 Raúl Sampieri
Raúl Sampieri Eridani Fuentes
Eridani Fuentes Jorge A. Sánchez
Jorge A. Sánchez
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 14 January 2020
Sec. Membrane Physiology and Membrane Biophysics
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01589
Voltage-dependent Ca2+ channels and store-operated Ca2+ channels (SOCs) are the major routes of Ca2+ entry into mammalian cells. Previously, we reported that pharmacological preconditioning (PPC) leads to a decrease in the amplitude of L-type calcium channel current in the heart. In this study, we examined PPC-associated changes in SOC function. We measured adult cardiomyocyte membrane currents using the whole-cell patch-clamp technique, and we evaluated reactive oxygen species (ROS) production and intracellular Ca2+ levels in cardiomyocytes using fluorescent probes. Diazoxide (Dzx) and thapsigargin (Tg) were used to induce PPC and to deplete internal stores of Ca2+, respectively. Ca2+ store depletion generated inward currents with strong rectification, which were suppressed by the SOC blocker GSK-7975-A. These currents were completely abolished by PPC, an effect that could be countered with 5-hydroxydecanoate (5-HD; a selective mitochondrial ATP-sensitive K+ channel blocker), an intracellular mitochondrial energizing solution, or Ni2+ [a blocker of sodium–calcium exchanger (NCX)]. Buffering of ROS and intracellular Ca2+ also prevented PPC effects on SOC currents. Refilling of intracellular stores was largely suppressed by PPC, as determined by measuring intracellular Ca2+ with a fluorescent Ca2+ indicator. These results indicate that influx of Ca2+ through SOCs is inhibited by their ROS and Ca2+-dependent inactivation during PPC and that NCX is a likely source of PPC-inactivating Ca2+. We further showed that NCX associates with Orai1. Down-regulation of SOCs by PPC may play a role in cardioprotection following ischemia–reperfusion.
Severe damage to heart muscle caused by ischemia and reperfusion (I/R) has been shown to involve Ca2+ overload during reperfusion (Murphy and Steenbergen, 2008). Mitochondria are key participants and regulators of myocardial injury during I/R. I-induced mitochondrial stress leads to the onset of mitochondrial permeability transition pore (MPTP) opening, release of cytochrome c through MPTPs, and, eventually, consequent cell death during reperfusion (Lesnefsky et al., 2017).
Ischemia and reperfusion injury can be mitigated by preconditioning with brief periods of ischemia (Murry et al., 1986) or by pharmacological preconditioning (PPC) with agents, such as diazoxide (Dzx), that open mitochondrial ATP-sensitive K+ channels (mitoKATPs) (Garlid et al., 1997; Pain et al., 2000). Blockade of mitoKATP channels prevents both ischemic preconditioning and PPC (Ardehali and O’Rourke, 2005; Halestrap et al., 2007). Pharmacological activation of mitoKATPs depends on reactive oxygen species (ROS) generated in mitochondria during preconditioning, which act to prevent mitochondrial Ca2+ overloading and MPTP opening (Lesnefsky et al., 2017). In addition to mitochondrial channels, L-type calcium channels play a role in PPC in adult rat cardiomyocytes (González et al., 2010). PPC reduces both Cav1.2 channel current amplitudes and action-potential produced surges in myoplasmic Ca2+ concentrations, which then attenuates I/R-induced damage (González et al., 2010). There has been little exploration of potential PPC-induced changes in the functions of other types of channels involved in Ca2+ homeostasis.
Ca2+ ions are critical mediators in cardiac excitation–contraction coupling, the process that enables the heart chambers to contract and relax. The development of contraction depends on a rise in myoplasmic Ca2+ concentration. During the cardiac action potential, the Ca2+ influx through depolarization-activated Cav1.2 channels in the cell’s plasma membrane triggers the release of Ca2+ from the sarcoplasmic reticulum (SR) (Bers, 2002). Subsequently, to enable relaxation, Ca2+ is removed from the cytosol via four transporters, namely the SR Ca2+ pump. the sarcolemmal Ca2+ ATPase, the sodium–calcium exchanger (NCX; Hinata et al., 2002), and the mitochondrial Ca2+ uniporter (Bers, 2002). Because NCX is electrogenic, it can transport Ca2+ out of or into (reverse mode) the myocyte, depending on the membrane potential (Bers, 2002). In the adult mammalian heart, the predominant two Ca2+ influx pathways are Cav1.2 channels and NCXs.
Sodium–calcium exchanger-mediated Ca2+ influx has a dual role in ischemic settings. That is, it has a deleterious role during I/R-induced damage as well as a beneficial role via triggering cardioprotective mechanisms during ischemic preconditioning (Castaldo et al., 2017). The mechanism by which NCX promotes ischemic tolerance is not yet known.
In non-excitable cells, Ca2+ influx is mediated primarily by a family of proteins known as store-operated Ca2+ channels (SOCs). As implied by their name, SOCs respond to intraluminal Ca2+ reductions in the endoplasmic reticulum. The stromal interaction molecule (STIM) and Orai proteins have been identified as Ca2+ sensors and store-operated channels, respectively. Their main role is to replenish internal Ca2+ stores (Prakriya and Lewis, 2015). In excitable cells, such as cardiomyocytes, store-operated Ca2+ entry (SOCE) has also been described (Hunton et al., 2004).
Although the role of SOCs in the adult heart is not well defined, SOC blockade has been shown to enhance functional recovery of heart muscle tissues following I/R (Liu et al., 2006; Collins et al., 2013), suggesting that SOCE may play a role in Ca2+ overload. In this study, we tested the hypotheses that (1) SOCE is downregulated during PPC, (2) downregulation of SOCE results from SOCs inactivation, and (3) that inactivation involves NCX and mitoKATP channels. Because PPC involves mitoKATP channel opening and increased ROS levels (González et al., 2010), and Orai1, but not Orai3, channels are inactivated by ROS (Bogeski et al., 2010), we examined how PPC cardiomyocyte physiology is affected by the ROS scavenger N-acetylcysteine (NAC). SOCs are inactivated by Ca2+ (Zweifach and Lewis, 1995). Thus, to test whether intracellular (cytosolic) Ca2+ concentration ([Ca2+]i) elevation contributes to SOC inactivation, in addition to ROS, we buffered myoplasmic Ca2+ with a Ca2+ chelator. SOC activation has been reported to require actively respiring mitochondria and mitochondrial Ca2+ uptake can be maintained when mitochondria are induced to remain in an energized state (Gilabert and Parekh, 2000; Glitsch et al., 2002). To test whether decreased SOC activation in PPC cardiomyocytes is related to de-energization of mitochondria, we examined whether the addition of a cocktail to the pipette solution that maintains mitochondria in an energized state restores SOC currents in PPC cardiomyocytes.
Experiments were conducted with male Wistar rats (300–350 g) employing protocols approved by the Division of Laboratory Animal Units and CINVESTAV-IPN, in compliance with federal law, federal statute, and Consejo Nacional de Ciencia y Tecnología regulations in Mexico. Rats were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneally), which was injected with sodium heparin (500 U/kg, intraperitoneally).
Ventricular myocytes were isolated as described previously (Narasimhan et al., 2018), with slight modifications. In brief, adult rat hearts were mounted in a Langendorff apparatus and perfused for 5 min at 37°C with Ca2+-free Tyrode’s solution containing 136 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose. Unless otherwise stated, all chemicals were purchased from Sigma–Aldrich (St. Louis, MO, United States). Hearts were recirculated for ∼60 min using Tyrode’s solution supplemented with 70 U/mL type II collagenase (Worthington, Lakewood, NJ, United States) and 0.5 mg/100 mL type XIV protease. Ventricles were minced and shaken two to three times at 2 × g for 7 min in the same solution. Dislodged cells were filtered through a cell strainer (100 mm nylon BD Falcon, Fisher Scientific, Waltham, MA, United States) and centrifuged at 72 × g for 2 min. The pellet was re-suspended in Tyrode’s solution and the cardiomyocytes thus harvested were used immediately.
We recorded membrane currents in dissociated adult rat ventricular myocytes using the whole-cell patch-clamp technique, as described previously (González et al., 2010). Currents were recorded using an Axopatch 200-A amplifier (Axon Instruments, Foster City, CA, United States). To measure membrane capacitance, 10 mV depolarizing pulses were applied. Current records lasting 100–300 s were digitized at a sampling interval of 120 ms via a Digidata interface (Axon Instruments, Foster City, CA, United States) at a 16-bit resolution. To measure the voltage dependence of membrane currents, ramps from +50 to −120 mv lasting 1 s were delivered every 10 s, and currents were sampled at 1-ms intervals. The holding potential (HP) was −80 mV. Data were analyzed using pCLAMP 8.0 (Axon Instruments, Foster City, CA, United States) and an in-house software. The standard pipette solution (pH 7.2) contained 137 mM cesium aspartate, 2 mM CsCl, 8 mM MgSO4, 1.8 mM MgCl2, 10 mM EGTA, and 15 mM HEPES. The bath solution (pH 7.4) contained 137 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose, 10 μM verapamil, 200 μM ouabain, and 10 mM HEPES. To deplete SR Ca2+ stores, we used the SR Ca2+-ATPase blocker thapsigargin (Tg) at a concentration of 2 μM from a 2-mM stock solution in dimethyl sulfoxide (DMSO). The ROS scavenger NAC was used at a concentration of 2 mM. NCX was blocked with 5 mM Ni2+ (Hinata et al., 2002). Orai1 channels were blocked with GSK-7975-A at a concentration of 10 μM that completely blocks Orai1/Orai3 channels (Derler et al., 2013).
Where indicated, the standard pipette solution was supplemented with a mitochondrial cocktail solution to maintain the mitochondria in an energized state (Gunter and Pfeiffer, 1990). This cocktail contained 2 mM pyruvic acid, 2 mM malic acid, 1 mM NaH2PO4, 0.5 mM cAMP, and 0.5 mM MgCl2. To observe the effect of intracellular Ca2+ on SOC inactivation, we added the cell-permeant Ca2+ chelator BAPTA-AM [1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) 30 μM, Molecular Probes/Thermo Fisher, Waltham, MA, United States, 30 min prior to electrophysiological recordings, as has been done previously (Yu et al., 2013).
Pharmacological preconditioning of cardiomyocytes was performed in Tyrode’s solution supplemented with 100 μM Dzx (Tocris Bioscience, Bristol, United Kingdom). Dzx was added from a 0.1-M stock solution in DMSO to achieve a final DMSO concentration <0.01%. At the concentration used in this study, Dzx is a selective opener of mitoKATP channels (Garlid et al., 1996). The selective mitoKATP channel inhibitor 5-hydroxydecanoate (5-HD; Hu et al., 1999) was used at a concentration of 100 μM. To characterize PPC effects on membrane currents, intact cardiomyocytes were incubated following the experimental protocols indicated in the figures and individual cardiomyocytes were patch-clamped to record whole-cell membrane currents. The experiments were performed at room temperature (23°C).
We measured [Ca2+]i from single cardiomyocytes as described previously (González et al., 2010) with minor modifications. Briefly, cardiomyocytes were loaded with the cell-permeant fluorescent Ca2+ indicator Fura-2 acetoxymethyl ester (AM; Molecular Probes/Thermo Fisher, Waltham, MA, United States) for 45 min at room temperature. Fura-2 AM was diluted in Tyrode’s solution to a final concentration of ∼5 μM from a stock solution in DMSO containing 9 mM Fura-2 AM and 25% w/v Pluronic F127 (Molecular Probes/Thermo Fisher, Waltham, MA, United States). The cells were washed in Tyrode’s solution and attached to laminin-coated coverslips for 20–30 min at room temperature before performing [Ca2+]i measurements. Ratiometric images of Fura-2 AM fluorescence were monitored with an Eclipse TE300 microscope (Nikon, Tokyo, Japan) equipped with a Polychrome V monochromator (TILL Photonics, Munich, Germany), allowing high speed changing between 340 and 380-nm excitation wavelengths. Fluorescence emissions were captured though a 510WB80 filter (Chroma Technology Corp., Bellows Falls, VT, United States) with an iXon EM + DU885 digital camera (Andor Technology, Belfast, United Kingdom). Image acquisition and ratio analysis were performed in Imaging Workbench 6.0 software (INDEC Biosystems, Los Altos, CA, United States). [Ca2+]i was estimated as the Fura-2 AM fluorescence ratio (340/380). Ca2+ SR stores were depleted with caffeine (10 mM) and Tg (2 μM). N-Methyl-D-glucamine replaced extracellular Na+ equimolarly (Na+-free solution).
Reactive oxygen species levels were measured as described by Narasimhan et al. (2018) with the cell-permeant fluorescent probe 5-(and 6)-chloromethyl-20,70 dichlorodihydrofluorescein diacetate acetylester (CM-H2DCFDA; Molecular Probes/Thermo Fisher, Waltham, MA, United States). Fluorescence (505-nm excitation and 545-nm emission) was measured in arbitrary units (AU) for 30 ms in user-defined segments of cardiomyocytes within images acquired at 5-min intervals. Dzx-group cardiomyocytes were treated the same as those in the control group, except that they were pre-incubated with Dzx (100 μM) for 90 min. For each experimental condition, ROS measurements were normalized to initial fluorescence values and fitted to a straight line.
To assess protection against I/R, hearts previously perfused in Tyrode’s solution with or without Dzx (100 μM) for 90 min were subjected to global ischemia for 30 min, followed by 2 h of reperfusion with the control solution. Hearts were dyed, as described elsewhere (González et al., 2010), with 1% 2,3,5-triphenyltetrazolium chloride (TTC) for 10 min, fixed overnight with paraformaldehyde (4%), cut into 500-μm slices, and photographed with a digital camera attached to a microscope (Olympus, Center Valley, PA, United States). TTC produces colored precipitates in the presence of dehydrogenase activity, which is severely reduced or non-existent in necrotic cells.
Membrane extracts from whole ventricles and ventricular cardiomyocytes were obtained. Isolated ventricular cardiomyocytes, preincubated in Tyrode or Dzx (PPC), were washed with ice cold PBS and sonicated (4 × 15 s) in buffer I containing (in mmol/L, pH = 7.5): 10 Tris–HCl, 1 Na+ vanadate, 1 phenylmethylsulfonyl fluoride (PMSF), 100 NaF, 1 EGTA, and a cocktail of protease inhibitors (1× ThermoFisher Scientific, Waltham, MA, United States). Buffer II containing (in mol/L, pH = 7.5): 10 Tris–HCl, 300 KCl, 1 Na+ vanadate, 1 PMSF, 100 NaF, 1 EGTA, 20% sucrose, and a cocktail of protease inhibitors (1× ThermoFisher Scientific, Waltham, MA, United States) was added and centrifugation at 10,000 × g for 10 min at 4°C followed. The supernatant was centrifuged at 100,000 × g for 1 h at 4°C. The pellet was washed and re-suspended in buffer III containing (in mmol/L, pH = 7.5): 50 Tris–HCl, 150 NaCl, 0.05% SDS, 1% TritonX-100, 0.5% deoxycholate with 0.5% CHAPS for IP:NCX1 and without CHAPS for IP:Orai1, and a cocktail of protease inhibitors (1× ThermoFisher Scientific, Waltham, MA, United States) and incubated for 30 min at 4°C. Whole ventricles were minced in liquid nitrogen, Buffers I and II were added (1v:1v) and the tissue was homogenized. Thereafter, an identical procedure used to obtain the membrane fraction from cardiomyocytes was followed. The membrane fraction from cardiomyocytes or ventricles was then used for immunoprecipitation experiments. Protein content was measured with Bradford assay. Solubilized proteins were pre-incubated with protein A/G-sepharose beads. 350–400 μg of precleared proteins were incubated with anti-NCX1 or anti-Orai1 antibodies overnight. Protein A/G-sepharose beads were added to the immuno-complex for 2 h. Control was performed by incubation with rabbit IgG. The beads were centrifuged, washed five times in PBS plus Tween 20 (0.15%), and warmed at 60°C for 10 min in Laemmli buffer to release the bound proteins. After SDS–PAGE and Western blotting, nitrocellulose membranes were blocked and treated with the first antibody against NCX1 (1:1000) or against Orai1 (1:500). Secondary antibodies were horseradish peroxidase conjugated anti-rabbit antibody (1:200,000; ThermoFisher Scientific, Waltham, MA, United States) or anti-mouse (1:400,000) horseradish peroxidase conjugated secondary antibody (ThermoFisher Scientific, Waltham, MA, United States) for experiments involving NCX1 and Orai1 primary antibodies, respectively. The blots were developed using a chemiluminescence detection system and were scanned at 600 dpi. The source of antibodies was: polyclonal anti-NCX1 antibody (Proteintech Group, Inc., Rosemont, IL, United States), and polyclonal anti-Orai1 (ThermoFisher Scientific, Waltham, MA, United States).
The data, which are expressed as means ± standard errors of the mean (SEMs), were tested for normal distribution, were analyzed with independent t-tests when two groups were compared or analyses of variance (ANOVAs), followed by multiple comparison Dunnett’s test, used to compare each of a number of treatments with a control. A significance criterion of p < 0.05 was used.
Depletion of cardiomyocyte intracellular Ca2+ stores with Tg led to the development of an inward current, which stabilized rapidly and was maintained throughout the recording period. The results from several experimental replicates are reported in Figure 1A. Membrane current values were normalized to unit capacitance to allow for comparisons among experiments. Mean membrane currents (±SEMs; n = 10) generated over time following addition of Tg are shown. To test whether SOCs may mediate the aforementioned currents, we blocked SOCs with 10 μM GSK-7975-A (Figure 1B). Depletion of intracellular Ca2+ stores with Tg produced inward currents (Figure 1A) that were completely inhibited by GSK-7975-A, followed by the development of a small outward current (Figure 1B).
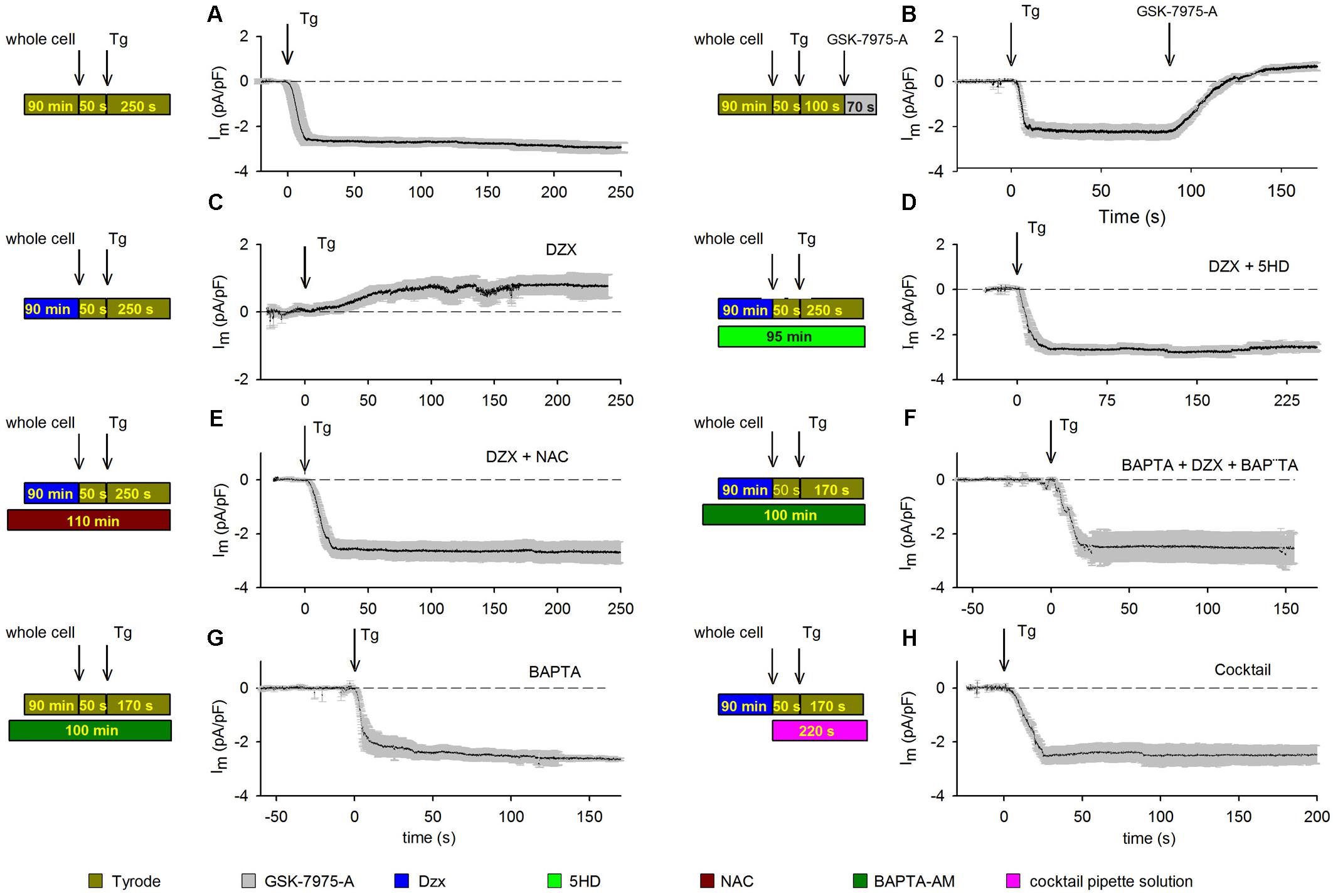
Figure 1. PPC inhibition of SOCs involves ROS and Ca2+-dependent inactivation. Visual summaries of experimental protocols are shown to the left of associated data graphs. Mean currents (±SEMs) are shown as a function of time and the HP was –80 mV in panels. (A) Membrane currents (n = 10) recorded from control cardiomyocytes treated with the SR Ca2+-ATPase blocker Tg (arrow). (B) The SOC inhibitor GSK-7975-A (arrow) blocked the inward current generated by Tg (n = 5); experiment otherwise as in panel (A). (C) Membrane currents recorded in Dzx-induced PPC in cardiomyocytes (n = 13). (D) Blockade of mitoKATP channels with pre/concurrent 5-HD treatment reinstated the inward-current response to Tg in PPC cardiomyocytes (n = 7). (E,F) Pre/concurrent treatment of PPC cardiomyocytes with the ROS scavenger NAC (n = 5) (E) or with BAPTA-AM (n = 5) (F) prevented the effect of Dzx on Tg-induced inward currents. (G) Pre/concurrent BAPTA-AM treatment of control cardiomyocytes (no PPC) did not alter currents alone (n = 6). (H) Supplementing the pipette solution with a cocktail that keeps mitochondria energized in cardiomyocytes pretreated with Dzx also prevented the effect of Dzx on Tg-induced inward currents (±SEM, n = 6).
We verified the ability of Dzx perfusion to produce PPC by exposing control and Dzx-pretreated hearts to severe ischemia (Supplementary Figure S1). Note that extensive areas of infarction are evident (light-colored areas) in an infarcted heart perfused with control solution before ischemia (representative cross-section in Supplementary Figure S1A). When an isolated heart was perfused with Dzx prior to ischemia, infarcted areas were greatly reduced (Supplementary Figure S1B). Similar results were obtained across experiments performed in triplicate.
Having confirmed our PPC model, we tested the effects of PPC on membrane currents. Tg-induced currents in cardiomyocytes were reversed with SOC blockade (Figures 1A,B). Dzx-induced PPC blocked Tg-generated currents; instead, we observed the development of a small outward current that did not inactivate (Figure 1C). In the presence of the mitoKATP channel inhibitor 5-HD, the PPC effect of Dzx was blocked and Tg-induced currents were re-enabled (Figure 1D), consistent with the hypothesis that mitoKATP channels are involved in the Tg response in PPC cardiomyocytes.
We tested the hypothesis that inhibition of SOC currents in PPC cardiomyocytes is due, at least in large part, to Orai1 inactivation by ROS. First, we verified that ROS production was indeed increased by Dzx treatment in cardiomyocytes under our experimental conditions (Supplementary Figure S2). Next, we found that in the presence of the ROS scavenger NAC, cardiomyocytes exhibit currents in response to Tg (Figure 1E) that were similar to that seen in control cardiomyocytes (Figures 1A,B), consistent with ROS involvement in the inhibition of SOCs in PPC cardiomyocytes.
In experiments testing whether PPC-related elevation of [Ca2+]i plays a role in SOC inactivation, we found that Tg-induced inward currents were not inhibited by PPC when myoplasmic Ca2+ was buffered with BAPTA-AM (Figure 1F). BAPTA-AM alone had negligible effects on currents generated in non-PPC cardiomyocytes (Figure 1G). When a cocktail that keeps mitochondria in an energized state was added to the pipette solution, SOC-mediated currents were not inhibited in PPC cardiomyocytes (Figure 1H).
Data obtained with treatments illustrated in Figures 1B–H were compared with results from control experiments (Figure 1A) and analyzed for significant differences at t = 150 s after the addition of Tg. Only the currents from PPC cardiomyocytes (Figure 1C) and those from cardiomyocytes treated with GSK-7975-A SOCs blocker (Figure 1B) had a statistically significant difference (p < 0.001).
When three ramps (a, b, and c) were delivered to a control cardiomyocyte held at −80 mV (Figure 2A) and Tg was added after the first ramp, an inward current developed but, in contrast to that in cardiomyocytes held continuously at −80 mV (Figure 1A), the current was not maintained and decreased spontaneously. Consistent with the results seen in Tg-treated PPC cardiomyocytes in the previous experiment (Figure 1C), no inward currents were recorded in the PPC cardiomyocytes, and ramps had little effect (Figure 2B).
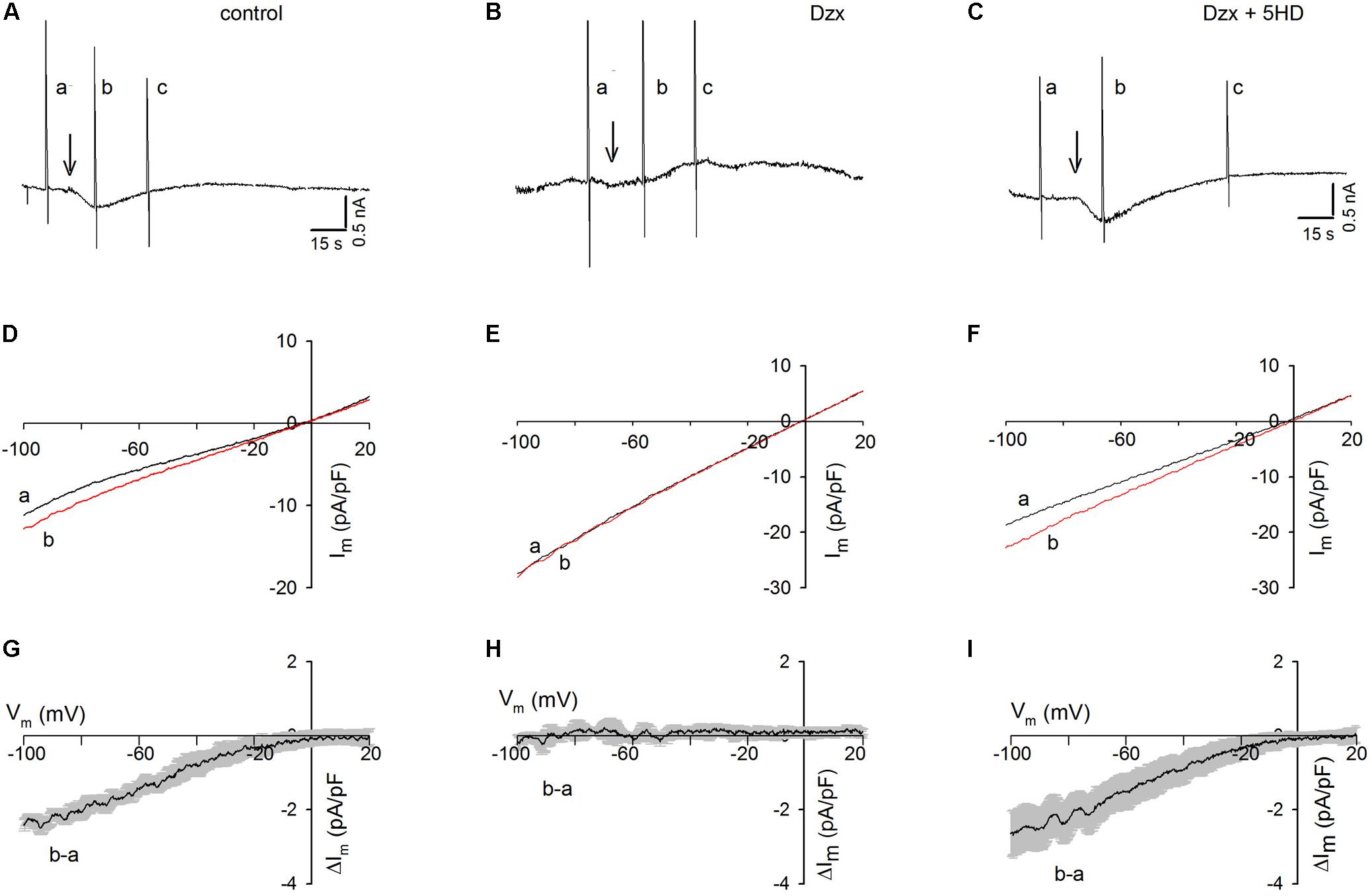
Figure 2. Dzx-induced PPC prevents SOC currents in cardiomyocytes upon application of voltage ramps. Black lines represent I–V curves during ramps before Tg; red lines represent I–V relationships during the first voltage ramp after Tg. (A–C) Continuous recordings of membrane currents under control (A), Dzx pretreatment (B), and Dzx + 5-HD pretreatment (C) conditions. Letters a–c indicate application of voltage ramps. Arrows indicate the addition of Tg. HP = –80 mV. (D–F) I–V curves correspond with the experiments illustrated above in panels (A–C). Notice that the addition of Tg increased membrane currents (D,F), as reflected by separation of the a and b curves, but had no effect when mitoKATP channels were opened by Dzx (E). (G–I) Mean values (±SEM, n = 4–8) of Tg-generated currents b–a as a function of membrane potential from experiments performed exactly as those in panels (D–F).
As shown in Figure 2C, in the presence of the mitoKATP channel inhibitor 5-HD, Tg administration resulted in the development of an inward current in PPC cardiomyocytes, which decreased spontaneously following the application of voltage ramps, as was seen in the non-PPC control cardiomyocytes (Figure 2A). The unsubtracted current–voltage (I–V) relationships during ramps of the experiments shown in Figures 2A–C are illustrated in Figures 2D–F. Estimates of the I–V relationships of Tg-activated SOC currents generated by subtracting currents during ramps before Tg from those recorded during ramps after Tg are summarized in Figures 2G–I.
In control experiments, Tg-induced SR Ca2+ depletion resulted in the activation of currents with strong inward rectification. In PPC cardiomyocytes, currents before and after Tg addition were almost identical, indicating complete inhibition of SOCs (Figure 2H), an effect that was blocked by 5-HD (Figure 2I).
To test whether changes in membrane potential during application of ramps would increase [Ca2+]i in the vicinity of SOCs, and thereby promote SOC inactivation, voltage ramps were delivered every 10 s, first in Tyrode’s solution and then after addition of Tg (representative experiment shown in Figure 3A). Tg generated an inward current like those seen in non-PPC control cardiomyocytes (Figure 2A), but that current was not maintained and it turned outward in response to successive ramps. When the same ramp protocol was applied to cardiomyocytes previously incubated in the Ca2+ chelator BAPTA-AM, the inward current did not become inactivated, affirming the involvement of Ca2+ in the inactivation (Figure 3B).
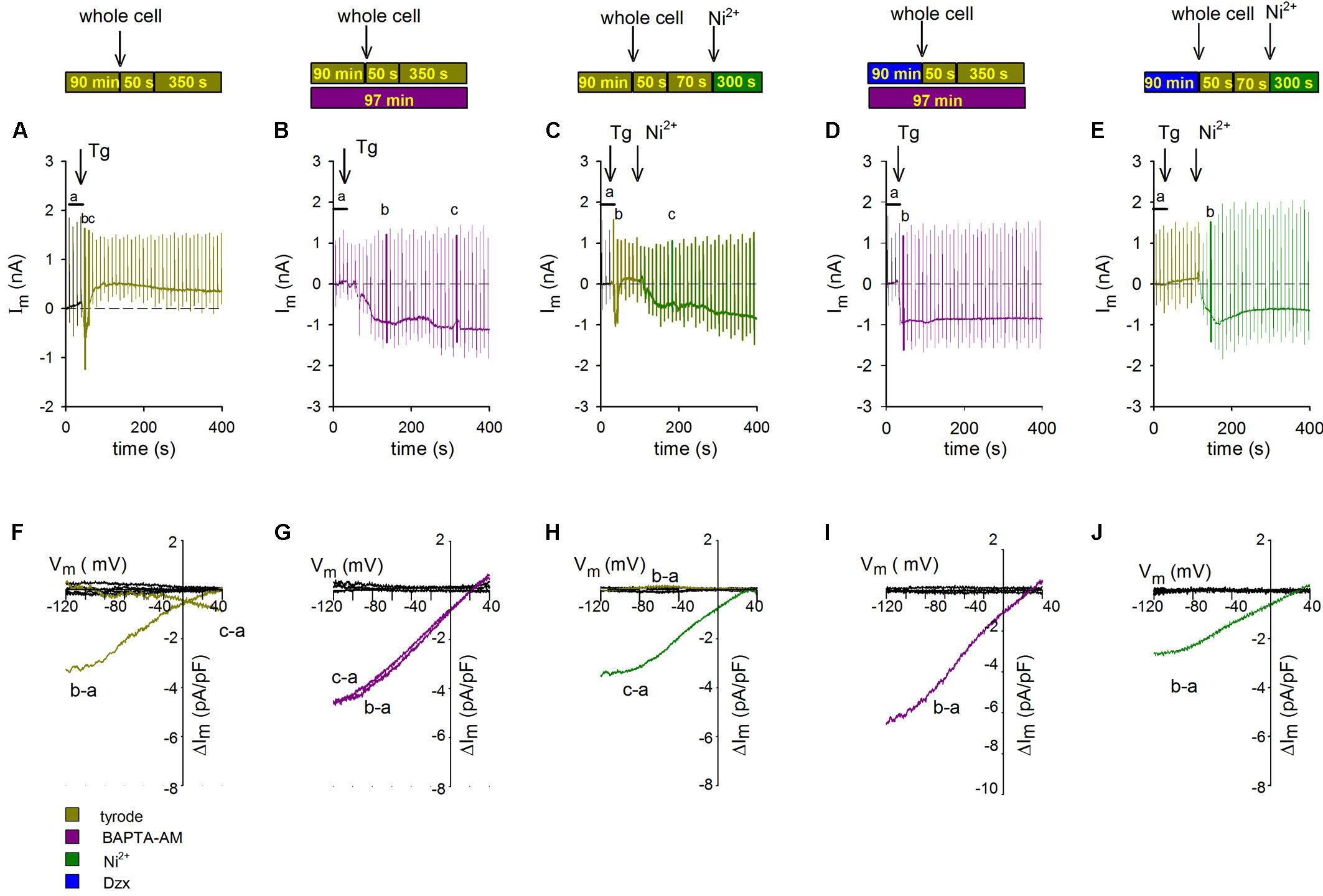
Figure 3. Inactivation of SOCs by voltage ramps is prevented by Ca2+ chelation with BAPTA-AM or NCX blockade with Ni2+. The experimental protocol employed for each experiment is summarized above the data associated with that experiment. Voltage ramps were delivered every 10 s with an HP of –80 mV. The additions of Tg and Ni2+ are indicated with arrows. (A–C) Representative continuous recordings of membrane currents in a control non-PPC cardiomyocyte (A), a non-PPC cardiomyocyte preincubated with BAPTA-AM (B), a non-PPC cardiomyocyte treated with Ni2+ (C), a PPC cardiomyocyte preincubated with BAPTA-AM (D), and a PPC cardiomyocyte treated with Tg and Ni2+ (E). (F–J) I–V relationships of subtracted currents from the experiments illustrated in panels (A–E). The average current upon control ramp application (indicated by the letter a) was subtracted from the currents upon the application of selected ramps, which are indicated by the letters b and c in panels (A–E). Average control currents during ramps (black lines) were also subtracted from each individual control current.
To test whether the source of Ca2+ mediating SOC inactivation upon application of ramps was NCX, we used the NCX blocker Ni2+. In cardiomyocytes held at −80 mV and administered a protocol identical to that used in the experiments shown in Figures 3A,B, Tg-induced inward currents through SOCs that inactivated completely in response to successive ramps, after which a small outward current developed. The addition of Ni2+ restored SOC currents (representative experiment in Figure 3C). The I–V relationships of SOCs from the experiments shown in Figures 3A–C, determined by subtracting the average of currents exhibited upon ramp application before Tg addition from selected currents generated upon ramp application after Tg, are illustrated in Figures 3F–H. In non-treated, non-PPC cardiomyocytes, only the first ramp generated a clear inward-rectifying SOC current (Figure 3F; replication of the effect in Figure 2G). In the presence of BAPTA-AM, SOC currents were generated in non-PPC cardiomyocytes throughout the whole recording period (Figure 3G). Meanwhile, in non-PPC cardiomyocytes, the addition of Ni2+ reversed SOC current inactivation generated by successive ramps (Figure 3H). Values of subtracted control currents were confirmed to be close to zero at all voltages. Similar results were obtained in five additional experiments.
Confirming the hypothesis that the Ca2+-dependent inactivation of SOCs is involved in the inhibition of currents upon ramp application in PPC cardiomyocytes, a ramp protocol applied in the presence of BAPTA-AM resulted in a non-inactivating inward currents (Figure 3D). In PPC cardiomyocytes, Tg-generated inward currents were absent (Figures 1C, 2B). SOC currents were restored in Tg-treated PPC cardiomyocytes following addition of Ni2+, suggesting that NCX may also participate in PPC-induced SOC inactivation (Figure 3E). The corresponding I–V relations of subtracted currents upon administration of ramps shown in Figures 3D,E are illustrated in Figures 3I,J, respectively. Similar results were obtained in five to eight additional experiments.
Given our finding that PPC cardiomyocytes with energized mitochondria exhibited currents that were similar to those in control cardiomyocytes under quiescent conditions (Figures 1A,H), we assessed whether this finding would hold true when voltage ramps are applied. As shown in Figure 4A, during continuous recording of membrane currents in a PPC cardiomyocyte, performed using the standard pipette solution, no inward currents were observed after Tg administration upon voltage ramp application (consistent with the results in Figure 2B). Conversely, in a parallel experiment performed with a mitochondria energizing solution, in place of the standard pipette solution, inward SOC currents were observed after Tg administration upon voltage ramp application (representative experiment in Figure 4B). Depletion of intracellular Ca2+ stores generated an inward current with an amplitude and time course that were similar to those recorded in non-PPC cardiomyocytes (Figure 2A). Average subtracted currents (b, a) from several experimental replicates recorded using the energizing pipette solution are presented as a function of membrane potential upon the application of voltage ramps to PPC cardiomyocytes in Figure 4C. These currents exhibited inward rectification with an I–V relationship similar to that observed for non-PPC cardiomyocytes (Figure 2G).
Figure 4. Activation of SOCs in PPC cardiomyocytes with energized mitochondria upon voltage ramp application. The experimental protocol employed for each experiment is summarized to the left of the data associated with that experiment. Ramps were applied at points a–c in the traces. Arrows show the addition of Tg; HP = –80 mV. (A,B) Representative continuous recordings of membrane currents in PPC cardiomyocytes (pretreated with Dzx) exposed to a standard pipette solution (A) or to a mitochondrion energizing cocktail pipette solution (B). (C) Mean current values (±SEMs, n = 6) of SOC currents b–a as a function of membrane potential from experiments performed as in panel (B).
To obtain further information on activation of SOCs in PPC cardiomyocytes, intracellular Ca2+ was measured. Figure 5A shows intracellular Ca2+ signals (visualized with Fura-2 AM and expressed as 340/380 ratios) associated with action potentials generated in a control cardiomyocyte. Figure 5C shows the change in intracellular Ca2+ for the same cardiomyocyte under conditions of emptied intracellular stores, as well as after Ca2+ was reintroduced into the external solution of the depleted cardiomyocyte, intracellular Ca2+ increased over time (Figure 5C; increase rate represented by the slope of the line fitted to the data points). When the same experiment was conducted in parallel in a PPC cardiomyocyte (Figures 5B,D), reintroduction of external Ca2+ produced only a subtle increase in intracellular Ca2+, indicating only a very low level of SOC activation (Figure 5D). Averaged results from several experiments like those shown in Figures 5C,D confirmed that the PPC-inducing Dzx treatment reduced the rate of intracellular Ca2+ rise (Figure 5E) and the peak amplitude of intracellular Ca2+ levels (Figure 5F) following reintroduction of external Ca2+, consistent with far fewer SOCs being open in PPC cardiomyocytes than in non-PPC control cardiomyocytes.
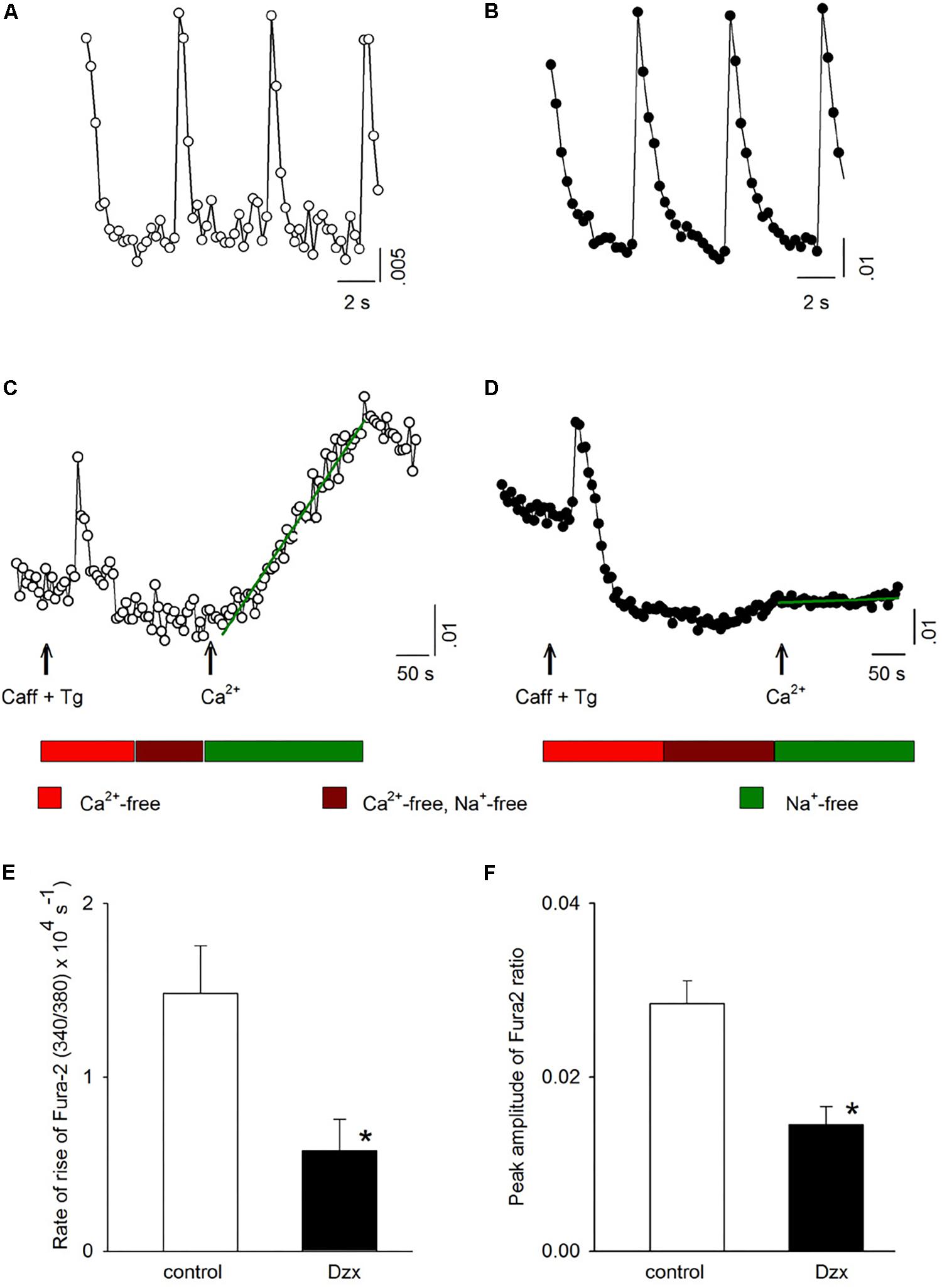
Figure 5. PPC with Dzx inhibits the influx of Ca2+ induced by SR depletion. (A,B) Ca2+ signals generated by action potentials in a control experiment (A) and in a PPC (Dzx-pretreated) cardiomyocyte (B). (C,D) Intracellular Ca2+ measurements from the same respective cardiomyocytes whose data are shown in panels (A) and (B), respectively, following the protocols illustrated below the associated data line graphs. The rate of the rise of intracellular Ca2+ associated with restoration of external Ca2+ was calculated on the basis of the slope of the straight lines superimposed over the data points following Ca2+ restoration (second arrows). (E,F) Mean values (±SEMs) of the rate of rise of intracellular Ca2+ (E) and corresponding intracellular Ca2+ level peaks (F) from replicates of the experiments shown in panels (C) and (D); (n = 10–14), ∗p < 0.05.
We next examined whether Orai1 interacts with NCX1. NCX1 was immunoprecipitated from membrane fractions of control and PPC preparations with the NCX1 antibody, followed by immunoblotting for Orai1. Upon immunoprecipitating NCX1, a band with a molecular weight corresponding to Orai1 (∼50 kDa) was detected in the membrane fraction of control and PPC ventricles (Figure 6B). We verified with the specific Orai1 antibody that these fractions contained Orai1 (Figure 6A). Solubilized proteins from membrane fractions of control and PPC ventricles were immunoblotted to detect NCX1. On SDS-PAGE, the NCX1 protein in the adult heart appears as a predominant band of ∼120 kDa (Shigekawa and Iwamoto, 2001), as shown in Figure 6C. After immunoprecipitating with the Orai1 antibody and immunoblotting with the NCX1 antibody, NCX1 was detected in control and PPC ventricles, confirming Orai1–NCX1 interaction (Figure 6D). Non-specific binding was determined using anti-rabbit IgG which failed to pull down any Orai1/NCX1 immunoreactivity (Figures 6B,D). Similar results were obtained in three experimental replicates.
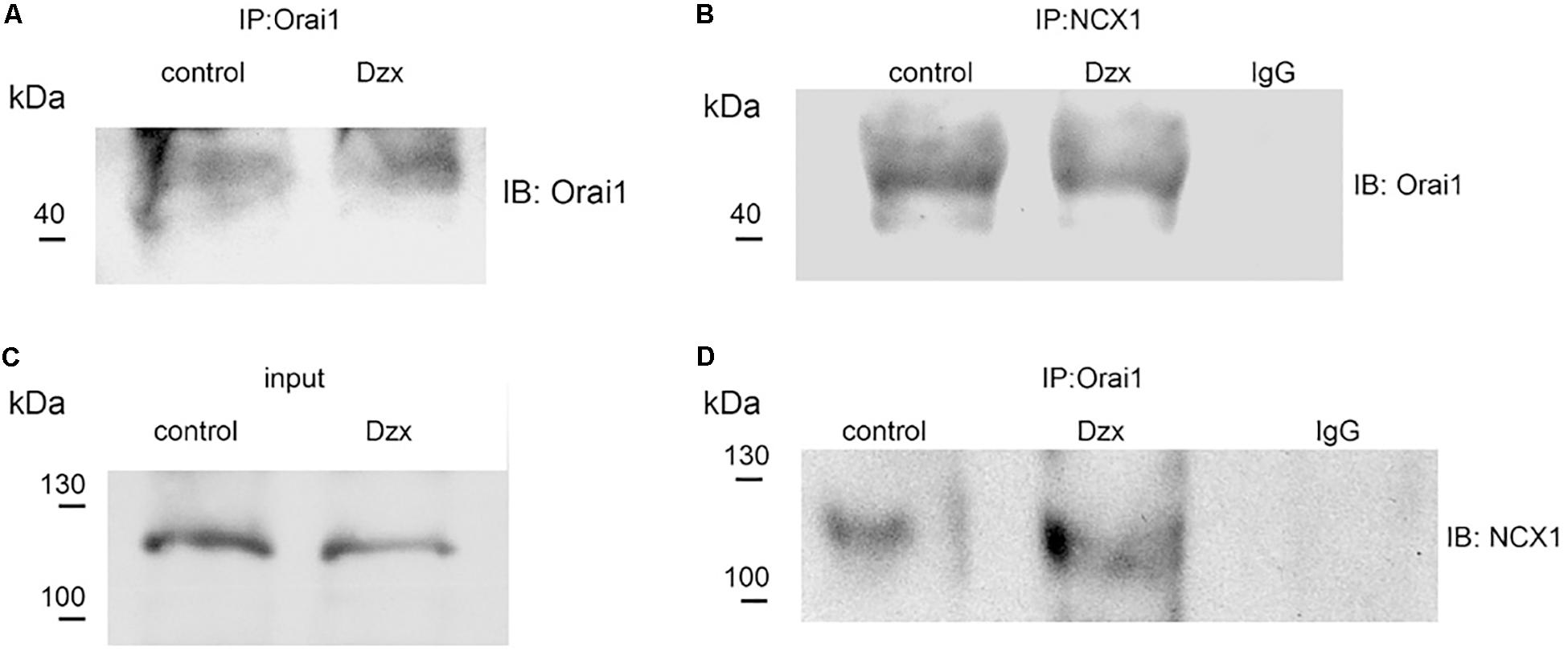
Figure 6. Orai1 associates with NCX1 in the adult rat heart. (A,B) Orai1 and NCX1 were immunoprecipitated from membrane fractions of control and PPC ventricles, respectively, and. immunoblotted with anti-Orai1 antibody. (C) NCX1 from membrane fractions of control and PPC ventricles was immunoblotted with anti-NCX1 antibody (input). (D) Orai1-immunoprecipitated proteins were immunoblotted with anti-NCX1 antibody.
Myocardial cell death associated with I/R is related to major alterations in Ca2+ handling (Murphy and Steenbergen, 2008). Protection against injury can be achieved by PPC (Sato et al., 2000; Wang et al., 2001; Pasdois et al., 2008; González et al., 2010), which involves opening of mitoKATP channels through the use of drugs. We present here, the novel observation that SOCs in adult cardiomyocytes are largely inhibited by PPC based on SOC current recordings via the whole-cell patch-clamp technique and quantitation of the Ca2+ levels with the Fura 2 AM indicator.
Store-operated Ca2+ entry, a ubiquitous and functionally important Ca2+ homeostasis pathway first recognized in non-excitable cells, was found to be present and functional in excitable cells as well (Prakriya and Lewis, 2015). The SOC Ca2+ sensor STIM1 is expressed in skeletal muscle, where its absence leads to decreased force, accelerated fatigue, and eventual perinatal death in mice due to skeletal myopathy (Stiber et al., 2008). STIM1 is also expressed in heart muscle (Ohba et al., 2009; Domínguez-Rodríguez et al., 2015; Saliba et al., 2015; Zhao et al., 2015) and its cardiac-restricted ablation leads to ventricular dilatation (Collins et al., 2014), reduced left ventricular contractility, and decreased shortening of isolated cardiomyocytes (Parks et al., 2016). Orai1 channels are also expressed in the heart muscle as revealed by western blotting (Ohba et al., 2009; Horton et al., 2014; Domínguez-Rodríguez et al., 2015; Saliba et al., 2015). In zebra fish, Orai1 deficiency in cardiomyocytes results in severe heart failure, reduced ventricular systolic function, and bradycardia (Völkers et al., 2012). SOCs in the heart have also been associated with cardiac disease, playing a role in the development of hypertrophy in neonatal and adult cardiomyocytes (Hunton et al., 2002; Ohba et al., 2009; Luo et al., 2012; Ji et al., 2017). Both STIM1 and Orai1 have been implicated in hypertrophic cardiac growth regulation. In mice, STIM1 overexpression enhances Ca2+ influx following Ca2+ depletion and also leads to the induction of the fetal gene program and sudden cardiac death or hypertrophy (Correll et al., 2015). Meanwhile, Orai1 knockdown has a beneficial effect, preventing hypertrophy in neonatal cardiomyocytes (Voelkers et al., 2010).
In heart muscle, SOCE differs over the course of development. In rat neonatal cardiomyocytes, SOCE is prominent after SR depletion, as evidenced by SOC current recordings and myoplasmic Ca2+ measurements (Ohba et al., 2009; Ji et al., 2017). In adult cardiomyocytes, SOCE and STIM1 expression are less prominent (Luo et al., 2012), though SOC currents showing inward rectification and corresponding changes in intracellular Ca2+ have been observed under Tg-induced SR depletion conditions (Hunton et al., 2004; Saliba et al., 2015; but see Zhao et al., 2015). Our experiments showed the presence of inward rectifying SOC currents as a result of SR Ca2+ depletion in adult cardiomyocytes but do not provide direct evidence regarding the identity of the channel responsible for SOCE.
Recently, canonical transient receptor potential channels (TRPC) have been implicated in SOCE in neonatal cardiomyocytes (He et al., 2017) and TRPC3/6 channels specifically have been associated with I/R tissue damage (He et al., 2017). Although a role of TRPC channels in our experiments cannot be ruled out entirely, several lines of evidence suggest that Orai1 channels play a leading role in PPC-associated SOC current inactivation. Firstly, we found that SOC currents were completely suppressed by GSK-7975-A, which, at the concentration used, produces a complete block of Orai1 and Orai3 channels (Derler et al., 2013; Molnár et al., 2016) while most TRP channels (IC50 > 10 μM) remain unblocked at this concentration (Derler et al., 2013). Secondly, the TRP blocker SKF-96365 has no effect on membrane currents in Tg-treated adult cardiomyocytes (Domínguez-Rodríguez et al., 2015). Thirdly, our experiments revealed that SOC currents were inactivated by an elevation in ROS levels, and Orai1 channels are inhibited by oxidation (Bogeski et al., 2010). Several members of the TRP channel family are activated by ROS, including TRPC3 and TRPC6 (Poteser et al., 2006; Ma et al., 2016). Additional evidence pointing to the Orai1 channel as the target of PPC is provided below.
In the present study, we confirmed that ROS production increases in response to PPC and found that inhibition of SOC currents can be overcome by administration of the general antioxidant NAC, which reacts with hydroxyl radicals and H2O2 (Aruoma et al., 1989), suggesting that PPC inhibits SOCs via ROS. MitoKATP channel opening has been shown previously to modulate the mitochondrial redox state and mitochondrial ROS production (Liu et al., 1998; Pasdois et al., 2008; González et al., 2010).
In addition to their well-studied involvement in pathological processes, ROS are also important for normal physiological phenomena (Dröge, 2002), including preconditioning (González et al., 2010). ROS have been shown to reduce Orai1 channel activity in T lymphocytes and Orai1 redox sensitivity has been shown to depend mainly on the extracellular reactive Cys195 locus, which is present in Orai1 but absent in Orai3 (Bogeski et al., 2010; Saul et al., 2016), though both isoforms are expressed in cardiomyocytes (Saliba et al., 2015). Meanwhile, Orai2 has an extremely low level of expression in heart tissues (Saliba et al., 2015). Together, such findings support a tentative conclusion that Orai1 channels are the primary, if not the sole, targets of ROS during PPC.
Because SOCs are strongly inactivated by Ca2+ (Prakriya and Lewis, 2015), the extent and duration of Ca2+ entry through SOCs depend on [Ca2+]i in their microdomains. Ca2+-dependent inactivation of SOCs develops over a period of seconds (Zweifach and Lewis, 1995). Among the three Orai isoforms that have been characterized, only Orai1 shows significant Ca2+-dependent inactivation (Lis et al., 2007). In our experiments, SOC currents remained active when cardiomyocytes were continuously held at −80 mV. However, when voltage ramps were applied, currents were inactivated within seconds, an effect that could be prevented by Ca2+ buffering, consistent with the involvement of Orai1 channels in the generation of SOC currents.
The present finding showing that NCX blockade with Ni2+ produced partial restoration of SOC currents in Tg-treated non-PPC cardiomyocytes suggests that the Ca2+ that inactivates SOCs upon ramp application is likely provided via NCX operating in the reverse mode (Khananshvili, 2014). Ca2+ influx via NCX is expected to inactivate Orai1 channels in their proximity, providing a functional link between NCX and SOCs. In PPC cardiomyocytes, we found that Tg-generated currents were restored by Ni2+, suggesting that NCX also plays a role in the inactivation of SOCs by PPC. The formation of a complex between Orai1 and NCX in the plasma membrane observed in our co-immunoprecipitation experiments further supports the notion that these proteins may be associated in a cardiac signaling complex promoting inactivation of SOCE during PPC. Because SOC inactivation reduces Ca2+ influx into cardiomyocytes, it could help to attenuate Ca2+ overload and thereby provide protection from I/R damage. In agreement with this possibility, inhibition of NCX has been shown to abolish ischemic tolerance in the heart induced by either the volatile anesthetic sevoflurane or ischemic preconditioning (Bouwman et al., 2006; Castaldo et al., 2017).
Our experiments suggest that a decreased Ca2+ buffering capacity by mitochondria during PPC contributes to the promotion of SOC inactivation. Mitochondria are active participants in intracellular Ca2+ signaling and oppose slow SOC inactivation by buffering Ca2+ and, thus, competing with SOC inactivation sites for Ca2+ (Gilabert and Parekh, 2000; Prakriya and Lewis, 2015). In non-excitable cells, mitochondria located near SOCs at the plasma membrane take up considerable amounts of Ca2+ through the mitochondrial Ca2+ uniporter and regulate Ca2+ microdomains (Hoth et al., 1997; Gilabert and Parekh, 2000; Gilabert et al., 2001). Consistent with this role of mitochondria, it has been reported that Ca2+ influx through SOCs in Jurkat cells is reduced when mitochondrial buffering capacity is compromised by mitochondrial membrane depolarization following Dzx-induced mitoKATP channel opening (Holmuhamedov et al., 1999, 2002; Valero et al., 2008) or when mitochondria are in a de-energized state (Hoth et al., 1997, 2000).
The use of an energizing cocktail solution in the patch pipette delays and attenuates slow SOC inactivation (Gilabert and Parekh, 2000). In PPC cardiomyocytes, we noted that with use of a similar pipette solution, SOC currents recovered from inactivation when held at −80 mV, even with the application of voltage ramps. Therefore, it is likely that mitochondria are also essential for SOCE activation in this preparation. In agreement with this possibility, mitochondria have been reported to occupy at least 30% of the cell volume in adult cardiomyocytes, and a significant fraction of mitochondria is located in the subsarcolemmal region (Piquereau et al., 2013), where they are able to interact with membrane channels. In fact, previous work has shown that mitochondria help to shape Ca2+-dependent inactivation of Cav1.2 channels in adult cardiomyocytes (Sánchez et al., 2001).
The SOC inhibitor glucosamine (Hunton et al., 2002; Nagy et al., 2006; Collins et al., 2013) has been shown to provide protection against I/R injury (Liu et al., 2006), suggesting that SOCs play a role in Ca2+ overload (Collins et al., 2013). A potential role for SOCs in I/R is further supported by the observation that SR Ca2+ contents are markedly depleted in intact hearts subjected to I/R (Valverde et al., 2010). The ensuing SR Ca2+ depletion would be expected to activate SOCE.
The main findings of this work are that PPC results in a large inhibition of SOC currents upon depletion of SR, and that this effect is due to ROS and Ca2+-dependent channel inactivation. Additionally, we showed that SOC currents can be restored by maintaining mitochondria in an energized state, which indicates that mitochondria play a key role in these observations, and that the NCX contributes as a source of channel-inactivating Ca2+. Hence, this study suggests that PPC mediated protection against I/R damage may be explained, in part, by decreased SOCE as a result of the largely abrogated influx of Ca2+ through SOCs. Although the present experiments were performed in single cells, it is plausible that PPC would produce similar changes in intact hearts. Future research is needed to test this prediction directly. Given the clinical importance of protecting the heart from I/R damage, a detailed understanding of the cellular mechanisms underlying PPC is relevant to the development of cardioprotective therapies.
The datasets generated for this study are available on request to the corresponding author.
The animal study was reviewed and approved by the Division of Laboratory Animal Units, CINVESTAV.
JS designed the study. JS, RS, EC, and MG planned the experiments. RS performed the patch-clamp experiments and ROS measurements. EF and MG performed the Ca2+ experiments. EC performed the co-immunoprecipitation experiments. AH assisted with the isolation of cardiomyocytes. JS and MG contributed reagents, material, and acquired financial support for the project. JS analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
This study was funded in part by the Consejo Nacional de Ciencia y Tecnología (CONACYT) (Grant Numbers 284053 to JS and 0250937 to MG) and by the SEP-Cinvestav (Grant Number 53-2018 to JS). RS and EF were supported by fellowships from CONACYT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Ivonne Lezama and Wilibaldo Orea for their assistance at all experimental stages.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.01589/full#supplementary-material
FIGURE S1 | Pharmacological preconditioning and cardioprotection. (A,B) Cross-sections of isolated ventricles under conditions indicated below panels. Light areas represent areas of infarction after severe ischemia.
FIGURE S2 | Pharmacological preconditioning and ROS production. The graph shows mean values (±SEM) of ROS production in control (open circles, n = 17) and PPC cardiomyocytes (filled circles, n = 10) as a function of time. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Ardehali, H., and O’Rourke, B. (2005). Mitochondrial K(ATP) channels in cell survival and death. J. Mol. Cell. Cardiol. 39, 7–16. doi: 10.1016/j.yjmcc.2004.12.003
Aruoma, O. I., Halliwell, B., Hoey, B. M., and Butler, J. (1989). The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 6, 593–597. doi: 10.1016/0891-5849(89)90066-x
Bers, D. M. (2002). Cardiac excitation–contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bogeski, I., Kummerow, C., Al-Ansary, D., Schwarz, E. C., Koehler, R., Kozai, D., et al. (2010). Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci. Signal. 3:ra24. doi: 10.1126/scisignal.2000672
Bouwman, R. A., Salic, K., Padding, F. G., Eringa, E. C., van Beek-Harmsen, B. J., Matsuda, T., et al. (2006). Cardioprotection via activation of protein kinase C-delta depends on modulation of the reverse mode of the Na+/Ca2+ exchanger. Circulation 114, I226–I232. doi: 10.1161/CIRCULATIONAHA.105.000570
Castaldo, P., Macrì, M. L., Lariccia, V., Matteucci, A., Maiolino, M., Gratteri, S., et al. (2017). Na+/Ca2+ exchanger 1 inhibition abolishes ischemic tolerance induced by ischemic preconditioning in different cardiac models. Eur. J. Pharmacol. 794, 246–256. doi: 10.1016/j.ejphar.2016.11.045
Collins, H. E., He, L., Zou, L., Qu, J., Zhou, L., Litovsky, S. H., et al. (2014). Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am. J. Physiol. Heart Circ. Physiol. 306, H1231–H1239. doi: 10.1152/ajpheart.00075.2014
Collins, H. E., Zhu-Mauldin, X., Marchase, R. B., and Chatham, J. C. (2013). STIM1/Orai1-mediated SOCE: current perspectives and potential roles in cardiac function and pathology. Am. J. Physiol. Heart Circ. Physiol. 305, H446–H458. doi: 10.1152/ajpheart.00104.2013
Correll, R. N., Goonasekera, S. A., van Berlo, J. H., Burr, A. R., Accornero, F., Zhang, H., et al. (2015). STIM1 elevation in the heart results in aberrant Ca2+ handling and cardiomyopathy. J. Mol. Cell. Cardiol. 87, 38–47. doi: 10.1016/j.yjmcc.2015.07.032
Derler, I., Schindl, R., Fritsch, R., Heftberger, P., Riedl, M. C., Begg, M., et al. (2013). The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 53, 139–151. doi: 10.1016/j.ceca.2012.11.005
Domínguez-Rodríguez, A., Ruiz-Hurtado, G., Sabourin, J., Gómez, A. M., Alvarez, J. L., and Benitah, J.-P. (2015). Proarrhythmic effect of sustained EPAC activation on TRPC3/4 in rat ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 87, 74–78. doi: 10.1016/j.yjmcc.2015.07.002
Dröge, W. (2002). Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95. doi: 10.1152/physrev.00018.2001
Garlid, K. D., Paucek, P., Yarov-Yarovoy, V., Murray, H. N., Darbenzio, R. B., D’Alonzo, A. J., et al. (1997). Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ. Res. 81, 1072–1082. doi: 10.1161/01.res.81.6.1072
Garlid, K. D., Paucek, P., Yarov-Yarovoy, V., Sun, X., and Schindler, P. A. (1996). The mitochondrial KATP channel as a receptor for potassium channel openers. J. Biol. Chem. 271, 8796–8799.
Gilabert, J. A., Bakowski, D., and Parekh, A. B. (2001). Energized mitochondria increase the dynamic range over which inositol 1,4,5-trisphosphate activates store-operated calcium influx. EMBO J. 20, 2672–2679. doi: 10.1093/emboj/20.11.2672
Gilabert, J. A., and Parekh, A. B. (2000). Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J. 19, 6401–6407. doi: 10.1093/emboj/19.23.6401
Glitsch, M. D., Bakowski, D., and Parekh, A. B. (2002). Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J. 21, 6744–6754. doi: 10.1093/emboj/cdf675
González, G., Zaldívar, D., Carrillo, E., Hernández, A., García, M., and Sánchez, J. (2010). Pharmacological preconditioning by diazoxide downregulates cardiac L-type Ca(2+) channels. Br. J. Pharmacol. 161, 1172–1185. doi: 10.1111/j.1476-5381.2010.00960.x
Gunter, T. E., and Pfeiffer, D. R. (1990). Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 258, C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755
Halestrap, A. P., Clarke, S. J., and Khaliulin, I. (2007). The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta 1767, 1007–1031. doi: 10.1016/j.bbabio.2007.05.008
He, X., Li, S., Liu, B., Susperreguy, S., Formoso, K., Yao, J., et al. (2017). Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc. Natl. Acad. Sci. U.S A. 114, E4582–E4591. doi: 10.1073/pnas.1621384114
Hinata, M., Yamamura, H., Li, L., Watanabe, Y., Watano, T., Imaizumi, Y., et al. (2002). Stoichiometry of Na+-Ca2+ exchange is 3:1 in guinea-pig ventricular myocytes. J. Physiol. 545, 453–461. doi: 10.1113/jphysiol.2002.025866
Holmuhamedov, E., Lewis, L., Bienengraeber, M., Holmuhamedova, M., Jahangir, A., and Terzic, A. (2002). Suppression of human tumor cell proliferation through mitochondrial targeting. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 16, 1010–1016. doi: 10.1096/fj.01-0996com
Holmuhamedov, E. L., Wang, L., and Terzic, A. (1999). ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J. Physiol. 519(Pt 2), 347–360. doi: 10.1111/j.1469-7793.1999.0347m.x
Horton, J. S., Buckley, C. L., Alvarez, E. M., Schorlemmer, A., and Stokes, A. J. (2014). The calcium release-activated calcium channel Orai1 represents a crucial component in hypertrophic compensation and the development of dilated cardiomyopathy. Channels Austin Tex 8, 35–48. doi: 10.4161/chan.26581
Hoth, M., Button, D. C., and Lewis, R. S. (2000). Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 97, 10607–10612. doi: 10.1073/pnas.180143997
Hoth, M., Fanger, C. M., and Lewis, R. S. (1997). Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J. Cell Biol. 137, 633–648. doi: 10.1083/jcb.137.3.633
Hu, H., Sato, T., Seharaseyon, J., Liu, Y., Johns, D. C., O’Rourke, B., et al. (1999). Pharmacological and histochemical distinctions between molecularly defined sarcolemmal KATP channels and native cardiac mitochondrial KATP channels. Mol. Pharmacol. 55, 1000–1005. doi: 10.1124/mol.55.6.1000
Hunton, D. L., Lucchesi, P. A., Pang, Y., Cheng, X., Dell’Italia, L. J., and Marchase, R. B. (2002). Capacitative calcium entry contributes to nuclear factor of activated T-cells nuclear translocation and hypertrophy in cardiomyocytes. J. Biol. Chem. 277, 14266–14273. doi: 10.1074/jbc.M107167200
Hunton, D. L., Zou, L., Pang, Y., and Marchase, R. B. (2004). Adult rat cardiomyocytes exhibit capacitative calcium entry. Am. J. Physiol. Heart Circ. Physiol. 286, H1124–H1132. doi: 10.1152/ajpheart.00162.2003
Ji, Y., Guo, X., Zhang, Z., Huang, Z., Zhu, J., Chen, Q.-H., et al. (2017). CaMKIIδ meditates phenylephrine induced cardiomyocyte hypertrophy through store-operated Ca2+ entry. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 27, 9–17. doi: 10.1016/j.carpath.2016.11.004
Khananshvili, D. (2014). Sodium-calcium exchangers (NCX): molecular hallmarks underlying the tissue-specific and systemic functions. Pflugers Arch. 466, 43–60. doi: 10.1007/s00424-013-1405-y
Lesnefsky, E. J., Chen, Q., Tandler, B., and Hoppel, C. L. (2017). Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu. Rev. Pharmacol. Toxicol. 57, 535–565. doi: 10.1146/annurev-pharmtox-010715-103335
Lis, A., Peinelt, C., Beck, A., Parvez, S., Monteilh-Zoller, M., Fleig, A., et al. (2007). CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. CB 17, 794–800. doi: 10.1016/j.cub.2007.03.065
Liu, J., Pang, Y., Chang, T., Bounelis, P., Chatham, J. C., and Marchase, R. B. (2006). Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J. Mol. Cell. Cardiol. 40, 303–312. doi: 10.1016/j.yjmcc.2005.11.003
Liu, Y., Sato, T., O’Rourke, B., and Marban, E. (1998). Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation 97, 2463–2469. doi: 10.1161/01.cir.97.24.2463
Luo, X., Hojayev, B., Jiang, N., Wang, Z. V., Tandan, S., Rakalin, A., et al. (2012). STIM1-dependent store-operated Ca2+ entry is required for pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 52, 136–147. doi: 10.1016/j.yjmcc.2011.11.003
Ma, R., Chaudhari, S., and Li, W. (2016). Canonical transient receptor potential 6 channel: a new target of reactive oxygen species in renal physiology and pathology. Antioxid. Redox Signal. 25, 732–748. doi: 10.1089/ars.2016.6661
Molnár, T., Yarishkin, O., Iuso, A., Barabas, P., Jones, B., Marc, R. E., et al. (2016). Store-Operated Calcium Entry in Müller Glia Is Controlled by Synergistic Activation of TRPC and Orai Channels. J. Neurosci. Off. J. Soc. Neurosci. 36, 3184–3198. doi: 10.1523/JNEUROSCI.4069-15.2016
Murphy, E., and Steenbergen, C. (2008). Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 88, 581–609. doi: 10.1152/physrev.00024.2007
Murry, C. E., Jennings, R. B., and Reimer, K. A. (1986). Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136. doi: 10.1161/01.cir.74.5.1124
Nagy, T., Champattanachai, V., Marchase, R. B., and Chatham, J. C. (2006). Glucosamine inhibits angiotensin II-induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-linked N-acetylglucosamine. Am. J. Physiol. Cell Physiol. 290, C57–C65. doi: 10.1152/ajpcell.00263.2005
Narasimhan, G., Carrillo, E. D., Hernández, A., García, M. C., and Sánchez, J. A. (2018). Protective action of diazoxide on isoproterenol-induced hypertrophy is mediated by reduction in MicroRNA-132 expression. J. Cardiovasc. Pharmacol. 72, 222–230. doi: 10.1097/FJC.0000000000000619
Ohba, T., Watanabe, H., Murakami, M., Sato, T., Ono, K., and Ito, H. (2009). Essential role of STIM1 in the development of cardiomyocyte hypertrophy. Biochem. Biophys. Res. Commun. 389, 172–176. doi: 10.1016/j.bbrc.2009.08.117
Pain, T., Yang, X. M., Critz, S. D., Yue, Y., Nakano, A., Liu, G. S., et al. (2000). Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ. Res. 87, 460–466. doi: 10.1161/01.res.87.6.460
Parks, C., Alam, M. A., Sullivan, R., and Mancarella, S. (2016). STIM1-dependent Ca(2+) microdomains are required for myofilament remodeling and signaling in the heart. Sci. Rep. 6:25372. doi: 10.1038/srep25372
Pasdois, P., Beauvoit, B., Tariosse, L., Vinassa, B., Bonoron-Adèle, S., and Dos Santos, P. (2008). Effect of diazoxide on flavoprotein oxidation and reactive oxygen species generation during ischemia-reperfusion: a study on Langendorff-perfused rat hearts using optic fibers. Am. J. Physiol. Heart Circ. Physiol. 294, H2088–H2097. doi: 10.1152/ajpheart.01345.2007
Piquereau, J., Caffin, F., Novotova, M., Lemaire, C., Veksler, V., Garnier, A., et al. (2013). Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front. Physiol. 4:102. doi: 10.3389/fphys.2013.00102
Poteser, M., Graziani, A., Rosker, C., Eder, P., Derler, I., Kahr, H., et al. (2006). TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol. Chem. 281, 13588–13595. doi: 10.1074/jbc.M512205200
Prakriya, M., and Lewis, R. S. (2015). Store-operated calcium channels. Physiol. Rev. 95, 1383–1436. doi: 10.1152/physrev.00020.2014
Saliba, Y., Keck, M., Marchand, A., Atassi, F., Ouillé, A., Cazorla, O., et al. (2015). Emergence of Orai3 activity during cardiac hypertrophy. Cardiovasc. Res. 105, 248–259. doi: 10.1093/cvr/cvu207
Sánchez, J. A., García, M. C., Sharma, V. K., Young, K. C., Matlib, M. A., and Sheu, S. S. (2001). Mitochondria regulate inactivation of L-type Ca2+ channels in rat heart. J. Physiol. 536, 387–396. doi: 10.1111/j.1469-7793.2001.0387c.xd
Sato, T., Sasaki, N., Seharaseyon, J., O’Rourke, B., and Marbán, E. (2000). Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation 101, 2418–2423. doi: 10.1161/01.cir.101.20.2418
Saul, S., Gibhardt, C. S., Schmidt, B., Lis, A., Pasieka, B., Conrad, D., et al. (2016). A calcium-redox feedback loop controls human monocyte immune responses: the role of ORAI Ca2+ channels. Sci. Signal. 9:ra26. doi: 10.1126/scisignal.aaf1639
Shigekawa, M., and Iwamoto, T. (2001). Cardiac Na(+)-Ca(2+) exchange molecular and pharmacological aspects. Circ Res. 88, 864–876. doi: 10.1161/hh0901.090298
Stiber, J., Hawkins, A., Zhang, Z.-S., Wang, S., Burch, J., Graham, V., et al. (2008). STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 10, 688–697. doi: 10.1038/ncb1731
Valero, R. A., Senovilla, L., Núñez, L., and Villalobos, C. (2008). The role of mitochondrial potential in control of calcium signals involved in cell proliferation. Cell Calcium 44, 259–269. doi: 10.1016/j.ceca.2007.12.002
Valverde, C. A., Kornyeyev, D., Ferreiro, M., Petrosky, A. D., Mattiazzi, A., and Escobar, A. L. (2010). Transient Ca2+ depletion of the sarcoplasmic reticulum at the onset of reperfusion. Cardiovasc. Rese. 85, 671–680. doi: 10.1093/cvr/cvp371
Voelkers, M., Salz, M., Herzog, N., Frank, D., Dolatabadi, N., Frey, N., et al. (2010). Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J. Mol. Cell. Cardiol. 48, 1329–1334. doi: 10.1016/j.yjmcc.2010.01.020
Völkers, M., Dolatabadi, N., Gude, N., Most, P., Sussman, M. A., and Hassel, D. (2012). Orai1 deficiency leads to heart failure and skeletal myopathy in zebrafish. J. Cell Sci. 125, 287–294. doi: 10.1242/jcs.090464
Wang, S., Cone, J., and Liu, Y. (2001). Dual roles of mitochondrial K(ATP) channels in diazoxide-mediated protection in isolated rabbit hearts. Am. J. Physiol. Heart Circ. Physiol. 280, H246–H255. doi: 10.1152/ajpheart.2001.280.1.H246
Yu, J., Deliu, E., Zhang, X.-Q., Hoffman, N. E., Carter, R. L., Grisanti, L. A., et al. (2013). Differential activation of cultured neonatal cardiomyocytes by plasmalemmal versus intracellular G protein-coupled receptor 55. J. Biol. Chem. 288, 22481–22492. doi: 10.1074/jbc.M113.456178
Zhao, G., Li, T., Brochet, D. X. P., Rosenberg, P. B., and Lederer, W. J. (2015). STIM1 enhances SR Ca2+ content through binding phospholamban in rat ventricular myocytes. Proc. Natl. Acad. Sci. U. S. A. 112, E4792–E4801. doi: 10.1073/pnas.1423295112
Keywords: Orai channels, STIM, SOCs, cardiomyocyte, pharmacological preconditioning, diazoxide, ROS, ischemia–reperfusion
Citation: Sampieri R, Fuentes E, Carrillo ED, Hernández A, García MC and Sánchez JA (2020) Pharmacological Preconditioning Using Diazoxide Regulates Store-Operated Ca2 + Channels in Adult Rat Cardiomyocytes. Front. Physiol. 10:1589. doi: 10.3389/fphys.2019.01589
Received: 03 September 2019; Accepted: 19 December 2019;
Published: 14 January 2020.
Edited by:
Tarik Smani, University of Seville, SpainReviewed by:
Sabrina Brechard, University of Luxembourg, LuxembourgCopyright © 2020 Sampieri, Fuentes, Carrillo, Hernández, García and Sánchez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorge A. Sánchez, anNhbmNoZXpAY2ludmVzdGF2Lm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.