 Brandon N. VanderVeen
Brandon N. VanderVeen E. Angela Murphy
E. Angela Murphy James A. Carson
James A. Carson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 31 August 2020
Sec. Striated Muscle Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.01037
This article is part of the Research Topic Skeletal Muscle Immunometabolism View all 18 articles
Progressive weight loss combined with skeletal muscle atrophy, termed cachexia, is a common comorbidity associated with cancer that results in adverse consequences for the patient related to decreased chemotherapy responsiveness and increased mortality. Cachexia’s complexity has provided a barrier for developing successful therapies to prevent or treat the condition, since a large number of systemic disruptions that can regulate muscle mass are often present. Furthermore, considerable effort has focused on investigating how tumor derived factors and inflammatory mediators directly signal skeletal muscle to disrupt protein turnover regulation. Currently, there is developing appreciation for understanding how cancer alters skeletal muscle’s complex microenvironment and the tightly regulated interactions between multiple cell types. Skeletal muscle microenvironment interactions have established functions in muscle response to regeneration from injury, growth, aging, overload-induced hypertrophy, and exercise. This review explores the growing body of evidence for immune cell modulation of the skeletal muscle microenvironment during cancer-induced muscle wasting. Emphasis is placed on the regulatory network that integrates physiological responses between immune cells with other muscle cell types including satellite cells, fibroblast cells, and endothelial cells to regulate myofiber size and plasticity. The overall goal of this review is to provide an understanding of how different cell types that constitute the muscle microenvironment and their signaling mediators contribute to cancer and chemotherapy-induced muscle wasting.
Cachexia is a perilous comorbidity occurring with many chronic diseases that are defined by progressive weight loss, skeletal muscle atrophy, and the inability to be fully prevented or treated by nutritional support (Fearon et al., 2011). Adverse consequences resulting from cachexia include loss of functional independence, poor treatment outcomes, and a reduced quality of life (Bruera, 1997, Fearon et al., 2011). Cancer cachexia is a complex condition that is the most common cancer-associated comorbidity and occurs in ~50–80% of cancer patients (Baracos et al., 2018). To this end, the cachectic condition is linked to reduced responsiveness to anticancer therapy and increased mortality (Kazemi-Bajestani et al., 2016; Baracos et al., 2018; Sealy et al., 2020). There is inherent difficulty to examining the mechanistic drivers of cachexia, namely a large number of disruptions to systemic homeostatic regulation that occur with cancer, the inherent heterogeneity of different cancers, and the patient’s underlying health condition. Moreover, skeletal muscle mass is sensitive to the several systemic disruptions associated with cancer including insulin resistance, chronic inflammation, anorexia, and hypogonadism (Baracos et al., 2018). There also has been significant interest in investigating tumor derived factors that can directly drive skeletal muscle catabolism, and numerous factors have been identified (Biswas and Acharyya, 2020). Despite our vastly improved mechanistic understanding of cancer-induced muscle wasting through established and well-defined preclinical cachexia and in vitro atrophy models, the complexity and heterogeneity of cancer cachexia have hindered the development of effective treatments for the cancer patient (Anderson et al., 2017). Additionally, mechanistic studies have not historically considered the potential additive effects of cancer and chemotherapy on the mechanisms inducing cachexia, and we are only beginning to understand the implications of this interaction for the management of cachexia (Barreto et al., 2016a,b; Bozzetti, 2020).
Systemic and local inflammation accompany many different conditions that produce skeletal muscle metabolic plasticity, growth, and atrophy, and a regulatory role for inflammation in these processes has been widely investigated for decades (Tidball, 1995; Deans and Wigmore, 2005). Additionally, transient increases in systemic inflammation and intrinsic skeletal muscle inflammatory signaling can occur with exercise and has been linked to many important muscle adaptations (Febbraio et al., 2004; Deyhle et al., 2015). Chronic systemic inflammation is a widely investigated driver of muscle wasting through its direct effects on skeletal muscle (Baracos et al., 2018), and its ability to induce other systemic disruptions that can ultimately regulate skeletal muscle mass, such as insulin resistance and hypogonadism (Wu and Ballantyne, 2017). The ability to regenerate from injury is a recognized property of healthy skeletal muscle, and immune cells have a well-established role in this regenerative process (Howard et al., 2020). While inflammation’s contribution to initiating and accelerating cancer cachexia has been widely investigated (Evans et al., 2008; Carson and Baltgalvis, 2010), a major focus of this research has centered on circulating inflammatory mediators and how they directly regulate muscle intracellular signaling to disrupt protein turnover and metabolism to drive wasting (Talbert et al., 2018). To this end, significant gaps remain in our understanding of other aspects of the complex relationship between the immune system and the regulation of skeletal muscle mass. Additional research is warranted to delineate the capacity for inflammation to regulate signaling between different cell types in skeletal muscle that is involved in maintaining metabolic and protein turnover homeostasis. Immune cells comprise 2–6% of skeletal muscle’s cell population, but maintain a well-established role in skeletal muscle homeostasis, especially macrophages (MΦ; Tidball, 2002; Reidy et al., 2019a). While the understanding of the MΦ’s role in skeletal muscle repair and remodeling is well-appreciated, there is strong evidence for both T-cells and neutrophils in the maintenance of skeletal muscle MΦ function and overall skeletal muscle plasticity (Frenette et al., 2002; Tidball, 2005; Dumont et al., 2008; Schiaffino et al., 2017; Tidball, 2017; Deyhle and Hyldahl, 2018). Despite the importance of immune cell activity in muscle plasticity and aging (Reidy et al., 2019a), our understanding of immune cell involvement in cancer‐ and chemotherapy-induced muscle wasting is just emerging.
The potential for cancer to disrupt tightly regulated interactions between cell types in the skeletal muscle microenvironment continues to develop and be appreciated (Talbert and Guttridge, 2016). Skeletal muscle microenvironment interactions have established functions in muscle response to regeneration from injury, growth, aging, overload-induced hypertrophy, and exercise (Morgan and Partridge, 2020). Furthermore, there has been extensive investigation into the importance and regulation of satellite cell proliferation and differentiation, angiogenesis, and extracellular matrix (ECM) remodeling after muscle injury and with aging (Tidball and Wehling-Henricks, 2007; Xiao et al., 2016; Ceafalan et al., 2018; Yang and Hu, 2018). These adaptive processes are often coupled to local inflammatory responses initiated by remodeling stimuli. These inflammatory responses are subjected to precise temporal regulation and if this response is altered, muscle remodeling can be either attenuated or blocked (Howard et al., 2020). Moreover, systemic and intrinsic stimuli can induce MΦs to initiate signaling that regulates muscle fibroblasts, satellite cells, endothelial/vascular cells, as well as within the myofiber (Tidball, 2002; Arnold et al., 2007; Fry et al., 2014). Inflammatory signaling can impact several cell types located in the muscle microenvironment leading to altered myofiber protein synthesis (Gao et al., 2017) and mitochondrial quality control (Gomez-Cabrera et al., 2016; Fix et al., 2018), which are known drivers of muscle wasting with cancer. Additionally, muscle fibrosis and dysregulation of the ECM, of which immune cells play a central regulatory role, have been reported in cachectic skeletal muscle from pancreatic cancer patients (Judge et al., 2018; Nosacka et al., 2020). Therefore, this review explores the growing body of evidence for immune cell modulation of the skeletal muscle microenvironment during cancer-induced muscle wasting. Emphasis is placed on the regulatory network that integrates physiological responses between immune cells with other muscle cell types including satellite cells, fibroblast cells, and endothelial cells to regulate myofiber size and plasticity. The overall goal of this review is to provide an understanding of the potential for different cell types that constitute the muscle microenvironment that can contribute to cancer-induced muscle wasting. Specific attention is given to MΦ, neutrophil, and T-cell’s regulation of the communication between several cell types in the muscle microenvironment that could promote cancer-induced myofiber catabolism and metabolic dysfunction. The current understanding of chemotherapy as an underlying pathology that could disrupt immune cell interaction with the skeletal muscle microenvironment also is discussed.
Skeletal muscle loss is a critical manifestation of cancer cachexia (Argiles et al., 2010). While our mechanistic understanding of cancer-induced muscle wasting continues to develop and potential therapeutic targets to treat the condition have been identified, both the complexity of the underlying disease and the multiple mechanisms responsible for the maintenance of skeletal muscle homeostasis have clouded our ability to understand the primary drivers of this catabolic condition. While there may still be hope for a “master regulator” of cancer-induced muscle atrophy (Lecker et al., 2004), the heterogeneity of cancer, pre-existing conditions of the patient, and equivocal findings between preclinical cachexia models have led to a host of potential drivers of the cachectic condition that are too broad to describe in detail here. However, this topic has been extensively reviewed (Argiles et al., 2014, 2015; Carson et al., 2016; Vanderveen et al., 2017; Baracos, 2018). Skeletal muscle responds to a multitude of systemic cues that are vital for whole body homeostasis, and the muscle fiber responds to global stimuli including disuse, increased activity, aging, and metabolic mediators (Atherton et al., 2016), which can all be altered with cancer and chemotherapy. Furthermore, due to these varied physiological and functional demands, muscle fibers differ in contractile and metabolic properties, and these properties can impact how the muscle fiber responds to the cancer environment (Carson et al., 2016; Vanderveen et al., 2017). The complexity of the muscle’s physiological response to cancer and chemotherapy is also impacted by the myofiber’s interaction with the ECM, vasculature, and response to local inflammation, which is examined in this review.
Despite the identified complexities in understanding muscle mass regulation with cancer, muscle mass maintenance requires a balanced regulation of protein turnover, which is maintained by anabolic and catabolic signaling pathways. Cancer induces fundamental disruptions to the homeostatic regulation of the protein turnover that has negative consequences for skeletal muscle function and cellular metabolism (Baracos et al., 2018; Counts et al., 2020). Cachexia drives overall muscle mass loss even with adequate nutrient intake (Fearon et al., 2012a; Porporato, 2016), and results in suppressed protein synthesis and/or increased degradation. Muscle protein synthesis and breakdown can be regulated by various stimuli including nutrient availability, hormones, mechanical loading, and metabolic stress, which can all be disrupted by cancer and potentially chemotherapy (Glass, 2005; Powers et al., 2005; Zhang et al., 2007; Egerman and Glass, 2014). Active areas of investigation for the disruption of protein turnover with cancer have focused on excessive protein breakdown by the ubiquitin proteasome system (UPS) and disrupted autophagy regulation (McClung et al., 2010; White et al., 2011; Fearon et al., 2012b; Penna et al., 2013; Luo et al., 2014; Aversa et al., 2016). Critical intracellular regulator networks in skeletal muscle exert tight control of these processes but can be disrupted by cancer and chemotherapy. This skeletal muscle regulatory nexus involves Akt, mTORC1, FOXO3A, and AMPK signaling, which responds to growth factors, mechanical stimuli (i.e., stretch and contraction), cellular energy levels (AMP: ATP), nutrients (amino acids), and inflammation (Carson, 1997; Carson and Wei, 2000; Glass, 2005; Judge et al., 2014). These mechanisms exert control over cell growth through regulation of protein synthesis (e.g., transcription and translation), protein degradation (e.g., UPS) autophagy, and metabolism (Laplante and Sabatini, 2009). Disruption to this signaling network in cachectic skeletal muscle also has been linked to cancer-induced mitochondrial dysfunction (Carson et al., 2016; Vanderveen et al., 2017, 2019; Fix et al., 2019). Muscle mitochondrial dysfunction continues to be extensively investigated as an underlying mechanism for the initiation and progression of cancer-induced muscle wasting. However, understanding how cancer disrupts the normal dynamic regulation of muscle protein turnover remains an active area of inquiry.
Cancer-induced systemic inflammation and the increased production of specific cytokines have been extensively investigated as an underlying driver of cancer cachexia (Argilés et al., 2019). Both adipose tissue and skeletal muscle catabolism can be induced by exacerbated inflammation. Examination of cachectic cancer patients and preclinical models of cachexia has identified interleukin 6 (IL-6), tumor necrosis factor α (TNF-α), TNF-like inducer of apoptosis (TWEAK), TNF receptor (TNFR)-associated factor 6 (TRAF6), interferon gamma (INF-γ), and leukemia inhibitory factor (LIF) as mediators of cancer-induced muscle wasting (White et al., 2013; Hetzler et al., 2015; Zimmers et al., 2016; Yakovenko et al., 2018; Argilés et al., 2019). These cytokines can induce several intracellular pathways including the nuclear factor-κB (NF-κB) pathway, p38 mitogen-activated protein kinase (MAPK) pathway, and the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. Notwithstanding the identification of several different inflammatory mediators in cachexia, we lack understanding of how these signaling pathways interact or overlap with other physiological signaling within the muscle microenvironment to regulate muscle catabolism with cancer. Due to this complexity barriers remain for implementing therapeutic strategies that target an individual signaling cascade to preserve skeletal muscle mass in the cancer patient. Moreover, there are currently no FDA-approved treatments for cancer cachexia. That said, there remains strong therapeutic potential for nutritional, anti-inflammatory, and lifestyle management strategies to mitigate cachexia severity and improve patient life quality.
Cachexia’s prevalence with cancer is often dependent on cancer type and stage of the disease; however, the functional deficits with chemotherapy are consistent and pervasive (Barreto et al., 2016a; Bozzetti, 2020). Along with nausea, emesis, and anorexia, weakness and fatigue remain among the most commonly reported off-target effects of chemotherapy (Dahele et al., 2007). Our understanding of chemotherapy’s deleterious off-target effects on skeletal muscle has improved over the last decade highlighting a key role for metabolic and DNA/cell stress (Gilliam et al., 2009; Barreto et al., 2016b; Morton et al., 2019; Sougiannis et al., 2019). Targeting the generation of mitochondrial reactive oxygen species (ROS), by exercise or antioxidants, has shown promise in mitigating Doxorubicin (DOX)-induced skeletal muscle dysfunction (Smuder, 2019; Montalvo et al., 2020), and targeting the activation of MAPKs with ACVR2B/Fc and MEK1 inhibitors has been proposed to alleviate 5-FU induced mitochondrial dysfunction (Barreto et al., 2016b, 2017). Inflammatory signaling has been demonstrated to regulate chemotherapy-induced E3 ligase activation through modulating TNF-α (Gilliam et al., 2009). Interestingly, while conclusive mechanisms involving intrinsic skeletal muscle inflammatory signaling with chemotherapy are lacking, leukopenia with chemotherapy has been well-established (Kvinnsland, 1999; Yamanaka et al., 2007; Shitara et al., 2009; Baechler et al., 2010; Han et al., 2012; Abraham et al., 2015). This naturally invites intrigue into understanding how chemotherapy-induced immunosuppression can converge with cancer-associated chronic inflammation to disrupt the skeletal muscle microenvironment, which can lead to wasting, reduced life quality, poor treatment outcomes, and subsequent reduced survival. While again there are currently no approved treatments for chemotherapy’s off-target effects on muscle, there remains similar strong therapeutic potential for nutritional, anti-inflammatory, antioxidant, and/or lifestyle management to mitigate chemotherapy-induced toxicities.
The identification of immune checkpoints, specifically PD-1/PDL1 and CTLA-4, has led to the use of specific inhibitors, which have greatly improved cancer therapies (Haanen and Robert, 2015). These drugs improve the anti-tumor capacity of T-cells and potentially natural killer (NK) cells by blocking the ligand/receptor binding induced by cancer cells that cause T-cell inactivation (Curran et al., 2010; Pesce et al., 2019). Similar to other cancer treatments, patients receiving immune check point inhibitors (ICPIs) that suffer from cachexia have reduced survival when compared to patients who maintain their weight (Chu et al., 2020; Roch et al., 2020). Whether ICPIs can induce cachexia has not been established; however, body weight loss in aged mice with mesothelioma tumors was exacerbated by immunotherapy, but whole body macrophage depletion reduced tumor mass and body weight loss (Duong et al., 2018). As using ICPIs become increasingly more common as a cancer treatment, understanding the impact of cachexia and the role of ICPI in cachexia progression remains an intriguing area for future inquiry.
Developing an understanding of the distinct contributions of cancer and chemotherapy to muscle wasting and dysfunction has been at the forefront of cachexia related mechanistic inquiry; however, understanding the potential synergism or negation of specific mechanisms with both cancer and chemotherapy would improve our understanding of cancer patient’s condition. To date, few studies have directly investigated the effect of cancer and chemotherapy on skeletal muscle which leads to cachexia. Barreto et al. (2016a) identified the common pathways activated by C26-induced cachexia and chemotherapeutic Folfiri [5-fluorouracil (5FU), leucovorin, irinotecan]-induced cachexia. This study highlighted a common downregulation of metabolic and structure proteins with particularly striking changes to mitochondrial function (Barreto et al., 2016a). Interestingly, only Folfiri induced neurological damage, while only C26 was associated with exercise intolerance. Others have demonstrated that TNF-α plays a key role in cancer and DOX-induced skeletal muscle dysfunction (Hardin et al., 2008; Gilliam et al., 2009, 2011). While disrupted protein turnover has been a central regulator of cancer-induced cachexia (White et al., 2013; Montalvo et al., 2018; Counts et al., 2020), there is little evidence to suggest chemotherapy impacts muscle in this same way.
The mononuclear phagocyte system (MPS) includes circulating monocytes, monocyte derived MΦs, and dendritic cells (DC; Gordon and Taylor, 2005; Hume, 2006). As their name implies, they share a mononuclear structure and play key roles in tissue defense and homeostasis through phagocytosis of old or damaged materials as well as invading pathogens (Murphy and Weaver, 2016). Upon insult, naïve monocytes proliferate in the bone marrow in response to chemokines, most notably monocyte chemoattractant 1 (MCP-1), to infiltrate the damaged/infected tissue to phagocytize pathogens or damaged material (Hume, 2006; Capoccia et al., 2008). Once outside vasculature, monocytes most commonly differentiate to either DC or MΦ; however, tissues also house resident, nascent, and self-renewing DCs and MΦs (Van Furth and Cohn, 1968; Hoeffel and Ginhoux, 2018). It is also important to note that MΦs have been demonstrated to play both an anti‐ and pro-inflammatory roles in both innate and adaptive immunity depending on the stimuli; however, MΦs are the primary phagocytic effector cell of the MPS (Gordon and Taylor, 2005). While DCs share a morphology with undifferentiated monocytes and MΦs, they primarily function as antigen presenting cells (APC) which activate naïve T-cells within the adaptive immune response (Mellman, 2013; Murphy and Weaver, 2016; Abbas et al., 2018; Figure 1).
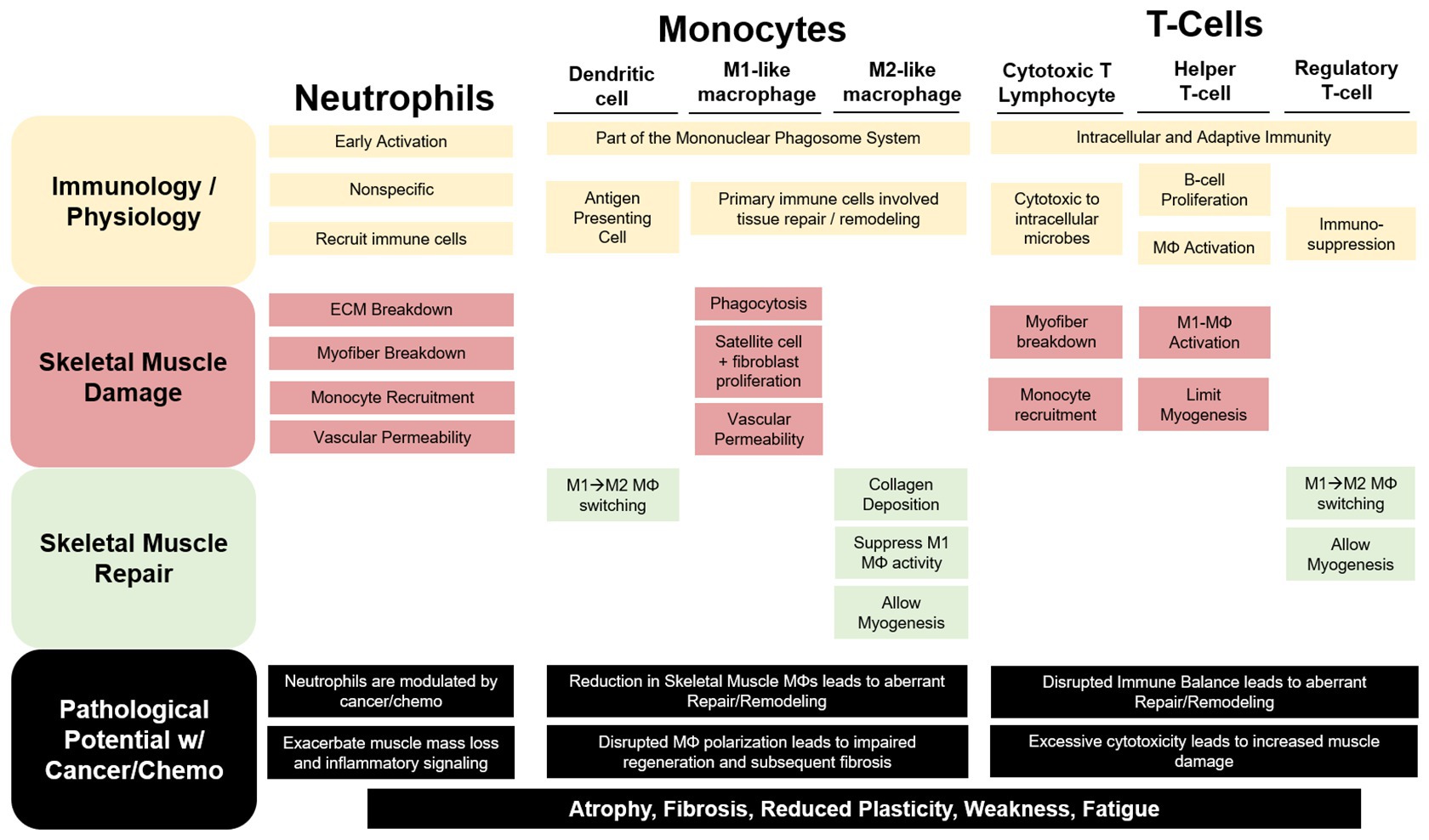
Figure 1. The contribution of immune cells to skeletal muscle repair and remodeling. Neutrophils, monocytes, and T-cells have both unique and redundant processes that aid in resolving a skeletal muscle insult. Neutrophils are recruited early and exact a robust, non-specific response that can exacerbate damage to the myofiber and extracellular matrix (ECM). Cytotoxic T-lymphocytes (CTLs) have been demonstrated to perform similar functions to neutrophils in the early stage of the damage response. Additionally, Neutrophils and CTLs recruit inflammatory (M1-like) macrophages (MΦ) to help phagocytize damaged tissue for removal while releasing pro-inflammatory mediators to stimulate satellite and fibroblast cell proliferation and increase vascular permeability. As repair progresses anti-inflammatory, profibrotic (M2-like), MΦs predominate skeletal muscle allowing for myogenesis and collagen deposition. Regulatory T-cells and dendritic cells (DC) aid in the MΦ phenotype switching contributing to the switch from a pro-inflammatory to an anti-inflammatory skeletal muscle microenvironment. Cancer and chemotherapy have been demonstrated to modulate each of these immune cells uniquely in circulation or other non-muscle tissue; however, we are limited in our understanding of their roles in cancer-associated wasting. We can speculate that these disruptions to these cells within the skeletal muscle microenvironment would lead to muscle mass loss and increased fibrosis contributing to reduced muscle plasticity, weakness, and fatigue.
Unfortunately, due to overlapping functions and shared cell surface markers, characterizing and assessing the different subsets of these monocytes are contentious and equivocal (Gordon and Taylor, 2005; Guilliams et al., 2014). The most conserved monocyte cell surface marker between humans and rodents are CX3C chemokine receptor (CX3CR1) and cluster of differentiation (CD) 11b. These markers vary in expression based on the phenotype of the monocyte. For example, resident or “patrolling” monocytes will have a higher expression of CX3CR1 compared to circulating or inflammatory monocytes, and the expression of Ly6C in mice is a spectrum from resident (low-none) to inflammatory or circulating (med-high; Gordon and Taylor, 2005). However, undifferentiated Ly6CHigh monocytes have been described in healthy uninjured tissue while Ly6CLow survey the endothelium (Guilliams et al., 2014). DCs share the CX3CR1 and CD11b receptors with other monocytes; however they differ in their expression of CD11c and major histocompatibility complex (MHC) II and can even express common T-lymphocyte markers CD4 and CD8 (Sato and Fujita, 2007). The varying degrees of these cell surface markers help define physiology and function as well as their myeloid or lymphoid origin/maturation (Sato and Fujita, 2007; Mellman, 2013). All murine MΦs are said to express CD68 and F4/80 irrespective of phenotype. Mature/differentiated MΦs are not common in the circulation but are found as naïve monocytes until they extravasate. Resident self-renewing MΦs are commonly considered M0 and lack, or have low expression of CD11c, MHCII, CD80, or CD206. These resident M0-like MΦs can either remain nascent or be polarized to increase expression of CD11c and/or MHCII to become a M1-like pro-inflammatory, phagocytic MΦ (Shapouri-Moghaddam et al., 2018; Orecchioni et al., 2019). While these resident cells are able to polarize to an M1-like phenotype, this classification is more common among infiltrating monocytes (Ly6CHigh). Furthermore, these M1-like MΦs are plastic and their expression of pro-inflammatory cell surface markers and release of pro-inflammatory cytokines can be suppressed with a corresponding increase in the expression of CD206 and/or CD163 and release of anti-inflammatory cytokines resulting in an M2-like anti-inflammatory, pro-fibrotic MΦ (Gordon and Taylor, 2005; Mosser and Edwards, 2008; Wynn et al., 2013). MΦs are plastic and responsive to extrinsic stimuli; however, autocrine/paracrine MΦ signaling can regulate their phenotype through feedback or feedforward mechanisms. Furthermore, the M1/M2 dichotomy does not adequately described the nature and function of the MΦ and should be considered a spectrum rather than an “either/or” (Mosser and Edwards, 2008). This classification of cell surface markers and function is not exhaustive and is often contentious, which has been previously reviewed (Hume, 2006; Mellman, 2013; Guilliams et al., 2014).
MΦs have received significant attention in skeletal muscle physiology given their central role in repair and remodeling, and thus our understanding of skeletal muscle MΦs primarily comes from studies investigating repair, regeneration, and remodeling following unloading/reloading-, eccentric contraction-, or cardiotoxin-induced damage (St Pierre and Tidball, 1994; Tidball, 2005, 2017; Cote et al., 2013). Early in skeletal muscle repair, there is an influx of circulating monocytes that differentiate to a phagocytic and pro-inflammatory M1-like MΦ to aide in the removal of damaged tissue. Additionally, they recruit additional inflammatory monocytes by MCP-1, promote vascularization by HIF-1α, stimulate satellite cells proliferation, and inhibit satellite cells differentiation by IL-6 and TNF-α (Frenette et al., 2003; Arnold et al., 2007; Capoccia et al., 2008; Novak et al., 2014; Liu et al., 2017; Ceafalan et al., 2018). The plasticity of MΦs, however, allows for phenotypic flexibility and within 7 days following damage a more M2-like profibrotic anti-inflammatory phenotype predominates (St Pierre and Tidball, 1994; Tidball and Wehling-Henricks, 2007; Hammers et al., 2015). These M2-like MΦs then promote angiogenesis by secreting vascular endothelial growth factor (VEGF) and ECM remodeling by secreting TGFβ and matrix metalloproteases (MMP) 2 and 9 while allowing for myogenesis through suppression of TNF-α release (De Santa et al., 2018).
Disrupted skeletal muscle repair with aging and chronic disease has been described, though mechanistic explanations often focus on sex hormones, satellite cells, or intrinsic structural dysmorphias. Despite the understood immune changes with aging and disease and the established roles of monocytes in skeletal muscle homeostasis, our understanding of the relationship between the immune system and musculoskeletal system under such conditions is in its infancy (Cui et al., 2019; Reidy et al., 2019a). Immunological investigation into muscular dystrophy demonstrated that dystrophic mice had profound increases in total muscle MΦs and targeting this infiltration can mitigate disease progression (Mojumdar et al., 2014; Villalta et al., 2015). Additionally, reloading aged skeletal muscle had a blunted hypertrophy response associated with a lower number of M1-like MΦs at baseline and blunted M1-like MΦ infiltration (early) and M2-like MΦ transition (late; Reidy et al., 2019b). To this end, it is important to note that the mean age of cancer patients is ~65 years and it may be that these changes with aging overlap with cancer and chemotherapy-induced changes to skeletal muscle MΦs (Dunne et al., 2019).
To date, few papers have directly investigated the changes to skeletal muscle monocytes or MΦs with cancer and/or cancer-associated wasting (Inaba et al., 2018; Costamagna et al., 2020). Interleukin-4 (IL-4), a potent stimulator of M2-like MΦs, was shown to attenuate skeletal muscle wasting in the C26 mouse model of cachexia associated with reduced satellite cell accumulation and increased skeletal muscle CD206 protein expression (Costamagna et al., 2020). Protein analysis showed no significant differences between control and C26-tumor bearing mice with F4/80, and CD206 (marker of M2-like MΦs) appears lower in C26 mice but did not achieve statistical significance. While elevated circulating IL-6 and LIF is diagnostic in the C26 model, Inaba et al. (2018) demonstrated reduced skeletal muscle MΦ and neutrophil number without the loss of satellite cell proliferative or differentiative abilities that others have demonstrated (Penna et al., 2010). Evidence of disrupted skeletal muscle regeneration – a process reliant on skeletal muscle MΦs – with cachexia has been demonstrated (Devine et al., 2015; Judge et al., 2018). Whether cachexia leads to muscle degeneration or fibrosis without a damaging insult is equivocal likely due to the heterogeneity of tumor type and preclinical model; however, reduced specific force (force per unit area) with cachexia has been demonstrated across several preclinical models, corresponding with the severity of weight loss (Jaweed et al., 1983; Gorselink et al., 2006; Murphy et al., 2012; Roberts et al., 2013; Ramage and Skipworth, 2018; Vanderveen et al., 2018). The role of monocytes/MΦs in these aberrations has been merely speculative highlighting a need for additional studies examining changes to MΦs in skeletal muscle with cachexia progression. There is, however, intriguing evidence suggesting the immunosuppressive nature of common chemotherapeutic DOX may be disrupting skeletal muscle MΦ activation and polarization leading to blunted skeletal muscle repair and remodeling (Huang et al., 2017). Interestingly, disrupted skeletal muscle inflammatory signaling with DOX and/or 5FU has not been evident, despite the cytotoxic nature of the drugs. Current evidence remains focused on DNA damage or stress sensors, p38 and Erk1/2, mitochondrial damage, oxidative stress, and autophagy with these chemotherapeutics (Barreto et al., 2016a; Ballaro et al., 2019; Hiensch et al., 2019; Bozzetti, 2020).
Neutrophils belong to a class of leukocytes characterized by their polymorphic nuclear shape and their early recruitment during acute inflammation (Kolaczkowska and Kubes, 2013; Stackowicz et al., 2019). Neutrophils make up roughly ~50% of circulating leukocytes in healthy individuals (~15% in mice) and are the first line of defense against bacterial infection (Kolaczkowska and Kubes, 2013). Interestingly, neutrophils share a precursor cell with monocytes and originate in the bone marrow but rely on granulocyte colony-stimulating factor (G-CSF) for simulation as opposed to monocyte colony-stimulating factor (M-CSF). Classifying neutrophils is much more straight forward compared to monocytes; however, there remains variety among immunologists. Neutrophils commonly express CD11b and CD16 (Fujimoto et al., 2000); however, recently the use of Ly6G and Gr-1 has been used to differentiate neutrophils from other leukocytes (Lee et al., 2013; Romee et al., 2013). Neutrophils kill/eliminate pathogens through phagocytosis, degranulation, ROS production, and/or neutrophil extracellular traps, which have been well-reviewed (Kolaczkowska and Kubes, 2013; Mayadas et al., 2014). The granules within neutrophils contain lysozymes, collagenases, and elastases, which serve the function of damaging/killing foreign material. Unfortunately, neutrophils are early and robust in their response and their lack of specificity often damages the host tissue along with the pathogen (Faurschou and Borregaard, 2003; Mayadas et al., 2014). While they play an important role in acute cytotoxicity, neutrophils release a myriad of cytokines and oxidative factors to initiate and potentiate the immune response depending on the stimuli. Neutrophils are often replaced by MΦs following the initial stages of damage or infection primarily through neutrophil release of pro-inflammatory cytokines and myeloperoxidases (MPOs).
The majority of studies investigating the musculoskeletal and immune system relationship have focused on monocytes/MΦs; however, there is evidence that neutrophils play a key role in the clearance of damaged/dead tissue and recruitment of MΦs (Tidball et al., 1999; Pizza et al., 2005). Historically, ischemia/reperfusion-induced skeletal muscle damage resulted in a dramatic increase influx of neutrophils while MΦs play a more minor role (Korthuis et al., 1988; Smith et al., 1989; Kanwar et al., 1998); however, the role for neutrophils in eccentric contraction-induced damaged also has been described (Frenette et al., 2002; Pizza et al., 2005). While the plasticity of MΦ polarization is a key regulator of the switch from the initial skeletal muscle damage response to repair/regeneration, neutrophils appear to play a significant role only in the initial damage response. To this end, persistent neutrophil infiltration has been demonstrated to delay skeletal muscle repair and absence of neutrophil accumulation mitigates signs of skeletal muscle damage (Pizza et al., 2005). Further, elevated neutrophils have been demonstrated to exacerbate joint damage with arthritis, and some evidence suggests that neutrophils can induce or exacerbate skeletal muscle damage (Dumont et al., 2008). The induction of neutrophils with exercise and ischemia/reperfusion-induced damage is transient, and the decrease in the relative abundance of neutrophils is accompanied by a polarization switch of MΦs to an M2-like profibrotic/anti-inflammatory phenotype for remodeling (Tidball et al., 1999; Frenette et al., 2003; Tidball, 2005; Tidball and Villalta, 2010). Interestingly, the acute changes in the genetic profile of skeletal muscle with exercise reflect similar changes in blood neutrophils and circulating inflammatory cytokines (Broadbent et al., 2017). While the role of neutrophils in skeletal muscle physiology requires significant work, there is evidence to support that neutrophils exacerbate the damage response to perpetuate the acute inflammatory stimuli necessary for repair and remodeling.
Neutropenia has been shown to be a strong prognostic indicator for survival in cancer patients undergoing chemotherapy (Kvinnsland, 1999; Yamanaka et al., 2007; Han et al., 2012; Abraham et al., 2015). Regarding cancer, the circulating neutrophil-to-lymphocyte ratio (NLR) is also a strong prognosticator for all-around survival (Grecian et al., 2018). In general, tumors produce G-CSF and can increase circulating neutrophils (Jablonska et al., 2017) while chemotherapy reduces circulating neutrophils (Shitara et al., 2011). These findings appear dependent on cancer type and chemotherapy as some investigators have reported decreases in neutrophils with cancer and increases with chemotherapy. To our knowledge, the impact of neutrophil changes with cancer and chemotherapy on skeletal muscle has not been investigated. We can speculate, however, that an increase in circulating neutrophils with cancer may initiate and/or potentiate skeletal muscle degeneration or mass loss while neutropenia may blunt the homeostatic balance of skeletal muscle repair and remodeling. It is understood that a proper balance of pro‐ and anti-inflammatory effectors is essential for skeletal muscle homeostasis and the undulating changes to neutrophils with cancer and cancer therapies warrants investigative inquiry.
Lymphocytes are key regulators of the immune response and play a central role in adaptive immunity. Lymphocytes are characterized as B (B-cells) or T lymphocytes (T-cells) based on their function and origin (B-Bone Marrow; T-Thymus) rather than their appearance as they both maintain a similar morphology. Similar to other immune cells, B-cells release inflammatory mediators to regulate an immune response, but their primary function is often focused on extracellular pathogens and antibody production (Murphy and Weaver, 2016). Their role as an immune modulator with skeletal muscle has not been well-characterized and requires additional work; therefore, this review will focus on the known roles of T-cells. T-cells are key regulators of cellular immunity either through eliminating intracellular microbes or the activation of other immune cells including MΦs (Abbas et al., 2018). The classification of T-cells is extensive and depends again on their cellular function and role in the immune response. Classic characterization aligns with CD3+CD4+ helper T-cells (Th), CD3+CD4+CD25+FoxP3+ T regulatory cells (Tregs), and CD3+CD4-CD8+ cytotoxic T lymphocytes (CTL). Historically, Th cells regulate B-cell differentiation in immune tissue and activate resident or local MΦs for cellular immunity and can be further classified into Th1, Th2, and Th17 (Th3, Th9, Th22, and TFH cells have also been described; Hirahara and Nakayama, 2016). Th1 cells regulate intracellular phagocytosis by activating MΦs through the release of pro-inflammatory IFNγ, while Th2 cells can release IL-4 and IL-13 to modulate the MΦ phenotype switching to an M2-like anti-inflammatory phenotype (Shapouri-Moghaddam et al., 2018; Mazzoni et al., 2019). Th17 cells are only beginning to be understood; however, they produce known pro-inflammatory cytokines IL-17 and IL-6, and have an emerging role in auto-immunity (Bi et al., 2007; Mazzoni et al., 2019). CD8+ CTL are aptly named as they provide intracellular immunity against pathogens by inducing cell death in cells that are not accessible or recognized by antibodies (Mittrucker et al., 2014). While Th cell activation and proliferation play a key role in antibody-mediated immune function in circulation, CTLs are the primary effectors of the intracellular immune response (Abbas et al., 2018). Tregs, as their name suggests, play a key role in regulating the immune response primarily through immune suppression and regulate the M1-like to M2-like MΦ phenotype switch during remodeling (Hori et al., 2003). The different subsets of lymphocytes are vast, and each play an important role in the immune system; however, for our purposes we will highlight the literature examining the roles of Th, Tregs, and CTLs in skeletal muscle physiology, repair, and remodeling and their potential role in modulating muscle wasting with cancer and chemotherapy.
The role of T-cells in skeletal muscle repair and remodeling has been recently reviewed and described briefly below (Schiaffino et al., 2017; Deyhle and Hyldahl, 2018). Tregs primarily function in immune suppression, which is necessary during the later stages of skeletal muscle repair/remodeling, while CTLs and Th cells may potentiate the early damage response (Zhang et al., 2014; Deyhle and Hyldahl, 2018). Tregs release anti-inflammatory cytokine IL-10 to regulate M1-to-M2 like MΦ phenotype switching during muscle repair following injury (Deng et al., 2012; Schiaffino et al., 2017; D’Alessio et al., 2019). Tregs also have been shown to stimulate proliferation of satellite cells through the release of Amphireglin (AREG) – a key regulator of immunity and tissue repair (Burzyn et al., 2013; Arpaia et al., 2015; Castiglioni et al., 2015; Zaiss et al., 2015). The mechanisms of Treg’s regulation of skeletal muscle repair are still being unearthed; however, the induction of IL-33 with injured skeletal muscle appears the likely candidate for the increased abundance of Tregs and Treg-associated AREG (Kuswanto et al., 2016). Our understanding of CD8+ CTLs role in skeletal muscle repair centers around potentiating the immune response through its own infiltration into damaged muscle and releasing pro-inflammatory cytokines, namely MCP-1 (Zhang et al., 2014). Rag1 deficient mice that lack both T‐ and B-cells, but maintain intact MΦs, had delayed skeletal muscle repair following cardiotoxin; however, reintroduction of both CD4+ and CD8+ T-cells rescued the repair process timeline (Fu et al., 2015). These investigations primarily focused on the impact of T-cells on the activation and proliferation of satellite cells, which is necessary for the repair process.
CTLs and Th cells appear to play a role in the pathological damage response in Duchenne’s muscular dystrophy (DMD), while there is promise for manipulating Tregs to mitigate excessive skeletal muscle damage (Spencer et al., 2001; Villalta et al., 2015). Similar to neutrophils, there has not been investigative inquiry into the role of T-cells in cancer or chemotherapy-induced skeletal muscle wasting; however, T-cells have been investigated extensively for their role in immune suppression during cancer progression and are promising therapeutic targets given their role in cytotoxicity/cellular immunity (Naito et al., 1998; Almand et al., 2001). To this end, tumor infiltrating lymphocytes (TILs) and ex vivo expansion of T-cells provides important prognosis information (Walsh et al., 2005; Morgan et al., 2006). Interestingly, CD8+ T-cells have been demonstrated to induce skeletal muscle wasting during infection-associated cachexia as shown by improved body weight maintenance in CD8+ T-cell null mice (Baazim et al., 2019). However, mechanisms underlying infection-associated cachexia may be distinct from cancer-associated cachexia. The most promising study was conducted by Wang et al. (2008) showing infusion of CD4+CD44Low naïve T-cells could attenuate muscle mass loss associated with ameliorating cancer-associated lymphopenia. Additionally, there is evidence of an inverse relationship between body weight and NLR – as NLR increases, body weight decreases – highlighting that this ratio may be an intriguing surrogate for the immune milieu responsible for cachexia progression (Derman et al., 2017). Given the important role of T-cells in cancer progression, treatment, and proposed role in muscle mass maintenance, studies aimed at understanding each T-cell’s role in cancer and chemotherapy-associated cachexia are needed.
While the primary focus of this review is to highlight the impact of monocytes, neutrophils, and T-cells on the skeletal muscle microenvironment, other immune cells involved in cancer’s immune disruptions should not be neglected as their role in skeletal muscle homeostasis is merely less understood. These cells include, but are not limited to, B-cells, NK cells, myeloid derived suppressor cells (MDSC), and mast cells (Murphy and Weaver, 2016). The role of these various cell types in the pathogenesis of DMD has been reviewed (Madaro and Bouche, 2014). B-cells were recently shown to modulate muscle weakness during dermatomyositis through the regulation of IFNγ (Radke et al., 2018). Mast cells aggregate and proliferated in sites of muscle necrosis following damage and can restore vascular permeability during myositis (Gorospe et al., 1996; Yokota et al., 2014); however, little is known about their role with muscle wasting with cancer and chemotherapy. Interestingly, MDSCs were shown to be increased with cancer thought to contribute to global disruptions in energy metabolism contributing to cachexia progression (Cuenca et al., 2014). A correlative increase of IL-17 and MDSCs were both associated with nutritional impairments with gastrointestinal cancer which was proposed to contribute to cachexia (Yazawa et al., 2013). Furthermore, the metabolic cost of myelopoiesis, mainly increased MDSCs, may also contribute to the metabolic abnormalities seen with cancer and chemotherapy (Sica and Strauss, 2017).
The myofiber response to cancer and the cachectic environment is considered to be central to understanding muscle wasting regulation. However, there is an emerging consideration for tumor derived factors and inflammatory mediators to go beyond targeting the myofiber directly (Biswas and Acharyya, 2020). The cachectic environment has the potential to also indirectly disrupt myofiber homeostasis through the altered regulation of other cell types in the muscle microenvironment (Talbert and Guttridge, 2016), highlighting a need to delineate the capacity for cancer and chemotherapy to disrupt myofiber interactions with other cell types in muscle (Figure 2). Furthermore, each cell type has a critical function within the skeletal muscle microenvironment, involving ECM remodeling and angiogenesis, which are established components of muscle plasticity in response to regeneration from injury, growth, aging, overload-induced hypertrophy, and exercise (Dennis et al., 2009; Morgan and Partridge, 2020). Since skeletal muscle fiber nuclei are post-mitotic, the role of the satellite cell in aging and overload hypertrophy has been actively investigated for over 30 years. This line of inquiry includes the role of myonuclear apoptosis in sarcopenia and myonuclear accretion in hypertrophy myofibers (McCarthy et al., 2011; Fry et al., 2014). As skeletal muscle remodeling is a critical property of muscle and is thought to be a continuous process over an individual’s life span, there is growing interest in how aging, disuse, cancer, and chemotherapy treatments can disrupt remodeling processes in skeletal muscle, which could serve to exacerbate the development and progression of cachexia.
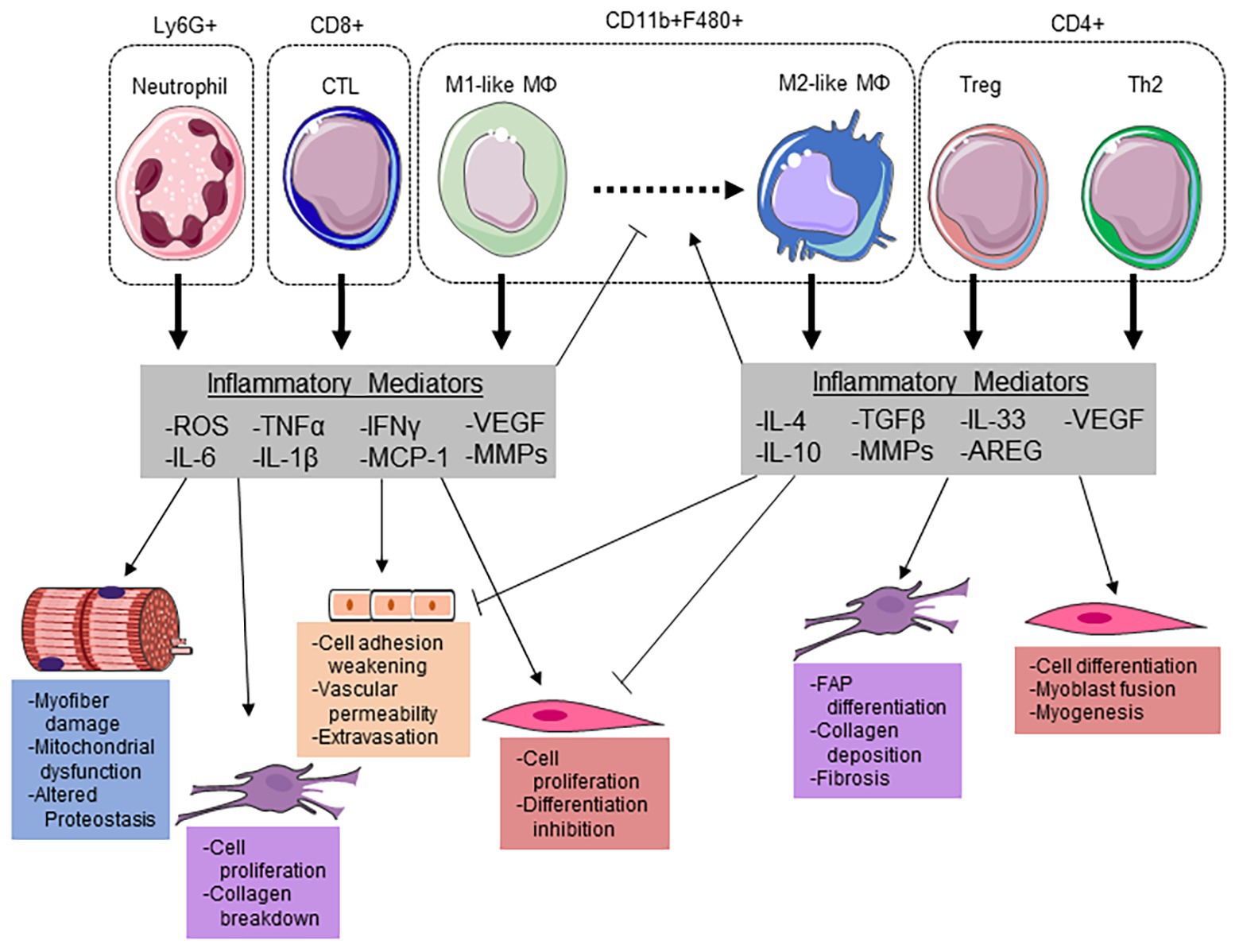
Figure 2. Immune cells modulate the myofiber microenvironment through releasing key inflammatory mediators. Neutrophils potentiate cellular damage through the release of reactive oxygen species (ROS), which can induce myofiber damage and increase vascular permeability for extravasation. Additionally, neutrophils release pro-inflammatory cytokines interleukin (IL)-6, IL-1β, and tumor necrosis factor α (TNF-α) to stimulate satellite cell proliferation and suppress differentiation. IL-6 and ROS have been demonstrated to induce mitochondrial dysfunction/damage and altered proteostasis within the myofiber. Along with cytotoxic T-lymphocytes (CTLs), neutrophils release monocyte chemoattractant protein (MCP) 1 to recruit inflammatory (M1-like) macrophages (MΦ). Both infiltrating monocytes and resident MΦs can polarize to an M1-like phenotype to aid in phagocytizing damaged myofibrillar proteins and the breakdown of collagen in the ECM through matrix metalloproteinases (MMPs). Along with an increase in Interleukin-4 (IL-4), regulatory T-cells (Tregs) can release IL-33 to stimulate amphiregulin (AREG) to promote MΦ polarization to a M2-like phenotype. These MΦs then release transforming growth factor (TGF) β and IL-10 to promote fibro-adipogenic progenitor (FAP) cell differentiation, ECM remodeling, and suppress the pro-inflammatory milieu. The reduction in pro-inflammatory cytokines allows for satellite cell differentiation and myogenesis. This figure was made with Servier Medical Art (http://www.servier.com/Powerpoint-image-bank).
The ECM surrounding each muscle fiber can contain an assortment of fibroblasts, endothelial cells, immune cells, and satellite cells which are located beneath the basal lamina (Forcina et al., 2019). These cells have individual and coordinated roles in the response to local inflammation that accompanies muscle remodeling to damage, increased use/activity, and overload (Morgan and Partridge, 2020). Furthermore, changes within the ECM serve to initiate intracellular myofiber signaling through receptors on the sarcolemma in response to damage and mechanical stretch (Carson and Wei, 2000). The ECM is a rich source of growth factors and cytokines that can be released to regulate myofiber proteostasis and metabolic function (Forcina et al., 2019). Muscle fibers interact with the fibrous elements of the matrix through integrin receptors to sense mechanical signaling, and shifts in the expression of fibrous matrix proteins such as collagens have potential to alter intracellular signaling in muscle fibers (Carson and Wei, 2000). Thus, the cancer systemic environment has the potential to dysregulate myofiber gene expression and metabolic signaling through alterations to the ECM. Muscle fibrosis and dysregulation of the ECM has been reported in cachectic skeletal muscle from pancreatic cancer patients (Judge et al., 2018; Nosacka et al., 2020), and fibrosis can negatively impact myofiber growth and healing from damage (Bedair et al., 2007; Li et al., 2016a). Muscle inflammatory processes are established modulators of the ECM for healing and wound repair and also critical for muscle remodeling to exercise and overload. In fact, disruptions to IL-6 expression during overload-induced hypertrophy and during the recovery from disuse-induced muscle atrophy in mice can alter the hypertrophy response, collagen composition within the ECM, and muscle fibrosis in mice (White et al., 2009; Washington et al., 2011). Furthermore, during the initiation of compensatory overload-induced hypertrophy in mice an inflammatory and ECM gene expression is strongly induced (Carson et al., 2002). These observations demonstrate the complexity of the ECM response and local inflammation, since they drive hypertrophy with overload and healing from damage, but are also implicated in cancer-induced muscle wasting (Forcina et al., 2019). To this end, local inflammatory responses are subjected to precise temporal regulation and if this response is altered muscle remodeling can be either attenuated or blocked (Howard et al., 2020), and further research is warranted to determine if cancer can disrupt the temporal aspects of local inflammation needed for successful remodeling. For example, inflammatory signaling can impact several cell types located in the muscle microenvironment leading to altered myofiber protein synthesis (Gao et al., 2017) and mitochondrial quality control (Gomez-Cabrera et al., 2016), which are known drivers of muscle wasting with cancer. In this section, three important cell types are highlighted that interact with the myofiber within the muscle microenvironment and their potential role in cancer and chemotherapy-induced muscle wasting is emphasized.
The role of the satellite cell in skeletal muscle physiological and biological responses to a variety of conditions, stimuli, and disease states has been extensively investigated since being first discovered by Mauro in 1961 (Mauro, 1961; Penna et al., 2010; He et al., 2013). The satellite cell is located between the myofiber sarcolemma and basal lamina and is not easily distinguished from a myonuclei with light microscopy. These cells remain in a quiescent state, but can be activated to proliferate and differentiate by a variety of growth factors and mitogenic stimuli (Bischoff, 1986a,b). Satellite cell differentiation can involve either fusion with existing myofibers to allow for myonuclear accretion or the formation of de novo myotubes. Therefore, the satellite cell has been extensively examined as a myogenic stem cell in skeletal muscle critical for muscle repair and growth processes. Both the regulation and disruption of satellite cell number, activation, proliferation potential, and differentiation have been widely investigated for 60 years (Forcina et al., 2019). Interestingly, one of the earliest described inducers of satellite cell proliferation was FGF, and this activation was also linked to damage disruption of the muscle ECM (Bischoff, 1986a,b). This line of inquiry coincides with investigation to this day involving satellite cell control by their microenvironment and the cells that are at the source of these mitogenic factors. There has been increasing interest in understanding the cancer environment’s effect on satellite cell activity, which could directly impact cachectic muscle’s diminished capacity for muscle regeneration (Schwarzkopf et al., 2006; Penna et al., 2010; He et al., 2013; Talbert and Guttridge, 2016). Deficits in the capacity for myogenesis, which is needed for successful muscle regeneration, have been reported in skeletal muscle from preclinical cachexia models and cancer patients, and involve an inability of satellite cells to effectively differentiate to complete the regenerative process (Penna et al., 2010; He et al., 2013). Due to the importance of the successful myogenic response in muscle regeneration, signaling pathways disrupting satellite cell activity have been actively investigated as potential therapeutic targets for cancer-induced muscle wasting (Schwarzkopf et al., 2006; He et al., 2013; Cerquone Perpetuini et al., 2018; Costamagna et al., 2020; Schmidt et al., 2020). To this end, skeletal muscle satellite cell differentiation has been reported to be rescued in colon-26 induced cachexia in mice through the inhibition or ERK signaling (Penna et al., 2010), and more recently IL-4 administration (Costamagna et al., 2020). Pax7 is a positive regulator of satellite cell proliferation, which has been reported to be disrupted in cachectic muscle from mice and cancer patients (He et al., 2013); dysregulation of Pax7 in cachectic muscle was driven by aberrant NF-κΒ signaling induced by circulating tumor derived factors. Wnt signaling is an established regulator of muscle satellite cell activity through the expression of myogenic regulatory factors and increased Wnt7a in colon-26 induced cachexia in mice can increase muscle regeneration by the improvement of muscle satellite regulation of differentiation and the induction of anabolic signaling through Akt/mTORC1 (Schmidt et al., 2020). Interestingly, mathematical modeling of colon-26 induced cachexia in mice recently predicted that blocking the myostatin/activin A pathway could attenuate cancer-induced muscle mass loss through the restoration of functional satellite cell differentiation (Farhang-Sardroodi and Wilkie, 2020). Depending on the extent of the damage, successful muscle regeneration including the initiation and resolution of local inflammatory processes involves infiltrating monocytes, resident MΦs, neutrophils, and T-cells (Morgan and Partridge, 2020). Local inflammatory responses are subjected to precise temporal regulation and if this response is altered muscle remodeling can be either attenuated or blocked (Howard et al., 2020), and further research is warranted to determine if cancer can disrupt the temporal aspects of local inflammation that contribute to disrupted satellite cell regulation of differentiation in cachectic muscle.
The satellite cell has been hypothesized to have critical roles in post-natal muscle growth, overload-induced hypertrophy, capacity of muscle regeneration after damage, and the development of sarcopenia. The development of modern molecular biology tools has shed light on the role of satellite depletion on these processes (McCarthy et al., 2011; Fry et al., 2014), but interesting gain of function experiments remain elusive. While the role of the satellite cell with some types of overload-induced growth and sarcopenia remains equivocal (Murach et al., 2018), the satellite cell has an established role in muscle regeneration from injury. Interestingly, there is emerging evidence that satellite cells have a regulatory role in ECM during overload-induced hypertrophy (Murach et al., 2018). Additionally, MΦs have been demonstrated to interact with satellite cells to regulate myogenesis (Dort et al., 2019). Pro-inflammatory monocytes and MΦs release cytokines, IL-6 and TNF-α, to stimulate satellite cell proliferation and inhibit differentiation until switching to an M2-like phenotype to promote differentiation and fusion (Arnold et al., 2007). T-cells have been demonstrated to maintain satellite cells myogenic potency in vitro (Fu et al., 2015), while T-cell deficient mice had reduced satellite cell proliferation contributing to blunted repair (Castiglioni et al., 2015; Deyhle and Hyldahl, 2018). There is significant interest in muscle microenvironment changes that occur with aging that could alter satellite cell regulation and serve to impede muscle regeneration from injury (Lee et al., 2019). Satellite cell number and activity are thought to be impacted by aging, and other regulators of satellite cell function such as the immune response are also aging targets (Forcina et al., 2019). Therefore, the impact of age on satellite cell activity could be a factor in the development and progression of cachexia due to the advanced age of many patients having cancers that are associated with high cachexia incidence. These facts could directly impact our current mechanistic understanding of cancer cachexia, as preclinical modeling that account for the effects of sarcopenia and aging on the skeletal muscle microenvironment are needed to answer these critical questions.
Fibroblasts have a central role in the formation and remodeling of the skeletal muscle ECM which provides stability for the myofiber, blood vessels, and nerves (Chapman et al., 2016; Mendias, 2017). Fibroblasts aid in the synthesis and deposition of collagen, which predominate skeletal muscle connective tissues. Matrix metalloproteinases (MMPs) are largely responsible to the breakdown and deposition of collagen in the ECM. MMPs are released by immune cells, myofibers, satellite cells, epithelial cells, and fibroblasts themselves and are transcriptionally regulated by classic cytokine signaling cascades (Thomas et al., 1998; Liu et al., 2006; Phatharajaree et al., 2007; Yndestad et al., 2007; Iyer et al., 2016). These cells release MMPs to degrade several components of the cytoskeleton and stimulate de novo collagen deposition by the fibroblast (Sundararaj et al., 2009). Additionally, fibroblasts and satellite cells coordinate during skeletal muscle regeneration to limit fibrosis and promote healthy hypertrophic repair (Murphy et al., 2011).
The role of several immune cell types and inflammatory mediators in skeletal muscle fibroblast proliferation and collagen deposition has been extensively studied due to their integral functions in tissue repair (Van Linthout et al., 2014). In addition to providing structural support, fibroblasts are sensitive to mechanical stretch and maintain the ECM by aiding in the breakdown and synthesis/deposition of skeletal muscle collagens (Tomasek et al., 2002). Additionally, this mechanical sensitivity allows fibroblasts to release their own cytokines/chemokines for early neutrophil recruitment to aid in the breakdown of the damaged ECM through the action of MMPs, collagenases, and elastases (Baici et al., 1982; Kim and Luster, 2015); however, this mechanistic link in skeletal muscle requires additional work. On the other hand, innate immune cells can induce re-expression of fetal genes, activation of MMPs, fibro-adipogenic progenitor (FAP) differentiation, proliferation of fibroblasts, and myofibroblast formation (Tomasek et al., 2002; Gentek and Hoeffel, 2017; Tidball, 2017). Pro-inflammatory cytokines TNF-α, IL-6, and IL-1β are produced and secreted by Th cells and M1-like MΦs to stimulate fibroblast proliferation, while the anti-inflammatory TGFβ and IL-10 are predominately produced by Tregs and M2-like MΦs to stimulate fibroblast collagen synthesis and deposition (Van Linthout et al., 2014; Jablonski et al., 2015). M2-like MΦ release of arginase 1 (Arg1) also can stimulate fibroblast proliferation through the production of polyamines from arginase metabolism (Tidball, 2017). While the proper coordination of fibroblast proliferation and collagen formation is necessary for healthy skeletal muscle repair and remodeling, both reduced or exacerbated fibroblast activity can result in fibrosis and reduced skeletal muscle plasticity and overall functional capacity (Chapman et al., 2016). Fibrosis during the progression of cancer cachexia has received significant attention due to its role in reduced skeletal muscle plasticity and the loss of strength disproportionate to mass (Judge et al., 2018; Ramage and Skipworth, 2018); however, evidence of impaired regeneration and increased fibrosis in cachectic skeletal muscle is limited to cancer type and severity of the disease (Nosacka et al., 2020).
Endothelial cells are cells that are associated with blood vessels, which vascularize skeletal muscle. Intuitively, skeletal muscle vascularization plays central roles in nutrient transportation (e.g., glucose delivery and CO2 removal) and global/systemic communication (e.g., hormones and cytokines/myokines). When necessary, the myofiber, resident fibroblasts, and/or resident immune cells secrete cytokines and pro-inflammatory mediators to activate nearby endothelial cells initiating circulating immune cell extravasation into the damaged tissue (Vestweber, 2015). As previously discussed, the influx of neutrophils, activated monocytes, and pro-inflammatory MΦs are central for skeletal muscle repair and remodeling. A key function of neutrophils in promoting cell death is the generation of ROS through NADPH oxygenase-dependent mechanisms (Kolaczkowska and Kubes, 2013). While ROS can be destructive to skeletal muscle in several ways, ROS is an important regulator of skeletal muscle vascular permeability (Korthuis et al., 1985). VEGF has received significant attention for its role in stimulating angiogenesis and promotion of vascular permeability is vital for skeletal muscle repair and remodeling (Arsic et al., 2004). Endothelial cells rely heavily on self VEGF production and autocrine signaling for vascular homeostasis and this autocrine signaling cascade differs from exogenous or paracrine VEGF signaling (Lee et al., 2007). Interestingly, VEGF has been implicated as a regulator of DC differentiation and MΦ polarization (Li et al., 2016b). While monocytes and MCP-1 stimulate the release of VEGF by endothelial cells to increase myofibrillar vascularization during the later stages of repair (Hong et al., 2005), tumor associated macrophages (TAMs) promote the release of VEGF to promote tumor vascularization (Wynn et al., 2013). However, IL-4 inhibits basic Fibroblast Growth Factor (bFGF) induced angiogenesis (Volpert et al., 1998). Therefore, it is vital to understand off-target effects of modulating immune cells and their inflammatory mediators on the endothelium within the skeletal muscle microenvironment.
Endothelial cells have sustained significant attention in tumor cell biology given the importance of tumor vascularization in tumor cell growth and metastasis (Dudley, 2012; Li et al., 2016b). Naturally, treatments targeting endothelial cells and mitigating vascularization has emerged as a promising therapy; however, given the importance of vascular homeostasis in maintaining healthy tissue, the off-target implications of these treatments invoke caution (Morabito et al., 2006). While the effects of cachexia and chemotherapy on the skeletal muscle endothelium or the relationship between immune cells and the skeletal muscle endothelium has not been directly studied, we can glean information from studies examining aging, non-cancer-induced cachexia, and established inflammatory signaling and nutrient availability mechanisms. For instance, COPD patients suffering from cachexia have reduced skeletal muscle capillary density partly caused by deconditioning (Jobin et al., 1998); given that cancer patients experience fatigue and reduced activity, we can expect that cachectic skeletal muscle with cancer has decreased vascularization. Reduced vascularization would diminish skeletal muscle’s regenerative capacity resulting in increased fibrosis, reduced strength, and fatigue (Arsic et al., 2004; Allen et al., 2008; Inaba et al., 2018; Judge et al., 2018; Vanderveen et al., 2018). Additionally, aging processes contributing to frailty has been associated with endothelial dysfunction (Argiles et al., 2016). Inhibition of angiogenesis is a critical target for some chemotherapeutics aimed at preventing vascularization of solid tumors through suppressing VEGF (Zhang et al., 2002; Shih and Lindley, 2006; Ooyama et al., 2008; Bozzetti, 2020). The impact on non-cancerous tissues continues to be unearthed; however, endothelial dysfunction has been demonstrated during chemotherapy driven cardiotoxicity (Brown et al., 2012). Additionally, anti-angiogenic therapies should aim to achieve vascular normalization in order to maintain the delivery and anti-tumor efficacy of chemotherapeutics as opposed to eliminating tumor vascularity completely (Viallard and Larrivee, 2017). Chemotherapy’s effect on skeletal muscle’s endothelium has not been directly investigated. Given the neutrophil’s role in ROS generation, the neutropenia associated with chemotherapy may inhibit the skeletal muscle’s ability to increase vascular permeability and immune cell extravasation. Further work is needed to understand the impact of cancer and its treatments on skeletal muscle’s vascular endothelium.
The nervous and musculoskeletal systems are inextricably linked through the motor unit and denervation, or the loss of the neuromuscular junction (NMJ), results in loss of function and rapid muscle atrophy (Bonaldo and Sandri, 2013). Denervation results in the affected myofibers losing their contractile capacity, which is then followed by a marked reduction in myofibrillar cross sectional area. If the NMJ is not restored, the affected myofibers will become unrecognizable, as they lose their sarcomeric structure and become fibrotic (Carlson, 2014). Similar to other atrophic conditions, MΦs have been identified as contributors to denervation-induced fibrosis (Mochizuki et al., 2005). Interestingly, denervation with cancer cachexia has been suggested in C26 mice (Daou et al., 2020), and neurotoxicity was observed with chemotherapeutic Folfiri (Barreto et al., 2016a); however, gastrointestinal cancer patients with cachexia had no significant loss of NMJs compared to weight stable or health controls (Boehm et al., 2020). Chemotherapeutic DOX disrupted the NMJ in sedentary rats while exercise was able to improve neuromuscular gene and protein expression (Huertas et al., 2020).
The cellular environment surrounding the myofiber, or the ECM, is a mixture of mononucleated satellite cells, fibroblasts, endothelial cells, and immune cells. Together these cells cooperate and aid in locomotion (e.g., tendon formation/strength), systemic and local metabolic homeostasis, structural support, repair/remodeling, and systemic communication (e.g., myokines). Coordination of this complex network relies on each cell functioning to communicate within endocrine, paracrine, and autocrine mechanisms given a multitude of stimuli (Meng et al., 2015; Bersini et al., 2018). The complex nature of the skeletal muscle microenvironment invites difficulty in identifying key regulators of muscle mass and function loss with cancer and chemotherapy; however, targeting inflammation and immune cells prior to‐ or during cachexia’s progression could promote widespread benefits given their regulatory roles in several skeletal muscle processes. While several inflammatory mediators (IL-6, TNF-α, TWEAK, TRAF6, INF-γ, and LIF) have been implicated as drivers of cancer-associated wasting, very little is known about their origin, whether skeletal muscle, immune cell, or tumor-derived (Flint et al., 2016; Jackman et al., 2017). In this review, we highlighted that resident and circulating immune cells can modulate several aspects of the skeletal muscle microenvironment through a multitude of mechanisms. To this end, omitting investigation of multiple cell types within the muscle’s microenvironment during cachexia has the potential to hamper therapeutic developments/advancements leading to the current treatments, which have been unable to improve muscle function. Additional mechanistic inquiry is needed to understand how immune cells contribute to cancer and chemotherapy associated muscle mass and function loss.
For decades now, endurance and resistance exercise have been proposed as promising therapies as they broadly impact chronic inflammation, metabolic homeostasis, and muscle protein turnover (Al-Majid and McCarthy, 2001; Montalvo et al., 2018). There are barriers to exercise as a therapeutic for cancer cachexia as it is unlikely that a level of exercise to induce muscle mass can be performed by patients with debilitating conditions like cancer. However, muscle contractions induced by neuromuscular electrical stimulation was recently shown to improve muscle mass in breast cancer patients receiving chemotherapy (Guigni et al., 2019; Toth et al., 2020). Both resistance (Bredahl et al., 2020) and endurance (Smuder, 2019; Huertas et al., 2020; Montalvo et al., 2020) exercise have shown promise in mitigating chemotherapy-induced skeletal muscle toxicities in preclinical rats. Additionally, resistance (Hardee et al., 2016, 2018) and endurance (Puppa et al., 2012; Vanderveen et al., 2020) exercise can improve muscle mass and function in cachectic mice. Exercise also showed promise in preventing the loss of muscle mass and strength in C26 mice given 5FU + Oxaliplatin (Ballaro et al., 2019). Exercise oncology continues to be an active and intriguing field of study aimed at improving cancer patient’s survival and quality of life (Schmitz et al., 2019). While traditional nutritional support has been unsuccessful, several nutraceuticals and nutritional anti-inflammatories have promise in mitigating cancer-induced wasting (Narsale et al., 2016). Moreover, there is growing interest in identifying plant-derived compounds to treat cachexia given that they can modulate multiple pathways, are inexpensive, and have low toxicity as prolonged treatments (Saklani and Kutty, 2008). Plant-derived compounds have been shown to regulate inflammation through the modulation of MΦ polarization with cancer (Jia et al., 2014; Hu et al., 2017); however, the therapeutic efficacy of modulating MΦ phenotype and function during cancer-associated wasting has not been investigated.
JC planned, wrote, and edited manuscript. BV planned, wrote, and edited manuscript, created figures. EM edited and wrote manuscript. All authors contributed to the article and approved the submitted version.
This work is supported by the National Institutes of Health (NCI) RO1CA121249 and R21CA231131 to JC and (NCCIH) R41AT009964 to EM.
BV and EM were employed by company AcePre, LLC.The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbas, A. K., Lichtman, A. H., Pillai, S., Baker, D. L., and Baker, A. (2018). Cellular and molecular immunology. Philadelphia, PA: Elsevier.
Abraham, J. E., Hiller, L., Dorling, L., Vallier, A. L., Dunn, J., Bowden, S., et al. (2015). A nested cohort study of 6,248 early breast cancer patients treated in neoadjuvant and adjuvant chemotherapy trials investigating the prognostic value of chemotherapy-related toxicities. BMC Med. 13:306. doi: 10.1186/s12916-015-0547-5
Allen, D. G., Lamb, G. D., and Westerblad, H. (2008). Skeletal muscle fatigue: cellular mechanisms. Physiol. Rev. 88, 287–332. doi: 10.1152/physrev.00015.2007
Al-Majid, S., and McCarthy, D. O. (2001). Cancer-induced fatigue and skeletal muscle wasting: the role of exercise. Biol. Res. Nurs. 2, 186–197. doi: 10.1177/109980040100200304
Almand, B., Clark, J. I., Nikitina, E., Van Beynen, J., English, N. R., Knight, S. C., et al. (2001). Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J. Immunol. 166, 678–689. doi: 10.4049/jimmunol.166.1.678
Anderson, L. J., Albrecht, E. D., and Garcia, J. M. (2017). Update on management of cancer-related cachexia. Curr. Oncol. Rep. 19:3. doi: 10.1007/s11912-017-0562-0
Argiles, J. M., Anker, S. D., Evans, W. J., Morley, J. E., Fearon, K. C., Strasser, F., et al. (2010). Consensus on cachexia definitions. J. Am. Med. Dir. Assoc. 11, 229–230. doi: 10.1016/j.jamda.2010.02.004
Argiles, J. M., Busquets, S., Stemmler, B., and Lopez-Soriano, F. J. (2014). Cancer cachexia: understanding the molecular basis. Nat. Rev. Cancer 14, 754–762. doi: 10.1038/nrc3829
Argiles, J. M., Campos, N., Lopez-Pedrosa, J. M., Rueda, R., and Rodriguez-Manas, L. (2016). Skeletal muscle regulates metabolism via interorgan crosstalk: roles in health and disease. J. Am. Med. Dir. Assoc. 17, 789–796. doi: 10.1016/j.jamda.2016.04.019
Argiles, J. M., Lopez-Soriano, F. J., and Busquets, S. (2015). Muscle wasting in cancer: the role of mitochondria. Curr. Opin. Clin. Nutr. Metab. Care 18, 221–225. doi: 10.1097/MCO.0000000000000164
Argilés, J. M., López-Soriano, F. J., and Busquets, S. (2019). Mediators of cachexia in cancer patients. Nutrition 66, 11–15. doi: 10.1016/j.nut.2019.03.012
Arnold, L., Henry, A., Poron, F., Baba-Amer, Y., Van Rooijen, N., Plonquet, A., et al. (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069. doi: 10.1084/jem.20070075
Arpaia, N., Green, J. A., Moltedo, B., Arvey, A., Hemmers, S., Yuan, S., et al. (2015). A distinct function of regulatory T cells in tissue protection. Cell 162, 1078–1089. doi: 10.1016/j.cell.2015.08.021
Arsic, N., Zacchigna, S., Zentilin, L., Ramirez-Correa, G., Pattarini, L., Salvi, A., et al. (2004). Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol. Ther. 10, 844–854. doi: 10.1016/j.ymthe.2004.08.007
Atherton, P. J., Greenhaff, P. L., Phillips, S. M., Bodine, S. C., Adams, C. M., and Lang, C. H. (2016). Control of skeletal muscle atrophy in response to disuse: clinical/preclinical contentions and fallacies of evidence. Am. J. Physiol. Endocrinol. Metab. 311, E594–E604. doi: 10.1152/ajpendo.00257.2016
Aversa, Z., Pin, F., Lucia, S., Penna, F., Verzaro, R., Fazi, M., et al. (2016). Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 6:30340. doi: 10.1038/srep30340
Baazim, H., Schweiger, M., Moschinger, M., Xu, H., Scherer, T., Popa, A., et al. (2019). CD8+ T cells induce cachexia during chronic viral infection. Nat. Immunol. 20, 701–710. doi: 10.1038/s41590-019-0397-y
Baechler, S., Hobbs, R. F., Jacene, H. A., Bochud, F. O., Wahl, R. L., and Sgouros, G. (2010). Predicting hematologic toxicity in patients undergoing radioimmunotherapy with 90Y-ibritumomab tiuxetan or 131I-tositumomab. J. Nucl. Med. 51, 1878–1884. doi: 10.2967/jnumed.110.079947
Baici, A., Salgam, P., Cohen, G., Fehr, K., and Boni, A. (1982). Action of collagenase and elastase from human polymorphonuclear leukocytes on human articular cartilage. Rheumatol. Int. 2, 11–16. doi: 10.1007/BF00541264
Ballaro, R., Beltra, M., De Lucia, S., Pin, F., Ranjbar, K., Hulmi, J. J., et al. (2019). Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 33, 5482–5494. doi: 10.1096/fj.201801862R
Baracos, V. E. (2018). Bridging the gap: are animal models consistent with clinical cancer cachexia? Nat. Rev. Clin. Oncol. 15, 197–198. doi: 10.1038/nrclinonc.2018.14
Baracos, V. E., Martin, L., Korc, M., Guttridge, D. C., and Fearon, K. C. H. (2018). Cancer-associated cachexia. Nat. Rev. Dis. Primers 4:17105. doi: 10.1038/nrdp.2017.105
Barreto, R., Kitase, Y., Matsumoto, T., Pin, F., Colston, K. C., Couch, K. E., et al. (2017). ACVR2B/Fc counteracts chemotherapy-induced loss of muscle and bone mass. Sci. Rep. 7:14470. doi: 10.1038/s41598-017-15040-1
Barreto, R., Mandili, G., Witzmann, F. A., Novelli, F., Zimmers, T. A., and Bonetto, A. (2016a). Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front. Physiol. 7:472. doi: 10.3389/fphys.2016.00472
Barreto, R., Waning, D. L., Gao, H., Liu, Y., Zimmers, T. A., and Bonetto, A. (2016b). Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 7, 43442–43460. doi: 10.18632/oncotarget.9779
Bedair, H., Liu, T. T., Kaar, J. L., Badlani, S., Russell, A. J., Li, Y., et al. (2007). Matrix metalloproteinase-1 therapy improves muscle healing. J. Appl. Physiol. 102, 2338–2345. doi: 10.1152/japplphysiol.00670.2006
Bersini, S., Gilardi, M., Ugolini, G. S., Sansoni, V., Talo, G., Perego, S., et al. (2018). Engineering an environment for the study of fibrosis: a 3D human muscle model with endothelium specificity and endomysium. Cell Rep. 25, 3858.e4–3868.e4. doi: 10.1016/j.celrep.2018.11.092
Bi, Y., Liu, G., and Yang, R. (2007). Th17 cell induction and immune regulatory effects. J. Cell. Physiol. 211, 273–278. doi: 10.1002/jcp.20973
Bischoff, R. (1986a). Proliferation of muscle satellite cells on intact myofibers in culture. Dev. Biol. 115, 129–139. doi: 10.1016/0012-1606(86)90234-4
Bischoff, R. (1986b). A satellite cell mitogen from crushed adult muscle. Dev. Biol. 115, 140–147. doi: 10.1016/0012-1606(86)90235-6
Biswas, A. K., and Acharyya, S. (2020). Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 20, 274–284. doi: 10.1038/s41568-020-0251-4
Boehm, I., Miller, J., Wishart, T. M., Wigmore, S. J., Skipworth, R. J., Jones, R. A., et al. (2020). Neuromuscular junctions are stable in patients with cancer cachexia. J. Clin. Invest. 130, 1461–1465. doi: 10.1172/JCI128411
Bonaldo, P., and Sandri, M. (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 6, 25–39. doi: 10.1242/dmm.010389
Bozzetti, F. (2020). Chemotherapy-induced sarcopenia. Curr. Treat. Options in Oncol. 21:7. doi: 10.1007/s11864-019-0691-9
Bredahl, E. C., Sharif, S., Siedlik, J. A., Wagner, M. K., Twaddell, M. D., Tigner, A. T., et al. (2020). Resistance training during chemotherapy with doxorubicin. Med. Sci. Sports Exerc. doi: 10.1249/MSS.0000000000002409 [Epub ahead of print]
Broadbent, J., Sampson, D., Sabapathy, S., Haseler, L. J., Wagner, K. H., Bulmer, A. C., et al. (2017). Gene networks in skeletal muscle following endurance exercise are coexpressed in blood neutrophils and linked with blood inflammation markers. J. Appl. Physiol. 122, 752–766. doi: 10.1152/japplphysiol.00114.2016
Brown, J. C., Winters-Stone, K., Lee, A., and Schmitz, K. H. (2012). Cancer, physical activity, and exercise. Compr. Physiol. 2, 2775–2809. doi: 10.1002/cphy.c120005
Bruera, E. (1997). ABC of palliative care. Anorexia, cachexia, and nutrition. BMJ 315, 1219–1222. doi: 10.1136/bmj.315.7117.1219
Burzyn, D., Kuswanto, W., Kolodin, D., Shadrach, J. L., Cerletti, M., Jang, Y., et al. (2013). A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295. doi: 10.1016/j.cell.2013.10.054
Capoccia, B. J., Gregory, A. D., and Link, D. C. (2008). Recruitment of the inflammatory subset of monocytes to sites of ischemia induces angiogenesis in a monocyte chemoattractant protein-1-dependent fashion. J. Leukoc. Biol. 84, 760–768. doi: 10.1189/jlb.1107756
Carlson, B. M. (2014). The biology of long-term denervated skeletal muscle. Eur. J. Transl. Myol. 24:3293. doi: 10.4081/ejtm.2014.3293
Carson, J. A. (1997). The regulation of gene expression in hypertrophying skeletal muscle. Exerc. Sport Sci. Rev. 25, 301–320. doi: 10.1249/00003677-199700250-00013
Carson, J. A., and Baltgalvis, K. A. (2010). Interleukin 6 as a key regulator of muscle mass during cachexia. Exerc. Sport Sci. Rev. 38, 168–176. doi: 10.1097/JES.0b013e3181f44f11
Carson, J. A., Hardee, J. P., and Vanderveen, B. N. (2016). The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Semin. Cell Dev. Biol. 54, 53–67. doi: 10.1016/j.semcdb.2015.11.005
Carson, J. A., Nettleton, D., and Reecy, J. M. (2002). Differential gene expression in the rat soleus muscle during early work overload-induced hypertrophy. FASEB J. 16, 207–209. doi: 10.1096/fj.01-0544fje
Carson, J. A., and Wei, L. (2000). Integrin signaling’s potential for mediating gene expression in hypertrophying skeletal muscle. J. Appl. Physiol. (1985) 88, 337–343. doi: 10.1152/jappl.2000.88.1.337
Castiglioni, A., Corna, G., Rigamonti, E., Basso, V., Vezzoli, M., Monno, A., et al. (2015). FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS One 10:e0128094. doi: 10.1371/journal.pone.0128094
Ceafalan, L. C., Fertig, T. E., Popescu, A. C., Popescu, B. O., Hinescu, M. E., and Gherghiceanu, M. (2018). Skeletal muscle regeneration involves macrophage-myoblast bonding. Cell Adhes. Migr. 12, 228–235. doi: 10.1080/19336918.2017.1346774
Cerquone Perpetuini, A., Re Cecconi, A. D., Chiappa, M., Martinelli, G. B., Fuoco, C., Desiderio, G., et al. (2018). Group I Paks support muscle regeneration and counteract cancer-associated muscle atrophy. J. Cachexia. Sarcopenia Muscle 9, 727–746. doi: 10.1002/jcsm.12303
Chapman, M. A., Meza, R., and Lieber, R. L. (2016). Skeletal muscle fibroblasts in health and disease. Differentiation 92, 108–115. doi: 10.1016/j.diff.2016.05.007
Chu, M. P., Li, Y., Ghosh, S., Sass, S., Smylie, M., Walker, J., et al. (2020). Body composition is prognostic and predictive of ipilimumab activity in metastatic melanoma. J. Cachexia. Sarcopenia Muscle 11, 748–755. doi: 10.1002/jcsm.12538
Costamagna, D., Duelen, R., Penna, F., Neumann, D., Costelli, P., and Sampaolesi, M. (2020). Interleukin-4 administration improves muscle function, adult myogenesis, and lifespan of colon carcinoma-bearing mice. J. Cachexia. Sarcopenia Muscle 11, 783–801. doi: 10.1002/jcsm.12539
Cote, C. H., Bouchard, P., Van Rooijen, N., Marsolais, D., and Duchesne, E. (2013). Monocyte depletion increases local proliferation of macrophage subsets after skeletal muscle injury. BMC Musculoskelet. Disord. 14:359. doi: 10.1186/1471-2474-14-359
Counts, B. R., Hardee, J. P., Fix, D. K., Vanderveen, B. N., Montalvo, R. N., and Carson, J. A. (2020). Cachexia disrupts diurnal regulation of activity, feeding, and muscle mechanistic target of rapamycin complex 1 in mice. Med. Sci. Sports Exerc. 52, 577–587. doi: 10.1249/MSS.0000000000002166
Cuenca, A. G., Cuenca, A. L., Winfield, R. D., Joiner, D. N., Gentile, L., Delano, M. J., et al. (2014). Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J. Immunol. 192, 6111–6119. doi: 10.4049/jimmunol.1302895
Cui, C. Y., Driscoll, R. K., Piao, Y., Chia, C. W., Gorospe, M., and Ferrucci, L. (2019). Skewed macrophage polarization in aging skeletal muscle. Aging Cell 18:e13032. doi: 10.1111/acel.13032
Curran, M. A., Montalvo, W., Yagita, H., and Allison, J. P. (2010). PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. U. S. A. 107, 4275–4280. doi: 10.1073/pnas.0915174107
Dahele, M., Skipworth, R. J., Wall, L., Voss, A., Preston, T., and Fearon, K. C. (2007). Objective physical activity and self-reported quality of life in patients receiving palliative chemotherapy. J. Pain Symptom Manag. 33, 676–685. doi: 10.1016/j.jpainsymman.2006.09.024
D’Alessio, F. R., Kurzhagen, J. T., and Rabb, H. (2019). Reparative T lymphocytes in organ injury. J. Clin. Invest. 129, 2608–2618. doi: 10.1172/JCI124614
Daou, N., Hassani, M., Matos, E., De Castro, G. S., Costa, R. G. F., Seelaender, M., et al. (2020). Displaced myonuclei in cancer cachexia suggest altered innervation. Int. J. Mol. Sci. 21:1092. doi: 10.3390/ijms21031092
De Santa, F., Vitiello, L., Torcinaro, A., and Ferraro, E. (2018). The role of metabolic remodeling in macrophage polarization and its effect on skeletal muscle regeneration. Antioxid. Redox Signal. 30, 1553–1598. doi: 10.1089/ars.2017.7420
Deans, C., and Wigmore, S. J. (2005). Systemic inflammation, cachexia and prognosis in patients with cancer. Curr. Opin. Clin. Nutr. Metab. Care 8, 265–269. doi: 10.1097/01.mco.0000165004.93707.88
Deng, B., Wehling-Henricks, M., Villalta, S. A., Wang, Y., and Tidball, J. G. (2012). IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 189, 3669–3680. doi: 10.4049/jimmunol.1103180
Dennis, R. A., Zhu, H., Kortebein, P. M., Bush, H. M., Harvey, J. F., Sullivan, D. H., et al. (2009). Muscle expression of genes associated with inflammation, growth, and remodeling is strongly correlated in older adults with resistance training outcomes. Physiol. Genomics 38, 169–175. doi: 10.1152/physiolgenomics.00056.2009
Derman, B. A., Macklis, J. N., Azeem, M. S., Sayidine, S., Basu, S., Batus, M., et al. (2017). Relationships between longitudinal neutrophil to lymphocyte ratios, body weight changes, and overall survival in patients with non-small cell lung cancer. BMC Cancer 17:141. doi: 10.1186/s12885-017-3122-y
Devine, R. D., Bicer, S., Reiser, P. J., Velten, M., and Wold, L. E. (2015). Metalloproteinase expression is altered in cardiac and skeletal muscle in cancer cachexia. Am. J. Physiol. Heart Circ. Physiol. 309, H685–H691. doi: 10.1152/ajpheart.00106.2015
Deyhle, M. R., Gier, A. M., Evans, K. C., Eggett, D. L., Nelson, W. B., Parcell, A. C., et al. (2015). Skeletal muscle inflammation following repeated bouts of lengthening contractions in humans. Front. Physiol. 6:424. doi: 10.3389/fphys.2015.00424
Deyhle, M. R., and Hyldahl, R. D. (2018). The role of T lymphocytes in skeletal muscle repair from traumatic and contraction-induced injury. Front. Physiol. 9:768. doi: 10.3389/fphys.2018.00768
Dort, J., Fabre, P., Molina, T., and Dumont, N. A. (2019). Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem Cells Int. 2019:4761427. doi: 10.1155/2019/4761427
Dudley, A. C. (2012). Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2:a006536. doi: 10.1101/cshperspect.a006536
Dumont, N., Bouchard, P., and Frenette, J. (2008). Neutrophil-induced skeletal muscle damage: a calculated and controlled response following hindlimb unloading and reloading. Am. J. Phys. Regul. Integr. Comp. Phys. 295, R1831–R1838. doi: 10.1152/ajpregu.90318.2008
Dunne, R. F., Loh, K. P., Williams, G. R., Jatoi, A., Mustian, K. M., and Mohile, S. G. (2019). Cachexia and sarcopenia in older adults with cancer: a comprehensive review. Cancers (Basel) 11:1861. doi: 10.3390/cancers11121861
Duong, L., Radley-Crabb, H. G., Gardner, J. K., Tomay, F., Dye, D. E., Grounds, M. D., et al. (2018). Macrophage depletion in elderly mice improves response to tumor immunotherapy, increases anti-tumor T cell activity and reduces treatment-induced cachexia. Front. Genet. 9:526. doi: 10.3389/fgene.2018.00526
Egerman, M. A., and Glass, D. J. (2014). Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 49, 59–68. doi: 10.3109/10409238.2013.857291
Evans, W. J., Morley, J. E., Argiles, J., Bales, C., Baracos, V., Guttridge, D., et al. (2008). Cachexia: a new definition. Clin. Nutr. 27, 793–799. doi: 10.1016/j.clnu.2008.06.013
Farhang-Sardroodi, S., and Wilkie, K. P. (2020). Mathematical model of muscle wasting in cancer cachexia. J. Clin. Med. 9:E2029. doi: 10.3390/jcm9072029
Faurschou, M., and Borregaard, N. (2003). Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 5, 1317–1327. doi: 10.1016/j.micinf.2003.09.008
Fearon, K., Arends, J., and Baracos, V. (2012a). Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 10, 90–99. doi: 10.1038/nrclinonc.2012.209
Fearon, K. C., Glass, D. J., and Guttridge, D. C. (2012b). Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 16, 153–166. doi: 10.1016/j.cmet.2012.06.011
Fearon, K., Strasser, F., Anker, S. D., Bosaeus, I., Bruera, E., Fainsinger, R. L., et al. (2011). Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495. doi: 10.1016/S1470-2045(10)70218-7
Febbraio, M. A., Hiscock, N., Sacchetti, M., Fischer, C. P., and Pedersen, B. K. (2004). Interleukin-6 is a novel factor mediating glucose homeostasis during skeletal muscle contraction. Diabetes 53, 1643–1648. doi: 10.2337/diabetes.53.7.1643
Fix, D. K., Hardee, J. P., Gao, S., Vanderveen, B. N., Velazquez, K. T., and Carson, J. A. (2018). Role of gp130 in basal and exercise-trained skeletal muscle mitochondrial quality control. J. Appl. Physiol. 124, 1456–1470. doi: 10.1152/japplphysiol.01063.2017
Fix, D. K., Vanderveen, B. N., Counts, B. R., and Carson, J. A. (2019). Regulation of skeletal muscle DRP-1 and FIS-1 protein expression by IL-6 Signaling. Oxidative Med. Cell. Longev. 2019:8908457. doi: 10.1155/2019/8908457
Flint, T. R., Janowitz, T., Connell, C. M., Roberts, E. W., Denton, A. E., Coll, A. P., et al. (2016). Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 24, 672–684. doi: 10.1016/j.cmet.2016.10.010
Forcina, L., Miano, C., Scicchitano, B. M., and Musaro, A. (2019). Signals from the niche: insights into the role of IGF-1 and IL-6 in modulating skeletal muscle fibrosis. Cell 8:232. doi: 10.3390/cells8030232
Frenette, J., Chbinou, N., Godbout, C., Marsolais, D., and Frenette, P. S. (2003). Macrophages, not neutrophils, infiltrate skeletal muscle in mice deficient in P/E selectins after mechanical reloading. Am. J. Phys. Regul. Integr. Comp. Phys. 285, R727–R732. doi: 10.1152/ajpregu.00175.2003
Frenette, J., St-Pierre, M., Cote, C. H., Mylona, E., and Pizza, F. X. (2002). Muscle impairment occurs rapidly and precedes inflammatory cell accumulation after mechanical loading. Am. J. Phys. Regul. Integr. Comp. Phys. 282, R351–R357. doi: 10.1152/ajpregu.00189.2001
Fry, C. S., Lee, J. D., Jackson, J. R., Kirby, T. J., Stasko, S. A., Liu, H., et al. (2014). Regulation of the muscle fiber microenvironment by activated satellite cells during hypertrophy. FASEB J. 28, 1654–1665. doi: 10.1096/fj.13-239426
Fu, X., Xiao, J., Wei, Y., Li, S., Liu, Y., Yin, J., et al. (2015). Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res. 25, 655–673. doi: 10.1038/cr.2015.58
Fujimoto, H., Sakata, T., Hamaguchi, Y., Shiga, S., Tohyama, K., Ichiyama, S., et al. (2000). Flow cytometric method for enumeration and classification of reactive immature granulocyte populations. Cytometry 42, 371–378. doi: 10.1002/1097-0320(20001215)42:6<371::AID-CYTO1004>3.0.CO;2-G
Gao, S., Durstine, J. L., Koh, H. J., Carver, W. E., Frizzell, N., and Carson, J. A. (2017). Acute myotube protein synthesis regulation by IL-6-related cytokines. Am. J. Phys. Cell Physiol. 313, C487–C500. doi: 10.1152/ajpcell.00112.2017
Gentek, R., and Hoeffel, G. (2017). The innate immune response in myocardial infarction, repair, and regeneration. Adv. Exp. Med. Biol. 1003, 251–272. doi: 10.1007/978-3-319-57613-8_12
Gilliam, L. A., Ferreira, L. F., Bruton, J. D., Moylan, J. S., Westerblad, H., St Clair, D. K., et al. (2009). Doxorubicin acts through tumor necrosis factor receptor subtype 1 to cause dysfunction of murine skeletal muscle. J. Appl. Physiol. 107, 1935–1942. doi: 10.1152/japplphysiol.00776.2009
Gilliam, L. A., Moylan, J. S., Ferreira, L. F., and Reid, M. B. (2011). TNF/TNFR1 signaling mediates doxorubicin-induced diaphragm weakness. Am. J. Phys. Lung Cell. Mol. Phys. 300, L225–L231. doi: 10.1152/ajplung.00264.2010
Glass, D. J. (2005). Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 37, 1974–1984. doi: 10.1016/j.biocel.2005.04.018
Gomez-Cabrera, M. C., Vina, J., and Ji, L. L. (2016). Role of redox signaling and inflammation in skeletal muscle adaptations to training. Antioxidants (Basel) 5:48. doi: 10.3390/antiox5040048
Gordon, S., and Taylor, P. R. (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964. doi: 10.1038/nri1733
Gorospe, J. R., Nishikawa, B. K., and Hoffman, E. P. (1996). Recruitment of mast cells to muscle after mild damage. J. Neurol. Sci. 135, 10–17. doi: 10.1016/0022-510x(95)00255-z
Gorselink, M., Vaessen, S. F., Van Der Flier, L. G., Leenders, I., Kegler, D., Caldenhoven, E., et al. (2006). Mass-dependent decline of skeletal muscle function in cancer cachexia. Muscle Nerve 33, 691–693. doi: 10.1002/mus.20467
Grecian, R., Whyte, M. K. B., and Walmsley, S. R. (2018). The role of neutrophils in cancer. Br. Med. Bull. 128, 5–14. doi: 10.1093/bmb/ldy029
Guigni, B. A., Fix, D. K., Bivona, J. J. 3rd, Palmer, B. M., Carson, J. A., and Toth, M. J. (2019). Electrical stimulation prevents doxorubicin-induced atrophy and mitochondrial loss in cultured myotubes. Am. J. Phys. Cell Physiol. 317, C1213–C1228. doi: 10.1152/ajpcell.00148.2019
Guilliams, M., Ginhoux, F., Jakubzick, C., Naik, S. H., Onai, N., Schraml, B. U., et al. (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol. 14, 571–578. doi: 10.1038/nri3712
Haanen, J. B., and Robert, C. (2015). Immune checkpoint inhibitors. Prog. Tumor. Res. 42, 55–66. doi: 10.1159/000437178
Hammers, D. W., Rybalko, V., Merscham-Banda, M., Hsieh, P. L., Suggs, L. J., and Farrar, R. P. (2015). Anti-inflammatory macrophages improve skeletal muscle recovery from ischemia-reperfusion. J. Appl. Physiol. 118, 1067–1074. doi: 10.1152/japplphysiol.00313.2014
Han, Y., Yu, Z., Wen, S., Zhang, B., Cao, X., and Wang, X. (2012). Prognostic value of chemotherapy-induced neutropenia in early-stage breast cancer. Breast Cancer Res. Treat. 131, 483–490. doi: 10.1007/s10549-011-1799-1
Hardee, J. P., Counts, B. R., Gao, S., Vanderveen, B. N., Fix, D. K., Koh, H. J., et al. (2018). Inflammatory signalling regulates eccentric contraction-induced protein synthesis in cachectic skeletal muscle. J. Cachexia. Sarcopenia Muscle 9, 369–383. doi: 10.1002/jcsm.12271
Hardee, J. P., Mangum, J. E., Gao, S., Sato, S., Hetzler, K. L., Puppa, M. J., et al. (2016). Eccentric contraction-induced myofiber growth in tumor-bearing mice. J. Appl. Physiol. 120, 29–37. doi: 10.1152/japplphysiol.00416.2015
Hardin, B. J., Campbell, K. S., Smith, J. D., Arbogast, S., Smith, J., Moylan, J. S., et al. (2008). TNF-alpha acts via TNFR1 and muscle-derived oxidants to depress myofibrillar force in murine skeletal muscle. J. Appl. Physiol. 104, 694–699. doi: 10.1152/japplphysiol.00898.2007
He, W. A., Berardi, E., Cardillo, V. M., Acharyya, S., Aulino, P., Thomas-Ahner, J., et al. (2013). NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Invest. 123, 4821–4835. doi: 10.1172/JCI68523
Hetzler, K. L., Hardee, J. P., Puppa, M. J., Narsale, A. A., Sato, S., Davis, J. M., et al. (2015). Sex differences in the relationship of IL-6 signaling to cancer cachexia progression. Biochim. Biophys. Acta 1852, 816–825. doi: 10.1016/j.bbadis.2014.12.015
Hiensch, A. E., Bolam, K. A., Mijwel, S., Jeneson, J. A. L., Huitema, A. D. R., Kranenburg, O., et al. (2019). Doxorubicin-induced skeletal muscle atrophy: elucidating the underlying molecular pathways. Acta Physiol. 229, 1–18. doi: 10.1111/apha.13400
Hirahara, K., and Nakayama, T. (2016). CD4+ T-cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm. Int. Immunol. 28, 163–171. doi: 10.1093/intimm/dxw006
Hoeffel, G., and Ginhoux, F. (2018). Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol. 330, 5–15. doi: 10.1016/j.cellimm.2018.01.001
Hong, K. H., Ryu, J., and Han, K. H. (2005). Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 105, 1405–1407. doi: 10.1182/blood-2004-08-3178
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061. doi: 10.1126/science.1079490
Howard, E. E., Pasiakos, S. M., Blesso, C. N., Fussell, M. A., and Rodriguez, N. R. (2020). Divergent roles of inflammation in skeletal muscle recovery from injury. Front. Physiol. 11:87. doi: 10.3389/fphys.2020.00087
Hu, L., Cui, R., Liu, H., and Wang, F. (2017). Emodin and rhein decrease levels of hypoxia-inducible factor-1alpha in human pancreatic cancer cells and attenuate cancer cachexia in athymic mice carrying these cells. Oncotarget 8, 88008–88020. doi: 10.18632/oncotarget.21330
Huang, S. C., Wu, J. F., Saovieng, S., Chien, W. H., Hsu, M. F., Li, X. F., et al. (2017). Doxorubicin inhibits muscle inflammation after eccentric exercise. J. Cachexia. Sarcopenia Muscle 8, 277–284. doi: 10.1002/jcsm.12148
Huertas, A. M., Morton, A. B., Hinkey, J. M., Ichinoseki-Sekine, N., and Smuder, A. J. (2020). Modification of neuromuscular junction protein expression by exercise and doxorubicin. Med. Sci. Sports Exerc. 52, 1477–1484. doi: 10.1249/MSS.0000000000002286
Hume, D. A. (2006). The mononuclear phagocyte system. Curr. Opin. Immunol. 18, 49–53. doi: 10.1016/j.coi.2005.11.008
Inaba, S., Hinohara, A., Tachibana, M., Tsujikawa, K., and Fukada, S. I. (2018). Muscle regeneration is disrupted by cancer cachexia without loss of muscle stem cell potential. PLoS One 13:e0205467. doi: 10.1371/journal.pone.0205467
Iyer, R. P., Jung, M., and Lindsey, M. L. (2016). MMP-9 signaling in the left ventricle following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 311, H190–H198. doi: 10.1152/ajpheart.00243.2016
Jablonska, J., Lang, S., Sionov, R. V., and Granot, Z. (2017). The regulation of pre-metastatic niche formation by neutrophils. Oncotarget 8, 112132–112144. doi: 10.18632/oncotarget.22792
Jablonski, K. A., Amici, S. A., Webb, L. M., de Dios Ruiz-Rosado, J., Popovich, P. G., Partida-Sanchez, S., et al. (2015). Novel markers to delineate murine M1 and M2 macrophages. PLoS One 10:e0145342. doi: 10.1371/journal.pone.0145342
Jackman, R. W., Floro, J., Yoshimine, R., Zitin, B., Eiampikul, M., El-Jack, K., et al. (2017). Continuous release of tumor-derived factors improves the modeling of cachexia in muscle cell culture. Front. Physiol. 8:738. doi: 10.3389/fphys.2017.00738
Jaweed, M. M., Herbison, G. J., Miller, E. E., and Ditunno, J. F. (1983). Compensatory hypertrophy of the soleus in tumor-bearing rats. J. Neurol. Sci. 61, 171–179. doi: 10.1016/0022-510x(83)90003-5
Jia, X., Yu, F., Wang, J., Iwanowycz, S., Saaoud, F., Wang, Y., et al. (2014). Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res. Treat. 148, 291–302. doi: 10.1007/s10549-014-3164-7
Jobin, J., Maltais, F., Doyon, J. F., Leblanc, P., Simard, P. M., Simard, A. A., et al. (1998). Chronic obstructive pulmonary disease: capillarity and fiber-type characteristics of skeletal muscle. J. Cardpulm. Rehabil. 18, 432–437. doi: 10.1097/00008483-199811000-00005
Judge, S. M., Nosacka, R. L., Delitto, D., Gerber, M. H., Cameron, M. E., Trevino, J. G., et al. (2018). Skeletal muscle fibrosis in pancreatic cancer patients with respect to survival. JNCI Cancer Spectr. 2:pky043. doi: 10.1093/jncics/pky043
Judge, S. M., Wu, C. L., Beharry, A. W., Roberts, B. M., Ferreira, L. F., Kandarian, S. C., et al. (2014). Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 14:997. doi: 10.1186/1471-2407-14-997
Kanwar, S., Smith, C. W., and Kubes, P. (1998). An absolute requirement for P-selectin in ischemia/reperfusion-induced leukocyte recruitment in cremaster muscle. Microcirculation 5, 281–287.
Kazemi-Bajestani, S. M. R., Mazurak, V. C., and Baracos, V. (2016). Computed tomography-defined muscle and fat wasting are associated with cancer clinical outcomes. Semin. Cell Dev. Biol. 54, 2–10. doi: 10.1016/j.semcdb.2015.09.001
Kim, N. D., and Luster, A. D. (2015). The role of tissue resident cells in neutrophil recruitment. Trends Immunol. 36, 547–555. doi: 10.1016/j.it.2015.07.007
Kolaczkowska, E., and Kubes, P. (2013). Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175. doi: 10.1038/nri3399
Korthuis, R. J., Granger, D. N., Townsley, M. I., and Taylor, A. E. (1985). The role of oxygen-derived free radicals in ischemia-induced increases in canine skeletal muscle vascular permeability. Circ. Res. 57, 599–609. doi: 10.1161/01.res.57.4.599
Korthuis, R. J., Grisham, M. B., and Granger, D. N. (1988). Leukocyte depletion attenuates vascular injury in postischemic skeletal muscle. Am. J. Phys. 254, H823–H827. doi: 10.1152/ajpheart.1988.254.5.H823
Kuswanto, W., Burzyn, D., Panduro, M., Wang, K. K., Jang, Y. C., Wagers, A. J., et al. (2016). Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44, 355–367. doi: 10.1016/j.immuni.2016.01.009
Kvinnsland, S. (1999). The leucocyte nadir, a predictor of chemotherapy efficacy? Br. J. Cancer 80:1681. doi: 10.1038/sj.bjc.6690583
Laplante, M., and Sabatini, D. M. (2009). mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594. doi: 10.1242/jcs.051011
Lecker, S. H., Jagoe, R. T., Gilbert, A., Gomes, M., Baracos, V., Bailey, J., et al. (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 18, 39–51. doi: 10.1096/fj.03-0610com
Lee, D. E., Bareja, A., Bartlett, D. B., and White, J. P. (2019). Autophagy as a therapeutic target to enhance aged muscle regeneration. Cell 8:183. doi: 10.3390/cells8020183
Lee, S., Chen, T. T., Barber, C. L., Jordan, M. C., Murdock, J., Desai, S., et al. (2007). Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703. doi: 10.1016/j.cell.2007.06.054
Lee, P. Y., Wang, J. X., Parisini, E., Dascher, C. C., and Nigrovic, P. A. (2013). Ly6 family proteins in neutrophil biology. J. Leukoc. Biol. 94, 585–594. doi: 10.1189/jlb.0113014
Li, H., Hicks, J. J., Wang, L., Oyster, N., Philippon, M. J., Hurwitz, S., et al. (2016a). Customized platelet-rich plasma with transforming growth factor beta1 neutralization antibody to reduce fibrosis in skeletal muscle. Biomaterials 87, 147–156. doi: 10.1016/j.biomaterials.2016.02.017
Li, Y. L., Zhao, H., and Ren, X. B. (2016b). Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol. Med. 13, 206–214. doi: 10.20892/j.issn.2095-3941.2015.0070
Liu, X., Liu, Y., Zhao, L., Zeng, Z., Xiao, W., and Chen, P. (2017). Macrophage depletion impairs skeletal muscle regeneration: the roles of regulatory factors for muscle regeneration. Cell Biol. Int. 41, 228–238. doi: 10.1002/cbin.10705
Liu, P., Sun, M., and Sader, S. (2006). Matrix metalloproteinases in cardiovascular disease. Can. J. Cardiol. 22, 25B–30B. doi: 10.1016/s0828-282x(06)70983-7
Luo, Y., Yoneda, J., Ohmori, H., Sasaki, T., Shimbo, K., Eto, S., et al. (2014). Cancer usurps skeletal muscle as an energy repository. Cancer Res. 74, 330–340. doi: 10.1158/0008-5472.CAN-13-1052
Madaro, L., and Bouche, M. (2014). From innate to adaptive immune response in muscular dystrophies and skeletal muscle regeneration: the role of lymphocytes. Biomed. Res. Int. 2014:438675. doi: 10.1155/2014/438675
Mauro, A. (1961). Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493–495. doi: 10.1083/jcb.9.2.493
Mayadas, T. N., Cullere, X., and Lowell, C. A. (2014). The multifaceted functions of neutrophils. Annu. Rev. Pathol. 9, 181–218. doi: 10.1146/annurev-pathol-020712-164023
Mazzoni, A., Maggi, L., Liotta, F., Cosmi, L., and Annunziato, F. (2019). Biological and clinical significance of T helper 17 cell plasticity. Immunology 158, 287–295. doi: 10.1111/imm.13124
McCarthy, J. J., Mula, J., Miyazaki, M., Erfani, R., Garrison, K., Farooqui, A. B., et al. (2011). Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 138, 3657–3666. doi: 10.1242/dev.068858
McClung, J. M., Judge, A. R., Powers, S. K., and Yan, Z. (2010). p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Phys. Cell Physiol. 298, C542–C549. doi: 10.1152/ajpcell.00192.2009
Mellman, I. (2013). Dendritic cells: master regulators of the immune response. Cancer Immunol. Res. 1, 145–149. doi: 10.1158/2326-6066.CIR-13-0102
Mendias, C. L. (2017). Fibroblasts take the Centre stage in human skeletal muscle regeneration. J. Physiol. 595:5005. doi: 10.1113/JP274403
Meng, J., Bencze, M., Asfahani, R., Muntoni, F., and Morgan, J. E. (2015). The effect of the muscle environment on the regenerative capacity of human skeletal muscle stem cells. Skelet. Muscle 5:11. doi: 10.1186/s13395-015-0036-8
Mittrucker, H. W., Visekruna, A., and Huber, M. (2014). Heterogeneity in the differentiation and function of CD8+ T cells. Arch. Immunol. Ther. Exp. 62, 449–458. doi: 10.1007/s00005-014-0293-y
Mochizuki, Y., Ojima, K., Uezumi, A., Masuda, S., Yoshimura, K., and Takeda, S. (2005). Participation of bone marrow-derived cells in fibrotic changes in denervated skeletal muscle. Am. J. Pathol. 166, 1721–1732. doi: 10.1016/S0002-9440(10)62482-0
Mojumdar, K., Liang, F., Giordano, C., Lemaire, C., Danialou, G., Okazaki, T., et al. (2014). Inflammatory monocytes promote progression of duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol. Med. 6, 1476–1492. doi: 10.15252/emmm.201403967
Montalvo, R. N., Doerr, V., Min, K., Szeto, H. H., and Smuder, A. J. (2020). Doxorubicin-induced oxidative stress differentially regulates proteolytic signaling in cardiac and skeletal muscle. Am. J. Phys. Regul. Integr. Comp. Phys. 318, R227–R233. doi: 10.1152/ajpregu.00299.2019
Montalvo, R. N., Hardee, J. P., Vanderveen, B. N., and Carson, J. A. (2018). Resistance exercise’s ability to reverse cancer-induced anabolic resistance. Exerc. Sport Sci. Rev. 46, 247–253. doi: 10.1249/JES.0000000000000159
Morabito, A., De Maio, E., Di Maio, M., Normanno, N., and Perrone, F. (2006). Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: current status and future directions. Oncologist 11, 753–764. doi: 10.1634/theoncologist.11-7-753
Morgan, R. A., Dudley, M. E., Wunderlich, J. R., Hughes, M. S., Yang, J. C., Sherry, R. M., et al. (2006). Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314, 126–129. doi: 10.1126/science.1129003
Morgan, J., and Partridge, T. (2020). Skeletal muscle in health and disease. Dis. Model. Mech. 13:dmm042192. doi: 10.1242/dmm.042192
Morton, A. B., Mor Huertas, A., Hinkley, J. M., Ichinoseki-Sekine, N., Christou, D. D., and Smuder, A. J. (2019). Mitochondrial accumulation of doxorubicin in cardiac and diaphragm muscle following exercise preconditioning. Mitochondrion 45, 52–62. doi: 10.1016/j.mito.2018.02.005
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Murach, K. A., Fry, C. S., Kirby, T. J., Jackson, J. R., Lee, J. D., White, S. H., et al. (2018). Starring or supporting role? Satellite cells and skeletal muscle fiber size regulation. Physiology (Bethesda) 33, 26–38. doi: 10.1152/physiol.00019.2017
Murphy, K. T., Chee, A., Trieu, J., Naim, T., and Lynch, G. S. (2012). Importance of functional and metabolic impairments in the characterization of the C-26 murine model of cancer cachexia. Dis. Model. Mech. 5, 533–545. doi: 10.1242/dmm.008839
Murphy, M. M., Lawson, J. A., Mathew, S. J., Hutcheson, D. A., and Kardon, G. (2011). Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138, 3625–3637. doi: 10.1242/dev.064162
Murphy, K., and Weaver, C. (2016). Janeway’s immunobiology. New York, NY: Garland Science/Taylor and Francis Group, LLC.
Naito, Y., Saito, K., Shiiba, K., Ohuchi, A., Saigenji, K., Nagura, H., et al. (1998). CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 58, 3491–3494.
Narsale, A. A., Puppa, M. J., Hardee, J. P., Vanderveen, B. N., Enos, R. T., Murphy, E. A., et al. (2016). Short-term pyrrolidine dithiocarbamate administration attenuates cachexia-induced alterations to muscle and liver in ApcMin/+ mice. Oncotarget 7, 59482–59502. doi: 10.18632/oncotarget.10699
Nosacka, R. L., Delitto, A. E., Delitto, D., Patel, R., Judge, S. M., Trevino, J. G., et al. (2020). Distinct cachexia profiles in response to human pancreatic tumours in mouse limb and respiratory muscle. J. Cachexia. Sarcopenia Muscle 11, 820–837. doi: 10.1002/jcsm.12550
Novak, M. L., Weinheimer-Haus, E. M., and Koh, T. J. (2014). Macrophage activation and skeletal muscle healing following traumatic injury. J. Pathol. 232, 344–355. doi: 10.1002/path.4301
Ooyama, A., Oka, T., Zhao, H. Y., Yamamoto, M., Akiyama, S., and Fukushima, M. (2008). Anti-angiogenic effect of 5-fluorouracil-based drugs against human colon cancer xenografts. Cancer Lett. 267, 26–36. doi: 10.1016/j.canlet.2008.03.008
Orecchioni, M., Ghosheh, Y., Pramod, A. B., and Ley, K. (2019). Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front. Immunol. 10:1084. doi: 10.3389/fimmu.2019.01084
Penna, F., Costamagna, D., Fanzani, A., Bonelli, G., Baccino, F. M., and Costelli, P. (2010). Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One 5:e13604. doi: 10.1371/journal.pone.0013604
Penna, F., Costamagna, D., Pin, F., Camperi, A., Fanzani, A., Chiarpotto, E. M., et al. (2013). Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 182, 1367–1378. doi: 10.1016/j.ajpath.2012.12.023
Pesce, S., Greppi, M., Grossi, F., Del Zotto, G., Moretta, L., Sivori, S., et al. (2019). PD/1-PD-Ls checkpoint: insight on the potential role of NK cells. Front. Immunol. 10:1242. doi: 10.3389/fimmu.2019.01242
Phatharajaree, W., Phrommintikul, A., and Chattipakorn, N. (2007). Matrix metalloproteinases and myocardial infarction. Can. J. Cardiol. 23, 727–733. doi: 10.1016/S0828-282X(07)70818-8
Pizza, F. X., Peterson, J. M., Baas, J. H., and Koh, T. J. (2005). Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J. Physiol. 562, 899–913. doi: 10.1113/jphysiol.2004.073965
Porporato, P. (2016). Understanding cachexia as a cancer metabolism syndrome. Oncogene 5:e200. doi: 10.1038/oncsis.2016.74
Powers, S. K., Kavazis, A. N., and Deruisseau, K. C. (2005). Mechanisms of disuse muscle atrophy: role of oxidative stress. Am. J. Phys. Regul. Integr. Comp. Phys. 288, R337–R344. doi: 10.1152/ajpregu.00469.2004
Puppa, M. J., White, J. P., Velazquez, K. T., Baltgalvis, K. A., Sato, S., Baynes, J. W., et al. (2012). The effect of exercise on IL-6-induced cachexia in the Apc (Min/+) mouse. J. Cachexia. Sarcopenia Muscle 3, 117–137. doi: 10.1007/s13539-011-0047-1
Radke, J., Koll, R., Preusse, C., Pehl, D., Todorova, K., Schonemann, C., et al. (2018). Architectural B-cell organization in skeletal muscle identifies subtypes of dermatomyositis. Neurol. Neuroimmunol. Neuroinflamm. 5:e451. doi: 10.1212/NXI.0000000000000451
Ramage, M. I., and Skipworth, R. J. E. (2018). The relationship between muscle mass and function in cancer cachexia: smoke and mirrors? Curr. Opin. Support. Palliat. Care 12, 439–444. doi: 10.1097/SPC.0000000000000381
Reidy, P. T., Dupont-Versteegden, E. E., and Drummond, M. J. (2019a). Macrophage regulation of muscle regrowth from disuse in aging. Exerc. Sport Sci. Rev. 47, 246–250. doi: 10.1249/JES.0000000000000201
Reidy, P. T., Mckenzie, A. I., Mahmassani, Z. S., Petrocelli, J. J., Nelson, D. B., Lindsay, C. C., et al. (2019b). Aging impairs mouse skeletal muscle macrophage polarization and muscle-specific abundance during recovery from disuse. Am. J. Physiol. Endocrinol. Metab. 317, E85–E98. doi: 10.1152/ajpendo.00422.2018
Roberts, B. M., Frye, G. S., Ahn, B., Ferreira, L. F., and Judge, A. R. (2013). Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochem. Biophys. Res. Commun. 435, 488–492. doi: 10.1016/j.bbrc.2013.05.018
Roch, B., Coffy, A., Jean-Baptiste, S., Palaysi, E., Daures, J. P., Pujol, J. L., et al. (2020). Cachexia ‐ sarcopenia as a determinant of disease control rate and survival in non-small lung cancer patients receiving immune-checkpoint inhibitors. Lung Cancer 143, 19–26. doi: 10.1016/j.lungcan.2020.03.003
Romee, R., Foley, B., Lenvik, T., Wang, Y., Zhang, B., Ankarlo, D., et al. (2013). NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 121, 3599–3608. doi: 10.1182/blood-2012-04-425397
Saklani, A., and Kutty, S. K. (2008). Plant-derived compounds in clinical trials. Drug Discov. Today 13, 161–171. doi: 10.1016/j.drudis.2007.10.010
Sato, K., and Fujita, S. (2007). Dendritic cells: nature and classification. Allergol. Int. 56, 183–191. doi: 10.2332/allergolint.R-06-139
Schiaffino, S., Pereira, M. G., Ciciliot, S., and Rovere-Querini, P. (2017). Regulatory T cells and skeletal muscle regeneration. FEBS J. 284, 517–524. doi: 10.1111/febs.13827
Schmidt, M., Poser, C., and Von Maltzahn, J. (2020). Wnt7a counteracts cancer cachexia. Mol. Ther. Oncolytics 16, 134–146. doi: 10.1016/j.omto.2019.12.011
Schmitz, K. H., Campbell, A. M., Stuiver, M. M., Pinto, B. M., Schwartz, A. L., Morris, G. S., et al. (2019). Exercise is medicine in oncology: engaging clinicians to help patients move through cancer. CA Cancer J. Clin. 69, 468–484. doi: 10.3322/caac.21579
Schwarzkopf, M., Coletti, D., Sassoon, D., and Marazzi, G. (2006). Muscle cachexia is regulated by a p53-PW1/Peg3-dependent pathway. Genes Dev. 20, 3440–3452. doi: 10.1101/gad.412606
Sealy, M. J., Dechaphunkul, T., Van Der Schans, C. P., Krijnen, W. P., Roodenburg, J. L. N., Walker, J., et al. (2020). Low muscle mass is associated with early termination of chemotherapy related to toxicity in patients with head and neck cancer. Clin. Nutr. 39, 501–509. doi: 10.1016/j.clnu.2019.02.029
Shapouri-Moghaddam, A., Mohammadian, S., Vazini, H., Taghadosi, M., Esmaeili, S. A., Mardani, F., et al. (2018). Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 233, 6425–6440. doi: 10.1002/jcp.26429
Shih, T., and Lindley, C. (2006). Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 28, 1779–1802. doi: 10.1016/j.clinthera.2006.11.015
Shitara, K., Matsuo, K., Oze, I., Mizota, A., Kondo, C., Nomura, M., et al. (2011). Meta-analysis of neutropenia or leukopenia as a prognostic factor in patients with malignant disease undergoing chemotherapy. Cancer Chemother. Pharmacol. 68, 301–307. doi: 10.1007/s00280-010-1487-6
Shitara, K., Matsuo, K., Takahari, D., Yokota, T., Inaba, Y., Yamaura, H., et al. (2009). Neutropaenia as a prognostic factor in metastatic colorectal cancer patients undergoing chemotherapy with first-line FOLFOX. Eur. J. Cancer 45, 1757–1763. doi: 10.1016/j.ejca.2009.01.019
Sica, A., and Strauss, L. (2017). Energy metabolism drives myeloid-derived suppressor cell differentiation and functions in pathology. J. Leukoc. Biol. 102, 325–334. doi: 10.1189/jlb.4MR1116-476R
Smith, J. K., Grisham, M. B., Granger, D. N., and Korthuis, R. J. (1989). Free radical defense mechanisms and neutrophil infiltration in postischemic skeletal muscle. Am. J. Phys. 256, H789–H793. doi: 10.1152/ajpheart.1989.256.3.H789
Smuder, A. J. (2019). Exercise stimulates beneficial adaptations to diminish doxorubicin-induced cellular toxicity. Am. J. Phys. Regul. Integr. Comp. Phys. 317, R662–R672. doi: 10.1152/ajpregu.00161.2019
Sougiannis, A. T., Vanderveen, B. N., Enos, R. T., Velazquez, K. T., Bader, J. E., Carson, M., et al. (2019). Impact of 5 fluorouracil chemotherapy on gut inflammation, functional parameters, and gut microbiota. Brain Behav. Immun. 80, 44–55. doi: 10.1016/j.bbi.2019.02.020
Spencer, M. J., Montecino-Rodriguez, E., Dorshkind, K., and Tidball, J. G. (2001). Helper (CD4+) and cytotoxic (CD8+) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. 98, 235–243. doi: 10.1006/clim.2000.4966
St Pierre, B. A., and Tidball, J. G. (1994). Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J. Appl. Physiol. 77, 290–297. doi: 10.1152/jappl.1994.77.1.290
Stackowicz, J., Jonsson, F., and Reber, L. L. (2019). Mouse models and tools for the in vivo study of neutrophils. Front. Immunol. 10:3130. doi: 10.3389/fimmu.2019.03130
Sundararaj, K. P., Samuvel, D. J., Li, Y., Sanders, J. J., Lopes-Virella, M. F., and Huang, Y. (2009). Interleukin-6 released from fibroblasts is essential for up-regulation of matrix metalloproteinase-1 expression by U937 macrophages in coculture: cross-talking between fibroblasts and U937 macrophages exposed to high glucose. J. Biol. Chem. 284, 13714–13724. doi: 10.1074/jbc.M806573200
Talbert, E. E., and Guttridge, D. C. (2016). Impaired regeneration: a role for the muscle microenvironment in cancer cachexia. Semin. Cell Dev. Biol. 54, 82–91. doi: 10.1016/j.semcdb.2015.09.009
Talbert, E. E., Lewis, H. L., Farren, M. R., Ramsey, M. L., Chakedis, J. M., Rajasekera, P., et al. (2018). Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naive pancreatic cancer patients. J. Cachexia. Sarcopenia Muscle 9, 358–368. doi: 10.1002/jcsm.12251
Thomas, C. V., Coker, M. L., Zellner, J. L., Handy, J. R., Crumbley, A. J. 3rd, and Spinale, F. G. (1998). Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with end-stage dilated cardiomyopathy. Circulation 97, 1708–1715. doi: 10.1161/01.CIR.97.17.1708
Tidball, J. G. (1995). Inflammatory cell response to acute muscle injury. Med. Sci. Sports Exerc. 27, 1022–1032. doi: 10.1249/00005768-199507000-00011
Tidball, J. G. (2002). Interactions between muscle and the immune system during modified musculoskeletal loading. Clin. Orthop. Relat. Res. (Suppl. 403), S100–S109. doi: 10.1097/00003086-200210001-00012
Tidball, J. G. (2005). Inflammatory processes in muscle injury and repair. Am. J. Phys. Regul. Integr. Comp. Phys. 288, R345–R653. doi: 10.1152/ajpregu.00454.2004
Tidball, J. G. (2017). Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 17, 165–178. doi: 10.1038/nri.2016.150
Tidball, J. G., Berchenko, E., and Frenette, J. (1999). Macrophage invasion does not contribute to muscle membrane injury during inflammation. J. Leukoc. Biol. 65, 492–498. doi: 10.1002/jlb.65.4.492
Tidball, J. G., and Villalta, S. A. (2010). Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Phys. Regul. Integr. Comp. Phys. 298, R1173–R1187. doi: 10.1152/ajpregu.00735.2009
Tidball, J. G., and Wehling-Henricks, M. (2007). Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol. 578, 327–336. doi: 10.1113/jphysiol.2006.118265
Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C., and Brown, R. A. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363. doi: 10.1038/nrm809
Toth, M. J., Voigt, T. B., Tourville, T. W., Prior, S. M., Guigni, B. A., Schlosberg, A. V., et al. (2020). Effect of neuromuscular electrical stimulation on skeletal muscle size and function in patients with breast cancer receiving chemotherapy. J. Appl. Physiol. (1985) 128, 1654–1665. doi: 10.1152/japplphysiol.00203.2020
Van Furth, R., and Cohn, Z. A. (1968). The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 128, 415–435. doi: 10.1084/jem.128.3.415
Van Linthout, S., Miteva, K., and Tschope, C. (2014). Crosstalk between fibroblasts and inflammatory cells. Cardiovasc. Res. 102, 258–269. doi: 10.1093/cvr/cvu062
Vanderveen, B. N., Fix, D. K., and Carson, J. A. (2017). Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxidative Med. Cell. Longev. 2017:3292087. doi: 10.1155/2017/3292087
Vanderveen, B. N., Fix, D. K., Counts, B. R., and Carson, J. A. (2020). The effect of wheel exercise on functional indices of cachexia in tumor-bearing mice. Med. Sci. Sports Exerc. doi: 10.1249/mss.0000000000002393 [Epub ahead of print]
Vanderveen, B. N., Fix, D. K., Montalvo, R. N., Counts, B. R., Smuder, A. J., Murphy, E. A., et al. (2019). The regulation of skeletal muscle fatigability and mitochondrial function by chronically elevated interleukin-6. Exp. Physiol. 104, 385–397. doi: 10.1113/EP087429
Vanderveen, B. N., Hardee, J. P., Fix, D. K., and Carson, J. A. (2018). Skeletal muscle function during the progression of cancer cachexia in the male Apc(Min/+) mouse. J. Appl. Physiol. 124, 684–695. doi: 10.1152/japplphysiol.00897.2017
Vestweber, D. (2015). How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 15, 692–704. doi: 10.1038/nri3908
Viallard, C., and Larrivee, B. (2017). Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis 20, 409–426. doi: 10.1007/s10456-017-9562-9
Villalta, S. A., Rosenberg, A. S., and Bluestone, J. A. (2015). The immune system in duchenne muscular dystrophy: friend or foe. Rare Dis. 3:e1010966. doi: 10.1080/21675511.2015.1010966
Volpert, O. V., Fong, T., Koch, A. E., Peterson, J. D., Waltenbaugh, C., Tepper, R. I., et al. (1998). Inhibition of angiogenesis by interleukin 4. J. Exp. Med. 188, 1039–1046. doi: 10.1084/jem.188.6.1039
Walsh, S. R., Cook, E. J., Goulder, F., Justin, T. A., and Keeling, N. J. (2005). Neutrophil-lymphocyte ratio as a prognostic factor in colorectal cancer. J. Surg. Oncol. 91, 181–184. doi: 10.1002/jso.20329
Wang, Z., Zhao, C., Moya, R., and Davies, J. D. (2008). A novel role for CD4+ T cells in the control of cachexia. J. Immunol. 181, 4676–4684. doi: 10.4049/jimmunol.181.7.4676
Washington, T. A., White, J. P., Davis, J. M., Wilson, L. B., Lowe, L. L., Sato, S., et al. (2011). Skeletal muscle mass recovery from atrophy in IL-6 knockout mice. Acta Physiol. 202, 657–669. doi: 10.1111/j.1748-1716.2011.02281.x
White, J. P., Baynes, J. W., Welle, S. L., Kostek, M. C., Matesic, L. E., Sato, S., et al. (2011). The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc min/+ mouse. PLoS One 6:e24650. doi: 10.1371/journal.pone.0024650
White, J. P., Puppa, M. J., Gao, S., Sato, S., Welle, S. L., and Carson, J. A. (2013). Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am. J. Physiol. Endocrinol. Metab. 304, E1042–E1052. doi: 10.1152/ajpendo.00410.2012
White, J. P., Reecy, J. M., Washington, T. A., Sato, S., Le, M. E., Davis, J. M., et al. (2009). Overload-induced skeletal muscle extracellular matrix remodelling and myofibre growth in mice lacking IL-6. Acta Physiol. 197, 321–332. doi: 10.1111/j.1748-1716.2009.02029.x
Wu, H., and Ballantyne, C. M. (2017). Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Invest. 127, 43–54. doi: 10.1172/JCI88880
Wynn, T. A., Chawla, A., and Pollard, J. W. (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. doi: 10.1038/nature12034
Xiao, W., Liu, Y., and Chen, P. (2016). Macrophage depletion impairs skeletal muscle regeneration: the roles of pro-fibrotic factors, inflammation, and oxidative stress. Inflammation 39, 2016–2028. doi: 10.1007/s10753-016-0438-8
Yakovenko, A., Cameron, M., and Trevino, J. G. (2018). Molecular therapeutic strategies targeting pancreatic cancer induced cachexia. World J. Gastrointest. Surg. 10, 95–106. doi: 10.4240/wjgs.v10.i9.95
Yamanaka, T., Matsumoto, S., Teramukai, S., Ishiwata, R., Nagai, Y., and Fukushima, M. (2007). Predictive value of chemotherapy-induced neutropenia for the efficacy of oral fluoropyrimidine S-1 in advanced gastric carcinoma. Br. J. Cancer 97, 37–42. doi: 10.1038/sj.bjc.6603831
Yang, W., and Hu, P. (2018). Skeletal muscle regeneration is modulated by inflammation. J. Orthop. Translat. 13, 25–32. doi: 10.1016/j.jot.2018.01.002
Yazawa, T., Shibata, M., Gonda, K., Machida, T., Suzuki, S., Kenjo, A., et al. (2013). Increased IL-17 production correlates with immunosuppression involving myeloid-derived suppressor cells and nutritional impairment in patients with various gastrointestinal cancers. Mol. Clin. Oncol. 1, 675–679. doi: 10.3892/mco.2013.134
Yndestad, A., Damas, J. K., Oie, E., Ueland, T., Gullestad, L., and Aukrust, P. (2007). Role of inflammation in the progression of heart failure. Curr. Cardiol. Rep. 9, 236–241. doi: 10.1007/BF02938356
Yokota, M., Suzuki, K., Tokoyoda, K., Meguro, K., Hosokawa, J., Tanaka, S., et al. (2014). Roles of mast cells in the pathogenesis of inflammatory myopathy. Arthritis Res. Ther. 16:R72. doi: 10.1186/ar4512
Zaiss, D. M. W., Gause, W. C., Osborne, L. C., and Artis, D. (2015). Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42, 216–226. doi: 10.1016/j.immuni.2015.01.020
Zhang, P., Chen, X., and Fan, M. (2007). Signaling mechanisms involved in disuse muscle atrophy. Med. Hypotheses 69, 310–321. doi: 10.1016/j.mehy.2006.11.043
Zhang, J., Xiao, Z., Qu, C., Cui, W., Wang, X., and Du, J. (2014). Ion and Gr1(high) macrophage infiltration. J. Immunol. 193, 5149–5160. doi: 10.4049/jimmunol.1303486
Zhang, L., Yu, D., Hicklin, D. J., Hannay, J. A., Ellis, L. M., and Pollock, R. E. (2002). Combined anti-fetal liver kinase 1 monoclonal antibody and continuous low-dose doxorubicin inhibits angiogenesis and growth of human soft tissue sarcoma xenografts by induction of endothelial cell apoptosis. Cancer Res. 62, 2034–2042.
Keywords: extracellular matrix, macrophage, satellite cell, fibroblast, endothelium
Citation: VanderVeen BN, Murphy EA and Carson JA (2020) The Impact of Immune Cells on the Skeletal Muscle Microenvironment During Cancer Cachexia. Front. Physiol. 11:1037. doi: 10.3389/fphys.2020.01037
Edited by:
Lykke Sylow, University of Copenhagen, DenmarkReviewed by:
Marilia Seelaender, University of São Paulo, BrazilCopyright © 2020 VanderVeen, Murphy and Carson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James A. Carson, amNhcnNvMTZAdXRoc2MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.