 Xinguo Jiang
Xinguo Jiang Wen Tian
Wen Tian Mark R. Nicolls
Mark R. Nicolls Stanley G. Rockson
Stanley G. Rockson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 14 November 2019
Sec. Lipid and Fatty Acid Research
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01402
This article is part of the Research TopicThe Role of the Lymphatic System in Lipid and Energy Metabolism, and Immune Homeostasis During Obesity and DiabetesView all 15 articles
Obesity, insulin resistance, dyslipidemia, and hypertension are fundamental clinical manifestations of the metabolic syndrome. Studies over the last few decades have implicated chronic inflammation and microvascular remodeling in the development of obesity and insulin resistance. Newer observations, however, suggest that dysregulation of the lymphatic system underlies the development of the metabolic syndrome. This review summarizes recent advances in the field, discussing how lymphatic abnormality promotes obesity and insulin resistance, and, conversely, how the metabolic syndrome impairs lymphatic function. We also discuss lymphatic biology in metabolically dysregulated diseases, including type 2 diabetes, atherosclerosis, and myocardial infarction.
Obesity, characterized by increased storage of fatty acids in expanded adipose tissues, is becoming a major health problem in modern society, as humans increasingly embrace a relatively sedentary lifestyle. The chronic obese status predisposes individuals to the development of the metabolic syndrome and increases the incidence of type 2 diabetes (T2D) and cardiovascular diseases (Kahn and Flier, 2000; O’Neill and O’Driscoll, 2015; Catrysse and van Loo, 2017). Dysregulated lipid metabolism and low-grade chronic inflammation are among the notable pathologies in obese adipose tissue (Ruotolo and Howard, 2002; Konner and Bruning, 2011). More recently, there is increasing evidence that dysfunction of the lymphatic vasculature is involved in the pathogenesis of obesity and obesity-associated dyslipidemia and low-grade chronic inflammation (Harvey et al., 2005; Aspelund et al., 2016), presumably because the lymphatic system is important for immune homeostasis and lipid transport (Jiang et al., 2018). This review provides an overview of the interplay between the function of lymphatic system and presence of obesity and insulin resistance. We also discuss how the lymphatic system may be harnessed to treat T2D and cardiovascular diseases associated with obesity and insulin resistance.
The lymphatic system is comprised of lymphatic vessels and the secondary lymphoid organs that include lymph nodes, spleen, tonsils, and Peyer’s patches (Ruddle and Akirav, 2009; Choi et al., 2012). The lymphatic vasculature is a unidirectional circulatory network that begins as blunt-ended capillaries composed of a single layer of lymphatic endothelial cells (LECs) that tether directly to the interstitial tissue through anchoring filaments, with discontinuous basement membrane coverage (Jiang et al., 2018). Capillary LECs are interconnected by unique button-like structures that are formed by discontinuous layers of junctional proteins such as VE-cadherin, claudin, Zonula occludens-1 (ZO-1), connexin, and occludin (Baluk et al., 2007). The presence of these buttons serves to create overlapping LEC flaps that serve as de facto primary valves. In response to interstitial fluid pressure fluctuation, these flaps can open and close to regulate fluid reabsorption as well as the uptake of macromolecules and immune cells (Yao et al., 2012; Jiang et al., 2018). The lymphatic capillaries converge into precollectors and these in turn coalesce into the collecting lymphatics, in which LECs are joined by zipper-like, continuous, seamless junctions and invested with basement membrane along with smooth muscle cell coverage. Intraluminal lymphatic valves divide the collecting lymphatics into contractile segments designated as lymphangions, thus providing a structural basis for the unidirectional lymph flow (Aspelund et al., 2016; Figure 1). The collecting lymphatics travel through chains of lymph nodes, which allows the delivery of free antigens and antigen-loaded dendritic cells (DCs) from interstitial tissue for immune priming; the central lymphatic vasculature eventually joins the subclavian veins via the thoracic duct(s) conveying the lymph node-filtered interstitial fluid back to the blood circulatory system (Thomas et al., 2016; Jiang et al., 2018).
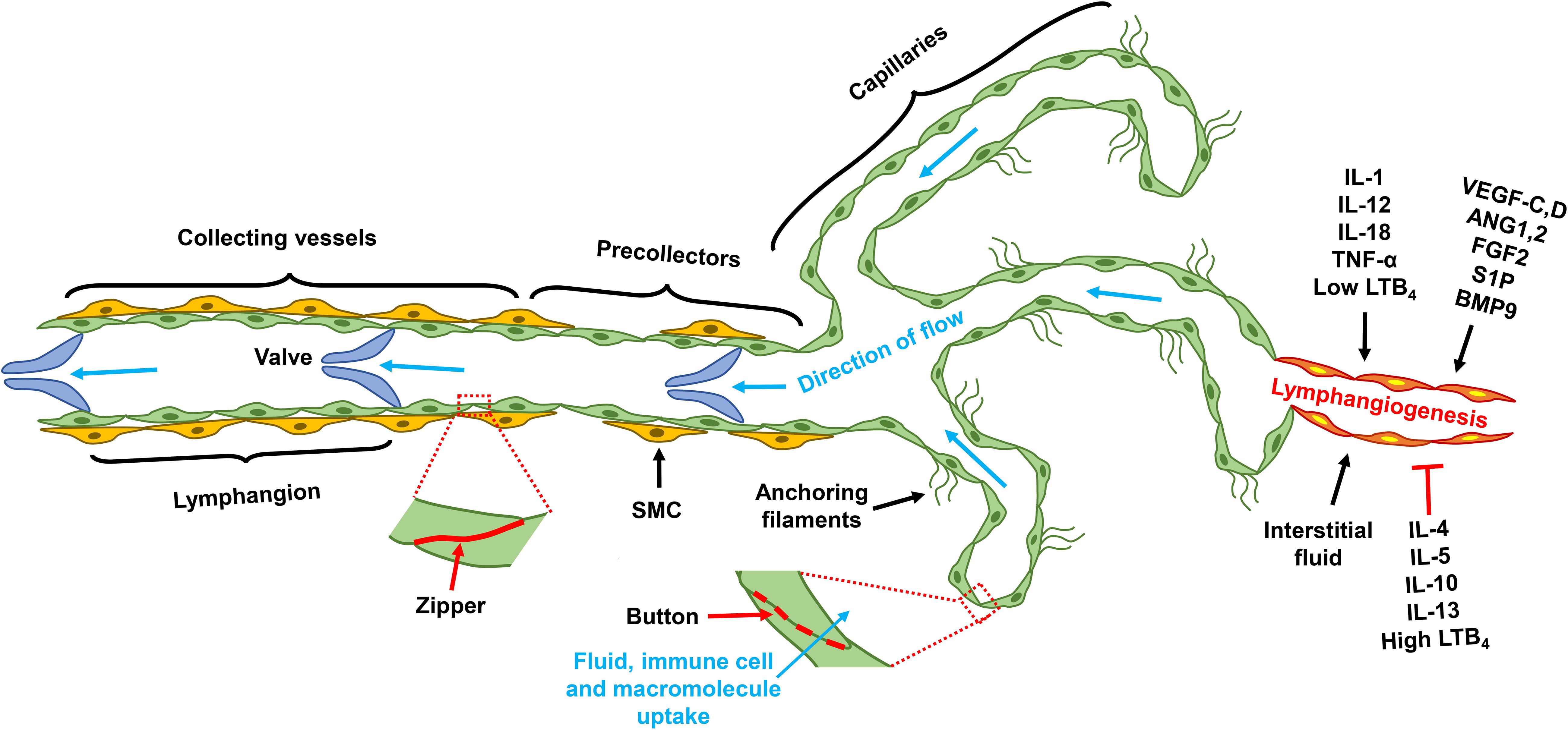
Figure 1. Lymphatic vascular tree and lymphangiogenesis. Lymphatic capillaries are comprised of single layer LECs that tether directly to the extra cellular matrix. Junctional proteins form button structure that interconnects capillary LECs; and those buttons allow the formation of overlapping LEC flap, through which interstitial fluid and macromolecule enter the blind-ends of the capillaries. Capillaries converge into pre-collectors, which in turn coalesces into collecting lymphatics. SMCs loosely cover the precollector, but invest collecting lymphatics more completely. Junctional proteins of the collecting lymphatics form a continuous structure, known as zipper. Collecting lymphatics are comprised of lymphangions that are demarcated by intravascular valves, the segment between two valves is designated as one lymphangion. Factor regulates lymphangiogenesis include pro-lymphangiogenic growth factors, pro-inflammatory cytokines, and interstitial fluid. Type 2 cytokines, and high concentrations of LTB4, are molecules that suppress lymphangiogenesis. LEC, lymphatic endothelial cell; VEGF, vascular endothelial growth factor; ANG, angiopoietin; FGF, fibroblast growth factor; S1P, sphingosine 1 phosphate; BMP, bone morphogenetic protein; IL, interleukin; LTB4, leukotriene B4; TNF, tumor necrosis factor; SMC, smooth muscle cell.
Lymphatic vasculature originates from the cardinal vein and subsequently develops independently from the blood circulatory system (Wigle and Oliver, 1999). The homeobox gene Prox-1 appears to be the master control gene for lymphatic differentiation and development (Choi et al., 2012). In E9.5 mouse embryos, Prox-1 expression begins in a subset of endothelial cells (ECs) of the cardinal vein and specifies them as LECs. Prox-1 then upregulates lymphatic markers such as LYVE-1, VEGFR3, and Chemokine (C–C motif) ligand (CCL)21, and concurrently downregulates blood vascular signature genes (Wigle et al., 2002). This molecular differentiation enables LEC budding from the cardinal vein to form the rudimentary lymphatic vessels, known as the jugular lymph sac, at E11.5 (Choi et al., 2012). Conditional Prox-1 downregulation reprograms LECs into blood endothelial cells (BECs) in both developing and adult mice as well as in cell culture (Johnson et al., 2008), supporting the notion that Prox-1 is an indispensable transcriptional factor for the maintenance of LEC identity. Following the initial specification and budding, the lymph sac then expands through lymphangiogenesis, a process of new lymphatic vessel sprouting from preexisting structures (Aspelund et al., 2016). VEGF-C/D-activated VEGFR3 signaling is the most central pathway for lymphangiogenesis (Zheng et al., 2014). VEGF-C deficiency leads to lymphatic insufficiency and lymphedema, a defect that can be rescued by VEGF-D during development (Karkkainen et al., 2004); VEGFR3 activation is also critical for pathophysiological lymphangiogenesis following lymphatic injury (Szuba et al., 2002). VEGFR3 missense mutations in the tyrosine kinase domains underlie the etiology of 70% cases of primary congenital lymphedema, known as Milroy disease (Karkkainen et al., 2000; Connell et al., 2009), indicating that the VEGFR3 signaling is important for the normal development of human lymphatic vasculature. The axon guidance protein neuropilin (NRP)-2 enhances VEGFR3 signaling by acting as its co-receptor (Yuan et al., 2002; Xu et al., 2010). By facilitating VEGF-C maturation, the collagen- and calcium-binding EGF domains 1 (CCBE1) protein also promotes lymphangiogenesis (Bos et al., 2011; Jha et al., 2017). Thus, both NRP2 and CCBE1 are factors involved in lymphangiogenesis by regulating the VEGF-C/VEGFR3 signaling.
Following the initial sprouting, lymphatic capillaries mature through activating the Notch signaling pathway (Zheng et al., 2011), resembling the well-known tip–stalk cell paradigm in angiogenesis (Potente et al., 2011). Maturation of collecting lymphatic vessels requires concerted smooth muscle cell recruitment and patterning as well as valve development. Although angiopoietin (ANG)2 antagonizes TIE2 receptor activation during blood angiogenesis, it appears to stimulate TIE2 signaling in LECs and to promote postnatal lymphatic remodeling (Gale et al., 2007); ANG2-deficient mice do not develop normal hierarchical lymphatic vascular system (Dellinger et al., 2008). The forkhead box protein FOXC2 modulates lymphatic capillary development by controlling SMC recruitment and basement membrane formation (Petrova et al., 2004). In coordination with Prox-1, FOXC2 senses lymph flow and induces the expression of gap junction protein connexin 37 (Cx37) and activates calcineurin/nuclear factor of activated T-cells (NFAT) signaling, which regulates lymphatic valve formation (Sabine et al., 2012). While FOXC2 controls ANG2 expression in angiogenesis (Xue et al., 2008), it induces TIE2 in LECs (Thomson et al., 2014), suggesting that FOXC2 may promote lymphatic maturation by regulating the ANG2/TIE2 signaling.
Other signaling pathways, including fibroblast growth factor (FGF)2/FGF receptor (FGFR)1, sphingosine-1-phosphate (S1P)/S1P receptor (S1PR)1, and bone morphogenetic protein (BMP)9/activin receptor-like kinase 1 (ALK1), also regulate lymphangiogenesis (Zheng et al., 2014). Additionally, pro-inflammatory cytokines influence lymphangiogenesis and affect immune activation and resolution. In general, type 1 cytokines including IL-1, IL12, IL-18, and TNF-α promote, and type 2 and anti-inflammatory cytokines such as IL-4, IL-5, IL13, and IL-10 suppress lymphangiogenesis (Sainz-Jaspeado and Claesson-Welsh, 2018). The pro-inflammatory lipid molecule leukotriene B4 (LTB4) promotes lymphatic regeneration at low concentration but suppresses lymphangiogenesis at high pathological concentrations (Tian et al., 2017). In summary, lymphangiogenesis is regulated by growth factors and cytokines, as well as by interstitial fluid flow (Figure 1).
Interaction of genetic, epigenetic, environmental, and psychological factors regulates the production of physiological mediators that control the balance of energy intake and expenditure (Gonzalez-Muniesa et al., 2017). When energy intake is in surplus, about 70–80% of the excessive intake is stored as fat, and the remainder is converted into glycogen or protein or lost as heat; long-term positive energy balance ultimately leads to obesity (Oussaada et al., 2019). Genetic mediation of obesity can be monogenic or polygenic. Monogenic mutation is relatively rare and primarily affects the genes involved in the leptin–melanocortin pathway, the central regulator of food intake and energy balance (Oussaada et al., 2019); as examples, the leptin receptor (LEPR) (Farooqi et al., 2007), pro-opiomelanocortin (POMC) (Krude et al., 2003), melanocortin 4 receptor (MC4R) (Krude et al., 2003), and MC3R (Lee et al., 2002) have all been linked to early onset of obesity in humans. Polygenic causation, however, is more common and accounts for >90% cases of childhood-onset obesity (Kleinendorst et al., 2018). Notably, common variants in certain loci within the fat mass and obesity-associated gene (FTO) have been linked to higher BMI in human populations (Claussnitzer et al., 2015; Oussaada et al., 2019). DNA methylation studies have identified epigenetic modification of several genes that are associated with obesity, as detailed elsewhere (Rohde et al., 2019). Whether obesity promotes epigenetic change and further deteriorates the imbalance between energy intake and expenditure to exacerbate obesity is an open question.
The lymphatic vasculature regulates both dietary lipid absorption and peripheral cholesterol removal. The intestinal lacteals are lymphatic vessels comprised of both capillary and collecting lymphatic elements. The lacteals absorb dietary lipids packaged as chylomicrons (Iqbal and Hussain, 2009; Randolph and Miller, 2014). Intravital imaging indicates that lacteals possess a spontaneous contractile feature; they actively absorb and transport enterocyte-processed lipids to the systemic circulation in concert with contractile forces produced by adjacent smooth muscle cells controlled by the autonomic nervous system (Choe et al., 2015). It was recently revealed that lacteal function controls dietary lipid absorption and, consequently, body weight, supporting the concept that the lacteals are the gatekeepers of lipid intake from the environment (McDonald, 2018; Zhang et al., 2018; Cifarelli and Eichmann, 2019).
In peripheral tissues, the lymphatic vasculature is generally considered to be the only route for the return of lipoprotein to the blood circulation (Cooke et al., 2004); removal of interstitial cholesterol by the lymphatic route is known as reverse cholesterol transport (RCT) (Huang et al., 2015). Although both lacteals and peripheral lymphatics may selectively uptake cargoes based on their size (Randolph and Miller, 2014), they may preferentially absorb lipoproteins depending on specific receptor expression. As an example, LEC expression of the scavenger receptor class B type I (SR-B1) is required for RCT in skin lymphatic capillaries but not for intestinal cholesterol absorption (Bura et al., 2013; Lim et al., 2013). Impaired RCT has been observed in ob/ob mice with induced obesity (Duong et al., 2018), suggesting that defective RCT may be a prerequisite for the development of obesity.
Several lines of evidence support the notion that lymphatic functionality impacts the pathogenesis of obesity. In patients with lymphatic injury-induced (secondary) lymphedema, fat hypertrophy in the lymphedematous tissues is prominent, accompanying tissue swelling and fibrosis (Jiang et al., 2018); the Chy mutant mouse, with its defective lymphatic development, also display abnormal lipid accumulation adjacent to affected hypoplastic lymphatic vessels (Karkkainen et al., 2001). Prox-1 haploinsufficiency causes lymphatic dysfunction and leads to adult-onset of obesity (Harvey et al., 2005), in those mice, lymphatic restoration rescues them from the development of obesity (Escobedo et al., 2016). Accumulated interstitial fluid, including retrograde lymph leakage from the dysfunctional lymphatics, may promote adipocyte differentiation and enhance local fat deposition (Harvey et al., 2005; Escobedo and Oliver, 2017). Increasing lymphatic density in adipose tissue by overexpressing VEGF-D reduces local immune cell accumulation and improves systemic metabolic responsiveness in high-fat diet (HFD)-induced obese mice (Chakraborty et al., 2019), although it cannot be excluded that VEGF-D may also exert effects on blood vascular cells because it also binds to VEGFR2 (Achen et al., 1998). Increased expression of Apelin, an endogenous peptide identified as a ligand of the orphan G protein-coupled receptor APJ, was shown to inhibit HFD-induced obesity by promoting both lymphatic and blood vascular integrity (Sawane et al., 2013). Collectively, these studies suggest that lymphatic dysfunction promotes obesity, and that improving lymphatic function inhibits the development of obesity and alleviates obesity-caused metabolic syndrome. Studies have also illustrated that obesity promotes lymphatic abnormalities, such as decreased initial lymphatic density, heightened lymphatic leakiness, impaired collecting lymphatic pumping, and diminished macromolecule transport; but those phenotypic and functional changes are reversible in response to dietary modification and weight control (Garcia Nores et al., 2016; Nitti et al., 2016). Decreased collecting lymphatic pumping may result from the perilymphatic accumulation of iNOS-expressing macrophages that can affect lymphatic contractility and damage LECs by conversion of NO to a powerful oxidant, peroxynitrite (Nitti et al., 2016; Torrisi et al., 2016). Consistent with experimental animal data, a clinical study showed that obesity is a risk factor for the development of lymphedema in post-surgical breast cancer patients (Helyer et al., 2010). Also, severely obese individuals often develop acquired lymphedema of the extremities (Greene et al., 2012). In vitro cell culture study showed that leptin compromises LEC proliferation and tube formation by enhancing STAT3 phosphorylation, although leptin also induces IL-6, which, on the other hand, promotes lymphatic tube formation. In aggregate, the net effect of high concentrations of leptin on lymphangiogenesis appears to be suppressive (Sato et al., 2016). These findings suggest that high concentrations of leptin produced by adipose tissue maybe responsible for suppressing lymphatic vasculature in obese individuals. In summary, lymphatic dysfunction sensitizes individuals to develop obesity, and obesity worsens lymphatic function.
Insulin signaling regulates glucose, lipid, and energy homeostasis, predominantly through its action on adipose tissues, liver, and skeletal muscles (Boucher et al., 2014). Insulin exerts its known function by binding to the insulin receptor (INSR) expressed on target cells; ligand engagement leads to INSR autophosphorylation, followed by recruitment of various phosphotyrosine-binding scaffold proteins, which in turn activates downstream effectors (Taniguchi et al., 2006). The most crucial INSR substrates for metabolic regulation are insulin receptor substrate (IRS)1 and IRS2 proteins. They exert downstream effects by activating the PI3K/AKT signaling pathway (Petersen and Shulman, 2018). Insulin signaling in skeletal muscle promotes glucose uptake and net glycogen synthesis; IRS1 appears to be the primary scaffold protein for this process (Bouzakri et al., 2006; Thirone et al., 2006). In the liver, insulin promotes the synthesis of major classes of metabolic macromolecules, including glycogen, lipids, and proteins, and, concurrently, it reduces hepatic glucose production by controlling the PI3K/AKT/GSK3α/β or mTORC1 or FOXO1 signaling cascades (Cherrington et al., 1998; Petersen and Shulman, 2018). In white adipose tissue (WAT), insulin signaling suppresses lipolysis; but its role in glucose uptake is relatively minor, accounting for about 5–10% of whole body glucose uptake (Virtanen et al., 2002; Ng et al., 2012). Attenuation or reversal of cAMP/PKA-mediated lipolysis induced by adrenergic signaling is the best understood mechanism for insulin suppression of lipolysis (Jaworski et al., 2007). Insulin resistance arises under the condition of chronic energy surplus (Samuel and Shulman, 2016). Continuous overnutrition with insulin resistance impairs insulin secretion by pancreatic β-cells, which eventually leads to overt T2D (DeFronzo et al., 2015). At the molecular level, both decreased INSR expression and impaired intracellular signaling transduction contribute to typical obesity-induced insulin resistance (Petersen and Shulman, 2018).
While WAT, liver, and skeletal muscles have been generally recognized as major insulin target tissues, ECs, including both BECs and LECs, are also sensitive to insulin. It was suggested that blood vascular ECs might act as “first-responders” to overnutrition (Barrett and Liu, 2013). Impaired insulin signaling in BECs diminishes AKT-dependent NO production and simultaneously increases Endothelin 1 (ET-1) activity, which leads to endothelial dysfunction (Mather et al., 2002; Federici et al., 2004; Okon et al., 2005; Shemyakin et al., 2006; Muniyappa and Sowers, 2013). Unhealthy microvasculature hampers insulin delivery to muscle and adipose tissue and affects glucose disposal and lipid homeostasis (Barrett and Liu, 2013). LEC insulin signaling has only recently been explored. One study showed that LECs derived from human dermal tissue (HDLECs) express much higher levels of INSR than that of adipose tissue microvascular ECs; an insulin level as low as 2.5 nM can induce AKT phosphorylation in HDLEC (Jaldin-Fincati et al., 2018), although circulating insulin concentrations in healthy individuals may still well-below the 2.5 nM range, which are around 100 pmol/L (Ford et al., 2006), whether similar responses can be induced at physiological conditions are unknown. Insulin-induced downstream signaling appears to be required for normal lymphatic vascular structure and function (Lee et al., 2018). Diminished LEC insulin signaling decreases eNOS phosphorylation and NO production, reduces mitochondria oxygen consumption, which alters LEC metabolism, and causes increased expression of proinflammatory molecules (Lee et al., 2018); these results suggest that physiological insulin signaling is essential for normal functioning of LECs. Supporting the role of insulin signaling in lymphatic function, blockade of IRS1 suppresses lymphangiogenesis (Hos et al., 2011). Collectively, these studies indicate that insulin signaling likely plays important roles in regulating both LEC metabolism and lymphangiogenesis (Figure 2); LEC insulin resistance diminishes lymphatic function, and exacerbates obesity and metabolic abnormality.
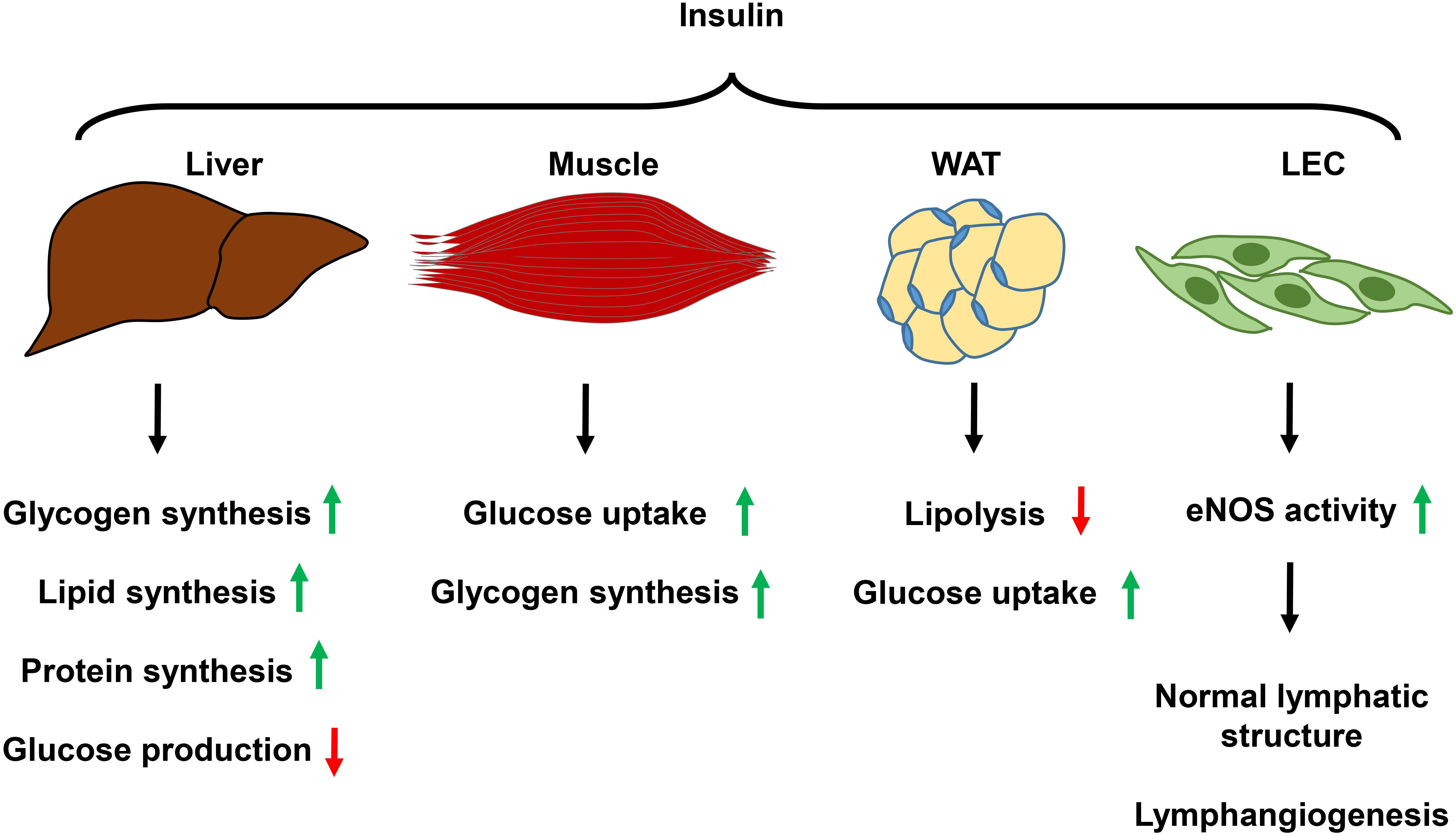
Figure 2. Summary of the function of insulin signaling in metabolic organs and LECs. Insulin signaling in the liver promotes glycogen, lipid, and protein synthesis while decreases glucose production. In the skeletal muscle, insulin mediates glucose uptake and glycogen synthesis. In white adipose tissue, insulin suppresses lipolysis and promotes glucose uptake. Insulin activation of its receptor in LECs promotes eNOS activity, which keeps normal lymphatic structure, and promotes lymphangiogenesis. WAT, white adipose tissue; LEC, lymphatic endothelial cell; eNOS, endothelial nitric oxide synthase.
Type 2 diabetes is characterized by dysregulation of carbohydrate, lipid, and protein metabolism caused by impaired insulin signaling resulting from reduced insulin secretion, insulin resistance, or the combination of both (DeFronzo et al., 2015). The lymphatic pathology likely co-evolves with the pathogenesis of diabetes. In an alloxan-induced rat diabetes model, the lymph flow through the thoracic duct in diabetic rats is significantly higher than in that of healthy controls, possibly because increased interstitial glucose levels increase tissue colloid pressure, which then enhances interstitial fluid absorption and lymph production. By contrast, lymph node uptake of 99mTc−dextran 500 is impaired in diabetic rats, a result that might explain the observation that patients with diabetes often have decreased function for immune priming (Moriguchi et al., 2005). Interestingly, insulin treatment normalizes both lymph flow and lymph node dextran retention, but glucose normalization through diet-control only corrects lymph flow (Moriguchi et al., 2005), suggesting that fluid transport and immune regulatory function of the lymphatics are regulated by discrete mechanisms. It is worth noting that the alloxan model simulates type 1 diabetes (Ighodaro et al., 2017), it nevertheless will provide insights about how hyperglycemia may impact the lymphatic system. In a clinical study, skin specimens of diabetic patients displayed increased lymphatic density, and transcriptional analysis of isolated dermal LECs indicated that cells from diabetic patients exhibit signatures of inflammation, adhesion, migration, growth, and lymphangiogenesis (Haemmerle et al., 2013). Increased lymphatic density found in human patient samples seemingly correlates with the phenomenon observed in diabetic rats that have enhanced lymphatic return to the systemic circulation. However, it is possible that the lymphatic function may decompensate after prolonged interstitial fluid overload caused by hyperglycemia (Kanapathy et al., 2015). Increased lymphatic collecting vessel permeability caused by diminished NO availability appears to be a contributing factor for lymphatic dysfunction (Scallan et al., 2015).
One common complication of diabetes is diabetic retinopathy, which is characterized by a pathology that involves vascular, glial, and neuronal components and causes significant visual loss (Duh et al., 2017). A recent study investigating the lymphatics in diabetic retinopathy detected the expression of LEC markers, such as VEGFR3 and Prox-1, in excised human specimens (Loukovaara et al., 2015), providing the first clinical evidence that abnormal lymphatic growth, in addition to pathological microvascular remodeling, occurs in diabetic retinopathy. A follow-up study demonstrated that soluble pro-growth factors in vitreous fluid promoted lymphatic sprouting of patient-derived tissues with diabetic retinopathy (Gucciardo et al., 2018a), further supporting the notion that the microenvironment of the eyes of diabetic retinopathy contains sufficient cue for abnormal lymphatic expansion. Pathological lymphangiogenesis observed in the eyes of diabetic retinopathy may, therefore, be regarded as a feature for therapeutic targeting (Yang et al., 2016; Gucciardo et al., 2018b). Ongoing clinical trials targeting VEGFR- or RTK-mediated signaling may provide possibilities to suppress pathological lymphatic overgrowth, but identification of novel lymphatic specific targets is probably necessary for better therapeutic outcomes (Williams et al., 2010).
The metabolic syndrome poses a significant risk for the development of cardiovascular diseases, such as atherosclerosis and its severe complication, myocardial infarction (MI) (Wilson et al., 2005; Mottillo et al., 2010). Following the revelation of the interplay between metabolic syndrome and lymphatic dysfunction, a multiplicity of research has shown that the lymphatic vasculature is also actively involved in the progression of atherosclerosis and development of MI (Aspelund et al., 2016). Atherosclerosis is a chronic inflammatory disease of the arterial wall with dyslipidemia as the root cause. The atherosclerotic lesion is characterized by a prominent population of lipid-filled foam cells, which are macrophages that contain an excess of plasma-derived, modified lipoproteins (Back et al., 2019). In this inflammatory pathology, the balance between pro- and anti-inflammatory molecules dictates whether a nascent atherosclerotic lesion will reverse course to normal or progress to more advanced stages. Among all the inflammatory cells that populate an atherosclerotic locus, the lipid-laden macrophage is central to disease progression. Macrophage uptake and modification of lipoprotein are physiologically important for lipid removal from intima, but uncontrolled, excessive lipid infiltration and inflammation exhausts macrophages and creates a hypoxic environment; this, in turn, promotes macrophage death and release of oxidized lipid and perpetuates the vicious cycle of foam cell formation, death, and intimal lipid accumulation (Tabas, 2010). Tipping the balance of local inflammation to an anti-inflammatory profile is, therefore, a therapeutic concept in treating atherosclerosis (Ruparelia et al., 2017). Recent studies have delved into lymphatic biology in the pathogenesis of atherosclerosis, presumably because the lymphatics play an essential role in immune trafficking and lipid transport (Milasan et al., 2015; Csanyi and Singla, 2019).
Lymphatic vessels are present in the adventitial and periadventitial regions of arterial walls (Sacchi et al., 1990; Martel et al., 2013; Milasan et al., 2015). Early observations that associated diminished lymphatic drainage to atherosclerosis were reported nearly three decades ago (Miller et al., 1992; Solti et al., 1994). Utilizing a mouse aorta transplantation model, the lymphatic vasculature was shown to be critical for RCT in arterial wall of the large vessel (Martel et al., 2013). Another study demonstrated that mice fed a HFD were prone to atheroma formation when the lymphatic vasculature is defective (Vuorio et al., 2014), supporting the notion that lymphatic dysfunction diminishes cholesterol removal and promotes atherosclerosis. In atherosclerosis-prone Ldlr–/–; ApoB100+/+ mice, lymphatic dysfunction, mainly of the collecting lymphatic vessels, occurs before the onset, and during the progression, of atherosclerosis (Milasan et al., 2016), suggesting that lymphatic vascular abnormalities likely promote atherosclerosis. In agreement with these hypotheses, the rescue of lymphatic vasculature during the early phase of atherogenesis retards disease progression, through reducing tissue inflammation and, likely also through increased lymphatic cholesterol transportation (Milasan et al., 2019). These studies in aggregate link lymphatic vascular functionality to atherosclerosis pathogenesis.
While the lymphatic vasculature is critical for immune trafficking and immune regulation, inflammatory molecules, produced by infiltrated local immune cells, also impact lymphatic vascular remodeling and function. Targeting inflammation may, therefore, not only ameliorate tissue inflammation but also improve lymphatic function. In a mouse tail surgery-induced tail lymphedema model, we have previously shown that high concentrations of LTB4 sustain local tissue inflammation, which is characterized by infiltration of cells of both innate and adaptive immunity; blockade of LTB4 signaling not only reduces tissue inflammation but also improves lymphatic function and alleviates lymphedema (Tian et al., 2017). In a series of in vivo and in vitro experiments, we showed that >100 nM LTB4 induces LEC apoptosis and suppresses lymphangiogenesis, while LTB4 in the 10 nM range enhances lymphatic regeneration; a physiological LTB4 level appears to be essential for surgery-induced wound healing (Tian et al., 2017); these experiments indicate that inflammation and lymphatic dysfunction are closely associated. LTB4 is an important proinflammatory molecule that promotes atherosclerosis by magnifying monocyte chemotaxis and foam cell formation (Subbarao et al., 2004), LTB4 may also directly impact endothelial survival and angiogenesis (Tian et al., 2013) and exacerbates insulin resistance (Li et al., 2015). It is therefore possible that targeting the LTB4 signaling pathway may simultaneously limit tissue inflammation, boost both lymphatic and blood vasculature, increase insulin sensitivity, and normalize cellular metabolism. Several Phase I atherosclerosis clinical trials targeting the LTB4 pathway are underway (Bhatt et al., 2017).
Myocardial infarction can be triggered by rupture or erosion of vulnerable atherosclerotic plaque, which results in blood clot formation by the exposure of thrombogenic core and matrix components of plaque (Anderson and Morrow, 2017). MI is followed by robust inflammatory responses that can be categorized into the initial pro-inflammatory responses that function to remove necrotic debris, and reparative anti-inflammatory responses poised to repair the damaged tissue (Ong et al., 2018). Targeting the inflammatory response in different phases of MI appears to be logical, but several such interventions have failed to improve patient outcomes (Ong et al., 2018), indicating additional mechanisms are likely at play for post-MI recovery. A recent study revealed significant lymphangiogenic responses following MI. Promotion of lymphangiogenesis by exogenous administration of VEGFC results in a transient improvement of post-MI myocardial function (Klotz et al., 2015). Improved outcome by VEGFC-induced lymphangiogenesis depends on lymphatic-mediated immune cell clearance through a pathway involving LEC expressed LYVE-1 (Vieira et al., 2018). Consistent with a protective role of the lymphatic vasculature in promoting post-MI recovery, downregulation of the LEC marker VEGFR3 alters cardiac lymphatic structure, increases lymphatic leakage, and raises MI-induced mortality (Vuorio et al., 2018). These recent studies suggest that the lymphatic vasculature might be a viable therapeutic target for post-MI cardiac repair (Vuorio et al., 2017).
Type 2 diabetes and cardiovascular diseases associated with obesity are the leading cause of death in the developed world (Benjamin et al., 2019). There is an unmet need to improve the medical care for these patients. Our understanding of mechanisms underlying these diseases has grown substantially but remains incomplete. Promoting lymphatic function has the apparent capacity to reduce pathology in preclinical obesity and cardiovascular disease models. More in-depth study of lymphatic biology is therefore urgently needed. Discoveries that derive from these investigations will likely provide novel therapeutic targets and improve disease survival.
XJ contributed to the writing and editing of the manuscript, and creation of figures. WT and MN contributed to the concepts and editing of the manuscript. SR contributed to the concepts, editing, and final formatting of the manuscript.
This work was supported by the Stanford Startup Funds and Endowed Chair Funds (to MN and SR) and the National Institutes of Health Grants K12 HL120001-05 (to XJ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Achen, M. G., Jeltsch, M., Kukk, E., Mäkinen, T., Vitali, A., Wilks, A. F., et al. (1998). Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. U.S.A. 95, 548–553. doi: 10.1073/pnas.95.2.548
Anderson, J. L., and Morrow, D. A. (2017). Acute myocardial infarction. N. Engl. J. Med. 376, 2053–2064.
Aspelund, A., Robciuc, M. R., Karaman, S., Makinen, T., and Alitalo, K. (2016). Lymphatic system in cardiovascular medicine. Circ. Res. 118, 515–530. doi: 10.1161/CIRCRESAHA.115.306544
Back, M., Yurdagul, A. Jr., Tabas, I., Öörni, K., and Kovanen, P. T. (2019). Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406. doi: 10.1038/s41569-019-0169-2
Baluk, P., Fuxe, J., Hashizume, H., Romano, T., Lashnits, E., Butz, S., et al. (2007). Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 204, 2349–2362. doi: 10.1084/jem.20062596
Barrett, E. J., and Liu, Z. (2013). The endothelial cell: an “early responder” in the development of insulin resistance. Rev. Endocr. Metab. Disord. 14, 21–27. doi: 10.1007/s11154-012-9232-6
Benjamin, E. J., Muntner, P., Alonso, A., Bittencourt, M. S., Callaway, C. W., Carson, A. P., et al. (2019). Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation 139, e56–e528. doi: 10.1161/CIR.0000000000000659
Bhatt, L., Roinestad, K., Van, T., and Springman, E. B. (2017). Recent advances in clinical development of leukotriene B4 pathway drugs. Semin. Immunol. 33, 65–73. doi: 10.1016/j.smim.2017.08.007
Bos, F. L., Caunt, M., Peterson-Maduro, J., Planas-Paz, L., Kowalski, J., Karpanen, T., et al. (2011). CCBE1 is essential for mammalian lymphatic vascular development and enhances the lymphangiogenic effect of vascular endothelial growth factor-C in vivo. Circ. Res. 109, 486–491. doi: 10.1161/CIRCRESAHA.111.250738
Boucher, J., Kleinridders, A., and Kahn, C. R. (2014). Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 6:a009191. doi: 10.1101/cshperspect.a009191
Bouzakri, K., Zachrisson, A., Al-Khalili, L., Zhang, B. B., Koistinen, H. A., Krook, A., et al. (2006). siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab. 4, 89–96. doi: 10.1016/j.cmet.2006.04.008
Bura, K. S., Lord, C., Marshall, S., McDaniel, A., Thomas, G., Warrier, M., et al. (2013). Intestinal SR-BI does not impact cholesterol absorption or transintestinal cholesterol efflux in mice. J. Lipid Res. 54, 1567–1577. doi: 10.1194/jlr.M034454
Catrysse, L., and van Loo, G. (2017). Inflammation and the metabolic syndrome: the tissue-specific functions of NF-kappaB. Trends Cell Biol. 27, 417–429. doi: 10.1016/j.tcb.2017.01.006
Chakraborty, A., Barajas, S., Lammoglia, G. M., Reyna, A. J., Morley, T. S., Johnson, J. A., et al. (2019). Vascular endothelial growth factor-D (VEGF-D) overexpression and lymphatic expansion in murine adipose tissue improves metabolism in obesity. Am. J. Pathol. 189, 924–939. doi: 10.1016/j.ajpath.2018.12.008
Cherrington, A. D., Edgerton, D., and Sindelar, D. K. (1998). The direct and indirect effects of insulin on hepatic glucose production in vivo. Diabetologia 41, 987–996.
Choe, K., Jang, J. Y., Park, I., Kim, Y., Ahn, S., Park, D. Y., et al. (2015). Intravital imaging of intestinal lacteals unveils lipid drainage through contractility. J. Clin. Invest. 125, 4042–4052. doi: 10.1172/JCI76509
Choi, I., Lee, S., and Hong, Y. K. (2012). The new era of the lymphatic system: no longer secondary to the blood vascular system. Cold Spring Harb. Perspect. Med. 2:a006445. doi: 10.1101/cshperspect.a006445
Cifarelli, V., and Eichmann, A. (2019). The intestinal lymphatic system: functions and metabolic implications. Cell. Mol. Gastroenterol. Hepatol. 7, 503–513. doi: 10.1016/j.jcmgh.2018.12.002
Claussnitzer, M., Dankel, S. N., Kim, K. H., Quon, G., Meuleman, W., Haugen, C., et al. (2015). FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 373, 895–907. doi: 10.1056/NEJMoa1502214
Connell, F. C., Ostergaard, P., Carver, C., Brice, G., Williams, N., Mansour, S., et al. (2009). Analysis of the coding regions of VEGFR3 and VEGFC in Milroy disease and other primary lymphoedemas. Hum. Genet. 124, 625–631. doi: 10.1007/s00439-008-0586-5
Cooke, C. J., Nanjee, M. N., Stepanova, I. P., Olszewski, W. L., and Miller, N. E. (2004). Variations in lipid and apolipoprotein concentrations in human leg lymph: effects of posture and physical exercise. Atherosclerosis 173, 39–45. doi: 10.1016/j.atherosclerosis.2003.07.004
Csanyi, G., and Singla, B. (2019). Arterial lymphatics in atherosclerosis: old questions, new insights, and remaining challenges. J. Clin. Med. 8:E495. doi: 10.3390/jcm8040495
DeFronzo, R. A., Ferrannini, E., Groop, L., Henry, R. R., Herman, W. H., Holst, J. J., et al. (2015). Type 2 diabetes mellitus. Nat. Rev. Dis. Primers. 1:15019. doi: 10.1038/nrdp.2015.19
Dellinger, M., Hunter, R., Bernas, M., Gale, N., Yancopoulos, G., Erickson, R., et al. (2008). Defective remodeling and maturation of the lymphatic vasculature in Angiopoietin-2 deficient mice. Dev. Biol. 319, 309–320. doi: 10.1016/j.ydbio.2008.04.024
Duh, E. J., Sun, J. K., and Stitt, A. W. (2017). Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight 2:93751. doi: 10.1172/jci.insight.93751
Duong, M., Uno, K., Nankivell, V., Bursill, C., and Nicholls, S. J. (2018). Induction of obesity impairs reverse cholesterol transport in ob/ob mice. PLoS One 13:e0202102. doi: 10.1371/journal.pone.0202102
Escobedo, N., and Oliver, G. (2017). The lymphatic vasculature: its role in adipose metabolism and obesity. Cell Metab. 26, 598–609. doi: 10.1016/j.cmet.2017.07.020
Escobedo, N., Proulx, S. T., Karaman, S., Dillard, M. E., Johnson, N., Detmar, M., et al. (2016). Restoration of lymphatic function rescues obesity in Prox1-haploinsufficient mice. JCI Insight 1:e85096. doi: 10.1172/jci.insight.85096
Farooqi, I. S., Bullmore, E., Keogh, J., Gillard, J., O’Rahilly, S., and Fletcher, P. C. (2007). Leptin regulates striatal regions and human eating behavior. Science 317:1355. doi: 10.1126/science.1144599
Federici, M., Pandolfi, A., De Filippis, E. A., Pellegrini, G., Menghini, R., Lauro, D., et al. (2004). G972R IRS-1 variant impairs insulin regulation of endothelial nitric oxide synthase in cultured human endothelial cells. Circulation 109, 399–405. doi: 10.1161/01.CIR.0000109498.77895.6F
Ford, E. S., Li, C., Imperatore, G., and Cook, S. (2006). Age, sex, and ethnic variations in serum insulin concentrations among U.S. youth: findings from the National Health and Nutrition Examination Survey 1999-2002. Diabetes Care 29, 2605–2611. doi: 10.2337/dc06-1083
Gale, N. W., Prevo, R., Espinosa, J., Ferguson, D. J., Dominguez, M. G., Yancopoulos, G. D., et al. (2007). Normal lymphatic development and function in mice deficient for the lymphatic hyaluronan receptor LYVE-1. Mol. Cell. Biol. 27, 595–604. doi: 10.1128/MCB.01503-06
Garcia Nores, G. D., Cuzzone, D. A., Albano, N. J., Hespe, G. E., Kataru, R. P., Torrisi, J. S., et al. (2016). Obesity but not high-fat diet impairs lymphatic function. Int. J. Obes. 40, 1582–1590. doi: 10.1038/ijo.2016.96
Gonzalez-Muniesa, P., Mártinez-González, M. A., Hu, F. B., Després, J. P., Matsuzawa, Y., Loos, R. J. F., et al. (2017). Obesity. Nat. Rev. Dis. Primers 3:17034. doi: 10.1038/nrdp.2017.34
Greene, A. K., Grant, F. D., and Slavin, S. A. (2012). Lower-extremity lymphedema and elevated body-mass index. N. Engl. J. Med. 366, 2136–2137.
Gucciardo, E., Loukovaara, S., Korhonen, A., Repo, P., Martins, B., Vihinen, H., et al. (2018a). The microenvironment of proliferative diabetic retinopathy supports lymphatic neovascularization. J. Pathol. 245, 172–185. doi: 10.1002/path.5070
Gucciardo, E., Loukovaara, S., Salven, P., and Lehti, K. (2018b). Lymphatic vascular structures: a new aspect in proliferative diabetic retinopathy. Int. J. Mol. Sci. 19:E4034. doi: 10.3390/ijms19124034
Haemmerle, M., Keller, T., Egger, G., Schachner, H., Steiner, C. W., Stokic, D., et al. (2013). Enhanced lymph vessel density, remodeling, and inflammation are reflected by gene expression signatures in dermal lymphatic endothelial cells in type 2 diabetes. Diabetes 62, 2509–2529. doi: 10.2337/db12-0844
Harvey, N. L., Srinivasan, R. S., Dillard, M. E., Johnson, N. C., Witte, M. H., Boyd, K., et al. (2005). Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat. Genet. 37, 1072–1081. doi: 10.1038/ng1642
Helyer, L. K., Varnic, M., Le, L. W., Leong, W., and McCready, D. (2010). Obesity is a risk factor for developing postoperative lymphedema in breast cancer patients. Breast J. 16, 48–54. doi: 10.1111/j.1524-4741.2009.00855.x
Hos, D., Regenfuss, B., Bock, F., Onderka, J., and Cursiefen, C. (2011). Blockade of insulin receptor substrate-1 inhibits corneal lymphangiogenesis. Invest. Ophthalmol. Vis. Sci. 52, 5778–5785. doi: 10.1167/iovs.10-6816
Huang, L. H., Elvington, A., and Randolph, G. J. (2015). The role of the lymphatic system in cholesterol transport. Front. Pharmacol. 6:182. doi: 10.3389/fphar.2015.00182
Ighodaro, O. M., Adeosun, A. M., and Akinloye, O. A. (2017). Alloxan-induced diabetes, a common model for evaluating the glycemic-control potential of therapeutic compounds and plants extracts in experimental studies. Medicina 53, 365–374. doi: 10.1016/j.medici.2018.02.001
Iqbal, J., and Hussain, M. M. (2009). Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 296, E1183–E1194. doi: 10.1152/ajpendo.90899.2008
Jaldin-Fincati, J. R., Pereira, R. V. S., Bilan, P. J., and Klip, A. (2018). Insulin uptake and action in microvascular endothelial cells of lymphatic and blood origin. Am. J. Physiol. Endocrinol. Metab. 315, E204–E217. doi: 10.1152/ajpendo.00008.2018
Jaworski, K., Sarkadi-Nagy, E., Duncan, R. E., Ahmadian, M., and Sul, H. S. (2007). Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G1–G4. doi: 10.1152/ajpgi.00554.2006
Jha, S. K., Rauniyar, K., Karpanen, T., Leppänen, V. M., Brouillard, P., Vikkula, M., et al. (2017). Efficient activation of the lymphangiogenic growth factor VEGF-C requires the C-terminal domain of VEGF-C and the N-terminal domain of CCBE1. Sci. Rep. 7:4916. doi: 10.1038/s41598-017-04982-1
Jiang, X., Nicolls, M. R., Tian, W., and Rockson, S. G. (2018). Lymphatic dysfunction, leukotrienes, and lymphedema. Annu. Rev. Physiol. 80, 49–70. doi: 10.1146/annurev-physiol-022516-034008
Johnson, N. C., Dillard, M. E., Baluk, P., McDonald, D. M., Harvey, N. L., Frase, S. L., et al. (2008). Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev. 22, 3282–3291. doi: 10.1101/gad.1727208
Kahn, B. B., and Flier, J. S. (2000). Obesity and insulin resistance. J. Clin. Invest. 106, 473–481.
Kanapathy, M., Portou, M. J., Tsui, J., and Richards, T. (2015). Diabetic foot ulcers in conjunction with lower limb lymphedema: pathophysiology and treatment procedures. Chronic Wound Care Manag. Res. 2, 129–136. doi: 10.2147/CWCMR.S62919
Karkkainen, M. J., Ferrell, R. E., Lawrence, E. C., Kimak, M. A., Levinson, K. L., McTigue, M. A., et al. (2000). Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat. Genet. 25, 153–159.
Karkkainen, M. J., Haiko, P., Sainio, K., Partanen, J., Taipale, J., Petrova, T. V., et al. (2004). Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 5, 74–80. doi: 10.1038/ni1013
Karkkainen, M. J., Saaristo, A., Jussila, L., Karila, K. A., Lawrence, E. C., Pajusola, K., et al. (2001). A model for gene therapy of human hereditary lymphedema. Proc. Natl. Acad. Sci. U.S.A. 98, 12677–12682.
Kleinendorst, L., Massink, M. P. G., Cooiman, M. I., Savas, M., van der Baan-Slootweg, O. H., Roelants, R. J., et al. (2018). Genetic obesity: next-generation sequencing results of 1230 patients with obesity. J. Med. Genet. 55, 578–586. doi: 10.1136/jmedgenet-2018-105315
Klotz, L., Norman, S., Vieira, J. M., Masters, M., Rohling, M., Dubé, K. N., et al. (2015). Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 522, 62–67. doi: 10.1038/nature14483
Konner, A. C., and Bruning, J. C. (2011). Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol. Metab. 22, 16–23.
Krude, H., Biebermann, H., and Gruters, A. (2003). Mutations in the human proopiomelanocortin gene. Ann. N. Y. Acad. Sci. 994, 233–239. doi: 10.1111/j.1749-6632.2003.tb03185.x
Lee, Y., Chakraborty, S., Meininger, C. J., and Muthuchamy, M. (2018). Insulin resistance disrupts cell integrity, mitochondrial function, and inflammatory signaling in lymphatic endothelium. Microcirculation 25:e12492. doi: 10.1111/micc.12492
Lee, Y. S., Poh, L. K., and Loke, K. Y. (2002). A novel melanocortin 3 receptor gene (MC3R) mutation associated with severe obesity. J. Clin. Endocrinol. Metab. 87, 1423–1426.
Li, P., Oh, D. Y., Bandyopadhyay, G., Lagakos, W. S., Talukdar, S., and Osborn, O. (2015). LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 21, 239–247. doi: 10.1038/nm.3800
Lim, H. Y., Thiam, C. H., Yeo, K. P., Bisoendial, R., Hii, C. S., McGrath, K. C., et al. (2013). Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 17, 671–684. doi: 10.1016/j.cmet.2013.04.002
Loukovaara, S., Gucciardo, E., Repo, P., Vihinen, H., Lohi, J., Jokitalo, E., et al. (2015). Indications of lymphatic endothelial differentiation and endothelial progenitor cell activation in the pathology of proliferative diabetic retinopathy. Acta Ophthalmol. 93, 512–523. doi: 10.1111/aos.12741
Martel, C., Li, W., Fulp, B., Platt, A. M., Gautier, E. L., Westerterp, M., et al. (2013). Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J. Clin. Invest. 123, 1571–1579. doi: 10.1172/JCI63685
Mather, K. J., Mirzamohammadi, B., Lteif, A., Steinberg, H. O., and Baron, A. D. (2002). Endothelin contributes to basal vascular tone and endothelial dysfunction in human obesity and type 2 diabetes. Diabetes 51, 3517–3523.
Milasan, A., Dallaire, F., Mayer, G., and Martel, C. (2016). Effects of LDL receptor modulation on lymphatic function. Sci. Rep. 6:27862. doi: 10.1038/srep27862
Milasan, A., Ledoux, J., and Martel, C. (2015). Lymphatic network in atherosclerosis: the underestimated path. Future Sci. OA 1:FSO61. doi: 10.4155/fso.15.61
Milasan, A., Smaani, A., and Martel, C. (2019). Early rescue of lymphatic function limits atherosclerosis progression in Ldlr(-/-) mice. Atherosclerosis 283, 106–119. doi: 10.1016/j.atherosclerosis.2019.01.031
Miller, A. J., DeBoer, A., and Palmer, A. (1992). The role of the lymphatic system in coronary atherosclerosis. Med. Hypotheses 37, 31–36.
Moriguchi, P., Sannomiya, P., Lara, P. F., Oliveira-Filho, R. M., Greco, K. V., and Sudo-Hayashi, L. S. (2005). Lymphatic system changes in diabetes mellitus: role of insulin and hyperglycemia. Diabetes Metab. Res. Rev. 21, 150–157. doi: 10.1002/dmrr.500
Mottillo, S., Filion, K. B., Genest, J., Joseph, L., Pilote, L., Poirier, P., et al. (2010). The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J. Am. Coll. Cardiol. 56, 1113–1132. doi: 10.1016/j.jacc.2010.05.034
Muniyappa, R., and Sowers, J. R. (2013). Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 14, 5–12. doi: 10.1007/s11154-012-9229-1
Ng, J. M., Azuma, K., Kelley, C., Pencek, R., Radikova, Z., Laymon, C., et al. (2012). PET imaging reveals distinctive roles for different regional adipose tissue depots in systemic glucose metabolism in nonobese humans. Am. J. Physiol. Endocrinol. Metab. 303, E1134–E1141. doi: 10.1152/ajpendo.00282.2012
Nitti, M. D., Hespe, G. E., Kataru, R. P., García Nores, G. D., Savetsky, I. L., Torrisi, J. S., et al. (2016). Obesity-induced lymphatic dysfunction is reversible with weight loss. J. Physiol. 594, 7073–7087. doi: 10.1113/JP273061
Okon, E. B., Chung, A. W., Rauniyar, P., Padilla, E., Tejerina, T., McManus, B. M., et al. (2005). Compromised arterial function in human type 2 diabetic patients. Diabetes 54, 2415–2423. doi: 10.2337/diabetes.54.8.2415
O’Neill, S., and O’Driscoll, L. (2015). Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes. Rev. 16, 1–12. doi: 10.1111/obr.12229
Ong, S. B., Hernández-Reséndiz, S., Crespo-Avilan, G. E., Mukhametshina, R. T., Kwek, X. Y., Cabrera-Fuentes, H. A., et al. (2018). Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 186, 73–87. doi: 10.1016/j.pharmthera.2018.01.001
Oussaada, S. M., van Galen, K. A., Cooiman, M. I., Kleinendorst, L., Hazebroek, E. J., van Haelst, M. M., et al. (2019). The pathogenesis of obesity. Metabolism 92, 26–36. doi: 10.1016/j.metabol.2018.12.012
Petersen, M. C., and Shulman, G. I. (2018). Mechanisms of insulin action and insulin resistance. Physiol. Rev. 98, 2133–2223. doi: 10.1152/physrev.00063.2017
Petrova, T. V., Karpanen, T., Norrmén, C., Mellor, R., Tamakoshi, T., Finegold, D., et al. (2004). Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 10, 974–981. doi: 10.1038/nm1094
Potente, M., Gerhardt, H., and Carmeliet, P. (2011). Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887. doi: 10.1182/blood-2010-11-317800
Randolph, G. J., and Miller, N. E. (2014). Lymphatic transport of high-density lipoproteins and chylomicrons. J. Clin. Invest. 124, 929–935. doi: 10.1172/JCI71610
Rohde, K., Keller, M., la Cour Poulsen, L., Blüher, M., Kovacs, P., and Böttcher, Y. (2019). Genetics and epigenetics in obesity. Metabolism 92, 37–50. doi: 10.1016/j.metabol.2018.10.007
Ruddle, N. H., and Akirav, E. M. (2009). Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J. Immunol. 183, 2205–2212. doi: 10.4049/jimmunol.0804324
Ruotolo, G., and Howard, B. V. (2002). Dyslipidemia of the metabolic syndrome. Curr. Cardiol. Rep. 4, 494–500.
Ruparelia, N., Chai, J. T., Fisher, E. A., and Choudhury, R. P. (2017). Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat. Rev. Cardiol. 14, 133–144. doi: 10.1038/nrcardio.2016.185
Sabine, A., Agalarov, Y., Maby-El Hajjami, H., Jaquet, M., Hägerling, R., Pollmann, C., et al. (2012). Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev. Cell. 22, 430–445. doi: 10.1016/j.devcel.2011.12.020
Sacchi, G., Weber, E., and Comparini, L. (1990). Histological framework of lymphatic vasa vasorum of major arteries: an experimental study. Lymphology 23, 135–139.
Sainz-Jaspeado, M., and Claesson-Welsh, L. (2018). Cytokines regulating lymphangiogenesis. Curr. Opin. Immunol. 53, 58–63. doi: 10.1016/j.coi.2018.04.003
Samuel, V. T., and Shulman, G. I. (2016). The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest. 126, 12–22. doi: 10.1172/JCI77812
Sato, A., Kamekura, R., Kawata, K., Kawada, M., Jitsukawa, S., Yamashita, K., et al. (2016). Novel mechanisms of compromised lymphatic endothelial cell homeostasis in obesity: the role of leptin in lymphatic endothelial cell tube formation and proliferation. PLoS One 11:e0158408. doi: 10.1371/journal.pone.0158408
Sawane, M., Kajiya, K., Kidoya, H., Takagi, M., Muramatsu, F., and Takakura, N. (2013). Apelin inhibits diet-induced obesity by enhancing lymphatic and blood vessel integrity. Diabetes 62, 1970–1980. doi: 10.2337/db12-0604
Scallan, J. P., Hill, M. A., and Davis, M. J. (2015). Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc. Res. 107, 89–97. doi: 10.1093/cvr/cvv117
Shemyakin, A., Böhm, F., Wagner, H., Efendic, S., Båvenholm, P., and Pernow, J. (2006). Enhanced endothelium-dependent vasodilatation by dual endothelin receptor blockade in individuals with insulin resistance. J. Cardiovasc. Pharmacol. 47, 385–390. doi: 10.1097/01.fjc.0000210070.47205.16
Solti, F., Lengyel, E., Jellinek, H., Schneider, F., Juhász-Nagy, A., and Kékesi, V. (1994). Coronary arteriopathy after lymphatic blockade: an experimental study in dogs. Lymphology 27, 173–180.
Subbarao, K., Jala, V. R., Mathis, S., Suttles, J., Zacharias, W., Ahamed, J., et al. (2004). Role of leukotriene B4 receptors in the development of atherosclerosis: potential mechanisms. Arterioscler. Thromb. Vasc. Biol. 24, 369–375. doi: 10.1161/01.ATV.0000110503.16605.15
Szuba, A., Skobe, M., Karkkainen, M. J., Shin, W. S., Beynet, D. P., Rockson, N. B., et al. (2002). Therapeutic lymphangiogenesis with human recombinant VEGF-C. FASEB J. 16, 1985–1987.
Tabas, I. (2010). Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 10, 36–46. doi: 10.1038/nri2675
Taniguchi, C. M., Emanuelli, B., and Kahn, C. R. (2006). Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell. Biol. 7, 85–96.
Thirone, A. C., Huang, C., and Klip, A. (2006). Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol. Metab. 17, 72–78.
Thomas, S. N., Rohner, N. A., and Edwards, E. E. (2016). Implications of lymphatic transport to lymph nodes in immunity and immunotherapy. Annu. Rev. Biomed. Eng. 18, 207–233. doi: 10.1146/annurev-bioeng-101515-014413
Thomson, B. R., Heinen, S., Jeansson, M., Ghosh, A. K., Fatima, A., Sung, H. K., et al. (2014). A lymphatic defect causes ocular hypertension and glaucoma in mice. J. Clin. Invest. 124, 4320–4324. doi: 10.1172/JCI77162
Tian, W., Jiang, X., Tamosiuniene, R., Sung, Y. K., Qian, J., and Dhillon, G. (2013). Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci. Transl. Med. 5:200ra117. doi: 10.1126/scitranslmed.3006674
Tian, W., Rockson, S. G., Jiang, X., Kim, J., Begaye, A., Shuffle, E. M., et al. (2017). Leukotriene B4 antagonism ameliorates experimental lymphedema. Sci. Transl. Med. 9:eaal3920. doi: 10.1126/scitranslmed.aal3920
Torrisi, J. S., Hespe, G. E., Cuzzone, D. A., Savetsky, I. L., Nitti, M. D., Gardenier, J. C., et al. (2016). Inhibition of inflammation and iNOS improves lymphatic function in obesity. Sci. Rep. 6:19817. doi: 10.1038/srep19817
Vieira, J. M., Norman, S., Villa Del Campo, C., Cahill, T. J., Barnette, D. N., Gunadasa-Rohling, M., et al. (2018). The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J. Clin. Invest. 128, 3402–3412. doi: 10.1172/JCI97192
Virtanen, K. A., Lönnroth, P., Parkkola, R., Peltoniemi, P., Asola, M., Viljanen, T., et al. (2002). Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. J. Clin. Endocrinol. Metab. 87, 3902–3910. doi: 10.1210/jcem.87.8.8761
Vuorio, T., Nurmi, H., Moulton, K., Kurkipuro, J., Robciuc, M. R., Ohman, M., et al. (2014). Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler. Thromb. Vasc. Biol. 34, 1162–1170. doi: 10.1161/ATVBAHA.114.302528
Vuorio, T., Tirronen, A., and Yla-Herttuala, S. (2017). Cardiac lymphatics - a new avenue for therapeutics? Trends Endocrinol Metab. 28, 285–296. doi: 10.1016/j.tem.2016.12.002
Vuorio, T., Ylä-Herttuala, E., Laakkonen, J. P., Laidinen, S., Liimatainen, T., and Ylä-Herttuala, S. (2018). Downregulation of VEGFR3 signaling alters cardiac lymphatic vessel organization and leads to a higher mortality after acute myocardial infarction. Sci Rep. 8:16709. doi: 10.1038/s41598-018-34770-4
Wigle, J. T., Harvey, N., Detmar, M., Lagutina, I., Grosveld, G., Gunn, M. D., et al. (2002). An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J. 21, 1505–1513.
Wigle, J. T., and Oliver, G. (1999). Prox1 function is required for the development of the murine lymphatic system. Cell 98, 769–778.
Williams, S. P., Karnezis, T., Achen, M. G., and Stacker, S. A. (2010). Targeting lymphatic vessel functions through tyrosine kinases. J. Angiogenes Res. 2:13. doi: 10.1186/2040-2384-2-13
Wilson, P. W., D’Agostino, R. B., Parise, H., Sullivan, L., and Meigs, J. B. (2005). Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation 112, 3066–3072. doi: 10.1161/CIRCULATIONAHA.105.539528
Xu, Y., Yuan, L., Mak, J., Pardanaud, L., Caunt, M., Kasman, I., et al. (2010). Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J. Cell Biol. 188, 115–130. doi: 10.1083/jcb.200903137
Xue, Y., Cao, R., Nilsson, D., Chen, S., Westergren, R., Hedlund, E.-M., et al. (2008). FOXC2 controls Ang-2 expression and modulates angiogenesis, vascular patterning, remodeling, and functions in adipose tissue. Proc. Natl. Acad. Sci. U.S.A. 105, 10167–10172. doi: 10.1073/pnas.0802486105
Yang, J. F., Walia, A., Huang, Y. H., Han, K. Y., Rosenblatt, M. I., Azar, D. T., et al. (2016). Understanding lymphangiogenesis in knockout models, the cornea, and ocular diseases for the development of therapeutic interventions. Surv. Ophthalmol. 61, 272–296. doi: 10.1016/j.survophthal.2015.12.004
Yao, L. C., Baluk, P., Srinivasan, R. S., Oliver, G., and McDonald, D. M. (2012). Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 180, 2561–2575. doi: 10.1016/j.ajpath.2012.02.019
Yuan, L., Moyon, D., Pardanaud, L., Bréant, C., Karkkainen, M. J., Alitalo, K., et al. (2002). Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development 129, 4797–4806.
Zhang, F., Zarkada, G., Han, J., Li, J., Dubrac, A., Ola, R., et al. (2018). Lacteal junction zippering protects against diet-induced obesity. Science 361, 599–603. doi: 10.1126/science.aap9331
Zheng, W., Aspelund, A., and Alitalo, K. (2014). Lymphangiogenic factors, mechanisms, and applications. J. Clin. Invest. 124, 878–887. doi: 10.1172/JCI71603
Keywords: lymphatic, LEC, insulin resistance, type 2 diabetes, atherosclerosis, myocardial infarction
Citation: Jiang X, Tian W, Nicolls MR and Rockson SG (2019) The Lymphatic System in Obesity, Insulin Resistance, and Cardiovascular Diseases. Front. Physiol. 10:1402. doi: 10.3389/fphys.2019.01402
Received: 28 August 2019; Accepted: 31 October 2019;
Published: 14 November 2019.
Edited by:
Joshua Scallan, University of South Florida, United StatesReviewed by:
Andrew Carley, The Ohio State University Wexner Medical Center, United StatesCopyright © 2019 Jiang, Tian, Nicolls and Rockson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinguo Jiang, eGluZ3VvakBzdGFuZm9yZC5lZHU=; Stanley G. Rockson, cm9ja3NvbkBzdGFuZm9yZC5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.