 Kirsten Sehlmeyer
Kirsten Sehlmeyer Jannik Ruwisch
Jannik Ruwisch Nuria Roldan
Nuria Roldan Elena Lopez-Rodriguez
Elena Lopez-Rodriguez
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 05 May 2020
Sec. Respiratory Physiology and Pathophysiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00386
This article is part of the Research TopicUnderstanding Lung Acinar Micromechanics in Health and Disease: Linking Quantitative Imaging and Organ Scale Mechanics by Computational ModelingView all 15 articles
Surfactant protein C (SP-C) is an important player in enhancing the interfacial adsorption of lung surfactant lipid films to the alveolar air-liquid interface. Doing so, surface tension drops down enough to stabilize alveoli and the lung, reducing the work of breathing. In addition, it has been shown that SP-C counteracts the deleterious effect of high amounts of cholesterol in the surfactant lipid films. On its side, cholesterol is a well-known modulator of the biophysical properties of biological membranes and it has been proven that it activates the inflammasome pathways in the lung. Even though the molecular mechanism is not known, there are evidences suggesting that these two molecules may interplay with each other in order to keep the proper function of the lung. This review focuses in the role of SP-C and cholesterol in the development of lung fibrosis and the potential pathways in which impairment of both molecules leads to aberrant lung repair, and therefore impaired alveolar dynamics. From molecular to cellular mechanisms to evidences in animal models and human diseases. The evidences revised here highlight a potential SP-C/cholesterol axis as target for the treatment of lung fibrosis.
As recently reviewed by Zuniga-Hertz and Patel (2019), 2.32 billion years ago, the atmospheric oxygen raised (Bekker et al., 2004) leading to many changes in life. One of these changes may have been the appearance of sterols, around 2.7 billion years ago (Brocks et al., 1999; French et al., 2015) after the GOE (Great Oxygen Event) as suggested by fossil evidences. However, which is the connection between oxygen and sterols? It has been proposed that sterols emerged as an evolutionary strategy to reduce oxygen diffusion through cellular membranes, probably in a world where organisms were not ready to respond to oxidative stress yet. Interestingly, sterol biosynthesis is highly dependent on oxygen (DeBose-Boyd, 2008).
How can sterols, such as cholesterol, regulate oxygen diffusion through biological membranes? The molecular mechanism by which cholesterol can influence small solute permeation and diffusion through lipid membranes is not well understood. However, it has been proposed that the interactions between cholesterol and the acyl carbon chains in phospholipids (stabilized by Van der Waals’ forces) (Wennberg et al., 2012) create tightly packed arrangements that limits small molecule diffusion (Zocher et al., 2013). By decreasing trans-gauche rotation of the phospholipid acyl chains, membrane rigidity increases (Cassera et al., 2002; Molugu and Brown, 2019) leading to less free volume and free pockets which may accommodate oxygen, therefore reducing its flux (its partition and diffusion) through membranes (Zuniga-Hertz and Patel, 2019).
Mammals developed sophisticated organs to optimize the uptake of oxygen during the life essential breathing cycle. From conducting airways to the second biggest surface exposed to the environment, the alveolar human surface (Ochs et al., 2004). In addition, the first membrane that oxygen encounters in the mammalian lungs is a complex mixture of lipids (mainly phosphatidylcholines (PC), such as dipalmitoylphosphatidylcholine (DPPC), up to a 90%) and proteins (10%) with a 5–10% of cholesterol (14–20% mol) (Zuo et al., 2008; Bernhard, 2016), called lung surfactant (surface active agent). Interestingly two surfaces in the human body present with abnormally higher cholesterol molar ratio, and both of them are in contact with the environment tightly regulating the uptake of oxygen. On the one hand, as previously explained, lung surfactant presents around a 14–20% mol cholesterol in its composition. On the other hand the eye lens surface contains up to 35% mol of cholesterol (Raguz et al., 2008; Mainali et al., 2013), compared to a normal cell plasma membrane with a 0.5 phospholipid to cholesterol molar ratio (van Meer et al., 2008; Widomska et al., 2017). Although the main function of lung surfactant has not been related to oxygen flux control, it has been described to accelerate oxygen diffusion through a water layer (Olmeda et al., 2010), reflecting the importance of lung surfactant as potential oxygen flux regulator. The main function of lung surfactant is to reduce surface tension in order to prevent alveolar collapse during expiration and therefore stabilizing open alveoli allowing oxygen to diffuse through the lung tissue to the blood (Knudsen et al., 2017; Bates and Smith, 2018; Knudsen and Ochs, 2018). Alveolar parenchyma is thin enough to allow oxygen diffusion and is composed of a minimum of three components: (1) alveolar epithelium, where alveolar epithelial type I cells (AE1C) cover 60% of the surface with a small cytoplasm but in a long and thin disposition, whereas alveolar type II cells (AE2C) are in charge of synthesizing, secreting and regulating surfactant composition; (2) both cells sit on a very thin basal membrane; (3) to which a thin endothelium is attached on the opposite side, creating a thin but extensive air-blood barrier. The last step oxygen encounters in its way to the rest of the organs is the erythrocyte membrane. Red blood cells present a rather high cholesterol content, which comprises a 1:1 phospholipid to cholesterol ratio (Zuniga-Hertz and Patel, 2019). In addition, it has been described that oxygen diffusion through red blood cell membranes is decreased in the presence of increased cholesterol content (Buchwald et al., 2000). And therefore, reduction of cholesterol content in those membranes (such as after the use of Simvastatin for 12 weeks) improves oxygen diffusion rate (Menchaca et al., 1998).
In the long way of oxygen through the human body this review focuses on the first barrier, lung surfactant and how its components may impact the normal function of the lung. There is a particular interest in the relevance of the role of cholesterol, and more specifically, in the long described potential relation between surfactant protein C (SP-C) and cholesterol content in lung surfactant. Here, we described the state of the art at the molecular level, contrasting with data from animal models and human patients, where lung mechanics and alveolar dynamics is affected during SP-C deficiency related disease.
SP-C is the smallest (4.2 kDa) and most hydrophobic protein in lung surfactant. It accounts for ∼1% of lung surfactant mass, becoming the most abundant protein in molar terms. SP-C appeared relatively late in evolution and its sequence has remained highly conserved among species (Potter et al., 2007). The lack of any known homologous protein and its confined expression (Korfhagen et al., 1990), makes it a specific marker associated with the differentiation of lung tissue, and particularly of AE2C. SP-C exists as a 35 amino acid transmembrane protein expressed as a larger precursor (21 kDa) in AE2C cells. Structurally, it adopts a metastable α-helical structure, although its high proportion of branched residues makes it prone to adopt β-sheet structures and fibrillogenic amyloid-like aggregates (Johansson, 2001; Johansson et al., 2004; Chou and Fasman, 2006), a common feature in several interstitial lung diseases such as pulmonary fibrosis. SP-C greatly alters lipid packing in membranes influencing lipid motion and lateral distribution (Morrow et al., 1993; Dico et al., 1997). As described before, this may also influence the availability of free volume and free pockets, which may accommodate oxygen. However, a direct study of SP-C content and oxygen diffusion through surfactant membranes has not been assessed so far. SP-C also increases membrane permeability (Plasencia et al., 2004; Parra et al., 2011, 2013) and promotes interfacial lipid adsorption and lipid transfer among different lipid structures (Creuwels et al., 1993; Possmayer et al., 2001; Wang et al., 2005). SP-C is responsible for the reversible formation of multilayered stacks connected to the interfacial monolayer (Amrein et al., 1997; Wang et al., 2005). Doing so, it allows the capture of surfactant material presumably squeezed out during exhalation (area compression) and surfactant re-spreading upon inhalation (area expansion). In this process, protein palmitoylation seems to be relevant to sustain protein association to the highly compressed interfacial films reached during exhalation (Plasencia et al., 2001; Lukovic et al., 2012).
SP-C-associated functions overlap in many cases with surfactant protein B (SP-B) activity, at least when assayed in different in vitro models. This includes facilitating lipid adsorption (Creuwels et al., 1993; Possmayer et al., 2001; Wang et al., 2005) into the air-liquid interface or generating 3D structures that serve as a surfactant reservoir (Amrein et al., 1997; Wang et al., 2005), which store newly secreted surfactant complexes and surfactant molecules squeezed out from the interface upon compression. In this context, SP-C could be figured as a supporting molecule for SP-B function rather than an element competent by itself to assist specific features of the complex and dynamic surfactant functionality. Nevertheless, considering the extensive processing of SP-C to its mature form, its extremely conserved sequence and tissue-specific localization, and the difficulties that a cell must overcome to produce and store such a hydrophobic molecule, it is unlikely that this peptide appeared evolutionarily just as an alternative strategy to assist SP-B activities.
Surfactant cholesterol represents a paradox regarding its origin (Orgeig and Daniels, 2001; Lopez-Rodriguez et al., 2017). Some works have suggested that it is supplied by the low and high-density lipoproteins present in blood circulation (Olmeda et al., 2017). However, other studies have failed to prove that circulating cholesterol ends up forming part of surfactant complexes (Orgeig and Daniels, 2001; Milos et al., 2016), suggesting that other possible sources must be taken into account. It is remarkable that cholesterol levels in surfactant are tightly regulated to ensure a proper breathing function, and they are able to increase and decrease extremely fast in response to changes in temperature or breathing rate (Doyle et al., 1994; Orgeig et al., 2011). This seems to imply that a cholesterol reservoir might exist in order to provide cholesterol at fast rates when an increase is required. A specific cell type, the lipofibroblast, has been suggested as a reservoir of cholesterol (Besnard et al., 2009; Torday and Rehan, 2011), although further validation is required to confirm its presence through different organisms and whether it constitutes a surfactant cholesterol storage. AE2C cells are able of producing cholesterol in peroxisomes (Batenburg and Haagsman, 1998), but alveolar macrophages (AMs) also exhibit enzymes involved in cholesterol synthesis (Baker et al., 2010b). Elucidating how cholesterol levels are regulated in the context of surfactant physiology is key to understand responses associated with several respiratory pathologies, especially those characterized by the incorporation in surfactant of abnormal cholesterol amounts such as the acute respiratory distress syndrome (ARDS) (Vockeroth et al., 2009) or pulmonary alveolar proteinosis (PAP).
The presence of cholesterol induces a marked segregation of fluid phases in surfactant (Bernardino de la Serna et al., 2004; Keating et al., 2007), and variations in cholesterol levels are known to adapt surfactant structures extremely fast to defined physiological situations (Doyle et al., 1994; Orgeig and Daniels, 2001). This evidence highlights cholesterol as a structural modulator of surfactant membranes and films. Besides, mechanisms involved in cholesterol sensing and mobilization may be evolutionary conserved to regulate cholesterol levels in surfactant. On the other hand, some studies suggest that SP-C might be involved in cholesterol regulation (Gómez-Gil et al., 2009a, b; Baumgart et al., 2010; Roldan et al., 2016, 2017), which could also be linked to the role of SP-C in lung homeostasis. Therefore, the role of SP-C in cholesterol mobilization and dynamics could be tracked back to a combined effect of protein- and cholesterol-induced alterations on membrane structure. In fact, an increase in cholesterol motion was described upon incorporation of SP-C into lung surfactant-derived vesicles (Roldan et al., 2016), an effect potentially associated to SP-C-promoted membrane-fragmenting effect (Parra et al., 2011, 2013; Roldan et al., 2016). Besides, SP-C and cholesterol have been also related to modulating membrane architecture responding in a coordinate manner to temperature changes (Roldan et al., 2017), suggesting that SP-C is involved in cholesterol mobilization by altering membrane structure (Roldan et al., 2016). In addition, taking into account that SP-C supplementation restores the functionality of cholesterol-containing films in a dynamic context (Gómez-Gil et al., 2009b; Baumgart et al., 2010), we could hypothesize that SP-C could be related to the compositional refinement of lung surfactant films from less surface-active molecules, involving cholesterol and other unsaturated phospholipids. Interestingly, observations made by Leonenko et al. (2007) could support this hypothesis. The incorporation of increasing amounts of cholesterol in a surfactant clinical preparation resulted in structurally and functionally different interfacial films (Gunasekara et al., 2005; Leonenko et al., 2007). Compression-expansion isotherms allowed the determination of lipid loss from the interfacial film upon compression, which were considerably low for physiological cholesterol amounts, permitting the re-establishment of a functional interfacial film (Gunasekara et al., 2005; Leonenko et al., 2007). Shifting that to SP-C function, lipid loss occurring for physiological cholesterol levels could be associated with a SP-C-dependent cholesterol refinement, in which SP-C-induced lipid reorganizations could be involved. However, lipid loss increased substantially for cholesterol concentrations beyond physiological levels, showing a markedly different film structure and impaired functionality (Gunasekara et al., 2005; Leonenko et al., 2007). For these supra-physiological cholesterol levels, the SP-C amount present in a clinical surfactant preparation like the one tested in this study (∼1 wt%), could be likely insufficient to overcome cholesterol impairing effects and the irreversible film collapse. Supplementation of these films with different SP-C amounts would provide valuable information to confirm SP-C effects on cholesterol mobilization.
SP-C/cholesterol relationships might be involved in surfactant refinement with a homeostatic purpose in the lung. Both, cholesterol-loaded vesicles, as well as their combination with SP-C, led to a higher lipid engulfment by AMs (Ruwisch et al., 2020). Increased cholesterol content, however, seemed to increase lipid uptake by AMs regardless of the presence of SP-C. Vesicle size and membrane fluidity can affect lipid uptake by AMs (Justice et al., 2014), and thus, the combined effect of cholesterol and SP-C on membrane structure and fluidity (Roldan et al., 2016, 2017) is a factor that must be taken into account. The study of the transcriptional response of MH-S cells to lipid administration revealed that genes associated to lipid metabolism and cholesterol transport were altered upon lipid uptake. Changes in gene expression appear to depend on lipid composition, including the presence or absence of SP-C and/or cholesterol. However, specific effects of cholesterol and SP-C require a thorough and extensive analysis, which also warrant a deeper exploration of the poorly studied pathways for cholesterol synthesis, degradation and mobilization in AMs.
SP-C could then be considered as a pivotal molecule linking cholesterol and lipid homeostasis, lung immunity, lung surfactant biophysical activity and potentially oxygen diffusion regulation, which would explain its particular features, lung tissue specific localization and sequence conservation along evolution. Several questions remain to be answered, including how SP-C effects are coordinated with SP-B function to modulate surfactant activity; which are the molecular mechanisms and SP-C structural determinants involved in membrane reorganizations and how cholesterol levels in surfactant are sensed and regulated regarding SP-C function by both AE2C and AMs. Future research assessing these questions would constitute an essential piece to understand the molecular mechanisms ruling lung surfactant behavior and its implication in respiratory pathologies, generating valuable information useful to improve the current clinical preparations applied for surfactant replacement therapy.
According to LaPlace law, the intra-alveolar pressure of and idealized isotropic alveolus is inversely proportional to its radius. Therefore, at end-expiration, the breathing stage in which the intra-alveolar volume reaches its minimum (ignoring rare occurring phenomena like Pendelluft; Tabuchi et al., 2016), the intra-alveolar pressure reaches its maximum. Under physiologic conditions, end-expiratory pressure is homogenously distributed among the alveoli and surface tension is low enough preventing alveolar collapse. To maintain alveolar pressure throughout the lung constant, surface tension also changes accordingly to alveolar radius (Figure 1).
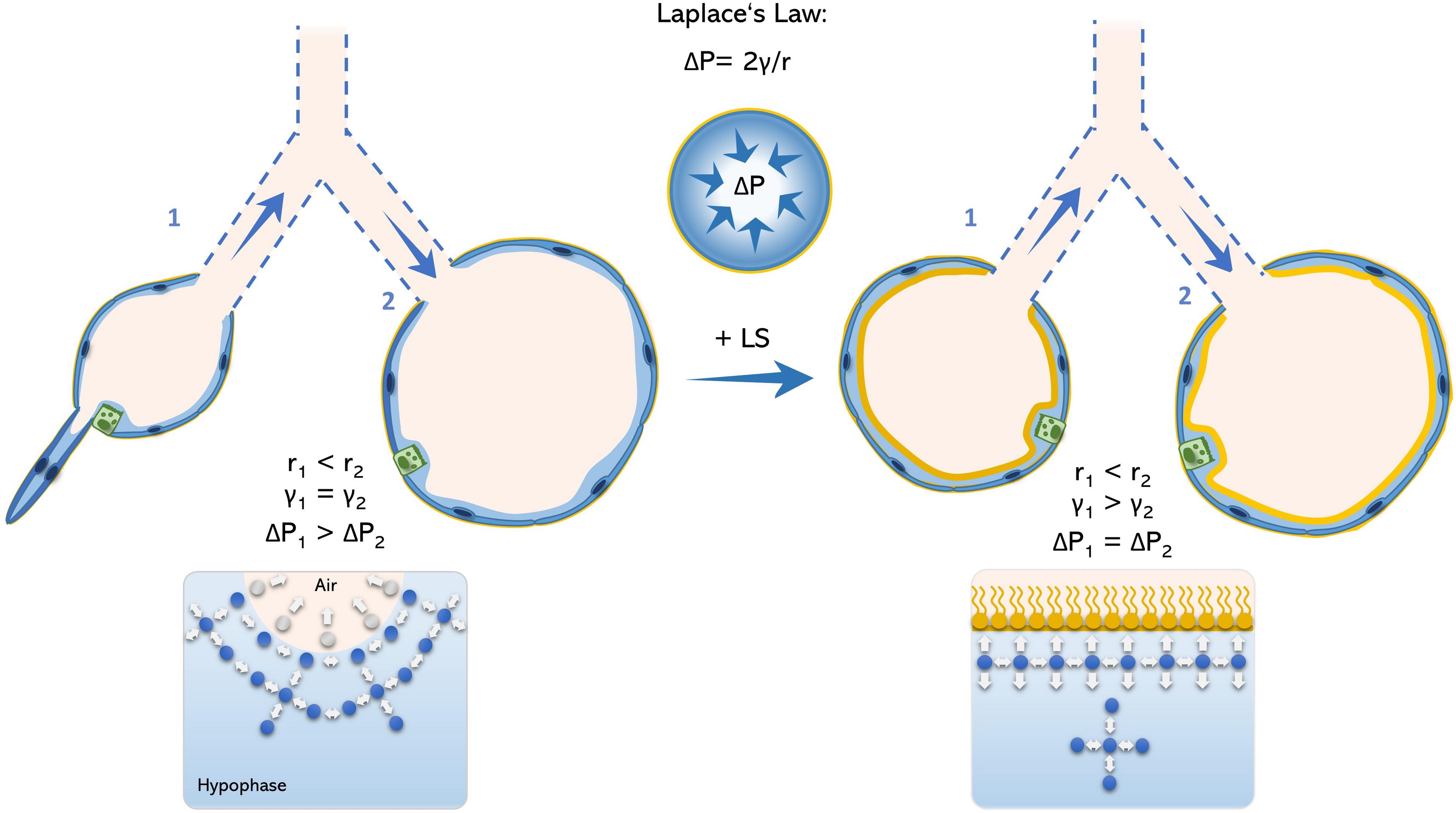
Figure 1. Schematic model of Young-La Place law in the acinar airways. The inner pressure of an idealized spherical alveolus is defined by the Young-La Place law as ΔP = 2γ/r. Therefore, alveoli with small radius (alveolus 1) comprise higher internal pressures in relation to alveoli with larger radius (alveolus 2). Accordingly, alveolus 1 would tend to collapse toward alveolus 2 under the absence of a functional surfactant film, what permits alveolar size heterogeneity (left panel). However, in the presence of surfactant, surface tension is reduced as intermolecular forces are homogenously generated between water-water- and water-air phase, avoiding bending of the air-liquid interface and avoiding alveolar collapse (right panel). LS, lung surfactant.
However, during expiration the surfactant film collapses and thus, end-expiratory surface tension may drastically increase and lead to higher intra-alveolar pressures as well as a rather heterogeneous inter-alveolar pressure distribution. As the intra-alveolar pressure is per se higher in small-radius alveoli (relative to its larger neighbors) by its anatomy, this may increase the likelihood of alveolar collapse. Hence, it has been described that the result of surfactant dysfunction in a murine model of AE2C apoptosis is ductal airspace over distension together with alveolar collapse (Mouded et al., 2009). On the one hand, this may explain the increased vulnerability of small subpleural alveoli to surfactant dysfunction. On the other hand, points toward an essential role of alveolar collapse in fibrotic remodeling, as collapsing alveoli was proposed to be the initiating hotspot in pulmonary fibrogenesis (Mai et al., 2016; Knudsen et al., 2017; Petroulia et al., 2018).
The central role of alveolar collapse in the development of diseases such as lung fibrosis is not a novel finding and was already stated by Myers and Katzenstein (1988). Moreover, alveolar collapse induced by surfactant dysfunction has also been well described in the field of ARDS and acute lung injury (ALI) (Bates and Smith, 2018; Nieman G.F. et al., 2018). Although the increase in surface tension and alveolar instability is much more pronounced in ARDS (Smith et al., 2017; Autilio and Pérez-Gil, 2018) than under SP-C deficiency, leading to a rather acute than chronic respiratory pathology, ARDS as well as rather chronic lung fibrosis may be associated with a similar underlying disease mechanism. This is highlighted by the fact that alveolar collapse, respiratory distress and concomitant ventilation in-homogeneities haven been demonstrated to be present in patients from both diseases (Todd et al., 2015; Al-Saiedy et al., 2018; Autilio and Pérez-Gil, 2018; Petroulia et al., 2018). Moreover, pulmonary fibrosis is also seen in later disease stage of ARDS patients, resembling an aberrant pulmonary repair mechanism in response to lung injury (Hasan et al., 2017), which has also been stated to be the major mechanism of disease in idiopathic pulmonary fibrosis (IPF) patients (Snijder et al., 2019).
Atelectrauma and volutrauma are two mechanism contributing to tissue injury in ARDS (Figure 2). In the injured lung, repetitive alveolar collapse (atelectrauma) is caused by increased surface tension (induced by various agents). During the inspiration phase, the transpulmonary pressure gradient increases until the alveolar pressure of the closed (de-recruited) alveolus is exceeded and is re-opened (recruited). If the maximal generated transpulmonary pressure fails to exceed the intra-alveolar pressure, the alveolus remains collapsed and its alveolar walls stay adjacent to each other. This model is supported by findings of Bachofen and Schürch, who described alveolar wall stretching and ductal airspace over distension on electron microscopic images resulting from surfactant inactivation-induced alveolar collapse (Bachofen and Schürch, 2001). Considering the mechanism of atelectrauma, the alveolar lining fluid should not be neglected. Weibel and Gil first described the alveolar lining lavage as an interfacial film and sub-interfacial liquid reservoir defined as the hypophase (Weibel and Gil, 1968). As the alveolus deflates during expiration, its defining septa start to fold. The folding septal branches are continuously filled with this alveolar lining fluid during expiration (Rühl et al., 2019). Under increased surface tensions the alveolus is further de-recruited leading to more pronounced septal folding and enhanced inter-septal liquid accumulation. During inspiration, an increasing transpulmonary pressure is generated by the respiratory muscles (Mead et al., 1970; Gattinoni et al., 2017). As soon as this pressure exceeds the end-expiratory intra-alveolar pressure, a bubble of air is forced through the lining fluid between the septal folds in order increase alveolar volume by forcing septal de-folding. During this process mechanical axial shear stresses are generated with the air-bubble moving toward the lining fluid, leading to injurious deformation of AEC2 (Bilek et al., 2003). Of note, the cell damaging force vectors strongly increase with rising surface tension at the interface of the lining fluid, as the speed of the bubble is highly reduced (Cassidy et al., 1999; Ghadiali and Gaver, 2008). Meanwhile, AEC2 injury impairs the epithelial functional integrity resulting in accumulation of alveolar edema and reduced rates of surfactant production. Thus, starting a vicious cycle leading to continuously increasing surface tension and epithelial injury, resulting in AEC2 hyperplasia and fibrosis (Fehrenbach, 2001; Mouded et al., 2009; Sisson et al., 2010) (Figure 2).
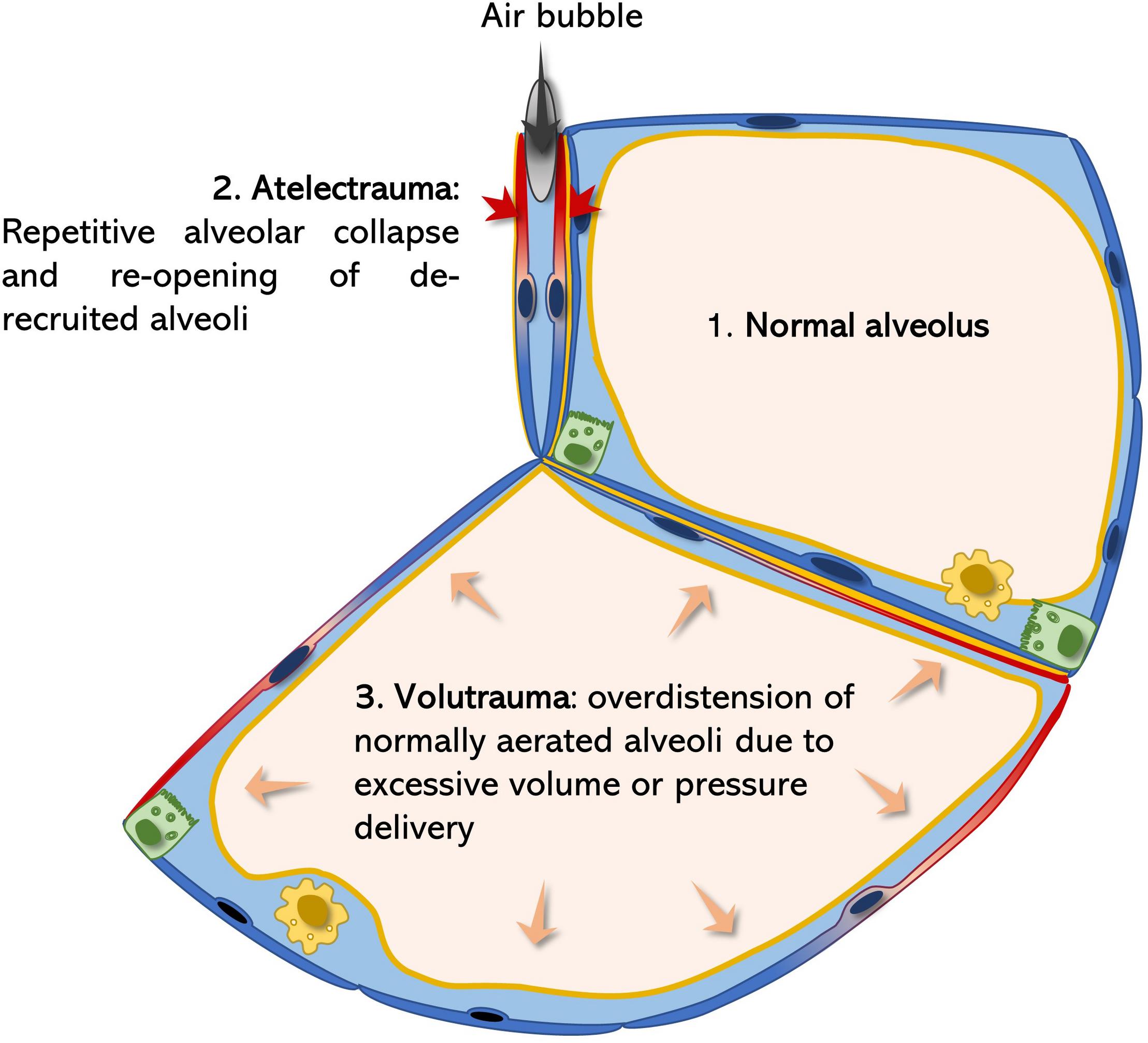
Figure 2. Surfactant dysfunction related atelectrauma and volutrauma. According to the La Place law (Figure 1) surfactant dysfunction predisposes alveoli for collapse. Alveolar micro-atelectasis causes septal distortion of neighboring alveoli due septal, interalveolar architectural dependence. Thereby the collapsed alveolus acts as a stress concentrator leading to injurious mechanical deformation of neighboring epithelial cells (blue-red color transition) during the breathing cycle (volutrauma). Meanwhile, every time a bubble of air is forced into the atelectatic, lining-fluid filled alveolus during inspiration, as a result of increasing transpulmonary pressures, extremely damaging shear stresses are generated on the epithelium (atelectrauma), further potentiating damage of the alveolar epithelium (Bates and Smith, 2018; Rühl et al., 2019).
In the healthy parenchymal lung architecture, tissue stress and stretch occurring during breathing are homogenously distributed. However, if certain alveolar regions become atelectatic, homogenous ventilation is significantly impaired. Alveolar collapse leads to microscale air redistribution, resulting in an inhomogeneous alveolar size distribution before inspiration (Mead et al., 1970). According to alveolar anatomy, single septal walls usually build the parenchymal border of two neighboring alveoli, also known as alveolar interdependence (Knudsen and Ochs, 2018). If one of these alveoli collapses, septal distortion of the adjacent alveoli leads to rising mechanical stresses (Mead et al., 1970). Hence, atelectatic alveoli act as stress concentrators and amplify the applied stresses and strains of their neighbors (Bates and Smith, 2018). During inspiration and increasing lung volumes, this phenomenon becomes even more relevant: transpulmonary pressures generated during inspiration will initially rather lead to over inflation of the already enlarged, distorted alveoli than reopening the atelectatic ones. This causes further alveolar over distension during inspiration. During over distension, an alveolus is first fully de-folded before its wall is stretched. Therefore, the effective septal deformation (= strain) strongly depends on the inspired/applied tidal volume and the alveolar “baseline” shape during resting expiratory position. Usually mechanical stretch primarily occurs under rather high pressures reaching over the upper inflection point of the pulmonary pressure-volume curve (Knudsen and Ochs, 2018). However, when inflating a healthy lung beyond total lung capacity (TLC) of 80% the extracellular matrix components of the alveolar septa, collagen and elastin, become the major stress bearing components, thus mechanical forces start acting on the septal interstitium (Bachofen et al., 1987; Knudsen and Ochs, 2018). Moreover, single vulnerable alveoli in proximity to stress concentrators, may already get stretched at physiologic low tidal volumes (Mead et al., 1970). In line with this, Kollisch-Singule et al. (2015) found pulmonary injury in surfactant depleted pig lungs already at physiologic tidal volumes (6 ml/kg bodyweight), while pigs with maintained surfactant function, required a drastically higher degree of volutrauma to induce relevant lung injury (30 ml/kg bodyweight). In the ARDS lung atelectrauma and volutrauma act synergistically to induce vascular leakage. Smith et al. (2013, 2017) elegantly demonstrated that initial epithelial injury by atelectrauma predisposes septal walls for volutrauma, leading to significant leakage of the gas-blood barrier. Ventilated mice lungs only developed high concentrations of BALF protein levels, an indicator for gas-blood barrier integrity, when ventilated at PEEP (positive end-expiratory pressure) of 0cmH2O (atelectrauma) in combination with mid or high tidal volumes (volutrauma) (see Figure 2). Meanwhile, neither atelectrauma nor volutrauma alone were sufficient to induce pulmonary edema (Smith et al., 2017). As alveolar epithelial cell (AEC) damage has been demonstrated to precede edema formation (Rühl et al., 2019), the absence of alveolar edema in the SP-C deficient lung (Ruwisch et al., 2020), does not rule out the relevance of this protein in preventing alveolar collapse and alveolar over distension, which commonly lead to microscale tissue injuries. Interestingly, hyperplastic AEC2 are present in a global SP-C knock out (KO) mouse model, which may resemble a reaction to chronic mechanical stress (Glasser et al., 2003; Ruwisch et al., 2020). Moreover, surfactant dysfunction in a chronic murine model of AEC injury impressively demonstrated the long-term effects of chronic airway instability to cause microarchitectural air re-distribution resulting in an emphysema-like phenotype, which is characterized by collapsed alveoli and enlarged alveolar ducts (Mouded et al., 2009). Furthermore, repetitive insults during dynamic strain have been related to more harmful properties on AEC integrity than constant high static strains (Nieman G. et al., 2018). This may further emphasize the relevance of airway instabilities, in the absence of volutrauma, in a rather chronic setting, as occurs in IPF patients, where recruitment/de-recruitment cycles take place over a long period of time. In line with this, “velcro-crackles” have been often described to precede computer tomographic changes in IPF lungs. Indeed, they are considered the auditory correlate of damaging energy-rich alveolar re-openings during inspiration, supporting the clinical relevance of impaired alveolar micromechanics in fibrosing lung diseases (Vyshedskiy et al., 2009).
At the lung mechanical macroscale level, alveolar collapse and surfactant dysfunction exhibit notable influence on the lung’s viscoelastic properties. Collapsed airways require high amounts of energy to be reopened, while dysfunctional surfactant increases the hysteresis effect in inflating lungs by rising surface tension. This effect results in parenchymal stiffening, increasing levels of pulmonary tissue elastance (Smith et al., 2013; Birkelbach et al., 2015). During inspiration, the energy applied is rather dissipated creating injurious axial force vectors on the airway epithelium than being stored in the septal elastic fiber network. Therefore, pulmonary tissue damping rises accordingly. In line with this, data from our group demonstrated a significant increase in tissue damping and tissue elastance in 10 weeks old SP-C deficient mice in contrast to age-matched control mice, supporting the concept of alveolar collapse as an initial trigger (Ruwisch et al., 2020). During aging, redistribution of air as well as ECM (Extracellular Matrix) remodeling may alter lung mechanics consistent with findings of Glasser et al. (2001, 2003). Indeed, this work emphasized aberrant tissue hysteresivity (the quotient of energy dissipative forces and lung elastance) at low PEEP, a condition where alveoli are prone to collapse. At those low lung volumes, surfactant becomes the major defining factor of pulmonary breathing mechanics (Bachofen et al., 1987). Moreover, pressure-volume loops demonstrated an increased pulmonary hysteresis, which also reflects increased energy dissipation during inflation of the lung. Gattinoni et al. (2017) already suggested this energy to be potentially harmful for the pulmonary tissue, as it is not reused for elastic recoil properties. Persistent septal micro injury may be, in turn, a relevant factor for altered AEC2 biology, aberrant wound repair, pulmonary inflammation and interstitial fibrotic remodeling. Phenomena described in a number of children suffering interstitial lung disease (chILD), who showed reduced or absent levels of mature SP-C (Cong et al., 2017), as described in the next sections.
Under healthy conditions and homogenously distributed lung stress, plasma membrane injury of AEC is normally seen. Indeed, AEC are highly capable of Ca2+ and lysosome dependent repair mechanisms as well as membrane folds for preventing cellular damage (Cong et al., 2017). However, lysosomal stress as seen in Hermansky-Pudlack Syndrome (HPS) patients may impair cellular repair, favoring profibrotic wound repair and the development of unusual interstitial pneumonia (UIP) (Ozyilmaz et al., 2013; Kook et al., 2016). Moreover, if high parenchymal stress leads to plasma membrane defects of more than 1 μm, aberrant AEC wound repair cascades are induced (Cong et al., 2017). Very high strains induced by pressure above 40cmH2O have been shown to be sufficient to cause membrane blebbing and cellular apoptosis (Dreyfuss et al., 1999).
Typically, AEC injury response includes signaling of several mechano-transduction pathways including the transforming growth factor β1 (TGF-β1), Wnt β-catenin, sonic hedgehog (Shh), and the Notch-midkine signaling pathways as well as induction of endoplasmic reticulum (ER) stress. Interestingly, all these pathways have been interlinked with the induction of profibrotic genes, epithelial-to-mesenchymal transition (EMT) or increased ECM deposition. Moreover, ER stress is known to compromise the secretory capacity of AEC2, impairing surfactant metabolism, which initiates a vicious cycle of alveolar instability and stress-mediated AEC injury (Cong et al., 2017). In a murine model of chloride acid (HCl) induced surfactant depletion, impaired lung mechanics was characterized by increased tissue elastance. This was followed by an increased expression of various mesenchymal markers, such as α-smooth muscle actin (α-SMA) and vimentin in AEC2, whereas the expression of epithelial cell markers, including pro-SP-B, were reduced (Cabrera-Benítez et al., 2012; Mao et al., 2017). Consequently, EMT may be a potent second hit even further compromising: (1) surfactant function and; (2) the regenerative capacitiy of the pulmonary epithelium further directing the damage response toward fibrosis. TGF-β1 is expressed by both AECs as well as resident inflammatory cells like AM (Saito et al., 2018). In addition, a large pool of latent-inactive TGF-β1 is located as a complex with latency associated peptide 1 (LAP-1) and latency TGF-β1 binding protein-1 (LTP-1) in the ECM (Hinz and Suki, 2016; Saito et al., 2018). In the healthy lung, during inspiration, ECM fiber stretching is not sufficient to release active TGF-β1 from its inhibitory binding complex. However, as soon as pulmonary tissue stiffening occurs, interstitial fibroblasts are primed toward myofibroblast differentiation by expressing contractile actin-myosin elements (Zhou et al., 2013). This drastically increases the likelihood of TGF-β1 release upon parenchymal strain, as myofibroblast are able to bind LAP-1 via αV-integrins (Hinz, 2012; Hinz and Suki, 2016). Interestingly, increased epithelial injury resulted in increased expression of epithelial αVß6-integrins, further potentiating the release of TGF-β1 by mechano-transduction (Sheppard, 2015). In line with this, Froese et al. (2016) showed increased expression and release of TGF- β1 in fibrotic IPF lungs, which are characterized by parenchymal stiffening. Importantly the pressures generated by Froese and his team rather reflected gradients comparable to pressure induced by spontaneous breathing than high unphysiological gradients, emphasizing the role of spontaneous breathing as a relevant source of mechanical stress-released active TGF-β1 (Hinz and Suki, 2016). Nevertheless, the role of non-fibrotic but rather collapse-induced parenchymal stiffening in the context of extracellular (EC) TGF-β1 release to date has not been clarified. Finally, TGF-β1 overexpressing mice have been shown to develop surfactant dysfunction and high surface tension (Lopez-Rodriguez et al., 2016) including down-regulation of SP-C expression, by interfering with its transcription factor activity. In this context, considering the afore mentioned section, it becomes clear that an EC TGF-β1 pool bears the potential of an additional devastating loop by further compromising lung mechanics, potentiating epithelial injury.
The role of inflammation and inflammatory cells in fibrogenesis has been controversial over the last decades. Although anti-inflammatory medications have produced devastating clinical results in a IPF clinical trials (Raghu et al., 2006; Idiopathic Pulmonary Fibrosis Clinical Research Network et al., 2012), the role of immune cells, like AMs, in lung fibrogenesis and pulmonary tissue repair has been repetitively underscored (Wynn and Barron, 2010; Wynn et al., 2013; Romero et al., 2014; Jin et al., 2018; Smigiel and Parks, 2018; Puttur et al., 2019). A promising link between mechanical stretch and inflammatory response may be the release of adenosine triphosphate (ATP) (Cong et al., 2017; Hasan et al., 2017). Physiologically, mechanical stretch of AEC1 transmits the release of ATP via P2XR7, functioning as a stretch sensor (Patel et al., 2005; Mishra et al., 2011). However, under prolonged alveolar injury concentrations of ATP and its related nucleotides sharply rise, finally overwhelming the degradative enzymatic machinery. Hence, levels of ATP continuously stay high in the early exudative stage of ARDS until the expression of its degrading ecto-enzyme catches up (Hasan et al., 2017). Under these circumstances, ATP and its metabolites act as damage associated molecular patterns (DAMPs). This results in pro-inflammatory signaling on the one hand, but interestingly also in the induction of various profibrotic pathways on the other hand. In fact, high levels of EC ATP enhanced TGF-β1 expression and deposition of collagen 1 and fibronectin in fibroblasts via P2XR7 signaling in vitro (Wang et al., 2003; Qu et al., 2009). Meanwhile, ATP signaling via the same receptor enhanced the release of profibrotic tissue-metalloprotease inhibitor 1 (TIMP1), which attenuates the function of ECM-degrading enzymes, resulting in increased interstitial amounts of ECM, a hallmark of lung fibrosis (Gu and Wiley, 2006). Interestingly, elevated levels of TIMP1 have also been observed in bronchoalveolar lavage fluid (BALF) of IPF patients, whereas expression of Timp1 was induced in 10 weeks old SP-C deficient mice (Beeh et al., 2003). In addition, both Ca2+ and ATP are well-known stimulating agents of surfactant secretion (Voyno-Yasenetskaya et al., 1991; Dico et al., 1997; Patel et al., 2005; Dietl et al., 2012). This may reflect a counteracting response, where surfactant secretion may be enhanced during high release of ATP and Ca2+, during injury.
Alternatively activated alveolar macrophages (aaAMs) are mainly characterized by expression of resistin-like alpha (Relm-α), chitinase-like proteins (YM1), inducible nitric oxide synthase (NOS-2) or arginase 1 (Arg-1) (Wynn and Barron, 2010). Their profibrotic potential derives primarily through expression of TGF-β1 and the activation of latent TGF-β1 via matrix-metalloproteases (e.g., MMP-9) (Xing et al., 1997; Schmid-Kotsas et al., 1999; Cameron et al., 2001; Karlmark et al., 2009; Lin et al., 2009). Moreover, aaAMs haven been shown in topographic proximity to myofibroblasts, being highly capable of collagen secretion (Ramm et al., 1998; Leicester et al., 2004; Thompson et al., 2008). Finally, aaAMs prime the alveolar micromilieu toward fibrosis by secreting various pro-fibrotic cytokines including IL-4, IL-10, IL-13 and the rather pro-inflammatory cytokine IL-1β. Of note, induced expression of YM1, a chitinase-like protein 1 (Bargagli et al., 2007; Terèelj et al., 2009; Wynn and Barron, 2010), but also in SP-C deficient mice (Glasser et al., 2003). Increased accumulation of macrophages with induced TGF-β1 expression was seen in bleomycin-challenged SP-C deficient mice in areas with significant parenchymal distortion. This further emphasizes the link between mechanical stress and macrophage regulation (Lawson et al., 2005). Interestingly, this misbalance between classical and alternative differentiation was not solely dependent on SP-C deficiency, but also specific to the genetic 129/Sv6 background. The 129/Sv6 strain is characterized by an increased dietary cholesterol accumulation and has been described to develop foamy, lipid-laden macrophages with highly induced YM1 expression with age (Jolley et al., 1999; Hoenerhoff et al., 2006). Curiously, SP-C deficient mice as well as patients suffering mutant SP-C derived interstitial lung diseases (ILDs) related to mutations of SP-C, showed similar foamy macrophages in BALF and tissue as well as abundant cholesterol clefts (Glasser et al., 2003; Hamvas et al., 2004; Abou Taam et al., 2009; Ruwisch et al., 2020). Taken together, this suggests a SP-C deficiency and cholesterol induced axis resulting in dysregulated macrophage metabolism and in misbalanced alternative differentiation (Figure 3). Cholesterol clefts are present in aging 129/Sv4 mice (Hoenerhoff et al., 2006), SP-C deficient mice (Glasser et al., 2003; Ruwisch et al., 2020) or aaAMs in human herpes virus (HHV)-induced lung fibrosis (Mora et al., 2006). Hence, this may be a potential morphological biomarker of dysregulated alternative activation and an indicator for lung fibrosis. However, the exact interplay between cholesterol and SP-C and macrophage regulation has not been yet elucidated. Nevertheless, studies from our group, suggested SP-C to be necessary for maintaining cholesterol homeostasis in an AM cell line (MHS cells) (Ruwisch et al., 2020). In line with this, Romero and colleagues recently proposed the profibrotic potential of a lipid related paracrine axis between macrophages and epithelial cells (Romero et al., 2014). Interestingly, their team demonstrated the presence of foamy, lipid-laden macrophages overexpressing YM1 and TGF-β1 in bleomycin induced lung injury, which has been already associated with reduced expression of the Sftpc and Sftpb gene (Schmidt et al., 2004; Lutz et al., 2015). Romero’s team further showed oxidized phospholipids to be another crucial inductor of foam cell formation and alternative differentiation. The role of oxidized lipid species to induce foam cell formation in macrophages has been extensively studied in the field of atherosclerosis (Ertunc and Hotamisligil, 2016). Circulatory macrophages located in atherosclerotic plaques are frequently challenged with oxidized low-density lipoprotein (LDL), oxysterols or cholesterol. These lipids are endocytosed via various receptors, including CD36 and scavenger receptors. Accumulation of toxic lipid species decompensates the metabolic capacity of those macrophages, leading to cholesterol cleft formation and induction of necrotic factor kappa beta (NFκB) related cellular stress pathways. One central element in the so called “metaflammatory” process. This is the formation of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, resulting in the production of pro-inflammatory cytokines including IL-1β and IL-18 (Duewell et al., 2010; Ramji and Davies, 2015). Importantly, macrophage derived IL-1β and inflammasome activation have been referred to as key features in various forms of lung fibrosis including IPF (Zhang et al., 1993; Kolb et al., 2001; Gasse et al., 2007; Wynn and Barron, 2010; Eldridge et al., 2017).
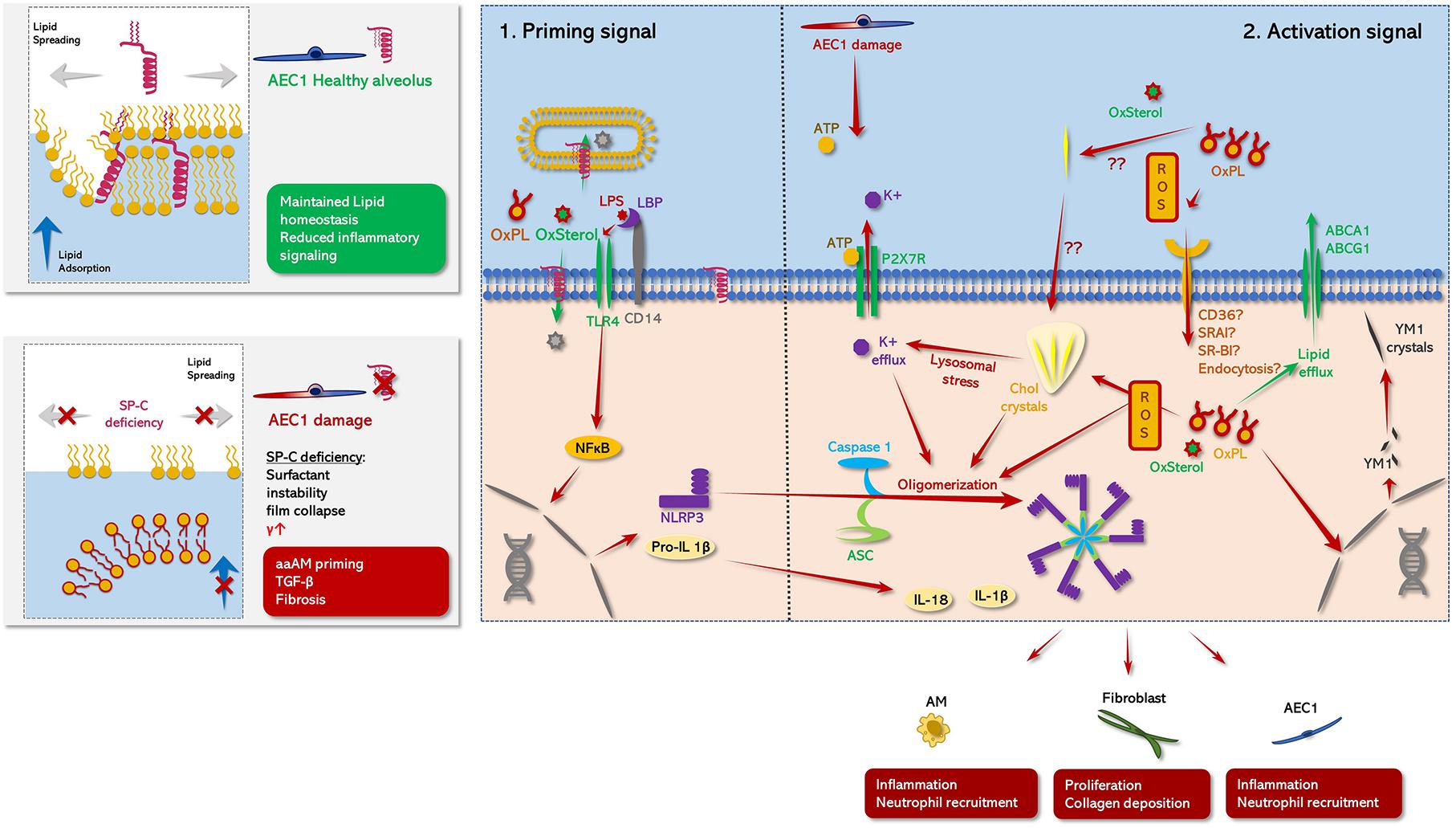
Figure 3. Schematic role of SP-C in pulmonary metaflammation. (Left panel) comparison between the status of AE1C in the presence (healthy alveolus, top panel) or absence (damaged alveolus, bottom panel) of SP-C. (Right panel: 1) Priming signal: TLR4 signaling is abrogated under the presence of mature SP-C. On the one hand, SP-C interferes with the complex formation of LBP and CD14 potentially attenuating TLR4 mediated NLPR3 priming in response DAMP/PAMP stimuli. This ant-inflammatory effect may be further potentiated by the N-terminal segment of SP-C transferring TLR4 activators either into neighboring phospholipid microsomes or directly to the cytoplasmic compartment of the macrophage (Chaby et al., 2005; Garcia-Verdugo et al., 2009). Moreover, maintained surfactant homoeostasis/surfactant catabolism counteracts the accumulation/formation of oxidized lipid species and cholesterol clefts on the one hand, and minimizes atelec- and volutrauma on the other hand (Ruwisch et al., 2020), likely diminishing the release of DAMPS like ATP on the other hand. Together this prevents the buildup of inflammasome oligomerizing stimuli, what may at least partly explain the aberrant inflammatory response of SP-C KO mice to various inflammatory stimuli in contrast to WT mice (Glasser et al., 2008, 2013a). (Right panel: 2) Activating signal: NLRP3 inflammasome comprises an initial NFκβ related priming phase, preceding a secondary activation signal (Swanson et al., 2019): In the first step various proinflammatory DAMPS (oxysterols, oxidized PL species) or PAMPS (LPS) activate macrophages via TLR-4 signaling via an either LPS binding protein (LBP)/CD14 dependent or independent way (Chaby et al., 2005; Garcia-Verdugo et al., 2009). Thereby, LBP facilitates the binding of e.g., LPS to CD14, what accelerates LPS related TLR4 signaling. TLR4 signaling in turn drives NFκB-mediated expression of NLRP3, pro-IL-β1 and proIL-18 (Priming phase) (Gasse et al., 2007; Swanson et al., 2019). In the second step, multiple potential hits including PAMPS like extracellular ATP (Mariathasan et al., 2006; Swanson et al., 2019), intracellular cholesterol clefts (Ertunc and Hotamisligil, 2016), oxysterols and oxidized phospholipid derived mitochondrial oxidative stress (Fessler, 2017; Manon et al., 2018) induce previously synthetized NLRP3 to form oligomers with caspase 1 and ASC leading to mature inflammasome formation. Thereby, increased level of extracellular ATP may be derived from mechanically stressed AEC1 (Hasan et al., 2017), leading to an inflammasome activating K+ efflux via P2X7R. Meanwhile, SP-C deficiency related dysfunctional surfactant catabolism and surfactant dysfunction may favor the generation of oxidized PL species and the formation of cholesterol clefts (Fessler and Summer, 2016; Ruwisch et al., 2020). Likewise, these cholesterol crystals cause lysosomal stress, which in turn resembles another potent driving factor of inflammasome formation via induction of a K+ efflux. Finally, the active NLRP3 inflammasome converts inactive proIL-1β and proIL-18 into their active form IL-1β and IL-18. IL-1β promotes fibrotic remodeling (Gasse et al., 2007; Cassel et al., 2008; Wree et al., 2014; Lv et al., 2018). Meanwhile, another effect of accumulation of injurious lipid species inside the macrophages may also prime their hosts toward a profibrotic aaAM-phenotype (Romero et al., 2014) via induction of several aaAM related genes including, chitinase-like-3 (YM1), which has also been described to form electron dense crystals in various alternative activation of macrophages-disease models (Hoenerhoff et al., 2006; Mora et al., 2006), what may result in a profibrotic feed-forward loop (Smigiel and Parks, 2018).
In addition, cholesterol-induced “metaflammation” in macrophages may also be not only important in development of atherosclerosis but seems to be also highly relevant in the lung. For example, AMs are central elements in surfactant catabolism (comprehensively reviewed in Lopez-Rodriguez et al., 2017), and are continuously challenged with various phospho- and neutral lipids. Therefore, knocking down cholesterol transporters, such as ATP binding cassette transporter A1 (ABCA1) and G1 (ABCG1) leads to impaired cellular cholesterol efflux and cholesterol accumulation. In addition, those animal models showed respiratory distress, impaired surfactant metabolism and cholesterol cleft containing, lipid-laden AMs (Thomassen et al., 2007; Baker et al., 2010a; Chai et al., 2017; Fessler, 2017). Moreover, impaired reverse cholesterol transport in apolipoprotein E (ApoE) and apolipoprotein A1 (ApoA1) deficient mice was also linked to increased pulmonary oxidative stress and inflammation (in ApoE KO mice) as well as to foam cell formation and lung fibrogenesis (in ApoA1 KO mice) (Yao et al., 2016). Interestingly, surfactant function as well as metabolic integrity of AM was restored after application of cyclodextrin, a cholesterol sequestering-drug, underlining the injurious potential of cholesterol for surfactant function and pulmonary lipid homeostasis (Gunasekara et al., 2010). The finding of cholesterol clefts and lipid laden macrophages in patients with SP-C mutations suggests impaired cholesterol homeostasis to be of relevance for development of lung disease in these patients (Hamvas et al., 2004; Abou Taam et al., 2009; Mechri et al., 2010; Cottin and Cordier, 2011; Litao et al., 2017).
Cholesterol overloaded macrophages exhibited an increased Toll-like receptor 4 (TLR-4) signaling mediated immune response as well as elevated expression levels of IL-1β, emphasizing a potential role of NLRP3 inflammasome activation in pulmonary macrophages (Fessler, 2017). The NLRP3-inflammasome consists of three elements including the NLRP3, procaspase 1 and the apoptosis speck-like protein (ASC) containing a caspase recruitment domain (CARD), which links NLRP3 with procaspase 1 (Robert et al., 2016). In order to generate full activation of the inflammasome complex usually two independent signals are needed (Figure 3): (1) a NFκβ priming signal TLR-4 signaling [enhanced by lipopolysaccharide (LPS) or oxidized phospholipid species] or the presence of cholesterol crystals (Bauernfeind et al., 2009; Ertunc and Hotamisligil, 2016; Fessler and Summer, 2016; Nakayama, 2018; Saito et al., 2019) and; (2) purinergic signaling as may result from mechanical stress induced AEC1 injury (Hasan et al., 2017). Elevated levels of ATP have been found in BALF of patients with lung fibrosis, further supporting the idea of EC ATP as a potential inflammasome co-activator (Riteau et al., 2010). Hence, the NLRP3 inflammasome may resemble a crucial link between impaired lung mechanics and aberrant lipid metabolism in fibrosing lung diseases. Interestingly, IPF patients have been described to have elevated levels of unsaturated PC, a PC variant which highly susceptible for oxidative stress (Fessler and Summer, 2016). In combination with the fact, that IPF occurs predominantly in the elderly and has been interlinked with cigarette smoking, mitochondrial stress and cellular senescence, factors driving the generation of reactive oxygen species (ROS). Thus, the IPF lung appears as a hotspot for lipid oxidation (Lederer and Martinez, 2018). Elevated levels of ROS and increased levels of cholesterol in surfactant are also present in bleomycin-induced lung fibrosis (Allawzi et al., 2019), highlighting the relevance of ROS, surfactant oxidation and foam cell formation in fibrogenesis. This is further supported by the presence foamy, Oil-Red-O positive cells in BALF of patients not only with Sftpc mutations, but also in patients with sporadic interstitial fibrosis (Basset-Léobon et al., 2010).
Nevertheless, SP-C deficiency may play a distinct role in foam cell formation. Not only is SP-C crucial for stabilizing cholesterol-containing surfactant films (Gómez-Gil et al., 2009a, b) and maintaining physiologic mechanical lung properties but also it seems to interfere with TLR-4 signaling. In vitro studies demonstrated that SP-C attenuates LPS induced cytokine production via TLR-4 signaling in a macrophage cell line, suggesting SP-C to play a role in innate immunity (Augusto et al., 2003). Thereby, SP-C seems to compromise the affinity of TLR-4 to LPS via a complex interplay with CD14 (Chaby et al., 2005). However, Garcia-Verdugo et al. (2009) also proposed a CD14 independent mechanism, by which the transfer of LPS into liposomes mediated by the N-terminal SP-C segment would prevent LPS binding to TLR4. This results in a reduced cytokine production upon LPS stimulation (Garcia-Verdugo et al., 2009). In line with these findings, SP-C KO mice exhibited an increased inflammatory response upon LPS challenge characterized by increased macrophage accumulation and higher expression levels of IL-1β, IL-6 and tumor necrosis factor alpha (TNF-α) (Glasser et al., 2013a). Interestingly, SP-C containing phospholipid vesicles reduced TLR-4 signaling in these mice. Noteworthy, this immunomodulatory role of SP-C seems to not to be restricted to LPS and bacteria-induced inflammation, as SP-C also demonstrated an anti-inflammatory role under pathogen-free conditions (Jin et al., 2018). Taken all together, this suggest that SP-C may not only orchestrate the activation of TLR-4 in an infectious setting, but it also has an impact on TLR-4 signaling in macrophages induced by oxidized surfactant lipids, a question that, to date, remains unclear.
Assuming this hypothesis (Figure 3), SP-C deficiency could predispose to TLR-4 activation by various oxidized cholesterol and PL species as well as their uptake. This results in proinflammatory signaling and NLRP3 inflammasome activation in macrophages (Ertunc and Hotamisligil, 2016). Impaired lung mechanics induced by SP-C deficiency may further promote inflammasome activation by ATP release and at the same time, lead to accelerated surfactant aggregate conversion (Veldhuizen et al., 1996, 1997; Vazquez De Anda et al., 2000; Maitra et al., 2002). Reduced amounts of surface-tension lowering large aggregates (LA) of surfactant, resulting from pronounced aggregate conversion to inactive small aggregates (SA), were accompanied by larger amounts of ROS susceptible phospholipid species (Fessler and Summer, 2016). Increased uptake of SA by AMs in combination with SP-C disrupted cholesterol metabolism, which may partly explain the presence of foamy macrophages in the SP-C deficient lung and lung diseases related to SP-C mutated forms (Glasser et al., 2003; Hamvas et al., 2004; Lawson et al., 2004; Stevens et al., 2005; Henderson et al., 2013; Liptzin et al., 2015; Salerno et al., 2016).
The relevance of inflammasome activation and IL-1β signaling in fibrotic parenchyma remodeling has been shown for silica induced- ILDs (Cassel et al., 2008), bleomycin-induced lung fibrosis (Gasse et al., 2007) and various other organ systems (Vilaysane et al., 2010; Wree et al., 2014; Lv et al., 2018). NLRP3 associated IL-1β release mediates NFκβ activation via IL-1R1/myeloid differentiation primary response 88 (MyD88) related signal transduction in fibroblasts. This results in a misbalance between TIMP-1 and matrix-metalloprotease 9 (MMP-9) and 13 (MMP-13) promoting interstitial collagen deposition and fibrosis (So et al., 2007; Ertunc and Hotamisligil, 2016). Furthermore, genetic deletion of NLRP3-inflammasome associated genes in a ventilated-induced fibrosis model resulted in decreased rates of stretch-induced EMT, which was accompanied by ameliorated fibrotic lung remodeling (Lv et al., 2018).
All the molecular and cellular mechanisms explained in the previous sections impact the health and mechanical properties of the lungs. We described in this section the results of deleting the expression or mutating SP-C in animal models (Table 1). In addition, we present here evidences from ILD patients, described to have different mutations in SP-C (Table 2).
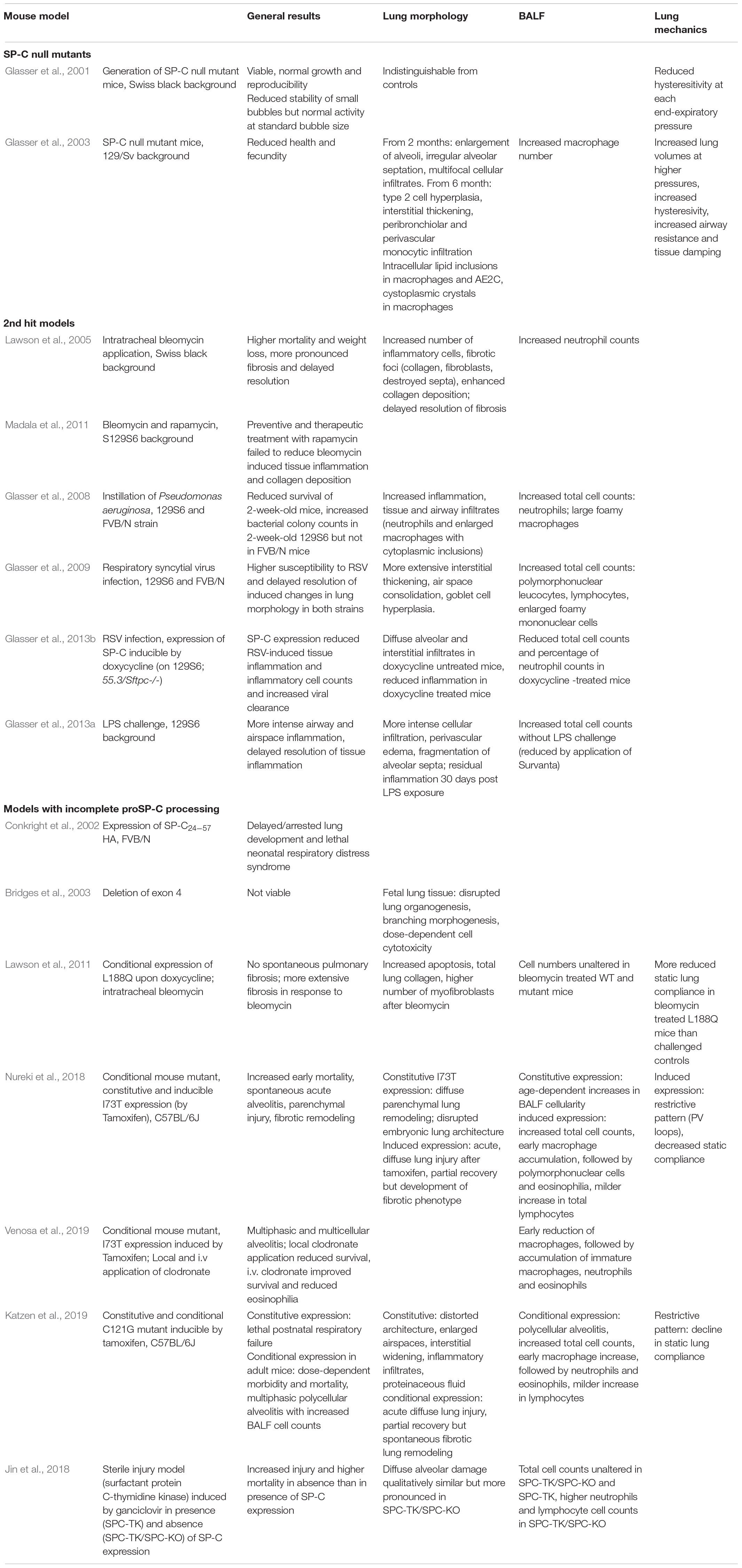
Table 1. SP-C related mouse models.

Table 2. Lung mechanics and BALF cells data from patients.
Glasser et al. (2001, 2003) reported the generation of SP-C null mutant mice deficient in the expression of Sftpc in a Swiss black background. SP-C deficient mice were viable at birth and did grow and reproduce normally with only mild alterations in lung mechanics. In addition, in vitro studies of the SP-C deficient derived surfactant proved reduced stability of small bubbles but normal activity at normal bubble size using a captive bubble surfactometer (Glasser et al., 2001, 2003). In contrast, in 129/Sv background, SP-C deficient mice presented with severe morphological changes including enlargement of alveoli, AE2C hyperplasia and interstitial thickening together with a peribronchiolar and perivascular monocyte infiltration in some animals. Although areas of thickened alveolar septa stained positive for alpha-smooth muscle actin and blue in trichrome staining, septa were thinner in regions of airspace enlargement and total lung hydroxyproline remained unaffected (Glasser et al., 2003; Ruwisch et al., 2020). Importantly, lung structure was normal at birth, but emphysema and remodeling developed upon aging suggesting a process of ongoing injury and repair. Investigation of pulmonary mechanics exhibited increased lung volumes at higher pressures as well as increased hysteresivity in SP-C deficient mice. Abnormal intracellular lipid inclusions and crystals were observed in macrophages and AE2C (Glasser et al., 2001, 2003). While phospholipid content was normal in SP-C deficient lungs on the swiss black background, it was 2-fold elevated in 129/Sv background. In addition, lung mechanics showed increased lung volume at high pressure, tissue damping and hysteresivity in 129Sv background. Together, both studies show strain specific influences on the severity of the pulmonary phenotype (Glasser et al., 2001, 2003). Interestingly, as mentioned before, the 129Sv strain demonstrates an increased absorption of dietary cholesterol (Jolley et al., 1999) which might also contribute to the strain specific phenotype in Sftpc null mice.
Further investigations included additional hits aggravating the effects of SP-C deficiency (Lawson et al., 2005; Glasser et al., 2008, 2009, 2013a,b). Intratracheal application of bleomycin led to higher mortality, more pronounced weight loss, increased neutrophil counts and enhanced collagen deposition in SP-C deficient mice compared to controls. Increased fibrosis as additionally indicated by morphological parameters went along with a delayed resolution of bleomycin-induced fibrosis (Lawson et al., 2005). Preventive and therapeutic treatment with rapamycin failed to reduce bleomycin induced tissue inflammation and collagen deposition in SP-C deficiency (Madala et al., 2011). Instillation of Pseudomonas aeruginosa increased pulmonary injury and bacterial colony counts in SP-C deficient mice compared to control mice. As for unchallenged SP-C deficient mice, sensitivity to Pseudomonas challenge seemed to be strain specific since bacterial cell counts were enhanced in 129/Sv6 strain but unaltered in age-matched FVB/N mice. Further, neutrophils and enlarged macrophages with cytoplasmic inclusions were observed upon Pseudomonas aeruginosa challenge in absence of SP-C whereby the phagocytic index of macrophages in the SP-C deficient mouse model was reduced (Glasser et al., 2008). Similarly, SP-C deficient mice were more susceptible to respiratory syncytial virus (RSV) infection and resolution of RSV-induced alterations in lung morphology was delayed. Mice exhibited more extensive interstitial thickening, air space consolidation and goblet cell hyperplasia. Polymorphonuclear and macrophage cell counts were increased in BALF obtained from SP-C deficient mice as well as viral titers in lung homogenate (Glasser et al., 2008, 2009). In contrast to challenge with Pseudomonas aeruginosa, increased susceptibility of SP-C deficient mice to RSV was not restricted to 129/Sv6 strain but also observed in FVB/N background (Glasser et al., 2008, 2009). However, induction of SP-C expression in compound transgenic mice could reduce RSV-induced tissue inflammation and inflammatory cell counts (Glasser et al., 2013b). LPS-challenged mice develop more intense airway and airspace inflammation in the absence of SP-C. While control mice demonstrate a rapid resolution of LPS-induced cellular and tissue inflammation, inflammation persisted longer in SP-C deficient mice. Cell culture experiments further demonstrated increased LPS-induced cytokine expression of SP-C deficient AE2C compared to those from controls (Glasser et al., 2013b).
The model studied by Glasser et al. (2001, 2008) represents a null mutant mouse model without sufficient Sftpc expression leading to the absence of SP-C and proSP-C. Other models include mutations that cause aberrant accumulation of SP-C precursor proteins. Deletion of exon 4 (g.1728 G3A), a mutation detected in human SFTPC, resulted in a truncated form of SP-C (SP-CΔexon4) and incomplete pro-protein processing. Affected mice exhibited disrupted lung organogenesis, branching morphogenesis and expression-depended epithelial cell cytotoxicity (Bridges et al., 2003). Further, transgenic mice that express the mutant L188Q SP-C did not develop pulmonary fibrosis spontaneously, despite induction of ER stress, but developed a more extensive pulmonary fibrosis with reduced compliance and enhanced AEC apoptosis in response to bleomycin and authors suggested alveolar epithelial ER stress to play a crucial role in enhanced disease development (Lawson et al., 2011). Recently, Nureki et al. (2018) developed a conditional mouse mutant in which the expression of I73T, the most common SFTPC mutation found in human patients, is regulated by tamoxifen. In their study, expression of the human mutation induced a spontaneous acute alveolitis, followed by parenchymal injury and fibrotic remodeling in mice (Nureki et al., 2018). Tamoxifen controlled expression of isoleucin to threonine substitution at codon 73 in Sftpc (I73T) results in misprocessed proSP-C and leads to increased mortality at day 7 to 14 and acute diffuse parenchymal lung injury with polycellular alveolitis. An early influx of polymorphonuclear cells and macrophages was followed by eosinophilia. Partial recovery from acute injury was followed by aberrant remodeling, collagen deposition and AE2C cell hyperplasia and mechanical abnormalities. Pressure-volume curves displayed a restrictive pattern at 4 to 6 weeks post-tamoxifen and static compliance was reduced at week 4 but rose with resolution of inflammation. However, the fibrotic phenotype was preceded by TGF-β1 (Nureki et al., 2018). A detailed analysis of the same model showed an early reduction of macrophages, followed by an accumulation of immature macrophages and neutrophils before onset of eosinophilia at week 2 in combination with shift from proinflammatory to anti-inflammatory/profibrotic activation state in mRNA BALF cell analysis (Venosa et al., 2019). Total inspiratory capacity and static compliance were reduced in transgenic mice following the induction of the mutant protein whereas tissue damping was elevated, all together suggesting an enhanced tissue stiffness. While local clodronate application diminishing resident macrophages led to reduced survival, intravenous application of clodronate decreased accumulation of immature macrophages, improved survival and reduced eosinophilia (Venosa et al., 2019). Expression of another mutant protein, C121G, resulted in postnatal respiratory failure due to disrupted lung morphogenesis with enlargement of airspaces and interstitial widening in mice constitutively expressing C121G whereas a tamoxifen-mediated expression in adult mice induced acute parenchymal lung injury and a multiphasic polycellular alveolitis with increased BALF cell counts. Similar to the I73T model, an early increase of macrophages and neutrophils was followed by a later influx of eosinophils. Overall, lung mechanics showed a restrictive impairment indicated by a decline in static lung compliance (Katzen et al., 2019).
Since Nogee et al., (2001) described the first case of a Sftpc mutation in an infant girl and her mother diagnosed with ILD (Nogee et al., 2001), several studies have been conducted to provide a link between variants in SFTPC and the manifestation of ILDs in adults or children. Since previous groups have already performed systematic reviews describing detected mutations, clinical parameters and radiographic as well as histological findings (Ono et al., 2011; Litao et al., 2017), we decided to focus on the work that reported either BALF cell counting or data on lung mechanics in their study (Table 2). In general, lung mechanics display a restrictive pattern (Thomas et al., 2002; Ono et al., 2011; Kuse et al., 2013; Avital et al., 2014; Hevroni et al., 2015). In particular, vital capacity, forced vital capacity were markedly reduced in some patients (Ono et al., 2011; Kuse et al., 2013; Avital et al., 2014; Hevroni et al., 2015) whereas mild reductions in forced expiratory pressure in 1 s (FEV1) (Cottin et al., 2011; Ono et al., 2011) were attributable to a generally reduced lung volume and not indicative of an obstruction. However, the reported residual volumes were high to normal (Cottin et al., 2011; Hevroni et al., 2015). Changes in functional residual capacity (FRC) were unspecific. While some patients demonstrated a reduced FRC, other studies reported elevated or normal values. Gas exchange as indicated by diffusing capacity for carbon monoxide exhibited distinct impairment in most (Thomas et al., 2002; Abou Taam et al., 2009; Cottin et al., 2011; Ono et al., 2011; Avital et al., 2014) resulting in a generally reduced oxygen (Thouvenin et al., 2010; Hevroni et al., 2015). However, Abou Taam and coworkers reported normal and increased diffusion capacity for carbon monoxide (DLCO) in two patients (Abou Taam et al., 2009) and oxygenation was sufficient in another patient reported by Avital et al. (2014). Hence, heterogeneity of the disease is also reflected in lung function. While the majority of studies linked SFTPC mutations to the familial form of pulmonary fibrosis, genetic mutations in SFTPC were also detected in a subset of patients suffering from sporadic IPF (Lawson et al., 2004). Interestingly, sporadic patients also demonstrate with mixed pattern as reported by Cottin et al. (2011). Indeed, they reported a case of combined pulmonary fibrosis and emphysema in a patient with the most common I73T mutation.
As explained before, pulmonary fibrosis and especially IPF was linked to an AE2C dysfunction and increased ER stress response due to aberrant protein accumulation. Hyperplastic AE2C in histological examination are a common finding (Nogee et al., 2001; Chibbar et al., 2004; Hamvas et al., 2004; Cameron et al., 2005; Stevens et al., 2005; Soraisham et al., 2006; Mechri et al., 2010; Thouvenin et al., 2010; Citti et al., 2013; Litao et al., 2017; Park et al., 2018) and enhanced ER stress is a widely acknowledged pathomechanism in IPF (Korfei et al., 2008). However, reduced transcription of SFTPC due to promotor variant were associated with neonatal respiratory distress syndrome in late preterm infants (Wambach et al., 2010) and SP-C was absent in BALF samples of a family with chronic ILDs, together with reduced pro-SP-C staining, though no mutation was identified (Amin et al., 2001). While the majority of mutations leads to the onset of ILDs already in childhood (Chibbar et al., 2004; Hamvas et al., 2004; Tredano et al., 2004; Rosen and Waltz, 2005; Stevens et al., 2005; Bullard and Nogee, 2007; Guillot et al., 2009; Mechri et al., 2010; Cottin et al., 2011; Citti et al., 2013; Henderson et al., 2013; Hepping et al., 2013; Turcu et al., 2013; Akimoto et al., 2014; Avital et al., 2014; van Hoorn et al., 2014; Kroner et al., 2015; Liptzin et al., 2015; Peca et al., 2015; Griese et al., 2016; Liu et al., 2016; Hayasaka et al., 2018) a subset of variants results in a manifestation in later child- or adulthood (Lawson et al., 2004; Setoguchi et al., 2006; Markart et al., 2007; van Moorsel et al., 2010; Cottin et al., 2011; Ono et al., 2011; Kuse et al., 2013) suggesting a chronic process.
Besides to AE2C hyperplasia, cholesterol clefts are frequently detected on lung biopsy in context of an SFTPC mutations (Hamvas et al., 2004; Abou Taam et al., 2009; Mechri et al., 2010; Cottin et al., 2011; Litao et al., 2017) or on lung specimen from ILD patients lacking SP-C (Amin et al., 2001). Macrophages were often described as foamy (Lawson et al., 2004; Stevens et al., 2005; Henderson et al., 2013) or lipid-laden (Liptzin et al., 2015; Salerno et al., 2016). When analyzing BALF cells, the majority of cells usually consists of macrophages (Abou Taam et al., 2009; Thouvenin et al., 2010; Ono et al., 2011). However, some samples showed a marked increase in neutrophil (Thouvenin et al., 2010; Cottin et al., 2011) or lymphocyte (Ono et al., 2011) cell counting in the absence of an infection. Whether immune cell infiltration occurs in reaction to abnormal histological findings or rather causes alterations remains unclear in view of single time point studies in human patients. In addition, a detailed characterization of macrophages was not provided by the majority of studies, but different activation could have distinct effects on fibrogenesis.
Recently, we have described the accumulation of cholesterol crystals in AMs of SP-C deficient animals (Ruwisch et al., 2020). We also showed similar crystals accumulated in the lung of an IPF patient. Looking through the literature this feature is commonly described (Glasser et al., 2003; Hoenerhoff et al., 2006; Mora et al., 2006) but no further investigated. Therefore, the role of cholesterol or accumulation of cholesterol in the form of crystals in cells is yet not well understood and often overlooked. As explained before, cholesterol is also involved in many processes apart from regulating membrane biophysical properties. Not many experimental data are available in the context of the influence of cholesterol in lung diseases. However, in the clinic settings, it is not surprising that certain patients are suffering from comorbidities that should be treated such as hypercholesterolemia. In fact, it is known that IPF increases the risk of heart diseases. Thus, it is common for IPF patients to receive medications aimed at reducing cardiovascular risk, including statins, which lower cholesterol levels (Kreuter et al., 2018). Data in this regard is still quite controversial. While, 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) aggravated pulmonary fibrosis in mice by increased production of mitochondrial ROS and NLRP3 inflammasome activation in macrophages. Statins, as potent inhibitors of the endogenous cholesterol synthesis have been associated with a lower decline in FVC in a cohort of untreated IPF patients (Kreuter et al., 2018). In line with this, diet induced systemic hyperlipidemia resulted in altered surfactant phospholipid composition leading to increased alveolar collapsibility, providing evidence that pulmonary lipid homeostasis is strongly related to the blood circulation (Baritussio et al., 1980). As approximately 83% of surfactant cholesterol has been stated to be derived from blood plasma (Turley et al., 1981), statins could contribute in preventing deleterious high levels of cholesterol in surfactant films, providing surfactant film stability and avoiding alveolar collapse. In addition, evaluation of lung function showed that patients receiving statin therapy and Ofev had less decline in pulmonary capacity, compared with a placebo (Kreuter et al., 2018). The yardstick that researchers used was patients’ annual rate of decline in forced vital capacity (FVC). The results suggested that statins do not diminish Ofev’s effectiveness (Kreuter et al., 2018).
Statins may also have an anti-inflammatory effect in the lung (Liao and Laufs, 2004; Jain and Ridker, 2005), thus a multifactorial mechanism may play an important role in their use. In conclusion, more experimental evidence is needed to support the use of cholesterol lowering drugs for the treatment of lung diseases such as fibrosis. In addition, the role of cholesterol and SP-C has to be further described and understood in order to develop new therapies. However, there is already promising data in this regard, opening a new pathway to target.
Summing up there are various potential either direct or indirect mechanisms how SP-C is able to interfere and modulate fibrogenesis in the lung. While SP-C deficiency mediated surfactant dysfunction and impaired lung mechanics are likely contributors in the generation of local mechanical stresses and strains, its biophysical interplay with cholesterol on the one hand, and its modulation of TLR-4 signaling on the other hand, highlights SP-C as a key element in a complex profibrotic network. Thereby SP-C deficiency comes into play not only in animal models, but also in patients with familial and non-familial forms of lung fibrosis such as IPF. Further research is needed in order to determine the potential of cholesterol lowering drugs as treatment or combined with anti-fibrotic drugs to find a better therapy for IPF patients.
KS, JR, NR, and EL-R contributed to the review, writing, and revising of the manuscript.
This research was funded by Biomedical Research in Endstage and Obstructive Lung Disease (BREATH), Member of the German Centre for Lung Research (DZL), Cluster of Excellence REBIRTH (Regenerative Biology to Reconstructive Therapy Hannover), and HilF program (Hochschulinterne Leistungsförderung, MHH).
NR was employed by the company AlveoliX AG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
α-SMA, alpha smooth muscle actin; aaAMs, alternatively activated alveolar macrophages; ABCA1, ATP binding cassette transporter A1; ABCG1, ATP binding cassette transporter G1; ADA, adenosine-deaminase; ADP, adenosine diphosphate; AEC, alveolar epithelial cell; AE1C, alveolar epithelial type 1 cell; AE2C, alveolar epithelial type 2 cell; ALI, acute lung injury; AM, alveolar macrophages; AMP, adenosine monophosphate; ApoA1, apolipoprotein A1; ApoE, apolipoprotein E; ARDS, acute respiratory distress syndrome; Arg-1, arginase 1; ASC, apoptosis speck-like protein; ATP, adenosine triphosphate; BALF, bronchoalveolar lavage fluid; Ca2+, calcium; caAMs, classically activated alveolar macrophages; CARD, caspase recruitment domain; chILD, Children Interstitial Lung Disease; DAMPs, damage associated molecular patterns; DLCO, diffusion capacity for carbon monoxide; DPPC, dipalmitoylphosphatidylcholine; EC, extracellular; ECM, extracellular matrix; EMT, epithelial to mesenchymal transition; ER, endoplasmic reticulum; FEV1, forced expiratory pressure in 1 second; FRC, forced residual capacity; GOE, great oxygen event; HCl, chloride acid; HPS, Hermansky–Pudlak syndrome; HRCT, high resolution computed tomography; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; LA, large aggregate; LAP-1, latency associated peptide 1; LDL, low-density lipoprotein; LPS, lipopolysaccharide; LTP-1, latency TGF- β 1 binding protein-1; mRNA, messenger ribonucleic acid; MMP-9, matrix-metalloprotease 9; MMP-13, matrix-metalloprotease 13; MyD88, myeloid differentiation primary response 88; NF κβ, necrosis factor-kappa-beta; NLRP3 NOD-, LRR- and pyrin domain-containing protein 3; NOS-2, inducible nitric oxide synthase; NRDS, neonatal respiratory distress syndrome; PAP, pulmonary alveolar proteinosis; PC, phosphatydilcholine; PEEP, positive end-expiratory pressure; P2XR7, P2X purinoreceptor 7; P2 × 4R, P2X purinoreceptor 4; P2Y2R, P2Y purinoreceptor 2; Relm- α, resistin-like alpha; ROS, reactive oxygen species; RSV, respiratory syncytial virus; SA, small aggregate; SHH, sonic hedgehog; SP-B, surfactant protein B; SP-C, surfactant protein C; SFTPC, surfactant protein C gene; TGF-b1, transforming growth factor beta 1; TIMP1, tissue-metalloprotease inhibitor 1; TLC, total lung capacity; TLR-4, Toll-like receptor 4; TNF- α, tumor necrosis factor alpha; UIP, unusual interstitial pneumonia; VILI, ventilation induced lung injury; YM1, chitinase-like protein 1.
Abou Taam, R., Jaubert, F., Emond, S., le Bourgeois, M., Epaud, R., Karila, C., et al. (2009). Familial interstitial disease with I73T mutation: a mid- and long-term study. Pediatr. Pulmonol. 44, 167–175. doi: 10.1002/ppul.20970
Akimoto, T., Cho, K., Hayasaka, I., Morioka, K., Kaneshi, Y., Furuta, I., et al. (2014). Hereditary interstitial lung diseases manifesting in early childhood in Japan. Pediatr. Res. 76, 453–458. doi: 10.1038/pr.2014.114
Allawzi, A., Elajaili, H., Redente, E. F., and Nozik-Grayck, E. (2019). Oxidative toxicology of bleomycin: role of the extracellular redox environment. Curr. Opin. Toxicol. 13, 68–73. doi: 10.1016/j.cotox.2018.08.001
Al-Saiedy, M., Gunasekara, L., Green, F., Pratt, R., Chiu, A., Yang, A., et al. (2018). Surfactant dysfunction in ARDS and bronchiolitis is repaired with cyclodextrins. Mil. Med. 183, 207–215. doi: 10.1093/milmed/usx204
Amin, R. S., Wert, S. E., Baughman, R. P., Tomashefski, J. F., Nogee, L. M., Brody, A. S., et al. (2001). Surfactant protein deficiency in familial interstitial lung disease. J. Pediatr. 139, 85–92. doi: 10.1067/mpd.2001.114545
Amrein, M., von Nahmen, A., and Sieber, M. (1997). A scanning force- and fluorescence light microscopy study of the structure and function of a model pulmonary surfactant. Eur. Biophys. J. 26, 349–357. doi: 10.1007/s002490050089
Augusto, L. A., Synguelakis, M., Espinassous, Q., Lepoivre, M., Johansson, J., and Chaby, R. (2003). Cellular antiendotoxin activities of lung surfactant protein C in lipid vesicles. Am. J. Respir. Crit. Care Med. 168, 335–341. doi: 10.1164/rccm.200212-1440OC
Autilio, C., and Pérez-Gil, J. (2018). Understanding the principle biophysics concepts of pulmonary surfactant in health and disease. Arch. Dis. Child Fetal Neonat. Ed. 104, F443–F451. doi: 10.1136/archdischild-2018-315413
Avital, A., Hevroni, A., Godfrey, S., Cohen, S., Maayan, C., Nusair, S., et al. (2014). Natural history of five children with surfactant protein C mutations and interstitial lung disease. Pediatr. Pulmonol. 49, 1097–1105. doi: 10.1002/ppul.22971
Bachofen, H., and Schürch, S. (2001). Alveolar surface forces and lung architecture. Compar. Biochem. Physiol. Mol. Integr. Physiol. 129, 183–193. doi: 10.1016/S1095-6433(01)00315-314
Bachofen, H., Schurch, S., Urbinelli, M., and Weibel, E. R. (1987). Relations among alveolar surface tension, surface area, volume, and recoil pressure. J. Appl. Physiol. 62, 1878–1887. doi: 10.1152/jappl.1987.62.5.1878
Baker, A. D., Malur, A., Barna, B. P., Ghosh, S., Kavuru, M. S., Malur, A. G., et al. (2010a). Targeted PPARγ deficiency in alveolar macrophages disrupts surfactant catabolism. J. Lipid Res. 51, 1325–1331. doi: 10.1194/jlr.m001651
Baker, A. D., Malur, A., Barna, B. P., Kavuru, M. S., Malur, A. G., and Thomassen, M. J. (2010b). PPARγ regulates the expression of cholesterol metabolism genes in alveolar macrophages. Biochem. Biophys. Res. Commun. 393, 682–687. doi: 10.1016/j.bbrc.2010.02.056
Bargagli, E., Margollicci, M., Luddi, A., Nikiforakis, N., Grazia Perari, M., Grosso, S., et al. (2007). Chitotriosidase activity in patients with interstitial lung diseases. Respirat. Med. 101, 2176–2181. doi: 10.1016/j.rmed.2007.05.008
Baritussio, A., Enzi, G., Inelmen, E. M., Schiavon, M., de Biasi, F., Allegra, L., et al. (1980). Altered surfactant synthesis and function in rats with diet-induced hyperlipidemia. Metabolism 29, 503–510. doi: 10.1016/0026-0495(80)90075-X
Basset-Léobon, C., Lacoste-Collin, L., Aziza, J., Bes, J. C., Jozan, S., and Courtade-Saïdi, M. (2010). Cut-off values and significance of Oil Red O-positive cells in bronchoalveolar lavage fluid. Cytopathology 21, 245–250. doi: 10.1111/j.1365-2303.2009.00677.x
Batenburg, J. J., and Haagsman, H. P. (1998). The lipids of pulmonary surfactant: dynamics and interactions with proteins. Progress Lipid Res. 37, 235–276. doi: 10.1016/S0163-7827(98)00011-13
Bates, J. H. T., and Smith, B. J. (2018). Ventilator-induced lung injury and lung mechanics. Ann. Transl. Med. 6:378. doi: 10.21037/atm.2018.06.29
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., et al. (2009). Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791. doi: 10.4049/jimmunol.0901363
Baumgart, F., Ospina, O. L., Mingarro, I., Rodríguez-Crespo, I., and Pérez-Gil, J. (2010). Palmitoylation of pulmonary surfactant protein SP-C Is critical for its functional cooperation with SP-B to sustain compression/expansion dynamics in cholesterol-containing surfactant films. Biophys. J. 99, 3234–3243. doi: 10.1016/J.BPJ.2010.08.070
Beeh, K. M., Beier, J., Kornmann, O., and Buhl, R. (2003). Sputum matrix metalloproteinase-9, tissue inhibitor of metalloprotinease-1, and their molar ratio in patients with chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and healthy subjects. Respirat. Med. 97, 634–639. doi: 10.1053/rmed.2003.1493
Bekker, A., Holland, H. D., Wang, P.-L., Rumble, D., Stein, H. J., Hannah, J. L., et al. (2004). Dating the rise of atmospheric oxygen. Nature 427, 117–120. doi: 10.1038/nature02260
Bernardino de la Serna, J., Perez-Gil, J., Simonsen, A. C., and Bagatolli, L. A. (2004). Cholesterol rules: direct observation of the coexistence of two fluid phases in native pulmonary surfactant membranes at physiological temperatures. J. Biol. Chem. 279, 40715–40722. doi: 10.1074/jbc.M404648200
Bernhard, W. (2016). Lung surfactant: function and composition in the context of development and respiratory physiology. Ann. Anat. Anatomischer Anzeiger 208, 146–150. doi: 10.1016/J.AANAT.2016.08.003
Besnard, V., Wert, S. E., Stahlman, M. T., Postle, A. D., Xu, Y., Ikegami, M., et al. (2009). Deletion of Scap in alveolar type II cells influences lung lipid homeostasis and identifies a compensatory role for pulmonary lipofibroblasts. J. Biol. Chem. 284, 4018–4030. doi: 10.1074/jbc.m805388200
Bilek, A. M., Dee, K. C., and Gaver, D. P. (2003). Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J. Appl. Physiol. 94, 770–783. doi: 10.1152/japplphysiol.00764.2002
Birkelbach, B., Lutz, D., Ruppert, C., Henneke, I., Lopez-Rodriguez, E., Günther, A., et al. (2015). Linking progression of fibrotic lung remodeling and ultrastructural alterations of alveolar epithelial type II cells in the amiodarone mouse model. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L63–L75.
Bridges, J. P., Wert, S. E., Nogee, L. M., and Weaver, T. E. (2003). Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J. Biol. Chem. 278, 52739–52746. doi: 10.1074/jbc.m309599200
Brocks, J. J., Logan, G. A., Buick, R., and Summons, R. E. (1999). Archean molecular fossils and the early rise of eukaryotes. Science 285, 1033–1036. doi: 10.1126/science.285.5430.1033
Buchwald, H., O’Dea, T. J., Menchaca, H. J., Michalek, V. N., and Rohde, T. D. (2000). Effect of plasma cholesterol on red blood cell oxygen transport. Clin. Exp. Pharmacol. Physiol. 27, 951–955. doi: 10.1046/j.1440-1681.2000.03383.x
Bullard, J. E., and Nogee, L. M. (2007). Heterozygosity for ABCA3 mutations modifies the severity of lung disease associated with a surfactant protein C gene (SFTPC) mutation. Pediatr. Res. 62, 176–179. doi: 10.1203/PDR.0b013e3180a72588
Cabrera-Benítez, N. E., Parotto, M., Post, M., Han, B., Spieth, P. M., Cheng, W. E., et al. (2012). Mechanical stress induces lung fibrosis by epithelial-mesenchymal transition. Crit. Care Med. 40, 510–517. doi: 10.1097/CCM.0b013e31822f09d7
Cameron, H. S., Somaschini, M., Carrera, P., Hamvas, A., Whitsett, J. A., Wert, S. E., et al. (2005). A common mutation in the surfactant protein C gene associated with lung disease. J. Pediatr. 146, 370–375. doi: 10.1016/j.jpeds.2004.10.028
Cameron, R. G., Blendis, L. M., and Neuman, M. G. (2001). Accumulation of macrophages in primary sclerosing cholangitis. Clin. Biochem. 34, 195–201. doi: 10.1016/S0009-9120(01)00215-216
Cassel, S. L., Eisenbarth, S. C., Iyer, S. S., Sadler, J. J., Colegio, O. R., Tephly, L. A., et al. (2008). The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. U.S.A. 105, 9035–9040. doi: 10.1073/pnas.0803933105
Cassera, M. B., Silber, A. M., and Gennaro, A. M. (2002). Differential effects of cholesterol on acyl chain order in erythrocyte membranes as a function of depth from the surface. An electron paramagnetic resonance (EPR) spin label study. Biophys. Chem. Biophys. Chem. 99, 117–127. doi: 10.1016/S0301-4622(02)00139-134
Cassidy, K. J., Halpern, D., Ressler, B. G., and Grotberg, J. B. (1999). Surfactant effects in model airway closure experiments. J. Appl. Physiol. 87, 415–427. doi: 10.1152/jappl.1999.87.1.415
Chaby, R., Garcia-Verdugo, I., Espinassous, Q., and Augusto, L. A. (2005). Interactions between LPS and lung surfactant proteins. J. Endotoxin. Res. 11, 181–185. doi: 10.1179/096805105X37358
Chai, A. B., Ammit, A. J., and Gelissen, I. C. (2017). Examining the role of ABC lipid transporters in pulmonary lipid homeostasis and inflammation. Respirat. Res. 18:41. doi: 10.1186/s12931-017-0526-529
Chibbar, R., Shih, F., Baga, M., Torlakovic, E., Ramlall, K., Skomro, R., et al. (2004). Nonspecific interstitial pneumonia and usual interstitial pneumonia with mutation in surfactant protein C in familial pulmonary fibrosis. Mod. Pathol. 17, 973–980. doi: 10.1038/modpathol.3800149
Chou, P. Y., and Fasman, G. D. (2006). Prediction of the secondary structure of proteins from their amino acid sequence. Adv. Enzymol. Relat. Areas Mol. Biol. 47, 45–48. doi: 10.1002/9780470122921.ch2
Citti, A., Peca, D., Petrini, S., Cutrera, R., Biban, P., Haass, C., et al. (2013). Ultrastructural characterization of genetic diffuse lung diseases in infants and children: a cohort study and review. Ultrastruct. Pathol. 37, 356–365. doi: 10.3109/01913123.2013.811454
Cong, X., Hubmayr, R. D., Li, C., and Zhao, X. (2017). Plasma membrane wounding and repair in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L371–L391. doi: 10.1152/ajplung.00486.2016
Conkright, J. J., Na, C.-L., and Weaver, T. E. (2002). Overexpression of surfactant protein-c mature peptide causes neonatal lethality in transgenic mice. Am. J. Respir. Cell Mol. Biol. 26, 85–90. doi: 10.1165/ajrcmb.26.1.4686
Cottin, V., and Cordier, J.-F. (2011). SFTPC mutations in patients with familial pulmonary fibrosis: combined with emphysema? Am. J. Respir. Crit. Med. 183:1113. doi: 10.1164/ajrccm.183.8.1113
Cottin, V., Reix, P., Khouatra, C., Thivolet-Béjui, F., Feldmann, D., and Cordier, J.-F. (2011). Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax 66, 918–919. doi: 10.1136/thx.2010.151407
Creuwels, L. A., Demel, R. A., van Golde, L. M., Benson, B. J., and Haagsman, H. P. (1993). Effect of acylation on structure and function of surfactant protein C at the air-liquid interface. J. Biol. Chem. 268, 26752–26758.
DeBose-Boyd, R. A. (2008). Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 18, 609–621. doi: 10.1038/cr.2008.61
Dico, A. S., Taneva, S., Morrow, M. R., and Keough, K. M. (1997). Effect of calcium on phospholipid interaction with pulmonary surfactant protein C. Biophys. J. 73, 2595–2602. doi: 10.1016/S0006-3495(97)78289-78284
Dietl, P., Haller, T., and Frick, M. (2012). Spatio-temporal aspects, pathways and actions of Ca(2+) in surfactant secreting pulmonary alveolar type II pneumocytes. Cell Calcium 52, 296–302. doi: 10.1016/j.ceca.2012.04.010
Doyle, I. R., Jones, M. E., Barr, H. A., Orgeig, S., Crockett, A. J., McDonald, C. F., et al. (1994). Composition of human pulmonary surfactant varies with exercise and level of fitness. Am. J. Respir. Crit. Med. 149, 1619–1627. doi: 10.1164/ajrccm.149.6.8004321
Dreyfuss, D., Martin-Lefèvre, L., and Saumon, G. (1999). Hyperinflation-induced lung injury during alveolar flooding in rats: effect of perfluorocarbon instillation. Am. J. Respir. Crit. Care Med. 159, 1752–1757. doi: 10.1164/ajrccm.159.6.9805018
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. doi: 10.1038/nature08938
Eldridge, W. B., Zhang, Q., Faro, A., Sweet, S. C., Eghtesady, P., Hamvas, A., et al. (2017). Outcomes of lung transplantation for infants and children with genetic disorders of surfactant metabolism. J. Pediatr. 184, 157–164. doi: 10.1016/J.JPEDS.2017.01.017
Ertunc, M. E., and Hotamisligil, G. S. (2016). Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 57, 2099–2114. doi: 10.1194/jlr.R066514
Fehrenbach, H. (2001). Alveolar epithelial type II cell: defender of the alveolus revisited. Respir. Res. 2, 33–46. doi: 10.1186/rr36
Fessler, M. B. (2017). A new frontier in immunometabolism cholesterol in lung health and disease. Ann. Am. Thorac. Soc. 14, S399–S405. doi: 10.1513/AnnalsATS.201702-136AW
Fessler, M. B., and Summer, R. S. (2016). Surfactant lipids at the host-environment interface. metabolic sensors, suppressors, and effectors of inflammatory lung disease. Am. J. Respir. Cell Mol. Biol. 54, 624–635. doi: 10.1165/rcmb.2016-0011PS
French, K. L., Hallmann, C., Hope, J. M., Schoon, P. L., Zumberge, J. A., Hoshino, Y., et al. (2015). Reappraisal of hydrocarbon biomarkers in archean rocks. Proc. Natl. Acad. Sci. U.S.A. 112, 5915–5920. doi: 10.1073/pnas.1419563112
Froese, A. R., Shimbori, C., Bellaye, P.-S., Inman, M., Obex, S., Fatima, S., et al. (2016). Stretch-induced activation of transforming growth factor-β1 in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 194, 84–96. doi: 10.1164/rccm.201508-1638OC
Garcia-Verdugo, I., Garcia de Paco, E., Espinassous, Q., Gonzalez-Horta, A., Synguelakis, M., Kanellopoulos, J., et al. (2009). Synthetic peptides representing the N-terminal segment of surfactant protein C modulate LPS-stimulated TNF-α production by macrophages. Innate Immun. 15, 53–62. doi: 10.1177/1753425908100500
Gasse, P., Mary, C., Guenon, I., Noulin, N., Charron, S., Schnyder-Candrian, S., et al. (2007). IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Invest. 117, 3786–3799. doi: 10.1172/JCI32285
Gattinoni, L., Marini, J. J., Collino, F., Maiolo, G., Rapetti, F., Tonetti, T., et al. (2017). The future of mechanical ventilation: Lessons from the present and the past. Crit. Care 21:183. doi: 10.1186/s13054-017-1750-x
Ghadiali, S. N., and Gaver, D. P. (2008). Biomechanics of liquid-epithelium interactions in pulmonary airways. Respir. Physiol. Neurobiol. 163, 232–243. doi: 10.1016/j.resp.2008.04.008
Glasser, S. W., Burhans, M. S., Korfhagen, T. R., Na, C.-L. L., Sly, P. D., Ross, G. F., et al. (2001). Altered stability of pulmonary surfactant in SP-C-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 6366–6371. doi: 10.1073/pnas.101500298
Glasser, S. W., Detmer, E. A., Ikegami, M., Na, C.-L., Stahlman, M. T., and Whitsett, J. A. (2003). Pneumonitis and emphysema in sp-C gene targeted mice. J. Biol. Chem. 278, 14291–14298. doi: 10.1074/jbc.m210909200
Glasser, S. W., Maxfield, M. D., Ruetschilling, T. L., Akinbi, H. T., Baatz, J. E., Kitzmiller, J. A., et al. (2013a). Persistence of LPS-induced lung inflammation in surfactant protein-C-deficient mice. Am. J. Respir. Cell Mol. Biol. 49, 845–854. doi: 10.1165/rcmb.2012-0374oc
Glasser, S. W., Senft, A. P., Maxfield, M. D., Ruetschilling, T. L., Baatz, J. E., Page, K., et al. (2013b). Genetic replacement of surfactant protein-C reduces respiratory syncytial virus induced lung injury. Respir. Res. 14:19. doi: 10.1186/1465-9921-14-19
Glasser, S. W., and Senft, A. (2009). “Pulmonary surfactant homeostasis and altered macrophage function,” in Lung Macrophages in Health And Disease, ed. S. Hodge (Bentham: Science Publisher Ltd), 1–13. doi: 10.2174/978160805020810901010001
Glasser, S. W., Senft, A. P., Whitsett, J. A., Maxfield, M. D., Ross, G. F., Richardson, T. R., et al. (2008). Macrophage dysfunction and susceptibility to pulmonary pseudomonas aeruginosa infection in surfactant protein C-deficient mice. J. Immunol. 181, 621–628. doi: 10.4049/jimmunol.181.1.621
Glasser, S. W., Witt, T. L., Senft, A. P., Baatz, J. E., Folger, D., Maxfield, M. D., et al. (2009). Surfactant protein C-deficient mice are susceptible to respiratory syncytial virus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L64–L72.
Gómez-Gil, L., Pérez-Gil, J., and Goormaghtigh, E. (2009a). Cholesterol modulates the exposure and orientation of pulmonary surfactant protein SP-C in model surfactant membranes. Biochim. Biophys. Acta Biomemb. 1788, 1907–1915. doi: 10.1016/J.BBAMEM.2009.05.011
Gómez-Gil, L., Schürch, D., Goormaghtigh, E., and Pérez-Gil, J. (2009b). Pulmonary surfactant protein SP-C counteracts the deleterious effects of cholesterol on the activity of surfactant films under physiologically relevant compression-expansion dynamics. Biophys. J. 97, 2736–2745. doi: 10.1016/J.BPJ.2009.08.045
Griese, M., Lorenz, E., Hengst, M., Schams, A., Wesselak, T., Rauch, D., et al. (2016). Surfactant proteins in pediatric interstitial lung disease. Pediatr. Res. 79, 34–41. doi: 10.1038/pr.2015.173
Gu, B. J., and Wiley, J. S. (2006). Rapid ATP-induced release of matrix metalloproteinase 9 is mediated by the P2X7 receptor. Blood 107, 4946–4953. doi: 10.1182/blood-2005-07-2994
Guillot, L., Epaud, R., Thouvenin, G., Jonard, L., Mohsni, A., Couderc, R., et al. (2009). New surfactant protein C gene mutations associated with diffuse lung disease. J. Med. Genet. 46, 490–494. doi: 10.1136/jmg.2009.066829
Gunasekara, L., Schürch, S., Schoel, W. M., Nag, K., Leonenko, Z., Haufs, M., et al. (2005). Pulmonary surfactant function is abolished by an elevated proportion of cholesterol. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1737, 27–35. doi: 10.1016/j.bbalip.2005.09.002
Gunasekara, L. C., Pratt, R. M., Schoel, W. M., Gosche, S., Prenner, E. J., and Amrein, M. W. (2010). Methyl-β-cyclodextrin restores the structure and function of pulmonary surfactant films impaired by cholesterol. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1798, 986–994. doi: 10.1016/J.BBAMEM.2009.12.003
Hamvas, A. (2010). Evaluation and management of inherited disorders of surfactant metabolism. Chin. Med. J. 123, 2943–2947.
Hamvas, A., Nogee, L. M., White, F., Schuler, P., Hackett, B. P., Huddleston, C. B., et al. (2004). Progressive lung disease and surfactant dysfunction with a deletion in surfactant protein C gene. Am. J. Respir. Cell Mol. Biol. 30, 771–776. doi: 10.1165/rcmb.2003-0323oc
Hasan, D., Blankman, P., and Nieman, G. F. (2017). Purinergic signalling links mechanical breath profile and alveolar mechanics with the pro-inflammatory innate immune response causing ventilation-induced lung injury. Puriner. Signal. 13, 363–386. doi: 10.1007/s11302-017-9564-9565
Hayasaka, I., Cho, K., Akimoto, T., Ikeda, M., Uzuki, Y., Yamada, M., et al. (2018). Genetic basis for childhood interstitial lung disease among Japanese infants and children. Pediatr. Res. 83, 477–483. doi: 10.1038/pr.2017.217
Henderson, L. B., Melton, K., Wert, S., Couriel, J., Bush, A., Ashworth, M., et al. (2013). Large ABCA3 and SFTPC deletions resulting in lung disease. Ann. Am. Thorac. Soc. 10, 602–607.
Hepping, N., Griese, M., Lohse, P., Garbe, W., and Lange, L. (2013). Successful treatment of neonatal respiratory failure caused by a novel surfactant protein C p.Cys121Gly mutation with hydroxychloroquine. J. Perinatol. 33:492. doi: 10.1038/jp.2012.131
Hevroni, A., Goldman, A., and Springer, C. (2015). Infant pulmonary function testing in chronic pneumonitis of infancy due to surfactant protein C mutation. Pediatr. Pulmonol. 50, E17–E23.
Hinz, B. (2012). Mechanical aspects of lung fibrosis: a spotlight on the myofibroblast. Proc. Am. Thorac. Soc. 9, 137–147. doi: 10.1513/pats.201202-017AW
Hinz, B., and Suki, B. (2016). Does breathing amplify fibrosis? Am. J. Respir. Crit. Care Med. 194, 9–11. doi: 10.1164/rccm.201601-0149ED
Hoenerhoff, M. J., Starost, M. F., and Ward, J. M. (2006). Eosinophilic crystalline pneumonia as a major cause of death in 129S4/SvJae mice. Vet. Pathol. 43, 682–688. doi: 10.1354/vp.43-5-682
Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu, G., Anstrom, K. J., King, T. E., Lasky, J. A., and Martinez, F. J. (2012). Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 366, 1968–1977. doi: 10.1056/NEJMoa1113354
Jain, M. K., and Ridker, P. M. (2005). Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat. Rev. Drug Discov. 4, 977–987. doi: 10.1038/nrd1901
Jin, H., Ciechanowicz, A. K., Kaplan, A. R., Wang, L., Zhang, P. X., Lu, Y. C., et al. (2018). Surfactant protein C dampens inflammation by decreasing JAK/STAT activation during lung repair. Am. J. Physiol. Lung. Cell Mol. Physiol. 314, L882–L892. doi: 10.1152/ajplung.00418.2017
Johansson, J. (2001). Membrane properties and amyloid fibril formation of lung surfactant protein C. Biochem. Soc. Trans. 29, 601–606. doi: 10.1042/bst0290601
Johansson, J., Weaver, T. E., and Tjernberg, L. O. (2004). Proteolytic generation and aggregation of peptides from transmembrane regions: lung surfactant protein C and amyloid β-peptide. Cell. Mol. Life Sci. CMLS 61, 326–335. doi: 10.1007/s00018-003-3274-3276
Jolley, C. D., Dietschy, J. M., and Turley, S. D. (1999). Genetic differences in cholesterol absorption in 129/Sv and C57BL/6 mice: effect on cholesterol responsiveness. Am. J. Physiolo. Gastrointest. Liver Physiol. 276, G1117–G1124. doi: 10.1152/ajpgi.1999.276.5.g1117
Justice, M. J., Petrusca, D. N., Rogozea, A. L., Williams, J. A., Schweitzer, K. S., Petrache, I., et al. (2014). Effects of lipid interactions on model vesicle engulfment by alveolar macrophages. Biophys. J. 106, 598–609. doi: 10.1016/J.BPJ.2013.12.036
Karlmark, K. R., Weiskirchen, R., Zimmermann, H. W., Gassler, N., Ginhoux, F., Weber, C., et al. (2009). Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 50, 261–274. doi: 10.1002/hep.22950
Katzen, J., Venosa, A., Tomer, Y., Kopp, M., Morely, M., Diwadkar, A., et al. (2019). Interstitial lung disease related surfactant protein-C mutations alter the transcriptome and progenitor cell function of alveolar epithelial cells in mice. Eur. Respirat. J. 54, A1291–A1291. doi: 10.1183/13993003.congress-2019.PA1291
Keating, E., Rahman, L., Francis, J., Petersen, A., Possmayer, F., Veldhuizen, R., et al. (2007). Effect of cholesterol on the biophysical and physiological properties of a clinical pulmonary surfactant. Biophys. J. 93, 1391–1401. doi: 10.1529/BIOPHYSJ.106.099762
Knudsen, L., and Ochs, M. (2018). The micromechanics of lung alveoli: structure and function of surfactant and tissue components. Histochem. Cell Biol. 150, 661–676. doi: 10.1007/s00418-018-1747-1749
Knudsen, L., Ruppert, C., and Ochs, M. (2017). Tissue remodelling in pulmonary fibrosis. Cell Tissue Res. 367, 607–626. doi: 10.1007/s00441-016-2543-2542
Kolb, M., Margetts, P. J., Anthony, D. C., Pitossi, F., and Gauldie, J. (2001). Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Invest. 107, 1529–1536. doi: 10.1172/JCI12568
Kollisch-Singule, M., Emr, B., Jain, S. V., Andrews, P., Satalin, J., Liu, J., et al. (2015). The effects of airway pressure release ventilation on respiratory mechanics in extrapulmonary lung injury. Intens. Care Med. Exper. 3:71. doi: 10.1186/s40635-015-0071-70
Kook, S., Wang, P., Young, L. R., Schwake, M., Saftig, P., Weng, X., et al. (2016). Impaired lysosomal integral membrane protein 2-dependent peroxiredoxin 6 delivery to lamellar bodies accounts for altered alveolar phospholipid content in adaptor protein-3-deficient pearl mice. J. Biol. Chem. 291, 8414–8427. doi: 10.1074/jbc.M116.720201
Korfei, M., Ruppert, C., Mahavadi, P., Henneke, I., Markart, P., Koch, M., et al. (2008). Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 838–846. doi: 10.1164/rccm.200802-313OC
Korfhagen, T. R., Glasser, S. W., Wert, S. E., Bruno, M. D., Daugherty, C. C., McNeish, J. D., et al. (1990). Cis-acting sequences from a human surfactant protein gene confer pulmonary-specific gene expression in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 87, 6122–6126. doi: 10.1073/pnas.87.16.6122
Kreuter, M., Costabel, U., Richeldi, L., Cottin, V., Wijsenbeek, M., Bonella, F., et al. (2018). Statin therapy and outcomes in trials of nintedanib in idiopathic pulmonary fibrosis. Respiration 95, 317–326. doi: 10.1159/000486286
Kroner, C., Reu, S., Teusch, V., Schams, A., Grimmelt, A.-C., Barker, M., et al. (2015). Genotype alone does not predict the clinical course of SFTPC deficiency in paediatric patients. Eur. Respir. J. 46, 197–206. doi: 10.1183/09031936.00129414
Kuse, N., Abe, S., Hayashi, H., Kamio, K., Saito, Y., Azuma, A., et al. (2013). Familial interstitial pneumonia in an adolescent boy with surfactant protein C gene (Y104H) mutation. Sarcoidos. Vasculit. Diffuse Lung Dis. 30, 73–77.
Lawson, W., Grant, S., Ambrosini, V., Womble, K., Dawson, E., Lane, K., et al. (2004). Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 59, 977–980. doi: 10.1136/thx.2004.026336
Lawson, W. E., Cheng, D.-S., Degryse, A. L., Tanjore, H., Polosukhin, V. V., Xu, X. C., et al. (2011). Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. U.S.A. 108, 10562–10567. doi: 10.1073/pnas.1107559108
Lawson, W. E., Polosukhin, V. V., Stathopoulos, G. T., Zoia, O., Han, W., Lane, K. B., et al. (2005). Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am. J. Pathol. 167, 1267–1277. doi: 10.1016/s0002-9440(10)61214-x
Lederer, D., and Martinez, F. J. (2018). Idiopathic pulmonary fibrosis. New Engl. J. Med. 379, 795–798.
Leicester, K. L., Olynyk, J. K., Brunt, E. M., Britton, R. S., and Bacon, B. R. (2004). CD14-positive hepatic monocytes/macrophages increase in hereditary hemochromatosis. Liver Intern.? 24, 446–451. doi: 10.1111/j.1478-3231.2004.0943.x
Leonenko, Z., Gill, S., Baoukina, S., Monticelli, L., Doehner, J., Gunasekara, L., et al. (2007). An elevated level of cholesterol impairs self-assembly of pulmonary surfactant into a functional film. Biophys. J. 93, 674–683. doi: 10.1529/biophysj.107.106310
Liao, J. K., and Laufs, U. (2004). Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 45, 89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748
Lin, S. L., Castaño, A. P., Nowlin, B. T., Lupher, M. L., and Duffield, J. S. (2009). Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J. Immunol. 183, 6733–6743. doi: 10.4049/jimmunol.0901473
Liptzin, D. R., Patel, T., and Deterding, R. R. (2015). Chronic ventilation in infants with surfactant protein C mutations: an alternative to lung transplantation. Am. J. Respir. Crit. Care Med. 191, 1338–1340. doi: 10.1164/rccm.201411-1955LE
Litao, M. K. S., Hayes, D., Chiwane, S., Nogee, L. M., and Kurland, G. (2017). A novel surfactant protein C gene mutation associated with progressive respiratory failure in infancy. Pediatr. Pulmonol. 52, 57–68. doi: 10.1002/ppul.23493
Liu, T., Sano, K., Ogiwara, N., and Kobayashi, N. (2016). A novel surfactant protein C L55F mutation associated with interstitial lung disease alters subcellular localization of proSP-C in A549 cells. Pediatr. Res. 79, 27–33. doi: 10.1038/pr.2015.178
Lopez-Rodriguez, E., Boden, C., Echaide, M., Perez-Gil, J., Kolb, M., Gauldie, J., et al. (2016). Surfactant dysfunction during overexpression of TGF-β1 precedes profibrotic lung remodeling in vivo. Am. J. Physiol. Lung. Cell Mol. Physiol. 310, L1260–L1271. doi: 10.1152/ajplung.00065.2016
Lopez-Rodriguez, E., Gay-Jordi, G., Mucci, A., Lachmann, N., and Serrano-Mollar, A. (2017). Lung surfactant metabolism: early in life, early in disease and target in cell therapy. Cell Tissue Res. 367, 721–735. doi: 10.1007/s00441-016-2520-9
Lukovic, D., Cruz, A., Gonzalez-Horta, A., Almlen, A., Curstedt, T., Mingarro, I., et al. (2012). Interfacial behavior of recombinant forms of human pulmonary surfactant protein SP-C. Langmuir 28, 7811–7825. doi: 10.1021/la301134v
Lutz, D., Gazdhar, A., Lopez-Rodriguez, E., Ruppert, C., Mahavadi, P., Günther, A., et al. (2015). Alveolar derecruitment and collapse induration as crucial mechanisms in lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 52, 232–243. doi: 10.1165/rcmb.2014-0078oc
Lv, Z., Wang, Y., Liu, Y.-J., Mao, Y.-F., Dong, W.-W., Ding, Z.-N., et al. (2018). NLRP3 inflammasome activation contributes to mechanical stretch-induced endothelial-mesenchymal transition and pulmonary fibrosis. Crit. Care Med. 46, e49–e58. doi: 10.1097/CCM.0000000000002799
Madala, S. K., Maxfield, M. D., Davidson, C. R., Schmidt, S. M., Garry, D., Ikegami, M., et al. (2011). Rapamycin regulates bleomycin-induced lung damage in SP-C-deficient mice. Pulmon. Med. 2011, 1–12. doi: 10.1155/2011/653524
Mai, C., Verleden, S. E., McDonough, J. E., Willems, S., de Wever, W., Coolen, J., et al. (2016). Thin-section CT features of idiopathic pulmonary fibrosis correlated with micro-CT and histologic analysis. Radiology 283, 252–263. doi: 10.1148/radiol.2016152362
Mainali, L., Raguz, M., O’Brien, W. J., and Subczynski, W. K. (2013). Properties of membranes derived from the total lipids extracted from the human lens cortex and nucleus. Biochim. Biophys. Acta 1828, 1432–1440. doi: 10.1016/j.bbamem.2013.02.006
Maitra, G., Inchley, K., Novick, R. J., Veldhuizen, R. A. W., Lewis, J. F., and Possmayer, F. (2002). Acute lung injury and lung transplantation influence in vitro subtype conversion of pulmonary surfactant. Am. J. Physiol. Lung. Cell Mol. Physiol. 282, L67–L74. doi: 10.1152/ajplung.2002.282.1.L67
Manon, V., Stoyan, I., Nemanja, V., Madalina, D.-M., Lazaro-Emilio, A., Thibault, B., et al. (2018). Lysosomal cholesterol hydrolysis couples efferocytosis to anti-inflammatory oxysterol production. Circ. Res. 122, 1369–1384. doi: 10.1161/CIRCRESAHA.117.312333
Mao, P., Li, J., Huang, Y., Wu, S., Pang, X., He, W., et al. (2017). MicroRNA-19b mediates lung epithelial-mesenchymal transition via phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase in response to mechanical stretch. Am. J. Respir. Cell Mol. Biol. 56, 11–19. doi: 10.1165/rcmb.2015-0377OC
Mariathasan, S., Weiss, D. S., Newton, K., McBride, J., O’Rourke, K., Roose-Girma, M., et al. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. doi: 10.1038/nature04515
Markart, P., Ruppert, C., Wygrecka, M., Schmidt, R., Korfei, M., Harbach, H., et al. (2007). Surfactant protein C mutations in sporadic forms of idiopathic interstitial pneumonias. Eur. Respir. J. 29, 134–137. doi: 10.1183/09031936.00034406
Mead, J., Takishima, T., and Leith, D. (1970). Stress distribution in lungs: a model of pulmonary elasticity. J. Appl. Physiol. 28, 596–608. doi: 10.1152/jappl.1970.28.5.596
Mechri, M., Epaud, R., Emond, S., Coulomb, A., Jaubert, F., Tarrant, A., et al. (2010). Surfactant protein C gene (SFTPC) mutation-associated lung disease: high-resolution computed tomography (HRCT) findings and its relation to histological analysis. Pediatr. Pulmonol. 45, 1021–1029. doi: 10.1002/ppul.21289
Menchaca, H. J., Michalek, V. N., Rohde, T. D., O’Dea, T. J., and Buchwald, H. (1998). Decreased blood oxygen diffusion in hypercholesterolemia. Surgery 124, 692–698. doi: 10.1067/MSY.1998.90944
Milos, S., Hiansen, J. Q., Yamashita, C., and Veldhuizen, R. (2016). The role of a high cholesterol diet on surfactant biophysics during acute lung injury. FASEB J. 30:1297.1. doi: 10.1096/fasebj.30.1_supplement.1297.1
Mishra, A., Chintagari, N. R., Guo, Y., Weng, T., Su, L., and Liu, L. (2011). Purinergic P2X7 receptor regulates lung surfactant secretion in a paracrine manner. J. Cell Sci. 124, 657–668. doi: 10.1242/jcs.066977
Molugu, T. R., and Brown, M. F. (2019). “Cholesterol effects on the physical properties of lipid membranes viewed by solid-state NMR spectroscopy,” in Cholesterol Modulation of Protein Function: Sterol Specificity and Indirect Mechanisms, eds A. Rosenhouse-Dantsker and A. N. Bukiya (Cham: Springer International Publishing), 99–133. doi: 10.1007/978-3-030-04278-3_5
Mora, A. L., Torres-González, E., Rojas, M., Corredor, C., Ritzenthaler, J., Xu, J., et al. (2006). Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am. J. Respir. Cell Mol. Biol. 35, 466–473. doi: 10.1165/rcmb.2006-0121OC
Morrow, M. R., Taneva, S., Simatos, G. A., Allwood, L. A., and Keough, K. M. W. (1993). Deuterium NMR studies of the effect of pulmonary surfactant SP-C on the 1,2-dipalmitoyl-sn-glycero-3-phosphocholine headgroup: a model for transbilayer peptides in surfactant and biological membranes. Biochemistry 32, 11338–11344. doi: 10.1021/bi00093a010
Mouded, M., Egea, E. E., Brown, M. J., Hanlon, S. M., Houghton, A. M. G., Tsai, L. W., et al. (2009). Epithelial cell apoptosis causes acute lung injury masquerading as emphysema. Am. J. Respir. Cell Mol. Biol. 41, 407–414. doi: 10.1165/rcmb.2008-0137OC
Myers, J. L., and Katzenstein, A. L. (1988). Ultrastructural evidence of alveolar epithelial injury in idiopathic bronchiolitis obliterans-organizing pneumonia. Am. J. Pathol. 132, 102–109.
Nakayama, M. (2018). Macrophage recognition of crystals and nanoparticles. Front. Immunol. 9:103. doi: 10.3389/fimmu.2018.00103
Nieman, G. F., Andrews, P., Satalin, J., Wilcox, K., Kollisch-Singule, M., Madden, M., et al. (2018). Acute lung injury: how to stabilize a broken lung. Crit. Care 22:136. doi: 10.1186/s13054-018-2051-2058
Nieman, G., Satalin, J., Andrews, P., Wilcox, K., Aiash, H., Baker, S., et al. (2018). Preemptive mechanical ventilation based on dynamic physiology in the alveolar microenvironment: novel considerations of time-dependent properties of the respiratory system. J. Trauma Acute Care Surg. 85, 1081–1091. doi: 10.1097/TA.0000000000002050
Nogee, L. M., Dunbar, A. E., Wert, S. E., Askin, F., Hamvas, A., and Whitsett, J. A. (2001). A mutation in the surfactant protein C gene associated with familial interstitial lung disease. New Engl. J. Med. 344, 573–579. doi: 10.1056/nejm200102223440805
Nureki, S.-I., Tomer, Y., Venosa, A., Katzen, J., Russo, S. J., Jamil, S., et al. (2018). Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Invest. 128, 4008–4024. doi: 10.1172/jci99287
Ochs, M., Nyengaard, J. R., Jung, A., Knudsen, L., Voigt, M., Wahlers, T., et al. (2004). The number of alveoli in the human lung. Am. J. Respir. Crit. Care Med. 169, 120–124. doi: 10.1164/rccm.200308-1107OC
Olmeda, B., Martínez-Calle, M., and Pérez-Gil, J. (2017). Pulmonary surfactant metabolism in the alveolar airspace: biogenesis, extracellular conversions, recycling. Ann. Anat. 209, 78–92. doi: 10.1016/j.aanat.2016.09.008
Olmeda, B., Villén, L., Cruz, A., Orellana, G., and Perez-Gil, J. (2010). Pulmonary surfactant layers accelerate O2 diffusion through the air-water interface. Biochim. Biophys. Acta Biomemb. 1798, 1281–1284. doi: 10.1016/J.BBAMEM.2010.03.008
Ono, S., Tanaka, T., Ishida, M., Kinoshita, A., Fukuoka, J., Takaki, M., et al. (2011). Surfactant protein C G100S mutation causes familial pulmonary fibrosis in Japanese kindred. Eur. Respir. J. 38, 861–869. doi: 10.1183/09031936.00143610
Orgeig, S., and Daniels, C. B. (2001). The roles of cholesterol in pulmonary surfactant: insights from comparative and evolutionary studies. Compar. Biochem. Physiol. Part A 129, 75–89. doi: 10.1016/s1095-6433(01)00307-5
Orgeig, S., Morrison, J. L., and Daniels, C. B. (2011). Evolution, development, and function of the pulmonary surfactant system in normal and perturbed environments. Compr. Physiol. 6, 363–422. doi: 10.1002/cphy.c150003
Ozyilmaz, E., Gunasti, S., Kuyucu, Y., Polat, S., Gumurdulu, D., Kuleci, S., et al. (2013). Hermansky Pudlak Syndrome and pulmonary alveolar proteinosis at the same patient: First case report in the world literature. Sarcoidos. Vasculit. Diffuse Lung Dis. 30, 217–220.
Park, J. S., Choi, Y. J., Kim, Y. T., Park, S., Chae, J.-H., Park, J. D., et al. (2018). Pediatric case report on an interstitial lung disease with a novel mutation of SFTPC successfully treated with lung transplantation. J. Korean Med. Sci. 33:e159.
Parra, E., Alcaraz, A., Cruz, A., Aguilella, V. M., and Pérez-Gil, J. (2013). Hydrophobic pulmonary surfactant proteins SP-B and SP-C induce pore formation in planar lipid membranes: evidence for proteolipid pores. Biophys. J. 104, 146–155. doi: 10.1016/J.BPJ.2012.11.014
Parra, E., Moleiro, L. H., López-Montero, I., Cruz, A., Monroy, F., and Pérez-Gil, J. (2011). A combined action of pulmonary surfactant proteins SP-B and SP-C modulates permeability and dynamics of phospholipid membranes. Biochem. J. 438, 555–564. doi: 10.1042/BJ20110681
Patel, A. S., Reigada, D., Mitchell, C. H., Bates, S. R., Margulies, S. S., and Koval, M. (2005). Paracrine stimulation of surfactant secretion by extracellular ATP in response to mechanical deformation. Am. J. Physiol. Lung. Cell Mol. Physiol. 289, L489–L496. doi: 10.1152/ajplung.00074.2005
Peca, D., Boldrini, R., Johannson, J., Shieh, J. T., Citti, A., Petrini, S., et al. (2015). Clinical and ultrastructural spectrum of diffuse lung disease associated with surfactant protein C mutations. Eur. J. Hum. Genet. 23, 1033–1041. doi: 10.1038/ejhg.2015.45
Petroulia, V., Funke, M., Zumstein, P., Berezowska, S., Ebner, L., Geiser, T., et al. (2018). Increased expiratory computed tomography density reveals possible abnormalities in radiologically preserved lung parenchyma in idiopathic pulmonary fibrosis. Invest. Radiol. 53, 45–51. doi: 10.1097/rli.0000000000000405
Plasencia, I., Cruz, A., Casals, C., and Pérez-Gil, J. (2001). Superficial disposition of the N-terminal region of the surfactant protein SP-C and the absence of specific SP-B-SP-C interactions in phospholipid bilayers. Biochem. J. 359, 651–659. doi: 10.1042/bj3590651
Plasencia, I., Rivas, L., Keough, K. M. W., Marsh, D., and Perez-Gil, J. (2004). The N-terminal segment of pulmonary surfactant lipopeptide SP-C has intrinsic propensity to interact with and perturb phospholipid bilayers. Biochem. J. 377, 183–193. doi: 10.1042/bj20030815
Possmayer, F., Nag, K., Rodriguez, K., Qanbar, R., and Schürch, S. (2001). Surface activity in vitro: role of surfactant proteins. Compar. Biochem. Physiol. Part A 129, 209–220. doi: 10.1016/S1095-6433(01)00317-318
Potter, S., Orgeig, S., Donnellan, S., and Daniels, C. B. (2007). Purifying selection drives the evolution of surfactant protein C (SP-C) independently of body temperature regulation in mammals. Compar. Biochem. Physiol. Part A 2, 165–176. doi: 10.1016/J.CBD.2007.02.003
Puttur, F., Gregory, L. G., and Lloyd, C. M. (2019). Airway macrophages as the guardians of tissue repair in the lung. Immunol. Cell Biol. 97, 246–257. doi: 10.1111/imcb.12235
Qu, L., Xue, H., Yuan, P., Zhou, L., Yao, T., Huang, Y., et al. (2009). Adenosine 5’-triphosphate stimulates the increase of TGF-beta1 in rat mesangial cells under high-glucose conditions via reactive oxygen species and ERK1/2. Acta Pharmacol. Sin. 30, 1601–1606. doi: 10.1038/aps.2009.155
Raghu, G., Weycker, D., Edelsberg, J., Bradford, W. Z., and Oster, G. (2006). Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 174, 810–816.
Raguz, M., Widomska, J., Dillon, J., Gaillard, E. R., and Subczynski, W. K. (2008). Characterization of lipid domains in reconstituted porcine lens membranes using EPR spin-labeling approaches. Biochim. Biophys. Acta Biomemb. 1778, 1079–1090. doi: 10.1016/J.BBAMEM.2008.01.024
Ramji, D. P., and Davies, T. S. (2015). Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth. Factor. Rev. 26, 673–685. doi: 10.1016/j.cytogfr.2015.04.003
Ramm, G. A., Nair, V. G., Bridle, K. R., Shepherd, R. W., and Crawford, D. H. (1998). Contribution of hepatic parenchymal and nonparenchymal cells to hepatic fibrogenesis in biliary atresia. Am. J. Pathol. 153, 527–535. doi: 10.1016/S0002-9440(10)65595-65592
Riteau, N., Gasse, P., Fauconnier, L., Gombault, A., Couegnat, M., Fick, L., et al. (2010). Extracellular ATP is a danger signal activating P2X 7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 182, 774–783. doi: 10.1164/rccm.201003-0359OC
Robert, S., Gicquel, T., Victoni, T., Valença, S., Barreto, E., Bailly-Maître, B., et al. (2016). Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci. Rep. 36:e00360. doi: 10.1042/BSR20160107
Roldan, N., Nyholm, T. K. M., Slotte, J. P., Pérez-Gil, J., and García-Álvarez, B. (2016). Effect of lung surfactant protein SP-C and SP-C-promoted membrane fragmentation on cholesterol dynamics. Biophys. J. 111, 1703–1713. doi: 10.1016/j.bpj.2016.09.016
Roldan, N., Pérez-Gil, J., Morrow, M. R., and García-Álvarez, B. (2017). Divide & conquer: surfactant protein SP-C and cholesterol modulate phase segregation in lung surfactant. Biophys. J. 113, 847–859. doi: 10.1016/j.bpj.2017.06.059
Romero, F., Shah, D., Duong, M., Penn, R. B., Fessler, M. B., Madenspacher, J., et al. (2014). A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am. J. Respir. Cell Mol. Biol. 53, 74–86. doi: 10.1165/rcmb.2014-0343OC
Rosen, D. M., and Waltz, D. A. (2005). Hydroxychloroquine and surfactant protein C deficiency. N. Engl. J. Med. 352, 207–208. doi: 10.1056/NEJM200501133520223
Rühl, N., Lopez-Rodriguez, E., Albert, K., Smith, B. J., Weaver, T. E., Ochs, M., et al. (2019). Surfactant protein B deficiency induced high surface tension: relationship between alveolar micromechanics, alveolar fluid properties and alveolar epithelial cell injury. Intern. J. Mol. Sci. 20:243. doi: 10.3390/ijms20174243
Ruwisch, J., Sehlmeyer, K., Roldan, N., Garcia-Alvarez, B., Perez-Gil, J., Weaver, T. E., et al. (2020). Air space distension precedes spontaneous fibrotic remodeling and impaired cholesterol metabolism in the absence of surfactant protein C. Am. J. Respir. Cell Mol. Biol. 62, 466–478. doi: 10.1165/rcmb.2019-0358OC
Saito, A., Horie, M., and Nagase, T. (2018). TGF-β signaling in lung health and disease. Intern. J. Mol. Sci. 19:2460. doi: 10.3390/ijms19082460
Saito, K., Tanaka, N., Ikari, J., Suzuki, M., Anazawa, R., Abe, M., et al. (2019). Comprehensive lipid profiling of bleomycin-induced lung injury. J. Appl. Toxicol. 39, 658–671. doi: 10.1002/jat.3758
Salerno, T., Peca, D., Menchini, L., Schiavino, A., Boldrini, R., Esposito, F., et al. (2016). Surfactant Protein C-associated interstitial lung disease; three different phenotypes of the same SFTPC mutation. Itali. J. Pediatr. 42:23.
Schmid-Kotsas, A., Gross, H. J., Menke, A., Weidenbach, H., Adler, G., Siech, M., et al. (1999). Lipopolysaccharide-activated macrophages stimulate the synthesis of collagen type I and C-fibronectin in cultured pancreatic stellate cells. Am. J. Pathol. 155, 1749–1758. doi: 10.1016/S0002-9440(10)65490-65499
Schmidt, R., Ruppert, C., Markart, P., Lübke, N., Ermert, L., Weissmann, N., et al. (2004). Changes in pulmonary surfactant function and composition in bleomycin-induced pneumonitis and fibrosis. Toxicol. Appl. Pharmacol. 195, 218–231. doi: 10.1016/j.taap.2003.11.011
Setoguchi, Y., Ikeda, T., and Fukuchi, Y. (2006). Clinical features and genetic analysis of surfactant protein C in adult-onset familial interstitial pneumonia. Respirology 11(Suppl.), S41–S45. doi: 10.1111/j.1440-1843.2006.00807.x
Sheppard, D. (2015). Epithelial-mesenchymal interactions in fibrosis and repair: transforming growth factor-β activation by epithelial cells and fibroblasts. Ann. Am. Thorac. Soc. 12, S21–S23. doi: 10.1513/AnnalsATS.201406-245MG
Sisson, T. H., Mendez, M., Choi, K., Subbotina, N., Courey, A., Cunningham, A., et al. (2010). Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 181, 254–263.
Smigiel, K. S., and Parks, W. C. (2018). Macrophages, wound healing, and fibrosis: recent insights. Curr. Rheumatol. Rep. 20:725. doi: 10.1007/s11926-018-0725-725
Smith, B. J., Bartolak-Suki, E., Suki, B., Roy, G. S., Hamlington, K. L., Charlebois, C. M., et al. (2017). Linking ventilator injury-induced leak across the blood-gas barrier to derangements in murine lung function. Front. Physiol. 8:466. doi: 10.3389/fphys.2017.00466
Smith, B. J., Grant, K. A., and Bates, J. H. T. (2013). Linking the development of ventilator-induced injury to mechanical function in the lung. Ann. Biomed. Eng. 41, 527–536. doi: 10.1007/s10439-012-0693-692
Snijder, J., Peraza, J., Padilla, M., Capaccione, K., and Salvatore, M. M. (2019). Pulmonary fibrosis: a disease of alveolar collapse and collagen deposition. Expert Rev. Respir. Med. 13, 615–619. doi: 10.1080/17476348.2019.1623028
So, A., de Smedt, T., Revaz, S., and Tschopp, J. (2007). A pilot study of IL-1 inhibition by anakinra in acute gout. Arthrit. Res. Ther. 9, R28–R28. doi: 10.1186/ar2143
Soraisham, A. S., Tierney, A. J., and Amin, H. J. (2006). Neonatal respiratory failure associated with mutation in the surfactant protein C gene. J. Perinatol. 26, 67–70. doi: 10.1038/sj.jp.7211417
Stevens, P. A., Pettenazzo, A., Brasch, F., Mulugeta, S., Baritussio, A., Ochs, M., et al. (2005). Nonspecific interstitial pneumonia, alveolar proteinosis, and abnormal proprotein trafficking resulting from a spontaneous mutation in the surfactant protein C gene. Pediatr. Res. 57:89. doi: 10.1203/01.pdr.0000147567.02473.5a
Swanson, K. V., Deng, M., and Ting, J. P.-Y. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. doi: 10.1038/s41577-019-0165-160
Tabuchi, A., Nickles, H. T., Kim, M., Semple, J. W., Koch, E., Brochard, L., et al. (2016). Acute lung injury causes asynchronous alveolar ventilation that can be corrected by individual sighs. Am. J. Respir. Crit. Care Med. 193, 396–406. doi: 10.1164/rccm.201505-0901OC
Terèelj, M., Salobir, B., Simcic, S., Wraber, B., Zupancic, M., and Rylander, R. (2009). Chitotriosidase activity in sarcoidosis and some other pulmonary diseases. Scand. J. Clin. Lab. Invest. 69, 575–578. doi: 10.1080/00365510902829362
Thomas, A. Q., Lane, K., Phillips, J., Prince, M., Markin, C., Speer, M., et al. (2002). Heterozygosity for a surfactant protein c gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 165, 1322–1328. doi: 10.1164/rccm.200112-123OC
Thomassen, M. J., Barna, B. P., Malur, A. G., Bonfield, T. L., Farver, C. F., Malur, A., et al. (2007). ABCG1 is deficient in alveolar macrophages of GM-CSF knockout mice and patients with pulmonary alveolar proteinosis. J. Lipid Res. 48, 2762–2768. doi: 10.1194/jlr.P700022-JLR200
Thompson, R. W., Pesce, J. T., Ramalingam, T., Wilson, M. S., White, S., Cheever, A. W., et al. (2008). Cationic amino acid transporter-2 regulates immunity by modulating arginase activity. PLoS Pathog. 4:e1000023. doi: 10.1371/journal.ppat.1000023
Thouvenin, G., Taam, R. A., Flamein, F., Guillot, L., le Bourgeois, M., Reix, P., et al. (2010). Characteristics of disorders associated with genetic mutations of surfactant protein C. Arch. Dis. Childhood 95, 449–454. doi: 10.1136/adc.2009.171553
Todd, N. W., Atamas, S. P., Luzina, I. G., and Galvin, J. R. (2015). Permanent alveolar collapse is the predominant mechanism in idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 9, 411–418. doi: 10.1586/17476348.2015.1067609
Torday, J., and Rehan, V. (2011). Neutral lipid trafficking regulates alveolar type II cell surfactant phospholipid and surfactant protein expression. Exper. Lung Res. 37, 376–386. doi: 10.3109/01902148.2011.580903
Tredano, M., Griese, M., Brasch, F., Schumacher, S., Blic, J., de Marque, S., et al. (2004). Mutation of SFTPC in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease. Am. J. Med. Genet. Part A 126A, 18–26. doi: 10.1002/ajmg.a.20670
Turcu, S., Ashton, E., Jenkins, L., Gupta, A., and Mok, Q. (2013). Genetic testing in children with surfactant dysfunction. Arch. Dis. Child. 98, 490–495. doi: 10.1136/archdischild-2012-303166
Turley, S. D., Andersen, J. M., and Dietschy, J. M. (1981). Rates of sterol synthesis and uptake in the major organs of the rat in vivo. J. Lipid. Res. 22, 551–569.
van Hoorn, J., Brouwers, A., Griese, M., and Kramer, B. (2014). Successful weaning from mechanical ventilation in a patient with surfactant protein C deficiency presenting with severe neonatal respiratory distress. BMJ Case Rep. 2014:203053. doi: 10.1136/bcr-2013-203053
van Meer, G., Voelker, D. R., and Feigenson, G. W. (2008). Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. doi: 10.1038/nrm2330
van Moorsel, C. H. M., van Oosterhout, M. F. M., Barlo, N. P., de Jong, P. A., van der Vis, J. J., Ruven, H. J. T., et al. (2010). Surfactant protein c mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 182, 1419–1425. doi: 10.1164/rccm.200906-0953oc
Vazquez De Anda, G. F., Gommers, D., Verbrugge, S. J. C., de Jaegere, A., and Lachmann, B. (2000). Mechanical ventilation with high positive end-expiratory pressure and small driving pressure amplitude is as effective as high-frequency oscillatory ventilation to preserve the function of exogenous surfactant in lung-lavaged rats. Crit. Care Med. 28, 2921–2925. doi: 10.1097/00003246-200008000-00039
Veldhuizen, R. A., Ito, Y., Marcou, J., Yao, L. J., McCaig, L., and Lewis, J. F. (1997). Effects of lung injury on pulmonary surfactant aggregate conversion in vivo and in vitro. Am. J. Physiol. 272, L872–L878. doi: 10.1152/ajplung.1997.272.5.L872
Veldhuizen, R. A., Marcou, J., Yao, L. J., McCaig, L., Ito, Y., and Lewis, J. F. (1996). Alveolar surfactant aggregate conversion in ventilated normal and injured rabbits. Am. J. Physiol. 270, L152–L158. doi: 10.1152/ajplung.1996.270.1.L152
Venosa, A., Katzen, J., Tomer, Y., Kopp, M., Jamil, S., Russo, S. J., et al. (2019). Epithelial expression of an interstitial lung disease-associated mutation in surfactant protein-c modulates recruitment and activation of key myeloid cell populations in mice. J. Immunol. 202, 2760–2771. doi: 10.4049/jimmunol.1900039
Vilaysane, A., Chun, J., Seamone, M. E., Wang, W., Chin, R., Hirota, S., et al. (2010). The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 21, 1732–1744. doi: 10.1681/ASN.2010020143
Vockeroth, D., Gunasekara, L., Amrein, M., Possmayer, F., Lewis, J. F., and Veldhuizen, R. A. W. (2009). Role of cholesterol in the biophysical dysfunction of surfactant in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L117–L125. doi: 10.1152/ajplung.00218.2009
Voyno-Yasenetskaya, T. A., Dobbs, L. G., and Williams, M. C. (1991). Regulation of ATP-dependent surfactant secretion and activation of second-messenger systems in alveolar type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 261, 105–109. doi: 10.1152/ajpheart.1991.261.4.105
Vyshedskiy, A., Alhashem, R. M., Paciej, R., Ebril, M., Rudman, I., Fredberg, J. J., et al. (2009). Mechanism of inspiratory and expiratory crackles. Chest 135, 156–164. doi: 10.1378/chest.07-1562
Wambach, J. A., Yang, P., Wegner, D. J., An, P., Hackett, B. P., Cole, F. S., et al. (2010). Surfactant Protein-C promoter variants associated with neonatal respiratory distress syndrome reduce transcription. Pediatr. Res. 68:216. doi: 10.1203/pdr.0b013e3181eb5d68
Wang, C.-M., Chang, Y.-Y., and Sun, S. H. (2003). Activation of P2X7 purinoceptor-stimulated TGF-beta 1 mRNA expression involves PKC/MAPK signalling pathway in a rat brain-derived type-2 astrocyte cell line, RBA-2. Cell. Signal. 15, 1129–1137. doi: 10.1016/s0898-6568(03)00112-118
Wang, L., Cai, P., Galla, H.-J., He, H., Flach, C. R., and Mendelsohn, R. (2005). Monolayer-multilayer transitions in a lung surfactant model: IR reflection-absorption spectroscopy and atomic force microscopy. Eur. Biophys. J. 34, 243–254. doi: 10.1007/s00249-004-0446-448
Weibel, E. R., and Gil, J. (1968). Electron microscopic demonstration of an extracellular duplex lining layer of alveoli. Respir. Physiol. 4, 42–57. doi: 10.1016/0034-5687(68)90006-90006
Wennberg, C. L., van der Spoel, D., and Hub, J. S. (2012). Large influence of cholesterol on solute partitioning into lipid membranes. J. Am. Chem. Soc. 134, 5351–5361. doi: 10.1021/ja211929h
Widomska, J., Subczynski, W. K., Mainali, L., and Raguz, M. (2017). Cholesterol bilayer domains in the eye lens health: a review. Cell Biochem. Biophys. 75, 387–398. doi: 10.1007/s12013-017-0812-817
Wree, A., McGeough, M. D., Peña, C. A., Schlattjan, M., Li, H., Inzaugarat, M. E., et al. (2014). NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 92, 1069–1082. doi: 10.1007/s00109-014-1170-1171
Wynn, T. A., and Barron, L. (2010). Macrophages: master regulators of inflammation and fibrosis. Semin. Liver Dis. 30, 245–257. doi: 10.1055/s-0030-1255354
Wynn, T. A., Chawla, A., and Pollard, J. W. (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. doi: 10.1038/nature12034
Xing, Z., Tremblay, G. M., Sime, P. J., and Gauldie, J. (1997). Overexpression of granulocyte-macrophage colony-stimulating factor induces pulmonary granulation tissue formation and fibrosis by induction of transforming growth factor-β1 and myofibroblast accumulation. Am. J. Pathol. 150, 59–66.
Xu, J.-F., Washko, G. R., Nakahira, K., Hatabu, H., Patel, A. S., Fernandez, I. E., et al. (2012). Statins and pulmonary fibrosis: the potential role of NLRP3 inflammasome activation. Am. J. Respir. Crit. Care Med. 185, 547–556. doi: 10.1164/rccm.201108-1574OC
Yao, X., Gordon, E. M., Figueroa, D. M., Barochia, A. V., and Levine, S. J. (2016). Emerging Roles of Apolipoprotein E and apolipoprotein A-I in the pathogenesis and treatment of lung disease. Am. J. Respir. Cell Mol. Biol. 55, 159–169. doi: 10.1165/rcmb.2016-0060TR
Zhang, Y., Lee, T. C., Guillemin, B., Yu, M. C., and Rom, W. N. (1993). Enhanced IL-1 beta and tumor necrosis factor-alpha release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J. Immunol. 150, 4188–4196.
Zhou, Y., Huang, X., Hecker, L., Kurundkar, D., Kurundkar, A., Liu, H., et al. (2013). Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Invest. 123, 1096–1108. doi: 10.1172/JCI66700
Zocher, F., van der Spoel, D., Pohl, P., and Hub, J. S. (2013). Local partition coefficients govern solute permeability of cholesterol-containing membranes. Biophys. J. 105, 2760–2770. doi: 10.1016/J.BPJ.2013.11.003
Zuniga-Hertz, J. P., and Patel, H. H. (2019). The evolution of cholesterol-rich membrane in oxygen adaption: the respiratory system as a model. Front. Physiol. 10:1340. doi: 10.3389/fphys.2019.01340
Keywords: surfactant protein C, pulmonary fibrosis, alveolar dynamics, lipid metabolism, alveolar macrophages, cholesterol, metaflammation
Citation: Sehlmeyer K, Ruwisch J, Roldan N and Lopez-Rodriguez E (2020) Alveolar Dynamics and Beyond – The Importance of Surfactant Protein C and Cholesterol in Lung Homeostasis and Fibrosis. Front. Physiol. 11:386. doi: 10.3389/fphys.2020.00386
Received: 31 January 2020; Accepted: 30 March 2020;
Published: 05 May 2020.
Edited by:
Yu Ru Kou, National Yang-Ming University, TaiwanReviewed by:
Bela Suki, Boston University, United StatesCopyright © 2020 Sehlmeyer, Ruwisch, Roldan and Lopez-Rodriguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Lopez-Rodriguez, ZWxlbmEubG9wZXotcm9kcmlndWV6QGNoYXJpdGUuZGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.