 Nensi Ikonomi
Nensi Ikonomi Silke D. Kühlwein
Silke D. Kühlwein Julian D. Schwab
Julian D. Schwab Hans A. Kestler
Hans A. Kestler
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 31 July 2020
Sec. Systems Biology Archive
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00848
This article is part of the Research TopicSystems Biology of Cell SignalingView all 16 articles
Hematopoietic stem cells (HSCs) provide all types of blood cells during the entire life of the organism. HSCs are mainly quiescent and can eventually enter the cell cycle to differentiate. HSCs are maintained and tightly regulated in a particular environment. The stem cell niche regulates dormancy and awakening. Deregulations of this interplay can lead to hematopoietic failure and diseases. In this paper, we present a Boolean network model that recapitulates HSC regulation in virtue of external signals coming from the niche. This Boolean network integrates and summarizes the current knowledge of HSC regulation and is based on extensive literature research. Furthermore, dynamic simulations suggest a novel systemic regulation of TP53 in homeostasis. Thereby, our model indicates that TP53 activity is balanced depending on external stimulations, engaging a regulatory mechanism involving ROS regulators and RAS activated transcription factors. Finally, we investigated different mouse models and compared them to in silico knockout simulations. Here, the model could recapitulate in vivo observed behaviors and thus sustains our results.
The hematopoietic system is composed of heterogenic populations. These populations comprise highly specialized cells with unique and peculiar functions. All these different blood cells, approximately 4 − 5 · 1011 in total, are produced hierarchically from a population of cells called hematopoietic stem cells (HSCs) (Kaushansky, 2006; Jagannathan-Bogdan and Zon, 2013). Nevertheless, recent results coming from single-cell RNA sequencing suggest that hematopoiesis is not necessarily hierarchical, but can also be radial (Macaulay et al., 2016; Athanasiadis et al., 2017; Laurenti and Göttgens, 2018; Zhang et al., 2018; Yokota, 2019).
HSCs are a particular population of cells characterized by the ability to self-renew and to generate differentiated blood cells (Pietras et al., 2011; Sun et al., 2014; Busch et al., 2015; Sawai et al., 2016). Under steady-state conditions, HSCs are maintained as a quiescent population of cells that can be activated in the presence of external differentiation stimuli (Fleming et al., 1993; Bradford et al., 1997; Cheshier et al., 1999; Passegué et al., 2005). This dormant state is believed to contribute to the long-term preservation of HSCs, mostly by minimizing replication and metabolic activity (Wilson et al., 2009; Warr et al., 2011; Bakker and Passegué, 2013). Fine-tuning of dormancy and the awakening mechanism of HSCs is of crucial relevance for correct hematopoiesis. In fact, not responsive dormant HSCs would lead to hematopoietic failure due to a lack of differentiated blood cells (Wilson et al., 2009). On the other hand, highly active HSCs would get to exhaustion of the population and lack of long-term maintenance of the hematopoietic system (Wilson et al., 2009).
Leading intrinsic players regulating HSC quiescence are polycomb ring finger protein (BMI1), TP53, reactive oxygen species (ROS) regulators and protein kinase B (AKT) signaling (Abbas et al., 2011; Pietras et al., 2011; Suda et al., 2011; Warr et al., 2011). Besides intrinsic factors, HSCs are regulated by the niche. The niche is defined as the specialized bone marrow environment where the HSCs reside (Schofield, 1978). Classically, two types of niches have been individuated for HSCs: the endosteal and the vascular niches. The endosteal niche is responsible for the maintenance of quiescence and is composed mainly of osteoblastic cells. In this context, hypoxic condition and signals promoting quiescence such as transforming growth factor beta (TGF-β) set HSCs in a dormant state (Zhang et al., 2003; Kopp et al., 2005; Sugiyama et al., 2006; Geiger et al., 2007; Jones and Wagers, 2008; Wagner et al., 2008; Guerrouahen et al., 2011). On the other hand, the vascular niche is characterized by endothelial and stromal cells and creates a milieu that supports proliferation, differentiation and trans-endothelial migration of HSCs (Zhang et al., 2003; Kopp et al., 2005; Sugiyama et al., 2006; Geiger et al., 2007; Jones and Wagers, 2008; Wagner et al., 2008; Guerrouahen et al., 2011). Here, a higher oxygen concentration and proliferating signals such as stem cell factor (SCF), fibroblast growth factor (FGF), and other growth factors are mostly present. Overall, these signals stimulate HSC activation and cycling (Coskun and Hirschi, 2010; Lilly et al., 2011). Moreover, HSCs and progenitors can be defined by surface markers as LSK (Lin− Sca+ cKit+) population (Spangrude et al., 1988). Additionally, HSCs can be further divided into long-term HSCs (LT-HSC) and short-term HSCs (ST-HSC) depending again on marker selection. In this context, LT-HSCs are defined as Lin− Sca+ cKit+CD150+CD48+ (Adolfsson et al., 2001; Kiel et al., 2005; Wilson and Trumpp, 2006; Challen et al., 2009; Foudi et al., 2009; Wilson et al., 2009). Phenotypically these two types of HSCs are distinguished based on their quiescence, long-term self-renewal ability, and engraftment. Hence, even the dormant state itself is regulated distinguishing between highly dormant and more active HSCs (Adolfsson et al., 2001; Kiel et al., 2005; Wilson and Trumpp, 2006; Challen et al., 2009; Foudi et al., 2009; Wilson et al., 2009).
Different models to unravel HSC fate commitment are available (Glauche et al., 2011; Krumsiek et al., 2011; Hamey et al., 2017; Olariu and Peterson, 2019). However, as it was underlined, HSC function does not only reside in its ability to differentiate. Unraveling mechanistic strategies by which HSCs are kept in quiescence and activeness is of crucial interest. Accordingly, the deregulation of HSC function is linked to cancerous conditions and aging (Warr et al., 2011; Bakker and Passegué, 2013). Even if a lot of effort has been implied in understanding the maintenance of HSCs, still many regulatory mechanisms are unknown. Crucial regulators of quiescence such as TP53, BMI1, ataxia-telangiectasia mutated (ATM) have been individuated. However, it is still not clear how they are differentially regulated in dormant (LT-HSC) and more active HSC (ST-HSC) (Warr et al., 2011; Bakker and Passegué, 2013). The regulation of these factors is often believed to be a result of crosstalk among different intrinsic and extrinsic pathways involved in HSC maintenance (Warr et al., 2011; Bakker and Passegué, 2013). Of interest in this context is the regulation of TP53.
TP53 is a ubiquitously expressed protein described to be activated in the context of DNA damage response and regulates cell survival (Toledo and Wahl, 2006; Vousden and Lane, 2007; Meek, 2009). In normal conditions, instead, TP53 is down-regulated by mouse double minute 2 homolog (MDM2) that causes its degradation (Jones et al., 1995; Montes de Oca Luna et al., 1995; Ringshausen et al., 2006). MDM2 expression is further sustained by TP53, causing negative feedback on its activity (Jones et al., 1995; Montes de Oca Luna et al., 1995; Ringshausen et al., 2006). Nevertheless, high levels of TP53 have been detected in HSCs. Its expression is linked to the maintenance of quiescence by induction of growth factor independent one transcriptional repressor (GFI1) (Forsberg et al., 2005; Liu Y. et al., 2009). How both intrinsic and extrinsic factors regulate TP53 in steady-state conditions is still an open question and a central point in further understanding of HSC maintenance (van Os et al., 2009; Pant et al., 2012).
To get new mechanistic insights about HSC maintenance and, in particular, on TP53 regulation, we constructed a Boolean network model describing quiescence and activation of HSCs.
Among dynamic modeling approaches, Boolean network models are used to describe the qualitative dynamic behavior of gene and protein regulatory networks (Kauffman, 1969; Hopfensitz et al., 2013). Boolean network models rely on the assumption of a switch-like behavior for each node (corresponding to a gene or a protein) that can be considered either active (1) or inactive (0) (Kauffman, 1969; Hopfensitz et al., 2013). Moreover, interactions are qualitatively described and summarized in Boolean functions, which do not require frequently unavailable kinetic parameters to be constructed. This peculiarity makes Boolean networks a powerful tool to describe large biological networks (Kauffman, 1969; Hopfensitz et al., 2013). Despite their simplicity, Boolean network models can faithfully represent the behavior of biological networks. In this context, they have been successfully applied to recapitulate different biological conditions such as cancer, aging, and senescence (Herrmann et al., 2012; Dahlhaus et al., 2016; Meyer et al., 2017; Siegle et al., 2018).
In the present work, we performed an extensive literature investigation to construct a Boolean network model recapitulating the regulation of HSC quiescence and cell cycle entry in the dependency on niche stimulation. Our aim is to unravel new mechanistic insights in the maintenance of hematopoietic stem cell quiescence, with particular attention to TP53 regulation. Strikingly, our model recapitulates the different statuses of the HSC, successfully describing dormant LT-HSC, activated ST-HSC, and cycling HSC as well as the progression from one to the other. Moreover, we could suggest a new general mechanism for TP53 regulation in homeostatic conditions. Finally, we tested our regulatory model by assessing its ability to recapitulate different knockout (K.O.) conditions. Here, the results support our model-based hypothesis of HSCs regulation.
Boolean networks are dynamic mathematical models to describe gene regulatory processes (Kauffman, 1969). These networks are defined as a set of n variables X = {x1, x2, …, xn}, xi ∈ 𝔹 and a corresponding set of transition functions . Boolean functions integrate information about regulatory interactions coming from literature statements or other data sources. Here, regulatory dependencies are expressed using logical operators (Kauffman, 1969). Each compound xi has its own Boolean function fi which defines whether xi is present or not. The state of the network at a specific point in time t is defined by a vector containing the assignments of all compounds xi within the network at this particular time step. Considering all possible combinations of assignments, this leads to a total number of 2n possible states. Using synchronous updates, transitions from states at one point in time to their successors, , are computed by updating all compounds at the same time. The transition of each compound is done by applying the corresponding Boolean function (Kauffman, 1969). Due to the deterministic nature of the state space, with synchronous updating, the model eventually enters a recurrent sequence of states called attractor (Kauffman, 1969). Attractors denote the long-term behavior of a system, and in a biological context, they are often related to phenotypes (Kauffman, 1993; Thomas and Kaufman, 2001). All states leading to the same attractor are part of its so-called basin of attraction (Hopfensitz et al., 2013; Schwab et al., 2020).
For the construction of the Boolean network model describing the maintenance of HSC in their niche, we performed extensive literature research collecting published papers from NCBI and Google Scholar. We collected data in the context of hematopoietic stem cell regulation. Moreover, the influences of niche factors on intrinsic regulators were considered. In the present work, studies from mouse models as well as from human HSCs when available were used. For all components of the model, different levels of regulation have been considered. Detailed descriptions about all interactions and resulting Boolean network functions can be found in Table 1. The java framework ViSiBooL was used for modeling (Schwab et al., 2017).

Table 1. Boolean functions for the HSC model.
The analysis of the dynamic behavior of the HSC model was performed by synchronous updates with the R package BoolNet (Müssel et al., 2010). Simulations were performed by applying the SAT-based attractor search algorithm.
First, analyses of the general network dynamics were performed by exhaustive attractor-search in the entire state space. Resulting attractors were compared to described HSC phenotypes. Here, one attractor was excluded from further analyses due to a complete lack of external stimulation. This does not correspond to a biologically plausible condition. In a further step, progressions from one attractor to the next were studied. We used the attractor pattern of each HSC phenotype as a starting state and altered the external niche stimuli to trigger the cascade. Finally, paths from one attractor to the next were examined. Altogether, we analyzed trajectories from:
• LT-HSC to ST-HSC by activating external cycling stimuli
• ST-HSC to cycling HSC by inactivating external quiescence stimuli
• LT-HSC to cycling HSC by combining activation of external cycling stimuli and inactivation of quiescence stimuli.
We simulated all possible single-compound perturbations (knockout and overexpression, see Supplementary Section 5). To validate the model, in silico intervention studies were performed and compared to known mouse phenotypes. Knockouts were obtained by fixing the state of desired node(s) constantly to 0. Dynamic analyses of the considered interventions were simulated exhaustively and attractors were analyzed.
All scripts to run the simulations are provided on github (https://github.com/sysbio-bioinf/HSC-boolean-network-model) and reported in the Supplementary Section 6.
We applied stochastic noise to our Boolean network model to evaluate the robustness—and thus the significance—of our model and the applied simulations. First, the ability of the network to compensate noise has been computed based on its attractors and their robustness against noise in the form of random bit flips in their basin of attraction. In our setup, we created a set of 1 million random initial states and computed the attractors for all of these states individually. Next, we created a perturbed copy of each drawn initial state and applied attractor search on all these perturbed states. For perturbation, we used random bit flips (assignments of selected nodes were toggled for 1 to 0 and vice versa). Flipping nodes in the networks' states is a common approach to simulate noise in Boolean networks (see, Qu et al., 2002; Aldana and Cluzel, 2003; Kauffman et al., 2004). To evaluate the robustness of the network, we compared the resulting attractor of each random state and its perturbed copy. Consequently, we could assess if this perturbation causes a switch of the attractors toward different basins. This procedure was exerted with toggling a different amount of random nodes to simulate an increasing amount of noise. We ran the simulation repeatedly, flipping 1, 2, and 3 nodes of each random state. For each number of bit flips, we repeated the simulation three times.
As a second measure for robustness, we analyzed the impact of noise in the model and compared it with randomly generated Boolean networks. Again, random bit flips are applied by adding noise to the network. Then, the corresponding successor state of the original one as well as of the perturbed state is computed. Then, the distance between the two successor states is measured using the normalized Hamming distance. The Hamming distance measures the number of differing bits across two state vectors. It can be formally described as . The normalization is done by dividing the measured Hamming distance by the number of bits in the vectors, . The distance is an indicator of the ability of the Boolean network to maintain its functionality under noisy conditions. A normalized Hamming distance of zero indicates that the mutation has no effect on evaluated network behavior. The procedure was repeatedly done for 1,000 randomly drawn states. The number of bit flips for the perturbation was set to one. Finally, results were compared to the ones obtained by 1,000 randomly generated Boolean networks. For this computerintensive test, we computed a p-value to evaluate if our null hypothesis “the hamming distance for the constructed network is larger or equal than the distance for the random networks” can be rejected, as follows : . Here HDrndi represents the mean Hamming distance over all states of one random network rndi and r is the total number of random networks tested. HDc represents the mean normalized Hamming distance over all measured states of the constructed network. We considered p < 0.001 to be significant. This test procedure is included in the BoolNet package (Müssel et al., 2010). The final aim of this analysis is to test the robustness of the constructed Boolean network model compared to a number of randomly generated ones.
The HSC regulation in the niche is of crucial relevance for the maintenance of the hematopoietic system. To unravel HSC maintenance mechanisms in homeostatic conditions, we constructed a Boolean network model based on extensive literature research (Table 1). A version of Table 1 endowed with references is available in our Supplementary Section 1.1.
Here we considered primary intrinsic regulators of HSC quiescence and niche external stimuli involved in their regulation. As intrinsic regulatory factors, we included cell cycle regulators (such as BMI1, MYC protoncogene (cMyc), TP53 together with cyclin-dependent kinases inhibitors (CDKN) and cyclins), proliferative pathways involving phosphoinositide 3-kinase (PI3K) and rat sarcoma (RAS), regulators of ROS, regulators of apoptosis and all potential cross-regulation among them. Furthermore, we also included a node describing autophagy in light of possible enlargement of our work toward aging studies (Ho et al., 2017). Extrinsic stimuli from the niche have been summarized as quiescence signals able to induce cell cycle inhibitors, or as cycling signals activating the RAS/PI3K axis. In total, the network consists of 37 nodes and 78 regulatory interactions (Figure 1).
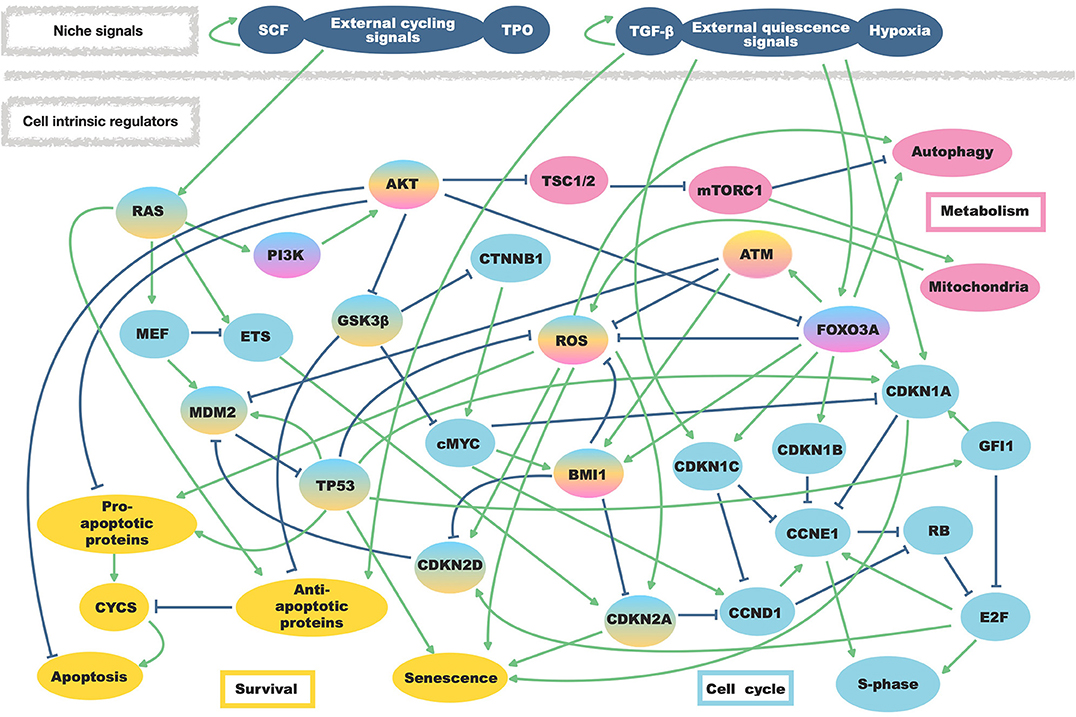
Figure 1. Regulatory interactions of the HSC model. External niche factors as well as regulations within the hematopoietic stem cells are summarized in one model. Regulatory activating interactions are depicted by arrows while bar-headed lines represent inhibiting regulations. Affected mechanisms and involved proteins are separated by colors.
First, we exhaustively simulated our model, obtaining four single state attractors. The network contains two external inputs (external quiescence and external cycling), resulting in four possible combinations. In accordance, each reached attractor shows a different combination of these nodes (Figure 2). Given that these input nodes represent niche stimuli, a condition with any stimulation from the environment is considered not realistic. Therefore, we do not consider this condition for further analyses.
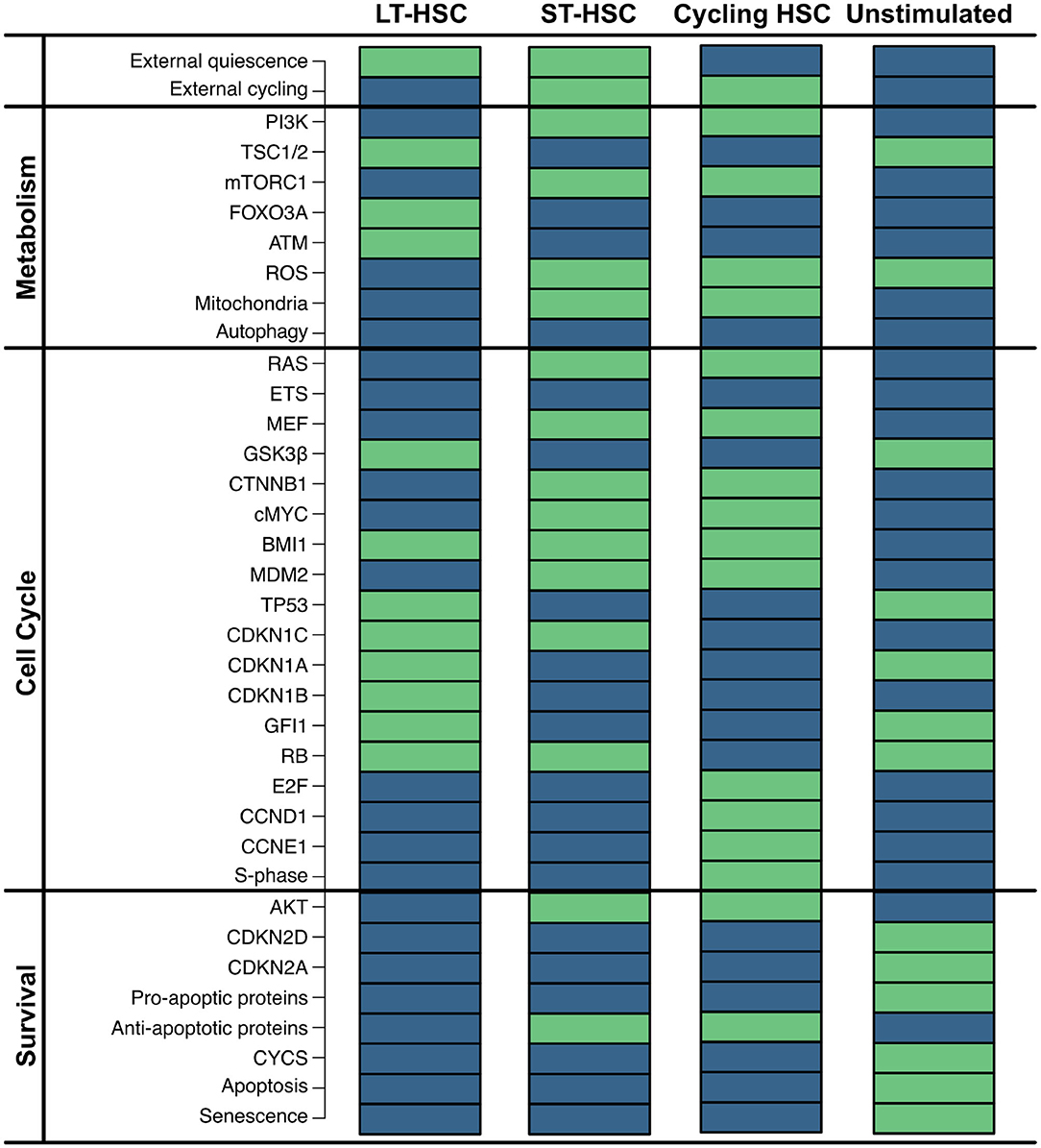
Figure 2. Impact of external stimulation. Simulation of the HSC model revealed four attractors that are dependent on the combinations of the two external inputs (external quiescence an external cycling). Components of the model are listed on the left separated into assigned pathways. The state of each component is depicted by colored rectangles. Green indicates active nodes, blue indicates inactive ones.
Attractor 1 is affected by quiescent external stimulation, while external cycling factors are absent. This attractor shows neither activation of the cell cycle [synthesis phase (S-phase) and cyclins] nor active metabolism or cell growth [mammalian target of rapamycin complex 1 (mTORC1) and ROS]. Instead, many inhibitors of the cell cycle [cyclin-dependent kinase inhibitor 1C (CDKN1C), cyclin-dependent kinase inhibitor 1A (CDKN1A), cyclin dependent kinase inhibitor 1B (CDKN1B), GFI1], as well as ROS and mTORC1 signaling [Forkhead box O3 (FOXO3A), ATM, Tuberous sclerosis protein 1/2 (TSC1/2)] are active. Furthermore, no apoptosis is observable, although TP53 is active. However, pro-apoptotic proteins and ROS that are required for apoptosis induction are inactive. This attractor pattern matches the behavior of a dormant LT-HSC which is considered as a reservoir of the HSCs and typically found in the endosteal region (Kopp et al., 2005; Sugiyama et al., 2006; Geiger et al., 2007; Jones and Wagers, 2008; Wagner et al., 2008; Guerrouahen et al., 2011). The attractor pattern was compared to the expression levels of quiescent HSCs. Here, low ROS (Chen et al., 2008, 2009; Ludin et al., 2014), AKT/mTORC1 (Jang and Sharkis, 2007; Chen et al., 2008, 2009; Ludin et al., 2014; Rodgers et al., 2014; Cabezas-Wallscheid et al., 2017; Baumgartner et al., 2018), RAS (Geest and Coffer, 2009), and cMYC (Wilson et al., 2004; Forsberg et al., 2005; Cabezas-Wallscheid et al., 2017) have also been observed. Additionally, quiescence is described to be maintained by expression of FOXO3A and ATM (Suda et al., 2011; Bakker and Passegué, 2013; Ludin et al., 2014) as well as TP53 activity (Forsberg et al., 2005; Liu Y. et al., 2009; Pant et al., 2012).
Attractor 2 is affected by the activity of both external stimuli. Similarly to attractor 1, the cell cycle (S-phase) is not active based on these external conditions. Hence, this attractor also shows quiescent behavior. However, this time RAS/PI3K signaling as well as mTORC1, ROS, and cMYC are active. Moreover, in attractor 2, many cell cycle inhibitors (CDKN1A, CDKN1B, GFI1) and ROS regulators (FOXO3A and ATM) are inactive. In comparison to attractor 1, apoptosis is still inactive. This might be due to inactive TP53 and the activation of the anti-apoptotic protein. Attractor 2 resembles the phenotype of an ST-HSC. The observed attractor pattern is comparable with the expression levels of ST-HSC (Wilson et al., 2004; Forsberg et al., 2005; Jang and Sharkis, 2007; Chen et al., 2008, 2009; Geest and Coffer, 2009; Liu Y. et al., 2009; Ludin et al., 2014; Rodgers et al., 2014; Cabezas-Wallscheid et al., 2017; Baumgartner et al., 2018). Moreover, in our attractor, the main cell cycle inhibitor CDKN1C is still active. It is also consistent with experimental observations indicating CDKN1C as the crucial regulator of quiescence in HSC (Umemoto et al., 2005; Matsumoto et al., 2011; Tesio and Trumpp, 2011; Zou et al., 2011) and is maintained by several quiescence-inducing external stimuli (Scandura et al., 2004; Qian et al., 2007; Blank and Karlsson, 2015).
The presence of cycling stimulation regulates attractor 3. Here, proliferative components such as cyclins are active, while cell cycle inhibitors are inactive. Thus, a proliferative phenotype can be assumed (Orford and Scadden, 2008; Pietras et al., 2011).
To sum up, our model is able to recapitulate the three main phenotypes observed for HSC, which we summarized again in Table 2. A version of Table 2 with references for each phenotype describes is provided in the Supplementary Section 1.2. Moreover, we are able to show that intrinsic stem cell behavior is affected by external stimulation. This indicates that complex niche-stem cell interactions determine the overall long-term behavior of HSCs.
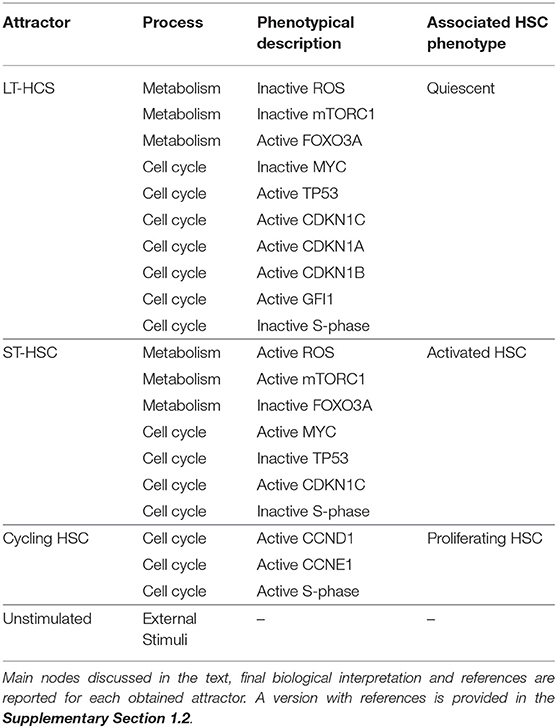
Table 2. Summary of analyzed attractors.
Above, we showed the presence of different individual phenotypes in the HSC model. Furthermore, we believe that distinct phenotypes depend on each other. To support this assumption, we investigated whether the single HSC entities transit into each other by alterations of external niche factors (Figure 3). This hypothesis is in line with previous findings indicating that LT-HSCs are activated and transit to ST-HSCs which further activated proliferation (Wilson and Trumpp, 2006; Wilson et al., 2007, 2009; Foudi et al., 2009; Pietras et al., 2011). Since we already excluded the possibility of no external stimulation, there are three remaining influences from the niche: only quiescent stimuli, only proliferative (cycling) stimuli, or both stimuli are present.
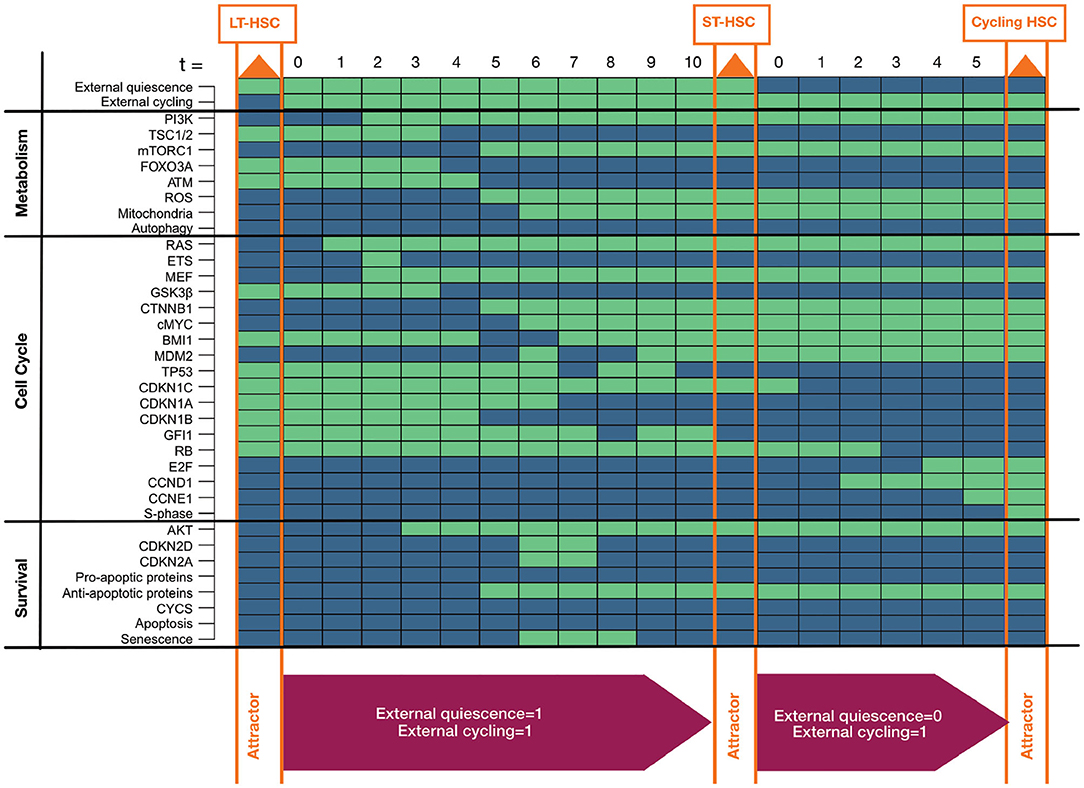
Figure 3. Progression toward awakening. The figure shows progression from LT to ST to cycling HSC depending on activation of external stimuli. Attractors are highlighted in orange in the progression. Activated external stimuli are depicted in magenta boxes below. Components of the model are listed on the left separated into assigned pathways. The state of each component is depicted by colored rectangles. Green indicates active components, blue indicates inactive ones.
Hence, we started our simulation by the LT-HSC attractor. We used this attractor state and further activated the external cycling input. This progression leads to a single state attractor describing the ST-HSC phenotype (Figure 3). Finally, form the obtained attractor pattern, we further inactivated the external quiescence node and simulated the progression toward the attractor again.
Our simulation of the awakening process started from the LT-HSC matched attractor. This attractor is characterized by active cell cycle inhibitors (CDKN1A, CDKN1B, CDKN1C, GFI1) and inactive RAS/PI3K pathways. The impact of both external stimuli was investigated. The progression toward the attractor is takes ten-time steps (Figure 3). First, the presence of external quiescent stimuli leads to up-regulation of cell cycle inhibitors (CDKN1C, CDKN1A, CDKN1B). However, the additional presence of the external cycling stimuli leads to a stabilization of RAS and PI3K signaling (time step zero to four). Furthermore, the stabilization of RAS and AKT induces the activation of cMYC by downregulation of GSK3β. In turn, cMYC leads to loss of cell cycle inhibitor CDKN1A (time step three to seven).
On the other hand, AKT activity induces mTORC1 signaling and represses FOXO3A (time steps three to seven). Thus, activation of mTORC1 directly activates mitochondria and the production of ROS. ROS activation is further sustained by the inhibition of FOXO3A and, consequently, ATM (time steps three to seven). Loss of ATM in the absence of cyclin-dependent kinase inhibitor 2D (CDKN2D) stabilizes MDM2 that, in turn, downregulates TP53 and its downstream target GFI1 (time steps six to ten). Stabilization of active MDM2 is further maintained by activation of myeloid elf-1-like factor (MEF) in response to RAS activation (from time step two). The finally reached attractor is the one that was previously connected to an ST-HSC. This cascade, including both external niche factors, indicates a destabilization of TP53 based on the activity of RAS/AKT and their downstream targets. Finally, starting from the ST-HSC attractor, we simulated our system by removing external quiescence stimuli. This time the system reaches a cycling HSC phenotype in five-time steps. Due to lack of external quiescence input, the phenotype switches toward further evolution of states leading to entry in the cell cycle. In fact, the lack of active cell cycle inhibitors leads to regulation of Retinoblastoma protein (RB), Cyclin D (CCND1) and Cyclin E (CCNE1), and finally to activation of the S-phase entry (time steps one to four) and thus proliferation.
Further, we considered the possibility that a highly quiescent HSC can be directly driven to enter the cell cycle. Hence we considered activation of external cycling stimulation starting from the LT-HSC attractor. The progression toward the cycling attractor presents a similar cascade, as shown in Figure 3, and is reported and briefly discussed in the Supplementary Section 2.
Summarizing these simulations, our model is not only able to recapitulate the different phenotypes of an HSC (Figure 2) but also shows that they depend on each other (Figure 3 and Supplementary Section 2). This indicates a dependency of HSC behavior from niche factors. However, we do not want to underestimate the impact of intrinsic rewiring regulations. The switching cascade between HSC entities that mTORC1 signaling and ROS regulation play a significant role in reasserting the final phenotypes. Besides, we observed that different mechanisms could govern the quiescence status of HSC, making them more or less reactive to external stimuli (Figure 3). Starting from an ST-HSC phenotype, the progression toward a cycling HSC happens in fewer time steps than considering an LT-HSC (Figure 3 and Supplementary Section 2). The results of our progressions are in accordance with the fact that ST-HSCs are thought to be more active HSCs: they preserve a quiescent state but can enter the cell cycle (Wilson et al., 2004, 2007, 2009; Foudi et al., 2009).
TP53 activity is known to be downregulated by MDM2. Furthermore, MDM2 expression is triggered by TP53 itself, setting a negative feedback loop in its own regulation (Jones et al., 1995; Montes de Oca Luna et al., 1995; Ringshausen et al., 2006). However, we assume that this general mechanism is somehow uneven in HSCs to allow TP53 activity and maintenance of quiescence.
In the previous section, we showed progression toward different HSC phenotypes (Figure 3 and Supplementary Section 2). In our simulation we observed alterations in the TP53 regulation during the cascades leading to the different attractors. In fact, in LT-HSC the activity of TP53 is maintained through a FOXO3A-ATM axis that destabilizes MDM2 activity. In ST-HSC, instead, external cycling signals cause an up regulation of the RAS/PI3K axis. This activation has two major effects that down regulate TP53. First, AKT activation causes a down regulation of FOXO3A and ATM (Figure 3, time steps three to seven). Second, RAS activation upregulates MEF, a transcriptional activator of MDM2. As a result, MDM2 and TP53 as well as its downstream target the cell cycle inhibitor GFI1 are downregulated. Hence, we believe that TP53 activity in steady state conditions is balanced by a crosstalk of intrinsic and extrinsic factors, finely tuning its activity in regulating HSC quiescence. This general regulatory hypothesis will be further discussed in the following section.
The hypothesized mechanism of TP53 regulation was further investigated by performing in silico knockouts. In the following, we compare our interventions to phenotypes of mouse models. To verify our model's prediction power, we assessed general long-term behavior and its impact on TP53.
FOXO3A and ATM are two significant regulators of ROS in quiescent HSCs (Bakker and Passegué, 2013; Ludin et al., 2014). Mouse models for FOXO3A knockout (Tothova and Gilliland, 2007; Tothova et al., 2007; Yalcin et al., 2008; Maryanovich et al., 2012) and ATM knockout (Ito et al., 2004, 2006; Maryanovich et al., 2012) show increased ROS, CDKN2D, and CDKN2A levels in the HSC compartment leading to senescence and impaired survival. This leads impaired self-renewal and repopulation ability of the LT-HSCs. However, it is still possible to observe a normal differentiation in hematopoiesis (Ito et al., 2004, 2006; Tothova and Gilliland, 2007; Tothova et al., 2007; Yalcin et al., 2008; Maryanovich et al., 2012). The observed phenotypes within the mouse models could be matched to our attractors (Figure 4). LT-HSC shows senescence and apoptosis activation and increased ROS levels due to the activation of CDKN2A and CDKN2D. In particular, activation of CDKN2D sustains TP53 activation. The latter, in the presence of ROS, can trigger apoptosis and senescence. ST-HSCs are less affected and do not undergo apoptosis due to the activation of anti-apoptotic proteins and already inactive FOXO3A and ATM. Also, the cycling HSC attractor is not altered. This result is also in line with our cascades showed in Figure 3. There it is shown that FOXO3A and ATM are already inactivated in ST-HSC after activation of PI3K/AKT signaling. To summarize, in the FOXO3A and ATM knockouts, our attractors show significant perturbations in the LT-HSC population. Here, activation of ROS, CDKN2A, CDKN2D leads depletion of the LT-HSC population. However, the other two attractors, namely the one representing ST-HSC and cycling HSC are not affected by the perturbation. This might explain the normal hematopoiesis seen in the FOXO3A and ATM mouse models (Ito et al., 2004; Yalcin et al., 2008). Moreover, in the FOXO3A depleted mice, it was shown that the depletion of the HSCs comes from the LT-HSC population (Yalcin et al., 2008), supporting our population-based results.
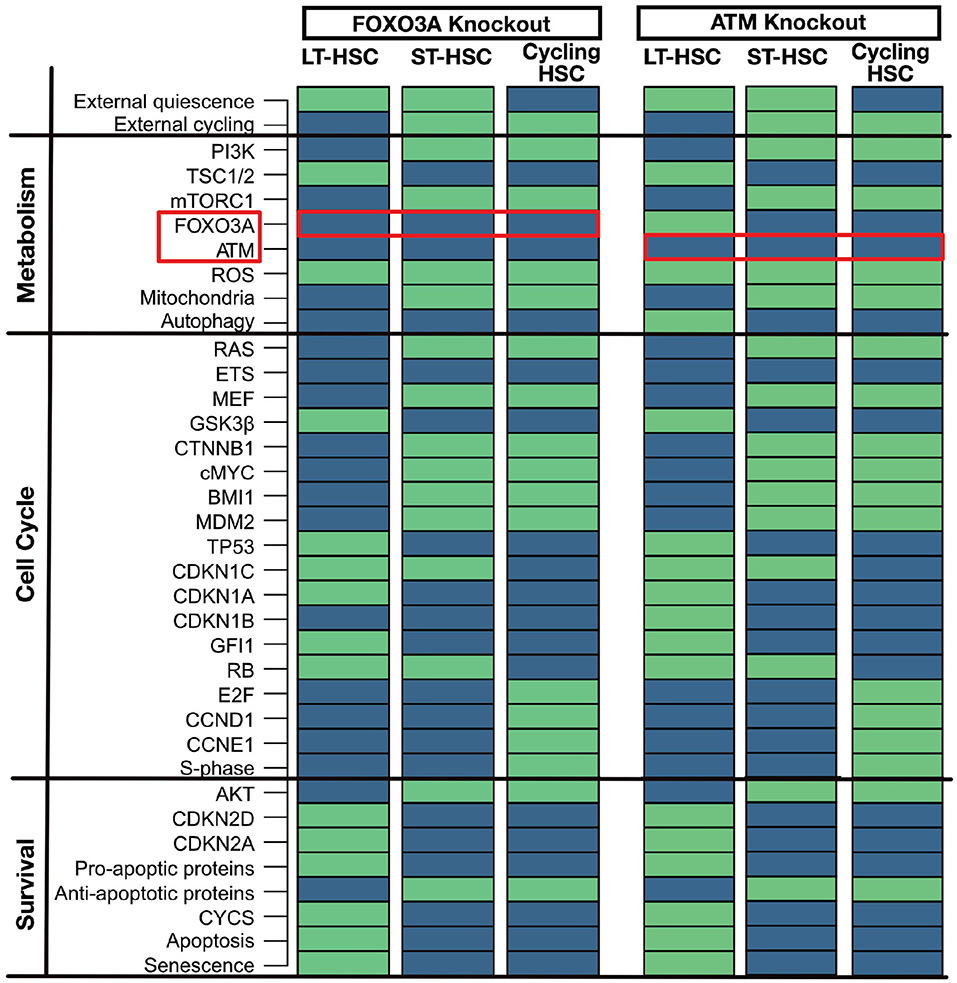
Figure 4. Similar phenotypes of ATM and FOXO3A knockout. An in silico knockout of FOXO3A or ATM increases ROS levels but only induces apoptosis and senescence in the LT-HSC population. Proteins are listed on the left with their pathway affiliations. The state of each protein is represented by colored rectangles. Activity is represented in light green and inhibition in dark blue. For simplicity due to the highly similar phenotype of the two in silico knockouts only FOXO3A is reported.
BMI1 is a crucial regulator of self-renewal and is widely expressed in the HSC compartment. Therefore, the BMI1 knockout simulation shows a huge effect on the phenotype in mice leading to defect in self-renewal, depletion of the HSC compartment, and shortened life-span (Park et al., 2003; Liu J. et al., 2009; Rizo et al., 2009). This effect is accompanied by increased ROS levels and increased CDKN2A and CDKN2D expression, which finally induces apoptosis and senescence (Park et al., 2003; Liu J. et al., 2009; Rizo et al., 2009). These huge implications are also observable in the attractor pattern. Here, BMI1 loss induces a substantial impairment of cell cycle entry for the HSC in all obtained attractors (Figure 5). All attractors show senescence, and the LT-HSC attractor shows apoptosis additionally. The attractor for the cycling HSC does not show activation of S-phase. Based on our cascades shown in Figure 3, loss of BMI1 upregulates CDKN2A and CDKN2D, causing impaired quiescence and activation maintenance among the three phenotypes.
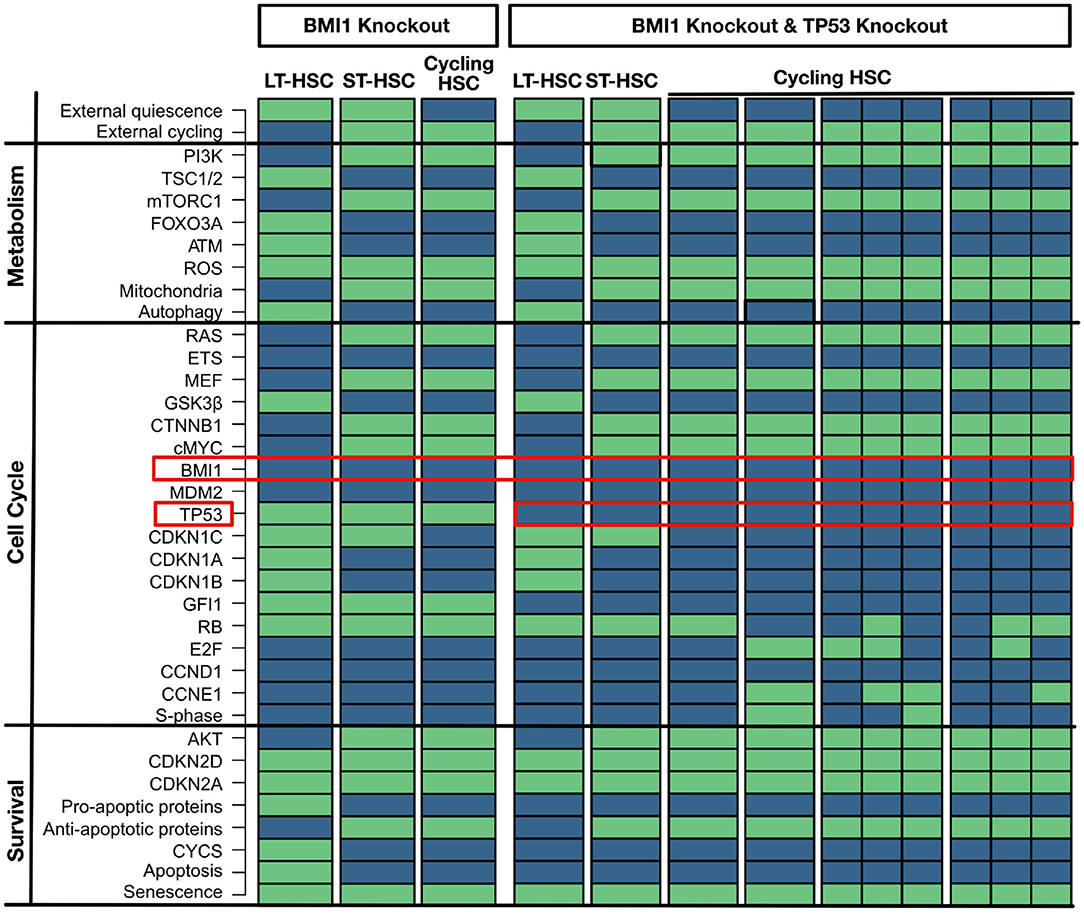
Figure 5. BMI1 loss impairs self-renewal (left part). An in silico knockout of BMI1 impairs cell cycle entry for all HSC populations. Proteins are listed on the left with their pathway affiliations. TP53 partially rescue a BMI1 knockout (right part). An in silico knockout of BMI1 and TP53 rescues a the impact of a loss of BMI1 by impairing apoptosis. Proteins are listed on the left with their pathway affiliations. The state of each protein is represented by colored rectangles. Activity is represented in light green and inhibition in dark blue.
Various possibilities to partially rescue the phenotype of BMI1 loss have been suggested. One of these suggestions is a loss of TP53 which impairs apoptosis (Akala et al., 2008; Warr et al., 2011). This behavior can also be seen in the attractor when TP53 is perturbed together with BMI1 (Figure 5). Again, these simulations show that an increase of ROS and CDKN2D activates TP53 and guide its activity toward pro-apoptotic behavior.
TP53 knockout has no major implications on hematopoiesis in mice (TeKippe et al., 2003; Liu Y. et al., 2009; Abbas et al., 2011; Pant et al., 2012). However, loss of GFI1 gives an advantage in competitive assay due to less regulated quiescent state (TeKippe et al., 2003; Liu Y. et al., 2009; Abbas et al., 2011; Pant et al., 2012). A matching attractor pattern is observable in the in-silico simulation with no significant induction of apoptosis (Figure 6). Moreover, even in our LT-HSC attractor GFI1 gets inactivated. The inactivation of GFI after TP53 was observed in our previous results in the transition between LT and ST-HSC. Hence, our LT-HSC TP53 knockout attractor matches now partially with the ST-HSC (Figure 2). Our attractors show also activation of ROS in line with literature results (Liu et al., 2008). Activation of ROS in HSCs has also been connected to a more active state, prone to enter the cell cycle (Bakker and Passegué, 2013; Ludin et al., 2014). Altogether, our results are following experimental observations on TP53 knockout models. After accessing the loss of TP53, we further analyzed the effects of its over stabilization in HSCs. For this reason, we selected another mouse model causing MDM2 downregulation via MEF deletion. MEF knockout mice show a stabilization of TP53 connected to an enhancement of quiescence (Lacorazza et al., 2006; Liu Y. et al., 2009). Likewise, attractor for cycling HSC showed enhanced quiescence due to loss of S-phase. In accordance also the ST-HSC attractor shows now TP53 and GFI1. The stabilization of TP53 affects the transition from one HSC phenotype to another, mainly by the maintenance of GFI1 activity. The unperturbed cascade shows that loss of GFI1 activity happens during the transition between LT- and ST-HSC (Figure 3). Hence, in our in silico simulations, stabilization of TP53 causes an enhanced quiescent phenotype (Figure 6).
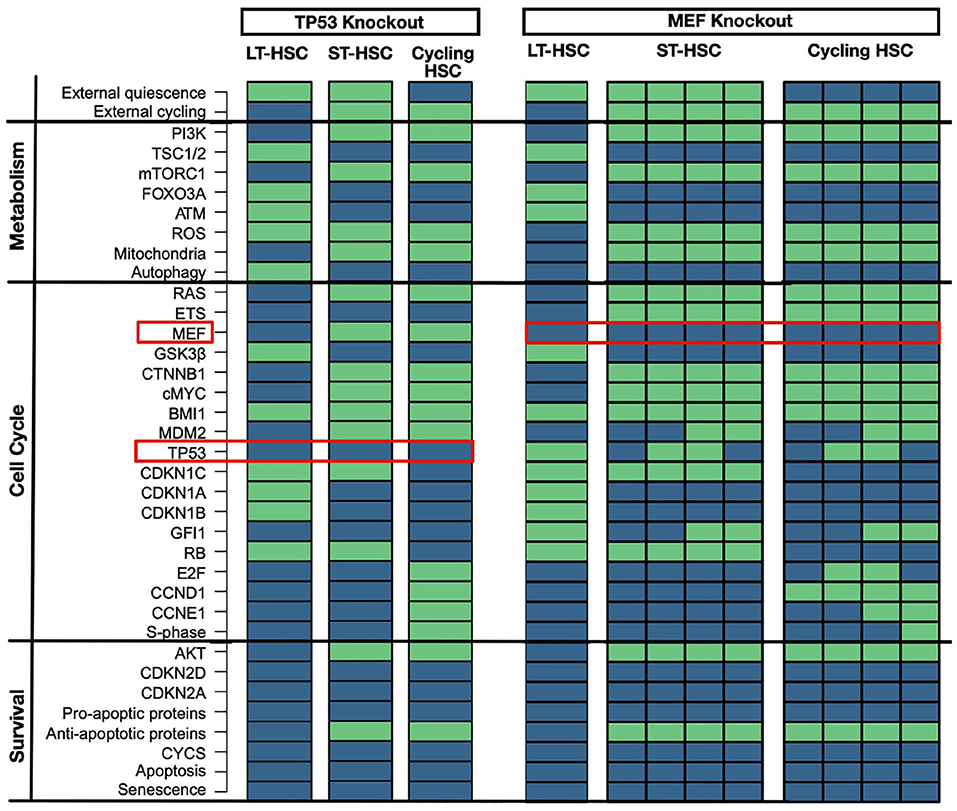
Figure 6. TP53 and MEF knockouts. TP53 knockout has minor impact on HSC populations. An in silico knockout of TP53 causes loss of GFI1 in LT-HSCs and increased ROS levels. ST-HSC and cycling ones are not impacted. Proteins are listed on the left with their pathway affiliations. The state of each protein is represented by colored rectangles. Activity is represented in light green and inhibition in dark blue. MEF knockout stabilizes TP53. An in silico knockout of MEF stabilized TP53 and enhanced quiescence. Here, ST-HSC and cycling HSCs show activity of GFI1 and cycling HSCs are impaired in entering S-phase. Proteins are listed on the left with their pathway affiliations. The state of each protein is represented by colored rectangles. Activity is represented in light green and inhibition in dark blue.
Finally, we showed that our model could recapitulate the phenotype of published mouse models. Moreover, perturbation results sustain our hypothesis concerning the general mechanism of regulation of TP53 in HSCs. When ATM, FOXO3A, or BMI1 are depleted, increased ROS and CDKN2D lead to activation of TP53. This effect switches its activity toward the induction of apoptosis and senescence. In homeostatic conditions, instead, they guarantee TP53 activity toward quiescence by downregulating ROS and CDKN2D. Our simulations also showed the relevance of the RAS/PI3K axis in down regulating TP53 when HSCs are activated. The loss of MEF down regulates MDM2 in ST- and cycling HSCs. This loss causes an impaired entry in the S-phase despite the presence of external cycling stimuli. Altogether, our results suggest a general mechanism of regulation of TP53 activity in HSCs. The described mechanism is summarized in Figure 7.
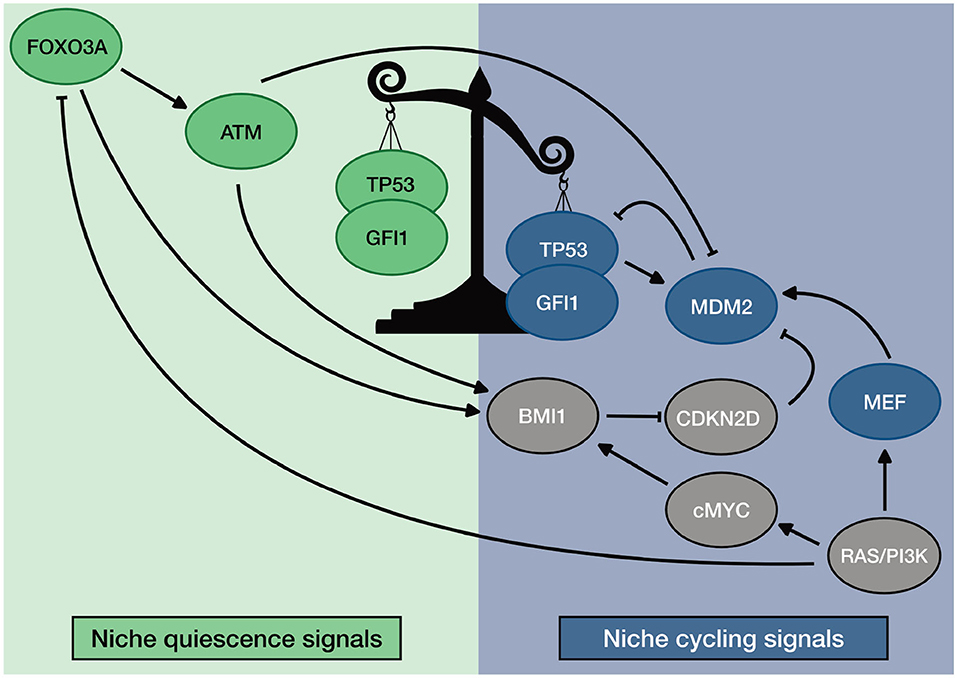
Figure 7. General description of TP53 regulation in HSC. Homeostatic activity of TP53 is promoted by ROS regulators ATM and FOXO3A. Unbalance of TP53 activity is promoted by MEF that stabilizes the transcription of MDM2. Furthermore, the unbalanced activity of TP53 is maintained by niche regulation: quiescence promoting environment favors ROS regulators; Cycling promoting environment promotes MEF expression.
We applied stochastic noise to our Boolean network model to evaluate the robustness—and thus the significance—of our model and the applied simulation. First, the robustness of the network has been computed based on its attractors. Their robustness is measured by the addition of noise by random bit flips in their basin of attraction. We ran the simulation repeatedly, flipping 1, 2, and 3 nodes of each random state. For each number of bit flips, we repeated the simulation three times. Results showed for equal attractors for perturbed and unperturbed states with a mean and standard deviation of 95% (±0.0003)/89% (±0.0001)/85% (±0.0005) for the simulation with 1/2/3 random bit flips per initial node respectively.
As a second measure for robustness, we analyzed the impact of noise on the state transitions in the model and compared it to randomly generated Boolean networks. As before, noise is simulated using random bit flips. We calculate the mean normalized Hamming distance between the successor states of the original state and perturbed copy. Smaller Hamming distance can be interpreted as a better ability to compensate noise. The aim of this analysis is to evaluate the transition robustness of the constructed Boolean network model compared to random networks. Results show a mean Hamming distance of 0.028 for the constructed HSC model. In contrast, the mean normalized Hamming distance over all random networks is 0.034. The calculated p-value is below 0.0001. Thus, we conclude that our established model is significantly more robust to noise than a set of randomly generated networks of the same size (Figure 8).
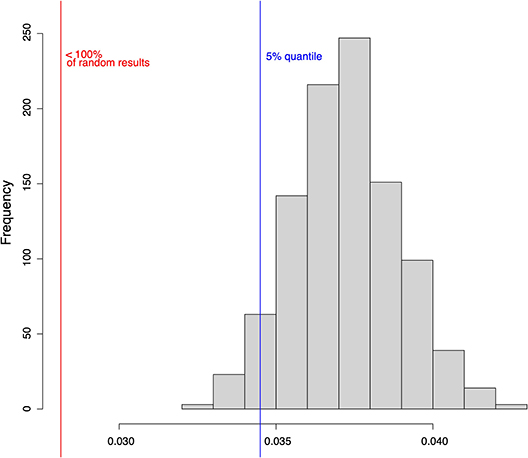
Figure 8. One thousand randomly drawn states of the HSC model were mutated by bit flip and their successor states were computed. The successor states of the mutated and the original states were then compared using the normalized Hamming distance (red line). The same analysis was performed for 1,000 randomly generated networks of the same size (histogram). The blue line shows the 5% quantile.
In the present work, we proposed an in silico Boolean network model depicting the unique wiring that guarantees the maintenance of HSCs in their niche. Our model is able to describe the quiescence mechanism of LT-HSC and ST-HSC. Moreover, we showed the progression toward entry in the cell cycle with respect to changes in external conditions. Additionally, our model strikingly recovered several phenotypes observed in HSCs of knockout mouse models for major intrinsic regulators. This evaluation might be quite challenging under in vitro conditions where both isolation and maintenance of HSC require much effort. Identifying methods of isolation and maintenance of HSCs is still challenging (Passegué et al., 2005; Weissman and Shizuru, 2008; Challen et al., 2009; Rossi et al., 2011; Frisch and Calvi, 2014; Jiang et al., 2018; Kobayashi et al., 2019). Hence, the possibility of having an in silico mathematical model for simulating the behavior of HSC is of great interest.
External stimuli coming from the niche were summarized as either quiescent or cycling inputs. This choice might be considered an over-simplification of the complex network of signals surrounding the HSC. However, it has been shown that, due to its crucial relevance, HSC maintenance is redundantly regulated by multiple sides. Exemplarily, CDKN1C which is considered the central gatekeeper of quiescence (Passegué et al., 2005; Umemoto et al., 2005; Matsumoto et al., 2011; Tesio and Trumpp, 2011; Zou et al., 2011), is regulated by a multiplicity of endosteal stimuli including TGF-β, TPO and hypoxia (Scandura et al., 2004; Qian et al., 2007; Eliasson et al., 2010; Blank and Karlsson, 2015). Following the idea of redundancy, the loss of hypoxic conditions will not lead to a complete lack of CDKN1C activity (Eliasson et al., 2010). Also, the loss of single developmental pathways has been shown not to affect HSC maintenance, indicating again that a multiplicity of signals conquer the task (Mancini et al., 2005; Maillard et al., 2008; Gao et al., 2009; Hofmann et al., 2009; Kabiri et al., 2015; Oostendorp, 2015). Nevertheless, dissecting too much single influence of each external signal might lead to contradictory results. For example, SCF and TPO have been associated with quiescence in vivo (Qian et al., 2007; Yoshihara et al., 2007; Thorén et al., 2008). However, these factors are commonly used to sustain the proliferation of HSC in vitro (Ema et al., 2000; Gammaitoni et al., 2003). This differential behavior suggests the possibility that cross-regulation among quiescence signals exists. It has been shown that TGF-β signaling, highly present in the endosteal niche (Yamazaki et al., 2009; Blank and Karlsson, 2015), is able to repress the cytokine-induced activation of PI3K by inhibiting the formation of lipid rafts (Yamazaki et al., 2009). Besides, other mathematical modeling approaches successfully described HSC differentiation by suggesting signaling cumulative environments (Roeder and Lorenz, 2006; Roeder et al., 2008; Glauche et al., 2011). Therefore, it is reasonable to consider an environment with predominant quiescence or cycling stimuli.
Besides single predominant stimuli, we also considered the co-presence of both quiescence and cycling inducing stimuli as sort of gray zones with a balance between the two signals. This consideration is actually in line with different niche models that suggest activated HSCs to be found in regions of the niche that are boundaries between two different environments (Murphy et al., 2005; Wilson and Trumpp, 2006). In this direction Murphy et al., describe HSCs able to self-renew and proliferate to be found in the connection between endosteal and stromal area (Murphy et al., 2005). These HSCs would then sense both quiescence signals coming from the endosteal niche and proliferation stimuli coming from the stroma (Murphy et al., 2005). Also, Trumpp et al. hypothezed a similar regulation, describing the presence of a reservoir quiescent compartment with specialized cells maintaining LT-HSC (Wilson and Trumpp, 2006; Trumpp et al., 2010). Again they characterize a “self-renewing endosteal niche” as the one present at the boundaries of this region when HSCs can get activated and more responsive (Wilson and Trumpp, 2006; Trumpp et al., 2010).
Nevertheless, our results suggest a balance of external signals that sets the instructions for the intrinsic rewiring of the HSC. This means that potentially an LT-HSC could be found in any region having quiescence inducing signals. Thereby expanding the possibility of finding niches for quiescence also in different regions of the bone marrow. This consideration is in accordance to further observations that describe the presence of quiescent HSCs for example in lowly oxygenized regions of sinusoids (Itkin et al., 2016; Pinho and Frenette, 2019).
Different phenotypic characterizations of LT- and ST-HSC describing differential expression patterns have been published (Wilson et al., 2004; Forsberg et al., 2005; Passegué et al., 2005; Jang and Sharkis, 2007). Our attractors could match the primary expression and activity features that identify the two HSC phenotypes. Moreover, we also suggested two different mechanisms of maintaining quiescence in these two subpopulations. Our model indicates that LT-HSC quiescence is preserved by tight regulation of ROS levels from a combination of intrinsic factors induced by external stimuli. It has been observed that keeping oxidative stress low in quiescent HSC is fundamental for preserving their integrity and survival (Jang and Sharkis, 2007; Suda et al., 2011; Ludin et al., 2014). In fact, despite the activity of TP53 and lack of survival stimuli, we do not detect activation of apoptosis due to the highly hampering of cellular stress.
On the other hand, it has been shown that HSCs have a high expression of induced myeloid leukemia cell differentiation protein (MCL1), an anti-apoptotic protein activated in the presence of survival stimuli (Opferman et al., 2005). However, this study was performed on the entire subset of HSC and progenitor cells. Similarly, if we consider our whole set of attractors, we also observe an expression of anti-apoptotic proteins. Sustaining our hypothesis of maintenance of LT-HSC, Kobayashi et al. recently published a study describing the ideal setting for in vitro maintenance of quiescent HSCs (Kobayashi et al., 2019). In their work, the authors actually show that hypoxic condition in the presence of residual cytokine stimuli sets the best environment for the maintenance of quiescence (Kobayashi et al., 2019). Our dynamic simulations also lead to a single state attractor depicting a cycling HSC. Initially, this result seems in contradiction with previously published mammalian cell cycle models (Fauré et al., 2006; Tanaka et al., 2017; Diop et al., 2019). In these studies, proliferating cells are represented by cycling attractors. However, we were interested in modeling exit from Gap phase zero (G0) and entering the S-phase. Hence, we selected cell cycle regulators involved in this switch. If we consider only this regulators, also previously published cell cycle models would show a single state attractor (Fauré et al., 2006; Tanaka et al., 2017; Diop et al., 2019).
Moreover, we inserted our starting states of cell cycle regulators from our cycling HSC attractor in the Fauré et al. model and obtained a cyclic attractor (see Supplementary Section 3). Altogether, this underlines the correctness of our approach and the extendability of our network model.
Notably, we could also show that LT-HSC and ST-HSC have different responsiveness in terms of time to enter the cell cycle once stimulated. This is also in line with the observation that actually ST-HSC is more reactive and can efficiently enter the cell cycle (Forsberg et al., 2005; Murphy et al., 2005; Wilson et al., 2007, 2008). Our hypothesis or regulatory wiring is further encouraged given the predictive power that our model has toward a variety of in silico knockouts that can faithfully recapitulate the phenotype of analog mouse models.
Furthermore, based on our simulation of the progression from a quiescent cell toward the entry in the cycle, we can suggest a mechanism for TP53 homeostatic regulation in HSCs. Here, we propose that the awakening of the HSC follows the initial activation of cellular metabolism. This will then induce the rewiring of the intrinsic network of ROS sensors (FOXO3A and ATM) and finally lead to a loss of cell cycle regulators connected to quiescence such as TP53, CDKN1A, and CDKN1B. In the end, with loss of quiescence signals, the HSC can rapidly enter the cell cycle. Stress sensors have been shown to be fundamental for regulating HSC quiescence (Suda et al., 2011; Bakker and Passegué, 2013; Ludin et al., 2014). The expression of FOXO3A is directly connected to the hypoxic environment, and this leads in turn to the upregulation of active ATM (Yalcin et al., 2008; Suda et al., 2011). The last, besides being an independent regulator of cellular stress, is also involved in the activation of TP53 (Yalcin et al., 2008; Suda et al., 2011; Bakker and Passegué, 2013; Ludin et al., 2014). Thus, our simulations indicate that in LT-HSC ATM is responsible for the regulation of active TP53. This activation is actually new to the HSC maintenance context. This is further enforced from the fact that ATM seems to be required for TP53 immediate regulation of irradiation in quiescent HSC (Mohrin et al., 2010; Kosan and Godman, 2016). Besides its well-defined role in DNA damage response, ATM has already been shown to regulate activation of TP53 under physiological conditions in the context of homeostatic cellular metabolism outside of the HSC context (Armata et al., 2010). Here, It has been suggested that activation of TP53 by ATM is involved in the reduction of ROS (Armata et al., 2010). When instead RAS and PI3K are activated by external stimuli, FOXO3A is downregulated by AKT/mTORC1 signaling potentially also downregulating ATM. This regulation is in accordance with the fact that already in ST-HSC, FOXO3A is excluded from the nucleus (Yalcin et al., 2008). Consequently, in this condition, TP53 is mainly regulated by the MEF/MDM2 and the BMI1/CDKN2D axis (Liu Y. et al., 2009; Abbas et al., 2010; Warr et al., 2011). These regulators set the downregulation of TP53 and GFI1 in the progression toward the ST-HSC phenotype. In accordance, TP53 is downregulated in ST-HSCs (Forsberg et al., 2005; Jang and Sharkis, 2007; Pant et al., 2012). Nevertheless, our model also maintains the hypothesis that TP53 activity is context-dependent (Asai et al., 2011) as shown by our knockout simulations. To further analyze the balanced regulation of TP53 under homeostatic conditions, we performed loss of interaction mutation simulations (see Supplementary Section 4). Here, we could show the loss of regulatory axes causing disruption of the original wildtype attractors, when removing the interaction between ATM and MDM2, MEF, and MDM2 or both. In addition, constitutive FOXO3A/ATM or RAS/PI3K in absence of external cycling signals can recapitulate the TP53 activation status (see Supplementary Section 4).
Our model of progression is also in accordance with the idea that mTORC1 signaling triggers cellular metabolism and prompts the HSC to enter the cell cycle (Chen et al., 2008, 2009; Rodgers et al., 2014; Baumgartner et al., 2018). Hence, we provide an overall mechanism of HSC awakening and a new general suggestion of TP53 regulation in homeostatic conditions. To our best knowledge, no previous models could show neither HSC maintenance nor a general mechanistic explanation of TP53 regulation able to summarize observed phenotypes in different experimental conditions.
In addition to our dynamic simulations, we also investigated the robustness of our established Boolean network. Biological networks are considered to be robust toward perturbations. This means that they can adapt to environmental changes, and their functions are resistant to random noise (Kitano, 2002; Greenbury et al., 2010; Graudenzi et al., 2011; Barabási, 2016; Schwab et al., 2020). Hence, we also evaluated the robustness of our network against noise. We both investigated the shift of attractor basin after perturbation and the normalized Hamming distance after bit flip perturbation. In the latter, we compared the performance of our models to one of randomly generated networks of the same size. Equivalent approaches to the ones we presented have been already applied to investigate the robustness of Boolean networks (Joo et al., 2018; Siegle et al., 2018). Altogether, we could show that our model is robust toward random noise.
In conclusion, we established a model able to faithfully recapitulate the regulation of HSC maintenance in the presence of external niche stimuli. Also, we simulated in silico the process of awakening of the HSC, giving a mechanistic and overall picture of the process. Further, we suggest a new general regulatory wiring responsible for the regulation of TP53 activity in HSCs. Moreover, we further validated our model by testing its predictive power in a variety of mouse models.
All datasets generated for this study are included in the article/Supplementary Material. All data are deposited in github.
HK, SK, and JS devised the project. NI and SK established the model. NI, SK, and JS performed simulations and were responsible for the visualization. All authors discussed the results and contributed to the final manuscript. HK provided funding and supervised the project.
HK acknowledges funding from the German Federal Ministery of Education and Research (BMBF) e:MED confirm (id 01ZX1708C). Furthermore, HK acknowledges funding from the German Science Foundation [DFG, 217328187 (SFB 1074) and 288342734 (GRK HEIST)].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.00848/full#supplementary-material
Abbas, H. A., Maccio, D. R., Coskun, S., Jackson, J. G., Hazen, A. L., Sills, T. M., et al. (2010). Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 7, 606–617. doi: 10.1016/j.stem.2010.09.013
Abbas, H. A., Pant, V., and Lozano, G. (2011). The ups and downs of p53 regulation in hematopoietic stem cells. Cell Cycle 10, 3257–3262. doi: 10.4161/cc.10.19.17721
Adolfsson, J., Borge, O. J., Bryder, D., Theilgaard-Mönch, K., Åstrand-Grundström, I., Sitnicka, E., et al. (2001). Upregulation of Flt3 expression within the bone marrow lin?Sca1+c-kit+ stem cell compartment is accompanied by loss of self-renewal capacity. Immunity 15, 659–669. doi: 10.1016/S1074-7613(01)00220-5
Akala, O. O., Park, I.-K., Qian, D., Pihalja, M., Becker, M. W., and Clarke, M. F. (2008). Long-term haematopoietic reconstitution by Trp53−/−p16Ink4a−/−p19Arf−/− multipotent progenitors. Nature 453, 228–232. doi: 10.1038/nature06869
Aldana, M., and Cluzel, P. (2003). A natural class of robust networks. Proc. Natl. Acad. Sci. U.S.A. 100, 8710–8714. doi: 10.1073/pnas.1536783100
Armata, H. L., Golebiowski, D., Jung, D. Y., Ko, H. J., Kim, J. K., and Sluss, H. K. (2010). Requirement of the atm/p53 tumor suppressor pathway for glucose homeostasis. Mol. Cell. Biol. 30, 5787–5794. doi: 10.1128/MCB.00347-10
Asai, T., Liu, Y., Bae, N., and Nimer, S. D. (2011). The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J. Cell. Physiol. 226, 2215–2221. doi: 10.1002/jcp.22561
Athanasiadis, E. I., Botthof, J. G., Andres, H., Ferreira, L., Lio, P., and Cvejic, A. (2017). Single-cell RNA-sequencing uncovers transcriptional states and fate decisions in haematopoiesis. Nat. Commun. 8, 1–11. doi: 10.1038/s41467-017-02305-6
Bakker, S. T., and Passegué, E. (2013). Resilient and resourceful: genome maintenance strategies in hematopoietic stem cells. Exp. Hematol. 41, 915–923. doi: 10.1016/j.exphem.2013.09.007
Baumgartner, C., Toifl, S., Farlik, M., Halbritter, F., Scheicher, R., Fischer, I., et al. (2018). An erk-dependent feedback mechanism prevents hematopoietic stem cell exhaustion. Cell Stem Cell 22, 879–892. doi: 10.1016/j.stem.2018.05.003
Blank, U., and Karlsson, S. (2015). TGF-β signaling in the control of hematopoietic stem cells. Blood 125, 3542–3550. doi: 10.1182/blood-2014-12-618090
Bradford, G. B., Williams, B., Rossi, R., and Bertoncello, I. (1997). Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp. Hematol. 25, 445–453.
Busch, K., Klapproth, K., Barile, M., Flossdorf, M., Holland-Letz, T., Schlenner, S. M., et al. (2015). Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518, 542–546. doi: 10.1038/nature14242
Cabezas-Wallscheid, N., Buettner, F., Sommerkamp, P., Klimmeck, D., Ladel, L., Thalheimer, F. B., et al. (2017). Vitamin a-retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell 169, 807–823. doi: 10.1016/j.cell.2017.04.018
Challen, G. A., Boles, N., Lin, K.-Y. K., and Goodell, M. A. (2009). Mouse hematopoietic stem cell identification and analysis. Cytometry A, 75A, 14–24. doi: 10.1002/cyto.a.20674
Chen, C., Liu, Y., Liu, R., Ikenoue, T., Guan, K.-L., Liu, Y., et al. (2008). TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 205, 2397–2408. doi: 10.1084/jem.20081297
Chen, C., Liu, Y., Liu, Y., and Zheng, P. (2009). The axis of mTOR-mitochondria-ROS and stemness of the hematopoietic stem cells. Cell Cycle 8, 1158–1160. doi: 10.4161/cc.8.8.8139
Cheshier, S. H., Morrison, S. J., Liao, X., and Weissman, I. L. (1999). In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 96, 3120–3125. doi: 10.1073/pnas.96.6.3120
Coskun, S., and Hirschi, K. K. (2010). Establishment and regulation of the HSC niche: roles of osteoblastic and vascular compartments. Birth Defects Res. 90, 229–242. doi: 10.1002/bdrc.20194
Dahlhaus, M., Burkovski, A., Hertwig, F., Müssel, C., Volland, R., Fischer, M., et al. (2016). Boolean modeling identifies Greatwall/MASTL as an important regulator in the AURKA network of neuroblastoma. Cancer Lett. 371, 79–89. doi: 10.1016/j.canlet.2015.11.025
Diop, O., Tourniel, L., and Fromion, V. (2019). “Summarizing complex asynchronous Boolean attractors, application to the analysis of a mammalian cell cycle model,” in 2019 18th European Control Conference (ECC), ed F. Garofolo (Naples), 1677–1682. doi: 10.23919/ECC.2019.8795712
Eliasson, P., Rehn, M., Hammar, P., Larsson, P., Sirenko, O., Flippin, L. A., et al. (2010). Hypoxia mediates low cell-cycle activity and increases the proportion of long-term-reconstituting hematopoietic stem cells during in vitro culture. Exp. Hematol. 38, 301–310.e2. doi: 10.1016/j.exphem.2010.01.005
Ema, H., Takano, H., Sudo, K., and Nakauchi, H. (2000). In vitro self-renewal division of hematopoietic stem cells. J. Exp. Med. 192, 1281–1288. doi: 10.1084/jem.192.9.1281
Fauré, A., Naldi, A., Chaouiya, C., and Thieffry, D. (2006). Dynamical analysis of a generic Boolean model for the control of the mammalian cell cycle. Bioinformatics 22, e124–e131. doi: 10.1093/bioinformatics/btl210
Fleming, W., Alpern, E., Uchida, N., Ikuta, K., Spangrude, G., and Weissman, I. (1993). Functional heterogeneity is associated with the cell cycle status of murine hematopoietic stem cells. J. Cell Biol. 122, 897–902. doi: 10.1083/jcb.122.4.897
Forsberg, E. C., Prohaska, S. S., Katzman, S., Heffner, G. C., Stuart, J. M., and Weissman, I. L. (2005). Differential expression of novel potential regulators in hematopoietic stem cells. PLoS Genet. 1:e28. doi: 10.1371/journal.pgen.0010028
Foudi, A., Hochedlinger, K., Van Buren, D., Schindler, J. W., Jaenisch, R., Carey, V., et al. (2009). Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 27, 84–90. doi: 10.1038/nbt.1517
Frisch, B. J., and Calvi, L. M. (2014). Hematopoietic stem cell cultures and assays. Methods Mol. Biol. 1130, 315–324. doi: 10.1007/978-1-62703-989-5_24
Gammaitoni, L., Bruno, S., Sanavio, F., Gunetti, M., Kollet, O., Cavalloni, G., et al. (2003). Ex vivo expansion of human adult stem cells capable of primary and secondary hemopoietic reconstitution. Exp. Hematol. 31, 261–270. doi: 10.1016/S0301-472X(02)01077-9
Gao, J., Graves, S., Koch, U., Liu, S., Jankovic, V., Buonamici, S., et al. (2009). Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell 4, 548–558. doi: 10.1016/j.stem.2009.03.015
Geest, C. R., and Coffer, P. J. (2009). MAPK signaling pathways in the regulation of hematopoiesis. J. Leukocyte Biol. 86, 237–250. doi: 10.1189/jlb.0209097
Geiger, H., Koehler, A., and Gunzer, M. (2007). Stem cells, aging, niche, adhesion and Cdc42: a model for changes in cell-cell interactions and hematopoietic stem cell aging. Cell Cycle 6, 884–887. doi: 10.4161/cc.6.8.4131
Glauche, I., Thielecke, L., and Roeder, I. (2011). Cellular aging leads to functional heterogeneity of hematopoietic stem cells: a modeling perspective. Aging Cell 10, 457–465. doi: 10.1111/j.1474-9726.2011.00692.x
Graudenzi, A., Serra, R., Villani, M., Colacci, A., and Kauffman, S. A. (2011). Robustness analysis of a boolean model of gene regulatory network with memory. J. Comput. Biol. 18, 559–577. doi: 10.1089/cmb.2010.0224
Greenbury, S. F., Johnston, I. G., Smith, M. A., Doye, J. P., and Louis, A. A. (2010). The effect of scale-free topology on the robustness and evolvability of genetic regulatory networks. J. Theor. Biol. 267, 48–61. doi: 10.1016/j.jtbi.2010.08.006
Guerrouahen, B. S., Al-Hijji, I., and Tabrizi, A. R. (2011). Osteoblastic and vascular endothelial niches, their control on normal hematopoietic stem cells, and their consequences on the development of leukemia. Stem Cells Int. 2011:375857-8. doi: 10.4061/2011/375857
Hamey, F. K., Nestorowa, S., Kinston, S. J., Kent, D. G., Wilson, N. K., and Göttgens, B. (2017). Reconstructing blood stem cell regulatory network models from single-cell molecular profiles. Proc. Natl. Acad. Sci. U.S.A. 114, 5822–5829. doi: 10.1073/pnas.1610609114
Herrmann, F., Groß, A., Zhou, D., Kestler, H. A., and Kühl, M. (2012). A Boolean model of the cardiac gene regulatory network determining first and second heart field identity. PLoS ONE 7:e46798. doi: 10.1371/journal.pone.0046798
Ho, T. T., Warr, M. R., Adelman, E. R., Lansinger, O. M., Flach, J., Verovskaya, E. V., et al. (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. doi: 10.1038/nature21388
Hofmann, I., Stover, E. H., Cullen, D. E., Mao, J., Morgan, K. J., Lee, B. H., et al. (2009). Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell 4, 559–567. doi: 10.1016/j.stem.2009.03.016
Hopfensitz, M., Müssel, C., Maucher, M., and Kestler, H. A. (2013). Attractors in Boolean networks: a tutorial. Comput. Stat. 28, 19–36. doi: 10.1007/s00180-012-0324-2
Itkin, T., Gur-Cohen, S., Spencer, J. A., Schajnovitz, A., Ramasamy, S. K., Kusumbe, A. P., et al. (2016). Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 532, 323–328. doi: 10.1038/nature17624
Ito, K., Hirao, A., Arai, F., Matsuoka, S., Takubo, K., Hamaguchi, I., et al. (2004). Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997–1002. doi: 10.1038/nature02989
Ito, K., Hirao, A., Arai, F., Takubo, K., Matsuoka, S., Miyamoto, K., et al. (2006). Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 12, 446–451. doi: 10.1038/nm1388
Jagannathan-Bogdan, M., and Zon, L. I. (2013). Hematopoiesis. Development 140, 2463–2467. doi: 10.1242/dev.083147
Jang, Y.-Y., and Sharkis, S. J. (2007). A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 110, 3056–3063. doi: 10.1182/blood-2007-05-087759
Jiang, M., Chen, H., Lai, S., Wang, R., Qiu, Y., Ye, F., et al. (2018). Maintenance of human haematopoietic stem and progenitor cells in vitro using a chemical cocktail. Cell Discov. 4:59. doi: 10.1038/s41421-018-0059-5
Jones, D. L., and Wagers, A. J. (2008). No place like home: anatomy and function of the stem cell niche. Nat. Rev. Mol. Cell Biol. 9, 11–21. doi: 10.1038/nrm2319
Jones, S. N., Roe, A. E., Donehower, L. A., and Bradley, A. (1995). Rescue of embryonic lethality in mdm2-deficient mice by absence of p53. Nature 378, 206–208. doi: 10.1038/378206a0
Joo, J. I., Zhou, J. X., Huang, S., and Cho, K.-H. (2018). Determining relative dynamic stability of cell states using boolean network model. Sci. Rep. 8, 1–14. doi: 10.1038/s41598-018-30544-0
Kabiri, Z., Numata, A., Kawasaki, A., Edison, B., Tenen, D. G., and Virshup, D. M. (2015). Wnts are dispensable for differentiation and self-renewal of adult murine hematopoietic stem cells. Blood 126, 1086–1094. doi: 10.1182/blood-2014-09-598540
Kauffman, S. A. (1969). Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 22, 437–467. doi: 10.1016/0022-5193(69)90015-0
Kauffman, S. A. (1993). The Origins of Order. Self Organization and Selection in Evolution. New York, NY: Oxford University Press. doi: 10.1007/978-94-015-8054-0_8
Kauffman, S. A., Peterson, C., Samuelsson, B., and Troein, C. (2004). Genetic networks with canalyzing Boolean rules are always stable. Proc. Natl. Acad. Sci. U.S.A. 101, 17102–17107. doi: 10.1073/pnas.0407783101
Kaushansky, K. (2006). Lineage-specific hematopoietic growth factors. N. Engl. J. Med. 354, 2034–2045. doi: 10.1056/NEJMra052706
Kiel, M. J., Yilmaz, O. H., Iwashita, T., Yilmaz, O. H., Terhorst, C., and Morrison, S. J. (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. doi: 10.1016/j.cell.2005.05.026
Kobayashi, H., Morikawa, T., Okinaga, A., Hamano, F., Hashidate-Yoshida, T., Watanuki, S., et al. (2019). Environmental optimization enables maintenance of quiescent hematopoietic stem cells ex vivo. Cell Rep. 28, 145–158.e9. doi: 10.1016/j.celrep.2019.06.008
Kopp, H.-G., Avecilla, S. T., Hooper, A. T., and Rafii, S. (2005). The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology 20, 349–356. doi: 10.1152/physiol.00025.2005
Kosan, C., and Godman, M. (2016). Genetic and epigenetic mechanisms that maintain hematopoietic stem cell function. Stem Cells Int. 2016:5178965. doi: 10.1155/2016/5178965
Krumsiek, J., Marr, C., Schroeder, T., and Theis, F. J. (2011). Hierarchical differentiation of myeloid progenitors is encoded in the transcription factor network. PLoS ONE 6:e22649. doi: 10.1371/journal.pone.0022649
Lacorazza, H. D., Yamada, T., Liu, Y., Miyata, Y., Sivina, M., Nunes, J., et al. (2006). The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell 9, 175–187. doi: 10.1016/j.ccr.2006.02.017
Laurenti, E., and Göttgens, B. (2018). From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426. doi: 10.1038/nature25022
Lilly, A. J., Johnson, W. E., and Bunce, C. M. (2011). The haematopoietic stem cell niche: new insights into the mechanisms regulating haematopoietic stem cell behaviour. Stem Cells Int. 2011, 274564–10. doi: 10.4061/2011/274564
Liu, B., Chen, Y., and Clair, D. K. S. (2008). Ros and p53: a versatile partnership. Free Radic. Biol. Med. 44, 1529–1535. doi: 10.1016/j.freeradbiomed.2008.01.011
Liu, J., Cao, L., Chen, J., Song, S., Lee, I. H., Quijano, C., et al. (2009). Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459, 387–392. doi: 10.1038/nature08040
Liu, Y., Elf, S. E., Miyata, Y., Sashida, G., Liu, Y., Huang, G., et al. (2009). p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4, 37–48. doi: 10.1016/j.stem.2008.11.006
Ludin, A., Gur-Cohen, S., Golan, K., Kaufmann, K. B., Itkin, T., Medaglia, C., et al. (2014). Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 21, 1605–1619. doi: 10.1089/ars.2014.5941
Macaulay, I. C., Svensson, V., Labalette, C., Ferreira, L., Hamey, F., Voet, T., et al. (2016). Single-cell RNA-sequencing reveals a continuous spectrum of differentiation in hematopoietic cells. Cell Rep. 14, 966–977. doi: 10.1016/j.celrep.2015.12.082
Maillard, I., Koch, U., Dumortier, A., Shestova, O., Xu, L., Sai, H., et al. (2008). Canonical notch signaling is dispensable for the maintenance of adult hematopoietic stem cells. Cell Stem Cell 2, 356–366. doi: 10.1016/j.stem.2008.02.011
Mancini, S. J. C., Mantei, N., Dumortier, A., Suter, U., MacDonald, H. R., and Radtke, F. (2005). Jagged1-dependent Notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood 105, 2340–2342. doi: 10.1182/blood-2004-08-3207
Maryanovich, M., Oberkovitz, G., Niv, H., Vorobiyov, L., Zaltsman, Y., Brenner, O., et al. (2012). The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 14, 535–541. doi: 10.1038/ncb2468
Matsumoto, A., Takeishi, S., Kanie, T., Susaki, E., Onoyama, I., Tateishi, Y., et al. (2011). p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell 9, 262–271. doi: 10.1016/j.stem.2011.06.014
Meek, D. W. (2009). Tumour suppression by p53: a role for the DNA damage response? Nat. Rev. Cancer 9, 714–723. doi: 10.1038/nrc2716
Meyer, P., Maity, P., Burkovski, A., Schwab, J., Müssel, C., Singh, K., et al. (2017). A model of the onset of the senescence associated secretory phenotype after DNA damage induced senescence. PLoS Comput. Biol. 13:e1005741. doi: 10.1371/journal.pcbi.1005741
Mohrin, M., Bourke, E., Alexander, D., Warr, M. R., Barry-Holson, K., Le Beau, M. M., et al. (2010). Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 7, 174–185. doi: 10.1016/j.stem.2010.06.014
Montes de Oca Luna, R., Wagner, D. S., and Lozano, G. (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206. doi: 10.1038/378203a0
Murphy, M. J., Wilson, A., and Trumpp, A. (2005). More than just proliferation: Myc function in stem cells. Trends Cell Biol. 15, 128–137. doi: 10.1016/j.tcb.2005.01.008
Müssel, C., Hopfensitz, M., and Kestler, H. A. (2010). BoolNet–an R package for generation, reconstruction and analysis of Boolean networks. Bioinformatics 26, 1378–1380. doi: 10.1093/bioinformatics/btq124
Olariu, V., and Peterson, C. (2019). Kinetic models of hematopoietic differentiation. WIREs Syst. Biol. Med. 11:e1424. doi: 10.1002/wsbm.1424
Oostendorp, R. A. J. (2015). Secretion of Wnts is dispensable for hematopoiesis. Blood 126, 1051–1052. doi: 10.1182/blood-2015-07-653402
Opferman, J. T., Iwasaki, H., Ong, C. C., Suh, H., Mizuno, S.-I., Akashi, K., et al. (2005). Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 307, 1101–1104. doi: 10.1126/science.1106114
Orford, K. W., and Scadden, D. T. (2008). Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet. 9, 115–128. doi: 10.1038/nrg2269
Pant, V., Quintás-Cardama, A., and Lozano, G. (2012). The p53 pathway in hematopoiesis: lessons from mouse models, implications for humans. Blood 120, 5118–5127. doi: 10.1182/blood-2012-05-356014
Park, I.-K., Qian, D., Kiel, M., Becker, M. W., Pihalja, M., Weissman, I. L., et al. (2003). BMI-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423, 302–305. doi: 10.1038/nature01587
Passegué, E., Wagers, A. J., Giuriato, S., Anderson, W. C., and Weissman, I. L. (2005). Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 202, 1599–1611. doi: 10.1084/jem.20050967
Pietras, E. M., Warr, M. R., and Passegué, E. (2011). Cell cycle regulation in hematopoietic stem cells. J. Cell Biol. 195, 709–720. doi: 10.1083/jcb.201102131
Pinho, S., and Frenette, P. S. (2019). Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 20, 303–320. doi: 10.1038/s41580-019-0103-9
Qian, H., Buza-Vidas, N., Hyland, C. D., Jensen, C. T., Antonchuk, J., Månsson, R., et al. (2007). Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell 1, 671–684. doi: 10.1016/j.stem.2007.10.008
Qu, X., Aldana, M., and Kadanoff, L. P. (2002). Numerical and theoretical studies of noise effects in the Kauffman model. J. Stat. Phys. 109, 967–986. doi: 10.1023/A:1020416308456
Ringshausen, I., O'Shea, C. C., Finch, A. J., Swigart, L. B., and Evan, G. I. (2006). MDM2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 10, 501–514. doi: 10.1016/j.ccr.2006.10.010
Rizo, A., Olthof, S., Han, L., Vellenga, E., de Haan, G., and Schuringa, J. J. (2009). Repression of BMI1 in normal and leukemic human CD34+ cells impairs self-renewal and induces apoptosis. Blood 114, 1498–1505. doi: 10.1182/blood-2009-03-209734
Rodgers, J. T., King, K. Y., Brett, J. O., Cromie, M. J., Charville, G. W., Maguire, K. K., et al. (2014). mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert. Nature 510, 393–396. doi: 10.1038/nature13255
Roeder, I., Horn, K., Sieburg, H.-B., Cho, R., Muller-Sieburg, C., and Loeffler, M. (2008). Characterization and quantification of clonal heterogeneity among hematopoietic stem cells: a model-based approach. Blood 112, 4874–4883. doi: 10.1182/blood-2008-05-155374
Roeder, I., and Lorenz, R. (2006). Asymmetry of stem cell fate and the potential impact of the niche. Stem Cell Rev. 2, 171–180. doi: 10.1007/s12015-006-0045-4
Rossi, L., Challen, G. A., Sirin, O., Lin, K. K.-Y., and Goodell, M. A. (2011). Hematopoietic stem cell characterization and isolation. Methods Mol. Biol. 750, 47–59. doi: 10.1007/978-1-61779-145-1_3
Sawai, C. M., Babovic, S., Upadhaya, S., Knapp, D. J. H. F., Lavin, Y., Lau, C. M., et al. (2016). Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity 45, 597–609. doi: 10.1016/j.immuni.2016.08.007
Scandura, J. M., Boccuni, P., Massagué, J., and Nimer, S. D. (2004). Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc. Natl. Acad. Sci. U.S.A. 101, 15231–15236. doi: 10.1073/pnas.0406771101
Schofield, R. (1978). The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 4, 7–25.
Schwab, J., Burkovski, A., Siegle, L., Müssel, C., and Kestler, H. A. (2017). ViSiBooL-visualization and simulation of Boolean networks with temporal constraints. Bioinformatics 33, 601–604. doi: 10.1093/bioinformatics/btw661
Schwab, J. D., Kühlwein, S. D., Ikonomi, N., Kühl, M., and Kestler, H. A. (2020). Concepts in Boolean network modeling: what do they all mean? Comput. Struct. Biotechnol. J. 18, 571–582. doi: 10.1016/j.csbj.2020.03.001
Siegle, L., Schwab, J. D., Kühlwein, S. D., Lausser, L., Tümpel, S., Pfister, A. S., et al. (2018). A Boolean network of the crosstalk between IGF and Wnt signaling in aging satellite cells. PLoS ONE 13:e0195126. doi: 10.1371/journal.pone.0195126
Spangrude, G., Heimfeld, S., and Weissman, I. (1988). Purification and characterization of mouse hematopoietic stem cells. Science 241, 58–62. doi: 10.1126/science.2898810
Suda, T., Takubo, K., and Semenza, G. L. (2011). Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9, 298–310. doi: 10.1016/j.stem.2011.09.010
Sugiyama, T., Kohara, H., Noda, M., and Nagasawa, T. (2006). Maintenance of the hematopoietic stem cell pool by CXCL14-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988. doi: 10.1016/j.immuni.2006.10.016
Sun, J., Ramos, A., Chapman, B., Johnnidis, J. B., Le, L., Ho, Y.-J., et al. (2014). Clonal dynamics of native haematopoiesis. Nature 514, 322–327. doi: 10.1038/nature13824
Tanaka, H., Fauré, A., and Matsuno, H. (2017). “Boolean modeling of mammalian cell cycle and cancer pathways,” in International Conference on Artificial Life and Robotics 2017, ed M. Sugisaka (Miyazaki), 507–510. doi: 10.5954/ICAROB.2017.GS4-3
TeKippe, M., Harrison, D. E., and Chen, J. (2003). Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp. Hematol. 31, 521–527. doi: 10.1016/S0301-472X(03)00072-9
Tesio, M., and Trumpp, A. (2011). Breaking the cell cycle of HSCs by p57 and friends. Cell Stem Cell 9, 187–192. doi: 10.1016/j.stem.2011.08.005
Thomas, R., and Kaufman, M. (2001). Multistationarity, the basis of cell differentiation and memory. II. Logical analysis of regulatory networks in terms of feedback circuits. Chaos 11, 180–195. doi: 10.1063/1.1349893
Thorén, L. A., Liuba, K., Bryder, D., Nygren, J. M., Jensen, C. T., Qian, H., et al. (2008). Kit regulates maintenance of quiescent hematopoietic stem cells. J. Immunol. 180, 2045–2053. doi: 10.4049/jimmunol.180.4.2045
Toledo, F., and Wahl, G. M. (2006). Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 6, 909–923. doi: 10.1038/nrc2012
Tothova, Z., and Gilliland, D. G. (2007). FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 1, 140–152. doi: 10.1016/j.stem.2007.07.017
Tothova, Z., Kollipara, R., Huntly, B. J., Lee, B. H., Castrillon, D. H., Cullen, D. E., et al. (2007). FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339. doi: 10.1016/j.cell.2007.01.003
Trumpp, A., Essers, M., and Wilson, A. (2010). Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 10, 201–209. doi: 10.1038/nri2726
Umemoto, T., Yamato, M., Nishida, K., Yang, J., Tano, Y., and Okano, T. (2005). p57Kip2 is expressed in quiescent mouse bone marrow side population cells. Biochem. Biophys. Res. Commun. 337, 14–21. doi: 10.1016/j.bbrc.2005.09.008
van Os, R., de Haan, G., and Dykstra, B. J. (2009). Hematopoietic stem cell quiescence: yet another role for p53. Cell Stem Cell 4, 7–8. doi: 10.1016/j.stem.2008.12.007
Vousden, K. H., and Lane, D. P. (2007). p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283. doi: 10.1038/nrm2147
Wagner, W., Horn, P., Bork, S., and Ho, A. D. (2008). Aging of hematopoietic stem cells is regulated by the stem cell niche. Exp. Gerontol. 43, 974–980. doi: 10.1016/j.exger.2008.04.007
Warr, M. R., Pietras, E. M., and Passegué, E. (2011). Mechanisms controlling hematopoietic stem cell functions during normal hematopoiesis and hematological malignancies. WIREs Syst. Biol. Med. 3, 681–701. doi: 10.1002/wsbm.145
Weissman, I. L., and Shizuru, J. A. (2008). The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood 112, 3543–3553. doi: 10.1182/blood-2008-08-078220
Wilson, A., Laurenti, E., Oser, G., van der Wath, R. C., Blanco-Bose, W., Jaworski, M., et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. doi: 10.1016/j.cell.2008.10.048
Wilson, A., Laurenti, E., and Trumpp, A. (2009). Balancing dormant and self-renewing hematopoietic stem cells. Curr. Opin. Genet. Dev. 19, 461–468. doi: 10.1016/j.gde.2009.08.005
Wilson, A., Murphy, M. J., Oskarsson, T., Kaloulis, K., Bettess, M. D., Oser, G. M., et al. (2004). c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 18, 2747–2763. doi: 10.1101/gad.313104
Wilson, A., Oser, G. M., Jaworski, M., Blanco-Bose, W. E., Laurenti, E., Adolphe, C., et al. (2007). Dormant and self-renewing hematopoietic stem cells and their niches. Ann. N. Y. Acad. Sci. 1106, 64–75. doi: 10.1196/annals.1392.021
Wilson, A., and Trumpp, A. (2006). Bone-marrow haematopoietic-stem-cell niches. Nat. Rev. Immunol. 6, 93–106. doi: 10.1038/nri1779
Yalcin, S., Zhang, X., Luciano, J. P., Mungamuri, S. K., Marinkovic, D., Vercherat, C., et al. (2008). Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J. Biol. Chem. 283, 25692–25705. doi: 10.1074/jbc.M800517200
Yamazaki, S., Iwama, A., Takayanagi, S.-I., Eto, K., Ema, H., and Nakauchi, H. (2009). TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 113, 1250–1256. doi: 10.1182/blood-2008-04-146480
Yokota, T. (2019). “Hierarchy” and “holacracy” a paradigm of the hematopoietic system. Cells 8:1138. doi: 10.3390/cells8101138
Yoshihara, H., Arai, F., Hosokawa, K., Hagiwara, T., Takubo, K., Nakamura, Y., et al. (2007). Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 1, 685–697. doi: 10.1016/j.stem.2007.10.020
Zhang, J., Niu, C., Ye, L., Huang, H., He, X., Tong, W.-G., et al. (2003). Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425, 836–841. doi: 10.1038/nature02041
Zhang, Y., Gao, S., Xia, J., and Liu, F. (2018). Hematopoietic hierarchy-an updated roadmap. Trends Cell Biol. 28, 976–986. doi: 10.1016/j.tcb.2018.06.001
Keywords: hematopoietic stem cell, Boolean network, dynamic modeling, TP53, stem cell awakening, maintenance of quiescence, niche interactions, homeostasis
Citation: Ikonomi N, Kühlwein SD, Schwab JD and Kestler HA (2020) Awakening the HSC: Dynamic Modeling of HSC Maintenance Unravels Regulation of the TP53 Pathway and Quiescence. Front. Physiol. 11:848. doi: 10.3389/fphys.2020.00848
Received: 30 April 2020; Accepted: 24 June 2020;
Published: 31 July 2020.
Edited by:
Zhike Zi, Max Planck Institute for Molecular Genetics, GermanyReviewed by:
Nathan Weinstein, Universidad Nacional Autónoma de México, MexicoCopyright © 2020 Ikonomi, Kühlwein, Schwab and Kestler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans A. Kestler, aGFucy5rZXN0bGVyQHVuaS11bG0uZGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.