 Christian U. Oeing
Christian U. Oeing Sumita Mishra
Sumita Mishra Brittany L. Dunkerly-Eyring
Brittany L. Dunkerly-Eyring Mark J. Ranek
Mark J. Ranek
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 28 July 2020
Sec. Striated Muscle Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00858
This article is part of the Research TopicTargeting Cardiac ProteotoxicityView all 14 articles
Impaired or insufficient protein kinase G (PKG) signaling and protein quality control (PQC) are hallmarks of most forms of cardiac disease, including heart failure. Their dysregulation has been shown to contribute to and exacerbate cardiac hypertrophy and remodeling, reduced cell survival and disease pathogenesis. Enhancement of PKG signaling and PQC are associated with improved cardiac function and survival in many pre-clinical models of heart disease. While many clinically used pharmacological approaches exist to stimulate PKG, there are no FDA-approved therapies to safely enhance cardiomyocyte PQC. The latter is predominantly due to our lack of knowledge and identification of proteins regulating cardiomyocyte PQC. Recently, multiple studies have demonstrated that PKG regulates PQC in the heart, both during physiological and pathological states. These studies tested already FDA-approved pharmacological therapies to activate PKG, which enhanced cardiomyocyte PQC and alleviated cardiac disease. This review examines the roles of PKG and PQC during disease pathogenesis and summarizes the experimental and clinical data supporting the utility of stimulating PKG to target cardiac proteotoxicity.
Protein kinase G (PKG) elicits cardioprotection during various forms of cardiac stress by transducing a vast array of beneficial processes (Dunkerly-Eyring and Kass, 2019; Pinilla-Vera et al., 2019). PKG stimulates left ventricular relaxation and counters pathological hypertrophy and remodeling (Dunkerly-Eyring and Kass, 2019). Insufficient PKG signaling has been implicated in the pathogenesis of cardiac disease toward heart failure, giving considerable interest to strategies to enhance PKG signaling (Kokkonen and Kass, 2017; Dunkerly-Eyring and Kass, 2019). New pharmacological approaches to stimulate PKG are being evaluated as therapy for heart failure and other forms of cardiac disease in clinical trials (Pinilla-Vera et al., 2019). However, a better understanding of the substrates targeted by PKG for cardioprotection is needed. Recently the activation of PKG was demonstrated to regulate protein homeostasis (proteostasis), to attenuate disease pathogenesis (Dunkerly-Eyring and Kass, 2019). The protein targets, underlying mechanisms, and therapeutic strategies to facilitate PKG regulation of proteostasis are only beginning to be identified.
Cardiomyocyte proteostasis is maintained by elaborate protein quality control (PQC) systems. These systems help fold polypeptide chains into properly functioning proteins, refold proteins that become misfolded or damaged during stress, and then remove terminally misfolded and/or aggregated proteins by degradation (Wang and Robbins, 2006; Wang et al., 2008). Cardiomyocyte PQC is maintained by three separate but interlinking systems: molecular chaperones (protein folding/refolding), the ubiquitin proteasome system (UPS) (proteasome mediated degradation of soluble proteins), and autophagy (lysosomal degradation of protein aggregates and organelles) (Wang et al., 2008). Similar to PKG signaling, cardiomyocyte PQC is also perturbed/insufficient during cardiac disease pathogenesis. This results in the intracellular accumulation of proteins targeted for degradation, aggregation of proteins, and subsequent declined cardiac function; a class of disorders termed cardiac proteotoxicity (Wang and Robbins, 2006; Willis and Patterson, 2013). The proteins and macromolecular structures that are responsible for maintaining cardiomyocyte PQC are known; however, only recently have we begun to understand the proteins and/or posttranslational modifications that regulate PQC. Pharmacological strategies to safely enhance PQC are beginning to be explored. This review focuses on one protein, PKG, which has shown promise as a cardiac PQC-enhancing therapy. We will detail the studies that demonstrate a role for PKG regulating PQC during physiological and pathological states, examine the therapeutic potential of targeting PKG, and discuss potential future directions.
The ubiquitin-proteasome system (UPS) removes damaged and/or misfolded proteins that are first tagged by ubiquitination for degradation by the proteasome (Wang et al., 2008). Ubiquitination is mediated by multiprotein ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). Ubiquitinated proteins are then translocated to the core proteasome for degradation (Amerik and Hochstrasser, 2004; Reyes-Turcu et al., 2009; Metzger et al., 2012). The mammalian proteasome is a multi-subunit protease complex composed of the 20S catalytic core particle and two 19S regulatory cap particles. The 19S regulatory subunits recognize the polyubiquitin chain and unfold the protein for subsequent degradation by the 20S catalytic core (Nussbaum et al., 1998; Lasker et al., 2012).
All forms of cardiac disease, including heart failure, present with an accumulation of ubiquitinated proteins, demonstrating the vital role the UPS has during pathogenesis (Drews et al., 2010; Su and Wang, 2010; Willis et al., 2010; Wang et al., 2011; Powell et al., 2012; Day, 2013). Indeed, dysfunctional UPS has been reported in human end-stage heart failure, ischemic heart disease, and cardiac hypertrophy (Hein et al., 2003; Weekes et al., 2003; Drews et al., 2010; Powell and Divald, 2010; Predmore et al., 2010). Weekes et al. first reported increased levels of ubiquitinated proteins in myocardial samples obtained from patients with familial dilated cardiomyopathies, which has been supported by others (Weekes et al., 2003; Chen et al., 2005; Liu et al., 2006a, b). Left ventricular unloading in humans with chronic heart failure leads to improved proteasome activity (Wohlschlaeger et al., 2010). Similar results were reported by Predmore et al. (2010), who demonstrated markedly reduced proteolytic activities in failing human hearts that was restored after LV unloading. The mechanism by which ventricular unloading stimulates proteasome activity remains unknown; however, there are multiple potential explanations: (1) LV unloading decreases intracellular ROS, thus oxidized proteins for proteasomal degradation along with less oxidation of the proteasome itself; (2) a separate post-translational modification(s) of the UPS by yet to be identified kinase(s) (Predmore et al., 2010). Proteasome inhibition in pre-clinical models is associated with exacerbated or accelerated pathogenesis of cardiac disease. Mice treated with bortezomib (proteasome inhibitor) worsened transaortic constriction (TAC)-induced cardiac hypertrophy, resulting in early heart failure and death in mice (Tang et al., 2010). These findings were supported by a study using a genetic cardiomyocyte restricted-proteasome inhibited mouse model (beta5T60A) following TAC surgery (Ranek et al., 2015). Similarly, models of myocardial ischemia demonstrated that pretreatment of isolated rat hearts with a proteasome inhibitor dose-dependently decreased post-ischemic cardiac function (Powell et al., 2005). Tian et al. (2012) reported mice that express a threonine 60 to alanine mutation on the proteasome subunit beta 5 (beta5T60A) to reduce the proteolytic activity of the proteasome had worsened myocardial ischemia-reperfusion injury. Impaired or insufficient proteasome activity has been associated with myocarditis (Szalay et al., 2006) and diabetic cardiomyopathy (Li and Wang, 2011). Genetic overexpression of key proteasome subunits enhances proteasome-mediated protein degradation and protects the heart against oxidative stress, proteotoxicity, and myocardial ischemia (Li et al., 2011a, b). Together, these studies demonstrate the prominent role the UPS has in degrading proteins to maintain proteostasis during cardiac disease.
The other primary mediator of cardiomyocyte proteostasis is autophagy, which is comprised of macroautophagy, microautophagy, chaperone-mediated autophagy, and organelle-specific autophagy (e.g., mitophagy) (Ghosh and Pattison, 2018). Macroautophagy involves the formation of an autophagosome which surrounds the cargo for degradation by the lysosome. Microautophagy is the direct engulfment of cellular debris by the lysosome. Chaperone-mediated autophagy degrades proteins containing a KFERQ pentapeptide motif that are translocated into the lysosome via a heat shock cognate 70 (Hsc70) chaperone complex (Dice, 1990). Autophagy is required for the development, differentiation, and function of cardiomyocytes (Nakai et al., 2007; Zhang et al., 2012; Ikeda et al., 2015) and has an important role in cardioprotection (Gustafsson and Gottlieb, 2008; Sciarretta et al., 2014).
Dysregulated autophagic flux is associated with and has been implicated in the pathogenesis of many forms of cardiac disease. Formation of autophagosomes in dilated cardiomyopathy patients has a positive correlation with better prognosis, indicating the protective role of autophagy in heart failure (Saito et al., 2016). Many pre-clinical models have associated reduced autophagy with heart failure and cardiac functional decline (Eisenberg et al., 2016; Shirakabe et al., 2016; Ghosh and Pattison, 2018). Mice with mutations in MYBPC3 (cardiac myosin-binding protein C) and in MYH7 (β-myosin heavy) forms of hypertrophic cardiomyopathy (HCM) present with an accumulation of autophagic vacuoles, suggesting impaired autophagic flux (Schlossarek et al., 2012; Song et al., 2014; Carrier et al., 2015; Singh et al., 2017). Further, deletion of Atg5 (autophagy related gene 5), a key protein involved in the extension of the phagophore membrane during autophagic vesicle formation, causes cardiac hypertrophy and left ventricular dysfunction (Nakai et al., 2007; Pattison et al., 2011). In a cardiac proteinopathy model (CryABR120G), cardiac-specific overexpression of Atg7 increased autophagic activity and improved cardiac function (Bhuiyan et al., 2013). Obese mice see the downregulation of several autophagic genes, including Atg7 (Sciarretta et al., 2012). The exact mechanism for the downregulation of Atg7 was not revealed in these studies; however, it is known that proteinopathy and obesity are characterized by increased mammalian (mechanistic) target of rapamycin complex 1 (mTORC1) activity, which suppresses Atg7 expression (Sciarretta et al., 2018). Interestingly the increased expression of Atg7 in the heart prevents the heart from hypertrophying in response to high-fat diet-induced obesity (Tong et al., 2018). Autophagic flux was reduced in both type 1 and 2 diabetic mouse models and in aged mice (Epstein et al., 1989; Lee et al., 1996; Xie et al., 2011; Kanamori et al., 2015; Munasinghe et al., 2016; Nakamura and Sadoshima, 2018).
Collectively, these studies demonstrate the pivotal role that proteostasis, specifically the UPS and autophagy, has during cardiac pathogenesis (Figure 1). To translate these findings to the clinic to directly target cardiac proteotoxicity, a better understanding of the mechanisms regulating PQC and identification of druggable targets is needed. This review describes exciting investigations into a potential target that has the ability to enhance cardiomyocyte PQC and has many therapeutic strategies available.
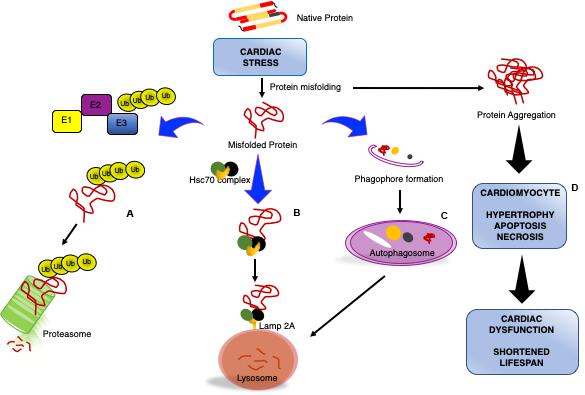
Figure 1. Impaired cardiomyocyte proteostasis results in cardiac dysfunction. Cardiac pathologic stress increases production/formation of misfolded proteins that if not removed form large, insoluble protein aggregates. Cardiomyocytes utilize various processes to maintain proteostasis: misfolded proteins will be catalyzed by the ubiquitin proteasome system (UPS) through ubiquitination via a series of enzymatic reactions involving an ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), and ubiquitin ligase (E3) for degradation by the proteasome (A). Chaperone-mediated autophagy is a process by which the heat shock cognate 70 (HSC70) complex recognizes and binds select protein targets for internalization and degradation to the lysosome through the lysosome-associated membrane protein 2A (LAMP2A) receptor (B). Macroautophagy is the bulk removal of proteins, protein aggregates, and organelles by first forming an autophagosome to surround the cargo followed by merging with the lysosome for degradation (C). The insufficiency or overwhelming of the protein degradation systems during cardiac stress results in an accumulation of aggregated proteins, culminating in reduced cardiac function and lifespan (D).
Protein kinase G is stimulated by cyclic guanosine monophosphate (cGMP). cGMP is generated downstream of nitric oxide (NO) or natriuretic peptide (NP) activation of guanylate cyclase (GC-1, formerly soluble—sGC) and GC-2 (formerly particulate—pGC), respectively (Dunkerly-Eyring and Kass, 2019). Phosphodiesterases (PDEs) selective for cGMP (PDEs 5 and 9 in the heart) negatively regulate PKG activity. PDE regulation of PKG activity is known to be highly compartmentalized within the cardiomyocyte, adding a level of regulation (Kokkonen and Kass, 2017; Dunkerly-Eyring and Kass, 2019). Indeed, PDE5A primarily regulates NO-generated cGMP, which tends to be dispersed throughout the cytosol of the cardiomyocyte, whereas PDE9A regulates the cGMP pool that is localized at the membrane (Kokkonen and Kass, 2017). PDE1, a dual cAMP and cGMP esterase, regulates PKA and PKG activity in an isoform-specific manner in the heart (Hashimoto et al., 2018; Kokkonen-Simon et al., 2018; Dunkerly-Eyring and Kass, 2019).
Multiple strategies of PKG activation have proven to be cardioprotective in response to various pathological stimuli: inhibition of PDE5 or PDE9 reduced cardiac hypertrophy and improved function following left ventricular pressure overload (PO) induced by TAC (Takimoto et al., 2005; Nagayama et al., 2009; Lee et al., 2015). Stimulating PKG directly reduced infarct size and myocardial fibrosis/remodeling following myocardial infarction and ischemia-reperfusion injury (Das et al., 2006; Salloum et al., 2008; Krieg et al., 2009). Neurohormonal stimulation of G-protein-coupled (Gq and Gi) receptor signaling is suppressed by PKG phosphorylation of and/or binding to the regulator of G protein signaling (RGS) proteins, RGS2 and RGS4 (Takimoto et al., 2009). PKG phosphorylates and inhibits the transient receptor potential cation channels type 6 (TRPC6) to block calcineurin/NFAT signaling (Kinoshita et al., 2010; Koitabashi et al., 2010; Nishida et al., 2010) and RhoA to decrease Rho-kinase (Sawada et al., 2001). PKG also regulates mechanosensing via phosphorylation of the sarcomeric proteins: myosin-binding protein C, phospholamban, TnI, and titin (Raeymaekers et al., 1988; Kruger et al., 2009; Thoonen et al., 2015; Rainer and Kass, 2016). Collectively, these studies demonstrate the ability of PKG to correct pathological imbalances, but evidence that PKG stimulation could restore proteostasis during cardiac disease is more recent and forthcoming.
With PKG acting as a brake on many pathological processes, the attention turned to a potential regulation of cardiomyocyte proteostasis. Over the last decade, multiple studies have indicated that PKG activation enhances PQC as a primary mechanism of action to protect the myocardium. Pioneering studies from the Wang lab discovered PKG positively regulates proteasome activity by phosphorylating key proteasome subunits, Rpt6 of the 19S cap and Beta5 of the 20S proteolytic core (Ranek et al., 2013). Intriguingly, activation of PKG pharmacologically (PDE5 inhibitor, sildenafil) or genetically (expression of a constitutively active PKG) in a proteinopathy model (CryABR120G) reduced the accumulation of ubiquitinated proteins and cleared the degradation of misfolded, but not normal, proteins (Ranek et al., 2013). Enhanced PQC with PKG stimulation was associated with reduced cytotoxicity (in vitro) and improved cardiac function (in vivo) (Ranek et al., 2013). Similarly, VerPlank et al. (2020) reported stimulation PKG with both a PDE5 inhibitor or GC-1 activator enhanced proteasome proteolytic activity, the degradation of short lived-and long lived proteins, and determined a direct relationship between PKG and purified proteasomes. Stimulation of PKG via activation of the muscarinic 2 receptor also increased proteasome activity (Ranek et al., 2014), suggesting both the plasma membrane localized and cytosolic localized pools of PKG can enhance proteasome activity. Inhibition of PKG or antagonizing muscarinic 2 receptors decreased the proteasome peptidase activities in both the absence or presence of ATP (Ranek et al., 2013, 2014), suggesting that PKG basally regulates proteasome peptidase activities. These data were supported by and expanded on by a recent study using a PDE1 inhibitor, IC86430 (Zhang et al., 2019). Here the authors utilized CryABR120G proteinopathy mice, which develop a heart failure with a preserved ejection fraction (HFpEF)-like phenotype, and show increased expression of the PDE1A isoform. Inhibition of PDE1 in these mice and cultured cardiomyocytes attenuated proteotoxic stress, increased proteasome activity, and extended mouse lifespan in a PKA and PKG-dependent manner (Zhang et al., 2019). Collectively these findings demonstrate that PKG regulates proteasome activities, proteasome-mediated degradation of misfolded proteins, and that pharmacological approaches can be utilized to elicit these responses.
Recently, it was reported that PKG can also enhance macroautophagy to enhance cardiomyocyte PQC to attenuate cardiac hypertrophy. Tuberous sclerosis complex 2 (TSC2, tuberin), an upstream negative regulator of mTORC1, is phosphorylated by various kinases that can either inhibit (Akt and ERK) or stimulate (AMPK and GSK-3β) its activity. The Kass lab recently reported a new signaling paradigm whereby PKG phosphorylates TSC2 at serine 1365 (1364 in humans) (Ranek et al., 2019). Interestingly, this regulation is itself dependent on the redox state of PKG with reduced phosphorylation of TSC2 detected with oxidation of PKG at cysteine 42 (Oeing et al., 2020). Unlike other TSC2 phosphorylation sites, the phosphorylation of 1365 did not affect basal mTORC1 activity. However, a potent inhibition of mTORC1 hyperactivity was observed once mouse hearts were subjected to hemodynamic (left ventricular pressure overload), or cardiomyocytes to hormonal, stress (endothelin-1). PQC was enhanced as evidenced by upregulation of autophagic flux, clearance of ubiquitinated proteins, and decreased protein aggregation (Ranek et al., 2019). TSC2 S1365 phospho-mimetic decreased, whereas phospho-null exacerbated cardiomyocyte cell size and cytotoxicity following endothelin-1 treatment. Phospho-mimetic mice had attenuated cardiac hypertrophy, improved function, and extended lifespan in response to pressure overload, with opposing findings yielded in phospho-null mice (Ranek et al., 2019). This phosphosite on TSC2 is unique in that there only appears to be mTORC1 regulation in the presence of a pathological co-stimulus, thereby not altering the physiological homeostatic role of mTORC1 (Manning, 2019). Considering the issues with chronic, broad mTORC1 inhibition, utilizing a pharmacological approach with a PKG stimulator represents a unique advantage.
Currently, there are no approved therapeutic strategies to enhance PQC. However, interest has increased as more and more studies implicate exacerbated disease pathogenesis with proteotoxicity, and the evidence in pre-clinical models that PQC enhancement strategies elicit cardioprotection. PKG is an attractive therapeutic target for multiple reasons: (1) PKG modulators have been used in clinic for decades, (2) PKG activators/stimulators boast an excellent safety profile and are well tolerated, and (3) there are many proteins available to interrogate the PKG pathway (Kokkonen-Simon et al., 2018; Dunkerly-Eyring and Kass, 2019; Pinilla-Vera et al., 2019). The first PKG activators were used in the 1800 s in the form of inhaled organic nitrates to treat angina pectoris (Kots et al., 2009; Daiber and Munzel, 2015). It would be roughly 100 years before it was discovered that these work by releasing NO and enhancing cGMP levels (Schlossmann and Schinner, 2012). Therapeutic strategies available to activate the PKG pathway either aim to promote cGMP synthesis (e.g., GC-1 stimulators and activators) or to inhibit cGMP degradation (e.g., PDE5 inhibitors), or both. The GC-1 stimulator, Riociguat, is approved for the treatment of pulmonary arterial hypertension (PAH) as well as inoperable chronic thromboembolic pulmonary hypertension. GC-1 stimulators are being tested in heart failure, diabetic nephropathy, systemic sclerosis, as well as sickle cell disease and central nervous system disease (Xiao et al., 2019). The GC-1 stimulator, vericiguat, was tested in clinical trials for both heart failure with a reduced ejection fraction (HFrEF) [SOCRATES-REDUCED NCT01951625 (Gheorghiade et al., 2015) and VICTORIA NCT02861534 (Armstrong et al., 2018)] and in HFpEF (SOCRATES-PRESERVE NCT01951638) (Filippatos et al., 2017; Pieske et al., 2017). Sacubitril/valsartan (entresto) that combines a neprilysin inhibitor with an angiotensin receptor blocker is increasingly popular as a heart failure therapy, as demonstrated in the PARADIGM-HF (NCT01035255) trial (McMurray et al., 2014; Fala, 2015). The ability of neprilysin inhibitors to increase the levels of natriuretic peptides make PKG a potential target of sacubitril/valsartan (Yan et al., 2003; Riddell and Vader, 2017). Indeed, a 2019 study determined that sacubitril/valsartan decreased cardiac fibrosis in a mouse cardiac hypertrophy model and protected cardiac fibroblasts from myofibroblast transition via PKG-dependent inhibition of RhoA activation (Burke et al., 2019).
These trials did not specifically implicate impairment in proteostasis; however, existing data from human heart tissue suggest that proteostasis is impaired in a disease specific manner. Understanding the PQC systems that are perturbed in different diseases will allow for selective therapeutic targeting. Polyubiquitinated proteins are increased in failing hearts in early as well as late stage disease, suggesting that accumulation of polyubiquitinated proteins occurs before the development of decompensated heart failure (Day, 2013). A histological study from human failing hearts due to idiopathic dilated cardiomyopathy (DCM) noted colocalization of ubiquitin with monodansylcadaverine, a specific marker for autophagic vacuoles, suggesting a link between ubiquitin conjugate accumulation and autophagy (Kostin et al., 2003). Patients with ischemic cardiomyopathy (ICM) and DCM show differential UPS activity patterns. Human ICM heart tissue exhibits reduced trypsin-like proteasomal activity compared to DCM, while both chymotrypsin- and caspase-like proteasomal activities were reduced in DCM and ICM hearts compared to non-failing controls (Spanig et al., 2019). HCM is also characterized by a reduction in chymotrypsin- and caspase-like activities compared to control hearts (Predmore et al., 2010). Failing human hearts exhibit reduced proteasome activity compared to donor controls, which is thought to be related to reduced docking of the 19S proteasome to the 20S proteasome and decreased phosphorylation of Rpt6 (Day, 2013), a potential target of PKG (Ranek et al., 2013). Right ventricular heart disease has not been focused on in clinical trials regarding proteostasis despite emerging data of its important role (Rajagopalan et al., 2013; Drews, 2014; Drews and Taegtmeyer, 2014). Hence, new insights into the regulation of proteostasis via PKG signaling in these diseases might help select the correct patient cohort for successful therapy.
Although the PKG pathway has long been a focus to treat cardiac disease, only recently has the stimulation of PKG been suggested as a new therapeutic strategy to treat cardiac proteinopathies (Figure 2). HFpEF is a lethal syndrome for which there are no evidence-based therapies, characterized by an imbalance in NO levels and low myocardial cGMP and PKG activity (Rainer and Kass, 2016; Schiattarella et al., 2019). A novel murine HFpEF model required metabolic stress accompanied by L-NAME, an NO synthase inhibitor, hence PKG inhibitor, to present some HFpEF symptoms (Schiattarella et al., 2019). These findings suggest that decreased PKG activity facilitates the development and pathogenesis of HFpEF. PDE5 inhibition reduced ER stress in isoproterenol-treated rats and pressure-overloaded mice (Rainer and Kass, 2016) and could successfully treat a desmin-related proteinopathy of the murine heart (Ranek et al., 2013). Xuejun Wang’s group reported that inhibiting PDE1, which hydrolyzes both cAMP and cGMP, increases 26S proteasome activity in a CryABR120G-based proteinopathy of the murine heart (Zhang et al., 2019). Treatment with PDE1 inhibitor IC86430 increased proteasome phosphorylation, reduced misfolded CryAB protein in the murine heart, attenuated HFpEF-like phenotype, and ultimately improved survival (Zhang et al., 2019). These studies further support the notion that activating PKG could be beneficial in HFpEF therapy, at least in part by enhancing PQC. Inhibitors of mTORC1 potently increase autophagic flux, attenuate cardiac hypertrophy, and enhance function; however, chronic use leads to cardiac dilation and failure along with immunosuppression and metabolic disturbances (Benjamin et al., 2011; Zeng et al., 2013). These deleterious side effects can be avoided by stimulating PKG to inhibit mTORC1, hence might present a better tool than mTOR inhibitors (Manning, 2019; Ranek et al., 2019). Collectively these studies demonstrate that (1) PKG is vital to maintain proteostasis basally, (2) many therapeutic targets (NO, NPs, PDEs) are available to stimulate PKG, and (3) PKG activators/stimulators could be the first therapy that enhances PQC, is cardioprotective, and does so without deleterious side effects.
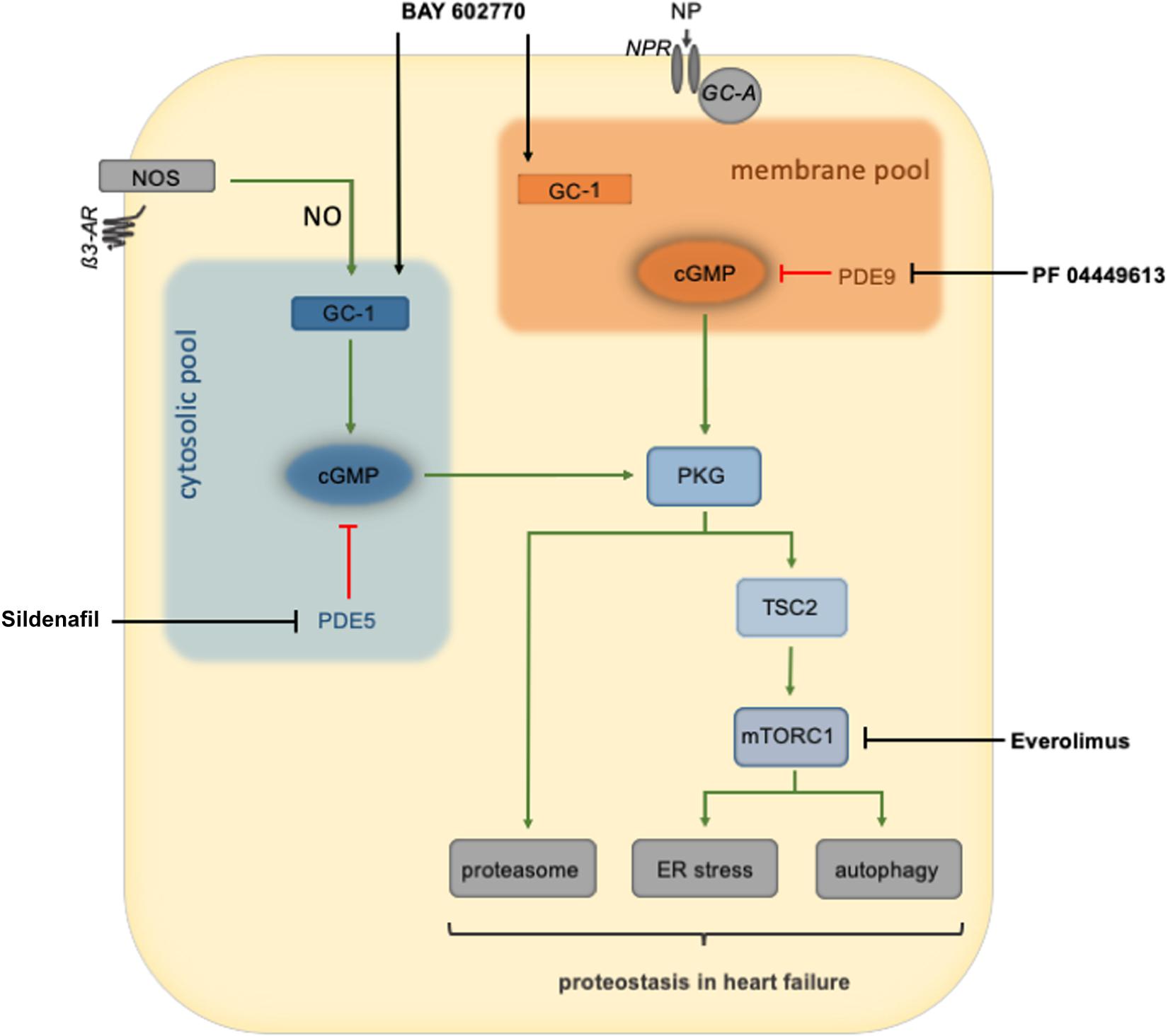
Figure 2. An overview of PKG regulation of cardiomyocyte protein quality control and therapeutic interventions to stimulate PKG activity. PKG is thought to be divided into two primary pools: the membrane and cytosolic pools. Natriuretic peptides binding to the natriuretic peptide receptor activate guanylate cyclase to produce cGMP and ultimately stimulate the membrane pool of PKG. Nitric oxide, produced by nitric oxide synthase, activates guanylate cyclase-1 to produce cGMP, culminating in the activation of the cytosolic PKG pool. Once activated, PKG can increase the activity of the proteasome or phosphorylation of TSC2 to inhibit mTORC1 and enhance autophagic flux. PKG stimulation of the proteasome or autophagy restores cardiomyocyte proteostasis during cardiac stress. AR, adrenoreceptor; GC-1, soluble guanylate cyclase 1; GC-A, guanylyl cyclase-A (receptor); GMP, cyclic guanylyl monophosphate; ER, endoplasmatic reticulum; mTORC1, mammalian target of rapamycin complex 1; NOS, nitric oxide synthase; NP, natriuretic peptide; NPR, NP receptor; PDE, phosphodiesterase; PKG, protein kinase G; PQC, protein quality control; TSC2, tuberin/tuberous sclerosis complex 2.
Our knowledge of the protein kinases that regulate cardiomyocyte PQC has vastly expanded. The discovery of new targets to pursue, pharmacological strategies to test, and increased understanding of the regulatory mechanisms are pivotal to translating successful experimental studies into efficacious clinical therapies. Several crucial hurdles remain. The first being the safety of a therapy, as agents may increase PQC but as a compensatory mechanism for damage induced by the therapy. Second issue is having druggable targets for therapeutic interventions. The third hurdle is to match the appropriate disease state to the PQC deficiency to the microdomain in which the kinase and PQC system reside. PKG activators/stimulators are safe and well tolerated and many are already in clinical use. Further research to gain a precise understanding of the microdomains these compounds work in and PQC machinery they associate with will identify the conditions and patient subsets that will likely benefit from specific PKG modulation. The final hurdle is the need for a blood biomarker capable of detecting impaired proteostasis in the heart, which to the best of our knowledge has not been verified. This would be essential to detect patients that might benefit from strategies to improve cardiomyocyte PQC and to monitor potential success of the therapy.
BD-E and MR are co-inventors on a patent application (PCT: 448070145WO1) that was filed in July 2018 (provisional filed in June 2017). The patent relates to the use of TSC2(S1365/S1364) modifications for immunological applications.
MR conceived and designed the framework for the review and supervised the writing. CO wrote the manuscript with assistance from SM and BD-E. All authors provided critical feedback while writing the manuscript.
This study was supported by the NIH – National Heart, Lung, and Blood Institute grant F31-HL143905 (BD-E), the Deutsche Forschungsgemeinschaft (German Research Foundation) OE 688/1-1 (CO), the BIH Charité Clinician Scientist Program (CO), Amyloidosis Foundation Donald C. Brockman Memorial Research Grant (MR), and an American Heart Association Career Development Award 18CDA34110140 (MR).
MR is a co-founder and shareholder of Meta-T Cellular, a start-up company that aims to develop applications of this intellectual property for immune therapy.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Amerik, A. Y., and Hochstrasser, M. (2004). Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 1695, 189–207. doi: 10.1016/j.bbamcr.2004.10.003
Armstrong, P. W., Roessig, L., Patel, M. J., Anstrom, K. J., Butler, J., Voors, A. A., et al. (2018). A multicenter, randomized, double-blind, placebo-controlled trial of the efficacy and safety of the oral soluble guanylate cyclase stimulator: the VICTORIA trial. JACC Heart Fail. 6, 96–104. doi: 10.1016/j.jchf.2017.08.013
Benjamin, D., Colombi, M., Moroni, C., and Hall, M. N. (2011). Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 10, 868–880. doi: 10.1038/nrd3531
Bhuiyan, M. S., Pattison, J. S., Osinska, H., James, J., Gulick, J., McLendon, P. M., et al. (2013). Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 123, 5284–5297. doi: 10.1172/jci70877
Burke, R. M., Lighthouse, J. K., Mickelsen, D. M., and Small, E. M. (2019). Sacubitril/valsartan decreases cardiac fibrosis in left ventricle pressure overload by restoring PKG signaling in cardiac fibroblasts. Circ. Heart Fail. 12: e005565.
Carrier, L., Mearini, G., Stathopoulou, K., and Cuello, F. (2015). Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 573, 188–197. doi: 10.1016/j.gene.2015.09.008
Chen, Q., Liu, J. B., Horak, K. M., Zheng, H., Kumarapeli, A. R., Li, J., et al. (2005). Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ. Res. 97, 1018–1026. doi: 10.1161/01.res.0000189262.92896.0b
Daiber, A., and Munzel, T. (2015). Organic nitrate therapy, nitrate tolerance, and nitrate-induced endothelial dysfunction: emphasis on redox biology and oxidative stress. Antioxid. Redox Signal. 23, 899–942. doi: 10.1089/ars.2015.6376
Das, A., Smolenski, A., Lohmann, S. M., and Kukreja, R. C. (2006). Cyclic GMP-dependent protein kinase Ialpha attenuates necrosis and apoptosis following ischemia/reoxygenation in adult cardiomyocyte. J. Biol. Chem. 281, 38644–38652. doi: 10.1074/jbc.m606142200
Day, S. M. (2013). The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am. J. Physiol. Heart Circ. Physiol. 304, H1283–H1293.
Dice, J. F. (1990). Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 15, 305–309. doi: 10.1016/0968-0004(90)90019-8
Drews, O. (2014). The left and right ventricle in the grip of protein degradation: similarities and unique patterns in regulation. J. Mol. Cell. Cardiol. 72, 52–55. doi: 10.1016/j.yjmcc.2014.02.016
Drews, O., and Taegtmeyer, H. (2014). Targeting the ubiquitin-proteasome system in heart disease: the basis for new therapeutic strategies. Antioxid. Redox Signal. 21, 2322–2343. doi: 10.1089/ars.2013.5823
Drews, O., Tsukamoto, O., Liem, D., Streicher, J., Wang, Y., and Ping, P. (2010). Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ. Res. 107, 1094–1101. doi: 10.1161/circresaha.110.222364
Dunkerly-Eyring, B., and Kass, D. A. (2019). Myocardial phosphodiesterases and their role in cGMP regulation. J. Cardiovasc. Pharmacol. 75, 483–493. doi: 10.1097/fjc.0000000000000773
Eisenberg, T., Abdellatif, M., Schroeder, S., Primessnig, U., Stekovic, S., Pendl, T., et al. (2016). Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438.
Epstein, P. N., Overbeek, P. A., and Means, A. R. (1989). Calmodulin-induced early-onset diabetes in transgenic mice. Cell 58, 1067–1073. doi: 10.1016/0092-8674(89)90505-9
Fala, L. (2015). Entresto (Sacubitril/Valsartan): first-in-class angiotensin receptor neprilysin inhibitor FDA approved for patients with heart failure. Am. Health Drug Benefits 8, 330–334.
Filippatos, G., Maggioni, A. P., Lam, C. S. P., Pieske-Kraigher, E., Butler, J., Spertus, J., et al. (2017). Patient-reported outcomes in the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED ejection fraction (SOCRATES-PRESERVED) study. Eur. J. Heart Fail. 19, 782–791. doi: 10.1002/ejhf.800
Gheorghiade, M., Greene, S. J., Butler, J., Filippatos, G., Lam, C. S., Maggioni, A. P., et al. (2015). Effect of vericiguat, a soluble Guanylate Cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: the socrates-reduced randomized trial. JAMA 314, 2251–2262.
Ghosh, R., and Pattison, J. S. (2018). Macroautophagy and chaperone-mediated autophagy in heart failure: the known and the unknown. Oxid. Med. Cell Longev. 2018:8602041.
Gustafsson, A. B., and Gottlieb, R. A. (2008). Recycle or die: the role of autophagy in cardioprotection. J. Mol. Cell. Cardiol. 44, 654–661. doi: 10.1016/j.yjmcc.2008.01.010
Hashimoto, T., Kim, G. E., Tunin, R. S., Adesiyun, T., Hsu, S., Nakagawa, R., et al. (2018). Acute enhancement of cardiac function by phosphodiesterase type 1 inhibition. Circulation 138, 1974–1987. doi: 10.1161/circulationaha.117.030490
Hein, S., Arnon, E., Kostin, S., Schonburg, M., Elsasser, A., Polyakova, V., et al. (2003). Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 107, 984–991. doi: 10.1161/01.cir.0000051865.66123.b7
Ikeda, Y., Shirakabe, A., Maejima, Y., Zhai, P., Sciarretta, S., Toli, J., et al. (2015). Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 116, 264–278. doi: 10.1161/circresaha.116.303356
Kanamori, H., Takemura, G., Goto, K., Tsujimoto, A., Mikami, A., Ogino, A., et al. (2015). Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 11, 1146–1160. doi: 10.1080/15548627.2015.1051295
Kinoshita, H., Kuwahara, K., Nishida, M., Jian, Z., Rong, X., Kiyonaka, S., et al. (2010). Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ. Res. 106, 1849–1860. doi: 10.1161/circresaha.109.208314
Koitabashi, N., Aiba, T., Hesketh, G. G., Rowell, J., Zhang, M., Takimoto, E., et al. (2010). Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J. Mol. Cell. Cardiol. 48, 713–724. doi: 10.1016/j.yjmcc.2009.11.015
Kokkonen, K., and Kass, D. A. (2017). Nanodomain regulation of cardiac cyclic nucleotide signaling by phosphodiesterases. Annu. Rev. Pharmacol. Toxicol. 57, 455–479. doi: 10.1146/annurev-pharmtox-010716-104756
Kokkonen-Simon, K. M., Saberi, A., Nakamura, T., Ranek, M. J., Zhu, G., Bedja, D., et al. (2018). Marked disparity of microRNA modulation by cGMP-selective PDE5 versus PDE9 inhibitors in heart disease. JCI Insight 3:e121739.
Kostin, S., Pool, L., Elsasser, A., Hein, S., Drexler, H. C., Arnon, E., et al. (2003). Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 92, 715–724. doi: 10.1161/01.res.0000067471.95890.5c
Kots, A. Y., Martin, E., Sharina, I. G., and Murad, F. (2009). A short history of cGMP, guanylyl cyclases, and cGMP-dependent protein kinases. Handb. Exp. Pharmacol. 191, 1–14. doi: 10.1007/978-3-540-68964-5_1
Krieg, T., Liu, Y., Rutz, T., Methner, C., Yang, X. M., Dost, T., et al. (2009). BAY 58-2667, a nitric oxide-independent guanylyl cyclase activator, pharmacologically post-conditions rabbit and rat hearts. Eur. Heart J. 30, 1607–1613. doi: 10.1093/eurheartj/ehp143
Kruger, M., Kotter, S., Grutzner, A., Lang, P., Andresen, C., Redfield, M. M., et al. (2009). Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 104, 87–94. doi: 10.1161/circresaha.108.184408
Lasker, K., Forster, F., Bohn, S., Walzthoeni, T., Villa, E., Unverdorben, P., et al. (2012). Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. U.S.A. 109, 1380–1387. doi: 10.1073/pnas.1120559109
Lee, D. I., Zhu, G., Sasaki, T., Cho, G. S., Hamdani, N., Holewinski, R., et al. (2015). Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 519, 472–476. doi: 10.1038/nature14332
Lee, G. H., Proenca, R., Montez, J. M., Carroll, K. M., Darvishzadeh, J. G., Lee, J. I., et al. (1996). Abnormal splicing of the leptin receptor in diabetic mice. Nature 379, 632–635. doi: 10.1038/379632a0
Li, J., Horak, K. M., Su, H., Sanbe, A., Robbins, J., and Wang, X. (2011a). Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J. Clin. Invest. 121, 3689–3700. doi: 10.1172/jci45709
Li, J., Powell, S. R., and Wang, X. (2011b). Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J. 25, 883–893. doi: 10.1096/fj.10-160895
Li, Y. F., and Wang, X. (2011). The role of the proteasome in heart disease. Biochim. Biophys. Acta 1809, 141–149. doi: 10.1016/j.bbagrm.2010.09.001
Liu, J., Chen, Q., Huang, W., Horak, K. M., Zheng, H., Mestril, R., et al. (2006a). Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J. 20, 362–364. doi: 10.1096/fj.05-4869fje
Liu, J., Tang, M., Mestril, R., and Wang, X. (2006b). Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J. Mol. Cell. Cardiol. 40, 451–454. doi: 10.1016/j.yjmcc.2005.12.011
Manning, B. D. (2019). Signalling protein protects the heart muscle from pressure-related stress. Nature 566, 187–188. doi: 10.1038/d41586-019-00245-3
McMurray, J. J., Packer, M., Desai, A. S., Gong, J., Lefkowitz, M. P., Rizkala, A. R., et al. (2014). Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 371, 993–1004.
Metzger, M. B., Hristova, V. A., and Weissman, A. M. (2012). HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell. Sci. 125, 531–537. doi: 10.1242/jcs.091777
Munasinghe, P. E., Riu, F., Dixit, P., Edamatsu, M., Saxena, P., Hamer, N. S., et al. (2016). Type-2 diabetes increases autophagy in the human heart through promotion of Beclin-1 mediated pathway. Int. J. Cardiol. 202, 13–20. doi: 10.1016/j.ijcard.2015.08.111
Nagayama, T., Hsu, S., Zhang, M., Koitabashi, N., Bedja, D., Gabrielson, K. L., et al. (2009). Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J. Am. Coll. Cardiol. 53, 207–215. doi: 10.1016/j.jacc.2008.08.069
Nakai, A., Yamaguchi, O., Takeda, T., Higuchi, Y., Hikoso, S., Taniike, M., et al. (2007). The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624. doi: 10.1038/nm1574
Nakamura, M., and Sadoshima, J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387–407. doi: 10.1038/s41569-018-0007-y
Nishida, M., Watanabe, K., Sato, Y., Nakaya, M., Kitajima, N., Ide, T., et al. (2010). Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J. Biol. Chem. 285, 13244–13253. doi: 10.1074/jbc.m109.074104
Nussbaum, A. K., Dick, T. P., Keilholz, W., Schirle, M., Stevanovic, S., Dietz, K., et al. (1998). Cleavage motifs of the yeast 20S proteasome beta subunits deduced from digests of enolase 1. Proc. Natl. Acad. Sci. U.S.A. 95, 12504–12509. doi: 10.1073/pnas.95.21.12504
Oeing, C. U., Nakamura, T., Pan, S., Mishra, S., Dunkerly-Eyring, B., Kokkonen-Simon, K. M., et al. (2020). PKG1alpha cysteine-42 redox state controls mTORC1 activation in pathological cardiac hypertrophy. Circ. Res. doi: 10.1161/CIRCRESAHA.119.315714 [Epub ahead of print].
Pattison, J. S., Osinska, H., and Robbins, J. (2011). Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ. Res. 109, 151–160. doi: 10.1161/circresaha.110.237339
Pieske, B., Maggioni, A. P., Lam, C. S. P., Pieske-Kraigher, E., Filippatos, G., Butler, J., et al. (2017). Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur. Heart J. 38, 1119–1127. doi: 10.1093/eurheartj/ehw593
Pinilla-Vera, M., Hahn, V. S., and Kass, D. A. (2019). Leveraging signaling pathways to treat heart failure with reduced ejection fraction. Circ. Res. 124, 1618–1632. doi: 10.1161/circresaha.119.313682
Powell, S. R., and Divald, A. (2010). The ubiquitin-proteasome system in myocardial ischaemia and preconditioning. Cardiovasc. Res. 85, 303–311. doi: 10.1093/cvr/cvp321
Powell, S. R., Herrmann, J., Lerman, A., Patterson, C., and Wang, X. (2012). The ubiquitin-proteasome system and cardiovascular disease. Prog. Mol. Biol. Transl. Sci. 109, 295–346.
Powell, S. R., Wang, P., Katzeff, H., Shringarpure, R., Teoh, C., Khaliulin, I., et al. (2005). Oxidized and ubiquitinated proteins may predict recovery of postischemic cardiac function: essential role of the proteasome. Antioxid. Redox Signal. 7, 538–546. doi: 10.1089/ars.2005.7.538
Predmore, J. M., Wang, P., Davis, F., Bartolone, S., Westfall, M. V., Dyke, D. B., et al. (2010). Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121, 997–1004. doi: 10.1161/circulationaha.109.904557
Raeymaekers, L., Hofmann, F., and Casteels, R. (1988). Cyclic GMP-dependent protein kinase phosphorylates phospholamban in isolated sarcoplasmic reticulum from cardiac and smooth muscle. Biochem. J. 252, 269–273. doi: 10.1042/bj2520269
Rainer, P. P., and Kass, D. A. (2016). Old dog, new tricks: novel cardiac targets and stress regulation by protein kinase G. Cardiovasc. Res. 111, 154–162. doi: 10.1093/cvr/cvw107
Rajagopalan, V., Zhao, M., Reddy, S., Fajardo, G., Wang, X., Dewey, S., et al. (2013). Altered ubiquitin-proteasome signaling in right ventricular hypertrophy and failure. Am. J. Physiol. Heart Circ. Physiol. 305, H551–H562.
Ranek, M. J., Kokkonen-Simon, K. M., Chen, A., Dunkerly-Eyring, B. L., Vera, M. P., Oeing, C. U., et al. (2019). PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 566, 264–269. doi: 10.1038/s41586-019-0895-y
Ranek, M. J., Kost, C. K. Jr., Hu, C., Martin, D. S., and Wang, X. (2014). Muscarinic 2 receptors modulate cardiac proteasome function in a protein kinase G-dependent manner. J. Mol. Cell. Cardiol. 69, 43–51. doi: 10.1016/j.yjmcc.2014.01.017
Ranek, M. J., Terpstra, E. J., Li, J., Kass, D. A., and Wang, X. (2013). Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation 128, 365–376. doi: 10.1161/circulationaha.113.001971
Ranek, M. J., Zheng, H., Huang, W., Kumarapeli, A. R., Li, J., Liu, J., et al. (2015). Genetically induced moderate inhibition of 20S proteasomes in cardiomyocytes facilitates heart failure in mice during systolic overload. J. Mol. Cell. Cardiol. 85, 273–281. doi: 10.1016/j.yjmcc.2015.06.014
Reyes-Turcu, F. E., Ventii, K. H., and Wilkinson, K. D. (2009). Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363–397. doi: 10.1146/annurev.biochem.78.082307.091526
Riddell, E., and Vader, J. M. (2017). Potential expanded indications for neprilysin inhibitors. Curr. Heart Fail. Rep. 14, 134–145. doi: 10.1007/s11897-017-0327-y
Saito, T., Asai, K., Sato, S., Hayashi, M., Adachi, A., Sasaki, Y., et al. (2016). Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy 12, 579–587. doi: 10.1080/15548627.2016.1145326
Salloum, F. N., Abbate, A., Das, A., Houser, J. E., Mudrick, C. A., Qureshi, I. Z., et al. (2008). Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am. J. Physiol. Heart Circ. Physiol. 294, H1398–H1406.
Sawada, N., Itoh, H., Yamashita, J., Doi, K., Inoue, M., Masatsugu, K., et al. (2001). cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem. Biophys. Res. Commun. 280, 798–805. doi: 10.1006/bbrc.2000.4194
Schiattarella, G. G., Altamirano, F., Tong, D., French, K. M., Villalobos, E., Kim, S. Y., et al. (2019). Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568, 351–356.
Schlossarek, S., Englmann, D. R., Sultan, K. R., Sauer, M., Eschenhagen, T., and Carrier, L. (2012). Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res. Cardiol. 107:235.
Schlossmann, J., and Schinner, E. (2012). cGMP becomes a drug target. Naunyn Schmiedebergs Arch. Pharmacol. 385, 243–252. doi: 10.1007/s00210-012-0730-6
Sciarretta, S., Forte, M., Frati, G., and Sadoshima, J. (2018). New insights into the role of mTOR signaling in the cardiovascular system. Circ. Res. 122, 489–505. doi: 10.1161/circresaha.117.311147
Sciarretta, S., Volpe, M., and Sadoshima, J. (2012). Is reactivation of autophagy a possible therapeutic solution for obesity and metabolic syndrome? Autophagy 8, 1252–1254. doi: 10.4161/auto.20670
Sciarretta, S., Yee, D., Shenoy, V., Nagarajan, N., and Sadoshima, J. (2014). The importance of autophagy in cardioprotection. High Blood Press. Cardiovasc. Prev. 21, 21–28. doi: 10.1007/s40292-013-0029-9
Shirakabe, A., Ikeda, Y., Sciarretta, S., Zablocki, D. K., and Sadoshima, J. (2016). Aging and autophagy in the heart. Circ. Res. 118, 1563–1576.
Singh, S. R., Zech, A. T. L., Geertz, B., Reischmann-Dusener, S., Osinska, H., Prondzynski, M., et al. (2017). Activation of autophagy ameliorates cardiomyopathy in Mybpc3-targeted Knockin mice. Circ. Heart Fail. 10:e004140.
Song, L., Su, M., Wang, S., Zou, Y., Wang, X., Wang, Y., et al. (2014). MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J. Cell. Mol. Med. 18, 2266–2274. doi: 10.1111/jcmm.12380
Spanig, S., Kellermann, K., Dieterlen, M. T., Noack, T., Lehmann, S., Borger, M. A., et al. (2019). The ubiquitin proteasome system in ischemic and dilated cardiomyopathy. Int. J. Mol. Sci. 20:6354. doi: 10.3390/ijms20246354
Su, H., and Wang, X. (2010). The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc. Res. 85, 253–262. doi: 10.1093/cvr/cvp287
Szalay, G., Meiners, S., Voigt, A., Lauber, J., Spieth, C., Speer, N., et al. (2006). Ongoing coxsackievirus myocarditis is associated with increased formation and activity of myocardial immunoproteasomes. Am. J. Pathol. 168, 1542–1552. doi: 10.2353/ajpath.2006.050865
Takimoto, E., Champion, H. C., Li, M., Belardi, D., Ren, S., Rodriguez, E. R., et al. (2005). Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 11, 214–222. doi: 10.1038/nm1175
Takimoto, E., Koitabashi, N., Hsu, S., Ketner, E. A., Zhang, M., Nagayama, T., et al. (2009). Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J. Clin. Invest. 119, 408–420.
Tang, M., Li, J., Huang, W., Su, H., Liang, Q., Tian, Z., et al. (2010). Proteasome functional insufficiency activates the calcineurin-NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc. Res. 88, 424–433. doi: 10.1093/cvr/cvq217
Thoonen, R., Giovanni, S., Govindan, S., Lee, D. I., Wang, G. R., Calamaras, T. D., et al. (2015). Molecular screen identifies cardiac myosin-binding protein-C as a protein kinase G-Ialpha substrate. Circ. Heart Fail. 8, 1115–1122. doi: 10.1161/circheartfailure.115.002308
Tian, Z., Zheng, H., Li, J., Li, Y., Su, H., and Wang, X. (2012). Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia-reperfusion injury in mice. Circ. Res. 111, 532–542. doi: 10.1161/circresaha.112.270983
Tong, M., Saito, T., Zhai, P., Oka, S., and Sadoshima, J. (2018). Atg7-dependent autophagy is essential for cardiac function with high fat diet. FASEB J. 32, 35.1–35.1.
VerPlank, J. J. S., Tyrkalska, S. D., Fleming, A., Rubinsztein, D. C., and Goldberg, A. L. (2020). cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc. Natl. Acad. Sci. U.S.A. 117, 14220–14230. doi: 10.1073/pnas.2003277117
Wang, X., Li, J., Zheng, H., Su, H., and Powell, S. R. (2011). Proteasome functional insufficiency in cardiac pathogenesis. Am. J. Physiol. Heart Circ. Physiol. 301, H2207–H2219.
Wang, X., and Robbins, J. (2006). Heart failure and protein quality control. Circ. Res. 99, 1315–1328. doi: 10.1161/01.res.0000252342.61447.a2
Wang, X., Su, H., and Ranek, M. J. (2008). Protein quality control and degradation in cardiomyocytes. J. Mol. Cell. Cardiol. 45, 11–27. doi: 10.1016/j.yjmcc.2008.03.025
Weekes, J., Morrison, K., Mullen, A., Wait, R., Barton, P., and Dunn, M. J. (2003). Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 3, 208–216. doi: 10.1002/pmic.200390029
Willis, M. S., and Patterson, C. (2013). Proteotoxicity and cardiac dysfunction–Alzheimer’s disease of the heart? N. Engl. J. Med. 368, 455–464. doi: 10.1056/nejmra1106180
Willis, M. S., Townley-Tilson, W. H., Kang, E. Y., Homeister, J. W., and Patterson, C. (2010). Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ. Res. 106, 463–478. doi: 10.1161/circresaha.109.208801
Wohlschlaeger, J., Sixt, S. U., Stoeppler, T., Schmitz, K. J., Levkau, B., Tsagakis, K., et al. (2010). Ventricular unloading is associated with increased 20s proteasome protein expression in the myocardium. J. Heart Lung Transplant 29, 125–132. doi: 10.1016/j.healun.2009.07.022
Xiao, S., Li, Q., Hu, L., Yu, Z., Yang, J., Chang, Q., et al. (2019). Soluble guanylate cyclase stimulators and activators: where are we and where to go? Mini Rev. Med. Chem. 19, 1544–1557. doi: 10.2174/1389557519666190730110600
Xie, Z., Lau, K., Eby, B., Lozano, P., He, C., Pennington, B., et al. (2011). Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60, 1770–1778. doi: 10.2337/db10-0351
Yan, C., Kim, D., Aizawa, T., and Berk, B. C. (2003). Functional interplay between angiotensin II and nitric oxide: cyclic GMP as a key mediator. Arterioscler Thromb Vasc. Biol. 23, 26–36. doi: 10.1161/01.atv.0000046231.17365.9d
Zeng, H., Yang, K., Cloer, C., Neale, G., Vogel, P., and Chi, H. (2013). mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490. doi: 10.1038/nature12297
Zhang, H., Pan, B., Wu, P., Parajuli, N., Rekhter, M. D., Goldberg, A. L., et al. (2019). PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci. Adv. 5:eaaw5870. doi: 10.1126/sciadv.aaw5870
Keywords: proteostasis, PKG, proteotoxicity, proteasome, autophagy, heart failure
Citation: Oeing CU, Mishra S, Dunkerly-Eyring BL and Ranek MJ (2020) Targeting Protein Kinase G to Treat Cardiac Proteotoxicity. Front. Physiol. 11:858. doi: 10.3389/fphys.2020.00858
Received: 15 February 2020; Accepted: 26 June 2020;
Published: 28 July 2020.
Edited by:
Julian Stelzer, Case Western Reserve University, United StatesReviewed by:
Robert Morris Blanton, Tufts Medical Center, United StatesCopyright © 2020 Oeing, Mishra, Dunkerly-Eyring and Ranek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark J. Ranek, bXJhbmVrMUBqaC5lZHU=; bXJhbmVrMUBqaG1pLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.