 Lingfei Li
Lingfei Li Qiong Zhang
Qiong Zhang Xia Lei1
Xia Lei1 Jiongyu Hu
Jiongyu Hu- 1Department of Dermatology, Daping Hospital, Third Military Medical University (Army Medical University), Chongqing, China
- 2Institute of Burn Research, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, China
- 3State Key Laboratory of Trauma, Burns and Combined Injury, Third Military Medical University (Army Medical University), Chongqing, China
- 4Department of Endocrinology, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, China
Microtubule and mitochondrial dysfunction have been implicated in the pathogenesis of cardiovascular diseases (CVDs), including cardiac hypertrophy, fibrosis, heart failure, and hypoxic/ischemic related heart dysfunction. Microtubule dynamics instability leads to disrupted cell homeostasis and cell shape, decreased cell survival, and aberrant cell division and cell cycle, while mitochondrial dysfunction contributes to abnormal metabolism and calcium flux, increased cell death, oxidative stress, and inflammation, both of which causing cell and tissue dysfunction followed by CVDs. A cytosolic skeleton protein, microtubule-associated protein 4 (MAP4), belonging to the family of microtubule-associated proteins (MAPs), is widely expressed in non-neural cells and possesses an important role in microtubule dynamics. Increased MAP4 phosphorylation results in microtubule instability. In addition, MAP4 also expresses in mitochondria and reveals a crucial role in maintaining mitochondrial homeostasis. Phosphorylated MAP4 promotes mitochondrial apoptosis, followed by cardiac injury. The aim of the present review is to highlight the novel role of MAP4 as a potential candidate in multiple cardiovascular pathologies.
Introduction
Cardiovascular disease (CVD) is the leading cause of mortality worldwide, accounting for approximately 40% of all deaths (Schwalm et al., 2016). Hypoxia/ischemia, hypertension, inflammation, diabetes, cardiomyopathies, cardiometabolic syndrome, and obesity are the common risk factors for CVD, and these multiple comorbidities elevate the rates of mortality and morbidity in CVD (Koene et al., 2016; Ceylan et al., 2018; Ren and Zhang, 2018; Zhang et al., 2018; Zhou et al., 2018a; Caporizzo et al., 2019). Despite the advent of a number of drugs and techniques, only limited efficacy has been reached in CVD treatment. Considering these facts, novel interventions, with a broad scope to cover these multiple comorbidities, are necessary.
Cardiovascular dysfunction leads to the development and progress of CVD. Cardiomyocytes and endothelial cells are mainly involved in the maintenance of normal cardiovascular function. Accumulating evidence suggests that damaged microtubules (MT) and mitochondria are the key players in the pathogenesis of cardiomyocytes and endothelial dysfunction. As an important cytoskeletal component, MTs are involved in cell death, maintenance of cellular homeostasis, and organelle transport, and the instability of MT dynamics induces cardiac hypertrophy/heart failure (Tagawa et al., 1996; Sato et al., 1997), myocardial ischemia–reperfusion injury (Sato et al., 1993), and catecholamine-induced myocardial injury. Since mitochondria are involved in cell metabolism, oxidative reactions, and energy supply, mitochondrial disruption reportedly leads to apoptosis, metabolic disorder, and excessive oxidative stress, which contribute to the dysfunction of cardiomyocytes and endothelial cells, and is followed by abnormal cardiovascular events (Bravo-San Pedro et al., 2017; Suomalainen and Battersby, 2018).
In the current review, we discuss a novel candidate intervention target in CVD and elucidate its evolutionary genetic history, basic characteristics, and function, and its correlation with MTs and mitochondria.
Microtubule-Associated Protein 4 (MAP4)
Gene Evolutionary History
In mammals, microtubule-associated proteins (MAPs) are mainly consisted of MAP2, tau (also known as MAPT), and MAP4, all of which are crucial cytosolic skeletal proteins. Both MAP2 and tau are mainly distributed in neurons and have been reviewed elsewhere (Dehmelt and Halpain, 2005). MAP4 is a unique member of MAPs that is widely expressed in various tissues and cells, and recently, an increasing number of studies have suggested that MAP4 might play an important role in cardiovascular disorders. MAP4 is encoded by a single gene located in 3p21 and consists of 23 exons. Moreover, MAP4 gene has many isoforms, as well as three to five MT binding repeats (Dehmelt and Halpain, 2005). Following isolation of the cDNA sequence encoding human and mouse MAP4, three distinct regions consisting of related sequences with different motifs were identified in its open reading frames. Analysis of its amino acid sequence suggests that human, mouse, and bovine MAP4 are close homologs and their sequences reach 0–75% similarity, and have highly conserved domains, including the amino terminus, MT binding domain, and carboxyl terminus, thus inferring that they are members of the same class (West et al., 1991).
Characteristic Structural Features
The MAP4 protein is a ubiquitous, heat-stable protein composed of an asymmetrical structure similar to other MAPs and consists of an amino terminal projection domain and a carboxyl terminal MT binding domain (Mangan and Olmsted, 1996). Accumulating studies have shown that the MT binding domain contains a conserved phosphorylated KXGS motif (Lewis et al., 1988; Drewes et al., 1995; Illenberger et al., 1996; Ozer and Halpain, 2000). It has been reported that the phosphorylation sites, such as Ser696, Ser768, and Ser787 (corresponding to Ser667, Ser737, and Ser760 in mouse) in the proline-rich region of the human MT binding domains, are involved in the dissociation of MAP4 from MTs and regulate MT dynamics instability (Srsen et al., 1999; Kitazawa et al., 2000; Hu et al., 2010). MT dynamic is a process that depends on α and β subunits of tubulin, their post-translational modifications, and interaction of MT with MAPs, and is regulated by the balance between MT polymerization and disassembly, the disruption of which could lead to a series of diseases. Although a number of studies have concentrated on this region, the detailed mechanism of the regulation of MT dynamics remains unclear (Chapin and Bulinski, 1991; Tokuraku et al., 1999). In contrast, the amino terminal projection domain, which protrudes from the MT wall and does not bind to MTs, varies in length among different MAPs. The projection domain of MAP4 consists of three regions, including the amino terminal acidic region, the KDM-repeat sequence, and the b-regions. The region containing KDM-repeat sequence is a representative structure of the projection domain, and is a highly conserved unit composed of an imperfect 14 amino acid repeat (KD(M/V)X(L/P)(P/L)XETEVALA) starting with KD(M/V) (Aizawa et al., 1990; Chapin and Bulinski, 1991; Permana et al., 2005). Recent studies have indicated that the projection domain of MAP4 allows the separation of individual MTs by inhibiting the bundle-forming ability of the MT binding domain. Further, this suppressive activity of the projection domain is correlated with its length rather than its amino acid sequence. However, the detail process of the regulation of MT dynamics or their organization by the projection domain remains elusive (Faruki and Karsenti, 1994; Tokuraku et al., 1999; Iida et al., 2002; Permana et al., 2005).
Distribution
Microtubule-associated protein 4 is reported to be widely expressed in non-neuronal cells and tissues, such as the adrenal gland, liver, lung, heart, vascular tissues, and skin (Parysek et al., 1985; Kotani et al., 1988; Matsushima et al., 2005; Hu et al., 2010; Li et al., 2015; Xia and He, 2018; Zhang et al., 2019a).
Function
MAP4 and Cytoskeleton
Microtubules dynamics is affected by the balance between tubulin polymerization and depolymerization. As a cytoskeleton regulator, MAP4 regulates MT dynamics through its posttranscriptional phosphorylation modification (Chapin and Bulinski, 1991; Nguyen et al., 1997). Under normal conditions, MAP4 combines with MTs, resulting in the stability of MTs. Once phosphorylated, MAP4 dissociates from MTs, leading to MT disassembly and dynamic instability (Ebneth et al., 1999; Hu et al., 2014). Previous studies indicated that the MT binding domain of MAP4 contains a repeat region with tandemly organized repeat sequences, and different types of MAP4 contain a different number and arrangement of these repeat sequences. However, different isoform fragments of MAP4 exhibit similar degrees of MT polymerization activity and MT binding affinity, suggesting that variations in the repeat region are not essential for the regulation of MT dynamics, which is believed to be the main function of MAP4. In contrast, the MT bundle-forming activity is reported to differ among MAP4 isoform fragments, with the bundle formation being augmented by increasing the number of repeat sequences in the fragments, indicating that the role of MAP4 isoforms is to regulate the surface charge of MTs (Tokuraku et al., 2003). In addition, Tokuraku et al. (2003) demonstrated that MAP4 isoforms may also affect kinesin motor activity by regulating MT surface properties. They analyzed MAP4 isoforms in an in vitro gliding assay and found that the five-repeat isoforms suppressed the movement of MTs in a concentration-dependent manner. Furthermore, they revealed that MAP4 isoforms did not compete with kinesin for binding to MTs in a sedimentation assay, suggesting that kinesin could bind to MAP4-bound MTs, although it could not move on them (Tokuraku et al., 2007).
Semenova et al. (2014) used Xenopus melanophores to test the regulation of MT transport by MAP4. They found that pigment granules moved along the MTs to the cell center or periphery by means of dynein and kinesin-2, respectively. They showed that aggregation signals induced phosphorylation of threonine residues in the MT binding domain; meanwhile, the binding of the protein to MTs was shown to be reduced. Whereas overexpression of Xenopus MAP4 suppressed pigment aggregation by shortening dynein-dependent MT runs of melanosomes, removal of Xenopus MAP4 from MTs was reported to decrease the length of kinesin-2-dependent runs, inhibiting pigment dispersion. Thus, their data indicated that the MAP4-MT binding promoted the kinesin-2-based movement in parallel with the suppression of dynein-dependent movement of melanosomes. In addition, phosphorylation led to the dissociation of Xenopus MAP4 from MTs, thereby raising dynein-dependent and reducing kinesin-2-dependent motility of melanosomes, which stimulates their accumulation in the cell center. Conversely, dephosphorylation of Xenopus MAP4 during dispersion triggered the opposite effect (Semenova et al., 2014).
On a separate note, Matsushima et al. (2012) tested the interaction of MAP4 with actin filaments. Their data demonstrated that MAP4 and its MT binding domain fragments bound to actin filaments under normal conditions. The apparent dissociation constant and binding stoichiometry of the fragments to actin have been demonstrated to be approximately 0.1 μM and 1:3 (MAP4/actin), respectively, with the actin binding site on MAP4 reported to be located at the carboxyl terminal part of the proline-rich region, consistent with the MT binding site. Besides, they showed that MAP4-bound actin filaments tended to be straighter and longer, with the number of actin bundles increasing when the MAP4 fragment was added, suggesting that MAP4 binding altered the properties of the actin filaments. A multiple sequence alignment of the proline-rich regions of MAP4 and tau also revealed the presence of two putative actin-binding consensus sequences (Matsushima et al., 2012).
MAP4 and Cell Cycle
Previous studies demonstrated that mutations in human MAP4 (Ser696 and Ser787) affect MT dynamics and subsequently mediate the progression of cell cycle, with p34cdc2/cyclin B being deemed an important kinase in this biological process. In addition, the phosphorylation of the human MAP4 at Ser787 is noted during mitosis, whereas the phosphorylation of MAP4 at Ser696 is shown to occur throughout the cell cycle in proliferating cells. Overexpression of dephosphorylated MAP4 did not affect cell doubling time; in contrast, expression of phosphorylated MAP4 may affect progression into or through cell division, indicating that MAP4 phosphorylation plays a crucial role in regulation of cell cycle (Tombes et al., 1991; Ookata et al., 1995, 1997; Nguyen et al., 1997; Chang et al., 2001).
MAP4 and Mitosis
The MAP4 protein is involved in the regulation of MT dynamics during the M-phase (Ookata et al., 1995; Dehmelt and Halpain, 2005). However, both the MT network and MT dynamics show little change when MAP4 is removed from MTs using a blocking antibody. In addition, absence of MAP4 binding to MTs results in maintenance of the normal structure of spindles in cells progressing to mitosis, and posttranslational modifications of tubulin subunits also remain unchanged, suggesting that MAP4 may just be a component of a functionally redundant system (Wang et al., 1996). Zahnleiter et al. (2015) found that MAP4 mutations lead to the development of a clinical spectrum of centrosomal defects and demonstrated that centrosomal proteins play a regulatory role in the centrosomes and ciliary and Golgi bodies related to severe short stature. Both MAP4 and MT rescue factor and cytoplasmic linker associated protein 1 (CLASP1) are necessary for maintaining spindle location and an accurate cell division axis. In human cells, CLASP1 is required for capturing astral MTs, whereas MAP4 prevents engagement of excess dynein motors (Samora et al., 2011). Consistent with this, MAP4 was observed to interact with dynein-dynactin, thereby suppressing dynein-induced MT sliding. In contrast, absence of MAP4 leads to spindle disorientation in the vertical plane, revealing that force generators are under spatial control. Collectively, these data showed an essential role of MAP4 during mitosis, as spindle positioning is considered to be crucial during embryogenesis and stem-cell homeostasis (Samora et al., 2011). Shiina and Tsukita (1999) reported that mutations of serine and threonine to alanine at p34cdc2 kinase-specific phosphorylation sites interfered with the mitosis-associated reduction in MT affinity of Xenopus MAP4, and their overexpression affected the chromosomal movement during anaphase A in mitotic cells.
Regulating Mechanisms
As already known, MT dynamics are affected by a variety of factors, including drugs (e.g., taxol and colchicine), temperature, GTP, free tubulin, and MAPs. Moreover, the effect of drugs and GTP on MT dynamics was also shown to be mediated by MAP4 (Holmfeldt et al., 2002, 2003; Delphin et al., 2012; Gombos et al., 2013; Ojeda-Lopez et al., 2014; Watanabe and Goshima, 2014).
Xiao et al. (2012) found that the MT binding domain of MAP4 binds to the outer and luminal surfaces of MTs, and taxol, an MT stabilizing drug, suppresses these interactions. Although taxol negatively regulated the interaction of MAP4 with MTs, its binding to the MT binding domain-MAP4-MT complex further decreased the overall deuterium incorporation, suggesting that a more stable complex was formed in the presence of the drug (Xiao et al., 2012). In addition, DNA-damaging agents could affect sensitivity to anti-MT drugs through the regulation of the expression of MAP4 (Zhang et al., 1998, 1999). Chapin and Bulinski (1994) revealed that MAP4 could bind to different types of MTs, including detyrosinated and tyrosinated tubulin, with the differential binding to these forms of tubulin not directly leading to a mechanism of segregation of MAP4 on MTs. Additionally, they also suggested that in TC-7 cells, the absence of MAP4 may affect rapid growth of MTs to which MAP4 was not yet bound, or the presence of other MAPs may compete with MAP4 for MT binding sites. These observations indicate that different binding states of MAP4 on MTs directly regulate MT dynamics within single cells, as well as other MT functions such as those involving MT motor activity (Chapin and Bulinski, 1994).
Septins are a conserved family of GTPases forming heterooligomers and interacting with the actin-based cytoskeleton and MTs. Ghossoub et al. (2013) explored the effect of septins in the primary cilium of retinal pigmented epithelial cells and showed that septin2, septin7, and septin9 formed a 1:1:1 complex, colocalizing along the length of the axoneme. Similarly, knockdown of cilium-localized septins were reported to suppress ciliogenesis in kidney epithelial cells. As a binding partner of septin2, MAP4 regulated the accessibility of septins to MTs and was also localized to the axoneme, where it appeared to negatively control ciliary length. This study provided new insights into the functions and regulations of septins and MAP4 in the organization of the primary cilium and MT-based activities in cells (Ghossoub et al., 2013). Kremer et al. (2005) found that reduced septin in Hela cells leads to a robust elevation in MT stability, and they also identified MAP4 as the septin binding partner using mass spectroscopic analysis. Septin2, spetin6, and septin7 were shown to directly bind to the proline-rich region of MAP4 to form a heterotrimer and this binding suppressed the ability of the MAP4 fragment to bind and bundle MTs. In addition, the absence of septin increased the number of abnormal cell, whereas this effect was suppressed when MAP4 was knocked down. Thus, these data reveal a novel role of septins in mediating MT dynamics through interaction with MAP4 (Kremer et al., 2005).
Hoshi et al. (1992) reported that MAP4 might be a good substrate for MAPK, with its phosphorylation leading to inactivation of tubulin polymerization. In addition, Mori et al. (1991) demonstrated that protein kinase C regulated the activity of MAP4 and subsequently led to changes in the MT dynamics.
Microtubules in CVD
Microtubules, the major component of the cytoskeleton, are composed of α/β tubulin dimers and exhibit a crucial role in regulating many processes, including cell survival and death, cell migration and proliferation, cell shape maintenance, and organelle transport (Walczak, 2000; Birukova et al., 2004; Hu et al., 2010; Zhang et al., 2013, 2019a). In particular, MTs target focal adhesions to regulate extracellular matrix adhesion, and the attachment of MTs to special cortical regions of cells is required for cell polarization (Small et al., 2002). The functions of MTs are affected by MT dynamics as active polymerization and depolymerization indicate MT growth and shortening (Mitchison and Kirschner, 1984). Thus, the growth, shortening, catastrophe, and rescue of MTs are reflected by MT instability, induced cellular homeostasis, division, and movement. Based on the essential functions of MTs, many researchers have investigated the role of MT dysfunction in CVD, including cardiac hypertrophy/heart failure (Tagawa et al., 1996; Sato et al., 1997), myocardial ischemia–reperfusion injury (Sato et al., 1993), and catecholamine-induced myocardial injury (Hori et al., 1994). In addition, desmin intermediate filaments have been identified as a key component of an anchoring complex that links MTs to the sarcomere and imparts structural organization to the MT network. Mutations in the human desmin gene lead to an autosomal dominant, autosomal recessive, and sporadic forms of protein aggregation myopathies and cardiomyopathies (Clemen et al., 2015; Robison et al., 2016). Previous studies have also reported that a complex receptor dependent or independent kinase cascade regulates the transcription of MT deformation-sensitive genes, such as PAI-1 and CTGF, which may provide novel targets to impede the development of CVD (Samarakoon and Higgins, 2018). These studies provided a phenomenological insight into the role of MTs in cardiovascular injury, but the detailed molecular perspective regarding their role has not yet been elucidated. In addition, other researchers have focused on tubulin posttranslational modifications, including detyrosination and tyrosination, which are currently emerging as key modulators of MTs. They demonstrated that tubulin detyrosination affected mechanotransduction in muscle cells and was crucial for load-bearing of buckling MTs during cardiomyocyte contraction (Kerr et al., 2015). Excess or reduced tubulin detyrosination was reported to affect the stiffness of cardiomyocytes, thereby contributing to cardiac dysfunction. Upregulation of tubulin detyrosination is also exhibited in patients with hypertrophic and dilated cardiomyopathies (Robison et al., 2016). These clinical links suggested that an imbalance in the levels of tubulin tyrosination and detyrosination may be a risk factor for heart failure, and could be linked to muscle dysfunctions in various other diseases. Thus, a better understanding of the posttranslational regulation of MTs might substantially change the way we perceive the roles of MTs in CVD (Magiera et al., 2018).
Mitochondria in CVD
Mitochondria are double-membrane bound intracellular organelles responsible for energy production and regulation of cellular metabolism, and occupy a core role in eukaryotic cells, particularly cells of the cardiovascular system (Galluzzi et al., 2012, 2018; Lopez-Crisosto et al., 2017; Mehta et al., 2017). Mitochondrial dynamics (both fusion and fission), mitophagy, and homeostasis are crucial factors in the maintenance of mitochondrial integrity. Mitochondria are more prone to stress, including hypoxia and ischemia–reperfusion. Several studies have shown that accumulation of reactive oxygen species, inflammatory damage, molecular defects, and abnormal signal transduction pathways, including AMPK/ULK1, PI3K/Akt, and p38/MAPK/MAP4, lead to mitochondrial dysfunction and are at the root of numerous diseases. Alterations in the endogenous apoptotic pathway, acute exercise, impaired mitophagy, excessive or untimely mitochondrial fission or fusion, and aging are also known to lead to mitochondrial dysfunction, followed by abnormal metabolism. Alteration in metabolism leads to defects in mitochondrial biogenesis and energy supply, cell death, and calcium fluxes, which are contributing factors toward the pathogenesis of multiple cardiovascular disorders such as myocardial infarction, cardiomyopathies of various etiologies, hypertension, and atherosclerosis (Hu et al., 2014; Bravo-San Pedro et al., 2017; Suomalainen and Battersby, 2018; Zhou et al., 2018b; Wu et al., 2019a, b). In addition, impaired mitochondria have been demonstrated to actively promote inflammatory responses and cell death, thereby contributing to the pathogenesis of CVD. Based on these studies, attempts were made to develop mitochondria-targeting drugs for the treatment of cardiovascular disorders in the clinical setting. The main direction of these interventions involved mitochondrial metabolism, mitochondrial dynamics, mitophagy, calcium homeostasis, oxidative stress, inflammation, regulated cell death, and mitochondrial microRNAs. Despite extraordinary efforts spanning three decades, results have been rather dismal and a small number of effective targeted drugs have been approved for the treatment of patients with CVD (Bonora et al., 2019). Thus, molecules with optimized pharmacological characteristics and precise mechanistic insights into mitochondrial processes, along with a reassessment of the pathogenesis of CVD, are essential for the development of new drugs with clinical utility.
Trends in MAP4-Mediated CVD
Accumulating evidence has suggested that MT and mitochondria are crucial subcellular components involved in the pathogenesis of CVD. Moreover, MAP4 is a key mediator in regulating MTs and mitochondria (Li et al., 2018, 2019; Zhang et al., 2019b). Fang et al. (2011) demonstrated that hypoxia-induced MAP4 phosphorylation in cardiomyocytes leads to MT disassembly, whereas MAP4 overexpression promotes MT polymerization leading to the stabilization of MT networks. Besides, they found that MAP4 overexpression increased viability and ATP content of cardiomyocytes under hypoxia. In addition, MAP4 was shown to inhibit the increased permeability of the mitochondrial membrane under hypoxia via regulating the interaction between dynein light chain tctex-type (DYNLT1) and voltage-dependent anion channel 1 (VDAC1) (Fang et al., 2011). Our previous study suggested that p38/MAPK affected MT dynamics and the viability of cardiomyocytes by regulating MAP4 phosphorylation under hypoxic conditions. Following hypoxia, p38/MAPK and MAP4 phosphorylation increased, with activation of p38/MAPK leading to MAP4 phosphorylation, which in turn contributed to MT disassembly and cell viability reduction. We also revealed that p38/MAPK interacts with MAP4, and thus administration of the p38/MAPK inhibitor could increase MT polymerization and enhance the viability of cardiomyocytes after hypoxia. In contrast, MKK6 (Glu), the upstream activator of p38/MAPK, was shown to reduce cell viability and promote MT disassembly under the same conditions (Hu et al., 2010). We further showed that in hypoxic cardiomyocytes, MAP4 phosphorylation was notably increased with MAP4-MT dissociation, and MAP4 translocated to mitochondria where it induced mitochondrial permeability transition pore opening and cytochrome c release followed by endogenous mitochondrial apoptosis in cardiomyocytes (Hu et al., 2014). In our recent study, we showed that MAP4 phosphorylation was evident not only in cardiac tissues in right ventricular hypertrophy in tetralogy of Fallot, but was also increased in the hearts of both myocardial infraction and transverse aortic constriction mouse models. Briefly, mice exhibiting MAP4 phosphorylation exerted age-dependent effects such as pathological cardiac remodeling, cardiomyocyte mitochondrial apoptosis and disruption, and systolic and diastolic dysfunction. As such, MT disassembly, translocation of phosphorylated MAP4, and cardiomyocyte apoptosis are deemed important factors in initiating cardiac remodeling, with the activation of p38/MAPK being suggested as the crucial upstream kinase involved in the MAP4 phosphorylation (Li et al., 2018) as shown in previous studies (Teiger et al., 1996; Choi et al., 2009; Dees et al., 2012). Interestingly, some other studies revealed that MAP4 (Ser472, Ser924, and Ser1056) phosphorylation increases in the model of pressure overload-induced cardiac hypertrophy. When these three sites were mutated to alanine and transfected into cardiomyocytes to obtain mutant MAP4 constructs, the dephosphorylated MAP4 (Ser924 and Ser1056) caused MT reorganization in the pressure overload model, similar to the hypertrophic cardiac MT phenotype, indicating that MAP4 (Ser924 and Ser1056) dephosphorylation might be the core process in mediating MT reorganization. Further, the underlying mechanism correlated with the activation of the serine/threonine phosphatase 1 and 2A, as well as the upstream p21 kinase (Cheng et al., 2010; Chinnakkannu et al., 2010). Takahashi et al. (2003) reported that MAP4 overexpression not only promoted tubulin expression and stability but also changed the characteristics of MT networks, which is considered a key factor in mediating the phenotype of the pressure overload-induced cardiac hypertrophy. Although there have been some inconsistencies in the proposed mechanisms of MAP4-mediated cardiac remodeling, this might be attributed to different models and phosphorylation sites used in these studies. Our focused sites were located in the proline-rich regions and were responsible for the longitudinal affinity between each tubulin dimer in a protofilament, whereas focused sites in other studies were located in the KXGS region, which is responsible for lateral protofilament–protofilament interactions (Katsuki et al., 1999). Even though both regions are located in the MT binding domain, the detailed functional mechanisms and resulting phenotypes may not be identical. Thus, further research is necessary to elucidate the mechanisms underlying these potential differences.
Another study revealed that the total expression of the cardiac MAP4 protein reached its highest level in the first week in neonatal rats, and then was shown to gradually reduce with aging. Likewise, MAP4 was also demonstrated to be involved in drug- and hypothermia-induced MT stability in cardiomyocytes of newborns, suggesting that the regulation of MAP4 plays an important role in MT functions during cardiac development (Webster and Bratcher, 2006). In addition, previous studies have demonstrated that MAP4 might be a potential regulator of vascular permeability. In microvascular endothelial cells, tumor necrosis-α (TNF-α)/lipopolysaccharides (LPS) induced MAP4 phosphorylation-dependent MT disassembly, followed by increased vascular permeability via activation of p38/MAPK (Li et al., 2015), providing a novel insight for MAP4 phosphorylation in regulating microvascular permeability. Furthermore, Zhou et al. (2015) found that LPS stimulation leads to disruption of MT dynamics through the activation of p38/MAPK in endothelial cells, which was shown to be blocked by a MT stabilizer. Moreover, p38/MAPK was observed to interact with MAP4 to form a complex, subsequently leading to MT disassembly, which could be suppressed by a p38/MAPK inhibitor (Zhou et al., 2015). These data indicate that MAP4 phosphorylation-dependent MT disassembly plays a key role in regulating the permeability of endothelial cells. Another study revealed that Down syndrome exhibits variable features, including heart defect, which is correlated with mitochondrial dysfunction. Overexpression of genes on chromosome-21 is responsible for the phenotypic features of Down syndrome either in a direct or in an indirect manner (Izzo et al., 2018). Our previous study showed that MAP4 phosphorylation is deemed a crucial factor in mediating mitochondrial apoptosis, morphological and functional disruption, and heart defects, which is partially similar to the mitochondrial phenotype of Down syndrome. However, whether MAP4 is involved in Down syndrome remains unclear and warrants further studies.
Future Research Focuses
Conclusively, MAP4 has been demonstrated to regulate MTs or mitochondria in cardiomyocytes and endothelial cells, both of which play a key role in the pathogenesis of CVD (Figure 1). The correlation and interactions between MAP4 and MT/mitochondria, and MT/mitochondrial-targeting drugs may provide useful tools to analyze potential MAP4-related signaling pathways or molecular interactions among p38/MAPK, septins, DYNLT1, and VDAC1. Moreover, the elucidation of their association with pathological outcomes may provide novel targets for the development of MAP4-targeting drugs. In addition, MAP4 is a unique member of MAPs, which is expressed in both vascular tissues and the heart. Recent studies have shown an important role of MAP4 in the cardiovascular system, and interventions targeting the function of MAP4 may provide protection against cardiomyocyte or endothelial cell dysfunction. For these reasons, MAP4 may serve as a potential target for pharmacological interventions in CVD.
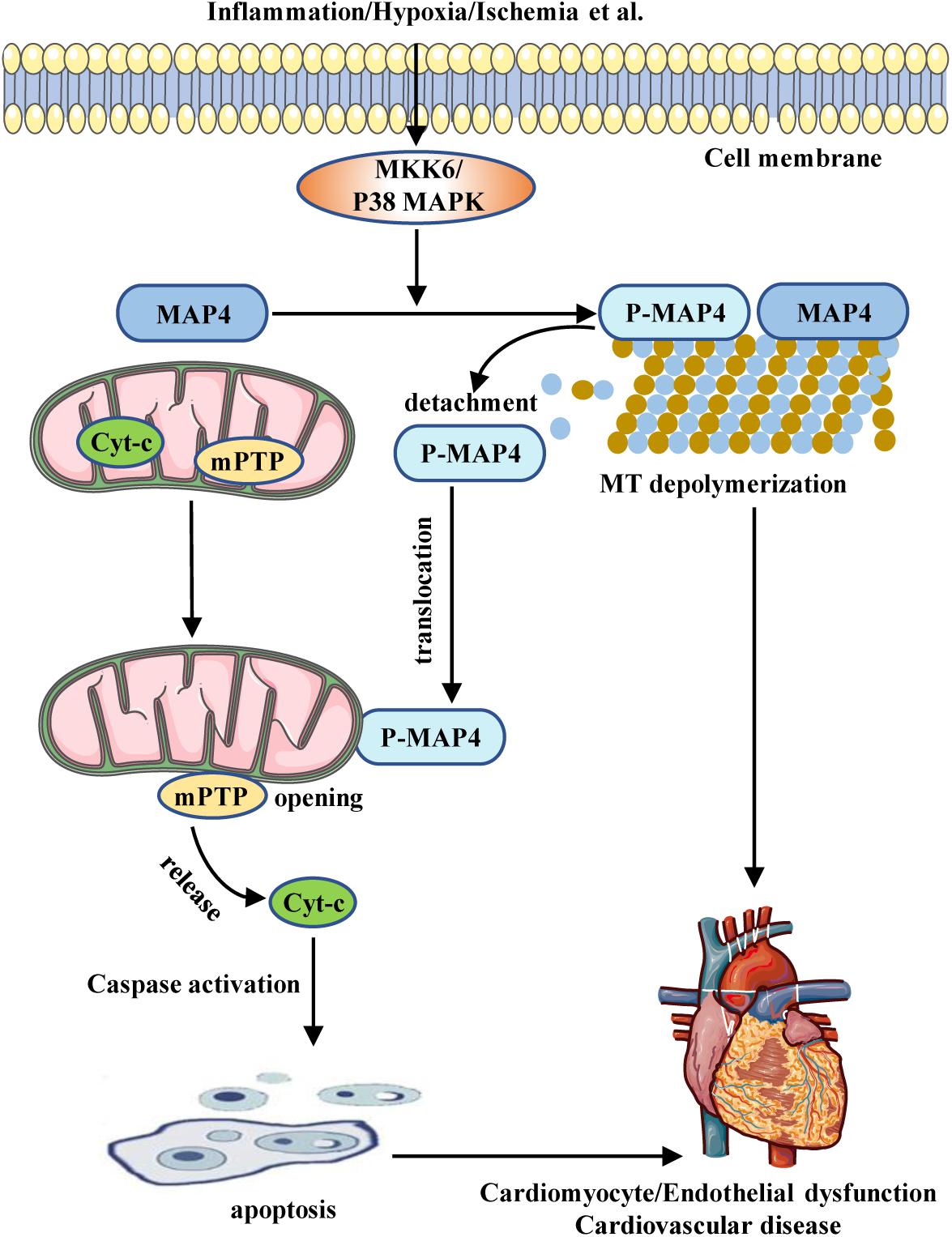
Figure 1. Schematic illustrating the role of MAP4 in CVD. Inflammation, hypoxia, ischemia, etc., stimulate the activation of the MKK6/p38 MAPK pathway. The activated p38 MAPK induces MAP4 phosphorylation and, sequentially, the depolymerization of MT. The phosphorylated MAP4 detaches from MT and translocates to mitochondria from cytosol, leading to mitochondrial permeability transition pore (mPTP) opening and cytochrome c (cyt-c) release, which activates caspase pathway and promotes mitochondrial apoptosis. Increased apoptosis together with MT depolymerization causes cardiomyocyte/endothelial cell dysfunction, which is involved in the pathogenesis of CVD.
In contrast, although there has been some progress regarding MAP4 in both basic and clinical studies, more information on MAP4, including its detailed structure, function, and regulatory mechanism, remains elusive. Thus, future studies should focus on these issues and elucidate the role of MAP4 specific to CVD both in vivo and in vitro. Meanwhile, unraveling of related pathways, molecule–molecule interactions, and the role of MAP4 in other systems may provide us with substantial evidence for the development of novel drugs targeting MAP4.
Author Contributions
LL, QZ, XL, and YH participated in research design. JH supervised in research design. LL, QZ, XL, YH, and JH wrote or contributed to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Military Medical Innovation Program in Daping Hospital (2019CXJSC014) and the Innovation Research Program in Southwest Hospital (SWH2018BJKJ-11).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aizawa, H., Emori, Y., Murofushi, H., Kawasaki, H., Sakai, H., and Suzuki, K. (1990). Molecular cloning of a ubiquitously distributed microtubule-associated protein with Mr 190,000. J. Biol. Chem. 265, 13849–13855.
Birukova, A. A., Birukov, K. G., Smurova, K., Adyshev, D., Kaibuchi, K., Alieva, I., et al. (2004). Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 18, 1879–1890. doi: 10.1096/fj.04-2328com
Bonora, M., Wieckowski, M. R., Sinclair, D. A., Kroemer, G., Pinton, P., and Galluzzi, L. (2019). Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat. Rev. Cardiol. 16, 33–55. doi: 10.1038/s41569-018-0074-0
Bravo-San Pedro, J. M., Kroemer, G., and Galluzzi, L. (2017). Autophagy and mitophagy in cardiovascular disease. Circ. Res. 120, 1812–1824. doi: 10.1161/circresaha.117.311082
Caporizzo, M. A., Chen, C. Y., and Prosser, B. L. (2019). Cardiac microtubules in health and heart disease. Exp. Biol. Med. 244, 1255–1272. doi: 10.1177/1535370219868960
Ceylan, A. F., Wang, S., Kandadi, M. R., Chen, J., Hua, Y., Pei, Z., et al. (2018). Cardiomyocyte-specific knockout of endothelin receptor a attenuates obesity cardiomyopathy. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3339–3352. doi: 10.1016/j.bbadis.2018.07.020
Chang, W., Gruber, D., Chari, S., Kitazawa, H., Hamazumi, Y., Hisanaga, S., et al. (2001). Phosphorylation of MAP4 affects microtubule properties and cell cycle progression. J. Cell Sci. 114, 2879–2887.
Chapin, S. J., and Bulinski, J. C. (1991). Non-neuronal 210 x 10(3) Mr microtubule-associated protein (MAP4) contains a domain homologous to the microtubule-binding domains of neuronal MAP2 and tau. J. Cell Sci. 98(Pt 1), 27–36.
Chapin, S. J., and Bulinski, J. C. (1994). Cellular microtubules heterogeneous in their content of microtubule-associated protein 4 (MAP4). Cell Motil. Cytoskeleton 27, 133–149. doi: 10.1002/cm.970270205
Cheng, G., Takahashi, M., Shunmugavel, A., Wallenborn, J. G., DePaoli-Roach, A. A., Gergs, U., et al. (2010). Basis for MAP4 dephosphorylation-related microtubule network densification in pressure overload cardiac hypertrophy. J. Biol. Chem. 285, 38125–38140. doi: 10.1074/jbc.m110.148650
Chinnakkannu, P., Samanna, V., Cheng, G., Ablonczy, Z., Baicu, C. F., Bethard, J. R., et al. (2010). Site-specific microtubule-associated protein 4 dephosphorylation causes microtubule network densification in pressure overload cardiac hypertrophy. J. Biol. Chem. 285, 21837–21848. doi: 10.1074/jbc.m110.120709
Choi, Y. H., Cowan, D. B., Moran, A. M., Colan, S. D., Stamm, C., Takeuchi, K., et al. (2009). Myocyte apoptosis occurs early during the development of pressure-overload hypertrophy in infant myocardium. J. Thorac. Cardiovasc. Surg. 1362, e1351–e1353.
Clemen, C. S., Stockigt, F., Strucksberg, K. H., Chevessier, F., Winter, L., Schutz, J., et al. (2015). The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 129, 297–315. doi: 10.1007/s00401-014-1363-2
Dees, E., Miller, P. M., Moynihan, K. L., Pooley, R. D., Hunt, R. P., Galindo, C. L., et al. (2012). Cardiac-specific deletion of the microtubule-binding protein CENP-F causes dilated cardiomyopathy. Dis. Model Mech. 5, 468–480. doi: 10.1242/dmm.008680
Dehmelt, L., and Halpain, S. (2005). The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 6:204.
Delphin, C., Bouvier, D., Seggio, M., Couriol, E., Saoudi, Y., Denarier, E., et al. (2012). MAP6-F is a temperature sensor that directly binds to and protects microtubules from cold-induced depolymerization. J. Biol. Chem. 287, 35127–35138. doi: 10.1074/jbc.m112.398339
Drewes, G., Trinczek, B., Illenberger, S., Biernat, J., Schmitt-Ulms, G., Meyer, H. E., et al. (1995). Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J. Biol. Chem. 270, 7679–7688. doi: 10.1074/jbc.270.13.7679
Ebneth, A., Drewes, G., Mandelkow, E. M., and Mandelkow, E. (1999). Phosphorylation of MAP2c and MAP4 by MARK kinases leads to the destabilization of microtubules in cells. Cell Motil. Cytoskeleton 44, 209–224. doi: 10.1002/(sici)1097-0169(199911)44:3<209::aid-cm6>3.0.co;2-4
Fang, Y. D., Xu, X., Dang, Y. M., Zhang, Y. M., Zhang, J. P., Hu, J. Y., et al. (2011). MAP4 mechanism that stabilizes mitochondrial permeability transition in hypoxia: microtubule enhancement and DYNLT1 interaction with VDAC1. PLoS One 6:e28052. doi: 10.1371/journal.pone.0028052
Faruki, S., and Karsenti, E. (1994). Purification of microtubule proteins from Xenopus egg extracts: identification of a 230K MAP4-like protein. Cell Motil. Cytoskeleton 28, 108–118. doi: 10.1002/cm.970280203
Galluzzi, L., Kepp, O., Trojel-Hansen, C., and Kroemer, G. (2012). Mitochondrial control of cellular life, stress, and death. Circ. Res. 111, 1198–1207. doi: 10.1161/circresaha.112.268946
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541.
Ghossoub, R., Hu, Q., Failler, M., Rouyez, M. C., Spitzbarth, B., Mostowy, S., et al. (2013). Septins 2, 7 and 9 and MAP4 colocalize along the axoneme in the primary cilium and control ciliary length. J. Cell Sci. 126, 2583–2594. doi: 10.1242/jcs.111377
Gombos, L., Neuner, A., Berynskyy, M., Fava, L. L., Wade, R. C., Sachse, C., et al. (2013). GTP regulates the microtubule nucleation activity of gamma-tubulin. Nat. Cell Biol. 15, 1317–1327. doi: 10.1038/ncb2863
Holmfeldt, P., Brattsand, G., and Gullberg, M. (2002). MAP4 counteracts microtubule catastrophe promotion but not tubulin-sequestering activity in intact cells. Curr. Biol. 12, 1034–1039. doi: 10.1016/s0960-9822(02)00897-7
Holmfeldt, P., Brattsand, G., and Gullberg, M. (2003). Interphase and monoastral-mitotic phenotypes of overexpressed MAP4 are modulated by free tubulin concentrations. J. Cell Sci. 116, 3701–3711.
Hori, M., Sato, H., Kitakaze, M., Iwai, K., Takeda, H., Inoue, M., et al. (1994). Beta-adrenergic stimulation disassembles microtubules in neonatal rat cultured cardiomyocytes through intracellular Ca2+ overload. Circ. Res. 75, 324–334. doi: 10.1161/01.res.75.2.324
Hoshi, M., Ohta, K., Gotoh, Y., Mori, A., Murofushi, H., Sakai, H., et al. (1992). Mitogen-activated-protein-kinase-catalyzed phosphorylation of microtubule-associated proteins, microtubule-associated protein 2 and microtubule-associated protein 4, induces an alteration in their function. Eur. J. Biochem. 203, 43–52.
Hu, J., Chu, Z., Han, J., Zhang, Q., Zhang, D., Dang, Y., et al. (2014). Phosphorylation-dependent mitochondrial translocation of MAP4 is an early step in hypoxia-induced apoptosis in cardiomyocytes. Cell Death Dis. 5:e1424. doi: 10.1038/cddis.2014.369
Hu, J. Y., Chu, Z. G., Han, J., Dang, Y. M., Yan, H., Zhang, Q., et al. (2010). The p38/MAPK pathway regulates microtubule polymerization through phosphorylation of MAP4 and Op18 in hypoxic cells. Cell Mol. Life Sci. 67, 321–333. doi: 10.1007/s00018-009-0187-z
Iida, J., Itoh, T. J., Hotani, H., Nishiyama, K., Murofushi, H., Bulinski, J. C., et al. (2002). The projection domain of MAP4 suppresses the microtubule-bundling activity of the microtubule-binding domain. J. Mol. Biol. 320, 97–106. doi: 10.1016/s0022-2836(02)00402-3
Illenberger, S., Drewes, G., Trinczek, B., Biernat, J., Meyer, H. E., Olmsted, J. B., et al. (1996). Phosphorylation of microtubule-associated proteins MAP2 and MAP4 by the protein kinase p110mark. Phosphorylation sites and regulation of microtubule dynamics. J. Biol. Chem. 271, 10834–10843. doi: 10.1074/jbc.271.18.10834
Izzo, A., Mollo, N., Nitti, M., Paladino, S., Cali, G., Genesio, R., et al. (2018). Mitochondrial dysfunction in down syndrome: molecular mechanisms and therapeutic targets. Mol. Med. 24:2.
Katsuki, M., Tokuraku, K., Murofushi, H., and Kotani, S. (1999). Functional analysis of microtubule-binding domain of bovine MAP4. Cell Struct. Funct. 24, 337–344. doi: 10.1247/csf.24.337
Kerr, J. P., Robison, P., Shi, G., Bogush, A. I., Kempema, A. M., Hexum, J. K., et al. (2015). Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat. Commun. 6:8526.
Kitazawa, H., Iida, J., Uchida, A., Haino-Fukushima, K., Itoh, T. J., Hotani, H., et al. (2000). Ser787 in the proline-rich region of human MAP4 is a critical phosphorylation site that reduces its activity to promote tubulin polymerization. Cell Struct. Funct. 25, 33–39. doi: 10.1247/csf.25.33
Koene, R. J., Prizment, A. E., Blaes, A., and Konety, S. H. (2016). Shared risk factors in cardiovascular disease and cancer. Circulation 133, 1104–1114. doi: 10.1161/circulationaha.115.020406
Kotani, S., Murofushi, H., Maekawa, S., Aizawa, H., and Sakai, H. (1988). Isolation of rat liver microtubule-associated proteins. Evidence for a family of microtubule-associated proteins with molecular mass of around 200,000 which distribute widely among mammalian cells. J. Biol. Chem. 263, 5385–5389.
Kremer, B. E., Haystead, T., and Macara, I. G. (2005). Mammalian septins regulate microtubule stability through interaction with the microtubule-binding protein MAP4. Mol. Biol. Cell 16, 4648–4659. doi: 10.1091/mbc.e05-03-0267
Lewis, S. A., Wang, D. H., and Cowan, N. J. (1988). Microtubule-associated protein MAP2 shares a microtubule binding motif with tau protein. Science 242, 936–939. doi: 10.1126/science.3142041
Li, L., Hu, J., He, T., Zhang, Q., Yang, X., Lan, X., et al. (2015). P38/MAPK contributes to endothelial barrier dysfunction via MAP4 phosphorylation-dependent microtubule disassembly in inflammation-induced acute lung injury. Sci.Rep. 5:8895.
Li, L., Zhang, J., Zhang, Q., Huang, Y., and Hu, J. (2019). Cardiac proteomics reveals the potential mechanism of microtubule associated protein 4 phosphorylation-induced mitochondrial dysfunction. Burns Trauma 7:8.
Li, L., Zhang, Q., Zhang, X., Zhang, J., Wang, X., Ren, J., et al. (2018). Microtubule associated protein 4 phosphorylation leads to pathological cardiac remodeling in mice. EBioMedicine 37, 221–235. doi: 10.1016/j.ebiom.2018.10.017
Lopez-Crisosto, C., Pennanen, C., Vasquez-Trincado, C., Morales, P. E., Bravo-Sagua, R., Quest, A. F. G., et al. (2017). Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 14, 342–360. doi: 10.1038/nrcardio.2017.23
Magiera, M. M., Singh, P., Gadadhar, S., and Janke, C. (2018). Tubulin posttranslational modifications and emerging links to human disease. Cell 173, 1323–1327. doi: 10.1016/j.cell.2018.05.018
Mangan, M. E., and Olmsted, J. B. (1996). A muscle-specific variant of microtubule-associated protein 4 (MAP4) is required in myogenesis. Development 122, 771–781.
Matsushima, K., Aosaki, M., Tokuraku, K., Hasan, M. R., Nakagawa, H., and Kotani, S. (2005). Identification of a neural cell specific variant of microtubule-associated protein 4. Cell Struct. Funct. 29, 111–124. doi: 10.1247/csf.29.111
Matsushima, K., Tokuraku, K., Hasan, M. R., and Kotani, S. (2012). Microtubule-associated protein 4 binds to actin filaments and modulates their properties. J. Biochem. 151, 99–108. doi: 10.1093/jb/mvr119
Mehta, M. M., Weinberg, S. E., and Chandel, N. S. (2017). Mitochondrial control of immunity: beyond ATP. Nat. Rev. Immunol. 17, 608–620. doi: 10.1038/nri.2017.66
Mitchison, T., and Kirschner, M. (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. doi: 10.1038/312237a0
Mori, A., Aizawa, H., Saido, T. C., Kawasaki, H., Mizuno, K., Murofushi, H., et al. (1991). Site-specific phosphorylation by protein kinase C inhibits assembly-promoting activity of microtubule-associated protein 4. Biochemistry 30, 9341–9346. doi: 10.1021/bi00102a029
Nguyen, H. L., Chari, S., Gruber, D., Lue, C. M., Chapin, S. J., and Bulinski, J. C. (1997). Overexpression of full- or partial-length MAP4 stabilizes microtubules and alters cell growth. J. Cell Sci. 110(Pt 2), 281–294.
Ojeda-Lopez, M. A., Needleman, D. J., Song, C., Ginsburg, A., Kohl, P. A., Li, Y., et al. (2014). Transformation of taxol-stabilized microtubules into inverted tubulin tubules triggered by a tubulin conformation switch. Nat. Mater. 13, 195–203. doi: 10.1038/nmat3858
Ookata, K., Hisanaga, S., Bulinski, J. C., Murofushi, H., Aizawa, H., Itoh, T. J., et al. (1995). Cyclin B interaction with microtubule-associated protein 4 (MAP4) targets p34cdc2 kinase to microtubules and is a potential regulator of M-phase microtubule dynamics. J. Cell Biol. 128, 849–862. doi: 10.1083/jcb.128.5.849
Ookata, K., Hisanaga, S., Sugita, M., Okuyama, A., Murofushi, H., Kitazawa, H., et al. (1997). MAP4 is the in vivo substrate for CDC2 kinase in HeLa cells: identification of an M-phase specific and a cell cycle-independent phosphorylation site in MAP4. Biochemistry 36, 15873–15883. doi: 10.1021/bi971251w
Ozer, R. S., and Halpain, S. (2000). Phosphorylation-dependent localization of microtubule-associated protein MAP2c to the actin cytoskeleton. Mol. Biol. Cell 11, 3573–3587. doi: 10.1091/mbc.11.10.3573
Parysek, L. M., del Cerro, M., and Olmsted, J. B. (1985). Microtubule-associated protein 4 antibody: a new marker for astroglia and oligodendroglia. Neuroscience 15, 869–875. doi: 10.1016/0306-4522(85)90084-3
Permana, S., Hisanaga, S., Nagatomo, Y., Iida, J., Hotani, H., and Itoh, T. J. (2005). Truncation of the projection domain of MAP4 (microtubule-associated protein 4) leads to attenuation of microtubule dynamic instability. Cell Struct. Funct. 29, 147–157. doi: 10.1247/csf.29.147
Ren, J., and Zhang, Y. (2018). New therapeutic approaches in the management of cardiometabolic diseases: bringing the concepts together. Curr. Drug Targets 19, 987–988. doi: 10.2174/138945011909180629095709
Robison, P., Caporizzo, M. A., Ahmadzadeh, H., Bogush, A. I., Chen, C. Y., Margulies, K. B., et al. (2016). Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352:aaf0659. doi: 10.1126/science.aaf0659
Samarakoon, R., and Higgins, P. J. (2018). The cytoskeletal network regulates expression of the profibrotic genes PAI-1 and CTGF in vascular smooth muscle cells. Adv. Pharmacol. 81, 79–94. doi: 10.1016/bs.apha.2017.08.006
Samora, C. P., Mogessie, B., Conway, L., Ross, J. L., Straube, A., and McAinsh, A. D. (2011). MAP4 and CLASP1 operate as a safety mechanism to maintain a stable spindle position in mitosis. Nat. Cell Biol. 13, 1040–1050. doi: 10.1038/ncb2297
Sato, H., Hori, M., Kitakaze, M., Iwai, K., Takashima, S., Kurihara, H., et al. (1993). Reperfusion after brief ischemia disrupts the microtubule network in canine hearts. Circ. Res. 72, 361–375. doi: 10.1161/01.res.72.2.361
Sato, H., Nagai, T., Kuppuswamy, D., Narishige, T., Koide, M., Menick, D. R., et al. (1997). Microtubule stabilization in pressure overload cardiac hypertrophy. J. Cell Biol. 139, 963–973. doi: 10.1083/jcb.139.4.963
Schwalm, J. D., McKee, M., Huffman, M. D., and Yusuf, S. (2016). Resource effective strategies to prevent and treat cardiovascular disease. Circulation 133, 742–755. doi: 10.1161/circulationaha.115.008721
Semenova, I., Ikeda, K., Resaul, K., Kraikivski, P., Aguiar, M., Gygi, S., et al. (2014). Regulation of microtubule-based transport by MAP4. Mol. Biol. Cell 25, 3119–3132. doi: 10.1091/mbc.e14-01-0022
Shiina, N., and Tsukita, S. (1999). Mutations at phosphorylation sites of Xenopus microtubule-associated protein 4 affect its microtubule-binding ability and chromosome movement during mitosis. Mol. Biol. Cell 10, 597–608. doi: 10.1091/mbc.10.3.597
Small, J. V., Geiger, B., Kaverina, I., and Bershadsky, A. (2002). How do microtubules guide migrating cells? Nat. Rev. Mol. Cell Biol. 3, 957–964. doi: 10.1038/nrm971
Srsen, V., Kitazawa, H., Sugita, M., Murofushi, H., Bulinski, J. C., Kishimoto, T., et al. (1999). Serum-dependent phosphorylation of human MAP4 at Ser696 in cultured mammalian cells. Cell Struct. Funct. 24, 321–327. doi: 10.1247/csf.24.321
Suomalainen, A., and Battersby, B. J. (2018). Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92. doi: 10.1038/nrm.2017.66
Tagawa, H., Rozich, J. D., Tsutsui, H., Narishige, T., Kuppuswamy, D., Sato, H., et al. (1996). Basis for increased microtubules in pressure-hypertrophied cardiocytes. Circulation 93, 1230–1243. doi: 10.1161/01.cir.93.6.1230
Takahashi, M., Shiraishi, H., Ishibashi, Y., Blade, K. L., McDermott, P. J., Menick, D. R., et al. (2003). Phenotypic consequences of beta1-tubulin expression and MAP4 decoration of microtubules in adult cardiocytes. Am. J. Physiol. Heart Circ. Physiol. 285, H2072–H2083.
Teiger, E., Than, V. D., Richard, L., Wisnewsky, C., Tea, B. S., Gaboury, L., et al. (1996). Apoptosis in pressure overload-induced heart hypertrophy in the rat. J. Clin. Invest. 97, 2891–2897. doi: 10.1172/jci118747
Tokuraku, K., Katsuki, M., Nakagawa, H., and Kotani, S. (1999). A new model for microtubule-associated protein (MAP)-induced microtubule assembly. The Pro-rich region of MAP4 promotes nucleation of microtubule assembly in vitro. Eur. J. Biochem. 259, 158–166. doi: 10.1046/j.1432-1327.1999.00019.x
Tokuraku, K., Matsushima, K., Matui, T., Nakagawa, H., Katsuki, M., Majima, R., et al. (2003). The number of repeat sequences in microtubule-associated protein 4 affects the microtubule surface properties. J. Biol. Chem. 278, 29609–29618. doi: 10.1074/jbc.m302186200
Tokuraku, K., Noguchi, T. Q., Nishie, M., Matsushima, K., and Kotani, S. (2007). An isoform of microtubule-associated protein 4 inhibits kinesin-driven microtubule gliding. J. Biochem. 141, 585–591. doi: 10.1093/jb/mvm063
Tombes, R. M., Peloquin, J. G., and Borisy, G. G. (1991). Specific association of an M-phase kinase with isolated mitotic spindles and identification of two of its substrates as MAP4 and MAP1B. Cell Regul. 2, 861–874. doi: 10.1091/mbc.2.11.861
Walczak, C. E. (2000). Microtubule dynamics and tubulin interacting proteins. Curr. Opin. Cell Biol. 12, 52–56. doi: 10.1016/s0955-0674(99)00056-3
Wang, X. M., Peloquin, J. G., Zhai, Y., Bulinski, J. C., and Borisy, G. G. (1996). Removal of MAP4 from microtubules in vivo produces no observable phenotype at the cellular level. J. Cell Biol. 132, 345–357. doi: 10.1083/jcb.132.3.345
Watanabe, S., and Goshima, G. (2014). Friction on MAP determines its traveling direction on microtubules. Dev. Cell 29, 5–6. doi: 10.1016/j.devcel.2014.03.022
Webster, D. R., and Bratcher, J. M. (2006). Developmental regulation of cardiac MAP4 protein expression. Cell Motil. Cytoskeleton 63, 512–522. doi: 10.1002/cm.20140
West, R. R., Tenbarge, K. M., and Olmsted, J. B. (1991). A model for microtubule-associated protein 4 structure. Domains defined by comparisons of human, mouse, and bovine sequences. J. Biol. Chem. 266, 21886–21896.
Wu, N. N., Tian, H., Chen, P., Wang, D., Ren, J., and Zhang, Y. (2019a). Physical exercise and selective autophagy: benefit and risk on cardiovascular health. Cells 8:1436. doi: 10.3390/cells8111436
Wu, N. N., Zhang, Y., and Ren, J. (2019b). Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid Med. Cell Longev. 2019:9825061.
Xia, X., and He, C. (2018). Microtubule-associated protein 4 is a prognostic factor and promotes tumor progression in lung adenocarcinoma. Dis. Markers 2018:8956072.
Xiao, H., Wang, H., Zhang, X., Tu, Z., Bulinski, C., Khrapunovich-Baine, M., et al. (2012). Structural evidence for cooperative microtubule stabilization by Taxol and the endogenous dynamics regulator MAP4. ACS Chem. Biol. 7, 744–752. doi: 10.1021/cb200403x
Zahnleiter, D., Hauer, N. N., Kessler, K., Uebe, S., Sugano, Y., Neuhauss, S. C., et al. (2015). MAP4-dependent regulation of microtubule formation affects centrosome, cilia, and Golgi architecture as a central mechanism in growth regulation. Hum. Mutat. 36, 87–97. doi: 10.1002/humu.22711
Zhang, C. C., Yang, J. M., Bash-Babula, J., White, E., Murphy, M., Levine, A. J., et al. (1999). DNA damage increases sensitivity to vinca alkaloids and decreases sensitivity to taxanes through p53-dependent repression of microtubule-associated protein 4. Cancer Res. 59, 3663–3670.
Zhang, C. C., Yang, J. M., White, E., Murphy, M., Levine, A., and Hait, W. N. (1998). The role of MAP4 expression in the sensitivity to paclitaxel and resistance to vinca alkaloids in p53 mutant cells. Oncogene 16, 1617–1624. doi: 10.1038/sj.onc.1201658
Zhang, J., Li, L., Zhang, Q., Wang, W., Zhang, D., Jia, J., et al. (2019a). Microtubule-associated protein 4 phosphorylation regulates epidermal keratinocyte migration and proliferation. Int. J. Biol. Sci. 15, 1962–1976. doi: 10.7150/ijbs.35440
Zhang, J., Li, L., Zhang, Q., Yang, X., Zhang, C., Zhang, X., et al. (2019b). Phosphorylation of microtubule- associated protein 4 promotes hypoxic endothelial cell migration and proliferation. Front. Pharmacol. 10:368. doi: 10.3389/fphar.2019.00368
Zhang, L., Shao, H., Zhu, T., Xia, P., Wang, Z., Liu, L., et al. (2013). DDA3 associates with microtubule plus ends and orchestrates microtubule dynamics and directional cell migration. Sci. Rep. 3:1681.
Zhang, Y., Whaley-Connell, A. T., Sowers, J. R., and Ren, J. (2018). Autophagy as an emerging target in cardiorenal metabolic disease: from pathophysiology to management. Pharmacol. Ther. 191, 1–22. doi: 10.1016/j.pharmthera.2018.06.004
Zhou, H., Wang, S., Hu, S., Chen, Y., and Ren, J. (2018a). ER-mitochondria microdomains in cardiac ischemia-reperfusion injury: a fresh perspective. Front. Physiol. 9:755. doi: 10.3389/fphys.2018.00755
Zhou, H., Wang, S., Zhu, P., Hu, S., Chen, Y., and Ren, J. (2018b). Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 15, 335–346. doi: 10.1016/j.redox.2017.12.019
Zhou, Z., Guo, F., Yi, L., Tang, J., Dou, Y., and Huan, J. (2015). The p38/mitogen-activated protein kinase pathway is implicated in lipopolysaccharide-induced microtubule depolymerization via up-regulation of microtubule-associated protein 4 phosphorylation in human vascular endothelium. Surgery 157, 590–598. doi: 10.1016/j.surg.2014.10.007
Keywords: microtubule-associated protein 4, microtubule-associated proteins, microtubule, mitochondria, cardiovascular disease
Citation: Li L, Zhang Q, Lei X, Huang Y and Hu J (2020) MAP4 as a New Candidate in Cardiovascular Disease. Front. Physiol. 11:1044. doi: 10.3389/fphys.2020.01044
Received: 21 May 2020; Accepted: 29 July 2020;
Published: 26 August 2020.
Edited by:
Jun Ren, University of Washington, United StatesReviewed by:
Ne Natalie Wu, Zhongshan Hospital, Fudan University, ChinaYing Tan, Nanfang Hospital, Southern Medical University, China
Copyright © 2020 Li, Zhang, Lei, Huang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiongyu Hu, jiongyuhu@163.com