 Eilidh A. MacDonald
Eilidh A. MacDonald Robert A. Rose
Robert A. Rose T. Alexander Quinn
T. Alexander Quinn
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 03 March 2020
Sec. Cardiac Electrophysiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00170
The sinoatrial node is perhaps one of the most important tissues in the entire body: it is the natural pacemaker of the heart, making it responsible for initiating each-and-every normal heartbeat. As such, its activity is heavily controlled, allowing heart rate to rapidly adapt to changes in physiological demand. Control of sinoatrial node activity, however, is complex, occurring through the autonomic nervous system and various circulating and locally released factors. In this review we discuss the coupled-clock pacemaker system and how its manipulation by neurohumoral signaling alters heart rate, considering the multitude of canonical and non-canonical agents that are known to modulate sinoatrial node activity. For each, we discuss the principal receptors involved and known intracellular signaling and protein targets, highlighting gaps in our knowledge and understanding from experimental models and human studies that represent areas for future research.
The heart’s natural pacemaker, the sinoatrial node (SAN), spontaneously initiates each heartbeat. Located adjacent to the crista terminalis at the junction of the right atrium and the superior vena cava, the SAN fires action potentials (AP) that propagate through the electrical conduction system and the myocardium, initiating contraction. This specialized group of cells is necessary for life, as its rhythmical firing leads to the pumping of blood to the rest of the body. Because its function is so essential, the SAN has many redundant systems in place to ensure it continues to consistently generate APs (Difrancesco and Noble, 2012; Maltsev and Lakatta, 2012; Rosen et al., 2012). The frequency of AP firing from the SAN determines heart rate (HR), and there are a multitude of factors that influence the systems that generate the SAN AP. Thus, SAN activity is tightly controlled to maintain normal heart rhythm and allow for adaption to changes in physiological demand.
Modulation of SAN activity occurs through intracellular signaling and its alteration by neurohumoral agents, including neurotransmitters and neuropeptides, and autocrine, paracrine, and endocrine factors (Irisawa et al., 1993; MacDonald et al., 2017), as well as response to changes in mechanical load (Quinn and Kohl, 2012). Neurohumoral agents primarily (although not exclusively) activate a variety of G-protein coupled receptors (GPCR) in the sarcolemma of SAN myocytes (Marin-Garcia, 2011). GPCR in turn activate second messenger cascades that target intracellular components responsible for SAN automaticity, causing a change in HR (Mangoni and Nargeot, 2008; Figure 1). Many of the factors that modulate SAN activity have been extensively studied and are understood to be principally important for the control of HR. Other factors, especially those with secondary effects on the SAN, are less well understood, complicated by contradictory results in the literature. A list of neurohumoral agents, their receptors, and their effect on HR is found in Table 1.
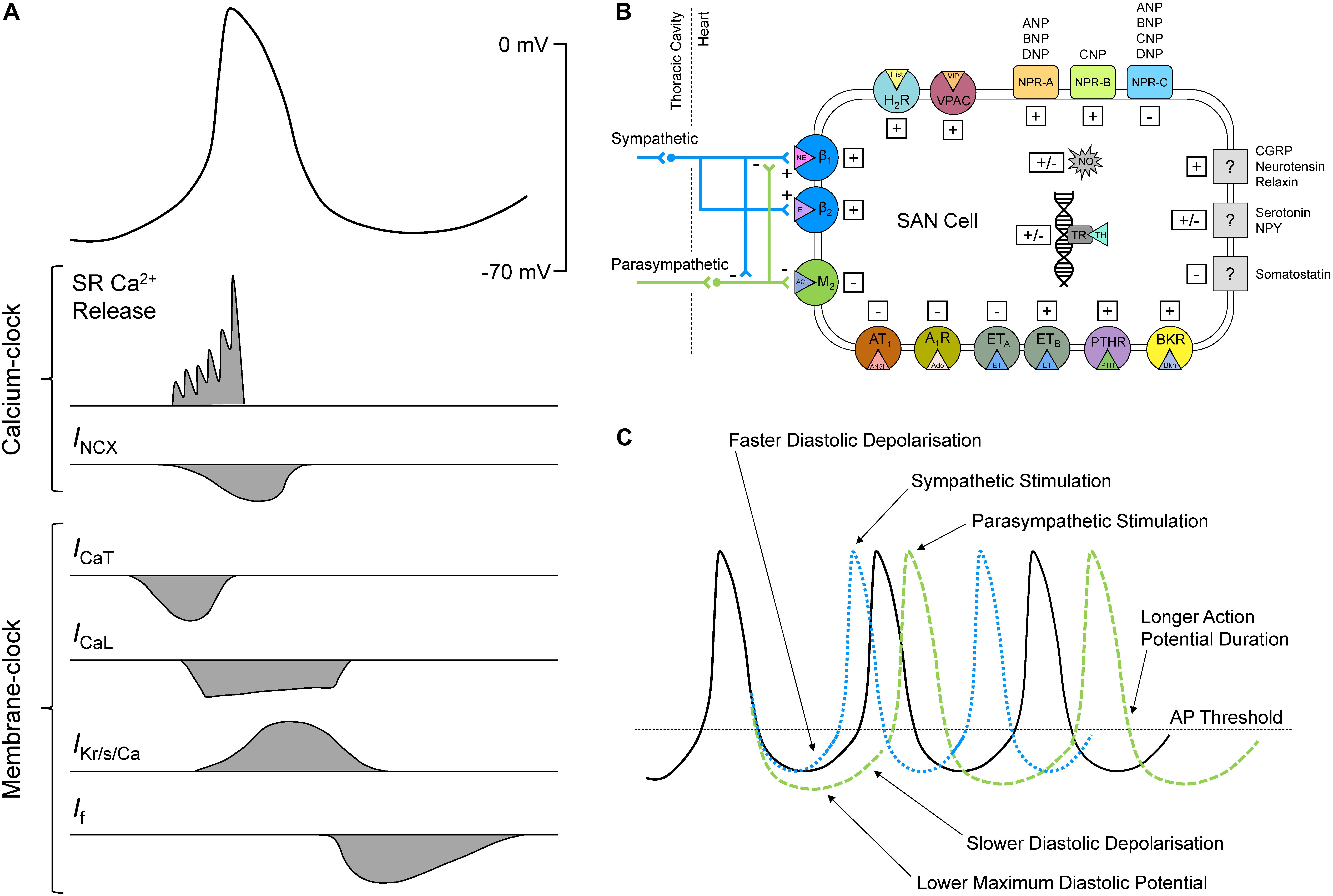
Figure 1. Sinoatrial node (SAN) cell action potential (AP) and ionic fluxes responsible for automaticity (A), neurotransmitters and neuropeptides, autocrine, paracrine, and endocrine factors, and other biologically active agents with known effects on SAN activity and heart rate (B), effect of sympathetic and parasympathetic stimulation on the SAN AP (C). A1R, adenosine receptor 1; ACh, acetylcholine; Ado, adenosine; ANGII, angiotensin II; ANP, atrial natriuretic peptide; AT1, angiotensin receptor 1; BNP, brain natriuretic peptide; Bkn, bradykinin; BKR, bradykinin receptor; Ca2+, calcium; CGRP, calcitonin gene-related peptide; CNP, C-type natriuretic peptide; DNP, Dendroaspis natriuretic peptide; E, epinephrine; ET, endothelin; ETA/B, endothelin receptor A/B; H2R, histamine H2 receptor; Hist, histamine; ICaL, long-lasting (L-type) Ca2+ current; ICaT, transient (T-type) Ca2+ current; If, “funny” current; IKr/s/Ca, rapid delayed rectifier / slow delayed rectifier / Ca2+-activated K+ currents; INCX, Na+-Ca2+ exchanger current; M2, muscarinic receptor 2; NE, norepinephrine; NO, nitric oxide; NPR, natriuretic peptide receptor; NPY, neuropeptide Y; PTH, parathyroid hormone; PTHR, PTH receptor; SR, sarcoplasmic reticulum; TH, thyroid hormone; TR, thyroid receptor; VIP, vasoactive intestinal polypeptide; VPAC, vasoactive intestinal polypeptide receptor.
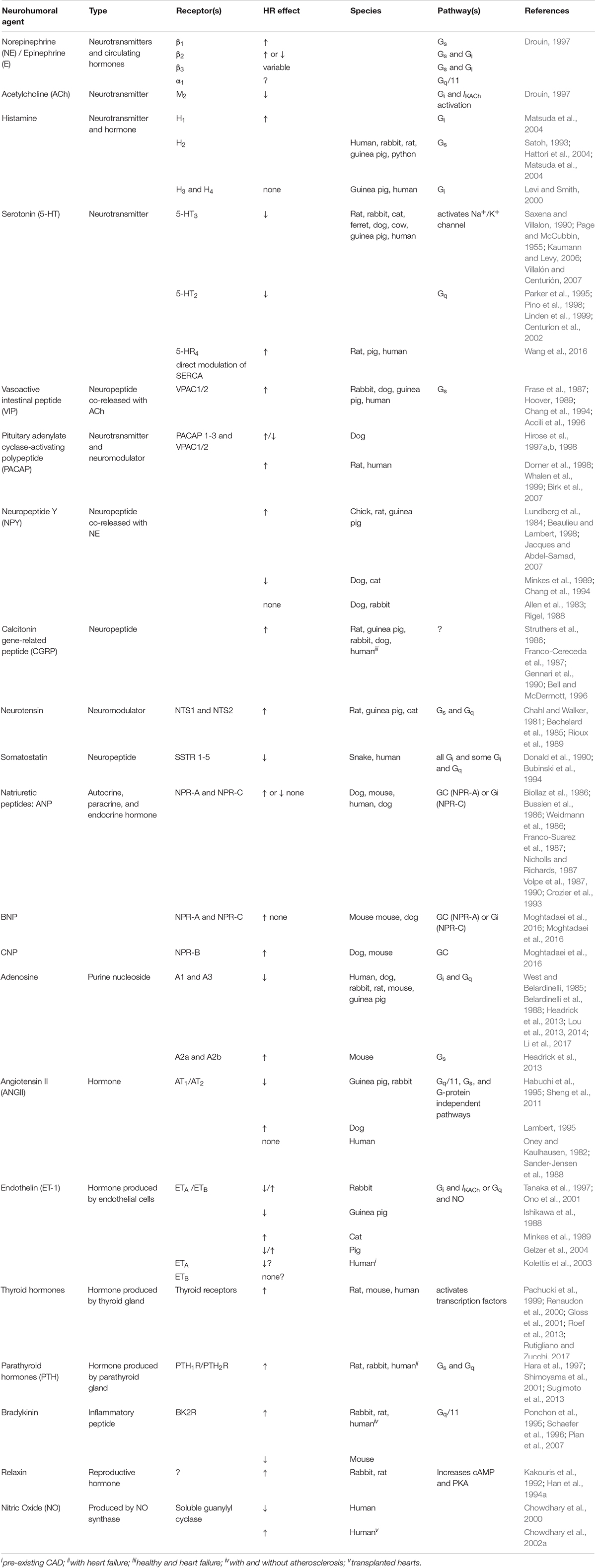
Table 1. Summary of neurohumoral agents with known effects on heart rate (HR).
In this review, we first provide a brief overview of the mechanisms that drive SAN automaticity, to introduce the principal targets for modulation of SAN activity by neurohumoral signaling. This is followed by consideration of neurotransmitters and neuropeptides, and autocrine, paracrine, and endocrine factors with known effects on SAN activity and HR, including their receptors, intracellular signaling, and protein targets, in particular highlighting what is known from experimental models, what has been confirmed in human studies, and where gaps in our current understanding exist, to identify areas for future research.
Cardiac automaticity is driven by depolarization of SAN membrane potential during diastole (spontaneous diastolic depolarization, SDD; Figure 1). The principal mechanisms responsible for SDD have been heavily debated (Rosen et al., 2012). The two concepts that have emerged are that of a “membrane-clock,” resulting from a flow of ions through trans-sarcolemmal channels, and a “Ca2+-clock,” resulting from intracellular Ca2+ cycling (Bartos et al., 2015). It is now generally accepted that these are in fact overlapping and redundant systems that work together to cause SAN automaticity, and form a combined system often referred to as the “coupled-clock” (Lakatta et al., 2010; Rosen et al., 2012).
During early SDD, the membrane-clock is driven by an inward cation current known as the “funny” current (If), passing through hyperpolarization-activated cyclic nucleotide-gated (HCN) channels (Difrancesco, 2010). There are four isoforms of HCN (HCN1-4), with HCN1, 2, and 4 expressed in the human heart, notably more prominently in the SAN than the atria, and with HCN1 specifically being expressed almost exclusively in the SAN (Li et al., 2015). Studies of HCN1 knock-out in mice support these findings, demonstrating high expression and critical importance of HCN1 for maintenance of HR and beat-to-beat stability (Fenske et al., 2013). Along with If, inward Ca2+ currents through both Cav3.1 transient (T-type, ICa,T) and Cav1.2/1.3 long-lasting (L-type, ICaL) channels are important for SAN firing (Mangoni and Nargeot, 2008). Cav3.1 and Cav1.3 activate at relatively low thresholds (∼ –70 and –55 mV, respectively), allowing them to contribute to the early portion of SDD (Mangoni and Nargeot, 2008). Once the threshold for Cav1.2 activation is reached (∼ –40 mV), ICaL generates the upstroke of the SAN AP [unlike working cardiomyocytes, in which the fast sodium (Na+) current (INa), passing through Nav1.5 channels, is responsible for the AP upstroke] (Mesirca et al., 2015). Nav1.5 channels are heterogeneously expressed in the SAN, and primarily at the periphery, so while they are not necessary for SAN automaticity, they can indirectly influence pacemaker rate (Lei et al., 2005). Other heterogeneously expressed currents have been suggested to also contribute to SAN automaticity, such as the current passed by transient receptor potential-canonical (TRPC) channels (Ju et al., 2007) and a sustained inward current (Ist) (Zhang et al., 2002), which has been postulated to represent the combined effects of ICaL and current through the Na+-Ca2+ exchanger (INCX) (Lakatta et al., 2010). In fact, the Na+-Ca2+ exchanger plays a fundamental role in SAN automaticity, and even though it could be included in the membrane clock as it generates a transmembrane current, it is fundamental in the “Ca2+-clock” system, so is discussed in the following section.
Another important consideration for SAN activity is the repolarizing currents, which determine the maximum diastolic membrane potential (MDP, the most negative occurring membrane potential), a key driver for If. The rapid and slow delayed rectifier K+ currents (IKr and IKs, respectively) are the primary repolarizing currents in SAN myocytes, with their decay at the end of the AP allowing for inward cation currents to cause SDD (Mangoni and Nargeot, 2008). Important for the occurrence of SDD, in primary SAN myocytes there is very little inward rectifier potassium (K+) current (IK1, passed by Kir2.1-2.4 channels) (Chandler et al., 2009), which in working myocytes stabilizes diastolic membrane potential at a resting value of ∼ –90 mV. It has also been suggested that the Ca2+-activated K+ channel current (IK,Ca) may be important for the early portion of SDD by maintaining MDP, and thus the driving force for If, as their blockade can result in bradycardia or complete suppression of excitation (Weisbrod et al., 2016). There is evidence of other K+ currents in SAN myocytes that may contribute to repolarization, including: the transient outward K+ current (Ito); the ultra-rapid delayed rectifier K+ current (IKur); the acetylcholine (ACh)-activated K+ current (IKACh); and the adenosine triphosphate (ATP)-sensitive K+ current (IK,ATP) (Chandler et al., 2009). In addition, the inwardly rectifying chloride current (ICl), which is activated late during the upstroke, as well as current through the Na+-K+ ATPase (which extrudes three Na+ ions while bringing two K+ ions into the cell, thus generating a net outward current) may contribute to SAN repolarization and the MDP (Mangoni and Nargeot, 2008).
Intracellular Ca2+ cycling has been shown to be a fundamental contributor to SAN automaticity. During late SDD in SAN myocytes, there are localized spontaneous and/or Cav1.3-triggered sub-sarcolemmal Ca2+ releases from the sarcoplasmic reticulum (SR) via ryanodine receptors (RyR), which are in close proximity to the Na+-Ca2+ exchanger (Lakatta et al., 2010; Torrente et al., 2016). These local Ca2+ releases are large in size and duration and rhythmic in nature (unlike Ca2+ sparks emerging from the SR in ventricular cells, which are smaller, stochastic events, Sirenko et al., 2013), and result in a depolarizing current as 1 Ca2+ ion is extruded from the cell in exchange for 3 Na+ ions through the Na+-Ca2+ exchanger (INCX) (Lakatta et al., 2008). The importance of this mechanism for SAN firing has been demonstrated by INCX blockade in guinea pig and rabbit SAN myocytes (Bogdanov et al., 2001, 2006; Sanders et al., 2006), as well as by inhibition of RyR in guinea pig (Rigg et al., 2000), rabbit (Hata et al., 1996; Satoh, 1997; Bogdanov et al., 2001; Bucchi et al., 2007; Lyashkov et al., 2007), mouse (Masumiya et al., 2003; Ju et al., 2007; Nikmaram et al., 2008; Wu et al., 2009), and dog (Vinogradova et al., 2006; Joung et al., 2009). As a result, the rate and size of local Ca2+ releases, and the balance between Ca2+ reuptake into the SR [through the SR Ca2+-ATPase (SERCA) pump] and extrusion through the Na+-Ca2+ exchanger are key determinants of the rate of diastolic depolarization. Importantly, SAN myocytes have a higher basal level of cyclic adenosine monophosphate (cAMP), and thus protein kinase A (PKA)-dependent phosphorylation of Ca2+-handling proteins than working myocytes, resulting in a greater level of SR Ca2+ cycling, which facilitates Ca2+ clock function (Vinogradova et al., 2006). It has also been shown that SAN myocytes have higher basal levels of Ca2+/calmodulin dependent protein kinase II (CaMKII) compared to working myocytes, resulting in higher basal phosphorylation of L-type Ca2+ channels, RyR, and phospholamban (PLB), which when unphosphorylated inhibits SERCA (Vinogradova et al., 2000; Li et al., 2016).
The membrane- and Ca2-clocks together form a cohesive and robust system for SAN automaticity. Their coupling occurs via effects of one on the other and through the mutual entrainment of sarcolemmal ion channel activity and Ca2+ cycling by intracellular regulatory mechanisms (Lakatta et al., 2008). For instance, clock-coupling occurs as membrane repolarization by K+ channels (key to the membrane-clock) both activates If and affects intracellular Ca2+ balance (and in turn the Ca2+ clock) via voltage-dependent effects on ICaL and INCX (Lakatta et al., 2010). Vice versa, INCX (essential for the Ca2+-clock) directly affects membrane potential (and thus voltage-dependent components of the membrane-clock) (Bogdanov et al., 2006). This also highlights that some currents, such as ICaL and INCX, are important components of both clocks, such that they are intricately linked. The fidelity of clock coupling is altered through simultaneous modifications of components within each system. For instance, neurohumoral alteration of PKA phosphorylation levels [through changes in adenylyl cyclase (AC) activity, the protein that converts ATP into cAMP] entrains the two clocks (Mattick et al., 2007; Vinogradova and Lakatta, 2009). In guinea pig and rabbit, this has been shown to be enhanced by Ca2+-activated isoforms of AC (important for basal AC activity in the absence of β-AR stimulation) found in SAN, but not working myocytes, which also acts to drive intracellular clock-coupling (Mattick et al., 2007; Younes et al., 2008).
And yet, one should note that oscillatory behavior, as occurs in the SAN, can result from a combination of activities that themselves are not oscillatory. In fact, neither If, nor INCX, nor the decay of IK can in itself produce the oscillations in membrane potential that are key to the rhythmic excitation of the SAN. It is only their combined, out-of-phase activity that allows for SAN automaticity. Even ion fluxes not typically considered “pacemaker currents” are essential coupled-clock components, such as the flux generated by the Na+-K+ ATPase, which helps maintain electrochemical gradients and ionic homeostasis, which are crucial for SAN pacemaking (Sirenko et al., 2016). Thus, SAN automaticity may continue even with the loss of one (or more) of the currents described above, although important for the subject of this review, this may result in the loss of important neurohumoral control mechanisms.
While the mechanisms of SAN automaticity described above are well conserved, there exist species-specific differences in SAN function potentially important for neurohumoral control of HR, which may provide some explanation for inconsistencies of experimental results. For instance, the range of basal HR across mammals is large, with an inverse relation to animal weight (Opthof, 2001). The normal resting HR of mouse is approximately 10-fold faster than human (∼700 vs. 70 beats/min), whereas rabbit falls somewhere between (∼250 beats/min). This is in part related to differences in the balance of sympathetic and parasympathetic nervous system control of the SAN. Although activation of both branches of the autonomic nervous system (ANS) alter HR in all species studied, there are species-differences in the relative contribution of the sympathetic and parasympathetic branches to basal HR. Specifically, in mouse and rat, resting HR is predominantly determined by sympathetic tone (Adachi et al., 2013), whereas in large animals, such as dog and human, HR is mostly determined by basal parasympathetic activity (Opthof, 2001). The predominance of sympathetic activity in mouse and rat is driven primarily by a need for non-shivering thermogenesis (via sympathetic stimulation of brown fat) when acclimated to room temperature, to maintain core body temperature (Axsom et al., 2019). At thermoneutral temperatures (∼30°C) sympathetic activity is reduced, unmasking parasympathetic input and significantly lowering HR (Axsom et al., 2019). Characteristics of the SAN AP also differ across species. In mouse, AP duration is ∼80 ms, in rabbit it is ∼200 ms, and in human it is ∼300 ms (Opthof, 2001). These differences in AP duration suggest relative differences in ionic current densities. It is known, for instance that while IK1 is almost entirely not present in rabbit SAN myocytes, it is active in some (but not all) mouse SAN pacemaker cells, although still with a much smaller current than in working cardiomyocytes (Cho et al., 2003). Further, differences in ionic current densities could relate to differences in post-translational modifications or activation and inactivation kinetics of various currents across species.
Important species-specific differences in intracellular Ca2+ handling also exist. In rabbit SAN myocytes it has been shown that Ca2+ release from the SR via RyR during SDD is spontaneous (Bogdanov et al., 2001), while in mouse Ca2+ release is instead trigged by Cav1.3-mediated ICaL (Torrente et al., 2016), and in cat by voltage-dependent activation of T-type Ca2+ channels (Hüser et al., 1996). In fact, α1D (which is the pore forming subunit of Cav1.3) L-type Ca2+ channels are present exclusively in the atria of mouse, rat, rabbit, and human and are thought to contribute to SDD, based on their activation at more negative membrane potentials than the most predominantly expressed L-type Ca2+ channel (with the α1C pore-forming subunit), which is found throughout the heart (Mangoni et al., 2003; Qu et al., 2004). Removal of Ca2+ from the cytosol also differs, as in mouse and rat ∼92% of Ca2+ is pumped back into the SR by SERCA and only ∼7% is extruded through the Na+-Ca2+ exchanger, whereas in rabbit and human only 70% returns to the SR and 28% is extruded from the cell (Bers, 2000). As a result, INCX and its contribution to SDD is greater in rabbit/human than mouse/rat.
The SAN is strongly influenced by the ANS, with a variety of neurotransmitters [catecholamines such as norepinephrine (NE) and epinephrine, ACh, histamine, serotonin (5-HT)], as well as a host of neuropeptides (vasoactive intestinal polypeptide, calcitonin gene-related peptide, neuropeptide Y, neurotensin, and somatostatin) released by neurons in the heart that alter SAN activity and thus HR (Gordan et al., 2015).
In general, these substances bind to a GPCR, causing a conformational change in its cytoplasmic domains, which activates its subunits (Gα and Gβγ) (Oldham and Hamm, 2008; Rosenbaum et al., 2009; Zhang et al., 2015). The Gα isoforms of primary importance in the SAN are Gαs, Gαi/o, and Gαq/11 (Marin-Garcia, 2011). In the “stimulatory” Gαs pathway, activation of AC increases cAMP, which directly or indirectly (via PKA activation and subsequent phosphorylation) activates downstream targets (Gordan et al., 2015). When the “inhibitory” Gαi/o pathway is initiated, either Gαi or Gαo binds and inhibits AC, decreasing cAMP levels and preventing PKA activation (Gordan et al., 2015). Activation of Gαq/11 on the other hand does not alter cAMP levels, but instead either Gαq or Gα11 activates phospholipase C (PLC), which breaks down phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol triphosphate (IP3), both of which affect multiple downstream targets (Marin-Garcia, 2011). And while the Gα subunits initiate the primary signaling cascades, Gβγ subunits may also be involved (Zhang et al., 2015). Another important signaling pathway in SAN myocytes occurs through particulate or soluble guanylyl cylcase (GC) activation, which results in cGMP activation and modulation of downstream targets by protein kinase G (PKG) or other downstream regulatory molecules (Zaccolo and Movsesian, 2007).
Catecholamines (NE and epinephrine) are released from sympathetic neurons of the ANS (Gordan et al., 2015) (although both are also produced by the adrenal gland and can act as circulating hormones, or can be intrinsically generated by cells within the SAN) (Marin-Garcia, 2011; Moen et al., 2019). NE and epinephrine bind to α- and β-adrenergic receptors (AR). There are four subtypes of β-AR: β1-AR are the most abundant in the heart (75–80%); β2-AR are expressed in the heart, but to a much smaller extent than β1-AR, and are mostly compartmentalized in caveolae (Rybin et al., 2000); β3-AR are only minimally expressed in the heart; and β4-AR have not yet been thoroughly characterized (Madamanchi, 2007).
β1-AR are coupled to Gs proteins, so initiate the stimulatory AC/cAMP/PKA cascade (Figure 2), which has been shown to be essential for the increase in HR during adrenergic stimulation in rabbit SAN myocytes (Behar et al., 2016). β1-AR stimulation in the SAN increases HR through Gαs-activation of AC, resulting in an increase in cAMP and PKA, which modulate the activity of various intracellular targets (Gordan et al., 2015). The combined action on these targets causes an increase in the rate of SDD, thus increasing SAN firing (Figure 1). For instance, in rabbit SAN myocytes the binding of cAMP to C-terminals of HCN channels causes a depolarizing shift in their activation kinetics, increasing open probability and If (DiFrancesco and Tortora, 1991; Barbuti and DiFrancesco, 2008). At the same time, PKA increases If by a phosphorylation-driven shift of its voltage-dependence of activation, as shown in mouse SAN myocytes (Liao et al., 2010). Although it is still not entirely clear whether cAMP and PKA work synergistically or independently on If during β1-AR stimulation, it is clear that they both cause an increase in HR (Larsson, 2010). Further, inhibition of If results in a moderate decrease in the effect of β-AR stimulation in rabbit (Noma et al., 1983; Vinogradova et al., 2002), mouse (Choate and Feldman, 2003), and rat (Yamamoto et al., 2006), demonstrating its key contribution to the chronotropic response. Yet, while it is apparent that an alteration in If during β1-AR stimulation modulates HR, it is still increased by β1-AR stimulation in animals in which HCN is deficient or absent, suggesting that other mechanisms are also involved (Vinogradova and Lakatta, 2009).
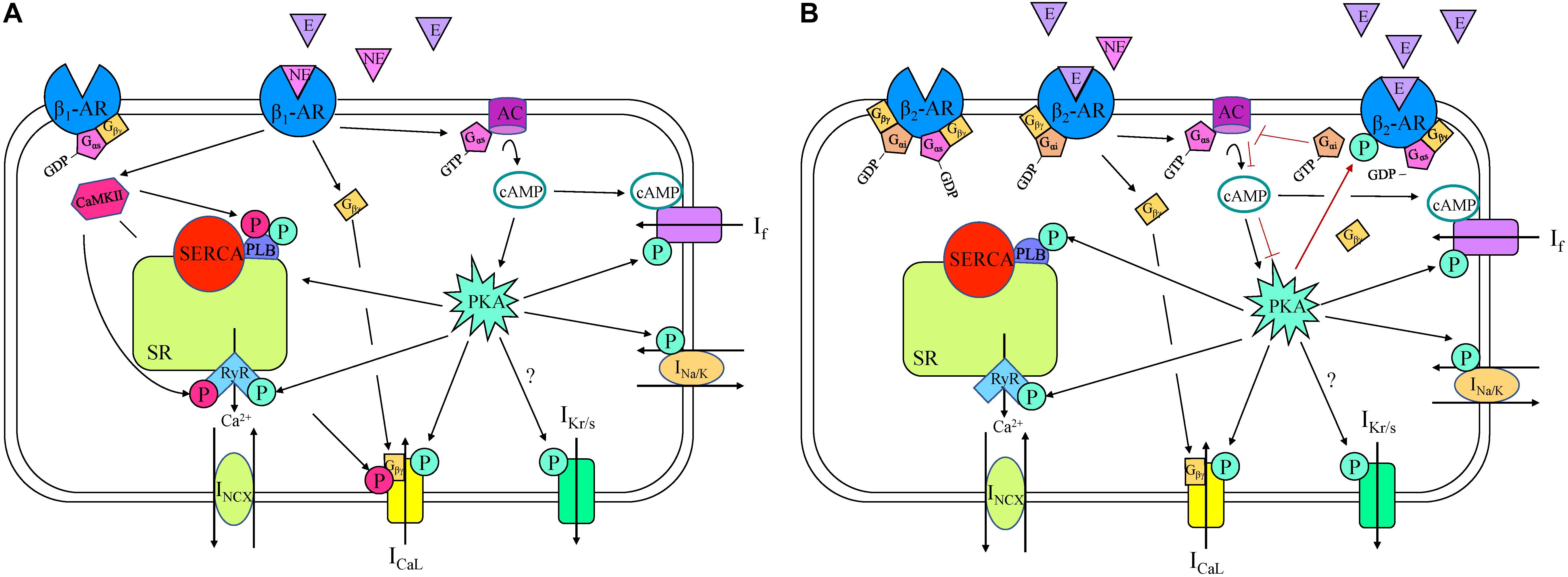
Figure 2. β1- (A) and β2- (B) adrenergic receptor (AR) signaling cascades in a sinoatrial node myocyte. AC, adenylyl cyclase; Ca2+, calcium; CaMKII, Ca2+/calmodulin-dependent protein kinase; cAMP, cyclic adenosine monophosphate; E, epinephrine; GDP, guanosine diphosphate; GTP, guanosine triphosphate; ICaL, long-lasting (L-type) Ca2+ current; If, “funny” current; IKr/s, rapid/slow delayed rectifier K+ current; INa/K, Na+-K+ ATPase current; INCX, Na+-Ca2+ exchanger current; NE, norepinephrine; P, phosphorylation; PKA, phosphokinase A; PLB, phospholamban; RyR, ryanodine receptor, SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum.
PKA also phosphorylates numerous other coupled-clock components contributing to SDD. Phosphorylation of the α1 subunit of L-type Ca2+ channels, purified from bovine hearts, increases their open probability, thus increasing current (De Jongh et al., 1996). It has also been shown that the Gαs subunit can directly stimulate L-type Ca2+ channels (Yatani et al., 1988) (T-type Ca2+ channels, on the other hand, are not affected by β1-AR stimulation in rabbit SAN myocytes, Hagiwara et al., 1988). Delayed-rectifier K+ channels show an increase in rate of deactivation and an increase in current amplitude following β1-AR stimulation in rabbit SAN myocytes, contributing to the increase in HR (Lei et al., 2000). β1-AR activation also increases Na+-K+ ATPase activity, which is an important compensatory mechanism for the maintenance of ionic homeostasis during increases in membrane currents (Shimoni, 1999) and it modulates INa and ICl, however, the mechanism and the significance of this is uncertain, and likely neither of these contribute in a significant way to the β1-AR-related increase in HR (Mangoni and Nargeot, 2008). It has also been shown that there are phosphorylation sites on the Na+-Ca2+ exchanger, yet whether its phosphorylation occurs or is functionally relevant following β1-AR stimulation in the SAN remains unknown (Vinogradova and Lakatta, 2009).
Although some groups exclusively emphasize the role of effects on sarcolemmal currents in increased HR during β1-AR activation (Bucchi et al., 2007; Himeno et al., 2008), there is also evidence demonstrating a role of alterations in Ca2+-clock components in the response (Lyashkov et al., 2018). Specifically, the effect of RyR inhibition by ryanodine on the chronotropic response following β1-AR stimulation has been extensively studied in a multitude of species. In rabbit SAN cells, β1-AR activation increases the frequency and amplitude of local Ca2+ releases during SDD, which increases INCX, the rate of SDD, and in turn, AP frequency. This effect is eliminated when RyR are inhibited by ryanodine, despite a preserved increase in L-type Ca2+ current amplitude (Vinogradova et al., 2002). Although this result suggests that the target of β1-AR activation is indeed the Ca2+-clock, another study in rabbit SAN has suggested that the alteration of Ca2+ homeostasis by RyR suppression has an indirect effect on HR, by disrupting the increase in cAMP near HCN channels (Bucchi et al., 2003). Either way, studies in the mouse (Wu et al., 2009), guinea pig (Rigg et al., 2000), and dog (Vinogradova et al., 2006; Joung et al., 2009) have all demonstrated that suppression of RyR with ryanodine diminishes the chronotropic response to β1-AR stimulation, indicating a critical role for Ca2+ release from the SR. β1-AR activation also increases PLB phosphorylation in SAN cells (Vinogradova et al., 2006), releasing its inhibition on SERCA and causing more rapid Ca2+ re-uptake into the SR (Marin-Garcia, 2011), which increases SR Ca2+ content.
Basal levels of CaMKII-dependent phosphorylation of RyR, PLB, and L-type Ca2+ channels are present in rabbit (but not mouse) SAN myocytes (Vinogradova et al., 2000; Wu and Anderson, 2014; Li et al., 2016), suggesting a species-dependence of the role that CaMKII plays in the maintenance of basal HR. In both mouse and rabbit, however, CAMKII is important under physiological stress, with CAMKII levels increased by sympathetic stimulation (Wu and Anderson, 2014). For instance, when CaMKII is inhibited, mice are less responsive to β1-AR stimulation (Wu et al., 2009).
While sympathetic stimulation in the SAN primarily acts through β1-AR, automaticity may also be affected by activation of β2-, β3-, and α1-AR (Marin-Garcia, 2011). β2-AR are coupled to both Gαs and Gαi subunits, activating the Gαs cascade under normal physiological conditions, but under other conditions can switch to the Gαi cascade (Paur et al., 2012; Machuki et al., 2018). For example, when high levels of epinephrine are released from the adrenal gland during stress, binding to β2-AR initially activates the Gαs subunit (as demonstrated in ventricular myocytes), triggering the same signaling pathways as β1-AR stimulation, thus increasing HR (Figure 2). The resulting PKA produced by this signaling cascade, however, phosphorylates the receptor, initiating a switch from Gαs to Gαi activation (Fischmeister et al., 2006). Once the Gαi subunit is activated, AC activity is inhibited, decreasing intracellular cAMP, PKA, and HR (Madamanchi, 2007). In the SAN, these actions are facilitated by the co-localization of β2-AR with HCN4 channels in caveolae, thus restricting the increase in cAMP to specifically modulate If [the close proximity of the receptor and the effector allows for this compartmentalization by phosphodiesterases (PDEs)] (Barbuti and DiFrancesco, 2008). In contrast, in the piglet and rat SAN it has been shown that β2-AR are important in mediating the response to NE and epinephrine independent of PDE compartmentalization (Christ et al., 2009; Galindo-Tovar et al., 2010).
β3-AR have also not been thoroughly studied in the SAN and therefore the role they play in HR modulation is uncertain. One complicating factor is that different isoforms of β3-AR have been shown to differentially couple to Gαs or Gαi, although the existence of β3-AR isoforms have not been characterized in the heart (Machuki et al., 2018). Furthermore, likely due to the variable G-protein binding abilities of β3-AR, responses to β3-AR agonists are variable throughout the heart (Dessy and Balligand, 2010).
Catecholamines also bind to α-AR in the heart. α-AR are coupled to Gq/11-proteins, which activate PLC (Jensen et al., 2014), causing hydrolysis of PIP2 into IP3 and DAG (Marin-Garcia, 2011). When IP3 binds to IP3R on the SR membrane, this causes release of Ca2+, increasing cytosolic Ca2+ levels, which causes the further release of Ca2+ from the SR through RyR (Marin-Garcia, 2011). Three IP3R isoforms have been identified in the mouse SAN, and it has been shown that their activation can modulate automaticity via Ca2+-dependent mechanisms (Ju et al., 2011, 2012; Kapoor et al., 2015). DAG, on the other hand activates PKC, which phosphorylates various targets such as L-type Ca2+ channels and the Na+-Ca2+ exchanger (Baker, 2014). Yet some studies of the effects of α-AR in the SAN are conflicting. For instance, in isolated SAN from both young and adult rabbits, phenylephrine (which activates α-AR) does not alter HR (Hewett and Rosen, 1985). Despite this result, greater expression of α-AR occurs in the SAN compared to surrounding myocardium in rat, suggesting a role for α-AR in HR control (Saito et al., 1994).
The control of HR by the sympathetic ANS is well documented in humans, as stimulation of sympathetic neurons in the human SAN increase HR and blockade of sympathetic receptors decreases HR (James et al., 1970), but many of the mechanistic details observed in experimental model systems have yet to be confirmed. β1- and β2-AR are both predominant in the human SAN, and β2-AR expression is more than two times higher in the SAN than the surrounding atrial tissue (Rodefeld et al., 1996). In the isolated human SAN isoproterenol has been shown to decrease cycle length and shorten AP duration by increasing the rate of repolarization, while also hyperpolarizing the MDP, consistent with what has been shown in animal models (Drouin, 1997). Yet, in contrast to what has been shown in animals, administration of phenylephrine in human causes either an increase (a response that is attenuated with age) (Saitoh et al., 1995) or a decrease in HR (Mary-Rabine et al., 1978; Drugge et al., 1985).
The parasympathetic branch of the ANS counterbalances sympathetic effects. Parasympathetic neurons release ACh, which binds to muscarinic receptors in the sarcolemma (Gordan et al., 2015). There are five muscarinic subtypes that have been identified, but it is the M2-receptor (M2R) that elicits the functional response in the human heart (Harvey, 2012). In the SAN, ACh activation of M2R (a GPCR) leads to decreased HR as it is coupled to Gi-proteins (Figure 3; Harvey, 2012). In rabbit SAN myocytes, this decrease in HR is associated with a negative shift of MDP, a decrease in SDD rate, and an increase in AP duration (Verkerk et al., 2012; Figure 1). Multiple M2R-mediated pathways have been shown in rabbit SAN cells to produce these effects over different time scales and with different ACh concentrations: activation of IKACh, inhibition of If, and inhibition of Ca2+-clock components (Accili et al., 1998). IKACh is the fastest-muscarinic pathway, with the response occurring in less than 2 s in rabbit SAN cells, as no cytosolic components are required for current activation (Accili et al., 1998). Specifically, again in rabbit SAN cells, when ACh binds M2R the Gβγ subunit activates G-protein regulated K+ (GIRK) channels, with the ensuing efflux of K+ causing a negative shift of MDP, slowing HR (Renaudon et al., 1997). GIRK1 and GIRK4 are both expressed in the heart and work together to form the channel (two α-subunits of each isoform) (Marin-Garcia, 2011), although in the human SAN GIRK expression is heterogeneous, with GIRK4 expression being highest in the center of the SAN (Li et al., 2017). Interestingly, SAN myocytes from GIRK4 knockout (KO) mice have a similar basal HR compared to control, but GIRK4 is indispensable for the generation of IKACh, which accounts for approximately half of the HR reduction provoked by vagal stimulation (Wickman et al., 1998).
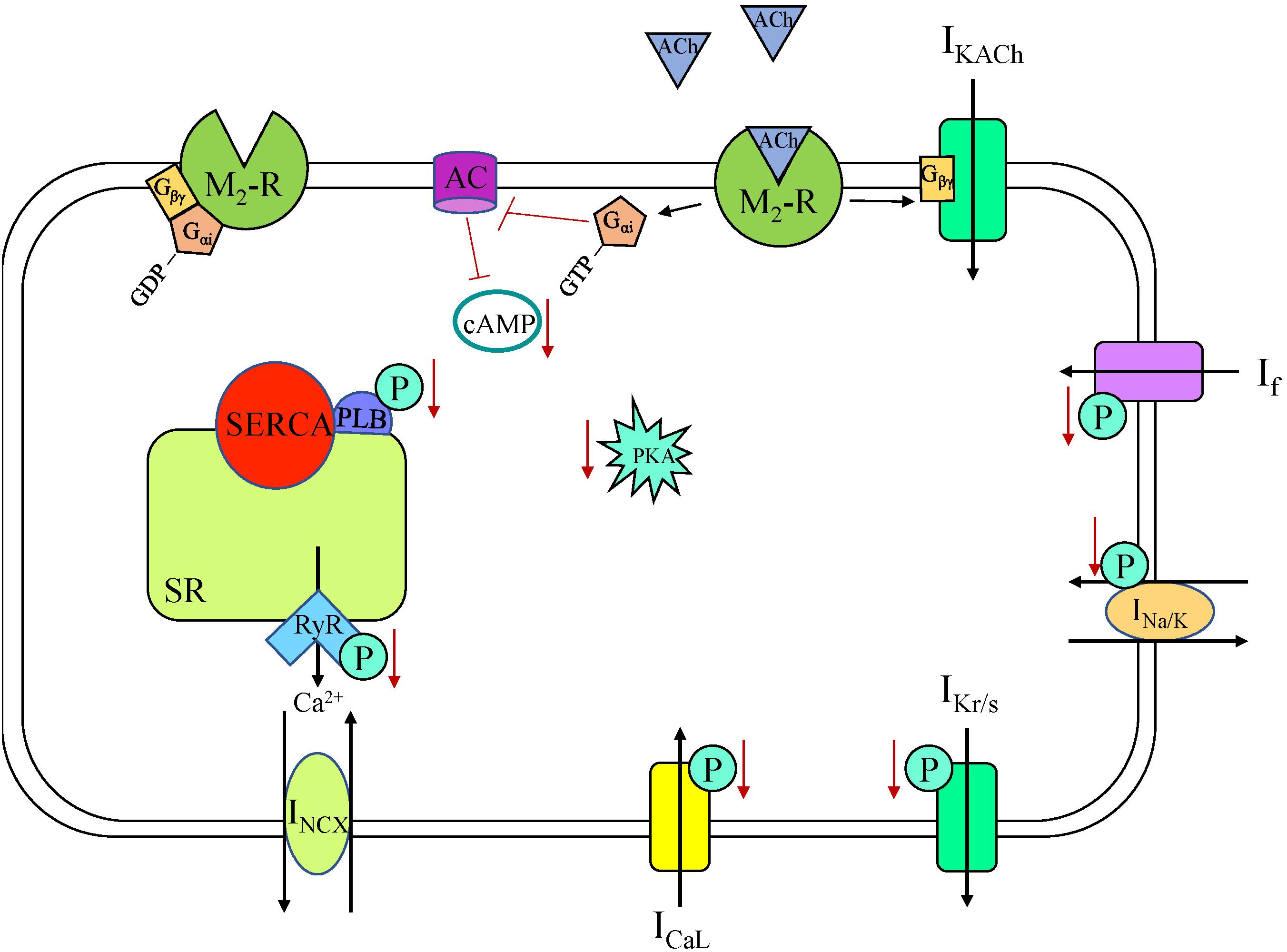
Figure 3. Muscarinic acetylcholine receptor (M2-R) signaling cascade in a sinoatrial node myocyte. AC, adenylyl cyclase; ACh, acetylcholine; cAMP, cyclic adenosine monophosphate; ICaL, long-lasting (L-type) Ca2+ current; If, “funny” current; IKACh, acetylcholine-activated K+ current; IKr/s, rapid/slow delayed rectifier K+ current; INa/K, Na+-K+ ATPase current; INCX, Na+-Ca2+ exchanger current; P, phosphorylation; PKA, phosphokinase A; PLB, phospholamban; RyR, ryanodine receptor, SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum.
While direct binding of the Gβγ subunit to GIRK elicits a response, multiple mechanisms for how this activates the channel have been proposed, based on data from Xenopus oocytes. One suggestion is that GIRK channels are intrinsically inhibited by a terminal domain of GIRK1 protein and binding of Gβγ removes inhibition and opens the channel (Reuveny et al., 1994). Another is that GIRK is activated by PIP2, but with a low sensitivity, and that Gβγ binding increases the affinity of GIRK for PIP2, thus increasing current activation (Huang et al., 1998). It is unclear, however, why the Gβγ subunit of Gs proteins do not activate IKACh (Marin-Garcia, 2011). This lack of response may be due to compartmentalization of M2R and GIRK channels, but this possibility requires further investigation.
The slower components causing decreased HR upon M2R activation are second messenger cascades. The activated Gαi subunit reduces AC activity in rabbit SAN cells, resulting in decreased cAMP levels, fewer active HCN channels (due to a negative shift in the voltage dependence of activation of If), and a reduction in the SDD slope (Accili et al., 1998; Van Borren et al., 2010). Activation of M2R with carbachol in rabbit SAN myocytes has been shown to have a dose-dependent effect on PLB phosphorylation and Ca2+-cycling via a Gi-dependent pathway (Lyashkov et al., 2009). The negative chronotropic effect of ACh was shown to be dependent upon cAMP/PKA-dependent phosphorylation both in single rabbit SAN cells and in a computational rabbit SAN cell model, through effects on multiple components of the coupled-clock system (Yaniv et al., 2013; Behar et al., 2016). At low concentrations carbachol has been shown to decrease HR without activating IKACh, acting only via a cAMP-mediated, PKA-dependent suppression of Ca2+ signaling (Lyashkov et al., 2009). With higher concentrations, carbachol additionally decreases HR via activation of IKACh (Lyashkov et al., 2009). These data demonstrate the importance of Ca2+ -clock suppression in the reduction in HR. Activation of M2R has also been shown to be coupled with NO synthesis, mediated by endothelial NO synthase (eNOS) (Han et al., 1994b). NO generation activates GC receptors, causing an increase in cGMP levels, PDE2 activation, inhibition of cAMP, reduced L-type Ca2+ channel phosphorylation, and ultimately decreased ICaL (Han et al., 1995). Overall, the combination of the direct effects of M2R stimulation, including a more negative MDP due to IKACh activation and a slower SDD due to reduced If and Ca2+ cycling, along with indirect NO/cGMP-mediated effects, leads to a decrease in HR in response to ACh release.
Although a vast amount of research has been done regarding the intracellular mechanisms for parasympathetic control of HR in the SAN of many experimental animals, the translation to humans is somewhat lacking. It is known that M2R density is nearly three times higher in the human SAN compared to surrounding atrial myocardium (Rodefeld et al., 1996) and that GIRK protein expression follows a similar trend (Li et al., 2017). In human isolated SAN, ACh increases cycle length and negatively shifts the MDP (Drouin, 1997). Direct stimulation of parasympathetic ganglia innervating the human SAN similarly causes a cycle length prolongation (Quan et al., 1999), and anticholinergic drugs slow HR (Nwazue et al., 2014). Although the HR effect of parasympathetic ANS activity is well characterized in human, some of the specific mechanisms demonstrated in animal models remain unconfirmed due to limitations of experimentation. This has been supplemented by the use of human-specific SAN cell computational models, which suggest that the voltage-dependence of M2R has significant effects on SDD, and that changes in the voltage-sensitivity of M2R may provide a mechanistic explanation for the increased sensitivity to changes in vagal tone in certain disease states (Moss et al., 2018).
Histamine released from neurons acts as a neurotransmitter but can also act as a hormone when released from mast cells and basophils in the heart (Marin-Garcia, 2011). There are four types of histamine receptors, all of which are GPCRs, with H1R coupled to the Gq/11 pathway activating PLC, H2R coupled to the Gs cascade activating cAMP and PKA, and both H3R and H4R coupled to Gi signaling (Hattori et al., 2004). Histamine receptor expression is highly variable across species and cardiac tissues, and although both H1R and H2R are expressed in the atria of rat, guinea pig, rabbit, and human hearts, H2R is most abundant and dominates cardiac responses (Matsuda et al., 2004), including in the SAN, where H2R stimulation increases HR via increased cAMP and PKA levels (Kevelaitis et al., 1994; Hattori et al., 2004). In rabbit SAN tissue this increase in HR is associated with a shift in the primary pacemaker site (Kevelaitis et al., 1994). In rabbit isolated SAN myocytes, histamine increases firing via H2R activation, and at high concentrations induces arrhythmias (Satoh, 1993). Interestingly, resident cardiac mast cells, the main supply of histamine in the heart, predominantly reside in close proximity to the SAN (Nistri et al., 2008). When these mast cells become activated due to an inflammatory response, such as during acute myocardial infarction or septic shock, they release histamine, which has been shown to cause severe tachyarrhythmias in pigs (Matsuda et al., 2004; Nistri et al., 2008). Yet, the role of histamine in the heart is complex, as activation of H3R can inhibit the arrhythmogenic increase in NE release from sympathetic nerve terminals that is associated with ischemia (Levi and Smith, 2000). Not only does histamine alter HR during an inflammatory response, it also has been shown to increase HR in the initial stages of digestion in pythons, a useful model system for postprandial tachycardia due to the magnitude and infrequency of their meals (Skovgaard et al., 2009). Although these studies and the varying receptor subtypes, signaling cascades, and receptor locations (both across tissue types and in nerve terminals) demonstrate the potential for a subtle and more complex role for histamine in HR control, the positive chronotropic effect via H2R stimulation is the most commonly reported finding, particularly in human (Wolff and Levi, 1986; Hu et al., 1997; Hattori et al., 2004; Matsuda et al., 2004). In fact, it has been suggested that that administration of H2R inhibitors could be used to decrease HR following autonomic blockade in people with SAN dysfunction (Hu et al., 1997).
The chronotropic response to serotonin (5-HT) is complex, as it binds to many different receptor types, both directly on cardiac tissue and also on autonomic nerve terminals, causing bradycardia, tachycardia, or sometimes both (Linden et al., 1999). There are at least fourteen different receptor types expressed in the heart which elicit inconsistent responses (Saxena and Villalon, 1990).
Many experimental studies have shown 5-HT to cause a decrease in HR. Intravenously administered 5-HT in the rat, rabbit, cat, ferret, dog, cow, and guinea pig causes an acute decrease in HR that can be blocked by vagotomy or atropine (James, 1964; Kaumann and Levy, 2006; Villalón and Centurión, 2007). This response is largely due to activation of 5-HT3-type receptors on parasympathetic neurons, which are coupled to a ligand-gated Na+/K+ channel. When opened, this channel causes depolarization, neurotransmitter release, and reflex bradycardia (Saxena and Villalon, 1990). Bradycardia can also be the result of 5-HT binding to 5-HT1 receptors on sympathetic neurons, causing their inhibition, or on parasympathetic neurons, causing their stimulation (Villalón and Centurión, 2007).
Conversely, an increase in HR with 5-HT has also been demonstrated experimentally. For example, in pithed rats, it has been shown that circulating 5-HT increases HR by binding to 5-HT2 receptors in the myocardium or on presynaptic sympathetic neurons (Gothert et al., 1986; Centurion et al., 2002). The 5-HT2 receptor is a Gq-protein coupled receptor, and activates the PLC signaling cascade (Villalón and Centurión, 2007). It was also recently shown in rat cardiomyocytes combined with computational modeling that SERCA can be serotonylated (receptor-independent activation directly by 5-HT), which alters SERCA’s affinity for Ca2+ and represents a potential mechanism for direct effects of 5-HT on SAN automaticity that warrants further investigation (Wang et al., 2016). It appears that intravenously administered 5-HT in pig, on the other hand, causes tachycardia via activation of 5-HT4 receptors, as inhibition of 5-HT4 abolished this response (Parker et al., 1995).
In humans, intravenously administered 5-HT has been shown to both increase or decrease HR, consistent with the variable response seen in animal models (Page and McCubbin, 1955; Kaumann and Levy, 2006). Although it has not been demonstrated experimentally, it is likely that bradycardia is the result of 5-HT3 receptor activation on parasympathetic neurons as demonstrated in animal models. Contrarily, the tachycardia is hypothesized to be due to activation of 5-HT4 receptors, as a number of studies have identified 5-HT4 antagonists that inhibit the 5-HT induced increase in contractility seen in the human right atrium (Kaumann et al., 1990, 1991, 1994; Kaumann, 1993; Parker et al., 1995). 5-HT4 receptor activation increases contractility via an increase in cAMP, and it is feasible that 5-HT would also increase HR in the SAN through a 5-HT4 receptor activation mediated increase in cAMP (Kaumann et al., 1991). This is further supported by the observation that 5-HT4 receptor activation increases If in human atrial myocytes, which might be expected to also occur in SAN myocytes (Pino et al., 1998). Interestingly, in humans it has also been shown that chronic treatment with β-blockers increases sensitivity to 5-HT in the atria, implicating cross-talk between 5-HT4 receptors, β-AR, and M2R (Sanders et al., 1995). 5-HT has also been implicated in a vast array of human pathological cardiac conditions, which is unsurprising given the large number of different receptor types and their locations (Kaumann and Levy, 2006). Overall, while many studies have assessed the effect of 5-HT on HR, a consistent effect on HR in humans and the mechanisms eliciting these responses remain to be clearly elucidated.
Vasoactive intestinal polypeptide (VIP) is a neuropeptide co-released with ACh from parasympathetic neurons, yet has the opposite effect of ACh, increasing HR (Mangoni and Nargeot, 2008). VIP binds to two receptor subtypes, VPAC1 and VPAC2, both of which are GPCRs that activate Gs-protein cascades, stimulating AC to produce cAMP, which triggers PKA (Marin-Garcia, 2011). VIP application on rabbit SAN myocytes and canine Purkinje fibers positively shifts the activation curve of If, which increases the rate of SDD and SAN firing (Chang et al., 1994; Accili et al., 1996). A dose-dependent increase in HR has been observed in guinea pig isolated hearts following bolus injections of VIP (Hoover, 1989). Similarly, when VIP is given intravenously to healthy humans, there is a dose-dependent increase in HR (Frase et al., 1987). Yet, still little is known about the role of VIP in human HR regulation (Henning and Sawmiller, 2001). Because VIP is released along with ACh but removed more slowly from the synapse, it is thought to be important for moderating the ACh-induced decrease in HR, contributing to the tachycardia seen after vagal stimulation (Mangoni and Nargeot, 2008), and possibly protecting against neutrally mediated bradycardia (Henning and Sawmiller, 2001). Interestingly, the affinity, density, responsiveness of VIP receptors changes in heart failure and hypertension, warranting further investigation of its role in disease progression and prevention in humans (Henning and Sawmiller, 2001).
Pituitary adenylate cyclase-activating polypeptide (PACAP) is a neurotransmitter and modulator which is very closely related to VIP (Henning and Sawmiller, 2001). It binds to three GPCR subtypes, two of which are identical to VPAC1 and VPAC2 (Hirose et al., 1997a). In a group of studies done in the dog (under anesthesia, in the isolated heart, and in the atria), PACAP caused a biphasic chronotropic response, with an initial increase, followed by a decrease in HR (Hirose et al., 1997a, b, 1998). In the anesthetized dog, the PACAP-induced decrease in HR is abolished following M2R blockade, indicating that the change in HR is likely due to activation of parasympathetic nerves (Hirose et al., 1997a). Interestingly, in the dog it appears that the increase in HR with PACAP is not mediated through β-ARs (as the response is not attenuated by β-AR blockade), but is due to activation of PACAP receptors directly on the tissue (Hirose et al., 1997a). The specific intracellular pathways eliciting this response remain unknown, however, in other tissues PACAP has been reported to increase cAMP, which could explain the positive chronotropic response. Conversely, in the anesthetized rat, HR increases with PACAP administration and the response is abolished by β-AR blockade, indicating that in this species the positive chronotropic response is mediated by neuromodulation of sympathetic neurons, rather than a direct effect on SAN tissue (Whalen et al., 1999). Although not much is known about the role of PACAP in the human SAN, in two separate studies on young, healthy humans, PACAP caused an increase in HR (Dorner et al., 1998; Birk et al., 2007).
Neuropeptide Y (NPY) is co-localized with NE and expressed by sympathetic neurons innervating the cardiovascular system (Marin-Garcia, 2011). NPY binds to a group of GPCRs (Y1, Y2, and Y5) which activate the Gi/o signaling cascade (Marin-Garcia, 2011). Chronotropic responses vary across species and experimental preparations, with NPY having been shown to cause inconsistent changes in HR (Beaulieu and Lambert, 1998). NPY pre-synaptically inhibits NE release from sympathetic neurons, but can also potentiate post-synaptic effects of NE, therefore working both with and against sympathetic nervous system stimulation of the SAN (Beaulieu and Lambert, 1998). In embryonic chick ventricular myocytes, NPY increases spontaneous firing frequency by increasing stimulation of L-type Ca2+ channels via activation of Y1 receptors (Jacques and Abdel-Samad, 2007). An increase in firing rate is observed with NPY application in guinea pig isolated atria and rat isolated hearts (Lundberg et al., 1984; Beaulieu and Lambert, 1998). Conversely, in canine Purkinje fibers, NPY reduces If (either via a decrease in maximal If conductance or via If current blockade, rather than altering the voltage-dependence) causing decreased HR (Chang et al., 1994). Similarly, in anesthetized cats, NPY injection causes a decrease in HR (Minkes et al., 1989). In Langendorff perfused rabbit hearts and in anesthetized dogs no change in HR is observed with NPY application (Allen et al., 1983; Rigel, 1988). Differences in experimental models might explain some of these differences, however, the specific actions of NPY on SAN myocytes requires further investigation. Very little is known about the impact of NPY on the human SAN. A correlation between endogenous NPY and tachycardia was found in adults with heart failure, however, whether this correlation is causative or the change in HR is a confounding variable is unknown (Hulting et al., 1990). Contrarily, in children with vasovagal syncope no correlation was found between NPY levels and HR (Liao et al., 2017). NPY has been investigated broadly for its role in many cardiovascular diseases, but regarding its effect on HR in human, further investigation is needed.
Calcitonin gene-related peptide (CGRP) has been shown to increase HR in many different expression systems, however, whether it is acting directly on the SAN to alter HR and/or as a neuromodulator with indirect chronotropic effects is unclear (Beaulieu and Lambert, 1998). Tachycardia has been observed in response to intravenous administration of CGRP in many species including the rat, rabbit, dog, and in humans (Bell and McDermott, 1996). In neonatal rat isolated cardiomyocytes and rat and guinea pig isolated right atria CGRP increases beating rate, and in rats intravenous CGRP increases HR even in the presence of autonomic antagonists, suggesting a direct action of CGRP on the SAN (Marshall et al., 1986; Fisher et al., 1988). Conversely, despite HR being increased following intravenous administration of CGRP in the rabbit, in rabbit isolated right atria no change in beating rate is observed, suggesting an effect on ANS ganglia rather than directly on SAN myocytes (Holman et al., 1986). In the dog, CGRP also seems to act as a neuromodulator, as when given intravenously it does not affect HR at baseline, but does attenuate the reduction of HR with vagal stimulation, a response that is abolished by sympathectomy and parasympathectomy (Rigel et al., 1989). In humans, a higher amount of CGRP is found in the atria than ventricles, and in the SAN than the rest of the atria, with CGRP binding sites found throughout the heart (Beaulieu and Lambert, 1998). In three different clinical studies, two involving healthy individuals and one with patients in heart failure, intravenously administered CGRP increased HR, even in the presence of adrenergic receptor blockade (Struthers et al., 1986; Franco-Cereceda et al., 1987; Gennari et al., 1990). However, whether the CGRP-mediated increase in HR in humans is exclusively due to the direct action on the SAN or if CGRP also modulates HR through neuromodulation of ANS ganglia requires further investigation.
Neurotensin is a neuromodulator with many targets within the brain, but also the heart, including the SAN (Marin-Garcia, 2011). Neurotensin receptor subtypes NTS1 and NTS2 are expressed in the heart and both are GPCRs (Osadchii, 2015). NTS1 has been shown to activate PLC and PKA through the Gq/11 and Gs signaling pathways, respectively (Marin-Garcia, 2011). It has been hypothesized that neurotensin modulates cardiovascular responses primarily via NTS1 (Osadchii, 2015). Neurotensin has been shown to increase HR in anesthetized rat, guinea pig, and cat, and in guinea pig isolated hearts and atria (Osadchii, 2015). The increase in HR in both the anesthetized guinea pig and the guinea pig isolated heart was not abolished even in the presence of a multitude of autonomic antagonists, suggesting that neurotensin can act directly on the SAN, not simply by altering neuronal activation (Bachelard et al., 1985; Rioux et al., 1989). Conversely, in the anesthetized rat the response was abolished by adrenergic blockade, suggesting the response may be neurally mediated (Chahl and Walker, 1981). Although it’s feasible to predict that neurotensin would also increase HR in humans, through either direct action on the SAN or on ANS ganglia within the atria, currently no data exists. As levels of endogenous neurotensin increase in some pathological conditions it is of potential importance to establish the impact of neurotensin on the human SAN.
Similar to the other neuropeptides described above, somatostatin may act both directly on cardiovascular tissue through its own receptors, while also modulating the release of catecholamines or ACh from autonomic neurons (Marin-Garcia, 2011). Somatostatin activates five GPCRs, all of which stimulate the Gi/o pathway, and some of which also activate Gq/11 cascades (Marin-Garcia, 2011). In snake isolated atrial preparations somatostatin causes a decrease in HR (Donald et al., 1990). Similarly, in a clinical study involving patients admitted for unexplained heart palpitations, intravenous somatostatin decreased HR (Bubinski et al., 1994). It is unclear if this effect is due to the direct action of somatostatin on SAN myocytes or through modulation of autonomic inputs to the SAN. No other data regarding the chronotropic effect of somatostatin on HR exists, and therefore this is an area requiring further investigation.
Various peptides, such as the neuropeptides described above, are important modulators of HR. Many are produced by cells within the heart, acting as autocrine and paracrine factors, while others are produced elsewhere (e.g., in the brain, kidneys, liver, or vasculature) and reach the heart through the circulation, thus acting as endocrine factors (Beaulieu and Lambert, 1998). Their importance for HR regulation is an active area of investigation, and crucial for a complete understanding of SAN control by neurohumoral agents.
Natriuretic peptides (NP) are a family of peptide hormones that are important in cardiovascular physiology and disease. There are four NP family members: atrial NP (ANP); brain NP (BNP); C-type NP (CNP), and Dendroaspis NP (DNP) (Moghtadaei et al., 2016). NP can be produced and stored in atrial myocytes and released during atrial stretch or produced and released from cardiac fibroblasts. All four NPs are present in the circulation, but NP concentrations are much higher in the myocardium than the circulation, due to their local production and perform autocrine and paracrine functions. NP bind to three NP receptors (NPR): NPR-A; NPR-B; and NPR-C, all of which are expressed in the SAN. ANP, BNP, and DNP all bind to NPR-A, whereas only CNP binds to NPR-B. Both NPR-A and NPR-B are particulate (membrane bound) GC receptors, meaning ligand binding to the extracellular NPR activates intracellular GC enzymes and increases intracellular cGMP concentrations (Figure 4). cGMP activates protein kinase G (PKG) and PDE2 and inhibits PDE3. PKG can phosphorylate many components of SAN automaticity, while PDEs hydrolyze cAMP and cGMP and are therefore important modulators of cyclic nucleotide activity. Unlike the other NPR subtypes, NPR-C is not a GC receptor but an atypical GPCR with a single transmembrane domain (unlike a classical GPCR with seven transmembrane domains) that activates a specific Gi-protein signaling cascade that inhibits AC, thus decreasing cAMP levels.
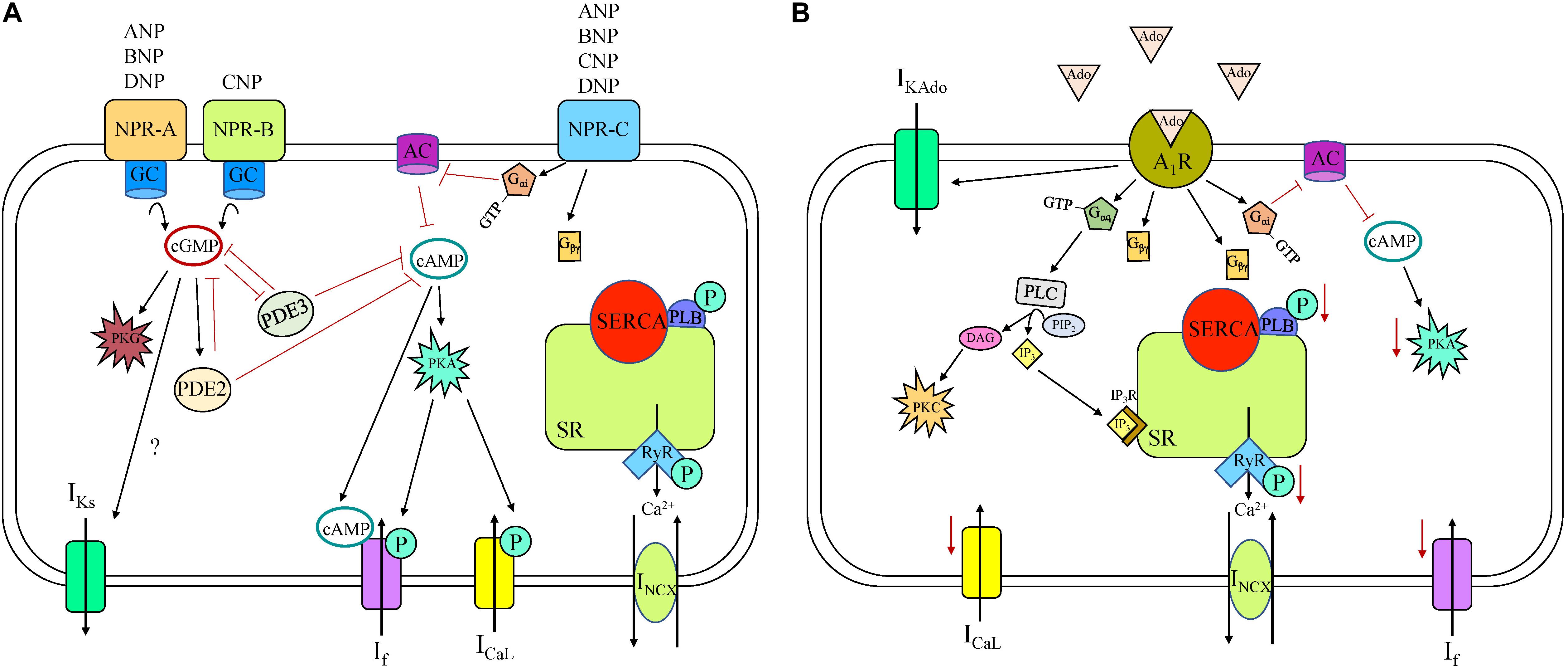
Figure 4. Natriuretic peptide receptor (NPR) (A) and adenosine (B) receptor signaling cascade in a sinoatrial node myocyte. A1R, adenosine receptor 1; AC, adenylyl cyclase; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; CNP, C-type natriuretic peptide; DAG, diacylglycerol; DNP, Dendroaspis natriuretic peptide; GC, guanylyl cyclase; ICaL, long-lasting (L-type) Ca2+ current; If, “funny” current; IKAdo, adenosine-induced inwardly rectifying K+ current; IKs, slow delayed rectifier K+ current; INCX, Na+-Ca2+ exchanger current; IP3, inositol triphosphate; IP3R, inositol triphosphate receptor; P, phosphorylation; PDE, phosphodiesterase; PIP2, Phosphatidylinositol 4,5-bisphosphate; PKA, phosphokinase A; PKC, phosphokinase C; PKG, phosphokinase G; PLB, phospholamban; PLC, phospholipase C; RyR, ryanodine receptor, SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum.
Numerous studies have been performed using various animal models to assess the chronotropic effects of NP. ANP has been shown to decrease HR in rat and not effect HR in dog, BNP has been shown to increase HR in mouse and not effect HR in mouse and dog, and CNP has been shown to increase HR in dog and mouse (Moghtadaei et al., 2016). BNP and CNP increase HR by increasing If and ICaL and shifting the voltage dependence of activation of both currents (Springer et al., 2012). Interestingly, the effects of BNP and CNP on If and ICaL are indistinguishable between NPR-C KO and wild-type mice, revealing that activation of NPR-C does not modulate automaticity under basal conditions. Conversely, blocking NPR-A or inhibiting PDE3 both abolish the effects of BNP or CNP on If and ICaL (Springer et al., 2012). These data indicate that under basal conditions, BNP and CNP increase HR by way of NPR-A and NPR-B (not NPR-C), by activating GC, increasing cytosolic cGMP, and inhibiting PDE3, all of which lead to higher cAMP concentrations. In guinea pig SAN myocytes, application of ANP causes an increase in IKs through an increase in cGMP (Shimizu et al., 2002). Together, these results indicate that under basal conditions, NPR-A and NPR-B activation have a greater influence on SAN automaticity than NPR-C.
Although under basal conditions NPR-C specific agonist cANF does not affect HR, in the presence of a β-AR agonist, NPR-C signaling becomes important. Specifically, activation of NPR-C by cANF decreases AP firing in a dose-dependent manner in SAN myocytes that have been first stimulated with isoproterenol (Azer et al., 2012). This response is due to modulation of ICaL by NPR-C/Gi activation causing a reduction in cAMP concentration in mouse SAN myocytes (Rose et al., 2004), although NPR-C activation does not modify If in any way (potentially due to compartmentalization of receptors and channels in the SAN). Interestingly, when β-agonist concentrations are high, activation of β-AR switches the actions of BNP and CNP from increasing HR to decreasing HR due to increasingly important contributions of NPR-C to the overall effects of NP (Moghtadaei et al., 2016).
In humans, the response to ANP has been variable. In some studies, ANP has had no effect on HR (Volpe et al., 1987, 1990; Crozier et al., 1993), while in others it has either increased or decreased HR in a dose-dependent manner (Biollaz et al., 1986; Bussien et al., 1986; Weidmann et al., 1986; Franco-Suarez et al., 1987; Nicholls and Richards, 1987). Although cellular and molecular mechanisms of ANP action have not been explored in the human SAN, in human atrial myocytes ANP has been shown to increase If and ICaL, likely via an increase in cytosolic cGMP and cAMP concentrations (Boixel et al., 2001; Lonardo et al., 2004). The influence, if any, of BNP or CNP on HR has not been shown in human, although they, along with ANP, are both found in the circulation and in the myocardium of humans (Moghtadaei et al., 2016).
So overall, NP plays an important role in controlling HR via specific modulations of pacemaker components in animal models, but how this translates to human remains uncertain. Responses are complicated because of the multiple receptors and the non-specificity of NPR binding by the various NP. Because endogenous levels of NP increase under certain pathological conditions, such as heart failure, it is important to understand the role they play in SAN function.
Adenosine is a purine nucleoside formed from the breakdown of ATP that controls a plethora of cellular functions in an autocrine and paracrine fashion in the cardiovascular system, including in the SAN (Mangoni and Nargeot, 2008). Adenosine shares a similar signaling pathway to M2R and causes decreased HR in many species including human, dog, rabbit, guinea pig, rat, and mouse (Mangoni and Nargeot, 2008). Adenosine binds to purinergic receptors (A1, A2A, A2B, and A3), all of which are found in the heart (Headrick et al., 2013). A1 and A3 receptors are coupled to Gi and Gq/11 pathways, causing a decrease in intracellular cAMP, whereas the A2 receptors are coupled to Gs and cause an increase (Headrick et al., 2013). However, it is the A1 receptor (A1R) that is attributed to modulating HR in the SAN (Figure 4). Adenosine application activates an inwardly rectifying K+ current (IKAdo) that is mediated by the same G-protein signaling and coupled K+ channels as IKACh (Kir3.1/3.4) (Belardinelli et al., 1988). This has been demonstrated in the human isolated SAN where adenosine application decreases HR via A1R-induced GIRK channel activation, an effect that is abolished by GIRK channel blockade, implicating that the chronotropic response may be predominantly due to IKAdo rather than alternative intracellular pathways and targets (Li et al., 2017). In humans there is greater expression of both A1R and GIRK4 in the SAN compared to the adjacent right atrium, suggesting the SAN might be more sensitive to adenosine than the surrounding tissue (Li et al., 2017). Interestingly, the sensitivity to adenosine-challenge is heart-specific, potentially explained by differences in A1R and GIRK protein expression between hearts. Although in humans GIRK channel activation is thought to be the main contributor to decreased HR, in animal models other mechanisms have been identified. Specifically, in rabbit SAN myocytes, adenosine also decreases HR through inhibition of ICaL and If, and causes a negative shift in the activation of If, leading to a negative shift of MDP and a reduced SDD rate (West and Belardinelli, 1985; Belardinelli et al., 1988). Although activation of A1R by adenosine reduces HR, it has been shown that A2A gene deletion in mice also reduces HR, implying that activation of A2A would instead increase HR. A3 receptor deletion in mice, on the other hand, has the opposite effect, pointing to a role for A3 receptors in reduced HR, consistent with A1R (Headrick et al., 2013). And not only does adenosine contribute to normal daily rhythm control, it has also been implicated in pathophysiological settings. Upregulation of A1R has been demonstrated in dogs with induced chronic heart failure compared to control, which may worsen conduction abnormalities, leading to SAN dysfunction (Lou et al., 2014). Adenosine is produced in greater amounts in ischemia and heart failure, and can cause SAN conduction block and tachy-bradyarrhythmia in the dog (Lou et al., 2013).
The renin-angiotensin system plays a critical role in cardiovascular and fluid homeostasis. Renin converts angiotensinogen to angiotensin I, which is then cleaved by angiotensin converting enzyme (ACE) into angiotensin II (ANGII), which is the main effector molecule (Mehta and Griendling, 2007). ANGII is an important and far reaching hormone, as it affects virtually all organs, including the heart, the kidneys, the vasculature, and the brain, yet knowledge of its modulatory effects on the SAN is limited and controversial (Mehta and Griendling, 2007; Sheng et al., 2011). Acutely, ANGII has been shown to decrease the firing rate of guinea pig SAN myocytes by a reduction in ICaL and an increase in IKs (Sheng et al., 2011). Similarly, in rabbit SAN myocytes ANGII decreases spontaneous firing rate by decreasing ICaL (Habuchi et al., 1995). However, in anesthetized dogs, ANGII injection increases HR (Lambert, 1995). This difference could be due to species-specific effects or related to differences in whole animal and single cell responses. ANGII acts on angiotensin (AT) receptors, AT1 and AT2, however, it is via AT1 that ANGII modulates the SAN (Sechi et al., 1992; Sheng et al., 2011). Signaling pathways downstream of AT1 are complex, and the mechanisms by which binding of ANGII to AT1 modulates automaticity is unclear. ANGII binding to AT1 can activate both Gq/11 and Gi, as well as G-protein independent pathways (Mehta and Griendling, 2007). AT1 activation also modulates other receptors in the plasma membrane and activates signaling molecules such as MAPK and arachidonic acid (Mehta and Griendling, 2007). Not only does ANGII act directly on myocytes, it can serve as a neuromodulator stimulating the release of catecholamines from sympathetic neurons (Marin-Garcia, 2011). Perhaps the multiple targets and complex signaling networks downstream of ANGII activation underlie the varying chronotropic effects seen in different experimental species and preparations. Interestingly, ANGII has not been shown to change HR in humans, despite its well documented role in regulation of blood pressure (Mehta and Griendling, 2007). Both AT receptors are present in the human heart, with higher AT2 expression in fetal development and in failing hearts compared to healthy adult hearts (Mehta and Griendling, 2007). Although no studies have been done in human tissue, when given ANGII intravenously no changes are observed in fetal or adult HR (Oney and Kaulhausen, 1982; Sander-Jensen et al., 1988). ANGII converting enzyme, however, is found in great quantity in the human SAN, perhaps indicating local synthesis of ANGII (Beaulieu and Lambert, 1998). ANGII is clearly a hormone that warrants further investigation regarding its role in HR control in human, especially as receptor expression and circulating ANGII concentration change substantially in pathology (Mehta and Griendling, 2007).
Endothelin peptide hormones, ET-1, ET-2, and ET-3, are produced in vascular endothelial cells, and act as vasoconstrictors (Gordan et al., 2015). However, they have also been shown to affect automaticity of the SAN. Experimental studies have shown varying chronotropic effects following ET-1 application. ET-1 application to guinea pig right atrial tissue increases HR (Ishikawa et al., 1988), whereas decreased HR occurs in rabbit SAN myocytes (Tanaka et al., 1997; Ono et al., 2001). In anesthetized cats, ET-1 application results in increased HR at low doses but a biphasic response at higher doses, whereas ET-3 application only increases HR (Minkes et al., 1989). It has been proposed that these differing responses are due to differential activation of the two endothelin receptors A and B (ETA and ETB). Both are expressed in the SAN, with ETA causing a decrease and ETB causing an increase in HR (Ono et al., 2001). ETA activation by ET-1 hyperpolarizes membrane potential by activating IKACh, and inhibits ICaL by way of Gi protein signaling (Ono et al., 2001). In pigs, intravenous infusion of an ETA inhibitor causes an increase in HR, suggesting a role for endogenous ET and basal activation of ETA receptors in the maintenance of HR (Gelzer et al., 2004). The pathway activated by ETB in the SAN that results in an increase in HR requires further investigation. ETB activation in endothelial cells causes NO production and activates the Gq protein cascade, so perhaps a similar pathway is activated in the SAN (Horinouchi et al., 2013). Minimal data exist regarding endothelin and its receptors in the SAN of human. In one study, patients with coronary artery disease were infused with the same ETA inhibitor as in the pig study above, yet no change in HR was observed (Kolettis et al., 2003). Although this could relate to a species difference, it may also be a consequence of altered levels of basal ET and other neurohumoral agents affecting HR with coronary artery disease. It has also been shown that ET-1 levels contribute to the reduced HR variability seen in heart failure patients, perhaps by modulating SAN function (Aronson et al., 2001).
Thyroid hormones are biogenic amines that are released from the thyroid gland and can influence HR over a longer time time scale than most agents (Mangoni and Nargeot, 2008). It is well documented in humans that high levels of thyroid hormone in the circulation (hyperthyroidism) lead to increased HR, whereas low levels of thyroid hormone (hypothyroidism) are associated with decreased HR. Thyroid hormones act on nuclear hormone receptors (thyroid receptors, TR) which are a group of intracellular ligand-dependent transcription factors of which there are two subtypes: TRα and TRβ (Mangoni and Nargeot, 2008). Their activation can alter the expression of different cardiac ion channels. In rabbit isolated SAN myocytes, incubation with triiodothyronine (T3) to mimic hyperthyroidism increases the amplitude of If without altering the voltage-dependence of the current (Renaudon et al., 2000). It has been shown in rats that T3 application significantly increases the mRNA expression level of HCN2 (Pachucki et al., 1999). Thus, it appears that an increase in If channel expression with T3 stimulation increases the density of If, causing an increase in HR. In mouse lines meant to mimic hypothyroidism (TRα and TRβ KO), TRβ KO mice demonstrated no alterations in HCN expression level or HR, whereas the TRα KO had decreased HR and a reduction in both HCN2 and HCN4 expression levels, implying that the effects of T3 on HCN channel expression occur through activation of TRα (Gloss et al., 2001). Expression levels of both the Na+-Ca2+ exchanger and Na+/K+-ATPase have been shown to change based on thyroid concentration in rats, which could also affect HR (Mangoni and Nargeot, 2008). Thyroid hormones can also indirectly modulate HR in rats by increasing the number of β1-AR in the plasma membrane, increasing the ability for NE or epinephrine to increase HR (Dillmann, 1989). Thyroid hormones have also been shown to change HR in rats on a much shorter times scale, implicating a non-genomic mechanism perhaps mediated by eNOS (Rutigliano and Zucchi, 2017). Although there is no clear data describing specific mechanisms for the chronotropic effect of thyroid hormones seen in humans, it was shown that even when within a normal range in healthy individuals, there is a positive correlation between circulating T3 concentration and resting HR (Roef et al., 2013). In a study to determine the effect of thyroid hormones on the ANS control of HR, basal HR was higher in people with hyperthyroidism, and following complete ANS blockade, HR values remained higher in people with hyperthyroidism, suggesting the tachycardia with hyperthyroidism may at least in part be due to direct effect of thyroid hormones on the SAN (Valcavi et al., 1992). However, at least some component of the response is likely via indirect action through the ANS, as blockade of the sympathetic nerves alone reduces tachycardia in patients with hyperthyroidism (Valcavi et al., 1992). This could be due to the upregulation of β1-AR expression, as in rats. Although the intracellular mechanisms(s) are unclear, there is clearly a role for thyroid hormones in control of the human SAN. Understanding the specific mechanisms by which this happens is important for people with hyperthyroidism not only because of the determinantal effects of chronic tachycardia, but also because it increases the likelihood of atrial fibrillation, atrioventricular conduction abnormalities, and sinus node dysfunction (Valcavi et al., 1992).
Parathyroid hormone (PTH) is synthesized by the parathyroid gland and modulates many organs and tissues via the circulation in an endocrine fashion. PTH-related protein does not normally circulate in the blood but is present in most cells. It is produced in cardiomyocytes and acts locally as an autocrine or paracrine hormone (Shimoni, 1999). There are two parathyroid receptors, PTH1R and PTH2R, both of which increase intracellular cAMP concentrations (Potthoff et al., 2011). PTH1R is coupled to both the Gs and Gq protein pathways, thus increasing intracellular cAMP, PKA, PLC, IP3, and PKC (Chorev, 2002). PTH2R activation increases intracellular cAMP and activates receptor internalization (Usdin et al., 2002). In rabbit SAN myocytes, PTH increases AP firing frequency by increasing the amplitude of If and in turn the slope of SDD (Hara et al., 1997). PTH has also been shown to increase HR in rat isolated hearts (Shimoyama et al., 2001). In a clinical study involving chronic heart failure patients, there was a positive correlation between endogenous PTH levels and HR, suggesting a role for PTH in the associated increased HR (Sugimoto et al., 2013). Similarly, in two studies of patients undergoing kidney dialysis, increased endogenous PTH was associated with a decrease in HR variability, implicating PTH in the ANS dysfunction that occurs in individuals with kidney failure (Ussawawongaraya et al., 2013; Poulikakos et al., 2014). Although both PTH receptors are expressed in human atria and thus PTH might increase HR by directly affecting SAN myocytes as it does rabbit myocytes, it is also possible that observed increases in HR in humans are due to indirect effects on the SAN via the ANS (Potthoff et al., 2011). Specifically, in human atrial tissue PTH causes release of NE from sympathetic nerve terminals, which could account for an increase in HR, although this effect has not yet been demonstrated experimentally (Potthoff et al., 2011). Although it seems apparent that PTH modulates HR, there are still some areas for investigation, including understanding the role of PTH in HR control in healthy humans compared to pathological settings, the intracellular pathway(s) by which PTH affects human SAN myocytes and/or sympathetic nerve terminals, and the relative contribution of each mechanism to the chronotropic response with PTH.
Bradykinin is an inflammatory mediator peptide primarily known for its role in regulating blood pressure (Golias et al., 2007). Bradykinin receptors are GPCRs and have been reported to exist in the heart (Golias et al., 2007). With co-expression of one of these receptors (BK2R) and HCN1 or HCN2 in Xenopus oocytes, a modulatory effect of bradykinin on HCN channel dynamics has been demonstrated (Pian et al., 2007). BK2R is coupled to a Gq/11 pathway that stimulates PLC activity, which causes a depolarizing shift in the activation of HCN channels (Pian et al., 2007). When bradykinin is applied to patch-clamped rabbit SAN myocytes, a similar depolarizing shift in If activation is observed (Pian et al., 2007), which would be expected to increase HR. This is supported by a study in anesthetized rats, in which intravenous bradykinin causes tachycardia (Ponchon et al., 1995). In contrast, however, BK2R knock-out mice have increased basal HR and bradykinin injections into the brainstem of the mouse decreases HR, suggesting that BK2R activation may in fact lower HR in mice (Madeddu et al., 1999). In patients both with and without atherosclerosis, bradykinin increases HR when injected into the left coronary artery, although no effect is seen when injected into the right coronary artery (Schaefer et al., 1996), which is a surprising result as the branch supplying blood to the SAN most commonly arises from the right coronary artery (Pejkovic et al., 2008). Clearly, our understanding of the role of bradykinin on HR regulation and SAN modulation is lacking, particularly in humans.
Relaxin is a reproductive hormone that is released during the menstrual cycle and during pregnancy, with many known effects on the reproductive (Bell et al., 1987) and cardiovascular systems (Han et al., 1994a). Experimentally, relaxin has been shown to increase HR in rat atria and rabbit SAN myocytes (Kakouris et al., 1992; Han et al., 1994a). In rabbit SAN myocytes, relaxin increases firing rate by enhancing ICaL in a cAMP/PKA dependent manner (Han et al., 1994a). Relaxin binds to GPCRs and increases intracellular cAMP, but the specific receptor subtypes and cascades in the SAN are unknown. Administration of relaxin has also been shown to have secondary therapeutic effects via inhibition of cardiac mast cell activation, preventing histamine release and subsequent tachyarrhythmias in the pig (Nistri et al., 2008). No studies have been reported specifically investigating the effects of relaxin on the SAN in humans, but it may be relevant in the context of cardiovascular complications that occur with pregnancy, as circulating relaxin levels are high during the first trimester (Bell et al., 1987). During this period there is also an increase in cardiac output (in preparation for the increased cardiac demand imposed from the development of the fetus) (Meah et al., 2016), so relaxin may play a role in increasing cardiac output during pregnancy (as has been shown in rats, through an increase in HR and contractility, Bell et al., 1987). Interestingly, relaxin can cross the placenta during the early stages of pregnancy, and circulating relaxin levels correlate with fetal HR, suggesting it may play a role in controlling HR of the developing fetus (Johnson et al., 1994). While it may be that relaxin’s influence on the SAN is an important normal control mechanism during pregnancy, in the absence of pregnancy it may have unwarranted effects on HR.
Nitric oxide (NO) is a gas produced in almost all cell types that has numerous cardiac effects (Massion et al., 2003). Even within the SAN itself, NO has diverse, and sometimes conflicting consequences, and is found as a component in many signaling pathways. NO production occurs in cardiomyocytes and in nearby endothelial cells, either by endothelial NO synthase (eNOS) or by neuronal NO synthase (nNOS), and in pathological situations NO can be produced by inducible NO synthase (iNOS) (Fischmeister et al., 2005). NO binds to soluble GC (sGC), which causes cGMP production, and therefore an increase in intracellular cGMP concentrations (Fischmeister et al., 2005). cGMP interacts with many targets to modulate HR, including PKG and PDEs. NO-activated cGMP attenuates ICaL in rat and guinea pig SAN myocytes by PKG-mediated channel phosphorylation (Fischmeister et al., 2005). NO also has conflicting downstream effects on ICaL, as cGMP activates PDE2 (which should decrease ICaL), but also inhibits PDE3 (which should increase ICaL) (Fischmeister et al., 2005). Not all effects of NO are due to cGMP, however. It has been shown that low concentrations of NO can activate Gs proteins (which would be expected to increase HR), and NO can activate Gi proteins (which would be expected to decrease HR) (Fischmeister et al., 2005). It has also been shown that NO can activate If (Massion et al., 2003). An additional complicating factor is that NO modulates release of neurotransmitters from autonomic neurons. NO can inhibit the presynaptic release of NE from sympathetic neurons and facilitate the release of ACh from vagal neurons (Paterson, 2001).
Ultimately, it would seem that the multitude of ways in which NO can influence SAN automaticity makes it difficult to discern its net effect on HR (Chowdhary and Townend, 1999). Yet a series of human studies may have helped provide insight into its role in modulating HR (Chowdhary et al., 2000, 2002a,b, 2004). In healthy individuals, intravenous administration of a NOS inhibitor, as well as a control agent, caused an increase in blood pressure and a subsequent baroreflex-mediated decrease in HR; however, NOS caused less bradycardia than the control agent (Chowdhary et al., 2000). This indicates that inhibition of NOS removes the tonic excitatory effect of NO on vagal activity. Further, administration of exogenous NO and a control agent caused decreased blood pressure and a subsequent baroreflex-mediated increase in HR, yet the exogenous NO caused less tachycardia than the control agent, demonstrating the preservation of vagal control (Chowdhary et al., 2000). Although NO may also directly act on SAN myocytes, these experiments suggest that any such effects are overwhelmed by the effect of NO on the ANS. This was demonstrated in a study in human heart transplant recipients, whose hearts are disconnected from the ANS. In these patients, exogenous NO increased HR and NOS inhibition decreased HR, changes that were not a reflex response, as the control agents increased blood pressure but did not change HR (Chowdhary et al., 2002a). During heart failure, baroreflex-mediated parasympathetic activation is still dependent on NO synthesis (Chowdhary et al., 2002b), and stimulation of the endogenous NO pathway may be a strategy for increasing parasympathetic activity under conditions of adverse sympathetic overactivity, such as with ventricular arrhythmias (Chowdhary et al., 2004).
As there are a multitude of different neurohumoral factors that influence SAN activity, but share common intracellular signaling pathways, understanding their integration is important for understanding the combined effect on the SAN and HR.
Intracellular compartmentalization of the various complex signaling cascades in the SAN is essential for its proper function (Steinberg and Brunton, 2001). Not only are compartments physically formed in the membrane, such as caveolae and lipid rafts, compartmentalization also occurs by intracellular biochemical modulators. PDEs degrade cAMP and/or cGMP and the variety and abundance of PDEs helps modulate the cellular responses by controlling the duration and amplitude of cAMP/cGMP responses, both at basal levels and in response to neurohumoral stimulation (Zaccolo and Movsesian, 2007), as well as creating spatially and temporally distinct pools of cAMP and cGMP by forming functional diffusion barriers (Steinberg and Brunton, 2001; Yaniv et al., 2015). There are 8 different PDE families that have been identified in the heart: PDE1, PDE2, and PDE3 hydrolyze both cAMP and cGMP; PDE4, PDE7, and PDE8 are cAMP-specific enzymes; and PDE5 and PDE9 are cGMP-specific (Zaccolo and Movsesian, 2007). Not only do each of these PDE families have differential ability to enzymatically degrade cAMP and/or cGMP, they also have distinct effects in different cardiac cell types (Hua et al., 2012). In mouse isolated SAN myocytes, PDEs regulate basal firing rate, which is increased with global or selective PDE inhibition (PDE2, PDE3, or PDE4) (Hua et al., 2012). In addition, when PDE2 in mouse or human SAN myocytes, or PDE3 or PDE4 in mouse SAN myocytes is inhibited, ICaL is activated at more negative membrane potentials and its current amplitude increases, accounting for the increase in firing rate (Rivet-Bastide et al., 1997; Hua et al., 2012). PDE3 also regulates IKs in guinea pig SAN myocytes via hydrolysis of cGMP, as its inhibition increases current amplitude (Shimizu et al., 2002).
Basal PDE degradation of cAMP in rabbit SAN myocytes has been shown to modulate and compartmentalize Ca2+-clock mechanisms such as RyR and PLB, as PDE inhibition increases the rate and amplitude of local Ca2+ releases (Vinogradova et al., 2008). This has been confirmed specifically for PDE1A in rabbit SAN myocytes, facilitated by its higher expression in the SAN compared to other regions of the heart (Lukyanenko et al., 2016). Although inhibition of single PDEs only moderately increases SAN myocyte firing, recently it has been shown that dual PDE3 and PDE4 inhibition synergistically increases basal SAN firing rate by ∼50% (Vinogradova et al., 2018a). Concurrent PDE3 and PDE4 activation modulates Ca2+-clock components, specifically via a decrease in cAMP and PKA phosphorylation, which suppresses local Ca2+ releases and decreases HR (Vinogradova et al., 2018b). Following blockade of RyR with ryanodine, dual PDE3 and PDE4 inhibition failed to increase spontaneous HR, implicating that modulation of the Ca2+-clock is necessary for basal synergistic effects of PDE3 and PDE4 on HR (Vinogradova et al., 2018b).
Not only do PDEs attenuate basal SAN firing, they also modulate the response to neurohumoral stimulation. Because so many of the neurohumoral pathways in the SAN activate AC and GC, without intracellular control these secondary messenger systems would be limited in their ability to specifically target a response (Fischmeister et al., 2006). In mouse SAN myocytes it has been shown that compartmentalization of HCN channels by PDEs may play an important role in regulating β1-AR modulation of If (St. Clair et al., 2017). Specifically, it appears that PDE4 acts as a barrier that isolates HCN channels from the rest of the cell, so that under basal conditions they cannot be accessed by cAMP, and that PDE3 interacts with PKA to favor the β1-AR/cAMP/PKA/HCN pathway (rather than activation by direct binding of cAMP to HCN) (St. Clair et al., 2017).
Another important consideration in the SAN’s complex signaling network is crosstalk between cAMP and cGMP. Negative feedback pathways exist for both cAMP and cGMP, such that when they activate PKA or PKG, these kinases activate PDEs that then degrade cAMP or cGMP. Specifically, cAMP-dependent PKA stimulates PDE3 and PDE4, which degrades cAMP, while cGMP-stimulated PKG activates PDE5, thus inhibiting cGMP. These signals are also interconnected, as cGMP can activate or inactivate PDEs that degrade cAMP. Specifically, cGMP activates PDE2 (decreasing levels of cAMP) and inactivates PDE3 (increasing cAMP). More cGMP is required to activate PDE2 than to inhibit PDE3, thus the latter occurs more readily and at lower cGMP concentrations. Conversely, cAMP can inhibit cGMP production through PKA-activated PDE5. An important aspect of the above responses is that the particular PDE activated or inhibited by cGMP or cAMP partly depends on their concentration. This interplay and its importance during neurohumoral stimulation has been demonstrated in mouse SAN tissue, as following a β-AR-induced increase in firing rate, application of a PDE5 inhibitor decreases frequency, a response that is attenuated by PDE2 inhibition (Isidori et al., 2015). This suggests that by preventing degradation of cGMP by PDE5, more PDE2 is activated by cGMP, which then degrades cAMP and decreases HR.
Neurohumoral signaling pathways are not only controlled by PDE hydrolysis of cAMP and cGMP, but also by tethering of PKA to specific intracellular locations by A-kinase-anchoring proteins (AKAP) (Zaccolo and Movsesian, 2007). Sequestering of PKA into localized pools by anchoring proteins allows for targeted substrate phosphorylation. AKAP not only spatially restricts and controls PKA but can also temporally modulate the actions of PKA (Dodge-Kafka et al., 2006). Although the intracellular distribution and dynamics of cGMP are not as well understood as cAMP, it is likely that proteins analogous to AKAP localize the actions of PKG to specific regions (Dodge-Kafka et al., 2006). It has been shown that PKG directly anchors to NPR-A, and this compartmentalization of cGMP helps modulate responses elicited by activation from NP versus NO (Airhart et al., 2003). Specific control of PKA and PKG by anchoring proteins in the SAN, however, requires further investigation.
Regulators of G-protein signaling (RGS) catalyze the hydrolysis of Gα-guanosine triphosphate (GTP) to Gα-guanosine diphosphate (GDP), effectively inactivating the GPCR (Riddle et al., 2005). GTP to GDP hydrolysis is a rate-limiting step for signal termination, so the longer the Gα subunit is in its active form (Gα-GTP) the longer a signal is activated in a cell (Mighiu and Heximer, 2012). RGS proteins inhibit G-protein signaling cascades by speeding up this intrinsically slow GTP hydrolysis.
Limited studies have been performed examining RGS proteins in the SAN, but it is clear that they are important for controlling the downstream effects of M2R activation in mouse SAN myocytes (Cifelli et al., 2008; Mighiu and Heximer, 2012; Stewart et al., 2012; Wydeven et al., 2014). RGS4 and RGS6 have been shown to be more abundantly expressed in the SAN than other cardiac tissues (Mighiu and Heximer, 2012; Wydeven et al., 2014). These endogenous RGS proteins oppose the decrease in HR following parasympathetic stimulation by accelerating GTPase activity to more rapidly terminate G protein signaling, thus impeding activation of hyperpolarizing GIRK channels (Berman et al., 1996). In cultured rabbit SAN cells, RGS2 overexpression has been shown to increase the phosphorylation of PLB along with HR, likely due to suppression of the Gi signaling cascade (Yang et al., 2012). Other RGS proteins are expressed in the heart, with important roles in controlling neurohumoral activation, but they have not been demonstrated specifically in SAN tissue or cells. For instance, in ventricular myocytes RGS2 proteins have been shown to be inhibitory downstream of catecholamines and ANGII activation by hydrolysis of β2-AR activated Gαi subunits, as well as α1-AR or AT activated Gαq subunits (Chakir et al., 2011).
G-protein coupled receptor kinases (GRK) contribute to β-AR desensitization. Seven characterized mammalian GRKs have been shown to be expressed in the heart, with GRK2 expression most prominent (Huang et al., 2014). When GPCRs are activated, GRK2 trans-locates from the cytosol to the plasma membrane by binding with the Gβγ subunit and phosphorylating the agonist-bound GPCR (Fu and Xiang, 2015). Following phosphorylation by GRK, the GPCR becomes a target for β-arrestin, which binds to the GRK-phosphorylated GPCR and physically inhibits its signaling cascade by sterically hindering interactions between the GPCR and the downstream G-proteins (a process called homologous desensitization) (Huang et al., 2014). Conversely, heterologous desensitization is initiated by PKA or PKC phosphorylation of the GPCR (Huang et al., 2014). GPCR phosphorylation increases the affinity of GPCR for adaptor proteins, which initiates clathrin-pit formation, which results in GPCR internalization (Fu and Xiang, 2015). Once internalized, the GPCR is then sorted to either recycling endosomes, to be trafficked back to the membrane, or to lysosomes for degradation (Fu and Xiang, 2015).
One could argue that the SAN is one of the most important tissues in the body, as it initiates each-and-every normal heartbeat. Because the SAN is so critical to life, it is not surprising that it is heavily controlled, ensuring that the heart can adapt and respond to meet physiological demands. There are numerous overlapping and redundant mechanisms that drive automaticity in the SAN, to ensure that it continues to fire under widely varying conditions. The combination of these pacemaking mechanisms and their control by a multitude of neurohumoral factors creates an intricate and complex system (Li et al., 2017).
Despite a solid foundation, our current understanding of the complexities of the neurohumoral control of SAN activity is lacking. Further characterization of the distribution and expression of the many receptor types in SAN myocytes may help provide insight into the comparative contribution of the different neurohumoral agents. Often, the missing link in our current understanding of how a specific neurohumoral agent modulates HR lies between receptor activation and its intracellular target; i.e., we know what receptor an agonist activates and what pacemaker mechanisms it ultimately modulates, but the pathways involved are not clear. Also, in many cases investigations focus on effects on If and/or ICaL and do not consider impacts on other important membrane currents such as IKr or IKs, or on Ca2+-clock mechanisms, leaving us with an incomplete understanding of subcellular mechanisms driving observed HR responses.
There are many discrepancies in the literature regarding the effects of specific neurohumoral factors on HR. This may relate to species differences or differences in experimental approaches or preparations. At the level of the whole SAN, however, there may also be an important contribution of cellular heterogeneity. While we have focused on the intracellular modulation of SAN activity, it is important to appreciate that the SAN is structurally and electrophysiologically heterogeneous (Mangoni and Nargeot, 2008), so effects will vary across it. The SAN is heavily innervated, with a complex network of nerve fibers and ganglia whose density is much higher than the surrounding myocardium (Roberts et al., 1989; Chow et al., 2001; Newton et al., 2014; Pauza et al., 2014; Stoyek et al., 2015; Inokaitis et al., 2016). Yet the SAN is not ubiquitously innervated and this, along with variable receptor densities throughout the node, make it functionally heterogeneous (MacKaay et al., 1980; Opthof, 2007). Sympathetic control will dominate in heavily innervated regions and areas with high receptor density, whereas these regions may be silenced by parasympathetic control, such that areas with fewer receptors or little innervation would then determine rate. Moreover, neurohumoral stimulation can shift the location of the leading pacemaker site in the SAN, which itself can result in a change in HR, as different SAN myocytes have different intrinsic cycle lengths (Opthof et al., 1987).
Overall, it is clear that further work is needed for a complete, comprehensive understanding of the complex mechanisms involved in neurohumoral control of SAN function. This will be driven by technological advances in experimental techniques (Kohl and Quinn, 2014), such as higher resolution imaging and cell-specific optogenetics (Quinn et al., 2016), and may be further enhanced by the use of alternative experimental models (Stoyek et al., 2016, 2018; Stoyek and Quinn, 2018) or computational modeling (Quinn and Kohl, 2011, 2013). Ultimately, by improving our knowledge of SAN control we will move toward a better understanding of how it responds to constantly changing physiological demands, and how we may better treat SAN dysregulation that leads to debilitating and often deadly cardiac diseases.
EM designed the figures and wrote the manuscript. RR and TQ revised the manuscript. All authors approved the final version of the manuscript.
This work was supported by the Natural Sciences and Engineering Research Council of Canada (RGPIN-2016-04879 to TQ), the Heart and Stroke Foundation of Canada (G-18-0022185 to TQ), and the Canadian Institutes of Health Research (MOP 342562 to TQ). Work in the laboratory of RR is supported by the Canadian Institutes of Health Research (MOP 142486 and PJT 166105) and the Heart and Stroke Foundation of Canada (G-18-0022148). TQ is a National New Investigator of the Heart and Stroke Foundation of Canada.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Accili, E. A., Redaelli, G., and DiFrancesco, D. (1996). Activation of the hyperpolarization-activated current (If) in sino-atriai node myocytes of the rabbit by vasoactive intestinal peptide. Eur. J. Physiol. 431, 803–805. doi: 10.1007/bf02253849
Accili, E. A., Redaelli, G., and DiFrancesco, D. (1998). Two distinct pathways of muscarinic current responses in rabbit sino- atrial node myocytes. Pflugers Arch. Eur. J. Physiol. 437, 164–167. doi: 10.1007/s004240050763
Adachi, T., Shibata, S., Okamoto, Y., Sato, S., Fujisawa, S., Ohba, T., et al. (2013). The mechanism of increased postnatal heart rate and sinoatrial node pacemaker activity in mice. J. Physiol. Sci. 63, 133–146. doi: 10.1007/s12576-012-0248-1
Airhart, N., Yang, Y., Roberts, C. T., and Silberbach, M. (2003). Atrial natriuretic peptide induces natriuretic peptide receptor-cGMP-dependent protein kinase interaction. J. Biol. Chem. 278, 38693–38698. doi: 10.1074/jbc.M304098200
Allen, J., Bircham, P., Edwards, A., Tatemoto, K., and Bloom, S. (1983). Neuropeptide Y (NPY) reduces myocardial perfusion and inhibits the force of contraction of the isolated perfused rabbit heart. Regul. Pept. 6, 247–253. doi: 10.1007/bf02253849
Aronson, D., Mittleman, M. A., and Burger, A. J. (2001). Role of endothelin in modulation of heart rate variability in patients with decompensated heart failure. Pacing Clin. Electrophysiol. 24, 1607–1615. doi: 10.1046/j.1460-9592.2001.01607.x
Axsom, J. E., Nanavati, A. P., Rutishauser, C. A., Bonin, J. E., Moen, J. M., and Lakatta, E. G. (2019). Acclimation to a thermoneutral environment abolishes age-associated alterations in heart rate and heart rate variability in conscious, unrestrained mice. Geroscience [Epub ahead of print], doi: 10.1007/s11357-019-00126-7
Azer, J., Hua, R., Vella, K., and Rose, R. A. (2012). Natriuretic peptides regulate heart rate and sinoatrial node function by activating multiple natriuretic peptide receptors. J. Mol. Cell. Cardiol. 53, 715–724. doi: 10.1016/j.yjmcc.2012.08.020
Bachelard, H., St-Pierre, S., and Rioux, F. (1985). The chronotropic action of neurotensin in the guinea pig isolated heart. Peptides 6, 841–845. doi: 10.1016/0196-9781(85)90311-0
Baker, A. J. (2014). Adrenergic signaling in heart failure: a balance of toxic and protective effects. Eur. J. Physiol. 466, 1139–1150. doi: 10.1007/s00424-014-1491-5
Barbuti, A., and DiFrancesco, D. (2008). Control of cardiac rate by “funny” channels in health and disease. Ann. N. Y. Acad. Sci. 1123, 213–223. doi: 10.1196/annals.1420.024
Bartos, D. C., Grandi, E., and Ripplinger, C. M. (2015). Ion channels in the heart. Compr. Physiol. 5, 1423–1464. doi: 10.1002/cphy.c140069.Ion
Beaulieu, P., and Lambert, C. (1998). Peptidic regulation of heart rate and interactions with the autonomic nervous system. Cardiovasc. Res. 37, 578–585. doi: 10.1016/S0008-6363(97)00305-2
Behar, J., Ganesan, A., Zhang, J., and Yaniv, Y. (2016). The autonomic nervous system regulates the heart rate through cAMP-PKA dependent and independent coupled-clock pacemaker cell mechanisms. Front. Physiol. 7:419. doi: 10.3389/fphys.2016.00419
Belardinelli, L., Giles, W. R., and West, A. (1988). Ionic mechanisms of adenosine actions in pacemaker cells from rabbit heart. J. Physiol. 405, 615–633. doi: 10.1113/jphysiol.1988.sp017352
Bell, D., and McDermott, B. J. (1996). Calcitonin gene-related peptide in the cardiovascular system: characterization of receptor populations and their (patho)physiological significance. Pharmacol. Rev. 48, 253–288.
Bell, R., Eddie, L., Lester, A., Wood, E., Johnston, P., and Niall, H. (1987). Relaxin in human pregnancy serum measured with an homologous radioimmunoassay. Obstet. Gynecol. 69, 585–589.
Berman, D. M., Wilkie, T. M., and Gilman, A. G. (1996). GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G-protein subunits. Cell 86, 445–452. doi: 10.1016/s0092-8674(00)80117-8
Bers, D. M. (2000). Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 87, 275–281. doi: 10.1161/01.res.87.4.275
Biollaz, J., Nussberger, J., Waeber, B., and Brunner, H. (1986). Clinical pharmacology of atrial natriuretic (3-28) eicosahexapeptide. J. Hypertens. Suppl. 4, 101–108.
Birk, S., Thomas, J., Ahrend, K., Sandor, P., Kruuse, C., Fahrenkrug, J., et al. (2007). The effect of intravenous PACAP38 on cerebral hemodynamics in healthy volunteers. Regul. Pept. 140, 185–191. doi: 10.1016/j.regpep.2006.12.010
Bogdanov, K. Y., Maltsev, V. A., Vinogradova, T. M., Lyashkov, A. E., Spurgeon, H. A., Stern, M. D., et al. (2006). Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ. Res. 99, 979–987. doi: 10.1161/01.RES.0000247933.66532.0b
Bogdanov, K. Y., Vinogradova, T. M., and Lakatta, E. G. (2001). Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger. Cell. Biol. 88, 1254–1258.
Boixel, C., Dinanian, S., Lang-Lazdunski, L., Mercadier, J.-J., and Hatem, S. (2001). Characterization of effects of endothelin-1 on the L-type Ca2+ current in human atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 281, 764–773. doi: 10.1152/ajpheart.2001.281.2.H764
Bubinski, R., Kus, W., and Goch, J. (1994). Effect of somatostatin on the conduction system of the heart. Kardiol. Pol. 47:177.
Bucchi, A., Baruscotti, M., Robinson, R. B., and DiFrancesco, D. (2003). I(f)-dependent modulation of pacemaker rate mediated by cAMP in the presence of ryanodine in rabbit sino-atrial node cells. J. Mol. Cell. Cardiol. 35, 905–913. doi: 10.1016/s0022-2828(03)00150-0
Bucchi, A., Baruscotti, M., Robinson, R. B., and DiFrancesco, D. (2007). Modulation of rate by autonomic agonists in SAN cells involves changes in diastolic depolarization and the pacemaker current. J. Mol. Cell. Cardiol. 43, 39–48. doi: 10.1016/j.yjmcc.2007.04.017
Bussien, J., Biollaz, J., Waeber, B., Nussberger, J., Turini, G., Brunner, H., et al. (1986). Dose-dependent effect of atrial natriuretic peptide on blood pressure, heart rate, and skin blood flow of normal volunteers. J. Cardiovasc. Pharmacol. 8, 216–220. doi: 10.1097/00005344-198601000-00031
Centurion, D., Ortiz, M. I., Saxena, P. R., and Villalon, C. M. (2002). The atypical 5-HT2 receptor mediating tachycardia in pithed rats: pharmacological correlation with the 5-HT2A receptor subtype. Br. J. Pharmacol. 135, 1531–1539. doi: 10.1038/sj.bjp.0704593
Chahl, L. A., and Walker, S. B. (1981). Responses of the rat cardiovascular system to substance P, neurotensin and bombesin. Life Sci. 29, 2009–2015. doi: 10.1016/0024-3205(81)90611-1
Chakir, K., Zhu, W., Tsang, S., Woo, A. Y., Yang, D., Wang, X., et al. (2011). RGS2 is a primary terminator of β2-adrenergic receptor-mediated Gi signaling. J. Mol. Cell. Cardiol. 50, 1000–1007. doi: 10.1016/j.yjmcc.2011.01.015
Chandler, N. J., Greener, I. D., Tellez, J. O., Inada, S., Musa, H., Molenaar, P., et al. (2009). Molecular architecture of the human sinus node insights into the function of the cardiac pacemaker. Circulation 119, 1562–1575. doi: 10.1161/CIRCULATIONAHA.108.804369
Chang, F., Yu, H., and Cohen, I. S. (1994). Actions of vasoactive intestinal peptide and neuropeptide Y on the pacemaker current in canine purkinje fibers. Circ. Res. 74, 157–162. doi: 10.1161/01.res.74.1.157
Cho, H., Takano, M., and Noma, A. (2003). The electrophysiological properties of spontaneously beating pacemaker cells isolated from mouse sinoatrial node. J. Physiol. 550, 169–180. doi: 10.1113/jphysiol.2003.040501
Choate, J. K., and Feldman, R. (2003). Neuronal control of heart rate in isolated mouse atria. Am. J. Physiol. Heart Circ. Physiol. 285, H1340–H1346. doi: 10.1152/ajpheart.01119.2002
Chorev, M. (2002). Parathyroid hormone-1 receptor: insights into structure and function. Recept. Channels 8, 219–242. doi: 10.1080/10606820290005119
Chow, L. T. C., Chow, S. S. M., Anderson, R. H., and Gosling, J. A. (2001). Autonomic innervation of the human cardiac conduction system: changes from infancy to senility - an immunohistochemical and histochemical analysis. Anat. Rec. 264, 169–182. doi: 10.1002/ar.1158
Chowdhary, S., Harrington, D., Bonser, R. S., Coote, J. H., and Townend, J. N. (2002a). Chronotropic effects of nitric oxide in the denervated human heart. J. Physiol. 541, 645–651. doi: 10.1113/jphysiol.2001.015107
Chowdhary, S., Marsh, A. M., Coote, J. H., and Townend, J. N. (2004). Nitric oxide and cardiac muscarinic control in humans. Hypertension 43, 1023–1028. doi: 10.1161/01.HYP.0000126171.46645.f9
Chowdhary, S., Ng, G. A., Nuttall, S. L., Coote, J. H., Ross, H. F., and Townend, J. N. (2002b). Nitric oxide and cardiac parasympathetic control in human heart failure. Clin. Sci. 402, 397–402. doi: 10.1042/cs1020397
Chowdhary, S., and Townend, J. N. (1999). Role of nitric oxide in the regulation of cardiovascular autonomic control. Clin. Sci. 17, 5–17.
Chowdhary, S., Vaile, J. C., Fletcher, J., Ross, H. F., Coote, J. H., and Townend, J. N. (2000). Nitric oxide and cardiac autonomic control in humans. Hypertension 36, 264–269.
Christ, T., Galindo-tovar, A., Thoms, M., Ravens, U., and Kaumann, A. J. (2009). Inotropy and L-type Ca2+ current, activated by β1- and β2-adrenoceptors, are differently controlled by phosphodiesterases 3 and 4 in rat heart. Br. J. Pharmacol. 156, 62–83. doi: 10.1111/j.1476-5381.2008.00015.x
Cifelli, C., Rose, R. A., Zhang, H., Voigtlaender-bolz, J., Bolz, S., Backx, P. H., et al. (2008). RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 103, 527–535. doi: 10.1161/CIRCRESAHA.108.180984
Crozier, I., Richards, A. M., Foy, S. G., and Ikram, H. (1993). Electrophysiological effects of atriai natriuretic peptide on the cardiac conduction system in man. PACE Pacing Clin. Electrophysiol. 16, 738–743. doi: 10.1111/j.1540-8159.1993.tb01653.x
Dessy, C., and Balligand, J. (2010). β3-adrenergic receptors in cardiac and vascular tissues?: emerging concepts and therapeutic perspectives. Adv. Pharmacol. 59, 135–163. doi: 10.1016/S1054-3589(10)59005-7
Difrancesco, D. (2010). The role of the funny current in pacemaker activity. Circ. Res. 106, 434–446. doi: 10.1161/CIRCRESAHA.109.208041
Difrancesco, D., and Noble, D. (2012). The funny current has a major pacemaking role in the sinus node. Hear. Rhythm 9, 299–301. doi: 10.1016/j.hrthm.2011.09.021
DiFrancesco, D., and Tortora, P. (1991). Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Lett. Nat. 351, 145–147. doi: 10.1038/351145a0
Dillmann, W. H. (1989). Diabetes and thyroid-hormone-induced changes in cardiac function and their molecular basis. Annu. Rev. Med. 40, 373–394. doi: 10.1146/annurev.me.40.020189.002105
Dodge-Kafka, K. L., Langeberg, L., and Scott, J. D. (2006). Compartmentation of cyclic nucleotide signaling in the heart the role of A-kinase anchoring proteins. Circ. Res. 98, 993–1001. doi: 10.1161/01.RES.0000218273.91741.30
Donald, J. A., O’Shea, J. E., and Lillywhite, H. B. (1990). Somatostatin and innervation of the snake elaphe obsoleta of the heart. Am. Physiol. Soc. 258, 1001–1007. doi: 10.1152/ajpregu.1990.258.4.R1001
Dorner, G. T., Wolzt, M., Hans-Georg, E., and Schmetterer, L. (1998). Effect of pituitary adenylate cyclase activating polypeptide 1–27 on ocular, cerebral and skin blood flow in humans. Naunyn Schmiedeberg Arch. Pharmacol. 358, 657–662. doi: 10.1007/pl00005308
Drouin, E. (1997). Electrophysiologic properties of the adult human sinus node. J. Cardiovasc. Electrophysiol. 8, 254–258. doi: 10.1111/j.1540-8167.1997.tb00788.x
Drugge, E. D., Rosen, M. R., and Robinson, R. B. (1985). Neuronal regulation of the development of the alpha-adrenergic chronotropic response in the rat heart. Circ. Res. 57, 415–423. doi: 10.1161/01.res.57.3.415
Fenske, S., Krause, S. C., Hassan, S. I., Becirovic, E., Auer, F., Bernard, R., et al. (2013). Sick sinus syndrome in HCN1-deficient mice. Circulation 128, 2585–2594. doi: 10.1161/CIRCULATIONAHA.113.003712
Fischmeister, R., Castro, L., Abi-Gerges, A., Rochais, F., and Vandecasteele, G. (2005). Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 142, 136–143. doi: 10.1016/j.cbpb.2005.04.012
Fischmeister, R., Castro, L. R. V., Abi-Gerges, A., Rochais, F., Jurevičius, J., Leroy, J., et al. (2006). Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ. Res. 99, 816–828. doi: 10.1161/01.RES.0000246118.98832.04
Fisher, R. A., Robertson, S. M., and Olson, M. S. (1988). Stimulation and homologous desensitization of calcitonin gene-related peptide receptors in cultured beating rat heart cells. Endocrinology 123, 106–112. doi: 10.1210/endo-123-1-106
Franco-Cereceda, A., Gennari, C., Nami, R., Agnusdei, D., Pernow, J., Lundberg, J. M., et al. (1987). Cardiovascular effects of calcitonin gene-related peptides I and II in man. Circ. Res. 60, 393–397. doi: 10.1161/01.res.60.3.393
Franco-Suarez, R., Somani, P., and Mulrow, P. J. (1987). Bradycardia after infusion of atrial natriuretic factor. Ann. Intern. Med. 107:594. doi: 10.7326/0003-4819-107-4-594-1
Frase, L. L., Gaffney, A. F., Lane, L. D., Buckey, J. C., Said, S. I., Blomqvist, C. G., et al. (1987). Cardiovascular effects of vasoactive intestinal peptide in healthy subjects. Am. J. Cardiol. 60, 1356–1361. doi: 10.1016/0002-9149(87)90619-9
Fu, Q., and Xiang, Y. K. (2015). Trafficking of β-Adrenergic Receptors: Implications in Intracellular Receptor Signaling, 1st Edn, Amsterdam: Elsevier Inc, doi: 10.1016/bs.pmbts.2015.03.008
Galindo-Tovar, A., Vargas, M. L., and Kaumann, A. J. (2010). Function of cardiac β1-and β2-adrenoceptors of newborn piglets: role of phosphodiesterases PDE3 and PDE4. Eur. J. Pharmacol. 638, 99–107. doi: 10.1016/j.ejphar.2010.04.013
Gelzer, A. R., Attmann, T., Radicke, D., Nydam, D., Candinas, R., and Lutter, G. (2004). Effects of acute systemic endothelin receptor blockade on cardiac electrophysiology in vivo. J. Cardiovasc. Pharmacol. 44, 564–570. doi: 10.1097/00005344-200411000-00008
Gennari, C., Narni, R., Agnusdei, D., and Fischer, J. (1990). Improved cardiac performance with human calcitonin gene related peptide in patients with congestive heart failure. Cardiovasc. Res. 24, 239–241. doi: 10.1093/cvr/24.3.239
Gloss, B., Trost, S. U., Bluhm, W. F., Swanson, E. A., Clark, R., Winkfein, R., et al. (2001). Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor alpha or beta. Endocrinology 142, 544–550. doi: 10.1210/endo.142.2.7935
Golias, C., Charalabopoulos, A., Stagikas, D., Charalabopoulos, K., and Batistatou, A. (2007). The kinin system - bradykinin: biological effects and clinical implications. Multiple role of the kinin system. Hippokratia 11, 124–128.
Gordan, R., Gwathmey, J. K., and Xie, L.-H. (2015). Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 7, 204–214. doi: 10.4330/wjc.v7.i4.204
Gothert, M., Schlicker, E., and Kollecker, P. (1986). Receptor-mediated effects of serotonin and 5-methoxytryptamine on noradrenaline release in the rat vena cava and in the heart of the pithed rat. Arch. Pharmacol. 332, 124–130. doi: 10.1007/bf00511401
Habuchi, Y., Lu, L., Morikawa, J., and Yoshimura, M. (1995). Angiotensin II inhibition of L-type Ca2+ current in sinoatrial node cells of rabbits. Am. J. Physiol. 268, 1053–1060. doi: 10.1152/ajpheart.1995.268.3.H1053
Hagiwara, N., Irisawa, H., and Kameyama, M. (1988). Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 395, 233–253. doi: 10.1113/jphysiol.1988.sp016916
Han, X., Habuchi, Y., and Giles, W. R. (1994a). Relaxin increases heart rate by modulating calcium current in cardiac pacemaker cells. Circ. Res. 74, 537–542. doi: 10.1161/01.res.74.3.537
Han, X., Shimoni, Y., and Giles, W. R. (1995). A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. J. Gen. Physiol. 106, 45–65. doi: 10.1085/jgp.106.1.45
Han, X., Shimoni, Y., Giles, W. R., and Tn, C. (1994b). An obligatory role for nitric oxide in autonomic control of mammalian heart rate. J. Physiol. 476, 309–314. doi: 10.1113/jphysiol.1994.sp020132
Hara, M., Liu, Y.-M., Zhen, L., Cohen, I. S., Yu, H., Danilo, P. J., et al. (1997). Positive chronotropic actions of parathyroid hormone and parathyroid hormone-related peptide are associated with increases in the current, If, and the slope of the pacemaker potential. Circulation 96, 3704–3709. doi: 10.1161/01.cir.96.10.3704
Harvey, R. D. (2012). Muscarinic receptor agonists and antagonists: effects on cardiovascular function. Muscar. Recept. 2012, 299–316. doi: 10.1007/978-3-642-23274-9_13
Hata, T., Noda, T., Nishimura, M., and Watanabe, Y. (1996). The role of Ca2+ release from sarcoplasmic reticulum in the regulation of sinoatrial node automaticity. Heart Vessels 11, 234–241. doi: 10.1007/bf01746203
Hattori, Y., Hattori, K., and Matsuda, N. (2004). Regulation of the cardiovascular system by histamine. Histamine Recept. Health Dis. 241, 239–258. doi: 10.1007/164-2016-15
Headrick, J. P., Ashton, K. J., Meyer, R. B., and Peart, J. N. (2013). Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacol. Ther. 140, 92–111. doi: 10.1016/j.pharmthera.2013.06.002
Henning, R. J., and Sawmiller, D. R. (2001). Vasoactive intestinal peptide: cardiovascular effects. Cardiovasc. Res. 49, 27–37. doi: 10.1016/s0008-6363(00)00229-7
Hewett, K., and Rosen, M. (1985). Developmental changes in the rabbit sinus node action potential and its response to adrenergic agonists. J. Pharmacol. Exp. Ther. 235, 308–312.
Himeno, Y., Sarai, N., Matsuoka, S., and Noma, A. (2008). Ionic mechanisms underlying the positive chronotropy induced by β1-adrenergic stimulation in guinea pig sinoatrial node cells: a simulation study. J. Physiol. Sci. 58, 53–65. doi: 10.2170/physiolsci.RP015207
Hirose, M., Furukawa, Y., Lakhe, M., and Chiba, S. (1998). Regional differences in cardiac effects of pituitary adenylate cyclase-activating polypeptide-27 in the isolated dog heart. Eur. J. Pharmacol. 349, 269–276. doi: 10.1016/s0014-2999(98)00210-6
Hirose, M., Furukawa, Y., Nagashima, Y., Lakhe, M., and Chiba, S. (1997a). Pituitary adenylate cyclase-activating polypeptide-27 causes a biphasic chronotropic effect and atrial fibrillation in autonomically decentralized, anesthetized dogs. J. Pharmacol. Exp. Ther. 283, 478–487.
Hirose, M., Furukawa, Y., Nagashima, Y., Yamazaki, K., Hoyano, Y., and Chiba, S. (1997b). Effects of PACAP-38 on the SA nodal pacemaker activity in autonomically decentralized hearts of anesthetized dogs. J. Cardiovasc. Pharmacol. 29, 216–221. doi: 10.1097/00005344-199702000-00010
Holman, J. J., Craig, R. K., and Marshall, I. (1986). Human α- and β-CGRP and rat α-CGRP are coronary vasodilators in the rat. Peptides 7, 231–235. doi: 10.1016/0196-9781(86)90218-4
Hoover, D. (1989). Effects of guinea pig vasoactive intestinal peptide on the isolated perfused guinea pig heart. Peptides 10, 343–347. doi: 10.1016/0196-9781(89)90041-7
Horinouchi, T., Terada, K., Higashi, T., and Miwa, S. (2013). Endothelin receptor signaling: new insight into its regulatory mechanisms. J. Pharmacol. Sci. 101, 85–101. doi: 10.1254/jphs.13R02CR
Hu, W.-H., Wang, K.-Y., Hwang, D.-S., Ting, C.-T., and Wu, T.-C. (1997). Histamine-2 receptor blocker-ranitidine and sinus node dysfunction. Chin. Med. J. 60, 1–5.
Hua, R., Adamczyk, A., Robbins, C., Ray, G., and Rose, R. A. (2012). Distinct patterns of constitutive phosphodiesterase activity in mouse sinoatrial node and atrial myocardium. PLoS One 7:47652. doi: 10.1371/journal.pone.0047652
Huang, C., Feng, S., and Hilgemann, D. W. (1998). Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by GBetaGamma. Nature 391, 803–806. doi: 10.1038/35882
Huang, Z. M., Gao, E., Chuprun, J. K., and Koch, W. J. (2014). GRK2 in the heart: a GPCR kinase and beyond. Antioxid. Redox Signal. 21, 2032–2043. doi: 10.1089/ars.2014.5876
Hulting, J., Sollevi, A., Ullman, B., Franco-cereceda, A., and Lundberg, J. M. (1990). Plasma neuropeptide Y on admission to a coronary care unit: raised levels in patients with left heart failure. Cardiovasc. Res. 24, 102–108. doi: 10.1093/cvr/24.2.102
Hüser, J., Lipsius, S. L., and Blatter, L. A. (1996). Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J. Physiol. 940, 641–651. doi: 10.1113/jphysiol.1996.sp021521
Inokaitis, H., Pauziene, N., Rysevaite-Kyguoliene, K., and Pauza, D. H. (2016). Innervation of sinoatrial nodal cells in the rabbit. Ann. Anat. 205, 113–121. doi: 10.1016/j.aanat.2016.03.007
Irisawa, H., Brown, H. F., and Giles, W. (1993). Cardiac pacemaking in the sinoatrial node. Physiol. Rev. 73, 197–227. doi: 10.1152/physrev.1993.73.1.197
Ishikawa, T., Yanagisawa, M., Kimura, S., Goto, K., and Masaki, T. (1988). Positive chronotropic effects of endothelin, a novel endothelium-derived vasoconstrictor peptide. Eur. J. Physiol. 413, 108–110. doi: 10.1007/bf00581239
Isidori, A. M., Cornacchione, M., Barbagallo, F., Di Grazia, A., Barrios, F., Fassina, L., et al. (2015). Inhibition of type 5 phosphodiesterase counteracts β2-adrenergic signalling in beating cardiomyocytes. Cardiovasc. Res. 106, 408–420. doi: 10.1093/cvr/cvv123
Jacques, D., and Abdel-Samad, D. (2007). Neuropeptide Y (NPY) and NPY receptors in the cardiovascular system: implication in the regulation of intracellular calcium. Can. J. Physiol. Pharmacol. 85, 43–53. doi: 10.1139/Y06-106
James, T. N. (1964). The chronotropic action of seroronin studied by direct perfusion of the sinus node. J. Pharmacol. Exp. Ther. 146, 209–214.
James, T. N., Bear, E. S., Lang, K. F., Green, E. W., and Winkler, H. H. (1970). Adrenergic mechanisms in the sinus node. Arch. Intern. Med. 125, 512–547. doi: 10.1001/archinte.1970.00310030123017
Jensen, B. C., Connell, T. D. O., and Simpson, P. C. (2014). Alpha-1-adrenergic receptors in heart failure: the adaptive arm of the cardiac response to chronic catecholamine stimulation. J. Cardiovasc. Pharmacol. 63, 291–301. doi: 10.1097/FJC.0000000000000032
Johnson, M., Jauniaux, E., Ramsay, B., Jurkovic, D., and Meuris, S. (1994). Maternal relaxin: a determinant of fetal heart rate? Br. J. Obstet. Gynecol. 101, 1003–1004. doi: 10.1111/j.1471-0528.1994.tb13048.x
De Jongh, K. S., Murphy, B. J., Colvin, A. A., Hell, J. W., Takahashi, M., Catterall, W. A., et al. (1996). Specific phosphorylation of a site in the full-length form of the alpha1 subunit of the cardiac L-type calcium channel by adenosine 3’, 5’-cyclic monophosphate - dependent protein kinase. Biochemistry 2960, 10392–10402. doi: 10.1021/bi953023c
Joung, B., Tang, L., Maruyama, M., Han, S., Chen, Z., Stucky, M., et al. (2009). Intracellular calcium dynamics and acceleration of sinus rhythm by beta-adrenergic stimulation. Circulation 119, 788–796. doi: 10.1161/CIRCULATIONAHA.108.817379
Ju, Y., Chu, Y., Chaulet, H., Lai, D., Gervasio, O. L., Graham, R. M., et al. (2007). Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ. Res. 100, 1605–1614. doi: 10.1161/CIRCRESAHA.107.152181
Ju, Y., Liu, J., Lee, B. H., Lai, D., Woodcock, E. A., Lei, M., et al. (2011). Distribution and functional role of inositol 1,4,5-trisphosphate receptors in mouse sinoatrial node. Circ. Res. 109, 848–857. doi: 10.1161/CIRCRESAHA.111.243824
Ju, Y., Woodcock, E. A., Allen, D. G., and Cannell, M. B. (2012). Inositol 1,4,5-trisphosphate receptors and pacemaker rhythms. J. Mol. Cell. Cardiol. 53, 375–381. doi: 10.1016/j.yjmcc.2012.06.004
Kakouris, H., Lawrence, E. W., and Summers, R. J. (1992). Cardiac effects of relaxin in rats. Lancet 339, 1076–1078. doi: 10.1016/0140-6736(92)90665-p
Kapoor, N., Tran, A., Kang, J., Zhang, R., Philipson, K. D., and Goldhaber, J. I. (2015). Regulation of calcium clock-mediated pacemaking by inositol-1,4,5-trisphosphate receptors in mouse sinoatrial nodal cells. J. Physiol. 593, 2649–2663. doi: 10.1113/JP270082
Kaumann, A. J. (1993). Blockade of human atrial 5-HT4 receptors by GR 113808. Br. J. Pharmacol. 110, 1172–1174. doi: 10.1111/j.1476-5381.1993.tb13937.x
Kaumann, A. J., Gaster, L. M., King, F. D., and Brown, A. M. (1994). Blockade of human atrial 5-HT4 receptors by SB 207710, a selective and high affinity 5-HT4 receptor antagonist. Arch. Pharmacol. 349, 546–548. doi: 10.1007/bf00169146
Kaumann, A. J., and Levy, O. F. (2006). 5-Hydroxytryptamine receptors in the human cardiovascular system. Pharmacol. Ther. 111, 674–706. doi: 10.1016/j.pharmthera.2005.12.004
Kaumann, A. J., Sanders, L., Brown, A. M., Murray, K. J., and Brown, M. J. (1990). A 5-hydroxytryptamine receptor in human atrium. Br. J. Pharmacol. 100, 879–885. doi: 10.1007/bf00176614
Kaumann, A. J., Sanders, L., Brown, A. M., Murray, K. J., and Brown, M. J. (1991). A 5-HT4-like receptor in human right atrium. Arch. Pharmacol. 344, 150–159. doi: 10.1007/bf00167212
Kevelaitis, E., Abraitis, R., and Lazhauskas, R. (1994). Histamine and pacemaker shift in the sinoatrial node. Agent Actions 88, 87–88. doi: 10.0065-4299/94/020C87-02
Kohl, P., and Quinn, T. A. (2014). Novel technologies as drivers of progress in cardiac biophysics. Prog. Biophys. Mol. Biol. 115, 69–70. doi: 10.1016/j.pbiomolbio.2014.08.014
Kolettis, T. M., Kyriakides, Z. S., Leftheriotis, D., Papalambrou, A., Kremastinos, D. T., and Webb, D. J. (2003). Electrophysiologic effects of endothelin receptor - a blockade in patients with coronary artery disease. J. Interv. Card. Electrophysiol. 8, 173–179. doi: 10.1023/a:1023961004016
Lakatta, E. G., Maltsev, V. A., and Vinogradova, T. M. (2010). A Coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 106, 659–673. doi: 10.1161/CIRCRESAHA.109.206078
Lakatta, E. G., Vinogradova, T. M., and Maltsev, V. A. (2008). The missing link in the mystery of normal automaticity of cardiac pacemaker cells. Ann. N. Y. Acad. Sci. 1123, 41–57. doi: 10.1196/annals.1420.006
Lambert, C. (1995). Mechanisms of angiotensin II chronotropic effect in anaesthetized dogs. Br. J. Pharmacol. 115, 795–800. doi: 10.1111/j.1476-5381.1995.tb15003.x
Larsson, H. P. (2010). How is the heart rate regulated in the sinoatrial node? Another piece to the puzzle. J. Gen. Physiol. 136, 237–241. doi: 10.1085/jgp.201010506
Lei, M., Brown, H. F., and Terrar, D. A. (2000). Modulation of delayed rectifier potassium current, Ik, by isoprenaline in rabbit isolated pacemaker cells. Exp. Physiol. 85. 1, 27–35. doi: 10.1111/j.1469-445x.2000.01915.x
Lei, M., Goddard, C., Liu, J., Leoni, A. L., Royer, A., Fung, S. S., et al. (2005). Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene SCN5A. J. Physiol. 567, 387–400. doi: 10.1113/jphysiol.2005.083188
Levi, R., and Smith, N. C. (2000). Histamine H3-receptors: a new frontier in myocardial ischemia. J. Pharmacol. Exp. Ther. 292, 825–830.
Li, N., Csepe, T. A., Hansen, B. J., Dobrzynski, H., Higgins, R. S. D., Kilic, A., et al. (2015). Molecular mapping of sinoatrial node HCN channel expression in the human heart. Circ. Arrhythmia Electrophysiol. 8, 1219–1227. doi: 10.1161/CIRCEP.115.003070
Li, N., Hansen, B. J., Csepe, T. A., Zhao, J., Ignozzi, A. J., Sul, L. V., et al. (2017). Redundant and diverse intranodal pacemakers and conduction pathways protect the human sinoatrial node from failure. Sci. Transl. Med. 9, 1–11. doi: 10.1126/scitranslmed.aam5607
Li, Y., Sirenko, S., Riordon, D. R., Yang, D., Spurgeon, H., Lakatta, E. G., et al. (2016). CaMKII-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local Ca 2+ releases. Am. J. Physiol. Heart Circ. Physiol. 14, 532–544. doi: 10.1152/ajpheart.00765.2015
Liao, Y., Xu, W.-R., Li, H.-X., Tang, C.-S., Jin, H.-F., and Du, J.-B. (2017). Plasma neuropeptide Y levels in vasovagal syncope in children. Chin. Med. J. 130, 2778–2784. doi: 10.4103/0366-6999.219157
Liao, Z., Lockhead, D., Larson, E. D., and Proenza, C. (2010). Phosphorylation and modulation of hyperpolarization-activated HCN4 channels by protein kinase A in the mouse sinoatrial node. J. Gen. Physiol. 136, 247–258. doi: 10.1085/jgp.201010488
Linden, A., Desmecht, D., Amory, H., and Lekeux, P. (1999). Cardiovascular response to intravenous sdministration of 5-hydroxytryptamine after type-2 receptor blockade, by metrenperone, in healthy calves. Vet. J. 157, 31–37. doi: 10.1053/tvjl.1998.0250
Lonardo, G., Cerbai, E., Casini, S., Giunti, G., Bonacchi, M., Battaglia, F., et al. (2004). Atrial natriuretic peptide modulates the hyperpolarization-activated current (If) in human atrial myocytes. Cardiovasc. Res. 63, 528–536. doi: 10.1016/j.cardiores.2004.03.004
Lou, Q., Glukhov, A. V., Hansen, B., Hage, L., Vargas-pinto, P., Billman, G. E., et al. (2013). Tachy-brady arrhythmias: the critical role of adenosine-induced sinoatrial conduction block in post-tachycardia pauses. Hear. Rhythm 10, 110–118. doi: 10.1016/j.hrthm.2012.09.012
Lou, Q., Hansen, B. J., Fedorenko, O., Csepe, T. A., Kalyanasundaram, A., Li, N., et al. (2014). Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities revealed by novel dual-sided intramural optical mapping. Circulation 130, 315–324. doi: 10.1161/CIRCULATIONAHA.113.007086
Lukyanenko, Y. O., Younes, A., Lyashkov, A. E., Tarasov, K. V., Riordon, D. R., Lee, J., et al. (2016). Ca 2+/calmodulin-activated phosphodiesterase 1A is highly expressed in rabbit cardiac sinoatrial nodal cells and regulates pacemaker function. J. Mol. Cell. Cardiol. 98, 73–82. doi: 10.1016/j.yjmcc.2016.06.064
Lundberg, J. A. N. M., Hua, X., and Franco-Cereceda, A. (1984). Effects of neuropeptide Y (NPY) on mechanical activity and neurotransmission in the heart, vas deferens and urinary bladder of the guinea-pig. Acta Physiol. Scand. 121, 325–332. doi: 10.1111/j.1748-1716.1984.tb07463.x
Lyashkov, A. E., Behar, J., Lakatta, E. G., Yaniv, Y., and Maltsev, V. A. (2018). Positive feedback mechanisms among local Ca releases, NCX, and ICaL ignite pacemaker action potentials. Biophys. J. 114, 1176–1189. doi: 10.1016/j.bpj.2017.12.043
Lyashkov, A. E., Juhaszova, M., Dobrzynski, H., Vinogradova, T. M., Maltsev, V. A., Juhasz, O., et al. (2007). Calcium cycling protein density and functional importance to automaticity of isolated sinoatrial nodal cells are independent of cell size. Circ. Res. 100, 1723–1731. doi: 10.1161/CIRCRESAHA.107.153676
Lyashkov, A. E., Vinogradova, T. M., Zahanich, I., Li, Y., Younes, A., Nuss, H. B., et al. (2009). Cholinergic receptor signaling modulates spontaneous firing of sinoatrial nodal cells via integrated effects on PKA-dependent Ca2+ cycling and IKACh. Am. J. Physiol. Heart Circ. Physiol. 297, 949–959. doi: 10.1152/ajpheart.01340.2008
MacDonald, E. A., Stoyek, M. R., Rose, R. A., and Quinn, T. A. (2017). Intrinsic regulation of sinoatrial node function and the zebrafish as a model of stretch effects on pacemaking. Prog. Biophys. Mol. Biol. 130, 198–211. doi: 10.1016/j.pbiomolbio.2017.07.012
Machuki, J. O., Zhang, H. Y., Harding, S. E., and Sun, H. (2018). Molecular pathways of oestrogen receptors and β-adrenergic receptors in cardiac cells: recognition of their similarities, interactions and therapeutic value. Acta Physiol. 222:e12978. doi: 10.1111/apha.12978
MacKaay, A. J., Opthof, T., Bleeker, W. K., Jongsma, H. J., and Bouman, L. N. (1980). Interaction of adrenaline and acetylcholine on cardiac pacemaker function - functional inhomogeneity of the rabbit sinus node. J. Pharmacol. Exp. Ther. 214, 417–422.
Madamanchi, A. (2007). Beta-adrenergic receptor signaling in cardiac function and heart failure. McGill J. Med. 10, 99–104.
Madeddu, P., Salis, M. B., and Emanueli, C. (1999). Altered baroreflex control of heart rate in bradykinin β2-receptor knockout mice. Immunopharmacology 45, 21–27. doi: 10.1016/s0162-3109(99)00053-3
Maltsev, V. A., and Lakatta, E. G. (2012). The funny current in the context of the coupled-clock pacemaker cell system. Hear. Rhythm 9, 457–458. doi: 10.1016/j.hrthm.2011.09.023
Mangoni, M. E., Couette, B., Bourinet, E., Platzer, J., and Reimer, D. (2003). Functional role of L-type Ca v1.3 Ca2+ channels in cardiac pacemaker activity. PNAS 100, 5543–5548.
Mangoni, M. E., and Nargeot, J. (2008). Genesis and regulation of the heart automaticity. Physiol. Rev. 88, 919–982. doi: 10.1152/physrev.00018.2007
Marin-Garcia, J. (2011). Rapid signaling pathways. Signal. Heart 45, 49–68. doi: 10.1007/978-1-4419-9461-5-4
Marshall, I., Al-Kazwini, S., Roberts, P., Shepperson, N., Adams, M., and Craig, R. (1986). Cardiovascular effects of human and rat CGRP compared in the rat and other species. Eur. J. Pharmacol. 123, 207–216. doi: 10.1016/0014-2999(86)90661-8
Mary-Rabine, L., Hordof, A. J., Bowman, F., Malm, J. R., and Rosen, M. R. (1978). Alpha and beta adrenergic effects on human atrial specialized conducting fibers. Circulation 57, 84–90. doi: 10.1161/01.cir.57.1.84
Massion, P. B., Feron, O., Dessy, C., and Ballingand, J. L. (2003). Nitric oxide and cardiac function. Circ. Res. 93, 388–398. doi: 10.1161/01.RES.0000088351.58510.21
Masumiya, H., Yamamoto, H., Hemberger, M., Tanaka, H., Shigenobu, K., Chen, S. R. W., et al. (2003). The mouse sino-atrial node expresses both the type 2 and type 3 Ca(2+) release channels/ryanodine receptors. FEBS Lett. 553, 141–144. doi: 10.1016/s0014-5793(03)00999-2
Matsuda, N., Jesmin, S., Takahashi, Y., Hatta, E., Kobayashi, M., Matsuyama, K., et al. (2004). Histamine H1 and H2 receptor gene and protein levels are differentially expressed in the hearts of rodents and humans. J. Pharmacol. Exp. Ther. 309, 786–795. doi: 10.1124/jpet.103.063065.tion
Mattick, P., Parrington, J., Odia, E., Simpson, A., Collins, T., and Terrar, D. (2007). Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the If pacemaker current. J. Physiol. 582, 1195–1203. doi: 10.1113/jphysiol.2007.133439
Meah, V. L., Cockcroft, J. R., Backx, K., Shave, R., and Stöhr, E. J. (2016). Cardiac output and related haemodynamics during pregnancy: a series of meta-analyses. Heart 102, 518–526. doi: 10.1136/heartjnl-2015-308476
Mehta, P. K., and Griendling, K. K. (2007). Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 292, C82–C97. doi: 10.1152/ajpcell.00287.2006
Mesirca, P., Torrente, A. G., and Mangoni, M. E. (2015). Functional role of voltage gated Ca2+ channels in heart automaticity. Front. Physiol. 6:19. doi: 10.3389/fphys.2015.00019
Mighiu, A. S., and Heximer, S. P. (2012). Controlling parasympathetic regulation of heart rate: a gatekeeper role for RGS proteins in the sinoatrial node. Front. Physiol. 3:204. doi: 10.3389/fphys.2012.00204
Minkes, A., Bellan, J., Saroyan, R., Kerstein, M., Coy, H., Murphy, W., et al. (1989). Analysis of cardiovascular and pulmonary responses to to endothelin-1 and endothelin-3 in the anesthetized cat. J. Pharmacol. Exp. Ther. 253, 1118–1125.
Moen, J. M., Matt, M. G., Ramirez, C., Tarasov, K. V., Chakir, K., Tarasov, Y. S., et al. (2019). Overexpression of a neuronal type adenylyl cyclase (type 8) in sinoatrial node markedly impacts heart rate and rhythm. Front. Neurosci. 13:615. doi: 10.3389/fnins.2019.00615
Moghtadaei, M., Polina, I., and Rose, R. A. (2016). Electrophysiological effects of natriuretic peptides in the heart are mediated by multiple receptor subtypes. Prog. Biophys. Mol. Biol. 120, 37–49. doi: 10.1016/j.pbiomolbio.2015.12.001
Moss, R., Sachse, F. B., Moreno-Galindo, E. G., Navarro-Polanco, R. A., Tristani-Firouzi, M., and Seemann, G. (2018). Modeling effects of voltage dependent properties of the cardiac muscarinic receptor on human sinus node function. PLoS Comput. Biol. 14:6438. doi: 10.1371/journal.pcbi.1006438
Newton, C. M., Stoyek, M. R., Croll, R. P., and Smith, F. M. (2014). Regional innervation of the heart in the goldfish, carassius auratus: a confocal microscopy study. J. Comp. Neurol. 478, 456–478. doi: 10.1002/cne.23421
Nicholls, M., and Richards, A. (1987). Human studies with atrial natriuretic factor. Endocrinol. Metab. Clin. North Am. 16, 199–223.
Nikmaram, M. R., Liu, J., Abdelrahman, M., Dobrzynski, H., Boyett, M. R., and Lei, M. (2008). Characterization of the effects of ryanodine, TTX, E-4031 and 4-AP on the sinoatrial and atrioventricular nodes. Prog. Biophys. Mol. Biol. 96, 452–464. doi: 10.1016/j.pbiomolbio.2007.07.003
Nistri, S., Cinci, L., Perna, A., Masini, E., Mastroianni, R., and Bani, D. (2008). Relaxin induces mast cell inhibition and reduces ventricular arrhythmias in a swine model of acute myocardial infarction. Pharmacol. Res. 57, 43–48. doi: 10.1016/j.phrs.2007.11.001
Noma, A., Morad, M., and Irisawa, H. (1983). Does the “pacemaker current” generate the diastolic depolarization in the rabbit SA node cells? Pflugers Arch. 397, 190–194. doi: 10.1007/bf00584356
Nwazue, V. C., Paranjape, S. Y., Black, B. K., Biaggioni, I., Dupont, W. D., Robertson, D., et al. (2014). Postural tachycardia syndrome and inappropriate sinus tachycardia: role of autonomic modulation and sinus node automaticity. J. Am. Heart Assoc. 3, 1–8. doi: 10.1161/JAHA.113.000700
Oldham, W. M., and Hamm, H. E. (2008). Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71. doi: 10.1038/nrm2299
Oney, T., and Kaulhausen, H. (1982). Effect of angiotensin infusion during pregnancy fetal heart rate and on fetal activity. Eur. J. Obstet. Gynecol. Reprod. Biol. 13, 133–137. doi: 10.1016/0028-2243(82)90023-5
Ono, K., Masumiya, H., Sakamoto, A., Christé, G., Shijuku, T., Tanaka, H., et al. (2001). Electrophysiological analysis of the negative chronotropic effect of endothelin-1 in rabbit sinoatrial node cells. J. Physiol. 537, 467–488. doi: 10.1111/j.1469-7793.2001.00467.x
Opthof, T. (2001). Function and structure of the mouse sinus node: nothing you can see that isn’t shown. Cardiovasc. Res. 52, 1–4. doi: 10.1016/s0008-6363(01)00417-5
Opthof, T. (2007). Embryological development of pacemaker hierarchy and membrane currents related to the function of the adult sinus node: implications for autonomic modulation of biopacemakers. Medicial Biol. Eng. Comput. 45, 119–132. doi: 10.1007/s11517-006-0138-x
Opthof, T., VanGinneken, A., Bouman, L. N., and Jongsma, H. J. (1987). The intrinsic cycle length in small pieces isolated from the rabbit sinoatrial node. J. Mol. Cell. Cardiol. 19, 923–934. doi: 10.1016/s0022-2828(87)80621-1
Osadchii, O. E. (2015). Emerging role of neurotensin in regulation of the cardiovascular system. Eur. J. Pharmacol. 762, 184–192. doi: 10.1016/j.ejphar.2015.05.025
Pachucki, J., Burmeister, L. A., and Larsen, P. R. (1999). Thyroid hormone regulates hyperpolarization-activated cyclic nucleotide-gated channel (HCN2) mRNA in the rat heart. Circ. Res. 85, 498–503. doi: 10.1161/01.res.85.6.498
Page, H., and McCubbin, J. W. (1955). Arterial pressure response to infused serotonin in normotensive dogs, cats, hypertensive dogs and man. Am. J. Physiol. 184, 265–270. doi: 10.1152/ajplegacy.1956.184.2.265
Parker, S. G., Taylor, E. M., Hamburger, S. A., Vimal, M., and Kaumann, A. J. (1995). Blockade of human and porcine myocardial 5-HT4 receptors by SB 203186. Naunyn Schmiedebergs Arch. Pharmacol. 353, 28–35. doi: 10.1007/bf00168912
Paterson, D. J. (2001). Nitric oxide and the autonomic regulation of cardiac excitability. Exp. Physiol. 86, 1–12. doi: 10.1113/eph8602169
Paur, H., Wright, P. T., Sikkel, M. B., Tranter, M. H., Mansfield, C., O’Gara, P., et al. (2012). High levels of circulating epinephrine trigger apical cardiodepression in a β2-adrenergic receptor/Gi-dependent manner: a new model of takotsubo cardiomyopathy. Circulation 126, 697–706. doi: 10.1161/CIRCULATIONAHA.112.111591
Pauza, D. H., Rysevaite, K., Inokaitis, H., Jokubauskas, M., Pauza, A. G., Brack, K. E., et al. (2014). Innervation of sinoatrial nodal cardiomyocytes in mouse. A combined approach using immunofluorescent and electron microscopy. J. Mol. Cell. Cardiol. 75, 188–197. doi: 10.1016/j.yjmcc.2014.07.016
Pejkovic, B., Krajnc, I., Anderhuber, F., and Kosutic, D. (2008). Anatomical aspects of the arterial blood supply to the sinoatrial and atrioventricular nodes of the human. J. Int. Med. Res. 36, 691–698. doi: 10.1177/147323000803600410
Pian, P., Bucchi, A., Decostanzo, A., Robinson, R. B., and Siegelbaum, S. A. (2007). Modulation of cyclic nucleotide-regulated HCN channels by PIP2 and receptors coupled to phospholipase C. Eur. J. Physiol. 455, 125–145. doi: 10.1007/s00424-007-0295-2
Pino, R., Cerbai, E., Calamai, G., Alajmo, F., Borgioli, A., Braconi, L., et al. (1998). Effect of 5-HT 4 receptor stimulation on the pacemaker current If in human isolated atrial myocytes. Cardiovasc. Res. 40, 516–522. doi: 10.1016/s0008-6363(98)00198-9
Ponchon, P., Grichois, M.-L., Girolami, J.-P., and Elghozi, J.-L. (1995). Effects of bradykinin on short-term variability in blood pressure and heart rate in rats: a spectral study. J. Cardiovasc. Pharmacol. 25, 914–923. doi: 10.1097/00005344-199506000-00009
Potthoff, S. A., Janus, A., Hoch, H., Frahnert, M., Tossios, P., Reber, D., et al. (2011). Regulatory peptides PTH-receptors regulate norepinephrine release in human heart and kidney. Regul. Pept. 171, 35–42. doi: 10.1016/j.regpep.2011.06.002
Poulikakos, D., Malik, M., and Banerjee, D. (2014). Parathyroid hormone and heart rate variability in haemodialysis patients. Nephron Clin. Pract. 126, 110–115. doi: 10.1159/000360542
Qu, Y., Baroudi, G., Yue, Y., El-sherif, N., and Boutjdir, M. (2004). Localization and modulation of alpha1D (Cav1.3) L-type Ca channel by protein kinase A. Am. J. Physiol. Heart Circ. Physiol. 288, 2123–2130. doi: 10.1152/ajpheart.01023.2004
Quan, K. J., Lee, J. H., Geha, A. S., Biblo, L. A., Van Hare, G. F., Mackall, J. A., et al. (1999). Characterization of sinoatrial parasympathetic innervation in humans. J. Cardiovasc. Electrophysiol. 10, 1060–1065. doi: 10.1111/j.1540-8167.1999.tb00278.x
Quinn, T. A., Camelliti, P., Rog-Zielinska, E. A., Siedlecka, U., Poggioli, T., O’Toole, E. T., et al. (2016). Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc. Natl. Acad. Sci. U.S.A. 113, 14852–14857. doi: 10.1073/pnas.1611184114
Quinn, T. A., and Kohl, P. (2011). Systems biology of the heart: hype or hope? Ann. N. Y. Acad. Sci. 1245, 40–43. doi: 10.1111/j.1749-6632.2011.06327.x
Quinn, T. A., and Kohl, P. (2012). Mechano-sensitivity of cardiac pacemaker function: pathophysiological relevance, experimental implications, and conceptual integration with other mechanisms of rhythmicity. Prog. Biophys. Mol. Biol. 110, 257–268. doi: 10.1016/j.pbiomolbio.2012.08.008
Quinn, T. A., and Kohl, P. (2013). Combining wet and dry research: experience with model development for cardiac mechano-electric structure-function studies. Cardiovasc. Res. 97, 601–611. doi: 10.1093/cvr/cvt003
Renaudon, B., Bois, P., Bescond, J., and Lenfant, J. (1997). Acetylcholine modulates If and IK (ACh) via different pathways in rabbit sino-atrial node cells. J. Mol. Cell. Cardiol. 29, 969–975. doi: 10.1006/jmcc.1996.0340
Renaudon, B., Lenfant, J., Decressac, S., and Bois, P. (2000). Thyroid hormone increases the conductance density of f-channels in rabbit sinoatrial node cells. Recept. Channels 7, 1–8.
Reuveny, E., Slesinger, P. A., Inglese, J., Morales, J. M., Inlguez-Liuhl, J. A., Lefkowitz, R. J., et al. (1994). Activation of the clone muscarinic potassium channel by G protein betagamma subunits. Lett. Nat. 370, 143–146. doi: 10.1038/370143a0
Riddle, E. L., Schwartzman, A., Bond, M., and Insel, P. A. (2005). Multi-tasking RGS proteins in the heart - the next therapeutic target? Circ. Res. 96, 401–411. doi: 10.1161/01.RES.0000158287.49872.4e
Rigel, D., Grupp, I., Balasubramaniam, A., and Grupp, G. (1989). Contractile effects of cardiac neuropeptides in isolated canine atrial and ventricular muscles. Hear. Circ. Physiol. 257, 1082–1087. doi: 10.1152/ajpheart.1989.257.4.H1082
Rigel, D. F. (1988). Effects of neuropeptides on heart rate in dogs: comparison of VIP, PHI, NPY, CGRP, and NT. Am. Physiol. Soc. 255, 311–317. doi: 10.1152/ajpheart.1988.255.2.H311
Rigg, L., Heath, B. M., Cui, Y., and Terrar, D. A. (2000). Localisation and functional significance of ryanodine receptors during β-adrenoceptor stimulation in the guinea-pig sino-atrial node. Cardiovasc. Res. 48, 254–264. doi: 10.1016/s0008-6363(00)00153-x
Rioux, F., Lemieux, M., and Roy, G. (1989). Capsaicin-sensitive primary afferents are involved in the hypotensive effect of neurotensin in ganglion-blocked guinea pigs. Peptides 10, 1033–1040. doi: 10.1016/0196-9781(89)90187-3
Rivet-Bastide, M., Vandecasteele, G., Hatem, S., Verde, I., and Bénardeau, A. (1997). cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J. Clin. Invest. 99, 2710–2718. doi: 10.1172/JCI119460
Roberts, L., Slocum, G., and Riley, D. (1989). Morphological study of the innervation pattern of the rabbit sinoatrial node. Am. J. Anat. 185, 74–88. doi: 10.1002/aja.1001850108
Rodefeld, M. D., Beau, S. L., Schuessler, R. B., Boineau, J. P., and Saffitz, J. E. (1996). β-adrenergic and muscarinic cholinergic receptor densities in the human sinoatrial node: identification of a high β2-adrenergic receptor density. J. Cardiovasc. Electrophysiol. 7, 1039–1049. doi: 10.1111/j.1540-8167.1996.tb00479.x
Roef, G., Taes, Y., Kaufman, J., Van Daele, C., De Buyzere, M., Gillebert, T., et al. (2013). Thyroid hormone levels within reference range are associated with heart rate, cardiac structure, and function in middle-aged men and women. Thyroid 23, 947–954. doi: 10.1089/thy.2012.0471
Rose, R. A., Lomax, A. E., Kondo, C. S., Anand-srivastava, M. B., Giles, W. R., Robert, A., et al. (2004). Effects of C-type natriuretic peptide on ionic currents in mouse sinoatrial node: a role for the NPR-C receptor. Am. J. Physiol. Heart Circ. Physiol. 286, 1970–1977. doi: 10.1152/ajpheart.00893.2003
Rosen, M. R., Nargeot, J., and Salama, G. (2012). The case for the funny current and the calcium clock. Hear. Rhythm 9, 616–618. doi: 10.1016/j.hrthm.2011.10.008
Rosenbaum, D. M., Rasmussen, S. G. F., and Kobilka, B. K. (2009). The structure and function of G-protein-coupled receptors. Nature 459, 356–363. doi: 10.1038/nature08144
Rutigliano, G., and Zucchi, R. (2017). Cardiac actions of thyroid hormone metabolites. Mol. Cell. Endocrinol. 458, 76–81. doi: 10.1016/j.mce.2017.01.003
Rybin, V. O., Xu, X., Lisanti, M. P., and Steinberg, S. F. (2000). Differential targeting of β-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. J. Biol. Chem. 275, 41447–41457. doi: 10.1074/jbc.M006951200
Saito, K., Suetsugu, T., Oku, Y., Kuroda, A., and Tanaka, H. (1994). alpha1-adrenoceptors in the conduction system of rat hearts. Br. J. Pharmacol. 111, 465–468. doi: 10.1111/j.1476-5381.1994.tb14759.x
Saitoh, H., Nomura, A., Osaka, M., Sasabe, N., Atarashi, H., and Hayakawa, H. (1995). Developmental changes of alpha1-adrenergic chronotropic action on human sinus node. Am. J. Cardiol. 76, 89–91. doi: 10.1016/S0002-9149(99)80811-X
Sander-Jensen, K., Secher, N., Astrup, A., Christensen, N., Damkjaer-Nielsen, M., Giese, J., et al. (1988). Angiotensin II attenuates reflex decrease in heart rate and sympathetic activity in man. Clin. Physiol. 8, 31–40. doi: 10.1111/j.1475-097x.1988.tb00259.x
Sanders, L., Lynham, J. A., Bond, B., de Monte, F., Harding, S. E., and Kaumann, A. J. (1995). Sensitization of human atrial 5-HT4 receptors by chronic β-blocker treatment. Circulation 92, 2526–2539. doi: 10.1161/01.cir.92.9.2526
Sanders, L., Rakovic, S., Lowe, M., Mattick, P. A. D., and Terrar, D. A. (2006). Fundamental importance of Na+–Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node. J. Physiol. 571, 639–649. doi: 10.1113/jphysiol.2005.100305
Satoh, H. (1993). Modulation of the automaticity by histamine and cimetidine in rabbit sino-atrial node cells. Gen. Pharmacol. 24, 1213–1222. doi: 10.1016/0306-3623(93)90371-4
Satoh, H. (1997). Electrophysiological actions of ryanodine on single rabbit sinoatrial nodal cells. Gen. Pharmacol. 28, 31–38. doi: 10.1016/s0306-3623(96)00182-6
Saxena, P. R., and Villalon, C. M. (1990). Cardiovascular effects of serotonin agonists and antagonists. J. Cardiovasc. Pharmacol. 15, 17–34.
Schaefer, S., Valenta, R., Laslett, L., and Longhurst, J. (1996). Cardiac reflex effects of intracoronary bradykinin in humans. J. Investig. Med. 44, 160–167.
Sechi, L. A., Griffin, C. A., Grady, E. F., Kalinyak, J. E., and Schambelan, M. (1992). Characterization of angiotensin II receptor subtpes in rat heart. Circ. Res. 71, 1482–1489. doi: 10.1161/01.res.71.6.1482
Sheng, J., Wang, W., and Xu, Y. (2011). Angiotensin II decreases spontaneous firing rate of guinea-pig sino-atrial node cells. Eur. J. Pharmacol. 660, 387–393. doi: 10.1016/j.ejphar.2011.04.005
Shimizu, K., Shintani, Y., Ding, W.-G., Matsuura, H., and Bamba, T. (2002). Potentiation of slow component of delayed rectifier K+ current by cGMP via two distinct mechanisms: inhibition of phosphodiesterase 3 and activation of protein kinase G. Br. J. Pharmacol. 137, 127–137. doi: 10.1038/sj.bjp.0704843
Shimoni, Y. (1999). Hormonal control of cardiac ion channels and transporters. Prog. Biophys. Mol. Biol. 72, 67–108. doi: 10.1016/s0079-6107(99)00005-x
Shimoyama, M., Ogino, K., Uchida, K., Furuse, Y., Kinugasa, Y., Taniguchi, S., et al. (2001). Fragment-specific actions of parathyroid hormone in isolated perfused rat hearts. Calcif. Tissue Int. 69, 88–93. doi: 10.1007/s002230010023
Sirenko, S., Yang, D., Li, Y., Lyashkov, A. E., Lukyanenko, Y. O., Lakatta, E. G., et al. (2013). Ca2+-dependent phosphorylation of Ca2+ cycling proteins generates robust rhythmic local Ca2+ releases in cardiac pacemaker cells. Sci. Signal. 6:ra6. doi: 10.1126/scisignal.2003391
Sirenko, S. G., Maltsev, V. A., Yaniv, Y., Bychkov, R., Yaeger, D., Vinogradova, T., et al. (2016). Electrochemical Na+ and Ca2+ gradients drive coupled-clock regulation of automaticity of isolated rabbit sinoatrial nodal pacemaker cells. Am. J. Physiol. Heart Circ. Physiol. 311, H251–H267. doi: 10.1152/ajpheart.00667.2015
Skovgaard, N., Møller, K., Gesser, H., and Wang, T. (2009). Histamine induces postprandial tachycardia through a direct effect on cardiac H2-receptors in pythons. Am. J. Physiol. Integr. Comp. Physiol. 296, 774–785. doi: 10.1152/ajpregu.90466.2008
Springer, J., Azer, J., Hua, R., Robbins, C., Adamczyk, A., McBoyle, S., et al. (2012). The natriuretic peptides BNP and CNP increase heart rate and electrical conduction by stimulating ionic currents in the sinoatrial node and atrial myocardium following activation of guanylyl cyclase-linked natriuretic peptide receptors. J. Mol. Cell. Cardiol. 52, 1122–1134. doi: 10.1016/j.yjmcc.2012.01.018
St. Clair, J. R., Larson, E. D., Sharpe, E. J., Liao, Z., and Proenza, C. (2017). Phosphodiesterases 3 and 4 differentially regulate the funny current, If, in mouse sinoatrial node myocytes. J. Cardiovasc. Dev. Dis. 4, 1–15. doi: 10.3390/jcdd4030010
Steinberg, S. F., and Brunton, L. L. (2001). Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu. Rev. Pharmacol. Toxicol. 41, 751–773. doi: 10.1146/annurev.pharmtox.41.1.751
Stewart, A., Huang, J., and Fisher, R. A. (2012). RGS proteins in heart: brakes on the vagus. Front. Physiol. 3:95. doi: 10.3389/fphys.2012.00095
Stoyek, M. R., Croll, R. P., and Smith, F. M. (2015). Intrinsic and Extrinsic Innervation of the Heart in Zebrafish (Danio rerio). J. Comp. Neurol. 1700, 1683–1700. doi: 10.1002/cne.23764
Stoyek, M. R., and Quinn, T. A. (2018). One fish, two fish, red fish, blue fish∗: zebrafish as a model for cardiac research. Prog. Biophys. Mol. Biol. 138, 1–2. doi: 10.1016/j.pbiomolbio.2018.11.003
Stoyek, M. R., Quinn, T. A., Croll, R. P., and Smith, F. M. (2016). Zebrafish heart as a model to study the integrative autonomic control of pacemaker function. Am. Physiol. Soc. 311, 676–688. doi: 10.1152/ajpheart.00330.2016
Stoyek, M. R., Rog-Zielinska, E. A., and Quinn, T. A. (2018). Age-associated changes in electrical function of the zebrafish heart. Prog. Biophys. Mol. Biol. 138, 91–104. doi: 10.1016/j.pbiomolbio.2018.07.014
Struthers, A., Brown, M., Macdonald, D., Beacham, J., Stevenson, J., Morris, H., et al. (1986). Human calcitonin gene related peptide: a potent endogenous vasodilator in man. Clin. Sci. 70, 389–393. doi: 10.1042/cs0700389
Sugimoto, T., Dohi, K., Onishi, K., Watanabe, K., Sato, Y., Sugiura, E., et al. (2013). Interrelationship between haemodynamic state and serum intact parathyroid hormone levels in patients with chronic heart failure. Heart 99, 111–115. doi: 10.1136/heartjnl-2012-302779
Tanaka, H., Habuchi, Y., Yamamoto, T., Nishio, M., Morikawa, J., and Yoshimura, M. (1997). Negative chronotropic actions of endothelin-1 on rabbit sinoatrial node pacemaker cells. Br. J. Pharmacol. 122, 321–329. doi: 10.1038/sj.bjp.0701370
Torrente, A. G., Mesirca, P., Neco, P., Rizzetto, R., Dubel, S., Barrere, C., et al. (2016). L-type Cav1.3 channels regulate ryanodine receptor-dependent Ca2+ release during sino-atrial node pacemaker activity. Cardiovasc. Res. 109, 451–461. doi: 10.1093/cvr/cvw006
Usdin, T., Bonner, T., and Hoare, S. (2002). The parathyroid hormone 2 (PTH2) receptor. Recept. Channels 2, 211–218. doi: 10.1080/10606820290005092
Ussawawongaraya, W., Spilles, N., Nilwarangkoon, S., and Jariyapongskul, A. (2013). The correlation of parathyroid hormone and heart rate variability in CAPD patients. J. Med. Assoc. Thail. 96, 595–602.
Valcavi, R., Menozzi, C., Roti, E., Zini, M., Lolli, G., Roti, S., et al. (1992). Sinus node function in hyperthyroid patients. J. Clin. Endocrinol. Metab. 75, 239–242. doi: 10.1210/jcem.75.1.1619016
Van Borren, M. M. G. J., Verkerk, A. O., Wilders, R., Hajji, N., Zegers, J. G., Bourier, J., et al. (2010). Effects of muscarinic receptor stimulation on Ca2+ transient, cAMP production and pacemaker frequency of rabbit sinoatrial node cells. Basic Res. Cardiol. 105, 73–87. doi: 10.1007/s00395-009-0048-9
Verkerk, A. O., Geuzebroek, G. S. C., Veldkamp, M. W., and Wilders, R. (2012). Effects of acetylcholine and noradrenalin on action potentials of isolated rabbit sinoatrial and atrial myocytes. Front. Physiol. 3:174. doi: 10.3389/fphys.2012.00174
Villalón, C. M., and Centurión, D. (2007). Cardiovascular responses produced by 5-hydroxytriptamine: a pharmacological update on the receptors/mechanisms involved and therapeutic implications. Naunyn Schmiedebergs Arch. Pharmacol. 376, 45–63. doi: 10.1007/s00210-007-0179-1
Vinogradova, T. M., Bogdanov, K. Y., and Lakatta, E. G. (2002). β-adrenergic stimulation modulates ryanodine receptor pacemaker activity in rabbit sinoatrial nodal cells. Circ. Res. 90, 73–79. doi: 10.1161/hh0102.102271
Vinogradova, T. M., Kobrinsky, E., and Lakatta, E. G. (2018a). Dual activation of phosphodiesterases 3 and 4 regulates basal spontaneous beating rate of cardiac pacemaker cells: role of compartmentalization. Front. Physiol. 9:1301. doi: 10.3389/fphys.2018.01301
Vinogradova, T. M., and Lakatta, E. G. (2009). Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP-mediated PKA-dependent Ca2+ cycling with surface membrane channels. J. Mol. Cell. Cardiol. 47, 456–474. doi: 10.1016/j.yjmcc.2009.06.014
Vinogradova, T. M., Lyashkov, A. E., Zhu, W., Ruknudin, A. M., Sirenko, S., Yang, D., et al. (2006). High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Cell. Biol. 98, 505–514. doi: 10.1161/01.RES.0000204575.94040.d1
Vinogradova, T. M., Sirenko, S., Lukyanenko, Y. O., Yang, D., Tarasov, K. V., Lyashkov, A. E., et al. (2018b). Basal spontaneous firing of rabbit sinoatrial node cells is regulated by dual activation of PDEs (Phosphodiesterases) 3 and 4. Circ. Arrhythmia Electrophysiol. 11:e005896. doi: 10.1161/CIRCEP.117.005896
Vinogradova, T. M., Sirenko, S., Lyashkov, A. E., Younes, A., Li, Y., Zhu, W., et al. (2008). Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ. Res. 102, 761–769. doi: 10.1161/CIRCRESAHA.107.161679
Vinogradova, T. M., Zhou, Y.-Y., Bogdanov, K. Y., Yang, D., Kuschel, M., Cheng, H., et al. (2000). Sinoatrial node pacemaker activity requires Ca2+/calmodulin-dependent protein kinase II activation. Circ. Res. 87, 760–767. doi: 10.1161/01.RES.87.9.760
Volpe, M., Alessandro, F., Indolfi, C., Luca, N. D. E., Lembo, G., Focaccio, A., et al. (1987). Hemodynamic and hormonal effects of atrial natriuretic factor in patients with essential hypertension. J. Am. Coll. Cardiol. 10, 787–793. doi: 10.1016/S0735-1097(87)80271-1
Volpe, M., Lembo, G., Condorelli, G., Luca, N., De, Lamenza, F., et al. (1990). Converting enzyme inhibition prevents the effects of atrial natriuretic factor on baroreflex responses in humans. Circulation 82, 21–25. doi: 10.1161/01.cir.82.4.1214
Wang, Q., Wang, D., Yan, G., Qiao, Y., Sun, L., Zhu, B., et al. (2016). Biochemical and biophysical research communications SERCA2A was serotonylated and may regulate sino-atrial node pacemaker activity. Biochem. Biophys. Res. Commun. 480, 492–497. doi: 10.1016/j.bbrc.2016.10.082
Weidmann, P., Hasler, L., Gnadinger, M., Lang, R., Uehlinger, D., Shaw, S., et al. (1986). Blood levels and renal effects of atrial natriuretic peptide in normal man. J. Clin. Invest. 77, 734–742. doi: 10.1172/JCI112368
Weisbrod, D., Khun, S. H., Bueno, H., Peretz, A., and Attali, B. (2016). Mechanisms underlying the cardiac pacemaker: the role of SK4 calcium-activated potassium channels. Acta Pharmacol. Sin. 37, 82–97. doi: 10.1038/aps.2015.135
West, G. A., and Belardinelli, L. (1985). Correlation of sinus slowing and hyperpolarization caused by adenosine in sinus node. Eur. J. Physiol. 403, 75–81. doi: 10.1007/bf00583285
Whalen, E. J., Johnson, A. K., and Lewis, S. J. (1999). Hemodynamic actions of systemically injected pituitary adenylate cyclase activating polypeptide-27 in the rat. Eur. J. Pharmacol. 365, 205–215. doi: 10.1016/s0014-2999(98)00852-8
Wickman, K., Nemec, J., Gendler, S. J., and Clapham, D. E. (1998). Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20, 103–114. doi: 10.1016/s0896-6273(00)80438-9
Wolff, A. A., and Levi, R. (1986). Histamine and cardiac arrhythmias. Circ. Res. 58, 1–16. doi: 10.1161/01.res.58.1.1
Wu, Y., and Anderson, M. E. (2014). CaMKII in sinoatrial node physiology and dysfunction. Front. Pharmacol. 5:48. doi: 10.3389/fphar.2014.00048
Wu, Y., Gao, Z., Chen, B., Koval, O. M., Singh, M. V., Guan, X., et al. (2009). Calmodulin kinase II is required for fight or flight sinoatrial node physiology. PNAS 106, 5972–5977. doi: 10.1073/pnas.0806422106
Wydeven, N., Posokhova, E., Xia, Z., Martemyanov, K. A., and Wickman, K. (2014). RGS6, but Not RGS4, Is the Dominant Regulator of G Protein Signaling (RGS) Modulator of the Parasympathetic Regulation of Mouse Heart Rate. J. Biol. Chem. 289, 2440–2449. doi: 10.1074/jbc.M113.520742
Yamamoto, M., Dobrzynski, H., Tellez, J., Niwa, R., Billeter, R., Honjo, H., et al. (2006). Extended atrial conduction system characterised by the expression of the HCN4 channel and connexin45. Cardiovasc. Res. 72, 271–281. doi: 10.1016/j.cardiores.2006.07.026
Yang, D., Lyashkov, A. E., Li, Y., Ziman, B. D., and Lakatta, E. G. (2012). RGS2 overexpression or G(i) inhibition rescues the impaired PKA signaling and slow AP firing of cultured adult rabbit pacemaker cells. J. Mol. Cell. Cardiol. 53, 687–694. doi: 10.1016/j.yjmcc.2012.08.007
Yaniv, Y., Ganesan, A., Yang, D., Ziman, B. D., Lyashkov, A. E., Levchenko, A., et al. (2015). Real-time relationship between PKA biochemical signal network dynamics and increased action potential firing rate in heart pacemaker cells. J. Mol. Cell. Cardiol. 86, 168–178. doi: 10.1007/s00018-015-1997-9
Yaniv, Y., Sirenko, S., Ziman, B. D., Spurgeon, H. A., Maltsev, V. A., and Lakatta, E. G. (2013). New evidence for coupled clock regulation of the normal automaticity of sinoatrial nodal pacemaker cells: bradycardic effects of ivabradine are linked to suppression of intracellular Ca2+ cycling. J. Mol. Cell. Cardiol. 62, 80–89. doi: 10.1016/j.yjmcc.2013.04.026
Yatani, A., Imotos, Y., Codinai, J., Hamilton, S. L., and Brown, A. M. (1988). The stimulatory G protein of adenylyl cyclase, Gs also stimulates dihydropyridine-sensitive Ca2+ channels. Prog. Biophys. Cell Biol. 20, 9887–9895.
Younes, A., Lyashkov, A. E., Graham, D., Sheydina, A., Volkova, M. V., Mitsak, M., et al. (2008). Ca2+-stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 283, 14461–14468. doi: 10.1074/jbc.M707540200
Zaccolo, M., and Movsesian, M. A. (2007). cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 100, 1569–1578. doi: 10.1161/CIRCRESAHA.106.144501
Zhang, H., Holden, A., and Boyett, M. (2002). Sustained inward current and pacemaker activity of mammalian sinoatrial node. J Cardiovasc Electrophysiol 13, 809–812. doi: 10.1046/j.15408167.2002.00809.x
Keywords: acetylcholine, adenosine, catecholamines, G-protein-coupled receptors, heart rate, intracellular signaling, natriuretic peptides, regulation
Citation: MacDonald EA, Rose RA and Quinn TA (2020) Neurohumoral Control of Sinoatrial Node Activity and Heart Rate: Insight From Experimental Models and Findings From Humans. Front. Physiol. 11:170. doi: 10.3389/fphys.2020.00170
Received: 03 June 2019; Accepted: 13 February 2020;
Published: 03 March 2020.
Edited by:
Tobias Opthof, Academic Medical Center, NetherlandsReviewed by:
Yael Yaniv, Technion - Israel Institute of Technology, IsraelCopyright © 2020 MacDonald, Rose and Quinn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: T. Alexander Quinn, YWxleC5xdWlubkBkYWwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.