 Gabriel T. Kelly1
Gabriel T. Kelly1 Yao Zhang
Yao Zhang Ting Wang
Ting Wang- 1Department of Internal Medicine, College of Medicine Phoenix, The University of Arizona, Phoenix, AZ, United States
- 2Department of Pediatrics, University of California, San Francisco, San Francisco, CA, United States
- 3Department of Medicine, College of Medicine, The University of Arizona, Tucson, AZ, United States
Patients with critical illness such as acute lung injury often undergo mechanical ventilation in the intensive care unit. Though lifesaving in many instances, mechanical ventilation often results in ventilator induced lung injury (VILI), characterized by overdistension of lung tissue leading to release of edemagenic agents, which further damage the lung and contribute to the mortality and progression of pulmonary inflammation. The endothelium is particularly sensitive, as VILI associated mechanical stress results in endothelial cytoskeletal rearrangement, stress fiber formation, and integrity loss. At the heart of these changes are integrin tethered focal adhesions (FAs) which participate in mechanosensing, structure, and signaling. Here, we present the known roles of FA proteins including c-Src, talin, FAK, paxillin, vinculin, and integrins in the sensing and response to cyclic stretch and VILI associated stress. Attention is given to how stretch is propagated from the extracellular matrix through integrins to talin and other FA proteins, as well as signaling cascades that include FA proteins, leading to stress fiber formation and other cellular responses. This unifying picture of FAs aids our understanding in an effort to prevent and treat VILI.
Introduction
Ventilator induced lung injury (VILI) is a clinical syndrome in the intensive care unit that results from mechanical ventilation. It is often associated with overdistension as well as vascular leak caused by edemagenic agents and inflammatory cytokines such as thrombin, histamine, tumor necrosis factor-α, interleukin-8, and interleukin-1 (Dos Santos and Slutsky, 2000; Lionetti et al., 2005; Birukova et al., 2006). VILI associated mechanical stress imposes severe pro-inflammatory lung endothelial injury, leading to endothelial integrity loss, cytokine secretion, and vascular leakage. It is well believed that the focal adhesion (FA)-integrin system, as the bridge between endothelial and basal matrix, serves as a principal mechanical stress sensing and transducing complex. This review will focus on the known roles of various FA proteins in endothelial cells (ECs) in response to VILI associated mechanical stress, specifically cyclic stretch (CS).
Endothelial Injury in VILI
VILI and Endothelial Injury
Acute lung injury (ALI) and its more severe form Acute Respiratory Distress Syndrome (ARDS) are devastating conditions with an unacceptable mortality of approximately 40% about 2 weeks after the onset of the syndrome (Villar et al., 2011). The recent “Berlin” definition defines ARDS as an acute primary pulmonary condition characterized by radiologic infiltrates and impaired oxygenation (Laffey and Kavanagh, 2017). This condition may be the result of any number of underlying causes including direct lung injury such as pneumonia, aspiration, or traumatic pulmonary contusion, or indirect injuries such as non-pulmonary sepsis or non-septic shock (Frutos-Vivar et al., 2004). These underlying causes lead to inflammatory, ischemic, mechanical, or infective insults on the lung, triggering damage to alveolar capillaries, interstitium, and epithelium, which leads to increased vascular permeability and results in subsequent interstitial and alveolar edema (Laffey and Kavanagh, 2017). Mechanical ventilation is one of the lifesaving strategies for ARDS, yet the mortality rate of ARDS patients remains high with ventilation associated persistent lung inflammatory injury, which is called Ventilator-Induced Lung Injury or VILI (Slutsky and Ranieri, 2014).
Similar to ARDS itself, VILI can induce a range of inflammatory responses such as increases in lung vascular permeability due to damage to the endothelial cell barrier and subsequent alveolar flooding. The development and course of VILI is associated with mechanical ventilator settings including dose and pattern (Gajic et al., 2005). Similar to ARDS, VILI directly leads to damages to the gas exchange barrier or complete dysfunction of alveoli leading to respiratory failure in patients (Villar et al., 2014). Particularly, mechanical ventilation increases in the alveolar epithelial cell surface area by 1/3 (Tschumperlin and Margulies, 1999), with a similar effect in capillary endothelium which forms tight contacts with alveolar epithelium. This longitudinal tension produced by the mechanical ventilator also induces various cellular responses including mechanical stress associated molecular signaling, ROS generation, gene expression, and cellular remodeling (Birukov, 2009), leading to damage directly to ECs, which can be observed at the ultrastructural level (Dreyfuss and Saumon, 1998). This persistent VILI associated mechanical stress during ventilation leads to further dysregulation of the pulmonary capillary endothelium, leading to protein rich fluid leakage from the capillaries to the interstitium and continuing into the alveoli, resulting in life-threatening pulmonary edema (Cruz et al., 2018). Once lung damage occurs, lung ECs express pro-inflammatory cytokines and signaling molecules to further exacerbate vascular permeability, vascular tone, leukocyte recruitment, and apoptosis (Villar et al., 2014).
Endothelial Mechanical Sensing in VILI
Ample evidence has been found to support that reorganization of the pulmonary endothelial cytoskeleton caused by mechanical stress leads to VILI (Lionetti et al., 2005). Many ARDS studies also suggest that transient receptor potential (TRP) channels are activated in lung injury induced by mechanical stress, and some certain types of TRP, including TRPV4, facilitate mechanical stress sensing (Parker et al., 1998; Alvarez et al., 2006). This is complicated, however, by the fact that these responses are post-cellular injury, and particularly since other cellular stresses including heat, osmolarity changes, and metabolites can also activate TRP channels in a similar pattern (Darby et al., 2016; Simonsen et al., 2017). Given the fact that mechanical stress originates from the misalignment between the basal membrane and the cytoplasmic membrane, linker complexes, including FAs, between the two parties, must be the first line mechanical stress sensors for the cells to initiate other cellular responses (Geiger and Bershadsky, 2002). Interestingly, FAs are also dynamic regulators of cytoskeletal remodeling, where assembly, disassembly, and structure alteration adjust the formation and displacement of actin fibers (Oakes and Gardel, 2014).
Endothelial Cell Cyclic Stretch Experimental Models
Measurements of mechanical stress in the mechanically ventilated lung are technically challenging due to the complexity of local distension patterns in the lung parenchyma, however, calculations have been made to suggest that if the lung volume increases by 40% of the total lung capacity, the alveolar epithelial cell basal surface area increases by 34% (Tschumperlin and Margulies, 1999; Tschumperlin et al., 2000; Wirtz and Dobbs, 2000). High tidal volume mechanical ventilation results in a 40–50% surface area increase as would be reflected in vitro by 18% CS or repeated stretch (Tschumperlin et al., 2000; Birukov, 2011), and spontaneous breathing with a 25% surface area increase can be reflected as 5% CS (Tschumperlin et al., 2000; Wirtz and Dobbs, 2000; Birukov, 2011). Here 18 and 5% CS is the measure of the cell length elongation in one dimension compared to resting conditions, although the ECs may be exposed to one or two dimensional stretch (Wang J.H. et al., 2001). CS is accomplished by growing monolayers of ECs to confluence on flexible membranes and stretching those membranes either on commercial systems (Wang et al., 2000; Colombo et al., 2008) or custom machines. Published in vitro experiments of VILI mainly use 10–20% CS with 0.4–2 Hz of frequency to reflect the 20–120 breaths per minute ventilation used in the intensive care unit (Rimensberger, 2003; Belteki and Morley, 2018). In this review, in order to clarify the different experimental outcomes with different mechanical stretch conditions, amplitude of stretch (% CS) and frequency are noted in each in vitro research data cited.
Focal Adhesions in VILI
Endothelial FA
Focal adhesions (FAs) may be described as discrete areas on a cell’s basal surface located at the end of “stress fibers,” prominent bundles of actin filaments, which contain integrins and a variety of associated proteins. These provide anchor points for cells to adhere to their substratum, as well as sense and transmit forces and biochemical signals between cells and matrix (Wu, 2005). Essential to FAs are integrins, transmembrane proteins that bind matrix ligands extracellularly and a series of structural proteins that link it to the cytoskeleton intracellularly. These structural proteins include talin, focal adhesion kinase, paxillin, tensin, filamin, and α-actinin, as well as many other associated linkers and signaling proteins (Figure 1). To date, over 900 proteins have been found in FAs (Kuo et al., 2011). In ECs, FAs play central roles in angiogenesis, wound healing, vascular remodeling, cytoskeletal arrangement, and barrier regulation (Wu, 2005).
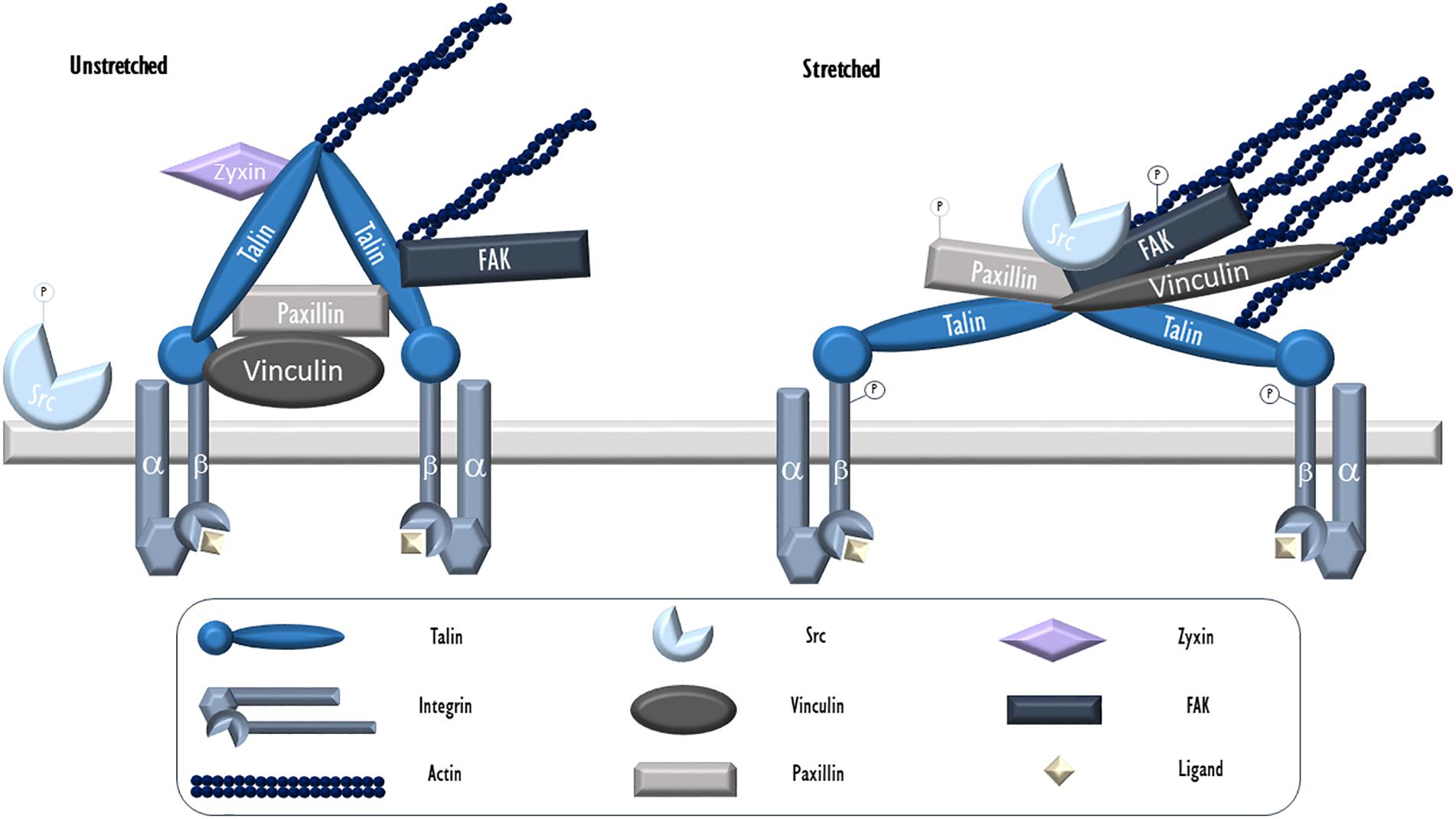
Figure 1. Endothelial focal adhesion in cyclic stretch. Integrins, heterodimers consisting of α and β subunits, serve as the key physical link between FAs and the ECM. During VILI, mechanical stress (cyclic stretch) causes calcium release from intracellular stores and c-Src activation via dephosphorylation. It then localizes to patches along the cytoskeleton and FAs where it targets and phosphorylates FA proteins including integrin β, paxillin, and FAK. Phosphorylated and activated FAK facilitates the formation of stress fibers. Talin is an adaptor protein essential for integrin connection to the cytoskeleton. In its activated form, talin dimers are bound to actin and likely assume a Y-shape with exposed vinculin binding sites. Vinculin is a cytosolic actin-binding protein that exists in a circular configuration localized close to integrins and binds paxillin. Upon activation, vinculin assumes an extended form and moves further toward actin fibers. Paxillin, another adaptor FA protein activated by c-Src mediated phosphorylation following stretch, allows for protein networking and signal transduction. Zyxin, a LIM protein, resides at unstretched FAs but dissociates and moves away from FAs and toward stress fibers during stretch. -P, phosphorylated.
During mechanical ventilation or related cellular stretch, like all other cell types, lung ECs respond to mechanical forces largely through the action of the actin cytoskeleton. Mechanical force associated cellular signals, often deleterious, rely on the contractile activity of F-actin associated actomyosin networks- interconnected two-dimensional contractile meshworks that include actomyosin fibers and their anchor points (Lecuit et al., 2011). This is especially true during mechanical ventilation when no pathologically high mechanical stress by blood flow (often seen in pulmonary hypertension or other pulmonary vascular diseases) is present. Located in association with the ECM, FA complexes are the main participants in ECs to receive mechanical stimuli and serves as key mechanical tension sensing and signaling hubs through complex signaling events such as post-translational modifications, binding to cytoskeletal proteins or kinases, and structural changes (Zaidel-Bar and Geiger, 2010). They play a central role in receiving and transducing mechanical stress to the cytoplasm, via associated transmembrane integrins (Figure 1).
SRC Proto-Oncogene, Non-receptor Tyrosine Kinase
One of the key components and the most critical kinase in FAs is cellular Src (c-Src), the prototypical member of the Src family of kinases (Kefalas et al., 1995). Under normal unstressed conditions, N-terminal myristoylation causes c-Src association with plasma membranes, but relocation occurs via cytoskeletal trafficking (Jones et al., 2000). C-Src has kinase activity to phosphorylate its substrates, including autophosphorylation on Y416 (Cooper and MacAuley, 1988). C-Src interacts with these targets (or other binding partners) through its SH2 and SH3 domains (Cooper and MacAuley, 1988). c-Src tertiary structure and activity can be regulated by the phosphorylation state of a tyrosine residue (Y527) at the C-terminal (Cooper and MacAuley, 1988). Phosphorylation at this site by c-terminal Src kinase (Csk) inactivates c-Src, while dephosphorylation by calcineurin, a Ca2+r/calmodulin-dependent protein phosphatase (PPIIB), activates it (Cooper and King, 1986).
During VILI, c-Src is activated through a variety of means. Mechanical stretching of ECs leads to Ca2+ influx through the opening of stretch-activated cation channels (SA channels), which in turn initiates Ca2+ release from intracellular calcium storage (Adams et al., 1989; Naruse and Sokabe, 1993; Naruse et al., 1998c). Histamine, whose levels rise in inflammatory lung injuries (Kim et al., 2005), also triggers [Ca2+] oscillations (Adams et al., 1989). This rise in intracellular [Ca2+] or [Ca2+]i activates c-Src, giving rise to many of the morphological changes seen in ECs during CS (Naruse et al., 1998a). Once c-Src is dephosphorylated and activated, it localizes to patches along cytoskeletal structures, as well as FAs where it targets several FA proteins including integrins (Hirst et al., 1986), FAK, and paxillin (Sokabe et al., 1997). Inhibition of c-Src activation or kinase activity prevents most of the effects of CS including cell alignment and formation of stress fibers that occurs with 20% CS at 1 Hz (Naruse et al., 1998a) and disassembly of adherans junctions that occurs with 18% CS at 0.4 Hz (Tian et al., 2016). C-Src activation subsequent to activation of VEGF receptor 2 (VEGFR2) leads to phosphorylation of cytoskeletal proteins outside of FAs by c-Src when subjected to 18% CS at 0.4 Hz for 120 min (Tian et al., 2016). Some of the effects of these phosphorylation events will be discussed later.
Talin
Talin is found universally in FAs and in fact is the only essential adaptor protein for integrin connection to the cytoskeleton (Jiang et al., 2003). Evolutionally, it is older than integrins, leading some to consider talin as the master of FAs (Klapholz and Brown, 2017). Talin is structurally split into a head domain, a short linker domain, and a long rod domain (Klapholz and Brown, 2017). The N-terminal FERM domain forms the head and is split into four subdomains termed F0-F3. F0 binds Rap1 small GTPase proteins. F1, F2, and F3 can bind directly to membranes. F2 and F3 can bind to actin via the actin binding site 1 (ABS1). The F3 domain can bind to the cytoplasmic domain of β integrin subunits at its integrin binding site 1 (IBS1) which is a variant of the canonical phosphotyrosine binding (PTB) domain (Garcia-Alvarez et al., 2003). F3 also can interact with many associated proteins including FAK (Klapholz and Brown, 2017). The rod domain is comprised of subunits R1-R13, and contains multiple sites for protein–protein interactions. Actin binding site 2 (ABS2) is located at R3-R8 and ABS3 is located at R13. The integrin binding site 2 (IBS2) falls within R11, and consists of two five-helix bundles connected by a kinked continuous helix (Gingras et al., 2009). Activation of integrin is required for this binding since the binding site on β integrin is on the same face that binds the tail of α integrin (Rodius et al., 2008). At least 18 vinculin binding sites have been found in the rod domain (Gingras et al., 2005). In addition, more binding sites for Rap1-GTP-interacting adaptor molecule (RIAM), paxillin, α-synemin, deleted in liver cancer 1 (DLC1), and KN motif and ankyrin repeat domain proteins Kank1 and Kank2 have been found within the rod domain. Talin’s only known activities are through binding other proteins, making it a classic adapter protein (Klapholz and Brown, 2017).
When bound to actin, talin is likely in its dimer form (Goldmann et al., 1994) and adopts a Y-shape (Winkler et al., 1997) or dumbbell shape (Hemmings et al., 1996). The dimerization domain is at the C-terminus, and just upstream are linker regions thought to be flexible. Molecular dynamics simulation in the Mofrad lab has shown that one of talin’s vinculin-binding sites (VBS1) is inactive unless pulled open by a stretching force (Lee et al., 2007). Cellular stretching causes hydrophobic residues on the VBS1 surface to rotate around a neighboring alpha-helix, exposing the hydrophobic vinculin binding pocket. This was shown to operate in vitro and was reversible (Yao et al., 2016). The same research group (Mofrad) that performed the molecular dynamics went on to confirm that the distance between integrins bound by a talin dimer alters the angle of the talin dimerization regions (Golji and Mofrad, 2014). The varying distance between integrins that occurs during stretching of the basement membrane during VILI makes this a likely method of mechanosensing within FAs. The stretch of talin modeled above is converted into a cellular response by vinculin (Elosegui-Artola et al., 2016).
Aside from stretch related stress, chemical factors released during VILI are capable of exerting effects on talin through vinculin. Although details have not been clarified, an active F-actin binding site on vinculin is required for production of thrombin induced talin positive FAs to cause increased endothelial monolayer permeability (Birukova et al., 2016).
Vinculin
Vinculin is a cytosolic actin-binding protein that is involved in stabilizing actin polymerization and recruiting actin remodeling proteins (Bays and DeMali, 2017). Like talin, it has no enzymatic activity. Its structure consists of a large head domain, a short linker, and a tail. When inactive in the cytosol, vinculin is in a circular configuration where the head and tail have a very tight bond, inhibiting interactions with other proteins. But when activated, such as in FAs, vinculin exists in an extended form (Chen et al., 2005). Many proposals have been presented on just how vinculin is opened, including multiple ligand interaction (Bois et al., 2006; Chen et al., 2006), single ligand displacement (Izard et al., 2004), phosphorylation (Golji et al., 2012), and stretching (Golji and Mofrad, 2010).
Vinculin requires a level of tension to remain in a FA and prevent disassembly of FAs, making it a likely mechanosensor by itself (Carisey et al., 2013). When inactive, vinculin is localized closer to integrins and binds paxillin. When activated by binding to talin, vinculin is seen to move further away from integrin where it interacts strongly with actin (Case et al., 2015). This seems to paint a picture where stretch induced FA rearrangement includes opening of vinculin so that it can stabilize stress fibers.
Focal Adhesion Kinase
Focal Adhesion Kinase (FAK) is an adapter protein, as well as an active kinase found at FAs and other locations throughout the cell. It is activated both through recruitment to FAs following integrin activation as well as by phosphorylation. Multiple tyrosine, serine, and threonine phosphorylation sites have been mapped by mass spectrometry (Grigera et al., 2005).
Exposure of human pulmonary ECs to 18% CS at 25 cycles per minute leads to FAK phosphorylation at Y397 and Y576 (Shikata et al., 2005). This CS also induces FAK re-distribution to the ends of newly formed stress fibers. Similar results were seen in bovine ECs. Besides phosphorylation and rearrangement of FAK, paxillin is phosphorylated and rearranges, events which were dependent on Rho/p21 activity (Yano et al., 1996a,b). Interestingly, some of these changes are mediated by SA channel opening and c-Src activation, while SA channel inhibition does not eliminate the changes to FAK and paxillin, suggesting other activation/inactivation mechanisms besides c-Src are at play (Sawada and Sheetz, 2002).
The pathophysiological changes caused by VILI are not mediated exclusively by tissue overdistension (by volume increase) or high frequency ventilation (by frequency increase). Agents such as thrombin and cytokines are also found at high levels in VILI patients and contribute to the pathology of this injury. Thrombin binds to its receptor PAR-1, thereby mediating FAK redistribution patterns similar to stretch (Shikata et al., 2003a). This is mediated by phosphorylation of the same residues (Y397 and Y576) as those seen in stretch but also at Y925. By contrast, c-Src phosphorylation of FAK at Y576 only is sufficient to cause many of the barrier protective effects seen in sphingosine-1-phosphate mediated endothelial barrier protection including peripheral translocation of FA proteins and cortical actin ring formation (Shikata et al., 2003a). This demonstrates the varying signaling pathways that can be activated by the phosphorylation profile of FAK.
When cells are cyclically stretched in only one direction, stress fibers form perpendicularly to the stretch direction. This alignment is dependent on the type of stretch applied. For example, when bovine aortic ECs are subjected to 10% CS, actin alignment is increased proportionally to the frequency (0.01 to 1 Hz) of CS (Shirinsky et al., 1989; Sokabe et al., 1997; Hsu et al., 2010). The involvement of FAK in stress fiber formation as a result of CS is controversial. Some reports show FAK phosphorylation, which occurred during 20% CS at 1 Hz, is required for this process (Sokabe et al., 1997), while others show that FAK/paxillin knockdown in ECs or overexpression in fibroblasts do not block stress fiber formation at 10% CS at 1 Hz (Hsu et al., 2010; Ngu et al., 2010).
More evidences have been generated to prove the role of FAK in endothelial signaling activated by mechanical stress. Uniaxial 10% CS at a rate of 3 cycles per minute activates cell proliferation in ECs (Sumpio et al., 1987). Additionally, FAK activation plays a role in cell proliferation in a variety of cell types including epithelial and fibroblasts. In epithelial cells, 10% CS at 20 cycles per minute activates Y418 phosphorylation of c-Src as well as the two tyrosine sites on FAK already mentioned (Y397 and Y576). These lead to downstream ERK1/2 activation and proliferation within 5 min after initiation of CS (Wang J.G. et al., 2001; Chaturvedi et al., 2007). Fibroblasts stretched 20% at 1 Hz activate the same pathway, though FAK is phosphorylated at Y397 and Y925 (Wang J.G. et al., 2001). Further light on the pathway of proliferation was gleaned in osteoblast-like cells where CS activated FAK, c-Src, and proline-rich tyrosine kinase 2 (PYK2). Although c-Src was not found to be necessary, FAK associated with PYK2 and led to ERK2 phosphorylation mediated by the Ras/Raf/MEK pathway (Boutahar et al., 2004). It is logical that the same pathway would function in pulmonary endothelia since 10% CS at 1 Hz activates ERK1/2 phosphorylation in bovine ECs, though the p21ras/PI3K pathway may also be involved (Ikeda et al., 1999). This PYK2 phosphorylation is mediated by c-Src (Cheng et al., 2002). Also, it appears that 12% CS at 1 Hz results in Ca2+ dependent PKCα activation leading to NADPH oxidase activity in ECs to produce ROS. This ROS is crucial for c-Src and PYK2 activation. Interestingly, pulmonary injury induced by 20% CS at 0.5 Hz-causes a reduction of FAK phosphorylation and activity following a transient increase in phosphorylation at 30 min of injury (Desai et al., 2009). This emphasizes the damaging effects of VILI on an already injured lung.
As discussed above, VILI leads to an increase in pulmonary vascular permeability. FAK plays a critical role in both initiation and integrating signaling pathways that regulate barrier function (Wu, 2005), though whether FAK causes barrier loss or protection is unclear. For example, in mice with ECs expressing defective FAK, pulmonary vascular permeability was severely compromised, an abnormal distribution of vascular endothelial cadherin (VE-cadherin) was observed, and reduced VE-cadherin Y658 phosphorylation was measured (Zhao et al., 2010). Additionally, FAK knockdown inhibits the normal barrier enhancement provided by sphongosine-1-phosphate (Zhao et al., 2009). On the other hand, FAK knockdown in pulmonary ECs exhibits a number of barrier enhanced phenomenon including stronger cell-cell contacts and a greater number and size of vinculin plaques (Arnold et al., 2013). All these data suggest that FAK is a key FA molecule regulating endothelial injury and signaling upon mechanical stress.
Paxillin
Paxillin is another adapter protein of FAs that allows for protein networking and signal transduction. Phosphorylation occurs primarily at Y31 and Y118 by FAK or Src family kinases (Schaller and Parsons, 1995) but also is targeted at Y40, Y88, and Y181 (Nakamura et al., 2000; Schaller and Schaefer, 2001). Five LD domains (LD1-5) near the N-terminus have been identified that function as protein interaction interfaces for actopaxin, ILK, vinculin, papillomavirus E6, FAK/PYK2, the Arf-GAPs p95PKL/GIT2/GIT1, and some evidence shows PAK3, clathrin, and PABP1 (Brown and Turner, 2004). Paxillin directly binds integrin β (Schaller et al., 1995).
During 10% CS at 1 Hz, paxillin levels remain unchanged, but phosphorylation is induced (Yano et al., 1996a). Whereas paxillin will exhibit a speckled pattern in static conditions, strain causes paxillin to align to the long axis of cells in parallel to F-actin. Inhibition of tyrosine phosphorylation blocks paxillin rearrangement, F-actin alignment, and cell elongation. This inhibition of tyrosine phosphorylation was indiscriminate for paxillin and FAK, so distinct roles for FAK versus paxillin could not be delineated. It is also interesting that cell migration is mediated in part by paxillin/FAK phosphorylation, and this is proportional to the degree of stretch. The upstream activator of phosphorylation in this case is not verified, but it is highly possible that activated c-Src (Sokabe et al., 1997) phosphorylates paxillin and leads to cytoskeletal rearrangement. Inhibition of FAK phosphorylation also prevents paxillin phosphorylation at 20% CS 1 Hz, demonstrating that paxillin phosphorylation occurs downstream of FAK activation (Naruse et al., 1998b).
Paxillin is not required for maintenance of a FA. In ECs, 20% CS at 0.5 Hz induced paxillin rearrangement is transient (Huang et al., 2012). Additionally, this study showed that paxillin only plays a role in the early portion of FA rearrangement; paxillin knockdown inhibited FA formation at 10 min CS, but did not inhibit FA formation at 30 min or longer.
Other FA Proteins
G protein-coupled receptor kinase-interacting target 1 (GIT1) has been proposed to be involved in FA disassembly (Zhao et al., 2000; Shikata et al., 2003b; van Nieuw Amerongen et al., 2004). GIT1 under normal conditions exhibits a cytoplasmic distribution. In ECs, some GIT1 is found weakly distributed to the end of stress fibers at FA following 18% CS (Shikata et al., 2005) and thrombin exposure (van Nieuw Amerongen et al., 2004) where it colocalizes with FAK and vinculin. RhoA and Rho kinase are required for this recruitment, providing evidence that the signal for GIT1 is derived outside of the FA. At the FA, GIT1 is phosphorylated either by Rho kinase or c-Src, and contributes to stretch or thrombin induced cell rounding and contraction, FA formation, and FAK phosphorylation.
Zyxin is a LIM protein that resides at FAs (Beckerle, 1997). In a number of cell types including umbilical vein ECs, under 15% CS at 0.5 Hz, zyxin moves away from FAs and associates with stress fibers (Yoshigi et al., 2005). The trigger for movement is derived at least in part by integrin activation and not by SA channels. The only known role of zyxin in these conditions is to reinforce the actin structure by organizing thicker filaments.
Integrins in VILI
Integrins are heterodimer transmembrane proteins that serve as the key physical anchor for the FA to the cytoplasmic membrane and the connection between FA and the ECM. They consist of an α and a β subunit. There are now eighteen identified α subunits and eight β subunits. Each subunit contains extracellular, transmembrane, and cytoplasmic domains. Based on the exact pairing, extracellularly, integrins bind particular ECM components such as collagen, laminin, fibronectin, and vitronectin, as well as some other cellular receptors and signals including E-cadherin, prothrombin, and von Willebrand factor (Plow et al., 2000; Figure 2). Intracellularly, β chains alone seem to define cytoskeletal interactions (Pan et al., 2016). Because of this, we will focus on the roles of β integrins with known roles in VILI, mentioning their binding pair when known.
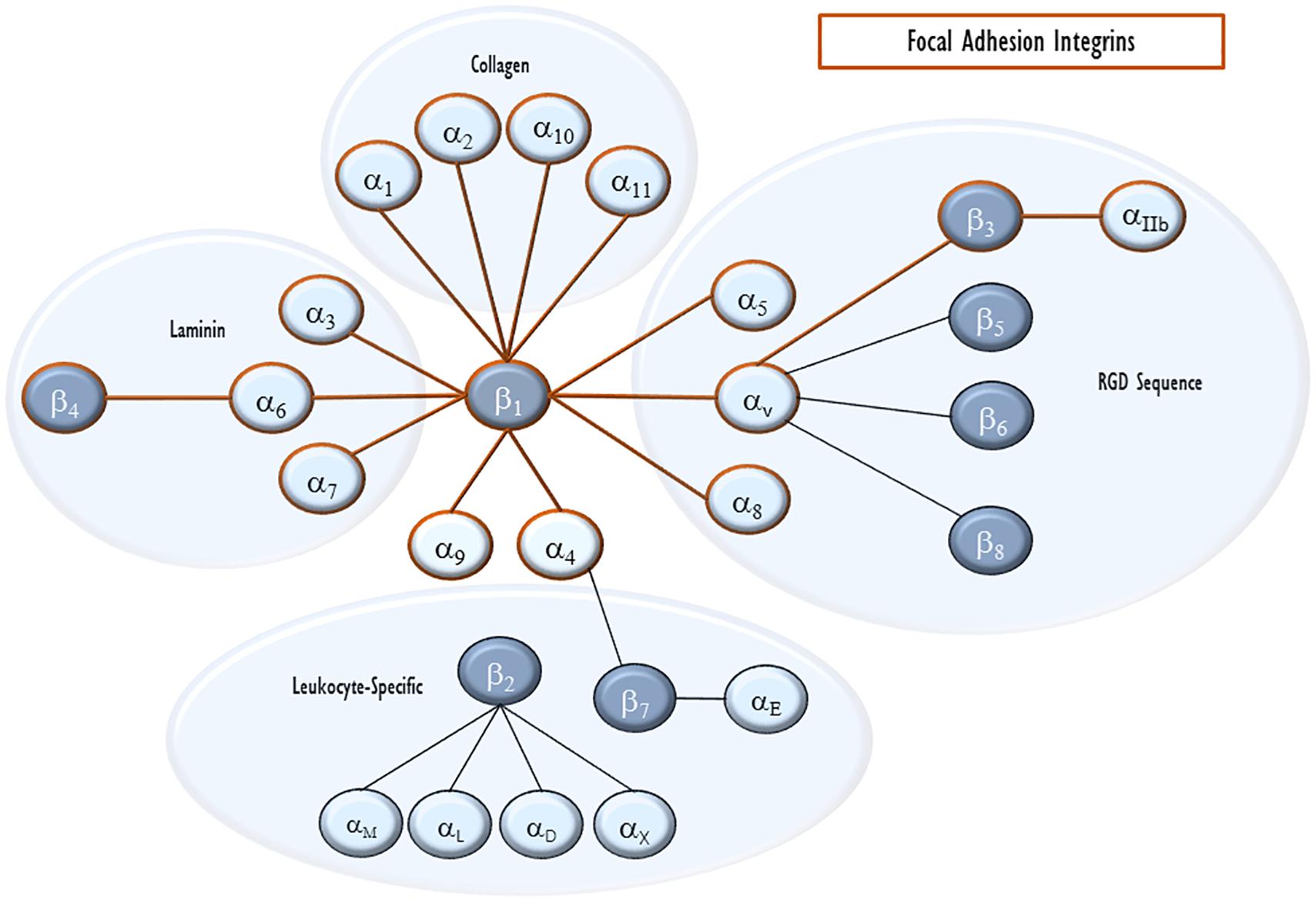
Figure 2. Integrin pairing. Integrins are heterodimeric cell-surface receptors for ECM proteins. Each heterodimer consists of an α and a β subunit. The eighteen α integrins and eight β integrins are shown with lines connecting pairs that are able to heterodimerize with each other. They are also clustered by their major ligand. Integrins that can enter endothelial focal adhesions are outlined in red.
β integrins, with the exception of β4, contain short cytoplasmic sequences (40–60 amino acids) (Sastry and Horwitz, 1993). Among these, the sequences of β1-3 and 5–7 are very similar. Adjacent to the transmembrane domain is a short sequence of 11 mostly charged amino acids. The second cluster contains a NPIY sequence that can be phosphorylated by c-Src and similar kinases (Hirst et al., 1986). An NPXY sequence comes next, though the spacing is variable between different β integrins. All three sequences, at least in integrin β1, are needed for full integration into FAs.
Because of their structure and location, integrins can mediate signaling in two directions (Hu and Luo, 2013). Binding of extracellular matrix proteins leads integrin heterodimers to undergo a conformational change including separation of cytoplasmic tails of the two integrin subunits, allowing for interactions between these tails and cytoplasmic proteins and thus propagating signals (Luo et al., 2007; Zhu et al., 2007). This has been termed outside-in signaling. Conversely, binding of intracellular proteins such as talin or kindlin result in integrin subunit separation (Anthis et al., 2009; Ye et al., 2011). This separation alters structural conformation such that the extracellular domains have an increased affinity for ligands (Hu and Luo, 2013). This has been termed inside-out signaling. β integrins in FAs are already bound to talin, FAK, and paxillin, which prevent association between subunits, keeping the integrin in an open conformation (Kim et al., 2011; Hu and Luo, 2013). It is imaginable, however, that during distension of the ECM during stretch, inactive integrins may come into contact with extracellular ligands leading to outside-in signaling and formation of new FAs. In our discussions of talin and vinculin, we saw that some of the events involved in FA activation during CS are dependent upon propagation of force from the ECM to these proteins. Obviously integrins, as the link across the plasma membrane, are central to this propagation (Geiger et al., 2009; Hu and Luo, 2013). This force propagation may be considered another outside-in mechanosensing function of integrins within the FA.
In most research on FAs, the exact integrin involved is not identified. In FAs of ECs, the most recognized β integrins are β1 and β3. It is also worth note that these two integrins are often interchangeable in which types of FAs they incorporate (Sastry and Horwitz, 1993).
Many of the studies used to analyze the role of integrins took little into account with regard to available ECM proteins and conditions. Since different αβ pairs have different ECM protein binding substrates, results of experiments can be misleading when a particular integrin is being measured and its substrate is not available on the culture surface (Hirayama and Sumpio, 2007). Detailed experiments have even showed that integrin heterodimer pairs switch depending on the availability of extracellular cations (Stuiver et al., 1996).
Integrin β1
Integrin β1 is referenced most often in the literature of FAs. It can be phosphorylated at T777, Y783, S785, T788, T789, and Y795, but only requires phosphorylation of T788 or T789 to be active (Wennerberg et al., 1998; Nilsson et al., 2006). This phosphorylation is achieved by PKCε (Stawowy et al., 2005).
Upon 10% CS at 1 Hz, integrin β1 redistributes to the ends of stress fibers, similar to other FA proteins FAK or paxillin. In capillary ECs, static stretch (15% elongation) induces integrin β1 phosphorylation at T788/T789 within 1 min of force application (Thodeti et al., 2009). Inhibition of T788/T789 phosphorylation prevents strain-induced cell reorientation, stress fiber alignment, and redistribution of FAs (Sastry and Horwitz, 1993; Hu and Luo, 2013). Upstream of phosphorylation was opening of the SA channel TRPV4 followed by PI3K activity.
One aspect not often studied with regard to CS is the influence of neighboring cells. One study addressed this by stretching isolated cells stretch (Huang et al., 2011). They noted that under static conditions, integrin β1 is loosely distributed throughout ECs. Following 20% stretch activation of confluent cells at a frequency of 0.5 Hz, integrin β1 levels rise and clustering is observed in lines perpendicular to the direction of stretch after 10 min. In isolated cells, however, integrin β1 failed to cluster or align in this fashion, showing that intercellular junctions play a role in endothelial cell cytoskeleton rearrangement and not just focal adhesion mechanosensing. Another report also recognized the redistribution of integrin β1 upon stretch to linear patterns, but did not see a rise in β1 levels at the mRNA level (Yano et al., 1997). The binding partner for integrin β1 is dependent on the coating of the stretched surface; when coated with fibronectin, the fibronectin receptor integrin α5β1 is found, and when coated with collagen, the collagen receptor α2β1 is found.
Integrin β3
Integrin β3 is a receptor for vitronectin whether dimerized with integrin αv or αIIb. Umbilical vein ECs exposed to 20% CS at a frequency of 1 Hz demonstrate significantly elevated integrin β3 levels of mRNA after 4 h and elevated protein expression at 12 h (Suzuki et al., 1997). Equally, the number of FAs containing integrin β3 increased. These results are debatable, however, because in similar experiments (at the less strenuous conditions of 10% CS at 1 Hz) using the same type of cells, integrin β3 did not follow FA rearrangement (Yano et al., 1997).
We have seen above that thrombin levels are elevated in VILI, and that thrombin regulates FAK and GIT1 distribution. Some evidence shows a secondary effect of thrombin to induce angiogenesis in ECs by directly binding to integrin αvβ3 (Tsopanoglou et al., 2002). However, these effects are overshadowed by the damage caused by overdistension and probably only plays a role during healthy physiologic breathing (Birukova et al., 2006).
Integrin β4
Integrin β4, with a very different cytoplasmic domain compared to other β integrins, does not incorporate into traditional FAs. Nevertheless, integrin β4 interacts with a number of FA proteins. And as we shall see, it has the most complex reaction to VILI of all the β integrins.
The only known partner for integrin β4 is integrin α6 (Hynes, 2002). Integrin α6β4’s extracellular target is laminin-5. Unlike other FA β integrins, β4’s cytoplasmic tail interacts with intermediate filaments through plectin. Syndecan-1, a cell surface proteoglycan which plays roles in FA structure, can bind integrin β4 as well as other intracellular and extracellular components (Altemeier et al., 2012; Wang et al., 2014).
The cytoplasmic tail of integrin β4 is 1088 amino acids long. This tail is composed of a proximal Calx Na-Ca exchanger domain followed by two pairs of fibronectin type II repeats with a tyrosine activation motif (TAM) between them (Hogervorst et al., 1990). Several tyrosines and serines can be phosphorylated; some of the tyrosines are targeted by Src family kinases (SFKs) including Fyn and Yes (Giancotti, 2007), and serines are targeted by epidermal growth factor (EGF) and/or PKCα (Rabinovitz et al., 2004). Phosphorylation by these kinases results in binding of FAK to integrin β4 (Tai et al., 2015). The exact point of interaction seems to be just proximal to the transmembrane domain of integrin β4 to a sequence of FAK just upstream of its Y397 autophosphorylation site (amino acids 376–386).
Integrin β4 is tyrosine phosphorylated on at least one of Y1440, Y1526, Y1640, or Y1422 within 30 min of exposure to 18% CS at 0.5 Hz (Chen et al., 2015). This phosphorylation is required for the full effects of CS induced inflammatory factor release into media, though it seems that at least one phosphotyrosine also provides protective effects. In a broader model, it seems that integrin β4’s cytoplasmic tail is responsible for many of the effects of VILI, as mutant mice lacking the cytoplasmic portion are almost completely protected from high tidal volume ventilation. Integrin β4 binding to laminin-5 does not seem to be required for the morphological changes seen in stretched ECs when other integrins are available (Hirayama and Sumpio, 2007).
Other Integrin β
Of the five remaining β integrins, β5 is the only one that is significantly expressed in the pulmonary endothelium, though data supporting localization within FAs is lacking (Wayner et al., 1991; Sastry and Horwitz, 1993). There is evidence, however, that integrin αvβ5 associates with FAK from c-Src activity in developing ECs (Eliceiri et al., 2002). This integrin certainly plays a role in VILI progression because knockout mice are protected from VILI and blocking integrin αvβ5 in vitro prevents thrombin injury (Su et al., 2007). And integrin β5 signaling is required for stretch induced changes in epithelial cells and associates with zyxin (Bianchi-Smiraglia et al., 2013). Integrin β6 is heavily concentrated in epithelium but is not normally expressed in endothelium (Tabata et al., 2008). Integrins β2 and β7 are principally expressed in leukocytes, but β7 is also found in some endothelia where it heterodimerizes with α4 and responds to a number of inflammatory agents (Brezinschek et al., 1996). Integrin β8, with a non-classical cytoplasmic domain compared to β1-3 and β5-7, was originally characterized as unimportant with regard to adhesion or the cytoskeleton (Nishimura et al., 1994). Later, however, it was found in ECs where it plays a role in angiogenesis (Giusti et al., 2013). Evidences of the involvement of these other types of integrin βs in FAs are very limited and require further experiments.
Summary and Conclusion
Ventilator induced lung injury is characterized by compromised vascular endothelial barrier protection and the production of edemagenic agents in response to mechanical stretch that may lead to overdistention depending upon tidal volume and ventilation frequency (Dos Santos and Slutsky, 2000; Lionetti et al., 2005; Birukova et al., 2006). This mechanical-force initiated cellular injury results in cytoskeletal rearrangement. FAs play a central role in mechanotransduction and cytoskeletal rearrangement (De et al., 2010; Ladoux and Nicolas, 2012; Iskratsch et al., 2014; De, 2018). Here, we present the known picture of the molecular changes that occur at FAs as a result of CS and other factors of VILI. Within the FA, force from stretching of the ECM is propagated through integrins into the FA (Geiger et al., 2009). This force is then exerted on adapter and signaling proteins such as talin and vinculin. Additionally, other receptors for stretch and agents involved in VILI result in phosphorylation events on c-Src and then on integrin, FAK, paxillin, and others. Together these lead to the characteristic stress fiber formation and cytoskeletal rearrangement seen in VILI.
The most essential proteins within the FA are integrins and talin (Jiang et al., 2003). These are the minimum structural components which link the ECM to the actin cytoskeleton. Talin may be considered the master regulator of FAs (Klapholz and Brown, 2017), but integrins anchor the FA to the plasma membrane, are the first proteins involved in a newly formed FA (Romer et al., 2006) and are the first link in the outside-in function of FA force propagation.
Ventilator induced lung injury remains a substantial health care burden with an obvious lack in therapies (Dos Santos and Slutsky, 2000; Plataki and Hubmayr, 2010). More research on the nature of VILI progression is essential in order to prevent and treat patients with this injury. This review focusses on individual FA proteins currently known to be involved in VILI related pathology. By integrating this knowledge with other VILI research on the cellular, tissue, organ, and individual level, it is hoped that new prevention and treatment methods will continue to reduce the impact of VILI. By investigating each individual protein as we have, we can better understand the basic mechanisms of mechanical stress on endothelial injury. This should help us identify more specific targets for drug therapies in VILI. Biological agents which target many of these proteins such as c-Src, FAK, paxillin, and integrins have been developed in other disease models (Paulhe et al., 2005; Infusino and Jacobson, 2012; Eke and Cordes, 2015), though currently none are approved for VILI therapy. The possibility that these agents would prove efficacious in VILI is difficult to determine because of the incomplete knowledge we have on the disease. Additionally, the various mathematical, in vitro, animal, and clinical data lead to many contradicting findings regarding the roles of individual proteins in VILI. Further research and understanding of individual proteins and sites within proteins should help develop a “more pure” understanding of the roles these proteins play and allow for more selective targeting and drug development.
More advanced tools are being developed to study VILI and related disease. One such tool is genetics. It is already common practice to personalize mechanical ventilation strategies using a variety of monitoring inputs (Nieman et al., 2017). Additionally, genetic influences may be at play, as health disparities are common in pulmonary injury (Moss and Mannino, 2002; Frutos-Vivar et al., 2006; Erickson et al., 2009), and genetic variants have been found to play a role in VILI and other pulmonary diseases (Barnes, 2005; Nonas et al., 2005; Gao et al., 2006). We are sure that variants in FA proteins will continue to be found and their function evaluated.
In summary, the FA-integrin complex is a key mechanical stress biosensor system in ECs in response to VILI. Although complex and sometimes controversial, the FA-integrin system modulates VILI associated endothelial injury and signaling, and obviously is a viable drug target for VILI. Our perspective is that the next breakthrough in FA-integrin research is the genetic influences on variable VILI outcome, which will lead to a better understanding of the pathobiology of endothelial mechanical stress sensing and signal transduction, thus more importantly providing a basis for personalized medicine.
Author Contributions
All authors wrote the text and edited the figures.
Funding
This study was supported in part by National Institutes of Health grant HL134610.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, D. J., Barakeh, J., Laskey, R., and Van Breemen, C. (1989). Ion channels and regulation of intracellular calcium in vascular endothelial cells. FASEB J. 3, 2389–2400. doi: 10.1096/fasebj.3.12.2477294
Altemeier, W. A., Schlesinger, S. Y., Buell, C. A., Parks, W. C., and Chen, P. (2012). Syndecan-1 controls cell migration by activating Rap1 to regulate focal adhesion disassembly. J. Cell Sci. 125(Pt 21), 5188–5195. doi: 10.1242/jcs.109884
Alvarez, D. F., King, J. A., Weber, D., Addison, E., Liedtke, W., and Townsley, M. I. (2006). Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ. Res. 99, 988–995. doi: 10.1161/01.res.0000247065.11756.19
Anthis, N. J., Wegener, K. L., Ye, F., Kim, C., Goult, B. T., Lowe, E. D., et al. (2009). The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 28, 3623–3632. doi: 10.1038/emboj.2009.287
Arnold, K. M., Goeckeler, Z. M., and Wysolmerski, R. B. (2013). Loss of focal adhesion kinase enhances endothelial barrier function and increases focal adhesions. Microcirculation 20, 637–649. doi: 10.1111/micc.12063
Barnes, K. C. (2005). Genetic determinants and ethnic disparities in sepsis-associated acute lung injury. Proc. Am. Thor. Soc. 2, 195–201. doi: 10.1513/pats.200502-013ac
Bays, J. L., and DeMali, K. A. (2017). Vinculin in cell-cell and cell-matrix adhesions. Cell. Mol. Life Sci. 74, 2999–3009. doi: 10.1007/s00018-017-2511-3
Beckerle, M. C. (1997). Zyxin: zinc fingers at sites of cell adhesion. Bioessays 19, 949–957. doi: 10.1002/bies.950191104
Belteki, G., and Morley, C. J. (2018). Frequency, duration and cause of ventilator alarms on a neonatal intensive care unit. Arch. Dis. Child. Fetal Neonatal Edn. 103, F307–F311. doi: 10.1136/archdischild-2017-313493
Bianchi-Smiraglia, A., Kunnev, D., Limoge, M., Lee, A., Beckerle, M. C., and Bakin, A. V. (2013). Integrin-beta5 and zyxin mediate formation of ventral stress fibers in response to transforming growth factor beta. Cell Cycle 12, 3377–3389. doi: 10.4161/cc.26388
Birukov, K. (2011). “Ventilator-induced mechancial stress and lung vascular dysfunction,” in Textbook of Pulmonary Vascular Disease, ed. J. X.-J. Yuan (Berlin: Springer), 913–934. doi: 10.1007/978-0-387-87429-6_64
Birukov, K. G. (2009). Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxid. Redox Signal. 11, 1651–1667. doi: 10.1089/ars.2008.2390
Birukova, A. A., Chatchavalvanich, S., Rios, A., Kawkitinarong, K., Garcia, J. G., and Birukov, K. G. (2006). Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am. J. Pathol. 168, 1749–1761. doi: 10.2353/ajpath.2006.050431
Birukova, A. A., Shah, A. S., Tian, Y., Gawlak, G., Sarich, N., and Birukov, K. G. (2016). Selective role of vinculin in contractile mechanisms of endothelial permeability. Am. J. Respir. Cell Mol. Biol. 55, 476–486. doi: 10.1165/rcmb.2015-0328oc
Bois, P. R., O’Hara, B. P., Nietlispach, D., Kirkpatrick, J., and Izard, T. (2006). The vinculin binding sites of talin and alpha-actinin are sufficient to activate vinculin. J. Biol. Chem. 281, 7228–7236. doi: 10.1074/jbc.m510397200
Boutahar, N., Guignandon, A., Vico, L., and Lafage-Proust, M. H. (2004). Mechanical strain on osteoblasts activates autophosphorylation of focal adhesion kinase and proline-rich tyrosine kinase 2 tyrosine sites involved in ERK activation. J. Biol. Chem. 279, 30588–30599. doi: 10.1074/jbc.m313244200
Brezinschek, R. I., Brezinschek, H. P., Lazarovits, A. I., Lipsky, P. E., and Oppenheimer-Marks, N. (1996). Expression of the beta 7 integrin by human endothelial cells. Am. J. Pathol. 149, 1651–1660.
Brown, M. C., and Turner, C. E. (2004). Paxillin: adapting to change. Physiol. Rev. 84, 1315–1339. doi: 10.1152/physrev.00002.2004
Carisey, A., Tsang, R., Greiner, A. M., Nijenhuis, N., Heath, N., Nazgiewicz, A., et al. (2013). Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner. Curr. Biol. 23, 271–281. doi: 10.1016/j.cub.2013.01.009
Case, L. B., Baird, M. A., Shtengel, G., Campbell, S. L., Hess, H. F., Davidson, M. W., et al. (2015). Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat. Cell Biol. 17, 880–892. doi: 10.1038/ncb3180
Chaturvedi, L. S., Marsh, H. M., and Basson, M. D. (2007). Src and focal adhesion kinase mediate mechanical strain-induced proliferation and ERK1/2 phosphorylation in human H441 pulmonary epithelial cells. Am. J. Physiol. Cell Physiol. 292, C1701–C1713.
Chen, H., Choudhury, D. M., and Craig, S. W. (2006). Coincidence of actin filaments and talin is required to activate vinculin. J. Biol. Chem. 281, 40389–40398. doi: 10.1074/jbc.m607324200
Chen, H., Cohen, D. M., Choudhury, D. M., Kioka, N., and Craig, S. W. (2005). Spatial distribution and functional significance of activated vinculin in living cells. J. Cell Biol. 169, 459–470. doi: 10.1083/jcb.200410100
Chen, W., Epshtein, Y., Ni, X., Dull, R. O., Cress, A. E., Garcia, J. G., et al. (2015). Role of integrin beta4 in lung endothelial cell inflammatory responses to mechanical stress. Sci. Rep. 5:16529. doi: 10.1038/srep16529
Cheng, J. J., Chao, Y. J., and Wang, D. L. (2002). Cyclic strain activates redox-sensitive proline-rich tyrosine kinase 2 (PYK2) in endothelial cells. J. Biol. Chem. 277, 48152–48157. doi: 10.1074/jbc.m110937200
Colombo, A., Cahill, P. A., and Lally, C. (2008). An analysis of the strain field in biaxial Flexcell membranes for different waveforms and frequencies. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 222, 1235–1245. doi: 10.1243/09544119jeim428
Cooper, J. A., and King, C. S. (1986). Dephosphorylation or antibody binding to the carboxy terminus stimulates pp60c-src. Mol. Cell. Biol. 6, 4467–4477. doi: 10.1128/mcb.6.12.4467
Cooper, J. A., and MacAuley, A. (1988). Potential positive and negative autoregulation of p60c-src by intermolecular autophosphorylation. Proc. Natl. Acad. Sci. U.S.A. 85, 4232–4236. doi: 10.1073/pnas.85.12.4232
Cruz, F. F., Rocco, P. R. M., and Pelosi, P. (2018). Role of the extracellular matrix in the genesis of ventilator-induced lung injury. Medizinische Klinik, Intensivmedizin und Notfallmedizin. 113(Suppl. 1), 2–6. doi: 10.1007/s00063-017-0376-8
Darby, W. G., Grace, M. S., Baratchi, S., and McIntyre, P. (2016). Modulation of TRPV4 by diverse mechanisms. Int. J. Biochem. Cell Biol. 78, 217–228. doi: 10.1016/j.biocel.2016.07.012
De, R. (2018). A general model of focal adhesion orientation dynamics in response to static and cyclic stretch. Commun. Biol. 1:81. doi: 10.1038/s42003-018-0084-9
De, R., Zemel, A., and Safran, S. A. (2010). Theoretical concepts and models of cellular mechanosensing. Methods Cell Biol. 98, 143–175. doi: 10.1016/S0091-679X(10)98007-2
Desai, L. P., White, S. R., and Waters, C. M. (2009). Mechanical stretch decreases FAK phosphorylation and reduces cell migration through loss of JIP3-induced JNK phosphorylation in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L520–L529. doi: 10.1152/ajplung.00076.2009
Dos Santos, C. C., and Slutsky, A. S. (2000). Invited review: mechanisms of ventilator-induced lung injury: a perspective. J. Appl. Physiol. 89, 1645–1655. doi: 10.1152/jappl.2000.89.4.1645
Dreyfuss, D., and Saumon, G. (1998). Ventilator-induced lung injury: lessons from experimental studies. Am. J. Respir. Crit. Care Med. 157, 294–323. doi: 10.1164/ajrccm.157.1.9604014
Eke, I., and Cordes, N. (2015). Focal adhesion signaling and therapy resistance in cancer. Semin. Cancer Biol. 31, 65–75. doi: 10.1016/j.semcancer.2014.07.009
Eliceiri, B. P., Puente, X. S., Hood, J. D., Stupack, D. G., Schlaepfer, D. D., Huang, X. Z., et al. (2002). Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J. Cell Biol. 157, 149–160. doi: 10.1083/jcb.200109079
Elosegui-Artola, A., Oria, R., Chen, Y., Kosmalska, A., Perez-Gonzalez, C., Castro, N., et al. (2016). Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 18, 540–548. doi: 10.1038/ncb3336
Erickson, S. E., Shlipak, M. G., Martin, G. S., Wheeler, A. P., Ancukiewicz, M., Matthay, M. A., et al. (2009). Racial and ethnic disparities in mortality from acute lung injury. Crit. Care Med. 37, 1–6. doi: 10.1097/CCM.0b013e31819292ea
Frutos-Vivar, F., Ferguson, N. D., and Esteban, A. (2006). Epidemiology of acute lung injury and acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 27, 327–336.
Frutos-Vivar, F., Nin, N., and Esteban, A. (2004). Epidemiology of acute lung injury and acute respiratory distress syndrome. Curr. Opin. Crit. Care 10, 1–6.
Gajic, O., Frutos-Vivar, F., Esteban, A., Hubmayr, R. D., and Anzueto, A. (2005). Ventilator settings as a risk factor for acute respiratory distress syndrome in mechanically ventilated patients. Intens. Care Med. 31, 922–926. doi: 10.1007/s00134-005-2625-1
Gao, L., Grant, A., Halder, I., Brower, R., Sevransky, J., Maloney, J. P., et al. (2006). Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am. J. Respir. Cell Mol. Biol. 34, 487–495. doi: 10.1165/rcmb.2005-0404oc
Garcia-Alvarez, B., de Pereda, J. M., Calderwood, D. A., Ulmer, T. S., Critchley, D., Campbell, I. D., et al. (2003). Structural determinants of integrin recognition by talin. Mol. Cell. 11, 49–58. doi: 10.1016/s1097-2765(02)00823-7
Geiger, B., and Bershadsky, A. (2002). Exploring the neighborhood: adhesion-coupled cell mechanosensors. Cell 110, 139–142. doi: 10.1016/s0092-8674(02)00831-0
Geiger, B., Spatz, J. P., and Bershadsky, A. D. (2009). Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 10, 21–33. doi: 10.1038/nrm2593
Giancotti, F. G. (2007). Targeting integrin beta4 for cancer and anti-angiogenic therapy. Trends Pharmacol Sci. 28, 506–511. doi: 10.1016/j.tips.2007.08.004
Gingras, A. R., Ziegler, W. H., Bobkov, A. A., Joyce, M. G., Fasci, D., Himmel, M., et al. (2009). Structural determinants of integrin binding to the talin rod. J. Biol. Chem. 284, 8866–8876. doi: 10.1074/jbc.M805937200
Gingras, A. R., Ziegler, W. H., Frank, R., Barsukov, I. L., Roberts, G. C., Critchley, D. R., et al. (2005). Mapping and consensus sequence identification for multiple vinculin binding sites within the talin rod. J. Biol. Chem. 280, 37217–37224. doi: 10.1074/jbc.m508060200
Giusti, B., Margheri, F., Rossi, L., Lapini, I., Magi, A., Serrati, S., et al. (2013). Desmoglein-2-integrin Beta-8 interaction regulates actin assembly in endothelial cells: deregulation in systemic sclerosis. PLoS One 8:e68117. doi: 10.1371/journal.pone.0068117
Goldmann, W. H., Bremer, A., Haner, M., Aebi, U., and Isenberg, G. (1994). Native talin is a dumbbell-shaped homodimer when it interacts with actin. J. Struct. Biol. 112, 3–10. doi: 10.1006/jsbi.1994.1002
Golji, J., and Mofrad, M. R. (2010). A molecular dynamics investigation of vinculin activation. Biophys. J. 99, 1073–1081. doi: 10.1016/j.bpj.2010.05.024
Golji, J., and Mofrad, M. R. K. (2014). The talin dimer structure orientation is mechanically regulated. Biophys. J. 107, 1802–1809. doi: 10.1016/j.bpj.2014.08.038
Golji, J., Wendorff, T., and Mofrad, M. R. (2012). Phosphorylation primes vinculin for activation. Biophys. J. 102, 2022–2030. doi: 10.1016/j.bpj.2012.01.062
Grigera, P. R., Jeffery, E. D., Martin, K. H., Shabanowitz, J., Hunt, D. F., and Parsons, J. T. (2005). FAK phosphorylation sites mapped by mass spectrometry. J. Cell Sci. 118(Pt 21), 4931–4935. doi: 10.1242/jcs.02696
Hemmings, L., Rees, D. J., Ohanian, V., Bolton, S. J., Gilmore, A. P., Patel, B., et al. (1996). Talin contains three actin-binding sites each of which is adjacent to a vinculin-binding site. J. Cell Sci. 109(Pt 11), 2715–2726.
Hirayama, Y., and Sumpio, B. E. (2007). Role of ligand-specific integrins in endothelial cell alignment and elongation induced by cyclic strain. Endothelium 14, 275–283. doi: 10.1080/10623320701746248
Hirst, R., Horwitz, A., Buck, C., and Rohrschneider, L. (1986). Phosphorylation of the fibronectin receptor complex in cells transformed by oncogenes that encode tyrosine kinases. Proc. Natl. Acad. Sci. U.S.A. 83, 6470–6474. doi: 10.1073/pnas.83.17.6470
Hogervorst, F., Kuikman, I., von dem Borne, A. E., and Sonnenberg, A. (1990). Cloning and sequence analysis of beta-4 cDNA: an integrin subunit that contains a unique 118 kd cytoplasmic domain. EMBO J. 9, 765–770. doi: 10.1002/j.1460-2075.1990.tb08171.x
Hsu, H. J., Lee, C. F., Locke, A., Vanderzyl, S. Q., and Kaunas, R. (2010). Stretch-induced stress fiber remodeling and the activations of JNK and ERK depend on mechanical strain rate, but not FAK. PLoS One 5:e12470. doi: 10.1371/journal.pone.0012470
Hu, P., and Luo, B. H. (2013). Integrin bi-directional signaling across the plasma membrane. J. Cell. Physiol. 228, 306–312. doi: 10.1002/jcp.24154
Huang, W., Sakamoto, N., Hanamura, K., Miyazawa, R., and Sato, M. (2011). Role of intercellular junctions in redistribution of focal adhesions and orientation of vascular endothelial cells exposed to cyclic stretching. Cell. Mol. Bioeng. 4:368. doi: 10.1007/s12195-011-0194-7
Huang, W., Sakamoto, N., Miyazawa, R., and Sato, M. (2012). Role of paxillin in the early phase of orientation of the vascular endothelial cells exposed to cyclic stretching. Biochem. Biophys. Res. Commun. 418, 708–713. doi: 10.1016/j.bbrc.2012.01.083
Ikeda, M., Kito, H., and Sumpio, B. E. (1999). Phosphatidylinositol-3 kinase dependent MAP kinase activation via p21ras in endothelial cells exposed to cyclic strain. Biochem. Biophys. Res. Commun. 257, 668–671. doi: 10.1006/bbrc.1999.0532
Infusino, G. A., and Jacobson, J. R. (2012). Endothelial FAK as a therapeutic target in disease. Microvasc. Res. 83, 89–96. doi: 10.1016/j.mvr.2011.09.011
Iskratsch, T., Wolfenson, H., and Sheetz, M. P. (2014). Appreciating force and shape-the rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 15, 825–833. doi: 10.1038/nrm3903
Izard, T., Evans, G., Borgon, R. A., Rush, C. L., Bricogne, G., and Bois, P. R. (2004). Vinculin activation by talin through helical bundle conversion. Nature 427, 171–175. doi: 10.1038/nature02281
Jiang, G., Giannone, G., Critchley, D. R., Fukumoto, E., and Sheetz, M. P. (2003). Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature 424, 334–337. doi: 10.1038/nature01805
Jones, R. J., Brunton, V. G., and Frame, M. C. (2000). Adhesion-linked kinases in cancer; emphasis on src, focal adhesion kinase and PI 3-kinase. Eur. J. Cancer 36, 1595–1606. doi: 10.1016/s0959-8049(00)00153-2
Kefalas, P., Brown, T. R., and Brickell, P. M. (1995). Signalling by the p60c-src family of protein-tyrosine kinases. Int. J. Biochem. Cell Biol. 27, 551–563. doi: 10.1016/1357-2725(95)00024-j
Kim, C., Ye, F., and Ginsberg, M. H. (2011). Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 27, 321–345. doi: 10.1146/annurev-cellbio-100109-104104
Kim, T. H., Yoon, H. J., Lim, C. M., Kim, E. K., Kim, M. J., and Koh, Y. (2005). The role of endogenous histamine on the pathogenesis of the lipopolysaccharide (LPS)-induced, acute lung injury: a pilot study. Inflammation 29, 72–80. doi: 10.1007/s10753-006-9001-3
Klapholz, B., and Brown, N. H. (2017). Talin - the master of integrin adhesions. J. Cell Sci. 130, 2435–2446. doi: 10.1242/jcs.190991
Kuo, J. C., Han, X., Hsiao, C. T., Yates, J. R. III, and Waterman, C. M. (2011). Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 13, 383–393. doi: 10.1038/ncb2216
Ladoux, B., and Nicolas, A. (2012). Physically based principles of cell adhesion mechanosensitivity in tissues. Rep. Progr. Phys. Phys. Soc. 75:116601. doi: 10.1088/0034-4885/75/11/116601
Laffey, J. G., and Kavanagh, B. P. (2017). Fifty years of research in ARDS. insight into acute respiratory distress syndrome. from models to patients. Am. J. Respir. Crit. Care Med. 196, 18–28. doi: 10.1164/rccm.201612-2415CI
Lecuit, T., Lenne, P. F., and Munro, E. (2011). Force generation, transmission, and integration during cell and tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 27, 157–184. doi: 10.1146/annurev-cellbio-100109-104027
Lee, S. E., Kamm, R. D., and Mofrad, M. R. (2007). Force-induced activation of talin and its possible role in focal adhesion mechanotransduction. J. Biomech. 40, 2096–2106. doi: 10.1016/j.jbiomech.2007.04.006
Lionetti, V., Recchia, F. A., and Ranieri, V. M. (2005). Overview of ventilator-induced lung injury mechanisms. Curr. Opin. Crit. Care 11, 82–86.
Luo, B. H., Carman, C. V., and Springer, T. A. (2007). Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647. doi: 10.1146/annurev.immunol.25.022106.141618
Moss, M., and Mannino, D. M. (2002). Race and gender differences in acute respiratory distress syndrome deaths in the United States: an analysis of multiple-cause mortality data (1979- 1996). Crit. Care Med. 30, 1679–1685. doi: 10.1097/00003246-200208000-00001
Nakamura, K., Yano, H., Uchida, H., Hashimoto, S., Schaefer, E., and Sabe, H. (2000). Tyrosine phosphorylation of paxillin alpha is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J. Biol. Chem. 275, 27155–27164.
Naruse, K., Sai, X., Yokoyama, N., and Sokabe, M. (1998a). Uni-axial cyclic stretch induces c-src activation and translocation in human endothelial cells via SA channel activation. FEBS Lett. 441, 111–115. doi: 10.1016/s0014-5793(98)01528-2
Naruse, K., Yamada, T., Sai, X. R., Hamaguchi, M., and Sokabe, M. (1998b). Pp125FAK is required for stretch dependent morphological response of endothelial cells. Oncogene 17, 455–463. doi: 10.1038/sj.onc.1201950
Naruse, K., Yamada, T., and Sokabe, M. (1998c). Involvement of SA channels in orienting response of cultured endothelial cells to cyclic stretch. Am. J. Physiol. 274(5 Pt 2), H1532–H1538. doi: 10.1152/ajpheart.1998.274.5.H1532
Naruse, K., and Sokabe, M. (1993). Involvement of stretch-activated ion channels in Ca2+ mobilization to mechanical stretch in endothelial cells. Am. J. Physiol. 264(4 Pt 1), C1037–C1044.
Ngu, H., Feng, Y., Lu, L., Oswald, S. J., Longmore, G. D., and Yin, F. C. (2010). Effect of focal adhesion proteins on endothelial cell adhesion, motility and orientation response to cyclic strain. Ann. Biomed. Eng. 38, 208–222. doi: 10.1007/s10439-009-9826-7
Nieman, G. F., Satalin, J., Andrews, P., Aiash, H., Habashi, N. M., and Gatto, L. A. (2017). Personalizing mechanical ventilation according to physiologic parameters to stabilize alveoli and minimize ventilator induced lung injury (VILI). Intens. Care Med. Exp. 5:8. doi: 10.1186/s40635-017-0121-x
Nilsson, S., Kaniowska, D., Brakebusch, C., Fassler, R., and Johansson, S. (2006). Threonine 788 in integrin subunit beta1 regulates integrin activation. Exp. Cell Res. 312, 844–853. doi: 10.1016/j.yexcr.2005.12.001
Nishimura, S. L., Sheppard, D., and Pytela, R. (1994). Integrin alpha v beta 8. Interaction with vitronectin and functional divergence of the beta 8 cytoplasmic domain. J. Biol. Chem. 269, 28708–28715.
Nonas, S. A., Finigan, J. H., Gao, L., and Garcia, J. G. (2005). Functional genomic insights into acute lung injury: role of ventilators and mechanical stress. Proc. Am. Thor. Soc. 2, 188–194. doi: 10.1513/pats.200501-005ac
Oakes, P. W., and Gardel, M. L. (2014). Stressing the limits of focal adhesion mechanosensitivity. Curr. Opin. Cell Biol. 30, 68–73. doi: 10.1016/j.ceb.2014.06.003
Pan, L., Zhao, Y., Yuan, Z., and Qin, G. (2016). Research advances on structure and biological functions of integrins. SpringerPlus 5:1094. doi: 10.1186/s40064-016-2502-0
Parker, J. C., Ivey, C. L., and Tucker, J. A. (1998). Gadolinium prevents high airway pressure-induced permeability increases in isolated rat lungs. J. Appl. Physiol. 84, 1113–1118. doi: 10.1152/jappl.1998.84.4.1113
Paulhe, F., Manenti, S., Ysebaert, L., Betous, R., Sultan, P., and Racaud-Sultan, C. (2005). Integrin function and signaling as pharmacological targets in cardiovascular diseases and in cancer. Curr. Pharm. Des. 11, 2119–2134. doi: 10.2174/1381612054065765
Plataki, M., and Hubmayr, R. D. (2010). The physical basis of ventilator-induced lung injury. Expert Rev. Respir. Med. 4, 373–385. doi: 10.1586/ers.10.28
Plow, E. F., Haas, T. A., Zhang, L., Loftus, J., and Smith, J. W. (2000). Ligand binding to integrins. J. Biol. Chem. 275, 21785–21788.
Rabinovitz, I., Tsomo, L., and Mercurio, A. M. (2004). Protein kinase C-alpha phosphorylation of specific serines in the connecting segment of the beta 4 integrin regulates the dynamics of type II hemidesmosomes. Mol. Cell. Biol. 24, 4351–4360. doi: 10.1128/mcb.24.10.4351-4360.2004
Rimensberger, P. C. (2003). ICU cornerstone: high frequency ventilation is here to stay. Crit. Care 7, 342–344.
Rodius, S., Chaloin, O., Moes, M., Schaffner-Reckinger, E., Landrieu, I., Lippens, G., et al. (2008). The talin rod IBS2 alpha-helix interacts with the beta3 integrin cytoplasmic tail membrane-proximal helix by establishing charge complementary salt bridges. J. Biol. Chem. 283, 24212–24223. doi: 10.1074/jbc.M709704200
Romer, L. H., Birukov, K. G., and Garcia, J. G. (2006). Focal adhesions: paradigm for a signaling nexus. Circ. Res. 98, 606–616. doi: 10.1161/01.res.0000207408.31270.db
Sastry, S. K., and Horwitz, A. F. (1993). Integrin cytoplasmic domains: mediators of cytoskeletal linkages and extra- and intracellular initiated transmembrane signaling. Curr. Opin. Cell Biol. 5, 819–831. doi: 10.1016/0955-0674(93)90031-k
Sawada, Y., and Sheetz, M. P. (2002). Force transduction by Triton cytoskeletons. J. Cell Biol. 156, 609–615. doi: 10.1083/jcb.200110068
Schaller, M. D., Otey, C. A., Hildebrand, J. D., and Parsons, J. T. (1995). Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J. Cell Biol. 130, 1181–1187. doi: 10.1083/jcb.130.5.1181
Schaller, M. D., and Parsons, J. T. (1995). pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol. Cell. Biol. 15, 2635–2645. doi: 10.1128/mcb.15.5.2635
Schaller, M. D., and Schaefer, E. M. (2001). Multiple stimuli induce tyrosine phosphorylation of the Crk-binding sites of paxillin. Biochem. J. 360(Pt 1), 57–66. doi: 10.1042/bj3600057
Shikata, Y., Birukov, K. G., Birukova, A. A., Verin, A., and Garcia, J. G. (2003a). Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J. 17, 2240–2249. doi: 10.1096/fj.03-0198com
Shikata, Y., Birukov, K. G., and Garcia, J. G. (2003b). S1P induces FA remodeling in human pulmonary endothelial cells: role of Rac, GIT1, FAK, and paxillin. J. Appl. Physiol. 94, 1193–1203. doi: 10.1152/japplphysiol.00690.2002
Shikata, Y., Rios, A., Kawkitinarong, K., DePaola, N., Garcia, J. G., and Birukov, K. G. (2005). Differential effects of shear stress and cyclic stretch on focal adhesion remodeling, site-specific FAK phosphorylation, and small GTPases in human lung endothelial cells. Exp. Cell Res. 304, 40–49. doi: 10.1016/j.yexcr.2004.11.001
Shirinsky, V. P., Antonov, A. S., Birukov, K. G., Sobolevsky, A. V., Romanov, Y. A., Kabaeva, N. V., et al. (1989). Mechano-chemical control of human endothelium orientation and size. J. Cell Biol. 109, 331–339. doi: 10.1083/jcb.109.1.331
Simonsen, U., Wandall-Frostholm, C., Olivan-Viguera, A., and Kohler, R. (2017). Emerging roles of calcium-activated K channels and TRPV4 channels in lung oedema and pulmonary circulatory collapse. Acta Physiol. 219, 176–187. doi: 10.1111/apha.12768
Slutsky, A. S., and Ranieri, V. M. (2014). Ventilator-induced lung injury. N. Engl. J. Med. 370:980.
Sokabe, M., Naruse, K., Sai, S., Yamada, T., Kawakami, K., Inoue, M., et al. (1997). Mechanotransduction and intracellular signaling mechanisms of stretch-induced remodeling in endothelial cells. Heart Vessels 12, 191–193.
Stawowy, P., Margeta, C., Blaschke, F., Lindschau, C., Spencer-Hansch, C., Leitges, M., et al. (2005). Protein kinase C epsilon mediates angiotensin II-induced activation of beta1-integrins in cardiac fibroblasts. Cardiovasc. Res. 67, 50–59. doi: 10.1016/j.cardiores.2005.03.002
Stuiver, I., Ruggeri, Z., and Smith, J. W. (1996). Divalent cations regulate the organization of integrins alpha v beta 3 and alpha v beta 5 on the cell surface. J. Cell. Physiol. 168, 521–531. doi: 10.1002/(sici)1097-4652(199609)168:3<521::aid-jcp4>3.0.co;2-r
Su, G., Hodnett, M., Wu, N., Atakilit, A., Kosinski, C., Godzich, M., et al. (2007). Integrin alphavbeta5 regulates lung vascular permeability and pulmonary endothelial barrier function. Am. J. Respir. Cell Mol. Biol. 36, 377–386. doi: 10.1165/rcmb.2006-0238oc
Sumpio, B. E., Banes, A. J., Levin, L. G., and Johnson, G. Jr. (1987). Mechanical stress stimulates aortic endothelial cells to proliferate. J. Vasc. Surg. 6, 252–256. doi: 10.1067/mva.1987.avs0060252
Suzuki, M., Naruse, K., Asano, Y., Okamoto, T., Nishikimi, N., Sakurai, T., et al. (1997). Up-regulation of integrin beta 3 expression by cyclic stretch in human umbilical endothelial cells. Biochem. Biophys. Res. Commun. 239, 372–376. doi: 10.1006/bbrc.1997.7364
Tabata, T., Kawakatsu, H., Maidji, E., Sakai, T., Sakai, K., Fang-Hoover, J., et al. (2008). Induction of an epithelial integrin alphavbeta6 in human cytomegalovirus-infected endothelial cells leads to activation of transforming growth factor-beta1 and increased collagen production. Am. J. Pathol. 172, 1127–1140. doi: 10.2353/ajpath.2008.070448
Tai, Y. L., Chu, P. Y., Lai, I. R., Wang, M. Y., Tseng, H. Y., Guan, J. L., et al. (2015). An EGFR/Src-dependent beta4 integrin/FAK complex contributes to malignancy of breast cancer. Sci. Rep. 5:16408. doi: 10.1038/srep16408
Thodeti, C. K., Matthews, B., Ravi, A., Mammoto, A., Ghosh, K., Bracha, A. L., et al. (2009). TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ. Res. 104, 1123–1130. doi: 10.1161/CIRCRESAHA.108.192930
Tian, Y., Gawlak, G., O’Donnell, J. J. III, Birukova, A. A., and Birukov, K. G. (2016). Activation of vascular endothelial growth factor (VEGF) receptor 2 mediates endothelial permeability caused by cyclic stretch. J. Biol. Chem. 291, 10032–10045. doi: 10.1074/jbc.M115.690487
Tschumperlin, D. J., and Margulies, S. S. (1999). Alveolar epithelial surface area-volume relationship in isolated rat lungs. J. Appl. Physiol. 86, 2026–2033. doi: 10.1152/jappl.1999.86.6.2026
Tschumperlin, D. J., Oswari, J., and Margulies, A. S. (2000). Deformation-induced injury of alveolar epithelial cells. Effect of frequency, duration, and amplitude. Am. J. Respir. Crit. Care Med. 162(2 Pt 1), 357–362. doi: 10.1164/ajrccm.162.2.9807003
Tsopanoglou, N. E., Andriopoulou, P., and Maragoudakis, M. E. (2002). On the mechanism of thrombin-induced angiogenesis: involvement of alphavbeta3-integrin. Am. J. Physiol. Cell Physiol. 283, C1501–C1510.
van Nieuw Amerongen, G. P., Natarajan, K., Yin, G., Hoefen, R. J., Osawa, M., Haendeler, J., et al. (2004). GIT1 mediates thrombin signaling in endothelial cells: role in turnover of RhoA-type focal adhesions. Circ. Res. 94, 1041–1049. doi: 10.1161/01.res.0000125627.77235.0c
Villar, J., Blanco, J., Anon, J. M., Santos-Bouza, A., Blanch, L., Ambros, A., et al. (2011). The ALIEN study: incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intens. Care Med. 37, 1932–1941. doi: 10.1007/s00134-011-2380-4
Villar, J., Sulemanji, D., and Kacmarek, R. M. (2014). The acute respiratory distress syndrome: incidence and mortality, has it changed? Curr. Opin. Crit. Care 20, 3–9. doi: 10.1097/MCC.0000000000000057
Wang, H., Jin, H., Beauvais, D. M., and Rapraeger, A. C. (2014). Cytoplasmic domain interactions of syndecan-1 and syndecan-4 with alpha6beta4 integrin mediate human epidermal growth factor receptor (HER1 and HER2)-dependent motility and survival. J. Biol. Chem. 289, 30318–30332. doi: 10.1074/jbc.M114.586438
Wang, J. G., Miyazu, M., Matsushita, E., Sokabe, M., and Naruse, K. (2001). Uniaxial cyclic stretch induces focal adhesion kinase (FAK) tyrosine phosphorylation followed by mitogen-activated protein kinase (MAPK) activation. Biochem. Biophys. Res. Commun. 288, 356–361. doi: 10.1006/bbrc.2001.5775
Wang, J. H., Goldschmidt-Clermont, P., Wille, J., and Yin, F. C. (2001). Specificity of endothelial cell reorientation in response to cyclic mechanical stretching. J. Biomech. 34, 1563–1572. doi: 10.1016/s0021-9290(01)00150-6
Wang, J. H., Goldschmidt-Clermont, P., and Yin, F. C. (2000). Contractility affects stress fiber remodeling and reorientation of endothelial cells subjected to cyclic mechanical stretching. Ann. Biomed. Eng. 28, 1165–1171. doi: 10.1114/1.1317528
Wayner, E. A., Orlando, R. A., and Cheresh, D. A. (1991). Integrins alpha v beta 3 and alpha v beta 5 contribute to cell attachment to vitronectin but differentially distribute on the cell surface. J. Cell Biol. 113, 919–929. doi: 10.1083/jcb.113.4.919
Wennerberg, K., Fassler, R., Warmegard, B., and Johansson, S. (1998). Mutational analysis of the potential phosphorylation sites in the cytoplasmic domain of integrin beta1A. Requirement for threonines 788-789 in receptor activation. J. Cell Sci. 111 (Pt 8), 1117–1126.
Winkler, J., Lunsdorf, H., and Jockusch, B. M. (1997). Energy-filtered electron microscopy reveals that talin is a highly flexible protein composed of a series of globular domains. Eur. J. Biochem. 243, 430–436. doi: 10.1111/j.1432-1033.1997.0430a.x
Wirtz, H. R., and Dobbs, L. G. (2000). The effects of mechanical forces on lung functions. Respir. Physiol. 119, 1–17. doi: 10.1016/s0034-5687(99)00092-4
Wu, M. H. (2005). Endothelial focal adhesions and barrier function. J. Physiol. 569(Pt 2), 359–366. doi: 10.1113/jphysiol.2005.096537
Yano, Y., Geibel, J., and Sumpio, B. E. (1996a). Tyrosine phosphorylation of pp125FAK and paxillin in aortic endothelial cells induced by mechanical strain. Am. J. Physiol. 271(2 Pt 1), C635–C649.
Yano, Y., Saito, Y., Narumiya, S., and Sumpio, B. E. (1996b). Involvement of rho p21 in cyclic strain-induced tyrosine phosphorylation of focal adhesion kinase (pp125FAK), morphological changes and migration of endothelial cells. Biochem. Biophys. Res. Commun. 224, 508–515. doi: 10.1006/bbrc.1996.1057
Yano, Y., Geibel, J., and Sumpio, B. E. (1997). Cyclic strain induces reorganization of integrin alpha 5 beta 1 and alpha 2 beta 1 in human umbilical vein endothelial cells. J. Cell. Biochem. 64, 505–513. doi: 10.1002/(sici)1097-4644(19970301)64:3<505::aid-jcb17>3.0.co;2-e
Yao, M., Goult, B. T., Klapholz, B., Hu, X., Toseland, C. P., Guo, Y., et al. (2016). The mechanical response of talin. Nat. Commun. 7:11966. doi: 10.1038/ncomms11966
Ye, F., Kim, C., and Ginsberg, M. H. (2011). Molecular mechanism of inside-out integrin regulation. J. Thromb. Haemost. 9(Suppl. 1), 20–25. doi: 10.1111/j.1538-7836.2011.04355.x
Yoshigi, M., Hoffman, L. M., Jensen, C. C., Yost, H. J., and Beckerle, M. C. (2005). Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J. Cell Biol. 171, 209–215. doi: 10.1083/jcb.200505018
Zaidel-Bar, R., and Geiger, B. (2010). The switchable integrin adhesome. J. Cell Sci. 123(Pt 9), 1385–1388. doi: 10.1242/jcs.066183
Zhao, J., Singleton, P. A., Brown, M. E., Dudek, S. M., and Garcia, J. G. (2009). Phosphotyrosine protein dynamics in cell membrane rafts of sphingosine-1-phosphate-stimulated human endothelium: role in barrier enhancement. Cell. Signal. 21, 1945–1960. doi: 10.1016/j.cellsig.2009.09.002
Zhao, X., Peng, X., Sun, S., Park, A. Y., and Guan, J. L. (2010). Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J. Cell Biol. 189, 955–965. doi: 10.1083/jcb.200912094
Zhao, Z. S., Manser, E., Loo, T. H., and Lim, L. (2000). Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol. 20, 6354–6363. doi: 10.1128/mcb.20.17.6354-6363.2000
Keywords: ARDS, VILI, focal adhesion, mechanical stress, cyclic stretch, integrin β4
Citation: Kelly GT, Faraj R, Zhang Y, Maltepe E, Fineman JR, Black SM and Wang T (2019) Pulmonary Endothelial Mechanical Sensing and Signaling, a Story of Focal Adhesions and Integrins in Ventilator Induced Lung Injury. Front. Physiol. 10:511. doi: 10.3389/fphys.2019.00511
Received: 16 December 2018; Accepted: 11 April 2019;
Published: 26 April 2019.
Edited by:
Andreea Trache, Texas A&M University, United StatesReviewed by:
Dan Predescu, Rush University, United StatesAndrea Moriondo, University of Insubria, Italy
Copyright © 2019 Kelly, Faraj, Zhang, Maltepe, Fineman, Black and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Wang, dHdhbmdAZW1haWwuYXJpem9uYS5lZHU=