 Chansik Hong1†
Chansik Hong1† Byeongseok Jeong1†
Byeongseok Jeong1† Hyung Joon Park1†
Hyung Joon Park1† Ji Yeon Chung2Jung Eun Lee3Jinsung Kim3Young-Cheul Shin4
Ji Yeon Chung2Jung Eun Lee3Jinsung Kim3Young-Cheul Shin4 Insuk So3*
Insuk So3*- 1Department of Physiology, Chosun University School of Medicine, Gwangju, South Korea
- 2Department of Neurology, Chosun University School of Medicine, Gwangju, South Korea
- 3Department of Physiology and Institute of Dermatological Science, Seoul National University College of Medicine, Seoul, South Korea
- 4Department of Cell Biology, Harvard Medical School, Boston, MA, United States
The development of treatment for neurodegenerative diseases (NDs) such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis is facing medical challenges due to the increasingly aging population. However, some pharmaceutical companies have ceased the development of therapeutics for NDs, and no new treatments for NDs have been established during the last decade. The relationship between ND pathogenesis and risk factors has not been completely elucidated. Herein, we review the potential involvement of transient receptor potential (TRP) channels in NDs, where oxidative stress and disrupted Ca2+ homeostasis consequently lead to neuronal apoptosis. Reactive oxygen species (ROS) -sensitive TRP channels can be key risk factors as polymodal sensors, since progressive late onset with secondary pathological damage after initial toxic insult is one of the typical characteristics of NDs. Recent evidence indicates that the dysregulation of TRP channels is a missing link between disruption of Ca2+ homeostasis and neuronal loss in NDs. In this review, we discuss the latest findings regarding TRP channels to provide insights into the research and quests for alternative therapeutic candidates for NDs. As the structures of TRP channels have recently been revealed by cryo-electron microscopy, it is necessary to develop new TRP channel antagonists and reevaluate existing drugs.
Introduction
Neurodegenerative disorders (NDs) are one of the most devastating types of chronic diseases and lead to a significant social and medical burden on society. With the growing elderly population, the number of patients with NDs is also increasing. Although many pharmaceutical companies are struggling to develop novel therapeutics for neurological diseases, some of the world’s leading pharmaceutical companies have declared their abandonment of the development of therapeutics for NDs. Alzheimer’s disease (AD) is the most common ND, which accounts for 60–70% of all dementia (Association, 2016). Nevertheless, no new treatments for AD have been developed in over a decade. Some of the reasons for the difficulty in treating NDs are the combination of complex causative factors and irreversible structural and functional damage of neurons.
Neurodegenerative disorders are typically progressive, late-onset disorders, and aging is the greatest risk factor (Wakabayashi et al., 2014). In addition, genetic and environmental factors not only contribute to their pathogenesis independently but also interact with each other to increase their effects. The pathogenesis of NDs involves an initial toxic insult and consequences of the secondary pathological damage. The most primary causative hypothesis of AD is the intraneuronal accumulation of amyloid-beta (Aβ) and hyperphosphorylated tau protein (Iqbal et al., 2010; Murphy and LeVine, 2010). Parkinson’s disease (PD) is caused by the degeneration of dopaminergic neurons in the substantia nigra par compacta (SNpc), with subsequent dopamine deficiency (Michel et al., 2013; Kalia and Lang, 2015). A type of hereditary ND, Huntington’s disease (HD) is an autosomal dominant disorder caused by CAG repeat expansion within the Huntington (HTT) gene (Kremer et al., 1994). The third common ND after AD and PD is amyotrophic lateral sclerosis (ALS) that is characterized by the deterioration of motor neurons.
Neurodegenerative disorders such as AD, PD, HD, and ALS are distinguished by clinical symptoms and specific neuronal sites with distinct pathology. However, apparent clinical symptoms are manifested only after extensive pathological damage, with significant neuronal and synaptic loss. Eventually, the contribution of individual insults reaches a common end state, which causes severe impairments in the function and plasticity of neuronal and glial cells (Rasband, 2016). Over the past few decades, there has been considerable effort to understand the pathogenesis of NDs. To date, a number of studies have reported that oxidative stress, ER (endoplasmic reticulum) stress, abnormal Ca2+ homeostasis, protein misfolding, aggregation, neuroinflammation, and mitochondrial dysfunction are highly related to neuronal damage. The relationship between them, however, has not been completely elucidated. Besides, even though Aβ is still a compelling candidate in the pathogenesis of AD, the latest experiments raise doubts about the Aβ hypothesis and Aβ -based drug development for AD (Du et al., 2018). Therefore, it is necessary to find the missing links amongst the risk factors of NDs and to discover new therapeutic targets based on novel mechanisms.
Ion channels are key determinants of brain function, since the physiological function of neurons is to carry information or impulses via electrical signals (action potentials) to communicate with each other at synapses. Thus, neurological channelopathies have been identified mainly in voltage-gated and ligand-gated channels or receptors that result from genetically determined defects in their function. However, based on patients with progressive NDs with adulthood manifestations, we have focused on the age-related susceptibility to environmental toxins and chemicals. Recently, emerging evidence has indicated that transient receptor potential (TRP) channels, ubiquitously expressed throughout the brain (Sawamura et al., 2017), play a significant role in the regulation of physiological functions, as well as in reactive oxygen species (RO)-related human diseases. Based on the polymodal activation of TRP channels acting as cellular sensors, many researchers are investigating their activation mechanisms (Takada et al., 2013). Here, we review the potential involvement of TRP channels in NDs, where oxidative stress and disrupted Ca2+ homeostasis have been characterized with respect to pathological consequences in neuronal apoptosis. Second, we discuss the latest findings in the field of TRP to provide insight into the research and quest for alternative therapeutic candidates for the treatment of NDs.
The Critical Role of Ca2+ in the Pathogenesis of Neurodegenerative Diseases
Ca2+ homeostasis is crucial to the normal physiological functions of neurons, such as neuronal survival, growth, and differentiation. Hence, long-lasting Ca2+ dyshomeostasis can eventually lead to neuronal loss. Accumulated evidence strongly implicates that abnormal Ca2+ levels stimulate dysregulation of intracellular signaling, which consequently induces neuronal cell death (Barnham et al., 2004). Therefore, disruption of Ca2+ homeostasis in neuronal cells leads to ROS generation and ATP depletion, following the mitochondrial dysfunction in NDs such as AD, PD, HD, and ALS. Interestingly, a close correlation between the increase in [Ca2+]i and other pathogenic mechanisms has been reported, such as Aβ deposition (Demuro et al., 2010), imbalance between ROS and antioxidant function (Gorlach et al., 2015), and mitochondrial dysfunction (Contreras et al., 2010; Pivovarova and Andrews, 2010).
Some reports have shown bidirectional crosstalk between amyloid pathology and the Ca2+ pathway. Most studies reported that Aβ increases intracellular Ca2+ levels by inducing ER Ca2+ depletion (Suen et al., 2003; Abramov et al., 2004b; Ferreiro et al., 2008). Abramov et al. (2004a) identified that Aβ causes Ca2+-dependent oxidative stress by the activation of NADPH oxidase in astrocytes and that the reduced antioxidant activity induces neuronal death. Recently, Calvo-Rodriguez et al. (2019) suggested that Aβ oligomers exacerbate Ca2+ remodeling from ER to mitochondria in aged neurons but not in young neurons. Conversely, Itkin et al. (2011) argued that Ca2+ stimulates the formation of Aβ oligomers, leading to neuronal toxicity in AD.
The imbalance between ROS production and antioxidant defenses results in the excessive accumulation of ROS and oxidative stress. Since aged neurons with low antioxidant capacity are more vulnerable to oxidative insults (Chen et al., 2012), ROS overproduction can chronically lead to irreversible oxidation (Ivanova et al., 2016). Oxidative stress also causes mitochondrial dysfunction, which itself aggravates ROS generation. Moreover, the opening of the mitochondrial permeability transition pore (mPTP) and the release of cytochrome c into cytoplasm activate pro-apoptotic caspases (Guo et al., 2013). Oxidative stress and mitochondrial dysfunction are well known to be related to an increase in cytosolic Ca2+ levels that underlies the pathogenesis of AD (Cenini and Voos, 2019), PD (Blesa et al., 2015), HD (Zheng et al., 2018), and ALS (Carri et al., 2015). Interestingly, the mitochondrial metabolic state can affect the Mg2+ concentration of both the matrix and the cytoplasm, where Mg2+ interferes with mitochondrial Ca2+ transport and mitochondrial ATP generation (Llorente-Folch et al., 2015). Since the efficient removal of [Ca2+]i requires ATP, impairment of mitochondrial ATP generation prevents Ca2 + pumps from operating both in the plasma membrane and in the ER (Ott et al., 2007). Thus, dysregulation of Ca2+ signaling is one of the key processes in early stage neuronal loss. In spite of its significance, the mediator of such aberrant Ca2+ increase and its source are not fully understood. To test the effectiveness of preventing Ca2+ overload for ND therapy, a variety of appropriate channel candidates should be further examined by developing channel-specific drugs for new channel targets. Last but not least, experimental data on existing drugs also need to be reevaluated.
Oxidative stress can directly modulate the gating properties of ion channel proteins. Pathological mechanisms underlying the dysregulation of ion channels by oxidation have been previously proposed in a variety of diseases, especially cancer (Reczek and Chandel, 2018) and NDs (Gorlach et al., 2015). Under normal conditions, defensive antioxidants can protect or repair the damage caused by oxidation. However, the target proteins are retained in their oxidized forms and are activated as long as the antioxidant activity is reduced. To date, several studies have shown that oxidative stress is involved in the modulation of activities of voltage-gated Ca2+ (Gorlach et al., 2015; Ramirez et al., 2016), Na+, and K+ (Sesti et al., 2010) channels and ligand-gated receptors such as NMDA (Kamat et al., 2016), AMPA (Joshi et al., 2015), GABA (Bradley and Steinert, 2016), and RyR (Zima and Mazurek, 2016). However, there is limited evidence that directly identifies the oxidative modification of a channel protein based on molecular mechanisms.
Since TRP channels are non-selective, Ca2+-permeable channels that can be opened at resting membrane potential in response to various stimuli, we focused on TRP channels. The activation of TRP channels consequently changes membrane depolarization toward the action potential threshold. When TRP channels open, they allow sodium and calcium into the cytoplasm, which subsequently triggers the opening of voltage-dependent Ca2+ channels. This is why TRP channels are upstream risk factors, ahead of voltage-dependent channels (Numata et al., 2011). Therefore, the hyperactivation of TRP channels is responsible for neuronal excitotoxicity, which is closely associated with NDs. In the following section, we will address the physiological and pathological roles of TRP channels in neurons through the recent studies related to TRP channels and our study of the TRPC5 channel.
The Physiological and Pathological Role of TRP Channels in Neurons
As mentioned above, TRP channels are widely expressed in almost every mammalian cell, predominantly in the brain. TRP channels can be activated by diverse stimuli ranging from temperature, mechanical or osmotic stress, chemical compounds, and redox modification (Sawamura et al., 2017; Samanta et al., 2018). Based on sequence homology, the TRP superfamily is divided into six subfamilies in mammals: TRPC (classical or canonical; seven sub-members), TRPM (melastatin; eight sub-members), TRPV (vanilloid; six sub-members), TRPA (ankyrin; one sub-member), TRPP (polycystin; three sub-members), and TRPML (mucolipin; three sub-members) (Figure 1). Notably, most TRP channels (except TRPM4 and TRPM5) are non-selective channels with consistent Ca2+ permeability (Guinamard et al., 2011). TRP channels are tetrameric protein complexes that can be assembled into homomeric or heteromeric channels, either with the same subfamily members or with the other subfamily members. Thus, when TRP channels assemble with different subunits, further heterogeneity diversifies their functions. In addition to the physiological roles of TRP channels in neurons, a number of studies regarding the pathological functions have been reported. Intracellular Ca2+ influx through TRP channels is involved either in neuronal survival or death and is discussed with respect to the different TRP channel families in the following sections (Bollimuntha et al., 2011).
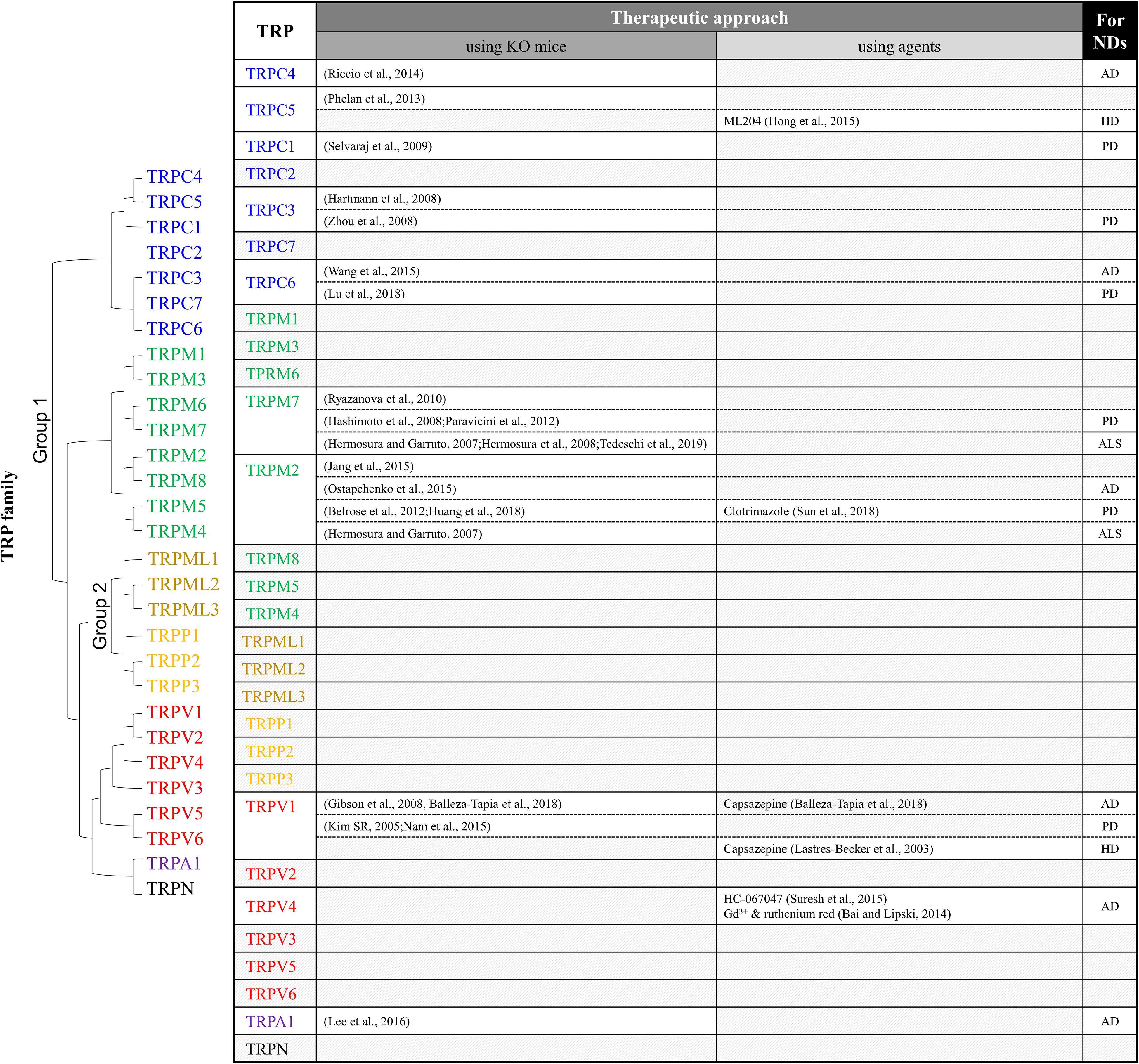
Figure 1. Summary of TRP studies using knockout mice or antagonists to investigate therapeutic targets of neurodegenerative diseases. ND, neurodegenerative diseases; AD, Alzheimer’s disease; PD, Parkinson’s disease; HD, Huntington’s disease; ALS, amyotrophic lateral sclerosis.
TRPC (Classical or Canonical)
TRPC was the first group of TRPs to be discovered in a mammal (Wes et al., 1995), and it shows the highest amino acid similarity to the Drosophila TRP channel. The TRPC subfamily is divided into seven subtypes, namely, TRPC1–7. Depending on amino acid similarities, the subtypes are divided into four groups: TRPC1, 2, 3/6/7, 4/5 (Venkatachalam and Montell, 2007). There is still disagreement over the mechanism of action of TRPC; TRPC has been reported to be involved in ion permeation as receptor operated channel (ROC) or to influence intracellular mechanisms of store-operated calcium entry (SOCE) (Vazquez et al., 2004). Recently, as the TRPC channel has been found to have regulation, structure, and novel small molecular probes, research is being actively conducted on it as a therapeutic target for various diseases (Wang et al., 2020).
TRPC1
In particular, there has been debate about the role or opening mechanisms of TRPC1. Initially, TRPC1 was claimed to take the role of a SOCE in regulating Orai1-mediated Ca2+ entry (Ambudkar et al., 2017). Consistent with this claim, the role of TRPC1 in AD has been reported by Linde et al. (2011). Knock-down (KD) of the amyloid precursor protein (APP) gene decreased store-operated Ca2+ channel-mediated Ca2+ entry and expression of TRPC1 and Orai1 in cultured astrocytes. However, overexpression of APP in TG5469 did not alter TRPC1/4/5 and stored Ca2+ level in astrocytes. In SH-SY5Y human neuroblastoma cells, TRPC1 has been reported to reduce expression levels by MPP+ (Bollimuntha et al., 2006). Activation of TRPC1 by TRPC1 overexpression or by ER depletion using thapsigargin (TG) ameliorates neurotoxicity. Selvaraj et al. (2009, 2012) showed that Ca2+ entry through the activation of store-operated channels (SOC) is important for the survival of dopaminergic neurons (Figure 2C). In the MPTP-induced PD model, TRPC1 expression was suppressed and induced the death of dopaminergic neurons in the substantia nigra. The authors suggested that the cause was reduced interaction with the SOCE modulator stromal interaction molecule 1 (STIM1) and decreased Ca2+ entry into the cell. However, our recent study showed that TRPC1 functions as a negative regulator of TRPC4 and TRPC5 (Figure 2C; Kim et al., 2019). Heterodimers of TRPC1/4 and TRPC1/5 suppressed inward current, which may reduce Ca2+ influx and Ca2+-dependent apoptosis in neurons. We identified that the expression level of endogenous TRPC1 in striatal cells of the HD model was decreased compared to wild-type cells, indicating that HD cells could be more susceptible to oxidative stress due to the activity of the dominant homomeric TRPC5 (Figure 2D; Hong et al., 2015).
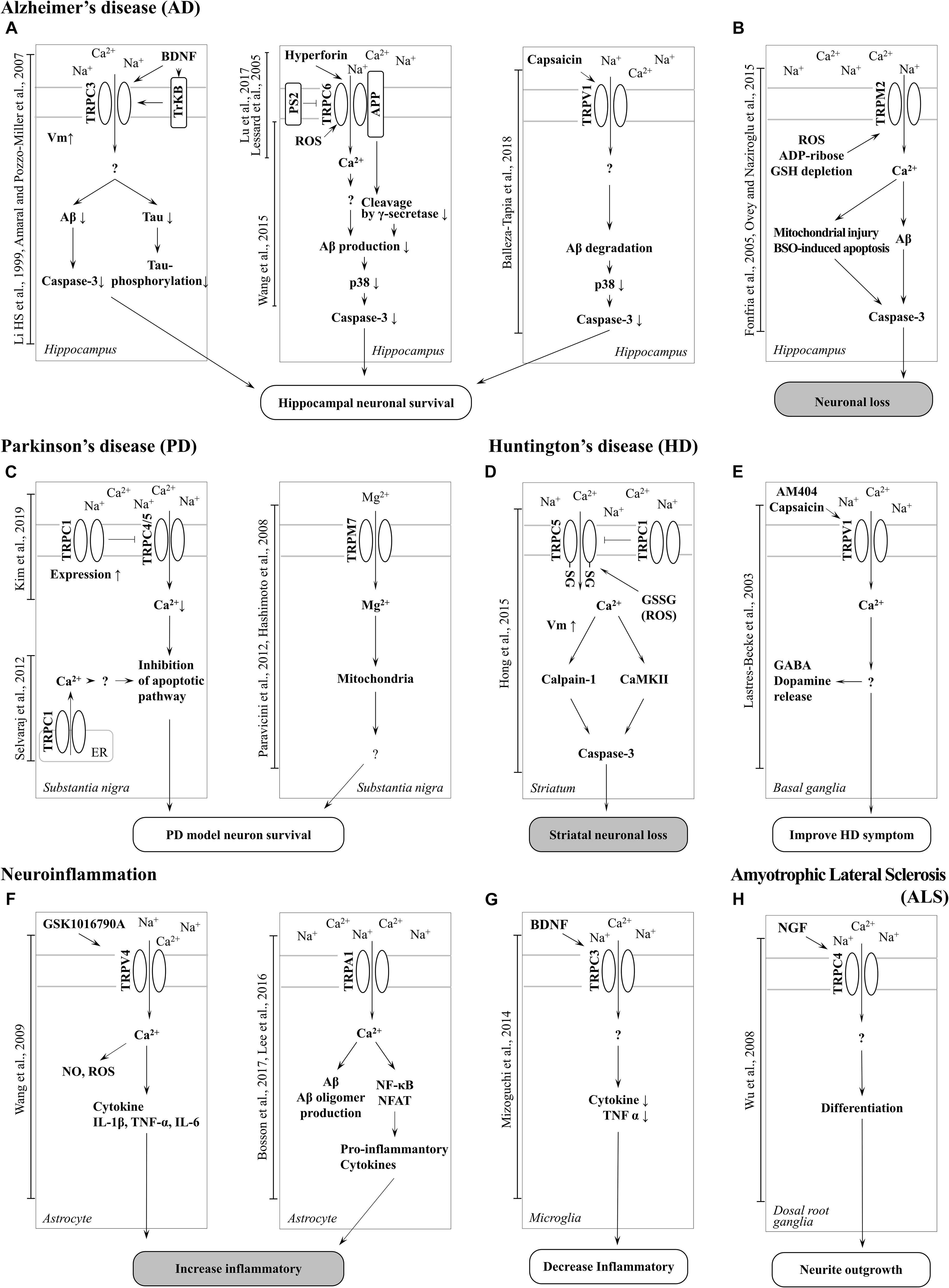
Figure 2. Schematic of TRP channel-mediated mechanisms in neurodegenerative diseases. (A) Activation of TRPC3, TRPC6 and TRPV1 channel increase neuronal survival in AD. (B) Neuronal loss can be induced by Aβ toxicity, ROS generation, and mitochondrial damage resulting from TRPM2 channel-mediated Ca2 + entry in AD. TRPA1 is also involved in neuroinflammation in AD. (C) Inhibition of TRPC4/5 by TRPC1 contribute to inhibition of apoptotic pathways and TRPM7-mediated Mg2 + influx is involved in neuronal survival in PD. (D) Increased activity of TRPC5 by oxidative stress induces striatal neuronal loss via Ca2 +-dependent pathways in HD. (E) Activation of TRPV1 by an agonist improves HD symptoms. (F) Activation of TRPV4 and TRPA1 induces a proinflammatory response in astrocytes (G) whereas upregulation of surface TRPC3 induced by BDNF regulates microglial functions and reduces inflammation. (H) Upregulation of TRPC4 promotes neurite outgrowth and differentiation in DRG (GTEx Consortium, 2013).
TRPC3
The important roles of TRPC3 in the hippocampus have been implicated in ND more than in other TRP channels with higher expression levels (Neuner et al., 2015). TRPC3 notably contributes to the maintenance of Ca2+ homeostasis and cell growth, such as differentiation and proliferation. In a study conducted by Wu et al. (2004) it was reported that switching proliferation to differentiation is related to TRPC3-induced Ca2+ influx and TRPC3-mediated SOCE in the H19-7 hippocampal cell line. Under cell differentiation conditions, TRPC3 expression and TRPC3-induced SOCE levels were increased. The differentiation was blocked by siRNA KD of TRPC3. In addition, TRPC3 is indirectly activated by BDNF. In a study conducted by Li et al. (1999) TRPC3 was activated by neurotrophin receptor TrkB, which is affected by BDNF. A non-selective cationic current was observed in CA1 pyramidal neurons treated with BDNF, although the current was inhibited by siRNA-mediated TRPC3 KD and TrkB-lgG (Amaral and Pozzo-Miller, 2007). This study suggests that BDNF-induced membrane current is due to stimulation of TRPC3 by TrkB (Figure 2A).
In a recent study by Mizoguchi et al. (2014) TRPC3 was found to be involved in the function of microglia, such as the release of cytokines and nitric oxide (NO). Treatment with BDNF rapidly increased the surface expression levels of TRPC3 in rodent microglial cells. In addition, pre-treatment with BDNF inhibited the release NO-induced tumor necrosis factor α (TNFα), which was rescued by treatment with TRPC3 inhibitor. This report suggests that Ca2+ influx and concentration maintenance by TRPC3 plays an important role in the improvement of NDs (Figure 2G).
Another characteristic of TRPC3 related to neurodegeneration is directly activated by oxidative stress. The change in Ca2+ influx by TRPC3 is associated with neuronal cell death (Selvaraj et al., 2010). Treatment with tertiary butyl hydroperoxide (tBHP) increased the Na+ current in HEK293T cells overexpressing TRPC3. Further, Rosker et al. (2004) reported the increase of Na+ influx by TRPC3-regulated Ca2+ influx in overexpressed HE293T cells. When the Na+ concentration of extracellular solution decreased to 5 mM, Ca2+ influx was increased by TRPC3 agonist. In addition, treatment of the inhibitor of the Na+/Ca2+ exchanger strongly inhibited Ca2+ influx but Na+ did not. This suggests that Ca2+ influx by TRPC3 is accompanied by Na+/Ca2+ exchange. Pesticides, such as rotenone and paraquat, are neurotoxins that induce PD by increasing intracellular oxidative stress. Moreover, both of these pesticides induce the loss of dopaminergic neurons in the SNpc. In a recent study by Roedding et al., chronic treatment of rotenone and paraquat dose-dependently reduced expression levels of TRPC3 and TRPC3-mediated Ca2+ influx in primary rat cortical neurons and astrocytes. In another report, OAG-induced Ca2+ transients were inhibited in MPP+-treated murine striatal astrocytes, and the same was observed in HEK293 cells overexpressing TRPC3 (Streifel et al., 2014). These studies suggest that an increased Na+ influx of TRPC3 due to oxidative stress may reduce Ca2+ influx and contribute to the treatment of PD. Inhibition of TRPC3 was shown to depolarize GABA neurons in the substantia nigra pars reticulate (SNpr), which are associated with parkinsonism (Zhou et al., 2008). In summary, AD symptoms are recovered by TRPC3 activation. In a previous study, BDNF protected neurons from the neurotoxicity of Aβ and tau (Arancibia et al., 2008; Jiao et al., 2016). TRPC3 activation may decrease Ca2+ concentration due to a change in the influx ratio of Na+/Ca2+. Insufficient Ca2+ concentration by TRPC3 activation may be involved in BDNF-induced interference of Aβ plaque formation and tau hyperphosphorylation (Figure 2A).
TRPC4 and TRPC5
TRPC4 and TRPC5, which share similar amino acid sequence identity, have important roles in the neuron (Tables 1, 2), in particular with respect to memory in the hippocampus. TRPC5 regulates synaptic plasticity by changing the presynaptic Ca2+ homeostasis of hippocampal neurons (Schwarz et al., 2019). TRPC1/4/5 knockout (KO) mice show reduced action potential-triggered excitatory postsynaptic currents (EPSCs) in hippocampal neurons and deficits in spatial working memory (Broker-Lai et al., 2017). However, comprehensive studies of TRPC4 in ND are yet to be undertaken. Only axonal regeneration is associated with TRPC4 expression in the dorsal root ganglia (DRG) (Wu et al., 2008). Neuron growth factor (NGF) and dibutyryl cAMP increase the expression level of TRPC4 in DRG differentiation. Improvement of TRPC4-siRNA reduces the length of neuritis. These results suggest a role for TRPC4 in ALS (Figure 2H). TRPC5, together with TRPC1, has high expression levels in the SN and an important role in dopaminergic neurons (De March et al., 2006). In rat PC12 cells, overexpression of TRPC5 inhibited the neurite outgrowth induced by NGF, and shRNA-mediated KD of TRPC5 enhanced outgrowth (Kumar et al., 2014). TRPC5 has also been reported to regulate neuronal growth cone morphology and nervous system development. In the downstream processes involving semaphorin 3A, growth cone collapse is induced through the cleavage and activation of TRPC5, using calpain (Kaczmarek et al., 2012). In neural progenitor cells, KD of TRPC5 using siRNA reduced the elevation of SOCE and blocked the switch between proliferation and neuronal differentiation (Shin et al., 2010). TRPC5 activity also inhibited neural migration and neurite extension (Tian et al., 2010). Similarly, in the striatum of both YAC128 HD transgenic (Tg) mice and patients, we identified that altered glutathione homeostasis, or increased oxidative potential, resulted in Ca2+-dependent apoptosis of striatal neurons, consistent with increased TRPC5 S-glutathionylation and hyperactivation (Figure 2D). Thus, downregulation of TRPC5 activity by siTRPC5 KD and ML204-specific blocker improved the survival of striatal neurons and behavioral motor symptoms (Hong et al., 2015). Furthermore, we recently reported that TRPC5 instability induced by depalmitoylation protects against neuronal death of HD striatal cells (Hong et al., 2019). S-palmitoylation is a reversible covalent lipid modification that promotes membrane trafficking and stability by anchoring the palmitoylated protein to the membrane (Greaves et al., 2009).
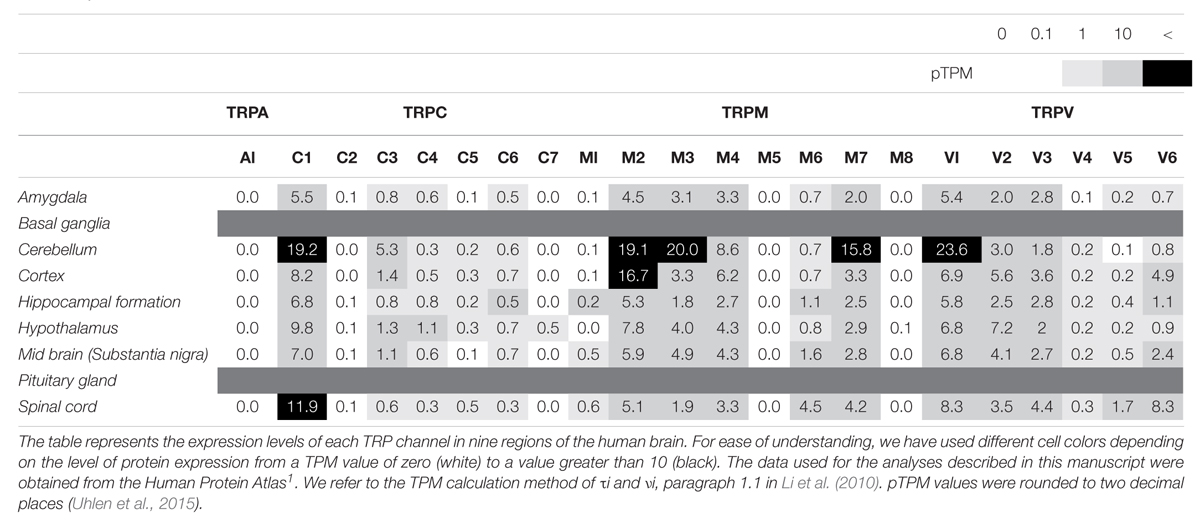
Table 1. Expression levels of TRP channels in human brain, as reported by the Human Protein Atlas.
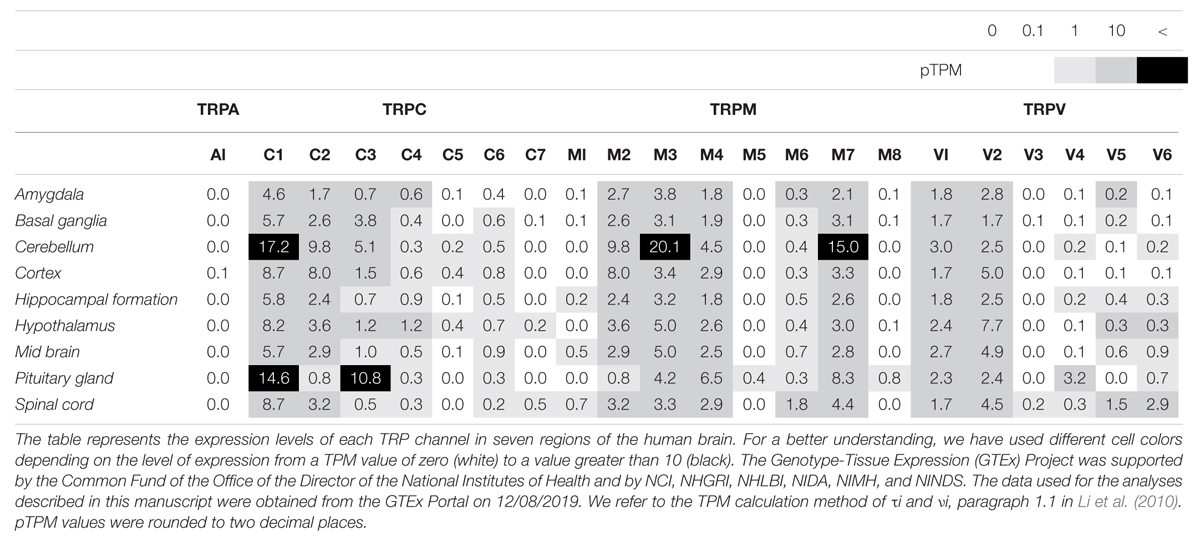
Table 2. Expression levels of TRP channels in the human brain, as reported by the GTEx project.
TRPC6
It is known that early onset dominant AD is caused by mutations in the APP (Dahlgren et al., 2002) or presenilin 1 (PS1) genes (Dillen and Annaert, 2006). APP is a precursor protein of Aβ, and PS1 is a key enzyme family that cleaves APP in complex with γ-secretase and cleavage of Notch. TRPC6 regulated the mechanism of the PS gene to prevent the progression of AD (Lu et al., 2017). Previous studies have reported that tetrahydrohyperforin, an agonist of TRPC6, lowers Aβ levels and ROS generation, also preventing learning and memory deficits in the AD model. In the research of Dinamarca and colleagues, tetrahydrohyperforin reduced amyloid deposition in rats injected with amyloid fibrils into the hippocampus, inhibited the neurotoxicity of amyloid fibrils and Aβ oligomers in hippocampal neurons, and improved neuropathological behavior in an amyloidosis rat model (Dinamarca et al., 2006). It has also been shown that interaction between TRPC6 and APP leads to inhibition of its cleavage by γ-secretase and a reduction in Aβ production (Wang et al., 2015). The authors also reported that expression of TRPC6 interferes with the interaction of APP (C99) with PS1 but does not interact with Notch. Crossing TRPC6 Tg mice and APP/PS1 model mice reduced plaque load and Aβ levels and improved cognition (Figure 2A). Lessard et al. (2005) reported the effect of PS2 on TRPC6-mediated Ca2+ entry. In this study, PS2 inhibited the influx of Ca2+ from TRPC6. Induction of Ca2+ was higher when FAD-linked PS2, a dominant negative form, was co-expressed with TRPC6 than with wild-type (WT) PS2 and TRPC6 (Figure 2A). However, TRPC6 was still activated by 1-oleoyl-2-acetyl-sn-glycerol (OAG), suggesting that it does not impair channel function1.
TRPM (Melastatin)
The TRPM subfamily has the highest expression level in the brain amongst the TRP channels, and there are many reports in relation to ND. One of the most distinctive features of TRPM ion channels is the high permeability to Ca2+ and Mg2+ (Hashimoto and Kambe, 2015). The important role of Mg2+ signaling in neuroprotection and neurodevelopment has been reported on (Lingam and Robertson, 2018). Thus, studies of TRPM in ND relate pathogenesis to Mg2+ signaling.
TRPM2
TRPM2 is an ion channel that is abundantly expressed in the brain (Tables 1, 2). TRPM2 has been reported to be activated by a wide range of factors, such as oxidative stress, NAD+-related metabolites, and ADP-ribose (Huang et al., 2018). In the study of Belrose et al. (2012) depletion of glutathione (GSH) was reported to be a factor in activating TRPM2. In hippocampal neurons, an increase in ROS due to GSH depletion activated TRPM2, and an increase in TRPM2-dependent Ca2+ influx induced neuronal apoptosis (Ovey and Naziroglu, 2015). In the study of Fonfria et al. (2005) an increase in intracellular Ca2+ and Aβ induced by TRPM2 activity induced neuronal cell death in rat striatal. Treatment with TRPM2 blocker or SB-750139, which inhibits the production of ADP-ribose, inhibited intracellular Ca2+ concentration and cell death via H2O2 and Aβ. In the study of Ostapchenko et al. (2015) an aged APP/PS1 AD mouse model showed increased ER stress and decreased presynaptic markers (Figure 2B). However, elimination of TRPM2 in APP/PS1 mice improved abnormal response regardless of plaque burden. Age-dependent spatial memory deficits were observed in APP/PS1 mice (Ostapchenko et al., 2015). However, the absence of TRPM2 in these mice attenuated synapse loss and spatial memory. In summary, GSH deficiency and ROS induction activate TRPM2, and Ca2+ influx by TRPM2 contributes to the neuronal toxicity of Aβ. TRPM2 may be an important therapeutic target for AD. In the study of PD, Ca2+ influx through the TRPM2 channel was induced by ROS and promoted the death of dopaminergic neurons in the SN (Sun et al., 2018). A variant of TRPM2 (P1018L) was found in a Guamanian ALS/PD patient. P1018L attenuates oxidative stress-induced Ca2+ influx through TRPM2 (Hermosura et al., 2008).
TRPM7
TRPM7 has the Mg2+ permeability to maintain the homeostasis of Mg2+. In HEK293 cells overexpressing TRPM7, H2O2 increased Ca2+ concentration and TRPM7 current (Nadler et al., 2001). In mouse cortical neurons, TRPM7-siRNA KD and treatment with TRPM7 inhibitors protected against neuronal cell damage (Coombes et al., 2011). In contrast, TRPM7-overexpressing HEK293 cells aggravated cell damage from H2O2, which was independent of the voltage-gated Ca2+ channel. Interestingly, in the study of Aarts et al. (2003) blocking of Ca2+-permeable non-selective cation conductance or KD of TRPM7 inhibited TRPM7 currents, anoxic Ca2+ uptake, ROS production, and anoxic death in cortical neurons. Mg2+ permeability of TRPM7 is implicated in PD (Figure 2C). Continuous administration of Mg2+ significantly inhibited the neurotoxicity of MPP+, reduced the death of dopaminergic neurons, and improved the length of dopaminergic neurites (Hashimoto et al., 2008). In a recent zebrafish study, TRPM7 mutation suppressed dopamine-dependent developmental transitions and increased sensitivity to the neurotoxicity of MPP+ (Decker et al., 2014), and expression of the channel-dead variant of TRPM7 in SH-SY5Y cells increased cell death. These studies suggest that the role of Mg2+ influx and TRPM7 in dopaminergic neurons is important and could be a therapeutic target for PD. A variant of TRPM7 (T1482I) was also found in Guamanian ALS/PD cases. Incidentally, mutant G93A-superoxide dismutase (SOD1) mice are used as an ALS model (Guo et al., 2009).
TRPV (Vanilloid)
The TRPV subfamily has been reported to have the highest number of sensory functions, such as nociception, mechano-sensing, osmolarity-sensing, and thermo-sensing. Usually, TRPV is expressed in peripheral sensory nerves, although pathological studies have also reported expression in the brain. The various antagonists of TRPV4 could protect damaged neurons and inhibit the production of ROS (Suresh et al., 2018; Wu et al., 2019).
TRPV1
TRPV1 is expressed not only in the plasma membrane but also in the ER and calcium storage vesicles (Marshall et al., 2003). TRPV1 is phosphorylated to enable translocation from the ER to the plasma membrane. TrkA activity due to NGF increases the surface expression level of TRPV1 located in the ER through PKC phosphorylation (Zhang et al., 2005). An increase in intracellular calcium levels due to TRPV1 activity may aggravate neuronal cell death. In microglia cells, TRPV1 activity by agonists such as capsaicin (CAP) and resiniferatoxin (RTX) induce apoptosis (Kim et al., 2006). Dopamine release is dependent on the mechanosensitive TRPV1 channels activated by cannabinoid receptor stimulation in dopaminergic neurons (Oakes et al., 2019). Capsaicin, a TRPV1 agonist, induces the death of mesencephalic dopaminergic neurons through the activation of TRPV1 and CB1 receptors. Activation of TRPV1 increases the release of the mitochondrial cytochrome C and caspase3 cleavage. Cell damage is attenuated by an intracellular Ca2+-chelator. In a recent study, the activity of TRPV1 was reported to decrease Aβ-induced cytotoxicity (Balleza-Tapia et al., 2018). Treatment with a TRPV1 agonist rescued Aβ-induced degradation of hippocampal neuron function (Figure 2A). In addition, it is suggested that TRPV1 contributes to the movement of patients with HD (Figure 2E). In the 3-nitropropionic acid-induced HD model, hyperkinesia was attenuated by administering AM404, an endocannabinoid reuptake inhibitor (Lastres-Becker et al., 2003). This phenomenon is reversed by the TRPV1 antagonist capsazepine, suggesting that TRPV1 activity may facilitate the movement of HD patients. Depending on the pathological mechanism, the role of TRPV1 activity can be distinguished accordingly and can be an important drug development target of NDs.
TRPV4
The activity of TRPV4 causes neuronal injury in pathological conditions. In many types of cells, TRPV4 activity increases the production of ROS and NO (Hong et al., 2016). GSK1016790A, an agonist of TRPV4, increased the concentration and NO in the hippocampus. TRPV4 agonist-induced neuronal cell death in hippocampal CA1 was inhibited by treatment with ROS scavengers such as Trolox or ARL-17477. In a recent report, TRPV4 enhanced neuronal inflammatory responses and pro-inflammatory cytokine release (Figure 2F; Wang et al., 2019). GSK1016790A-injected mice also showed increased levels of the pro-inflammatory cytokines IL-1β, TNF-α, and IL-6 and showed TRPV4-mediated microglial and astrocyte activation. Although direct evidence linking TRPV4 to NDs has not been reported, these results suggest a clear association between neuronal cell death and ROS.
TRPA
The TRP ankyrin 1 (TRPA1) channel is a non-selective transmembrane cation channel with multiple ankyrin repeats at its N-terminal. TRPA1 is mainly expressed in primary sensory neurons and non-neuronal cells (Jo et al., 2013). According to RNA-seq ALTAS data (Tables 1, 2), the expression level of TRPA1 in the brain is low, but various functions have been reported in recent studies. Reported TRPA1 functions are mainly the detection of pain, cold temperature, and cannabinoids, in addition to noxious compounds that elicit pain and neurogenic inflammation (Paulsen et al., 2015).
TRPA1
Until now, the role of TRPA1 in neurons has only been reported on with respect to pain and inflammation, although recent studies have revealed a potential involvement in AD pathogenesis. Deposition of Aβ is an important factor in the exacerbation of AD, and soluble Aβ oligomers mediate fast and widespread Ca2+ influx in astrocytes (Bosson et al., 2017). TRPA1 was first identified in mouse hippocampal astrocytes and associated with Aβ-mediated Ca2+ signaling (Figure 2F; Lee et al., 2016). The cause of Aβ oligomer-mediated fast Ca2+ signaling appears to be the hyperactivation of TRPA1 (Bosson et al., 2017). TRPA1-induced Ca2+ signaling initiates the release of inflammatory factors such as PP2B, NF-κB, and NFAT (Figure 2F). APP/PS1 Tg mice, an AD mouse model, have increased expression of TRPA1 in hippocampal astrocytes. Loss of function of TRPA1 channels improves spatial learning, memory and cognition, and decreases Aβ deposition in APP/PS1 Tg mouse also (Lee et al., 2016). In summary, TRPA1-induced Ca2+ influx in astrocytes may be evidence of the critical role of Aβ in inflammatory processes and AD progression. Drug development focused on TRPA1 could be a novel target for treating dysfunction in AD.
TRP Channels: Novel Therapeutic Candidate for Neurodegenerative Disease
Various causes of NDs have been reported recently. However, most of the brain lesions in ND present alongside several pathological environments, such as the presence of ROS, impaired antioxidant systems, and disrupted Ca2+ homeostasis (Di Meo et al., 2016). In consideration of the results discussed above, the regulation of TRP channels plays a key role in the Ca2+-dependent neuronal death in NDs. The involvement of TRP channels in NDs is summarized in Figure 1. To investigate the role of TRP channels in NDs, TRP channel antagonists and TRP-KO mice have been generated and utilized. TRP KO mice exhibit behavioral and neurological phenotypes.
TRPC3 is required for slow excitatory postsynaptic potential in cerebellar Purkinje cell synapses, and consequently, severe ataxic phenotypes have been shown in TRPC3 KO mice. In contrast to TRPC1/4 double-KO or TRPC1/4/6 triple-KO mice, TRPC3 KO mice show movement deficits of the hind-paws (Hartmann et al., 2008). In addition, TRPC4 plays a role in fear and anxiety-related behaviors. TRPC4 KO mice show innate fear responses in elevated plus maze and open-field tests. These fear responses result from mGluR-mediated EPSC in the lateral nucleus of the amygdala neurons (Riccio et al., 2014). TRPC5 also plays an essential role in fear-related behavior. In addition, disruption of burst firing in the potent muscarinic antagonist pilocarpine-induced seizure in TRPC5 KO mice was reduced. Seizure-induced neuronal loss in the hippocampal region was also reduced in TRPC5 KO mice (Phelan et al., 2013). TRPM2 KO mice exhibited disturbed EEG rhythms and bipolar disease-related behavior, including impairment of social behavior and increased anxiety (Jang et al., 2015). TRPM7 KO mice exhibited clasping, tremors, and slow movement associated with Mg2+ deficiency (Ryazanova et al., 2010).
TRPC KO mice combined with ND models have also been generated. In models of AD, TRPC6 modulates cleavage of APP by gamma secretase and APP (C99) interaction with PS1 (Wang et al., 2015). TRPC6 overexpression in APP/PS1 mice results in a reduction of Aβ accumulation in the hippocampus (Table 3). Therefore, TRPPC6 overexpression improves spatial learning and memory in APP/PS1 mice. In addition, the expression of the inflammatory factors TNF-α, IL-1β, COX-2, and IL-6 is regulated by levels of TRPC6 via Aβ, and levels of TRPC6 are increased by Aβ via NF-κB in BV-2 microglia cells (Lu et al., 2018). TRPM2 expression is involved in synapse loss, microglial activation, and spatial memory deficits in APP/PS1 mice (Ostapchenko et al., 2015). Activation of TRPV1 channels is required to trigger long-term depression at interneuronal synapses (Gibson et al., 2008) and prevents Aβ-involved impairment of functional networks in the hippocampus (Balleza-Tapia et al., 2018). Astrocytic Ca2+ hyperactivity is induced by Aβ oligomers via TRPA1 in the hippocampus. Moreover, astrocyte hyper-excitability is replaced by CA1 neuronal activity in APP/PS1 mice (Lee et al., 2016). Moreover, TRPA1 regulates astrocyte-derived inflammation in APP/PS1 mice. TRP channel antagonists regulate the production of ROS, APP processing, and Aβ accumulation. The TRPV4 antagonist HC-067047 attenuates the H2O2-induced Ca2+ influx (Suresh et al., 2015). Aβ-mediated cell damage was attenuated by treatment with TRPV4 blockers ruthenium red and gadolinium chloride (Bai and Lipski, 2014).
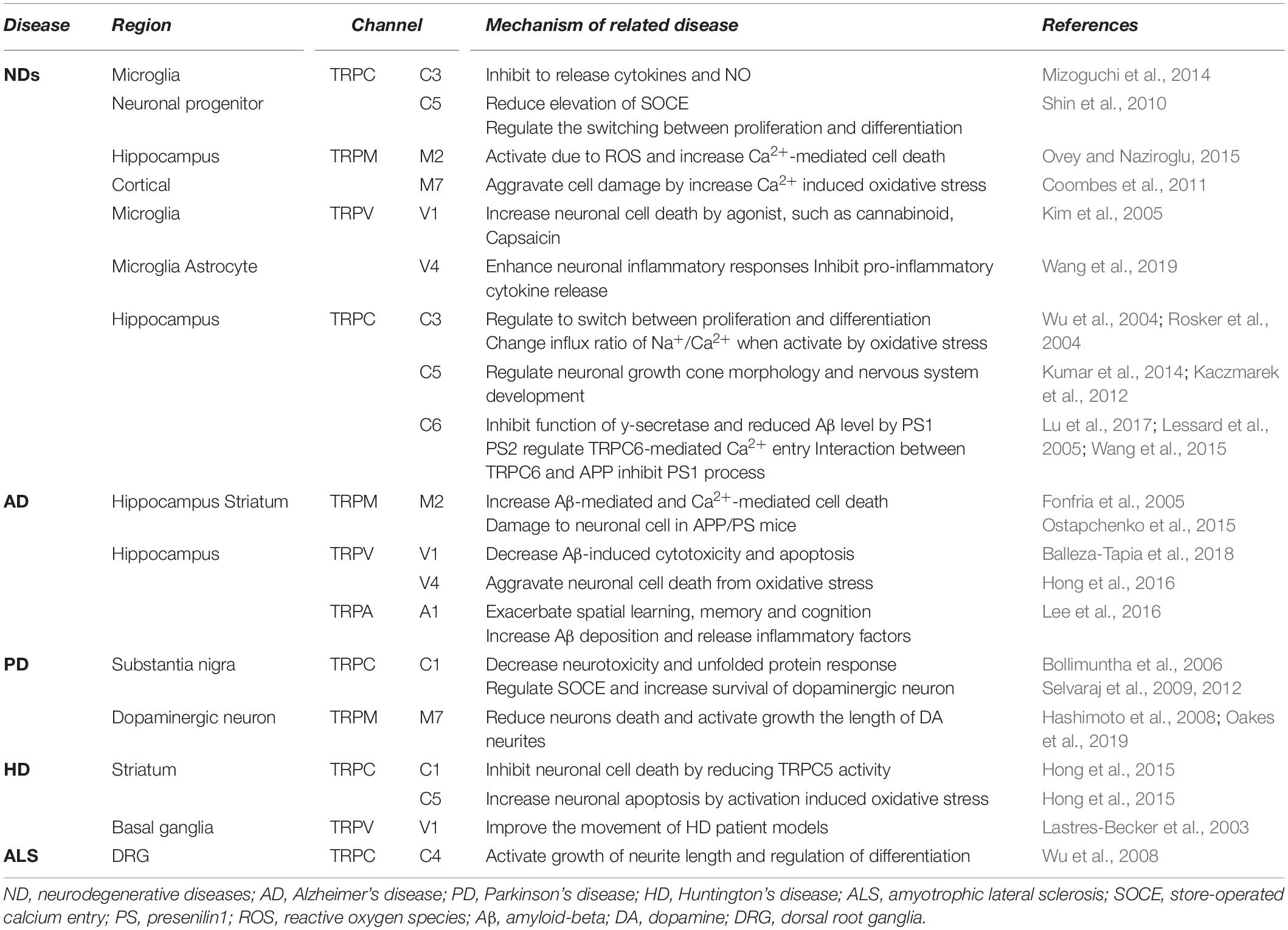
Table 3. Disease-related functions of TRP channels.
TRPC1, TRPC3, TRPM2, TRPM7, and TRPV1 have been shown to be involved in PD. TRPC1 activation reduces dopaminergic neuronal death (Selvaraj et al., 2009). TRPC3-mediated Ca2+ influx contributes to the survival of neurons in the SN (Zhou et al., 2008). Additionally, MPP+-induced oxidative stress increases intracellular Ca2+ via TRPM2 activation (Sun et al., 2018). TRPM7 channels regulate magnesium homeostasis in cells, and the presence of Mg ameliorates MPP+ toxicity (Hashimoto et al., 2008; Paravicini et al., 2012). Ca2+ influx via TRPV1 in dopaminergic neurons mediates mitochondrial dysfunction, microglial activation, ROS generation, and cell death (Kim et al., 2005; Nam et al., 2015). TRPV1 or TRPN-like channel-dependent dopamine release is mediated by CB1 stimulation (Table 3; Oakes et al., 2019). In addition, regulation of TRPV1 activity is closely related to the survival of dopaminergic neurons. TRPM2 is controlled by oxidative stress (Belrose et al., 2012; Huang et al., 2018). Therefore, the regulation of TRP channels contributes to overcoming dopamine depletion and the loss of dopaminergic neurons in PD patients. Likewise, oxidizing modulation of posttranslational modification (glutathionylation) of TRPC5 leads to apoptosis in an HD model (Figure 2D; Hong et al., 2015). Attenuation of TRPC5 activity by KD, blocker, or depalmitoylation shows therapeutic effects against oxidative stress by lowering TRPC5 toxicity (Hong et al., 2019). Additionally, regulation of TRPML1, TRPM2, and TRPM7 activity might also be a therapeutic strategy for ALS (Hermosura and Garruto, 2007; Hermosura et al., 2008; Tedeschi et al., 2019). The TRPV1 antagonist capsazepine has antihyperkinetic effects in a model of HD (Lastres-Becker et al., 2003).
As previously reported, TRP channels can be assembled as homo- or heteromeric complexes in nature. However, individual TRP channel KO models that lack phenotypes are limited in their ability to determine the cause of the functional compensation of each TRP channel. Moreover, in vivo studies regarding chronological changes in TRP channel expression patterns in the brain are needed for NDs. The regulation of TRP channels can be a novel therapeutic target for NDs. Nevertheless, a limitation to the development of TRP channel-specific antagonists is that their structures remain unknown. Therefore, structural analyses must precede pathological and clinical studies.
Conclusion
Transient receptor potential channels may not be the only, or main, pathogenic factors contributing to the pathogenesis of ND. Research in the field of ND is challenging; it is necessary to either conclusively prove a relationship between pathogenic factors or identify new therapeutic targets. In achieving one of these two possibilities, it is important not to underestimate the potential of TRP channels, based on the physiological and pathological functions of TRP channels discovered so far. Through the interrelationship between disruption of Ca2+ homeostasis and the development of NDs, lowering the activity of TRP channels is sufficient to enable expectations for new therapeutic strategies. As we have discussed, increased TRP channel activity has been widely observed in NDs, and model studies have shown that abnormal function due to upregulation of TRP channels can be controlled by drug treatments. Since many drugs have been reported to be TRP channel inhibitors, understanding binding modes will provide deep insight for pharmacological application in NDs. However, direct evidence for drug binding to TRP channels is unavailable. To address this issue, the most effective approach for understanding drug binding would be a structural study.
Possibilities of Drug-Bound Structure
For several decades, the detailed structures and topologies of TRP channels were not understood, although the first high-resolution structure of TRPV1 was recently resolved by a single particle cryo-EM (Liao et al., 2013). Since TRPV1 was the first membrane protein to be characterized from single particle cryo-EM and the biochemical methods were relatively similar for other family members, most follow-up studies concentrated on TRP channel structure. However, though high-resolution structures have been resolved for most ND-related TRP channels, the structures of the drug-bound forms are mostly unknown. Here we discuss the technical possibilities for determining the drug-bound structure of the TRP channel family.
For analysis of drug-channel binding, which would improve our understanding of the inhibition mechanisms and would provide clues for developing drug design, the determination of the high-resolution structure is absolutely necessary. In the TRP family, the resolutions of most ND-related TRP structures are above 3.5 Å, with a few exceptions such as TRPC6, TRPA1, and TRPV4 (3.8, 4.24, and 3.8 Å, respectively) (Paulsen et al., 2015; Deng et al., 2018; Tang et al., 2018). TRPC5 broke the 3 Å barrier (Duan et al., 2019), and TRPM2 also came close to hitting the barrier (3.07 Å) (Zhang et al., 2018). The structures of other members of the TRP family that are associated with NDs, such as TRPC3, TRPC4, TRPM7, and TRPV1, were determined at ∼3.3 Å (Liao et al., 2013; Duan et al., 2018; Fan et al., 2018; Liu et al., 2018). Therefore, there is still room for improvement in their resolutions, possibly by biochemical techniques such as nanodisc reconstruction. The resolution of the drug-bound form does not always guarantee higher resolution than that of the apo structure, but generally, the occupation of an inhibitor in the binding cavity facilitates conformational stabilization, resulting in higher resolution. Therefore, since there is optimism regarding drug-binding studies, we suggest that ongoing attention and efforts be focused on this area of therapeutic target research.
Author Contributions
CH, BJ, HP, and IS wrote and discussed the review at almost all stages. JL professionally edited the manuscript for English language. JC, JK, and Y-CS analyzed representative tables presenting TRP channel expression data and also provided technical support. IS and CH supervised the entire writing process.
Funding
This study was supported by the National Research Foundation (NRF) of Korea, which is funded by the Ministry of Science, ICT (Information and Communication Technology), and Future Planning (MSIP) (2018R1A4A1023822), and by the Ministry of Education (2015R1A6A3A04058395) of the Korean Government. JL was supported by Brain Korea 21 plus and the MSIP.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Aarts, M., Iihara, K., Wei, W. L., Xiong, Z. G., Arundine, M., Cerwinski, W., et al. (2003). A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877.
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004a). Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565–575. doi: 10.1523/jneurosci.4042-03.2004
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004b). Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta 1742, 81–87. doi: 10.1016/j.bbamcr.2004.09.006
Amaral, M. D., and Pozzo-Miller, L. (2007). TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J. Neurosci. 27, 5179–5189. doi: 10.1523/jneurosci.5499-06.2007
Ambudkar, I. S., De Souza, L. B., and Ong, H. L. (2017). TRPC1, Orai1, and STIM1 in SOCE: friends in tight spaces. Cell Calcium 63, 33–39. doi: 10.1016/j.ceca.2016.12.009
Arancibia, S., Silhol, M., Mouliere, F., Meffre, J., Hollinger, I., Maurice, T., et al. (2008). Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol. Dis. 31, 316–326. doi: 10.1016/j.nbd.2008.05.012
Association, A. S. (2016). 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12, 459–509. doi: 10.1016/j.jalz.2016.03.001
Bai, J. Z., and Lipski, J. (2014). Involvement of TRPV4 channels in Abeta(40)-induced hippocampal cell death and astrocytic Ca(2+) signalling. Neurotoxicology 41, 64–72. doi: 10.1016/j.neuro.2014.01.001
Balleza-Tapia, H., Crux, S., Andrade-Talavera, Y., Dolz-Gaiton, P., Papadia, D., Chen, G., et al. (2018). TrpV1 receptor activation rescues neuronal function and network gamma oscillations from Abeta-induced impairment in mouse hippocampus in vitro. eLife 7:e37703. doi: 10.7554/eLife.37703
Barnham, K. J., Masters, C. L., and Bush, A. I. (2004). Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214.
Belrose, J. C., Xie, Y. F., Gierszewski, L. J., Macdonald, J. F., and Jackson, M. F. (2012). Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol. Brain 5:11. doi: 10.1186/1756-6606-5-11
Blesa, J., Trigo-Damas, I., Quiroga-Varela, A., and Jackson-Lewis, V. R. (2015). Oxidative stress and Parkinson’s disease. Front. Neuroanat. 9:91. doi: 10.3389/fnana.2015.00091
Bollimuntha, S., Ebadi, M., and Singh, B. B. (2006). TRPC1 protects human SH-SY5Y cells against salsolinol-induced cytotoxicity by inhibiting apoptosis. Brain Res. 1099, 141–149. doi: 10.1016/j.brainres.2006.04.104
Bollimuntha, S., Selvaraj, S., and Singh, B. B. (2011). Emerging roles of canonical TRP channels in neuronal function. Adv. Exp. Med. Biol. 704, 573–593. doi: 10.1007/978-94-007-0265-3_31
Bosson, A., Paumier, A., Boisseau, S., Jacquier-Sarlin, M., Buisson, A., and Albrieux, M. (2017). TRPA1 channels promote astrocytic Ca(2+) hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-beta peptide. Mol. Neurodegener. 12:53. doi: 10.1186/s13024-017-0194-8
Bradley, S. A., and Steinert, J. R. (2016). Nitric oxide-mediated posttranslational modifications: impacts at the synapse. Oxid. Med. Cell. Longev. 2016:5681036. doi: 10.1155/2016/5681036
Broker-Lai, J., Kollewe, A., Schindeldecker, B., Pohle, J., Nguyen Chi, V., Mathar, I., et al. (2017). Heteromeric channels formed by TRPC1, TRPC4 and TRPC5 define hippocampal synaptic transmission and working memory. EMBO J. 36, 2770–2789. doi: 10.15252/embj.201696369
Calvo-Rodriguez, M., Hernando-Perez, E., Nunez, L., and Villalobos, C. (2019). Amyloid beta oligomers increase ER-mitochondria Ca(2+) cross talk in young hippocampal neurons and exacerbate aging-induced intracellular Ca(2+) remodeling. Front. Cell. Neurosci. 13:22. doi: 10.3389/fncel.2019.00022
Carri, M. T., Valle, C., Bozzo, F., and Cozzolino, M. (2015). Oxidative stress and mitochondrial damage: importance in non-SOD1 ALS. Front. Cell. Neurosci. 9:41. doi: 10.3389/fncel.2015.00041
Cenini, G., and Voos, W. (2019). Mitochondria as potential targets in alzheimer disease therapy: an update. Front. Pharmacol. 10:902. doi: 10.3389/fphar.2019.00902
Chen, X., Guo, C., and Kong, J. (2012). Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 7, 376–385. doi: 10.3969/j.issn.1673-5374.2012.05.009
Contreras, L., Drago, I., Zampese, E., and Pozzan, T. (2010). Mitochondria: the calcium connection. Biochim. Biophys. Acta 1797, 607–618. doi: 10.1016/j.bbabio.2010.05.005
Coombes, E., Jiang, J., Chu, X. P., Inoue, K., Seeds, J., Branigan, D., et al. (2011). Pathophysiologically relevant levels of hydrogen peroxide induce glutamate-independent neurodegeneration that involves activation of transient receptor potential melastatin 7 channels. Antioxid. Redox Signal. 14, 1815–1827. doi: 10.1089/ars.2010.3549
Dahlgren, K. N., Manelli, A. M., Stine, W. B. Jr., Baker, L. K., Krafft, G. A., and LaDu, M. J. (2002). Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. doi: 10.1074/jbc.m201750200
De March, Z., Giampa, C., Patassini, S., Bernardi, G., and Fusco, F. R. (2006). Cellular localization of TRPC5 in the substantia nigra of rat. Neurosci. Lett. 402, 35–39. doi: 10.1016/j.neulet.2006.03.061
Decker, A. R., Mcneill, M. S., Lambert, A. M., Overton, J. D., Chen, Y. C., Lorca, R. A., et al. (2014). Abnormal differentiation of dopaminergic neurons in zebrafish trpm7 mutant larvae impairs development of the motor pattern. Dev. Biol. 386, 428–439. doi: 10.1016/j.ydbio.2013.11.015
Demuro, A., Parker, I., and Stutzmann, G. E. (2010). Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 285, 12463–12468. doi: 10.1074/jbc.R109.080895
Deng, Z., Paknejad, N., Maksaev, G., Sala-Rabanal, M., Nichols, C. G., Hite, R. K., et al. (2018). Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nat. Struct. Mol. Biol. 25, 252–260. doi: 10.1038/s41594-018-0037-5
Di Meo, S., Reed, T. T., Venditti, P., and Victor, V. M. (2016). Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell. Longev. 2016:1245049. doi: 10.1155/2016/1245049
Dillen, K., and Annaert, W. (2006). A two decade contribution of molecular cell biology to the centennial of Alzheimer’s disease: are we progressing toward therapy? Int. Rev. Cytol. 254, 215–300. doi: 10.1016/s0074-7696(06)54005-7
Dinamarca, M. C., Cerpa, W., Garrido, J., Hancke, J. L., and Inestrosa, N. C. (2006). Hyperforin prevents beta-amyloid neurotoxicity and spatial memory impairments by disaggregation of Alzheimer’s amyloid-beta-deposits. Mol. Psychiatry 11, 1032–1048. doi: 10.1038/sj.mp.4001866
Du, X., Wang, X., and Geng, M. (2018). Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 7:2. doi: 10.1186/s40035-018-0107-y
Duan, J., Li, J., Chen, G. L., Ge, Y., Liu, J., Xie, K., et al. (2019). Cryo-EM structure of TRPC5 at 2.8-A resolution reveals unique and conserved structural elements essential for channel function. Sci. Adv. 5:eaaw7935. doi: 10.1126/sciadv.aaw7935
Duan, J., Li, J., Zeng, B., Chen, G. L., Peng, X., Zhang, Y., et al. (2018). Structure of the mouse TRPC4 ion channel. Nat. Commun. 9:3102. doi: 10.1038/s41467-018-05247-9
Fan, C., Choi, W., Sun, W., Du, J., and Lu, W. (2018). Structure of the human lipid-gated cation channel TRPC3. eLife 7:e36852. doi: 10.7554/eLife.36852
Ferreiro, E., Oliveira, C. R., and Pereira, C. M. (2008). The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 30, 331–342. doi: 10.1016/j.nbd.2008.02.003
Fonfria, E., Marshall, I. C., Boyfield, I., Skaper, S. D., Hughes, J. P., Owen, D. E., et al. (2005). Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 95, 715–723. doi: 10.1111/j.1471-4159.2005.03396.x
Gibson, H. E., Edwards, J. G., Page, R. S., Van Hook, M. J., and Kauer, J. A. (2008). TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron 57, 746–759. doi: 10.1016/j.neuron.2007.12.027
Gorlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Greaves, J., Prescott, G. R., Gorleku, O. A., and Chamberlain, L. H. (2009). The fat controller: roles of palmitoylation in intracellular protein trafficking and targeting to membrane microdomains (Review). Mol. Membr. Biol. 26, 67–79. doi: 10.1080/09687680802620351
Guinamard, R., Salle, L., and Simard, C. (2011). The non-selective monovalent cationic channels TRPM4 and TRPM5. Adv. Exp. Med. Biol. 704, 147–171. doi: 10.1007/978-94-007-0265-3_8
Guo, H., Shen, X., Xu, Y., Yuan, J., Zhao, D., and Hu, W. (2013). Emodin prevents hypoxic-ischemic neuronal injury: involvement of the activin A pathway. Neural Regen. Res. 8, 1360–1367. doi: 10.3969/j.issn.1673-5374.2013.15.002
Guo, Y. S., Wu, D. X., Wu, H. R., Wu, S. Y., Yang, C., Li, B., et al. (2009). Sensory involvement in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Exp. Mol. Med. 41, 140–150. doi: 10.3858/emm.2009.41.3.017
Hartmann, J., Dragicevic, E., Adelsberger, H., Henning, H. A., Sumser, M., Abramowitz, J., et al. (2008). TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 59, 392–398. doi: 10.1016/j.neuron.2008.06.009
Hashimoto, A., and Kambe, T. (2015). Mg, Zn and Cu transport proteins: a brief overview from physiological and molecular perspectives. J. Nutr. Sci. Vitaminol. 61(Suppl.), S116–S118. doi: 10.3177/jnsv.61.S116
Hashimoto, T., Nishi, K., Nagasao, J., Tsuji, S., and Oyanagi, K. (2008). Magnesium exerts both preventive and ameliorating effects in an in vitro rat Parkinson disease model involving 1-methyl-4-phenylpyridinium (MPP+) toxicity in dopaminergic neurons. Brain Res. 1197, 143–151. doi: 10.1016/j.brainres.2007.12.033
Hermosura, M. C., Cui, A. M., Go, R. C., Davenport, B., Shetler, C. M., Heizer, J. W., et al. (2008). Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc. Natl. Acad. Sci. U.S.A. 105, 18029–18034. doi: 10.1073/pnas.0808218105
Hermosura, M. C., and Garruto, R. M. (2007). TRPM7 and TRPM2-Candidate susceptibility genes for Western Pacific ALS and PD? Biochim. Biophys. Acta 1772, 822–835. doi: 10.1016/j.bbadis.2007.02.008
Hong, C., Choi, S. H., Kwak, M., Jeong, B., Ko, J., Park, H. J., et al. (2019). TRPC5 channel instability induced by depalmitoylation protects striatal neurons against oxidative stress in Huntington’s disease. Biochim. Biophys. Acta 1867:118620. doi: 10.1016/j.bbamcr.2019.118620
Hong, C., Seo, H., Kwak, M., Jeon, J., Jang, J., Jeong, E. M., et al. (2015). Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain 138, 3030–3047. doi: 10.1093/brain/awv188
Hong, Z., Tian, Y., Yuan, Y., Qi, M., Li, Y., Du, Y., et al. (2016). Enhanced oxidative stress is responsible for TRPV4-induced neurotoxicity. Front. Cell. Neurosci. 10:232. doi: 10.3389/fncel.2016.00232
Huang, Y., Winkler, P. A., Sun, W., Lu, W., and Du, J. (2018). Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 562, 145–149. doi: 10.1038/s41586-018-0558-4
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi: 10.2174/156720510793611592
Itkin, A., Dupres, V., Dufrene, Y. F., Bechinger, B., Ruysschaert, J. M., and Raussens, V. (2011). Calcium ions promote formation of amyloid beta-peptide (1-40) oligomers causally implicated in neuronal toxicity of Alzheimer’s disease. PLoS One 6:e18250. doi: 10.1371/journal.pone.0018250
Ivanova, D., Zhelev, Z., Aoki, I., Bakalova, R., and Higashi, T. (2016). Overproduction of reactive oxygen species - obligatory or not for induction of apoptosis by anticancer drugs. Chin. J. Cancer Res. 28, 383–396. doi: 10.21147/j.issn.1000-9604.2016.04.01
Jang, Y., Lee, S. H., Lee, B., Jung, S., Khalid, A., Uchida, K., et al. (2015). TRPM2, a susceptibility gene for bipolar disorder, regulates glycogen synthase kinase-3 activity in the brain. J. Neurosci. 35, 11811–11823. doi: 10.1523/JNEUROSCI.5251-14.2015
Jiao, S. S., Shen, L. L., Zhu, C., Bu, X. L., Liu, Y. H., Liu, C. H., et al. (2016). Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 6:e907. doi: 10.1038/tp.2016.186
Jo, K. D., Lee, K. S., Lee, W. T., Hur, M. S., and Kim, H. J. (2013). Expression of transient receptor potential channels in the ependymal cells of the developing rat brain. Anat. Cell Biol. 46, 68–78. doi: 10.5115/acb.2013.46.1.68
Joshi, D. C., Tewari, B. P., Singh, M., Joshi, P. G., and Joshi, N. B. (2015). AMPA receptor activation causes preferential mitochondrial Ca(2)(+) load and oxidative stress in motor neurons. Brain Res. 1616, 1–9. doi: 10.1016/j.brainres.2015.04.042
Kaczmarek, J. S., Riccio, A., and Clapham, D. E. (2012). Calpain cleaves and activates the TRPC5 channel to participate in semaphorin 3A-induced neuronal growth cone collapse. Proc. Natl. Acad. Sci. U.S.A. 109, 7888–7892. doi: 10.1073/pnas.1205869109
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kamat, P. K., Kalani, A., Rai, S., Swarnkar, S., Tota, S., Nath, C., et al. (2016). Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: understanding the therapeutics strategies. Mol. Neurobiol. 53, 648–661. doi: 10.1007/s12035-014-9053-6
Kim, J., Ko, J., Myeong, J., Kwak, M., Hong, C., and So, I. (2019). TRPC1 as a negative regulator for TRPC4 and TRPC5 channels. Pflugers Arch. 471, 1045–1053. doi: 10.1007/s00424-019-02289-w
Kim, S. R., Kim, S. U., Oh, U., and Jin, B. K. (2006). Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitro via Ca2+-mediated mitochondrial damage and cytochrome c release. J. Immunol. 177, 4322–4329. doi: 10.4049/jimmunol.177.7.4322
Kim, S. R., Lee, D., Chung, E. S., Oh, U. T., Kim, S. U., and Jin, B. K. (2005). Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J. Neurosci. 25, 662–671. doi: 10.1523/jneurosci.4166-04.2005
Kremer, B., Goldberg, P., Andrew, S. E., Theilmann, J., Telenius, H., Zeisler, J., et al. (1994). A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 330, 1401–1406. doi: 10.1056/nejm199405193302001
Kumar, S., Chakraborty, S., Barbosa, C., Brustovetsky, T., Brustovetsky, N., and Obukhov, A. G. (2014). Mechanisms controlling neurite outgrowth in a pheochromocytoma cell line: the role of TRPC channels. J. Cell Physiol. 227, 1408–1419. doi: 10.1002/jcp.22855
Lastres-Becker, I., De Miguel, R., De Petrocellis, L., Makriyannis, A., Di Marzo, V., and Fernandez-Ruiz, J. (2003). Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington’s disease. J. Neurochem. 84, 1097–1109. doi: 10.1046/j.1471-4159.2003.01595.x
Lee, K. I., Lee, H. T., Lin, H. C., Tsay, H. J., Tsai, F. C., Shyue, S. K., et al. (2016). Role of transient receptor potential ankyrin 1 channels in Alzheimer’s disease. J. Neuroinflammation 13:92. doi: 10.1186/s12974-016-0557-z
Lessard, C. B., Lussier, M. P., Cayouette, S., Bourque, G., and Boulay, G. (2005). The overexpression of presenilin2 and Alzheimer’s-disease-linked presenilin2 variants influences TRPC6-enhanced Ca2+ entry into HEK293 cells. Cell. Signal. 17, 437–445. doi: 10.1016/j.cellsig.2004.09.005
Li, B., Ruotti, V., Stewart, R. M., Thomson, J. A., and Dewey, C. N. (2010). RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500. doi: 10.1093/bioinformatics/btp692
Li, H. S., Shawn, X. X., and Montell, C. (1999). Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron 24, 261–273. doi: 10.1016/s0896-6273(00)80838-7
Liao, M., Cao, E., Julius, D., and Cheng, Y. (2013). Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112. doi: 10.1038/nature12822
Linde, C. I., Baryshnikov, S. G., Mazzocco-Spezzia, A., and Golovina, V. A. (2011). Dysregulation of Ca2+ signaling in astrocytes from mice lacking amyloid precursor protein. Am. J. Physiol. Cell Physiol. 300, C1502–C1512. doi: 10.1152/ajpcell.00379.2010
Lingam, I., and Robertson, N. J. (2018). Magnesium as a neuroprotective agent: a review of its use in the fetus, term infant with neonatal encephalopathy, and the adult stroke patient. Dev. Neurosci. 40, 1–12. doi: 10.1159/000484891
Liu, Y., Chen, C., Liu, Y., Li, W., Wang, Z., Sun, Q., et al. (2018). TRPM7 is required for normal synapse density, learning, and memory at different developmental stages. Cell Rep. 23, 3480–3491. doi: 10.1016/j.celrep.2018.05.069
Llorente-Folch, I., Rueda, C. B., Pardo, B., Szabadkai, G., Duchen, M. R., and Satrustegui, J. (2015). The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 593, 3447–3462. doi: 10.1113/jp270254
Lu, R., He, Q., and Wang, J. (2017). TRPC channels and Alzheimer’s disease. Adv. Exp. Med. Biol. 976, 73–83. doi: 10.1007/978-94-024-1088-4_7
Lu, R., Wang, J., Tao, R., Wang, J., Zhu, T., Guo, W., et al. (2018). Reduced TRPC6 mRNA levels in the blood cells of patients with Alzheimer’s disease and mild cognitive impairment. Mol. Psychiatry 23, 767–776. doi: 10.1038/mp.2017.136
Marshall, I. C., Owen, D. E., Cripps, T. V., Davis, J. B., Mcnulty, S., and Smart, D. (2003). Activation of vanilloid receptor 1 by resiniferatoxin mobilizes calcium from inositol 1,4,5-trisphosphate-sensitive stores. Br. J. Pharmacol. 138, 172–176. doi: 10.1038/sj.bjp.0705003
Michel, P. P., Toulorge, D., Guerreiro, S., and Hirsch, E. C. (2013). Specific needs of dopamine neurons for stimulation in order to survive: implication for Parkinson disease. FASEB J. 27, 3414–3423. doi: 10.1096/fj.12-220418
Mizoguchi, Y., Kato, T. A., Seki, Y., Ohgidani, M., Sagata, N., Horikawa, H., et al. (2014). Brain-derived neurotrophic factor (BDNF) induces sustained intracellular Ca2+ elevation through the up-regulation of surface transient receptor potential 3 (TRPC3) channels in rodent microglia. J. Biol. Chem. 289, 18549–18555. doi: 10.1074/jbc.M114.555334
Murphy, M. P., and LeVine, H. III (2010). Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 19, 311–323. doi: 10.3233/JAD-2010-1221
Nadler, M. J., Hermosura, M. C., Inabe, K., Perraud, A. L., Zhu, Q., Stokes, A. J., et al. (2001). LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 411, 590–595. doi: 10.1038/35079092
Nam, J. H., Park, E. S., Won, S. Y., Lee, Y. A., Kim, K. I., Jeong, J. Y., et al. (2015). TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson’s disease via CNTF. Brain 138, 3610–3622. doi: 10.1093/brain/awv297
Neuner, S. M., Wilmott, L. A., Hope, K. A., Hoffmann, B., Chong, J. A., Abramowitz, J., et al. (2015). TRPC3 channels critically regulate hippocampal excitability and contextual fear memory. Behav. Brain Res. 281, 69–77. doi: 10.1016/j.bbr.2014.12.018
Numata, T., Kiyonaka, S., Kato, K., Takahashi, N., and Mori, Y. (2011). “Activation of TRP channels in mammalian systems,” in TRP Channels, ed. M. X. Zhu (Boca Raton, FL: CRC Press).
Oakes, M., Law, W. J., and Komuniecki, R. (2019). Cannabinoids stimulate the TRP channel-dependent release of both serotonin and dopamine to modulate behavior in C. elegans. J. Neurosci. 39, 4142–4152. doi: 10.1523/JNEUROSCI.2371-18.2019
Ostapchenko, V. G., Chen, M., Guzman, M. S., Xie, Y. F., Lavine, N., Fan, J., et al. (2015). The transient receptor potential melastatin 2 (TRPM2) channel contributes to beta-amyloid oligomer-related neurotoxicity and memory impairment. J. Neurosci. 35, 15157–15169. doi: 10.1523/JNEUROSCI.4081-14.2015
Ott, M., Gogvadze, V., Orrenius, S., and Zhivotovsky, B. (2007). Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922. doi: 10.1007/s10495-007-0756-2
Ovey, I. S., and Naziroglu, M. (2015). Homocysteine and cytosolic GSH depletion induce apoptosis and oxidative toxicity through cytosolic calcium overload in the hippocampus of aged mice: involvement of TRPM2 and TRPV1 channels. Neuroscience 284, 225–233. doi: 10.1016/j.neuroscience.2014.09.078
Paravicini, T. M., Chubanov, V., and Gudermann, T. (2012). TRPM7: a unique channel involved in magnesium homeostasis. Int. J. Biochem. Cell. Biol. 44, 1381–1384. doi: 10.1016/j.biocel.2012.05.010
Paulsen, C. E., Armache, J. P., Gao, Y., Cheng, Y., and Julius, D. (2015). Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517. doi: 10.1038/nature14367
Phelan, K. D., Shwe, U. T., Abramowitz, J., Wu, H., Rhee, S. W., Howell, M. D., et al. (2013). Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol. Pharmacol. 83, 429–438. doi: 10.1124/mol.112.082271
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x
Ramirez, A., Vazquez-Sanchez, A. Y., Carrion-Robalino, N., and Camacho, J. (2016). Ion channels and oxidative stress as a potential link for the diagnosis or treatment of liver diseases. Oxid. Med. Cell. Longev. 2016:3928714. doi: 10.1155/2016/3928714
Rasband, M. N. (2016). Glial contributions to neural function and disease. Mol. Cell. Proteomics 15, 355–361. doi: 10.1074/mcp.R115.053744
Reczek, C. R., and Chandel, N. S. (2018). ROS promotes cancer cell survival through calcium signaling. Cancer Cell 33, 949–951. doi: 10.1016/j.ccell.2018.05.010
Riccio, A., Li, Y., Tsvetkov, E., Gapon, S., Yao, G. L., Smith, K. S., et al. (2014). Decreased anxiety-like behavior and Galphaq/11-dependent responses in the amygdala of mice lacking TRPC4 channels. J. Neurosci. 34, 3653–3667. doi: 10.1523/JNEUROSCI.2274-13.2014
Rosker, C., Graziani, A., Lukas, M., Eder, P., Zhu, M. X., Romanin, C., et al. (2004). Ca(2+) signaling by TRPC3 involves Na(+) entry and local coupling to the Na(+)/Ca(2+) exchanger. J. Biol. Chem. 279, 13696–13704. doi: 10.1074/jbc.m308108200
Ryazanova, L. V., Rondon, L. J., Zierler, S., Hu, Z., Galli, J., Yamaguchi, T. P., et al. (2010). TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat. Commun. 1:109. doi: 10.3390/cells7080109
Samanta, A., Hughes, T. E. T., and Moiseenkova-Bell, V. Y. (2018). Transient receptor potential (TRP) channels. Subcell. Biochem. 87, 141–165. doi: 10.1007/978-981-10-7757-9_6
Sawamura, S., Shirakawa, H., Nakagawa, T., Mori, Y., Kaneko, S., and Emir, T. L. R. (eds). (2017). “TRP channels in the brain: what are they there for?,” in Neurobiology of TRP Channels, 2nd Edn, (Boca Raton, FL: CRC Press), 295–322. doi: 10.1201/9781315152837-17
Schwarz, Y., Oleinikov, K., Schindeldecker, B., Wyatt, A., Weissgerber, P., Flockerzi, V., et al. (2019). TRPC channels regulate Ca2+-signaling and short-term plasticity of fast glutamatergic synapses. PLoS Biol. 17:e3000445. doi: 10.1371/journal.pbio.3000445
Selvaraj, S., Sun, Y., and Singh, B. B. (2010). TRPC channels and their implication in neurological diseases. CNS Neurol. Disord. Drug Targets 9, 94–104. doi: 10.2174/187152710790966650
Selvaraj, S., Sun, Y., Watt, J. A., Wang, S., Lei, S., Birnbaumer, L., et al. (2012). Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 122, 1354–1367. doi: 10.1172/JCI61332
Selvaraj, S., Watt, J. A., and Singh, B. B. (2009). TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP(+). Cell Calcium 46, 209–218. doi: 10.1016/j.ceca.2009.07.008
Sesti, F., Liu, S., and Cai, S. Q. (2010). Oxidation of potassium channels by ROS: a general mechanism of aging and neurodegeneration? Trends Cell Biol. 20, 45–51. doi: 10.1016/j.tcb.2009.09.008
Shin, H. Y., Hong, Y. H., Jang, S. S., Chae, H. G., Paek, S. L., Moon, H. E., et al. (2010). A role of canonical transient receptor potential 5 channel in neuronal differentiation from A2B5 neural progenitor cells. PLoS One 5:e10359. doi: 10.1371/journal.pone.0010359
Streifel, K. M., Gonzales, A. L., De Miranda, B., Mouneimne, R., Earley, S., and Tjalkens, R. (2014). Dopaminergic neurotoxicants cause biphasic inhibition of purinergic calcium signaling in astrocytes. PLoS One 9:e110996. doi: 10.1371/journal.pone.0110996
Suen, K. C., Lin, K. F., Elyaman, W., So, K. F., Chang, R. C., and Hugon, J. (2003). Reduction of calcium release from the endoplasmic reticulum could only provide partial neuroprotection against beta-amyloid peptide toxicity. J. Neurochem. 87, 1413–1426. doi: 10.1111/j.1471-4159.2003.02259.x
Sun, Y., Sukumaran, P., Selvaraj, S., Cilz, N. I., Schaar, A., Lei, S., et al. (2018). TRPM2 promotes neurotoxin MPP(+)/MPTP-induced cell death. Mol. Neurobiol. 55, 409–420. doi: 10.1007/s12035-016-0338-9
Suresh, K., Servinsky, L., Jiang, H., Bigham, Z., Yun, X., Kliment, C., et al. (2018). Reactive oxygen species induced Ca(2+) influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 314, L893–L907. doi: 10.1152/ajplung.00430.2017
Suresh, K., Servinsky, L., Reyes, J., Baksh, S., Undem, C., Caterina, M., et al. (2015). Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L1467–L1477. doi: 10.1152/ajplung.00275.2015
Takada, Y., Numata, T., and Mori, Y. (2013). Targeting TRPs in neurodegenerative disorders. Curr. Top. Med. Chem. 13, 322–334. doi: 10.2174/1568026611313030009
Tang, Q., Guo, W., Zheng, L., Wu, J. X., Liu, M., Zhou, X., et al. (2018). Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res. 28, 746–755. doi: 10.1038/s41422-018-0038-2
Tedeschi, V., Petrozziello, T., Sisalli, M. J., Boscia, F., Canzoniero, L. M. T., and Secondo, A. (2019). The activation of Mucolipin TRP channel 1 (TRPML1) protects motor neurons from L-BMAA neurotoxicity by promoting autophagic clearance. Sci. Rep. 9:10743. doi: 10.1038/s41598-019-46708-5
Tian, D., Jacobo, S. M., Billing, D., Rozkalne, A., Gage, S. D., Anagnostou, T., et al. (2010). Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci. Signal. 3:ra77. doi: 10.1126/scisignal.2001200
Uhlen, M., Fagerberg, L., Hallstrom, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419
Vazquez, G., Wedel, B. J., Aziz, O., Trebak, M., and Putney, J. W. Jr. (2004). The mammalian TRPC cation channels. Biochim. Biophys. Acta 1742, 21–36. doi: 10.1016/j.bbamcr.2004.08.015
Wakabayashi, T., Hidaka, R., Fujimaki, S., Asashima, M., and Kuwabara, T. (2014). MicroRNAs and epigenetics in adult neurogenesis. Adv. Genet. 86, 27–44. doi: 10.1016/B978-0-12-800222-3.00002-4
Wang, H., Cheng, X., Tian, J., Xiao, Y., Tian, T., Xu, F., et al. (2020). TRPC channels: structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. doi: 10.1016/j.pharmthera.2020.107497 [Epub ahead of print].
Wang, J., Lu, R., Yang, J., Li, H., He, Z., Jing, N., et al. (2015). TRPC6 specifically interacts with APP to inhibit its cleavage by gamma-secretase and reduce Abeta production. Nat. Commun. 6:8876. doi: 10.1038/ncomms9876
Wang, Z., Zhou, L., An, D., Xu, W., Wu, C., Sha, S., et al. (2019). TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death Dis. 10:386. doi: 10.1038/s41419-019-1612-3
Wes, P. D., Chevesich, J., Jeromin, A., Rosenberg, C., Stetten, G., and Montell, C. (1995). TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. U.S.A. 92, 9652–9656. doi: 10.1073/pnas.92.21.9652
Wu, D., Huang, W., Richardson, P. M., Priestley, J. V., and Liu, M. (2008). TRPC4 in rat dorsal root ganglion neurons is increased after nerve injury and is necessary for neurite outgrowth. J. Biol. Chem. 283, 416–426. doi: 10.1074/jbc.m703177200
Wu, Q., Lu, K., Zhao, Z., Wang, B., Liu, H., Zhang, S., et al. (2019). Blockade of transient receptor potential vanilloid 4 enhances antioxidation after myocardial ischemia/reperfusion. Oxid. Med. Cell. Longev. 2019:7283683. doi: 10.1155/2019/7283683
Wu, X., Zagranichnaya, T. K., Gurda, G. T., Eves, E. M., and Villereal, M. L. (2004). A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J. Biol. Chem. 279, 43392–43402. doi: 10.1074/jbc.m408959200
Zhang, X., Huang, J., and Mcnaughton, P. A. (2005). NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 24, 4211–4223. doi: 10.1038/sj.emboj.7600893
Zhang, Z., Toth, B., Szollosi, A., Chen, J., and Csanady, L. (2018). Structure of a TRPM2 channel in complex with Ca(2+) explains unique gating regulation. eLife 7:e36409. doi: 10.7554/eLife.36409
Zheng, J., Winderickx, J., Franssens, V., and Liu, B. (2018). A mitochondria-associated oxidative stress perspective on Huntington’s disease. Front. Mol. Neurosci. 11:329. doi: 10.3389/fnmol.2018.00329
Zhou, F. W., Matta, S. G., and Zhou, F. M. (2008). Constitutively active TRPC3 channels regulate basal ganglia output neurons. J. Neurosci. 28, 473–482. doi: 10.1523/JNEUROSCI.3978-07.2008
Keywords: TRP, Ca2+, ROS, cell death, neurodegeneration, AD, PD, HD
Citation: Hong C, Jeong B, Park HJ, Chung JY, Lee JE, Kim J, Shin Y-C and So I (2020) TRP Channels as Emerging Therapeutic Targets for Neurodegenerative Diseases. Front. Physiol. 11:238. doi: 10.3389/fphys.2020.00238
Received: 20 December 2019; Accepted: 02 March 2020;
Published: 15 April 2020.
Edited by:
Mustafa Naziroglu, Süleyman Demirel University, TurkeyReviewed by:
Patricia Hidalgo, Forschungszentrum Jülich, GermanyMarco Segatto, University of Molise, Italy
Copyright © 2020 Hong, Jeong, Park, Chung, Lee, Kim, Shin and So. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Insuk So, aW5zdWtAc251LmFjLmty
†These authors have contributed equally to this work and share first authorship