
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 30 October 2020
Sec. Clinical and Translational Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.538701
 Juhoon So1†
Juhoon So1† Mylarappa Ningappa2†Joseph Glessner3†
Mylarappa Ningappa2†Joseph Glessner3† Jun Min4†Chethan Ashokkumar2
Jun Min4†Chethan Ashokkumar2 Sarangarajan Ranganathan5Brandon W. Higgs2
Sarangarajan Ranganathan5Brandon W. Higgs2 Dong Li3Qing Sun2Lori Schmitt6Amy C. Biery5Steven Dobrowolski5Christine Trautz2Leah Fuhrman2Molly Christine Schwartz7
Dong Li3Qing Sun2Lori Schmitt6Amy C. Biery5Steven Dobrowolski5Christine Trautz2Leah Fuhrman2Molly Christine Schwartz7 Nikolai Thomas Klena7Joseph Fusco8Krishna Prasadan8Morayooluwa Adenuga2Nada Mohamed8Qi Yan9Wei Chen9William Horne10Anil Dhawan11Khalid Sharif12
Nikolai Thomas Klena7Joseph Fusco8Krishna Prasadan8Morayooluwa Adenuga2Nada Mohamed8Qi Yan9Wei Chen9William Horne10Anil Dhawan11Khalid Sharif12 Deirdre Kelly12Robert H Squires13
Deirdre Kelly12Robert H Squires13 George K. Gittes8
George K. Gittes8 Hakon Hakonarson3Victor Morell14
Hakon Hakonarson3Victor Morell14 Cecilia Lo7
Cecilia Lo7 Shankar Subramaniam4*
Shankar Subramaniam4* Donghun Shin1*
Donghun Shin1* Rakesh Sindhi2*
Rakesh Sindhi2*Background/Aims: Infectious and genetic factors are invoked, respectively in isolated biliary atresia (BA), or syndromic BA, with major extrahepatic anomalies. However, isolated BA is also associated with minor extrahepatic gut and cardiovascular anomalies and multiple susceptibility genes, suggesting common origins.
Methods: We investigated novel susceptibility genes with genome-wide association, targeted sequencing and tissue staining in BA requiring liver transplantation, independent of BA subtype. Candidate gene effects on morphogenesis, developmental pathways, and ciliogenesis, which regulates left-right patterning were investigated with zebrafish knockdown and mouse knockout models, mouse airway cell cultures, and liver transcriptome analysis.
Results: Single nucleotide polymorphisms in Mannosidase-1-α-2 (MAN1A2) were significantly associated with BA and with other polymorphisms known to affect MAN1A2 expression but were not differentially enriched in either BA subtype. In zebrafish embryos, man1a2 knockdown caused poor biliary network formation, ciliary dysgenesis in Kupffer’s vesicle, cardiac and liver heterotaxy, and dysregulated egfra and other developmental genes. Suboptimal man1a2 knockdown synergized with suboptimal EGFR signaling or suboptimal knockdown of the EGFR pathway gene, adenosine-ribosylation-factor-6, which had minimal effects individually, to reproduce biliary defects but not heterotaxy. In cultured mouse airway epithelium, Man1a2 knockdown arrested ciliary development and motility. Man1a2–/– mice, which experience respiratory failure, also demonstrated portal and bile ductular inflammation. Human BA liver and Man1a2–/– liver exhibited reduced Man1a2 expression and dysregulated ciliary genes, known to cause multisystem human laterality defects.
Conclusion: BA requiring transplantation associates with sequence variants in MAN1A2. man1a2 regulates laterality, in addition to hepatobiliary morphogenesis, by regulating ciliogenesis in zebrafish and mice, providing a novel developmental basis for multisystem defects in BA.
Biliary atresia (BA) accounts for 30–50% of all liver transplants (LTx), worldwide, and has a major public health impact which can be alleviated with a better understanding of pathogenesis (Otte et al., 1994). The heterogeneous BA phenotype includes cardiovascular or gastrointestinal anomalies in addition to atretic extrahepatic bile ducts and defies a unifying explanation (Schwarz et al., 2013). Surgical reconstruction restores bile drainage from the liver and prevents certain death during infancy, but fails over time leading to LTx in three–fourths of all children (Karrer et al., 1996). Major extrahepatic anomalies including laterality defects, which are characteristic of syndromic BA, suggest a multifactorial origin. However, minor extrahepatic anomalies in some children with isolated BA indicate that syndromic and isolated BA may also share common origins (Schwarz et al., 2013). As evidence, abnormal cilia are present on cholangiocytes in both syndromic and non-syndromic BA (Chu et al., 2012) Ciliary dysfunction predisposes to laterality defects (Afzelius, 1995). Although genetic factors are invoked in syndromic BA and remain to be proved in a controlled study, genome-wide association studies (GWAS) of cohorts predominantly comprised of isolated BA have identified multiple susceptibility genes strengthening the evidence for a common basis. The three susceptibility genes, adducin-3, glypican-1 (GPC1), and adenosine-ribosylation-factor-6 (ARF6), all cause biliary dysgenesis upon knockdown in zebrafish embryos likely by altering cell mobility during liver development (Cheng et al., 2013; Cui et al., 2013; Ningappa et al., 2015a,b). Additional susceptibility genes must also explain the extrahepatic anomalies of BA.
Here, we evaluate susceptibility to end-stage liver disease, which requires LTx in children with BA, independent of BA subtype. This restricts our GWAS to a cohort with homogeneous end-stage liver disease, without the heterogeneity introduced by including children without LTx, and in whom the liver disease has likely arrested at various stages. We identify MAN1A2 as a susceptibility gene, which explains the multisystem involvement in BA. Our experimental approach combines GWAS in children with LTx for BA, zebrafish and mouse models, and liver transcriptome analysis in human BA and animal models.
All human subjects and animal studies were performed under protocols approved by the University of Pittsburgh’s Institutional Review Board and Animal Care and Use Committee, respectively. We first used GWAS to identify SNPs associated with BA independent of BA subtype in 137 Caucasian LTx recipients at our single-center (Ningappa et al., 2015a). Targeted next-generation sequencing of the candidate, MAN1A2, identified potentially causal SNPs in LD with BA-associated SNPs. RT-qPCR and immunohistochemistry characterized expression of the MAN1A2 gene and protein in affected BA liver. The effect of MAN1A2 on biliary morphogenesis, left-right patterning, and ciliogenesis was evaluated with knockdown in zebrafish and mouse airway epithelial cell cultures, respectively (Li et al., 2015; Ningappa et al., 2015a,b). For further corroboration, we evaluated whether liver histology was abnormal in Man1a2–/– mice, which are known to experience respiratory failure, and in mice with heterotaxy due to knockout of Dnah11 (Bartoloni et al., 2002). Dnah11 is representative of dysregulated ciliary genes in the liver transcriptome of human BA and Man1a2–/– mice and man1a2 morphant zebrafish. Details are provided in Supplementary Material.
Self-reported Caucasian BA cases and healthy controls were genotyped in three batches. The first two batches of 52 discovery cases and 1969 controls, and 53 replication cases and 400 controls, respectively, were genotyped with the HumHap Infinium 550K (Illumina) and Quad Infinium 660k (Illumina) at the Center for Applied Genomics at the Children’s Hospital of Philadelphia. Replicated candidate SNPs were assayed with TaqMan SNP Genotyping Assays in the third batch of 32 BA cases. Genotyping calls with Taqman assays were identical to those called with SNP arrays in a subset of 19 randomly selected cases from the first two batches.
A final list of 507540 and 522193 SNPs in both SNP array versions of HumHap Infinium 550K and Quad Infinium 660k were identified, respectively, after excluding missing genotype data (>0.10), Hardy-Weinberg equilibrium (HWE) test (p = 0.00005), and low minor allele frequency (MAF < 0.01) as described in Supplementary Material. The genotyping success rate for SNPs was 0.973456 and 0.851671 in the discovery and replication cohorts, respectively, and one BA discovery case was excluded for missing genotype data above the threshold. After multidimensional scaling analysis (MDS) to select cases and controls with similar genetic substructure, association testing was performed in 44 of 51 BA cases and 1,713 of 1,969 controls in the discovery cohort, and 45 of 53 BA cases and 347 of 400 controls in the replication cohort with the Cochran-Armitage trend test in PLINK (Supplementary Figure 1; Purcell et al., 2007). MAF were compared between cases and controls in each cohort. The candidate gene was one which contained the highest ranked two or more SNPs in the same gene (p < 0.005), and which retained this level of significance in the replication cohort.
In association testing, 3,322 SNPs achieved significance (P = 0.005) in the discovery cohort with 24 of these SNPs also remaining significant in the replication cohort (Supplementary Tables 1, 2). The top ranked flanking SNP rs6657965 in the MAN1A2 gene demonstrated significantly higher MAF in the discovery cohort (p = 7.18E-05) and replication cohort (p = 9.73E-04) and in the 89 combined BA cases (p = 1.72E-08). The two top-ranked intronic SNPs rs12131109 and rs7531715 in LD (r2 > 0.8) in the MAN1A2 gene demonstrated significantly higher MAF in the discovery cohort (p = 2.14E-04 and p = 1.89E-04, respectively), replication cohort (p = 9.73E-04 and p = 9.73E-04, respectively) and in 89 combined BA cases (4.59E-08 and p = 4.59E-08, respectively) (Table 1). Genome-wide, regional Manhattan plots and QQ plots illustrate these SNPs in the MAN1A2 locus (Supplementary Table 1 and Supplementary Figures 2, 3). In the combined analysis, we excluded the 32 BA cases that were genotyped with Taqman assays because beyond self-reported ethnicity, we have no data regarding where these 32 cases may fall in the MDS space. Therefore, these p-values are a bit less significant than p-values if all 121 cases and all 2060 controls with self-reported Caucasian ethnicity are compared, for example, p = 4.59E-08 (n = 89 vs. 2060) vs. p = 6.97E-10 (n = 121 vs. 2060), respectively, for rs753175. However, the odds ratio estimate remains approximately the same and SNPs achieved genome-wide significance after adjusting for False Discovery Rate using Benjamini Y method (FDR_BY) (Table 1).
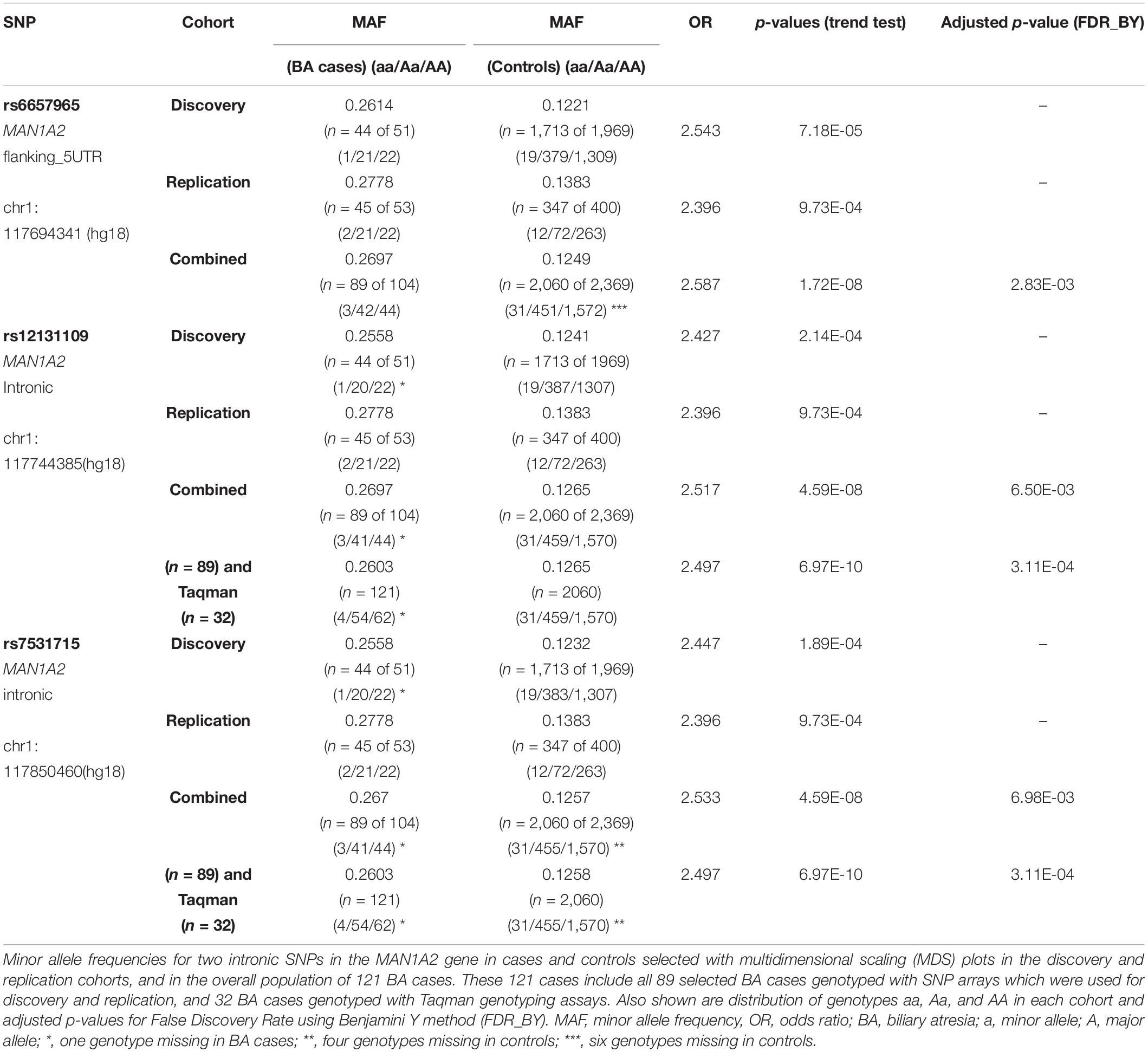
Table 1. SNPs associated with BA.
Interestingly, our BA cohort of 121 children included 98 with biliary involvement only and 23 children with extrahepatic abdominal and cardiovascular anomalies (Supplementary Table 3). The MAF of rs7531715 were 0.265 and 0.239, respectively, and not significantly different (p = 0.859, NS, test of proportions).
In 43 children with BA, targeted NGS of the MAN1A2 gene inclusive of the 20 kb upstream and downstream sequences identified 498 SNP variants. Genotyping calls for rs7531715 and rs12131109 with NGS were identical to those determined with SNP arrays (n = 38) and Taqman assay (n = 2) for 40 of 43 BA cases (Supplementary Material). For three of 43 BA cases, genotyping calls made with SNP arrays and Taqman assays for the two BA-associated SNPs were identical but did not agree with genotype calls made with NGS. Nineteen SNPs in LD (r2≥ 0.8) with the two BA-associated SNPs are negatively correlated with MAN1A2 expression at p ≤ 1.2067E-09 in Caucasian populations (Supplementary Table 4; Stranger et al., 2007). Among these SNPs, rs10923326 is strongly associated with MAN1A2 expression (p = 3.42e-19) in the liver (Figure 1A; Schadt et al., 2008). Novel missense, nonsense, or splice-altering variants were not identified.
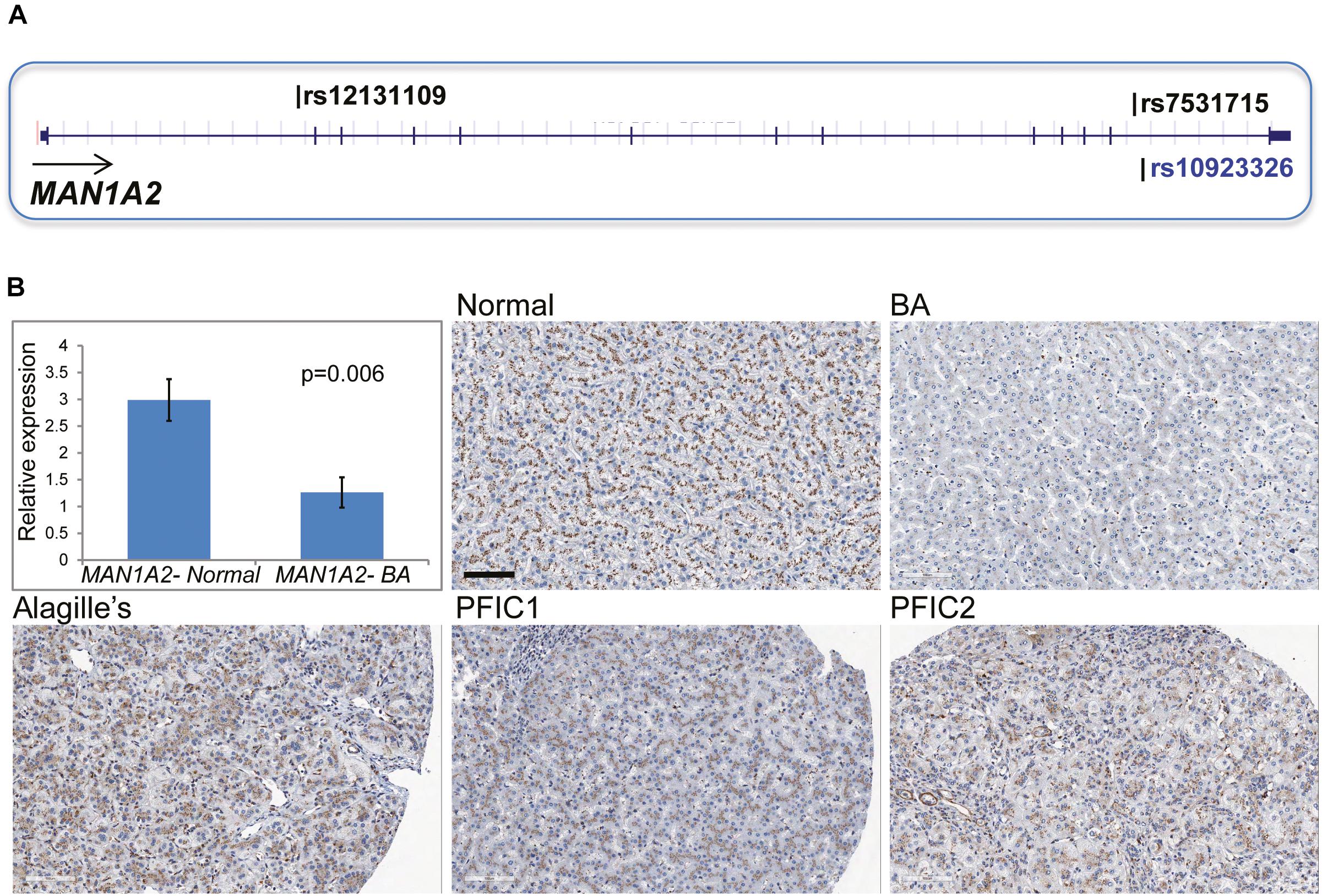
Figure 1. (A) Location of MAN1A2 SNPs associated with BA. The SNPs rs7531715 and rs12131109 are associated with BA in GWAS. The SNP rs10923326 in LD (r2 = 1) with rs7531715 is significantly associated with gene expression in the liver. (B) MAN1A2 expression in diseased BA liver. Bar diagram shows expression of the exon 6 and 7 region of MAN1A2 in liver tissue from BA cases [male: female 1:5, mean (SEM) age at LTx, 1.33 (0.55) years] and healthy controls. The remaining five panels show diffuse strong (3+) granular immunostaining of MAN1A2 in the hepatocyte cytoplasm along pericanalicular membranes in normal human allografts, and liver from children with Alagille’s syndrome and types 1 and 2 progressive familial intrahepatic cholestasis (PFIC). Reduced MAN1A2 immunostaining is seen in BA liver (x200).
Compared with 6 normal human liver samples, liver samples from 6 children with BA demonstrated lower expression of the MAN1A2 exon 6–7 junction (2.4-fold, p = 0.006, Figure 1B) and exon 10 (2.2-fold, p = 0.009, Supplementary Table 5) with reverse transcription quantitative PCR. MAN1A2 immunostaining revealed a diffuse cytoplasmic, peri-canalicular granular staining pattern in normal liver tissue and in diseased liver with Alagille’s syndrome and familial intrahepatic cholestasis, examples of childhood cholestatic diseases (Figure 1B). In contrast, MAN1A2 expression was markedly decreased in BA liver tissue. Staining characteristics for two normal and five BA liver samples are summarized in Supplementary Table 6.
To determine if MAN1A2 is required for proper biliary development, we first examined and found that man1a2 is expressed in the liver of developing zebrafish embryos/larvae from 48 h post-fertilization (hpf) at least to 96 hpf (Figure 2A, arrowheads). To perform knockdown experiments, we designed two different MOs, ATG and splicing MOs, and validated them (Supplementary Figures 4A–C). To examine whether bile secretion from hepatocytes into biliary ducts was defective in man1a2 morphants, we used the fluorescently labeled fatty acid reporter PED6, which is metabolized into bile in hepatocytes and accumulates in the gallbladder (Colland et al., 2004) PED6 accumulation in the gallbladder was greatly reduced in the ATG or splicing morphants compared to controls (Figure 2B and Supplementary Figure 4D). We further examined this potential biliary defect by evaluating the intrahepatic ductal network with another fluorescent lipid reporter, BODIPY C5 (Farber et al., 2001). Biliary ductules were well connected to each other in controls, frequently disconnected in man1a2 morphants (Figure 2C), and barely connected to each other in severe cases (Figure 2C). Hepatocyte bile canaliculi were also abnormal and were very short and rounded in the morphants compared with thin and elongated canaliculi in controls (Figure 2C, arrowheads). This was further confirmed by the abnormal expression pattern of Abcb11, a bile salt pump located in bile canaliculi (Figure 2D). Moreover, BECs appeared more clustered in the morphants than in controls. Analyses of the distribution of Tp1:H2B-mCherry+ nuclei of BECs in the entire liver revealed that the percentage of BECs in cluster of four or more cells was significantly higher in the morphants than in controls (Supplementary Figure 5A). Since BECs are dispersed between 60 hpf and 5 days post-fertilization (dpf) and BEC filopodia actively protrude between 70 and 96 hpf (Ningappa et al., 2015b), this clustering phenotype observed in man1a2 morphants suggests reduced filopodial protrusion in the morphants. In the Tg(Tp1:GFP) line that expresses GFP in BECs (Lorent et al., 2010; Delous et al., 2012), the length of BEC filopodia at 76 and 96 hpf and the length of interconnecting bile preductules at 5 dpf were significantly reduced in man1a2 morphants compared to controls (Supplementary Figure 5B). Interestingly, despite the intrahepatic biliary defects, the gallbladder and the extrahepatic duct appeared morphologically normal in man1a2 morphants (Supplementary Figure 6). Collectively, these data indicate the essential role of man1a2 in the proper formation of hepatocyte bile canaliculi and the intrahepatic biliary network.
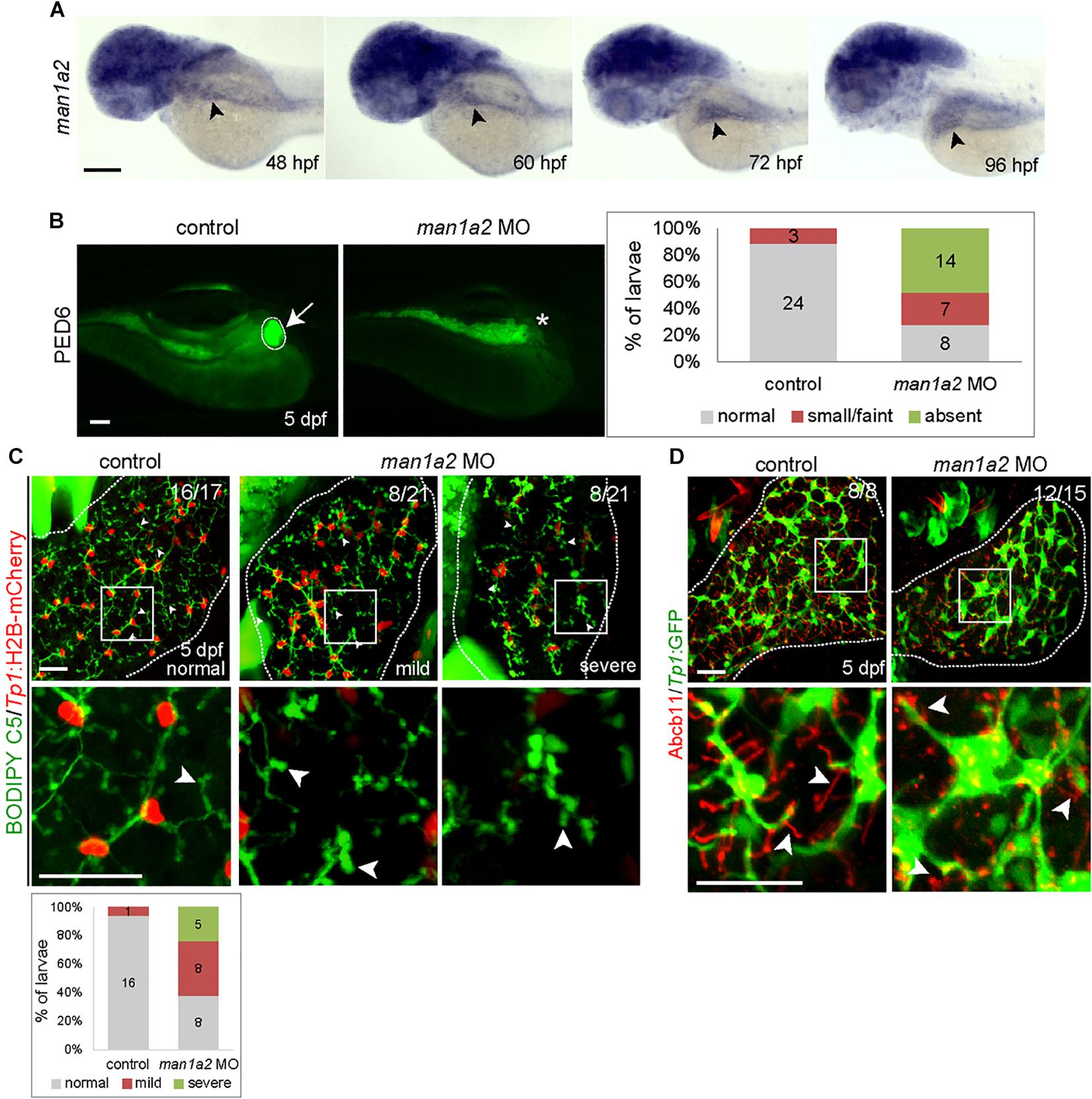
Figure 2. man1a2 knockdown results in developmental biliary defects in zebrafish. (A) Whole-mount in situ hybridization image showing man1a2 expression in developing embryos/larvae. Arrowheads point to the liver. (B) Epifluorescence images showing PED6 accumulation in the gallbladder. The arrow points to the gallbladder outlined by a dotted line; the asterisk denotes gallbladder location. Based on PED6 levels in the gallbladder, larvae were divided into three groups: normal, small/faint, and absent. Graph showing the percentage of larvae in each group. Numbers in the graph indicate the number of larvae in each group. (C) Confocal images of the liver showing the intrahepatic biliary network, as revealed by BODIPY C5 labeling (green, biliary ductal network) and Tp1:H2B-mCherry (red, BEC nuclei) expression. Based on the severity of biliary defects, larvae were divided into three groups: normal, mild, and severe. Higher magnification images of the square regions are also shown. Dotted lines outline the liver Graph shows the percentage of larvae in each group. Numbers in the graph indicate the number of larvae in each group. Arrows point to bile canaliculi; dotted lines outline the liver. (D) Confocal images of the liver showing the expression of Tp1:GFP (green) and Abcb11 (red) for biliary structure and hepatocyte bile canaliculi, respectively. Higher magnification images of the square regions are also shown. Arrows point to bile canaliculi; Dotted lines outline the liver. Numbers in the upper right corner are the fraction of larvae exhibiting the representative phenotype shown. Scale bars: 100 (A,B), 25 (C,D) μm.
While analyzing biliary morphogenesis, we noticed that liver position was reversed in a significant portion of man1a2 morphants. Since BA is associated with laterality defects (Schwarz et al., 2013), we investigated whether man1a2 also regulates laterality. Whole-mount in situ hybridization with 4 different hepatic markers confirmed that liver position was reversed in 40% of man1a2 morphants (n = 70, Supplementary Figure 7). Cardiac looping direction, as assessed by the expression of the cardiomyocyte marker myl7, was also reversed in 37% of the morphants (n = 38, Figure 3A). Double whole-mount in situ hybridization with myl7 and foxa3 probes revealed that the position of endoderm-derived organs, such as the liver and pancreas, was also reversed in embryos with reversed or no cardiac looping (Figure 3A). This left-right asymmetry prompted us to examine motile cilia with anti-acetylated tubulin antibody labeling in Kupffer’s vesicle in man1a2 morphants, because cilia create a directional fluid flow which regulates left-right patterning in zebrafish (Essner et al., 2005). We used the Tg(dusp6:d2GFP) line to visualize Kupffer’s vesicle cells (Molina et al., 2007). Both cilia length and number were significantly reduced in the morphants at 12 hpf compared to controls indicating the essential role of man1a2 in left-right patterning (Figure 3B).
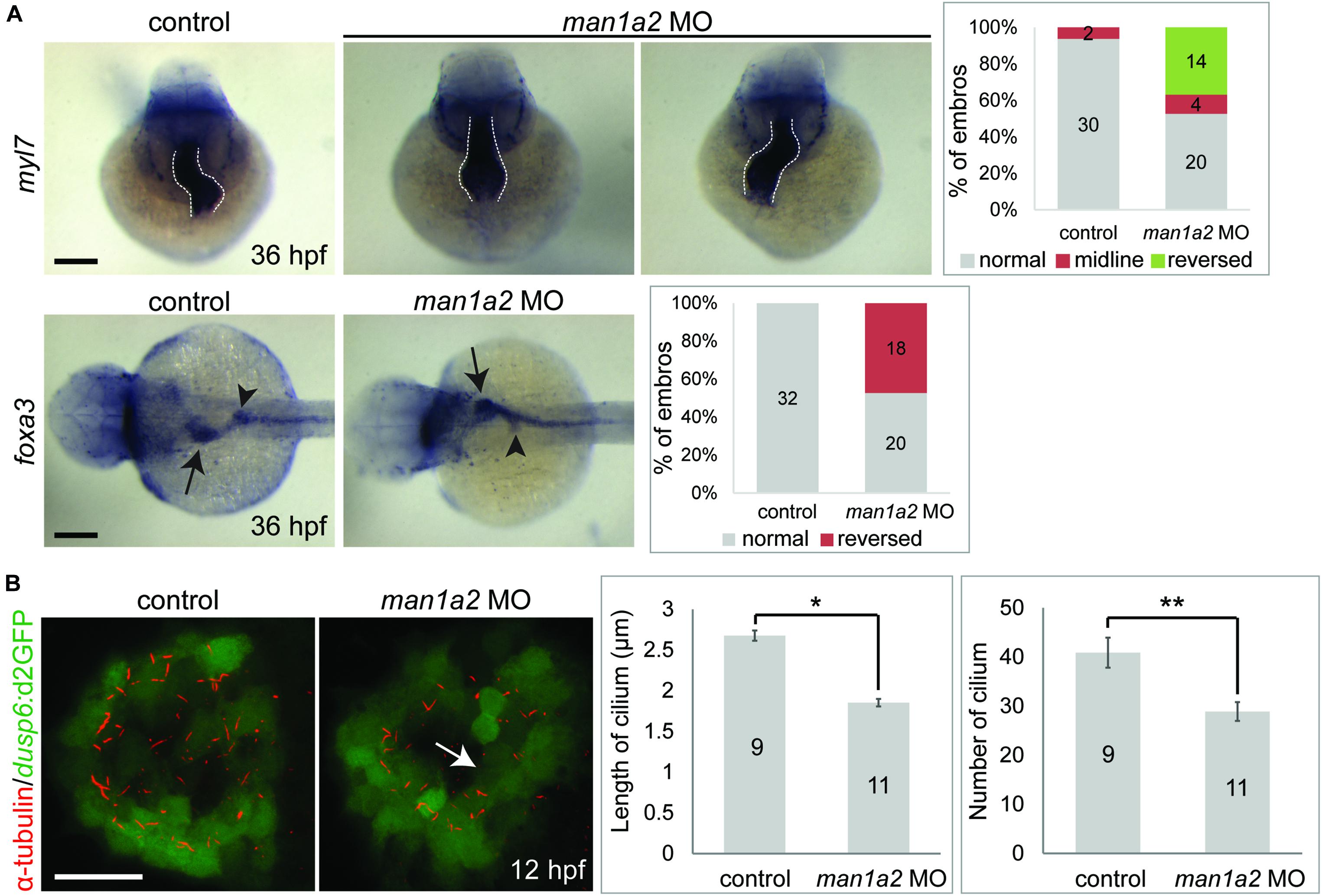
Figure 3. man1a2 knockdown results in laterality defects in zebrafish. (A) Whole-mount in situ hybridization images showing myl7 (cardiomyocytes) and foxa3 (endodermal cells) expression at 36 hpf, which was used to determine the laterality of embryos. Three patterns of heart looping (normal, reversed, and midline) and two patterns of visceral organ position (normal and reversed) were detected and were quantified in graphs. Numbers in the graph indicate the number of larvae in each group. Arrows and arrowheads point to the liver and the dorsal pancreas, respectively; dotted lines outline the heart. Scale bars: 100 μm. (B) Confocal images of Kupffer’s vesicle showing the expression of dusp6:d2GFP (green, Kupffer’s vesicle cells) and acetylated tubulin (red, cilia). Both cilia length and number were significantly reduced in man1a2 MO-injected larvae (n = 11) compared to controls (n = 9), as shown in graphs. Scale bar: 50 μm. Error bars: ± SEM. *p < 0.001; **p < 0.005.
To determine whether man1a2 knockdown affects biliary morphogenesis via known developmental pathways, we evaluated the expression of key genes in the EGFR (egfra, egfrb, arf6a, and arf6b), Hedgehog (ptch1, gli1, and gli2a), and the TGF-β (tgfb1a, tgfb1b, smad7, and smad9) signaling pathways. In previous zebrafish studies, biliary dysgenesis was induced with aberrant signaling of either the EGFR or Hedgehog pathway (Cui et al., 2013; Ningappa et al., 2015b). MAN1A2 can bind to SMAD9, which can mediate TGF-β signaling (Colland et al., 2004). SMAD7 promotes cross-talk between EGFR and TGF-β signaling (Kang et al., 2012). man1a2 knockdown induced consistent downregulation of egfra and upregulation of gli1, smad7, and smad9 in all batches of zebrafish embryos that were tested (Supplementary Table 7), indicating the involvement of EGFR and TGF-β signaling in the effect of man1a2 knockdown in the liver. RNA sequencing of pooled batches of liver tissue, one from untreated and one from morphant zebrafish revealed additional differentially expressed genes in these pathways (Table 2).

Table 2. Selected differentially expressed liver genes from human BA, man1a2 morphant zebrafish and Man1a2–/– mouse (see Supplementary Tables 9–12 for complete list).
We hypothesized that man1a2 can genetically interact with other known genes or pathways implicated in BA (Ningappa et al., 2015a), and because man1a2 knockdown affects EGFR and TGF-β signaling (Supplementary Table 7). We first tested if man1a2 and arf6 can synergistically influence proper intrahepatic biliary network formation by co-injecting suboptimal doses of man1a2 and arf6 MOs, which alone did not cause any obvious biliary defects (1.5 and 0.5 ng, respectively). Compared to single-injected larvae, co-injected larvae demonstrated greatly reduced PED6 accumulation in the gallbladder (Figure 4A) and a severely impaired intrahepatic ductal network in 57% of co-injected larvae on BODIPY C5 labeling (n = 14; Figure 4B). Intriguingly, liver position was not reversed in the co-injected larvae (data not shown). Next, we tested if man1a2 and EGFR signaling can synergistically influence biliary morphogenesis. Previously, blocking EGFR signaling in zebrafish larvae with the EGFR inhibitor AG1478 resulted in biliary morphogenesis defects (Ningappa et al., 2015b). Zebrafish embryos were first injected with the suboptimal dose of man1a2 MO and then treated from 60 hpf with a suboptimal dose of AG1478 (1 μM). This treatment impaired proper biliary network formation, as accessed by PED6 (Figure 4C) and BODIPY C5 (Figure 4D) labeling. Altogether, these data suggest that man1a2 can regulate intrahepatic biliary network formation via interactions with arf6, another BA susceptibility gene, and EGFR signaling.
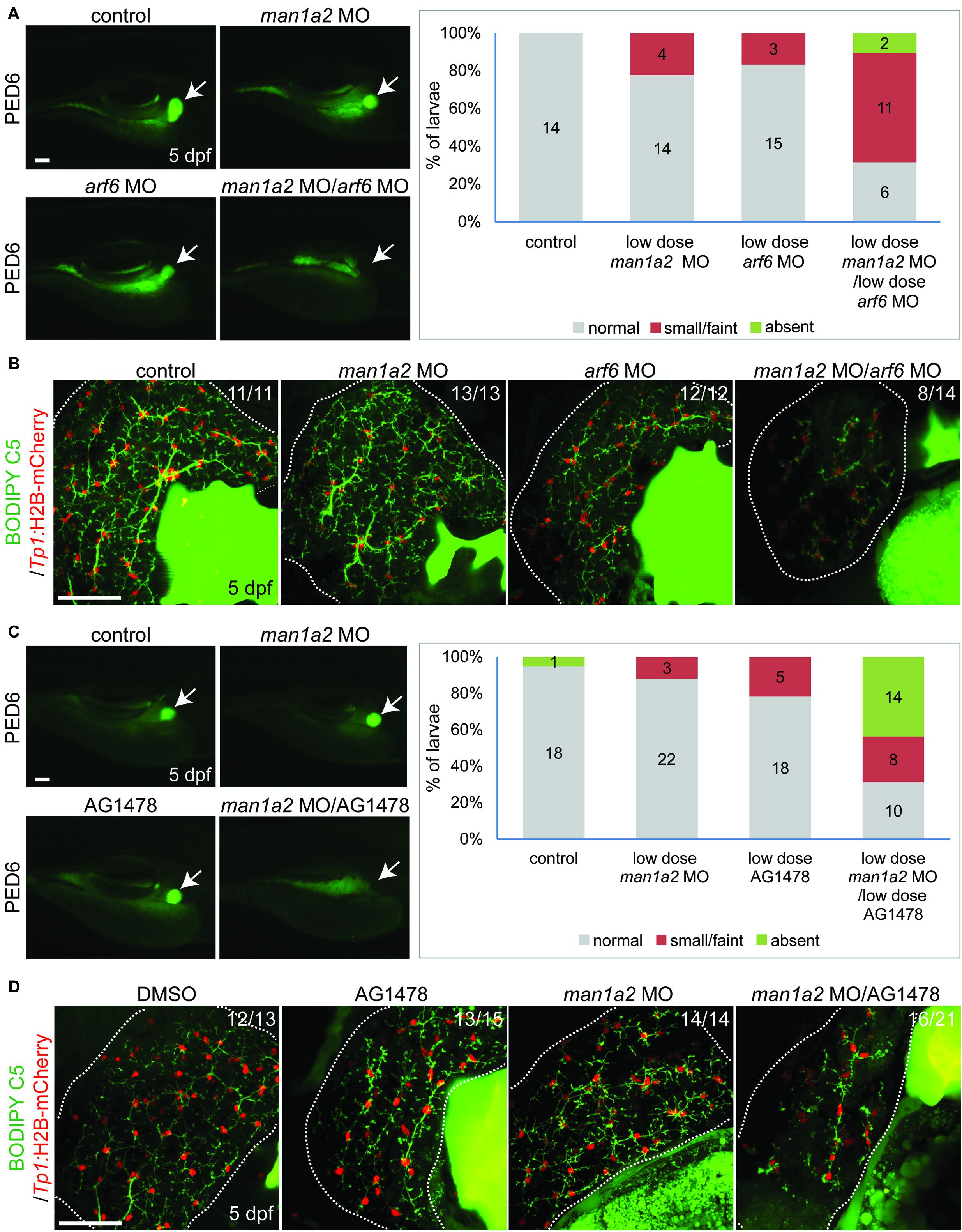
Figure 4. Genetic interaction among man1a2, arf6 and EGFR signaling in intrahepatic biliary network formation. (A,C) Epifluorescence images of larvae treated with PED6 and its quantification. Suboptimal doses of man1a2 (1.5 ng) and arf6 (0.5 ng) MOs were singly or in combination injected into embryos at the one-cell stage (A). Embryos were first injected with the suboptimal dose of man1a2 MO (1.5 ng) and from 60 hpf treated with a suboptimal dose of AG1478 (1 μM) (C). Arrows point to the gallbladder. Graphs show the percentage of larvae in each group. Numbers in the graph indicate the number of larvae in each group. (B,D) Confocal images of the liver showing the intrahepatic biliary network, as revealed by BODIPY-FL C5 feeding (green, biliary ductal network) and Tp1:H2B-mCherry (red, BEC nuclei) expression. Single injection of suboptimal doses of either man1a2 or arf6 MO did not impair proper intrahepatic biliary network formation, whereas their co-injection resulted in defective biliary network formation (B). The biliary network formation was not impaired in larvae only treated with AG1478, whereas it was impaired in AG1478-treated larvae with man1a2 partially knockdown (D). Numbers in the upper right corner are the fraction of larvae exhibiting the representative phenotype shown. Dotted lines outline the liver. Scale bars: 50 μm.
Because specification of left-right patterning requires motile cilia function in the embryonic node in mice or Kupffer’s vesicle in zebrafish embryos, we evaluated Man1a2 knockdown effects in reciliating mouse respiratory epithelia as a proxy for the embryonic node. In our previous studies, genes required for motile cilia function in the trachea are also usually required for motile cilia function for left-right patterning in the embryonic node (Li et al., 2015). Many such genes are known to cause human primary ciliary dyskinesia (PCD) (Bergström et al., 2012). Transfection of Man1a2 siRNA in the respiratory epithelia before placing cells into suspension culture for reciliation showed more than 80% reduction of Man1a2 expression by qPCR (Figure 5A), and marked inhibition of ciliogenesis resulting in very short cilia and markedly reduced percent ciliation in the Man1a2 siRNA treated cultures (Figure 5C). Videomicroscopy showed that most of these cilia were immotile, with only a few areas exhibiting slow dyskinetic ciliary motion in transfected cells (Supplementary Video 2). In comparison, scrambled siRNA knockdown had no effect on ciliogenesis and the ciliary motion remained robust with normal pattern of ciliary beat (Figure 5B and Supplementary Video 1). Together these findings indicate an important role for Man1a2 in regulating motile cilia function.
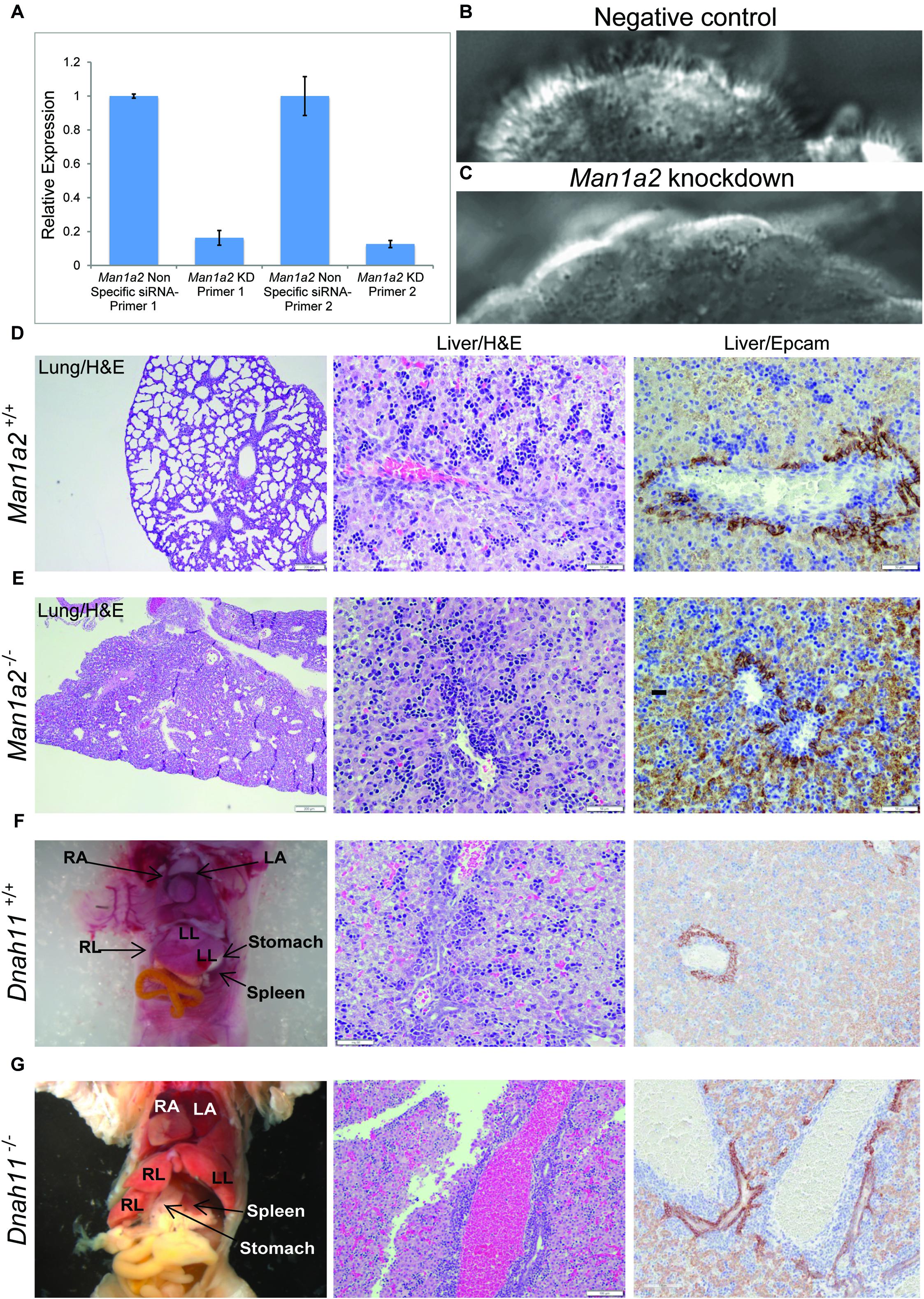
Figure 5. Man1a2 knockdown produces cilia defects in reciliating mouse respiratory epithelia. (A) Relative expression of Man1a2 in respiratory epithelia after siRNA transfection. (B) Control epithelia are well ciliated with well-coordinated ciliary beat with a full stroke (Supplementary Video 1). (C) Man1a2-siRNA-transfected respiratory epithelia exhibited very short cilia that were mostly immotile except for a few areas with slow dyskinetic ciliary beat with incomplete stroke (Supplementary Video 2). (D,E) Man1a2 knockout mice with lung involvement also exhibit liver inflammation. (D) Man1a2+/+ liver shows a portal area with indistinct bile ducts, abundant extramedullary hematopoiesis (EMH, H&E x200), and minimal ductular reaction (Epcam x200). The lung shows expanded alveoli and thin interalveolar septa. (E) Man1a2–/– liver shows expanded portal areas with inflammation including EMH (H&E) and ductular reaction (Epcam x 200). The lung shows thick septa with sparse, unexpanded alveoli. The Man1a2–/– liver transcriptome shows several dysregulated ciliary genes, including Dnah11 which causes PCD (Table 2 and Supplementary Table 10). (F,G). Dnah11–/– mice with situs inversus also exhibit biliary inflammation. (F) Dnah11+/+ mouse with normal location of heart and abdominal organs which includes two left and one right liver lobes, normal liver histology with EMH and some bile ducts in portal area. (G) Dnah11–/– null mouse shows situs inversus with cardiac heterotaxy, inverted lobation of the liver (two right lobes), right-sided stomach and more centrally located spleen. Liver shows portal congestion and bile duct proliferation. All experiments were performed in triplicate.
To corroborate the effects of Man1a2 on liver development, we bred Man1a2+/– mice with heterozygous exon 2 deletion (Tremblay et al., 2007). We confirmed that compared with Man1a2+/+ WT mice, Man1a2–/– null mice exhibit unexpanded alveoli, thick interalveolar lung septa, and respiratory failure, and a grossly normal liver as reported previously. However, upon detailed histological evaluation, the Man1a2–/– liver showed portal expansion, inflammation and ductular reaction, suggestive of biliary inflammation, compared with Man1a2+/+ WT liver (Figure 5). The reverse transcription quantitative PCR analysis showed complete loss of exon 2 expression in the lung and the liver. Reduced expression of mid-level (exon 5–6 junction) and terminal (exon 11–12 junction) exons in both organs in null mice, compared with WT achieved significance in the lung, and not the liver (Supplementary Table 8).
To evaluate cilia formation, immunostaining of the ciliary marker Arl13b was performed in explanted lung and liver tissue from WT and null pups. Explanted lung tissue from Null Man1a2 pups demonstrated 15.96% Arl13b+ cells compared with 86.5% Arl13b+ cells in WT Man1a2 pups (P = 3.05E-05) (Figure 6). Arl13b staining protocols showed inconsistent staining in explanted liver tissue.
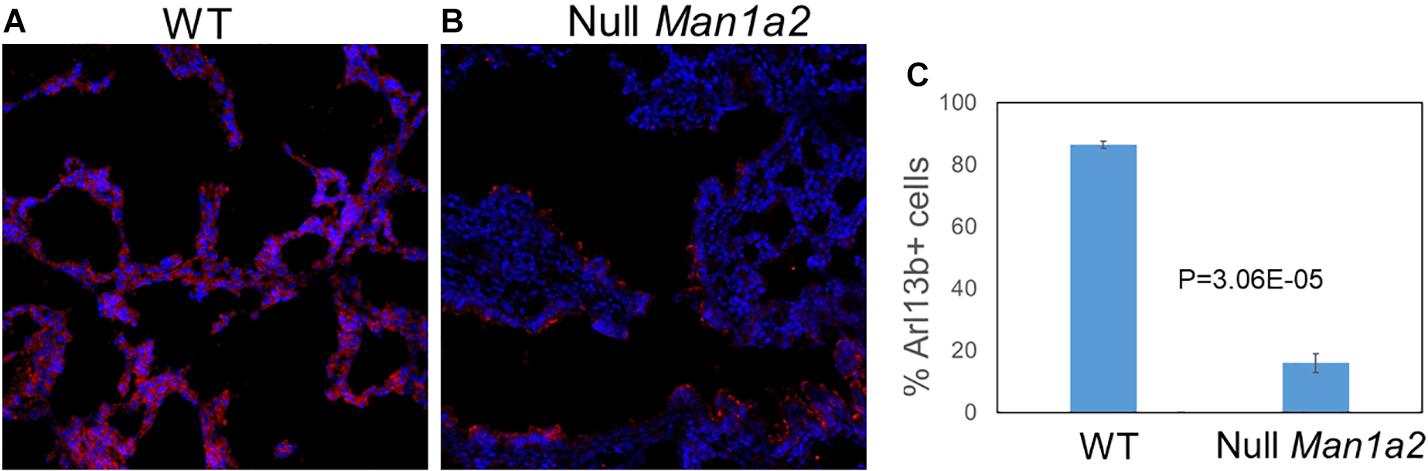
Figure 6. Man1a2 knockdown in mouse leads to decreased cilia development in lung. Confocal microscopy showed the cilia marker Arl13b (red) and nuclei (blue, Dapi) in the lung from the (A) WT (Man1a2+/+) and (B) Null Man1a2(−/−) pups. Scale bars: 50 μm. (C) The bar diagram showed the percent Arl13b+ cells (% Dapi) in lung from four WT and four Null Man1a2 pups. Error bars: ± SEM.
Given dysregulated developmental pathways in man1a2 morphant zebrafish liver, we asked whether developmental and cilia pathways, and genes associated with aberrant laterality in humans were also dysregulated in the whole transcriptome of Man1a2–/– liver and lung and human BA liver (De Luca et al., 2010; Shiraishi and Ichikawa, 2012; Van Dam et al., 2013; Andersen et al., 2014; Bergström et al., 2012; Cerami et al., 2011). Reverse transcription qPCR showed reduced MAN1A2 expression (Supplementary Tables 5, 8) and RNA sequencing showed dysregulated genes in EGF, TGF, hedgehog and ciliogenesis pathways in human BA and Man1a2–/– mouse liver and lung compared with corresponding controls (Table 2 and Supplementary Tables 9–11). Additionally, of 101 genes reportedly associated with PCD or human congenital heart disease with extracardiac and laterality defects, dysregulation of 7, 29, and 25 genes, respectively, was seen in human BA liver, Man1a2–/– liver and Man1a2–/– lung (Supplementary Tables 9–11; De Luca et al., 2010; Shiraishi and Ichikawa, 2012; Andersen et al., 2014; Bergström et al., 2012). In the BA liver, dysregulated DNAH11 is illustrative of ciliary genes which are implicated in multisystem laterality defects in PCD and heterotaxy (Bartoloni et al., 2002). Therefore, we asked whether previously reported situs inversus in the Dnah11–/– null mouse was accompanied by biliary pathology. Detailed histologic evaluation revealed bile ductular proliferation consistent with bile duct inflammation which is also seen in BA in Dnah11–/– mice, compared with a Dnah11+/+ WT mouse (Figure 5). Therefore, a dysregulated MAN1A2 gene can contribute to multisystem developmental defects directly, via differential expression of exon-level transcripts in different tissues, or via genes which regulate ciliogenesis and left-right patterning.
Conducted in an exploratory cohort of 137 Caucasian children with BA, all of whom received LTx at our center, GWAS and biological interrogation of the MAN1A2 locus in zebrafish and mouse models implicates ciliary dysgenesis as a common potential contributor to the syndromic and non-syndromic subtypes of BA. Three SNPs in this gene locus, two intronic and the third in the 5′prime region emerged as the top ranked SNP trio in screening and replication cohorts, each tested with a different version of the SNP array. In the combined cohort, these SNPs achieved genome-wide significance with OR exceeding 2 and p < 10E-08, statistical attributes which have characterized previous GWAS conducted in modest cohorts. Because of these results, the association of MAN1A2 with BA can at first be dismissed as one of many, for example, GPC1, ADD3, EFEMP1, and ARF6, which have failed exact replication in multiple studies. However, the ADD3 locus has been replicated on the basis of SNPs that are intronic in Chinese and 3′ regulatory region in Caucasian children. Thus, the metric for replication may have to evolve to include gene function and its associated mechanism or pathway, and biological validation. Remarkably, each of these five potential susceptibility genes are members of the ciliogenesis and planar polarity effector network of proteins, or CPLANE network. The CPLANE proteins interact with the ciliopathy-associated protein Jbts17 at the base of cilia to recruit intraflagellar transport (IFT-A) proteins. Sequence variants in genes for CPLANE proteins induce ciliopathies in mice and humans.
This association between MAN1A2 and BA is applicable to Caucasians but not East Asians, in whom the SNP locus is monomorphic. Further, although the associated SNPs do not affect MAN1A2 expression in public databases, both are in LD (r2 > 0.8) with several others identified by targeted sequencing of subject samples, which are known MAN1A2 eQTLs (Supplementary Table 4; Stranger et al., 2007). Of these SNPs, rs10923326 is strongly associated with MAN1A2 gene expression (p = 3.42E-19) in previous human liver studies (Schadt et al., 2008). This eQTL function explains decreased expression of MAN1A2 in the BA liver (2.4-fold, p = 0.006) (Figure 1). This translates into loss of peri-canalicular distribution of the MAN1A2 protein in BA compared with normal liver and liver with other childhood cholestatic diseases (Figure 1B). These findings have functional relevance because man1a2 knockdown impairs intrahepatic biliary network formation and bile drainage from the liver in zebrafish larvae (Figures 2, 3A). The accompanying heterotaxy of the liver, the pancreas and the heart is a unique effect of the man1a2 gene, which has not been reported for any BA susceptibility gene.
This novel effect of man1a2 knockdown, i.e., regulating left-right patterning in addition to biliary morphogenesis is independent of its effect on developmental pathways such as EGF (Supplementary Table 7; Molina et al., 2007). We evaluated whether the EGFR pathway, implicated in BA in our previous work is a representative downstream target of man1a2 knockdown. We find that suboptimal man1a2 knockdown synergizes with suboptimal MO-mediated knockdown of arf6, or suboptimal AG1478-mediated inhibition of EGFR signaling to produce biliary dysgenesis and inhibit bile drainage in zebrafish larvae (Figure 5). Therefore, man1a2 dysregulation alters EGFR signaling or can work synergistically with other BA susceptibility genes such as arf6, to limit bile duct development in BA. Because neither experiment combining suboptimal knockdown produced heterotaxy, man1a2 is the sole contributor to heterotaxy via its effect on cilia, which regulates normal left-right patterning during development (Afzelius, 1995). man1a2 knockdown reduces the length and numbers of cilia in Kupffer’s vesicles of zebrafish (Figure 3B). Further, siRNA-mediated Man1a2 knockdown arrests ciliary development and motility in mouse airway epithelia (Figure 5C and Supplementary Videos 1, 2) and in line with the decreased staining of the ciliary protein Arl13b in lungs of Null Man1a2 mice pups (Figure 6).
Corroborative evidence comes from detailed evaluation of the liver from previously described mouse models with known cardiothoracic defects. Null mice with Man1a2–/– exon 2 deletion which are known to die of respiratory failure due to poorly expanded alveoli also show portal expansion, inflammation and ductular reaction, suggestive of biliary inflammation in our studies (Figure 5E; Bergström et al., 2012). Despite loss of exon 2 expression in both organs, the liver phenotype is not as pronounced as would be expected from the lethal pulmonary lesions, partly because the accompanying reduced expression of mid-level and terminal Man1a2 exons is less pronounced in the liver than the lung in null mice. MAN1A2 is highly prone to differential splicing (Al-Balool et al., 2011). A unifying feature is that differentially expressed genes in the Man1a2–/– liver and lung as well as liver from man1a2 morphant zebrafish and the human BA liver participate in EGF, TGF and hedgehog signaling pathways (Table 2 and Supplementary Tables 9–12). The human BA and Man1a2–/– transcriptomes also demonstrate dysregulated ciliary genes, many of which cause human PCD, or congenital heart disease with laterality and/or extracardiac defects (De Luca et al., 2010; Shiraishi and Ichikawa, 2012; Andersen et al., 2014; Bergström et al., 2012). The final evidence includes features of bile duct inflammation in the liver from DNAH11–/– mice, which are known to exhibit situs inversus (Bartoloni et al., 2002). Dnah11 is differentially expressed along with other ciliary genes in the human BA liver.
Other mechanisms by which MAN1A2 may contribute to BA include the role of the MAN1A2 enzyme in removing excess mannose residues during maturation of glycoproteins (Tremblay and Herscovics, 2000). In experimental models, reduced MAN1A2 enzyme activity is associated with uncleaved mannose residues in EGF receptors, which resist phosphorylation and bind poorly to downstream targets (Kawashima et al., 2009). This effect can limit EGFR-dependent branching morphogenesis, which is essential for the proper development of epithelial tubular networks such as the biliary tree (Cabernard and Affolter, 2005). On the endothelial cell surface, glycoproteins with high mannose content promote inflammation via enhanced leukocyte adhesion (Chacko et al., 2011). Loss of extrahepatic bile ducts in children with BA is associated with a fibroinflammatory process (Schwarz et al., 2013).
A provocative inference from zebrafish experiments is that multisystem biliary network and laterality defects can arise from dysregulation of single gene such as MAN1A2, dysregulation of multiple genes with each contributing small effects, such as ARF6 and MAN1A2, or via downstream effects cascading through several interacting pathways, e.g., the EGF and perhaps the hedgehog and the TGF-β pathways. The implications for the heterogeneous BA phenotype and multiple associated susceptibility genes are that non-syndromic and syndromic BA also represent a phenotypic continuum arising from common mechanisms mediated by multiple genes, for example, ciliary dysgenesis, which affects human cholangiocytes from both disease variants (Afzelius, 1995). The lack of differential enrichment of rs7531715 in BA with or without extrahepatic anomalies supports this contention.
To evaluate the role of the CPLANE network and the MAN1A2 gene, we have recently initiated a large scale GWAS with 756 Caucasian BA patients and 4k plus matched controls. A targeted analysis reveals that the MAN1A2 SNP rs12131109 achieves a p-value of 0.05417. This is not surprising because most associations identified in large cohorts have odds ratios in the 1.1–1.2 range. Our ongoing investigations represent the largest GWAS study ever initiated for this disease using a 10-year collection from the US-based CHiLDReN network and our center. The strength of our hypothesis that ciliary dysgenesis is a common basis of BA resides in the fact that each previously reported susceptibility gene from three different groups worldwide, independently points to the CPLANE network, achieving a replication that cannot be dismissed as coincidental.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE138251, GSE138399, and GSE159720, embargoed for 6 months.
The studies involving human participants were reviewed and approved by the Institutional Review Board, University of Pittsburgh. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by Animal Care and Use Committee, University of Pittsburgh.
JS and MN conducted, described zebrafish, and human genomic studies, respectively, AB, SD, and BH conducted, supervised targeted NGS. JG, JM, DL, QS, QY, and BH designed, implemented, cross-checked genotyping, and sequencing analyses. LS and SR analyzed immunohistochemistry. MS, NK, and CL designed, conducted, described mouse airway cell culture, and Dnah11–/– mouse experiments. JF, KP, GG, MA, and NM conducted, described Man1a2–/– mouse studies. AB, CA, CT, and LF recruited subjects, coordinated study, prepared genomic material. AD, RHS, VM, HH, SS, KS, DK, CL, and WC interpreted findings for respective human therapeutic areas, edited manuscript. SS supervised the bioinformatics and systems biology analysis. DS designed, described, supervised zebrafish experiments. RS designed, supervised the entire project and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Partial support DK101426 (DS), DK109365 (RS and SS), Hillman Foundation of Pittsburgh, the Children’s Hospital of Pittsburgh Research Foundation, the Szalay Family Foundation, the Herridge, Giventer-Braff families, and Jane’t family (RS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Michael Tsang for the Tg(dusp6:d2EGFP) line, and Michael Pack for the Tg(krt18:EGFP) line. Zebrafish experiments were supported in part by NIH (DK101426) to DS. We thank Ms. Dale Zecca for manuscript preparation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.538701/full#supplementary-material
ARF6, adenosine-ribosylation-factor-6; BA, biliary atresia; GPC1, glypican-1; GWAS, genome-wide association study; LD, linkage disequilibrium; LTx, Liver transplantation; MAN1A2, Mannosidase-1- α -2; NGS, next-generation sequencing; PCD, Primary ciliary dyskinesia; RTqPCR, Reverse transcription quantitative polymerase chain reaction; SNP, single nucleotide polymorphisms.
Afzelius, B. A. (1995). Situs inversus and ciliary abnormalities. What is the connection? Int. J. Dev. Biol. 39, 839–844.
Al-Balool, H. H., Weber, D., Liu, Y., Wade, M., Guleria, K., Nam, P. L. P., et al. (2011). Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 21, 1788–1799. doi: 10.1101/gr.116442.110
Andersen, T. A., Troelsen Kde, L., and Larsen, L. A. (2014). Of mice and men: molecular genetics of congenital heart disease. Cell Mol. Life Sci. 71, 1327–1352.
Bartoloni, L., Blouin, J. L., Pan, Y., Gehrig, C., Maiti, A. K., Scamuffa, N., et al. (2002). Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. U.S.A. 99, 10282–10286. doi: 10.1073/pnas.152337699
Bergström, S.-E., King, T. E. Jr., and Hollingsworth, H. (2012). Primary Ciliary Dyskinesia (Immotile-Cilia Syndrome). UpToDate.com. Available online at: http://www.uptodate.com/contents/primary-ciliary-dyskinesia-immotile-cilia-syndrome (accessed September 8, 2016).
Cabernard, C., and Affolter, M. (2005). Distinct roles for two receptor tyrosine kinases in epithelial branching morphogenesis in Drosophila. Dev. Cell 9, 831–842. doi: 10.1016/j.devcel.2005.10.008
Cerami, E. G., Gross, B. E., Demir, E., Rodchenkov, I., Babur, O., Anwar, N., et al. (2011). Pathway commons, a web resource for biological pathway data. Nucleic Acids Res. 2011, D685–D690.
Chacko, B. K., Scott, D. W., Chandler, R. T., and Patel, R. P. (2011). Endothelial surface N-glycans mediate monocyte adhesion and are targets for anti-inflammatory effects of peroxisome proliferator-activated receptor γ ligands. J. Biol. Chem. 286, 38738–38747. doi: 10.1074/jbc.m111.247981
Cheng, G., Tang, C. S., Wong, E. H., Cheng, W. W., So, M. T., Miao, X., et al. (2013). Common genetic variants regulating ADD3 gene expression alter biliary atresia risk. J. Hepatol. 59, 1285–1291. doi: 10.1016/j.jhep.2013.07.021
Chu, A. S., Russo, P. A., and Wells, R. G. (2012). Cholangiocyte cilia are abnormal in syndromic and non- syndromic biliary atresia. Mod. Pathol. 25, 751–757. doi: 10.1038/modpathol.2011.212
Colland, F., Jacq, X., Trouplin, V., Mougin, C., Groizeleau, C., Hamburger, A., et al. (2004). Functional proteomics mapping of a human signaling pathway. Genome Res. 14, 1324–1332. doi: 10.1101/gr.2334104
Cui, S., Leyva-Vega, M., Tsai, E. A., EauClaire, S. F., Glessner, J. T., Hakonarson, H., et al. (2013). Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 144, 1107–1115. doi: 10.1053/j.gastro.2013.01.022
De Luca, A., Sarkozy, A., Consoli, F., Ferese, R., Guida, V., Dentici, M. L., et al. (2010). Familial transposition of the great arteries caused by multiple mutations in laterality genes. Heart 96, 673–677. doi: 10.1136/hrt.2009.181685
Delous, M., Yin, C., Shin, D., Ninov, N., Carten, J. D., Pan, L., et al. (2012). Sox9b is a key regulator of pancreaticobiliary ductal system development. PLoS Genet. 8:e1002754. doi: 10.1371/journal.pone.1002754
Essner, J. J., Amack, J. D., Nyholm, M. K., Harris, E. B., and Yost, J. (2005). Kupffer’s vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development 132, 1247–1260. doi: 10.1242/dev.01663
Farber, S. A., Pack, M., Ho, S. Y., Johnson, I. D., Wagner, D. S., Dosch, R., et al. (2001). Genetic analysis of digestive physiology using fluorescent phospholipid reporters. Science 292, 1385–1388. doi: 10.1126/science.1060418
Kang, M., Choi, S., Jeong, S. J., Lee, S. A., Kwak, T. K., Kim, H., et al. (2012). Cross-talk between TGFβ1 and EGFR signalling pathways induces TM4SF5 expression and epithelial-mesenchymal transition. Biochem. J. 443, 691–700. doi: 10.1042/bj20111584
Karrer, F. M., Price, M. R., Bensard, D. D., Sokol, R. J., Narkewicz, M. R., Smith, D. J., et al. (1996). Long-term results with the Kasai operation for biliary atresia. Arch. Surg. 131, 493–496. doi: 10.1001/archsurg.1996.01430170039006
Kawashima, N., Yoon, S. J., Itoh, K., and Nakayama, K. (2009). Tyrosine kinase activity of epidermal growth factor receptor is regulated by GM3 binding through carbohydrate to carbohydrate interactions. J. Biol. Chem. 284, 6147–6155. doi: 10.1074/jbc.m808171200
Li, Y., Klena, N., Gabriel, G., Liu, X., Kim, A. J., Lemke, K., et al. (2015). Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 521, 520–524. doi: 10.1038/nature14269
Lorent, K., Moore, J. C., Siekmann, A. F., Lawson, N., and Pack, M. (2010). Reiterative use of the notch signal during zebrafish intrahepatic biliary development. Dev. Dyn. 239, 855–864. doi: 10.1002/dvdy.22220
Molina, G. A., Watkins, S. C., and Tsang, M. (2007). Generation of FGF reporter transgenic zebrafish and their utility in chemical screens. BMC Dev. Biol. 7:62. doi: 10.1186/1471-213X-7-62
Ningappa, M., Min, J., Higgs, B. W., Ashokkumar, C., Ranganathan, S., and Sindhi, R. (2015a). Genome-wide association (studies) in biliary atresia. Wiley Interdiscip. Rev. Syst. Biol. Med. 7, 267–273. doi: 10.1002/wsbm.1303
Ningappa, M., So, J., Glessner, J., Ashokkumar, C., Ranganathan, S., Min, J., et al. (2015b). The role of ARF6 in biliary atresia. PLoS One 10:e0138381. doi: 10.1371/journal.pone.0138381
Otte, J. B., de Ville, de Goyet, J., Reding, R., Hausleithner, V., Sokal, E., et al. (1994). Sequential treatment of biliary atresia with Kasai portoenterostomy and liver transplantation: a review. Hepatology 20(1 Pt 2), 41S–48S.
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Schadt, E. E., Molony, C., Chudin, E., Hao, K., Yang, X., Lum, P. Y., et al. (2008). Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 6:e107. doi: 10.1371/journal.pbio.0060107
Schwarz, K. B., Haber, B. H., Rosenthal, P., Mack, C. L., Moore, J., Bove, K., et al. (2013). Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology 58, 1724–1731. doi: 10.1002/hep.26512
Shiraishi, I., and Ichikawa, H. (2012). Human heterotaxy syndrome - from molecular genetics to clinical features, management, and prognosis. Circ. J. 76, 2066–2075.
Stranger, B. E., Nica, A. C., Forrest, M. S., Dimas, A., Bird, C. P., Beazley, C., et al. (2007). Population genomics of human gene expression. Nat. Genet. 39, 1217–1224.
Tremblay, L. O., and Herscovics, A. (2000). Characterization of a cDNA encoding a novel human Golgi alpha 1, 2-mannosidase (IC) involved in N-glycan biosynthesis. J. Biol. Chem. 275, 31655–31660. doi: 10.1074/jbc.m004935200
Tremblay, L. O., Nagy Kovács, E., and Daniels, E. (2007). Respiratory distress and neonatal lethality in mice lacking Golgi alpha1,2-mannosidase IB involved in N-glycan maturation. J. Biol. Chem. 82, 2558–2566. doi: 10.1074/jbc.m608661200
Keywords: biliary atresia, liver transplantation, cilia, biliary morphogenesis, laterality
Citation: So J, Ningappa M, Glessner J, Min J, Ashokkumar C, Ranganathan S, Higgs BW, Li D, Sun Q, Schmitt L, Biery AC, Dobrowolski S, Trautz C, Fuhrman L, Schwartz MC, Klena NT, Fusco J, Prasadan K, Adenuga M, Mohamed N, Yan Q, Chen W, Horne W, Dhawan A, Sharif K, Kelly D, Squires RH, Gittes GK, Hakonarson H, Morell V, Lo C, Subramaniam S, Shin D and Sindhi R (2020) Biliary-Atresia-Associated Mannosidase-1-Alpha-2 Gene Regulates Biliary and Ciliary Morphogenesis and Laterality. Front. Physiol. 11:538701. doi: 10.3389/fphys.2020.538701
Received: 03 June 2020; Accepted: 23 September 2020;
Published: 30 October 2020.
Edited by:
Dechun Feng, National Institute on Alcohol Abuse and Alcoholism (NIAAA), United StatesReviewed by:
Paul Tam, The University of Hong Kong, Hong KongCopyright © 2020 So, Ningappa, Glessner, Min, Ashokkumar, Ranganathan, Higgs, Li, Sun, Schmitt, Biery, Dobrowolski, Trautz, Fuhrman, Schwartz, Klena, Fusco, Prasadan, Adenuga, Mohamed, Yan, Chen, Horne, Dhawan, Sharif, Kelly, Squires, Gittes, Hakonarson, Morell, Lo, Subramaniam, Shin and Sindhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shankar Subramaniam, c2hhbmthckB1Y3NkLmVkdQ==; Donghun Shin, ZG9uZ2h1bnNAcGl0dC5lZHU=; Rakesh Sindhi, cmFrZXNoLnNpbmRoaUBjaHAuZWR1
†These authors have contributed equally to the work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.