/OSR1 signaling pathway, which ultimately regulates NCC activity. The importance of [Cl–]i as a second messenger that responds to changes in [K+]e and translates them into modulation of NCC activity has been demonstrated in vivo, for example, by showing that hypomorphic Barttin mice do not upregulate NCC in the face of decreased K+ intake (Nomura et al., 2018).
In mice and humans, mutations in WNK4 that cause kinase overexpression are the cause of Familial Hyperkalemic Hypertension (FHHt), a disease that is mainly the consequence of the upregulation of NCC activity (Wilson et al., 2001; Lalioti et al., 2006; Yang et al., 2007; Shibata et al., 2013). WNK4 expression has been reported in different tissues (Kahle et al., 2004; Murillo-de-Ozores et al., 2018) and in different renal cell types (Ohno et al., 2011), although in some reports definitive proof of antibody’s signal specificity by comparison with WNK4–/– samples was lacking. The strictly renal origin of the FHHt phenotype suggests that absence of WNK4 activity in extrarenal tissues can be compensated probably by the activity of other WNK kinases.
Within the DCT WNK4 appears to be the major active WNK kinase. Recent evidence suggests that under basal, physiologic conditions WNK4 and KS-WNK1 (the truncated, kinase inactive version of WNK1) are probably the only WNK kinases expressed in DCT cells. Thus, WNK4 is the only WNK kinase that can phosphorylate SPAK and OSR1 in these cells. For instance, in WNK4–/– mice NCC phosphorylation levels are completely ablated and a Gitelman-like syndrome is developed (Castaneda-Bueno et al., 2012). Accordingly, immunofluorescent staining using an antibody that detects phosphorylation at the S-motif serine of all WNK kinases shows that no signal is observed in kidney sections from WNK4–/– mice (Thomson et al., 2020). This suggests that the catalytically active WNK in DCT is WNK4 and that no other catalytically active WNK kinase becomes activated to compensate for its absence. Additionally, in mice carrying the FHHt mutation R528H in KLHL3 that prevents WNK degradation, knocking down WNK4 completely impairs NCC phosphorylation, even when WNK1 expression levels observed in Western blot (probably KS-WNK1 in DCT and perhaps L-WNK1 in other nephron segments) remain upregulated (Susa et al., 2017).
WNK4 is also essential for low K+-mediated activation of NCC. No upregulation of NCC phosphorylation is observed in WNK4–/– mice (Castaneda-Bueno et al., 2014; Yang et al., 2018) and, consequently, mice develop severe hypokalemia when maintained on a low K+ diet (Castaneda-Bueno et al., 2014).
DCT’s [Cl–]i has been estimated to be relatively low, ranging between 10 and 20 mM (Beck et al., 1988; Boettger et al., 2002; Weinstein, 2005; Terker et al., 2015b). Works by Bazua-Valenti et al. (2015) and Terker et al. (2015a) have shown that WNK4 is more sensitive to inhibition by Cl– than its related kinases WNK1 and WNK3. In in vitro kinase assays performed by Terker et al. (2015a) it was shown that WNK4 activity was inhibited even by the lowest [Cl–] tested which was 10 mM, whereas inhibition of WNK1 and WNK3 only began to be observed when [Cl–] reached 112 and 150 mM, respectively. These observations indicate that indeed WNK4’s Cl– sensitivity lies within the range observed for [Cl–]i in the DCT, and thus, makes it the appropriate WNK to be expressed in this cell type to allow modulation of WNK activity in response to changes in [Cl–]i.
As mentioned before, mutations in the Cl– binding domain of WNK kinases (L369F/L371F and L322F/L324F mutations in human WNK1 and human WNK4, respectively) make them insensitive to Cl– and constitutively active (Piala et al., 2014; Bazua-Valenti et al., 2015; Terker et al., 2015a). Introduction of these mutations in a genetic mouse model has provided the definitive proof that under basal conditions WNK4 is indeed inhibited by Cl– within DCT cells, because these mice display increased NCC phosphorylation and an FHHt-like phenotype (Chen et al., 2019). Interestingly, administering a low K+ diet or an acute K+ load by oral gavage did not promote the expected increase or decrease, respectively, in NCC phosphorylation levels, supporting the idea that modulation of WNK4 kinase activity by intracellular Cl– is behind the signaling mechanism implicated in such regulation.
In cultured HEK293 cells, incubation with low [K+] media promotes an increase in pSPAK/OSR1 levels. This increase is secondary to the intracellular Cl– depletion that is induced by low [K+]e (Terker et al., 2015b). In mice, dietary K+ restriction induces an increase in renal SPAK/OSR1 phosphorylation levels. In DCT, apical localization of SPAK, OSR1, and pSPAK/OSR1 increases, as well as localization in cytoplasmic puncta (WNK bodies) whose formation is induced in conditions that promote pathway activation (Thomson et al., 2020). The increase in renal pSPAK/OSR1 is also observed when kidney slices are incubated on a low [K+] medium, suggesting that a direct effect of extracellular [K+] on DCT cells is implicated (Penton et al., 2016).
SPAK–/– mice and SPAK knockin mice carrying a mutation that prevents phosphorylation of the T-loop’s Thr243 that is essential for kinase’s activation, both display lower levels of expression and phosphorylation of NCC and a Gitelman-like syndrome (Rafiqi et al., 2010; Yang et al., 2010; Lin et al., 2011; McCormick et al., 2011; Grimm et al., 2012). In contrast, kidney specific OSR1–/– mice display normal to higher levels of pNCC (Lin et al., 2011; Ferdaus et al., 2016) and a Bartter-like phenotype with reduced pNKCC2 levels. These observations led to the idea that SPAK mainly participates in NCC regulation, while OSR1 may be more important for NKCC2 regulation. However, even though OSR1 cannot fully compensate to maintain NCC activity in the absence of active SPAK, several observations support the notion that OSR1 is also a physiological modulator of NCC.
First, Terker et al. (2014) showed that OSR1, but not SPAK is essential for β-adrenergic stimulation of NCC. Second, Chiga et al. (2011) showed that the FHHt phenotype of WNK4D561A/+ mice was not fully corrected by inactivation of SPAK (in WNK4D561A/+SPAKT243A/T243A mice). However, inactivation of one copy SPAK and one copy of OSR1 (WNK4D561A/+ SPAKT243A/+ OSR1T185A/+ mice) did normalize blood pressure, plasma [K+], and pNCC levels (Chiga et al., 2011). Third, Ferdaus et al. (2016) showed that, whereas in SPAK–/– mice and kidney specific OSR1–/– mice an increase in pNCC was observed when placed on K+ deficient diet, double knockout mice were unable to upregulate NCC phosphorylation under this condition. Accordingly, plasma [K+] levels were significantly lower in the double mutants than in the single mutants. The severe hypokalemia developed in the double knockouts under dietary K+ restriction was reminiscent to the one observed in WNK4–/– mice on this same condition (Castaneda-Bueno et al., 2014). These results highlight the importance of the WNK4-SPAK/OSR1 signaling pathway for NCC activation and maintenance of K+ homeostasis under K+ deprivation. Finally, supporting this view, Grimm et al. (2017) recently showed that constitutive activation of SPAK exclusively in the DCT is sufficient to develop hyperkalemia, secondary to the activation of NCC.
The [Cl–]i and its regulation by a diverse family of Cl– transporters is a crucial factor affecting GABAergic transmission during brain development and in the mature nervous system. In most neurons, [Cl–]i concentration is largely dependent on the activity of two cotransporters of the CCC family: KCC2 and NKCC1, although KCC3 is also an important cotransporter in the CNS. The activity of these transporters can decrease or increase neuronal [Cl–]i, respectively, and therefore they can alter the polarity (inhibitory or excitatory) and the magnitude of GABAergic transmission. The abnormal function of these transporters can lead to neurologic disorders, including developmental disorders, epilepsy, schizophrenia, and autism. Knowledge of the expression levels and functional regulation of these Cl– transporters in the central nervous system is crucial to understand the basis for Cl– homeostasis under normal and pathological conditions. For reviews see Ben-Ari et al. (2007); Blaesse et al. (2009), Kaila et al. (2014); Titz et al. (2015), Kahle and Delpire (2016), and Ben-Ari (2017).
The ability of neurons in the CNS to inhibit each other is just as important as the ability to excite each other. While several excitatory neurotransmitters exist (glutamate, acetylcholine, ATP etc.), neuronal inhibition is mainly mediated by γ- aminobutyric acid (GABA) and to lesser extent by glycine. GABA binds to Cl–-permeable GABAA receptors (GABAAR) and their resultant activation leads to the opening of the receptor’s ion channel, resulting in Cl– movement. The direction of the ion flux depends on the electrochemical driving force acting on the Cl– ions, which is the difference between the cell’s membrane potential (Vm) and the Cl– equilibrium potential (ECl–). The latter depends on the [Cl–] gradient across the cell membrane. With a [Cl–]i of 8 mM, the ECl– is about -70 mV (Figure 6). This value is often more negative than the neuron resting Vm (Kakazu et al., 1999; Rivera et al., 1999; Yamada et al., 2004; Glykys et al., 2014).
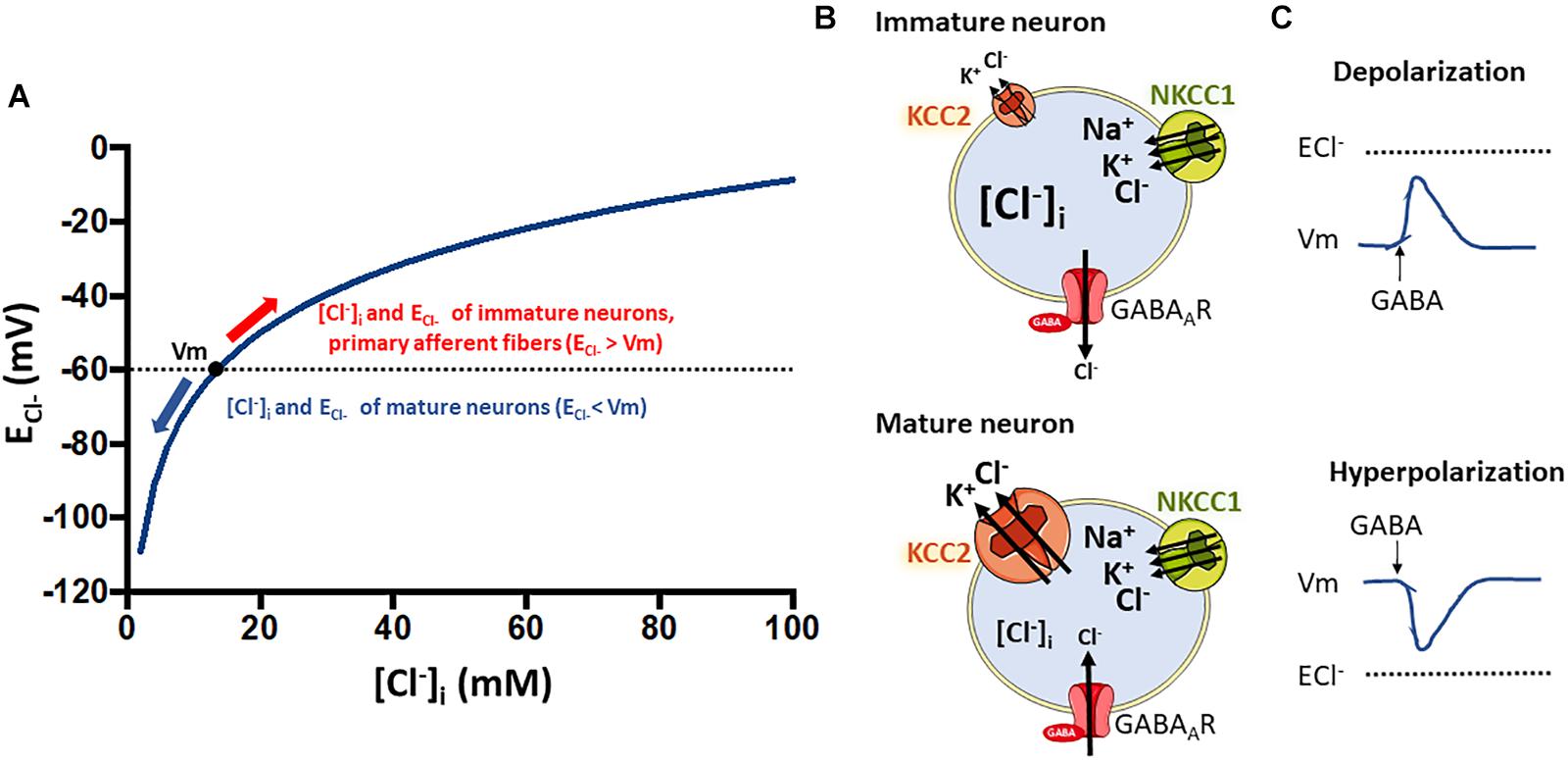
Figure 6. Relationship between intracellular chloride concentration ([Cl–]i) and the equilibrium potential for chloride (ECl–) that determine the type of response to GABA stimulation in neurons. (A) Shows how the ECl– is affected by changes in [Cl–]i according to the Nernst equation. Resting membrane potential in neurons at physiological conditions is around –60 mV. The [Cl–]i of neurons changes along development with higher levels in immature neurons that decrease as they develop into mature cells (Ben-Ari, 2002). This decrease is coupled to the developmental upregulation of KCC2 expression (Rivera et al., 1999). The low KCC2 expression and activity in immature neurons (B), and thus the high NKCC1 to KCC2 activity ratio, is responsible for the observed higher [Cl–]i levels (around 20–40 mM) (Kakazu et al., 1999; Ben-Ari, 2002; Yamada et al., 2004). At these levels of [Cl–]i, ECl– is higher than Vm (A) and GABA stimulation of GABAAR receptors promote Cl– influx and neuronal depolarization (C). Conversely, in mature neurons the upregulation of KCC2 expression decreases the NKCC2 to KCC2 activity ratio (B). This sets the [Cl–]i at a lower value (around 5–12 mM) and thus, ECl– is now lower than the Vm and GABA becomes an inhibitory neurotransmitter. As an exception, primary afferent neurons conserve a high NKCC1 to KCC2 activity ratio all the way through adulthood (Alvarez-Leefmans, 2010). Thus, GABA stimulation of their terminal synapses produce a depolarizing response that is responsible for pre-synaptic inhibition, a mechanism that modulates the input of painful signals from the periphery.
The regulation of neuronal [Cl–]i by CCCs is essential for the normal activity of many neural circuits. In the mature brain, neuronal [Cl–]i is low, and GABA binding to their postsynaptic receptors leads to Cl– influx and post-synaptic hyperpolarization, which moves Vm away from the firing threshold, causing inhibition of excitability. Conversely, in the immature brain, [Cl–]i is significantly higher, so ECl– is more positive than Vm, and GABA produces Cl– efflux, depolarizing responses, and increased excitability by moving Vm closer to the firing threshold (Ben-Ari, 2002).
The transition of GABAA responses, from excitatory in immature neurons and neurons precursors to inhibitory in mature neurons, occurs because [Cl–]i decreases and ECl– shifts in the negative direction due to the high expression of NKCC1 (mediating Cl– influx) and low expression of KCC2 (mediating Cl– efflux) in immature neurons and the strong developmental upregulation of KCC2 in mature ones (Rivera et al., 1999; Ben-Ari et al., 2007). Onset of developmental upregulation of KCC2 seems to be species-specific, for example, occurring postnatally in rats (Rivera et al., 1999), and during the second half of gestation in humans (Vanhatalo et al., 2005; Sedmak et al., 2016) (for review see Kaila et al., 2014). In the mature mammalian nervous system, KCC2 is highly expressed in most central neurons, but absent or expressed at low levels in peripheral neurons and in other nervous cell-types (Payne et al., 1996; Li et al., 2002).
Another example of reversal of GABAergic responses, but that occur in a shorter timescale, has been reported in neurons from the suprachiasmatic nuclei (SCN). The reversal potential of GABAergic postsynaptic currents of these cells (the potential at which GABA responses changes from hyperpolarizing to depolarizing), displays diurnal variations of about 30 mV, suggesting daytime versus nighttime differences of [Cl–]i levels (Irwin and Allen, 2009). Recent works have shown that NKCC1 expression in the SCN of the Syrian hamster is regulated by environmental light and displays circadian changes, suggesting that this may determine GABA polarity in a circadian manner (McNeill et al., 2020).
Similarly, in serotonergic neurons of the dorsal raphe nucleus (DRN), which participate in the sleep-wake cycle, GABAergic inhibition displays circadian variations. At daytime, hyperpolarizing responses to the GABAAR agonist muscimol are larger, and their equilibrium potential more negative compared to those measured at nighttime. Coincidently, the expression of KCC2 (mediating Cl– efflux) is higher during daytime than that during nighttime, with no changes in expression pattern of NKCC1 (mediating Cl– influx). Expression levels of the neuronal NO synthase (nNOS), present in most serotonergic DRN neurons, are higher at daytime than at night-time, and in brain slices treated with the NO donor sodium nitroprusside (SNP) the expression of KCC2, WNK1, WNK2, WNK3, SPAK, and OSR1 in the DRN increased, whereas phosphorylated SPAK decreased. Together, these results suggest that modulation of GABAergic inhibition of wake-inducing DRN neurons during the sleep-wake cycle is regulated by circadian variations in nNOS-derived NO concentration that in turn affect the WNK-SPAK/OSR1-KCC2 signaling (Kim et al., 2018).
KCC2–/– mice, lacking both KCC2a and KCC2b isoforms, exhibit elevated [Cl–]i and GABA-induced neuronal excitation throughout the nervous system (Hubner et al., 2001) (Table 3). They die shortly after birth due to severe motor abnormalities that cause respiratory failure. Brainstem preparations of E18.5 KCC2–/– failed to show respiratory-related motor output of the pre-Bötzinger complex, a cluster of interneurons in the medulla that participate in the generation of respiratory rhythm. Treatment of medullary slices from newborn (P0–P7) WT mice with the KCC2 inhibitor (Dihydroindenyl)oxy alkanoic acid (DIOA) has also been shown to decrease the frequency of the respiration-related rhythmic activity (Okabe et al., 2015). KCC2b–/– mice (the most abundant isoform in the nervous system) exhibit frequent generalized seizures that cause their death between postnatal days 12 and 17 (Woo et al., 2002). In these mice, KCC2a expression is intact (this isoform is produced from an alternative promoter and has an alternative exon 1 that was not targeted by the knockout strategy) and is estimated to represent between 5-10% of the normal total KCC2 expression in the mature brain cortex (Uvarov et al., 2007). Thus, the residual KCC2 activity is thought to explain the slight phenotypic differences between the two knockout models (Gagnon and Delpire, 2013). KCC2b+/– mice show reduced KCC2 expression and can reach adulthood but are prone to suffer epileptic seizures (Woo et al., 2002).
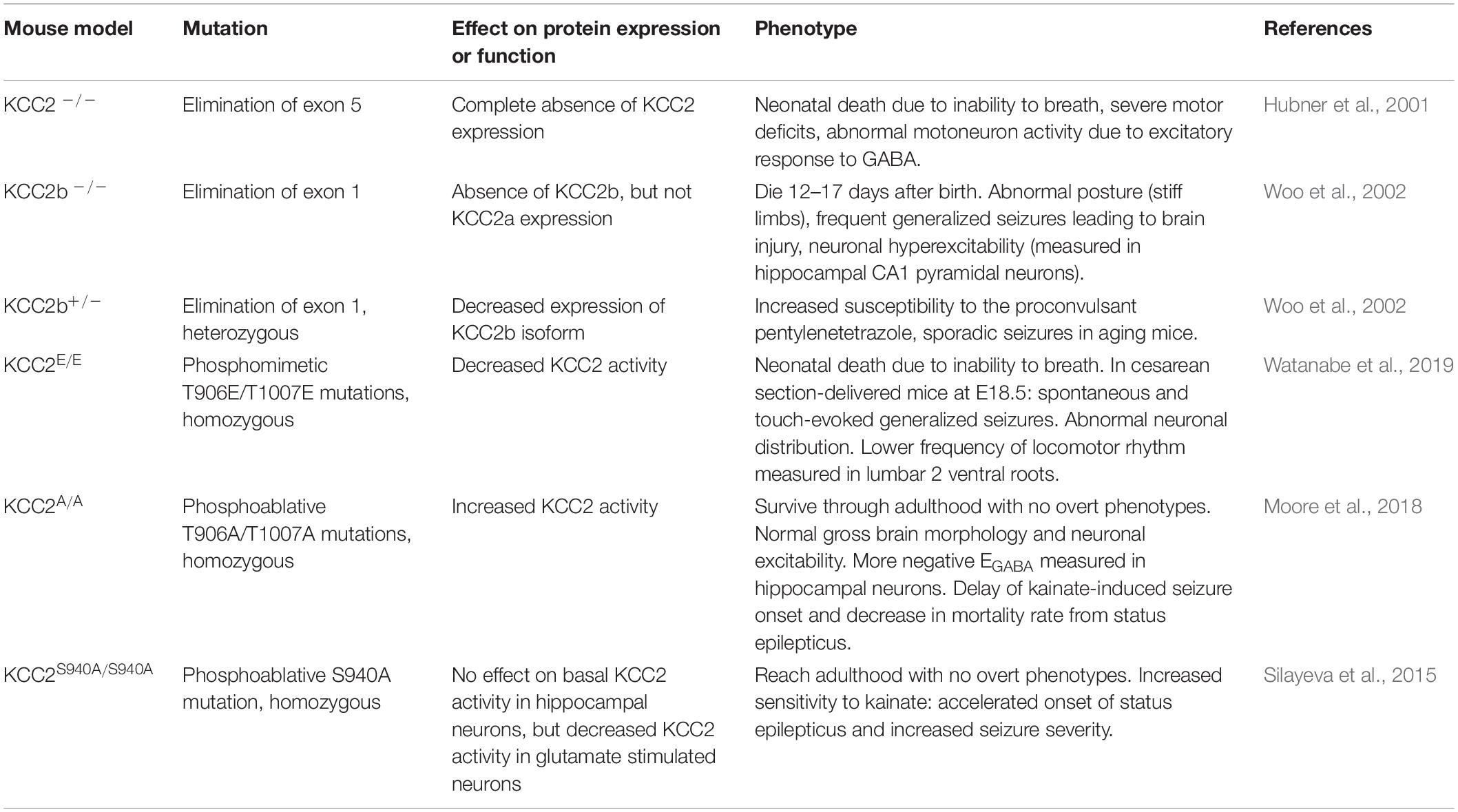
Table 3. Genetically engineered mouse models with mutations in KCC2.
Phosphorylation of KCC2 by the WNK-SPAK/OSR1 downregulates its activity, reducing the rate of Cl– extrusion. Thus, if KCC2 phosphorylation is stimulated, GABAergic inhibition is expected to be weaker or null and the polarity of GABAergic responses could even reverse from inhibitory to excitatory. For instance, KCC2 phosphorylation in Thr906/Thr1007 by WNK1 decreased Cl– extrusion and promoted GABAergic depolarization in cultured mature neurons (Inoue et al., 2012). Moreover, dephosphorylation of KCC2 in brain mouse has been shown to parallel the reversal of GABAergic responses (Friedel et al., 2015; Watanabe et al., 2019).
In the mouse model KCC2E/E that expresses a KCC2 cotransporter harboring the phosphomimetic T906E/T1007E mutations in both alleles, touch-evoked epilepsy, disrupted locomotor rhythmicity, absence of spontaneous respiratory discharges in cervical spinal cord neurons, and early death due to respiratory arrest were reported (Watanabe et al., 2019). It has been shown that the disruption in the developmental switch in polarity of GABAergic transmission can affect normal neuronal proliferation, migration, and dendritic spine maturation (Li et al., 2007; Ben-Ari et al., 2012). Accordingly, KCC2E/E mice presented anomalous neuronal distribution, but dendritic spine morphology was normal.
In contrast, in KCC2T906A/T1007A mice, in which mutations mimic a permanent dephosphorylated (hyperactive) state of KCC2, a reduction in kainate-induced epileptic seizures was observed, possibly due to stronger inhibitory GABAergic synapsis throughout the CNS (Moore et al., 2018). Altogether, these results suggest that adequate KCC2-dependent Cl– extrusion is essential for the correct function of a variety of neuronal circuits, and that its impairment or dysfunction causes inappropriate neuronal locomotor rhythmogenesis and touch-evoked epileptic seizures.
In humans, mutations that indirectly impair KCC2-Ser940 phosphorylation (R952H and R1049C) have been associated with idiopathic epilepsy (Kahle et al., 2014) and familial febrile seizures (Puskarjov et al., 2014). This latter condition associates with abnormal dendritic spine formation. While KCC2 phosphorylation in Thr residues (Rinehart et al., 2009; de los Heros et al., 2014) and Tyr residues (Watanabe et al., 2009; Lee et al., 2010) decrease its membrane availability and rate of ion transport, KCC2 phosphorylation in Ser940, mediated by protein kinase C (PKC), is associated with KCC2 stability in the plasma membrane and increased Cl– transport (Lee et al., 2007). Thus, KCC2 phosphorylation in Ser940 leads to reduced [Cl–]i and stronger GABAergic inhibition (Lee et al., 2007, 2011). In cultured cortical rat neurons, glutamate-mediated NMDA receptor activation triggers Ca2+ influx and PP1 activation that in turn mediates KCC2-Ser940 dephosphorylation (Lee et al., 2011). This leads to diminished Cl– extrusion and weaker GABAergic inhibition. However, if Ser940 dephosphorylation is blocked by treatment of cultured neurons with okadaic acid, the downregulation of KCC2 is prevented and the strength of GABAergic inhibition is unaffected. When Ser940 phosphorylation is prevented in vivo in the KCC2S940A/S940A mouse, no effect is observed on basal Cl– extrusion in hippocampal cultured neurons, but decreased KCC2 activity is observed in glutamate stimulated neurons. Onset of kainate-induced status epilepticus is accelerated and seizure severity is increased in these mice (Silayeva et al., 2015).
In mature neurons in culture, GABAAR activation correlates with increased expression of KCC2 in the plasma membrane and KCC2 dephosphorylation at Thr906/Thr1007 (Heubl et al., 2017). Activation of GABAAR allows Cl– influx that significantly increases [Cl–]i. This in turn, as a homeostatic mechanism, turns off the WNK-SPAK/OSR1 pathway leading to increased membrane KCC2 expression, thus, its activity, favoring Cl– extrusion. The opposite effect occurs when an antagonist blocks GABA transmission and produces activation of the WNK-SPAK cascade. [Cl–]i depletion induces activating phosphorylation of WNK1 at Ser382, SPAK at Ser373, and inactivating phosphorylation of KCC2 at Thr906/Thr1007. This also increases NKCC1 phosphorylation at sites Thr203, Thr207, and Thr212. All these events promote net Cl– influx. Thus, modulation of the WNK-SPAK/OSR1 pathway by [Cl–]i is an important mechanism for restoration of [Cl–]i levels after GABAAR activation or blockade.
The dysregulation of NKCC1 and KCC2 activity with the consequent altered [Cl–]i homeostasis has been related to psychiatric disorders like autism and schizophrenia in which GABA induced inhibition is altered. Rare genetic variants in SLC12A5 (encoding KCC2) that decrease the KCC2-mediated Cl– extrusion have been linked to autism (R952H and R1049C) and schizophrenia (R952H) (Merner et al., 2015) and a gain of function missense variant in SLC12A2 (encoding NKCC1; Y199C) has been reported in a large cohort of schizophrenic patients (Merner et al., 2016). In addition, Hyde et al. (2011) showed that the NKCC1/KCC2 expression ratio in the hippocampal formation of human brains from schizophrenic patients is higher than that observed in non-schizophrenic subjects. Furthermore, post-mortem analyzed brains from schizophrenic patients show higher transcript levels of OXSR1 (coding OSR1 kinase) and WNK3 (Arion et al., 2011). A role of NKCC1 in the pathophysiology of these brain diseases is also evidenced by the beneficial effects observed with bumetanide (an NKCC1 inhibitor) administration (Lemonnier et al., 2016, 2017; Merner et al., 2016; Ben-Ari, 2017; Rahmanzadeh et al., 2017; Kharod et al., 2019; Mollajani et al., 2019; Zhang et al., 2020). In three different clinical trials, bumetanide administered to autistic patients showed significant improvement of the autistic symptoms and signs (Lemonnier et al., 2017; Zhou et al., 2020). These effects correlated with an increase in the GABA/Glutamate synapsis ratio recorded using magnetic resonance microscopy in the human insular cortex (Zhang et al., 2020). Encouraging effects of bumetanide have also been reported in schizophrenic patients (Lemonnier et al., 2016; Merner et al., 2016; Rahmanzadeh et al., 2017).
CCCs are also expressed in neurons of the spinal cord and peripheral nervous system, where they play an important role in the processing of somatosensory information (Kahle et al., 2008; Alvarez-Leefmans, 2010). The observed effects of altered CCC function in different models suggest a particularly important role in the inhibition of spinal nociceptive processing (Sung et al., 2000; Coull et al., 2003; Laird et al., 2004; Tornberg et al., 2005; Gagnon and Delpire, 2013). The peripheral processes of primary sensory neurons (PSN), whose soma is located in the dorsal root ganglia (DRG), collect information from nociceptive receptors throughout the body. Their central processes conduct sensory signals into the dorsal horn of the spinal cord, the first relay in the central nervous system where nociceptive information is integrated and then transmitted to the brain through nociceptive specific projection neurons. Integration of these signals in the brain is necessary for conscious pain perception. GABAergic inhibition is an important mechanism for modulation of pain input from the periphery. Both, primary sensory neurons of the DRG and dorsal horn neurons are subject to GABAergic inhibition (Figure 7). GABA released from synaptic terminals of inhibitory interneurons of the dorsal horn and inhibitory descending fibers can modulate spinal nociceptive processing via two mechanisms: presynaptic inhibition of PSN and post-synaptic inhibition of spinal cord projection neurons (Rudomin and Schmidt, 1999; Alvarez-Leefmans, 2010; Guo and Hu, 2014).
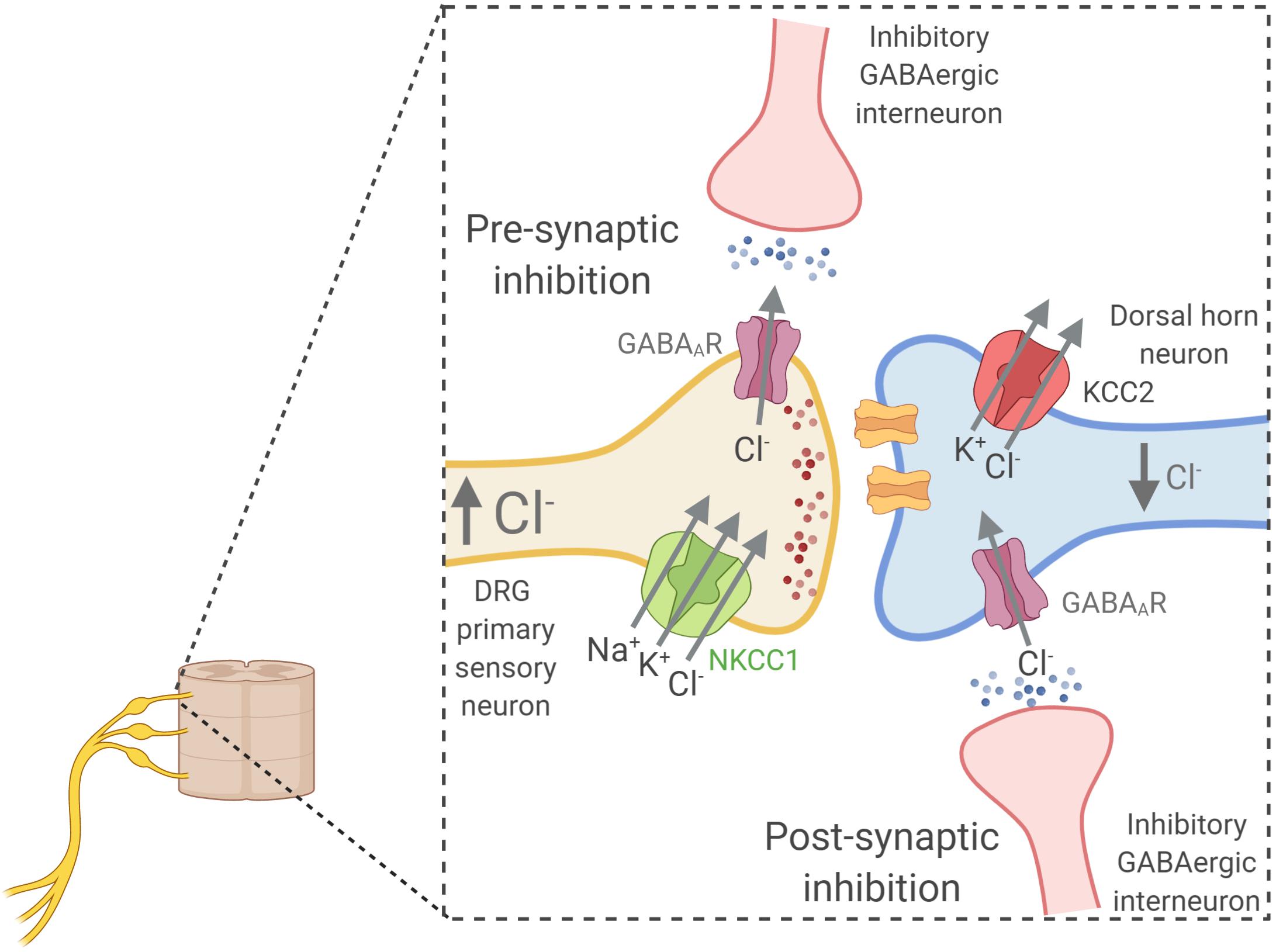
Figure 7. Simplified model of the role of CCCs in the modulation of pain perception. The high NKCC1 to KCC activity ratio in dorsal root ganglia (DRG) primary sensory neurons sets the [Cl–]i at high levels (∼40 mM) (Alvarez-Leefmans, 2010). Thus, in these cells, the ECl– is less negative than the Vm and stimulation of GABAAR located in their synaptic terminals by GABA released from terminals of inhibitory interneurons leads to Cl– efflux and primary afferent depolarization (PAD). PAD inhibits glutamate release preventing excitation of dorsal horn neurons. This mechanism is known as pre-synaptic inhibition. In contrast, dorsal horn neurons, like other CNS neurons, have a low [Cl–]i due in large part to a high KCC2 activity. Thus, GABA released from terminals of inhibitory interneurons stimulate GABAAR on the post-synaptic membranes of dorsal horn neurons and this leads to GABAAR mediated Cl– influx and hyperpolarization of the post synaptic membrane, reducing its excitability. This mechanism is known as post-synaptic inhibition. Pre- and post-synaptic inhibition are particularly important mechanisms for the modulation of spinal nociceptive processing, and thus, altered function of NKCC1 or KCC2 can lead to phenotypes of altered pain perception. Created with BioRender.com.
The pre-synaptic inhibition of PSN involves GABA-induced depolarization of their excitatory synaptic terminals that causes a reduction in neurotransmitter release due to still controversial mechanisms (Guo and Hu, 2014). This phenomenon has been called primary afferent depolarization. The depolarizing effect of GABA is possible because PSN, unlike mature neurons from the CNS, maintain [Cl–]i above electrochemical equilibrium due to a high NKCC1/KCC2 activity ratio (Sung et al., 2000; Alvarez-Leefmans et al., 2001; Price et al., 2006; Rocha-González et al., 2008; Mao et al., 2012). On the other hand, in post-synaptic inhibition, GABAA receptor stimulation in spinal cord neurons induce hyperpolarization and thus reduce their excitability. Like in other mature CNS neurons, [Cl–]i of these neurons is low due to greater KCC2 activity than NKCC1 activity (Price et al., 2005).
Dysregulation of CCCs function in PNS and spinal cord neurons that affect both inhibitory mechanisms has been associated to the pathogenesis of neuropathic pain. Regarding pre-synaptic inhibition, given that the high expression and activity of NKCC1 in DRG neurons is responsible for the high [Cl–]i that facilitates GABA-induced depolarization (Rocha-González et al., 2008; Alvarez-Leefmans, 2010), it has been proposed that the decreased sensitivity to pain observed in NKCC1–/– mice might be explained by absence of pre-synaptic inhibition (Sung et al., 2000; Laird et al., 2004; Gagnon and Delpire, 2013).
On the other hand, altered KCC2 activity in dorsal horn pain neurons has been associated to neuropathic pain related to altered post-synaptic GABAergic inhibition. Mice with reduced expression of KCC2 showed reduced sensitivity to tactile and noxious thermal stimuli (Tornberg et al., 2005). Coull et al. (2003) showed that KCC2 is highly expressed in lamina I dorsal horn pain neurons and that pharmacological blockade or knockdown of spinal KCC2 in rats reduced the nociceptive threshold. Additionally, in the model of neuropathic pain induced by peripheral nerve injury Coull et al. (2003) observed a reduction in KCC2 expression in lamina I neurons that altered the ECl– (making it less negative) and shifted the response to GABA stimulation from inhibitory to excitatory. Nomura et al. (2006) also showed a decrease in KCC2 expression in lamina I and lamina II neurons in a model of tissue injury-induced inflammatory pain. Thus, loss of post-synaptic inhibition due to KCC2 dysregulation seems to be a common mechanism underlying neuropathic and inflammatory pain. Supporting this, it has been shown that the CLP257 compound that restores Cl– transport and rescues KCC2 membrane expression in the dorsal horn following nerve injury, normalized stimulus-evoked responses in spinal nociceptive pathways and alleviated hypersensitivity (Gagnon et al., 2013).
Altered regulation of KCC2 activity has also been implicated in Hereditary Sensory and Autonomic Neuropathy type 2 (HSAN2, OMIM 201300). Patients with HSAN2 suffer from severe sensory loss of heat, touch or pain perception with a partial loss of peripheral sensory nerves (Rahmani et al., 2018). This is an autosomal recessive Mendelian disease caused by mutations in the HSN2 exon of WNK1 (Shekarabi et al., 2008; Pacheco-Cuellar et al., 2011). Interestingly, the case of a female patient has been reported who is a compound heterozygote for a 1 bp deletion in the HSN2 exon and a 2 bp deletion in exon 6 of the WNK1 gene (Shekarabi et al., 2008). The WNK1 gene encodes different WNK1 isoforms due to tissue-specific alternative splicing (Vidal-Petiot et al., 2012). Transcripts containing the HSN2 exon (located between exons 8 and 9) have been shown to be abundant in the dorsal horn, DRG, and peripheral nerves (Vidal-Petiot et al., 2012; Shekarabi et al., 2013). In a mouse model in which HSN2-containing WNK1 transcripts are absent due to the introduction of loxP recombination sites flanking this exon, the HASN2 phenotype is not fully recapitulated, but the mice show reduced pain hypersensitivity after peripheral nerve injury (PNI) (Kahle et al., 2016). PNI increased WNK1-HSN2 expression and KCC2 phosphorylation (at Thr906 and Thr1007) in spinal cord homogenates of wild-type mice, but this increase was blunted in WNK1ΔHSN2/ΔHSN2 mice. Accordingly, the reversal potential for GABA-induced currents (EGABA) measured in lamina II neurons of spinal cord slices was less negative in slices from WT mice with PNI than in those from sham operated mice. EGABA was restored to more negative values by incubation of slices with the WNK-SPAK pathway inhibitor STOCK1S-50699. Moreover, EGABA of lamina II neurons from WNK1ΔHSN2/ΔHSN2 mice was not altered after PNI. Thus, the PNI-induced increase in KCC2 phosphorylation is dependent on WNK1-HSN2 activity and the resulting decrease in KCC2 activity promotes an increase in [Cl–]i of spinal cord neurons that shifts the EGABA to more positive values.
Finally, there is a growing interest in understanding the importance of GABAergic transmission in regulating body weight balance (Tong et al., 2008; Kim et al., 2015; Sohrabipour et al., 2018). GABAergic transmission in hypothalamic areas and in the brainstem participates in a neuroendocrine network that modulates caloric intake, energy expenditure, and thermogenesis according to the level of food intake (Pigeyre et al., 2016). Neurons in the arcuate nucleus (ARC) express leptin receptors (LepR), whose activation triggers GABAergic inhibition by ARC neurons of PVN neurons (Kong et al., 2012). This in turn relieves the tonic inhibition that PVN neurons exert on brown adipose tissue (BAT), thus increasing energy expenditure through BAT-dependent thermogenesis. The effects of leptin on energy expenditure and thermogenesis are mediated by RIP neurons of the ARC (Tong et al., 2008; Vong et al., 2011; Kim et al., 2015). In mice in which GABA production is specifically impaired in RIP neurons, decreased oxygen consumption and BAT activity was observed, and they gained more body weight and fat mass than their wild type littermates when placed on a high fat diet (Kong et al., 2012). Given that in most neurons a key determinant of [Cl–]i, and thus of GABAergic response, is the NKCC1/KCC2 activity ratio, it is likely that this mechanism plays also an important role in GABA-sensitive neurons of the PVN. Interestingly, we have recently observed that mice expressing an inactive form of SPAK that is unable to phosphorylate NKCC1 and KCC2 (SPAKT243A/T243A mice), are resistant to developing obesity when placed on a high-fat diet despite similar levels of food intake (Torre-Villalvazo et al., 2018). These mice show higher energy expenditure, reflected by an increase in thermogenesis in the brown adipose tissue, a higher muscle mitochondrial activity, and a lower hepatic steatosis than their wild-type littermates. Thus, it is possible that this phenotype could be related to altered [Cl–]i and response to GABA in PVN neurons.
Even though WNK kinases direct regulation by the anion Cl– is clear now, it is likely that alternate mechanisms by which cells are able to respond to changes in [Cl–]i exist, given that WNK kinases are not found in Bacteria and Archaea domains (Cao-Pham et al., 2018). While mammalian cells (Eagle, 1956) and some species of bacteria require Cl– in order to grow (MacLeod and Onofrey, 1957; Roeßler and Müller, 1998), some other species of bacteria require Cl– in order to adapt to different conditions in the environment, such as acidity or hyperosmolarity (Jordan and Davies, 2001; Roeßler et al., 2003). Cl– also plays a role in signal transduction in bacteria, as it can modulate gene expression (Roeßler and Müller, 2002; Sewald et al., 2007) and enzymatic activity (Gut et al., 2006).
Accordingly, several studies have reported different genes whose expression is regulated in response to changes in [Cl–]i in eukaryotes, such as SCNN1A (Niisato et al., 2004), SCNN1B, SCNN1C (Niisato et al., 2007), COX2 (Cheng et al., 2000), GLRX5 and RPS27 (Valdivieso et al., 2016). GABAAR receptor subunits levels are also regulated by [Cl–]i (Succol et al., 2012), The detailed mechanisms responsible for these different phenomena are still unknown, even though the kinase p38 might be involved in the regulation of SCNN1B, SCNN1G (Niisato et al., 2007) and COX2 (Cheng et al., 2000). Interestingly, the transcription factor RUNX1 has been shown to directly bind Cl– ions (Bäckström et al., 2002), but further work will be necessary to elucidate its role in the context of intracellular Cl– handling, or if there are other transcription factors that respond directly or indirectly to changes in [Cl–]i.
Finally, it is not clear if other kinases might function as direct Cl– sensors. While the phosphorylation of different MAP kinases, such as JNK, p38, and MEK6 is increased in response to decreased [Cl–]i (Ohsawa et al., 2010; Wu et al., 2016), it is still unknown if these proteins are able to directly bind Cl– anions, or whether its activation depends on upstream activators that sense [Cl–]i. In the case of the kinase SGK1, in vitro kinase activity assays suggest that SGK1 can be directly activated by increasing [Cl–]i, as incubating recombinant SGK1 with increasing concentrations of NaCl or KCl (but not Na-gluconate or K-gluconate) promotes its phosphorylation (Zhang et al., 2018). Further analysis will be helpful to determine if SGK1 is indeed able to directly bind Cl– anions. Additionally, the opposite regulation of WNK kinases and SGK1 by Cl– is puzzling, and it will be an interesting avenue for future investigations, especially regarding epithelial physiology, where both kinases play important roles in fluid secretion and intracellular signaling.
In the present manuscript we reviewed the role of the Cl– anion as a second messenger participating in the modulation of several physiological processes, specifically by regulating the activity of WNK kinases and their downstream signaling pathway, comprising the kinases SPAK and OSR1, as well as the cation-coupled Cl– cotransporters (CCCs). Dynamic activation and inactivation of these kinases modulate the phosphorylation, and therefore activity of the CCCs. While initial description of the physiological roles of some of these proteins was facilitated by their association to genetic diseases, further investigations have shed additional light about their role in physiological and pathophysiological processes. For example, regulation of CCCs is relevant in maintaining normal cell volume in response to changes in extracellular osmolarity. In particular, the WNK3-SPAK complex seems to play an important role in cell volume regulation within the brain. Moreover, a complex [Cl–]i-sensitive signaling pathway involving basolateral ion channels and the kinases WNK4, SPAK, and OSR1 is responsible for NCC regulation by physiological plasma [K+], an essential process in the homeostatic modulation of renal K+ excretion. Finally, the WNK-SPAK/OSR1-CCC pathway has also been described to modulate GABAergic neuronal responses, where baseline [Cl–]i dictates the direction of Cl– currents elicited by binding of GABA to its receptor.
AM-d-O, MC-C, PH, GG, and MC-B wrote the manuscript, made the figures, edited the manuscript, revised and approved the final version. All the authors contributed to the article and approved the submitted version.
AM-d-O is a graduate student from the “Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM)” and received a fellowship 606808 from CONACYT. The work in the researcher’s lab is possible due to grant support no. DK51496 from NIH to GG, A1-S-8290, 283555, from Conacyt Mexico to GG and PH, respectively, IN201519, IN222320, IA203620, from PAPIIT UNAM to GG, PH, and MC-C, respectively, and RA202718 from Loreal L’Oréal–UNESCO-AMC-CONALMEX “For Women in Science, 2019” to MC-C.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alessi, D. R., Zhang, J., Khanna, A., Hochdorfer, T., Shang, Y., and Kahle, K. T. (2014). The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci. Signal. 7:re3. doi: 10.1126/scisignal.2005365
Alvarez-Leefmans, F. J. (2010). “Chloride transporters in presynaptic inhibition, pain and neurogenic inflammation,” in Physiology and Pathology of Chloride Transporters and Channels in the Nervous System: From Molecules to Diseases, eds F. J. Alvarez-Leefmans and E. Delpire (Amsterdam: Elsevier Inc), 439–470. doi: 10.1016/B978-0-12-374373-2.00022-4
Alvarez-Leefmans, F. J., León-Olea, M., Mendoza-Sotelo, J., Alvarez, F. J., Antón, B., and Garduo, R. (2001). Immunolocalization of the Na+-K+-2Cl- cotransporter in peripheral nervous tissue of vertebrates. Neuroscience 104, 569–582. doi: 10.1016/S0306-4522(01)00091-4
Anselmo, A. N., Earnest, S., Chen, W., Juang, Y.-C., Kim, S. C., Zhao, Y., et al. (2006). WNK1 and OSR1 regulate the Na+, K+, 2Cl- cotransporter in HeLa cells. Proc. Natl. Acad. Sci. U.S.A. 103, 10883–10888. doi: 10.1073/pnas.0604607103
Arion, D., and Lewis, D. A. (2011). Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch. Gen. Psychiatry 68, 21–31. doi: 10.1001/archgenpsychiatry.2010.114
Arroyo, J. P., Kahle, K. T., and Gamba, G. (2013). The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol. Aspects Med. 34, 288–298. doi: 10.1016/j.mam.2012.05.002
Bäckström, S., Wolf-Watz, M., Grundström, C., Härd, T., Grundström, T., and Sauer, U. H. (2002). The RUNX1 Runt domain at 1.25 Å resolution: a structural switch and specifically bound chloride ions modulate DNA binding. J. Mol. Biol. 322, 259–272. doi: 10.1016/S0022-2836(02)00702-7
Bazúa-Valenti, S., Castañeda-Bueno, M., and Gamba, G. (2016). Physiological role of SLC12 family members in the kidney. Am. J. Physiol. Ren. Physiol. 311, F131–F144. doi: 10.1152/ajprenal.00071.2016
Bazua-Valenti, S., Chavez-Canales, M., Rojas-Vega, L., Gonzalez-Rodriguez, X., Vazquez, N., Rodriguez-Gama, A., et al. (2015). The effect of WNK4 on the Na+-Cl- cotransporter is modulated by intracellular chloride. J. Am. Soc. Nephrol. 26, 1781–1786. doi: 10.1681/ASN.2014050470
Beck, F.-X., Dörge, A., Rick, R., Schramm, M., and Thurau, K. (1988). The distribution of potassium, sodium and chloride across the apical membrane of renal tubular cells: effect of acute metabolic alkalosis. Pflügers Arch. Eur. J. Physiol. 411, 259–267. doi: 10.1007/BF00585112
Begum, G., Yuan, H., Kahle, K. T., Li, L., Wang, S., Shi, Y., et al. (2015). Inhibition of WNK3 kinase signaling reduces brain damage and accelerates neurological recovery after stroke. Stroke 46, 1956–1965. doi: 10.1161/STROKEAHA.115.008939
Ben-Ari, Y. (2002). Excitatory actions of gaba during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. doi: 10.1038/nrn920
Ben-Ari, Y. (2017). NKCC1 chloride importer antagonists attenuate many neurological and psychiatric disorders. Trends Neurosci. 40, 536–554. doi: 10.1016/j.tins.2017.07.001
Ben-Ari, Y., Gaiarsa, J.-L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Ben-Ari, Y., Khalilov, I., Kahle, K. T., and Cherubini, E. (2012). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486. doi: 10.1177/1073858412438697
Birkenhager, R., Otto, E., Schurmann, M. J., Vollmer, M., Ruf, E. M., Maier-Lutz, I., et al. (2001). Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 29, 310–314. doi: 10.1038/ng752
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838. doi: 10.1016/j.neuron.2009.03.003
Bockenhauer, D., Feather, S., Stanescu, H. C., Bandulik, S., Zdebik, A. A., Reichold, M., et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 360, 1960–1970. doi: 10.1056/NEJMoa0810276
Boettger, T., Hubner, C., Maier, H., Rust, M., Beck, F., and Jentsch, T. (2002). Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nat. Lett. 416, 13445–13452. doi: 10.1038/416874a
Boettger, T., Rust, M. B., Maier, H., Seidenbecher, T., Schweizer, M., Keating, D. J., et al. (2003). Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 22, 5422–5434. doi: 10.1093/emboj/cdg519
Boscardin, E., Perrier, R., Sergi, C., Maillard, M., Loffing, J., Loffing-Cueni, D., et al. (2017). Severe hyperkalemia is rescued by low-potassium diet in renal βENaC-deficient mice. Pflugers Arch. Eur. J. Physiol. 469, 1387–1399. doi: 10.1007/s00424-017-1990-2
Boscardin, E., Perrier, R., Sergi, C., Maillard, M. P., Loffing, J., Loffing-Cueni, D., et al. (2018). Plasma potassium determines NCC abundance in adult kidney-specific γ ENaC Knockout. J. Am. Soc. Nephrol. 29, 977–990. doi: 10.1681/ASN.2017030345
Boyden, L. M., Choi, M., Choate, K. A., Nelson-Williams, C. J., Farhi, A., Toka, H. R., et al. (2012). Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482, 98–102. doi: 10.1038/nature10814
Breitwieser, G. E., Altamirano, A. A., and Russell, J. M. (1990). Osmotic stimulation of Na(+)-K(+)-Cl- cotransport in squid giant axon is [Cl-]i dependent. Am. J. Physiol. Physiol. 258, C749–C753. doi: 10.1152/ajpcell.1990.258.4.C749
Cao-Pham, A. H., Urano, D., Ross-Elliott, T. J., and Jones, A. M. (2018). Nudge-nudge, WNK-WNK (kinases), say no more? New Phytol. 220, 35–48. doi: 10.1111/nph.15276
Castañeda-Bueno, M., Arroyo, J. P., Zhang, J., Puthumana, J., Yarborough, O., Shibata, S., et al. (2017). Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc. Natl. Acad. Sci. U.S.A. 114, E879–E886. doi: 10.1073/pnas.1620315114
Castaneda-Bueno, M., Cervantes-Perez, L. G., Rojas-Vega, L., Arroyo-Garza, I., Vazquez, N., Moreno, E., et al. (2014). Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am. J. Physiol. Ren. Physiol. 306, F1507–F1519. doi: 10.1152/ajprenal.00255.2013
Castaneda-Bueno, M., Cervantes-Perez, L. G., Vazquez, N., Uribe, N., Kantesaria, S., Morla, L., et al. (2012). Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc. Natl. Acad. Sci. U.S.A. 109, 7929–7934. doi: 10.1073/pnas.1200947109
Chang, H., Tashiro, K., Hirai, M., Ikeda, K., Kurokawa, K., and Fujita, T. (1996). Identification of a cDNA encoding a thiazide-sensitive sodium-chloride cotransporter from the human and its mRNA expression in various tissues. Biochem. Biophys. Res. Commun. 223, 324–328. doi: 10.1006/bbrc.1996.0893
Chen, J.-C., Lo, Y.-F., Lin, Y.-W., Lin, S.-H., Huang, C.-L., and Cheng, C.-J. (2019). WNK4 kinase is a physiological intracellular chloride sensor. Proc. Natl. Acad. Sci. U.S.A. 116, 4502–4507. doi: 10.1073/pnas.1817220116
Chen, W., Yazicioglu, M., and Cobb, M. H. (2004). Characterization of OSR1, a member of the mammalian Ste20p/Germinal center kinase subfamily. J. Biol. Chem. 279, 11129–11136. doi: 10.1074/jbc.M313562200
Cheng, H., Wang, J., Zhang, M., McKanna, J., and Harris, R. (2000). Role of p38 in the regulation of renal cortical cyclooxygenase-2 expression by extracellular chloride. J. Clin. Invest. 106, 681–688. doi: 10.1172/JCI10318
Chiga, M., Rafiqi, F. H., Alessi, D. R., Sohara, E., Ohta, A., Rai, T., et al. (2011). Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J. Cell Sci. 124, 1391–1395. doi: 10.1242/jcs.084111
Coull, J. A. M., Boudreau, D., Bachand, K., Prescott, S. A., Nault, F., Sík, A., et al. (2003). Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942. doi: 10.1038/nature01868
Cruz, A. J., and Castro, A. (2013). Gitelman or bartter type 3 syndrome? A case of distal convoluted tubulopathy caused by CLCNKB gene mutation. Case Rep. 2013:bcr2012007929 doi: 10.1136/bcr-2012-007929
Cruz-Rangel, S., Gamba, G., Ramos-Mandujano, G., and Pasantes-Morales, H. (2012). Influence of WNK3 on intracellular chloride concentration and volume regulation in HEK293 cells. Pflugers Arch. Eur. J. Physiol. 464, 317–330. doi: 10.1007/s00424-012-1137-4
Csanády, L., Vergani, P., and Gadsby, D. C. (2019). Structure, gating, and regulation of the CFTR anion channel. Physiol. Rev. 99, 707–738. doi: 10.1152/physrev.00007.2018
Cuevas, C. A., Su, X.-T., Wang, M.-X., Terker, A. S., Lin, D.-H., McCormick, J. A., et al. (2017). Potassium sensing by renal distal tubules requires Kir4.1. J. Am. Soc. Nephrol. 28, 1814–1825. doi: 10.1681/ASN.2016090935
Darman, R. B., and Forbush, B. (2002). A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter. NKCC1. J. Biol. Chem. 277, 37542–37550. doi: 10.1074/jbc.M206293200
de los Heros, P., Alessi, D. R., Gourlay, R., Campbell, D. G., Deak, M., Macartney, T. J., et al. (2014). The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+–Cl- co-transporters. Biochem. J. 458, 559–573. doi: 10.1042/BJ20131478
de Los Heros, P., Kahle, K. T., Rinehart, J., Bobadilla, N. A., Vázquez, N., San Cristobal, P., et al. (2006). WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 1976–1981. doi: 10.1073/pnas.0510947103
de los Heros, P., Pacheco-Alvarez, D., and Gamba, G. (2018). Role of WNK kinases in the modulation of cell volume. Curr. Top. Membr. 81, 207–235. doi: 10.1016/bs.ctm.2018.08.002
Delaloy, C., Lu, J., Houot, A.-M., Disse-Nicodeme, S., Gasc, J.-M., Corvol, P., et al. (2003). Multiple promoters in the WNK1 gene: one controls expression of a kidney-specific kinase-defective isoform. Mol. Cell. Biol. 23, 9208–9221. doi: 10.1128/MCB.23.24.9208-9221.2003
Delpire, E., and Gagnon, K. B. (2018). Water homeostasis and cell volume maintenance and regulation. Curr. Top. Membr. 81, 3–52. doi: 10.1016/bs.ctm.2018.08.001
Delpire, E., Lu, J., England, R., Dull, C., and Thorne, T. (1999). Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 22, 192–195. doi: 10.1038/9713
Delpire, E., and Mount, D. B. (2002). Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu. Rev. Physiol. 64, 803–843. doi: 10.1146/annurev.physiol.64.081501.155847
Delpire, E., Rauchman, M. I., Beier, D. R., Hebert, S. C., and Gullans, S. R. (1994). Molecular cloning and chromosome localization of a putative basolateral Na+-K+-2Cl- cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J. Biol. Chem. 269, 25677–25683.
Delpire, E., Wolfe, L., Flores, B., Koumangoye, R., Schornak, C. C., Omer, S., et al. (2016). A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Mol. Case Stud. 2:a001289. doi: 10.1101/mcs.a001289
Dixon, M. J., Gazzard, J., Chaudhry, S. S., Sampson, N., Schulte, B. A., and Steel, K. P. (1999). Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum. Mol. Genet. 8, 1579–1584. doi: 10.1093/hmg/8.8.1579
Dowd, B. F. X., and Forbush, B. (2003). Pask (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1). J. Biol. Chem. 278, 27347–27353. doi: 10.1074/jbc.M301899200
Dvorak, M. M., De Joussineau, C., Carter, D. H., Pisitkun, T., Knepper, M. A., Gamba, G., et al. (2007). Thiazide diuretics directly induce osteoblast differentiation and mineralized nodule formation by interacting with a sodium chloride co-transporter in bone. J. Am. Soc. Nephrol. 18, 2509–2516. doi: 10.1681/ASN.2007030348
Eagle, H. (1956). The salt requirements of mammalian cells in tissue culture. Arch. Biochem. Biophys. 61, 356–366. doi: 10.1016/0003-9861(56)90358-7
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Estévez, R., Boettger, T., Stein, V., Birkenhäger, R., Otto, E., Hildebrandt, F., et al. (2001). Barttin is a Cl- channel β-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature 414, 558–561. doi: 10.1038/35107099
Evans, R. L., Park, K., James Turner, R., Watson, G. E., Nguyen, V. H., Dennett, M. R., et al. (2000). Severe impairment of salivation in Na+/K+/2Cl- cotransporter (NKCC1)-deficient mice. J. Biol. Chem. 275, 26720–26726. doi: 10.1074/jbc.M003753200
Ferdaus, M. Z., Barber, K. W., López-Cayuqueo, K. I., Terker, A. S., Argaiz, E. R., Gassaway, B. M., et al. (2016). SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J. Physiol. 594, 4945–4966. doi: 10.1113/JP272311
Flagella, M., Clarke, L. L., Miller, M. L., Erway, L. C., Giannella, R. A., Andringa, A., et al. (1999). Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 274, 26946–26955. doi: 10.1074/jbc.274.38.26946
Foskett, J. K. (1990). [Ca2+]i modulation of Cl- content controls cell volume in single salivary acinar cells during fluid secretion. Am. J. Physiol. Cell Physiol. 259, C998–C1004. doi: 10.1152/ajpcell.1990.259.6.c998
Friedel, P., Kahle, K. T., Zhang, J., Hertz, N., Pisella, L. I., Buhler, E., et al. (2015). WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal. 8:ra65. doi: 10.1126/scisignal.aaa0354
Funabashi, K., Fujii, M., Yamamura, H., Ohya, S., and Imaizumi, Y. (2010). Contribution of chloride channel conductance to the regulation of resting membrane potential in chondrocytes. J. Pharmacol. Sci. 113, 94–99. doi: 10.1254/jphs.10026SC
Gagnon, K. B., and Delpire, E. (2012). Molecular physiology of SPAK and OSR1: two ste20-related protein kinases regulating ion transport. Physiol. Rev. 92, 1577–1617. doi: 10.1152/physrev.00009.2012
Gagnon, K. B., and Delpire, E. (2013). Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am. J. Physiol. Physiol. 304, C693–C714. doi: 10.1152/ajpcell.00350.2012
Gagnon, K. B. E., England, R., and Delpire, E. (2006). Volume sensitivity of cation-Cl - cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am. J. Physiol. Physiol. 290, C134–C142. doi: 10.1152/ajpcell.00037.2005
Gagnon, M., Bergeron, M. J., Lavertu, G., Castonguay, A., Tripathy, S., Bonin, R. P., et al. (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 19, 1524–1528. doi: 10.1038/nm.3356
Gamba, G. (2005). Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493. doi: 10.1152/physrev.00011.2004
Gamba, G., Miyanoshita, A., Lombardi, M., Lytton, J., Lee, W. S., Hediger, M. A., et al. (1994). Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 269, 17713–17722.
Gandolfi, B., Gruffydd-jones, T. J., Malik, R., Cortes, A., Jones, B. R., Helps, C. R., et al. (2012). First WNK4-hypokalemia animal model identified by genome-wide association in burmese cats. PLoS One 7:e0053173. doi: 10.1371/journal.pone.0053173
Gillen, C. M., and Forbush, B. (1999). Functional interaction of the K-Cl cotransporter (KCC1) with the Na-K-Cl cotransporter in HEK-293 cells. Am. J. Physiol. Physiol. 276, C328–C336. doi: 10.1152/ajpcell.1999.276.2.C328
Glykys, J., Dzhala, V., Egawa, K., Balena, T., Saponjian, Y., Kuchibhotla, K. V., et al. (2014). Local impermeant anions establish the neuronal chloride concentration. Science 343, 670–675. doi: 10.1126/science.1245423
Grill, A., Schießl, I. M., Gess, B., Fremter, K., Hammer, A., and Castrop, H. (2016). Salt-losing nephropathy in mice with a null mutation of the Clcnk2 gene. Acta Physiol. 218, 198–211. doi: 10.1111/apha.12755
Grimm, P. R., Coleman, R., Delpire, E., and Welling, P. A. (2017). Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J. Am. Soc. Nephrol. 28, 2597–2606. doi: 10.1681/ASN.2016090948
Grimm, P. R., Taneja, T. K., Liu, J., Coleman, R., Chen, Y. Y., Delpire, E., et al. (2012). SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J. Biol. Chem. 287, 37673–37690. doi: 10.1074/jbc.M112.402800
Gruber, M., Söding, J., and Lupas, A. N. (2006). Comparative analysis of coiled-coil prediction methods. J. Struct. Biol. 155, 140–145. doi: 10.1016/j.jsb.2006.03.009
Guo, D., and Hu, J. (2014). Spinal presynaptic inhibition in pain control. Neuroscience 283, 95–106. doi: 10.1016/j.neuroscience.2014.09.032
Gut, H., Pennacchietti, E., John, R. A., Bossa, F., Capitani, G., De Biase, D., et al. (2006). Escherichia coli acid resistance: pH-sensing, activation by chloride and autoinhibition in GadB. EMBO J. 25, 2643–2651. doi: 10.1038/sj.emboj.7601107
Hennings, J. C., Andrini, O., Picard, N., Paulais, M., Huebner, A. K., Cayuqueo, I. K. L., et al. (2017). The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J. Am. Soc. Nephrol. 28, 209–217. doi: 10.1681/ASN.2016010085
Heubl, M., Zhang, J., Pressey, J. C., Al Awabdh, S., Renner, M., Gomez-Castro, F., et al. (2017). GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl–sensitive WNK1 kinase. Nat. Commun. 8:1776. doi: 10.1038/s41467-017-01749-0
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366. doi: 10.1152/physrev.00021.2009
Hoffmann, E. K., Lambert, I. H., and Pedersen, S. F. (2009). Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89, 193–277. doi: 10.1152/physrev.00037.2007
Holden, S., Cox, J., and Raymond, F. L. (2004). Cloning, genomic organization, alternative splicing and expression analysis of the human gene WNK3 (PRKWNK3). Gene 335, 109–119. doi: 10.1016/j.gene.2004.03.009
Howard, H. C., Mount, D. B., Rochefort, D., Byun, N., Dupré, N., Lu, J., et al. (2002). The K–Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat. Genet. 32, 384–392. doi: 10.1038/ng1002
Hubner, C. A., Stein, V., Hermans-Borgmeyer, I., Meyer, T., Ballanyi, K., and Jentsch, T. J. (2001). Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30, 515–524. doi: 10.1016/s0896-6273(01)00297-5
Hunter, R. W., Craigie, E., Homer, N. Z. M., Mullins, J. J., and Bailey, M. A. (2014). Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am. J. Physiol. Ren. Physiol. 306, F457–F467. doi: 10.1152/ajprenal.00339.2013
Hutter, O. F. (2017). A personal historic perspective on the role of chloride in skeletal and cardiac muscle. Physiol. Rep. 5, 1–7. doi: 10.14814/phy2.13165
Hyde, T. M., Lipska, B. K., Ali, T., Mathew, S. V., Law, A. J., Metitiri, O. E., et al. (2011). Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 31, 11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011
Inoue, K., Furukawa, T., Kumada, T., Yamada, J., Wang, T., Inoue, R., et al. (2012). Taurine inhibits K +-Cl - cotransporter KCC2 to regulate embryonic Cl - homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J. Biol. Chem. 287, 20839–20850. doi: 10.1074/jbc.M111.319418
Irwin, R. P., and Allen, C. N. (2009). GABAergic signaling induces divergent neuronal Ca2+ responses in the suprachiasmatic nucleus network. Eur. J. Neurosci. 30, 1462–1475. doi: 10.1111/j.1460-9568.2009.06944.x
Jentsch, T. J., and Pusch, M. (2018). CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol. Rev. 98, 1493–1590. doi: 10.1152/physrev.00047.2017
Jordan, K. N., and Davies, K. W. (2001). Sodium chloride enhances recovery and growth of acid-stressed E. Coli O157:H7. Lett. Appl. Microbiol. 32, 312–315. doi: 10.1046/j.1472-765X.2001.00911.x
Kahle, K. T., and Delpire, E. (2016). Kinase-KCC2 coupling: Cl- rheostasis, disease susceptibility, therapeutic target. J. Neurophysiol. 115, 8–18. doi: 10.1152/jn.00865.2015
Kahle, K. T., Gimenez, I., Hassan, H., Wilson, F. H., Wong, R. D., Forbush, B., et al. (2004). WNK4 regulates apical and basolateral Cl- flux in extrarenal epithelia. Proc. Natl. Acad. Sci. U.S.A. 101, 2064–2069. doi: 10.1073/pnas.0308434100
Kahle, K. T., Khanna, A. R., Alper, S. L., Adragna, N. C., Lauf, P. K., Sun, D., et al. (2015). K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 21, 513–523. doi: 10.1016/j.molmed.2015.05.008
Kahle, K. T., Merner, N. D., Friedel, P., Silayeva, L., Liang, B., Khanna, A., et al. (2014). Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 15, 766–774. doi: 10.15252/embr.201438840
Kahle, K. T., Rinehart, J., de los Heros, P., Louvi, A., Meade, P., Vazquez, N., et al. (2005). WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 102, 16783–16788. doi: 10.1073/pnas.0508307102
Kahle, K. T., Schmouth, J. F., Lavastre, V., Latremoliere, A., Zhang, J., Andrews, N., et al. (2016). Inhibition of the kinase WNK1/HSN2 ameliorates neuropathic pain by restoring GABA inhibition. Sci. Signal. 9, 1–9. doi: 10.1126/scisignal.aad0163
Kahle, K. T., Staley, K. J., Nahed, B. V., Gamba, G., Hebert, S. C., Lifton, R. P., et al. (2008). Roles of the cation–chloride cotransporters in neurological disease. Nat. Clin. Pract. Neurol. 4, 490–503. doi: 10.1038/ncpneuro0883
Kaila, K., Price, T. J., Payne, J. A., Puskarjov, M., and Voipio, J. (2014). Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654. doi: 10.1038/nrn3819
Kakazu, Y., Akaike, N., Komiyama, S., and Nabekura, J. (1999). Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J. Neurosci. 19, 2843–2851. doi: 10.1523/jneurosci.19-08-02843.1999
Kharod, S. C., Kang, S. K., and Kadam, S. D. (2019). Off-label use of bumetanide for brain disorders: an overview. Front Neurosci 13:310. doi: 10.3389/fnins.2019.00310
Kieferle, S., Fong, P., Bens, M., Vandewalle, A., and Jentsch, T. J. (1994). Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc. Natl. Acad. Sci. U.S.A. 91, 6943–6947. doi: 10.1073/pnas.91.15.6943
Kim, E. R., Wu, Z., Sun, H., Xu, Y., Mangieri, L. R., Xu, Y., et al. (2015). Hypothalamic Non-AgRP, Non-POMC GABAergic neurons are required for postweaning feeding and NPY hyperphagia. J. Neurosci. 35, 10440–10450. doi: 10.1523/JNEUROSCI.1110-15.2015
Kim, G. H., Masilamani, S., Turner, R., Mitchell, C., Wade, J. B., and Knepper, M. A. (1998). The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc. Natl. Acad. Sci. U.S.A. 95, 14552–14557. doi: 10.1073/pnas.95.24.14552
Kim, M. J., Yang, H. J., Kim, Y., Kang, I., Kim, S. S., and Cho, Y. W. (2018). Role of nitric oxide and WNK-SPAK/OSR1-KCC2 signaling in daily changes in GABAergic inhibition in the rat dorsal raphe neurons. Neuropharmacology 135, 355–367. doi: 10.1016/j.neuropharm.2018.03.035
Koivusalo, M., Kapus, A., and Grinstein, S. (2009). Sensors, transducers, and effectors that regulate cell size and shape. J. Biol. Chem. 284, 6595–6599. doi: 10.1074/jbc.R800049200
Kong, D., Tong, Q., Ye, C., Koda, S., Fuller, P. M., Krashes, M. J., et al. (2012). GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. Cell 151, 645–657. doi: 10.1016/j.cell.2012.09.020
Konrad, M., Vollmer, M., Lemmink, H. H., van den Heuvel, L. P., Jeck, N., Vargas-Poussou, R., et al. (2000). Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J. Am. Soc. Nephrol. 11, 1449–1459.
Koumangoye, R., Omer, S., and Delpire, E. (2018). Mistargeting of a truncated Na-K-2Cl cotransporter in epithelial cells. Am. J. Physiol. Cell Physiol. 315, C258–C276. doi: 10.1152/ajpcell.00130.2018
Koumangoye, R., Omer, S., Kabeer, M. H., and Delpire, E. (2020). Novel human NKCC1 mutations cause defects in goblet cell mucus secretion and chronic inflammation. Cell. Mol. Gastroenterol. Hepatol. 9, 239–255. doi: 10.1016/j.jcmgh.2019.10.006
Kumar, M., Gouw, M., Michael, S., Sámano-Sánchez, H., Pancsa, R., Glavina, J., et al. (2020). ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 48, D296–D306. doi: 10.1093/nar/gkz1030
Kursan, S., McMillen, T. S., Beesetty, P., Dias-Junior, E., Almutairi, M. M., Sajib, A. A., et al. (2017). The neuronal K+Cl- co-transporter 2 (Slc12a5) modulates insulin secretion. Sci. Rep. 7:1732. doi: 10.1038/s41598-017-01814-0
Lai, Z. F., Chen, Y. Z., and Nishi, K. (2003). Modulation of intracellular Cl- homeostasis by lectin-stimulation in Jurkat T lymphocytes. Eur. J. Pharmacol. 482, 1–8. doi: 10.1016/S0014-2999(03)02076-4
Laird, J. M. A., García-Nicas, E., Delpire, E. J., and Cervero, F. (2004). Presynaptic inhibition and spinal pain processing in mice: a possible role of the NKCC1 cation-chloride co-transporter in hyperalgesia. Neurosci. Lett. 361, 200–203. doi: 10.1016/j.neulet.2003.12.015
Lalioti, M. D., Zhang, J., Volkman, H. M., Kahle, K. T., Hoffmann, K. E., Toka, H. R., et al. (2006). Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat. Genet. 38, 1124–1132. doi: 10.1038/ng1877
Lea, I. A., Borghoff, S. J., and Travlos, G. S. (2018). “Electrolytes, blood gases, and acid-base balance,” in The Clinical Chemistry of Laboratory Animals, eds D. M. Kurtz and G. S. Travlos (Boca Raton: CRC Press Taylor & Francis group), 873–927.
Lee, H. H., Deeb, T. Z., Walker, J. A., Davies, P. A., and Moss, S. J. (2011). NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 14, 736–743. doi: 10.1038/nn.2806
Lee, H. H., Jurd, R., and Moss, S. J. (2010). Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell Neurosci. 45, 173–179. doi: 10.1016/j.mcn.2010.06.008
Lee, H. H. C., Walker, J. A., Williams, J. R., Goodier, R. J., Payne, J. A., and Moss, S. J. (2007). Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 282, 29777–29784. doi: 10.1074/jbc.M705053200
Lemonnier, E., Lazartigues, A., and Ben-Ari, Y. (2016). Treating schizophrenia with the diuretic bumetanide: a case report. Clin. Neuropharmacol. 39, 115–117. doi: 10.1097/wnf.0000000000000136
Lemonnier, E., Villeneuve, N., Sonie, S., Serret, S., Rosier, A., Roue, M., et al. (2017). Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7:e1124. doi: 10.1038/tp.2017.101
Li, H., Khirug, S., Cai, C., Ludwig, A., Blaesse, P., Kolikova, J., et al. (2007). KCC2 Interacts with the dendritic cytoskeleton to promote spine development. Neuron 56, 1019–1033. doi: 10.1016/j.neuron.2007.10.039
Li, H., Tornberg, J., Kaila, K., Airaksinen, M. S., and Rivera, C. (2002). Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur. J. Neurosci. 16, 2358–2370. doi: 10.1046/j.1460-9568.2002.02419.x
Limbutara, K., Chou, C.-L., and Knepper, M. A. (2020). Quantitative proteomics of all 14 renal tubule segments in rat. J. Am. Soc. Nephrol. 31, 1255–1266. doi: 10.1681/ASN.2020010071
Lin, S.-H., Yu, I.-S., Jiang, S.-T., Lin, S.-W., Chu, P., Chen, A., et al. (2011). Impaired phosphorylation of Na+-K+-2Cl- cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 17538–17543. doi: 10.1073/pnas.1107452108
Loffing, J., Vallon, V., Loffing-Cueni, D., Aregger, F., Richter, K., Pietri, L., et al. (2004). Altered renal distal tubule structure and renal Na+ and Ca2+ handling in a mouse model for gitelman’s syndrome. J. Am. Soc. Nephrol. 15, 2276–2288. doi: 10.1097/01.ASN.0000138234.18569.63
Louis-Dit-Picard, H., Barc, J., Trujillano, D., Miserey-Lenkei, S., Bouatia-Naji, N., Pylypenko, O., et al. (2012). KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 44, 456–460. doi: 10.1038/ng.2218
Lourdel, S., Paulais, M., Cluzeaud, F., Bens, M., Tanemoto, M., Kurachi, Y., et al. (2002). An inward rectifier K(+) channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J. Physiol. 538, 391–404. doi: 10.1013/jphysiol.2001.012961
Lüscher, B. P., Vachel, L., Ohana, E., and Muallem, S. (2020). Cl - as a bona fide signaling ion. Am. J. Physiol. Physiol. 318, C125–C136. doi: 10.1152/ajpcell.00354.2019
Lytle, C., and Forbush, B. (1992). The Na-K-Cl cotransport protein of shark rectal gland. II. Regulation by direct phosphorylation. J. Biol. Chem. 267, 25438–25443.
Lytle, C., and Forbush, B. (1996). Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: modulation by cytoplasmic Cl. Am. J. Physiol. Physiol. 270, C437–C448. doi: 10.1152/ajpcell.1996.270.2.C437
MacLeod, R. A., and Onofrey, E. (1957). Nutrition and metabolism of marine bacteria: VI. Quantitative requirement for halides, magnesium, calcium, and iron. Can. J. Microbiol. 3, 753–759. doi: 10.1139/m57-085
Macnamara, E. F., Koehler, A. E., D’Souza, P., Estwick, T., Lee, P., Vezina, G., et al. (2019). Kilquist syndrome: a novel syndromic hearing loss disorder caused by homozygous deletion of SLC12A2. Hum. Mutat. 40, 532–538. doi: 10.1002/humu.23722
Malik, S., Lambert, E., Zhang, J., Wang, T., Clark, H. L., Cypress, M., et al. (2018). Potassium conservation is impaired in mice with reduced renal expression of Kir4.1. Am. J. Physiol. Ren. Physiol. 315, F1271–F1282. doi: 10.1152/ajprenal.00022.2018
Mao, S., Garzon-Muvdi, T., Di Fulvio, M., Chen, Y., Delpire, E., Alvarez, F. J., et al. (2012). Molecular and functional expression of cation-chloride cotransporters in dorsal root ganglion neurons during postnatal maturation. J. Neurophysiol. 108, 834–852. doi: 10.1152/jn.00970.2011
Matsumura, Y., Uchida, S., Kondo, Y., Miyazaki, H., Ko, S. B., Hayama, A., et al. (1999). Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat. Genet. 21, 95–98. doi: 10.1038/5036
McCormick, J. A., and Ellison, D. H. (2011). The WNKs: atypical protein kinases with pleiotropic actions. Physiol. Rev. 91, 177–219. doi: 10.1152/physrev.00017.2010
McCormick, J. A., Mutig, K., Nelson, J. H., Saritas, T., Hoorn, E. J., Yang, C. L., et al. (2011). A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 14, 352–364. doi: 10.1016/j.cmet.2011.07.009
McNeill, J. K., Walton, J. C., Ryu, V., and Albers, H. E. (2020). The excitatory effects of GABA within the suprachiasmatic nucleus: regulation of Na-K-2Cl Cotransporters (NKCCs) by environmental lighting conditions. J. Biol. Rhythms 35, 275–286. doi: 10.1177/0748730420924271
Meneton, P., Loffing, J., and Warnock, D. G. (2004). Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am. J. Physiol. Renal Physiol. 287, F593–F601. doi: 10.1152/ajprenal.00454.2003
Merner, N. D., Chandler, M. R., Bourassa, C., Liang, B., Khanna, A. R., Dion, P., et al. (2015). Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front. Cell. Neurosci. 9:386. doi: 10.3389/fncel.2015.00386
Merner, N. D., Mercado, A., Khanna, A. R., Hodgkinson, A., Bruat, V., Awadalla, P., et al. (2016). Gain-of-function missense variant in SLC12A2, encoding the bumetanide-sensitive NKCC1 cotransporter, identified in human schizophrenia. J. Psychiatr. Res. 77, 22–26. doi: 10.1016/j.jpsychires.2016.02.016
Mollajani, R., Joghataei, M. T., and Tehrani-Doost, M. (2019). Bumetanide therapeutic effect in children and adolescents with autism spectrum disorder: a review study. Basic Clin. Neurosci. 10, 433–441. doi: 10.32598/bcn.9.10.380
Moon, T. M., Correa, F., Kinch, L. N., Piala, A. T., Gardner, K. H., and Goldsmith, E. J. (2013). Solution structure of the WNK1 autoinhibitory domain, a WNK-specific PF2 domain. J. Mol. Biol. 425, 1245–1252. doi: 10.1016/j.jmb.2013.01.031
Moore, Y. E., Deeb, T. Z., Chadchankar, H., Brandon, N. J., and Moss, S. J. (2018). Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. U.S.A. 115, 10166–10171. doi: 10.1073/pnas.1810134115
Moriguchi, T., Urushiyama, S., Hisamoto, N., Iemura, S. I., Uchida, S., Natsume, T., et al. (2005). WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases. SPAK and OSR1. J. Biol. Chem. 280, 42685–42693. doi: 10.1074/jbc.M510042200
Morris, R. G., Hoorn, E. J., and Knepper, M. A. (2006). Hypokalemia in a mouse model of Gitelman’s syndrome. Am. J. Physiol. Renal Physiol. 290, F1416–F1420. doi: 10.1152/ajprenal.00421.2005
Mount, D. B., Mercado, A., Song, L., Jason, X., George, A. L., Delpire, E., et al. (1999). Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J. Biol. Chem. 274, 16355–16362. doi: 10.1074/jbc.274.23.16355
Murillo-de-Ozores, A. R., Gamba, G., and Castañeda-Bueno, M. (2019). Molecular mechanisms for the regulation of blood pressure by potassium. Curr. Top. Membr. 83, 285–313. doi: 10.1016/bs.ctm.2019.01.004
Murillo-de-Ozores, A. R., Rodríguez-Gama, A., Bazúa-Valenti, S., Leyva-Ríos, K., Vázquez, N., Pacheco-Álvarez, D., et al. (2018). C-terminally truncated, kidney-specific variants of the WNK4 kinase lack several sites that regulate its activity. J. Biol. Chem. 293, 12209–12221. doi: 10.1074/jbc.RA118.003037
Murthy, M., and O’Shaughnessy, K. M. (2019). Modified HEK cells simulate DCT cells in their sensitivity and response to changes in extracellular K. Physiol. Rep. 7, 1–10. doi: 10.14814/phy2.14280
Na, T., Wu, G., and Peng, J.-B. (2012). Disease-causing mutations in the acidic motif of WNK4 impair the sensitivity of WNK4 kinase to calcium ions. Biochem. Biophys. Res. Commun. 419, 293–298. doi: 10.1016/j.bbrc.2012.02.013
Naguro, I., Umeda, T., Kobayashi, Y., Maruyama, J., Hattori, K., Shimizu, Y., et al. (2012). ASK3 responds to osmotic stress and regulates blood pressure by suppressing WNK1-SPAK/OSR1 signaling in the kidney. Nat. Commun. 3:1285. doi: 10.1038/ncomms2283
Niisato, N., Eaton, D. C., and Marunaka, Y. (2004). Involvement of cytosolic Cl- in osmoregulation of α-ENaC gene expression. Am. J. Physiol. Ren. Physiol. 287, F932–F939. doi: 10.1152/ajprenal.00131.2004
Niisato, N., Taruno, A., and Marunaka, Y. (2007). Involvement of p38 MAPK in hypotonic stress-induced stimulation of β- and γ-ENaC expression in renal epithelium. Biochem. Biophys. Res. Commun. 358, 819–824. doi: 10.1016/j.bbrc.2007.04.192
Nomura, H., Sakai, A., Nagano, M., Umino, M., and Suzuki, H. (2006). Expression changes of cation chloride cotransporters in the rat spinal cord following intraplantar formalin. Neurosci. Res. 56, 435–440. doi: 10.1016/j.neures.2006.08.012
Nomura, N., Shoda, W., Wang, Y., Mandai, S., Furusho, T., Takahashi, D., et al. (2018). Role of ClC-K and barttin in low-potassium induced sodium-chloride cotransporter activation and hypertension in mouse kidney. Biosci. Rep. 38:BSR20171243. doi: 10.1042/BSR20171243
Nomura, N., Tajima, M., Sugawara, N., Morimoto, T., Kondo, Y., Ohno, M., et al. (2011). Generation and analyses of R8L barttin knockin mouse. Am. J. Physiol. Renal Physiol. 301, F297–F307. doi: 10.1152/ajprenal.00604.2010
O’Doherty, J., and Stark, R. J. (1983). A transcellular route for Na-coupled Cl transport in secreting pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 8, 499–503. doi: 10.1152/ajpgi.1983.245.4.g499
Ohno, M., Uchida, K., Ohashi, T., Nitta, K., Ohta, A., Chiga, M., et al. (2011). Immunolocalization of WNK4 in mouse kidney. Histochem. Cell Biol. 136, 25–35. doi: 10.1007/s00418-011-0827-x
Ohsawa, R., Miyazaki, H., Niisato, N., Shiozaki, A., Iwasaki, Y., Otsuji, E., et al. (2010). Intracellular chloride regulates cell proliferation through the activation of stress-activated protein kinases in MKN28 human gastric cancer cells. J. Cell. Physiol. 223, 764–770. doi: 10.1002/jcp.22088
Ohta, A., Schumacher, F., Mehellou, Y., Johnson, C., Knebel, A., Macartney, T. J., et al. (2013). The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem. J. 451, 111–122. doi: 10.1042/bj20121903
Oi, K., Sohara, E., Rai, T., Misawa, M., Chiga, M., Alessi, D. R., et al. (2012). A minor role of WNK3 in regulating phosphorylation of renal NKCC2 and NCC co-transporters in vivo. Biol. Open 1, 120–127. doi: 10.1242/bio.2011048
Okabe, A., Shimizu-Okabe, C., Arata, A., Konishi, S., Fukuda, A., and Takayama, C. (2015). KCC2-mediated regulation of respiration-related rhythmic activity during postnatal development in mouse medulla oblongata. Brain Res. 1601, 31–39. doi: 10.1016/j.brainres.2015.01.007
Osei-Owusu, J., Yang, J., Vitery, M., del, C., and Qiu, Z. (2018). “Molecular biology and physiology of volume-regulated anion channel (VRAC),” in Current Topics in Membranes, eds I. Levitane, E. Delpire, and H. Rasgado-Flores (Cambridge, MA: Academic Press Inc.), 177–203. doi: 10.1016/bs.ctm.2018.07.005
Pace, A. J., Lee, E., Athirakul, K., Coffman, T. M., O’Brien, D. A., and Koller, B. H. (2000). Failure of spermatogenesis in mouse lines deficient in the Na+-K+- 2Cl- cotransporter. J. Clin. Invest. 105, 441–450. doi: 10.1172/JCI8553
Pacheco-Alvarez, D., Carrillo-Perez, D. L., Mercado, A., Leyva-Ríos, K., Moreno, E., Hernandez-Mercado, E., et al. (2020). WNK3 and WNK4 exhibit opposite sensitivity with respect to cell volume and intracellular chloride concentration. Am. J. Physiol. Physiol. 19, C371–C380. doi: 10.1152/ajpcell.00488.2019
Pacheco-Alvarez, D., San Cristóbal, P., Meade, P., Moreno, E., Vazquez, N., Muñoz, E., et al. (2006). The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 281, 28755–28763. doi: 10.1074/jbc.M603773200
Pacheco-Cuellar, G., González-Huerta, L. M., Valdés-Miranda, J. M., Peláez-González, H., Zenteno-Bacheron, S., Cazarin-Barrientos, J., et al. (2011). Hereditary sensory and autonomic neuropathy II due to novel mutation in the HSN2 gene in Mexican families. J. Neurol. 258, 1890–1892. doi: 10.1007/s00415-011-6025-x
Paulais, M., Bloch-Faure, M., Picard, N., Jacques, T., Ramakrishnan, S. K., Keck, M., et al. (2011). Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 10361–10366. doi: 10.1073/pnas.1101400108
Payne, J. A., Stevenson, J., and Donaldson, L. (1996). Molecular characterization of putative K - Cl cotransporter in rat brain. J. Biol. Chem. 271, 16245–16252. doi: 10.1074/jbc.271.27.16245
Pearson, M., Lu, J., Mount, D., and Delpire, E. (2001). Localization of the K+–Cl- cotransporter, KCC3, in the central and peripheral nervous systems: expression in the choroid plexus, large neurons and white matter tracts. Neuroscience 103, 481–491. doi: 10.1016/S0306-4522(00)00567-4
Pedemonte, N., and Galietta, L. J. V. (2014). Structure and function of tmem16 proteins (anoctamins). Physiol. Rev. 94, 419–459. doi: 10.1152/physrev.00039.2011
Penton, D., Czogalla, J., Wengi, A., Himmerkus, N., Loffing-Cueni, D., Carrel, M., et al. (2016). Extracellular K + rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl - -dependent and independent mechanisms. J. Physiol. 594, 6319–6331. doi: 10.1113/JP272504
Perrier, R., Boscardin, E., Malsure, S., Sergi, C., Maillard, M. P., Loffing, J., et al. (2016). Severe Salt-losing syndrome and hyperkalemia induced by adult nephron-specific knockout of the epithelial sodium channel a-subunit. J. Am. Soc. Nephrol. 27, 2309–2318. doi: 10.1681/ASN.2015020154
Pessia, M., Tucker, S. J., Lee, K., Bond, C. T., and Adelman, J. P. (1996). Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 15, 2980–2987. doi: 10.1002/j.1460-2075.1996.tb00661.x
Piala, A. T., Moon, T. M., Akella, R., He, H., Cobb, M. H., and Goldsmith, E. J. (2014). Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci. Signal. 7:ra41. doi: 10.1126/scisignal.2005050
Piechotta, K., Lu, J., and Delpire, E. (2002). Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J. Biol. Chem. 277, 50812–50819. doi: 10.1074/jbc.M208108200
Pigeyre, M., Yazdi, F. T., Kaur, Y., and Meyre, D. (2016). Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin. Sci. 130, 943–986. doi: 10.1042/CS20160136
Ponce-Coria, J., San-Cristobal, P., Kahle, K. T., Vazquez, N., Pacheco-Alvarez, D., de los Heros, P., et al. (2008). Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 8458–8463. doi: 10.1073/pnas.0802966105
Price, T. J., Cervero, F., and de Koninck, Y. (2005). Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr. Top. Med. Chem. 5, 547–555. doi: 10.2174/1568026054367629
Price, T. J., Hargreaves, K. M., and Cervero, F. (2006). Protein expression and mRNA cellular distribution of the NKCC1 cotransporter in the dorsal root and trigeminal ganglia of the rat. Brain Res. 1112, 146–158. doi: 10.1016/j.brainres.2006.07.012
Puskarjov, M., Seja, P., Heron, S. E., Williams, T. C., Ahmad, F., Iona, X., et al. (2014). A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep. 15, 723–729. doi: 10.1002/embr.201438749
Rafiqi, F. H., Zuber, A. M., Glouer, M., Richardson, C., Fleming, S., Jouanouić, S., et al. (2010). Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol. Med. 2, 63–75. doi: 10.1002/emmm.200900058
Rahmani, B., Fekrmandi, F., Ahadi, K., Ahadi, T., Alavi, A., Ahmadiani, A., et al. (2018). A novel nonsense mutation in WNK1/HSN2 associated with sensory neuropathy and limb destruction in four siblings of a large Iranian pedigree. BMC Neurol. 18:195. doi: 10.1186/s12883-018-1201-6
Rahmanzadeh, R., Eftekhari, S., Shahbazi, A., Khodaei Ardakani, M. R., Rahmanzade, R., Mehrabi, S., et al. (2017). Effect of bumetanide, a selective NKCC1 inhibitor, on hallucinations of schizophrenic patients; a double-blind randomized clinical trial. Schizophr. Res. 184, 145–146. doi: 10.1016/j.schres.2016.12.002
Reuter, D., Zierold, K., Schröder, W. H., and Frings, S. (1998). A depolarizing chloride current contributes to chemoelectrical transduction in olfactory sensory neurons in situ. J. Neurosci. 18, 6623–6630. doi: 10.1523/JNEUROSCI.18-17-06623.1998
Richardson, C., and Alessi, D. R. (2008). The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J. Cell Sci. 121, 3293–3304. doi: 10.1242/jcs.029223
Rickheit, G., Maier, H., Strenzke, N., Andreescu, C. E., De Zeeuw, C. I., Muenscher, A., et al. (2008). Endocochlear potential depends on Cl- channels: mechanism underlying deafness in Bartter syndrome IV. EMBO J. 27, 2907–2917. doi: 10.1038/emboj.2008.203
Rinehart, J., Kahle, K. T., de Los Heros, P., Vazquez, N., Meade, P., Wilson, F. H., et al. (2005). WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl- cotransporters required for normal blood pressure homeostasis. Proc. Natl. Acad. Sci. U.S.A. 102, 16777–16782. doi: 10.1073/pnas.0508303102
Rinehart, J., Maksimova, Y. D., Tanis, J. E., Stone, K. L., Hodson, C. A., Zhang, J., et al. (2009). Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536. doi: 10.1016/j.cell.2009.05.031
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rocha-González, H. I., Mao, S., and Alvarez-Leefmans, F. J. (2008). Na+,K+,2Cl- cotransport and intracellular chloride regulation in rat primary sensory neurons: thermodynamic and kinetic aspects. J. Neurophysiol. 100, 169–184. doi: 10.1152/jn.01007.2007
Roeßler, M., and Müller, V. (2002). Chloride, a new environmental signal molecule involved in gene regulation in a moderately halophilic bacterium, halobacillus halophilus. J. Bacteriol. 184, 6207–6215. doi: 10.1128/JB.184.22.6207-6215.2002
Roeßler, M., and Muüller, V. (1998). Quantitative and physiological analyses of chloride dependence of growth of halobacillus halophilus. Appl. Environ. Microbiol. 64, 3813–3817. doi: 10.1128/AEM.64.10.3813-3817.1998
Roeßler, M., Sewald, X., and Müller, V. (2003). Chloride dependence of growth in bacteria. FEMS Microbiol. Lett. 225, 161–165. doi: 10.1016/S0378-1097(03)00509-3
Rozansky, D. J., Cornwall, T., Subramanya, A. R., Rogers, S., Yang, Y. F., David, L. L., et al. (2009). Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J. Clin. Invest. 119, 2601–2612. doi: 10.1172/JCI38323
Rudomin, P., and Schmidt, R. F. (1999). Presynaptic inhibition in the vertebrate spinal cord revisited. Exp. Brain Res. 129, 1–37. doi: 10.1007/s002210050933
Rust, M. B., Alper, S. L., Rudhard, Y., Shmukler, B. E., Vicente, R., Brugnara, C., et al. (2007). Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J. Clin. Invest. 117, 1708–1717. doi: 10.1172/JCI30630
Saritas, T., Puelles, V. G., Su, X. T., McCormick, J. A., Welling, P. A., and Ellison, D. H. (2018). Optical clearing in the kidney reveals potassium-mediated tubule remodeling. Cell Rep. 25, 2668.e3–2675.e3. doi: 10.1016/j.celrep.2018.11.021
Sasaki, S., Hasegawa, K., Higashi, T., Suzuki, Y., Sugano, S., Yasuda, Y., et al. (2016). A missense mutation in solute carrier family 12, member 1 (SLC12A1) causes hydrallantois in Japanese Black cattle. BMC Genomics 17:724. doi: 10.1186/s12864-016-3035-1
Scholl, U. I., Choi, M., Liu, T., Ramaekers, V. T., Hausler, M. G., Grimmer, J., et al. (2009). Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. U.S.A. 106, 5842–5847. doi: 10.1073/pnas.0901749106
Schultheis, P. J., Lorenz, J. N., Meneton, P., Nieman, M. L., Riddle, T. M., Flagella, M., et al. (1998). Phenotype resembling gitelman’s syndrome in mice lacking the apical Na + -Cl - cotransporter of the distal convoluted tubule. J. Biol. Chem. 273, 29150–29155. doi: 10.1074/jbc.273.44.29150
Sedmak, G., Jovanov-Milošević, N., Puskarjov, M., Ulamec, M., Krušlin, B., Kaila, K., et al. (2016). Developmental expression patterns of KCC2 and functionally associated molecules in the human brain. Cereb. Cortex 26, 4574–4589. doi: 10.1093/cercor/bhv218
Seifter, J. L., and Chang, H.-Y. (2016). Disorders of acid-base balance: new perspectives. Kidney Dis. 2, 170–186. doi: 10.1159/000453028
Sewald, X., Saum, S. H., Palm, P., Pfeiffer, F., Oesterhelt, D., and Müller, V. (2007). Autoinducer-2-producing protein LuxS, a novel salt- and chloride-induced protein in the moderately halophilic bacterium halobacillus halophilus. Appl. Environ. Microbiol. 73, 371–379. doi: 10.1128/AEM.01625-06
Seyberth, H. W., and Schlingmann, K. P. (2011). Bartter- and gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr. Nephrol. 26, 1789–1802. doi: 10.1007/s00467-011-1871-4
Shekarabi, M., Girard, N., Rivière, J.-B., Dion, P., Houle, M., Toulouse, A., et al. (2008). Mutations in the nervous system–specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J. Clin. Invest. 118, 2496–2505. doi: 10.1172/JCI34088
Shekarabi, M., Lafrenière, R., Gaudet, R., Laganière, J., Marcinkiewicz, M., Dion, P., et al. (2013). Comparative analysis of the expression profile of Wnk1 and Wnk1/Hsn2 splice variants in developing and adult mouse tissues. PLoS One 8:e0057807. doi: 10.1371/journal.pone.0057807
Shibata, S., Zhang, J., Puthumana, J., Stone, K. L., and Lifton, R. P. (2013). Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc. Natl. Acad. Sci. U.S.A. 110, 7838–7843. doi: 10.1073/pnas.1304592110
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Silayeva, L., Deeb, T. Z., Hines, R. M., Kelley, M. R., Munoz, M. B., Lee, H. H. C., et al. (2015). KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. U.S.A. 112, 3523–3528. doi: 10.1073/pnas.1415126112
Simon, D. B., Bindra, R. S., Mansfield, T. A., Nelson-Williams, C., Mendonca, E., Stone, R., et al. (1997). Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 17, 171–178. doi: 10.1038/ng1097-171
Simon, D. B., Karet, F. E., Hamdan, J. M., Di Pietro, A., Sanjad, S. A., and Lifton, R. P. (1996a). Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na–K–2CI cotransporter NKCC2. Nat. Genet. 13, 183–188. doi: 10.1038/ng0696-183
Simon, D. B., Nelson-Williams, C., Bia, M. J., Ellison, D., Karet, F. E., Molina, A. M., et al. (1996b). Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 12, 24–30. doi: 10.1038/ng0196-24
Sohrabipour, S., Sharifi, M. R., Talebi, A., Sharifi, M., and Soltani, N. (2018). GABA dramatically improves glucose tolerance in streptozotocin-induced diabetic rats fed with high-fat diet. Eur. J. Pharmacol. 826, 75–84. doi: 10.1016/j.ejphar.2018.01.047
Sorensen, M. V., Grossmann, S., Roesinger, M., Gresko, N., Todkar, A. P., Barmettler, G., et al. (2013). Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 83, 811–824. doi: 10.1038/ki.2013.14
Stödberg, T., McTague, A., Ruiz, A. J., Hirata, H., Zhen, J., Long, P., et al. (2015). Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 6:8038. doi: 10.1038/ncomms9038
Su, G., Kintner, D. B., Flagella, M., Shull, G. E., and Sun, D. (2002). Astrocytes from Na(+)-K(+)-Cl(-) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am. J. Physiol. Cell Physiol. 282, C1147–C1160. doi: 10.1152/ajpcell.00538.2001
Su, X.-T., and Wang, W.-H. (2016). The expression, regulation, and function of Kir4.1 (Kcnj10) in the mammalian kidney. Am. J. Physiol. Ren. Physiol. 311, F12–F15. doi: 10.1152/ajprenal.00112.2016
Subramanya, A. R., and Ellison, D. H. (2014). Distal convoluted tubule. Clin. J. Am. Soc. Nephrol. 9, 2147–2163. doi: 10.1002/cphy.c140002
Succol, F., Fiumelli, H., Benfenati, F., Cancedda, L., and Barberis, A. (2012). Intracellular chloride concentration influences the GABA A receptor subunit composition. Nat. Commun. 3:738. doi: 10.1038/ncomms1744
Sun, Q., Wu, Y., Jonusaite, S., Pleinis, J. M., Humphreys, J. M., He, H., et al. (2018). Intracellular chloride and scaffold protein Mo25 cooperatively regulate transepithelial ion transport through WNK signaling in the malpighian tubule. J. Am. Soc. Nephrol. 29, 1449–1461. doi: 10.1681/ASN.2017101091
Sung, K. W., Kirby, M., McDonald, M. P., Lovinger, D. M., and Delpire, E. (2000). Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J. Neurosci. 20, 7531–7538.
Susa, K., Sohara, E., Takahashi, D., Okado, T., Rai, T., and Uchida, S. (2017). WNK4 is indispensable for the pathogenesis of pseudohypoaldosteronism type II caused by mutant KLHL3. Biochem. Biophys. Res. Commun. 491, 727–732. doi: 10.1016/j.bbrc.2017.07.121
Takahashi, N., Chernavvsky, D. R., Gomez, R. A., Igarashi, P., Gitelman, H. J., and Smithies, O. (2000). Uncompensated polyuria in a mouse model of Bartter’s syndrome. Proc. Natl. Acad. Sci. U.S.A 97, 5434–5439. doi: 10.1073/pnas.090091297
Tamari, M., Daigo, Y., and Nakamura, Y. (1999). Isolation and characterization of a novel serine threonine kinase gene on chromosome 3p22-21.3. J. Hum. Genet. 44, 116–120. doi: 10.1007/s100380050121
Tanemoto, M., Abe, T., and Ito, S. (2005). PDZ-Binding and Di-Hydrophobic motifs regulate distribution of Kir4.1 channels in renal cells. J. Am. Soc. Nephrol. 16, 2608–2614. doi: 10.1681/ASN.2005030266
Tanemoto, M., Kittaka, N., Inanobe, A., and Kurachi, Y. (2000). In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J. Physiol. 525, 587–592. doi: 10.1111/j.1469-7793.2000.00587.x
Terker, A. S., Yang, C. L., McCormick, J. A., Meermeier, N. P., Rogers, S. L., Grossmann, S., et al. (2014). Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension 64, 178–184. doi: 10.1161/HYPERTENSIONAHA.114.03335
Terker, A. S., Yarbrough, B., Ferdaus, M. Z., Lazelle, R. A., Erspamer, K. J., Meermeier, N. P., et al. (2016). Direct and indirect mineralocorticoid effects determine distal salt transport. J. Am. Soc. Nephrol. 27, 2436–2445. doi: 10.1681/ASN.2015070815
Terker, A. S., Zhang, C., Erspamer, K. J., Gamba, G., Yang, C.-L., and Ellison, D. H. (2015a). Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 89, 1–8. doi: 10.1038/ki.2015.289
Terker, A. S., Zhang, C., McCormick, J. A., Lazelle, R. A., Zhang, C., Meermeier, N. P., et al. (2015b). Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 21, 39–50. doi: 10.1016/j.cmet.2014.12.006
Thastrup, J. O., Rafiqi, F. H., Vitari, A. C., Pozo-Guisado, E., Deak, M., Mehellou, Y., et al. (2012). SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem. J. 441, 325–337. doi: 10.1042/BJ20111879
Thomson, M. N., Cuevas, C. A., Bewarder, T. M., Dittmayer, C., Miller, L. N., Si, J., et al. (2020). WNK bodies cluster WNK4 and SPAK/OSR1 to promote NCC activation in hypokalemia. Am. J. Physiol. Physiol. 318, F216–F228. doi: 10.1152/ajprenal.00232.2019
Titz, S., Sammler, E. M., and Hormuzdi, S. G. (2015). Could tuning of the inhibitory tone involve graded changes in neuronal chloride transport? Neuropharmacology 95, 321–331. doi: 10.1016/j.neuropharm.2015.03.026
Tong, Q., Ye, C. P., Jones, J. E., Elmquist, J. K., and Lowell, B. B. (2008). Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 11, 998–1000. doi: 10.1038/nn.2167
Tornberg, J., Voikar, V., Savilahti, H., Rauvala, H., and Airaksinen, M. S. (2005). Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur. J. Neurosci. 21, 1327–1337. doi: 10.1111/j.1460-9568.2005.03959.x
Torre-Villalvazo, I., Cervantes-Pérez, L. G., Noriega, L. G., Jiménez, J. V., Uribe, N., Chávez-Canales, M., et al. (2018). Inactivation of SPAK kinase reduces body weight gain in mice fed a high-fat diet by improving energy expenditure and insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 314, E53–E65. doi: 10.1152/ajpendo.00108.2017
Tucker, S. J., Imbrici, P., Salvatore, L., D’Adamo, M. C., and Pessia, M. (2000). pH Dependence of the inwardly rectifying potassium channel. Kir5.1, and localization in renal tubular epithelia. J. Biol. Chem. 275, 16404–16407. doi: 10.1074/jbc.C000127200
Ushiro, H., Tsutsumi, T., Suzuki, K., Kayahara, T., and Nakano, K. (1998). Molecular cloning and characterization of a novel Ste20-Related protein kinase enriched in neurons and transporting epithelia. Arch. Biochem. Biophys. 355, 233–240. doi: 10.1006/abbi.1998.0736
Uvarov, P., Ludwig, A., Markkanen, M., Pruunsild, P., Kaila, K., Delpire, E., et al. (2007). A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J. Biol. Chem. 282, 30570–30576. doi: 10.1074/jbc.M705095200
Valdivieso, ÁG., Clauzure, M., Massip-Copiz, M., and Santa-Coloma, T. A. (2016). The chloride anion acts as a second messenger in mammalian cells - modifying the expression of specific genes. Cell. Physiol. Biochem 38, 49–64. doi: 10.1159/000438608
Valdivieso, ÁG., and Santa-Coloma, T. A. (2019). The chloride anion as a signalling effector. Biol. Rev. 94, 1839–1856. doi: 10.1111/brv.12536
Vallon, V., Schroth, J., Lang, F., Kuhl, D., and Uchida, S. (2009). Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am. J. Physiol. Physiol. 297, F704–F712. doi: 10.1152/ajprenal.00030.2009
Van Poucke, M., Stee, K., Sonck, L., Stock, E., Bosseler, L., Van Dorpe, J., et al. (2019). Truncating SLC12A6 variants cause different clinical phenotypes in humans and dogs. Eur. J. Hum. Genet. 27, 1561–1568. doi: 10.1038/s41431-019-0432-3
Vanhatalo, S., Matias Palva, J., Andersson, S., Rivera, C., Voipio, J., and Kaila, K. (2005). Slow endogenous activity transients and developmental expression of K +-Cl- cotransporter 2 in the immature human cortex. Eur. J. Neurosci. 22, 2799–2804. doi: 10.1111/j.1460-9568.2005.04459.x
Veríssimo, F., and Jordan, P. (2001). WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene 20, 5562–5569. doi: 10.1038/sj.onc.1204726
Vidal-Petiot, E., Cheval, L., Faugeroux, J., Malard, T., Doucet, A., Jeunemaitre, X., et al. (2012). A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms. PLoS One 7:e0037751. doi: 10.1371/journal.pone.0037751
Villa, F., Goebel, J., Rafiqi, F. H., Deak, M., Thastrup, J., Alessi, D. R., et al. (2007). Structural insights into the recognition of substrates and activators by the OSR1 kinase. EMBO Rep. 8, 839–845. doi: 10.1038/sj.embor.7401048
Vitari, A. C., Deak, M., Morrice, N. A., and Alessi, D. R. (2005). The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 391, 17–24. doi: 10.1042/BJ20051180
Vitzthum, H., Castrop, H., Meier-Meitinger, M., Riegger, G. A. J., Kurtz, A., Krämer, B. K., et al. (2002). Nephron specific regulation of chloride channel CLC-K2 mRNA in the rat. Kidney Int. 61, 547–554. doi: 10.1046/j.1523-1755.2002.00165.x
Vong, L., Ye, C., Yang, Z., Choi, B., Chua, S. Jr., and Lowell, B. B. (2011). Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154. doi: 10.1016/j.neuron.2011.05.028
Wakabayashi, M., Mori, T., Isobe, K., Sohara, E., Susa, K., Araki, Y., et al. (2013). Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 3, 858–868. doi: 10.1016/j.celrep.2013.02.024
Waldegger, S., Jeck, N., Barth, P., Peters, M., Vitzthum, H., Wolf, K., et al. (2002). Barttin increases surface expression and changes current properties of CLC-K channels. Pflugers Arch. Eur. J. Physiol. 444, 411–418. doi: 10.1007/s00424-002-0819-8
Waldegger, S., and Jentsch, T. J. (2000). Functional and structural analysis of ClC-K chloride channels involved in renal disease. J. Biol. Chem. 275, 24527–24533. doi: 10.1074/jbc.M001987200
Wang, M. X., Cuevas, C. A., Su, X. T., Wu, P., Gao, Z. X., Lin, D. H., et al. (2018). Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 93, 893–902. doi: 10.1016/j.kint.2017.10.023
Watanabe, M., Wake, H., Moorhouse, A. J., and Nabekura, J. (2009). Clustering of neuronal K+-Cl- cotransporters in lipid rafts by tyrosine phosphorylation. J. Biol. Chem. 284, 27980–27988. doi: 10.1074/jbc.M109.043620
Watanabe, M., Zhang, J., Shahid Mansuri, M., Duan, J., Karimy, J. K., Delpire, E., et al. (2019). Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 12:eaaw9315. doi: 10.1126/scisignal.aaw9315
Weinstein, A. M. (2005). A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am. J. Physiol. Physiol. 289, F699–F720. doi: 10.1152/ajprenal.00043.2005
Wilson, C. S., and Mongin, A. A. (2019). The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci. Lett. 689, 33–44. doi: 10.1016/j.neulet.2018.01.012
Wilson, F. H., Disse-Nicodèm, S., Choate, K. A., Ishikawa, K., Nelson-Williams, C., Desitter, I., et al. (2001). Human hypertension caused by mutations in WNK Kinases. Science 293, 1107–1112. doi: 10.1126/science.1062844
Wilson, F. H., Kahle, K. T., Sabath, E., Lalioti, M. D., Rapson, A. K., Hoover, R. S., et al. (2003). Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc. Natl. Acad. Sci. U.S.A. 100, 680–684. doi: 10.1073/pnas.242735399
Woo, N. S., Lu, J., England, R., McClellan, R., Dufour, S., Mount, D. B., et al. (2002). Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus 12, 258–268. doi: 10.1002/hipo.10014
Wu, P., Gao, Z. X., Zhang, D. D., Su, X. T., Wang, W. H., and Lin, D. H. (2019). Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J. Am. Soc. Nephrol. 30, 1425–1438. doi: 10.1681/ASN.2019010025
Wu, Q., Delpire, E., Hebert, S. C., and Strange, K. (1998). Functional demonstration of Na + -K + -2Cl - cotransporter activity in isolated, polarized choroid plexus cells. Am. J. Physiol. Physiol. 275, C1565–C1572. doi: 10.1152/ajpcell.1998.275.6.C1565
Wu, Q. Q., Liu, X. Y., Xiong, L. X., Shang, J. Y., Mai, X. Y., Pang, R. P., et al. (2016). Reduction of intracellular chloride concentration promotes foam cell formation. Circ. J. 80, 1024–1033. doi: 10.1253/circj.CJ-15-1209
Xie, J., Wu, T., Xu, K., Huang, I. K., Cleaver, O., and Huang, C. L. (2009). Endothelial-specific expression of WNK1 kinase is essential for angiogenesis and heart development in mice. Am. J. Pathol. 175, 1315–1327. doi: 10.2353/ajpath.2009.090094
Xie, J., Yoon, J., Yang, S., Lin, S., and Huang, C. (2013). WNK1 protein kinase regulates embryonic cardiovascular development through the OSR1 signaling cascade. J. Biol. Chem. 288, 8566–8574. doi: 10.1074/jbc.M113.451575
Xie, Y., and Schafer, J. A. (2004). Inhibition of ENaC by intracellular Cl- in an MDCK clone with high ENaC expression. Am. J. Physiol. Ren. Physiol. 287, F722–F731.
Xu, B., English, J. M., Wilsbacher, J. L., Stippec, S., Goldsmith, E. J., and Cobb, M. H. (2000). WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem. 275, 16795–16801. doi: 10.1074/jbc.275.22.16795
Xu, B. E., Min, X., Stippec, S., Lee, B. H., Goldsmith, E. J., and Cobb, M. H. (2002). Regulation of WNK1 by an autoinhibitory domain and autophosphorylation. J. Biol. Chem. 277, 48456–48462. doi: 10.1074/jbc.M207917200
Xue, H., Liu, S., Ji, T., Ren, W., Zhang, X. H., Zheng, L. F., et al. (2009). Expression of NKCC2 in the rat gastrointestinal tract. Neurogastroenterol. Motil. 21, 1068–e89. doi: 10.1111/j.1365-2982.2009.01334.x
Yamada, J., Okabe, A., Toyoda, H., Kilb, W., Luhmann, H. J., and Fukuda, A. (2004). Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 557, 829–841. doi: 10.1113/jphysiol.2004.062471
Yang, C. L., Angell, J., Mitchell, R., and Ellison, D. H. (2003). WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J. Clin. Invest. 111, 1039–1045. doi: 10.1172/JCI200317443
Yang, S., Lo, Y., Wu, C., Lin, S., Yeh, C., Chu, P., et al. (2010). SPAK-knockout mice manifest gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 21, 1868–1877. doi: 10.1681/ASN.2009121295
Yang, S. S., Morimoto, T., Rai, T., Chiga, M., Sohara, E., Ohno, M., et al. (2007). Molecular pathogenesis of pseudohypoaldosteronism Type II: generation and analysis of a Wnk4D561A/+ knockin mouse model. Cell Metab. 5, 331–344. doi: 10.1016/j.cmet.2007.03.009
Yang, Y. S., Xie, J., Yang, S. S., Lin, S. H., and Huang, C. L. (2018). Differential roles of WNK4 in regulation of NCC in vivo. Am. J. Physiol. Ren. Physiol. 314, F999–F1007. doi: 10.1152/ajprenal.00177.2017
Zagórska, A., Pozo-Guisado, E., Boudeau, J., Vitari, A. C., Rafiqi, F. H., Thastrup, J., et al. (2007). Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J. Cell Biol. 176, 89–100. doi: 10.1083/jcb.200605093
Zambrowicz, B. P., Abuin, A., Ramirez-Solis, R., Richter, L. J., Piggott, J., BeltrandelRio, H., et al. (2003). Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc. Natl. Acad. Sci. U.S.A. 100, 14109–14114. doi: 10.1073/pnas.2336103100
Zhang, C., Wang, L., Su, X.-T., Lin, D.-H., and Wang, W.-H. (2015). KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am. J. Physiol. - Ren. Physiol. 308, F1288–F1296. doi: 10.1152/ajprenal.00687.2014
Zhang, J., Siew, K., Macartney, T., O’Shaughnessy, K. M., and Alessi, D. R. (2015). Critical role of the SPAK protein kinase CCT domain in controlling blood pressure. Hum. Mol. Genet. 24, 4545–4558. doi: 10.1093/hmg/ddv185
Zhang, C., Wang, L., Zhang, J., Su, X. T., Lin, D. H., Scholl, U. I., et al. (2014). KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc. Natl. Acad. Sci. U.S.A. 111, 11864–11869. doi: 10.1073/pnas.1411705111
Zhang, C., Wang, Z., Xie, J., Yan, F., Wang, W., Feng, X., et al. (2011). Identification of a novel WNK4 mutation in chinese patients with pseudohypoaldosteronism Type II. Nephron Physiol. 118, 53–61. doi: 10.1159/000321879
Zhang, J., Gao, G., Begum, G., Wang, J., Khanna, A. R., Shmukler, B. E., et al. (2016). Functional kinomics establishes a critical node of volume-sensitive cation-Cl- cotransporter regulation in the mammalian brain. Sci. Rep. 6, 1–19. doi: 10.1038/srep35986
Zhang, L., Huang, C. C., Dai, Y., Luo, Q., Ji, Y., Wang, K., et al. (2020). Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 10:63. doi: 10.1038/s41398-020-0692-2
Zhang, Y. L., Chen, P. X., Guan, W. J., Guo, H. M., Qiu, Z. E., Xu, J. W., et al. (2018). Increased intracellular Cl- concentration promotes ongoing inflammation in airway epithelium article. Mucosal Immunol. 11, 1149–1157. doi: 10.1038/s41385-018-0013-8
Zhao, H., Nepomuceno, R., Gao, X., Foley, L. M., Wang, S., Begum, G., et al. (2017). Deletion of the WNK3-SPAK kinase complex in mice improves radiographic and clinical outcomes in malignant cerebral edema after ischemic stroke. J. Cereb. Blood Flow Metab. 37, 550–563. doi: 10.1177/0271678X16631561
Keywords: distal convoluted tubule, GABAergic activity, cell volume regulation, intracellular chloride concentration, arterial blood pressure, potassium
Citation: Murillo-de-Ozores AR, Chávez-Canales M, de los Heros P, Gamba G and Castañeda-Bueno M (2020) Physiological Processes Modulated by the Chloride-Sensitive WNK-SPAK/OSR1 Kinase Signaling Pathway and the Cation-Coupled Chloride Cotransporters. Front. Physiol. 11:585907. doi: 10.3389/fphys.2020.585907
Received: 21 July 2020; Accepted: 29 September 2020;
Published: 20 October 2020.
Edited by:
Alexander A. Mongin, Albany Medical College, United StatesReviewed by:
Dandan Sun, University of Pittsburgh, United StatesCopyright © 2020 Murillo-de-Ozores, Chávez-Canales, de los Heros, Gamba and Castañeda-Bueno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Castañeda-Bueno, bWFyaWEuY2FzdGFuZWRhYkBpbmNtbnN6Lm14; bWNhc3RhODVAeWFob28uY29tLm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.