 Luca Soattin
Luca Soattin Anniek Frederike Lubberding
Anniek Frederike Lubberding Bo Hjorth Bentzen
Bo Hjorth Bentzen Torsten Christ
Torsten Christ Thomas Jespersen
Thomas Jespersen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 12 June 2020
Sec. Cardiac Electrophysiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00493
Background: Adenosine leads to atrial action potential (AP) shortening through activation of adenosine 1 receptors (A1-R) and subsequent opening of G-protein-coupled inwardly rectifying K+ channels. Extracellular production of adenosine is drastically increased during stress and ischemia.
Objective: The aim of this study was to address whether the pharmacological blockade of endogenous production of adenosine and of its signaling prevents atrial fibrillation (AF).
Methods: The role of A1-R activation on atrial action potential duration, refractoriness, and AF vulnerability was investigated in rat isolated beating heart preparations (Langendorff) with an A1-R agonist [2-chloro-N6-cyclopentyladenosine (CCPA), 50 nM] and antagonist [1-butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36), 40 nM]. Furthermore, to interfere with the endogenous adenosine release, the ecto-5′-nucleotidase (CD73) inhibitor was applied [5′-(α,β-methylene) diphosphate sodium salt (AMPCP), 500 μM]. Isolated trabeculae from human right atrial appendages (hRAAs) were used for comparison.
Results: As expected, CCPA shortened AP duration at 90% of repolarization (APD90) and effective refractory period (ERP) in rat atria. PSB36 prolonged APD90 and ERP in rat atria, and CD73 inhibition with AMPCP prolonged ERP in rats, confirming that endogenously produced amount of adenosine is sufficiently high to alter atrial electrophysiology. In human atrial appendages, CCPA shortened APD90, while PSB36 prolonged it. Rat hearts treated with CCPA are prone to AF. In contrast, PSB36 and AMPCP prevented AF events and reduced AF duration (vehicle, 11.5 ± 2.6 s; CCPA, 40.6 ± 16.1 s; PSB36, 6.5 ± 3.7 s; AMPCP, 3.0 ± 1.4 s; P < 0.0001).
Conclusion: A1-R activation by intrinsic adenosine release alters atrial electrophysiology and promotes AF. Inhibition of adenosine pathway protects atria from arrhythmic events.
G-protein-coupled inwardly rectifying K+ channels (GIRK) play a key role in the physiological regulation of the heart. GIRK channels are in part responsible for the vagus-induced negative chronotropy upon muscarinic receptor (M2-R) activation (Pfaffinger et al., 1985; Sato et al., 1990), and their current is therefore referred to as IK,ACh. Constitutive activation of these channels in the absence of muscarinic agonists has been implicated in human persistent AF (Dobrev et al., 2005). Apart from being activated through muscarinic receptors, recent evidence has shown that adenosine, via the A1-receptor (A1-R), also increases GIRK channel conductance (Wang et al., 2013; Li et al., 2016). In the atrium, GIRK channel activation causes resting membrane potential (RMP) hyperpolarization and shortens both action potential duration (APD) and effective refractory period (ERP). Since the shortening of APD and refractoriness represents hallmarks of pro-arrhythmicity, adenosine can evoke atrial arrhythmias in humans, as well as in animal models (Drury and Szent-Gyorgyi, 1929; Belhassen et al., 1984; Kabell et al., 1994; Bertolet et al., 1997; Strickberger et al., 1997; Tebbenjohanns et al., 1997). For this reason, adenosine can be used as a tool to detect the location of possible AF foci during ablation procedures (Li et al., 2016; Letsas et al., 2017).
Adenosine is a purine nucleoside produced from adenosine monophosphate (AMP) by the glycosyl phosphatidylinositol-anchored membrane ectonucleotidase CD73, catalyzing the dephosphorylation of AMP to adenosine (Colgan et al., 2006). CD73 has been associated with cardioprotection during ischemia and hypoxia (Eckle et al., 2007), as extracellular adenosine functions as a protective metabolic signal following oxygen demand (Fox et al., 1974; Haneda et al., 1989). Adenosine is released upon energy perturbations, such as ischemia or hypoxia, and its extracellular levels are subsequently fine-tuned by the activity of transporters and enzymatic cascades (Eltzschig et al., 2013). Adenosine levels are limited in time and space by a fast conversion to the metabolite inosine by adenosine deaminase, as well as adenosine reuptake and subsequent phosphorylation to AMP by the equilibrative nucleoside transporters and adenosine kinase, respectively (Moser et al., 1989; Sheth et al., 2014). During normoxia, systemic adenosine levels have been reported to be stable around ∼21 nM (Saito et al., 1999), while during cardiac injury and heart failure, adenosine levels can increase sevenfold (Funaya et al., 1997). Purinergic signaling is under control of the transcription factor hypoxia inducible factor-1α (HIF-1α) (Thompson et al., 2004; Eltzschig et al., 2013). In normoxic conditions, prolyl hydroxylases degrade HIF-1α via ubiquitine-proteasome degradation consuming oxygen, resulting in low adenosine levels (Semenza, 2011). During hypoxia, low oxygen levels reduce the activity of prolyl hydroxylases, stabilizing HIF-1α, which translocates into the cell nucleus and upregulates CD73 (Eltzschig and Carmeliet, 2011). Moreover, HIF-1α downregulates transcription of the nucleoside equilibrative transporters and adenosine deaminase (Eltzschig et al., 2013). Therefore, hypoxia increases adenosine availability, slows adenosine metabolism rate, and, thereby, increases adenosine receptor activation.
Four types of adenosine receptors are expressed in the heart (A1, A2A, A2B, and A3), which can also function as heterodimers in both physiological and pathological conditions (Headrick et al., 2011). A1-R is a Gi-protein-coupled receptor and is predominantly expressed in atrial and nodal tissue (Pelleg et al., 1987; Belardinelli et al., 1995). In the atrioventricular (AV) node, adenosine slows the heart rate. This is partly accomplished by the activation of GIRK channels through the Giβγ-subunits and partly by the reduction in cyclic adenosine monophosphate (cAMP) through the Giα-subunit. Reduced cAMP levels decrease hyperpolarization-activated cyclic nucleotide-gated (HCN) channel activity in pacemaker cells and decrease protein kinase A activity, thereby reducing L-type calcium current (Belardinelli and Isenberg, 1983; Belardinelli et al., 1989, 1995; Pelleg et al., 1990). This process leads to a slowing of AV conduction (Belardinelli et al., 1995). For this reason, adenosine is used in clinical practice to terminate supraventricular tachycardia, such as atrioventricular nodal reentrant tachycardia (AVNRT) (DiMarco et al., 1990; Tebbenjohanns et al., 1997). However, the upregulation of A1-R in the failing sinoatrial (SA) node has been associated with an increase in AF in a canine heart failure model (Lou et al., 2014). AF drivers anchor to heterogeneity regions, which can also be anatomically determined (Haissaguerre et al., 2016). A large area of structural and electrical heterogeneity has been identified between the SA node complex and the surrounding right atrial tissue (Swarup et al., 2014). In addition, impairment in SA node conduction causes electrical remodeling and fibrosis, leading to micro-reentry circuits, which promote initiation and maintenance of AF (Thery et al., 1977; Kistler et al., 2004; Sanders et al., 2004).
AF is associated with heart failure and thromboembolic diseases (Rienstra et al., 2004). Moreover, AF patients’ daily life is often affected due to exertional dyspnea and reduced exercise tolerance (Hagens et al., 2004; Rienstra et al., 2004). In fact, it has been shown that oxygen uptake is significantly lowered in AF patients during exercise (Takano et al., 2019). Normoxic conditions play a fundamental role in atrial physiological function (Schuttler et al., 1983). Reduced supply of oxygen in localized atrial regions of a canine acute model showed a profound conduction slowing, which promoted reentry mechanisms (Sinno et al., 2003). In addition, more evidence highlight the role of hypoxia/ischemia in AF. In a case study of inferior infarction, AF spontaneously terminated after atrial branch reperfusion (Bunc et al., 2001). Isolated atrial tissues from AF patients showed a higher degree of fibrosis and vessel density (Gramley et al., 2010). Moreover, AF tissues express higher amount of typical hypoxic markers, such as HIF-1α and vascular endothelial growth factor (VEGF) (Gramley et al., 2010). It has also been proposed that fibrosis, upon structural remodeling in AF, might increase the diffusion distance of oxygen through the tissue and chronically trigger hypoxic and ischemic signaling (Mihm et al., 2001; Gramley et al., 2010).
In this study, we scrutinized the electrophysiological effects of intrinsic adenosine release, as well as extrinsic A1-R activation, in rat heart and human atrial tissue. We show that the activation of A1-R not only shortens atrial APD and ERP but also promotes AF susceptibility and duration. In contrast, antagonizing the activation of A1-R, or pharmacologically impairing the intrinsic adenosine release by inhibition of CD73, protects against arrhythmias.
Animal experiments were performed according to the ethical standards of the Danish Research Animal Committee (license no. 2012/152934-00345) and in accordance with the Danish legislations. A total of 38 male Wistar rats, 300–400 g, were anesthetized with sodium pentobarbital (40 mg/kg IP). By means of a tracheostomy, the rats were ventilated (4 ml/60 strokes/min) through a ventilator (7025 Rodent ventilator, Ugo Basile, Comerio VA, Italy). To remove the hearts, a thoracotomy was performed. Through a small aortic transection close to the aortic arch, the hearts were cannulated and connected to the Langendorff retrograde perfusion system (Hugo Sachs Elektronik -Harvard Apparatus GmbH, March-Hugstetten, Germany). The hearts were perfused at a constant pressure of 80 mmHg with a 37°C, pH 7.4, Krebs–Henseleit buffer (in mmol/L) NaCl 120.0, NaHCO3 25.0, KCl 4.0, MgSO4 0.6, NaH2PO4 0.6, CaCl2 2.5, glucose 11.0, at constant oxygenation with a O2/CO2 mixture of 95/5%. The aforementioned devices were connected to an amplifier (Hugo Sachs Elektronik -Harvard Apparatus GmbH, Germany). Epicardial monophasic action potential (MAP) electrodes (Hugo Sachs Elektronik -Harvard Apparatus GmbH, March-Hugstetten, Germany) were placed on the left and right atria. The electrocardiogram (ECG) was obtained with three ECG electrodes (Hugo Sachs Elektronik -Harvard Apparatus GmbH, March-Hugstetten, Germany) placed in close proximity to the heart. The hearts were immersed into a temperature-controlled bath containing the above-mentioned buffer. The pacing stimuli were twice the diastolic threshold with a duration of 2 ms. A pacing electrode was placed on the atrium, and the heart was allowed to stabilize for 30 min before recording the electrophysiological parameters. After baseline measurements, each group was perfused with a drug [final concentration, 2-chloro-N6-cyclopentyladenosine (CCPA) 50 nM, 1-butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36) 40 nM, 5′-(α, β-methylene) diphosphate sodium salt (AMPCP) 500 μM]. After stabilization, hearts were paced for 5 min with a cycle length (CL) of 200, 150, and 100 ms, respectively. At each pacing rate, diastolic threshold (DT)—applied field stimulation to elicit an action potential—was evaluated and doubled. To measure the ERP at different pacing rates, the delay (ΔS2 – S1) between the normal electrical stimulus (S1) and an additional stimulus (S2) was increased until an AP occurred. To measure the atrioventricular Wenckebach point—block of AV nodal conduction—the pacing rate was increased until second- and/or third-degree AV block occurred. To evaluate intrinsic rhythm, pacing was switched off for 3 min. At a CL of 100 ms, high-frequency electrical pacing (CL of 10 ms, for 500–1,000 ms, twice the DT) for inducing AF was applied up to 20 times every 3–5 s. AF episode duration was calculated from the end of electrical stimulation to the termination of the arrhythmic event—evaluated as a rapid atrial firing longer than 0.5 s. All data were acquired using the 16-channel PowerLab system (ADInstruments Ltd., Dunedin, New Zealand) (Figures 1A,B).
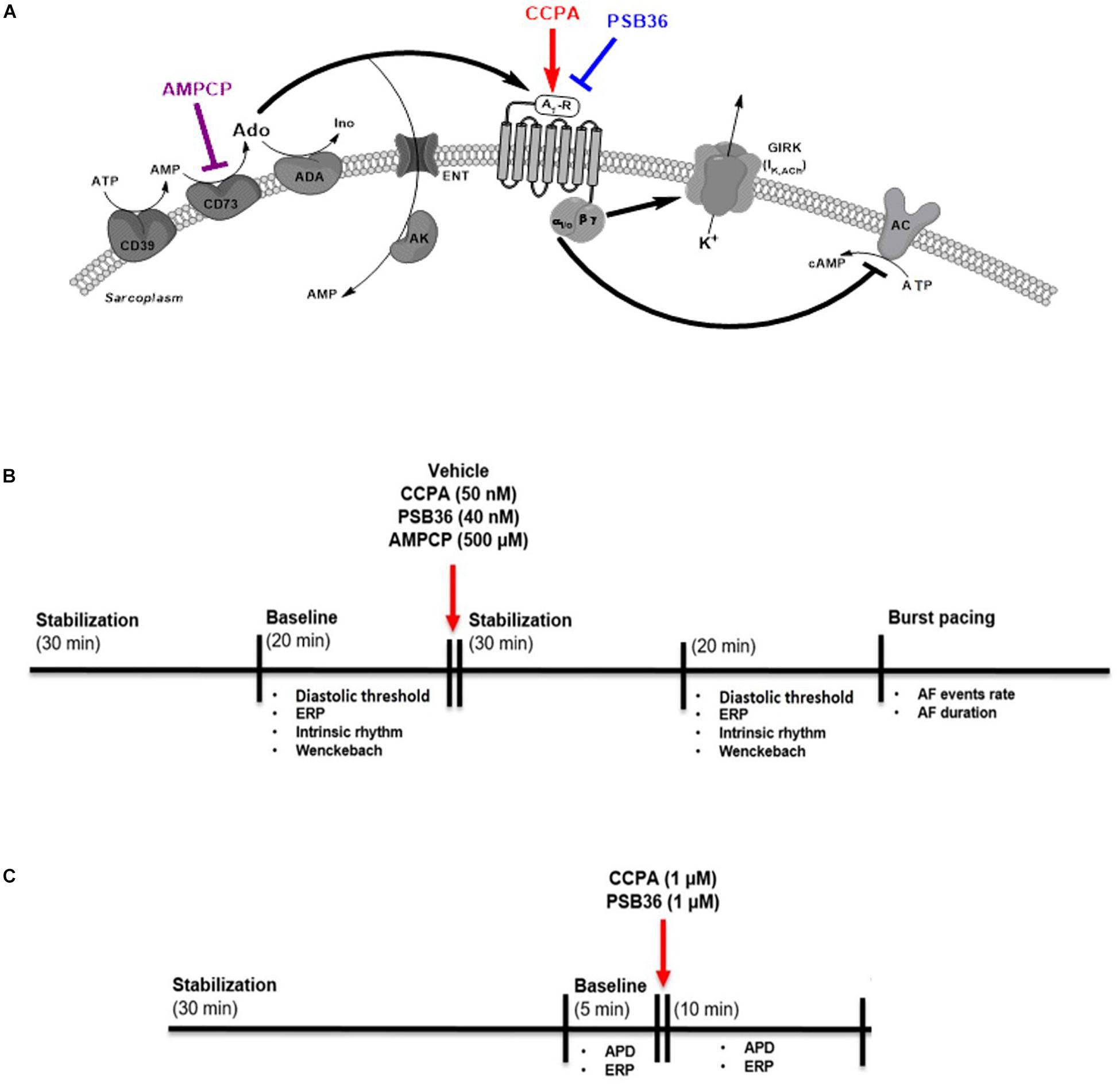
Figure 1. (A) Adenosine (Ado) pathway in cardiomyocytes. Adenosine release is regulated by the ectonucleotidases CD39 and CD73. CD39 converts ATP and ADP into AMP, while CD73 converts AMP into adenosine. Extracellular adenosine half-life is determined by adenosine deaminase (ADA) conversion to inosine (Ino) or internalization by specific equilibrative nucleoside transporters (ENT). A1-receptor activation increases K+ permeability for the G-protein-coupled inwardly rectifying K+ (GIRK) channel through the Gβγ and inhibits the adenylyl cyclase (AC) through Giα subunit. 2-Chloro-N6-cyclopentyladenosine (CCPA) is a potent A1-R agonist; 1-butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36) specifically antagonizes A1-R signaling. 5′-(α,β-methylene) diphosphate sodium salt (AMPCP) inhibits CD73. (B) Experimental protocol on isolated perfused male Wistar rat hearts. Hearts were removed and placed on the Langendorff configuration. After stabilization phase and baseline, hearts were perfused with vehicle, CCPA, PSB36, or AMPCP, respectively. Each group was paced at CL of 200, 150, and 100 ms. At the end of each experiment, the hearts were exposed to high-frequency pacing events in order to induce AF. (C) Experimental protocol on intact contracting muscles from human right atrial appendages (hRAAs). Human atrial tissues were paced at CL of 1.0 s. After stabilization phase, baseline was recorded for 5 min. After that, hRRAs were exposed to CCPA or PSB36, respectively, for 10 min.
The investigation on human samples conforms to all principles outlined by the Declaration of Helsinki and the Medical Association of Hamburg (Germany). According to the guidelines of the ethical review committee of the Medical Association of Hamburg, there is no need for a specific approval in this case, since patient data were used anonymized. All materials from patients were taken with informed consent of the donors. Eleven intact cardiac muscles from the right atrial appendages (RAAs) were obtained from 11 patients in sinus rhythm (SR) undergoing coronary artery bypass surgery and/or valve replacement (Supplementary Material 1). Atrial APs were recorded with intracellular microelectrodes in the right atrial trabeculae (microelectrode tip resistance, 10–25 MΩ; 1-ms stimulus, 25% above DT). The bath solution contained (in mmol/L): NaCl 127.0, KCl 4.5, MgCl2 1.5, CaCl2 1.8, glucose 10.0, NaHCO3 22.0, and NaH2PO4 0.42, equilibrated with O2/CO2 mixture of 95/5% at 37°C, pH 7.4. All data were acquired using the PowerLab system (ADInstruments Ltd., Dunedin, New Zealand). Drugs were applied using a fast perfusion system. Tissues were allowed to stabilize for 30 min at least. Afterward, the trabeculae were electrically paced during the entire experiment at CL of 1,000 ms. Baseline was recorded for 5 min before the administration of 1 μM CCPA or 1 μM PSB36. ERP was measured at baseline and after 10 min of drug superfusion (Figure 1C).
Experiments were conducted using the A1-R agonist CCPA (Tocris Bioscience Cat. No. 1705, Abingdon, United Kingdom) dissolved in a mixture of dimethyl sulfoxide (DMSO—Sigma Aldrich Cat. No. PHR1309) and distilled water (ratio 1:100), the A1-R antagonist PSB36 (Tocris Bioscience Cat. No. 2019, Abingdon, United Kingdom) dissolved in a mixture of DMSO and distilled water (ratio 1:100), the CD73 inhibitor AMPCP (Tocris Bioscience Cat. No. 3633, Abingdon, United Kingdom) dissolved in distilled water. DMSO (1%) was used as vehicle (Figure 1A).
Data are presented as mean ± SEM. All Langendorff and human AP measurements data were analyzed using Labchart 7 (ADInstruments, Ltd., Dunedin, New Zealand).
Statistical analysis was performed in GraphPad Prism 7 (GraphPad Software, La Jolla, CA, United States). Two-tailed paired t-tests were used to compare baseline with the effect of vehicle, CCPA, PSB36, and AMPCP on intrinsic rhythm, Wenckebach point APD90, and ERP. All detailed statistics of isolated heart data are presented in the Supplementary Material 2. AF duration values of each group were analyzed through a relative cumulative distribution (bin center, 1.0 s) by non-parametric Kruskal–Wallis test. Following, the Dunn’s multiple comparisons posttest was used to compare vehicle with the treated groups. P < 0.05 were considered statistically significant (denoted by asterisks in figures). Values of P < 0.01 and P < 0.001 are denoted by ∗∗ and ∗∗∗, respectively. Schemes were drawn with ChemBioDraw Ultra 12.0 (PerkinElmer, Waltham, MA, United States).
Isolated heart studies in Langendorff configuration allow for exploration of the cardiac effects of a biochemical pathway in isolation from the rest of the body. To study the effects of adenosine signaling in the atrium, we exposed the heart to vehicle (n = 10), the specific A1-R agonist CCPA (n = 11), the A1-R antagonist PSB36 (n = 10), and the specific CD73 antagonist AMPCP (n = 7) (Figure 1).
The intrinsic rhythm was measured by electrocardiogram (RR on ECG) (Figures 2A,B). The administration of DMSO did not affect heart rate. As expected, the A1-R agonist CCPA produced a profound negative chronotropic effect. In contrast, perfusion with the specific A1-R antagonist PSB36 produced a non-significant positive chronotropic effect (P = 0.056). Increased concentration of both CCPA (>50 nM) and PSB36 (>80 nM) produced runs of tachycardia as well as other arrhythmic activities (data not shown). No significant difference in beating rate was detected when the 5′-ecto-nucleotidase CD73 was inhibited with AMPCP (Figures 2A,B).
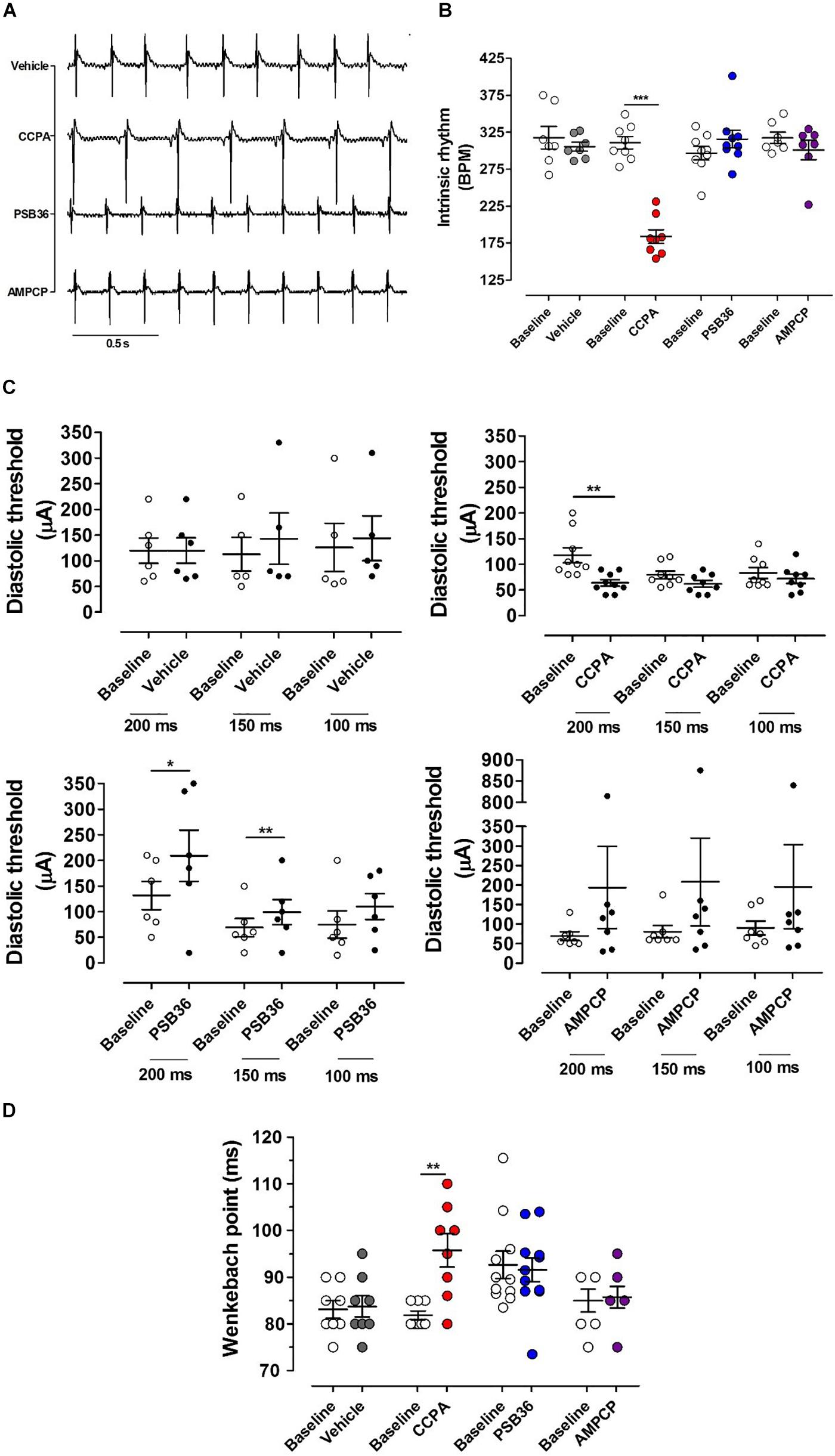
Figure 2. Chronotropic and dromotropic effects in rat hearts. (A) ECG lead II recordings under intrinsic rhythm. The R–R interval has been used to calculate heart rate. Paired t-tests between the baseline and the drugs were performed to evaluate significant differences. (B) Chronotropic effect of vehicle, 2-chloro-N6-cyclopentyladenosine (CCPA), 1-butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36), and 5′-(α,β-methylene) diphosphate sodium salt (AMPCP) (gray, red, blue, and purple dot plots, respectively). White dot plots represent the baseline. Values are presented as mean ± SEM. (C) Effect of vehicle, CCPA, PSB36, and AMPCP on the diastolic threshold (DT), respectively. (D) Effect of vehicle, CCPA, PSB36, and AMPCP on Wenckebach’s point, respectively. Vehicle did not affect atrio-ventricular node conduction. CCPA significantly increased Wenckebach point. PSB36 slightly reduced Wenckebach’s point. AMPCP had no effect on AV node conduction. *p < 0.05, **p < 0.01, ***p < 0.001.
The diastolic threshold (DT) is a measure of excitability of the atrial tissue (Figure 2C). While DT did not change in the vehicle group, the perfusion with CCPA increased membrane excitability, showing a significant DT reduction when the heart was paced at cycle length (CL) of 200 ms and a tendency toward reduction in DT at CL of 150 and 100 ms. By contrast, PSB36 increased DT at CLs of 200 and 150 ms. AMPCP-treated atria showed a tendency toward increased DT; however, this did not reach significance.
To detect whether targeting of the purinergic system affects the electrical conduction between atria and ventricles, we increased the pacing rate until the atrioventricular Wenckebach point was reached. Hearts treated with vehicle did not show any difference from baseline. Not surprisingly, the activation of A1-R with CCPA prolonged the Wenckebach point. PSB36, antagonizing A1-R, showed a slight tendency in reducing the Wenckebach point, while no effect was observed on the atrioventricular conduction when CD73 was inhibited with AMPCP (Figure 2D).
To investigate the effect on APD at 90% of repolarization (APD90) in the atria, the hearts were paced at CLs of 200, 150, and 100 ms for 5 min (Figure 3A). As expected, APD90 moderately decreased as pacing decreased (Supplementary Material 3). Atrial APD was not altered by the administration of the vehicle (Figure 3B). CCPA provoked a profound APD90 reduction at CL of 200, 150, and 100 ms (Figure 3C and Supplementary Material 4). CCPA showed a maximum APD90 shortening of ∼51% in dose–response relationship tests (Supplementary Material 5).
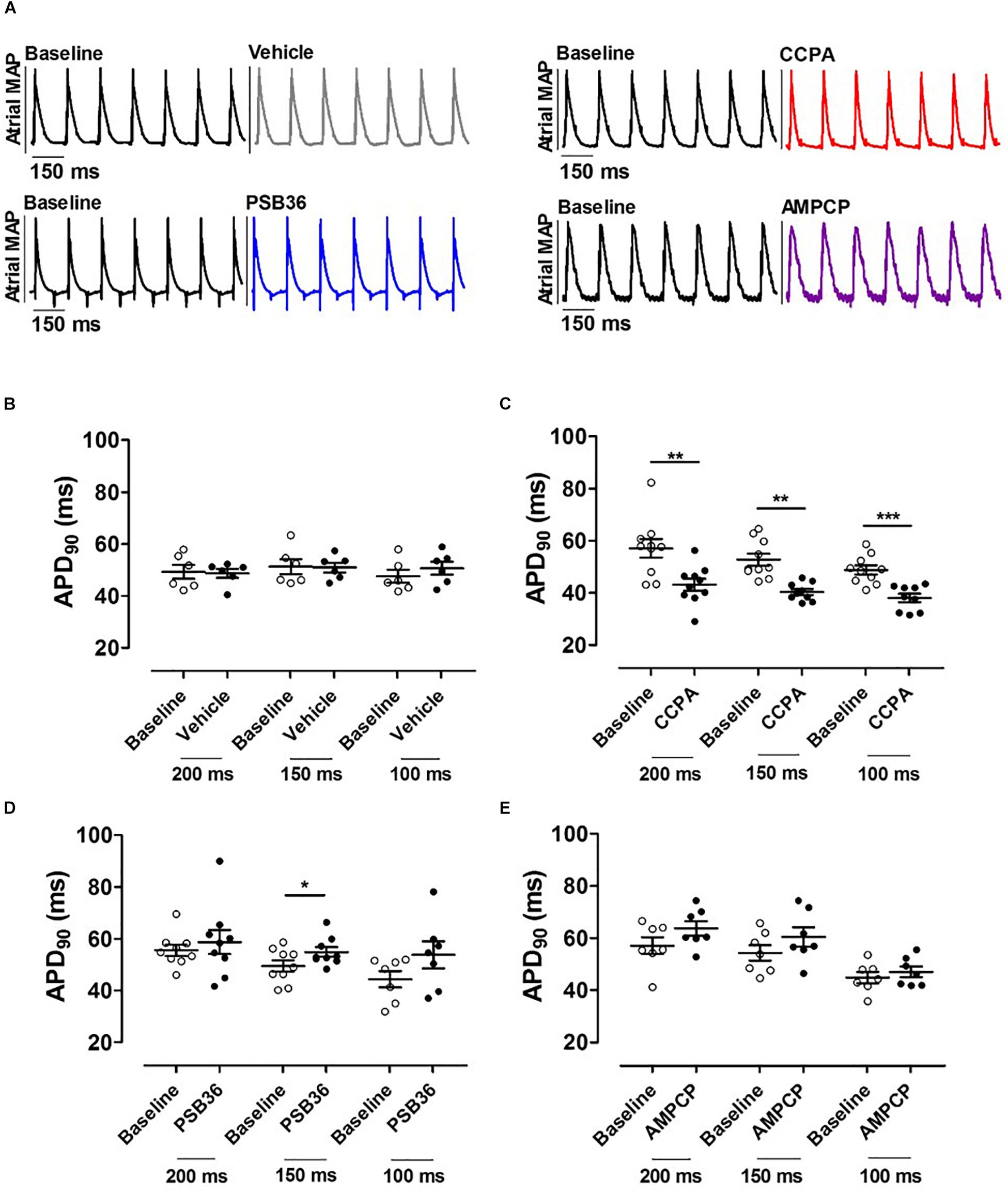
Figure 3. (A) Atrial monophasic action potentials (MAPs) paced at cycle length (CL) of 150 ms before and after the administration of vehicle, 2-chloro-N6-cyclopentyladenosine (CCPA), 1-butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36), or 5′-(α,β-methylene) diphosphate sodium salt (AMPCP); black MAPs refer to the baseline, while gray, red, blue, and purple MAPs represent hearts perfused with vehicle, CCPA, PSB36, and AMPCP, respectively. (B) Effect of vehicle on APD90 at CL of 200, 150, and 100 ms. (C) CCPA shortened APD90 at CL of 200, 150, and 100 ms, respectively. (D) PSB36 showed a prolongation of APD90 at CL of 200, 150, and 100 ms. (E) Effect of AMPCP on APD90 at CL of 200, 150, and 100 ms. *p < 0.05, **p < 0.01, ***p < 0.001.
On the other hand, PSB36 showed a tendency to prolong the atrial APD90 in a rate-dependent manner (Supplementary Material 4). The prolongation was significant at CL of 150 ms (Figure 3D). The inhibition of CD73 showed a tendency in APD90 increase (Figure 3E).
Hearts treated with vehicle did not change ERP over time, while CCPA significantly shortened ERP at CLs of 150 and 100 ms (Figures 4A–C). A1-R inhibition by PSB36 showed the opposite effect, prolonging ERP significantly in a rate-dependent manner at CLs of 200 and 150 ms, and with a tendency at CL of 100 ms (Figure 4D). AMPCP induced a significant prolongation of refractoriness at CLs of 200 and 150 ms, but not at CL of 100 ms (Figure 4E).
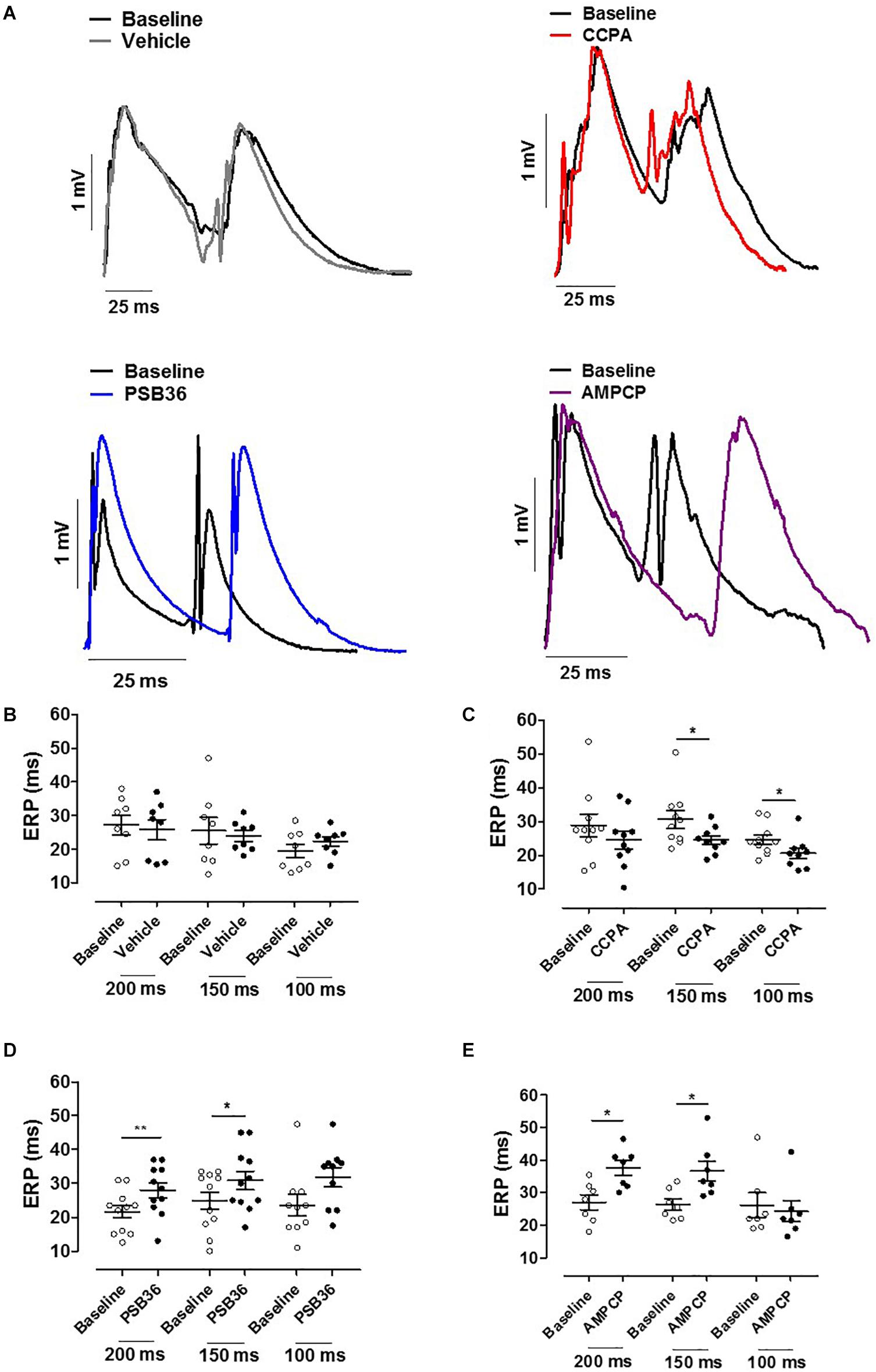
Figure 4. (A) Effective refractory period (ERP) monophasic action potentials (MAPs) of baseline and the relative treated hearts are shown superimposed. (B) Effect of vehicle on ERP at cycle length (CL) 0.20, 0.15, and 0.10 s. (C) 2-Chloro-N6-cyclopentyladenosine (CCPA) shortened refractoriness at CL of 200 ms, significantly at CL of 150 and 100 ms. (D) 1-Butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36) prolonged ERP at CL of 200 and 150 ms. (E) 5′-(α,β-Methylene) diphosphate sodium salt (AMPCP) prolonged ERP at CL of 200 and 150 ms, but not at 100 ms. *p < 0.05, **p < 0.01.
To induce AF, the hearts were exposed to fast electrical pacing (Figure 5A). AF duration for hearts treated with vehicle (11.5 ± 2.6 s, n = 9) were compared to CCPA (40.6 ± 16.1 s, n = 10), PSB36 (6.5 ± 3.7 s, n = 10), and AMPCP (3.0 ± 1.4 s, n = 7). PSB36 and AMPCP significantly shortened the duration of AF events compared to vehicle, while CCPA increased the duration of AF. The relative cumulative stratification (in %) of AF event duration frequencies for each group showed that ∼90% of PSB36 and 85% of AMPCP-treated hearts had AF events lasting <2 s, in contrast to ∼72% of vehicle and only ∼53% of CCPA hearts (P < 0.0001 within the groups) (Figure 5B and Supplementary Material 6). In addition, spontaneous AF (sAF) events occurred more frequently in CCPA-treated hearts than in the PSB36 and AMPCP groups (Supplementary Material 7). Moreover, when atria were not paced, the duration of sAF events in PSB36 and AMPCP groups was drastically reduced and significantly lower compared to CCPA-treated hearts (P < 0.05) (Supplementary Material 8).
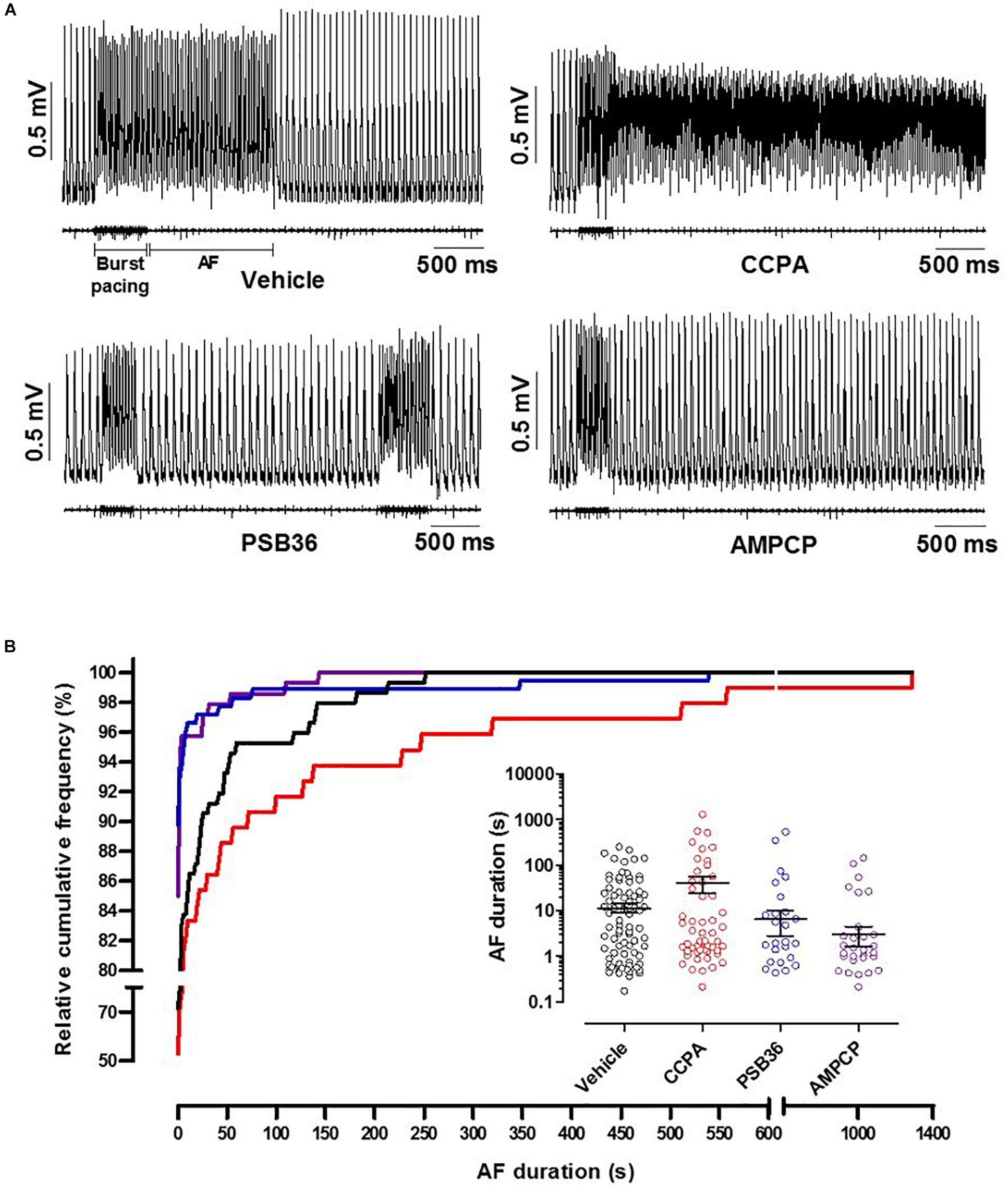
Figure 5. Induction of atrial fibrillation (AF) through high-frequency electrical pacing. (A) Atrial monophasic action potentials (MAPs) and pacing traces are shown. Vehicle produced short AF events; by contrasts, 2-chloro-N6-cyclopentyladenosine (CCPA) made the heart prone to sustained AF events. 1-Butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36) and 5′-(α,β-methylene) diphosphate sodium salt (AMPCP) reduced atrial sensitivity to high frequency electrical pacing (Supplementary Material 6; p < 0.0001). (B) Relative cumulative distribution (in%) of AF duration of each group and dot plot of AF duration events on a logarithmic scale, which shows the range of AF duration among groups when events are different from 0 s. Vehicle, CCPA, PSB36, and AMPCP are depicted in black, red, blue, and purple, respectively.
Action potential measurements were conducted on intact contracting muscles isolated from the right atrium appendage of patients in SR. After stabilization, 1 μM CCPA or PSB36 were superfused for 10 min (Figure 1C). CCPA produced a relative APD90 shortening of ∼12% (Supplementary Material 9) when paced at CL of 1,000 ms (baseline 364.9 ± 25.3 ms vs. CCPA 320.5 ± 20.2 ms, P < 0.01) (Figures 6A–C). A1-R inhibition by PSB36 showed a relative APD90 prolongation of ∼9% (baseline 383.6 ± 23.5 ms vs. PSB36 416.9 ± 24.5 ms, P < 0.001; Figures 6D–F). ERP recordings on isolated human trabeculae are technically demanding, as extra stimuli often result in loss of seal. Collected data and statistics are shown in Supplementary Material 10.
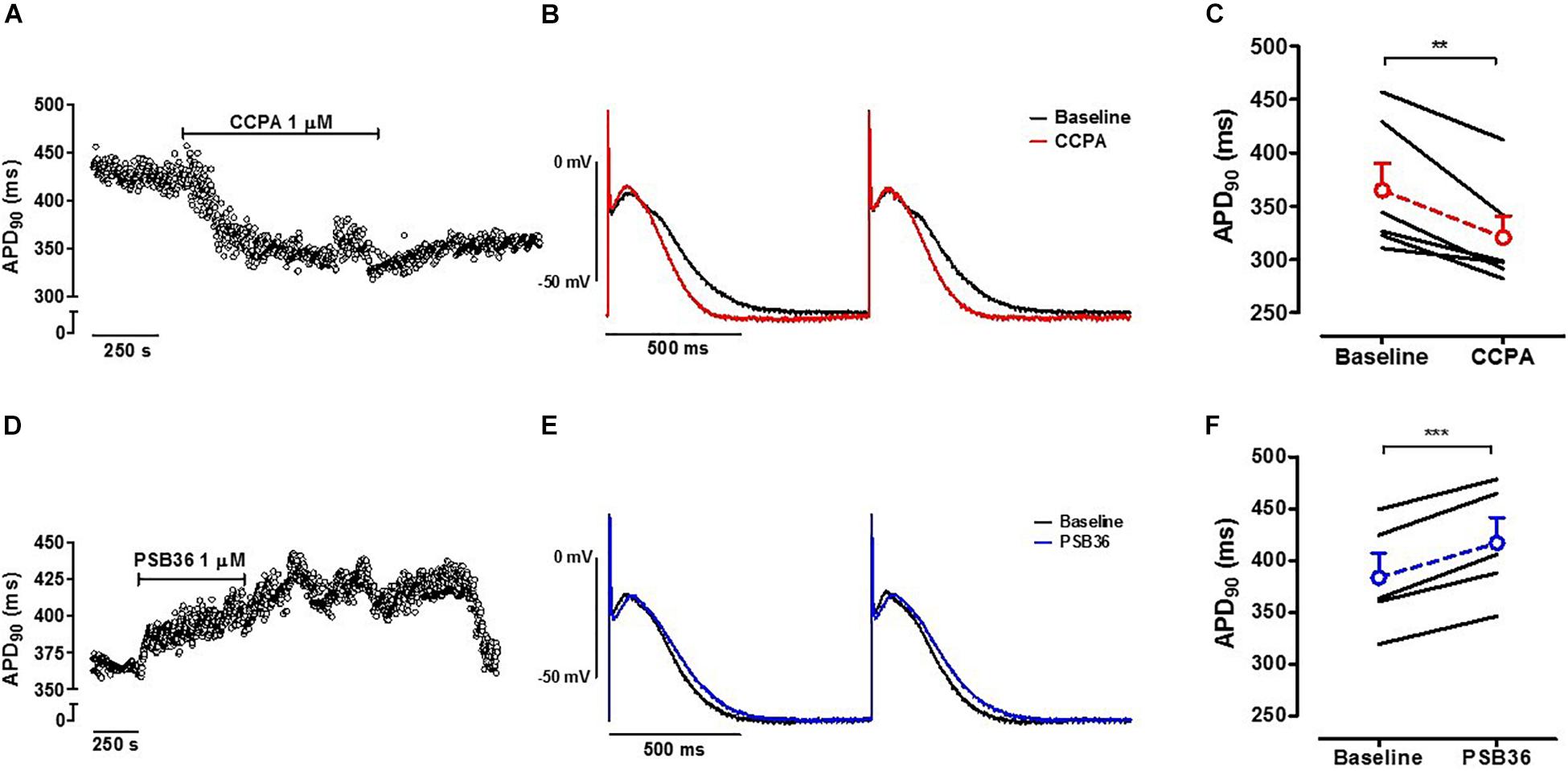
Figure 6. (A) A1-R activation in human right atrial appendages (hRAAs). APD90 measurements are shown over time when an isolated contracting atrial muscle was superfused with 1 μM 2-chloro-N6-cyclopentyladenosine (CCPA). (B) Baseline and CCPA-treated hRAA action potentials (APs) were superimposed to show the APD shortening due to A1-R activation. hRAA was paced at cycle length (CL) of 1.0 s. (C) CCPA shortened APD90 in hRAAs. (D) A1-R blockade in hRAAs. APD90 measurements are shown over time when an isolated contracting atrial muscle was superfused with 1 μM 1-butyl-3-(3-hydroxypropyl)-8-(3-noradamantyl)xanthine (PSB36). hRAA was paced at CL of 1,000 ms. (E) Baseline and PSB36-treated hRAA APs were superimposed to show the APD prolongation due to A1-R blockade. (F) PSB36 prolonged APD90 in hRAAs. Resting membrane potential (RMP) in representative panels (B,E) was normalized to show change in AP duration. **p < 0.01, ***p < 0.001.
Adenosine plays a key role in cardiac electrophysiology, which becomes especially prominent during hypoxia or ischemia. In the heart, adenosine activity is mediated by the aforementioned four types of adenosine receptors (Headrick et al., 2011). Although A2A-R also has been associated with AF (Hove-Madsen et al., 2006; Llach et al., 2011; Molina et al., 2016), the present manuscript focuses on A1-R as the most likely candidate involved in the relation between adenosine and AF (Belardinelli and Isenberg, 1983; Visentin et al., 1990; Kabell et al., 1994; Atienza et al., 2006; Ip et al., 2013; Li et al., 2016). In fact, A1-R has a higher expression in the atrium and nodal tissue compared to the other adenosine receptors (Headrick et al., 2011; Lou et al., 2014). Generally, A1-R and A2A-R are activated at nanomolar concentrations of adenosine and therefore show the highest affinity for adenosine compared to A2B-R and A3-R (low-affinity receptors), which exhibit their activity during hypoxic stress, when adenosine levels elevate drastically (Fredholm et al., 2001; Latini and Pedata, 2001; Pedata et al., 2001). In addition, it has also been shown that A1-R and A3-R stimulation would neutralize A2A-R signaling in the heart (Talukder et al., 2002; Headrick et al., 2011).
We used the specific A1-R agonist (CCPA) and antagonist (PSB36) that have potent and strong selectivity for A1-R and have very poor or no selectivity for the other adenosine receptors (Muller and Jacobson, 2011). Off-target effects through the other adenosine receptors are therefore unlikely in the present study.
Activation of A1-R increases potassium conductance through GIRK channels. These channels also play a pivotal role in the nodal tissue. Our experiments confirm that A1-R activation also activates GIRK channels in the nodes, as A1-R stimulation with CCPA showed a clear negative chronotropic effect and an increase in the Wenckebach point in rat isolated hearts. As previously shown, when A1-R is genetically deleted, the drop in heart rate normally induced by adenosine infusion is abolished (Koeppen et al., 2009). In our work, the inhibition of A1-R by PSB36 did not produce neither significant nor biologically relevant elevation in intrinsic rate (P = 0.056) of isolated perfused hearts, indicating a minor, or absent, effect of endogenous produced adenosine on the sinus node function. In addition, the Wenckebach point was not altered following PSB36 infusion, indicating that the AV node is also not affected by endogenous adenosine.
In the atria, we found that the activation of A1-R with CCPA profoundly shortened APD90 in both explanted rat hearts and human trabeculae. ERP was also shortened by CCPA in rat hearts. Several studies have shown that the prolonged stimulation of A1-R triggers desensitization of the receptors due to inactivation and internalization mechanisms (Saura et al., 1998; Klaasse et al., 2008). However, in our experiments, we did not observe effects indicating A1-R desensitization (Supplementary Material 11). Although it is not possible to exclude the role of other potassium channels, the observed effects at the beginning of the action potential might reflect changes in early IK,ACh (Tang et al., 2015). For instance, activation or inhibition of A1-R could also trigger fluctuations of cAMP levels, which might affect L-type calcium channels (Headrick et al., 2011). However, minor changes were detected on the action potentials in our experiments. Moreover, it has been shown that the adenosine-induced decrease in ICa currents, via cAMP, would have a limited or no effect on the shortening of the action potential (Visentin et al., 1990).
In our work, the relative APD90 shortening appeared more profound in rats than in humans. Recently, Li et al. (2016) showed that in humans A1-R and GIRK4 protein expression follows a gradient with a maximum level in the superior lateral region of the right atrium. While we only had access to the right atrial appendage from SR patients, exposure to CCPA revealed a significant AP shortening. Interestingly, PSB36 prolonged the APD90 in both rat and human atria. In addition, the direct antagonism of A1-R prolonged ERP in isolated rat hearts. This suggests that adenosine is intrinsically released and contributes to fine-tuning of the repolarization phase of the atrial action potential. Several groups have already described intrinsic and endogenous release of adenosine (Mubagwa et al., 1996; Headrick et al., 2003; Jammes et al., 2015). To confirm the electrophysiological effect of this phenomenon, we targeted the upstream adenosine releasing mechanism with the selective CD73 inhibitor (AMPCP). AMPCP had a tendency to prolong APD90 and significantly prolonged ERP, suggesting that adenosine is intrinsically released under the chosen experimental conditions.
Atrial fibrillation events were induced with an average duration of ∼12 s in rat hearts treated with vehicle. The A1-R agonist (CCPA) made the hearts more prone to AF, increasing the duration of AF to ∼41 s. In addition, spontaneous AF events occurred more frequently in CCPA-treated hearts rather than in the PSB36 and AMPCP groups. Both the direct antagonism of the A1-R with PSB36 and the inhibition of the CD73 with AMPCP profoundly reduced the ability to induce sustained AF events.
Interestingly, the activation of A1-R with CCPA significantly lowered the DT. This could be explained by an increased GIRK channel activity, hyperpolarizing the resting membrane potential (Supplementary Material 12A), thereby increasing the fraction of available sodium channels. Such an increased sodium current, combined with shorter refractory periods, would be expected to increase vulnerability and sustainability of AF. In contrast, antagonizing A1-R with PSB36 produced a profound reduction in atrial excitability, represented by an increase in DT. This could suggest an increased protection from ectopic firing or focal partial depolarization. Thus, antagonizing A1-R with PSB36 produces an antiarrhythmic state by prolonging APD and ERP, potentially supported by a depolarization of RMP (Supplementary Material 12B), thereby reducing the availability of sodium channels and thereby susceptibility to triggered activity (Wang et al., 2013).
During hypoxic and ischemic injury, adenosine reduces energy consumption and adapts the tissue to hypoxic conditions (Eltzschig et al., 2013). Based on this, it can be speculated that the shortening of APD in the atrium, evoked by A1-R activation resembles a protective mechanism to avoid calcium overload during cardiac metabolic stress (Cole et al., 1991; Findlay, 1994). However, even though adenosine protects from ischemic and hypoxic heart injuries in the ventricles, hypoxic conditions in the atria may constitute a substrate for AF through A1-R activation. Hypoxia and ischemia in the heart are cause of excessive energy consumption. To make matters worse, the high frequency of atrial excitation and contraction in AF increases oxygen consumption while limiting oxygen supply to the tissue (Harada et al., 2017). In this setting, atrial A1-R activation, especially in regions with high density of GIRK channels, may constitute an important promoting and sustaining factor for AF. Recent clinical trials with IK,ACh blockers did not show a reduction in paroxysmal AF (Podd et al., 2016). However, it has been shown that obstructive or central sleep apnea causes AF and atrial remodeling. The reiterated oxygen drop due to sleep apnea may recruit the purinergic pathway leading to A1-R stimulation in the atrium as AF substrate (Caples and Somers, 2009; Zhang et al., 2015). Thus, blocking endogenous adenosine production or inhibiting A1-R activation could be a potential treatment modality of AF. Pilot studies in rat Langendorff hearts indicated that, during AF, the acute A1-R blockade might restore the sinus rhythm (Supplementary Material 13). However, due to its relatively ubiquitous expression, targeting the purinergic signaling may have undesirable consequences on multiple related pathways and organs (Jacobson and Gao, 2006). Therefore, thorough electrophysiological investigations of adenosine block and A1-R inhibition are needed in intact AF animal models. In addition, further studies on energy consumption and calcium handling upon A1-R stimulation could better clarify the substantial role of the purinergic system in atrial electrophysiology.
Targeting CD73 for AF treatment appears to be promising. CD73 inhibition prolonged refractoriness and protected rat atria from sustained arrhythmogenic events. CD73 has been also identified as a target for pharmacotherapy strategies of breast cancer (Stagg et al., 2010), and it plays critical role during injury protection in the renal proximal tubule as well as in immune processes, such as inflammation (Antonioli et al., 2013; Sung et al., 2017). Furthermore, adenosine modulates vascular homeostasis, also in the heart (Koszalka et al., 2004; Headrick et al., 2011). Hence, non-beneficial side effects might limit the efficacy of CD73 as a target for AF treatment.
The use of rodents as translational model for AF carries study limitations regarding differences with human physiology. To repolarize the action potential, unlike humans, rodents rely on IK,to currents, which accelerate the repolarization rate and shorten the plateau phase (Roden, 1998; Guo et al., 1999). However, sufficient parallels in pathophysiology of cardiac diseases have been found between rodents and humans, so that rodents can be considered a valid model for cardiac electrophysiology and AF (Nishida et al., 2010; Diness et al., 2011; Farraj et al., 2011; Skibsbye et al., 2011, 2015).
Although the number of patients represents an important limitation to this study in terms of statistical and clinical significance, human trabeculae in SR were a useful investigation tool to validate data obtained in rat isolated perfused hearts showing an important translational level of confidence between rodents and human physiology.
The use of Krebs–Henseleit and Tyrode buffers for Langendorff and Steiert organ bath experiments, respectively, may have generated a slight hypoxic environment over time. This may have contributed to the intrinsic release of adenosine. However, hypoxic conditions cannot be assessed in the systems used in this study. During perfusion and superfusion experiments of vehicle, CCPA, or PSB36, the effect of the intrinsic adenosine release on A1-R was not measured. However, adenosine’s half-life is extremely short, and the dissociation equilibrium constants between adenosine, CCPA, and PSB36 for A1-R (700, 0.4, and 0.7 nM, respectively) (Lohse et al., 1988; Bilkei-Gorzo et al., 2008; de Lera Ruiz et al., 2014) would favor the binding of CCPA or PSB36 to A1-R rather than the natural occurring purine nucleoside. The choice of drugs concentration was based on previous laboratory data (not shown) and recent literature.
In this study, we highlighted the role of adenosine signaling in atrial electrophysiology and AF susceptibility. Reducing A1-R activation, either by direct inhibition or by interfering with endogenous adenosine release, produced an antiarrhythmic state of the atria. These data reveal CD73 as a potential pharmacological target in the setting of AF.
The datasets analyzed in this article are not publicly available. Requests to access the datasets should be directed to bHVjYS5zb2F0dGluMTk4NkBnbWFpbC5jb20=; dGhvamVzQHN1bmQua3UuZGsu
The studies involving human participants were reviewed and approved by Declaration of Helsinki and the Medical Association of Hamburg (Germany). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Danish Research Animal Committee (license n. 2012/152934-00345).
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 675351.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to express thankfulness to Prof. Hermann Reichenspurner, MD, Ph.D. of the Universitäres Herzzentrum Hamburg GmbH (UHZ) and Klinik und Poliklinik für Herz- und Gefäßchirurgie of Hamburg (Germany) for giving access to tissues from the surgery.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.00493/full#supplementary-material
Antonioli, L., Pacher, P., Vizi, E. S., and Hasko, G. (2013). CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 19, 355–367. doi: 10.1016/j.molmed.2013.03.005
Atienza, F., Almendral, J., Moreno, J., Vaidyanathan, R., Talkachou, A., Kalifa, J., et al. (2006). Activation of inward rectifier potassium channels accelerates atrial fibrillation in humans: evidence for a reentrant mechanism. Circulation 114, 2434–2442. doi: 10.1161/circulationaha.106.633735
Belardinelli, L., and Isenberg, G. (1983). Isolated atrial myocytes: adenosine and acetylcholine increase potassium conductance. Am. J. Physiol. 244, H734–H737.
Belardinelli, L., Linden, J., and Berne, R. M. (1989). The cardiac effects of adenosine. Prog. Cardiovasc. Dis. 32, 73–97. doi: 10.1016/0033-0620(89)90015-7
Belardinelli, L., Shryock, J. C., Song, Y., Wang, D., and Srinivas, M. (1995). Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J. 9, 359–365. doi: 10.1096/fasebj.9.5.7896004
Belhassen, B., Pelleg, A., Shoshani, D., and Laniado, S. (1984). Atrial fibrillation induced by adenosine triphosphate. Am. J. Cardiol. 53, 1405–1406. doi: 10.1016/0002-9149(84)90104-8
Bertolet, B. D., Hill, J. A., Kerensky, R. A., and Belardinelli, L. (1997). Myocardial infarction related atrial fibrillation: role of endogenous adenosine. Heart 78, 88–90. doi: 10.1136/hrt.78.1.88
Bilkei-Gorzo, A., Abo-Salem, O. M., Hayallah, A. M., Michel, K., Muller, C. E., and Zimmer, A. (2008). Adenosine receptor subtype-selective antagonists in inflammation and hyperalgesia. Naunyn. Schmiedebergs Arch. Pharmacol. 377, 65–76. doi: 10.1007/s00210-007-0252-9
Bunc, M., Starc, R., Podbregar, M., and Brucan, A. (2001). Conversion of atrial fibrillation into a sinus rhythm by coronary angioplasty in a patient with acute myocardial infarction. Eur. J. Emerg. Med. 8, 141–145. doi: 10.1097/00063110-200106000-00011
Caples, S. M., and Somers, V. K. (2009). Sleep-disordered breathing and atrial fibrillation. Prog. Cardiovasc. Dis. 51, 411–415. doi: 10.1016/j.pcad.2008.06.004
Cole, W. C., McPherson, C. D., and Sontag, D. (1991). ATP-regulated K+ channels protect the myocardium against ischemia/reperfusion damage. Circ. Res. 69, 571–581. doi: 10.1161/01.res.69.3.571
Colgan, S. P., Eltzschig, H. K., Eckle, T., and Thompson, L. F. (2006). Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic. Signal. 2, 351–360. doi: 10.1007/s11302-005-5302-5
de Lera Ruiz, M., Lim, Y. H., and Zheng, J. (2014). Adenosine A2A receptor as a drug discovery target. J. Med. Chem. 57, 3623–3650. doi: 10.1021/jm4011669
DiMarco, J. P., Miles, W., Akhtar, M., Milstein, S., Sharma, A. D., Platia, E., et al. (1990). Adenosine for paroxysmal supraventricular tachycardia: dose ranging and comparison with verapamil. Assessment in placebo-controlled, multicenter trials. the adenosine for PSVT study group. Ann. Intern. Med. 113, 104–110.
Diness, J. G., Skibsbye, L., Jespersen, T., Bartels, E. D., Sorensen, U. S., Hansen, R. S., et al. (2011). Effects on atrial fibrillation in aged hypertensive rats by Ca(2+)-activated K(+) channel inhibition. Hypertension 57, 1129–1135. doi: 10.1161/HYPERTENSIONAHA.111.170613
Dobrev, D., Friedrich, A., Voigt, N., Jost, N., Wettwer, E., Christ, T., et al. (2005). The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706. doi: 10.1161/circulationaha.105.575332
Drury, A. N., and Szent-Gyorgyi, A. (1929). The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J. Physiol. 68, 213–237. doi: 10.1113/jphysiol.1929.sp002608
Eckle, T., Krahn, T., Grenz, A., Kohler, D., Mittelbronn, M., Ledent, C., et al. (2007). Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 115, 1581–1590.
Eltzschig, H. K., Bonney, S. K., and Eckle, T. (2013). Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends Mol. Med. 19, 345–354. doi: 10.1016/j.molmed.2013.02.005
Eltzschig, H. K., and Carmeliet, P. (2011). Hypoxia and inflammation. N. Engl. J. Med. 364, 656–665.
Farraj, A. K., Hazari, M. S., and Cascio, W. E. (2011). The utility of the small rodent electrocardiogram in toxicology. Toxicol. Sci. 121, 11–30. doi: 10.1093/toxsci/kfr021
Findlay, I. (1994). The ATP sensitive potassium channel of cardiac muscle and action potential shortening during metabolic stress. Cardiovasc. Res. 28, 760–761. doi: 10.1093/cvr/28.6.760
Fox, A. C., Reed, G. E., Glassman, E., Kaltman, A. J., and Silk, B. B. (1974). Release of adenosine from human hearts during angina induced by rapid atrial pacing. J. Clin. Invest. 53, 1447–1457. doi: 10.1172/jci107693
Fredholm, B. B., Ap, I. J., Jacobson, K. A., Klotz, K. N., and Linden, J. (2001). International union of pharmacology. XXV. nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552.
Funaya, H., Kitakaze, M., Node, K., Minamino, T., Komamura, K., and Hori, M. (1997). Plasma adenosine levels increase in patients with chronic heart failure. Circulation 95, 1363–1365. doi: 10.1161/01.cir.95.6.1363
Gramley, F., Lorenzen, J., Jedamzik, B., Gatter, K., Koellensperger, E., Munzel, T., et al. (2010). Atrial fibrillation is associated with cardiac hypoxia. Cardiovasc. Pathol. 19, 102–111. doi: 10.1016/j.carpath.2008.11.001
Guo, W., Xu, H., London, B., and Nerbonne, J. M. (1999). Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J. Physiol. 521(Pt 3), 587–599. doi: 10.1111/j.1469-7793.1999.00587.x
Hagens, V. E., Ranchor, A. V., Van Sonderen, E., Bosker, H. A., Kamp, O., Tijssen, J. G., et al. (2004). Effect of rate or rhythm control on quality of life in persistent atrial fibrillation. results from the rate control versus electrical cardioversion (RACE) study. J. Am Coll. Cardiol. 43, 241–247.
Haissaguerre, M., Shah, A. J., Cochet, H., Hocini, M., Dubois, R., Efimov, I., et al. (2016). Intermittent drivers anchoring to structural heterogeneities as a major pathophysiological mechanism of human persistent atrial fibrillation. J. Physiol. 594, 2387–2398. doi: 10.1113/JP270617
Haneda, T., Ichihara, K., Abiko, Y., and Onodera, S. (1989). Release of adenosine and lactate from human hearts during atrial pacing in patients with ischemic heart disease. Clin. Cardiol. 12, 76–82. doi: 10.1002/clc.4960120203
Harada, M., Melka, J., Sobue, Y., and Nattel, S. (2017). Metabolic considerations in atrial fibrillation- mechanistic insights and therapeutic opportunities. Circ. J. 81, 1749–1757. doi: 10.1253/circj.CJ-17-1058
Headrick, J. P., Peart, J. N., Reichelt, M. E., and Haseler, L. J. (2011). Adenosine and its receptors in the heart: regulation, retaliation and adaptation. Biochim. Biophys. Acta. 1808, 1413–1428. doi: 10.1016/j.bbamem.2010.11.016
Headrick, J. P., Willems, L., Ashton, K. J., Holmgren, K., Peart, J., and Matherne, G. P. (2003). Ischaemic tolerance in aged mouse myocardium: the role of adenosine and effects of A1 adenosine receptor overexpression. J. Physiol. 549, 823–833. doi: 10.1113/jphysiol.2003.041541
Hove-Madsen, L., Prat-Vidal, C., Llach, A., Ciruela, F., Casado, V., Lluis, C., et al. (2006). Adenosine A2A receptors are expressed in human atrial myocytes and modulate spontaneous sarcoplasmic reticulum calcium release. Cardiovasc. Res. 72, 292–302. doi: 10.1016/j.cardiores.2006.07.020
Ip, J. E., Cheung, J. W., Chung, J. H., Liu, C. F., Thomas, G., Markowitz, S. M., et al. (2013). Adenosine-induced atrial fibrillation: insights into mechanism. Circ. Arrhythm. Electrophysiol. 6, e34–e37.
Jacobson, K. A., and Gao, Z. G. (2006). Adenosine receptors as therapeutic targets. Nat. Rev. Drug. Discov. 5, 247–264.
Jammes, Y., Joulia, F., Steinberg, J. G., Ravailhe, S., Delpierre, S., Condo, J., et al. (2015). Endogenous adenosine release is involved in the control of heart rate in rats. Can. J. Physiol. Pharmacol. 93, 667–675. doi: 10.1139/cjpp-2015-0042
Kabell, G., Buchanan, L. V., Gibson, J. K., and Belardinelli, L. (1994). Effects of adenosine on atrial refractoriness and arrhythmias. Cardiovasc. Res. 28, 1385–1389. doi: 10.1093/cvr/28.9.1385
Kistler, P. M., Sanders, P., Fynn, S. P., Stevenson, I. H., Spence, S. J., Vohra, J. K., et al. (2004). Electrophysiologic and electroanatomic changes in the human atrium associated with age. J. Am. Coll. Cardiol. 44, 109–116. doi: 10.1016/j.jacc.2004.03.044
Klaasse, E. C., Ijzerman, A. P., de Grip, W. J., and Beukers, M. W. (2008). Internalization and desensitization of adenosine receptors. Purinergic. Signal. 4, 21–37. doi: 10.1007/s11302-007-9086-7
Koeppen, M., Eckle, T., and Eltzschig, H. K. (2009). Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PLoS One 4:e6784. doi: 10.1371/journal.pone.0006784
Koszalka, P., Ozuyaman, B., Huo, Y., Zernecke, A., Flogel, U., Braun, N., et al. (2004). Targeted disruption of cd73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ. Res. 95, 814–821. doi: 10.1161/01.res.0000144796.82787.6f
Latini, S., and Pedata, F. (2001). Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J. Neurochem. 79, 463–484. doi: 10.1046/j.1471-4159.2001.00607.x
Letsas, K. P., Georgopoulos, S., Efremidis, M., Liu, T., Bazoukis, G., Vlachos, K., et al. (2017). Adenosine-guided radiofrequency catheter ablation of atrial fibrillation: a meta-analysis of randomized control trials. J. Arrhythm. 33, 247–255. doi: 10.1016/j.joa.2017.02.002
Li, N., Csepe, T. A., Hansen, B. J., Sul, L. V., Kalyanasundaram, A., Zakharkin, S. O., et al. (2016). Adenosine-induced atrial fibrillation: localized reentrant drivers in lateral right atria due to heterogeneous expression of adenosine A1 receptors and GIRK4 subunits in the human Heart. Circulation 134, 486–498. doi: 10.1161/CIRCULATIONAHA.115.021165
Llach, A., Molina, C. E., Prat-Vidal, C., Fernandes, J., Casado, V., Ciruela, F., et al. (2011). Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine A2A receptors. Eur. Heart. J. 32, 721–729. doi: 10.1093/eurheartj/ehq464
Lohse, M. J., Klotz, K. N., Schwabe, U., Cristalli, G., Vittori, S., and Grifantini, M. (1988). 2-Chloro-N6-cyclopentyladenosine: a highly selective agonist at A1 adenosine receptors. Naunyn. Schmiedebergs. Arch. Pharmacol. 337, 687–689.
Lou, Q., Hansen, B. J., Fedorenko, O., Csepe, T. A., Kalyanasundaram, A., Li, N., et al. (2014). Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities revealed by novel dual-sided intramural optical mapping. Circulation 130, 315–324. doi: 10.1161/CIRCULATIONAHA.113.007086
Mihm, M. J., Yu, F., Carnes, C. A., Reiser, P. J., McCarthy, P. M., Van Wagoner, D. R., et al. (2001). Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 104, 174–180. doi: 10.1161/01.cir.104.2.174
Molina, C. E., Llach, A., Herraiz-Martinez, A., Tarifa, C., Barriga, M., Wiegerinck, R. F., et al. (2016). Prevention of adenosine A2A receptor activation diminishes beat-to-beat alternation in human atrial myocytes. Basic. Res. Cardiol. 111:5. doi: 10.1007/s00395-015-0525-2
Moser, G. H., Schrader, J., and Deussen, A. (1989). Turnover of adenosine in plasma of human and dog blood. Am. J. Physiol. 256, C799–C806. doi: 10.1002/9780470724378.ch2
Mubagwa, K., Mullane, K., and Flameng, W. (1996). Role of adenosine in the heart and circulation. Cardiovasc. Res. 32, 797–813. doi: 10.1016/s0008-6363(96)00140-x
Muller, C. E., and Jacobson, K. A. (2011). Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta. 1808, 1290–1308. doi: 10.1016/j.bbamem.2010.12.017
Nishida, K., Michael, G., Dobrev, D., and Nattel, S. (2010). Animal models for atrial fibrillation: clinical insights and scientific opportunities. Europace 12, 160–172. doi: 10.1093/europace/eup328
Pedata, F., Corsi, C., Melani, A., Bordoni, F., and Latini, S. (2001). Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Ann. N. Y. Acad. Sci. 939, 74–84. doi: 10.1111/j.1749-6632.2001.tb03614.x
Pelleg, A., Hurt, C., Miyagawa, A., Michelson, E. L., and Dreifus, L. S. (1990). Differential sensitivity of cardiac pacemakers to exogenous adenosine in vivo. Am. J. Physiol. 258, H1815–H1822.
Pelleg, A., Mitsuoka, T., Michelson, E. L., and Menduke, H. (1987). Adenosine mediates the negative chronotropic action of adenosine 5′-triphosphate in the canine sinus node. J. Pharmacol. Exp. Ther. 242, 791–795.
Pfaffinger, P. J., Martin, J. M., Hunter, D. D., Nathanson, N. M., and Hille, B. (1985). GTP-binding proteins couple cardiac muscarinic receptors to a K channel. Nature 317, 536–538. doi: 10.1038/317536a0
Podd, S. J., Freemantle, N., Furniss, S. S., and Sulke, N. (2016). First clinical trial of specific IKACh blocker shows no reduction in atrial fibrillation burden in patients with paroxysmal atrial fibrillation: pacemaker assessment of BMS 914392 in patients with paroxysmal atrial fibrillation. Europace 18, 340–346. doi: 10.1093/europace/euv263
Rienstra, M., Hagens, V. E., Van Veldhuisen, D. J., Bosker, H. A., Tijssen, J. G., Kamp, O., et al. (2004). Van Gelder IC and Group RACvECfPAFS. Clinical characteristics of persistent lone atrial fibrillation in the RACE study. Am. J. Cardiol. 94, 1486–1490. doi: 10.1016/j.amjcard.2004.08.024
Roden, D. M. (1998). Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin. Electrophysiol. 21, 1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x
Saito, H., Nishimura, M., Shinano, H., Makita, H., Tsujino, I., Shibuya, E., et al. (1999). Plasma concentration of adenosine during normoxia and moderate hypoxia in humans. Am. J. Respir. Crit. Care Med. 159, 1014–1018. doi: 10.1164/ajrccm.159.3.9803100
Sanders, P., Kistler, P. M., Morton, J. B., Spence, S. J., and Kalman, J. M. (2004). Remodeling of sinus node function in patients with congestive heart failure: reduction in sinus node reserve. Circulation 110, 897–903. doi: 10.1161/01.cir.0000139336.69955.ab
Sato, R., Hisatome, I., Wasserstrom, J. A., Arentzen, C. E., and Singer, D. H. (1990). Acetylcholine-sensitive potassium channels in human atrial myocytes. Am. J. Physiol. 259, H1730–H1735.
Saura, C. A., Mallol, J., Canela, E. I., Lluis, C., and Franco, R. (1998). Adenosine deaminase and A1 adenosine receptors internalize together following agonist-induced receptor desensitization. J. Biol. Chem. 273, 17610–17617. doi: 10.1074/jbc.273.28.17610
Schuttler, K., Nilius, B., and Boldt, W. (1983). Rabbit atrial myocardium in hypoxic conditions: rate dependent variation of action potential. Biomed. Biochim. Acta. 42, 83–94.
Semenza, G. L. (2011). Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 365, 537–547. doi: 10.1056/nejmra1011165
Sheth, S., Brito, R., Mukherjea, D., Rybak, L. P., and Ramkumar, V. (2014). Adenosine receptors: expression, function and regulation. Int. J. Mol. Sci. 15, 2024–2052. doi: 10.3390/ijms15022024
Sinno, H., Derakhchan, K., Libersan, D., Merhi, Y., Leung, T. K., and Nattel, S. (2003). Atrial ischemia promotes atrial fibrillation in dogs. Circulation 107, 1930–1936. doi: 10.1161/01.cir.0000058743.15215.03
Skibsbye, L., Diness, J. G., Sorensen, U. S., Hansen, R. S., and Grunnet, M. (2011). The duration of pacing-induced atrial fibrillation is reduced in vivo by inhibition of small conductance Ca(2+)-activated K(+) channels. J. Cardiovasc. Pharmacol. 57, 672–681. doi: 10.1097/FJC.0b013e318217943d
Skibsbye, L., Wang, X., Axelsen, L. N., Bomholtz, S. H., Nielsen, M. S., Grunnet, M., et al. (2015). Antiarrhythmic mechanisms of SK channel inhibition in the rat atrium. J. Cardiovasc. Pharmacol. 66, 165–176. doi: 10.1097/FJC.0000000000000259
Stagg, J., Divisekera, U., McLaughlin, N., Sharkey, J., Pommey, S., Denoyer, D., et al. (2010). Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. U.S.A. 107, 1547–1552. doi: 10.1073/pnas.0908801107
Strickberger, S. A., Man, K. C., Daoud, E. G., Goyal, R., Brinkman, K., Knight, B. P., et al. (1997). Adenosine-induced atrial arrhythmia: a prospective analysis. Ann. Intern. Med. 127, 417–422.
Sung, S. J., Li, L., Huang, L., Lawler, J., Ye, H., Rosin, D. L., et al. (2017). Proximal tubule CD73 is critical in renal ischemia-reperfusion injury protection. J. Am. Soc. Nephrol. 28, 888–902. doi: 10.1681/ASN.2016020229
Swarup, V., Baykaner, T., Rostamian, A., Daubert, J. P., Hummel, J., Krummen, D. E., et al. (2014). Stability of rotors and focal sources for human atrial fibrillation: focal impulse and rotor mapping (FIRM) of AF sources and fibrillatory conduction. J. Cardiovasc. Electrophysiol. 25, 1284–1292. doi: 10.1111/jce.12559
Takano, N., Amiya, E., Oguri, G., Nakayama, A., Taya, M., Nakajima, T., et al. (2019). Influence of atrial fibrillation on oxygen uptake and exercise tolerance in cardiovascular patients; close association with heart rate response. Int. J. Cardiol. Heart Vasc. 22, 84–91. doi: 10.1016/j.ijcha.2018.12.014
Talukder, M. A., Morrison, R. R., Jacobson, M. A., Jacobson, K. A., Ledent, C., and Mustafa, S. J. (2002). Targeted deletion of adenosine A(3) receptors augments adenosine-induced coronary flow in isolated mouse heart. Am. J. Physiol. Heart Circ. Physiol. 282, H2183–H2189.
Tang, C., Skibsbye, L., Yuan, L., Bentzen, B. H., and Jespersen, T. (2015). Biophysical characterization of inwardly rectifying potassium currents (I(K1) I(K,ACh), I(K,Ca)) using sinus rhythm or atrial fibrillation action potential waveforms. Gen. Physiol. Biophys. 34, 383–392.
Tebbenjohanns, J., Schumacher, B., Pfeiffer, D., Jung, W., and Luderitz, B. (1997). Dose and rate-dependent effects of adenosine on atrial action potential duration in humans. J. Interv. Card. Electrophysiol. 1, 33–37.
Thery, C., Gosselin, B., Lekieffre, J., and Warembourg, H. (1977). Pathology of sinoatrial node. Correlations with electrocardiographic findings in 111 patients. Am. Heart J. 93, 735–740. doi: 10.1016/s0002-8703(77)80070-7
Thompson, L. F., Eltzschig, H. K., Ibla, J. C., Van De Wiele, C. J., Resta, R., Morote-Garcia, J. C., et al. (2004). Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 200, 1395–1405. doi: 10.1084/jem.20040915
Visentin, S., Wu, S. N., and Belardinelli, L. (1990). Adenosine-induced changes in atrial action potential: contribution of Ca and K currents. Am. J. Physiol. 258, H1070–H1078.
Wang, X., Liang, B., Skibsbye, L., Olesen, S. P., Grunnet, M., and Jespersen, T. (2013). GIRK channel activation via adenosine or muscarinic receptors has similar effects on rat atrial electrophysiology. J. Cardiovasc. Pharmacol. 62, 192–198. doi: 10.1097/FJC.0b013e3182965221
Keywords: adenosine, A1-R, arrhythmias, CD73, hypoxia, translational models
Citation: Soattin L, Lubberding AF, Bentzen BH, Christ T and Jespersen T (2020) Inhibition of Adenosine Pathway Alters Atrial Electrophysiology and Prevents Atrial Fibrillation. Front. Physiol. 11:493. doi: 10.3389/fphys.2020.00493
Received: 10 January 2020; Accepted: 23 April 2020;
Published: 12 June 2020.
Edited by:
Carol Ann Remme, University of Amsterdam, NetherlandsReviewed by:
Constanze Schmidt, Heidelberg University Hospital, GermanyCopyright © 2020 Soattin, Lubberding, Bentzen, Christ and Jespersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Soattin, bHNvYXR0aW5Ac3VuZC5rdS5kaw==; bHVjYS5zb2F0dGluMTk4NkBnbWFpbC5jb20=; Thomas Jespersen, dGhvamVzQHN1bmQua3UuZGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.