
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 16 June 2020
Sec. Clinical and Translational Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00661
 Dong Fan1*
Dong Fan1* Zamaneh Kassiri2,3*
Zamaneh Kassiri2,3*Tissue inhibitor of metalloproteinase 3 (TIMP3) is unique among the four TIMPs due to its extracellular matrix (ECM)-binding property and broad range of inhibitory substrates that includes matrix metalloproteinases (MMPs), a disintegrin and metalloproteinases (ADAMs), and ADAM with thrombospondin motifs (ADAMTSs). In addition to its metalloproteinase-inhibitory function, TIMP3 can interact with proteins in the extracellular space resulting in its multifarious functions. TIMP3 mRNA has a long 3’ untranslated region (UTR) which is a target for numerous microRNAs. TIMP3 levels are reduced in various cardiovascular diseases, and studies have shown that TIMP3 replenishment ameliorates the disease, suggesting a therapeutic potential for TIMP3 in cardiovascular diseases. While significant efforts have been made in identifying the effector targets of TIMP3, the regulatory mechanism for the expression of this multi-functional TIMP has been less explored. Here, we provide an overview of TIMP3 gene structure, transcriptional and post-transcriptional regulators (transcription factors and microRNAs), protein structure and partners, its role in cardiovascular pathology and its application as therapy, while also drawing reference from TIMP3 function in other diseases.
The family of tissue inhibitor of metalloproteinases (TIMPs) is composed of 4 members, TIMP1 to TIMP4, which are the physiological inhibitors of matrix metalloproteinases (MMPs). The balance between MMPs and TIMPs plays a key role in the stability and normal function of the extracellular matrix (ECM). A disruption in this balance can cause fibrosis, excess accumulation of matrix proteins, or ECM degeneration, although the complexity of this process extends beyond the ability of the MMPs degrading the ECM proteins and TIMPs inhibiting this process (Takawale et al., 2015). TIMP3 is unique among the four TIMPs because it is the only one with a high affinity for the proteoglycans in the ECM, as well as its broadest range of substrates, including all MMPs, and a number of ADAMs (a disintegrin and metalloproteinases) and ADAMTSs (ADAM with thrombospondin motifs) (Staskus et al., 1991; Pavloff et al., 1992; Leco et al., 1994; Apte et al., 1995; Brew and Nagase, 2010; Moore et al., 2012; Jackson et al., 2017). Nevertheless, increasing number of reports show that TIMPs have more diverse functions beyond MMP inhibition. A variety of molecular partners for TIMP3 have also been reported that can mediate its MMP-independent functions (Qi et al., 2003; Klenotic et al., 2004; Nour et al., 2005; Kang et al., 2008; Scilabra et al., 2013; Huttlin et al., 2015, 2017; Kwasna et al., 2018).
Cardiovascular diseases continue to be a leading cause of morbidity and mortality (GBD 2017 Causes of Death Collaborators, 2018; GBD 2017 Disease and Injury Incidence and Prevalence Collaborators, 2018). Cardiac remodeling is a key characteristic of heart disease; it is the cumulative outcome of myocyte death (apoptosis and/or necrosis), myocyte hypertrophy, and fibrosis, and plays a pivotal role in progression to heart failure (Cohn et al., 2000; Fan et al., 2012; Moore et al., 2012). TIMP3 is involved in various process of cardiac remodeling and its level is reduced in ischemic and dilated cardiomyopathy (Li et al., 1998; Moore et al., 2012). Translational studies on the treatment of heart diseases such as myocardial infarction (MI) with TIMP3 replenishment suggest the therapeutic potential of TIMP3 in patients with heart failure (Spinale and Villarreal, 2014; Takawale et al., 2017). Recently, Su et al. (2019) summarized the roles and therapeutic potential of TIMP3 in cancer. Therefore, it will be valuable to compile the knowledge and advances in TIMP3 structure and function which will support the basis for further investigations in understanding its clinical application in cardiovascular diseases. In this review, we will focus on the roles of TIMP3 in cardiomyopathies, on its partners and applications.
Tissue inhibitor of metalloproteinase 3 is widely expressed in various tissues at a relatively high level (Fagerberg et al., 2014; Uhlen et al., 2015). The genomic organization of TIMP3 contains 5 exons (Apte et al., 1995; Stohr et al., 1995). In human and mouse, TIMP3 gene is located in chromosome 10 and 22, respectively (Apte et al., 1994a, b). The TIMP3 gene (Timp3) is nested in an intron of the synapsin-3 gene (Syn3), but Timp3 and Syn3 are transcribed from different DNA strands, like most nested genes, and there is no evidence showing coordination of expression or biological function between these two genes (reviewed in Candiani et al., 2010; Brew, 2019). TIMP3 expression can be regulated at both transcriptional and post-transcriptional levels.
Transcriptional regulations are mainly mediated by transcription factors binding to cis-regulatory DNA elements in the promoter region of a gene (Inukai et al., 2017). The transcription initiation site is commonly referred to as the +1 position, and promoters can be located in its upstream (labeled as minus) and downstream (positive) sequence (Butler and Kadonaga, 2002). There are three conserved putative Spl consensus binding sites in both human and murine Timp3 promoter, and a number of not highly conserved sites such as AP1, PEA3, and CTF/NF-1 binding sites (Stohr et al., 1995). Timp3 expression can be upregulated by multiple cytokines and growth factors such as phorbol ester (PMA), epidermal growth factor (EGF), transforming growth factor-β1 (TGF-β1) (Leco et al., 1994), and bovine lactoferricin, a peptide derived from bovine lactoferrin (Yan et al., 2013). TGF-β1 and bovine lactoferricin induce Timp3 expression through ERK1/2-activated Sp1 binding to its regulatory element in the Timp3 promoter (Qureshi et al., 2005; Yan et al., 2013). TGF-β1 can also stimulate transcription of Timp3 through activation of smad2/3/4 which bind to Timp3 promoter regions −828 to −333 bp and −333 to +1 bp (Qureshi et al., 2008). Overexpression of Elf3a and Elf3b (two splice variants of ETS-related transcription factor Elf-3) enhances the activity of Timp3 promoter by binding to the regions corresponding to −177 to −147 bp (human promoter) and −174 to −144 bp (murine promoter). However, Elf3a and Elf3b are normally expressed in rat lung, kidney, liver, and retina, but not in the heart (Jobling et al., 2002), therefore, their contribution to cardiac Timp3 overexpression may be limited.
Among the four TIMPs, Timp3 has the longest 3’ untranslated region (UTR) contained in the exon 5 (Leco et al., 1994; Apte et al., 1995), which is a target for multiple microRNAs. MicroRNAs (miRNA, miR) are a group of short, non-coding RNAs with 18−25 bases which can regulate gene expression at the transcriptional and post-transcriptional stages (Mohr and Mott, 2015; Dutka et al., 2019; Yao et al., 2019). MiRNAs have been shown widely to be involved in cardiovascular diseases (Barwari et al., 2016; De Rosa et al., 2017; Kir et al., 2018; Dutka et al., 2019). MiR-17-92 cluster is able to induce cardiomyocyte proliferation in postnatal and adult heart as well as in response to myocardial infarction (Chen et al., 2013), which is partly caused by binding of miR-17 to 3’ UTR of Timp3 and suppressing Timp3 expression (Shi et al., 2017; Liu et al., 2018). Through binding to, and inhibiting Timp3 expression, miR-21 enhances survival, migration and capillary formation ability of human umbilical vein endothelial cell lines (HUVECs) (Hu et al., 2016). miR-181b targets Timp3 and reduces apoptosis of cardiomyocytes (Gao et al., 2017), but enhances gelatinases/MMP activity in human aortic valve endothelial cells which may contribute to calcific aortic valve disease (Heath et al., 2018). MiR-181b is increased in human atherosclerotic plaques and abdominal aortic aneurysms (AAA), and inhibition of miR-181b suppressed the development and progression of atherosclerosis and aneurysms through increasing expression of TIMP3, elastin, and collagen (Di Gregoli et al., 2017). Upregulation of miR-181b by TGF-β can also promote hepatocarcinogenesis via targeting TIMP3 (Wang et al., 2010). MiR-222 is required for exercise-induced cardiac growth and can protect the heart against adverse remodeling (Liu et al., 2015). MiR-222 has also been reported to target Timp3, thereby promoting the proliferation of pulmonary arterial smooth muscle cells (Xu et al., 2017). MiR-323a binds to and down-regulates Timp3 which was associated with increased fibrosis following cardiac pressure overload (Zhang J. et al., 2018). Timp3 expression can also be negatively regulated by miR-712 (human homolog, miR-205) (Son et al., 2013; Kim et al., 2014). And inhibition of miR-712 by Anti-miR-712 has been shown to suppress atherosclerosis, inflammation, and AAA induced by angiotensin II (Ang II) (Son et al., 2013; Kim et al., 2014), implying a protective role for TIMP3 in these processes. A summary of all microRNAs reported to date to target Timp3 with specific binding sites, and the corresponding cell types are listed in Table 1.
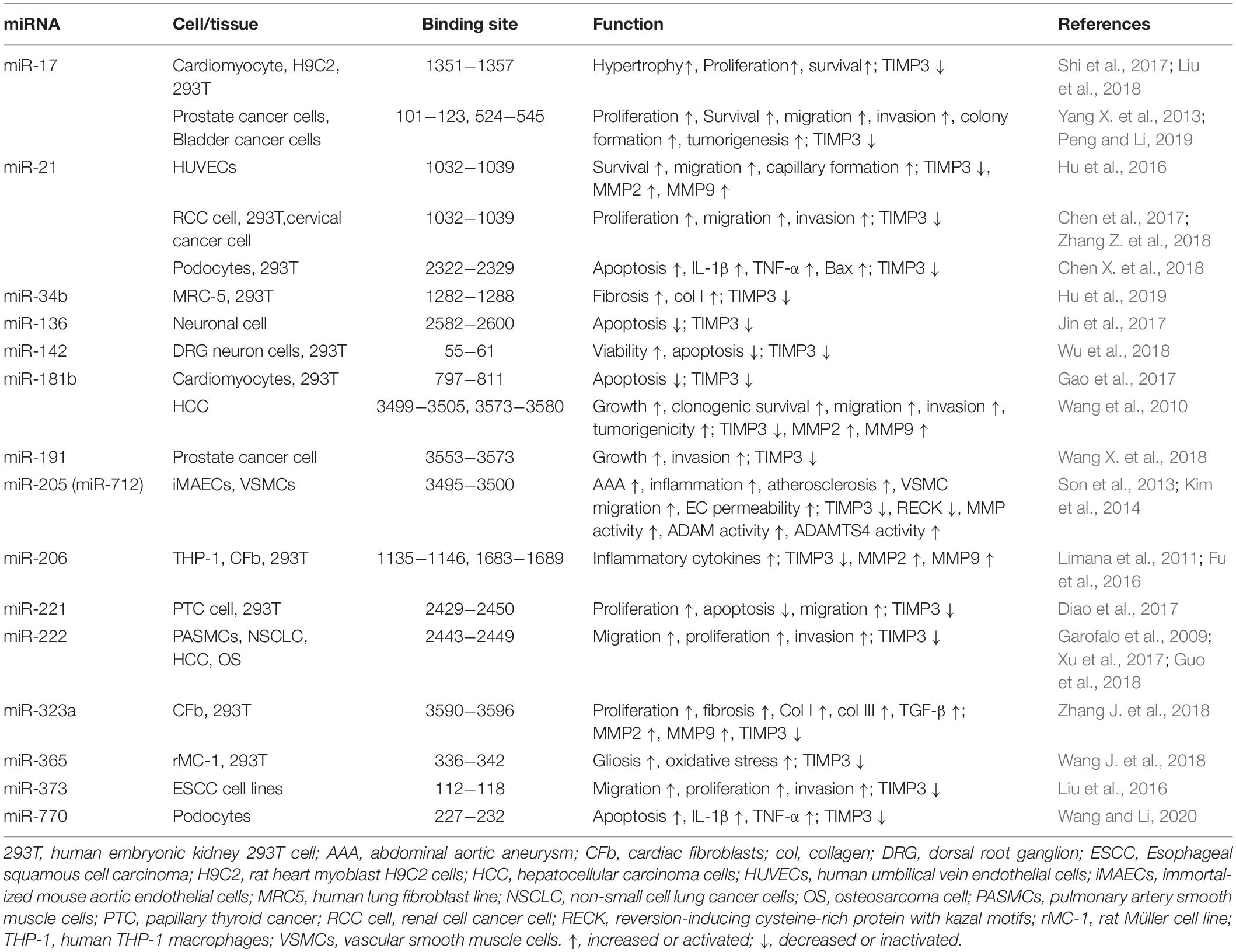
Table 1. Functions of miRNAs targeting Timp3 and their binding sites in 3′ UTR of Timp3 mRNA.
Human and mouse TIMP3 protein precursor is composed of 211 amino acids with a secretion signal peptide (1−23 aa) followed by the mature TIMP3 protein sequence (Leco et al., 1994; Wilde et al., 1994). The mature TIMP3 protein is about 24 kDa (Figure 1), and has an N-linked glycosylation site (Asn184) near the carboxyl terminus, while the size of the glycosylated TIMP3 is about 27 kDa (Leco et al., 1994; Wilde et al., 1994; Apte et al., 1995; Langton et al., 1998). Two-thirds of the mature TIMP3 (∼120 aa) forms the N-terminal domain, and the remaining one-third constitutes the C-terminal domain. The secondary structure of TIMP3 is composed of 6 loops stabilized by 6 disulfide bonds formed by 12 conserved cysteine residues (Figure 1), which is a common feature in all four TIMPs (Wisniewska et al., 2008; Moore et al., 2012). The N- and C-terminal domains contain 3 loops each. The 3-dimentional structure of the N-terminal TIMP3 (N-TIMP3, Cys1-Asn121) exhibits a wedge-like shape and contains an oligosaccharide/oligonucleotide binding (OB) fold (consisting of 5 β–pleated strands, sA-sE), an N-terminal and a C-terminal α-helix (hI-hII, Figure 1; Wisniewska et al., 2008; Brew and Nagase, 2010; Brew, 2019).
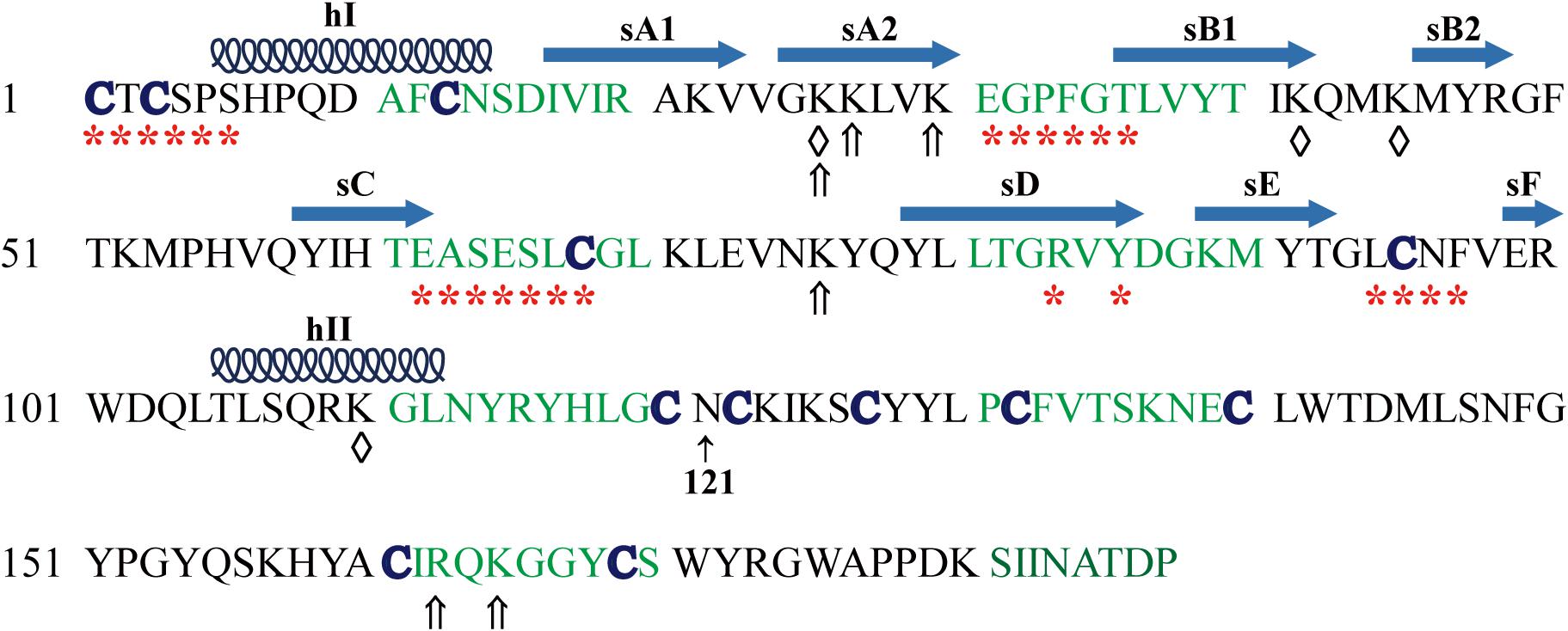
Figure 1. Mature human TIMP3 protein sequence and putative binding sites (domain) of TIMP3 with its interactors. hI-II, sA-F, location and extent of α-helices and β-strands as described (Wisniewska et al., 2008). Asn121 separates the N- and C-terminal domains of TIMP3. Partners of N-terminal TIMP3: ADAM17 (a disintegrin and metalloproteinase 17, *), ADAM12, MMPs (matrix metalloproteinases), ADAMTS (ADAM with thrombospondin motifs)-2, -4, -5, AT2R (angiotensin II type 2 receptor), GAGs (glycosaminoglycans, ⇑); LRP-1 [Low-density lipoprotein (LDL) receptor-related protein-1, ♢]. Partners of C-terminal TIMP3: MMP-2, -3, proMMP-2, -9, GAGs (⇑), fibulin-3, PC5A (a soluble isoform of proprotein convertase 5), VEGFR2 (vascular endothelial growth factor receptor 2).
With the widest inhibitory spectrum against metalloproteinases, TIMP3 can suppress all MMPs, a number of ADAMs (−10, −12, −17, −28, and −33) and ADAMTS (−1, −2, −4, and −5) (Brew and Nagase, 2010; Moore et al., 2012; Jackson et al., 2017; Brew, 2019). ADAM17 is inhibited by TIMP3 when present as a dimer; and the N-terminal domain of TIMP4 can interact with and inhibit ADAM17 in vitro, suggesting that the C-terminal domain of TIMP4 may disrupt this interaction in vivo (Amour et al., 1998; Lee et al., 2005; Brew and Nagase, 2010). Full-length and N-terminal (1−115 aa) TIMP3 demonstrate equal binding and inhibitory efficacy toward ADAM17 (Lee et al., 2001), suggesting that there is little influence of C-terminal domain of TIMP3 on its binding to ADAM17. TIMP3 interacts with the catalytic domain of ADAM17 (Arg215-Val477) via five segments as follows (Figure 1; Wisniewska et al., 2008): (1) major contacts by binding of Cys1-Ser6 of TIMP3 into the non-primed side of the active-site cleft of ADAM17, and the Cys1 directly above the catalytic zinc ion of ADAM17; (2) sA-sB hairpin loop of N-TIMP3 (Glu31-Gly32-Pro33-Phe34-Gly35- Thr36) extending into a hydrophobic groove formed by the side chains of ADAM17 residues Tyr352, Val353, Tyr369 and Leu380, which maintains the stability of TIMP3∗ADAM17 complex; (3) the sC-connector loop (Glu62-Ala63-Ser64-Glu65- Ser66-Leu67-Cys68) of TIMP3 with ADAM17 residues Lys315, Leu350-Tyr352 and His415; (4) Arg84 and Tyr86 of the sD-sE loop of TIMP3 with Asp313, Met316/Pro305 of ADAM17; (5) Multiple contacts of the sE-sF loop of N-TIMP3 with the catalytic domain of ADAM17 (Pro437 and His415). However, the extracellular form of ADAM17 (215−651 aa) is more resilient to N-TIMP3 than the catalytic domain of ADAM17 (215−477 aa), indicating that C-terminal domains of ADAM17, which contain disintegrin, EGF-like and Crambin-like domains, significantly weaken the inhibitory effect of N-TIMP3 (Lee et al., 2002).
Xu et al. (2012) reported that ADAM17 dimer has a greater capacity to interact with TIMP3, whereas activation of p38 or ERK1/2 reduces the dimerization of ADAM17 and its association with TIMP3, therefore increasing activation of ADAM17. Since a 1:1 stoichiometry has been reported for the TIMP3∗ADAM17 complex (Wisniewska et al., 2008), two TIMP3 protein would interact with the ADAM17 dimer. Because ADAM17 is a very important proteinase that can cleave numerous membrane-bound cytokines and growth factors (or their receptors) including the full length TNF-α (reviewed in Zhang et al., 2016; Zunke and Rose-John, 2017), TIMP3 has a variety of functions besides the anti-inflammatory effect through inhibition of ADAM17. The N-terminal domain of TIMP3 (N-TIMP3) also binds to the catalytic domain of ADAM12 and deletion of the disintegrin and cysteine-rich domains in ADAM12 enhanced the inhibitory effect of TIMP3 against this ADAM (Jacobsen et al., 2008; Kveiborg et al., 2010). However, N-TIMP3 is insufficient for binding to and inhibiting ADAM10, whereas full length TIMP3 is an efficient inhibitor of ADAM10 (Rapti et al., 2008).
The interaction and inhibitory effect of TIMP3 on MMPs are similar to the N-TIMP3∗ADAM17 and MMP∗TIMP1 complex, involving the ridge formed by N-TIMP3 residues Cys1-Thr2-Cys3-Ser4-Pro5, sC-sD loop (Ser64-Glu-Ser-Leu- Cys68) and the Cys1-Cys68 disulfide bond, which inserts into the MMP active site and the Cys1 lies above the catalytic zinc ion of MMPs (Wisniewska et al., 2008; Brew and Nagase, 2010). Mutation of Cys1 to Ser in TIMP3 results in loss of its MMP-inhibitory activity (Bond et al., 2000).
In the C-terminal domain of TIMP3, a mutation of Ser181 to Cys seen in Sorby’s fundus dystrophy (SFD), or N-glycosylation (Asn184) have no effect on TIMP3 inhibitory function against MMPs (Langton et al., 1998). In addition, the C-terminal domain of TIMP3 can interact with MMP2 and MMP3 which assists the MMP-inhibitory effects of TIMP3 (Troeberg et al., 2009). The C-terminal domain of TIMP3 can also bind to the hemopexin domains of proMMP2 and proMMP9 (Butler et al., 1999). TIMP3 has been reported to assist the activation of proMMP2 by membrane-type-3 MMP (MT3-MMP) through forming a trimolecular complex with proMMP2 and MT3-MMP (Zhao et al., 2004). The activation of proMMP2 by MT3-MMP is enhanced by TIMP3 in a dose-dependent fashion occurring at 100 nM, but not at 1−30 nM which in fact inhibited formation of the intermediate form of MMP2 (Zhao et al., 2004). TIMP3 has been reported not to affect the activation of proMMP2 by MT1-MMP (Zhao et al., 2004), while TIMP3-deficiency promotes activation of proMMP2, which can be inhibited by N-TIMP3 (10−50 nM) but not matrix-associated TIMP3 (English et al., 2006).
The N-terminal domain of TIMP3 can also interact with and inhibit ADAMTS-2, -4 and -5 (Kashiwagi et al., 2001; Gendron et al., 2003; Wang et al., 2006). The TIMP3 inhibitory site interacting with ADAMTS4 is composed of N-terminal tail (Cys1-Thr2-Cys3-Ser4-Pro5), an inhibitory loop (sC-connector loop) formed by Glu62-Ala63-Ser64-Glu65-Ser66-Leu67-Cys68, and the Cys1-Cys68 disulfide bond (Zhang W. et al., 2018). The inhibitory loop directly inserts into the catalytic center of ADAMTS4 which plays the primary role, whereas the N-terminal tail assists the interaction and forms a coordinate bond between the carbonyl group of Cys1 and the zinc ion chelator (Zhang W. et al., 2018). The C-terminal domains of ADAMTS4 and ADAMTS5 can enhance their association with N-TIMP3 (Troeberg et al., 2009).
Low-density lipoprotein (LDL) receptor-related protein-1 (LRP-1) belongs to the LDL receptor family which mediate endocytosis and degradation of multiple ligands (Strickland et al., 2002). It has been reported that LRP-1 could directly bind to TIMP-1, -2, and -3, and mediate their endocytosis and degradation (Emonard et al., 2004; Troeberg et al., 2008; Scilabra et al., 2013; Thevenard et al., 2014). Heparin, pentosan polysulfate, or receptor-associated protein (RAP), an antagonist of ligands binding to LRP family members, result in accumulation of TIMP3 in the culture medium (Troeberg et al., 2008). Scilabra et al. (2013) reported that TIMP3 binds to the extracellular domain of LRP-1, while a soluble form of LRP-1 (sLRP-1) shed from the cell membrane can also bind to TIMP3 leading to its accumulation in the medium since it cannot be endocytosed. This group further reported that TIMP3 binds to the N-terminal domain of the ligand-binding cluster II of LRP-1 (sLRP2N), which is not influenced by interaction of LRP-1 with MMP-13 or ADAMTS-5 (Yamamoto et al., 2016; Scilabra et al., 2017). LRP-1 can mediate the endocytosis of TIMP3, MMP-13 and ADAMTS-5 at the same time. The sLRP2N without the EGF-like repeat can be used as a TIMP3 trap to sequester TIMP3 in the culture medium (in vitro) or in the extracellular space (in vivo) which supresses metalloproteinase-mediated shedding of various cytokines and receptors (Scilabra et al., 2017). Blocking LRP-1 with RAP or an LRP-1 blocking antibody could also raise the level of TIMP-3 on the surface of endotoxin-activated macrophages and suppress the release of TNF by ADAM17 (Schubert et al., 2019). Mutation of two lysine pairs of TIMP3 (K26A/K45A or K42A/K110A, Figure 1) disrupts its interaction with LRP-1 and prevents its endocytosis (Doherty et al., 2016). This can be employed as an approach to accumulate TIMP3 in the extracellular space and thereby inhibit excess metalloproteinase activity.
Wildtype TIMP3 and a TIMP2-TIMP3 chimera created by fusing the N-terminal domain of TIMP2 to the C-terminal domain of TIMP3 are predominantly found in the ECM, whereas N-TIMP3 without the C-terminal domain was almost undetectable in the COS-7 cell ECM (Leco et al., 1994; Langton et al., 1998). Later, it was demonstrated that both N- (19-32 aa and 41-52 aa) and C-terminal domains participate in the binding of TIMP3 to ECM proteins (Yu et al., 2000; Lee et al., 2007). The residues of TIMP3 involved in this binding are mainly basic amino acids: Lys26, Lys27, Lys30, and Lys76 of the N-terminal domain, Arg163 and Lys165 of the C-terminal domain (Figure 1). Replacement of these residues with Glu and Gln deprives TIMP3 of its ECM-binding ability and renders it soluble (Lee et al., 2007). Both glycosylated and non-glycosylated TIMP3 can bind to ECM (Apte et al., 1995; Langton et al., 1998). The specific ECM proteins interacting with TIMP3 are mainly several glycosaminoglycans (GAGs): heparin, heparan sulfate (HS), chondroitin sulfates, and sulfated compounds such as suramin and pentosan, but not dermatan sulfate, aggrecan or hyaluronic acid (Yu et al., 2000; Troeberg et al., 2014). The C-terminal domain of TIMP3 can also bind to another matrix protein fibulin-3, known as EGF-containing fibulin-like extracellular matrix protein 1 (Klenotic et al., 2004).
Preincubation of TIMP3 with heparin, HS or chondroitin sulfate E (CSE) inhibits its binding to the ectodomain of LRP-1, and subsequently its endocytosis by LRP-1 (Troeberg et al., 2008, 2014; Scilabra et al., 2013). Treating cells in culture with HS or CSE also leads to the accumulation of TIMP3 in culture media. HS and CSE can enhance the inhibitory effects of TIMP3 on ADAMTS-5 by increasing TIMP3 affinity for ADAMTS-5 and reducing the dissociation rate (Troeberg et al., 2014). Therefore, ECM is not only a depot for TIMP3, but also protects TIMP3 against endocytosis and degradation, enhances the binding of TIMP3 to metalloproteinases and facilitates its inhibitory function.
Tissue inhibitor of metalloproteinase 3 has been reported to suppress vascular endothelial growth factor (VEGF)-mediated angiogenesis through directly binding to VEGF receptor 2 (VEGFR2) and disrupting the interaction of VEGF with VEGFR2 and the downstream signaling (Qi et al., 2003; Janssen et al., 2008). TIMP3 can also bind to soluble VEGFR2 (sVEGFR2), suggesting that TIMP3 binds to the extracellular domain of VEGFR2 (Qi et al., 2003). Two synthetic peptides derived from the loop 6 (aa 137−166) and tail region (aa 167−188) or C-terminal domain (aa 122−188) of TIMP3 mimic the interaction of TIMP3 and VEGFR2 (Janssen et al., 2008; Qi et al., 2013). Therefore, the C-terminal domain of TIMP3 has been proposed to be responsible for its interaction with VEGFR2. An SFD mutation S156C did not alter the interaction between TIMP3 and VEGFR2, or its anti-angiogenic effect (Fogarasi et al., 2008). However, TIMP3 (S156C) expression in porcine aortic endothelial cells enhanced VEGF binding to VEGFR2 and VEGF-induced angiogenesis through up-regulation of VEGFR2 (Qi et al., 2009), which might lead to choroidal neovascularization in SFD. The discrepancy between these two studies is probably due to the up-regulation of VEGFR2 in porcine aortic endothelial cells expressed with TIMP3 (S156C) and the levels of TIMP3 (S156C) involved. The up-regulation of VEGFR2 was also seen in human eyes with SFD (Qi et al., 2009). Meanwhile, this TIMP3-VEGR2 interaction has been reported to occur only at high concentrations of TIMP3; such that at physiological concentrations, TIMP3 does not interact with VEGFR2, and in fact promotes angiogenesis, but at high concentrations, TIMP3 can suppress angiogenesis by binding to VEGR2 and blocking the downstream pathway (Takawale et al., 2017). TIMP3 inhibits VEGF binding to VEGFR2 with a half-maximal inhibitory concentration (IC50) of 3.3−4.5 μg/ml, whereas cold VEGF competes with labeled VEGF for binding to VEGFR2 with an IC50 of 8.5−15 ng/ml (Qi et al., 2003). Therefore, the affinity of VEGFR2 for TIMP3 is much lower than its affinity for VEGF. However, the reason of pro-angiogenesis by low concentration of TIMP3 remains unknown.
Both Ang II type 2 (AT2R) and type 1 (AT1R) receptors contain seven-transmembrane domains and belong to the G-protein-coupled receptor (GPCR) family (Verdonk et al., 2012). TIMP3 can interact with AT2R but not AT1R (Kang et al., 2008). The interaction is mediated by the N-terminal domain of TIMP3 and the C-terminal sequence (aa 235−363) of AT2R (Kang et al., 2008). Overexpression of TIMP3 or AT2R have no obvious effect on normal cells (HEK 293 cells or HUVECs), but each of them can induce the apoptosis of SKOV-3 ovarian cancer cells through activation of caspase-3 and -9, and collectively they have additive effects (Kang et al., 2008). Overexpression of TIMP3 or AT2R inhibits VEGF-induced angiogenesis, and their combined overexpression has an additive effect; however, silencing AT2R with siRNA does not block the effect of TIMP3, and vice versa (Kang et al., 2008), which suggest that the anti-angiogenic effect of TIMP3 may not be mediated by AT2R. As discussed above, TIMP3 may suppress VEGF-induced angiogenesis through binding to and inhibiting VEGFR2 signaling. Although the specific binding site for TIMP3 on AT2R requires further investigation, AT2R binds to the N-terminal domain of TIMP3, while VEGFR2 binds to the C-terminal domain of TIMP3, therefore, TIMP3 may be able to bind to VEGFR2 and AT2R at the same time. The functional outcome of TIMP3 binding to AT2R remains largely unknown. Due to vital roles of AT2R and AT1R in the cardiovascular system, it is worth exploring the implications of this interaction in cardiovascular diseases.
The proprotein convertase 5 (PC5) is a serine proteinase that cleaves numerous proproteins at paired basic amino acids within the secretory pathway (Seidah and Chretien, 1999). The C-terminal cysteine-rich domain (CRD) of PC5A, a soluble isoform of PC5, can bind to the C-terminal domain of all four TIMPs (Nour et al., 2005). TIMP3 can also interact with a de-ubiquitinating enzyme ZUFSP which plays an important role in genomic stability (Kwasna et al., 2018). By using high-throughput affinity purification-mass spectrometry methodology, Huttlin et al. (2015, 2017) found other TIMP3 partners such as C-reactive protein (CRP), TIMP2, ADP-dependent glucokinase (ADPGK), Asialoglycoprotein receptor 2 (ASGR2), C-C motif chemokine 3-like 1 (CCL3L1), collectin-11, Dickkopf-related protein 3 (DKK3), neutrophil defensin 1 (DEFA1), gamma-interferon-inducible lysosomal thiol reductase (IFI30), Insulin-like growth factor-binding protein 1 (IGFBP1), von Willebrand factor C and EGF domain-containing protein (VWCE). However, the specific binding sites and functional consequences demand further investigation.
In summary, in addition to inhibiting metalloproteinases, TIMP3 has several important roles in regulation of multiple molecules in the extracellular space and on the cell membrane, and the corresponding downstream intracellular signaling pathways, which could explain the diverse functions of this molecule in various pathologies. Here, we will summarize the reports on the role of TIMP3 in cardiovascular diseases and its potential as a therapeutic factor.
Tissue inhibitor of metalloproteinase three levels have been reported to be altered in patients with different cardiomyopathies. In patients with aortic stenosis, myocardial TIMP3 mRNA and protein, TIMP2 mRNA and TIMP4 protein are increased, while TIMP1 mRNA and protein decreased (Fielitz et al., 2004). In patients with atrial fibrillation, the myocardial TIMP3 protein level is increased (Mukherjee et al., 2006) or unchanged in another study (Polyakova et al., 2008), while the myocardial TIMP1 protein level is increased in both studies. In myocarditis, only the myocardial levels of TIMP3 (among the four TIMPs) are significantly increased (Polyakova et al., 2011). In the failing heart of patients with ischemic cardiomyopathy (ICM) or dilated cardiomyopathy (DCM), TIMP1 and TIMP3 levels were reduced with no change in TIMP2 and TIMP4, except TIMP4 protein decreased in ICM myocardium (Li et al., 1998); however, myocardial levels of all four TIMPs were increased in another study (Polyakova et al., 2011). Following mechanical unloading of the ICM or DCM failing hearts with LV assist device, myocardial levels of TIMP1 and TIMP3 were increased while no change in TIMP2 and TIMP4 (Li et al., 2001). Therefore, there is some discrepancy in how TIMP levels are altered in patients with myocardial disease, which could be related to the stage of disease, the region of the heart where the specimen was collected, the ethnic background of the patients, to name a few.
The causal role of TIMP3 in progression of myocardial disease has been extensively explored in animal models (Figure 2). Timp3 deficiency results in DCM in aged mice due to enhanced MMP9 activity and the activation of the TNF-α cytokine system (Fedak et al., 2004). Compared with wild-type (WT) mice, cardiac pressure overload in Timp3-knockout (Timp3–/–) mice leads to more severe LV dilation, systolic and diastolic dysfunction, cardiac hypertrophy and fibrosis, with early heart failure (Kassiri et al., 2005, 2009). This phenotype is caused by excess ADAM17 and TNF-α, as well as activation of MMP2 and MT1-MMP. Suppression of both TNF-α and MMPs was found to be necessary for the complete prevention of DCM in Timp3–/– mice following pressure overload (Kassiri et al., 2005). Simultaneous activation of TNF-α and TGF-β1, and reciprocally stimulatory effects between these two factors, also played important roles in TIMP3-deficiency-induced adverse cardiac remodeling in the pressure-overload mice model (Kassiri et al., 2009). Other studies (Inagaki et al., 1995; Bitzer et al., 2000) had shown that TNF-α could inhibit TGF-β1 signaling, which might restrain signaling thresholds of TGF-β1, depending on physiological requirements. In addition to pressure overload, Ang II infusion induces severe fibrosis in Timp3–/– mice, although in the absence of hypertrophy, mainly due to post-translational stabilization and deposition of collagen by matricellular proteins (Fan et al., 2014). Following myocardial infarction (MI), TIMP3 levels are decreased in the infarct and peri-infarct myocardium (Kandalam et al., 2010; Takawale et al., 2017). Timp3-deficiency exacerbates cardiac remodeling, dysfunction, and survival following MI (Figure 2), partly through increased MMP activity, TNF-α signaling, and apoptosis in the non-infarcted myocardium, while coronary density was not reduced in the myocardium (Tian et al., 2007; Kandalam et al., 2010). Further, a marked but transient rise in MMP activity in the early post-MI phase was found to be responsible for the adverse remodeling and increased rate of LV rupture in Timp3–/– mice, since early treatment with a broad-spectrum MMP inhibitor ameliorated the adverse LV remodeling and rupture (Kandalam et al., 2010). On the other hand, targeted overexpression of TIMP3 or delivery of recombinant TIMP3 protein into the myocardium also improved the infarcted myocardium and heart function through suppression of the MMP activity and inflammation, and promoting angiogenesis (Eckhouse et al., 2014; Purcell et al., 2014, 2018; Barlow et al., 2017; Takawale et al., 2017; Chintalgattu et al., 2018; Thorn et al., 2019).
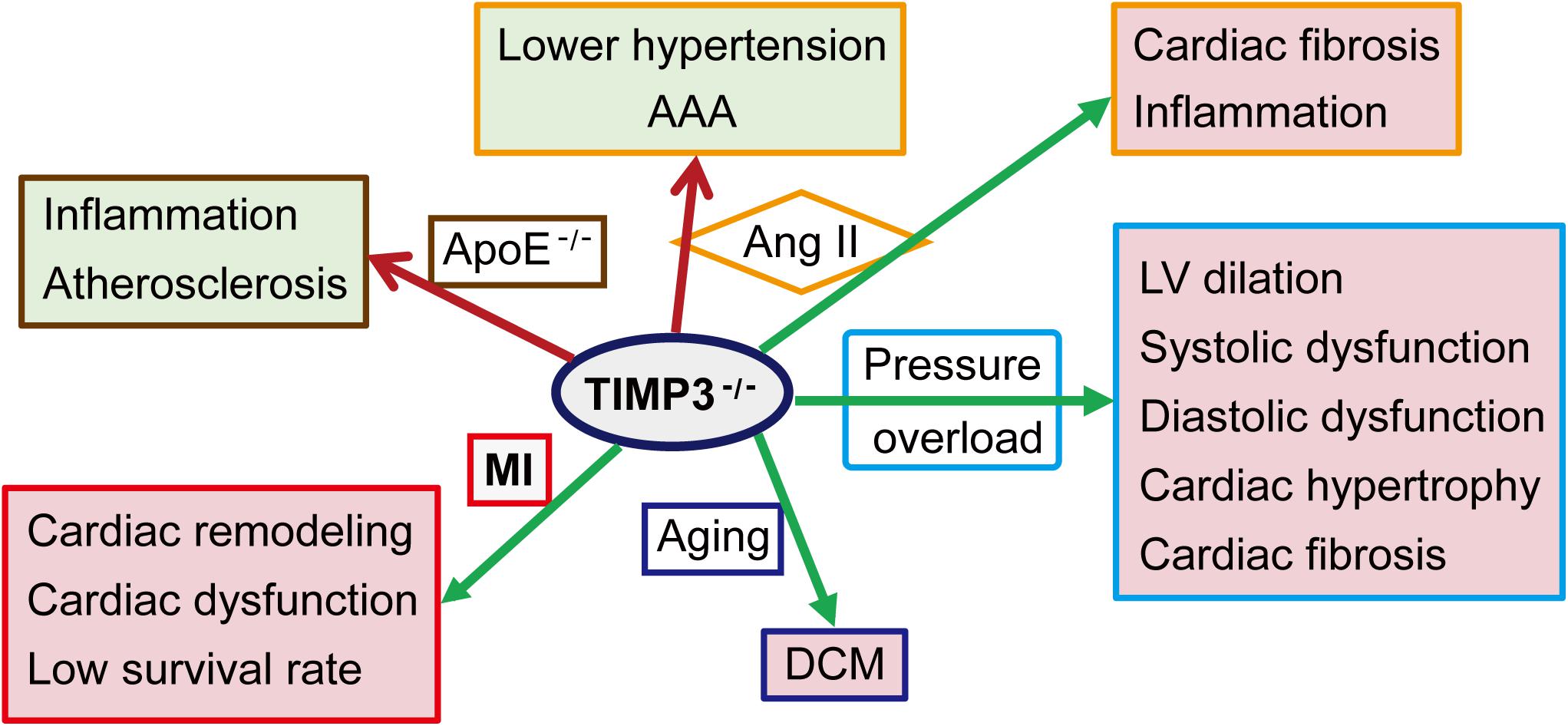
Figure 2. Role of TIMP3 in cardiovascular pathologies. TIMP3 deficiency (TIMP3– /–) exacerbates the structure change and dysfunction of the heart and/or the artery in a variety of animal models. AAA, abdominal aortic aneurysm; Ang II, Angiotensin II; ApoE– /–, apolipoprotein E-deficient mice; DCM, dilated cardiomyopathy; LV, left ventricular; MI, myocardial infarction.
Since in vitro experiments lack the multicellular interactions that exist in vivo, the results from in vitro studies may differ from those in vivo. An in vitro study showed that silencing TIMP3 reduces apoptosis of cardiomyocytes isolated from 5 to 6 week-old female rats (Gao et al., 2017). However, deficiency or inhibition of TIMP3 enhances neonatal mouse cardiomyocyte proliferation in vitro and in vivo, which can be inhibited by treatment with recombinant TIMP3, through a mechanism involving inhibition of MMP activity and EGFR/JNK/SP-1 signaling pathway (Hammoud et al., 2009; Shi et al., 2017). The different findings from these studies could be caused by the difference in the age of the animals used for cardiomyocyte isolation. TIMP3 deficiency also promotes Ang II- or phenylephrine-induced fibrotic response in the co-culture of neonatal cardiac myocytes and fibroblasts, which may result from elevated levels and activity of metalloproteinases, and the subsequent proteolytic processing and activation of TGF-β1 and TNF-α (Kassiri et al., 2009).
Tissue inhibitor of metalloproteinase 3 has been proposed to function as a physiological anti-inflammatory molecule primarily through its inhibitory ability against ADAM17, whereby it reduces the ADAM17-mediated release of soluble TNF-α, an important inflammatory cytokine (Khokha et al., 2013). Reduced MMP inhibition also contributes to migration of inflammatory cells in TIMP3 deficient mice (Moore et al., 2012; Khokha et al., 2013). TIMP3 deficiency enhances TNF-α levels and infiltration of neutrophils in the infarcted myocardium (Tian et al., 2007; Kandalam et al., 2010). Myocardial neutrophils and macrophages are also increased in TIMP3-deficient mice following Ang II infusion (Figure 2), which could be the cell source for the elevated matricellular proteins in these mice (Fan et al., 2014).
Tissue inhibitor of metalloproteinase 3 levels in the aorta are decreased in patients with thoracic aortic aneurysm (Blunder et al., 2012; Lee et al., 2014), and in patients with acute type A aortic dissection (Kimura et al., 2017). Timp3-deficiency suppressed the hypertensive response to Ang II (Basu et al., 2013), however induced abdominal aortic aneurysm (AAA) after 4 weeks of Ang II infusion (Figure 2), due to degradation of the elastin fibers and adverse remodeling of the aortic wall (Basu et al., 2012). Ang II-mediated AAA was characterized by enlarged lumen, reduced aortic wall thickness due to loss of elastic lamellae, and inflammation (Basu et al., 2012). On the other hand, Ang II can suppress the expression of TIMP3 and reversion-inducing cysteine-rich protein with Kazal motifs (RECK) by upregulating miR-712 (human/murine homolog miR-205) (Kim et al., 2014). Silencing miR-712 and miR-205 suppressed MMP activity and inflammation, preventing Ang II-induced AAA. Therefore, TIMP3 and targeting miR-712 (miR-205) could be potential therapeutic strategies for AAA.
Recombinant TIMP3 protein suppressed angiogenesis by binding to and inhibiting VEGFR2 with an IC50 of 2.9 μg/ml (Qi et al., 2003; Janssen et al., 2008). However, this function of TIMP3 was later found to be dose-dependent such that at lower (close to physiological) concentrations (0.01 and 0.1 μg/ml), rTIMP3 promoted endothelial sprouting and angiogenesis, but at higher concentrations (>1 μg/ml), rTIMP3 blocked angiogenesis by binding to VEGFR2 and blocking the downstream signaling pathways (Takawale et al., 2017). Overexpression of TIMP3 and AT2R can additively inhibit VEGF-induced angiogenesis (Kang et al., 2008), implying the importance of both VEGFR2 and AT2R in angiogenesis.
Atherosclerosis is described as a chronic inflammatory condition of the arterial system, and the thrombotic complications can cause MI, ischemic stroke, and peripheral arterial diseases, which can be potentially prevented by anti-inflammatory therapy (Libby et al., 2002; Geovanini and Libby, 2018). In patients with atherosclerosis, elevated TIMP3 levels in the plasma have been reported (Zheng et al., 2018), but TIMP3 is reduced in carotid atherosclerotic plaques from patients with type 2 diabetes (Cardellini et al., 2009). Deficiency of TIMP3 enhances inflammation and aggravates atherosclerosis in ApoE-knockout mice (Figure 2; Stohr et al., 2014), while overexpression of TIMP3 in macrophages decreases atherosclerosis (Casagrande et al., 2012). TIMP3 and RECK have also been reported to mediate the inhibitory effects of interleukin-32α (IL-32α) on endothelial inflammation, smooth muscle cell activation, and atherosclerosis development (Son et al., 2017). IL-32α upregulates TIMP3 and RECK by inhibiting miR-205 biogenesis. These data suggest that TIMP3 could exert anti-atherosclerotic effects.
Due to the important role of TIMP3 in cardiac structure and function, and the severe adverse outcomes of Timp3-deficiency in heart disease (Fedak et al., 2004; Kassiri et al., 2005; Kandalam et al., 2010; Basu et al., 2012, 2013; Moore et al., 2012; Fan et al., 2014), treatment with TIMP3 could be a potential therapeutic method in these diseases. In fact, there have been several studies on the application of TIMP3 in heart disease (Table 2). Various delivery routes have been employed to replenish the TIMP3 levels in the diseased heart, each with advantages and disadvantages as described below.
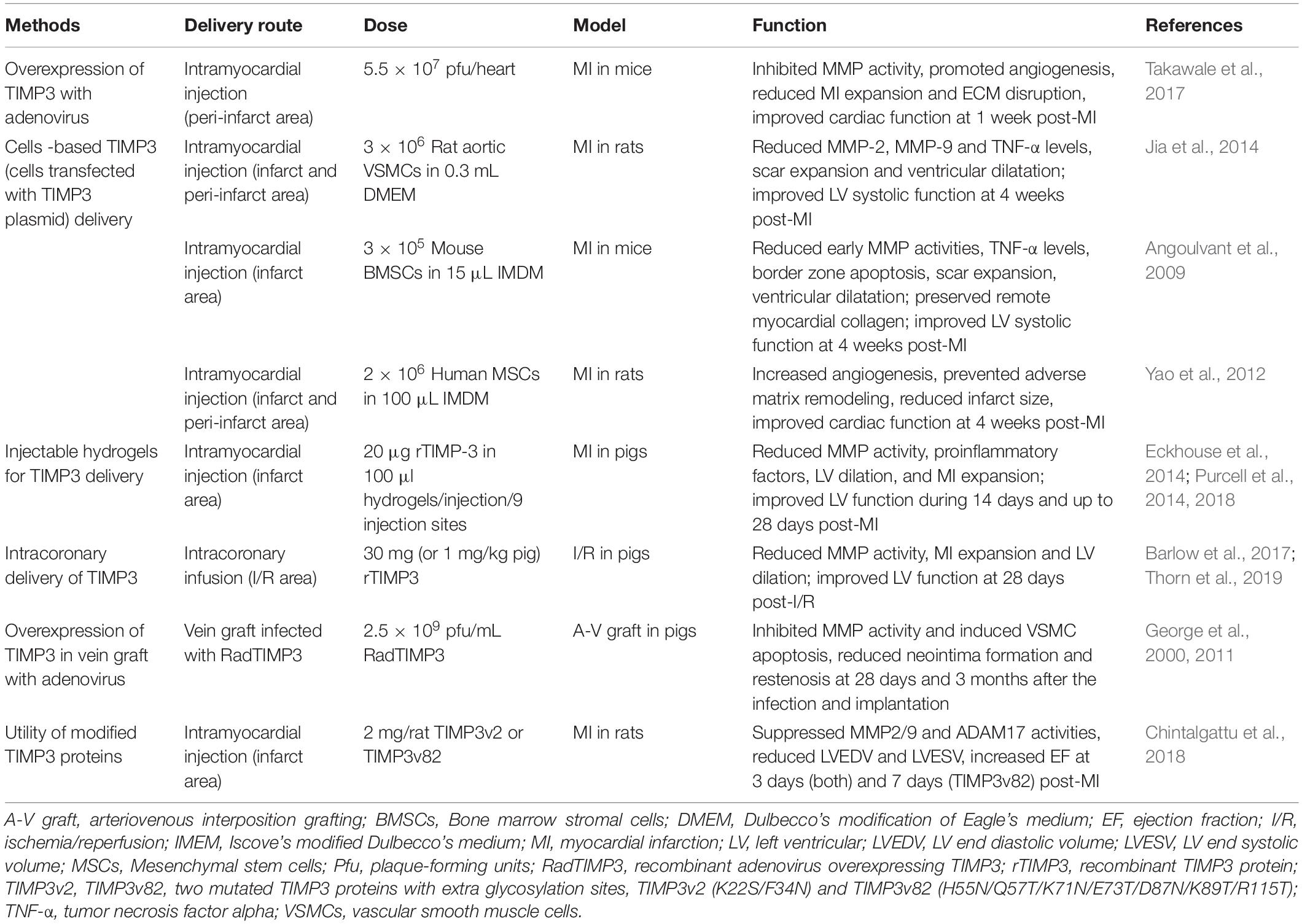
Table 2. Application of TIMP3 in cardiovascular diseases.
Viral delivery of a gene of interest has been achieved by adenovirus, adeno-associated virus (AAV), and lentivirus vectors as they allow for a broad host range. In a mouse model of MI, human TIMP3-expressing adenovirus (Ad-hTIMP3) was introduced into the peri-infarct myocardium by direct injections using a 36-gauge needle, increased TIMP3 mRNA and protein could be detected in the infarct and peri-infarcted myocardium (Takawale et al., 2017). This TIMP3 replenishment ameliorated cardiac dysfunction and the adverse remodeling post-MI, probably through inhibition of MMP activity and promotion of angiogenesis at 1 week post-MI, which is unexpected given the reports on anti-angiogenic functions of TIMP3 (Qi et al., 2003, 2013). Interestingly, an increased dose of Ad-hTIMP3 (5.5 × 108 pfu/heart) expression exerted negative effects on myocardial remodeling and function post-MI (Takawale et al., 2017). This observation demonstrates the important notion that attempts toward overexpressing TIMP3, or any other TIMP, should be only to achieve their physiological levels and it should not be in excess.
Compared with the adenovirus-mediated overexpression, AAV vectors mediate long term (more than 1 year in non-proliferating cells), cardiac-specific gene expression, with much less to no pathogenic response and cytotoxicity in human hosts (Chu et al., 2003; Belke et al., 2006; Bera and Sen, 2017; Bass-Stringer et al., 2018; Ishikawa et al., 2018; Ambrosi et al., 2019). Systemic delivery of the AAV serotype 9 (AAV9) resulted in transduction of myocytes throughout the myocardium, but caused cardiac fibrosis and dysfunction to some degree (Merentie et al., 2016), suggesting that targeted myocardial injection is perhaps a safer mode of delivery, although given the invasive nature of this approach which can pose limitations.
Cell-based gene therapy for MI is a valid approach since cardiomyocytes and vascular cells are lost in the process of infarct formation (Chen Z. et al., 2018). Intramyocardial injections of rat aortic VSMCs, mouse bone marrow stromal cells, or human mesenchymal stem cells transfected with a TIMP3 plasmid have been reported to be more effective in improving cardiac structure and function post-MI than the cells without transfection (Angoulvant et al., 2009; Yao et al., 2012; Jia et al., 2014). Therefore, cell-based TIMP3 delivery could be an efficient approach for MI since TIMP3 can inhibit metalloproteinases and activation of TNF-α, as well as promote angiogenesis, and further improve the structure and function of the heart post-MI. However, transplantation with exogenous cells may provoke immune reaction in the host, whereas employing autologous cells transfected with the gene of interest in vitro prior to transplantation has been proposed to provide a better option, but it requires 3 or more days for the newly introduced genes to be expressed before the transplant procedure can be completed (Jia et al., 2014; Melly et al., 2018). Thus it could be used for long term treatment effects to prevent progression of infarct expansion and cardiac dysfunction post-MI. However, the long term effects of this approach needs to be determined.
Local application of recombinant TIMP3 protein (rTIMP3) with hydrogels can also reduce myocardial remodeling and improve heart function post-MI (Eckhouse et al., 2014; Purcell et al., 2014, 2018). Hydrogels based on hyaluronic acid (HA) containing full-length human rTIMP3 were injected into the myocardium in an MI model in pigs (Eckhouse et al., 2014). When not bound to HA (Troeberg et al., 2014), TIMP3 can be released from the hydrogel at a nearly uniform rate for up to 14 days. After 7 days post-injection, human TIMP3 protein could be detected in the targeted myocardium, but not in the non-target myocardium or other organs (Eckhouse et al., 2014). An injectable and MMP degradable hydrogel has also been developed to contain rTIMP3 for “on-demand” MMP inhibition (Purcell et al., 2014). Dextran sulphated (DS), a heparin mimetic sulfated GAGs which can bind to TIMP3, crosslinks with HA hydrogels via MMP-cleavable peptide, so that rTIMP3 is encapsulated in the hydrogels until MMPs present and degrade the hydrogel (Purcell et al., 2014).
With their dual forms, liquid at room temperature (20−25°C) and gelatin-like at body temperature (37°C), the injectable hydrogels are attractive media for drug release and tissue engineering, as the hydrogel itself does not exert negative effects on the heart, while the bioresponsive injectable hydrogels provide optimal “on-demand” releasing media (Ifkovits et al., 2010; Eckhouse et al., 2014; Purcell et al., 2014; Vashist et al., 2017). Since most hydrogels contain ammonium persulfate (APS) and tetramethylethylene diamine (TEMED) (Ifkovits et al., 2010; Mironi-Harpaz et al., 2012; Yang H. et al., 2013; Eckhouse et al., 2014), some risk could be associated with using hydrogels as media for drug delivery. However, no significant cytotoxicity has been reported for the hydrogels in a short time frame (Ifkovits et al., 2010), and strategies have been developed to wash out APS and TEMED to minimize their potential toxic effects (Yang H. et al., 2013). HA hydrogels do not exacerbate heart function in MI animal models either (Ifkovits et al., 2010; Eckhouse et al., 2014).
In a model of cardiac ischemia/reperfusion (I/R) injury in pigs, intracoronary infusion of full-length human TIMP3 protein reduced infarct size and LV end-diastolic volume, and increased ejection fraction at 28 days post-I/R (Barlow et al., 2017; Thorn et al., 2019). Clearly, intracoronary delivery will require a larger amount of rTIMP3 compared to local myocardium injection in order to acquire the cardiac protective effect, while intracoronary infusion could result in off-target effects of TIMP3. However, it is easier to be translated from bench to bedside, because percutaneous coronary intervention (PCI) is an efficient and frequently used clinical therapeutic intervention for MI patients (Bates et al., 2016; Mehta et al., 2019).
Although PCI is a safe and minimally invasive way for treating acute coronary syndrome, there are risks of intima hyperplasia and restenosis after the procedure, and TIMP3 has been employed to prevent and reduce the problems (Jukema et al., 2011; Hall and Agrawal, 2018). Implantation of the stent coated with recombinant adenovirus overexpressing TIMP3 (RadTIMP3, 20 μL of 109 pfu/mL) in pig coronary arteries demonstrated localized overexpression of TIMP3 around the stent and increased apoptosis in the neointima and media (Johnson et al., 2005). RadTIMP3 decreased neointima area and expanded luminal area at 28 days after implantation. Late graft failure because of neointimal thickening and atherosclerosis is a serious problem in autologous saphenous vein coronary artery bypass graft surgery (CABG) (Wadey et al., 2018). Saphenous veins infected with RadTIMP3 (2.5 × 109 pfu/mL) showed enhanced TIMP3 expression in the intimal and upper medial layers, and suppressed neointimal formation in human saphenous veins in organ culture and in pig interposition grafts in vivo (George et al., 2000). RadTIMP3-infected grafts had decreased intimal thickness at 28 days, and 3 months after the infection and implantation in autologous porcine arteriovenous interposition grafting (George et al., 2000, 2011). This function of TIMP3 in reducing the neointima formation and restenosis could be through inhibition of metalloproteinases, and induction of VSMC apoptosis through activation of FAS (George et al., 2000; Johnson et al., 2005; English et al., 2018). Therefore, TIMP3 holds the potential as therapy for restenosis and vein graft intimal thickening. Interestingly, a novel adenoviral vector with augmented vascular transduction and enhanced resistance to human serum neutralization has been developed (White et al., 2013), which could facilitate the translational and therapeutic application of TIMP3 in vascular diseases.
Besides the recombinant wild-type TIMP3 protein, utility of engineered TIMP3 molecule has also been explored. These modifications include addition of extra glycosylation sites, PEGylation (polyethylene glycol conjugation), and fusion with Fc, human serum albumin (HSA), antibody (Ab), or latency-associated peptide (LAP) (Chintalgattu et al., 2018; Alberts et al., 2019). To sequester TIMP3 in the extracellular space, TIMP3 could also be mutated (K26A/K45A and K42A/K110A) to disrupt its interaction with LRP-1 and the subsequent endocytosis (Doherty et al., 2016). Two engineered mutant TIMP3 molecules with extra glycosylation sites, TIMP3v2 (K22S/F34N) and TIMP3v82 (H55N/Q57T/K71N/E73T/D87N/K89T/R115T), have a higher expression yield in a Chinese hamster ovary cell line and longer rat serum half-life than the wild-type TIMP3 (Chintalgattu et al., 2018). TIMP3v2 or TIMP3v82 were injected into the infarcted myocardium after 3 h of LAD ligation in rats. Both could improve cardiac function at 3 days post-MI, but only TIMP3v82 had lasting effects until 7 days post-MI (Chintalgattu et al., 2018).
Tissue inhibitor of metalloproteinase 3 fused with LAP (LAP-TIMP3) has been shown to have a higher expression yield in 293T cells than wild-type TIMP3 (Alberts et al., 2019). The LAP-TIMP3 was designed with an MMP cleavage site (PLGL) in between LAP and TIMP3; therefore, the engineered TIMP3 was latent until it was released from LAP-TIMP3 by MMP1 (Alberts et al., 2019). The released TIMP3, containing a residual leucine at its N-terminal, could inhibit ADAM17 and ADAMTS4 but not MMPs, although in synovial fluid from patients with osteoarthritis, there was sufficient MMP activity to cleave LAP-TIMP3 and release active TIMP3 (Alberts et al., 2019), suggesting that LAP-TIMP3 may be useful for anti-inflammatory therapy.
Other peptides or synthetic peptides derived from TIMP3 could also be potential tools for treatment of diseases. A cyclic peptide derived from TIMP3 (61EASESLAG70 enclosed by a Cys at each end) targeting ADAMTS4 has been explored as a treatment for osteoarthritis (Zhang W. et al., 2018), further investigation is required for its potential usage in patients. Synthetic peptides corresponding to the loop 6 (aa 137−166) and tail region (aa 167−188) of TIMP3 are efficient in inhibiting angiogenesis which may be employed to treat diseases with uncontrolled neovascularization, such as choroidal neovascularization (Qi et al., 2013). Therefore, TIMP3 can be engineered to enhance the expression yield and to prolong its effects, as well as to inhibit specific metalloproteinases.
In summary, application of TIMP3 has been shown to be beneficial and effective in inhibiting adverse remodeling and improving heart function in MI and I/R animal models ranging from mice to pigs (Table 2). Overexpression of TIMP3 can also prevent and reduce restenosis and neointima hyperplasia after PCI or CABG. However, increased levels of TIMP3 resulted in considerable change in the cellular secretome through inhibition of ADAM10 and ligand-binding of LRP-1 (Scilabra et al., 2018). These suggest that the level of TIMP3 supplemented and the side effects related to the secretome change need to be monitored when TIMP3 overexpression or supplementation is applied. Local supplementation of TIMP3 has little to no side effects in non-target organs, however, most of these studies have been short-term, and the long-term effects need to be investigated, particularly those involving AAV-mediated overexpression. Intramyocardial injections can be done during the CABG procedure, by closed-chest myocardial injections guided by high-resolution echocardiography (Springer et al., 2005; Mattila et al., 2016), or catheter-based intracoronary delivery.
The reports on the application of TIMP3 in vascular diseases are limited to those pertaining to restenosis after PCI or CABG, as discussed above. TIMP3 can counteract inflammation and atherosclerosis in mouse models (Casagrande et al., 2012; Son et al., 2017); therefore, TIMP3 holds the potential as treatment for patients with atherosclerosis, although this requires further translational and clinical studies. In addition, given the protective role of TIMP3 in aortic aneurysm, and its dose-dependent function in angiogenesis, many potential therapeutic functions of TIMP3 remain to be explored. For instance, while pro-angiogenic function of TIMP3 would be beneficial in post-MI scenarios, its anti-angiogenic property at higher doses would limit the uncontrolled neovascularization in tumor growth and likely limit metastasis.
In addition to the well-known MMP-inhibitory function of TIMP3, a variety of partners have been identified for TIMP3 revealing the diverse roles of this protein in various cellular and molecular functions, although the full spectrum of TIMP3 partners and the resulting outcomes requires further investigation. TIMP3 levels are mostly decreased in patients with cardiovascular conditions and deficiency of TIMP3 causes or exacerbates cardiovascular diseases partly mediated by elevated proteolytic activities (MMPs, ADAMs, and ADAMTSs). Supplementing TIMP3 in the myocardium can improve cardiovascular structure and function, although the mode, dose and site of administration will require rigorous monitoring and verification since excess TIMP3 can lead to undesirable outcomes.
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the National Natural Science Foundation of China (81860046) to DF. ZK holds a Canada Research Chair (Tier 1) in Cardiovascular Extracellular Matrix.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AT declared a past collaboration with one of the authors ZK to the handling editor.
Alberts, B. M., Sacre, S. M., Bush, P. G., and Mullen, L. M. (2019). Engineering of TIMP-3 as a LAP-fusion protein for targeting to sites of inflammation. J. Cell. Mol. Med. 23, 1617–1621. doi: 10.1111/jcmm.14019
Ambrosi, C. M., Sadananda, G., Han, J. L., and Entcheva, E. (2019). Adeno-associated virus mediated gene delivery: implications for scalable in vitro and in vivo cardiac optogenetic models. Front. Physiol. 10:168. doi: 10.3389/fphys.2019.00168
Amour, A., Slocombe, P. M., Webster, A., Butler, M., Knight, C. G., Smith, B. J., et al. (1998). TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 435, 39–44. doi: 10.1016/s0014-5793(98)01031-x
Angoulvant, D., Fazel, S., Weisel, R. D., Lai, T. Y., Fedak, P. W., Chen, L., et al. (2009). Cell-based gene therapy modifies matrix remodeling after a myocardial infarction in tissue inhibitor of matrix metalloproteinase-3-deficient mice. J. Thorac. Cardiovasc. Surg. 137, 471–480. doi: 10.1016/j.jtcvs.2008.08.031
Apte, S. S., Hayashi, K., Seldin, M. F., Mattei, M. G., Hayashi, M., and Olsen, B. R. (1994a). Gene encoding a novel murine tissue inhibitor of metalloproteinases (TIMP), TIMP-3, is expressed in developing mouse epithelia, cartilage, and muscle, and is located on mouse chromosome 10. Dev. Dyn. 200, 177–197. doi: 10.1002/aja.1002000302
Apte, S. S., Mattei, M. G., and Olsen, B. R. (1994b). Cloning of the cDNA encoding human tissue inhibitor of metalloproteinases-3 (TIMP-3) and mapping of the TIMP3 gene to chromosome 22. Genomics 19, 86–90. doi: 10.1006/geno.1994.1016
Apte, S. S., Olsen, B. R., and Murphy, G. (1995). The gene structure of tissue inhibitor of metalloproteinases (TIMP)-3 and its inhibitory activities define the distinct TIMP gene family. J. Biol. Chem. 270, 14313–14318. doi: 10.1074/jbc.270.24.14313
Barlow, S. C., Doviak, H., Jacobs, J., Freeburg, L. A., Perreault, P. E., Zellars, K. N., et al. (2017). Intracoronary delivery of recombinant TIMP-3 after myocardial infarction: effects on myocardial remodeling and function. Am. J. Physiol. Heart Circ. Physiol. 313, H690–H699. doi: 10.1152/ajpheart.00114.2017
Barwari, T., Joshi, A., and Mayr, M. (2016). MicroRNAs in cardiovascular disease. J. Am. Coll. Cardiol. 68, 2577–2584. doi: 10.1016/j.jacc.2016.09.945
Bass-Stringer, S., Bernardo, B. C., May, C. N., Thomas, C. J., Weeks, K. L., and McMullen, J. R. (2018). Adeno-associated virus gene therapy: translational progress and future prospects in the treatment of heart failure. Heart Lung Circ. 27, 1285–1300. doi: 10.1016/j.hlc.2018.03.005
Basu, R., Fan, D., Kandalam, V., Lee, J., Das, S. K., Wang, X., et al. (2012). Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II. J. Biol. Chem. 287, 44083–44096. doi: 10.1074/jbc.M112.425652
Basu, R., Lee, J., Morton, J. S., Takawale, A., Fan, D., Kandalam, V., et al. (2013). TIMP3 is the primary TIMP to regulate agonist-induced vascular remodelling and hypertension. Cardiovasc. Res. 98, 360–371. doi: 10.1093/cvr/cvt067
Bates, E. R., Tamis-Holland, J. E., Bittl, J. A., O’Gara, P. T., and Levine, G. N. (2016). PCI strategies in patients with ST-segment elevation myocardial infarction and multivessel coronary artery disease. J. Am. Coll. Cardiol. 68, 1066–1081. doi: 10.1016/j.jacc.2016.05.086
Belke, D. D., Gloss, B., Hollander, J. M., Swanson, E. A., Duplain, H., and Dillmann, W. H. (2006). In vivo gene delivery of HSP70i by adenovirus and adeno-associated virus preserves contractile function in mouse heart following ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 291, H2905–H2910. doi: 10.1152/ajpheart.00323.2006
Bera, A., and Sen, D. (2017). Promise of adeno-associated virus as a gene therapy vector for cardiovascular diseases. Heart Fail. Rev. 22, 795–823. doi: 10.1007/s10741-017-9622-7
Bitzer, M., von Gersdorff, G., Liang, D., Dominguez-Rosales, A., Beg, A. A., Rojkind, M., et al. (2000). A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 14, 187–197. doi: 10.1101/gad.14.2.187
Blunder, S., Messner, B., Aschacher, T., Zeller, I., Turkcan, A., Wiedemann, D., et al. (2012). Characteristics of TAV- and BAV-associated thoracic aortic aneurysms–smooth muscle cell biology, expression profiling, and histological analyses. Atherosclerosis 220, 355–361. doi: 10.1016/j.atherosclerosis.2011.11.035
Bond, M., Murphy, G., Bennett, M. R., Amour, A., Knauper, V., Newby, A. C., et al. (2000). Localization of the death domain of tissue inhibitor of metalloproteinase-3 to the N terminus. Metalloproteinase inhibition is associated with proapoptotic activity. J. Biol. Chem. 275, 41358–41363. doi: 10.1074/jbc.M007929200
Brew, K. (2019). Reflections on the evolution of the vertebrate tissue inhibitors of metalloproteinases. FASEB J. 33, 71–87. doi: 10.1096/fj.201801262R
Brew, K., and Nagase, H. (2010). The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803, 55–71. doi: 10.1016/j.bbamcr.2010.01.003
Butler, G. S., Apte, S. S., Willenbrock, F., and Murphy, G. (1999). Human tissue inhibitor of metalloproteinases 3 interacts with both the N- and C-terminal domains of gelatinases A and B. Regulation by polyanions. J. Biol. Chem. 274, 10846–10851. doi: 10.1074/jbc.274.16.10846
Butler, J. E., and Kadonaga, J. T. (2002). The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev. 16, 2583–2592. doi: 10.1101/gad.1026202
Candiani, S., Moronti, L., Pennati, R., De Bernardi, F., Benfenati, F., and Pestarino, M. (2010). The synapsin gene family in basal chordates: evolutionary perspectives in metazoans. BMC Evol. Biol. 10:32. doi: 10.1186/1471-2148-10-32
Cardellini, M., Menghini, R., Martelli, E., Casagrande, V., Marino, A., Rizza, S., et al. (2009). TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SirT1. Diabetes 58, 2396–2401. doi: 10.2337/db09-0280
Casagrande, V., Menghini, R., Menini, S., Marino, A., Marchetti, V., Cavalera, M., et al. (2012). Overexpression of tissue inhibitor of metalloproteinase 3 in macrophages reduces atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 32, 74–81. doi: 10.1161/ATVBAHA.111.238402
Chen, J., Gu, Y., and Shen, W. (2017). MicroRNA-21 functions as an oncogene and promotes cell proliferation and invasion via TIMP3 in renal cancer. Eur. Rev. Med. Pharmacol. Sci. 21, 4566–4576.
Chen, J., Huang, Z. P., Seok, H. Y., Ding, J., Kataoka, M., Zhang, Z., et al. (2013). mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 112, 1557–1566. doi: 10.1161/CIRCRESAHA.112.300658
Chen, X., Zhao, L., Xing, Y., and Lin, B. (2018). Down-regulation of microRNA-21 reduces inflammation and podocyte apoptosis in diabetic nephropathy by relieving the repression of TIMP3 expression. Biomed. Pharmacother. 108, 7–14. doi: 10.1016/j.biopha.2018.09.007
Chen, Z., Chen, L., Zeng, C., and Wang, W. E. (2018). Functionally improved mesenchymal stem cells to better treat myocardial infarction. Stem Cells Int. 2018:7045245. doi: 10.1155/2018/7045245
Chintalgattu, V., Greenberg, J., Singh, S., Chiueh, V., Gilbert, A., O’Neill, J. W., et al. (2018). Utility of Glycosylated TIMP3 molecules: inhibition of MMPs and TACE to improve cardiac function in rat myocardial infarct model. Pharmacol. Res. Perspect. 6:e00442. doi: 10.1002/prp2.442
Chu, D., Sullivan, C. C., Weitzman, M. D., Du, L., Wolf, P. L., Jamieson, S. W., et al. (2003). Direct comparison of efficiency and stability of gene transfer into the mammalian heart using adeno-associated virus versus adenovirus vectors. J. Thorac. Cardiovasc. Surg. 126, 671–679. doi: 10.1016/s0022-5223(03)00082-5
Cohn, J. N., Ferrari, R., and Sharpe, N. (2000). Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an international forum on cardiac remodeling. J. Am. Coll. Cardiol. 35, 569–582. doi: 10.1016/s0735-1097(99)00630-0
De Rosa, R., Polito, M. V., Benvenga, R., De Angelis, E., Piscione, F., and Galasso, G. (2017). Micrornas and cardiovascular diseases: from bench to bedside. Transl. Med. UniSa 17, 12–18.
Di Gregoli, K., Mohamad Anuar, N. N., Bianco, R., White, S. J., Newby, A. C., George, S. J., et al. (2017). MicroRNA-181b controls atherosclerosis and aneurysms through regulation of TIMP-3 and elastin. Circ. Res. 120, 49–65. doi: 10.1161/CIRCRESAHA.116.309321
Diao, Y., Fu, H., and Wang, Q. (2017). MiR-221 exacerbate cell proliferation and invasion by targeting TIMP3 in papillary thyroid carcinoma. Am. J. Ther. 24, e317–e328. doi: 10.1097/MJT.0000000000000420
Doherty, C. M., Visse, R., Dinakarpandian, D., Strickland, D. K., Nagase, H., and Troeberg, L. (2016). Engineered tissue inhibitor of metalloproteinases-3 variants resistant to endocytosis have prolonged chondroprotective activity. J. Biol. Chem. 291, 22160–22172. doi: 10.1074/jbc.M116.733261
Dutka, M., Bobinski, R., and Korbecki, J. (2019). The relevance of microRNA in post-infarction left ventricular remodelling and heart failure. Heart Fail. Rev. 24, 575–586. doi: 10.1007/s10741-019-09770-9
Eckhouse, S. R., Purcell, B. P., McGarvey, J. R., Lobb, D., Logdon, C. B., Doviak, H., et al. (2014). Local hydrogel release of recombinant TIMP-3 attenuates adverse left ventricular remodeling after experimental myocardial infarction. Sci. Transl. Med. 6:223ra21. doi: 10.1126/scitranslmed.3007244
Emonard, H., Bellon, G., Troeberg, L., Berton, A., Robinet, A., Henriet, P., et al. (2004). Low density lipoprotein receptor-related protein mediates endocytic clearance of pro-MMP-2. TIMP-2 complex through a thrombospondin-independent mechanism. J. Biol. Chem. 279, 54944–54951. doi: 10.1074/jbc.M406792200
English, J. L., Kassiri, Z., Koskivirta, I., Atkinson, S. J., Di Grappa, M., Soloway, P. D., et al. (2006). Individual TIMP deficiencies differentially impact pro-MMP-2 activation. J. Biol. Chem. 281, 10337–10346. doi: 10.1074/jbc.M512009200
English, W. R., Ireland-Zecchini, H., Baker, A. H., Littlewood, T. D., Bennett, M. R., and Murphy, G. (2018). Tissue Inhibitor of Metalloproteinase-3 (TIMP-3) induces FAS dependent apoptosis in human vascular smooth muscle cells. PLoS One 13:e0195116. doi: 10.1371/journal.pone.0195116
Fagerberg, L., Hallstrom, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. doi: 10.1074/mcp.M113.035600.
Fan, D., Takawale, A., Basu, R., Patel, V., Lee, J., Kandalam, V., et al. (2014). Differential role of TIMP2 and TIMP3 in cardiac hypertrophy, fibrosis, and diastolic dysfunction. Cardiovasc. Res. 103, 268–280. doi: 10.1093/cvr/cvu072
Fan, D., Takawale, A., Lee, J., and Kassiri, Z. (2012). Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 5:15. doi: 10.1186/1755-1536-5-15
Fedak, P. W., Smookler, D. S., Kassiri, Z., Ohno, N., Leco, K. J., Verma, S., et al. (2004). TIMP-3 deficiency leads to dilated cardiomyopathy. Circulation 110, 2401–2409. doi: 10.1161/01.CIR.0000134959.83967.2D
Fielitz, J., Leuschner, M., Zurbrugg, H. R., Hannack, B., Pregla, R., Hetzer, R., et al. (2004). Regulation of matrix metalloproteinases and their inhibitors in the left ventricular myocardium of patients with aortic stenosis. J. Mol. Med. 82, 809–820. doi: 10.1007/s00109-004-0606-4
Fogarasi, M., Janssen, A., Weber, B. H., and Stohr, H. (2008). Molecular dissection of TIMP3 mutation S156C associated with Sorsby fundus dystrophy. Matrix Biol. 27, 381–392. doi: 10.1016/j.matbio.2008.01.008
Fu, X., Zeng, L., Liu, Z., Ke, X., Lei, L., and Li, G. (2016). MicroRNA-206 regulates the secretion of inflammatory cytokines and MMP9 expression by targeting TIMP3 in mycobacterium tuberculosis-infected THP-1 human macrophages. Biochem. Biophys. Res. Commun. 477, 167–173. doi: 10.1016/j.bbrc.2016.06.038
Gao, Z., Wang, L., Wang, J., Yang, F., and Qu, J. (2017). Molecular mechanism of miR-181b in heart disease due to pregnancy-induced hypertension syndrome. Exp. Ther. Med. 14, 2953–2959. doi: 10.3892/etm.2017.4882
Garofalo, M., Di Leva, G., Romano, G., Nuovo, G., Suh, S. S., Ngankeu, A., et al. (2009). miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 16, 498–509. doi: 10.1016/j.ccr.2009.10.014
GBD 2017 Causes of Death Collaborators (2018). Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1736–1788. doi: 10.1016/S0140-6736(18)32203-7
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators (2018). Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet 392, 1789–1858. doi: 10.1016/S0140-6736(18)32279-7
Gendron, C., Kashiwagi, M., Hughes, C., Caterson, B., and Nagase, H. (2003). TIMP-3 inhibits aggrecanase-mediated glycosaminoglycan release from cartilage explants stimulated by catabolic factors. FEBS Lett. 555, 431–436. doi: 10.1016/s0014-5793(03)01295-x
George, S. J., Lloyd, C. T., Angelini, G. D., Newby, A. C., and Baker, A. H. (2000). Inhibition of late vein graft neointima formation in human and porcine models by adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-3. Circulation 101, 296–304. doi: 10.1161/01.cir.101.3.296
George, S. J., Wan, S., Hu, J., MacDonald, R., Johnson, J. L., and Baker, A. H. (2011). Sustained reduction of vein graft neointima formation by ex vivo TIMP-3 gene therapy. Circulation 124(11 Suppl), S135–S142. doi: 10.1161/circulationaha.110.012732
Geovanini, G. R., and Libby, P. (2018). Atherosclerosis and inflammation: overview and updates. Clin. Sci. 132, 1243–1252. doi: 10.1042/CS20180306
Guo, J., Liu, Q., Li, Z., Guo, H., Bai, C., and Wang, F. (2018). miR-222-3p promotes osteosarcoma cell migration and invasion through targeting TIMP3. Onco Targets Ther. 11, 8643–8653. doi: 10.2147/OTT.S175745
Hall, S., and Agrawal, D. K. (2018). Delivery of viral vectors for gene therapy in intimal hyperplasia and restenosis in atherosclerotic swine. Drug Deliv. Transl. Res. 8, 918–927. doi: 10.1007/s13346-017-0409-0
Hammoud, L., Burger, D. E., Lu, X., and Feng, Q. (2009). Tissue inhibitor of metalloproteinase-3 inhibits neonatal mouse cardiomyocyte proliferation via EGFR/JNK/SP-1 signaling. Am. J. Physiol. Cell Physiol. 296, C735–C745. doi: 10.1152/ajpcell.00246.2008
Heath, J. M., Fernandez Esmerats, J., Khambouneheuang, L., Kumar, S., Simmons, R., and Jo, H. (2018). Mechanosensitive microRNA-181b regulates aortic valve endothelial matrix degradation by targeting TIMP3. Cardiovasc. Eng. Technol. 9, 141–150. doi: 10.1007/s13239-017-0296-z
Hu, J., Ni, S., Cao, Y., Zhang, T., Wu, T., Yin, X., et al. (2016). The angiogenic effect of microRNA-21 targeting TIMP3 through the regulation of MMP2 and MMP9. PLoS One 11:e0149537. doi: 10.1371/journal.pone.0149537
Hu, R. P., Lu, Y. Y., and Zhang, X. J. (2019). MiR-34b-5p knockdown attenuates bleomycin-induced pulmonary fibrosis by targeting tissue inhibitor of metalloproteinase 3 (TIMP3). Eur. Rev. Med. Pharmacol. Sci. 23, 2273–2279. doi: 10.26355/eurrev_201903_17276
Huttlin, E. L., Bruckner, R. J., Paulo, J. A., Cannon, J. R., Ting, L., Baltier, K., et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. doi: 10.1038/nature22366
Huttlin, E. L., Ting, L., Bruckner, R. J., Gebreab, F., Gygi, M. P., Szpyt, J., et al. (2015). The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440. doi: 10.1016/j.cell.2015.06.043
Ifkovits, J. L., Tous, E., Minakawa, M., Morita, M., Robb, J. D., Koomalsingh, K. J., et al. (2010). Injectable hydrogel properties influence infarct expansion and extent of postinfarction left ventricular remodeling in an ovine model. Proc. Natl. Acad. Sci. U.S.A. 107, 11507–11512. doi: 10.1073/pnas.1004097107
Inagaki, Y., Truter, S., Tanaka, S., Di Liberto, M., and Ramirez, F. (1995). Overlapping pathways mediate the opposing actions of tumor necrosis factor-alpha and transforming growth factor-beta on alpha 2(I) collagen gene transcription. J. Biol. Chem. 270, 3353–3358. doi: 10.1074/jbc.270.7.3353
Inukai, S., Kock, K. H., and Bulyk, M. L. (2017). Transcription factor-DNA binding: beyond binding site motifs. Curr. Opin. Genet. Dev. 43, 110–119. doi: 10.1016/j.gde.2017.02.007
Ishikawa, K., Weber, T., and Hajjar, R. J. (2018). Human cardiac gene therapy. Circ. Res. 123, 601–613. doi: 10.1161/CIRCRESAHA.118.311587
Jackson, H. W., Defamie, V., Waterhouse, P., and Khokha, R. (2017). TIMPs: versatile extracellular regulators in cancer. Nat. Rev. Cancer 17, 38–53. doi: 10.1038/nrc.2016.115
Jacobsen, J., Visse, R., Sorensen, H. P., Enghild, J. J., Brew, K., Wewer, U. M., et al. (2008). Catalytic properties of ADAM12 and its domain deletion mutants. Biochemistry 47, 537–547. doi: 10.1021/bi701629c
Janssen, A., Hoellenriegel, J., Fogarasi, M., Schrewe, H., Seeliger, M., Tamm, E., et al. (2008). Abnormal vessel formation in the choroid of mice lacking tissue inhibitor of metalloprotease-3. Invest. Ophthalmol. Vis. Sci. 49, 2812–2822. doi: 10.1167/iovs.07-1444
Jia, Z. B., Tian, H., Kang, K., Miao, H. Z., Liu, K. Y., Jiang, S. L., et al. (2014). Expression of the tissue inhibitor of metalloproteinase-3 by transplanted VSMCs modifies heart structure and function after myocardial infarction. Transpl. Immunol. 30, 149–158. doi: 10.1016/j.trim.2014.03.006
Jin, R., Xu, S., Lin, X., and Shen, M. (2017). MiR-136 controls neurocytes apoptosis by regulating Tissue Inhibitor of Metalloproteinases-3 in spinal cord ischemic injury. Biomed. Pharmacother. 94, 47–54. doi: 10.1016/j.biopha.2017.07.053
Jobling, A. I., Fang, Z., Koleski, D., and Tymms, M. J. (2002). Expression of the ETS transcription factor ELF3 in the retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 43, 3530–3537.
Johnson, T. W., Wu, Y. X., Herdeg, C., Baumbach, A., Newby, A. C., Karsch, K. R., et al. (2005). Stent-based delivery of tissue inhibitor of metalloproteinase-3 adenovirus inhibits neointimal formation in porcine coronary arteries. Arterioscler. Thromb. Vasc. Biol. 25, 754–759. doi: 10.1161/01.ATV.0000157582.33180.a9
Jukema, J. W., Ahmed, T. A., Verschuren, J. J., and Quax, P. H. (2011). Restenosis after PCI. Part 2: prevention and therapy. Nat. Rev. Cardiol. 9, 79–90. doi: 10.1038/nrcardio.2011.148
Kandalam, V., Basu, R., Abraham, T., Wang, X., Awad, A., Wang, W., et al. (2010). Early activation of matrix metalloproteinases underlies the exacerbated systolic and diastolic dysfunction in mice lacking TIMP3 following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 299, H1012–H1023. doi: 10.1152/ajpheart.00246.2010
Kang, K. H., Park, S. Y., Rho, S. B., and Lee, J. H. (2008). Tissue inhibitor of metalloproteinases-3 interacts with angiotensin II type 2 receptor and additively inhibits angiogenesis. Cardiovasc. Res. 79, 150–160. doi: 10.1093/cvr/cvn072
Kashiwagi, M., Tortorella, M., Nagase, H., and Brew, K. (2001). TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J. Biol. Chem. 276, 12501–12504. doi: 10.1074/jbc.C000848200
Kassiri, Z., Defamie, V., Hariri, M., Oudit, G. Y., Anthwal, S., Dawood, F., et al. (2009). Simultaneous transforming growth factor beta-tumor necrosis factor activation and cross-talk cause aberrant remodeling response and myocardial fibrosis in Timp3-deficient heart. J. Biol. Chem. 284, 29893–29904. doi: 10.1074/jbc.M109.028449
Kassiri, Z., Oudit, G. Y., Sanchez, O., Dawood, F., Mohammed, F. F., Nuttall, R. K., et al. (2005). Combination of tumor necrosis factor-alpha ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase-3 knock-out mice. Circ. Res. 97, 380–390. doi: 10.1161/01.RES.0000178789.16929.cf
Khokha, R., Murthy, A., and Weiss, A. (2013). Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 13, 649–665. doi: 10.1038/nri3499
Kim, C. W., Kumar, S., Son, D. J., Jang, I. H., Griendling, K. K., and Jo, H. (2014). Prevention of abdominal aortic aneurysm by anti-microRNA-712 or anti-microRNA-205 in angiotensin II-infused mice. Arterioscler. Thromb. Vasc. Biol. 34, 1412–1421. doi: 10.1161/ATVBAHA.113.303134
Kimura, N., Futamura, K., Arakawa, M., Okada, N., Emrich, F., Okamura, H., et al. (2017). Gene expression profiling of acute type A aortic dissection combined with in vitro assessment. Eur. J. Cardiothorac. Surg. 52, 810–817. doi: 10.1093/ejcts/ezx095
Kir, D., Schnettler, E., Modi, S., and Ramakrishnan, S. (2018). Regulation of angiogenesis by microRNAs in cardiovascular diseases. Angiogenesis 21, 699–710. doi: 10.1007/s10456-018-9632-7
Klenotic, P. A., Munier, F. L., Marmorstein, L. Y., and Anand-Apte, B. (2004). Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a binding partner of epithelial growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1). Implications for macular degenerations. J. Biol. Chem. 279, 30469–30473. doi: 10.1074/jbc.M403026200
Kveiborg, M., Jacobsen, J., Lee, M. H., Nagase, H., Wewer, U. M., and Murphy, G. (2010). Selective inhibition of ADAM12 catalytic activity through engineering of tissue inhibitor of metalloproteinase 2 (TIMP-2). Biochem. J. 430, 79–86. doi: 10.1042/BJ20100649
Kwasna, D., Abdul Rehman, S. A., Natarajan, J., Matthews, S., Madden, R., De Cesare, V., et al. (2018). Discovery and characterization of ZUFSP/ZUP1, a distinct deubiquitinase class important for genome stability. Mol. Cell 70, 150–164.e6. doi: 10.1016/j.molcel.2018.02.023
Langton, K. P., Barker, M. D., and McKie, N. (1998). Localization of the functional domains of human tissue inhibitor of metalloproteinases-3 and the effects of a Sorsby’s fundus dystrophy mutation. J. Biol. Chem. 273, 16778–16781. doi: 10.1074/jbc.273.27.16778
Leco, K. J., Khokha, R., Pavloff, N., Hawkes, S. P., and Edwards, D. R. (1994). Tissue inhibitor of metalloproteinases-3 (TIMP-3) is an extracellular matrix-associated protein with a distinctive pattern of expression in mouse cells and tissues. J. Biol. Chem. 269, 9352–9360.
Lee, J., Shen, M., Parajuli, N., Oudit, G. Y., McMurtry, M. S., and Kassiri, Z. (2014). Gender-dependent aortic remodelling in patients with bicuspid aortic valve-associated thoracic aortic aneurysm. J. Mol. Med. 92, 939–949. doi: 10.1007/s00109-014-1178-6
Lee, M. H., Atkinson, S., and Murphy, G. (2007). Identification of the extracellular matrix (ECM) binding motifs of tissue inhibitor of metalloproteinases (TIMP)-3 and effective transfer to TIMP-1. J. Biol. Chem. 282, 6887–6898. doi: 10.1074/jbc.M610490200
Lee, M. H., Knauper, V., Becherer, J. D., and Murphy, G. (2001). Full-length and N-TIMP-3 display equal inhibitory activities toward TNF-alpha convertase. Biochem. Biophys. Res. Commun. 280, 945–950. doi: 10.1006/bbrc.2000.4192
Lee, M. H., Rapti, M., and Murphy, G. (2005). Total conversion of tissue inhibitor of metalloproteinase (TIMP) for specific metalloproteinase targeting: fine-tuning TIMP-4 for optimal inhibition of tumor necrosis factor-{alpha}-converting enzyme. J. Biol. Chem. 280, 15967–15975. doi: 10.1074/jbc.M500897200
Lee, M. H., Verma, V., Maskos, K., Becherer, J. D., Knauper, V., Dodds, P., et al. (2002). The C-terminal domains of TACE weaken the inhibitory action of N-TIMP-3. FEBS Lett. 520, 102–106. doi: 10.1016/s0014-5793(02)02776-x
Li, Y. Y., Feldman, A. M., Sun, Y., and McTiernan, C. F. (1998). Differential expression of tissue inhibitors of metalloproteinases in the failing human heart. Circulation 98, 1728–1734. doi: 10.1161/01.cir.98.17.1728
Li, Y. Y., Feng, Y., McTiernan, C. F., Pei, W., Moravec, C. S., Wang, P., et al. (2001). Downregulation of matrix metalloproteinases and reduction in collagen damage in the failing human heart after support with left ventricular assist devices. Circulation 104, 1147–1152. doi: 10.1161/hc3501.095215
Libby, P., Ridker, P. M., and Maseri, A. (2002). Inflammation and atherosclerosis. Circulation 105, 1135–1143. doi: 10.1161/hc0902.104353
Limana, F., Esposito, G., D’Arcangelo, D., Di Carlo, A., Romani, S., Melillo, G., et al. (2011). HMGB1 attenuates cardiac remodelling in the failing heart via enhanced cardiac regeneration and miR-206-mediated inhibition of TIMP-3. PLoS One 6:e19845. doi: 10.1371/journal.pone.0019845
Liu, W., Li, M., Chen, X., Zhang, D., Wei, L., Zhang, Z., et al. (2016). MicroRNA-373 promotes migration and invasion in human esophageal squamous cell carcinoma by inhibiting TIMP3 expression. Am. J. Cancer. Res. 6, 1–14.
Liu, X., Xiao, J., Zhu, H., Wei, X., Platt, C., Damilano, F., et al. (2015). miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 21, 584–595. doi: 10.1016/j.cmet.2015.02.014
Liu, Z., Zhang, Z., Yao, J., Xie, Y., Dai, Q., Zhang, Y., et al. (2018). Serum extracellular vesicles promote proliferation of H9C2 cardiomyocytes by increasing miR-17-3p. Biochem. Biophys. Res. Commun. 499, 441–446. doi: 10.1016/j.bbrc.2018.03.157
Mattila, M., Koskenvuo, J., Söderström, M., Eerola, K., and Savontaus, M. (2016). Intramyocardial injection of SERCA2a-expressing lentivirus improves myocardial function in doxorubicin-induced heart failure. J. Gene Med. 18, 124–133. doi: 10.1002/jgm.2885
Mehta, S. R., Wood, D. A., Storey, R. F., Mehran, R., Bainey, K. R., Nguyen, H., et al. (2019). Complete revascularization with multivessel PCI for myocardial infarction. N. Engl. J. Med. 381, 1411–1421. doi: 10.1056/NEJMoa1907775
Melly, L., Cerino, G., Frobert, A., Cook, S., Giraud, M. N., Carrel, T., et al. (2018). Myocardial infarction stabilization by cell-based expression of controlled vascular endothelial growth factor levels. J. Cell. Mol. Med. 22, 2580–2591. doi: 10.1111/jcmm.13511
Merentie, M., Lottonen-Raikaslehto, L., Parviainen, V., Huusko, J., Pikkarainen, S., Mendel, M., et al. (2016). Efficacy and safety of myocardial gene transfer of adenovirus, adeno-associated virus and lentivirus vectors in the mouse heart. Gene Ther. 23, 296–305. doi: 10.1038/gt.2015.114
Mironi-Harpaz, I., Wang, D. Y., Venkatraman, S., and Seliktar, D. (2012). Photopolymerization of cell-encapsulating hydrogels: crosslinking efficiency versus cytotoxicity. Acta Biomater. 8, 1838–1848. doi: 10.1016/j.actbio.2011.12.034
Mohr, A. M., and Mott, J. L. (2015). Overview of microRNA biology. Semin. Liver Dis. 35, 3–11. doi: 10.1055/s-0034-1397344
Moore, L., Fan, D., Basu, R., Kandalam, V., and Kassiri, Z. (2012). Tissue inhibitor of metalloproteinases (TIMPs) in heart failure. Heart Fail. Rev. 17, 693–706. doi: 10.1007/s10741-011-9266-y
Mukherjee, R., Herron, A. R., Lowry, A. S., Stroud, R. E., Stroud, M. R., Wharton, J. M., et al. (2006). Selective induction of matrix metalloproteinases and tissue inhibitor of metalloproteinases in atrial and ventricular myocardium in patients with atrial fibrillation. Am. J. Cardiol. 97, 532–537. doi: 10.1016/j.amjcard.2005.08.073
Nour, N., Mayer, G., Mort, J. S., Salvas, A., Mbikay, M., Morrison, C. J., et al. (2005). The cysteine-rich domain of the secreted proprotein convertases PC5A and PACE4 functions as a cell surface anchor and interacts with tissue inhibitors of metalloproteinases. Mol. Biol. Cell 16, 5215–5226. doi: 10.1091/mbc.e05-06-0504
Pavloff, N., Staskus, P. W., Kishnani, N. S., and Hawkes, S. P. (1992). A new inhibitor of metalloproteinases from chicken: ChIMP-3. A third member of the TIMP family. J. Biol. Chem. 267, 17321–17326.
Peng, H., and Li, H. (2019). The encouraging role of long noncoding RNA small nuclear RNA host gene 16 in epithelial-mesenchymal transition of bladder cancer via directly acting on miR-17-5p/metalloproteinases 3 axis. Mol. Carcinog. 58, 1465–1480. doi: 10.1002/mc.23028
Polyakova, V., Loeffler, I., Hein, S., Miyagawa, S., Piotrowska, I., Dammer, S., et al. (2011). Fibrosis in endstage human heart failure: severe changes in collagen metabolism and MMP/TIMP profiles. Int. J. Cardiol. 151, 18–33. doi: 10.1016/j.ijcard.2010.04.053
Polyakova, V., Miyagawa, S., Szalay, Z., Risteli, J., and Kostin, S. (2008). Atrial extracellular matrix remodelling in patients with atrial fibrillation. J. Cell. Mol. Med. 12, 189–208. doi: 10.1111/j.1582-4934.2008.00219.x
Purcell, B. P., Barlow, S. C., Perreault, P. E., Freeburg, L., Doviak, H., Jacobs, J., et al. (2018). Delivery of a matrix metalloproteinase-responsive hydrogel releasing TIMP-3 after myocardial infarction: effects on left ventricular remodeling. Am. J. Physiol. Heart Circ. Physiol. 315, H814–H825. doi: 10.1152/ajpheart.00076.2018
Purcell, B. P., Lobb, D., Charati, M. B., Dorsey, S. M., Wade, R. J., Zellars, K. N., et al. (2014). Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat. Mater. 13, 653–661. doi: 10.1038/nmat3922
Qi, J. H., Dai, G., Luthert, P., Chaurasia, S., Hollyfield, J., Weber, B. H., et al. (2009). S156C mutation in tissue inhibitor of metalloproteinases-3 induces increased angiogenesis. J. Biol. Chem. 284, 19927–19936. doi: 10.1074/jbc.M109.013763
Qi, J. H., Ebrahem, Q., Ali, M., Cutler, A., Bell, B., Prayson, N., et al. (2013). Tissue inhibitor of metalloproteinases-3 peptides inhibit angiogenesis and choroidal neovascularization in mice. PLoS One 8:e55667. doi: 10.1371/journal.pone.0055667
Qi, J. H., Ebrahem, Q., Moore, N., Murphy, G., Claesson-Welsh, L., Bond, M., et al. (2003). A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 9, 407–415. doi: 10.1038/nm846
Qureshi, H. Y., Ricci, G., and Zafarullah, M. (2008). Smad signaling pathway is a pivotal component of tissue inhibitor of metalloproteinases-3 regulation by transforming growth factor beta in human chondrocytes. Biochim. Biophys. Acta 1783, 1605–1612. doi: 10.1016/j.bbamcr.2008.04.005
Qureshi, H. Y., Sylvester, J., El Mabrouk, M., and Zafarullah, M. (2005). TGF-beta-induced expression of tissue inhibitor of metalloproteinases-3 gene in chondrocytes is mediated by extracellular signal-regulated kinase pathway and Sp1 transcription factor. J. Cell. Physiol. 203, 345–352. doi: 10.1002/jcp.20228
Rapti, M., Atkinson, S. J., Lee, M. H., Trim, A., Moss, M., and Murphy, G. (2008). The isolated N-terminal domains of TIMP-1 and TIMP-3 are insufficient for ADAM10 inhibition. Biochem. J. 411, 433–439. doi: 10.1042/BJ20071430
Schubert, K., Collins, L. E., Green, P., Nagase, H., and Troeberg, L. (2019). LRP1 controls TNF release via the TIMP-3/ADAM17 axis in endotoxin-activated macrophages. J. Immunol. 202, 1501–1509. doi: 10.4049/jimmunol.1800834
Scilabra, S. D., Pigoni, M., Pravata, V., Schatzl, T., Muller, S. A., Troeberg, L., et al. (2018). Increased TIMP-3 expression alters the cellular secretome through dual inhibition of the metalloprotease ADAM10 and ligand-binding of the LRP-1 receptor. Sci. Rep. 8:14697. doi: 10.1038/s41598-018-32910-4
Scilabra, S. D., Troeberg, L., Yamamoto, K., Emonard, H., Thogersen, I., Enghild, J. J., et al. (2013). Differential regulation of extracellular tissue inhibitor of metalloproteinases-3 levels by cell membrane-bound and shed low density lipoprotein receptor-related protein 1. J. Biol. Chem. 288, 332–342. doi: 10.1074/jbc.M112.393322
Scilabra, S. D., Yamamoto, K., Pigoni, M., Sakamoto, K., Muller, S. A., Papadopoulou, A., et al. (2017). Dissecting the interaction between tissue inhibitor of metalloproteinases-3 (TIMP-3) and low density lipoprotein receptor-related protein-1 (LRP-1): development of a “TRAP” to increase levels of TIMP-3 in the tissue. Matrix Biol. 59, 69–79. doi: 10.1016/j.matbio.2016.07.004
Seidah, N. G., and Chretien, M. (1999). Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain Res. 848(1-2), 45–62. doi: 10.1016/s0006-8993(99)01909-5
Shi, J., Bei, Y., Kong, X., Liu, X., Lei, Z., Xu, T., et al. (2017). miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. Theranostics 7, 664–676. doi: 10.7150/thno.15162
Son, D. J., Jung, Y. Y., Seo, Y. S., Park, H., Lee, D. H., Kim, S., et al. (2017). Interleukin-32alpha inhibits endothelial inflammation, vascular smooth muscle cell activation, and atherosclerosis by upregulating Timp3 and Reck through suppressing microRNA-205 Biogenesis. Theranostics 7, 2186–2203. doi: 10.7150/thno.18407
Son, D. J., Kumar, S., Takabe, W., Kim, C. W., Ni, C. W., Alberts-Grill, N., et al. (2013). The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat. Commun. 4:3000. doi: 10.1038/ncomms4000
Spinale, F. G., and Villarreal, F. (2014). Targeting matrix metalloproteinases in heart disease: lessons from endogenous inhibitors. Biochem. Pharmacol. 90, 7–15. doi: 10.1016/j.bcp.2014.04.011
Springer, M. L., Sievers, R. E., Viswanathan, M. N., Yee, M. S., Foster, E., Grossman, W., et al. (2005). Closed-chest cell injections into mouse myocardium guided by high-resolution echocardiography. Am. J. Physiol. Heart Circ. Physiol. 289, H1307–H1314. doi: 10.1152/ajpheart.00164.2005
Staskus, P. W., Masiarz, F. R., Pallanck, L. J., and Hawkes, S. P. (1991). The 21-kDa protein is a transformation-sensitive metalloproteinase inhibitor of chicken fibroblasts. J. Biol. Chem. 266, 449–454.
Stohr, H., Roomp, K., Felbor, U., and Weber, B. H. (1995). Genomic organization of the human tissue inhibitor of metalloproteinases-3 (TIMP3). Genome Res. 5, 483–487. doi: 10.1101/gr.5.5.483
Stohr, R., Cavalera, M., Menini, S., Mavilio, M., Casagrande, V., Rossi, C., et al. (2014). Loss of TIMP3 exacerbates atherosclerosis in ApoE null mice. Atherosclerosis 235, 438–443. doi: 10.1016/j.atherosclerosis.2014.05.946
Strickland, D. K., Gonias, S. L., and Argraves, W. S. (2002). Diverse roles for the LDL receptor family. Trends Endocrinol. Metab. 13, 66–74. doi: 10.1016/s1043-2760(01)00526-4
Su, C. W., Lin, C. W., Yang, W. E., and Yang, S. F. (2019). TIMP-3 as a therapeutic target for cancer. Ther. Adv. Med. Oncol. 11:1758835919864247. doi: 10.1177/1758835919864247
Takawale, A., Sakamuri, S. S., and Kassiri, Z. (2015). Extracellular matrix communication and turnover in cardiac physiology and pathology. Compr. Physiol. 5, 687–719. doi: 10.1002/cphy.c140045
Takawale, A., Zhang, P., Azad, A., Wang, W., Wang, X., Murray, A. G., et al. (2017). Myocardial overexpression of TIMP3 after myocardial infarction exerts beneficial effects by promoting angiogenesis and suppressing early proteolysis. Am. J. Physiol. Heart Circ. Physiol. 313, H224–H236. doi: 10.1152/ajpheart.00108.2017
Thevenard, J., Verzeaux, L., Devy, J., Etique, N., Jeanne, A., Schneider, C., et al. (2014). Low-density lipoprotein receptor-related protein-1 mediates endocytic clearance of tissue inhibitor of metalloproteinases-1 and promotes its cytokine-like activities. PLoS One 9:e103839. doi: 10.1371/journal.pone.0103839
Thorn, S. L., Barlow, S. C., Feher, A., Stacy, M. R., Doviak, H., Jacobs, J., et al. (2019). Application of hybrid matrix metalloproteinase-targeted and Dynamic 201 Tl single-photon emission computed tomography/computed tomography imaging for evaluation of early post-myocardial infarction remodeling. Circ. Cardiovasc. Imaging 12:e009055. doi: 10.1161/CIRCIMAGING.119.009055
Tian, H., Cimini, M., Fedak, P. W., Altamentova, S., Fazel, S., Huang, M. L., et al. (2007). TIMP-3 deficiency accelerates cardiac remodeling after myocardial infarction. J. Mol. Cell. Cardiol. 43, 733–743. doi: 10.1016/j.yjmcc.2007.09.003
Troeberg, L., Fushimi, K., Khokha, R., Emonard, H., Ghosh, P., and Nagase, H. (2008). Calcium pentosan polysulfate is a multifaceted exosite inhibitor of aggrecanases. FASEB J. 22, 3515–3524. doi: 10.1096/fj.08-112680
Troeberg, L., Fushimi, K., Scilabra, S. D., Nakamura, H., Dive, V., Thogersen, I. B., et al. (2009). The C-terminal domains of ADAMTS-4 and ADAMTS-5 promote association with N-TIMP-3. Matrix Biol 28, 463–469. doi: 10.1016/j.matbio.2009.07.005
Troeberg, L., Lazenbatt, C., Anower, E. K. M. F., Freeman, C., Federov, O., Habuchi, H., et al. (2014). Sulfated glycosaminoglycans control the extracellular trafficking and the activity of the metalloprotease inhibitor TIMP-3. Chem. Biol. 21, 1300–1309. doi: 10.1016/j.chembiol.2014.07.014
Uhlen, M., Fagerberg, L., Hallstrom, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419
Vashist, A., Kaushik, A., Alexis, K., Dev Jayant, R., Sagar, V., and Nair, M. (2017). Bioresponsive injectable hydrogels for on-demand drug release and tissue engineering. Curr. Pharm. Des. 23, 3595–3602. doi: 10.2174/1381612823666170516144914
Verdonk, K., Danser, A. H., and van Esch, J. H. (2012). Angiotensin II type 2 receptor agonists: where should they be applied? Expert Opin. Investig. Drugs 21, 501–513. doi: 10.1517/13543784.2012.664131
Wadey, K., Lopes, J., Bendeck, M., and George, S. (2018). Role of smooth muscle cells in coronary artery bypass grafting failure. Cardiovasc. Res. 114, 601–610. doi: 10.1093/cvr/cvy021
Wang, B., Hsu, S. H., Majumder, S., Kutay, H., Huang, W., Jacob, S. T., et al. (2010). TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene 29, 1787–1797. doi: 10.1038/onc.2009.468
Wang, J., Zhang, J., Chen, X., Yang, Y., Wang, F., Li, W., et al. (2018). miR-365 promotes diabetic retinopathy through inhibiting Timp3 and increasing oxidative stress. Exp. Eye Res. 168, 89–99. doi: 10.1016/j.exer.2017.11.006
Wang, L., and Li, H. (2020). MiR-770-5p facilitates podocyte apoptosis and inflammation in diabetic nephropathy by targeting TIMP3. Biosci. Rep. 40:BSR20193653. doi: 10.1042/bsr20193653
Wang, W. M., Ge, G., Lim, N. H., Nagase, H., and Greenspan, D. S. (2006). TIMP-3 inhibits the procollagen N-proteinase ADAMTS-2. Biochem. J. 398, 515–519. doi: 10.1042/BJ20060630
Wang, X., Shi, Z., Liu, X., Su, Y., Li, W., Dong, H., et al. (2018). Upregulation of miR-191 promotes cell growth and invasion via targeting TIMP3 in prostate cancer. J. BUON 23, 444–452.
White, K. M., Alba, R., Parker, A. L., Wright, A. F., Bradshaw, A. C., Delles, C., et al. (2013). Assessment of a novel, capsid-modified adenovirus with an improved vascular gene transfer profile. J. Cardiothorac. Surg. 8:183. doi: 10.1186/1749-8090-8-183
Wilde, C. G., Hawkins, P. R., Coleman, R. T., Levine, W. B., Delegeane, A. M., Okamoto, P. M., et al. (1994). Cloning and characterization of human tissue inhibitor of metalloproteinases-3. DNA Cell Biol 13, 711–718. doi: 10.1089/dna.1994.13.711
Wisniewska, M., Goettig, P., Maskos, K., Belouski, E., Winters, D., Hecht, R., et al. (2008). Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J. Mol. Biol. 381, 1307–1319. doi: 10.1016/j.jmb.2008.06.088
Wu, D. M., Wen, X., Han, X. R., Wang, S., Wang, Y. J., Shen, M., et al. (2018). MiR-142-3p enhances cell viability and inhibits apoptosis by targeting CDKN1B and TIMP3 following sciatic nerve injury. Cell. Physiol. Biochem. 46, 2347–2357. doi: 10.1159/000489626
Xu, P., Liu, J., Sakaki-Yumoto, M., and Derynck, R. (2012). TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 5:ra34. doi: 10.1126/scisignal.2002689
Xu, Y., Bei, Y., Shen, S., Zhang, J., Lu, Y., Xiao, J., et al. (2017). MicroRNA-222 promotes the proliferation of pulmonary arterial smooth muscle cells by targeting P27 and TIMP3. Cell. Physiol. Biochem. 43, 282–292. doi: 10.1159/000480371
Yamamoto, K., Okano, H., Miyagawa, W., Visse, R., Shitomi, Y., Santamaria, S., et al. (2016). MMP-13 is constitutively produced in human chondrocytes and co-endocytosed with ADAMTS-5 and TIMP-3 by the endocytic receptor LRP1. Matrix Biol. 56, 57–73. doi: 10.1016/j.matbio.2016.03.007
Yan, D., Chen, D., Hawse, J. R., van Wijnen, A. J., and Im, H. J. (2013). Bovine lactoferricin induces TIMP-3 via the ERK1/2-Sp1 axis in human articular chondrocytes. Gene 517, 12–18. doi: 10.1016/j.gene.2013.01.001
Yang, H., Zheng, Y., Zhao, B., Shao, T., Shi, Q., Zhou, N., et al. (2013). Encapsulation of liver microsomes into a thermosensitive hydrogel for characterization of drug metabolism and toxicity. Biomaterials 34, 9770–9778. doi: 10.1016/j.biomaterials.2013.09.025
Yang, X., Du, W. W., Li, H., Liu, F., Khorshidi, A., Rutnam, Z. J., et al. (2013). Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res. 41, 9688–9704. doi: 10.1093/nar/gkt680
Yao, J., Jiang, S. L., Liu, W., Liu, C., Chen, W., Sun, L., et al. (2012). Tissue inhibitor of matrix metalloproteinase-3 or vascular endothelial growth factor transfection of aged human mesenchymal stem cells enhances cell therapy after myocardial infarction. Rejuvenation Res. 15, 495–506. doi: 10.1089/rej.2012.1325
Yao, Q., Chen, Y., and Zhou, X. (2019). The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 51, 11–17. doi: 10.1016/j.cbpa.2019.01.024
Yu, W. H., Yu, S., Meng, Q., Brew, K., et al. (2000). TIMP-3 binds to sulfated glycosaminoglycans of the extracellular matrix. J. Biol. Chem. 275, 31226–31232. doi: 10.1074/jbc.M000907200
Zhang, J., Lang, Y., Guo, L., Pei, Y., Hao, S., Liang, Z., et al. (2018). MicroRNA-323a-3p promotes pressure overload-induced cardiac fibrosis by targeting TIMP3. Cell. Physiol. Biochem. 50, 2176–2187. doi: 10.1159/000495059
Zhang, P., Shen, M., Fernandez-Patron, C., and Kassiri, Z. (2016). ADAMs family and relatives in cardiovascular physiology and pathology. J. Mol. Cell. Cardiol. 93, 186–199. doi: 10.1016/j.yjmcc.2015.10.031
Zhang, W., Zhong, B., Zhang, C., Wang, Y., Guo, S., Luo, C., et al. (2018). Structural modeling of osteoarthritis ADAMTS4 complex with its cognate inhibitory protein TIMP3 and rational derivation of cyclic peptide inhibitors from the complex interface to target ADAMTS4. Bioorg. Chem. 76, 13–22. doi: 10.1016/j.bioorg.2017.10.017
Zhang, Z., Wang, J., Wang, X., Song, W., Shi, Y., and Zhang, L. (2018). MicroRNA-21 promotes proliferation, migration, and invasion of cervical cancer through targeting TIMP3. Arch. Gynecol. Obstet. 297, 433–442. doi: 10.1007/s00404-017-4598-z
Zhao, H., Bernardo, M. M., Osenkowski, P., Sohail, A., Pei, D., Nagase, H., et al. (2004). Differential inhibition of membrane type 3 (MT3)-matrix metalloproteinase (MMP) and MT1-MMP by tissue inhibitor of metalloproteinase (TIMP)-2 and TIMP-3 rgulates pro-MMP-2 activation. J. Biol. Chem. 279, 8592–8601. doi: 10.1074/jbc.M308708200
Zheng, Z., He, X., Zhu, M., Jin, X., Li, C., Zhu, F., et al. (2018). Tissue inhibitor of the metalloproteinases-3 gene polymorphisms and carotid plaque susceptibility in the Han Chinese population. Int. J. Neurosci. 128, 920–927. doi: 10.1080/00207454.2018.1436544
Keywords: tissue inhibitor of metalloproteinases, TIMP3, metalloproteinase, myocardial infarction, cardiovascular diseases, microRNA, extracellular matrix, gene therapy
Citation: Fan D and Kassiri Z (2020) Biology of Tissue Inhibitor of Metalloproteinase 3 (TIMP3), and Its Therapeutic Implications in Cardiovascular Pathology. Front. Physiol. 11:661. doi: 10.3389/fphys.2020.00661
Received: 27 March 2020; Accepted: 25 May 2020;
Published: 16 June 2020.
Edited by:
George Grant, University of Aberdeen, United KingdomReviewed by:
Abhijit Takawale, Montreal Heart Institute, CanadaCopyright © 2020 Fan and Kassiri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Fan, ZG9uZy1mYW4yMDEwQGhvdG1haWwuY29t; Zamaneh Kassiri, ei5rYXNzaXJpQHVhbGJlcnRhLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.