 Mingyue Zhao
Mingyue Zhao Dennis Ding-Hwa Wang
Dennis Ding-Hwa Wang Xiaojing Liu
Xiaojing Liu Rong Tian
Rong Tian- 1Laboratory of Cardiovascular Diseases, Regenerative Medicine Research Center, West China Hospital, Sichuan University, Chengdu, China
- 2Mitochondria and Metabolism Center, Department of Anesthesiology and Pain Medicine, University of Washington, Seattle, WA, United States
- 3Division of Cardiology, Department of Medicine, University of Washington, Seattle, WA, United States
- 4Department of Cardiology, West China Hospital, Sichuan University, Chengdu, China
Macrophages are key components of innate immunity, and they play critical roles in heart health and diseases. Following acute myocardial infarction (MI), infiltrating macrophages undergo drastic phenotypic transition from pro-inflammatory in the early stage to pro-healing in the late stage. Transcriptome analyses of macrophage in the infarct zone show a time-dependent reprogramming of mitochondrial and metabolic functions, which parallels the changes of macrophage function. These observations suggest that mitochondrial and metabolic targets could be exploited for therapeutic opportunities. In this article, we reviewed the recent work on immunometabolic features of macrophage over the MI time continuum. In addition, we summarized currently proposed mitochondrial pathways involved in the functional polarization of macrophage and discussed their potential relevance to the outcome of MI. We expect that these findings will stimulate further investigations in metabolic modulation of innate immunity in the post-MI setting, which could ultimately lead to new strategies for therapy.
Introduction
Ischemic heart disease is a leading cause of morbidity and mortality in developed countries. Cell death caused by myocardial infarction (MI) or ischemic/reperfusion (IR) injury invokes the recruitment of immune cells to the infarcted myocardium, which triggers inflammatory response in the early stage followed by wound healing and eventual scar formation (Van der Borght and Lambrecht, 2018; Bajpai et al., 2019). It has been shown that excessive inflammation and/or inadequate wound healing leads to poor clinical outcome after MI (Jia et al., 2019). Innate immune cells, particularly macrophages and monocytes, play a primary role in the tissue damage/repair process. Emerging studies suggest that macrophage function is intricately linked to its metabolic profile (Palmieri et al., 2017; Yurdagul et al., 2020). After acute MI (AMI), the majority of resident macrophages in the infarct zone die and are subsequently replenished by macrophages derived from circulating monocytes (Heidt et al., 2014; Ma et al., 2018). These macrophages undergo complex phenotypic changes that are central to post-MI healing. For example, infiltration and activation of macrophages in the infarcted region lead to strong inflammatory response, cytokine release, and further cell death immediately after MI (Mezzaroma et al., 2011; Chen et al., 2019). As the tissue repair commences, macrophage population evolves from pro-inflammatory to pro-healing, which promotes collagen deposition and extracellular matrix remodeling and scar formation (DeLeon-Pennell et al., 2017; Honold and Nahrendorf, 2018). Moreover, recent studies suggest that macrophages are critical mediators of the functional benefit associated with adjunctive cell therapy post-MI (de Couto et al., 2015; Vagnozzi et al., 2020). Therefore, regulatory mechanisms governing the transitions of macrophage functions during the post-MI period has recently become a fervid area of investigation. In this mini review, we will summarize the recent advances regarding the role of mitochondrial metabolism in regulating the phenotypic changes of macrophage post-MI.
Metabolic Reprogramming of Macrophage Post-MI
A global rewiring of metabolic pathways takes place in macrophages during the transition from a quiescent state to an activated state (Kelly and O’Neill, 2015). A hallmark of pro-inflammatory macrophage is a metabolic shift from oxidative metabolism toward glycolysis to meet the bioenergetic and biosynthetic demand (Rodriguez-Prados et al., 2010; Figure 1). In the case of pathogen invasion, upregulation of glycolysis rapidly generates ATP for activated macrophages. Furthermore, increased glycolytic intermediates feed into the pentose phosphate pathway (PPP) to produce NADPH, which is used to generate reactive oxygen species (ROS), through NADPH oxidase reaction, for bactericidal function (West et al., 2011a). On the other hand, mitochondrial oxidative metabolism in pro-inflammatory macrophages is suppressed, and the tricaboxylic acid (TCA) cycle flux is disrupted and rewired (Figure 1; Jha et al., 2015; Lampropoulou et al., 2016). In a sterile condition, infiltrating monocyte/macrophage in the infarct zone post-MI mounts a similar inflammatory response in the early stage post-MI (Lee et al., 2012). For instance, damage-associated molecular patterns (DAMPs) released by the dead cells activate toll-like receptors (TLRs) and promote glycolytic polarization of macrophages within the injured heart (Williams et al., 2018). The metabolic profile of macrophages in the infarct zone is not well described. However, a recent study examining macrophage transcriptome in the post-MI hearts showed a robust reprogramming of mitochondrial genes during the transition from tissue injury to repair (Mouton et al., 2018), suggesting that mitochondrial function could also be central to macrophage phenotype and cardiac remodeling post-MI.
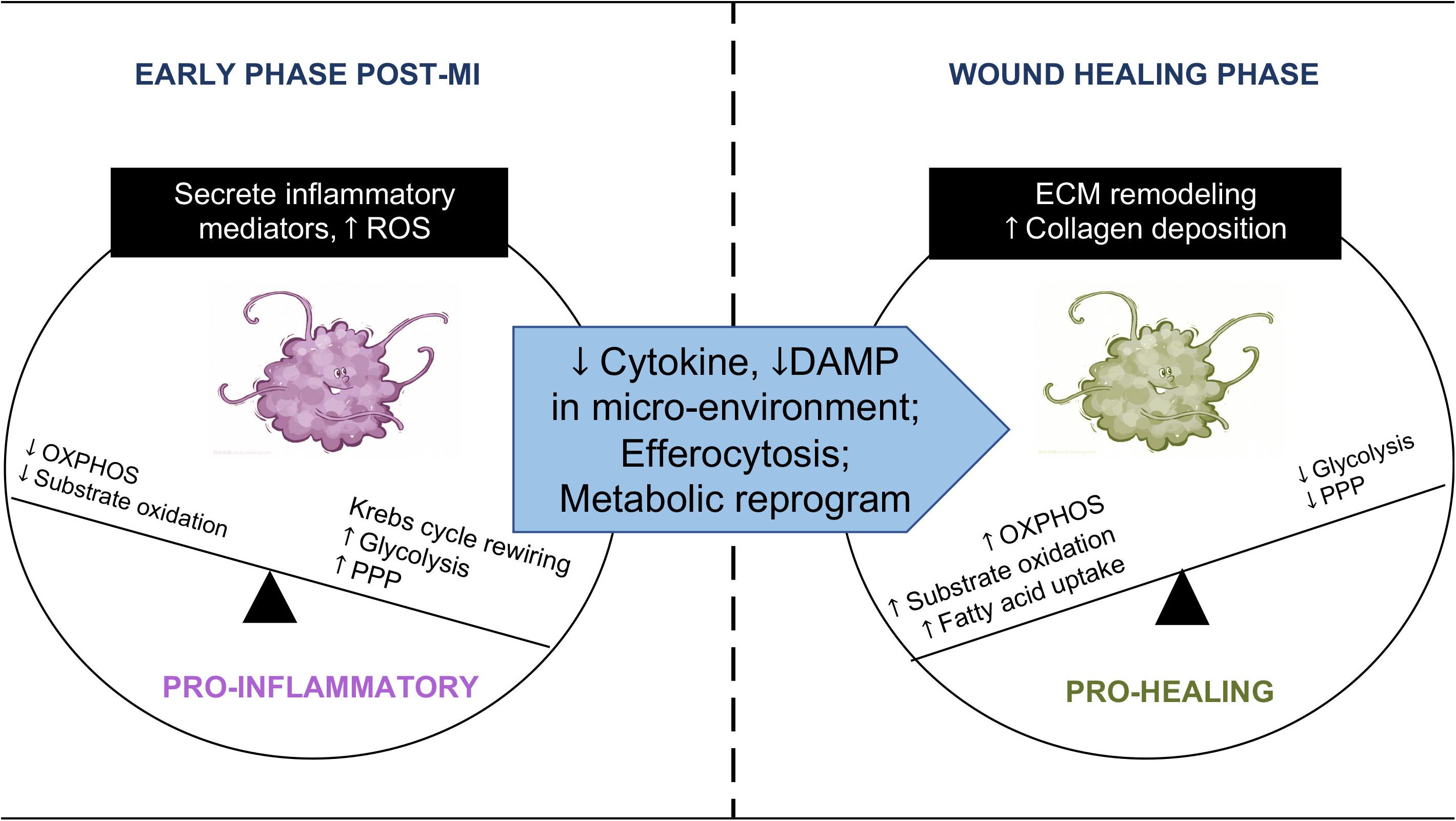
Figure 1. Immunometabolic transition of macrophage post-MI. Macrophages undergo an immunometabolic transition post-MI. At the early phase, macrophages secrete pro-inflammatory cytokines in the injured myocardium. Their energy metabolism shifts toward glycolysis and increased flux through the pentose phosphate pathway (PPP), accompanied by TCA cycle rewiring and a high level of mtROS. Substrate oxidation and ATP synthesis through OXPHOS are reduced. When the infarcted region enters the wound healing phase, macrophages become phagocytotic. Changes in the microenvironment, efferocytosis, and metabolic reprogramming promote the phenotype transition to pro-healing. Macrophages at this stage revert to oxidative metabolism and engage in collagen deposition and remodeling of the extracellular matrix (ECM).
Following MI, clearance of apoptotic cells by immune cells, i.e., efferocytosis, is a critical step leading to inflammation resolution and tissue repair (Serhan and Savill, 2005; Wan et al., 2013; Gordon, 2016). Distinct from microbial phagocytosis, efferocytosis after MI has been shown to fill the phagocytic macrophage with a heavy substrate load almost equal to the phagocyte itself (Zhang et al., 2019). The presence of metabolic cargo is accompanied by a marked elevation of oxygen consumption in phagocytic macrophages relative to non-efferocytes. In parallel, fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) are upregulated in phagocytic macrophages, while glycolysis decreases as cytokines and DAMPs decline in the microenvironment. These changes constitute a reversal of metabolic profile as the inflammation resolves (Park et al., 2011; Figure 1). The importance of mitochondria in macrophage is supported by a recent study showing poor wound healing and increased cardiac rupture in mice with myeloid-specific deficiency of Complex III in the electron transport chain (ETC) (Zhang et al., 2018). While these findings introduce a new role of mitochondria in cardiac repair and remodeling, they raise many questions in regard to mechanisms and therapeutic implications. In the section below, we will discuss briefly the proposed mechanisms linking mitochondrial metabolism to macrophage phenotype.
Role of Mitochondrial Metabolism in Modulating Macrophage Function
Besides producing ATP for the cell, mitochondria play multiple regulatory roles in cellular signaling, redox balance, cell growth, and survival (Zhou and Tian, 2018; Ritterhoff et al., 2020). Changes in mitochondrial function have been observed in activated macrophages, but the molecular mechanisms connecting mitochondrial function and macrophage phenotypes are not fully understood. In this section, we will summarize emerging findings on the role of mitochondrial ROS, intermediary metabolism, and NAD(H) redox state in modulating macrophage function, as well as the role of mitochondrial DNA (mtDNA) in triggering inflammatory response (Figure 2).
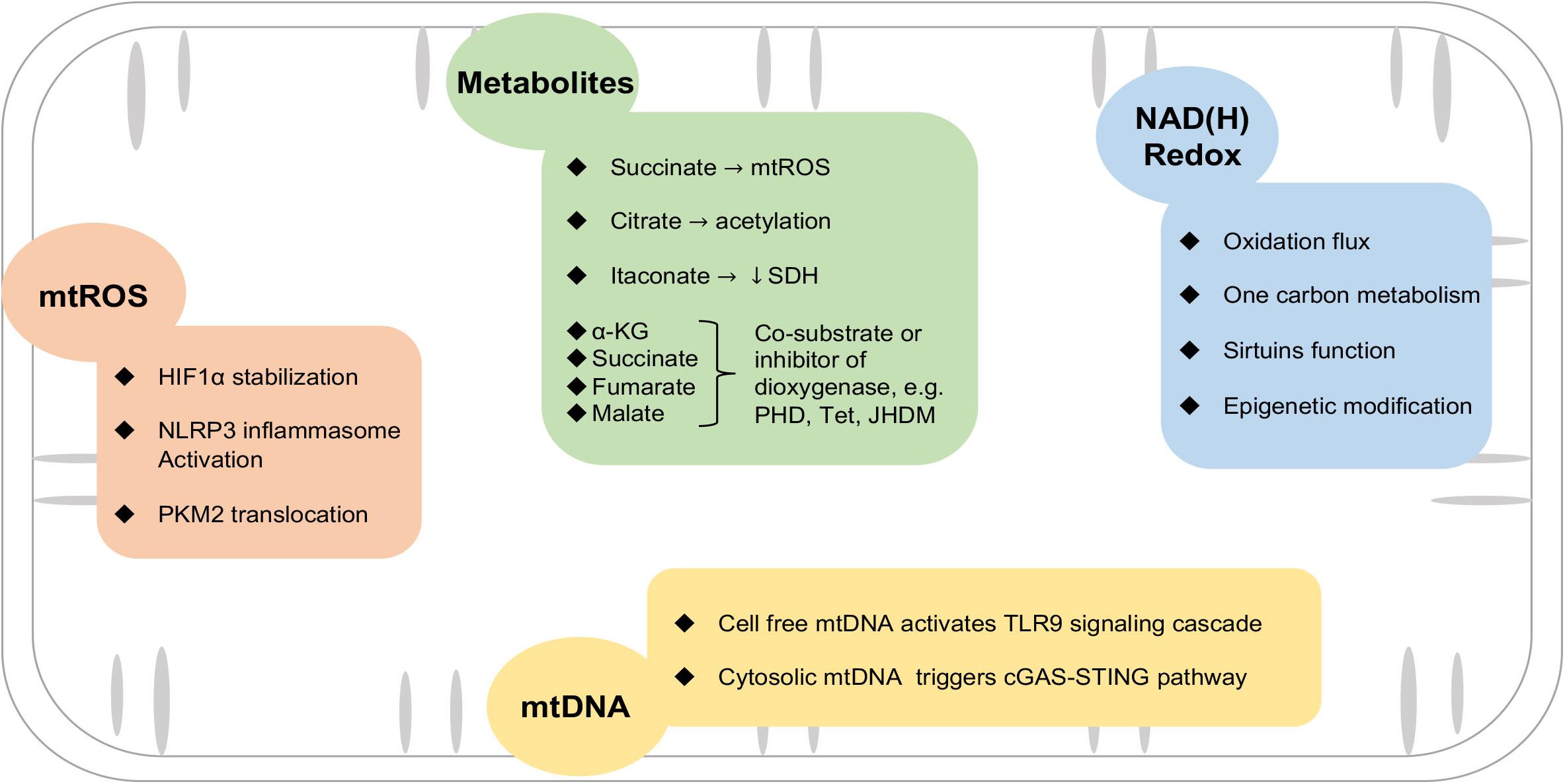
Figure 2. Mitochondrial mechanisms involved in macrophage activation. Mitochondria provide triggers and mediators of the inflammatory response in macrophages via mtROS, metabolites, NAD(H) redox balance, and leakage of mtDNA. See text for details. HIF1α, hypoxia-inducible factor 1α; SDH, succinate dehydrogenase; NLRP3, nod-like receptor family pyrin domain containing 3; cGAS-STING, GMP-AMP synthase/stimulator of interferon genes; PKM2, pyruvate kinase M2; JHDM, Jumonji domain-containing histone demethylase; PHD, prolyl hydroxylases; Tet, T5-methylcytosine hydroxylases.
Mitochondrial ROS
Reactive oxygen species is a by-product of mitochondrial respiratory activity. Under physiologic conditions, electron leakage in Complexes I and III of the ETC is a major source of ROS in mitochondria (mtROS) (Wong et al., 2017). Enzymes localized on the outer membrane of mitochondria, such as monoamine oxidase A, also generate ROS in activated macrophages, which has been thoroughly reviewed (Cathcart and Bhattacharjee, 2014). In pro-inflammatory macrophages, parallel studies show that impairment of TCA flux results in succinate accumulation via succinate oxidation by succinate dehydrogenase (SDH). Under conditions of reduced OXPHOS, succinate oxidation generates high mitochondrial membrane potential and drives the reverse electron transport (RET) at Complex I to produce ROS (Mills et al., 2016; Robb et al., 2018). In macrophages stimulated by lipopolysaccharide (LPS), succinate oxidation boosts mtROS generation and enhances interleukin-1β (IL-1β) production (Mills et al., 2016). It has been reported that the NOTCH signaling pathway is required to reprogram mitochondrial metabolism, thus leading to mtROS generation and pro-inflammatory gene expression (Xu et al., 2015). Blocking Complex I by alternative oxidase (AOX) or rotenone or scavenging mtROS by Mito-TEMPO inhibits inflammatory phenotype in macrophages (Jin et al., 2014; Scialo et al., 2017). Interestingly, blocking mtROS also reduces the population of anti-inflammatory macrophages with isoprenaline treatment (Shan et al., 2017), suggesting mtROS may modulate macrophage phenotype transition in a context-dependent fashion.
How does mtROS modulate inflammation? Increased mtROS is associated with increased gene expressions of pro-inflammatory cytokines, such as tumor necrosis factor α (TNFα) and IL-1β (van de Veerdonk et al., 2010; O’Neill and Pearce, 2016). mtROS could stabilize hypoxia-inducible factor 1α (HIF1α) through inhibiting the activity of prolyl hydroxylases (PHDs). Enhanced HIF1α is considered central to the upregulation of glycolysis and inflammatory cytokine expression (Calvani et al., 2012; Kelly and O’Neill, 2015). In monocytes and macrophages derived from atherosclerotic coronary artery disease patients, mtROS promotes dimerization and nuclear translocation of the glycolytic enzyme pyruvate kinase M2 (PKM2), phosphorylating the transcription factor signal transducer and activator of transcription (STAT3), thus boosting IL-6 and IL-1β production (Shirai et al., 2016). mtROS also serves as a key regulator of nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation (Afonina et al., 2017; Chen et al., 2017). In the early phase after AMI, NLRP3 senses danger signals both extracellularly and intracellularly, such as DAMPs, and assembles an inflammasome to mediate the downstream response (Mauro et al., 2019). On the other hand, mitochondrial antiviral protein (MAVS) facilitates the recruitment of NLRP3 to the mitochondria and hence enhances its oligomerization and activation by bringing it close to mtROS (Park et al., 2013; Figure 2).
Intermediary Metabolism
Disruption of the TCA cycle in the pro-inflammatory macrophages resulted in accumulations of metabolic intermediates such as citrate, succinate, fumarate, and malate (Ryan and O’Neill, 2017). As alluded to earlier, accumulation of succinate promotes mtROS generation via SDH. Succinate has also been shown to stabilize HIF1α by inhibiting the activity of PHD, an α-ketoglutarate (α-KG)-dependent dioxygenase. α-KG, on the contrary, depletes HIF1α by promoting the activity of PHD (Tannahill et al., 2013). The activity of other dioxygenases, e.g., Jumonji domain-containing histone demethylase (JHDM) or T5-methylcytosine hydroxylases (Tet), also requires α-KG as a co-substrate and can be inhibited by succinate, fumarate, or malate (Xu et al., 2011; Sinton et al., 2019; Figure 2). A recent study showed that increased glutaminolysis drove fumarate production via the TCA cycle, leading to enhanced TNFα and IL-6 expressions via boosting H3K4 trimethylation (H3K4me3) at the respective promoter regions (Arts et al., 2016). Moreover, α-KG derived from glutaminolysis is shown to be important for the alternative activation of macrophage via epigenetic regulations of pro-reparative genes (Liu et al., 2017).
Citrate transported from mitochondria can be converted to acetyl-CoA by ATP-citrate lyase (ACLY) in the cytosol. This mechanism has been implicated in the regulation of histone acetylation of cancer cells (Wellen et al., 2009). While upregulation of ACLY is observed in pro-inflammatory macrophages, it remains to be determined whether increased histone acetylation is responsible for the associated increase of inflammatory mediators, e.g., nitric oxide (NO), ROS, and prostaglandin E2 (PGE2) (Infantino et al., 2013). Another metabolite derived from the TCA cycle is itaconate, which is converted from aconitate (O’Neill and Artyomov, 2019). A recent study suggests that itaconate functions as an anti-inflammatory mediator in metabolic remodeling of macrophages via inhibition of SDH during IR injury (Lampropoulou et al., 2016).
In contrast to the pro-inflammatory macrophages, oxidative metabolism is robust in alternatively activated macrophages which phenotypically resemble reparative macrophages. During the clearance of injured tissues, phagocytes gain lipid load via efferocytosis, which likely stimulates fatty acid utilization. A recent study suggests that fatty acid is the preferred substrate in pro-healing macrophages after cardiac injury (Zhang et al., 2019). Increased fatty acid metabolism and associated activation of peroxisome proliferator-activated receptor (PPAR α/β/δ) in macrophages appear to play an important role in polarizing macrophages into a pro-reparative phenotype (Chawla, 2010). This led to the conjecture that the lipid load macrophages acquired via efferocytosis may potentially stimulate fatty acid utilization. Interestingly, elevated utilization of glucose is also necessary for pro-reparative macrophage polarization, and this may be mediated through increased production of UDP-GlcNAc (Jha et al., 2015; Huang et al., 2016). A major limitation in interpreting these observations is that most studies are done in vitro. Technical innovations, such as isolation procedures that preserve in vivo phenotype or genetic strategies for specific lineage tracing or perturbations, are required to allow for metabolic analysis of macrophages in vivo.
NAD(H) Redox Balance
Nicotinamide adenine dinucleotide (NAD+) accepts electrons from glycolysis, TCA cycle, and β-oxidation to form NADH, which then feeds its reducing potential to the ETC for OXPHOS. Therefore, NAD(H) redox is a major determinant of mitochondrial function and vice versa. Decreased OXPHOS lowers the NAD+/NADH ratio and reduces the availability of NAD+ for other NAD+-dependent proteins, such as poly ADP-ribose polymerase (PARP), cyclic ADP-ribose synthase, and sirtuins (Verdin, 2015; Katsyuba and Auwerx, 2017; Figure 2). Recently, Zhang et al. demonstrated that mitochondrial Complex III deficiency in macrophages impaired transcriptional activation of IL-10 induced by efferocytosis post-MI, resulting in poor healing and rupture of the infarcted ventricle. The defect was attributed to impaired Sirt1 function and could be rescued by supplementing nicotinamide mononucleotide (NMN), a NAD+ precursor (Zhang et al., 2019), suggesting that NAD(H) redox imbalance caused by mitochondrial dysfunction is an important regulatory mechanism of immune response. Impairment of the Sirt2 function due to NAD+ depletion leads to the hyperacetylation of α-tubulin, which in turn promotes colocalization of apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and NLRP3, and drives IL-1β production in macrophages (Zhang et al., 2018). NAD(H) redox balance is also a powerful regulator of epigenetics, likely via supporting the activity of sirtuin deacetylases (Class III HADCs) on histone (Halili et al., 2010; Ciarlo and Roger, 2016). Recent studies show that DNA and histone methylation also regulate macrophage function (Yu et al., 2019; Li et al., 2020). One carbon metabolism, which supplies the methyl group for methylation, is regulated by mitochondrial function and the NAD+/NADH ratio (Tibbetts and Appling, 2010; Kuhl et al., 2017). Together, these observations substantiate the role of NAD(H) redox in the modulation of innate immunity (Tibbetts and Appling, 2010; Zhang et al., 2019).
mtDNA Is a Potential Pro-inflammatory Mediator of Post-MI Macrophage
Increased levels of circulating cell-free mtDNA (CCF-DNA) have been observed in a number of disease conditions including MI (West and Shadel, 2017; Nakayama and Otsu, 2018). Due to the structural features resembling prokaryotic DNA, mtDNA is a potent pro-inflammatory trigger for immune cells in sterile inflammation (Collins et al., 2004) and is widely accepted as an integral member of the DAMPs. Hypomethylated CpG-rich mtDNA serves as an endogenous ligand for TLR9 to trigger the innate immune system governed by the NLRP3 inflammasome in response to cellular stress, infection, and AMI injury (West et al., 2011b). In the canonical pathway of sterile inflammation, upon sensing extracellular mtDNA released during necrotic tissue death, TLR9 recruits adaptor proteins MyD88 and TRIF to activate transcription factors NF-kB and NLRP3 to promote production, maturation, and secretion of pro-inflammatory cytokines, such as IL-1β, IL-6, and IL-18, to amplify the inflammatory signals (Chen and Nunez, 2010; Nakayama and Otsu, 2018).
The presence of mtDNA in the cytosol also triggers inflammation. Defective autophagy in bone-marrow-derived macrophages (BMDMs) resulted in enhanced cytosolic translocation of mtDNA via the mitochondrial membrane permeability transition pore (MPTP) in an mtROS- and NLRP3-dependent fashion to activate caspase 1, promoting IL-1β and IL-18 secretions (Nakahira et al., 2011). Key players in autophagy, Beclin-1 and LC3, have been found critical in the regulation of macrophage phagocytosis and efferocytosis, as well as the clearance of apoptotic cells, inflammatory resolution, and tissue repair following MI (Konishi et al., 2012; Heckmann et al., 2017; Heckmann and Green, 2019). More recently, cytidine/uridine monophosphate kinase 2 (CMPK2)-dependent mtDNA synthesis was shown to be required for NLRP3 inflammasome activation in LPS-primed macrophages, in that the newly synthesized mtDNA was oxidized by mtROS and translocated to the cytosol to activate NLRP3 inflammasome via direct binding (Zhong et al., 2018). Together, these studies demonstrate the co-dependence of mtDNA’s cytosolic translocation and the activation of NLRP3/caspase I axis of macrophage in a pro-inflammatory milieu such as the early phase of the post-MI myocardium.
GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) is another cytosolic mtDNA-sensing pathway which governs the gene expressions of interferons, such as CD14, CXCL10, and IRFs. In dendritic cells and macrophages, cGAS can be activated by cytoplasmic mtDNA to resist infection (Carroll et al., 2016; Chen et al., 2016). In the context of mtDNA stress induced by transcription factor A, mitochondrial (TFAM) deficiency in BMDMs, aberrant mtDNA packaging results in the cytosolic translocation of mtDNA. Upon sensing cytosolic fragmented mtDNA, cGAS activates STING, which then activates TBK1, resulting in the translation of interferon genes (West et al., 2015). Recently, cGAS-dependent mtDNA sensing has been demonstrated to mediate macrophage polarization. cGAS–/– mice displayed an improved outcome after MI, which is associated with an augmented reparative macrophage population. However, a loss of cGAS does not alter the mRNA levels of canonical cytokines (Cao et al., 2018). Together, these findings suggest that cGAS/STING is a pro-inflammatory pathway parallel to the TLR9/NF-kB axis to sense aberrant cytosolic mtDNA in post-MI macrophages.
Conclusion and Perspectives
As discussed above, mitochondrial metabolism is a key regulator of macrophage response. Substrate metabolism, mtROS, and NAD(H) redox state influence macrophage phenotype through epigenetic, transcriptional, and posttranscriptional mechanisms. Microenvironment, such as oxygen and nutrient availability, DAMPs, and metabolic cargo from cell debris, also modulates functional properties of macrophage via mitochondria and metabolic mechanisms. These observations not only provide mechanistic links between mitochondrial responses and the functional transition of macrophages during the remodeling after MI but also suggest a novel class of targets for therapy. Given the broad implication of innate immunity in human diseases, these findings will drive future work to identify molecular mechanisms connecting mitochondria and immune cell functions in a wide variety of diseases.
Author Contributions
MZ wrote the manuscript. DW and RT edited the manuscript. RT and XL supervised the work and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
MZ was supported by a scholarship from the Chinese Scholarship Council. DW was supported by a postdoc fellowship from the American Heart Association 18POST34030098.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Afonina, I. S., Zhong, Z., Karin, M., and Beyaert, R. (2017). Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat. Immunol. 18, 861–869. doi: 10.1038/ni.3772
Arts, R. J., Novakovic, B., Ter Horst, R., Carvalho, A., Bekkering, S., Lachmandas, E., et al. (2016). Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 24, 807–819. doi: 10.1016/j.cmet.2016.10.008
Bajpai, G., Bredemeyer, A., Li, W., Zaitsev, K., Koenig, A. L., Lokshina, I., et al. (2019). Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 124, 263–278. doi: 10.1161/CIRCRESAHA.118.314028
Calvani, M., Comito, G., Giannoni, E., and Chiarugi, P. (2012). Time-dependent stabilization of hypoxia inducible factor-1alpha by different intracellular sources of reactive oxygen species. PLoS One 7:e38388. doi: 10.1371/journal.pone.0038388
Cao, D. J., Schiattarella, G. G., Villalobos, E., Jiang, N., May, H. I., Li, T., et al. (2018). Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 137, 2613–2634. doi: 10.1161/CIRCULATIONAHA.117.031046
Carroll, E. C., Jin, L., Mori, A., Munoz-Wolf, N., Oleszycka, E., Moran, H. B. T., et al. (2016). The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS-STING-dependent induction of type I interferons. Immunity 44, 597–608. doi: 10.1016/j.immuni.2016.02.004
Cathcart, M. K., and Bhattacharjee, A. (2014). Monoamine oxidase A (MAO-A): a signature marker of alternatively activated monocytes/macrophages. Inflamm. Cell Signal. 1:e161. doi: 10.14800/ics.161
Chawla, A. (2010). Control of macrophage activation and function by PPARs. Circ. Res. 106, 1559–1569. doi: 10.1161/CIRCRESAHA.110.216523
Chen, B., Huang, S., Su, Y., Wu, Y. J., Hanna, A., Brickshawana, A., et al. (2019). Macrophage Smad3 protects the infarcted heart, stimulating phagocytosis and regulating inflammation. Circ. Res. 125, 55–70. doi: 10.1161/CIRCRESAHA.119.315069
Chen, G. Y., and Nunez, G. (2010). Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837. doi: 10.1038/nri2873
Chen, M. L., Zhu, X. H., Ran, L., Lang, H. D., Yi, L., and Mi, M. T. (2017). Trimethylamine-N-Oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J. Am. Heart Assoc. 6:e006347. doi: 10.1161/JAHA.117.006347
Chen, Q., Sun, L., and Chen, Z. J. (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149. doi: 10.1038/ni.3558
Ciarlo, E., and Roger, T. (2016). Screening the impact of sirtuin inhibitors on inflammatory and innate immune responses of macrophages and in a mouse model of endotoxic shock. Methods Mol. Biol. 1436, 313–334. doi: 10.1007/978-1-4939-3667-0_21
Collins, L. V., Hajizadeh, S., Holme, E., Jonsson, I. M., and Tarkowski, A. (2004). Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 75, 995–1000. doi: 10.1189/jlb.0703328
de Couto, G., Liu, W., Tseliou, E., Sun, B., Makkar, N., Kanazawa, H., et al. (2015). Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J. Clin. Invest. 125, 3147–3162. doi: 10.1172/JCI81321
DeLeon-Pennell, K. Y., Iyer, R. P., Ero, O. K., Cates, C. A., Flynn, E. R., Cannon, P. L., et al. (2017). Periodontal-induced chronic inflammation triggers macrophage secretion of Ccl12 to inhibit fibroblast-mediated cardiac wound healing. JCI Insight 2:e94207. doi: 10.1172/jci.insight.94207
Gordon, S. (2016). Phagocytosis: the legacy of metchnikoff. Cell 166, 1065–1068. doi: 10.1016/j.cell.2016.08.017
Halili, M. A., Andrews, M. R., Labzin, L. I., Schroder, K., Matthias, G., Cao, C., et al. (2010). Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 87, 1103–1114. doi: 10.1189/jlb.0509363
Heckmann, B. L., Boada-Romero, E., Cunha, L. D., Magne, J., and Green, D. R. (2017). LC3-associated phagocytosis and inflammation. J. Mol. Biol. 429, 3561–3576. doi: 10.1016/j.jmb.2017.08.012
Heckmann, B. L., and Green, D. R. (2019). LC3-associated phagocytosis at a glance. J. Cell Sci. 132:jcs222984. doi: 10.1242/jcs.222984
Heidt, T., Courties, G., Dutta, P., Sager, H. B., Sebas, M., Iwamoto, Y., et al. (2014). Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 115, 284–295. doi: 10.1161/CIRCRESAHA.115.303567
Honold, L., and Nahrendorf, M. (2018). Resident and monocyte-derived macrophages in cardiovascular disease. Circ. Res. 122, 113–127. doi: 10.1161/CIRCRESAHA.117.311071
Huang, S. C., Smith, A. M., Everts, B., Colonna, M., Pearce, E. L., Schilling, J. D., et al. (2016). Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity 45, 817–830. doi: 10.1016/j.immuni.2016.09.016
Infantino, V., Iacobazzi, V., Palmieri, F., and Menga, A. (2013). ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 440, 105–111. doi: 10.1016/j.bbrc.2013.09.037
Jha, A. K., Huang, S. C., Sergushichev, A., Lampropoulou, V., Ivanova, Y., Loginicheva, E., et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. doi: 10.1016/j.immuni.2015.02.005
Jia, D., Jiang, H., Weng, X., Wu, J., Bai, P., Yang, W., et al. (2019). Interleukin-35 promotes macrophage survival and improves wound healing after myocardial infarction in mice. Circ. Res. 124, 1323–1336. doi: 10.1161/CIRCRESAHA.118.314569
Jin, Z., Wei, W., Yang, M., Du, Y., and Wan, Y. (2014). Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 20, 483–498. doi: 10.1016/j.cmet.2014.07.011
Katsyuba, E., and Auwerx, J. (2017). Modulating NAD(+) metabolism, from bench to bedside. EMBO J. 36, 2670–2683. doi: 10.15252/embj.201797135
Kelly, B., and O’Neill, L. A. (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 25, 771–784. doi: 10.1038/cr.2015.68
Konishi, A., Arakawa, S., Yue, Z., and Shimizu, S. (2012). Involvement of Beclin 1 in engulfment of apoptotic cells. J. Biol. Chem. 287, 13919–13929. doi: 10.1074/jbc.M112.348375
Kuhl, I., Miranda, M., Atanassov, I., Kuznetsova, I., Hinze, Y., Mourier, A., et al. (2017). Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 6:e30952. doi: 10.7554/eLife.30952
Lampropoulou, V., Sergushichev, A., Bambouskova, M., Nair, S., Vincent, E. E., Loginicheva, E., et al. (2016). Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166. doi: 10.1016/j.cmet.2016.06.004
Lee, W. W., Marinelli, B., van der Laan, A. M., Sena, B. F., Gorbatov, R., Leuschner, F., et al. (2012). PET/MRI of inflammation in myocardial infarction. J. Am. Coll. Cardiol. 59, 153–163. doi: 10.1016/j.jacc.2011.08.066
Li, T., Garcia-Gomez, A., Morante-Palacios, O., Ciudad, L., Ozkaramehmet, S., Van Dijck, E., et al. (2020). SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Res. 48, 665–681. doi: 10.1093/nar/gkz1127
Liu, P. S., Wang, H., Li, X., Chao, T., Teav, T., Christen, S., et al. (2017). alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18, 985–994. doi: 10.1038/ni.3796
Ma, Y., Mouton, A. J., and Lindsey, M. L. (2018). Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 191, 15–28. doi: 10.1016/j.trsl.2017.10.001
Mauro, A. G., Bonaventura, A., Mezzaroma, E., Quader, M., and Toldo, S. (2019). NLRP3 inflammasome in acute myocardial infarction. J. Cardiovasc. Pharmacol. 74, 175–187. doi: 10.1097/FJC.0000000000000717
Mezzaroma, E., Toldo, S., Farkas, D., Seropian, I. M., Van Tassell, B. W., Salloum, F. N., et al. (2011). The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. U.S.A. 108, 19725–19730. doi: 10.1073/pnas.1108586108
Mills, E. L., Kelly, B., Logan, A., Costa, A. S. H., Varma, M., Bryant, C. E., et al. (2016). Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e13. doi: 10.1016/j.cell.2016.08.064
Mouton, A. J., DeLeon-Pennell, K. Y., Rivera Gonzalez, O. J., Flynn, E. R., Freeman, T. C., Saucerman, J. J., et al. (2018). Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 113:26. doi: 10.1007/s00395-018-0686-x
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
Nakayama, H., and Otsu, K. (2018). Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem. J. 475, 839–852. doi: 10.1042/BCJ20170714
O’Neill, L. A., and Pearce, E. J. (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213, 15–23. doi: 10.1084/jem.20151570
O’Neill, L. A. J., and Artyomov, M. N. (2019). Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 19, 273–281. doi: 10.1038/s41577-019-0128-5
Palmieri, E. M., Menga, A., Martin-Perez, R., Quinto, A., Riera-Domingo, C., De Tullio, G., et al. (2017). Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 20, 1654–1666. doi: 10.1016/j.celrep.2017.07.054
Park, D., Han, C. Z., Elliott, M. R., Kinchen, J. M., Trampont, P. C., Das, S., et al. (2011). Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 477, 220–224. doi: 10.1038/nature10340
Park, S., Juliana, C., Hong, S., Datta, P., Hwang, I., Fernandes-Alnemri, T., et al. (2013). The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 191, 4358–4366. doi: 10.4049/jimmunol.1301170
Ritterhoff, J., Young, S., Villet, O., Shao, D., Neto, F. C., Bettcher, L. F., et al. (2020). Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ. Res. 126, 182–196. doi: 10.1161/CIRCRESAHA.119.315483
Robb, E. L., Hall, A. R., Prime, T. A., Eaton, S., Szibor, M., Viscomi, C., et al. (2018). Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 293, 9869–9879. doi: 10.1074/jbc.RA118.003647
Rodriguez-Prados, J. C., Traves, P. G., Cuenca, J., Rico, D., Aragones, J., Martin-Sanz, P., et al. (2010). Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J. Immunol. 185, 605–614. doi: 10.4049/jimmunol.0901698
Ryan, D. G., and O’Neill, L. A. J. (2017). Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 591, 2992–3006. doi: 10.1002/1873-3468.12744
Scialo, F., Fernandez-Ayala, D. J., and Sanz, A. (2017). Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front. Physiol. 8:428. doi: 10.3389/fphys.2017.00428
Serhan, C. N., and Savill, J. (2005). Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197. doi: 10.1038/ni1276
Shan, M., Qin, J., Jin, F., Han, X., Guan, H., Li, X., et al. (2017). Autophagy suppresses isoprenaline-induced M2 macrophage polarization via the ROS/ERK and mTOR signaling pathway. Free Radic. Biol. Med. 110, 432–443. doi: 10.1016/j.freeradbiomed.2017.05.021
Shirai, T., Nazarewicz, R. R., Wallis, B. B., Yanes, R. E., Watanabe, R., Hilhorst, M., et al. (2016). The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 213, 337–354. doi: 10.1084/jem.20150900
Sinton, M. C., Hay, D. C., and Drake, A. J. (2019). Metabolic control of gene transcription in non-alcoholic fatty liver disease: the role of the epigenome. Clin. Epigenetics 11:104. doi: 10.1186/s13148-019-0702-5
Tannahill, G. M., Curtis, A. M., Adamik, J., Palsson-McDermott, E. M., McGettrick, A. F., Goel, G., et al. (2013). Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242. doi: 10.1038/nature11986
Tibbetts, A. S., and Appling, D. R. (2010). Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81. doi: 10.1146/annurev.nutr.012809.104810
Vagnozzi, R. J., Maillet, M., Sargent, M. A., Khalil, H., Johansen, A. K. Z., Schwanekamp, J. A., et al. (2020). An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577, 405–409. doi: 10.1038/s41586-019-1802-2
van de Veerdonk, F. L., Smeekens, S. P., Joosten, L. A., Kullberg, B. J., Dinarello, C. A., van der Meer, J. W., et al. (2010). Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc. Natl. Acad. Sci. U.S.A. 107, 3030–3033. doi: 10.1073/pnas.0914795107
Van der Borght, K., and Lambrecht, B. N. (2018). Heart macrophages and dendritic cells in sickness and in health: a tale of a complicated marriage. Cell Immunol. 330, 105–113. doi: 10.1016/j.cellimm.2018.03.011
Verdin, E. (2015). NAD(+) in aging, metabolism, and neurodegeneration. Science 350, 1208–1213. doi: 10.1126/science.aac4854
Wan, E., Yeap, X. Y., Dehn, S., Terry, R., Novak, M., Zhang, S., et al. (2013). Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 113, 1004–1012. doi: 10.1161/CIRCRESAHA.113.301198
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R., and Thompson, C. B. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. doi: 10.1126/science.1164097
West, A. P., Brodsky, I. E., Rahner, C., Woo, D. K., Erdjument-Bromage, H., Tempst, P., et al. (2011a). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480. doi: 10.1038/nature09973
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
West, A. P., and Shadel, G. S. (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375. doi: 10.1038/nri.2017.21
West, A. P., Shadel, G. S., and Ghosh, S. (2011b). Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402. doi: 10.1038/nri2975
Williams, A. L., Khadka, V., Tang, M., Avelar, A., Schunke, K. J., Menor, M., et al. (2018). HIF1 mediates a switch in pyruvate kinase isoforms after myocardial infarction. Physiol. Genomics 50, 479–494. doi: 10.1152/physiolgenomics.00130.2017
Wong, H. S., Dighe, P. A., Mezera, V., Monternier, P. A., and Brand, M. D. (2017). Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809. doi: 10.1074/jbc.R117.789271
Xu, J., Chi, F., Guo, T., Punj, V., Lee, W. N., French, S. W., et al. (2015). NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J. Clin. Invest. 125, 1579–1590. doi: 10.1172/JCI76468
Xu, W., Yang, H., Liu, Y., Yang, Y., Wang, P., Kim, S. H., et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. doi: 10.1016/j.ccr.2010.12.014
Yu, W., Wang, Z., Zhang, K., Chi, Z., Xu, T., Jiang, D., et al. (2019). One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol Cell 75, 1147–1160.e5. doi: 10.1016/j.molcel.2019.06.039
Yurdagul, A. Jr., Subramanian, M., Wang, X., Crown, S. B., Ilkayeva, O. R., Darville, L., et al. (2020). Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. 31, 518–533.e10. doi: 10.1016/j.cmet.2020.01.001
Zhang, B., Xu, D., She, L., Wang, Z., Yang, N., Sun, R., et al. (2018). Silybin inhibits NLRP3 inflammasome assembly through the NAD(+)/SIRT2 pathway in mice with nonalcoholic fatty liver disease. FASEB J. 32, 757–767. doi: 10.1096/fj.201700602R
Zhang, S., Weinberg, S., DeBerge, M., Gainullina, A., Schipma, M., Kinchen, J. M., et al. (2019). Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. 29, 443–456.e5. doi: 10.1016/j.cmet.2018.12.004
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., Lin, X. J., et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. doi: 10.1038/s41586-018-0372-z
Keywords: myocardial infarction, mitochondrial function, immunometabolism, macrophage, monocyte
Citation: Zhao M, Wang DD-H, Liu X and Tian R (2020) Metabolic Modulation of Macrophage Function Post Myocardial Infarction. Front. Physiol. 11:674. doi: 10.3389/fphys.2020.00674
Received: 16 March 2020; Accepted: 26 May 2020;
Published: 30 June 2020.
Edited by:
Paolo Bernardi, University of Padua, ItalyReviewed by:
Nina Kaludercic, National Research Council (CNR), ItalyJeffrey J. Saucerman, University of Virginia, United States
Copyright © 2020 Zhao, Wang, Liu and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojing Liu, bGl1eHFAc2N1LmVkdS5jbg==; Rong Tian, cm9uZ3RpYW5AdS53YXNoaW5ndG9uLmVkdQ==; cm9uZ3RpYW5AdXcuZWR1