 Marilén Federico
Marilén Federico Carlos A. Valverde
Carlos A. Valverde Alicia Mattiazzi
Alicia Mattiazzi Julieta Palomeque
Julieta Palomeque- 1Centro de Investigaciones Cardiovasculares “Dr. Horacio E. Cingolani”, CCT-La Plata/CONICET, Facultad de Cs. Médicas, Universidad Nacional de La Plata, La Plata, Argentina
- 2Centro de Altos Estudios en Ciencias Humanas y de la Salud, Universidad Abierta Interamericana, Buenos Aires, Argentina
The present review focusses on the regulation and interplay of cardiac SR Ca2+ handling proteins involved in SR Ca2+ uptake and release, i.e., SERCa2/PLN and RyR2. Both RyR2 and SERCA2a/PLN are highly regulated by post-translational modifications and/or different partners’ proteins. These control mechanisms guarantee a precise equilibrium between SR Ca2+ reuptake and release. The review then discusses how disruption of this balance alters SR Ca2+ handling and may constitute a first step toward cardiac damage and malignant arrhythmias. In the last part of the review, this concept is exemplified in different cardiac diseases, like prediabetic and diabetic cardiomyopathy, digitalis intoxication and ischemia-reperfusion injury.
Introduction
Cardiovascular diseases are the leading cause of morbidity and mortality worldwide, being Ca2+ mishandling one of the most striking abnormalities in the setting of a wide spectrum of pathologies, including cardiac hypertrophy, heart failure, DCM, and I/R damage. Indeed, alterations that constitute the hallmark of these diseases, like contractile dysfunction, cardiac arrhythmias or cell death are in great part a reflection of the impairment in Ca2+ handling and the altered function of the SR, a pivotal responsible of Ca2+ cycling within cardiac myocytes.
The purpose of this review is: (1) To summarize the regulation of the main cardiac SR Ca2+ handling proteins involved in SR Ca2+ uptake and release, i.e., SERCA2a/PLN and RyR2. Both proteins are highly regulated by additional partners’ proteins and/or PTMs that may increase or decrease their activity. (2) To describe how the disruption of the interplay among these proteins may constitute a key determinant of Ca2+ triggered arrhythmias and cardiac damage. (3) To recapitulate experimental evidence in which alterations of the balance between these key players contribute to SR Ca2+ mishandling with the ensuing production of Ca2+ triggered arrhythmias and cell death in different cardiac diseases.
Excitation-Contraction Coupling
Cardiomyocyte excitation-contraction coupling (ECC) is the process that links membrane depolarization, at the surface cell level, with myofilament interaction that drives contraction, inside the cell. Ca2+ ions are the link between the two processes (Figure 1).
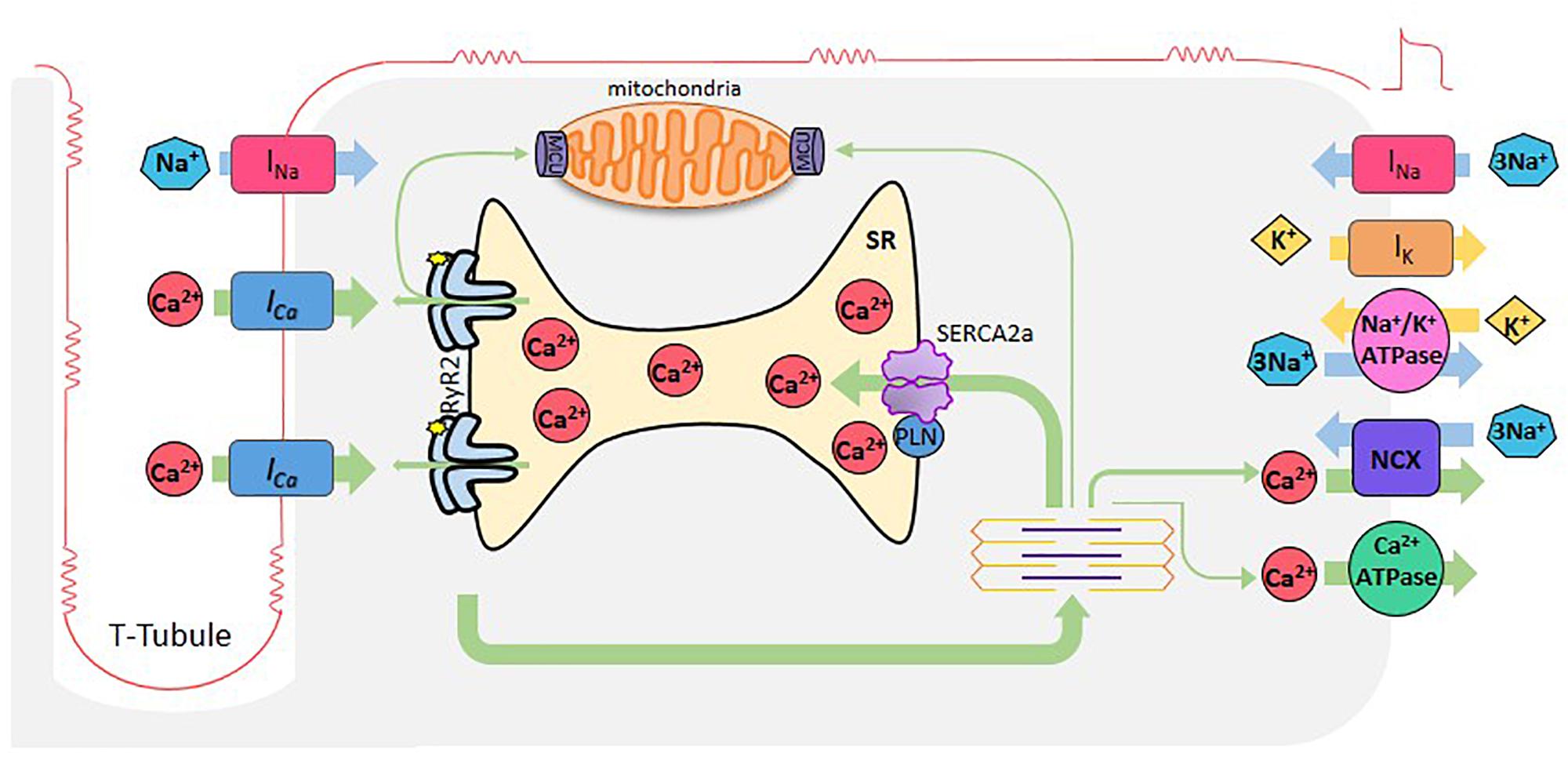
Figure 1. Cardiac excitation-contraction coupling. When cardiomyocytes are reached by an AP, depolarization of the plasma membrane by Na+ entry (INa), induces the opening of L-type Ca2+ channels. Ca2+ entering through these channels (Ca2+ current, ICa), induces Ca2+ release by the RyR2s (Ca2+-induced- Ca2+-release), which subsequently activates myofilaments for muscle contraction. Relaxation occurs when SR -Ca2+ ATPase (SERCA2a) reuptakes Ca2+, lowering cytosolic Ca2+ concentration in combination with Ca2+ extrusion via the Na+- Ca2+ exchanger (NCX) working in reverse mode and the sarcolemma Ca2+-ATPase. Mitochondria also participate taking and extruding Ca2+ from the cytosol during Ca2+ cycle. Green arrows depict Ca2+ fluxes.
Regulation of Ca2+ Handling Proteins Involved in Sr Ca2+ Uptake and Release
The SR orchestrates the ECC, being Ca2+ ions the main players. Ryanodine Receptors 2 (RyR2) and SERCA2a are highly and precisely regulated by several proteins and kinases, which allow a fine-tuned synchronization of Ca2+ cycling and therefore of cardiac contraction and relaxation processes. We will mention here several key modulators and PTMs of these two main proteins. For a further review see for instance (Kranias and Hajjar, 2012; Haghighi et al., 2014; Mattiazzi and Kranias, 2014; Eisner et al., 2017; Meissner, 2017; Santulli et al., 2017a, b).
Regulation of SR Ca2+ Uptake
SERCa2a Post-translational Modifications
Redox regulations appear to play an important role in SERCA2a function, both in health and disease. In cardiac myocytes it has been shown that oxidative stress reduces contractility with depletion of SR Ca2+ stores, due to SERCA2a inhibition (Morris and Sulakhe, 1997; Xu et al., 1997; Kaplan et al., 2003; Kuster et al., 2010). Indeed, several pathologies like metabolic syndrome (Balderas-Villalobos et al., 2013) or atherosclerosis (Cohen and Adachi, 2006), which are associated with an increase in oxidative stress, reduced SERCA2a activity and contractility.
Nitric oxide activates SERCA2a activity by a cGMP-independent pathway which involves the direct modification of reactive thiol groups on the protein, not only of vascular smooth muscle but also of cardiac and skeletal muscle. On its own, NO is a weak SH-group oxidant. However, in the presence of O2– it results in an increased ONOO– production. Under physiological conditions, the ONOO– produced may react with the thiol groups of proteins producing S-nitrosylation and S-gluthationylation of cysteine residues. S-gluthationylation increases the activity of the pump. The SERCA2a residue mainly involved in this reaction is the reactive thiol group Cys674 (Adachi et al., 2004). However, an exacerbated increase of ONOO– nitrosylates the hydroxyl groups of SERCA2a, producing impairment of cardiac relaxation (Braun et al., 2019).
Sarcoplasmic reticulum Ca2+-ATPase is also regulated by the SUMO1 (sumoylation), by AGEs (glycation) and by acetylation/deacetylation processes. SERCA2a sumoylation appears to prolong the lifetime of SERCA2a as well as to increase its intrinsic activity by SUMO1 binding to Lys480 and Lys585 residues. Indeed, increasing SUMO1 expression restores SERCA2a levels, improves hemodynamic performance, and reduces mortality in heart failure (Kho et al., 2011). AGEs complexes can compromise the pump activity by altering the structural movements required for translocating Ca2+ from the cytosol to the lumen of the SR (Bidasee et al., 2004). Finally, recent experiments indicated that the acetylation of SERCA2a at K492 site was significantly increased in heart failure (HF) in association with a reduction of SIRT1, a class III histone deacetylase. Acetylation of K492 significantly diminished SERCA2a activity, possibly by interfering with the binding of ATP. Activation of SIRT1 restored SERCA2a activity. This strategy may, therefore, be useful for HF treatment (Gorski et al., 2019).
PLN Post-translational Modifications and the PLN Interactome
Undoubtedly, the main regulator of SERCA2a activity is PLN (Tada et al., 1975). PLN is a small protein (52 amino acid residues) that binds to and allosterically inhibits SERCA2a (MacLennan and Kranias, 2003). Dephosphorylated PLN reduces the affinity of SERCA2a for Ca2+ whereas PLN phosphorylation increases SERCA2a pump activity. There are two PLN phosphorylation sites that are physiologically relevant: Ser16 residue, phosphorylated by PKA and Thr17 site, phosphorylated by the Ca2+-calmodulin-dependent protein kinase II (CaMKII) (Figure 2A). Phosphorylation of these sites increases the affinity of SERCA2a for Ca2+ and the rate of SR Ca2+ uptake. This, in turn, leads to increases in SR Ca2+ load, SR Ca2+ release and myocardial contractility (Lindemann et al., 1983; Lindemann and Watanabe, 1985; Mundina de Weilenmann et al., 1987; Mundina-Weilenmann et al., 1996).
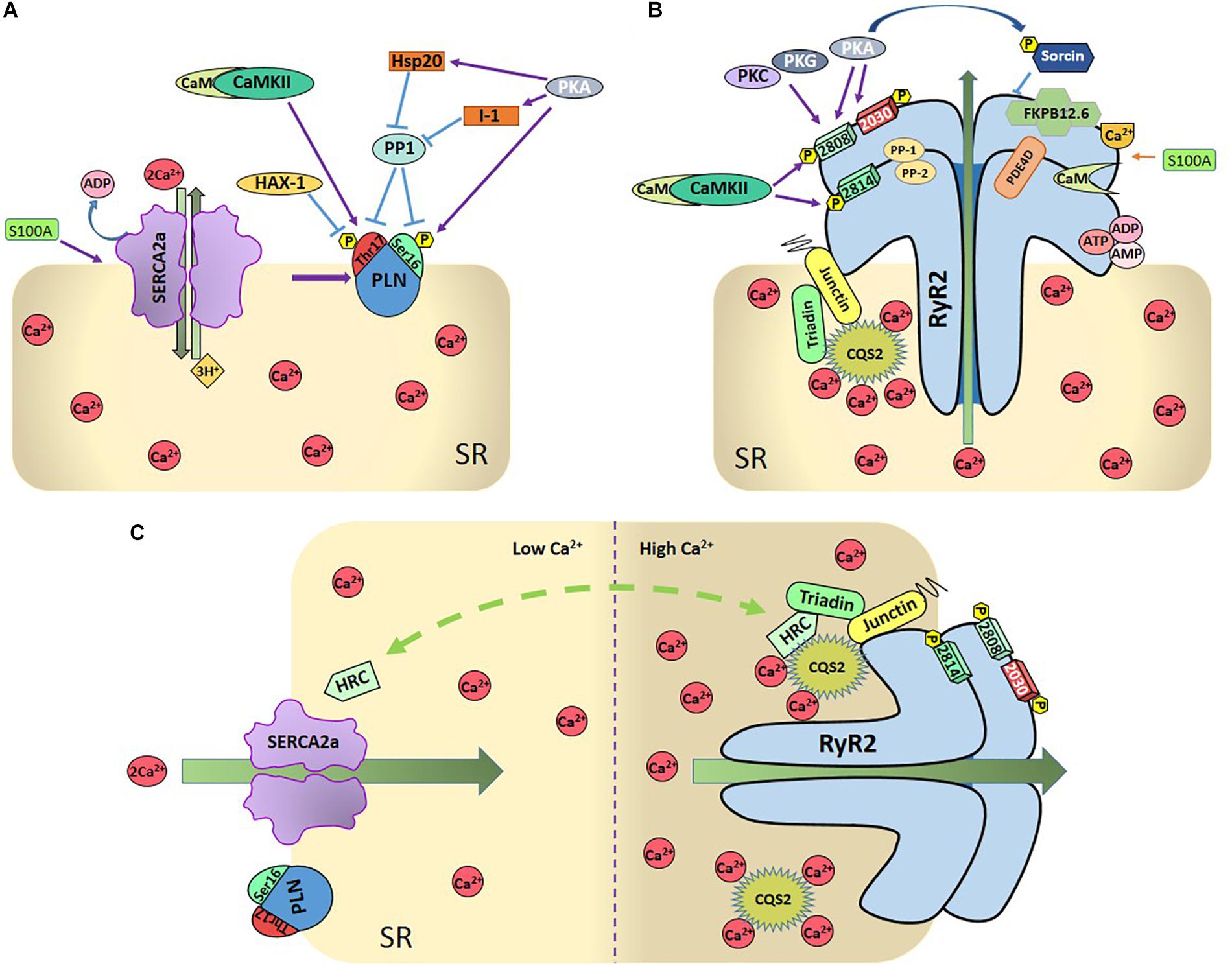
Figure 2. (A) Regulation of SR-Ca2+ uptake. SR- Ca2+ uptake takes place through the SR- Ca2+ ATPase (SERCA2a), being PLN the main regulatory protein of SERCA2a. PLN can be phosphorylated by PKA at Ser16 residue and by Ca2+-calmodulin-dependent protein kinase (CaMKII) at Thr17 site. Protein phosphatase 1 (PP1) dephosphorylates both sites. Either one or both phosphorylation sites relieve PLN inhibition on SERCA2a. Several other proteins regulate SERCA2 either directly, like S100A or Sumo1, or indirectly, through the regulation of PLN (like the hematopoietic lineage cell-specific protein-1 (HS-1) associated protein X-1 (Hax-1), the Heat-shot protein 20 (Hsp20), and the inhibitor 1 (I-1). As described in the text, PLN and SERCA2a are also regulated by redox processes and SERCA by acetylation and glycation (Not shown in the Figure for the sake of clarity). (B) Regulation of SR-Ca2+ release: SR-Ca2+ release occurs mainly through the RyR2 which are also highly regulated. CaMKII phosphorylates RyR2 at Ser2814 and S2808 sites and PKA at Ser2808 and 2030 sites. Ser2808 is also target of protein-kinase C (PKC) and protein-kinase G (PKG). PP1 and PP2 are the phosphatases that dephosphorylate RyR2. Other regulatory proteins are sorcin, S-100 at the cytosolic side, FKB12.6 and calmodulin (Cm), bounded to RyR2, CASQ2, triadin, and juntin at the cytosolic site. Phosphodiesterase (PDE), ions, and nucleoside phosphates (AMP, ADP, and ATP) are also bounded to RyR2. (C) Dual role of HRC: The intra-luminal protein histidine-rich calcium binding protein (HRC) interacts with SERCA2a as well as with triadin in a Ca2+-dependent fashion, increasing RyR2 Ca2+ release and SERCA2a Ca2+-uptake when Ca2+ increases in SR.
The status of PLN phosphorylation, as is the case of any other protein, depends on the dynamic balance between the activity of kinases and phosphatases that phosphorylate and dephosphorylate the protein, respectively. PP1, is the major SR phosphatase that specifically dephosphorylates PLN (Steenaart et al., 1992). Inhibition of PP1 results in increased phosphorylation of PLN and SERCA2a activation (Haghighi et al., 2015; Figure 2A). During β-ARS, PKA phosphorylates PLN at Ser16 site and simultaneously inhibits PP1 through the PKA dependent phosphorylation of two additional proteins, inhibitor-1 (I-1) and the small heat shock protein 20 (Hsp20) (Qian et al., 2011). Under β-ARS, PLN-Thr17 site is also phosphorylated by CaMKII activation due to the increase in intracellular Ca2+ and the inhibition of PP1, produced by the activity of PKA. In contrast, phosphorylation of Thr17 of PLN does not occur when only intracellular Ca2+ was increased without PKA activation, which is necessary to inhibit PP1 (Mundina-Weilenmann et al., 1996).
Post-translational modification of phospholamban by reactive oxygen and nitrogen species (ROS and RNS, respectively) may also influence SR Ca2+ uptake (Bigelow and Squier, 2005; Froehlich et al., 2008; Lancel et al., 2009; Ha et al., 2011). Among these PTM it is interesting to mention the one-electron reduction product of NO, nitroxyl (HNO). This molecule has received special attention not only as a possible signaling molecule in the cardiovascular system but also as a potential therapeutic strategy for HF treatment due to its positive inotropic and lusitropic effects in normal and failing canine hearts (Paolocci et al., 2001, 2003; Sivakumaran et al., 2013). PLN played a central role in these effects of HNO, by enhancing SERCA2a activity (Froehlich et al., 2008). Moreover, it has been suggested that S-nitrosylation of PLN at Cys36 and Cys41 modulates the PLN-dependent regulation of SERCA2a during β-ARS, i.e., S-nitrosylation of PLN is required for stabilization of the pentameric form of PLN, and consequent SERCA2a activation (Irie et al., 2015). Several additional regulatory proteins are associated with PLN and SERCA2a and contribute to the control of SR Ca2+-transport. These include the hematopoietic lineage cell-specific protein-1 (HS-1) associated protein X-1 (HAX1), a ∼35 kDa protein, which was identified forming a complex with HS-1 in lymphocytes (Suzuki et al., 1997), the intra-luminal histidine-rich Ca2+ binding protein (HRC), which has been shown to interact with both SERCA2a and triadin on the SR luminal side (see below and Figure 2C) and S100A1 on the cytosolic side (Kiewitz et al., 2003). Ca2+-dependent S100A1 binding to SERCA2a results in an increased enzymatic activity which is associated with enhanced SR Ca2+ uptake and load (Most et al., 2001; Kiewitz et al., 2003; Kettlewell et al., 2005). As will be discussed below, HRC and S100A1 also interact and regulate RyR2 and SR Ca2+ release (Völkers et al., 2007; for review, see Haghighi et al., 2014; Kranias and Hajjar, 2017; Arvanitis et al., 2018).
Regulation of SR Ca2+ Release
Ryanodine receptor 2 is the largest ion channel known in nature and one of the most relevant Ca2+ handling proteins. RyR forms a homotetrameric assembly comprising four monomers of 565 kDa each (Van Petegem, 2015). There are three mammalian isoforms that share 65% sequence identity: RyR1, predominantly expressed in skeletal muscle; RyR2, the cardiac isoform and RyR3, expressed in several tissues including the brain (Capes et al., 2011). While at the cytosolic portion, the channel contains multiple regulatory domains, such as binding sites for energy sensors (ATP, ADP, and AMP) (Figure 2B), and inorganic phosphate, metabolites such as pyruvate, fatty acids and polyamines, and ions (Mg2+, H+, and Cl–, not shown in the Figure for the sake of clarity) (Zucchi and Ronca-Testoni, 1997; Fill and Copello, 2002; Meissner, 2004); the Ca2+ binding site is located in the core domain of the channel just above the transmembrane domain an involves de carboxyl-terminal domain region (Murayama et al., 2018). This complex is also regulated and modulated by a diverse array of RyR2-interacting proteins which involve PKA, CaMKII, phosphatases (i.e., phosphatase 1 and 2A), and phosphodiesterase (PDE4D) which are tethered to the channel and held near their target sites by means of anchoring proteins (Marks, 2002; Lehnart et al., 2005). This allows for a tight and spatially confined homeostatic regulation of the balance between RyR2 phosphorylation and phosphatase dependent dephosphorylation.
Post-translational Modifications of RyR2
Phosphorylation is possibly the most studied and controversial PTM modification of RyR2. Phosphorylation of the channel modulates the effect of Ca2+ on the RyR2 without having the inherent ability to open or close the channel per se (Camors and Valdivia, 2014). Until now three phosphorylation sites in the RyR2 have been identified: Ser2808, Ser2814, and Ser2030 (Figure 2B). Serine 2808 (Ser2808, mouse, and Ser2809 in human and canine RyR2 nomenclature) was first described by Witcher and collaborators as a CaMKII site (Witcher et al., 1991). Further in-depth studies of this phospho-site indicated that Ser2808 is a target for PKA, CaMKII and possibly PKG (Jiang et al., 2002; Rodriguez et al., 2003; Stange et al., 2003; Currie et al., 2004; Ai et al., 2005; Xiao et al., 2005; Carter et al., 2006; Kohlhaas et al., 2006; Ferrero et al., 2007; Huke and Bers, 2008; MacDonnell et al., 2008; Fischer et al., 2013). Experiments by Marx et al. (2000) indicated that PKA-dependent phosphorylation of RyR2 at Ser2808 site under β-ARS, increases P0 and SR Ca2+ release. However, this contention was not supported by different studies and the functional meaning of this phosphorylation is not clear yet (Xiao et al., 2006; Ferrero et al., 2007; Huke and Bers, 2008). This is in part due to the fact that most studies found that Ser2808 is constitutively phosphorylated under basal conditions (Jiang et al., 2002; Rodriguez et al., 2003; Carter et al., 2006; Ferrero et al., 2007; Huke and Bers, 2008), generating doubts about the relevance of “extra” phosphorylation on this site. Moreover, it was also showed that RyR2s were hyperphosphorylated in failing hearts from humans and dogs at Ser2809, which was attributed at least in part to a decrease in the amount of PP1 associated to RyR2 (Marx et al., 2000; Wehrens et al., 2006). However, several subsequent experiments by other groups failed to reproduce these findings (Li et al., 2002), turning the attention to serine 2814 (Ser2814) site as the primary phosphorylation site responsible for SR Ca2+ leak and arrhythmogenic events in HF (Li et al., 2002; Respress et al., 2012). The role of Ser2808 phosphorylation was further complicated by the finding that both, minimum and maximum RyR2 phosphorylation at Ser2808, increase RyR2 activity, suggesting a U-shaped of RyR2 activity according to the PKA phosphorylation level (Carter et al., 2006). A clear revision of these controversial results is given by Bers (Bers, 2012) and Camors and Valdivia (Camors and Valdivia, 2014).
Ser2814 site was described by Wehrens et al. (2004) as a CaMKII site and further evidence confirmed that this site seems to be exclusively phosphorylated by CaMKII. In single-channel experiments, the P0 of the RyR2s was generally found to be increased upon phosphorylation by CaMKII (Lokuta et al., 1995; Wehrens et al., 2004; Yang et al., 2007). In line with these results, either activation or overexpression of CaMKII was associated with the positive inotropic effect of β-ARS (Ferrero et al., 2007), an increase of Ca2+ spark frequency (Guo et al., 2006) and the susceptibility to arrhythmias (Maier et al., 2003; Dybkova et al., 2011; Respress et al., 2012; Mazzocchi et al., 2016; Valverde et al., 2019). In contrast, animals in which Ser2814 was replaced by Alanine (S2814A mice) were protected from arrhythmias and cardiac dysfunction induced by several diseases (van Oort et al., 2010; Di Carlo et al., 2014; Mazzocchi et al., 2016).
Serine 2030 (Ser2030) was characterized as a PKA phosphorylation site using classical phospho-epitope mapping (Xiao et al., 2005). Whereas in quiescent cardiac myocytes the RyR2 appears to be completely unphosphorylated (Huke and Bers, 2008), this site has been suggested as the major phosphorylation site in RyR2 responding to PKA activation upon β-ARS in normal and failing hearts (Xiao et al., 2006). In this context, it has recently been described that phosphorylation of RyR2 at Ser2030 is required for a complete effect of β-ARS (Potenza et al., 2019) in mouse lines with genetic ablation of this site (RyR2-S2030A).
Interestingly, recent work reports crystal structures of the RyR2 phosphorylation domain with the PKA catalytic subunit (PKAc), showing Ser2808 captured within the active site of PKA. The results further demonstrated that the addition of a phosphomimetic at the CaMKII site (S2814D), results in structural changes in the RyR2 phosphorylation domain that enhance the interaction with PKAc. These findings strongly suggest that phosphorylation of Ser2814 site may affect the activity of PKA and impact on Ser2808, i.e., nearby phosphorylation sites might influence one each other (Haji-Ghassemi et al., 2019). This possible interaction among the different residues sharing the phosphorylation “hotspot” region of RyR2, might clarify previous controversial findings on the role of Ser2808 site on different physiological and disease situations. Since RyR2 phosphorylation by PKA and CaMKII may not be independent, the authors suggest that the phosphorylation status of Ser2808 may be altered in studies that have used S2814D mice. Of note, in contrast with this prediction, previous experiments indicate that isoproterenol-induced phosphorylation of RyR2-Ser2808 site did not vary when isoproterenol was administrated in the absence and presence of a CaMKII inhibitor (KN-93), to avoid the simultaneous phosphorylation of Ser2814 residue (Ferrero et al., 2007), i.e., phosphorylation of Ser2814 did not influence the extent of phosphorylation of Ser2808 site. However, the isoproterenol concentration used in these experiments was rather high and may not allow any further PKA-dependent phosphorylation of this site. Therefore, it would be important to perform similar experiments in the presence of lower isoproterenol concentrations to investigate the possible influence of Ser2814 phosphorylation on the isoproterenol-induced phosphorylation of Ser2808 site predicted by the crystal structure studies.
The role of phosphatases activity on RyR2 phosphorylation was recently emphasized. It was shown that PP1 activation counteracts the increased kinase activity in human heart failure reducing SR Ca2+ leak as well as cellular arrhythmias without significant changes in SR Ca2+ load and contractility (Fischer et al., 2018).
Oxidative conditions generally increase the RyR2 P0, while reducing agents do the opposite (Marengo et al., 1998; Xu et al., 1998; Salama et al., 2000; Sun et al., 2008). The functional consequence of a moderate cellular oxidative/nitrosative stress could result in an immediate enhancement of Ca2+ release from the SR in response to a given physiological trigger. However, severe oxidative stress can cause irreversible and persistent activation of RyR2s (Xu et al., 1998), increasing SR Ca2+ leak. It has been reported that NADPH oxidase 2 (NOX2) is the predominant isoform expressed in T-tubules and SR membranes of adult cardiomyocytes. Therefore, it is strategically positioned to modulate the activity of the RyR2s. ROS produced by NOX2 stimulates SR Ca2+ release via at least two pathways: direct oxidation or S-glutathionylation of RyR2s or indirectly through CaMKII activation (Palomeque et al., 2009), followed by phosphorylation of the RyR2s. In healthy cardiac muscle neuronal nitric oxide synthase (nNOS) is mainly located in the SR membrane, linked to the RyR2s, which would favor direct RyR2 nitrosation. The role of this PTM of RyR2 on cardiac ECC has been previously reviewed (Lim et al., 2008; Gonzalez et al., 2009).
The RyR2 Complex
Ryanodine receptor 2 modulation has been shown to involve also several key proteins (Figure 2B), most importantly CASQ2. CASQ2 not only acts as a Ca2+ buffer, but it also mediates the responsiveness of the RyR2 channel to luminal Ca2+ by serving as a Ca2+ sensor (Gyorke et al., 2004; Gyorke and Terentyev, 2008). This function is effective through protein-protein interactions, with junctin and triadin (Zhang et al., 1997; Shin et al., 2000). However, a direct interaction between CASQ2 and RyR2 has recently been described (Handhle et al., 2016). Junctin is a 26 kDa transmembrane protein forming a complex with triadin, CASQ2, and RyR2. It has been proposed that junctin is in direct contact with RyR2 and works as an anchor for CASQ2 (Zhang et al., 1997; Gyorke et al., 2004).
Calstabin 2 or FKBP12.6, is a peptidyl-prolyl cis/trans-isomerase of 12.6 kDa, associated with RyR2 with a stoichiometry of 4:1 (Figure 2B). The role of FKBP12.6 has been a matter of controversy for over the past two decades (Kaftan et al., 1996; Timerman et al., 1996; Barg et al., 1997; Marks, 2000; Doi et al., 2002; Jiang et al., 2002; Stange et al., 2003; Wehrens et al., 2003; Xiao et al., 2007; Guo et al., 2010). FKBP12.6 has been considered as RyR2 “stabilizer,” since some results indicate that FKBP12.6 dissociation from RyR2 produces RyR2 sub-conductance states and increase the Po of the channel. However, other piece of evidence indicates that FKBP12.6 failed to show any effect of FKBP12.6 on RyR2 gating. The recent identification of FKB12 binding site on RyR2 may help to understand the controversial matters encompassing the FKBP-RyR2 interactions. FKBP12 binds to RyR2 with lower affinity than FKBP12.6, but has a higher cardiac expression level (Jeyakumar et al., 2001). Recent results revealed that only 20% of RyR2 proteins are associated with FKBP12.6 in myocytes (Guo et al., 2010) which could explain why RyR2 is unaffected in FKBP12.6-KO mice (Xiao et al., 2007). Acute overexpression of FKBP12 in adult rabbit ventricular myocytes showed a reduction in the gain of ECC and a decrease in Ca2+ spark frequency, suggesting that FKBP12 reduces RyR2 sensitivity to cytosolic Ca2+(Seidler et al., 2007). In contrast, more recent results show that FKBP12 activates the RyR2 and competes with FKBP12.6. The last study, would suggest that rather than a direct stabilization of the channel, an increase in FKBP12.6-RyR2 binding competes with FKBP12 at the same binding site and blunts the activation of RyR2 promoted by FKBP12 (Galfre et al., 2012).
Apo-Calmodulin or Ca2+-free CaM has an inhibitory effect on RyR2 channel. The Ca2+-bound CaM is named Ca2+-CaM. Although Ca2+-CaM is the usual form that binds to target proteins, CaM can also bind to RyR2. CaM shifts the Ca2+-dependence of RyR2 activation to higher Ca2+ concentrations (Fruen et al., 2000; Balshaw et al., 2001; Yamaguchi et al., 2005). The role of CaM on RyR2 regulation was highlighted by results that indicate that mutations in CaM are associated with RyR2-mediated cardiac arrhythmias (Nomikos et al., 2014; Sondergaard et al., 2017). High resolution cryo-electron microscopy recently provided new insights into the modulation of RyR2 channel gating by CaM (Gong et al., 2019). These data indicate that Ca2+-CaM changes RyR2 conformation differently under different situations. Whereas Ca2+-CaM can reverse RyR2 opening by Ca2+ and PCB95, a potent channel opener (Samso et al., 2009), it cannot counteract the activation of the channel by a mixture of Ca2+, ATP and caffeine. These results emphasize that the P0 of RyR2 is critically determined by a strict balance between different activators and inhibitors of the channel (Van Petegem, 2019).
Several proteins that interact with SERCA2a regulating SR Ca2+ uptake, also modify the RyR2 function. As already mentioned, the HRC protein is not only associated with SERCA2a but also with triadin. The interaction of HRC with triadin increases with increasing Ca2+ concentration (Sacchetto et al., 1999; Arvanitis et al., 2007). This interaction is believed to modulate RyR2 function and SR Ca2+ release by conferring refractoriness to SR Ca2+ release. In turn, HRC-SERCA2a interaction is also Ca2+-dependent. In this case, the maximal HRC-SERCA2a association occurs at low Ca2+ concentration and diminishes with increasing Ca2+ concentrations (Arvanitis et al., 2007). The different Ca2+ dependence of the interaction HRC-SERCA2a and HRC-triadin determines the HRC effects on SR Ca2+ handling (Figure 2C): At low SR Ca2+, HRC interacts with SERCA2a inhibiting SR Ca2+ uptake. When SR Ca2+ concentration increases, HRC dissociates from SERCA2a and enhances its binding to triadin, regulating SR Ca2+ release (for review, see Arvanitis et al., 2018).
Sorcin is a 22 kDa penta-EF hand Ca2+-binding protein expressed in many tissues, including the heart. Single-channel studies indicated that when applied to the cytoplasmic region of RyR2s, sorcin inhibits RyR2 activity in a dose-dependent manner by prolonging the mean close time without modifying single-channel conductance, an effect that is abrogated when sorcin is phosphorylated by PKA. More recent experiments (Farrell et al., 2003) demonstrated that sorcin significantly inhibits both the spontaneous activity of RyR2s in quiescent cells and the Ca2+ current (ICa)-triggered activity of RyR2s. Moreover, it decreased the amplitude of the Ca2+ transient without affecting the amplitude or kinetics of ICa, reducing the “gain” of ECC mechanism. Sorcin seems to be a key RyR2-associated protein under stress conditions since its ablation displayed a significantly higher incidence of cardiac arrhythmias and sudden death in sorcin-KO mice when subjected to acute or chronic stress challenge (Chen et al., 2018). It has also been shown that sorcin increases SR Ca2+ uptake (Matsumoto et al., 2005) and interacts with NCX (Zamparelli et al., 2010) and L-type Ca2+ channels (LTCC) (Fowler et al., 2008). All these interactions point to an important role of this protein in ECC regulation.
Also, as in the case of SERCA2a, S100A1 modulates RyR2 function under both diastolic and systolic conditions (Kiewitz et al., 2003; Most et al., 2004; Völkers et al., 2007, Volkers et al., 2010). Most et al. (2004), first demonstrated that addition of S100A1 to isolated SR vesicles resulted in diminished 3H-ryanodine ([3H]Ry) binding to RyR2 at free Ca2+ concentrations of about 150 nM, while a significantly increased [3H]Ry binding occurred at Ca2+ concentrations greater than 300 nM. Hypothesizing a reduced RyR2 P0 at diastolic cytoplasmic Ca2+ levels, S100A1 would reduce SR Ca2+ leak in quiescent cardiomyocytes (Völkers et al., 2007). Moreover, S100A1 increases fractional SR Ca2+ release in voltage-clamped rabbit cardiomyocytes, suggesting that S100A1 enhances the ECC gain under systolic conditions (Kettlewell et al., 2005).
Cytosolic and Luminal Ca2+ Regulation of RyR2
Both cytosolic and luminal Ca2+ regulate RyR2. It has long been known that the release of SR Ca2+ in cardiac muscle during ECC is graded by the amount of activating Ca2+ outside the SR by the Ca2+-induced Ca2+ release (CICR) process (Fabiato and Fabiato, 1977). Experimental evidence suggested the presence of high and low affinity Ca2+ binding sites in the cytosolic region of RyR2 and luminal Ca2+-binding sites, whose luminal occupancy depends on SR Ca2+ load (Fabiato and Fabiato, 1979; Shannon et al., 2000). RyR2s are normally closed at low cytosolic Ca2+ (100–200 nM); channel activity is maximal at 10–100 μM cytosolic Ca2+, while elevating cytosolic Ca2+ beyond this point leads to a reduction in P0 (Xu and Meissner, 1998). This biphasic behavior implies there are at least two classes of Ca2+ binding sites: high-affinity activation and low-affinity inactivation sites. The P0 steep dependence of RyR2 on cytoplasmic Ca2+, which typically exhibits Hill coefficients of 2–4 (Sitsapesan and Williams, 1994) indicates that RyR2 activation resulted from cooperative involvement of one high-affinity (∼1 μM) Ca2+ binding site on each subunit of the homotetrameric channel (Zahradnik et al., 2005). RyR2s also possess two inhibitory sites in their cytoplasmic domains with Ca2+ affinities of the order of 1 μM and 1 mM. Mg2+ competes with Ca2+ at these sites to inhibit RyR2s (for instance, see Laver, 2018).
In 1994, Sitsapesan and Williams were the first to show that luminal Ca2+ could directly activate RyR2 (Sitsapesan and Williams, 1994). Since then, different type of evidence supports the notion that luminal Ca2+ controls RyR2 function (i.e., Bassani et al., 1995; Gyorke and Gyorke, 1998; Shannon et al., 2000; Kong et al., 2007; Gyorke and Terentyev, 2008). Two different mechanisms have been proposed to explain luminal Ca2+ regulation of RyR2: The “feed-through” hypothesis suggests that luminal Ca2+ acts on its own cytosolic Ca2+ binding site during or after Ca2+ passage through an open RyR2 (Herrmann-Frank and Lehmann-Horn, 1996; Xu and Meissner, 1998). A second mechanism proposes that luminal Ca2+ regulation of RyR2 is mediated by a Ca2+ sensing mechanism inside the SR (Gyorke and Gyorke, 1998; Ching et al., 2000; Jiang et al., 2007; Gyorke and Terentyev, 2008). Although CASQ2, may serve as a key SR luminal Ca2+ sensor (Gyorke et al., 2004), experiments in CASQ2-null mice (Knollmann et al., 2006) and in purified native and recombinant RyR2s that lack CASQ2 (Xu and Meissner, 1998; Kong et al., 2007), indicate that RyR2s are also regulated by a luminal Ca2+ sensing mechanism that does not require CASQ2. Indeed, a point mutation in RyR2 (RyR2-E4872A) which eliminates Ca2+ regulation by luminal but not by cytosolic Ca2+ was recently identified (Chen et al., 2014). Structural analysis, at near-atomic resolution, suggests that in addition to E4872, the E4878 residue may also be involved in luminal Ca2+ activation of RyR2 (Peng et al., 2016), although the precise mechanism by which each of these different sites promotes luminal Ca2+ activation of RyR2 is not clear yet.
How Can the Disruption of the Normal Interplay Among Sr Ca2+ Handling Proteins Evoke Ca2+ Triggered Arrhythmias and Apoptosis?
The normal interplay among the different proteins responsible for the release and reuptake of Ca2+ by the SR is regulated by different mechanisms as reviewed above. This regulation may be altered and evolve toward different types of cardiac disorders, which include arrhythmias and cell death through apoptotic and necrotic processes. Therefore, regulation and/or alteration of SR Ca2+ handling proteins (for instance by phosphorylation, redox changes or mutations), have received great attention from physiologists and clinicians. We will describe below two main consequences of the unbalance of SR Ca2+ uptake and release, i.e., Ca2+ triggered arrhythmias and cellular apoptosis and necroptosis, with a main focus on those produced by PTM of Ca2+ handling proteins.
Ca2+ Triggered Arrhythmias
Mechanisms
As stated above, RyR2s are highly regulated molecules. Genetic or PTM of RyR2 are a main cause of Ca2+ triggered arrhythmias, i.e., arrhythmias that are originated due to abnormal Ca2+ handling.
Triggered activity describes impulse initiation that is dependent on the so-called afterdepolarizations, which are oscillations in membrane potential that follow the primary depolarization phase (0) of an AP. Afterdepolarizations are divided into early and delayed afterdepolarizations, EAD, and DAD, respectively. EADs are defined as a slowing or reversal of normal repolarization that occurs before completion of AP, usually in phases 2 and 3 of human AP, whereas DADs occur after AP completion (Figure 3). These mechanisms may produce sustained arrhythmias by reentry circuits (Anderson, 2007). EADs occurs usually in the setting of prolonged repolarization and are classically attributed to reactivation of ICa (January and Riddle, 1989; Nuss et al., 1999). However, a second major current that facilitates EADs formation is NCX. Indeed, experimental evidence indicates that these two currents act synergistically to generate EADs, with their relative contributions varying under specific conditions (Weiss et al., 2010). The late component of the Na+ current (INa), has been recognized as an important player to set up the conditions for EADs, by producing SR Ca2+ overload, via the reduction of repolarization reserve and the increase in intracellular Na+ concentration. In addition, experimental evidence and modeling studies indicate that EADs may directly arise from Na+ channel reactivation (Horvath et al., 2013; Sato et al., 2017; Figure 3A).
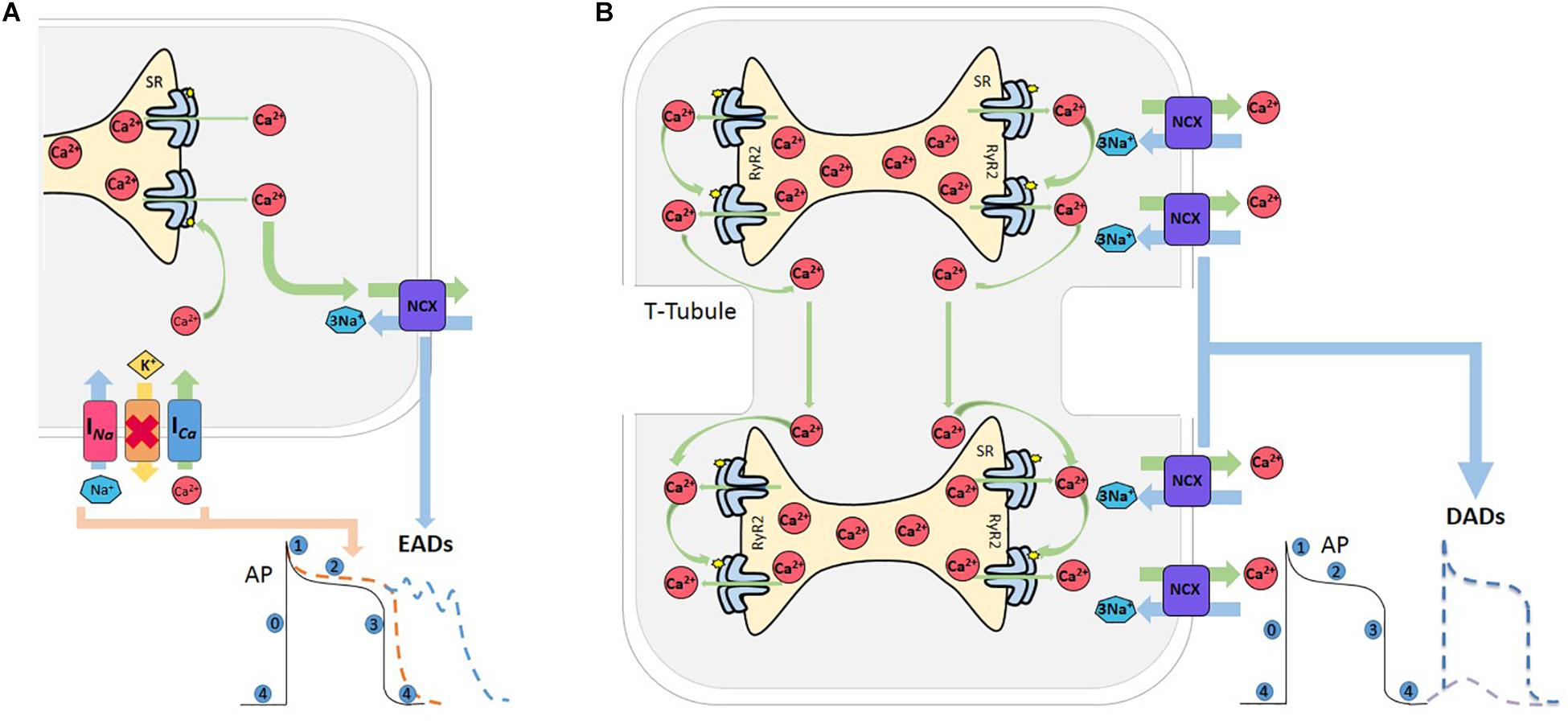
Figure 3. Afterdepolarizations. (A) Reactivation of L-type Ca2+ current (ICa) during prolongation of the AP, mainly during phase 2 or 3, increases the propensity for EADs. The Na+-Ca2+ current (NCX) also meets the criterion needed for the positive feedback required for an EAD to occur. Both currents can act synergistically facilitating EAD formation, and thus increasing the probability of an EAD-triggered AP. Red cross indicates inhibition of K+ channels. (B) Various conditions which increase SR-Ca2+ and/or sensitize RyR2 can induce spontaneous Ca2+ release from the SR. When this release achieves certain critical mass, Ca2+ can induce a saltatory propagation along cell sarcomeres, known as Ca2+ waves, which in turn activates the NCX that extrude 2 Ca2+ and enter 3 Na+, depolarizing the cell membrane and producing a delay after depolarization (DAD). If this depolarization attains the excitability threshold, a spontaneous AP occurs.
Delay afterdepolarizations are caused by spontaneous Ca2+ releases from the SR (Bers, 2006). Under conditions of SR Ca2+ overload and/or in circumstances which sensitize the RyR2s, the Ca2+ released by a group of RyR2 activates neighboring RyR2, in such a way that Ca2+ propagates in a regenerative way traveling along the myocytes in a saltatory fashion from sarcomere to sarcomere (Cheng et al., 1996). Ca2+ waves have substantial arrhythmogenic potential, since they may trigger Ca2+ activated currents, such as the NCX current (INCX). This promotes a transient Na+ current (Iti), that depolarizes cell membrane and may eventually trigger a spontaneous AP (Bers, 2006), which is referred to as triggered AP, leading to spontaneous contraction (Spencer and Sham, 2003; Fujiwara et al., 2008; Mazzocchi et al., 2016; Figure 3B).
When referring to a multicellular tissue, e.g., whole heart, spontaneous Ca2+ releases synchronized in a small group of cells is not enough for triggering an AP. 3D modeling (Xie et al., 2010) estimated that about 800,000 cells are required to trigger a premature ventricular complex, being able to bring the sink (adjacent tissue in basal conditions) to its activation threshold. A sort of synchronization mechanism must exist for EADs and DADs to overcome the source-sink mismatch. Normally, in an intact tissue, the source-sink mismatch is the main mechanism protecting the heart against spontaneous Ca2+ release-induced arrhythmias.
The Threshold Concept
In the context of Ca2+ triggered arrhythmias, an intriguing issue to consider is the SR Ca2+ threshold. As mentioned above, spontaneous SR Ca2+ leak can occur in the absence of membrane depolarization. It has long been known that several conditions that increase SR Ca2+ load increase SR Ca2+ waves and spontaneous contractions (Orchard et al., 1983; Stern et al., 1983; Wier et al., 1987). Diaz et al. (1997) showed that increasing extracellular Ca2+ in quiescent cells produced spontaneous Ca2+ release associated with increased SR Ca2+ content (Diaz et al., 1997). Once spontaneous Ca2+ release arose, further increase in extracellular Ca2+ did not affect SR Ca2+ content because of the proportional increase in SR Ca2+ leak. The authors conclude “It appears there is a maximum level of SR Ca2+ content, perhaps because spontaneous release results when the content reaches a threshold”(Diaz et al., 1997). Due to its dependence on the SR Ca2+ store, this depolarization–independent SR Ca2+ release has been called “Store Overload-Induced Ca2+ Release (SOICR)”(Jiang et al., 2004). It has been further shown that the threshold level also depends on the activity of RyR2. One important premise of this mechanism is that “…once a threshold level of SR Ca2+ content is reached, SOICR occurs”(Jiang et al., 2004). More recent experiments by Belevych et al. (2012) challenged the idea of immediacy that encompasses the last concept. These authors demonstrated that SR Ca2+ leak occurs with a substantial time delay after the attainment of diastolic SR Ca2+ level, i.e., the attainment of a certain SR Ca2+ level is not sufficient for spontaneous Ca2+ release and waves generation. The time factor is necessary. Interestingly, the post-refilling refractory period was shorter in myocytes from infarcted hearts than in control myocytes, even though the rate of SR Ca2+ content recovery after the stimulus-induced SR Ca2+ release was similar. Based on these and other results (Sobie et al., 2005), it was concluded that the probability of spontaneous Ca2+ triggering also depends on the recovery of RyR2 from refractoriness (time and Ca2+ store–dependent properties of RyR2). In post-infarction myocytes the post-refilling refractory period was reduced, an effect attributed to CaMKII phosphorylation and redox modifications of RyR2. Figure 4 schematizes a possible interpretation of the experimental results.
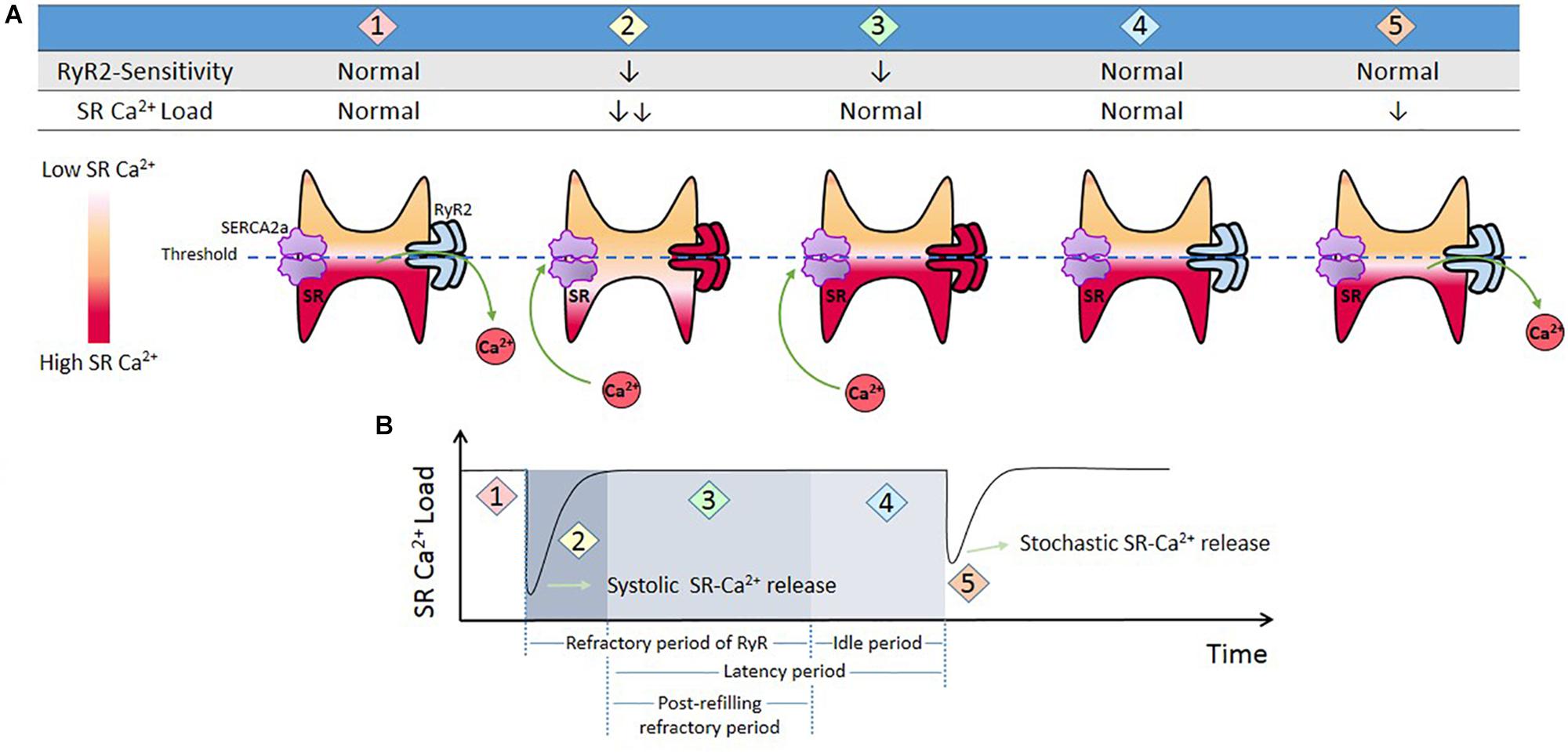
Figure 4. Threshold concept. (A) Cartoon that represents different conditions of RyR2 sensitivity and SR-Ca2+ load during Ca2+ release from SR. When the cell triggers a Ca2+ transient (1), SR Ca2+ load diminishes and RyR2s enter in a refractory period (2). SR Ca2+ uptake produces SR Ca2+ recovery (3). During this period there are no spontaneous releases, i.e., RyR2 remained refractory in spite of the fact that SR level has recovered completely (SR threshold). This period is followed by what has been named an “idle” period, by Belevych et al., 2012, (4) in which, after SR-Ca2+ load and RyR2s recovering, a stochastic activation of RyR2 may trigger a spontaneous release. (B) Schematic representation of time dependent changes of SR Ca2+ t (1 and 2), and refractoriness (2 and 3) after a stimulus-induced Ca2+ transient. (4) “Idle” period during which a spontaneous SR-Ca2+ release occurs. We called post-refilling refractory period to the period of time needed for refractoriness to recover after SR Ca2+ threshold was reached.
Ca2+-Induced Apoptosis and Necroptosis
It is generally accepted that mitochondria are at the central stage of cell death (Finkel, 2001; Dorn and Maack, 2013; Pan et al., 2013). Indeed, numerous recent investigations revealed the mitochondria are effectors of programed apoptosis or necrosis and sources of damaging ROS.
Mitochondria are organelle in close association with the SR. This proximity allows a cross-talk between mitochondria and SR which is extremely valuable under normal conditions: Mitochondrial ATP production is crucial for modulation of oxidative phosphorylation and therefore essential to maintain myocyte activity, including SR Ca2+ cycling, contraction, and relaxation (Denton, 2009). The physical contact between SR membranes and mitochondria are known as sarco/endoplasmic reticulum (SR/ER)/mitochondria microdomains or mitochondria-associated SR/ER membranes (MAMs). A sufficient local Ca2+ concentration may be achieved in specialized microdomains created by the close association of mitochondria and the SR/ER (Szabadkai et al., 2003). In these microdomains, cytosolic Ca2+ is predicted to transiently rise to micromolar concentrations, consequently allowing significant Ca2+ uptake via the MCU. Physical proximity and functional interplay between mitochondria and SR is maintained in part through tethering of these two organelles by different linkers that may contribute to either decrease or maintain the physical gap between the SR and the mitochondria. For an extensive review see Csordas et al. (2018). On the other hand, perturbation of Ca2+ handling may alter mitochondrial-SR Ca2+ crosstalk and excessive Ca2+ can go to the mitochondria which may contribute to apoptosis and necroptosis in different diseases. Stress conditions that lead to Ca2+ or ROS overload trigger mPTP opening, i.e., the mitochondrial membrane becomes permeable to any molecule less than 1.5 kDa in size. Consequent dissipation of the membrane potential (ΔΨm) leads to mitochondrial membrane depolarization, failure to produce ATP and release of mitochondrial proteins such as cytochrome c, which initiate cell death pathways (Bernardi and Di Lisa, 2015). Importantly, it has been shown that mitochondria-initiated cell death is one main mechanism in HF (Nakayama et al., 2007).
The Interplay Between SR Ca2+ Uptake and Leak
As discussed above, the properties of RyR2 are a main factor in determining the magnitude of SR Ca2+ leak. However, can an increase in RyR2 P0, increase SR Ca2+ leak by itself? Experiments by Venetucci et al., clearly demonstrated that the potentiation of RyR2 produces only a transient increase in SR Ca2+ leak, because once SR Ca2+ leak initiates, SR Ca2+ load decreases below the threshold for SR Ca2+ leak (Venetucci et al., 2007). Only a simultaneous enhancement of SR Ca2+ uptake would be able to maintain the necessary level of SR Ca2+ content to attain the threshold for SR Ca2+ leak. This conclusion is in agreement with clinical facts showing that patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) due to RyR2 mutation only suffer arrhythmias after β-ARS. Experiments performed in S2814D myocytes, in which Ser2814 was mutated to aspartic acid and behaves as pseudo constitutively phosphorylated (van Oort et al., 2010), also showed that an increase in the P0 of RyR2 produced by CaMKII phosphorylation was not able to evoke an SR Ca2+ leak higher than the one observed in wild type (WT) myocytes unless they are challenged by increasing extracellular Ca2+ or β-ARS (Mazzocchi et al., 2016).
Sarcoplasmic reticulum Ca2+ leak is also critically dependent on SR Ca2+ load. As stated above, SR Ca2+ overload triggers spontaneous Ca2+ release via the activation of the RyR2 luminal Ca2+ sensor (Gyorke and Terentyev, 2008). Therefore, the potential effect of increasing SR Ca2+ load on Ca2+ triggered arrhythmias, seems obvious. However, PLN knock-out mice (PLNKO), which have a fully loaded SR, have not proven to be prone to arrhythmias under basal conditions (Santana et al., 1997). Although at first sight these results might suggest that the increase in SR Ca2+ load per se does not increase arrhythmia propensity, several studies have provided evidence that the increase in SR Ca2+ load produced by PLN ablation does produce a dramatic increase SR Ca2+ leak (Santana et al., 1997; Huser et al., 1998; Mazzocchi et al., 2016). Unexpectedly, it hardly evokes SR Ca2+ waves (Huser et al., 1998; Mazzocchi et al., 2016). Figure 5A shows the results of experiments performed in S2814D mice. As discussed above, this mutation confers the hearts a high propensity to SR Ca2+ waves and arrhythmias when submitted to stress (van Oort et al., 2010), and in double mutant mice resulting from cross-breeding S2814D mice with PLNKO mice, to increase SR Ca2+ reuptake (SDKO mice) (Mazzocchi et al., 2016; Valverde et al., 2019). In the presence of high extracellular Ca2+, the frequency of Ca2+ sparks and SR Ca2+ leak were higher in SDKO than in S2814D myocytes, consistent with the overall higher SR Ca2+ load in SDKO cells (Mazzocchi et al., 2016). Similar results were obtained when SR Ca2+ load was increased in both strains subjected to an I/R protocol (Mazzocchi et al., 2016; Valverde et al., 2019; Figure 5B). Unexpectedly, whereas S2814D myocytes displayed full propagating Ca2+ waves when exposed to high extracellular Ca2+ (Figure 6A) or I/R (Figure 6B), SDKO myocytes mostly show non-propagating Ca2+ events, known as mini-waves. Thus, in spite of the higher SR Ca2+ leak observed in SDKO myocytes vs. S2814D, the proportion of fully propagating events in SDKO myocytes was significantly less (Mazzocchi et al., 2016). This seeming paradox can be clarified by the acknowledgment that two main factors intervene in the production of arrhythmogenic Ca2+ waves: 1. Increased SR Ca2+ leak and 2. Cytosolic Ca2+ wave propagation.
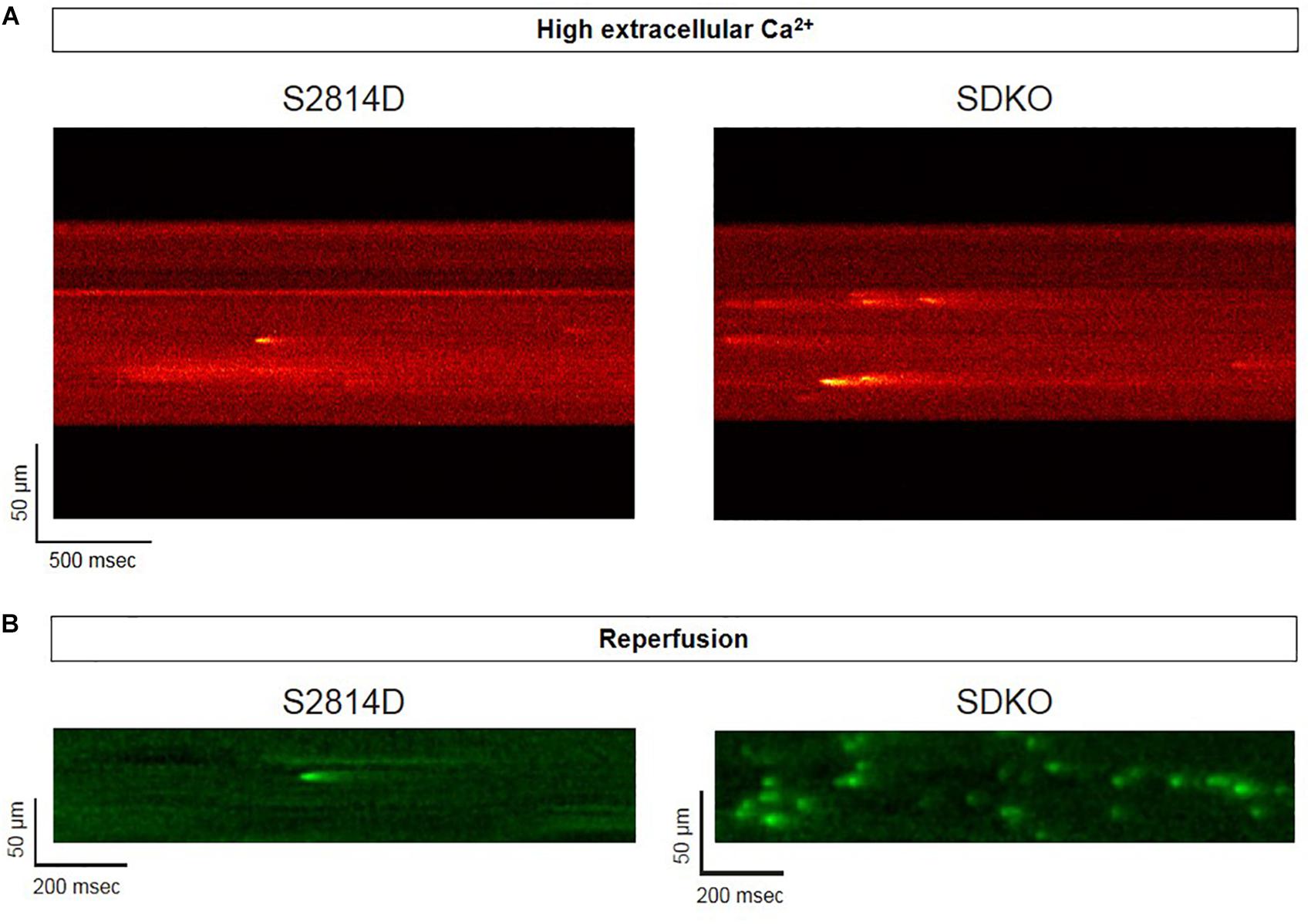
Figure 5. Confocal assessment of spontaneous Ca2+ release. (A) Representative confocal images showing that Ca2+ spark frequency is higher in isolated cardiomyocytes from SDKO mice in comparison to S2814D myocytes (unpublished confocal microscopy records representative of overall data shown in Mazzocchi et al., 2016). (B) Similar results were obtained in fluo-4 loaded intact isolated hearts from S2814D and SDKO mice under a confocal microscope, during reperfusion of the hearts submitted to a period of ischemia (unpublished confocal microscopy records representative of overall data shown in Valverde et al., 2019). In both examples, ablation of PLN increases Ca2+ spark frequency respect to hearts from S2814D mice.
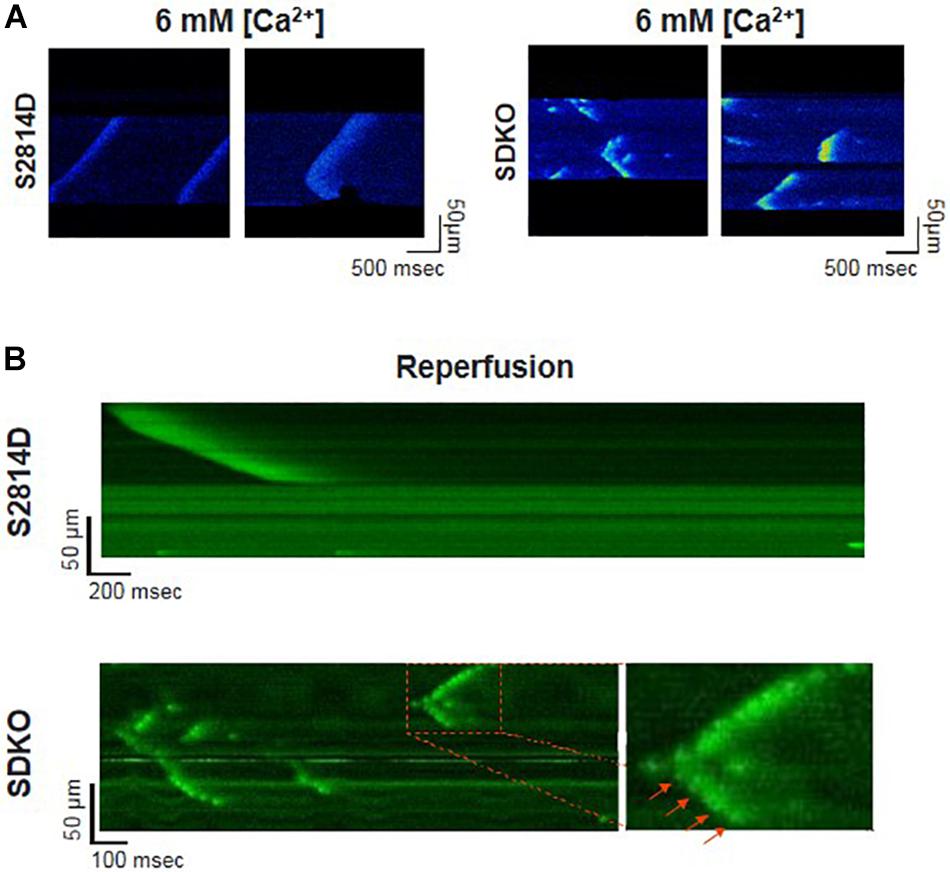
Figure 6. Confocal assessment of spontaneous SR Ca2+ waves. (A) Representative confocal images showing Ca2+ waves in isolated cardiomyocytes from S2814D myocytes (left panel) when exposed to 6 mM extracellular Ca2+. When SDKO isolated myocytes are exposed to the same condition, they exhibit miniwaves instead of full propagating Ca2+ waves (Reproduced with permission from Mazzocchi et al., 2016). (B) Representative confocal images of epicardial Ca2+ waves in S2814D and SDKO intact isolated hearts during reperfusion after an ischemic period. SDKO hearts display fragmented Ca2+ waves or mini-waves. Red arrows indicate a series of mini-events conserving a Ca2+ wave pattern (reproduced with permission from Valverde et al., 2019).
An increase in SR Ca2+ leak associated with a decrease in propagating SR Ca2+ waves indicates a limitation in cytosolic Ca2+ diffusion (Figure 7). PLN ablation interrupts cell-wide propagating Ca2+ waves, converting them into non-propagated events, like mini-waves or groups of Ca2+ sparks (Huser et al., 1998; Mazzocchi et al., 2016; Valverde et al., 2019), supporting the contention that by decreasing cytosolic Ca2+, PLN ablation would increase cytosolic Ca2+ buffer capacity, hampering Ca2+ wave propagation and preventing the arrhythmogenic susceptibility produced by an enhanced SR Ca2+ load. Further support to this idea is given by the experiments in which decreasing SR Ca2+ uptake by the SERCA2a inhibitor CPA, converts non-propagating mini-waves into full propagating Ca2+ waves (Valverde et al., 2019). These results indicate therefore that an increase in SR Ca2+ content does increase the propensity to arrhythmias. However, the mechanism by which the increase in SR Ca2+ occurred may conspire against the arrhythmogenic effect of the high SR Ca2+ load, i.e., an enhancement of SR Ca2+ sequestration, if high enough, would increase SR Ca2+ load and leak but also preclude Ca2+ wave propagation (Huser et al., 1998; Mazzocchi et al., 2016; Valverde et al., 2019).
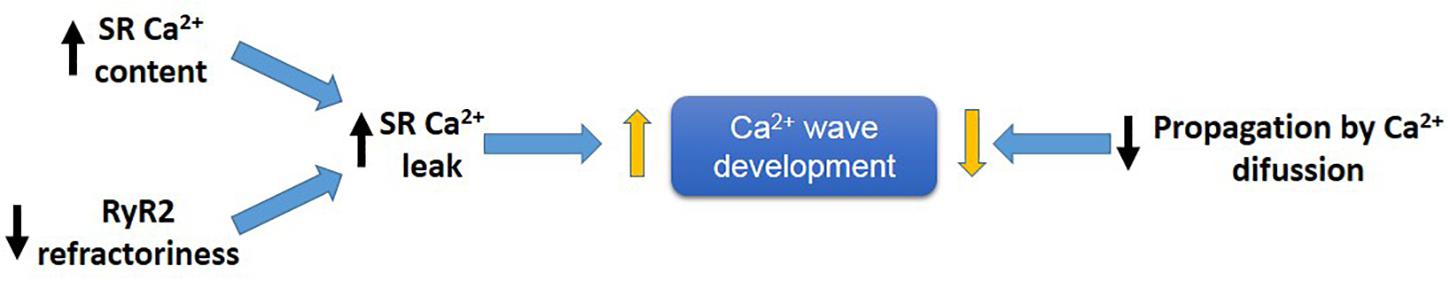
Figure 7. Main factors developing arrhythmogenic Ca2+ waves. An increase in SR-Ca2+ content and/or a decrease in RyR2 refractoriness favor SR-Ca2+ leak and Ca2+ wave generation, meanwhile a decrease in propagation of Ca2+ by diffusion (e.g., like that produce by PLN ablation), will prevent Ca2+ wave propagation by conversion of Ca2+ waves into non-propagable Ca2+ miniwaves.
In contrast, the increase in SR Ca2+ leak evoked by increasing SR Ca2+ uptake was unable to prevent but rather enhanced heart attack. It was speculated that increasing SR Ca2+ uptake was not efficient to hamper the excessive flow of SR Ca2+ to the mitochondria, aggravating cardiac damage (Valverde et al., 2019).
Experimental Evidence
In the following sections, we will give experimental evidence that highlight the importance of Ca2+ handling misbalance in the production of Ca2+ triggered arrhythmias and cell death. Although the mechanisms of cardiac arrhythmias and apoptosis/necroptosis are usually multifactorial, we will concentrate on experimental examples that emphasize the role of PTM of SR Ca2+ handling proteins. When possible, the interplay between Ca2+ uptake and release in determining arrhythmias and cardiac damage will be also discussed.
Abnormal RyR2 Regulation in the Development of Diabetic Cardiomyopathy
The current typical definition of DCM comprises structural and functional abnormalities of the myocardium in diabetic patients independently of other risk factors, as coronary artery disease or hypertension (Aneja et al., 2008). DCM is the last stage of cardiac damage in Type 1 and 2 Diabetes Mellitus (T1DM and T2DM, respectively) and of metabolic syndrome associated with insulin resistance. The subcellular mechanisms involved in this last stage of DCM have been discussed in several previous reviews (Bugger and Abel, 2014; Cox and Marsh, 2014; Fuentes-Antras, Picatoste et al. 2015, Fuentes-Antras, Picatoste et al. 2015). Most of them concluded that mayor alterations of Ca2+ handling, protein expressions, and activities in overt DCM mimic those of HF from different etiologies, including a decrease in SERCA2a activity, SR Ca2+ load, systolic Ca2+, rate of Ca2+ decay, and increase in SR Ca2+ leak (Lebeche et al., 2008). Protein glycosylation (Clark et al., 2003) and oxidized CaMKII are significantly up-regulated in the DCM (Jay et al., 2006), therefore CaMKII activation and RyR2 phosphorylation has been proposed as a potential mechanism of heart failure, ventricular arrhythmias and apoptosis in this disease (Daniels et al., 2015). In rats with T2DM, the opening of mPTP in ventricular myocytes was shown to be mainly influenced by the increased ROS and decreased ATP content. It was suggested that Ca2+ mishandling due to the slow rate of SR Ca2+ uptake could play a role in increasing mPTP opening that might further exacerbate mitochondrial dysfunction and induce cell death (Riojas-Hernandez et al., 2015). Moreover, ROS derived from hyperglycemia trigger myocardial apoptosis by mitochondrial cytochrome c release and consequent activation of the caspase-3 pathway (Cai et al., 2002). Although the underlying mechanism of cell death in DCM is not clear yet, these results suggest that a possible pathway underlying apoptosis in DMC is linked to Ca2+ mishandling and mitochondrial-SR Ca2+ crosstalk, as described above (For further review about the role of mitochondria on cardiac arrhythmogenesis in DCM see (El Hadi et al., 2019).
Much less is known about Ca2+ handling and mishandling and the potential occurrence of arrhythmias and apoptosis in the first stages of this disease, in which subclinical events develop for years before the clear emergence of HF symptoms. A possible cause for this lack of information may lie in the poor diagnosis of this stage of the disease and in the different models used to study prediabetic molecular events (King, 2012; Graham and Schuurman, 2015; King and Bowe, 2016). Indeed, the results that reveal the underlying mechanisms of the pathogenesis of DCM, are different according to the model, the degree of evolution of the disease before reaching DCM and the gender explored. Examples of conflicting results at the level of Ca2+ handling and particularly of RyR2 are observed in the metabolic syndrome model, (db/db mice), which lacks leptin receptors (Chen et al., 1996). In male db/db mouse hearts, the levels of RyR2 were found to be depressed and the RyR2 phosphorylation at the CaMKII site was not altered. However, RyR2 phosphorylation at the PKA site was found to be increased (Pereira et al., 2006, 2014). Intriguingly, db/db female mouse hearts showed no changes in RyR2 expression associated with a decrease in PKA and CaMKII RyR2 phosphorylation sites (Pereira et al., 2014). In a model of fructose-rich diet (FRD) applied to male rats and mice, our laboratory has described that this hypercaloric diet, which also induces insulin resistance, increases CaMKII activation and RyR2 dysfunction due to Ser2814 phosphorylation (Sommese et al., 2016; Federico et al., 2017). On the other hand, experiments in vitro revealed that during acute hyperglycemia, RyR2 activity can also be altered. Hyperglycemia leads to O-Glc-NAcylation of proteins such as CaMKII. Erickson et al. elegantly showed that the acute increase of glucose or O-linked N-acetylglucosamine is directly responsible for CaMKII-dependent diastolic RyR2 Ca2+ leak and SR Ca2+ load depletion in hyperglycemia (Erickson et al., 2013). Additionally, we recently demonstrated that in an early diabetic stage, prevention of CaMKII activation by ROS avoided SR Ca2+ leak evoked by CaMKII-dependent phosphorylation of RyR2 (Sommese et al., 2016). Thus, the available results indicate that a main disturbance of Ca2+ handling in prediabetic hearts occurs at the level of RyR2 phosphorylation with the consequent increase in SR Ca2+ leak and the possibility of triggered arrhythmias and cell death. Indeed, during prediabetic states, the risk of cardiovascular events is already increased and myocardial abnormalities might appear before the diagnosis of T2DM. These alterations are thought to root irregularities at the cardiac myocyte level, ultimately contributing to structural and functional anomalies observed in DCM (Hajat et al., 2004). Actually, in the mouse model of impaired glucose tolerance (IGT) mentioned above (FRD-induced insulin resistance), we found serious cardiac disorders (Sommese et al., 2016; Federico et al., 2017). The fact that this model lacks the more frequent co-morbidities of DCM, supports the metabolic origin of the alterations at the cell level. These animals develop remarkable cardiac remodeling (Sommese et al., 2016). Also, ventricular myocytes exhibit cardiac arrhythmogenic events leading to ventricular arrhythmias which can be prevented in transgenic mice expressing the CaMKII inhibitor AIP targeted to the SR membranes, avoiding phosphorylation of SR proteins (PLN and RyR2) by CaMKII (SR-AIP mice) (Figure 8). Moreover, a ROS scavenger as tempol could avoid RyR2 phosphorylation and SR Ca2+ leak, preventing the arrhythmogenic pattern of the prediabetic cells (Sommese et al., 2016). Of note, the increase in RyR2 phosphorylation observed in FRD myocytes decreases SR Ca2+ content. This decrease occurs in spite of the increase in SERCA2a activity which contributes to preserving SR Ca2+ load. This increase, which may be due to the CaMKII-dependent increase in Thr17 phosphorylation of PLN, would contribute to avoiding a further decrease in SR Ca2+ load but would also favor SR Ca2+ leak and arrhythmogenic Ca2+ waves.
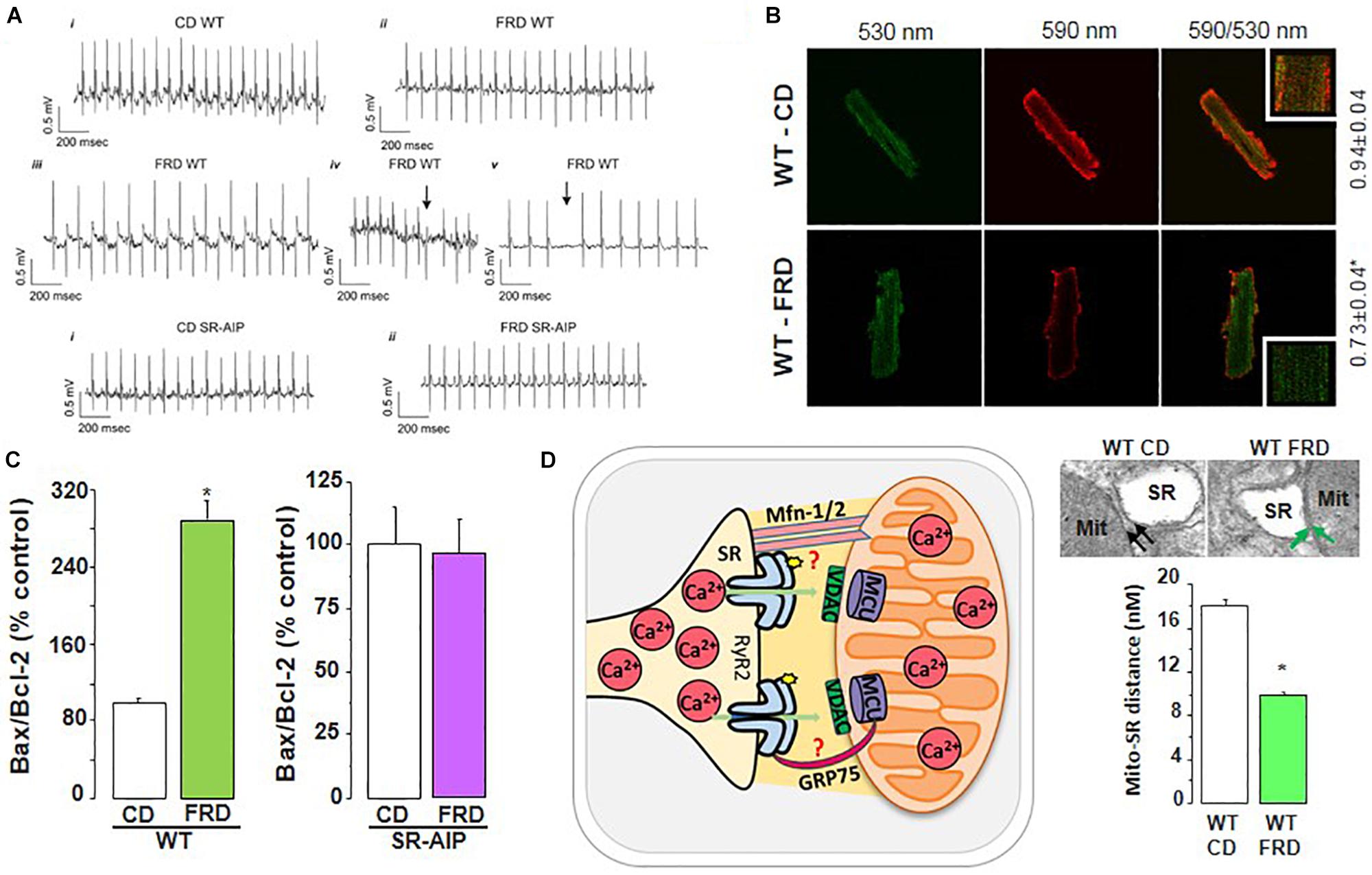
Figure 8. RyR2 dysregulation in DCM development. (A) Representative ECG recordings showing that fructose-rich diet (FRD) induces cardiac arrhythmias in WT but not in SR-AIP mice (CD, control diet). WT mice exhibit bradycardia (ii), bidirectional tachycardia (iii), ventricular ectopic beats (iv, arrow), and AV block (v, arrow) when treated with FRD. (B) FRD deteriorates mitochondria membrane potential, as shown by an increase in JC-1 green/red fluorescence ratio. (C) This alteration in FRD mitochondria is in accordance with a higher apoptotic ratio (Bax/Bcl-2 proteins), which can be prevented in mice expressing the CaMKII inhibitory peptide at the SR level (SR-AIP). (D) Histological samples (upper right) show a closer distance between mitochondria and SR in FRD vs. CD treated mice, which might be due to an alteration in structural proteins involved in SR-mitochondria communication. The cartoon in the left side of (D) represents this SR-mito interaction (reproduced with permission from Federico et al., 2017). *p < 0.05.
Apoptosis is also an early sign of myocardial dysfunction in the evolution of the diabetic disease, preceding the increase in collagen which may lead to structural and irreversible alterations (Federico et al., 2017). We described a cascade of events initiated by a CaMKII-induced increase in SR Ca2+ leak which is linked to mitochondrial membrane depolarization and cardiac damage. A particularly striking finding was the CaMKII-induced remodeling of SR-mitochondria microdomains. The latter would strongly support SR–mitochondria dialogue, facilitating Ca2+ drain to the mitochondria and cell death, in the scenario of an increased SR Ca2+ leak (Federico et al., 2017). The elucidation of the intracellular signaling pathway of this altered SR-mitochondria relationship would further contribute to the knowledge of DCM molecular alterations. Further investigations are needed to examine the proteins involved in the SR-mitochondria communication (like mitofusin-2, Mfn2, and the chaperone glucose-related protein 75, GRP-75), a completely unexplored field in this disease.
Ca2+ Triggered Arrhythmias Induced by Digitalis Intoxication
Cardiac glycosides have been used for the treatment of HF over the last 200 years due to their inotropic properties (Altamirano et al., 2006; Gonano et al., 2011). Although many doubts about their safety in HF treatment have emerged mainly at the end of the last millennium, it is still considered a valuable cardiac tool in some particular scenarios (see for review Whayne, 2018). Unfortunately, these compounds have a very narrow therapeutic range due to their toxic effects that include an enhanced propensity to arrhythmias. The arrhythmic effects of cardiac glycosides have been traditionally attributed to an increase in SR Ca2+ load which, by leading to an increase in Ca2+ leak, would evoke cytosolic Ca2+ waves and triggered arrhythmias (Wier and Hess, 1984; Fujiwara et al., 2008; Eisner et al., 2009; Weiss et al., 2011). More recent experiments indicated that a change in RyR2 may be also involved in cardiac glycosides–induced arrhythmias. Experiments by Gyorke’s group (Ho et al., 2011) indicate that the arrhythmogenic effect of cardiotonic glycosides is linked to NOX2-dependent ROS release from mitochondria. The increase in ROS was initially thought to produce RyR2 thiol oxidation that would increase the sensitivity of the channel to luminal Ca2+, thus lowering the critical SR Ca2+ content at which spontaneous Ca2+ waves occur (Terentyev et al., 2008). However, simultaneous experiments by Gonano et al., indicated that ouabain-induced arrhythmias requires CaMKII activation: Chronic administration or high–toxic doses of ouabain administered acutely, increased CaMKII activity in mouse hearts (Gonano et al., 2011). Moreover, inhibition of CaMKII was able to prevent spontaneous contractions in isolated myocytes and arrhythmias in intact mouse hearts, without affecting ouabain inotropic action. These experiments also showed that CaMKII phosphorylates both, RyR2 and PLN, which would increase SR Ca2+ leak and SERCA2a activity. Although this later effect would add to the increase in SR Ca2+ load resulting from Na+-K+-ATPase inhibition, these experiments concluded that CaMKII-dependent PLN phosphorylation might not contribute to ouabain–induced increase in SR Ca2+ content and inotropic effect since they were of similar magnitude in the absence and presence of CaMKII inhibition. However, since CaMKII dependent phosphorylation of RyR2 was also inhibited, it might be that the resultant similar SR Ca2+ load observed after CaMKII inhibition was due to the prevention of SR Ca2+ leak. Moreover, the fact that CaMKII inhibition prevents arrhythmias without affecting the ouabain-induced increase in SR Ca2+ load would suggest that the increase in SR Ca2+ content produced by the drug is not enough to reach the necessary threshold to trigger, by itself, ouabain-induced arrhythmias (Gonano et al., 2011). An increase in RyR2 sensitivity is needed.
The role of CaMKII-dependent induced increase in SR Ca2+ leak and ventricular arrhythmias was later confirmed by experiments by Gyorke’s group, which revealed that replacement of Ser2814 site of RyR2 by Ala -a non-phosphorylatable amino acid-prevents ouabain-induced Ca2+ leak and arrhythmias. These results definitively confirmed that phosphorylation, rather than RyR2 oxidation, was required for the increase in channel spontaneous activity and arrhythmogenesis in the context of digitalis toxicity. Instead, the increase in ROS would contribute to CaMKII activation that in turn produces the observed RyR2 phosphorylation (Palomeque et al., 2009; Ho et al., 2014).
Importantly, other studies have shown that mitochondria are also involved in the toxic and arrhythmogenic effects of cardiotonic glycosides. The results of these investigations indicate that ouabain-induced increase in cytoplasmic Na+ compromises mitochondrial energetics and redox balance by blunting mitochondrial Ca2+ accumulation. Improving mitochondrial Ca2+ retention by inhibition of mitochondrial NCX, can mitigate these effects, suppress Ca2+-triggered arrhythmias and improve the positive inotropic effects of cardiac glycosides (Liu et al., 2010).
Abnormal Ca2+ Handling in Ischemia/Reperfusion
Ischemic heart disease is a leading cause of mortality worldwide. Cardiac ischemia reduces cardiac output and promotes arrhythmias and cell death. Reperfusion therapies are the standard treatment for patients suffering myocardial infarction, however, re-establishing blood flow is associated with additional cell damage (I/R injury), and exacerbating the effect of the preceding ischemia. Indeed, it was shown that reperfusion may trigger life-threatening arrhythmias, accounting for up to half of the total I/R-induced infarcts (Braunwald and Kloner, 1985; Yellon and Hausenloy, 2007; Said et al., 2011; Garcia-Dorado et al., 2012). Although the factors contributing to I/R injury are complex (see for review (Murphy and Steenbergen, 2008). I/R injury constitutes another example in which experimental evidence reveals that disturbed Ca2+ handling and mitochondria ROS production are main responsible for reperfusion arrhythmias and cardiac damage (Valverde et al., 2006, 2010; Mattiazzi et al., 2015; Bagheri et al., 2016). The increase in cytosolic Ca2+ during ischemia was associated with an enhancement of SR Ca2+ load (Valverde et al., 2010), which is released at the onset of reperfusion and produces an abrupt rise in cytosolic Ca2+ (Ca2+ bump) and the consequent decrease in SR Ca2+ content and Ca2+ transient. Moreover, a major mechanism for the ischemia-induced increase in diastolic Ca2+ is an increase in the frequency of Ca2+ sparks which may switch to arrhythmogenic Ca2+ waves during reperfusion (Mattiazzi et al., 2015).
Reactive oxygen species/reactive nitrogen species are generated during reperfusion by several different cellular sources, being the mitochondria the more important one. Mitochondrial Ca2+ overload and subsequently ROS production trigger mitochondrial permeability transition pore and ROS production via ROS-induced ROS release mechanisms (Zorov et al., 2000). Both, Ca2+ mishandling and ROS production set an ideal intracellular milieu for activation of CaMKII, which play a main role in I/R arrhythmias, apoptosis, and necroptosis. Ex vivo and in vivo experiments described an increase in phosphorylated-CaMKII (p-CaMKII) and oxidized-CaMKII (ox-CaMKII) at the onset of reperfusion (Said et al., 2011; Becerra et al., 2016), which was associated to a significant increase in the phosphorylation of Thr17 site and RyR2 Ser2814 (Vittone et al., 2002; Salas et al., 2010; Ling et al., 2013), as well as redox changes of RyR2 (Becerra et al., 2016). Reperfusion arrhythmias are largely dependent on SR Ca2+ leak evoked by these PTM of RyR2 (Said et al., 2008; Becerra et al., 2016). In this scenario the role played by a substantial increase in SR Ca2+ uptake was similar to that observed in stress-induced Ca2+-triggered arrhythmias, i.e., increasing SR Ca2+ uptake by PLN ablation protects against reperfusion arrhythmias. This protection was achieved by alteration of Ca2+ wave propagation, which were transformed in non-arrhythmogenic mini-waves and reconverted in full Ca2+ waves in the presence of SERCA2a inhibition (Figure 6C; Valverde et al., 2019). Of note, PLN ablation is equivalent to a situation of permanent maximal PLN phosphorylation as stated above. In ischemic reperfused WT hearts, PLN phosphorylation is highly but transiently increased at the onset of reperfusion (Vittone et al., 2002; Said et al., 2008). Under these conditions, we did not observe Ca2+ mini-waves (Valverde et al., 2010). This would mean that Thr17 phosphorylation at the onset of reperfusion is unable to prevent SR Ca2+ waves progression. In contrast, it might help to increase SR Ca2+ load and maintain SR Ca2+ leak. This contention is supported by experiments in which hearts were subjected to a short I/R protocol to produce stunning. In the stunned heart, the transient phosphorylation of Thr17 of PLN is essential for contractile recovery upon reperfusion, even though phosphorylation of RyR2 also occurs and induces reperfusion arrhythmias (Said et al., 2003, 2011).
After a prolonged ischemic period, reperfusion evokes irreversible cardiac injury. Under these conditions, myocytes die by apoptosis, autophagy, and necrosis. The rise in Ca2+ during I/R leads to mitochondrial Ca2+ accumulation, which is greatly favored by the close association between mitochondria and SR and constitutes a main event in the initiation of cell death (Rizzuto and Pozzan, 2006). Experimental evidence consistently indicates that CaMKII inhibition is protective in the irreversible I/R injury (Zhang et al., 2005; Vila-Petroff et al., 2007; Salas et al., 2010). Although the mechanisms for myocardial protection by CaMKII inhibition are still unclear, one of the CaMKII deleterious pathway in I/R certainly involves the SR and the mitochondria (Vila-Petroff et al., 2007; Salas et al., 2010; Valverde et al., 2010; Joiner et al., 2012). In previous experiments, we showed that I/R damage was diminished in hearts from S2814A mice. Conversely, in the hearts of S2814D mice (constitutively pseudo-phosphorylated), cardiac damage increased (Di Carlo et al., 2014). A decrease in the expression of RyR2 described in I/R (Salas et al., 2010), compatible with a degradation/damage of these channels (Pedrozo et al., 2010) and changes in RyR2 activity induced by redox alterations, may contribute to increase SR Ca2+ leak (Hidalgo et al., 2004; Said et al., 2011; Di Carlo et al., 2014). These alterations in RyR2 would add to the deleterious action of RyR2 phosphorylation favoring the increase in mitochondria Ca2+ content and greatly contributing to necroptosis and apoptosis in reperfusion cardiac damage (Salas et al., 2010; Di Carlo et al., 2014). This cascade would be further stimulated by the increase in CaMKII-dependent phosphorylation of MCU described by Joiner, Koval et al.(Joiner et al., 2012). However, recent experiments do not support a relevant role of CaMKII for mitochondrial Ca2+ uptake in cardiac myocytes at least under physiological conditions (Nickel et al., 2019).
As stated above, the transient increase in CaMKII-dependent PLN phosphorylation plays a beneficial role in the stunned heart. The role of PLN phosphorylation in I/R injury is less clear. After prolonged ischemia, we showed that preventing PLN phosphorylation exacerbates the functional and structural heart damage after myocardial infarction, suggesting that CaMKII-dependent phosphorylation of PLN observed during reperfusion favors post-ischemic recovery and protects from I/R cardiac damage (Di Carlo et al., 2014). However, the fact that I/R does produce cardiac damage indicates that even being beneficial, CaMKII-dependent PLN phosphorylation results insufficient to counteract the effect of simultaneous detrimental mechanisms that take place during I/R. On the other hand, several groups have tested the effects of increasing SR Ca2+ uptake on cardiac damage during I/R injury, by different maneuvers. The outcome of these experiments is controversial. For instance, Yang et al. (2006) demonstrated that the protective effect of chronic CaMKII inhibition in AC3I mice was lost when they were interbred with PLNKO mice and submitted to myocardial infarction, supporting a detrimental effect of enhancing of SR Ca2+ uptake. Similar results were obtained in our laboratory. The ablation of PLN in SDKO mice increases Ca2+ leak upon reperfusion (Figure 6A). This increase was associated with an increase in infarct size and mitochondrial dysfunction. Therefore, these experiments demonstrated that an important increase in SR Ca2+ uptake as that produced by PLN ablation, was able to prevent reperfusion arrhythmias, but failed to prevent, and even enhance, cardiac damage (Valverde et al., 2019). The important increase in SR Ca2+ uptake would favor the unbalance between SR Ca2+ uptake and leak, promoting mitochondrial Ca2+ overload and cell death. Other studies demonstrated that accelerating SR Ca2+ uptake by different means (i.e., overexpressing SERCA1a, with higher kinetics than SERCA2a, or expressing a repressor of PLN activity, PP1 H-1), alleviated post-ischemic cardiac injury (Talukder et al., 2007, 2008; Nicolaou et al., 2009a), supporting a beneficial effect of accelerating SR Ca2+ uptake. These controversial results seem not to arise from species differences since most of the experiments mentioned above referred to rodents. It is possible that the final beneficial or detrimental outcome of increasing SR Ca2+ uptake might tightly depend on the extent of Ca2+ uptake and SR Ca2+ load achieved during ischemia and at the onset of reperfusion. For instance, moderate increases in SR Ca2+ content have been associated with beneficial effects (Nicolaou et al., 2009b), whereas more important increases, like those expected in PLNKO mice, were associated with detrimental actions (Valverde et al., 2019).
Is RyR2 Activation Always Detrimental?
We have previously associated the increase in RyR2 activation, for instance by CaMKII-dependent phosphorylation, with cardiac damage and arrhythmias due to exacerbated diastolic SR Ca2+ leak, as discussed above. However, it is important to bear in mind that potentiation of RyR2 activity persists during systole and enhances systolic fractional Ca2+ release, bringing the heart to a new state of higher efficiency. This would allow the heart to maintain a given contractility despite a decrease in SR Ca2+ content or to enhance contractility if SR Ca2+ content is simultaneously preserved (for further discussion, see Lascano et al., 2017).
Concluding Remarks
The normal interplay among the proteins involved in SR Ca2+ uptake and release is a main determinant of the regular beat to beat contractile function of cardiac myocytes. Regulation and deregulation of these proteins are crucial to understanding the balance between SR Ca2+ uptake and leak, responsible for SR Ca2+ content and myocardial contractility, as well as its unbalance, which determines an excess of SR Ca2+ leak, able to produce arrhythmias and cardiac damage. Post-translational enhancements of SR Ca2+ uptake have a beneficial effect resulting in a detectable increase in contractility when the unbalance between SR Ca2+ uptake and leak favors the uptake. This is the case of PLNKO mice and stunned hearts. When SR Ca2+ leak is increased by the enhancement of RyR2 activity, the increase in SR Ca2+ uptake may not be enough to counteract SR Ca2+ leak, resulting in two opposite effects: (a) beneficial, by opposing to SR Ca2+ leak and rescuing at least in part SR Ca2+ load and contractility; (b) detrimental, because the increase in SR Ca2+ load would favor SR Ca2+ leak, arrhythmias, and cardiac damage. Interestingly, a greatly exacerbated increase in SR Ca2+ uptake, as that produced by PLN ablation, does contribute to increasing SR Ca2+ leak and cardiac damage by incrementing SR Ca2+ load and leak. Paradoxically, and in spite of the exacerbated SR Ca2+ leak, this increase may prevent Ca2+ triggered arrhythmias, by a different mechanism, i.e., diminishing cytosolic Ca2+ and avoiding Ca2+ wave propagation. Post-translational activation of RyR2 activity would produce a deleterious effect by increasing SR Ca2+ leak and predisposing to cardiac damage and arrhythmias. When this modification occurs only at the level of SR Ca2+ release/leak, the short-lived enhancement of SR Ca2+ leak may produce a decrease in contractility due to the decrease in SR Ca2+ load, without any further detrimental effect. Moreover, the increased activity of RyR2 during systole tends to preserve contractility, even at lower SR Ca2+ loads. The detrimental effect of RyR2 activation (i.e., arrhythmias and cardiac damage) can only take place when it occurs associated to an increase in SR Ca2+ uptake, able to maintain SR Ca2+ leak. This is the typical case of Ser2814D myocytes, with constitutive pseudo-phosphorylation of RyR2 at the CaMKII site.
Author Contributions
JP and AM generated the idea. JP, AM, MF, and CV wrote the manuscript, designed, and edited the figures.
Funding
This work was supported by PICT 2015-3009 (FONCyT, Argentina) and PS-1 (UAI, Argentina) to JP, PICT 2015-3210 (FONCyT, Argentina) to CV, and PICT 2524 (FONCyT, Argentina) and PIP 0305 (CONICET, Argentina) to AM. MF is a doctoral fellow from CONICET.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
β-ARS, β-adrenergic stimulation; AGEs, advanced glycation end products; AP, action potential; Ca2+, calcium; CaM, calmodulin; CaMKII, Ca2+-calmodulin dependent Kinase II; CASQ2, calsequestrin; CPA, cyclopiazonic acid; DAD, delay afterdepolarizations; DCM, diabetic cardiomyopathy; EAD, early afterdepolarizations; ECC, excitation contraction coupling; ER, endoplasmic reticulum; HRC, histidine-rich Ca2+binding protein; I/R, ischemia/reperfusion; MCU, mitochondrial Ca2+ uniporter; Na+, sodium; NCX, Na+/Ca2+ exchanger; NO, nitric oxide; ONOO–, peroxynitrite; P0, open probability; PKA, protein kinase A; PLN, phospholamban; PP1, type 1 phosphatase; PTM, post-translational modification; RNS, reactive nitrogen species; ROS, reactive oxygen species; RyR2, ryanodine receptor 2; SERCA2a, sarcoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum; SUMO1, small ubiquitin-like modifier type 1; T2DM, type 2 diabetes mellitus.
References
Adachi, T., Weisbrod, R. M., Pimentel, D. R., Ying, J., Sharov, V. S., Schoneich, C., et al. (2004). S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 10, 1200–1207. doi: 10.1038/nm1119
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.res.0000194329.41863.89
Altamirano, J., Li, Y., DeSantiago, J., Piacentino, V. III, Houser, S. R., and Bers, D. M. (2006). The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J. Physiol. 575(Pt 3), 845–854. doi: 10.1113/jphysiol.2006.111252
Anderson, M. E. (2007). Multiple downstream proarrhythmic targets for calmodulin kinase II: moving beyond an ion channel-centric focus. Cardiovasc. Res. 73, 657–666. doi: 10.1016/j.cardiores.2006.12.009
Aneja, A., Tang, W. H., Bansilal, S., Garcia, M. J., and Farkouh, M. E. (2008). Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am. J. Med. 121, 748–757. doi: 10.1016/j.amjmed.2008.03.046
Arvanitis, D. A., Vafiadaki, E., Fan, G. C., Mitton, B. A., Gregory, K. N., Del Monte, F., et al. (2007). Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. Am. J. Physiol. Heart Circ. Physiol. 293, H1581–H1589.
Arvanitis, D. A., Vafiadaki, E., Johnson, D. M., Kranias, E. G., and Sanoudou, D. (2018). The histidine-rich calcium binding protein in regulation of cardiac rhythmicity. Front. Physiol. 9:1379. doi: 10.3389/fphys.2018.01379
Bagheri, F., Khori, V., Alizadeh, A. M., Khalighfard, S., Khodayari, S., and Khodayari, H. (2016). Reactive oxygen species-mediated cardiac-reperfusion injury: mechanisms and therapies. Life Sci. 165, 43–55. doi: 10.1016/j.lfs.2016.09.013
Balderas-Villalobos, J., Molina-Munoz, T., Mailloux-Salinas, P., Bravo, G., Carvajal, K., and Gomez-Viquez, N. L. (2013). Oxidative stress in cardiomyocytes contributes to decreased SERCA2a activity in rats with metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 305, H1344–H1353. doi: 10.1152/ajpheart.00211.2013
Balshaw, D. M., Xu, L., Yamaguchi, N., Pasek, D. A., and Meissner, G. (2001). Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 276, 20144–20153. doi: 10.1074/jbc.m010771200
Barg, S., Copello, J. A., and Fleischer, S. (1997). Different interactions of cardiac and skeletal muscle ryanodine receptors with FK-506 binding protein isoforms. Am. J. Physiol. 272(5 Pt 1), C1726–C1733.
Bassani, J. W., Yuan, W., and Bers, D. M. (1995). Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 268(5 Pt 1), C1313–C1319.
Becerra, R., Roman, B., Di Carlo, M. N., Mariangelo, J. I., Salas, M., Sanchez, G., et al. (2016). Reversible redox modifications of ryanodine receptor ameliorate ventricular arrhythmias in the ischemic-reperfused heart. Am. J. Physiol. Heart Circ. Physiol. 311, H713–H724. doi: 10.1152/ajpheart.00142.2016
Belevych, A. E., Terentyev, D., Terentyeva, R., Ho, H. T., Gyorke, I., Bonilla, I. M., et al. (2012). Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 110, 569–577. doi: 10.1161/CIRCRESAHA.111.260455
Bernardi, P., and Di Lisa, F. (2015). The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J. Mol. Cell Cardiol. 78, 100–106. doi: 10.1016/j.yjmcc.2014.09.023
Bers, D. M. (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiology 21, 380–387. doi: 10.1152/physiol.00019.2006
Bers, D. M. (2012). Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ. Res. 110, 796–799. doi: 10.1161/circresaha.112.265579
Bidasee, K. R., Zhang, Y., Shao, C. H., Wang, M., Patel, K. P., Dincer, U. D., et al. (2004). Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes Metab. Res. Rev. 53, 463–473. doi: 10.2337/diabetes.53.2.463
Bigelow, D. J., and Squier, T. C. (2005). Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim. Biophys. Acta 1703, 121–134. doi: 10.1016/j.bbapap.2004.09.012
Braun, J. L., Hamstra, S. I., Messner, H. N., and Fajardo, V. A. (2019). SERCA2a tyrosine nitration coincides with impairments in maximal SERCA activity in left ventricles from tafazzin-deficient mice. Physiol. Rep. 7, e14215. doi: 10.14814/phy2.14215
Braunwald, E., and Kloner, R. A. (1985). Myocardial reperfusion: a double-edged sword? J. Clin. Invest. 76, 1713–1719. doi: 10.1172/jci112160
Bugger, H., and Abel, E. D. (2014). Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57, 660–671. doi: 10.1007/s00125-014-3171-6
Cai, L., Li, W., Wang, G., Guo, L., Jiang, Y., and Kang, Y. J. (2002). Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes Metab. Res. Rev. 51, 1938–1948. doi: 10.2337/diabetes.51.6.1938
Camors, E., and Valdivia, H. H. (2014). CaMKII regulation of cardiac ryanodine receptors and inositol triphosphate receptors. Front. Pharmacol. 5:101. doi: 10.3389/fphar.2014.00101
Capes, E. M., Loaiza, R., and Valdivia, H. H. (2011). Ryanodine receptors. Skelet Muscle 1:18. doi: 10.1186/2044-5040-1-18
Carter, S., Colyer, J., and Sitsapesan, R. (2006). Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase a produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ. Res. 98, 1506–1513. doi: 10.1161/01.res.0000227506.43292.df
Chen, H., Charlat, O., Tartaglia, L. A., Woolf, E. A., Weng, X., Ellis, S. J., et al. (1996). Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84, 491–495. doi: 10.1016/s0092-8674(00)81294-5
Chen, W., Wang, R., Chen, B., Zhong, X., Kong, H., Bai, Y., et al. (2014). The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat. Med. 20, 184–192. doi: 10.1038/nm.3440
Chen, X., Weber, C., Farrell, E. T., Alvarado, F. J., Zhao, Y. T., Gomez, A. M., et al. (2018). Sorcin ablation plus beta-adrenergic stimulation generate an arrhythmogenic substrate in mouse ventricular myocytes. J. Mol. Cell Cardiol. 114, 199–210. doi: 10.1016/j.yjmcc.2017.11.017
Cheng, H., Lederer, M. R., Lederer, W. J., and Cannell, M. B. (1996). Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am. J. Physiol. 270(1 Pt 1), C148–C159.
Ching, L. L., Williams, A. J., and Sitsapesan, R. (2000). Evidence for Ca(2+) activation and inactivation sites on the luminal side of the cardiac ryanodine receptor complex. Circ. Res. 87, 201–206. doi: 10.1161/01.res.87.3.201
Clark, R. J., McDonough, P. M., Swanson, E., Trost, S. U., Suzuki, M., Fukuda, M., et al. (2003). Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J. Biol. Chem. 278, 44230–44237. doi: 10.1074/jbc.m303810200
Cohen, R. A., and Adachi, T. (2006). Nitric-oxide-induced vasodilatation: regulation by physiologic s-glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends Cardiovasc. Med. 16, 109–114. doi: 10.1016/j.tcm.2006.02.001
Cox, E. J., and Marsh, S. A. (2014). A systematic review of fetal genes as biomarkers of cardiac hypertrophy in rodent models of diabetes. PLoS One 9:e92903. doi: 10.1371/journal.pone.0092903
Csordas, G., Weaver, D., and Hajnoczky, G. (2018). Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell. Biol. 28, 523–540. doi: 10.1016/j.tcb.2018.02.009
Currie, S., Loughrey, C. M., Craig, M. A., and Smith, G. L. (2004). Calcium/calmodulin-dependent protein kinase IIdelta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem. J. 377(Pt 2), 357–366. doi: 10.1042/bj20031043
Daniels, L., Bell, J. R., Delbridge, L. M., McDonald, F. J., Lamberts, R. R., and Erickson, J. R. (2015). The role of CaMKII in diabetic heart dysfunction. Heart Fail. Rev. 20, 589–600. doi: 10.1007/s10741-015-9498-3
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316. doi: 10.1016/j.bbabio.2009.01.005
Di Carlo, M. N., Said, M., Ling, H., Valverde, C. A., De Giusti, V. C., Sommese, L., et al. (2014). CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J. Mol. Cell Cardiol. 74, 274–283. doi: 10.1016/j.yjmcc.2014.06.004
Diaz, M. E., Trafford, A. W., O’Neill, S. C., and Eisner, D. A. (1997). Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J. Physiol. 501(1 Pt 1), 3–16. doi: 10.1111/j.1469-7793.1997.003bo.x
Doi, M., Yano, M., Kobayashi, S., Kohno, M., Tokuhisa, T., Okuda, S., et al. (2002). Propranolol prevents the development of heart failure by restoring FKBP12.6-mediated stabilization of ryanodine receptor. Circulation 105, 1374–1379. doi: 10.1161/hc1102.105270
Dorn, G. W. II, and Maack, C. (2013). SR and mitochondria: calcium cross-talk between kissing cousins. J. Mol. Cell Cardiol. 55, 42–49. doi: 10.1016/j.yjmcc.2012.07.015
Dybkova, N., Sedej, S., Napolitano, C., Neef, S., Rokita, A. G., Hunlich, M., et al. (2011). Overexpression of CaMKIIdeltac in RyR2R4496C+/- knock-in mice leads to altered intracellular Ca2+ handling and increased mortality. J. Am. Coll. Cardiol. 57, 469–479. doi: 10.1016/j.jacc.2010.08.639
Eisner, D. A., Caldwell, J. L., Kistamas, K., and Trafford, A. W. (2017). Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195.
Eisner, D. A., Kashimura, T., O’Neill, S. C., Venetucci, L. A., and Trafford, A. W. (2009). What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J. Mol. Cell Cardiol. 46, 474–481. doi: 10.1016/j.yjmcc.2008.12.005
El Hadi, H., Vettor, R., and Rossato, M. (2019). Cardiomyocyte mitochondrial dysfunction in diabetes and its contribution in cardiac arrhythmogenesis. Mitochondrion 46, 6–14. doi: 10.1016/j.mito.2019.03.005
Erickson, J. R., Pereira, L., Wang, L., Han, G., Ferguson, A., Dao, K., et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376. doi: 10.1038/nature12537
Fabiato, A., and Fabiato, F. (1977). Calcium release from the sarcoplasmic reticulum. Circ. Res. 40, 119–129.
Fabiato, A., and Fabiato, F. (1979). Calcium and cardiac excitation-contraction coupling. Annu. Rev. Physiol. 41, 473–484. doi: 10.1146/annurev.ph.41.030179.002353
Farrell, E. F., Antaramian, A., Rueda, A., Gomez, A. M., and Valdivia, H. H. (2003). Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J. Biol. Chem. 278, 34660–34666. doi: 10.1074/jbc.m305931200
Federico, M., Portiansky, E. L., Sommese, L., Alvarado, F. J., Blanco, P. G., Zanuzzi, C. N., et al. (2017). Calcium-calmodulin-dependent protein kinase mediates the intracellular signalling pathways of cardiac apoptosis in mice with impaired glucose tolerance. J. Physiol. 595, 4089–4108. doi: 10.1113/JP273714
Ferrero, P., Said, M., Sanchez, G., Vittone, L., Valverde, C., Donoso, P., et al. (2007). Ca2+/calmodulin kinase II increases ryanodine binding and Ca2+-induced sarcoplasmic reticulum Ca2+ release kinetics during beta-adrenergic stimulation. J. Mol. Cell Cardiol. 43, 281–291. doi: 10.1016/j.yjmcc.2007.05.022
Fill, M., and Copello, J. A. (2002). Ryanodine receptor calcium release channels. Physiol. Rev. 82, 893–922. doi: 10.1152/physrev.00013.2002
Finkel, E. (2001). The mitochondrion: is it central to apoptosis? Science 292, 624–626. doi: 10.1126/science.292.5517.624
Fischer, T. H., Eiringhaus, J., Dybkova, N., Saadatmand, A., Pabel, S., Weber, S., et al. (2018). Activation of protein phosphatase 1 by a selective phosphatase disrupting peptide reduces sarcoplasmic reticulum Ca(2+) leak in human heart failure. Eur. J. Heart Fail. 20, 1673–1685. doi: 10.1002/ejhf.1297
Fischer, T. H., Herting, J., Tirilomis, T., Renner, A., Neef, S., Toischer, K., et al. (2013). Ca2+/calmodulin-dependent protein kinase II and protein kinase a differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 128, 970–981. doi: 10.1161/CIRCULATIONAHA.113.001746
Fowler, M. R., Colotti, G., Chiancone, E., Smith, G. L., and Fearon, I. M. (2008). Sorcin modulates cardiac L-type Ca2+ current by functional interaction with the alpha1C subunit in rabbits. Exp. Physiol. 93, 1233–1238. doi: 10.1113/expphysiol.2008.043497
Froehlich, J. P., Mahaney, J. E., Keceli, G., Pavlos, C. M., Goldstein, R., Redwood, A. J., et al. (2008). Phospholamban thiols play a central role in activation of the cardiac muscle sarcoplasmic reticulum calcium pump by nitroxyl. Biochemistry 47, 13150–13152. doi: 10.1021/bi801925p
Fruen, B. R., Bardy, J. M., Byrem, T. M., Strasburg, G. M., and Louis, C. F. (2000). Differential Ca(2+) sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am. J. Physiol. Cell Physiol. 279, C724–C733.
Fuentes-Antras, J., Picatoste, B., Gomez-Hernandez, A., Egido, J., Tunon, J., and Lorenzo, O. (2015). Updating experimental models of diabetic cardiomyopathy. J. Diabetes Res. 2015:656795. doi: 10.1155/2015/656795
Fuentes-Antras, J., Picatoste, B., Ramirez, E., Egido, J., Tunon, J., and Lorenzo, O. (2015). Targeting metabolic disturbance in the diabetic heart. Cardiovasc. Diabetol. 14:17. doi: 10.1186/s12933-015-0173-8
Fujiwara, K., Tanaka, H., Mani, H., Nakagami, T., and Takamatsu, T. (2008). Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ. Res. 103, 509–518. doi: 10.1161/CIRCRESAHA.108.176677
Galfre, E., Pitt, S. J., Venturi, E., Sitsapesan, M., Zaccai, N. R., Tsaneva-Atanasova, K., et al. (2012). FKBP12 activates the cardiac ryanodine receptor Ca2+-release channel and is antagonised by FKBP12.6. PLoS One 7:e31956. doi: 10.1371/journal.pone.0031956
Garcia-Dorado, D., Ruiz-Meana, M., Inserte, J., Rodriguez-Sinovas, A., and Piper, H. M. (2012). Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 94, 168–180. doi: 10.1093/cvr/cvs116
Gonano, L. A., Sepulveda, M., Rico, Y., Kaetzel, M., Valverde, C. A., Dedman, J., et al. (2011). Calcium-calmodulin kinase II mediates digitalis-induced arrhythmias. Circ. Arrhythm. Electrophysiol. 4, 947–957. doi: 10.1161/CIRCEP.111.964908
Gong, D., Chi, X., Wei, J., Zhou, G., Huang, G., Zhang, L., et al. (2019). Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature 572, 347–351. doi: 10.1038/s41586-019-1377-y
Gonzalez, D. R., Treuer, A., Sun, Q. A., Stamler, J. S., and Hare, J. M. (2009). S-Nitrosylation of cardiac ion channels. J. Cardiovasc. Pharmacol. 54, 188–195. doi: 10.1097/FJC.0b013e3181b72c9f
Gorski, P. A., Jang, S. P., Jeong, D., Lee, A., Lee, P., Oh, J. G., et al. (2019). Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca(2+)-ATPase in heart failure. Circ. Res. 124, e63–e80. doi: 10.1161/CIRCRESAHA.118.313865
Graham, M. L., and Schuurman, H. J. (2015). Validity of animal models of type 1 diabetes, and strategies to enhance their utility in translational research. Eur. J. Pharmacol. 759, 221–230. doi: 10.1016/j.ejphar.2015.02.054
Guo, T., Cornea, R. L., Huke, S., Camors, E., Yang, Y., Picht, E., et al. (2010). Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ. Res. 106, 1743–1752. doi: 10.1161/CIRCRESAHA.110.219816
Guo, T., Zhang, T., Mestril, R., and Bers, D. M. (2006). Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 99, 398–406. doi: 10.1161/01.res.0000236756.06252.13
Gyorke, I., and Gyorke, S. (1998). Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys. J. 75, 2801–2810. doi: 10.1016/s0006-3495(98)77723-9
Gyorke, I., Hester, N., Jones, L. R., and Gyorke, S. (2004). The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 86, 2121–2128. doi: 10.1016/s0006-3495(04)74271-x
Gyorke, S., and Terentyev, D. (2008). Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc. Res. 77, 245–255. doi: 10.1093/cvr/cvm038
Ha, K. N., Masterson, L. R., Hou, Z., Verardi, R., Walsh, N., Veglia, G., et al. (2011). Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase. Proc. Natl. Acad. Sci. U.S.A. 108, 2735–2740. doi: 10.1073/pnas.1013987108
Haghighi, K., Bidwell, P., and Kranias, E. G. (2014). Phospholamban interactome in cardiac contractility and survival: a new vision of an old friend. J. Mol. Cell Cardiol. 77, 160–167. doi: 10.1016/j.yjmcc.2014.10.005
Haghighi, K., Pritchard, T. J., Liu, G. S., Singh, V. P., Bidwell, P., Lam, C. K., et al. (2015). Human G109E-inhibitor-1 impairs cardiac function and promotes arrhythmias. J. Mol. Cell Cardiol. 89(Pt B), 349–359. doi: 10.1016/j.yjmcc.2015.10.004
Hajat, C., Tilling, K., Stewart, J. A., Lemic-Stojcevic, N., and Wolfe, C. D. (2004). Ethnic differences in risk factors for ischemic stroke: a European case-control study. Stroke 35, 1562–1567. doi: 10.1161/01.str.0000131903.04708.b8
Haji-Ghassemi, O., Yuchi, Z., and Van Petegem, F. (2019). The cardiac ryanodine receptor phosphorylation hotspot embraces PKA in a phosphorylation-dependent manner. Mol. Cell. 75, 39–52 e4. doi: 10.1016/j.molcel.2019.04.019
Handhle, A., Ormonde, C. E., Thomas, N. L., Bralesford, C., Williams, A. J., Lai, F. A., et al. (2016). Calsequestrin interacts directly with the cardiac ryanodine receptor luminal domain. J. Cell Sci. 129, 3983–3988.
Herrmann-Frank, A., and Lehmann-Horn, F. (1996). Regulation of the purified Ca2+ release channel/ryanodine receptor complex of skeletal muscle sarcoplasmic reticulum by luminal calcium. Pflugers. Arch. 432, 155–157. doi: 10.1007/s004240050117
Hidalgo, C., Bull, R., Behrens, M. I., and Donoso, P. (2004). Redox regulation of RyR-mediated Ca2+ release in muscle and neurons. Biol. Res. 37, 539–552.
Ho, H. T., Liu, B., Snyder, J. S., Lou, Q., Brundage, E. A., Velez-Cortes, F., et al. (2014). Ryanodine receptor phosphorylation by oxidized CaMKII contributes to the cardiotoxic effects of cardiac glycosides. Cardiovasc. Res. 101, 165–174. doi: 10.1093/cvr/cvt233
Ho, H. T., Stevens, S. C., Terentyeva, R., Carnes, C. A., Terentyev, D., and Gyorke, S. (2011). Arrhythmogenic adverse effects of cardiac glycosides are mediated by redox modification of ryanodine receptors. J. Physiol. 589(Pt 19), 4697–4708. doi: 10.1113/jphysiol.2011.210005
Horvath, B., Banyasz, T., Jian, Z., Hegyi, B., Kistamas, K., Nanasi, P. P., et al. (2013). Dynamics of the late Na(+) current during cardiac action potential and its contribution to afterdepolarizations. J. Mol. Cell Cardiol. 64, 59–68. doi: 10.1016/j.yjmcc.2013.08.010
Huke, S., and Bers, D. M. (2008). Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem. Biophys. Res. Commun. 376, 80–85. doi: 10.1016/j.bbrc.2008.08.084
Huser, J., Bers, D. M., and Blatter, L. A. (1998). Subcellular properties of [Ca2+]i transients in phospholamban-deficient mouse ventricular cells. Am. J. Physiol. 274, H1800–H1811.
Irie, T., Sips, P. Y., Kai, S., Kida, K., Ikeda, K., Hirai, S., et al. (2015). S-Nitrosylation of calcium-handling proteins in cardiac adrenergic signaling and hypertrophy. Circ. Res. 117, 793–803. doi: 10.1161/CIRCRESAHA.115.307157
January, C. T., and Riddle, J. M. (1989). Early afterdepolarizations: mechanism of induction and block. a role for L-type Ca2+ current. Circ. Res. 64, 977–990. doi: 10.1161/01.res.64.5.977
Jay, D., Hitomi, H., and Griendling, K. K. (2006). Oxidative stress and diabetic cardiovascular complications. Free Radic. Biol. Med. 40, 183–192. doi: 10.1016/j.freeradbiomed.2005.06.018
Jeyakumar, L. H., Ballester, L., Cheng, D. S., McIntyre, J. O., Chang, P., Olivey, H. E., et al. (2001). FKBP binding characteristics of cardiac microsomes from diverse vertebrates. Biochem. Biophys. Res. Commun. 281, 979–986. doi: 10.1006/bbrc.2001.4444
Jiang, D., Chen, W., Wang, R., Zhang, L., and Chen, S. R. (2007). Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc. Natl. Acad. Sci. U.S.A. 104, 18309–18314. doi: 10.1073/pnas.0706573104
Jiang, D., Xiao, B., Yang, D., Wang, R., Choi, P., Zhang, L., et al. (2004). RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. U.S.A. 101, 13062–13067. doi: 10.1073/pnas.0402388101
Jiang, M. T., Lokuta, A. J., Farrell, E. F., Wolff, M. R., Haworth, R. A., and Valdivia, H. H. (2002). Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ. Res. 91, 1015–1022. doi: 10.1161/01.res.0000043663.08689.05
Joiner, M. L., Koval, O. M., Li, J., He, B. J., Allamargot, C., Gao, Z., et al. (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. doi: 10.1038/nature11444
Kaftan, E., Marks, A. R., and Ehrlich, B. E. (1996). Effects of rapamycin on ryanodine receptor/Ca(2+)-release channels from cardiac muscle. Circ. Res. 78, 990–997. doi: 10.1161/01.res.78.6.990
Kaplan, P., Babusikova, E., Lehotsky, J., and Dobrota, D. (2003). Free radical-induced protein modification and inhibition of Ca2+-ATPase of cardiac sarcoplasmic reticulum. Mol. Cell. Biochem. 248, 41–47.
Kettlewell, S., Most, P., Currie, S., Koch, W. J., and Smith, G. L. (2005). S100A1 increases the gain of excitation-contraction coupling in isolated rabbit ventricular cardiomyocytes. J. Mol. Cell Cardiol. 39, 900–910. doi: 10.1016/j.yjmcc.2005.06.018
Kho, C., Lee, A., Jeong, D., Oh, J. G., Chaanine, A. H., Kizana, E., et al. (2011). SUMO1-dependent modulation of SERCA2a in heart failure. Nature 477, 601–605. doi: 10.1038/nature10407
Kiewitz, R., Acklin, C., Schafer, B. W., Maco, B., Uhrik, B., Wuytack, F., et al. (2003). Ca2+ -dependent interaction of S100A1 with the sarcoplasmic reticulum Ca2+ -ATPase2a and phospholamban in the human heart. Biochem. Biophys. Res. Commun. 306, 550–557. doi: 10.1016/s0006-291x(03)00987-2
King, A., and Bowe, J. (2016). Animal models for diabetes: understanding the pathogenesis and finding new treatments. Biochem. Pharmacol. 99, 1–10. doi: 10.1016/j.bcp.2015.08.108
King, A. J. (2012). The use of animal models in diabetes research. Br. J. Pharmacol. 166, 877–894. doi: 10.1111/j.1476-5381.2012.01911.x
Knollmann, B. C., Chopra, N., Hlaing, T., Akin, B., Yang, T., Ettensohn, K., et al. (2006). Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Invest. 116, 2510–2520.
Kohlhaas, M., Zhang, T., Seidler, T., Zibrova, D., Dybkova, N., Steen, A., et al. (2006). Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ. Res. 98, 235–244. doi: 10.1161/01.res.0000200739.90811.9f
Kong, H., Wang, R., Chen, W., Zhang, L., Chen, K., Shimoni, Y., et al. (2007). Skeletal and cardiac ryanodine receptors exhibit different responses to Ca2+ overload and luminal ca2+. Biophys. J. 92, 2757–2770. doi: 10.1529/biophysj.106.100545
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi: 10.1161/CIRCRESAHA.111.259754
Kranias, E. G., and Hajjar, R. J. (2017). The phospholamban journey 4 decades after setting out for ithaka. Circ. Res. 120, 781–783. doi: 10.1161/circresaha.116.310007
Kuster, G. M., Lancel, S., Zhang, J., Communal, C., Trucillo, M. P., Lim, C. C., et al. (2010). Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic. Biol. Med. 48, 1182–1187. doi: 10.1016/j.freeradbiomed.2010.01.038
Lancel, S., Zhang, J., Evangelista, A., Trucillo, M. P., Tong, X., Siwik, D. A., et al. (2009). Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ. Res. 104, 720–723. doi: 10.1161/CIRCRESAHA.108.188441
Lascano, E., Negroni, J., Vila Petroff, M., and Mattiazzi, A. (2017). Impact of RyR2 potentiation on myocardial function. Am. J. Physiol. Heart Circ. Physiol. 312, H1105–H1109. doi: 10.1152/ajpheart.00855.2016
Laver, D. R. (2018). Regulation of the RyR channel gating by Ca(2+) and Mg(2). Biophys. Rev. 10, 1087–1095. doi: 10.1007/s12551-018-0433-4
Lebeche, D., Davidoff, A. J., and Hajjar, R. J. (2008). Interplay between impaired calcium regulation and insulin signaling abnormalities in diabetic cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 5, 715–724. doi: 10.1038/ncpcardio1347
Lehnart, S. E., Wehrens, X. H., Reiken, S., Warrier, S., Belevych, A. E., Harvey, R. D., et al. (2005). Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123, 25–35. doi: 10.1016/j.cell.2005.07.030
Li, Y., Kranias, E. G., Mignery, G. A., and Bers, D. M. (2002). Protein kinase a phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ. Res. 90, 309–316. doi: 10.1161/hh0302.105660
Lim, G., Venetucci, L., Eisner, D. A., and Casadei, B. (2008). Does nitric oxide modulate cardiac ryanodine receptor function? implications for excitation-contraction coupling. Cardiovasc. Res. 77, 256–264. doi: 10.1093/cvr/cvm012
Lindemann, J. P., Jones, L. R., Hathaway, D. R., Henry, B. G., and Watanabe, A. M. (1983). beta-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J. Biol. Chem. 258, 464–471.
Lindemann, J. P., and Watanabe, A. M. (1985). Phosphorylation of phospholamban in intact myocardium. Role of Ca2+-calmodulin-dependent mechanisms. J. Biol. Chem. 260, 4516–4525.
Ling, H., Gray, C. B., Zambon, A. C., Grimm, M., Gu, Y., Dalton, N., et al. (2013). Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ. Res. 112, 935–944. doi: 10.1161/CIRCRESAHA.112.276915
Liu, T., Brown, D. A., and O’Rourke, B. (2010). Role of mitochondrial dysfunction in cardiac glycoside toxicity. J. Mol. Cell Cardiol. 49, 728–736. doi: 10.1016/j.yjmcc.2010.06.012
Lokuta, A. J., Rogers, T. B., Lederer, W. J., and Valdivia, H. H. (1995). Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J. Physiol. 487(Pt 3), 609–622. doi: 10.1113/jphysiol.1995.sp020904
MacDonnell, S. M., Garcia-Rivas, G., Scherman, J. A., Kubo, H., Chen, X., Valdivia, H., et al. (2008). Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ. Res. 102, e65–e72.
MacLennan, D. H., and Kranias, E. G. (2003). Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566–577. doi: 10.1038/nrm1151
Maier, L. S., Zhang, T., Chen, L., DeSantiago, J., Brown, J. H., and Bers, D. M. (2003). Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92, 904–911. doi: 10.1161/01.res.0000069685.20258.f1
Marengo, J. J., Hidalgo, C., and Bull, R. (1998). Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys. J. 74, 1263–1277. doi: 10.1016/s0006-3495(98)77840-3
Marks, A. R. (2000). Cardiac intracellular calcium release channels: role in heart failure. Circ. Res. 87, 8–11. doi: 10.1161/01.res.87.1.8
Marks, A. R. (2002). Ryanodine receptors, FKBP12, and heart failure. Front. Biosci. 7:d970–d977. doi: 10.2741/a822
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/s0092-8674(00)80847-8
Matsumoto, T., Hisamatsu, Y., Ohkusa, T., Inoue, N., Sato, T., Suzuki, S., et al. (2005). Sorcin interacts with sarcoplasmic reticulum Ca(2+)-ATPase and modulates excitation-contraction coupling in the heart. Basic Res. Cardiol. 100, 250–262. doi: 10.1007/s00395-005-0518-7
Mattiazzi, A., Argenziano, M., Aguilar-Sanchez, Y., Mazzocchi, G., and Escobar, A. L. (2015). Ca2+ Sparks and Ca2+ waves are the subcellular events underlying Ca2+ overload during ischemia and reperfusion in perfused intact hearts. J. Mol. Cell Cardiol. 79, 69–78. doi: 10.1016/j.yjmcc.2014.10.011
Mattiazzi, A., and Kranias, E. G. (2014). The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 5:5. doi: 10.3389/fphar.2014.00005
Mazzocchi, G., Sommese, L., Palomeque, J., Felice, J. I., Di Carlo, M. N., Fainstein, D., et al. (2016). Phospholamban ablation rescues the enhanced propensity to arrhythmias of mice with CaMKII-constitutive phosphorylation of RyR2 at site S2814. J. Physiol. 594, 3005–3030. doi: 10.1113/JP271622
Meissner, G. (2004). Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 35, 621–628. doi: 10.1016/j.ceca.2004.01.015
Meissner, G. (2017). The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 149, 1065–1089. doi: 10.1085/jgp.201711878
Morris, T. E., and Sulakhe, P. V. (1997). Sarcoplasmic reticulum Ca(2+)-pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic. Biol. Med. 22, 37–47. doi: 10.1016/s0891-5849(96)00238-9
Most, P., Bernotat, J., Ehlermann, P., Pleger, S. T., Reppel, M., Borries, M., et al. (2001). S100A1: a regulator of myocardial contractility. Proc. Natl. Acad. Sci. U.S.A. 98, 13889–13894. doi: 10.1073/pnas.241393598
Most, P., Pleger, S. T., Volkers, M., Heidt, B., Boerries, M., Weichenhan, D., et al. (2004). Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J. Clin. Invest. 114, 1550–1563. doi: 10.1172/jci21454
Mundina de Weilenmann, C., Vittone, L., de Cingolani, G., and Mattiazzi, A. (1987). Dissociation between contraction and relaxation: the possible role of phospholamban phosphorylation. Basic Res. Cardiol. 82, 507–516. doi: 10.1007/bf01907220
Mundina-Weilenmann, C., Vittone, L., Ortale, M., de Cingolani, G. C., and Mattiazzi, A. (1996). Immunodetection of phosphorylation sites gives new insights into the mechanisms underlying phospholamban phosphorylation in the intact heart. J. Biol. Chem. 271, 33561–33567. doi: 10.1074/jbc.271.52.33561
Murayama, T., Ogawa, H., Kurebayashi, N., Ohno, S., Horie, M., and Sakurai, T. (2018). A tryptophan residue in the caffeine-binding site of the ryanodine receptor regulates Ca(2+) sensitivity. Commun. Biol. 1:98. doi: 10.1038/s42003-018-0103-x
Murphy, E., and Steenbergen, C. (2008). Ion transport and energetics during cell death and protection. Physiology 23, 115–123. doi: 10.1152/physiol.00044.2007
Nakayama, H., Chen, X., Baines, C. P., Klevitsky, R., Zhang, X., Zhang, H., et al. (2007). Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Invest. 117, 2431–2444. doi: 10.1172/jci31060
Nickel, A. G., Kohlhaas, M., Bertero, E., Wilhelm, D., Wagner, M., Sequeira, V., et al. (2019). CaMKII does not control mitochondrial Ca(2+) uptake in cardiac myocytes. J. Physiol. [Epub ahead of print].
Nicolaou, P., Hajjar, R. J., and Kranias, E. G. (2009a). Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J. Mol. Cell Cardiol. 47, 365–371. doi: 10.1016/j.yjmcc.2009.05.010
Nicolaou, P., Rodriguez, P., Ren, X., Zhou, X., Qian, J., Sadayappan, S., et al. (2009b). Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ. Res. 104, 1012–1020. doi: 10.1161/CIRCRESAHA.108.189811
Nomikos, M., Thanassoulas, A., Beck, K., Vassilakopoulou, V., Hu, H., Calver, B. L., et al. (2014). Altered RyR2 regulation by the calmodulin F90L mutation associated with idiopathic ventricular fibrillation and early sudden cardiac death. FEBS Lett. 588, 2898–2902. doi: 10.1016/j.febslet.2014.07.007
Nuss, H. B., Kaab, S., Kass, D. A., Tomaselli, G. F., and Marban, E. (1999). Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am. J. Physiol. 277, H80–H91. doi: 10.1152/ajpheart.1999.277.1.H80
Orchard, C. H., Eisner, D. A., and Allen, D. G. (1983). Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature 304, 735–738. doi: 10.1038/304735a0
Palomeque, J., Rueda, O. V., Sapia, L., Valverde, C. A., Salas, M., Petroff, M. V., et al. (2009). Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ. Res. 105, 1204–1212. doi: 10.1161/CIRCRESAHA.109.204172
Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. doi: 10.1038/ncb2868
Paolocci, N., Katori, T., Champion, H. C., St John, M. E., Miranda, K. M., Fukuto, J. M., et al. (2003). Positive inotropic and lusitropic effects of HNO/NO- in failing hearts: independence from beta-adrenergic signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 5537–5542. doi: 10.1073/pnas.0937302100
Paolocci, N., Saavedra, W. F., Miranda, K. M., Martignani, C., Isoda, T., Hare, J. M., et al. (2001). Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 10463–10468. doi: 10.1073/pnas.181191198
Pedrozo, Z., Sanchez, G., Torrealba, N., Valenzuela, R., Fernandez, C., Hidalgo, C., et al. (2010). Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim. Biophys. Acta 1802, 356–362. doi: 10.1016/j.bbadis.2009.12.005
Peng, W., Shen, H., Wu, J., Guo, W., Pan, X., Wang, R., et al. (2016). Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354:aah5324. doi: 10.1126/science.aah5324
Pereira, L., Matthes, J., Schuster, I., Valdivia, H. H., Herzig, S., Richard, S., et al. (2006). Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes Metab. Res. Rev. 55, 608–615. doi: 10.2337/diabetes.55.03.06.db05-1284
Pereira, L., Ruiz-Hurtado, G., Rueda, A., Mercadier, J. J., Benitah, J. P., and Gomez, A. M. (2014). Calcium signaling in diabetic cardiomyocytes. Cell Calcium 56, 372–380. doi: 10.1016/j.ceca.2014.08.004
Potenza, D. M., Janicek, R., Fernandez-Tenorio, M., Camors, E., Ramos-Mondragon, R., Valdivia, H. H., et al. (2019). Phosphorylation of the ryanodine receptor 2 at serine 2030 is required for a complete beta-adrenergic response. J. Gen. Physiol. 151, 131–145. doi: 10.1085/jgp.201812155
Qian, J., Vafiadaki, E., Florea, S. M., Singh, V. P., Song, W., Lam, C. K., et al. (2011). Small heat shock protein 20 interacts with protein phosphatase-1 and enhances sarcoplasmic reticulum calcium cycling. Circ. Res. 108, 1429–1438. doi: 10.1161/CIRCRESAHA.110.237644
Respress, J. L., van Oort, R. J., Li, N., Rolim, N., Dixit, S. S., deAlmeida, A., et al. (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 110, 1474–1483. doi: 10.1161/CIRCRESAHA.112.268094
Riojas-Hernandez, A., Bernal-Ramirez, J. D., Rodriguez-Mier, F. E., Morales-Marroquin, E. M., Dominguez-Barragan, C., Borja-Villa, I., et al. (2015). Enhanced oxidative stress sensitizes the mitochondrial permeability transition pore to opening in heart from zucker Fa/fa rats with type 2 diabetes. Life Sci. 141, 32–43. doi: 10.1016/j.lfs.2015.09.018
Rizzuto, R., and Pozzan, T. (2006). Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 86, 369–408. doi: 10.1152/physrev.00004.2005
Rodriguez, P., Bhogal, M. S., and Colyer, J. (2003). Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase. J. Biol. Chem. 278, 38593–38600. doi: 10.1074/jbc.m301180200
Sacchetto, R., Turcato, F., Damiani, E., and Margreth, A. (1999). Interaction of triadin with histidine-rich Ca(2+)-binding protein at the triadic junction in skeletal muscle fibers. J. Muscle Res. Cell Motil. 20, 403–415.
Said, M., Becerra, R., Palomeque, J., Rinaldi, G., Kaetzel, M. A., Diaz-Sylvester, P. L., et al. (2008). Increased intracellular Ca2+ and SR Ca2+ load contribute to arrhythmias after acidosis in rat heart. Role of Ca2+/calmodulin-dependent protein kinase II. Am. J. Physiol. Heart Circ. Physiol. 295, H1669–H1683. doi: 10.1152/ajpheart.00010.2008
Said, M., Becerra, R., Valverde, C. A., Kaetzel, M. A., Dedman, J. R., Mundina-Weilenmann, C., et al. (2011). Calcium-calmodulin dependent protein kinase II (CaMKII): a main signal responsible for early reperfusion arrhythmias. J. Mol. Cell Cardiol. 51, 936–944. doi: 10.1016/j.yjmcc.2011.08.010
Said, M., Vittone, L., Mundina-Weilenmann, C., Ferrero, P., Kranias, E. G., and Mattiazzi, A. (2003). Role of dual-site phospholamban phosphorylation in the stunned heart: insights from phospholamban site-specific mutants. Am. J. Physiol. Heart Circ. Physiol. 285, H1198–H1205.
Salama, G., Menshikova, E. V., and Abramson, J. J. (2000). Molecular interaction between nitric oxide and ryanodine receptors of skeletal and cardiac sarcoplasmic reticulum. Antioxid. Redox. Signal. 2, 5–16. doi: 10.1089/ars.2000.2.1-5
Salas, M. A., Valverde, C. A., Sanchez, G., Said, M., Rodriguez, J. S., Portiansky, E. L., et al. (2010). The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J. Mol. Cell Cardiol. 48, 1298–1306. doi: 10.1016/j.yjmcc.2009.12.015
Samso, M., Feng, W., Pessah, I. N., and Allen, P. D. (2009). Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 7:e85. doi: 10.1371/journal.pbio.1000085
Santana, L. F., Kranias, E. G., and Lederer, W. J. (1997). Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J. Physiol. 503(Pt 1), 21–29. doi: 10.1111/j.1469-7793.1997.021bi.x
Santulli, G., Lewis, D. R., and Marks, A. R. (2017a). Physiology and pathophysiology of excitation-contraction coupling: the functional role of ryanodine receptor. J. Muscle Res. Cell Motil. 38, 37–45. doi: 10.1007/s10974-017-9470-z
Santulli, G., Nakashima, R., Yuan, Q., and Marks, A. R. (2017b). Intracellular calcium release channels: an update. J. Physiol. 595, 3041–3051. doi: 10.1113/JP272781
Sato, D., Clancy, C. E., and Bers, D. M. (2017). Dynamics of sodium current mediated early afterdepolarizations. Heliyon 3:e00388. doi: 10.1016/j.heliyon.2017.e00388
Seidler, T., Loughrey, C. M., Zibrova, D., Kettlewell, S., Teucher, N., Kogler, H., et al. (2007). Overexpression of FK-506 binding protein 12.0 modulates excitation contraction coupling in adult rabbit ventricular cardiomyocytes. Circ. Res. 101, 1020–1029. doi: 10.1161/circresaha.107.154609
Shannon, T. R., Ginsburg, K. S., and Bers, D. M. (2000). Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys. J. 78, 334–343. doi: 10.1016/s0006-3495(00)76596-9
Shin, D. W., Ma, J., and Kim, D. H. (2000). The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca(2+) and interacts with triadin. FEBS Lett. 486, 178–182. doi: 10.1016/s0014-5793(00)02246-8
Sitsapesan, R., and Williams, A. J. (1994). Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+. J. Membr. Biol. 137, 215–226.
Sivakumaran, V., Stanley, B. A., Tocchetti, C. G., Ballin, J. D., Caceres, V., Zhou, L., et al. (2013). HNO enhances SERCA2a activity and cardiomyocyte function by promoting redox-dependent phospholamban oligomerization. Antioxid. Redox. Signal. 19, 1185–1197. doi: 10.1089/ars.2012.5057
Sobie, E. A., Song, L. S., and Lederer, W. J. (2005). Local recovery of Ca2+ release in rat ventricular myocytes. J. Physiol. 565(Pt 2), 441–447. doi: 10.1113/jphysiol.2005.086496
Sommese, L., Valverde, C. A., Blanco, P., Castro, M. C., Rueda, O. V., Kaetzel, M., et al. (2016). Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca(2+) release events in a rodent model of early stage diabetes: the arrhythmogenic substrate. Int. J. Cardiol. 202, 394–406. doi: 10.1016/j.ijcard.2015.09.022
Sondergaard, M. T., Liu, Y., Larsen, K. T., Nani, A., Tian, X., Holt, C., et al. (2017). The Arrhythmogenic calmodulin p.Phe142 leu mutation impairs C-domain Ca2+ binding but not calmodulin-dependent inhibition of the cardiac ryanodine receptor. J. Biol. Chem. 292, 1385–1395. doi: 10.1074/jbc.M116.766253
Spencer, C. I., and Sham, J. S. (2003). Effects of Na+/Ca2+ exchange induced by SR Ca2+ release on action potentials and afterdepolarizations in guinea pig ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 285, H2552–H2562.
Stange, M., Xu, L., Balshaw, D., Yamaguchi, N., and Meissner, G. (2003). Characterization of recombinant skeletal muscle (Ser-2843) and cardiac muscle (Ser-2809) ryanodine receptor phosphorylation mutants. J. Biol. Chem. 278, 51693–51702. doi: 10.1074/jbc.m310406200
Steenaart, N. A., Ganim, J. R., Di Salvo, J., and Kranias, E. G. (1992). The phospholamban phosphatase associated with cardiac sarcoplasmic reticulum is a type 1 enzyme. Arch. Biochem. Biophys. 293, 17–24. doi: 10.1016/0003-9861(92)90359-5
Stern, M. D., Kort, A. A., Bhatnagar, G. M., and Lakatta, E. G. (1983). Scattered-light intensity fluctuations in diastolic rat cardiac muscle caused by spontaneous Ca++-dependent cellular mechanical oscillations. J. Gen. Physiol. 82, 119–153. doi: 10.1085/jgp.82.1.119
Sun, J., Yamaguchi, N., Xu, L., Eu, J. P., Stamler, J. S., and Meissner, G. (2008). Regulation of the cardiac muscle ryanodine receptor by O(2) tension and S-nitrosoglutathione. Biochemistry 47, 13985–13990. doi: 10.1021/bi8012627
Suzuki, Y., Demoliere, C., Kitamura, D., Takeshita, H., Deuschle, U., and Watanabe, T. (1997). HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J. Immunol. 158, 2736–2744.
Szabadkai, G., Simoni, A. M., and Rizzuto, R. (2003). Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J. Biol. Chem. 278, 15153–15161. doi: 10.1074/jbc.m300180200
Tada, M., Kirchberger, M. A., and Katz, A. M. (1975). Phosphorylation of a 22,000-dalton component of the cardiac sarcoplasmic reticulum by adenosine 3’:5’-monophosphate-dependent protein kinase. J. Biol. Chem. 250, 2640–2647.
Talukder, M. A., Kalyanasundaram, A., Zhao, X., Zuo, L., Bhupathy, P., Babu, G. J., et al. (2007). Expression of SERCA isoform with faster Ca2+ transport properties improves postischemic cardiac function and Ca2+ handling and decreases myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 293, H2418–H2428.
Talukder, M. A., Kalyanasundaram, A., Zuo, L., Velayutham, M., Nishijima, Y., Periasamy, M., et al. (2008). Is reduced SERCA2a expression detrimental or beneficial to postischemic cardiac function and injury? evidence from heterozygous SERCA2a knockout mice. Am. J. Physiol. Heart Circ. Physiol. 294, H1426–H1434. doi: 10.1152/ajpheart.01016.2007
Terentyev, D., Gyorke, I., Belevych, A. E., Terentyeva, R., Sridhar, A., Nishijima, Y., et al. (2008). Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 103, 1466–1472. doi: 10.1161/CIRCRESAHA.108.184457
Timerman, A. P., Onoue, H., Xin, H. B., Barg, S., Copello, J., Wiederrecht, G., et al. (1996). Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J. Biol. Chem. 271, 20385–20391.
Valverde, C. A., Kornyeyev, D., Ferreiro, M., Petrosky, A. D., Mattiazzi, A., and Escobar, A. L. (2010). Transient Ca2+ depletion of the sarcoplasmic reticulum at the onset of reperfusion. Cardiovasc. Res. 85, 671–680. doi: 10.1093/cvr/cvp371
Valverde, C. A., Mazzocchi, G., Di Carlo, M. N., Ciocci Pardo, A., Salas, N., Ragone, M. I., et al. (2019). Ablation of phospholamban rescues reperfusion arrhythmias but exacerbates myocardium infarction in hearts with Ca2+/calmodulin kinase II constitutive phosphorylation of ryanodine receptors. Cardiovasc. Res. 115, 556–569. doi: 10.1093/cvr/cvy213
Valverde, C. A., Mundina-Weilenmann, C., Reyes, M., Kranias, E. G., Escobar, A. L., and Mattiazzi, A. (2006). Phospholamban phosphorylation sites enhance the recovery of intracellular Ca2+ after perfusion arrest in isolated, perfused mouse heart. Cardiovasc. Res. 70, 335–345. doi: 10.1016/j.cardiores.2006.01.018
van Oort, R. J., McCauley, M. D., Dixit, S. S., Pereira, L., Yang, Y., Respress, J. L., et al. (2010). Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 122, 2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298
Van Petegem, F. (2015). Ryanodine receptors: allosteric ion channel giants. J. Mol. Biol. 427, 31–53. doi: 10.1016/j.jmb.2014.08.004
Van Petegem, F. (2019). Slaying a giant: structures of calmodulin and protein kinase a bound to the cardiac ryanodine receptor. Cell Calcium 83:102079. doi: 10.1016/j.ceca.2019.102079
Venetucci, L. A., Trafford, A. W., and Eisner, D. A. (2007). Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ. Res. 100, 105–111. doi: 10.1161/01.res.0000252828.17939.00
Vila-Petroff, M., Salas, M. A., Said, M., Valverde, C. A., Sapia, L., Portiansky, E., et al. (2007). CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc. Res. 73, 689–698. doi: 10.1016/j.cardiores.2006.12.003
Vittone, L., Mundina-Weilenmann, C., Said, M., Ferrero, P., and Mattiazzi, A. (2002). Time course and mechanisms of phosphorylation of phospholamban residues in ischemia-reperfused rat hearts. dissociation of phospholamban phosphorylation pathways. J. Mol. Cell. Cardiol. 34, 39–50. doi: 10.1006/jmcc.2001.1488
Völkers, M., Loughrey, C. M., Macquaide, N., Remppis, A., DeGeorge, BR. Jr., Wegner, F. V., et al. (2007). S100A1 decreases calcium spark frequency and alters their spatial characteristics in permeabilized adult ventricular cardiomyocytes. Cell Calcium 41, 135–143. doi: 10.1016/j.ceca.2006.06.001
Volkers, M., Rohde, D., Goodman, C., and Most, P. (2010). S100A1: a regulator of striated muscle sarcoplasmic reticulum Ca2+ handling, sarcomeric, and mitochondrial function. J. Biomed. Biotechnol. 2010, 178614. doi: 10.1155/2010/178614
Wehrens, X. H., Lehnart, S. E., Huang, F., Vest, J. A., Reiken, S. R., Mohler, P. J., et al. (2003). FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 113, 829–840. doi: 10.1016/s0092-8674(03)00434-3
Wehrens, X. H., Lehnart, S. E., Reiken, S., Vest, J. A., Wronska, A., and Marks, A. R. (2006). Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc. Natl. Acad. Sci. U.S.A. 103, 511–518. doi: 10.1073/pnas.0510113103
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70.
Weiss, J. N., Garfinkel, A., Karagueuzian, H. S., Chen, P. S., and Qu, Z. (2010). Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm 7, 1891–1899. doi: 10.1016/j.hrthm.2010.09.017
Weiss, J. N., Nivala, M., Garfinkel, A., and Qu, Z. (2011). Alternans and arrhythmias: from cell to heart. Circ. Res. 108, 98–112. doi: 10.1161/CIRCRESAHA.110.223586
Whayne, T. F. Jr. (2018). Clinical Use of Digitalis: a State of the art review. Am. J. Cardiovasc. Drugs 18, 427–440. doi: 10.1007/s40256-018-0292-1
Wier, W. G., Cannell, M. B., Berlin, J. R., Marban, E., and Lederer, W. J. (1987). Cellular and subcellular heterogeneity of [Ca2+]i in single heart cells revealed by fura-2. Science 235, 325–328. doi: 10.1126/science.3798114
Wier, W. G., and Hess, P. (1984). Excitation-contraction coupling in cardiac Purkinje fibers. effects of cardiotonic steroids on the intracellular [Ca2+] transient, membrane potential, and contraction. J. Gen. Physiol. 83, 395–415. doi: 10.1085/jgp.83.3.395
Witcher, D. R., Kovacs, R. J., Schulman, H., Cefali, D. C., and Jones, L. R. (1991). Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 266, 11144–11152.
Xiao, B., Jiang, M. T., Zhao, M., Yang, D., Sutherland, C., Lai, F. A., et al. (2005). Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ. Res. 96, 847–855. doi: 10.1161/01.res.0000163276.26083.e8
Xiao, B., Zhong, G., Obayashi, M., Yang, D., Chen, K., Walsh, M. P., et al. (2006). Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase a activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem. J. 396, 7–16. doi: 10.1042/bj20060116
Xiao, J., Tian, X., Jones, P. P., Bolstad, J., Kong, H., Wang, R., et al. (2007). Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J. Biol. Chem. 282, 34828–34838. doi: 10.1074/jbc.m707423200
Xie, Y., Sato, D., Garfinkel, A., Qu, Z., and Weiss, J. N. (2010). So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys. J. 99, 1408–1415. doi: 10.1016/j.bpj.2010.06.042
Xu, K. Y., Zweier, J. L., and Becker, L. C. (1997). Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ. Res. 80, 76–81. doi: 10.1161/01.res.80.1.76
Xu, L., Eu, J. P., Meissner, G., and Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237. doi: 10.1126/science.279.5348.234
Xu, L., and Meissner, G. (1998). Regulation of cardiac muscle Ca2+ release channel by sarcoplasmic reticulum lumenal Ca2+. Biophys. J. 75, 2302–2312. doi: 10.1016/s0006-3495(98)77674-x
Yamaguchi, N., Xu, L., Pasek, D. A., Evans, K. E., Chen, S. R., and Meissner, G. (2005). Calmodulin regulation and identification of calmodulin binding region of type-3 ryanodine receptor calcium release channel. Biochemistry 44, 15074–15081. doi: 10.1021/bi051251t
Yang, D., Zhu, W. Z., Xiao, B., Brochet, D. X., Chen, S. R., Lakatta, E. G., et al. (2007). Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ. Res. 100, 399–407. doi: 10.1161/01.res.0000258022.13090.55
Yang, Y., Zhu, W. Z., Joiner, M. L., Zhang, R., Oddis, C. V., Hou, Y., et al. (2006). Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am. J. Physiol. Heart Circ. Physiol. 291, H3065–H3075.
Yellon, D. M., and Hausenloy, D. J. (2007). Myocardial reperfusion injury. N. Engl. J. Med. 357, 1121–1135.
Zahradnik, I., Gyorke, S., and Zahradnikova, A. (2005). Calcium activation of ryanodine receptor channels–reconciling RyR gating models with tetrameric channel structure. J. Gen. Physiol. 126, 515–527. doi: 10.1085/jgp.200509328
Zamparelli, C., Macquaide, N., Colotti, G., Verzili, D., Seidler, T., Smith, G. L., et al. (2010). Activation of the cardiac Na(+)-Ca(2+) exchanger by sorcin via the interaction of the respective Ca(2+)-binding domains. J. Mol. Cell Cardiol. 49, 132–141. doi: 10.1016/j.yjmcc.2010.03.003
Zhang, L., Kelley, J., Schmeisser, G., Kobayashi, Y. M., and Jones, L. R. (1997). Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 272, 23389–23397. doi: 10.1074/jbc.272.37.23389
Zhang, R., Khoo, M. S., Wu, Y., Yang, Y., Grueter, C. E., Ni, G., et al. (2005). Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 11, 409–417. doi: 10.1038/nm1215
Zorov, D. B., Filburn, C. R., Klotz, L. O., Zweier, J. L., and Sollott, S. J. (2000). Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014.
Keywords: sarcoplasmic reticulum Ca2+-ATPase, ryanodine receptor 2, arrhythimas, phospholamban, apoptosis, diabetic myocardiopathy, isquemia-reperfusión
Citation: Federico M, Valverde CA, Mattiazzi A and Palomeque J (2020) Unbalance Between Sarcoplasmic Reticulum Ca2 + Uptake and Release: A First Step Toward Ca2 + Triggered Arrhythmias and Cardiac Damage. Front. Physiol. 10:1630. doi: 10.3389/fphys.2019.01630
Received: 28 August 2019; Accepted: 24 December 2019;
Published: 23 January 2020.
Edited by:
Maria Fernandez-Velasco, La Paz University Hospital, SpainReviewed by:
Sandor Gyorke, The Ohio State University, United StatesAgustín Guerrero-Hernández, Center for Research and Advanced Studies (CINVESTAV), Mexico
Copyright © 2020 Federico, Valverde, Mattiazzi and Palomeque. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julieta Palomeque, anBhbG9tZXF1ZUBjaWNsYXBsYXRhLm9yZy5hcg==; cGEubG9tZXF1ZUBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work