
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 07 July 2020
Sec. Vascular Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00742
This article is part of the Research TopicCell Communication in Vascular BiologyView all 16 articles
 Meiling Su1,2†
Meiling Su1,2† Shunyang Fan3†Zhenwei Ling1,2†
Shunyang Fan3†Zhenwei Ling1,2† Xuejiao Fan1,2
Xuejiao Fan1,2 Luoxing Xia1,2
Luoxing Xia1,2 Yingying Liu1,2
Yingying Liu1,2 Shaoying Li1,2
Shaoying Li1,2 Yuan Zhang1,2
Yuan Zhang1,2 Zhi Zeng1,2*
Zhi Zeng1,2* Wai Ho Tang1,2*
Wai Ho Tang1,2*Platelet hyperactivity is the hallmark of diabetes, and platelet activation plays a crucial role in diabetic vascular complications. Recent studies have shown that upon activation, platelet-derived miRNAs are incorporated into vascular smooth muscle cells (VSMCs), regulating the phenotypic switch of VSMC. Under diabetes, miRNA deficiency in platelets fails to regulate the VSMC phenotypic switch. Therefore, manipulation of platelet-derived miRNAs expression may provide therapeutic option for diabetic vascular complications. We seek to investigate the effect of calpeptin (calpain inhibitor) on the expression of miRNAs in diabetic platelets, and elucidate the downstream signaling pathway involved in protecting from neointimal formation in diabetic mice with femoral wire injury model. Using human cell and platelet coculture, we demonstrate that diabetic platelet deficient of miR-223 fails to suppress VSMC proliferation, while overexpression of miR-223 in diabetic platelets suppressed the proliferation of VSMC to protect intimal hyperplasia. Mechanistically, miR-223 directly targets the insulin-like growth factor-1 receptor (IGF-1R), which inhibits the phosphorylation of GSK3β and activates the phosphorylation of AMPK, resulting in reduced VSMC dedifferentiation and proliferation. Using a murine model of vascular injury, we show that calpeptin restores the platelet expression of miR-223 in diabetes, and the horizontal transfer of platelet miR-223 into VSMCs inhibits VSMC proliferation in the injured artery by targeting the expression of IGF-1R. Our data present that the platelet-derived miR-223 suppressed VSMC proliferation via the regulation miR-223/IGF-1R/AMPK signaling pathways, and inhibition of calpain alleviates neointimal formation by restoring the expression of miR-223 in diabetic platelet.
Diabetes mellitus (DM) is an important risk factor of intima hyperplasia and vascular restenosis, resulting in diabetic vascular complications (Kornowski et al., 1997; Davi and Patrono, 2007). It is known that platelet hyperreactivity is the hallmark of DM, and the activated platelets (APs) release thromboxane and PDGF, which induce vascular smooth muscle cells (VSMCs) switching from a quiescent contractile phenotype to a highly synthetic and proliferating phenotype upon vascular injury and repair (Millette et al., 2005). Excessive proliferation of VSMCs leads to intimal hyperplasia in DM (Faries et al., 2001). Therefore, the regulation of VSMC phenotypic switch plays a crucial role in maintaining the vascular stability.
Although platelets are anucleate cells, they contain enriched and diverse miRNAs and mRNAs (Landry et al., 2009; Nagalla et al., 2011). We and others have demonstrated that the platelet-derived miRNAs can be transferred and incorporated into recipient cells such as VSMCs, endothelial cells, and macrophages (Risitano et al., 2012; Kirschbaum et al., 2015; Zeng et al., 2019). Our study has also shown that the horizontal transfer of platelet miRNA (miR-143/145 and miR-223) inhibits VSMC dedifferentiation and proliferation, but the altered miRNA expression of diabetic platelets fails to inhibit VSMC dedifferentiation, consequently leading to increased neointimal formation (Zeng et al., 2019). Recent report has shown that Dicer is a calpain substrate, and calpain-dependent cleavage of Dicer reduced the expression of miRNAs in diabetic platelets (Elgheznawy et al., 2015). The altered expression of platelet miRNAs such as miR-223 can be restored by treatment of a calpain inhibitor in diabetic mice (Elgheznawy et al., 2015), suggesting that calpain inhibitor may provide therapeutic adjunct for platelet-associated vascular disease in diabetes.
In the present study, we seek to elucidate the mechanism by which inhibition of calpain regulated VSMC phenotypic switch via upregulating the expression of platelet-derived miRNAs, and the potential application of calpeptin on platelet-associated vascular disease in diabetes. Here we demonstrate that diabetic platelet deficient of miR-223 fails to suppress VSMC proliferation, while overexpression of miR-223 in diabetic platelets suppressed the proliferation of VSMC to protect intimal hyperplasia. Mechanistically, miR-223 directly targets the insulin-like growth factor-1 receptor (IGF-1R), which inhibits the phosphorylation of GSK3β and activates the phosphorylation of AMPK, resulting in reduced VSMC dedifferentiation and proliferation. Using a murine model of vascular injury, we show that calpeptin restores the platelet expression of miR-223 in diabetes, and the horizontal transfer of platelet miR-223 into VSMCs inhibits VSMC proliferation in the injured artery by targeting the expression of IGF-1R. Collectively, our results demonstrate that the platelet-derived miR-223 inhibits VSMC proliferation via miR-223/IGF-1R/AMPK signaling axis, and inhibition of calpain alleviates neointimal formation by restoring the expression of miR-223 in diabetic platelet.
Human peripheral blood was obtained from Guangzhou Women and Children’s Medical Center. Informed consent of underage subjects was obtained from their parents. Our study was compiled and approved by the Institutional Review Board of Guangzhou Women and Children’s Medical Center (Human Investigation Committee No. 201944101). Venous blood samples (approximately 4 ml) were drawn from Non-DM and DM subjects from Guangzhou Women and Children’s Medical Center. Venous blood samples (approximately 4 ml) were drawn into blood collection tubes containing 3.8% trisodium citrate (w/v). Platelet-rich plasma (PRP) was centrifuged at 1200 rpm at 25°C for 15 min. PRP were then treated with 100 nM Prostaglandin E1 (PGE1, Sigma) and centrifuged at 2500 rpm for another 5 min. After discarding the supernatant, the platelet pellet was resuspended with 3 ml Hank’s Balanced Salt Solution (HBSS, NaCl 135 mM, MgCl2 1 mM, NaHCO3 12 mM, CaCl2 1 mM, KCl 2.9 mM, Glucose 5 mM, BSA 0.35%, HEPES 10 mM).
For murine platelets, blood was drawn from the left ventricle after anesthesia. Platelet-rich plasma (PRP) was prepared by centrifuging at 1200 rpm for 15 min, and blood coagulation was inhibited by adding the anticoagulant citrate dextrose solution. By centrifuging at 2600 rpm for 5 min, washed platelets were prepared from PRP and were resuspended in HBSS. Platelet concentration was adjusted at 2 × 108 cells/mL using a platelet counter. To prevent platelet activation, the centrifuge temperature was kept at room temperature during the washing step.
The male C57BL/6 mice (8 weeks old) were purchased from Guangzhou Medical University. Procedures were approved by the Institutional Animal Care and Use Committee of Guangzhou Medical University (No. SYXK2016-0168). Eight week old mice were injected with 50mg/kg streptozotocin (STZ, Sigma) to induce diabetes for 5 consecutive days. The mice were randomly divided into four groups and were intraperitoneally injected PBS or calpeptin (calpain inhibitor) (30 mg/kg/day) diluted in DMSO in PBS every other day. The mice in the AgomiR group were administered with Agomir-223 200 μl at a concentration of 20 mM via tail vein injection. The calpeptin ware purchased from the SelleckChemicals Co, while the AgomiRs and miRNA mimic from Shanghai GenePharma Co.
The endothelial injury in the right femoral artery was done by guidewire abrasion. After anesthetizing the experimental mice, the right thigh was depilated from hair and disinfected with alcohol. The proximal part of the femoral artery and saphenous artery were dissected under the microscope, followed by a temporary stop of blood flow with a silk thread. Then the 0.25 mm diameter guidewire was inserted into the femoral artery from the saphenous artery, inserted and withdrawn 3 times to sufficiently create endothelial wear. The saphenous artery was ligated and the wound was sutured to allow the mice to enter a recovery phase. The contralateral femoral artery was sham-operated without guidewires injured as control. Four weeks after injury, the mice were anesthetized, sacrificed for harvesting the femoral, and fixed in 4% paraformaldehyde. After 30% sucrose dehydration, the arterial tissue was embedded in the Tissue-Tek O.C.T (SAKURA, United States) agent and performed a frozen section.
Human coronary artery smooth muscle cells (HCASMC) were purchased from Cell Application at passage 4, cultured in High Glucose DMEM (Gibco, United States) supplemented with 10% fetal bovine serum (Gibco, United States), 100 U/ml penicillin (Gibco, United States), and 100 μg/ml streptomycin (Gibco, United States). The cells were cultured in the incubator with 5% CO2 and 37°C. HCASMCs were cocultured with or without resting platelets (RPs) or 0.1 U/ml thrombin activated platelets (APs). After isolation and purification, platelets from healthy subject (HS) and DM patients were counted and added directly to the cultured VSMC (1: 100) for 48 h.
HCASMCs were transfected with miR-223 mimic or miR negative control (miR-NC, GenePharma, China) at 100 nM by using Lipofectamine RNAiMAX reagent (Invitrogen, United States). Cells were seeded to be 60–80% confluent before transfection. Lipofectamine reagent and miRNA mimic were diluted in Opti-MEM Medium (Gibco, United States) and mixed for 5 min before added to HCASMCs. Also, HCASMCs were transfected with miR-223 antagomir and miR-NC (Shanghai GenePharma Co) for 24 h, and then VSMCs were cocultured with the purified platelets from HS. After cultured for 48 h, the cells were harvested for subsequent experiments.
Cell Counting Kit-8 Kits (CCK8; Dojindo, Japan) were used to evaluate cell proliferation. HCASMCs were seeded into a 96-well plate at a concentration of 1.0 × 104 cells/well. Post-transfection 48 h, HCASMCs were incubated with 10 μl CCK8 in 100 μl DMEM for 2 h. The absorbance was detected at 450 nm with a microplate reader (Thermo Fisher Scientific).
The HCASMCs were starved in the DMEM without serum and then were transfected with miR-223 mimic and miR-NC as described above. Forty-eight hours after transfection, cell proliferation was determined by using the BrdU Cell Proliferation Assay Kit (Millipore, United States) according to the manufacturer’s protocol. Before measurement, BrdU was added to the cultured cells and incubated for 12 h. After washing with PBS, the cells were fixed and DNA denatured with 4% paraformaldehyde. After fixation, cells were labeled with peroxidase-conjugated BrdU antibody and then incubated with peroxidase substrate. Finally, a stop solution was added to stop the reaction. The absorbance (A450–A550 nm) representing the cell proliferation was measured on a microplate reader.
Cell proliferation rate was measured with Click-iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen, United States). HCASMCs were seeded into glass-bottom culture dishes at a concentration of 1.0 × 104 cells/ml and transfection experiments were performed as described above. Before termination of cell culture, EdU was added at a final transformation concentration 10 μg/L and incubated with cells at 37°C for 1 h. After incubation, cells were washed with PBS, fixed with 3.7% paraformaldehyde for 15 min, and permeabilized with 0.5% Triton®X-100 for 20 min. The Click-iT® reaction cocktail prepared 15 min in advance was added to the notch of each well and incubated at room temperature for 30 min, protected from light. After washing, DNA staining was performed using Hoechst 33342 solution at a concentration of 1: 2000. The EdU positive cells were observed and captured under the inverted fluorescence microscope (Leica), and the cell count was calculated by using Image J.
IGF-1R 3′-UTR plasmid was obtained from Biolink Technology Co. The HEK-293 cells were plated in a 24-well plate in a concentration of 50%. After 24 h plated, the 1μg plasmid and 50 nM miR-223 mimic and miR-NC were transfected by using Lipofectamine RNAiMAX reagent (Invitrogen, United States). Luciferase reporter analyses were performed according to the manufacturer’s instructions after 48 h of transfection by using Dual Luciferase Assay Kit (Promega, United States). The Firefly and Renilla fluorescence values were detected by the microplate infinite M1000 (Tecan) and the firefly fluorescence normalized by Renilla was served as luciferase activity.
The frozen sections of the tissues were dried at room temperature for 30 min and placed in PBS for 10 min. According to the following sequence, the tissues were placed in hematoxylin for 1 min, hydrochloric acid alcohol for 2,3 sec, and then moved to eosin for 8–10 sec. Finally, the tissues were rinsed in running water and then mounted with neutral gum.
Cells and tissues were fixed and permeabilized by 4% paraformaldehyde (Servicebio, China) and 0.2% Triton-100X (Solarbio, China) in PBS, respectively; staining was with antibodies and their corresponding secondary antibodies. Primary antibodies used include IGF-1R (1:100, ab182408, Abcam, United States) and α-smooth muscle actin (Acta2; 1:100, ab119952, Abcam, United States). After 24 h, sections were incubated with the Alexa Fluor 594-conjugated anti-rabbit IgG secondary antibody and Alexa Fluor 488-conjugated anti-goat IgG secondary antibody (Thermo Fisher Scientific) (1:200) for 60 min and stained with the chromatin-specific dye Hoechst for 5 min at RT. The confocal images were taken with a Leica SP8 (Leica) confocal microscope.
The cells were harvested with RIPA lysate containing protease inhibitors or phosphatase inhibitor cocktail. Extract protein by centrifugation at 4°C at 12,000 rpm and heated at 95°C with loading buffer. The protein samples were resolved in 12% SDS-PAGE gels and transferred to PVDF membranes (Millipore, United States). PVDF membranes were incubated with 5% skim milk powder (Biofroxx, Germany) for 2 h and then incubated with primary antibodies used including: IGF-1R (1:1000, ab182408, Abcam, United States); ACTA2 (1:2000, ab119952, Abcam, United States); calponin (1:2000, CNN1; ab46794, Abcam, United States); transgelin (TAGLAN; 1:2000, ab14106, Abcam, United States); KLF4 (1:2000, 101508, GenetTex, United States); KLF5 (1:2000, ab137676, Abcam, United States); osteopontin (1:2000, OPN; ab91655, Abcam, United States); Akt (1:1000, 9272S, CST, United States); Phospho-Akt (1:500, Ser473) (4060S, CST, United States); GSK3β (1:1000, 9315S, CST, United States); Phospho-GSK3β (Ser9) (1:500, 9336S, CST, United States); Src (1:1000, MAB3389, R&D, United States); Phospho-Src (Y419) (1:500, AF2685, R&D, United States); JNK (1:1000, 9252S, CST, United States); Phospho-JNK (Thr172) (1:500, 4668S, CST, United States); AMPK (1:1000, 2532S, CST, United States); Phospho-AMPK (Thr183/Tyr185) (1:500, 2535S, CST, United States); Acetyl-CoA Carboxylase (1:1000, ACC; 3662S, CST, United States); Phospho-ACC (Ser79) (1:500, 3661S, CST, United States); p21 (1:1000, 2847, CST, United States); P27 (1:1000, 3688, CST, United States); GAPDH (1:3000, ab125247, Abcam, United States); α-tubulin (1:3000, ab52866, Abcam, United States); α-actinin (1:3000, ab18061, Abcam, United States). The next day, the PVDF membranes were incubated with a secondary antibody (1:2000) dissolved in 5% nonfat milk after three times of washing with TBST. The specific band for the target protein was detected by ChemiDoc XRS+ system, and the band intensities were calculated using the Image Lab software.
The cell lysate was extracted from the cells after the treatment as described above, and the protein was quantified with a bicinchoninic acid kit. According to the manufacturer’s instructions, after blocking with Array Buffer 1, the phosphokinase array membrane was incubated with diluted cell lysate overnight. After washing three times with Wash Buffer to remove unbound protein, the array was incubated with a cocktail of biotinylated detection antibodies for 2 h at room temperature. Streptavidin-HRP and chemiluminescent detection reagents were applied, and the signal was detected by ChemiDoc XRS+ system. Refer to the coordinates of analytes and controls provided in the appendix of the kit instructions, the signal at each capture spot corresponding to the amount of phosphorylated protein bound was analyzed.
Total RNA purification kit (Axygen, United States) was used to extract and purify miRNA including miRNA from cells, and purification of miRNA were reverse transcribed with miRNeasy Serum/Plasma kit (Qiagen, Germany) and U6 snRNA Normalization RT-PCR Quantitation Kit (GenePharma). After normalizing the nucleic acid concentration by quantification, reverse transcription PCR was performed to obtain stable cDNA by using the PrimeScript RT reagent Kit (Takara, Japan). Transcript levels of the target genes were analyzed by qPCR using the 2–ΔΔCt method. The primer sequences were listed in Supplementary Table 1.
All data are presented as mean ± SD. All analysis was performed using Graph Pad Prism v.5.0 (GraphPad, La Jolla, CA, United States). Comparisons between two groups were analyzed using unpaired nonparametric Student’s t-test. Comparisons between more than two groups were analyzed using one-way ANOVA analysis with the Dunnett’s multiple correction. P < 0.05 was considered statistically significant.
Consistent to our previous (Zeng et al., 2019), VSMCs cocultured with DM-APs showed downregulation of VSMCs differentiation markers α-SMA (ACTA2), Calponin (CNN1), and SM22α (TAGLN) compared to HS-APs group (Figure 1A). Also, the expression of VSMCs dedifferentiation markers Osteopontin (OPN), MYH10, KLF4, and KLF5 was significantly increased in VSMCs cocultured with DM-APs (Figure 1A). Pretreatment with miR-223 antagomir, we found that the SMCs differentiation markers ACTA2 and CNN1 of VSMCs cocultured with HS-APs treated by miR-223 antagomir was significantly inhibited than VSMCs cocultured with HS-APs. Also, the expression of VSMCs dedifferentiation markers KLF4 was significantly increased in VSMCs cocultured with HS-APs treated by miR-223 antagomir (Supplementary Figure S1). Pretreatment with RNase A (degrades the RNA), we found that the proliferation rate of VSMCs cocultured with APs treated by RNase A was significantly higher than VSMCs cocultured with APs (Figure 1B). Also, overexpression of miR-223 significantly suppressed VSMC proliferation (Figures 1C,D). These results suggested that platelet-derived miR-223 was transferred and incorporated into VSMCs, inhibiting VSMC proliferation. Deficiency in diabetic platelet fails to suppress VSMC proliferation.
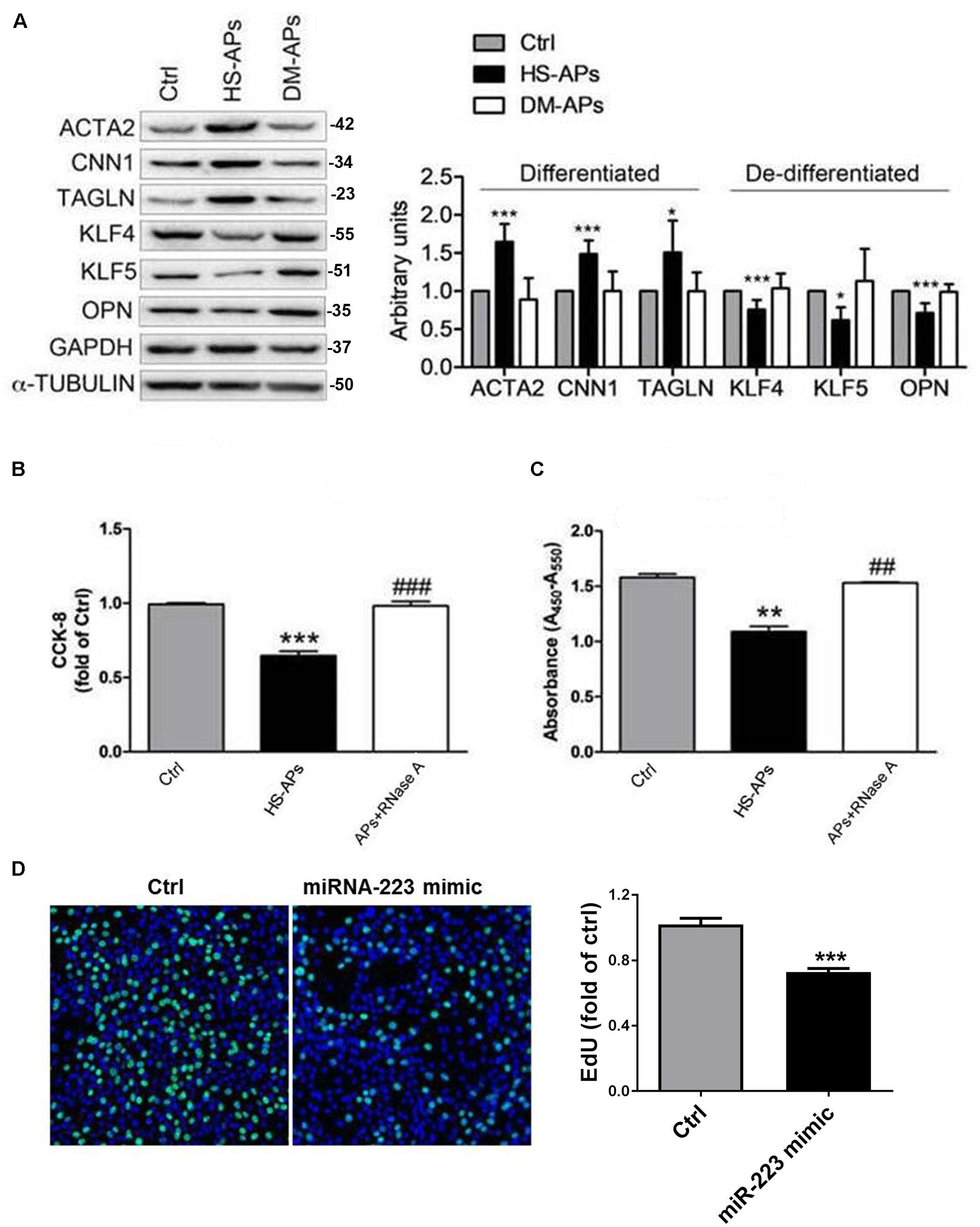
Figure 1. MiR-223 deficiency in diabetic platelet fails to suppress VSMC proliferation. Activated platelets (APs) from healthy subject (HS) and DM patients were cocultured with human VSMCs for 48 h. (A) Expression of differentiation markers ACTA2, CNN1, and TAGLN and dedifferentiation markers OPN, MYH10, KLF4, and KLF5 in control (Ctrl) VSMCs and APs cocultured with VSMCs (HS-APs, DM-APs) was detected with western blot analysis. (B,C) The rate of proliferation was determined by CCK8 assay and Brdu assay in VSMCs treated with APs pretreated by RNase A (degrades the RNA). Data are presented as mean ± SD (n = 6). Comparisons between more than two groups were analyzed using one-way ANOVA analysis with the Dunnett’s multiple correction. *P < 0.05, **P < 0.01, ***P < 0.001 vs Ctrl (VSMCs without platelets); #P < 0.05, ##P < 0.01, ###P < 0.001 vs HS-APs. (D) The rate of proliferation was determined by EdU incorporation assay in VSMCs transfected with miR-223 mimic. Data are presented as mean ± SD (n = 6). Comparisons between two groups were analyzed using unpaired nonparametric Student’s t-test.*P < 0.05 vs Ctrl (miR-NC).
Bioinformatics analysis identified a binding site for miR-223 in the 3′-UTR of IGF-1R. When we cloned IGF-1R 3′ UTR into luciferase report, we found that a miR-223 mimic significantly inhibited the luciferase activity; however, the inhibition of luciferase activity was abrogated by mutated control group (Figure 2A). To identify whether miR-223 could bind to IGF-1R and inhibited the expression, we performed that VSMC was treated with a miR-223 mimic. As expected, the expression of IGF-1R in VSMC was significantly decreased by overexpression of miR-223 compared to control group. The data suggest miR-223 can directly target the 3′-UTR of IGF-1R and inhibit its expression in VSMC (Figures 2A–C). Also, we performed overexpression of IGF-1R in VSMCs. We found that VSMC proliferation was significantly increased in overexpression of IGF-1R of VSMCs compared with control group using CCK8 (Figure 2D). The binding of IGF-1 to its receptor (IGF-1R) can activate the cascade signals including IRS family, thereby activating phosphatidyl inositol 3-kinase (PI3K) and Akt signaling pathway (Delafontaine et al., 2004). Thus we determined the downstream signal pathway involved in inhibiting VSMC proliferation treated with a miR-223 mimic. Figures 2E,F showed that the phosphorylation of Akt and GSK3β was inhibited by the miR-223 mimic than that of control group. Our data demonstrate that miR-223 directly targets IGF-1R in VSMC, which inhibits the phosphorylation of Akt and GSK3β, resulting in reduced VSMC dedifferentiation and proliferation.
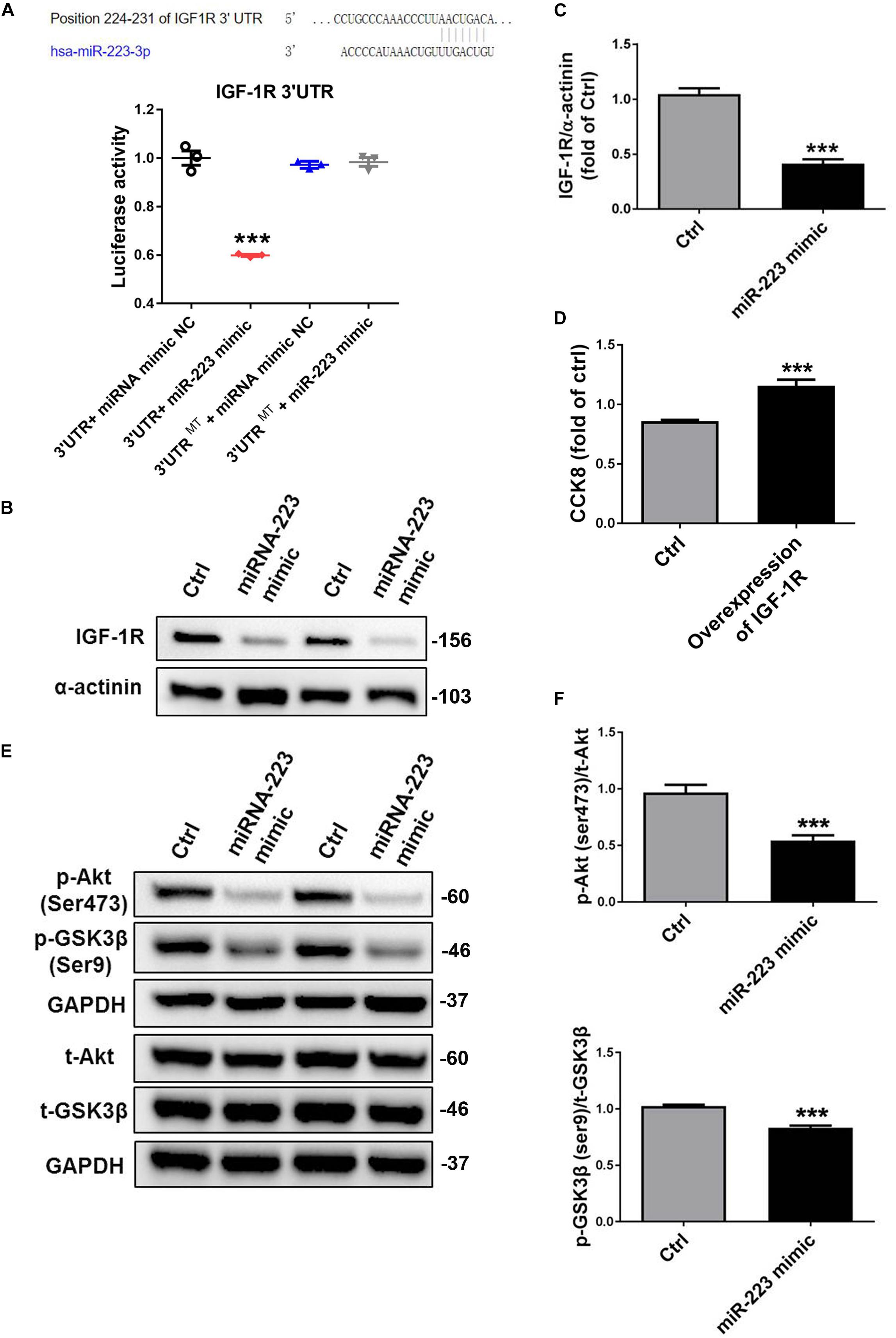
Figure 2. MiR-223 directly targets the IGF-1R, which inhibits the phosphorylation of Akt and GSK3β. (A) Bioinformatics analysis identified a binding site for miR-223 in the 3′-UTR of IGF-1R. Luciferase activity assay in VSMCs following introduction of IGF-1R 3′ UTR or mutant 3′ UTR (mut) with or without miR-223 mimic (n = 3). VSMCs were transfected with miR-223 mimic for 48 h. (B,C) The expression of IGF-1R in VSMC was detected with western blot analysis. (D) The rate of proliferation was determined by CCK8 assay in VSMCs transfected with overexpression of IGF-1R. (E,F) The phosphorylation of Akt and GSK3β in VSMC was determined with western blot analysis. Data are presented as mean ± SD (n = 6). Comparisons between two groups were analyzed using unpaired nonparametric Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001 vs Ctrl (miR-NC).
To further determine the downstream signal pathway of Akt/GSK3β involved in inhibiting VSMC proliferation, we determined that the phosphorylation of AMPKα, Src and Jnk were assessed by proteomics analysis of phosphokinase arrays in control (Ctrl) VSMCs and APs cocultured with VSMCs. As shown in Figures 3A,B, the phosphorylation of AMPKα, Src and Jnk in VSMCs treated with APs were significantly increased compared to the control group. Using western blot analysis, we further confirmed that the phosphorylation of AMPKα in VSMCs treated with APs was significantly upregulated compared to the control group (Figure 3C). However, we found that the phosphorylation of Src and Jnk did not change in VSMCs cocultured with resting platelets (RPs) or APs (Figures 3D,E). These results suggest that APs activate the AMPKα signal pathway to suppress VSMC proliferation.
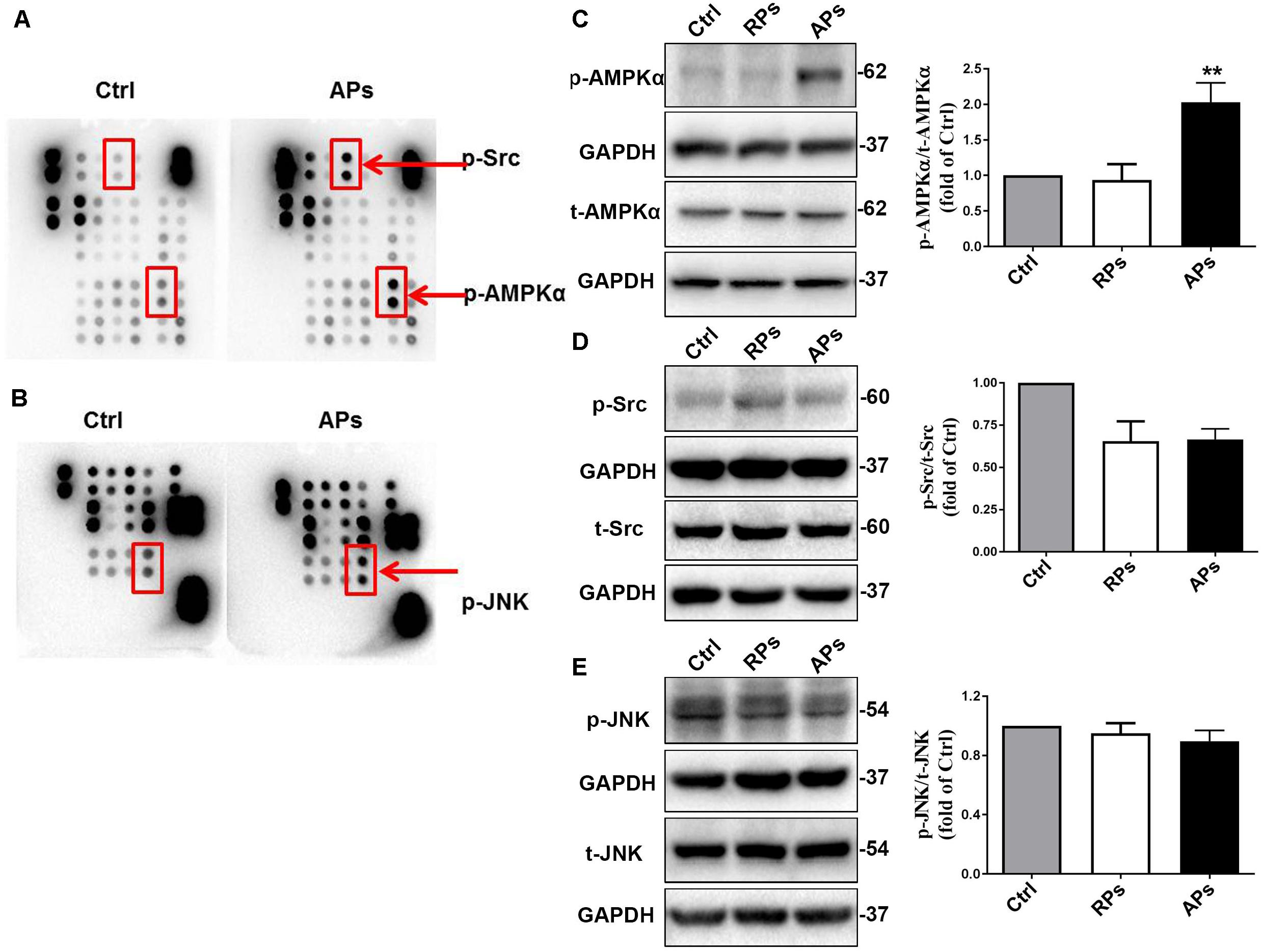
Figure 3. APs activate the AMPKα signal pathway to suppress VSMC proliferation. (A,B) The phosphorylation of AMPKα, Src and Jun were assessed by proteomics analysis of phosphokinase arrays in control (Ctrl) VSMCs and APs cocultured with VSMCs. The phosphorylation of (C) AMPKα, (D) Src, and (E) Jnk was determined with western blot analysis in control (Ctrl) VSMCs, RPs, and APs cocultured with VSMCs. Data are presented as mean ± SD (n = 6). Comparisons between more than two groups were analyzed using one-way ANOVA analysis with the Dunnett’s multiple correction. **P < 0.01 vs Ctrl (VSMCs without platelets).
To further investigate whether platelet-derived miR-223 suppressed VSMC proliferation through AMPKα signal pathway, we treated with a miR-223 mimic or miR negative control (Ctrl) in VSMCs. Similarly, we found that the phosphorylation of AMPKα in VSMCs with treatment of a miR-223 mimic was significantly increased compared to that of the Ctrl group (Figure 4A). As expected, we found that the phosphorylation of AMPKα in VSMCs cocultured with DM-APs was significantly inhibited compared to that in VSMCs with treatment of HS-APs (Supplementary Figure S2). We found that the phosphorylation of acetyl-CoA Carboxylase (ACC), the substrate of AMPKα, was significantly higher in VSMCs treated with a miR-223 mimic than Ctrl group (Figure 4B). We also found that treatment with miR-223 mimic significantly increased the expression of p27 and p21 (Figures 4C,D), suggesting that VSMC proliferation was suppressed by miR-223. Taken together, these results indicated that the platelet-derived miR-223 suppressed VSMC proliferation via the regulation of miR-223/IGF-1R/AMPK signaling pathways.
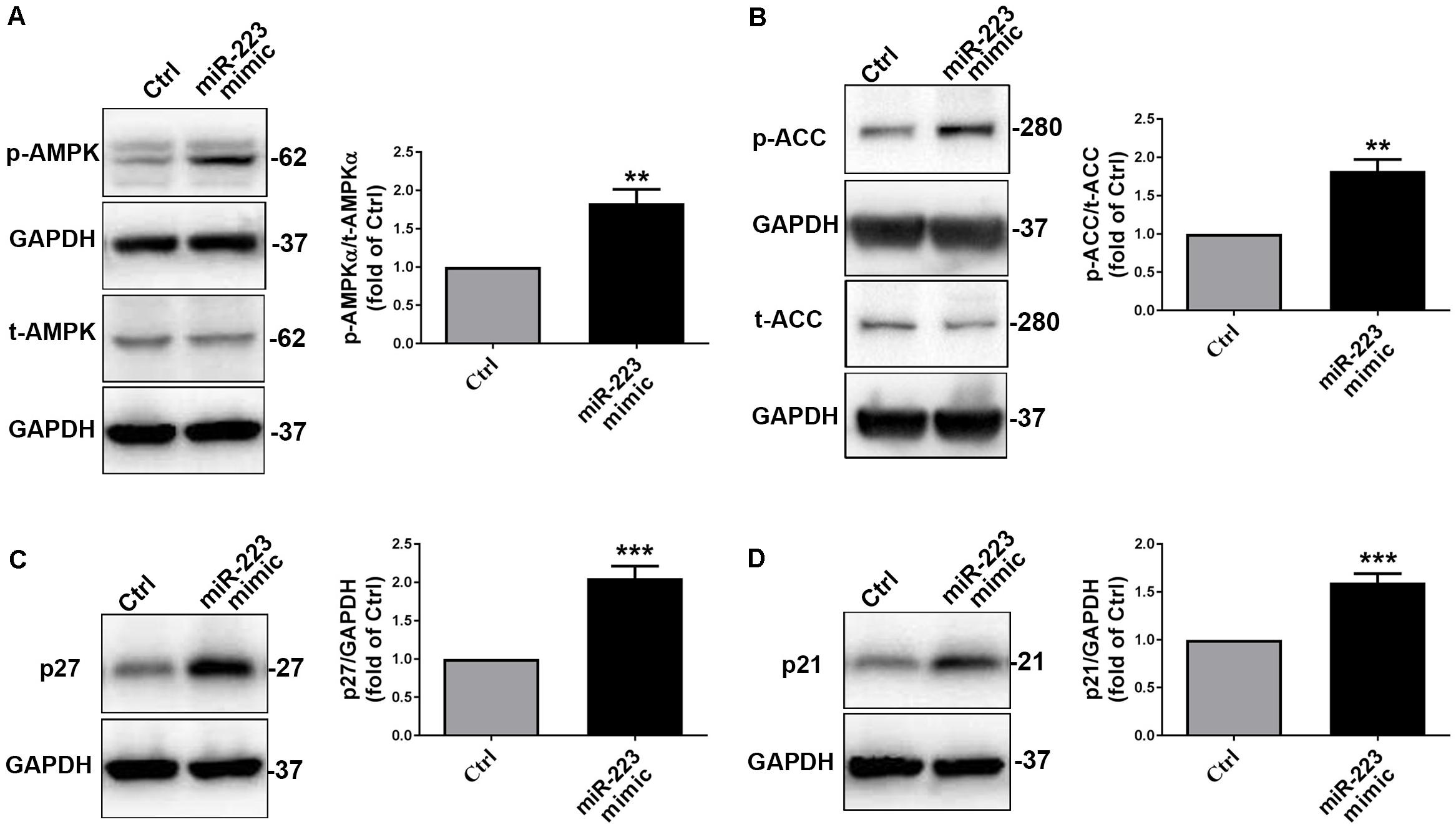
Figure 4. The platelet-derived miR-223 suppressed VSMC proliferation via the regulation AMPK signaling pathways. VSMCs were transfected with miR-223 mimic for 48 h. (A,B) The phosphorylation of AMPKα and ACC were assessed by western blot analysis in VSMCs transfected with miR-223 mimic. (C,D) The expression of cell cycle marks including p27 and p21 was determined with western blot analysis in VSMCs transfected with miR-223 mimic. Data are presented as mean ± SD (n = 6). Comparisons between two groups were analyzed using unpaired nonparametric Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001 vs Ctrl (miR-NC).
Our previous study has demonstrated a significant upregulation of miRNAs including miR-223, miR-143, and miR-145 in VSMCs cocultured with APs compared to VSMCs group. As shown in Figures 5A,B, the expression of miR-223 was downregulated in mouse and human diabetic platelets compared with non-DM group. We found that miR-223 level was significantly higher in diabetic platelets treated with a calpeptin (calpain inhibitor) than that of diabetic platelets (Figures 5C,D), suggesting that inhibition of calpain restores the expression of miR-223 in diabetic platelets. After restoring the expression of miR-223 in diabetic platelets and then coculturing with HCASMCs, we found the inhibition of Calpain restored the expression of miR-223 in diabetic platelets suppressed the proliferation of HCASMCs (Figures 5E,F). These data demonstrate that calpeptin restores the expression of miR-223 in diabetic platelets and transferred more platelet-derived miR-223 into VSMC, thereby suppressing VSMC proliferation in vitro.
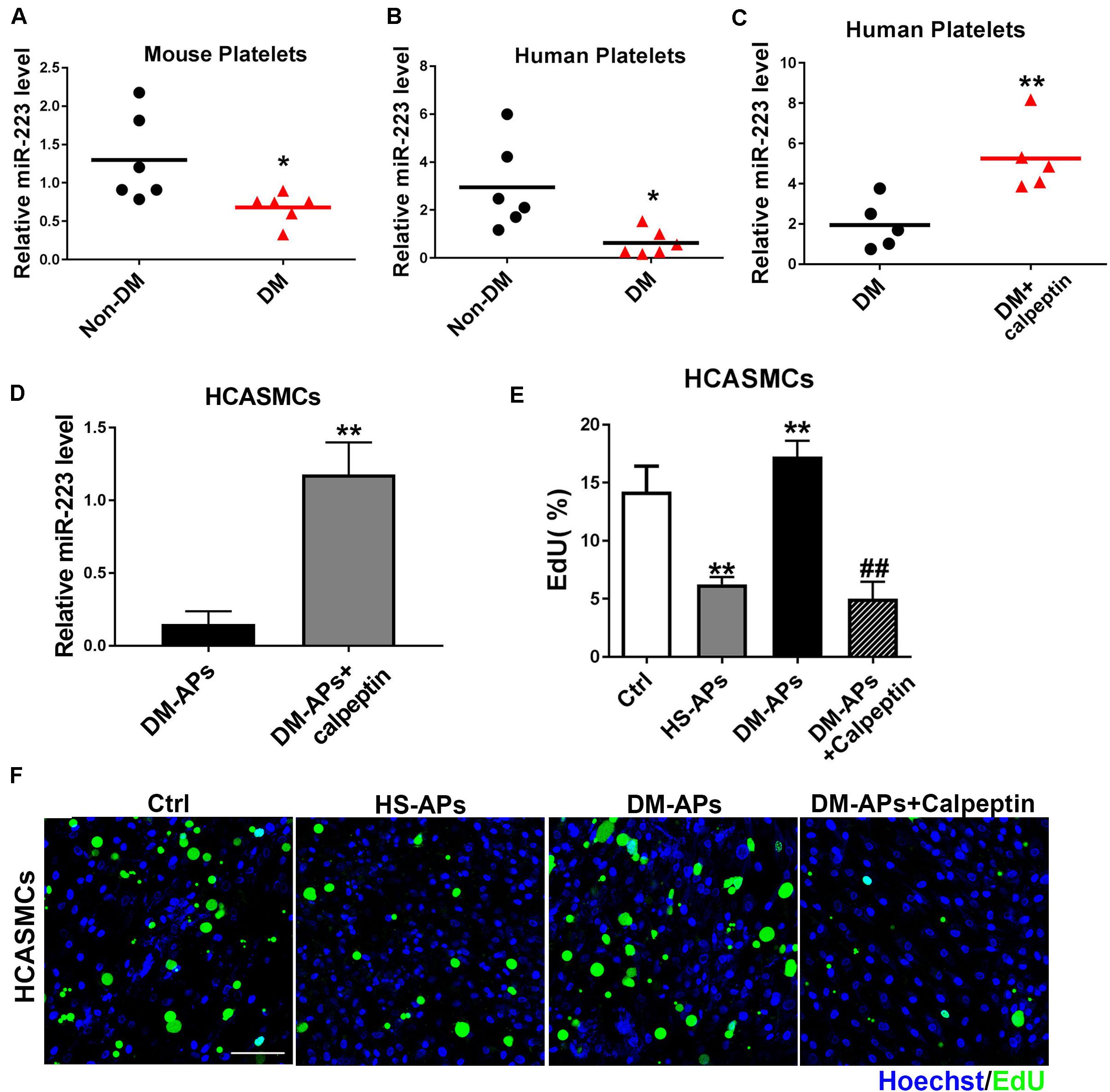
Figure 5. Treatment of a calpeptin restored the expression of miR-223 in diabetic platelets. (A,B) The miR-223 level in mouse and human platelets was detected by qRT-PCR assays. Data are presented as mean ± SD (n = 6). Comparisons between two groups were analyzed using unpaired nonparametric Student’s t-test. *P < 0.05 vs Non-DM. (C,D) Diabetic platelets were treated with a calpeptin for 24 h in vitro. The expression of miR-223 in human platelets and HCASMCs was detected by qRT-PCR assays. Data are presented as mean ± SD (n = 6). Comparisons between two groups were analyzed using unpaired nonparametric Student’s t-test. **P < 0.01 vs DM. (E,F) The rate of proliferation was determined by EdU incorporation assay in VSMCs treated with calpeptin. Data are presented as mean ± SD (n = 6). Comparisons between more than two groups were analyzed using one-way ANOVA analysis with the Dunnett’s multiple correction. **P < 0.01 vs Ctrl (VSMCs without platelets); ##P < 0.01 vs DM-APs.
Calpeptin is a specific pharmacological inhibitor for calpain activation, and administration of calpeptin restored the expression of miR-223 in diabetic murine platelet (Elgheznawy et al., 2015). To restore the downregulated miR-223 in diabetic platelet, we used streptozotocin induced (STZ-DM) mice and rescued the condition with a calpeptin and AgomiR-223 by femoral wire injury. Four weeks after injury, we found the expression of IGF-1R in uninjured vessels was reduced, while the expression of IGF-1R was upregulated in injured vessels of STZ-DM mice treated with miR-NC (miR negative control) (Figures 6A,C). Treatment with AgomiR-223 and calpeptin significantly suppressed the expression of IGF-1R in STZ-DM mice (Figure 6A). The results suggest that calpeptin and AgomiR-223 restored the expression of miR-223 in diabetic platelets to suppress VSMC proliferation via targeting IGF-1R in vivo. To verify the effect of calpeptin and AgomiR-223 on neointimal formation, the intimal to media (I/M ration) was detected in STZ-DM and Non-DM mice. As shown in Figures 6B,D, the neointimal hyperplasia and intimal size were increased in STZ-DM mice after 4 weeks of femoral arterial injury; however, the effect was rescued by treatment with calpeptin and AgomiR-223. We found that the expression of miR-223 was significantly downregulated in diabetic platelets of injured mice compared with non-DM injured group. However, the expression of miR-223 was restored by treatment with calpeptin and AgomiR-223, suggesting that inhibition of calpain restores the expression of miR-223 in injured DM mice (Figure 6E).Collectively, these data demonstrate that calpeptin restores the downregulated miR-223 in diabetic platelets and transferred more platelet-derived miR-223 into VSMC, thereby suppressing VSMC proliferation through targeting IGF-1R in the injured vessel, resulting in alleviation of neointimal formation in diabetes.
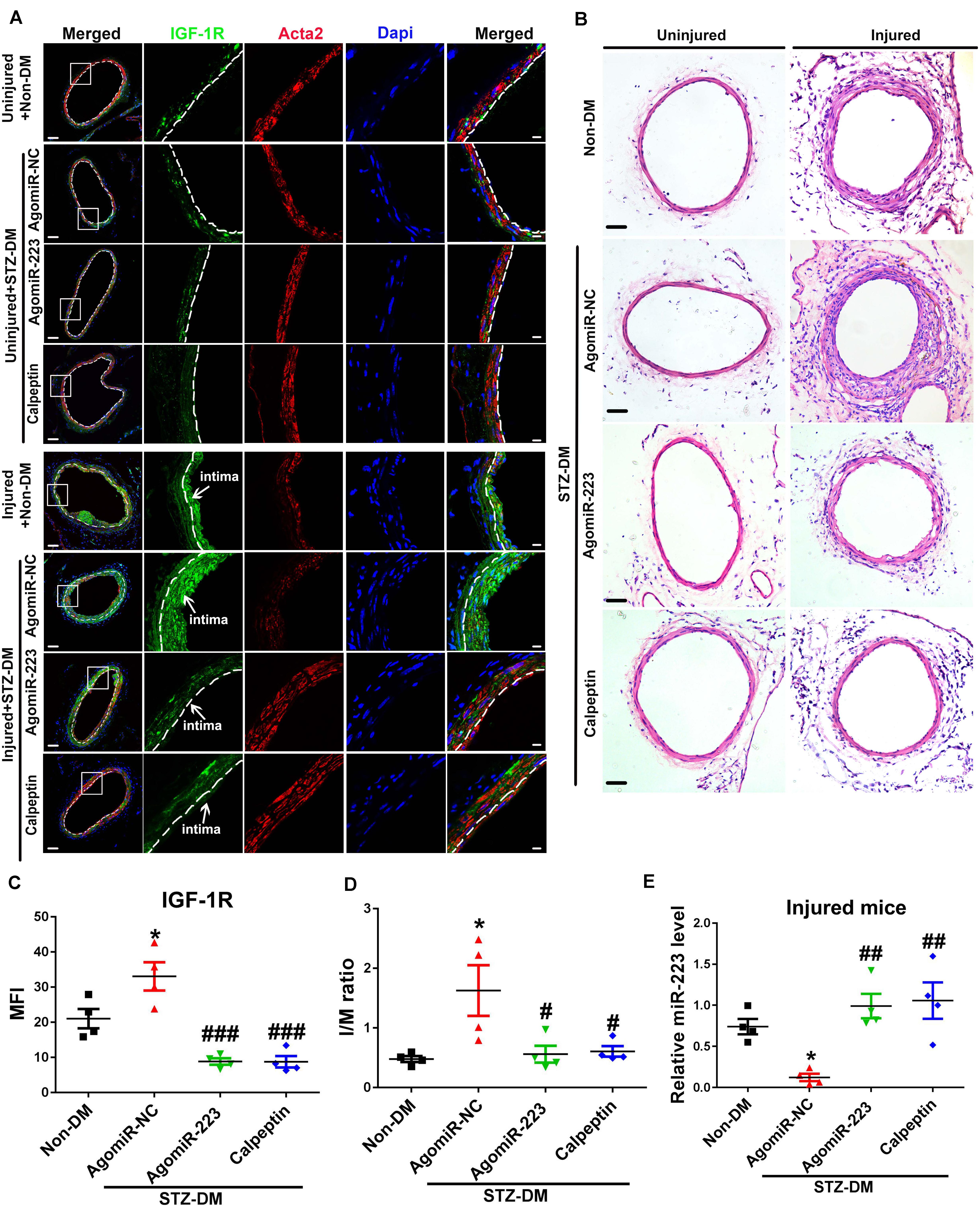
Figure 6. Inhibition of calpain alleviates neointima formation by restoring the expression of miR-223 in diabetic platelet. (A) Immunofluorescence analysis of IGF-1R in uninjured or injured femoral arteries from Non-DM mice, and STZ-DM mice treated with AgomiR-NC or AgomiR-223 or calpeptin at 4 weeks after wire injury (n = 4). Green, IGF-1R; red, Acta2 blue; Dapi nuclear staining in VSMCs. Scale bars: 20 μm. (B) H&E staining of serial cross sections of femoral arteries from Non-DM mice, and STZ-DM mice treated with AgomiR-NC or AgomiR-223 or calpeptin at 4 weeks after wire injury (n = 4). Scale bars: 50 μm. (C) Quantification of IGF-1R expression in VSMCs in injured DM mice. (D) The intima size (I/M ration) in the injured femoral arterial sections has been quantified. (E) The miR-223 level in injured DM mice was detected by qRT-PCR assays. Data are presented as mean ± SD (n = 4). Comparisons between more than two groups were analyzed using one-way ANOVA analysis with the Dunnett’s multiple correction. *P < 0.05 vs non-DM; #P < 0.05, ##P < 0.01, ###P < 0.001 vs STZ-DM mice treated with AgomiR-NC.
In summary, our data manifested that the platelet-derived miR-223 suppressed VSMC proliferation via the regulation IGF-1R/AMPK signaling pathway. However, treatment with calpeptin (calpain inhibitor) restored the expression of miR-223 in diabetic platelets, which was then transferred into VSMCs and inhibited VSMC proliferation at the site of vascular injury. Thus calpeptin may serve as therapeutic adjunct for playing a protective role in diabetic neointimal hyperplasia (Figure 7).
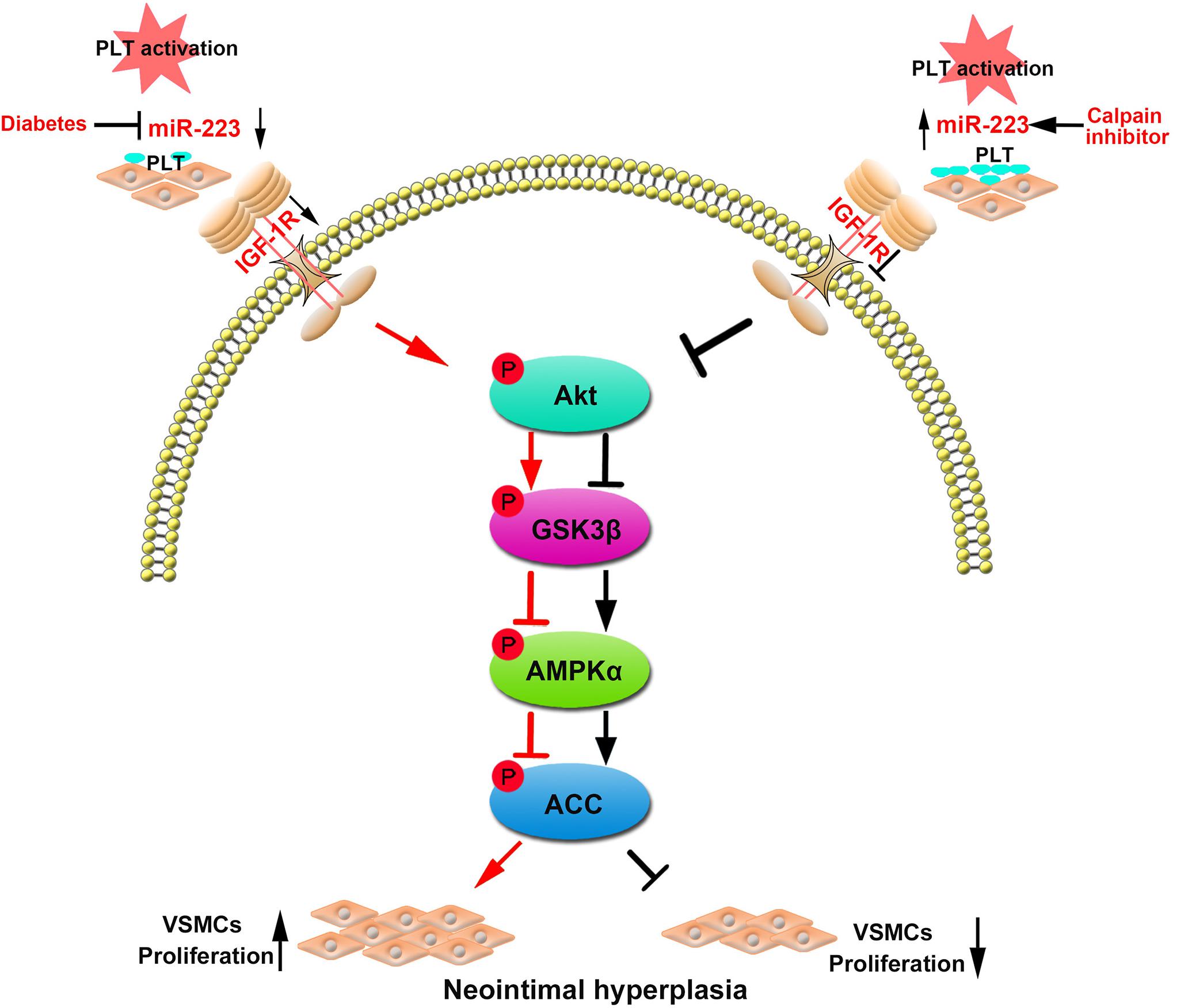
Figure 7. Restoring the platelet miR-223 by calpeptin alleviates the neointimal hyperplasia in diabetes through miR-223/IGF-1R/AMPK signaling pathway. MiR-223 deficiency in diabetic platelet failed to suppress VSMC proliferation during vascular injury. The platelet-derived miR-223 suppressed VSMC proliferation via the regulation IGF-1R/AMPK signaling pathways. Interestingly, treating with a calpeptin restores the downregulated expression of miR-223 in diabetic platelets to suppress VSMC proliferation via targeting IGF-1R and plays a protective role in diabetic neointimal hyperplasia.
In the present study, we demonstrate that diabetic platelet deficient of miR-223 fails to suppress VSMC phenotypic switch. The incorporated platelet miR-223 directly targets the insulin-like growth factor-1 receptor (IGF-1R) in VSMCs, which inhibits the phosphorylation of GSK3β and AMPK, resulting in reduced VSMC dedifferentiation and proliferation. In murine model of vascular injury, we show that calpeptin restores the platelet expression of miR-223 in diabetes, and the horizontal transfer of platelet miR-223 into VSMCs inhibits VSMC proliferation in the injured artery by targeting the expression of IGF-1R. Collectively, our results demonstrate that inhibition of calpain regulated VSMC phenotypic switch via miR-223/IGF-1R/AMPK signaling axis, supporting the potential application of calpeptin on platelet-associated vascular disease in diabetes.
Diabetes mellitus is an important risk factor of intimal hyperplasia and restenosis (Davi and Patrono, 2007). The phenotypic switch of VSMCs plays a crucial role in the onset of intimal hyperplasia in DM. The process of VSMC phenotypic switch (dedifferentiation and proliferation) was regulated by cytokines including insulin-like growth factor-1 (IGF-1), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF) (Delafontaine and Lou, 1993). Previous studies have reported miR-223 inhibited VSMC proliferation via target ITGB3 or RhoB/MLC2 in VSMCs (Zeng et al., 2016; Liu et al., 2019). Recently, our previous studies have demonstrated PDGFRβ is a key target of platelet-derived miR-223, which is incorporated into VSMCs and suppressed VSMC proliferation (Zeng et al., 2019). Previous studies have demonstrated treating with IGF-1 antibody inhibited VSMC proliferation, which was induced by angiotensin and thrombin (Delafontaine et al., 2004; Ma et al., 2006), suggesting that IGF-1 also plays a crucial role in VSMC proliferation (Jones and Kazlauskas, 2001). The binding of IGF-1 to its receptor (IGF-1R) can activate the cascade signals including IRS family, thereby activating phosphatidyl inositol 3-kinase (PI3K) and Akt signaling pathway (Saltiel and Kahn, 2001; Delafontaine et al., 2004). In the present study, we identified that IGF-1R was the direct target of miR-223, and platelet-derived miR-223 was incorporated into VSMCs thereby inhibiting IGF-1R, leading to the phosphorylation of Akt and GSK3β. Report showed PI3K-Akt signaling pathway could suppress glycogen synthase kinase 3 (GSK3) activity, leading to inhibit the function of AMPK via interactions with AMPKβ (Fukushima et al., 2012; Suzuki et al., 2013). It has been reported that activated phosphorylation of AMPKα to inhibit cells proliferation (Igata et al., 2005; Lee et al., 2015). Our phosphoproteomics array results showed that the incorporated platelet miR-223 significantly increased the phosphorylation of AMPKα and its substrate ACC in VSMCs cocultured with activated platelets. Therefore, platelet-derived miR-223 after incorporated into VSMCs at least in part suppress VSMC proliferation via targeting IGF-1R and activating AMPK to reduce neointimal hyperplasia in DM.
In diabetic platelets, the reduction of miRNAs leads to the upregulation of protein expression such as β1 integrin, resulting in increased platelet aggregation (Elgheznawy et al., 2015). Calpain family is intracellular Ca2+-activated cysteine proteases, which involved in cellular signaling, cell growth (Salimi et al., 2018), apoptosis (Storr et al., 2011), and platelet activation in DM (Elgheznawy et al., 2015). Increasing evidence has demonstrated calpain activation involved in cardiovascular complication of DM such as increased vascular permeability (Scalia et al., 2007), atherosclerosis (Miyazaki et al., 2011), and heart failure (Letavernier et al., 2012). The activation of calpain in DM is closely associated with the loss of platelet dicer, resulting in the significant dysregulation of platelet miRNA levels. Calpeptin is a specific pharmacological inhibitor for calpain activation, and administration of calpeptin restored the expression of miR-223 in diabetic murine platelet (Elgheznawy et al., 2015). Moreover, calpeptin was found to be an effective therapeutic drug for preventing endothelial dysfunction of mesenteric arteries in DM (Randriamboavonjy et al., 2017). Consistent with these studies, our results showed that the expression of miR-223 in diabetic platelets was restored by treatment of calpeptin. Thus, we employed calpeptin to treat STZ-DM murine model of vascular injury. We also found that calpeptin has inhibited the activative effect calpain on neointima hyperplasia in STZ-DM murine model of vascular injury. Both treatment with AgomiR-223 and calpeptin reduced neointimal hyperplasia and intimal size. At the site of injury, the increased IGF-1R expression in VSMCs was attenuated by treatment with AgomiR-223 and calpeptin. These results suggest that calpeptin may serve as therapeutic adjunct for intimal hyperplasia via the upregulation of platelet miR-223 in diabetes.
In summary, we provide evidence that the horizontal transfer of platelet miR-223 suppresses VSMC proliferation via targeting IGF-1R and activating AMPK. However, in diabetes, calpain activation contributes to the reduced expression of dicer, resulting in the deficiency of platelet miR-223. Treatment with calpeptin restored the expression of miR-223 in diabetic platelets, which was then transferred into VSMCs and inhibited VSMC proliferation at the site of vascular injury. Therefore, calpeptin may serve as therapeutic adjunct for platelet and VSMC associated vascular diseases in DM.
The datasets generated for this study are available on request to the corresponding author.
The studies involving human participants were reviewed and approved by Institutional Review Board of Guangzhou Women and Children’s Medical Center (Human Investigation Committee No. 2018022605). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by Institutional Animal Care and Use Committee of Guangzhou Medical University (No. SYXK2016-0168).
MS and SF performed the experimental design, the majority of experiments, analyzed majority of the data, and drafted manuscript. ZL and LX performed part of experiments and the data analyses. XF, YL, YZ, and SL performed animal experiments and image analyses. ZZ and WT directed all aspects of the experiments, the data analyses, manuscript editing and review. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81670117, 81970437, 81903605, and 81902144), the Guangdong Province Science and Technology Project (Grant No. 2017A050506008), the Guangdong Outstanding Young Scientist Funding (Grant No. 2016A030306049), and the fund from Guangzhou Women and Children’s Medical Center/Guangzhou Institute of Pediatrics (Grant No. IP-2018-022).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.00742/full#supplementary-material
Davi, G., and Patrono, C. (2007). Platelet activation and atherothrombosis. N. Engl. J. Med. 357, 2482–2494. doi: 10.1056/NEJMra071014
Delafontaine, P., and Lou, H. (1993). Angiotensin II regulates insulin-like growth factor I gene expression in vascular smooth muscle cells. J. Biol. Chem. 268, 16866–16870.
Delafontaine, P., Song, Y. H., and Li, Y. (2004). Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arterioscler. Thromb. Vasc. Biol. 24, 435–444. doi: 10.1161/01.ATV.0000105902.89459.09
Elgheznawy, A., Shi, L., Hu, J., Wittig, I., and Laban, H. (2015). Dicer cleavage by calpain determines platelet microRNA levels and function in diabetes. Circ. Res. 117, 157–165. doi: 10.1161/CIRCRESAHA.117.305784
Faries, P. L., Rohan, D. I., Takahara, H., Wyers, M. C., and Contreras, M. A. (2001). Human vascular smooth muscle cells of diabetic origin exhibit increased proliferation, adhesion, and migration. J. Vasc. Surg. 33, 601–607. doi: 10.1067/mva.2001.111806
Fukushima, T., Nakamura, Y., Yamanaka, D., Shibano, T., and Chida, K. (2012). Phosphatidylinositol 3-kinase (PI3K) activity bound to insulin-like growth factor-I (IGF-I) receptor, which is continuously sustained by IGF-I stimulation, is required for IGF-I-induced cell proliferation. J. Biol. Chem. 287, 29713–29721. doi: 10.1074/jbc.M112.393074
Igata, M., Motoshima, H., Tsuruzoe, K., Kojima, K., and Matsumura, T. (2005). Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ. Res. 97, 837–844. doi: 10.1161/01.RES.0000185823.73556.06
Jones, S. M., and Kazlauskas, A. (2001). Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat. Cell Biol. 3, 165–172. doi: 10.1038/35055073
Kirschbaum, M., Karimian, G., Adelmeijer, J., Giepmans, B. N., Porte, R. J., and Lisman, T. (2015). Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation. Blood 126, 798–806. doi: 10.1182/blood-2014-09-600312
Kornowski, R., Mintz, G. S., Kent, K. M., Pichard, A. D., and Satler, L. F. (1997). Increased restenosis in diabetes mellitus after coronary interventions is due to exaggerated intimal hyperplasia. A serial intravascular ultrasound study. Circulation 95, 1366–1369. doi: 10.1161/01.cir.95.6.1366
Landry, P., Plante, I., Ouellet, D. L., Perron, M. P., Rousseau, G., and Provost, P. (2009). Existence of a microRNA pathway in anucleate platelets. Nat. Struct. Mol. Biol. 16, 961–966. doi: 10.1038/nsmb.1651
Lee, K. Y., Lee, D. H., and Choi, H. C. (2015). Mesoglycan attenuates VSMC proliferation through activation of AMP-activated protein kinase and mTOR. Clin. Hypertens. 22:2. doi: 10.1186/s40885-016-0037-x
Letavernier, E., Zafrani, L., Perez, J., Letavernier, B., Haymann, J. P., and Baud, L. (2012). The role of calpains in myocardial remodelling and heart failure. Cardiovasc. Res. 96, 38–45. doi: 10.1093/cvr/cvs099
Liu, A., Liu, Y., Li, B., Yang, M., Liu, Y., and Su, J. (2019). Role of miR-223-3p in pulmonary arterial hypertension via targeting ITGB3 in the ECM pathway. Cell Prolif. 52:e12550. doi: 10.1111/cpr.12550
Ma, Y., Zhang, L., Peng, T., Cheng, J., and Taneja, S. (2006). Angiotensin II stimulates transcription of insulin-like growth factor I receptor in vascular smooth muscle cells: role of nuclear factor-kappaB. Endocrinology 147, 1256–1263. doi: 10.1210/en.2005-0888
Millette, E., Rauch, B. H., Defawe, O., Kenagy, R. D., Daum, G., and Clowes, A. W. (2005). Platelet-derived growth factor-BB-induced human smooth muscle cell proliferation depends on basic FGF release and FGFR-1 activation. Circ. Res. 96, 172–179. doi: 10.1161/01.RES.0000154595.87608.db
Miyazaki, T., Taketomi, Y., Takimoto, M., Lei, X. F., and Arita, S. (2011). m-Calpain induction in vascular endothelial cells on human and mouse atheromas and its roles in VE-cadherin disorganization and atherosclerosis. Circulation 124, 2522–2532. doi: 10.1161/CIRCULATIONAHA.111.021675
Nagalla, S., Shaw, C., Kong, X., Kondkar, A. A., and Edelstein, L. C. (2011). Platelet microRNA-mRNA coexpression profiles correlate with platelet reactivity. Blood 117, 5189–5197. doi: 10.1182/blood-2010-09-299719
Randriamboavonjy, V., Kyselova, A., Elgheznawy, A., Zukunft, S., Wittig, I., and Fleming, I. (2017). Calpain 1 cleaves and inactivates prostacyclin synthase in mesenteric arteries from diabetic mice. Basic Res. Cardiol. 112:10. doi: 10.1007/s00395-016-0596-8
Risitano, A., Beaulieu, L. M., Vitseva, O., and Freedman, J. E. (2012). Platelets and platelet-like particles mediate intercellular RNA transfer. Blood 119, 6288–6295. doi: 10.1182/blood-2011-12-396440
Salimi, R., Bandaru, S., Devarakonda, S., Gokalp, S., and Ala, C. (2018). Blocking the cleavage of filamin A by calpain inhibitor decreases tumor cell growth. Anticancer. Res. 38, 2079–2085. doi: 10.21873/anticanres.12447
Saltiel, A. R., and Kahn, C. R. (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806. doi: 10.1038/414799a
Scalia, R., Gong, Y., Berzins, B., Zhao, L. J., and Sharma, K. (2007). Hyperglycemia is a major determinant of albumin permeability in diabetic microcirculation: the role of mu-calpain. Diabetes Metab. Res. Rev. 56, 1842–1849. doi: 10.2337/db06-1198
Storr, S. J., Carragher, N. O., Frame, M. C., Parr, T., and Martin, S. G. (2011). The calpain system and cancer. Nat. Rev. Cancer 11, 364–374. doi: 10.1038/nrc3050
Suzuki, T., Bridges, D., Nakada, D., Skiniotis, G., and Morrison, S. J. (2013). Inhibition of AMPK catabolic action by GSK3. Mol. Cell. 50, 407–419. doi: 10.1016/j.molcel.2013.03.022
Zeng, Y., Zhang, X., Kang, K., Chen, J., and Wu, Z. (2016). MicroRNA-223 attenuates hypoxia-induced vascular remodeling by targeting RhoB/MLC2 in pulmonary arterial smooth muscle cells. Sci. Rep. 6:24900. doi: 10.1038/srep24900
Keywords: diabetes mellitus, platelet, vascular smooth muscle cells, miR-223, calpain, neointimal hyperplasia
Citation: Su M, Fan S, Ling Z, Fan X, Xia L, Liu Y, Li S, Zhang Y, Zeng Z and Tang WH (2020) Restoring the Platelet miR-223 by Calpain Inhibition Alleviates the Neointimal Hyperplasia in Diabetes. Front. Physiol. 11:742. doi: 10.3389/fphys.2020.00742
Received: 28 February 2020; Accepted: 08 June 2020;
Published: 07 July 2020.
Edited by:
Xavier Figueroa, Pontificia Universidad Católica de Chile, ChileReviewed by:
Ling-Qing Yuan, Second Xiangya Hospital, Central South University, ChinaCopyright © 2020 Su, Fan, Ling, Fan, Xia, Liu, Li, Zhang, Zeng and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi Zeng, emVuZ3poaUBnd2NtYy5vcmc=; Wai Ho Tang, d2FpaG8udGFuZ0B5YWhvby5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.