
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 18 December 2019
Sec. Integrative Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01523
This article is part of the Research TopicAdvances in Metabolic Mechanisms of Aging and Its Related DiseasesView all 11 articles
 Jacopo Sabbatinelli1
Jacopo Sabbatinelli1 Francesco Prattichizzo2
Francesco Prattichizzo2 Fabiola Olivieri1,3
Fabiola Olivieri1,3 Antonio Domenico Procopio1,3
Antonio Domenico Procopio1,3 Maria Rita Rippo1*
Maria Rita Rippo1* Angelica Giuliani1
Angelica Giuliani1Despite the decline in their proliferative potential, senescent cells display a high metabolic activity. Senescent cells have been shown to acquire a more glycolytic state even in presence of high oxygen levels, in a way similar to cancer cells. The diversion of pyruvate, the final product of glycolysis, away from oxidative phosphorylation results in an altered bioenergetic state and may occur as a response to the enhanced oxidative stress caused by the accumulation of dysfunctional mitochondria. This metabolic shift leads to increased AMP/ATP and ADP/ATP ratios, to the subsequent AMPK activation, and ultimately to p53-mediated growth arrest. Mounting evidences suggest that metabolic reprogramming is critical to direct considerable amounts of energy toward specific activities related to the senescent state, including the senescence-associated secretory phenotype (SASP) and the modulation of immune responses within senescent cell tissue microenvironment. Interestingly, despite the relative abundance of oxygen in the vascular compartment, healthy endothelial cells (ECs) produce most of their ATP content from the anaerobic conversion of glucose to lactate. Their high glycolytic rate further increases during senescence. Alterations in EC metabolism have been identified in age-related diseases (ARDs) associated with a dysfunctional vasculature, including atherosclerosis, type 2 diabetes and cardiovascular diseases. In particular, higher production of reactive oxygen species deriving from a variety of enzymatic sources, including uncoupled endothelial nitric oxide synthase and the electron transport chain, causes DNA damage and activates the NAD+-consuming enzymes polyADP-ribose polymerase 1 (PARP1). These non-physiological mechanisms drive the impairment of the glycolytic flux and the diversion of glycolytic intermediates into many pathological pathways. Of note, accumulation of senescent ECs has been reported in the context of ARDs. Through their pro-oxidant, pro-inflammatory, vasoconstrictor, and prothrombotic activities, they negatively impact on vascular physiology, promoting both the onset and development of ARDs. Here, we review the current knowledge on the cellular senescence-related metabolic changes and their contribution to the mechanisms underlying the pathogenesis of ARDs, with a particular focus on ECs. Moreover, current and potential interventions aimed at modulating EC metabolism, in order to prevent or delay ARD onset, will be discussed.
Aging is the leading single risk factor for the development of most, if not all, major age-related chronic diseases, such as neurodegenerative, cancer, metabolic and cardiovascular diseases (CVDs). Aging and age-related diseases (ARDs) share a common set of basic biological mechanisms, such as inflammation, the accumulation of macromolecular damage, adaptation to molecular and psychological stressors, epigenetic changes, metabolic dysfunction, loss of proteostasis, and defective stem cell function (Kennedy et al., 2014). Major ARDs include type 2 diabetes (T2DM), cardiovascular diseases (CVDs), osteoporosis and certain types of cancers (Olivieri et al., 2018; Prattichizzo, 2019). The low-grade, chronic, and systemic inflammation underlying the aging process and ARDs was called “inflammaging” (Franceschi et al., 2000; Fulop et al., 2018).
The notion that the lifespan of many species can be extended through reduction of energy intake (Speakman, 2005) suggests a critical role of macronutrient metabolism in the control of regulatory processes influencing proliferation, survival (Redman et al., 2018; Mitchell et al., 2019), and ARD development (Fontana et al., 2004). Accordingly, obesity is a risk factor for many ARDs and carries out a life-shortening action (Poirier et al., 2006). The onset of ARDs can be counteracted through overweight reduction by decreasing the energetic food and by increasing energy expenditure with physical activity (Stubbs and Lee, 2004; Everitt and Le Couteur, 2007; Fontana and Partridge, 2015). Notably, centenarians, individuals capable of reaching the extreme limit of human life and characterized by an exceptionally healthy phenotype, share features observed in human adult volunteers who followed caloric restriction (CR) regimens (Franceschi et al., 2018). All these evidences suggest that major pathways driving organismal aging are intimately connected with metabolism (Lopez-Otin et al., 2016), a hypothesis also confirmed by the fact that dysfunctional mitochondria are a common feature of aged cells and major ARDs (Lane et al., 2015; Correia-Melo et al., 2016).
Within the cells, energy is generated in the form of adenosine triphosphate (ATP), mainly through mitochondrial oxidative phosphorylation (OXPHOS) in the presence of oxygen, and through anaerobic glycolysis in its absence (Saraste, 1999). This “general rule” could be subjected to modifications during particular conditions, such as cellular senescence (Wiley and Campisi, 2016). Senescent cells (SCs) exhibit some peculiar characteristics, including growth arrest, telomere shortening, enhanced senescence associated (SA) β-Galactosidase activity, and the acquisition of a pro-inflammatory senescence-associated secretory phenotype (SASP), which is responsible for both inflammaging and the spreading of senescence to neighboring cells. Beyond the exhaustion of replicative potential, cellular senescence can also be triggered by different stressors, including hypoxia, endotoxin, and reactive oxygen species (ROS) (Campisi, 2013).
Endothelial cells (ECs) form the inner lining of blood vessels (Fishman, 1982). In adult organisms they are supposed to remain in a quiescent state, but they can be rapidly activated by a variety of stimuli (Schlereth et al., 2018). They provide a significant contribution to the transduction of signals between blood and tissues (Munoz-Chapuli et al., 2004); moreover, by the release of the gaseous mediator nitric oxide (NO), ECs play a crucial role in maintaining the vascular tone and in preventing platelet aggregation (Huang et al., 1995; Sabbatinelli et al., 2017). The integrity of the EC monolayer is a critical requisite to allow blood flow and avoid uncontrolled thrombosis (Giannotta et al., 2013). Growing evidence show that dysfunctional senescent ECs can play a key role in instigating the morphological and biochemical changes that accompany vascular dysfunction, thus not a mere epiphenomenon in pathogenesis of CVDs (Childs et al., 2014; Tian and Li, 2014). For this reason, senescence of ECs has become a central focus of the investigations on ARDs.
Here, we review the latest findings on the metabolic changes that occur during the aging process of endothelial cells. Moreover, due to EC strategic location in the human organism, here we support the hypothesis of a potential role of senescence-associated metabolic alterations of ECs in the onset and development of ARDs. Finally, the potential relevance of targeting specific EC metabolic features to counteract ARDs will be discussed.
A peculiar feature of SCs is that they remain metabolically active, despite their growth arrest. Their high metabolic rate is intimately linked to SASP acquisition, but whether it is a cause or an effect of the inflammatory phenotype and altered proliferative status of SCs has yet to be established (Wiley and Campisi, 2016).
Recent literature confers to metabolic reprogramming a deterministic role in modulating the inflammatory responses of the innate immune cells (Van den Bossche et al., 2017): in macrophages, metabolic pathways not only provide energy but also regulate their phenotype and function. Lipopolysaccharide (LPS)(+IFNγ)-activated proinflammatory (i.e., M1) macrophages mediate host defense through an enhanced glycolytic metabolism and impaired mitochondrial OXPHOS. On the contrary, interleukin (IL)-4(13)-activated (i.e., M2) macrophages, which promote wound healing and Th2-mediated responses, mainly rely on OXPHOS for the synthesis of ATP. These data suggest that metabolic changes may also underlie acquisition of the SASP in senescent cells.
In the 1980s, the first attempts to analyze energy metabolism in aging cells showed that glucose consumption and lactate production are elevated in replicative senescent human diploid fibroblasts (HDFs) (Goldstein et al., 1982; Bittles and Harper, 1984). This “glycolytic state,” accompanied by an imbalance of the activity of the glycolytic enzymes, results in a less energetic state, mirrored by the drop of ATP and GTP intracellular levels as cell cultures enter in replicative senescence (Zwerschke et al., 2003). More recently, metabolomic approaches on extracellular metabolites released by senescent fibroblasts confirmed a shift toward glycolysis compared to young cells (James et al., 2015). This general mechanism, however, is more or less pronounced depending on the cell type and shows some exceptions, such as senescent human mammary epithelial cells where glucose consumption and lactate secretion do not increase (Delfarah et al., 2019).
The role of malic enzyme (ME) in senescent cells has been extensively investigated due to its role in maintaining cellular redox homeostasis. The two ME isoforms ME1 and ME2 catalyze the decarboxylation of malate to pyruvate with concomitant formation NADPH or NADH, respectively. Regeneration of reducing equivalents for anabolic processes in form of NADPH can be achieved either through the “malate oxidation shunt” or through the pentose phosphate pathway (PPP) (Fox et al., 1994). In the context of aging cell, p53 by binding to specific response elements, attenuates ME1 and ME2 transcription. At the same time ME depletion induces senescence by stabilizing p53, suggesting that also ME exerts an inhibitory effect on p53 (Jiang et al., 2013). Moreover, expression of both malate dehydrogenase (MDH)1, a mitochondrial tricarboxylic acid (TCA) cycle enzyme catalyzing malate oxidation to oxaloacetate, and MDH2, the cytosolic enzyme of malate-aspartate shuttle, declines during aging, leading to an impaired transfer of reducing equivalents into the mitochondria. In HDF, the subsequent decrease of cytosolic NAD+/NADH ratio is accompanied by the induction of replicative senescence (Zwerschke et al., 2003; Lee et al., 2012; Wiley et al., 2016). The need to regenerate NAD+ explains the upregulation of lactate dehydrogenase (LDH) in senescent HDFs, which release lactate to avoid the arrest of glycolysis due to both product accumulation and excessively low intracellular pH (Lang et al., 2003; Zwerschke et al., 2003) (Figure 1). These observations on the role of malate prompted the hypothesis that malate supplementation could delay senescence and extend lifespan (Edwards et al., 2013). Similar effects have been shown in C. elegans and D. melanogaster for α-ketoglutarate and oxaloacetate, two other TCA cycle intermediates (Williams et al., 2009; Chin et al., 2014; Su et al., 2019). However, such observation needs to be corroborated by evidence in mammals.
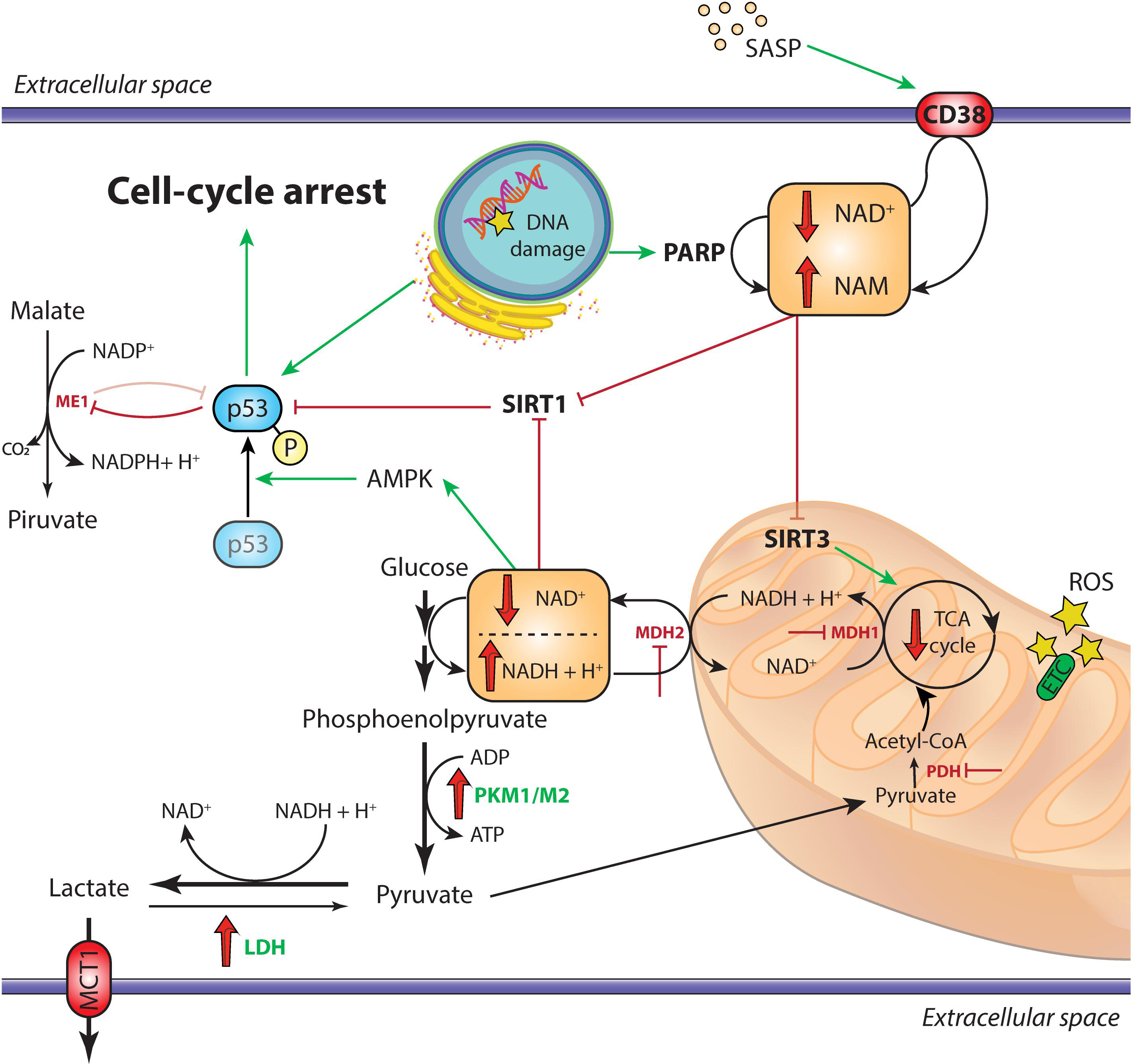
Figure 1. Overview of the metabolic alterations driving cellular senescence. In cells undergoing replicative senescence, the upregulation of LDH and the inhibition of both PDH and the malate-aspartate shuttle (MDH1 and MDH2) result in the diversion of pyruvate away from oxidative phosphorylation and toward aerobic glycolysis. This leads to the decrease of cytosolic NAD+/NADH ratio, which triggers the activation of the energy sensor AMPK. Moreover, the accumulation of DNA damage, also mediated by ROS in the dysfunctional mitochondria and the SASP, activate the NAD+-consuming enzymes PARP1 and CD38, respectively. The subsequent impairment of SIRT1 deacetylase activity, coupled with the AMPK-mediated phosphorylation of p53, triggers the arrest of cell replication and the establishment of irreversible senescence. Moreover, activated p53 inhibits the activity of the malic enzymes ME1 and ME2, further impairing the cellular antioxidant mechanisms through reduction of NADPH levels. Downregulated enzymes are in red, upregulated enzymes are in green; red and green arrows indicate repression or induction, respectively. AMPK, adenosine monophosphate-activated protein kinase; ETC, electron transport chain; LDH, lactate dehydrogenase; MCT1, monocarboxylate transporter 1; MDH1/MDH2, malate dehydrogenase 1/2; ME1, malic enzyme 1; PARP, poly (ADP-ribose) polymerase; PDH, pyruvate dehydrogenase; PKM1/PKM2, pyruvate kinase M1/M2; ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype; SIRT, sirtuin; TCA, tricarboxylic acid.
The role of PPP, the alternative source of NADPH, has been explored in cellular senescence. In fact, even if SCs have a lower demand of deoxyribonucleotides (dNTPs), they still require NADPH to allow the activity of the reactive oxygen species (ROS)-detoxifying thioredoxins, glutaredoxins and peroxiredoxins. Importantly, replicative senescence can be triggered by a lack of dNTPs due to reduced substrate availability. The activity of the first enzyme of the oxidative branch of PPP, namely glucose-6-phosphate dehydrogenase (G6PDH), is decreased during senescence. Accordingly, G6PDH-deficient cells exhibit accelerated oxidant-induced senescence (Ho et al., 2000; Cheng et al., 2004), a process that can be partially rescued by telomerase ectopic expression (Wu et al., 2009). Notably, transgenic mice overexpressing G6PDH display extended lifespan through increased NADPH levels (Nobrega-Pereira et al., 2016) and knockdown of the tumor suppressor ataxia-telangiectasia mutated (ATM) gene restores glucose flux throughout the PPP and allows cells to overcome senescence (Aird et al., 2015).
The findings on the metabolic features of replicative SCs have been only in part confirmed in oncogene-induced senescence (OIS). Seminal studies reported that cells undergoing OIS have high glycolysis rate, along with an elevated OXPHOS activity, when compared to proliferating cells (Quijano et al., 2012; Dorr et al., 2013; Kaplon et al., 2013; Takebayashi et al., 2015). In line with these findings, both pyruvate kinase (PK) isoforms, i.e., PKM1 and PKM2, and pyruvate dehydrogenase (PDH), two enzymes with a key role in glycolysis and in the conversion of pyruvate to acetyl-CoA, are upregulated in SCs. This leads to an enhanced use of pyruvate for the TCA cycle causing increased cellular respiration and redox stress (Zwerschke et al., 2003; Dorr et al., 2013; Kaplon et al., 2013). The TCA cycle could be also fueled by metabolites from fatty acid catabolism. Indeed, Ras-induced senescent cells manifest a decline in lipid synthesis and an increase in fatty acid oxidation (FAO), which results in a higher rate of basal oxygen consumption (Quijano et al., 2012). Inhibition of carnitine palmitoyltransferase 1A (CPT1A), the key rate-limiting enzyme for oxidation of free fatty acids (FFA) into the mitochondria, prevented senescence and SASP establishment (Quijano et al., 2012). In a recent work, Fafián-Labora et al. demonstrated that fatty acid synthase (FASN) activity is important for mitochondrial bioenergetics in the initial phases of senescence. FASN is an enzyme that catalyzes de novo synthesis of fatty acids by combining malonyl-CoA to the acetyl-CoA derived from glycolysis-produced pyruvate. Indeed, inhibition of FASN activity prevented the p53-mediated induction of senescence, the secretion of the canonical SASP factors IL-1α, IL-1β, IL-6, and the release of extracellular vesicles (EVs) mediating the spread of pro-senescence signals at the paracrine level (Borghesan et al., 2019; Fafian-Labora et al., 2019). Notably, other studies reported that p53 activation inhibits FASN, suggesting a negative feedback loop (Ford, 2010).
High lipogenesis would explain the progressive accumulation of membranous organelles during cell senescence (Kim et al., 2010). It is well known that lysosome mass expands during senescence, recently an increase in mitochondrial mass was also established (Correia-Melo et al., 2016; Fafian-Labora et al., 2019). A considerable number of senescent−associated changes are dependent on mitochondria. Indeed, mitochondrial dysfunction can elicit a specific type of proinflammatory phenotype, defined as mitochondrial dysfunction-associated senescence (MiDAS) (Wiley et al., 2016). MiDAS differs from the prototypical SASP for the lack of an IL-1/NF-κB-dependent mechanism. In MiDAS, a reduced NAD+/NADH ratio is believed to trigger adenosine monophosphate-activated protein kinase (AMPK) and p53 activation (Wiley et al., 2016; Giuliani et al., 2017).
NAD+/NADH ratio is one of the most reliable markers of the redox state of the cell. Its decrease has been reportedly linked to cellular senescence (Son et al., 2016; Zhang et al., 2016). NAD+ decline in aging is extensively reviewed by Verdin (Verdin, 2015). Low levels of NAD+ were also reported in several tissues (Massudi et al., 2012; Zhu et al., 2015), and supplementation of NAD+ precursors increased life span in different species (Fang et al., 2016; Zhang et al., 2016). Interestingly, SCs were shown to induce the SASP-mediated expression of CD38 – an ectoenzyme with a high NADase activity – in non-senescent cells, such as endothelial cells and bone marrow-derived macrophages (Chini et al., 2019; Covarrubias et al., 2019). CD38 inhibitors rescued NAD+ decline and ameliorated a number of age-related metabolic outcomes in mice (Tarrago et al., 2018). A similar beneficial effect on mouse lifespan has been described also for the EV-mediated cell-to-cell transfer of the extracellular isoform of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of NAD+ salvage pathway (Yoshida et al., 2019). EVs are membrane-coated nanoparticles actively released by almost all cell types, including ECs (Jansen et al., 2017). EVs are usually categorized according to their size and surface markers, and are able to shuttle and deliver functional proteins and nucleic acids in a paracrine and systemic manner (Thery et al., 2018).
Recently, Nacarelli et al. (2019) demonstrated a direct link between intracellular NAD+ levels and the SASP in OIS. High SA-chromatin remodeling is related to the upregulation chromatin-binding protein High-Mobility Group A1 (HMGA1) which binds genomic A + T reach regions to increase chromatin accessibility (Nacarelli et al., 2019). Interestingly, inhibition or silencing of NAMPT, one of the targets of HMGA1, decreased glycolysis, mitochondrial respiration, and oxygen consumption, along with NAD+/NADH ratio, which is normally elevated in OIS (Nacarelli et al., 2019; Søgaard and Gil, 2019). The subsequent increase of ADP/ATP ratio activates AMPK, which leads to p53-mediated inhibition of p38MAPK. By activating NF-κB, p38MAPK acts as an important up-stream effector of the SASP (Freund et al., 2011). Therefore, in OIS a high NAD+/NADH ratio finally results in the activation of the SASP. The apparently contrasting observation in different models of senescence lends support to the hypothesis that NAD+ metabolism specifically controls distinct subsets of SASP factors. Indeed, MiDAS is triggered by a drop of NAD+/NADH and leads to IL-10 and TNF-α production (Wiley et al., 2016), whereas OIS boosts NAD+ levels, in contrast with the general decline of NAD+/NADH ratio observed in aging and confirmed in replicative senescence (Zwerschke et al., 2003).
We can conclude that dramatic alterations of carbohydrate and lipid metabolism occur in senescent cells, with divergent outcomes partially depending on senescence trigger. In general, senescent cells acquire a “glycolytic state,” but the fate of the resulting pyruvate changes between replicative and oncogene/therapy induced senescence. Notably, the pyruvate “hub” has been promoted as a druggable target for treatment of many diseases, including diabetes, ischemic heart disease, and cancer (Roche and Hiromasa, 2007; Olenchock and Vander Heiden, 2013). By directing specific substrates to one pathway or another, the entire metabolic set-up of the cell can be affected, in some cases reaching a desirable effect.
Although they share common characteristics, ECs can exhibit several phenotypic differences, depending on the specific chemical and physical characteristics of the vascular districts in which they are living (Dejana et al., 2017). ECs in the microvasculature are involved in the bidirectional exchange of gases, macromolecules, and cells between tissues and blood, and can also perform enzymatic modifications of circulating mediators, such as lipoproteins and angiotensin I (Chi et al., 2003). ECs actively participate in angiogenesis, i.e., the generation of new capillaries from existing vessels; in the adult physiological angiogenesis occurs mainly in the female reproductive system and in wound healing, however it plays a significant role in promotion and progression of many pathological conditions, including cancer and chronic inflammation (Potente and Carmeliet, 2017). Angiogenesis implies a switch of selected ECs toward a proliferative and migratory phenotype (Jimenez-Valerio and Casanovas, 2017). These ECs are known as tip and stalk cells, the first guiding the migration of the latter to achieve the elongation of the developing vessel (Jakobsson et al., 2010). Angiogenesis is a high energy-demanding process, therefore, from their original quiescent state, ECs must undergo a substantial reprogramming of their metabolism.
Quiescent ECs rely mainly on glycolysis for their energy demanding, despite the high abundance of oxygen in the vascular compartment. Accordingly, even if the mitochondrial mass in ECs is variable depending to the vascular district; it is generally lower than other cell types (Eelen et al., 2018). During angiogenesis, vascular endothelial growth factor (VEGF) signaling induces in ECs a further boost of glycolysis, with the upregulation of the GLUT-1 transporter and an increased synthesis of the allosteric modulator fructose-2,6-bisphosphate (F2,6BP) by the enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), a master regulator of glycolysis (De Bock et al., 2013). In this context, EC mitochondrial metabolism remains largely underexplored. While a substantial amount of evidence is available on the impairment of angiogenesis under glucose-deprivation conditions (He et al., 2013; Terashima et al., 2013; Garcia et al., 2015; Gu et al., 2019), only one very recent report demonstrated that also the inhibition of electron transport chain decreases EC proliferation. Interestingly, this effect is related to a lowered NAD+/NADH ratio, rather than to a decreased availability of ATP (Diebold et al., 2019).
The notion that ECs exhibit different glycolytic rates according to their specialization or proliferation rate is quite intuitive, however, glycolysis is not only an energy supplier for these cells but can modulate their function and phenotype. Under hypoxic conditions, the hypoxia inducible factor 1-alpha (HIF-1α)-mediated upregulation of the glycolytic pathway, coupled with the downregulation of PDH, induces an accumulation of lactate, which in turn i) stabilizes HIF-1α and mediates a paracrine proangiogenic effect on neighboring ECs (Sonveaux et al., 2012; Lee et al., 2015), ii) affects the functional polarization of tumor-associated macrophages toward the pro-tumoral M2 phenotype (Colegio et al., 2014). EC phenotype can be changed through targeting enzymes or regulators of the glycolysis. The forkhead box O transcription factor 1 (FOXO1) plays a major role in maintaining ECs in a quiescent state by suppressing c-myc signaling and reducing glycolysis (Wilhelm et al., 2016). Similarly, the downregulation of hexokinase 2 (HK2) via the disruption of the fibroblast growth factor receptor (FGFR)/c-myc axis impairs proliferation and migration of ECs (Yu et al., 2017). The interplay between the two PK isoforms represents an important crossroad in determining the fate of the resulting pyruvate. In a model of pulmonary arterial hypertension, the increased PKM2 expression fosters aerobic glycolysis and induces a more proliferative state (Caruso et al., 2017). On the contrary, under physiological conditions, the laminar blood flow promotes the NO-mediated S-nitrosylation of PKM2, which results in a reduced glycolysis and in an enhanced funneling of substrates through the PPP (Siragusa et al., 2019). In cells with a low replication rate, like quiescent ECs, a balanced PPP activity promotes antioxidant responses through synthesis of reducing equivalents in form of NADPH.
The metabolic features of senescent ECs are still poorly explored. Moreover, whether the effects of EC senescence, including the SASP, could be mediated by metabolic changes is still being debated. An emerging role for SIRT3, a member of the sirtuin family mainly localized in the mitochondria, in cellular aging has been outlined by recent studies (Ansari et al., 2017). Through its NAD-dependent deacetylase activity, SIRT3 regulates the mitochondrial metabolic pathways, including FAO (Kanwal, 2018). Specifically, SIRT3 promotes the influx of substrates into the TCA cycle by promoting the activity of PDH and acyl-CoA dehydrogenase (Kincaid and Bossy-Wetzel, 2013). Moreover, SIRT3 seems to exert a positive regulator effect on the TCA cycle, even if results are still contradictory (Verdin et al., 2010). Similarly to the other members of the sirtuin family, depletion of SIRT3 has been shown to reduce human lifespan (Bellizzi et al., 2005; Brown et al., 2013). Notably, strategies aimed to upregulate SIRT3 in endothelial cells resulted in a higher protection against stress-induced premature senescence, an effect mediated by the deacetylation of FoxO3 (Liu et al., 2015; Xing et al., 2018). For some aspects, ECs seem to escape the rules describing the senescence-associated metabolic shift that have been postulated from studies in other cell lineages. For example, the observation that HDF senescence is accompanied by an upregulation of the whole glycolytic machinery was not confirmed in HUVECs (Unterluggauer et al., 2008). Conversely, a recent report showed a senescence-associated decline in EC glycolysis, which is mediated by a reduced PFKFB3 activity. This trend, but not senescence, was reverted by nuclear factor erythroid 2-related factor 2 (NRF2) overexpression (Kuosmanen et al., 2018). Another study on EC replicative senescence revealed that a NAMPT/SIRT1/FoxO1-mediated slight increase in aerobic glycolysis exerts a protective effect by limiting ROS production (Borradaile and Pickering, 2009).
Glutamine represents another important metabolite for ECs. It can be used as an energy source, via deamination and subsequent transamination to form α-ketoglutarate which enters the TCA cycle, to produce the antioxidant peptide glutathione, or to provide substrates for nucleotide biosynthesis (Eelen et al., 2015). Glutamine was shown to be required for vessel sprouting, even if the impaired angiogenesis during glutamine starvation could be rescued by asparagine supplementation (Huang H. et al., 2017). The notion that senescent ECs strongly rely on glutaminolysis for their energy demand comes from the observation in these cells of an increased lactate synthesis independent from glycolysis. Remarkably, the inhibition of glutaminase 1 (GLS1) is able to induce apoptosis and senescence even in young ECs (Unterluggauer et al., 2008).
Although ECs obtain a minor amount of ATP from oxidative phosphorylation, they efficiently oxidize fatty acids: ECs are endowed with all the proteins required for the uptake and intracellular transport of fatty acids (Hagberg et al., 2013). Indeed, in the microcirculation, ECs can extract fatty acids from circulating lipoproteins through lipoprotein lipase (LPL). Fatty acids are then absorbed through the fatty acid transport proteins (FATP)3 and FATP4 and bound by the intracellular fatty acid binding protein FABP4. Fatty acids can then be oxidized to provide carbons to replenish TCA cycle, thus allowing the synthesis of dNTP precursors, or directed to the surrounding tissues (Mehrotra et al., 2014). In a recent report, Kalucka et al. showed that fatty acid oxidation is the only upregulated metabolic pathway in quiescent ECs. Notably, beta-oxidation is increased neither for bioenergetic purposes nor to meet the anabolic demands of the cells. Rather, fatty acids are oxidized to increase NADPH regeneration by the malic enzyme once their carbons enter the TCA cycle in form of acetyl-CoA. As a result, quiescent ECs have higher amounts of reduced glutathione and thus are more protected against oxidative stress (Kalucka et al., 2018). Interestingly, this beneficial effect is mediated by Notch1 signaling, which exerts also a central role during the earlier phases of senescence, by switching the secretome of ECs away from the pro-inflammatory SASP and toward the TGF-β-mediated release of immunosuppressive and fibrogenic factors (Hoare et al., 2016).
Cellular senescence also involves endothelial progenitor cells (EPCs), a population of circulating CD34+ cells participating in new vessel formation and in vascular remodeling through their ability to differentiate into ECs. The age-related decline in EPC survival and regeneration could precipitate endothelial dysfunction, thus triggering the onset of CVDs (Olivieri et al., 2016). It has been demonstrated that the increased susceptibility of elderly individuals to ischemic disorders could be mediated, at least in part, by a blunted response of senescent EPCs to hypoxia, which fail to upregulate selected genes related to HIF-1 signaling and glucose uptake (Wu et al., 2018). To this regard, the upregulation of SIRT1 and NRF2 have both been shown to delay EPC senescence and to preserve the proliferative, migratory, and angiogenic activities in senescent EPCs (Lamichane et al., 2019; Wang et al., 2019).
Investigations on EC metabolic features could take advantage of their peculiar localization and lend important insight into the pathogenesis of CVDs. Targeted metabolomics of large cohorts of samples are required to get a more comprehensive point of view on mechanisms which were extensively characterized in in vitro models. While multiomics approaches proved useful to identify circulating signatures with a diagnostic and/or prognostic role for many conditions (Zierer et al., 2015; Correia et al., 2017; Niewczas et al., 2019), their role in providing ex vivo mechanistic clues is hampered by the lack of information on the relative contribution of various tissues to the circulating metabolome. To address this issue, the field is moving toward more specific approaches allowing the deconvolution of complex circulating signatures, including the application of machine learning tools and the study of EVs (Heitzer et al., 2019). A growing body of evidence is focusing on the characterization of endothelial EVs isolated from plasma (Igami et al., 2019; Mork et al., 2019; Oliveira et al., 2019; Utermohlen et al., 2019). Even if a consensus on the methods for isolation and characterization of these EVs is far from being reached, it is important to remark that the profiling of the cargo of circulating EVs sorted according to the parent cells allows enhanced specificity compared to single molecular markers in the blood.
Figure 2 provides an overview of the most relevant physiologic and pathologic pathways in ECs. Understanding the mutual influence between EC senescence and the complex network of metabolic pathways could provide valuable insight into the pathogenesis of many ARDs. This will be the focus of the next sections.
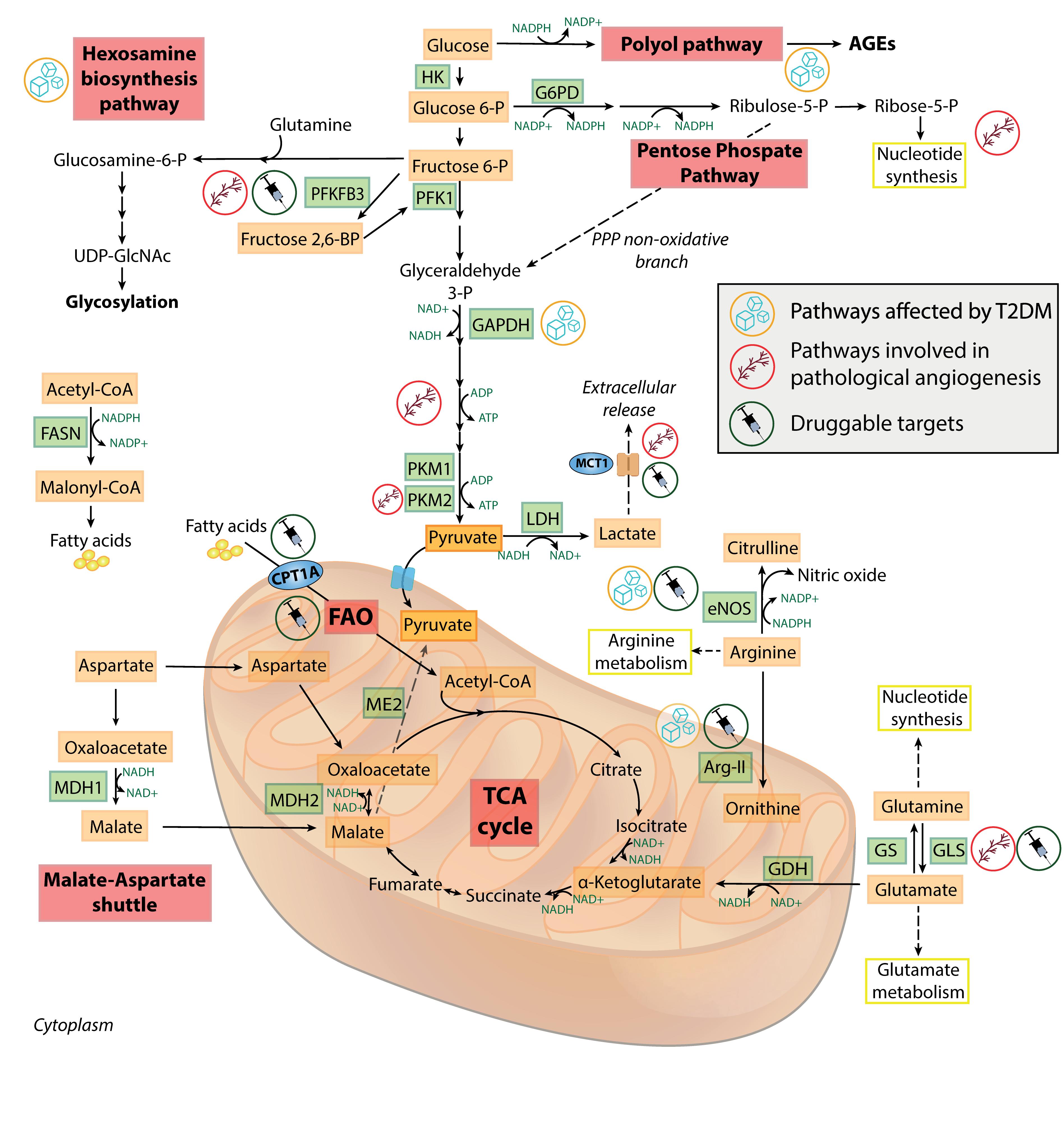
Figure 2. Metabolic features of healthy and dysfunctional endothelial cells. Schematic overview of the most relevant metabolic pathways in endothelial cells. Pathways, enzymes and metabolites affected by T2DM or involved in pathological angiogenesis, and the druggable targets discussed in the text, are labeled by specific icons. AGEs, advanced glycation end-products; Arg-II, arginase II; CPT1A, carnitine palmitoyltransferase 1A; eNOS, endothelial nitric oxide synthase; FAO, fatty acid oxidation; FASN, fatty acid synthase; G6PD, glucose 6-phosphate dehydrogenase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GDH, glutamate dehydrogenase; GLS, glutaminase; GS, glutamine synthetase; HK, hexokinase; LDH, lactate dehydrogenase; MCT1, monocarboxylate transporter 1; MDH1/MDH2, malate dehydrogenase 1/2; ME2, malic enzyme 2; PFK1, phosphofructokinase 1; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; PKM1/PKM2, pyruvate kinase 1/2; PPP, pentose phosphate pathway; T2DM, type 2 diabetes; TCA, tricarboxylic acid; UDP-GlcNAc, uridine diphosphate N-acetylglucosamine.
Increasing evidence is suggesting a bidirectional interplay among EC metabolism and ARDs (Graupera and Claret, 2018). One of the most characterized metabolic alteration in ECs exposed to hyperglycemia, i.e., a known senescence-promoting stimulus (Prattichizzo et al., 2018a), is the diversion of glycolytic intermediates into alternate pathways (Figure 2). This is the result of the activation of the enzyme polyADP-ribose polymerase 1 (PARP1) following the ROS-induced DNA damage. PARP1 mediates the ribosylation of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and depletes NAD+ intracellular levels, further inhibiting the glycolysis flux (Du et al., 2003). Moreover, entry of glucose 6-phosphate into the PPP is also restricted, due to a cAMP-mediated impairment of glucose 6-phosphate dehydrogenase during hyperglycemia (Zhang et al., 2010). The reduced PPP activity leads to lowered NADPH levels, causing both a reduced endothelial nitric oxide synthase (eNOS) activity and a blunted antioxidant response. In addition, excessive glucose is metabolized into the pathologic polyol and hexosamine biosynthetic pathways, which are the major contributors to the generation of advanced glycation end-products in diabetes (Berrone et al., 2006). The aberrant non-enzymatic glycation of circulating and intracellular proteins is a key determinant of T2DM cardiovascular complications (Mapanga and Essop, 2016). Endothelial dysfunction (ED) and senescence are “historically” considered two consequences of T2DM (Bonfigli et al., 2016; Prattichizzo et al., 2018a, d), possibly as a result of the overabundance of senescence-promoting, endothelium-damaging factors in the bloodstream of these patients, e.g., pro-oxidant molecules, glucose, and lipids (Prattichizzo et al., 2018c). In particular, the role of EC senescence has been emphasized in both the development of T2DM and of its CV complications. Indeed, EC senescence has been found in the adipose tissue of obese subjects preceding the development of T2DM (Minamino et al., 2009; Villaret et al., 2010), as well as in other vascular districts in diabetic mice and humans (Orimo et al., 2009; Prattichizzo et al., 2018a; Yokoyama et al., 2019). On the other side, the observation that ED development often precedes the appearance of the T2DM phenotype in obese subjects (Hadi and Suwaidi, 2007), coupled by the intuitive assumption that the endothelium represents the biggest organ regulating nutrient availability across the whole organism, has prompted the research toward the study of the effect of selective gain/loss of function of metabolic genes in ECs (Graupera and Claret, 2018).
A seminal paper suggested that deletion of insulin receptor (IR) in ECs is sufficient to alter the expression of eNOS and endothelin-1 in mice treated with a high-salt diet, providing the first link among EC metabolism and general vascular tone (Vicent et al., 2003). Later, another group showed that EC-specific Irs2 KO mice have a blunted insulin-dependent glucose uptake in the muscles when fed with HFD (Kubota et al., 2011). Beyond insulin signaling, also the selective modulation in ECs of genes involved in angiogenesis, i.e., VEGF/VEGFR2, ANG2/TIE2, and DLL4/NOTCH1 (Robciuc et al., 2016; Seki et al., 2018), as well as in fatty acids transport, i.e., VEGF-B/NRP1 and CD36, affect whole body metabolism (Hagberg et al., 2010; Son et al., 2018). Interestingly, both promotion and inhibition of angiogenesis have been shown to foster insulin sensitivity, suggesting a context-dependent effect (Sun et al., 2012). Proposed mechanistic explanations include the induction of apoptosis in dysfunctional adipocyte (for restricted angiogenesis) and “healthy” expansion of adipose tissue (for enhanced angiogenesis) (Graupera and Claret, 2018).
While the role of senescent EC metabolism has not been studied in relation to specific ARDs, a key role for an alteration of metabolism in senescent ECs may be inferred by studies modulating key factors involved in the senescence process. Indeed, mice with EC-specific p53 deficiency fed a high fat diet (HFD) showed improvement of insulin sensitivity and less fat accumulation compared to WT mice (Yokoyama et al., 2014). Of note, p53 is a master transcription factor promoting senescence in virtually all cell types (Hafner et al., 2019). Similarly, NF-κB activation is held to be a cornerstone of SASP development in a plethora of different SCs, including ECs (Salminen et al., 2012). Transgenic mice expressing dominant-negative IκB under the Tie2 promoter/enhancer (thus with functional inhibition of NF-κB signaling specifically in ECs) were protected from the development of insulin resistance associated with both genetic and diet-induced obesity. Strikingly, these mice showed also an increase in lifespan, coupled by a decreased age-related insulin resistance and vascular senescence (Hasegawa et al., 2012). The relevance of these findings for human ARDs remains to be tested. However, an increased expression of p53, along with an increased activation of NF-κB has been observed in aged arteries from human subjects (Donato et al., 2008; Morgan et al., 2013).
Beyond T2DM and vascular function, also heart remodeling has been shown to be affected by metabolic alterations in ECs. Indeed, genetic ablation of Rbp-jκ (Notch signaling) in ECs promoted alterations in fatty acid metabolism in the whole organism, followed by heart hypertrophy and failure (Jabs et al., 2018). The deprivation of available FFAs as substrate prompted the use of glucose in cardiomyocytes and the consequent activation of the mTOR pathway, a master metabolic rheostat regulating both cellular senescence and organismal aging (Weichhart, 2018). Accordingly, a ketogenic diet was able to restore normal cardiac function in this mouse model (Jabs et al., 2018), suggesting the potential of dietary intervention to treat (and not only prevent) also life-threating ARDs. A similar framework has been proposed also to explain the cardioprotective properties of sodium-glucose cotransporter (SGLT)-2 inhibitors (i) (Ferrannini et al., 2016). SGLT-2i are a recently introduced class of glucose-lowering drugs inhibiting the reabsorption of glucose in the proximal convoluted tubule, thus promoting glucose elimination through the kidneys (Santos-Gallego et al., 2019). Clinical trials have shown a striking benefit in terms of CV mortality and worsening of heart failure in diabetic patients treated with SGLT-2i, an effect not ascribable to an improved glycemic control (Prattichizzo et al., 2018b). It has been hypothesized that the decreased availability of glucose, coupled by an increase in the circulating levels of ketone bodies (KB), improves the energetic function of the heart (Ferrannini et al., 2016; Prattichizzo et al., 2018b). This hypothesis has been recently tested in porcine, non-diabetic hearts, where the treatment with a SGLT-2i switches myocardial fuel utilization away from glucose toward KB, FFA, and branched chain amino acids, thereby improving myocardial energetics, enhancing left ventricular systolic function, and ameliorating adverse left ventricle remodeling (Santos-Gallego et al., 2019).
The promotion of myocardial function by shifting toward FFA utilization is likely dependent on the function of the liver, rather than EC metabolism. However, EC metabolism may influence the pathobiology of different organs by at least 3 mechanisms: (i) by releasing active secondary mediators, such as NO (Kubota et al., 2011; Yokoyama et al., 2014); (ii) by regulating the flow of lipids from bloodstream to organs (Hagberg et al., 2010; Jabs et al., 2018); (iii) by regulating vessel density, and thus indirectly nutrients availability and interstitial insulin levels (Robciuc et al., 2016). Beyond adipose tissue and muscle insulin sensitivity, the latter mechanism has been shown to be crucial for the development of age-induced osteoporosis (Ramasamy et al., 2016). Indeed, skeletal blood flow and endothelial Notch activity are reduced in aged mice, leading to decreased angiogenesis and osteogenesis, which can be reverted by genetic reactivation of Notch (Ramasamy et al., 2016). As mentioned above, Notch is regarded as a central metabolic sensor and regulator in multiple cell types.
Regarding possible secondary mediators released by ECs, the role of EVs deserves particular attention. The effects of EVs are now attracting intense interest also in the context of aging and ARDs (Prattichizzo et al., 2019). For instance, senescent ECs secrete miR-31 enriched EVs to inhibit mesenchymal stem cells osteogenic differentiation, possibly contributing to age-induced osteoporosis (Weilner et al., 2016). Of note, miR-31 is upregulated by high-glucose and inhibits cell differentiation also in other contexts (Zhen et al., 2017).
Overall, increasing evidence is suggesting a key role for EC metabolism in a plethora of ARDs, while less studies are available regarding the effect of the specific alterations of senescent EC metabolism in the whole organisms. However, the emerging roles of endothelial p53 and NF-κB, two cornerstones of the senescence process, in the regulation of both cellular and organismal metabolism suggest that metabolic alterations in specific senescent cells, and in particular ECs, can affect whole body metabolism and ARD development, a hypothesis deserving exploration in the future.
Thirty years ago, the identification and cloning of VEGF paved the way to the development of novel strategies aimed to treat those conditions in which angiogenesis plays a dominant role (Apte et al., 2019). Agents targeting members of the VEGF family and their receptors are currently routinely employed in the treatment of many solid malignancies, including colorectal cancer, renal cell carcinoma, and non-small cell lung carcinoma (Tirumani et al., 2015), and in non-neoplastic conditions with a recognized angiogenic component, such as proliferative diabetic retinopathy and age-related macular degeneration (Virgili et al., 2018). Investigations on the mechanism of action of anti-VEGF agents helped to highlight the aforementioned interesting connections between angiogenesis and EC metabolism and offered a number of novel druggable targets that could prove useful, for example, in the case of failure of anti-VEGF treatments.
Targeting the high-glycolytic state of ECs within tumor vessels is emerging as an anti-cancer therapeutic strategy (Fitzgerald et al., 2018). In a murine model, pharmacological inhibition of glycolysis activator PFKFB3 promotes the normalization of tumor vessels and facilitates delivery of chemotherapeutic drugs. Moreover, PFKFB3 tightened ECs by upregulating vascular endothelium (VE)-cadherin, thus reducing the passage of cancer cells across the EC monolayer (Cantelmo et al., 2016). Interestingly, inhibition of PFKFB3 lowered the expression of adhesion molecules in ECs treated with IL-1β, suggesting that limiting glycolysis could represent a feasible approach to prevent the SASP-mediated spreading of EC senescence. PFKFB3 inhibitors recently reached the clinical trial phase, with promising results from a phase 1 multi-center study conducted on patients with solid tumors (Redman et al., 2015).
Given the proangiogenic role of lactate in ECs, several studies focused on target the monocarboxylate transporter 1 (MCT1) to avoid lactate exchange across ECs. Lactate mediates angiogenesis through the activation of the NF-κB/IL-8 pathway and the stabilization of HIF-1α (Vegran et al., 2011; Sonveaux et al., 2012). Evidence on cell and animal models revealed that MCT1 inhibition in ECs can drive direct anti-angiogenic effects through the enhanced degradation of HIF-1α. Drugs targeting MCT1 in a non-selective manner are currently been tested in small-scale trials (Kershaw et al., 2015). On the other hand, administration of the telomerase activator TA-65 contributes to improve blood flow recovery through increasing expression of HIF-1α, VEGF-A, and peroxisome proliferator-activated receptor (PPAR)-γ coactivator 1-alpha (PGC-1α), indicating that telomerase activation could prove a valuable therapeutic option to rescue ischemic tissues in elderly individuals (Kokubun et al., 2019).
Targeting EC metabolism could prove useful in treating a plethora of conditions sharing endothelial dysfunction as a pathogenic mechanism. Evidence of an age-related impairment of endothelial function dates back to 1990s. The identification of eNOS as the enzyme responsible for NO synthesis prompted the supplementation of its precursor L-arginine to reverse endothelial dysfunction (Chauhan et al., 1996). However, the enthusiasm for this essential amino acid as an easy therapeutic option in the prevention of many CVDs was tempered by the observation that L-arginine could even exert detrimental effects on vascular function through an inductive effect on arginase, which competes with NOSs for their common substrate (Hayashi et al., 2006; Schulman et al., 2006; Caldwell et al., 2015). For this reason, attention has moved to arginase as a putative target to ameliorate age-related endothelial dysfunction. A major contribution of its activity in determining microvascular dysfunction and remodeling has been outlined in obesity (Chung et al., 2014; Masi et al., 2018), arterial hypertension (Michell et al., 2011), and T2DM (Shemyakin et al., 2012). Interestingly, knockout of the gene encoding for the mitochondrial arginase type II (Arg-II) has been shown to restore eNOS function, to counteract the SASP in senescent ECs (Wu et al., 2015), and to extend lifespan in mice through the inhibition of mTOR signaling (Xiong et al., 2017). Moreover, mTOR blockade with rapamycin decreases the expression of the arterial senescence marker p19 and ameliorates oxidative stress- mediated endothelial dysfunction in old mice, suggesting a possible role for the rapamycin analogs, i.e., rapalogs, in the treatment of age-related CVDs (Lesniewski et al., 2017).
Fenofibrate, a PPAR-α agonist, is a common lipid-lowering drug exerting a number of interesting pleiotropic effects. By increasing EC eNOS expression and lowering circulating oxidized LDL, fenofibrate ameliorated the age-related endothelial dysfunction in a cohort of healthy individuals (Walker et al., 2012). Further mechanistic studies on animal models revealed that the eNOS stimulation, along with other fenofibrate effects, is mediated by enhanced AMPK activity following liver kinase B1 (LKB1) translocation from the nucleus to the cytoplasm (Sohn et al., 2017; Xin et al., 2019; Xu et al., 2019). Additionally, fenofibrate lowered the cyclooxygenase 2-mediated production of vasoconstrictor prostaglandins (Xu et al., 2019). By restoring the balance between vascular relaxation and contractility, fenofibrate could represent a feasible preventive approach for the vascular complications of T2DM. Notably, fenofibrate ameliorated osteoarthritis in elderly patients by selectively clearing senescent chondrocytes (Nogueira-Recalde et al., 2019). The observation of a similar senolytic effect on ECs would provide additional benefits for this largely employed drug also in the treatment of a number of age-related vascular conditions.
Investigations into a variety of diseases highlighted a role for CPT1, the rate-limiting enzyme for FAO, as a druggable target (Dai et al., 2018; Melone et al., 2018). Inhibition of endothelial CPT1a impairs EC proliferation and activates the endothelial-to-mesenchymal transition, which plays a role in the pathogenesis of pulmonary arterial hypertension, atherosclerosis, and tumor spreading (Schoors et al., 2015; Xiong et al., 2018). On the contrary, stimulation of CPT1 activity by chronic L-carnitine administration improved endothelial function in an animal model of congenital heart defect (Sharma et al., 2013). In light of the crucial role of FAO in maintaining EC quiescence and redox balance, L-carnitine supplementation, by counteracting the age-related decline in CPT1a activity (Gomez et al., 2012), could prove useful to delay the onset of age-related endothelial dysfunction. The encouraging evidence from several in vitro and animal studies still needs to be supported by large clinical trials (Bueno et al., 2005; Miguel-Carrasco et al., 2010; Ning and Zhao, 2013).
Glutamine, the most abundant circulating amino acid, has been extensively studied in aging research. Its blood levels decline during acute illness are increased in healthy centenarians (Montoliu et al., 2014; Meynial-Denis, 2016). A seminal study showed that glutamine administration in rats improves eNOS activity and reduces the endothelial inflammatory response following cardiopulmonary bypass (Hayashi et al., 2002). The activity of glutamine synthetase is impaired during endothelial dysfunction, due to peroxynitrite-mediated nitration of its active site (Gorg et al., 2005). This could lead to reduced TCA cycle anaplerosis, which results in the impairment of EC antioxidant system (see above) (Addabbo et al., 2013). Of note, restoring the glutamine-dependent anaplerosis through GLS overexpression has already proved beneficial in ameliorating the age-related bone loss (Huang T. et al., 2017), and in delaying EC senescence (Unterluggauer et al., 2008). Finally, the evidence that glutamine metabolism is also involved in EC proliferation supports the hypotheses that targeting GLS1 could represent a feasible approach to treat diseases associated with an aberrant EC proliferation (Peyton et al., 2018), and that glutamine supplementation can foster endothelial progenitor cell mobilization and promote vascular endothelium repair in diabetes-related ischemic injury (Su et al., 2017).
The observations discussed in the present section are summarized in Table 1. Altogether, these evidences reinforce the notion that alterations in EC metabolism are rather primary drivers than consequences of disease (Goveia et al., 2014). The development of drugs capable of targeting specific enzymes or pathways at the endothelial level is still in its infant phase. However, their progression into large clinical trials is imminent and could represent a turning point in the treatment of CVDs.
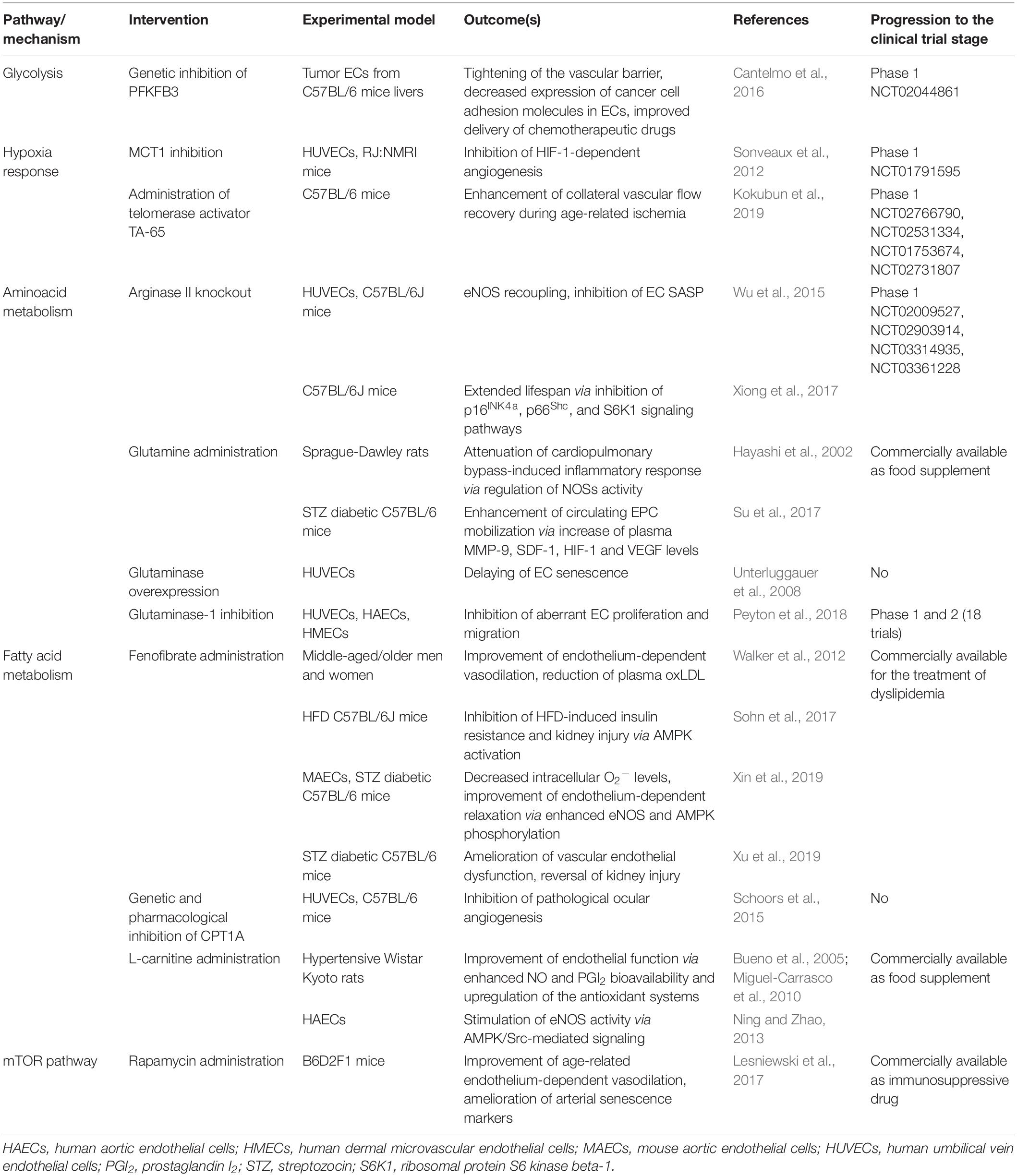
Table 1. Summary of the interventions targeting endothelial cell metabolism with a potential role in the treatment of age-related diseases.
Accumulating evidence is suggesting that SCs are characterized by a deep reshaping of metabolic pathways. The overall picture appears highly complex, considering that specific metabolic alterations characterize different cell types and various pro-senescence stimuli. However, a general trend toward a more glycolytic state in senescent cells has been reproduced with different stimuli and in a wide range of cell types, including ECs. The role of EC metabolism in the development of T2DM and CVDs is clearly emerging, mainly thank to specific mouse models with tissue-selective deletion of metabolic genes (Graupera and Claret, 2018). However, less information is available regarding the role of senescent EC metabolism in ARDs development, even though preliminary findings suggest a key role for senescence EC related genes, e.g., p53 and NF-κB, in the regulation of organismal aging (Hasegawa et al., 2012; Yokoyama et al., 2014). Disentangling the effective contribution of altered EC metabolism in humans remains challenging, as well as the impact of dietary or pharmacological intervention on this very specific process. Studies focusing on senescent EC metabolism are warranted to clarify the disease-modifying/preventive potential of both these approaches. In fact, interventions aimed at modifying diet and metabolism have already proven to be potentially effective strategies in the prevention and treatment of ARDs (Fontana and Partridge, 2015; Brandhorst and Longo, 2019). In particular, the preventive and curative roles of specific diets have now been demonstrated by placebo-controlled, randomized clinical trials showing a cardioprotective role for Mediterranean diet (Estruch et al., 2018) and the reduction of multiple cardiometabolic risk factors with CR (Kraus et al., 2019). While future studies will define to what extent these effects can be obtained with specific molecules/nutrients, the quality of evidence regarding these dietary patterns and ARD prevention should prompt the adoption of a Mediterranean-based, low-calories dietetic regimen at the population level.
JS, FP, and AG collected relevant literature and wrote the manuscript. JS and AG prepared the figures. FO, AP, and MR made a substantial, direct, and intellectual contribution through their experience in the field. MR critically advised and reviewed the manuscript.
This study was supported by grants from Università Politecnica delle Marche (Scientific Research Grant, year 2019 to MR; MIR-AGE UNIVPM to MR, FO, and AP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Addabbo, F., Chen, Q., Patel, D. P., Rabadi, M., Ratliff, B., Zhang, F., et al. (2013). Glutamine supplementation alleviates vasculopathy and corrects metabolic profile in an in vivo model of endothelial cell dysfunction. PLoS One 8:e65458. doi: 10.1371/journal.pone.0065458
Aird, K. M., Worth, A. J., Snyder, N. W., Lee, J. V., Sivanand, S., Liu, Q., et al. (2015). ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 11, 893–901. doi: 10.1016/j.celrep.2015.04.014
Ansari, A., Rahman, M. S., Saha, S. K., Saikot, F. K., Deep, A., and Kim, K. H. (2017). Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 16, 4–16. doi: 10.1111/acel.12538
Apte, R. S., Chen, D. S., and Ferrara, N. (2019). VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264. doi: 10.1016/j.cell.2019.01.021
Bellizzi, D., Rose, G., Cavalcante, P., Covello, G., Dato, S., De Rango, F., et al. (2005). A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 85, 258–263. doi: 10.1016/j.ygeno.2004.11.003
Berrone, E., Beltramo, E., Solimine, C., Ape, A. U., and Porta, M. (2006). Regulation of intracellular glucose and polyol pathway by thiamine and benfotiamine in vascular cells cultured in high glucose. J. Biol. Chem. 281, 9307–9313. doi: 10.1074/jbc.M600418200
Bittles, A. H., and Harper, N. (1984). Increased glycolysis in ageing cultured human diploid fibroblasts. Biosci. Rep. 4, 751–756. doi: 10.1007/bf01128816
Bonfigli, A. R., Spazzafumo, L., Prattichizzo, F., Bonafe, M., Mensa, E., Micolucci, L., et al. (2016). Leukocyte telomere length and mortality risk in patients with type 2 diabetes. Oncotarget 7, 50835–50844. doi: 10.18632/oncotarget.10615
Borghesan, M., Fafian-Labora, J., Eleftheriadou, O., Carpintero-Fernandez, P., Paez-Ribes, M., Vizcay-Barrena, G., et al. (2019). Small extracellular vesicles are key regulators of non-cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Rep. 27, 3956–3971.e6. doi: 10.1016/j.celrep.2019.05.095
Borradaile, N. M., and Pickering, J. G. (2009). Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 8, 100–112. doi: 10.1111/j.1474-9726.2009.00453.x
Brandhorst, S., and Longo, V. D. (2019). Dietary restrictions and nutrition in the prevention and treatment of cardiovascular disease. Circ. Res. 124, 952–965. doi: 10.1161/CIRCRESAHA.118.313352
Brown, K., Xie, S., Qiu, X., Mohrin, M., Shin, J., Liu, Y., et al. (2013). SIRT3 reverses aging-associated degeneration. Cell Rep. 3, 319–327. doi: 10.1016/j.celrep.2013.01.005
Bueno, R., Alvarez de Sotomayor, M., Perez-Guerrero, C., Gomez-Amores, L., Vazquez, C. M., and Herrera, M. D. (2005). L-carnitine and propionyl-L-carnitine improve endothelial dysfunction in spontaneously hypertensive rats: different participation of NO and COX-products. Life Sci. 77, 2082–2097. doi: 10.1016/j.lfs.2005.01.035
Caldwell, R. B., Toque, H. A., Narayanan, S. P., and Caldwell, R. W. (2015). Arginase: an old enzyme with new tricks. Trends Pharmacol. Sci. 36, 395–405. doi: 10.1016/j.tips.2015.03.006
Campisi, J. (2013). Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705. doi: 10.1146/annurev-physiol-030212-183653
Cantelmo, A. R., Conradi, L. C., Brajic, A., Goveia, J., Kalucka, J., Pircher, A., et al. (2016). Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 30, 968–985. doi: 10.1016/j.ccell.2016.10.006
Caruso, P., Dunmore, B. J., Schlosser, K., Schoors, S., Dos Santos, C., Perez-Iratxeta, C., et al. (2017). Identification of MicroRNA-124 as a major regulator of enhanced endothelial cell glycolysis in pulmonary arterial hypertension via PTBP1 (Polypyrimidine Tract Binding Protein) and pyruvate kinase M2. Circulation 136, 2451–2467. doi: 10.1161/CIRCULATIONAHA.117.028034
Chauhan, A., More, R. S., Mullins, P. A., Taylor, G., Petch, C., and Schofield, P. M. (1996). Aging-associated endothelial dysfunction in humans is reversed by L-arginine. J. Am. Coll. Cardiol. 28, 1796–1804.
Cheng, M. L., Ho, H. Y., Wu, Y. H., and Chiu, D. T. (2004). Glucose-6-phosphate dehydrogenase-deficient cells show an increased propensity for oxidant-induced senescence. Free Radic. Biol. Med. 36, 580–591. doi: 10.1016/j.freeradbiomed.2003.11.031
Chi, J. T., Chang, H. Y., Haraldsen, G., Jahnsen, F. L., Troyanskaya, O. G., Chang, D. S., et al. (2003). Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. U.S.A. 100, 10623–10628. doi: 10.1073/pnas.1434429100
Childs, B. G., Baker, D. J., Kirkland, J. L., Campisi, J., and van Deursen, J. M. (2014). Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 15, 1139–1153. doi: 10.15252/embr.201439245
Chin, R. M., Fu, X., Pai, M. Y., Vergnes, L., Hwang, H., Deng, G., et al. (2014). The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510, 397–401. doi: 10.1038/nature13264
Chini, C., Hogan, K. A., Warner, G. M., Tarrago, M. G., Peclat, T. R., Tchkonia, T., et al. (2019). The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD(+) decline. Biochem. Biophys. Res. Commun. 513, 486–493. doi: 10.1016/j.bbrc.2019.03.199
Chung, J. H., Moon, J., Lee, Y. S., Chung, H. K., Lee, S. M., and Shin, M. J. (2014). Arginase inhibition restores endothelial function in diet-induced obesity. Biochem. Biophys. Res. Commun. 451, 179–183. doi: 10.1016/j.bbrc.2014.07.083
Colegio, O. R., Chu, N. Q., Szabo, A. L., Chu, T., Rhebergen, A. M., Jairam, V., et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. doi: 10.1038/nature13490
Correia, C. N., Nalpas, N. C., McLoughlin, K. E., Browne, J. A., Gordon, S. V., MacHugh, D. E., et al. (2017). Circulating microRNAs as potential biomarkers of infectious disease. Front. Immunol. 8:118. doi: 10.3389/fimmu.2017.00118
Correia-Melo, C., Marques, F. D., Anderson, R., Hewitt, G., Hewitt, R., Cole, J., et al. (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742. doi: 10.15252/embj.201592862
Covarrubias, A. J., Lopez-Dominguez, J. A., Perrone, R., Kale, A., Newman, J., Iyer, S. S., et al. (2019). Aging-related inflammation driven by cellular senescence enhances NAD consumption via activation of CD38+ macrophages. bioRxiv [Preprint]. doi: 10.1101/609438
Dai, J., Liang, K., Zhao, S., Jia, W., Liu, Y., Wu, H., et al. (2018). Chemoproteomics reveals baicalin activates hepatic CPT1 to ameliorate diet-induced obesity and hepatic steatosis. Proc. Natl. Acad. Sci. U.S.A. 115, E5896–E5905. doi: 10.1073/pnas.1801745115
De Bock, K., Georgiadou, M., Schoors, S., Kuchnio, A., Wong, B. W., Cantelmo, A. R., et al. (2013). Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663. doi: 10.1016/j.cell.2013.06.037
Dejana, E., Hirschi, K. K., and Simons, M. (2017). The molecular basis of endothelial cell plasticity. Nat. Commun. 8:14361. doi: 10.1038/ncomms14361
Delfarah, A., Parrish, S., Junge, J. A., Yang, J., Seo, F., Li, S., et al. (2019). Inhibition of nucleotide synthesis promotes replicative senescence of human mammary epithelial cells. J. Biol. Chem. 294, 10564–10578. doi: 10.1074/jbc.RA118.005806
Diebold, L. P., Gil, H. J., Gao, P., Martinez, C. A., Weinberg, S. E., and Chandel, N. S. (2019). Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 1, 158–171. doi: 10.1038/s42255-018-0011-x
Donato, A. J., Black, A. D., Jablonski, K. L., Gano, L. B., and Seals, D. R. (2008). Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 7, 805–812. doi: 10.1111/j.1474-9726.2008.00438.x
Dorr, J. R., Yu, Y., Milanovic, M., Beuster, G., Zasada, C., Dabritz, J. H., et al. (2013). Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425. doi: 10.1038/nature12437
Du, X., Matsumura, T., Edelstein, D., Rossetti, L., Zsengeller, Z., Szabo, C., et al. (2003). Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Invest. 112, 1049–1057. doi: 10.1172/JCI18127
Edwards, C. B., Copes, N., Brito, A. G., Canfield, J., and Bradshaw, P. C. (2013). Malate and fumarate extend lifespan in Caenorhabditis elegans. PLoS One 8:e58345. doi: 10.1371/journal.pone.0058345
Eelen, G., de Zeeuw, P., Simons, M., and Carmeliet, P. (2015). Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 116, 1231–1244. doi: 10.1161/CIRCRESAHA.116.302855
Eelen, G., de Zeeuw, P., Treps, L., Harjes, U., Wong, B. W., and Carmeliet, P. (2018). Endothelial cell metabolism. Physiol. Rev. 98, 3–58. doi: 10.1152/physrev.00001.2017
Estruch, R., Ros, E., Salas-Salvado, J., Covas, M. I., Corella, D., Aros, F., et al. (2018). Primary prevention of cardiovascular disease with a Mediterranean diet supplemented with extra-virgin olive oil or nuts. N. Engl. J. Med. 378:e34. doi: 10.1056/NEJMoa1800389
Everitt, A. V., and Le Couteur, D. G. (2007). Life extension by calorie restriction in humans. Ann. N. Y. Acad. Sci. 1114, 428–433. doi: 10.1196/annals.1396.005
Fafian-Labora, J., Carpintero-Fernandez, P., Jordan, S. J. D., Shikh-Bahaei, T., Abdullah, S. M., Mahenthiran, M., et al. (2019). FASN activity is important for the initial stages of the induction of senescence. Cell Death Dis. 10:318. doi: 10.1038/s41419-019-1550-0
Fang, E. F., Kassahun, H., Croteau, D. L., Scheibye-Knudsen, M., Marosi, K., Lu, H., et al. (2016). NAD+ replenishment improves lifespan and healthspan in Ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581. doi: 10.1016/j.cmet.2016.09.004
Ferrannini, E., Mark, M., and Mayoux, E. (2016). CV Protection in the EMPA-REG OUTCOME trial: a “Thrifty Substrate” hypothesis. Diabetes Care 39, 1108–1114. doi: 10.2337/dc16-0330
Fishman, A. P. (1982). Endothelium: a distributed organ of diverse capabilities. Ann. N. Y. Acad. Sci. 401, 1–8. doi: 10.1111/j.1749-6632.1982.tb25702.x
Fitzgerald, G., Soro-Arnaiz, I., and De Bock, K. (2018). The Warburg effect in endothelial cells and its potential as an anti-angiogenic target in cancer. Front. Cell. Dev. Biol. 6:100. doi: 10.3389/fcell.2018.00100
Fontana, L., Meyer, T. E., Klein, S., and Holloszy, J. O. (2004). Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc. Natl. Acad. Sci. U.S.A. 101, 6659–6663. doi: 10.1073/pnas.0308291101
Fontana, L., and Partridge, L. (2015). Promoting health and longevity through diet: from model organisms to humans. Cell 161, 106–118. doi: 10.1016/j.cell.2015.02.020
Ford, J. H. (2010). Saturated fatty acid metabolism is key link between cell division, cancer, and senescence in cellular and whole organism aging. Age 32, 231–237. doi: 10.1007/s11357-009-9128-x
Fox, R. E., Kingwell, K. G., and Tildon, J. T. (1994). Malic enzyme activity in adult and newborn rat lung. Pediatr. Res. 35, 589–593.
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Franceschi, C., Ostan, R., and Santoro, A. (2018). Nutrition and inflammation: are centenarians similar to individuals on calorie-restricted diets? Annu. Rev. Nutr. 38, 329–356. doi: 10.1146/annurev-nutr-082117-051637
Freund, A., Patil, C. K., and Campisi, J. (2011). p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548. doi: 10.1038/emboj.2011.69
Fulop, T., Witkowski, J. M., Olivieri, F., and Larbi, A. (2018). The integration of inflammaging in age-related diseases. Semin. Immunol. 40, 17–35. doi: 10.1016/j.smim.2018.09.003
Garcia, N. A., Ontoria-Oviedo, I., Gonzalez-King, H., Diez-Juan, A., and Sepulveda, P. (2015). Glucose starvation in cardiomyocytes enhances exosome secretion and promotes angiogenesis in endothelial cells. PLoS One 10:e0138849. doi: 10.1371/journal.pone.0138849
Giannotta, M., Trani, M., and Dejana, E. (2013). VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev. Cell 26, 441–454. doi: 10.1016/j.devcel.2013.08.020
Giuliani, A., Prattichizzo, F., Micolucci, L., Ceriello, A., Procopio, A. D., and Rippo, M. R. (2017). Mitochondrial (Dys) function in inflammaging: do MitomiRs influence the energetic, oxidative, and inflammatory status of senescent cells? Mediators Inflamm. 2017:2309034. doi: 10.1155/2017/2309034
Goldstein, S., Ballantyne, S. R., Robson, A. L., and Moerman, E. J. (1982). Energy metabolism in cultured human fibroblasts during aging in vitro. J. Cell. Physiol. 112, 419–424. doi: 10.1002/jcp.1041120316
Gomez, L. A., Heath, S. H., and Hagen, T. M. (2012). Acetyl-L-carnitine supplementation reverses the age-related decline in carnitine palmitoyltransferase 1 (CPT1) activity in interfibrillar mitochondria without changing the L-carnitine content in the rat heart. Mech. Ageing Dev. 133, 99–106. doi: 10.1016/j.mad.2012.01.007
Gorg, B., Wettstein, M., Metzger, S., Schliess, F., and Haussinger, D. (2005). Lipopolysaccharide-induced tyrosine nitration and inactivation of hepatic glutamine synthetase in the rat. Hepatology 41, 1065–1073. doi: 10.1002/hep.20662
Goveia, J., Stapor, P., and Carmeliet, P. (2014). Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med. 6, 1105–1120. doi: 10.15252/emmm.201404156
Graupera, M., and Claret, M. (2018). Endothelial cells: new players in obesity and related metabolic disorders. Trends Endocrinol. Metab. 29, 781–794. doi: 10.1016/j.tem.2018.09.003
Gu, N., Wang, J., Di, Z., Liu, Z., Jia, X., Yan, Y., et al. (2019). The effects of intelectin-1 on antioxidant and angiogenesis in HUVECs exposed to oxygen glucose deprivation. Front. Neurol. 10:383. doi: 10.3389/fneur.2019.00383
Hadi, H. A., and Suwaidi, J. A. (2007). Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 3, 853–876.
Hafner, A., Bulyk, M. L., Jambhekar, A., and Lahav, G. (2019). The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210. doi: 10.1038/s41580-019-0110-x
Hagberg, C., Mehlem, A., Falkevall, A., Muhl, L., and Eriksson, U. (2013). Endothelial fatty acid transport: role of vascular endothelial growth factor B. Physiology 28, 125–134. doi: 10.1152/physiol.00042.2012
Hagberg, C. E., Falkevall, A., Wang, X., Larsson, E., Huusko, J., Nilsson, I., et al. (2010). Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464, 917–921. doi: 10.1038/nature08945
Hasegawa, Y., Saito, T., Ogihara, T., Ishigaki, Y., Yamada, T., Imai, J., et al. (2012). Blockade of the nuclear factor-kappaB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation 125, 1122–1133. doi: 10.1161/CIRCULATIONAHA.111.054346
Hayashi, T., Esaki, T., Sumi, D., Mukherjee, T., Iguchi, A., and Chaudhuri, G. (2006). Modulating role of estradiol on arginase II expression in hyperlipidemic rabbits as an atheroprotective mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 10485–10490. doi: 10.1073/pnas.0603918103
Hayashi, Y., Sawa, Y., Fukuyama, N., Nakazawa, H., and Matsuda, H. (2002). Preoperative glutamine administration induces heat-shock protein 70 expression and attenuates cardiopulmonary bypass-induced inflammatory response by regulating nitric oxide synthase activity. Circulation 106, 2601–2607. doi: 10.1161/01.cir.0000035651.72240.07
He, Q. W., Xia, Y. P., Chen, S. C., Wang, Y., Huang, M., Huang, Y., et al. (2013). Astrocyte-derived sonic hedgehog contributes to angiogenesis in brain microvascular endothelial cells via RhoA/ROCK pathway after oxygen-glucose deprivation. Mol. Neurobiol. 47, 976–987. doi: 10.1007/s12035-013-8396-8
Heitzer, E., Haque, I. S., Roberts, C. E. S., and Speicher, M. R. (2019). Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 20, 71–88. doi: 10.1038/s41576-018-0071-5
Ho, H. Y., Cheng, M. L., Lu, F. J., Chou, Y. H., Stern, A., Liang, C. M., et al. (2000). Enhanced oxidative stress and accelerated cellular senescence in glucose-6-phosphate dehydrogenase (G6PD)-deficient human fibroblasts. Free Radic. Biol. Med. 29, 156–169.
Hoare, M., Ito, Y., Kang, T. W., Weekes, M. P., Matheson, N. J., Patten, D. A., et al. (2016). NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 18, 979–992. doi: 10.1038/ncb3397
Huang, H., Vandekeere, S., Kalucka, J., Bierhansl, L., Zecchin, A., Bruning, U., et al. (2017). Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 36, 2334–2352. doi: 10.15252/embj.201695518
Huang, T., Liu, R., Fu, X., Yao, D., Yang, M., Liu, Q., et al. (2017). Aging reduces an ERRalpha-directed mitochondrial glutaminase expression suppressing glutamine anaplerosis and osteogenic differentiation of mesenchymal stem cells. Stem Cells 35, 411–424. doi: 10.1002/stem.2470
Huang, P. L., Huang, Z., Mashimo, H., Bloch, K. D., Moskowitz, M. A., Bevan, J. A., et al. (1995). Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377, 239–242. doi: 10.1038/377239a0
Igami, K., Uchiumi, T., Ueda, S., Kamioka, K., Setoyama, D., Gotoh, K., et al. (2019). Characterization and function of medium and large extracellular vesicles from plasma and urine by surface antigens and Annexin V. bioRxiv [Preprint]. doi: 10.1101/623553
Jabs, M., Rose, A. J., Lehmann, L. H., Taylor, J., Moll, I., Sijmonsma, T. P., et al. (2018). Inhibition of endothelial notch signaling impairs fatty acid transport and leads to metabolic and vascular remodeling of the adult heart. Circulation 137, 2592–2608. doi: 10.1161/CIRCULATIONAHA.117.029733
Jakobsson, L., Franco, C. A., Bentley, K., Collins, R. T., Ponsioen, B., Aspalter, I. M., et al. (2010). Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 12, 943–953. doi: 10.1038/ncb2103
James, E. L., Michalek, R. D., Pitiyage, G. N., de Castro, A. M., Vignola, K. S., Jones, J., et al. (2015). Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 14, 1854–1871. doi: 10.1021/pr501221g
Jansen, F., Li, Q., Pfeifer, A., and Werner, N. (2017). Endothelial- and immune cell-derived extracellular vesicles in the regulation of cardiovascular health and disease. JACC Basic Transl. Sci. 2, 790–807. doi: 10.1016/j.jacbts.2017.08.004
Jiang, P., Du, W., Mancuso, A., Wellen, K. E., and Yang, X. (2013). Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493, 689–693. doi: 10.1038/nature11776
Jimenez-Valerio, G., and Casanovas, O. (2017). Angiogenesis and metabolism: entwined for therapy resistance. Trends Cancer 3, 10–18. doi: 10.1016/j.trecan.2016.11.007
Kalucka, J., Bierhansl, L., Conchinha, N. V., Missiaen, R., Elia, I., Bruning, U., et al. (2018). Quiescent endothelial cells upregulate fatty acid beta-oxidation for vasculoprotection via redox homeostasis. Cell Metab. 28, 881–894.e13. doi: 10.1016/j.cmet.2018.07.016
Kanwal, A. (2018). Functional and therapeutic potential of mitochondrial SIRT3 deacetylase in disease conditions. Expert Rev. Clin. Pharmacol. 11, 1151–1155. doi: 10.1080/17512433.2018.1546119
Kaplon, J., Zheng, L., Meissl, K., Chaneton, B., Selivanov, V. A., Mackay, G., et al. (2013). A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112. doi: 10.1038/nature12154
Kennedy, B. K., Berger, S. L., Brunet, A., Campisi, J., Cuervo, A. M., Epel, E. S., et al. (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. doi: 10.1016/j.cell.2014.10.039
Kershaw, S., Cummings, J., Morris, K., Tugwood, J., and Dive, C. (2015). Optimisation of immunofluorescence methods to determine MCT1 and MCT4 expression in circulating tumour cells. BMC Cancer 15:387. doi: 10.1186/s12885-015-1382-y
Kim, Y. M., Shin, H. T., Seo, Y. H., Byun, H. O., Yoon, S. H., Lee, I. K., et al. (2010). Sterol regulatory element-binding protein (SREBP)-1-mediated lipogenesis is involved in cell senescence. J. Biol. Chem. 285, 29069–29077. doi: 10.1074/jbc.M110.120386
Kincaid, B., and Bossy-Wetzel, E. (2013). Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 5:48. doi: 10.3389/fnagi.2013.00048
Kokubun, T., Saitoh, S. I., Miura, S., Ishida, T., and Takeishi, Y. (2019). Telomerase plays a pivotal role in collateral growth under ischemia by suppressing age-induced oxidative stress, expression of p53, and pro-apoptotic proteins. Int. Heart J. 60, 736–745. doi: 10.1536/ihj.18-564
Kraus, W. E., Bhapkar, M., Huffman, K. M., Pieper, C. F., Krupa Das, S., Redman, L. M., et al. (2019). 2 years of calorie restriction and cardiometabolic risk (CALERIE): exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol. 7, 673–683. doi: 10.1016/S2213-8587(19)30151-2
Kubota, T., Kubota, N., Kumagai, H., Yamaguchi, S., Kozono, H., Takahashi, T., et al. (2011). Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 13, 294–307. doi: 10.1016/j.cmet.2011.01.018
Kuosmanen, S. M., Sihvola, V., Kansanen, E., Kaikkonen, M. U., and Levonen, A. L. (2018). MicroRNAs mediate the senescence-associated decline of NRF2 in endothelial cells. Redox Biol. 18, 77–83. doi: 10.1016/j.redox.2018.06.007
Lamichane, S., Baek, S. H., Kim, Y.-J., Park, J. H., Dahal Lamichane, B., Jang, W. B., et al. (2019). MHY2233 attenuates replicative cellular senescence in human endothelial progenitor cells via SIRT1 signaling. Oxid. Med. Cell. Longev. 2019:6492029. doi: 10.1155/2019/6492029
Lane, R. K., Hilsabeck, T., and Rea, S. L. (2015). The role of mitochondrial dysfunction in age-related diseases. Biochim. Biophys. Acta 1847, 1387–1400. doi: 10.1016/j.bbabio.2015.05.021
Lang, K. S., Mueller, M. M., Tanneur, V., Wallisch, S., Fedorenko, O., Palmada, M., et al. (2003). Regulation of cytosolic pH and lactic acid release in mesangial cells overexpressing GLUT1. Kidney Int. 64, 1338–1347. doi: 10.1046/j.1523-1755.2003.00213.x
Lee, D. C., Sohn, H. A., Park, Z. Y., Oh, S., Kang, Y. K., Lee, K. M., et al. (2015). A lactate-induced response to hypoxia. Cell 161, 595–609. doi: 10.1016/j.cell.2015.03.011
Lee, S. M., Dho, S. H., Ju, S. K., Maeng, J. S., Kim, J. Y., and Kwon, K. S. (2012). Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology 13, 525–536. doi: 10.1007/s10522-012-9397-0
Lesniewski, L. A., Seals, D. R., Walker, A. E., Henson, G. D., Blimline, M. W., Trott, D. W., et al. (2017). Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 16, 17–26. doi: 10.1111/acel.12524
Liu, H., Chen, T., Li, N., Wang, S., and Bu, P. (2015). Role of SIRT3 in Angiotensin II-induced human umbilical vein endothelial cells dysfunction. BMC Cardiovasc. Disord. 15:81. doi: 10.1186/s12872-015-0075-4
Lopez-Otin, C., Galluzzi, L., Freije, J. M. P., Madeo, F., and Kroemer, G. (2016). Metabolic control of longevity. Cell 166, 802–821. doi: 10.1016/j.cell.2016.07.031
Mapanga, R. F., and Essop, M. F. (2016). Damaging effects of hyperglycemia on cardiovascular function: spotlight on glucose metabolic pathways. Am. J. Physiol. Heart Circ. Physiol. 310, H153–H173. doi: 10.1152/ajpheart.00206.2015
Masi, S., Colucci, R., Duranti, E., Nannipieri, M., Anselmino, M., Ippolito, C., et al. (2018). Aging modulates the influence of arginase on endothelial dysfunction in obesity. Arterioscler. Thromb. Vasc. Biol. 38, 2474–2483. doi: 10.1161/ATVBAHA.118.311074
Massudi, H., Grant, R., Braidy, N., Guest, J., Farnsworth, B., and Guillemin, G. J. (2012). Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7:e42357. doi: 10.1371/journal.pone.0042357
Mehrotra, D., Wu, J., Papangeli, I., and Chun, H. J. (2014). Endothelium as a gatekeeper of fatty acid transport. Trends Endocrinol. Metab. 25, 99–106. doi: 10.1016/j.tem.2013.11.001
Melone, M. A. B., Valentino, A., Margarucci, S., Galderisi, U., Giordano, A., and Peluso, G. (2018). The carnitine system and cancer metabolic plasticity. Cell Death Dis. 9:228. doi: 10.1038/s41419-018-0313-7
Meynial-Denis, D. (2016). Glutamine metabolism in advanced age. Nutr. Rev. 74, 225–236. doi: 10.1093/nutrit/nuv052
Michell, D. L., Andrews, K. L., and Chin-Dusting, J. P. (2011). Endothelial dysfunction in hypertension: the role of arginase. Front. Biosci. 3, 946–960. doi: 10.2741/199
Miguel-Carrasco, J. L., Monserrat, M. T., Mate, A., and Vazquez, C. M. (2010). Comparative effects of captopril and l-carnitine on blood pressure and antioxidant enzyme gene expression in the heart of spontaneously hypertensive rats. Eur. J. Pharmacol. 632, 65–72. doi: 10.1016/j.ejphar.2010.01.017
Minamino, T., Orimo, M., Shimizu, I., Kunieda, T., Yokoyama, M., Ito, T., et al. (2009). A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 15, 1082–1087. doi: 10.1038/nm.2014
Mitchell, S. J., Bernier, M., Mattison, J. A., Aon, M. A., Kaiser, T. A., Anson, R. M., et al. (2019). Daily fasting improves health and survival in male mice independent of diet composition and calories. Cell Metab. 29, 221–228.e3. doi: 10.1016/j.cmet.2018.08.011
Montoliu, I., Scherer, M., Beguelin, F., DaSilva, L., Mari, D., Salvioli, S., et al. (2014). Serum profiling of healthy aging identifies phospho- and sphingolipid species as markers of human longevity. Aging 6, 9–25. doi: 10.18632/aging.100630
Morgan, R. G., Ives, S. J., Lesniewski, L. A., Cawthon, R. M., Andtbacka, R. H., Noyes, R. D., et al. (2013). Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am. J. Physiol. Heart Circ. Physiol. 305, H251–H258. doi: 10.1152/ajpheart.00197.2013
Mork, M., Andreasen, J. J., Rasmussen, L. H., Lip, G. Y. H., Pedersen, S., Baek, R., et al. (2019). Elevated blood plasma levels of tissue factor-bearing extracellular vesicles in patients with atrial fibrillation. Thromb. Res. 173, 141–150. doi: 10.1016/j.thromres.2018.11.026
Munoz-Chapuli, R., Quesada, A. R., and Angel Medina, M. (2004). Angiogenesis and signal transduction in endothelial cells. Cell. Mol. Life Sci. 61, 2224–2243.
Nacarelli, T., Lau, L., Fukumoto, T., Zundell, J., Fatkhutdinov, N., Wu, S., et al. (2019). NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 21, 397–407. doi: 10.1038/s41556-019-0287-4
Niewczas, M. A., Pavkov, M. E., Skupien, J., Smiles, A., Md Dom, Z. I., Wilson, J. M., et al. (2019). A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 25, 805–813. doi: 10.1038/s41591-019-0415-5
Ning, W. H., and Zhao, K. (2013). Propionyl-L-carnitine induces eNOS activation and nitric oxide synthesis in endothelial cells via PI3 and Akt kinases. Vascul. Pharmacol. 59, 76–82. doi: 10.1016/j.vph.2013.07.001
Nobrega-Pereira, S., Fernandez-Marcos, P. J., Brioche, T., Gomez-Cabrera, M. C., Salvador-Pascual, A., Flores, J. M., et al. (2016). G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 7:10894. doi: 10.1038/ncomms10894
Nogueira-Recalde, U., Lorenzo-Gomez, I., Blanco, F. J., Loza, M. I., Grassi, D., Shirinsky, V., et al. (2019). Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 45, 588–605. doi: 10.1016/j.ebiom.2019.06.049
Olenchock, B. A., and Vander Heiden, M. G. (2013). Pyruvate as a pivot point for oncogene-induced senescence. Cell 153, 1429–1430. doi: 10.1016/j.cell.2013.06.001
Oliveira, S. D. S., Chen, J., Castellon, M., Mao, M., Raj, J. U., Comhair, S., et al. (2019). Injury-induced shedding of extracellular vesicles depletes endothelial cells of Cav-1 (Caveolin-1) and enables TGF-beta (transforming growth factor-beta)-dependent pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 39, 1191–1202. doi: 10.1161/ATVBAHA.118.312038
Olivieri, F., Pompilio, G., and Balistreri, C. R. (2016). Endothelial progenitor cells in ageing. Mech. Ageing Dev. 159, 1–3. doi: 10.1016/j.mad.2016.09.002
Olivieri, F., Prattichizzo, F., Grillari, J., and Balistreri, C. R. (2018). Cellular senescence and inflammaging in age-related diseases. Mediators Inflamm. 2018:9076485. doi: 10.1155/2018/9076485
Orimo, M., Minamino, T., Miyauchi, H., Tateno, K., Okada, S., Moriya, J., et al. (2009). Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 29, 889–894. doi: 10.1161/ATVBAHA.109.185694
Peyton, K. J., Liu, X. M., Yu, Y., Yates, B., Behnammanesh, G., and Durante, W. (2018). Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem. Pharmacol. 156, 204–214. doi: 10.1016/j.bcp.2018.08.032
Poirier, P., Giles, T. D., Bray, G. A., Hong, Y., Stern, J. S., Pi-Sunyer, F. X., et al. (2006). Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 113, 898–918. doi: 10.1161/CIRCULATIONAHA.106.171016
Potente, M., and Carmeliet, P. (2017). The Link between angiogenesis and endothelial metabolism. Annu. Rev. Physiol. 79, 43–66. doi: 10.1146/annurev-physiol-021115-105134
Prattichizzo, F. (2019). Ageing as a druggable process: moving forward. EBioMedicine 40, 15–16. doi: 10.1016/j.ebiom.2019.01.025
Prattichizzo, F., De Nigris, V., Mancuso, E., Spiga, R., Giuliani, A., Matacchione, G., et al. (2018a). Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 15, 170–181. doi: 10.1016/j.redox.2017.12.001
Prattichizzo, F., De Nigris, V., Micheloni, S., La Sala, L., and Ceriello, A. (2018b). Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: is low-grade inflammation the neglected component? Diabetes Obes. Metab. 20, 2515–2522. doi: 10.1111/dom.13488
Prattichizzo, F., De Nigris, V., Spiga, R., Mancuso, E., La Sala, L., Antonicelli, R., et al. (2018c). Inflammageing and metaflammation: the yin and yang of type 2 diabetes. Ageing Res. Rev. 41, 1–17. doi: 10.1016/j.arr.2017.10.003
Prattichizzo, F., Giuliani, A., Mensa, E., Sabbatinelli, J., De Nigris, V., Rippo, M. R., et al. (2018d). Pleiotropic effects of metformin: shaping the microbiome to manage type 2 diabetes and postpone ageing. Ageing Res. Rev. 48, 87–98. doi: 10.1016/j.arr.2018.10.003
Prattichizzo, F., Giuliani, A., Sabbatinelli, J., Mensa, E., De Nigris, V., La Sala, L., et al. (2019). Extracellular vesicles circulating in young organisms promote healthy longevity. J. Extracell. Vesicles 8:1656044. doi: 10.1080/20013078.2019.1656044
Quijano, C., Cao, L., Fergusson, M. M., Romero, H., Liu, J., Gutkind, S., et al. (2012). Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 11, 1383–1392. doi: 10.4161/cc.19800
Ramasamy, S. K., Kusumbe, A. P., Schiller, M., Zeuschner, D., Bixel, M. G., Milia, C., et al. (2016). Blood flow controls bone vascular function and osteogenesis. Nat. Commun. 7:13601. doi: 10.1038/ncomms13601
Redman, L. M., Smith, S. R., Burton, J. H., Martin, C. K., Il’yasova, D., and Ravussin, E. (2018). Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab. 27, 805–815.e4. doi: 10.1016/j.cmet.2018.02.019
Redman, R., Pohlmann, P., Kurman, M., Tapolsky, G. H., and Chesney, J. (2015). Abstract CT206: PFK-158, first-in-man and first-in-class inhibitor of PFKFB3/glycolysis: a phase I, dose escalation, multi-center study in patients with advanced solid malignancies. Cancer Res. 75(Suppl. 15):CT206. doi: 10.1158/1538-7445.Am2015-ct206
Robciuc, M. R., Kivela, R., Williams, I. M., de Boer, J. F., van Dijk, T. H., Elamaa, H., et al. (2016). VEGFB/VEGFR1-induced expansion of adipose vasculature counteracts obesity and related metabolic complications. Cell Metab. 23, 712–724. doi: 10.1016/j.cmet.2016.03.004
Roche, T. E., and Hiromasa, Y. (2007). Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell. Mol. Life Sci. 64, 830–849. doi: 10.1007/s00018-007-6380-z
Sabbatinelli, J., Vignini, A., Salvolini, E., Nanetti, L., Mazzanti, L., and Anna Rabini, R. (2017). Platelet-derived no in subjects affected by type 2 diabetes with and without complications: is there any relationship with their offspring? Exp. Clin. Endocrinol. Diabetes 125, 290–296. doi: 10.1055/s-0043-102578
Salminen, A., Kauppinen, A., and Kaarniranta, K. (2012). Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 24, 835–845. doi: 10.1016/j.cellsig.2011.12.006
Santos-Gallego, C. G., Requena-Ibanez, J. A., San Antonio, R., Ishikawa, K., Watanabe, S., Picatoste, B., et al. (2019). Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J. Am. Coll. Cardiol. 73, 1931–1944. doi: 10.1016/j.jacc.2019.01.056
Saraste, M. (1999). Oxidative phosphorylation at the fin de siecle. Science 283, 1488–1493. doi: 10.1126/science.283.5407.1488
Schlereth, K., Weichenhan, D., Bauer, T., Heumann, T., Giannakouri, E., Lipka, D., et al. (2018). The transcriptomic and epigenetic map of vascular quiescence in the continuous lung endothelium. eLife 7:e34423. doi: 10.7554/eLife.34423
Schoors, S., Bruning, U., Missiaen, R., Queiroz, K. C., Borgers, G., Elia, I., et al. (2015). Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520, 192–197. doi: 10.1038/nature14362
Schulman, S. P., Becker, L. C., Kass, D. A., Champion, H. C., Terrin, M. L., Forman, S., et al. (2006). L-arginine therapy in acute myocardial infarction: the vascular interaction with age in myocardial infarction (VINTAGE MI) randomized clinical trial. JAMA 295, 58–64. doi: 10.1001/jama.295.1.58
Seki, T., Hosaka, K., Fischer, C., Lim, S., Andersson, P., Abe, M., et al. (2018). Ablation of endothelial VEGFR1 improves metabolic dysfunction by inducing adipose tissue browning. J. Exp. Med. 215, 611–626. doi: 10.1084/jem.20171012
Sharma, S., Aramburo, A., Rafikov, R., Sun, X., Kumar, S., Oishi, P. E., et al. (2013). L-carnitine preserves endothelial function in a lamb model of increased pulmonary blood flow. Pediatr. Res. 74, 39–47. doi: 10.1038/pr.2013.71
Shemyakin, A., Kovamees, O., Rafnsson, A., Bohm, F., Svenarud, P., Settergren, M., et al. (2012). Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation 126, 2943–2950. doi: 10.1161/CIRCULATIONAHA.112.140335
Siragusa, M., Thole, J., Bibli, S. I., Luck, B., Loot, A. E., de Silva, K., et al. (2019). Nitric oxide maintains endothelial redox homeostasis through PKM2 inhibition. EMBO J. 38:e100938. doi: 10.15252/embj.2018100938
Søgaard, P. P., and Gil, J. (2019). NAD+: a metabolic knob fine-tuning inflammation during senescence. Nat. Metab. 1, 310–311.
Sohn, M., Kim, K., Uddin, M. J., Lee, G., Hwang, I., Kang, H., et al. (2017). Delayed treatment with fenofibrate protects against high-fat diet-induced kidney injury in mice: the possible role of AMPK autophagy. Am. J. Physiol. Renal Physiol. 312, F323–F334. doi: 10.1152/ajprenal.00596.2015
Son, M. J., Kwon, Y., Son, T., and Cho, Y. S. (2016). Restoration of mitochondrial NAD+ levels delays stem cell senescence and facilitates reprogramming of aged somatic cells. Stem Cells 34, 2840–2851. doi: 10.1002/stem.2460
Son, N. H., Basu, D., Samovski, D., Pietka, T. A., Peche, V. S., Willecke, F., et al. (2018). Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Invest. 128, 4329–4342. doi: 10.1172/JCI99315
Sonveaux, P., Copetti, T., De Saedeleer, C. J., Vegran, F., Verrax, J., Kennedy, K. M., et al. (2012). Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 7:e33418. doi: 10.1371/journal.pone.0033418
Speakman, J. R. (2005). Body size, energy metabolism and lifespan. J. Exp. Biol. 208(Pt 9), 1717–1730. doi: 10.1242/jeb.01556
Stubbs, C. O., and Lee, A. J. (2004). The obesity epidemic: both energy intake and physical activity contribute. Med. J. Aust. 181, 489–491.
Su, S. T., Yeh, C. L., Hou, Y. C., Pai, M. H., and Yeh, S. L. (2017). Dietary glutamine supplementation enhances endothelial progenitor cell mobilization in streptozotocin-induced diabetic mice subjected to limb ischemia. J. Nutr. Biochem. 40, 86–94. doi: 10.1016/j.jnutbio.2016.10.010
Su, Y., Wang, T., Wu, N., Li, D., Fan, X., Xu, Z., et al. (2019). Alpha-ketoglutarate extends Drosophila lifespan by inhibiting mTOR and activating AMPK. Aging 11, 4183–4197. doi: 10.18632/aging.102045
Sun, K., Wernstedt Asterholm, I., Kusminski, C. M., Bueno, A. C., Wang, Z. V., Pollard, J. W., et al. (2012). Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc. Natl. Acad. Sci. U.S.A. 109, 5874–5879. doi: 10.1073/pnas.1200447109
Takebayashi, S., Tanaka, H., Hino, S., Nakatsu, Y., Igata, T., Sakamoto, A., et al. (2015). Retinoblastoma protein promotes oxidative phosphorylation through upregulation of glycolytic genes in oncogene-induced senescent cells. Aging Cell 14, 689–697. doi: 10.1111/acel.12351
Tarrago, M. G., Chini, C. C. S., Kanamori, K. S., Warner, G. M., Caride, A., de Oliveira, G. C., et al. (2018). A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab. 27, 1081–1095.e10. doi: 10.1016/j.cmet.2018.03.016
Terashima, J., Tachikawa, C., Kudo, K., Habano, W., and Ozawa, S. (2013). An aryl hydrocarbon receptor induces VEGF expression through ATF4 under glucose deprivation in HepG2. BMC Mol. Biol. 14:27. doi: 10.1186/1471-2199-14-27
Thery, C., Witwer, K. W., Aikawa, E., Alcaraz, M. J., Anderson, J. D., Andriantsitohaina, R., et al. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7:1535750. doi: 10.1080/20013078.2018.1535750
Tian, X. L., and Li, Y. (2014). Endothelial cell senescence and age-related vascular diseases. J. Genet. Genomics 41, 485–495. doi: 10.1016/j.jgg.2014.08.001
Tirumani, S. H., Fairchild, A., Krajewski, K. M., Nishino, M., Howard, S. A., Baheti, A. D., et al. (2015). Anti-VEGF molecular targeted therapies in common solid malignancies: comprehensive update for radiologists. Radiographics 35, 455–474. doi: 10.1148/rg.352140119
Unterluggauer, H., Mazurek, S., Lener, B., Hutter, E., Eigenbrodt, E., Zwerschke, W., et al. (2008). Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology 9, 247–259. doi: 10.1007/s10522-008-9134-x
Utermohlen, O., Jakobshagen, K., Blissenbach, B., Wiegmann, K., Merz, T., Hefti, J. P., et al. (2019). Emergence of AnnexinVpos CD31neg CD42blow/neg extracellular vesicles in plasma of humans at extreme altitude. PLoS One 14:e0220133. doi: 10.1371/journal.pone.0220133
Van den Bossche, J., O’Neill, L. A., and Menon, D. (2017). Macrophage immunometabolism: where are we (going)? Trends Immunol. 38, 395–406. doi: 10.1016/j.it.2017.03.001
Vegran, F., Boidot, R., Michiels, C., Sonveaux, P., and Feron, O. (2011). Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 71, 2550–2560. doi: 10.1158/0008-5472.CAN-10-2828
Verdin, E. (2015). NAD+ in aging, metabolism, and neurodegeneration. Science 350, 1208–1213. doi: 10.1126/science.aac4854
Verdin, E., Hirschey, M. D., Finley, L. W., and Haigis, M. C. (2010). Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem. Sci. 35, 669–675. doi: 10.1016/j.tibs.2010.07.003
Vicent, D., Ilany, J., Kondo, T., Naruse, K., Fisher, S. J., Kisanuki, Y. Y., et al. (2003). The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J. Clin. Invest. 111, 1373–1380. doi: 10.1172/JCI15211
Villaret, A., Galitzky, J., Decaunes, P., Esteve, D., Marques, M. A., Sengenes, C., et al. (2010). Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 59, 2755–2763. doi: 10.2337/db10-0398
Virgili, G., Parravano, M., Evans, J. R., Gordon, I., and Lucenteforte, E. (2018). Anti-vascular endothelial growth factor for diabetic macular oedema: a network meta-analysis. Cochrane Database Syst. Rev. 10:CD007419. doi: 10.1002/14651858.CD007419.pub6
Walker, A. E., Kaplon, R. E., Lucking, S. M., Russell-Nowlan, M. J., Eckel, R. H., and Seals, D. R. (2012). Fenofibrate improves vascular endothelial function by reducing oxidative stress while increasing endothelial nitric oxide synthase in healthy normolipidemic older adults. Hypertension 60, 1517–1523. doi: 10.1161/HYPERTENSIONAHA.112.203661
Wang, R., Liu, L., Liu, H., Wu, K., Liu, Y., Bai, L., et al. (2019). Reduced NRF2 expression suppresses endothelial progenitor cell function and induces senescence during aging. Aging 11, 7021–7035. doi: 10.18632/aging.102234
Weichhart, T. (2018). mTOR as regulator of lifespan, aging, and cellular senescence: a mini-review. Gerontology 64, 127–134. doi: 10.1159/000484629
Weilner, S., Schraml, E., Wieser, M., Messner, P., Schneider, K., Wassermann, K., et al. (2016). Secreted microvesicular miR-31 inhibits osteogenic differentiation of mesenchymal stem cells. Aging Cell 15, 744–754. doi: 10.1111/acel.12484
Wiley, C. D., and Campisi, J. (2016). From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 23, 1013–1021. doi: 10.1016/j.cmet.2016.05.010
Wiley, C. D., Velarde, M. C., Lecot, P., Liu, S., Sarnoski, E. A., Freund, A., et al. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314. doi: 10.1016/j.cmet.2015.11.011
Wilhelm, K., Happel, K., Eelen, G., Schoors, S., Oellerich, M. F., Lim, R., et al. (2016). FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529, 216–220. doi: 10.1038/nature16498
Williams, D. S., Cash, A., Hamadani, L., and Diemer, T. (2009). Oxaloacetate supplementation increases lifespan in Caenorhabditis elegans through an AMPK/FOXO-dependent pathway. Aging Cell 8, 765–768. doi: 10.1111/j.1474-9726.2009.00527.x
Wu, T.-W., Liu, C.-C., Hung, C.-L., Yen, C.-H., Wu, Y.-J., Wang, L.-Y., et al. (2018). Genetic profiling of young and aged endothelial progenitor cells in hypoxia. PLoS One 13:e0196572. doi: 10.1371/journal.pone.0196572
Wu, Y. H., Cheng, M. L., Ho, H. Y., Chiu, D. T., and Wang, T. C. (2009). Telomerase prevents accelerated senescence in glucose-6-phosphate dehydrogenase (G6PD)-deficient human fibroblasts. J. Biomed. Sci. 16:18. doi: 10.1186/1423-0127-16-18
Wu, Z., Yu, Y., Liu, C., Xiong, Y., Montani, J. P., Yang, Z., et al. (2015). Role of p38 mitogen-activated protein kinase in vascular endothelial aging: interaction with Arginase-II and S6K1 signaling pathway. Aging 7, 70–81. doi: 10.18632/aging.100722
Xin, R., An, D., Li, Y., Fu, J., Huang, F., and Zhu, Q. (2019). Fenofibrate improves vascular endothelial function in diabetic mice. Biomed. Pharmacother. 112:108722. doi: 10.1016/j.biopha.2019.108722
Xing, S. S., Li, J., Chen, L., Yang, Y. F., He, P. L., Li, J., et al. (2018). Salidroside attenuates endothelial cellular senescence via decreasing the expression of inflammatory cytokines and increasing the expression of SIRT3. Mech. Ageing Dev. 175, 1–6. doi: 10.1016/j.mad.2017.12.005
Xiong, J., Kawagishi, H., Yan, Y., Liu, J., Wells, Q. S., Edmunds, L. R., et al. (2018). A metabolic basis for endothelial-to-mesenchymal transition. Mol. Cell 69, 689–698.e7. doi: 10.1016/j.molcel.2018.01.010
Xiong, Y., Yepuri, G., Montani, J. P., Ming, X. F., and Yang, Z. (2017). Arginase-II deficiency extends lifespan in mice. Front. Physiol. 8:682. doi: 10.3389/fphys.2017.00682
Xu, N., Wang, Q., Jiang, S., Wang, Q., Hu, W., Zhou, S., et al. (2019). Fenofibrate improves vascular endothelial function and contractility in diabetic mice. Redox Biol. 20, 87–97. doi: 10.1016/j.redox.2018.09.024
Yokoyama, M., Okada, S., Nakagomi, A., Moriya, J., Shimizu, I., Nojima, A., et al. (2014). Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Rep. 7, 1691–1703. doi: 10.1016/j.celrep.2014.04.046
Yokoyama, M., Shimizu, I., Nagasawa, A., Yoshida, Y., Katsuumi, G., Wakasugi, T., et al. (2019). p53 plays a crucial role in endothelial dysfunction associated with hyperglycemia and ischemia. J. Mol. Cell. Cardiol. 129, 105–117. doi: 10.1016/j.yjmcc.2019.02.010
Yoshida, M., Satoh, A., Lin, J. B., Mills, K. F., Sasaki, Y., Rensing, N., et al. (2019). Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab. 30, 329–342.e5.
Yu, P., Wilhelm, K., Dubrac, A., Tung, J. K., Alves, T. C., Fang, J. S., et al. (2017). FGF-dependent metabolic control of vascular development. Nature 545, 224–228. doi: 10.1038/nature22322
Zhang, H., Ryu, D., Wu, Y., Gariani, K., Wang, X., Luan, P., et al. (2016). NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. doi: 10.1126/science.aaf2693
Zhang, Z., Liew, C. W., Handy, D. E., Zhang, Y., Leopold, J. A., Hu, J., et al. (2010). High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. FASEB J. 24, 1497–1505. doi: 10.1096/fj.09-136572
Zhen, L., Jiang, X., Chen, Y., and Fan, D. (2017). MiR-31 is involved in the high glucose-suppressed osteogenic differentiation of human periodontal ligament stem cells by targeting Satb2. Am. J. Transl. Res. 9, 2384–2393.
Zhu, X. H., Lu, M., Lee, B. Y., Ugurbil, K., and Chen, W. (2015). In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. U.S.A. 112, 2876–2881. doi: 10.1073/pnas.1417921112
Zierer, J., Menni, C., Kastenmuller, G., and Spector, T. D. (2015). Integration of ‘omics’ data in aging research: from biomarkers to systems biology. Aging Cell 14, 933–944. doi: 10.1111/acel.12386
Keywords: cellular senescence, endothelial cells, age-related diseases, type 2 diabetes, metabolism, glycolysis
Citation: Sabbatinelli J, Prattichizzo F, Olivieri F, Procopio AD, Rippo MR and Giuliani A (2019) Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 10:1523. doi: 10.3389/fphys.2019.01523
Received: 27 September 2019; Accepted: 03 December 2019;
Published: 18 December 2019.
Edited by:
Katia Aquilano, University of Rome Tor Vergata, ItalyReviewed by:
Ippei Shimizu, Niigata University, JapanCopyright © 2019 Sabbatinelli, Prattichizzo, Olivieri, Procopio, Rippo and Giuliani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Rita Rippo, bS5yLnJpcHBvQHN0YWZmLnVuaXZwbS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.