
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 24 January 2019
Sec. Gastrointestinal Sciences
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01918
 Amr A. Waly1†
Amr A. Waly1† Nada El-Ekiaby2†
Nada El-Ekiaby2† Reem A. Assal1Mohamed M. Abdelrahman1Karim A. Hosny3Hend M. El Tayebi1Gamal Esmat4
Reem A. Assal1Mohamed M. Abdelrahman1Karim A. Hosny3Hend M. El Tayebi1Gamal Esmat4 Kai Breuhahn5
Kai Breuhahn5 Ahmed I. Abdelaziz1,2*
Ahmed I. Abdelaziz1,2*miR-let-7a is a tumor suppressor miRNA with reduced expression in most cancers. Methylation of MIRLET7A3 gene was reported to be the cause of this suppression in several cancers; however, it was not explicitly investigated in hepatocellular carcinoma (HCC). We aimed at investigating miR-let-7a expression and molecular mode in HCC, identifying drug-targetable networks, which might be affected by its abundance. Our results illustrated a significant repression of miR-let-7a, which correlated with hypermethylation of its gene of origin MIRLRT7A3. This was further supported by the induction of miR-let-7a expression upon treatment of HCC cells with a DNA-methyltransferase inhibitor. Using a computational approach, insulin-like growth factor (IGF)-II and IGF-2 mRNA binding proteins (IGF2BP)-2/-3 were identified as potential targets for miR-let-7a that was further confirmed experimentally. Indeed, miR-let-7a mimics diminished IGF-II as well as IGF2BP-2/-3 expression. Direct binding of miR-let-7a to each respective transcript was confirmed using a luciferase reporter assay. In conclusion, this study suggests that DNA hypermethylation leads to epigenetic repression of miR-let-7a in HCC cells, which induces the oncogenic IGF-signaling pathway.
MicroRNA lethal-7 (miR-let-7), has been identified as the second earliest discovered microRNA (miR) in the development of the nematode Caenorhabditis elegans, the organism where miRs were first discovered (Reinhart et al., 2000). The naming of let-7 was based on early studies highlighting its crucial role in guiding C. elegans differentiation program, and the larval lethality that occurs upon its embryonic inactivation (Rougvie and Ambros, 1995; Slack et al., 2000). In humans, let-7 miRs comprise a family of 13 members and were the first miRs discovered in humans (Roush and Slack, 2008). Let-7 miRs are not expressed in human embryonic stem cells; however, their expression rises gradually upon differentiation and reaches a state of maintained high expression in many adult tissues (Sempere et al., 2004; Schulman et al., 2005). On the contrary, let-7 expression levels drop in various human cancers (Park et al., 2007).
Within the context of cancer, miR-let-7a is mostly regarded as a tumor suppressor miRNA with reduced expression in numerous cancers such as lymphoma, Kaposi sarcoma, lung and ovarian cancers (Takamizawa et al., 2004; Dahiya et al., 2008; O’Hara et al., 2009). Thus, the question on how miR-let-7a expression becomes restricted in cancer was insistently discussed. Of the three genomic origins of the mature miR-let-7a: MIRLET7A1, MIRLET7A2, and MIRLET7A3, the latter is of special importance owing a well-defined CpG island (Lu et al., 2007). This ∼700 bp long domain in the genomic region 22q12.31 is indicating that miR-let-7a could be epigenetically regulated via DNA methylation. In fact, methylation of MIRLET7A3 gene has been reported in acute myeloid leukemia, ovarian and breast cancer (Lu et al., 2007, 2011; Vrba et al., 2013; Ko et al., 2014). In hepatocellular carcinoma (HCC), induction of miR-let-7a expression was reported to exert a potent tumor-suppressive role both in vitro as well as in mouse xenograft models (Liu et al., 2014). Yet, the molecular mechanisms behind the role of miR-let-7a as well as its compromised activity in HCC are not well understood.
Hepatocellular carcinoma is characterized by dysregulation of many oncogenic signaling pathways including the Insulin-like Growth Factor (IGF) pathway. The IGF pathway includes two ligands; IGF-I and IGF-II, where binding of either ligands to the IGF-1 receptor (IGF-1R) stimulates the induction of RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways leading to increased cell growth, proliferation, survival and migration (Wu and Zhu, 2011). IGF signaling overstimulation in HCC is in part due to abnormally high expression of IGF-II by means of loss of imprinting (Poirier et al., 2003) and correlates with increased HCC cells proliferation (Bae et al., 1998) and tumor neovascularization (Lahm et al., 2002). IGF signaling can be tuned by IGF-II mRNA binding proteins (IGF2BPs) which are reported to influence the fate of IGF-II mRNA (Liao et al., 2011). IGF2BP-2 and 3 are well-established oncogenes whose aberrant expression in HCC leads to excessive cell proliferation and invasion, which culminates in poor prognosis (Jeng et al., 2008; Wachter et al., 2012; Kessler et al., 2013). In silico analysis led us to investigate the Insulin-like Growth Factor (IGF) oncogenic pathway as a possible sovereign area for miR-let-7a with IGF-II as well as IGF2BP-2 and 3 as possible downstream targets of miR-let-7a.
In this study we first show that DNA methylation is one possible mode of negative miR-let-7a regulation in HCC cells. Secondly, our data illustrate that miR-let-7a regulates the abundance of tumor-supporting insulin-like growth factor IGF-II through the coordinated regulation of IGF-II itself and probably its stabilizing interaction partners IGF2BP-2/3.
Liver tissues were obtained from 16 HCV-induced HCC patients and 9 healthy tissues obtained from liver donors (“healthy controls”) during liver transplantation at the Kasr Al-Ainy Hospital, Cairo University, Egypt. Fresh liver samples were snap frozen by liquid nitrogen and stored at −80°C. All subjects gave their written informed consent and the Cairo University ethical review committee approved the study. The study followed the ethical guidelines of the 1975 Declaration of Helsinki. According to the hospital’s pathology report, 66.6% of the patients had more than one focal lesion. Patients’ clinical parameters are presented in Table 1.

Table 1. Clinical parameters of patients.
Huh7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Lonza, Basel, Switzerland) supplemented with 4.5 g/L glucose, 4 mmol/L L-glutamine, 10% fetal bovine serum (Lonza, Verviers, Belgium), 1% Penicillin/Streptomycin/MycoZap (Lonza, Basel, Switzerland) at 37°C in 5% CO2. Huh7 cells were transfected with miR-let-7a mimics or antagomirs (MIMAT0000062: Syn-hsa-let-7a-5p miScript miRNA Mimic and MIMAT0000062: Anti-hsa-let-7a-5p miScript miRNA Inhibitor, respectively, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Briefly, Huh7 cells were seeded in 6-well plates (2.5 × 105 cells/well). At 60% confluence cells were treated with the transfection complex (150 ng oligonucleotides mixed in 6 μl of HiPerFect Transfection Reagent (Qiagen, Hilden, Germany) and 100 μl DMEM). Cells treated with transfection complex lacking oligonucleotides were used as negative controls (mock). Total RNA or proteins were extracted 48 h after transfection. All transfections were done in quadruples and the experiment was done twice for confirmation. For drug-induced DNA demethylation experiments, Huh7 cells were treated with 5 μM decitabine (Sigma-Aldrich, St. Louis, MO, United States) in three biological replicates for 5 days followed by total RNA or genomic DNA extraction.
Hundred milligram of frozen liver tissue specimens were pulverized. RNA was then extracted using mirVana miRNA Isolation Kit (Ambion, Foster City, CA, United States) according to the manufacturer’s protocol. Huh7 cells were directly lysed in culture plate using Biozol reagent according to the manufacturer’s protocol (Invitrogen, Burlington, ON, Canada). The RNA pellet was washed twice with 75% ethanol then dissolved in DEPC-treated water.
miR-let-7a was reverse transcribed from the extracted RNA samples into complementary DNA using the MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, United States). mRNAs of IGF-II, IGF2BP-2/-3 were reverse transcribed using the High-capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s instruction. Expression of the housekeeping gene RNU6B was measured and used for miR-let-7a normalization. Beta-2-microglobulin was used for normalization of mRNA expression levels. TaqMan probe-based quantification of gene expression was performed using the following ready-made assays as per the manufacturer’s protocol (hsa-let-7a-5p, 478575_mir; RNU6B, 001093; IGF-II: Hs04188276_m1; IGF2BP2, Hs00538954_g1; IGF2BP3, Hs00559907_g1; and B2M’ Hs00187842_m1), StepOneTM Real-Time PCR instrument (Applied Biosystems) and StepOneTM Software (Applied Biosystems). Gene expression is presented as relative quantitation, which was calculated as follows:
where ΔCT is the difference in threshold cycles for target gene and endogenous control and ΔΔCT is the difference in ΔCT for target (patients or Huh7 cells or transfected Huh7 cells) and reference (healthy controls or mock cells).
Lysis of cells for protein extraction was done using Cell Lysis buffer (Cell Signaling Technology, Frankfurt, Germany) with PhosSTOP (Roche, Mannheim, Germany) and Protease Inhibitor Cocktail Mix G (Serva, Heidelberg, Germany). Cells were scraped off culture dishes, transferred to 1.5 ml tubes, sonicated 3× for 30 s. followed by 10 min centrifugation at 14,000 rpm, 4°C. Supernatants were then transferred to clean tubes and protein concentrations were measured using Bradford Assay (Bio-Rad, Munich, Germany) according to manufacturer’s instructions.
Forty microgram of total protein lysates were loaded and run on a 10% SDS-polyacrylamide gel and subsequently transferred to nitrocellulose membrane. Membrane was blocked using 5% skim-milk powder in TBST for 1 h followed by incubation with the following primary antibodies at 4°C overnight: mouse anti-IMP-1/2/3 (A2), which detects the three IGF2BP isoforms, namely IGF2BP1, 2, and 3 (anti-IGF2BP antibody, sc-271785, Santa Cruz Biotechnology, Heidelberg, Germany) diluted 1:200 and chicken anti-GAPDH diluted 1:10,000 (AB2302 EMD Millipore, Darmstadt, Germany) in 5% skim-milk powder. Membrane was washed 3× with TBST, incubated with the fluorescent secondary antibodies donkey-anti-mouse and donkey-anti-chicken diluted 1:2,000 in 5% skim-milk powder (IRDye coupled, 800 CW, LI-COR biosciences). Protein bands were quantified by measuring relative fluorescence units using Image StudioTM Lite software. The western blots were performed in two biological and two technical replicates.
Genomic DNA was extracted from Huh7 cells, HCC and healthy liver tissues using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s protocol. Methylation-sensitive restriction enzymes coupled with quantitative PCR (MSRE-qPCR) was performed using the OneStep qMethylTM Kit (Zymo Research, Irvine, CA, United States). In brief, two reaction mixes were prepared for each DNA sample: the test reaction mix and reference reaction mix. Each reaction mix contained 20 ng DNA, 10 μM primers (Supplementary Table 1) and twofold concentrated Test or Reference Reaction PreMix. Reaction tubes were placed in a StepOne PCR machine with the following thermal conditions: 2 h at 37°C; 10 min at 5°C; 40 cycles 30 s at 95°C, 1 min 60°C, 1 min at 72°C followed by a melting curve analysis to check specificity of the amplicon. For each sample, the percentage of DNA methylation was calculated using the following formula: Methylation (%) = 100 × 2−ΔCt; where ΔCt = Ct(test) – Ct(reference). This test was performed for three independent passages of Huh7, 4 HCC and 4 healthy liver DNA samples in duplicates.
Online miRNA target prediction software such as Miranda1, miRDB2, RNA hybrid3, and miRTarBase4 were used for target mRNA prediction (Figure 2). Sense and antisense Oligonucleotides corresponding to the identified target sites were designed in a way that, when annealed, to be flanked by sticky ends resembling those of SacI and XbaI digestion (Supplementary Table 2). Target sites were cloned in the pmirGLO Dual-Luciferase miRNA Target Expression vector (Promega, Madison, WI, United States) downstream of the firefly luciferase gene. In brief, the vector was double digested by SacI and XbaI restriction enzymes (Thermo Scientific, United States) and the sticky ended annealed oligonucleotides were inserted into the vector using T4 DNA ligase with 2 h incubation at 16°C. FuGENE HD transfection reagent (Promega) was used to transfect 0.5 μg of each vector in Huh7 cells alone or in combination with 150 ng of miR-let-7a mimics. Forty-eight hours after transfection, luciferase assay was performed using Dual-Luciferase Reporter Assay System (Promega, Mannheim, Germany) according to manufacturer’s instructions. To test for correctness of insert, the constructs were sequenced (SeqLab, Microsynth, Heidelberg, Germany).
Data are expressed as mean ± standard error of the mean and were analyzed using unpaired Student t-test (GraphPad Prism 5). P-values lower than 0.05 were considered statistically significant.
MIRLET7A3 gene sequence was retrieved using UCSC Genome browser5 and the CpG islands were identified using the online tool Sequence Manipulation Suite6.
This tool identifies potential CpG island regions using the method described by Gardiner-Garden and Frommer (1987). Sequence ranges where the Obs/Exp value is larger than 0.6 and the GC content is more than 50% are defined as CpG islands. The expected number of CpG dimers in a window is calculated as the number of ‘C’s multiplied by the number of ‘G’s in the window, divided by the window length.
To first compare the expression levels in a human HCC cell line, HCC and healthy liver tissues, qRT-PCR for miR-let-7a was performed (Figure 1A). The analysis showed that in both HCC tissues and HuH7 cells the levels of miR-let-7a were significantly lower compared to healthy liver tissues (p = 0.02 and p = 0.01, respectively). These results indicate that HuH7 cells may represent a suitable in vitro model for further analyses.
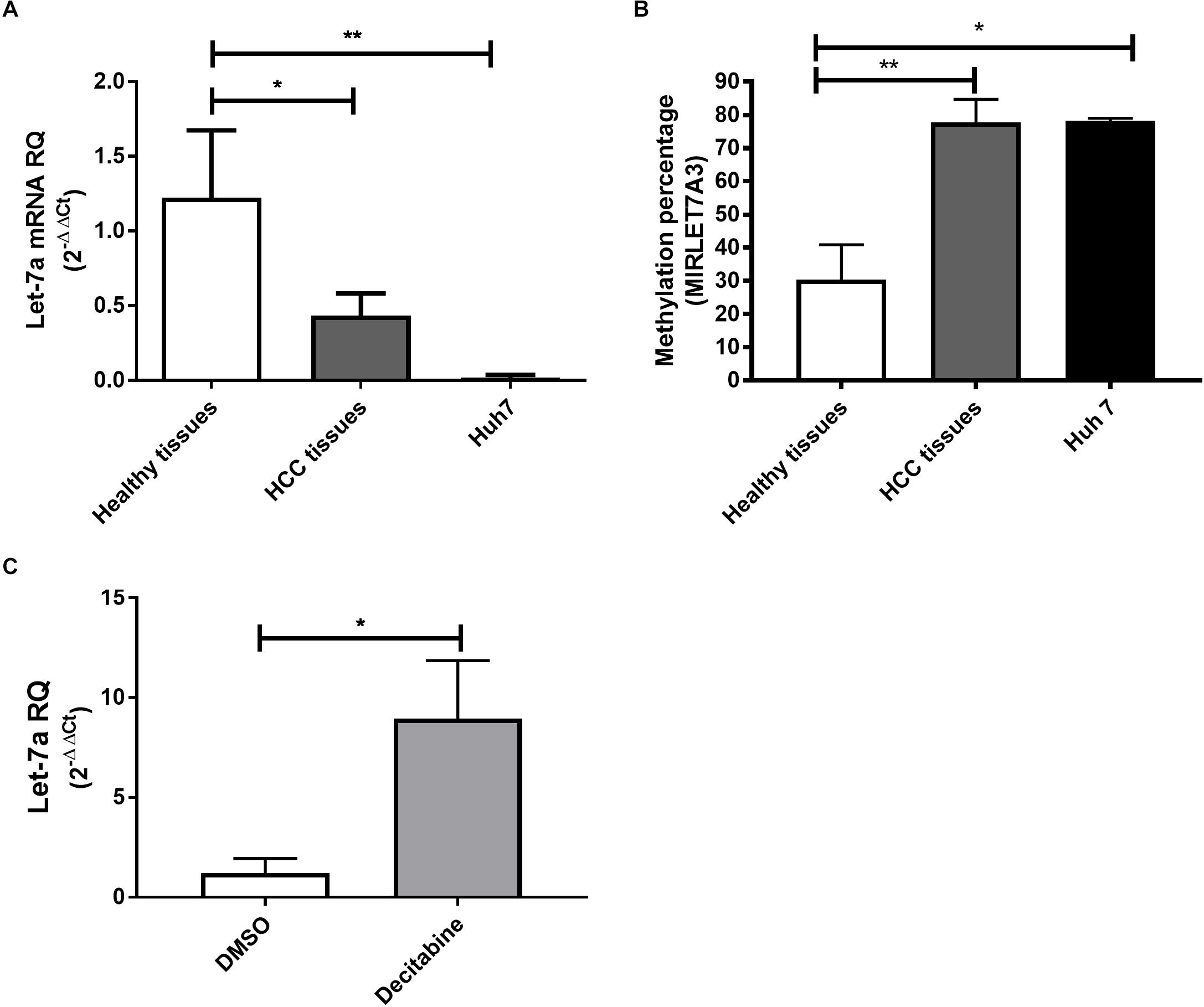
Figure 1. Expression pattern and methylation of miR-let-7a in liver tissues and cell lines. The expression levels of miR-let-7a were found to be significantly down-regulated in HCC tissues (P < 0.05) and cell lines (P < 0.01) compared to healthy controls (A). MSRE-qPCR analysis revealed significantly higher methylation levels in HCC tissues and Huh7 cell line with average methylation percentage of 77.5% (P < 0.01) and 78.15% (P < 0.05), respectively, compared to healthy liver tissues 28.2% (B). Treatment of Huh7 cells with decitabine, significantly increased miR-let-7a expression compared to untreated controls (P < 0.05) (C). RQ, relative quantitation; ∗∗P < 0.01; ∗P < 0.05.
After our observation that miR-let-7a is reduced in HCC cells, we decided to investigate the underlying regulatory mechanism for this possible repression. For this, the sequence of MIRLET7A3 gene (22q12.31, 26,753–26,823) located within the MIRLET7BHG (46,085,997–46,113,928) was obtained from UCSC Genome Browser and the presence of CpG islands was analyzed using the software CpG Islands-Bioinformatics.org. A well-defined CpG island with about 800 base pairs on chromosome 22q12.31 (26,118–26,919) was detected in the middle of the MIRLET7A gene a few base pairs upstream of the sequence of the precursor-miR-let-7a-3 (Supplementary Figure 1). MSRE-qPCR analysis was performed to detect DNA methylation of the identified region in DNA extracted from healthy, HCC tissues and Huh7 cells, which revealed high degree of methylation in HCC tissues with average methylation percentage of 77.5% ranging from 60 to 98.6%. The presence of high methylation levels was also observed in HCC cell lines with a methylation percentage average of 78.15% and a range from 77.3 to 79%. Methylation could be also detected in healthy liver tissues but at much lower levels (average methylation percentage of 28.2% and range from 7 to 56.6%) (Figure 1B). These results would suggest that the miR-let-7a is repressed in HCC cells on the transcriptional level via MIRLET7A3 gene hypermethylation.
Having first evidence that miR-let-7a levels are low in HCC cells due to epigenetic DNA methylation, we aimed at confirming this molecular mechanism experimentally. Therefore, Huh7 cells were treated with the DNA Methyltransferase (DNMT) inhibitor decitabine followed by assessment of changes in miR-let-7a expression levels. Indeed, decitabine-treatment significantly increased miR-let-7a levels in HCC cells (p = 0.028) (Figure 1C).
Together, these results illustrate that genomic hypermethylation of miR-let-7a is one possible mechanism for its reduced expression in HCC cells.
In order to understand the molecular mechanisms by which miR-let-7a performs its tumor-suppressive function in HCC, we intended to define networks of miR-let-7a targets, which might be involved in the regulation of tumor-relevant pathways. Since we are mainly interested in the IGF signaling pathway and its role in hepatocarcinogenesis (El Tayebi et al., 2011, 2015; Assal et al., 2015; Fawzy et al., 2016; Habashy et al., 2016; Youness et al., 2016; Rahmoon et al., 2017), we selected IGF-axis members potentially targeted by miR-let-7a using in silico microRNA target prediction softwares. This analysis yielded IGFII, IGF2BP2 as well as IGF2BP3 as potential miR-let-7a targets (Figure 2).
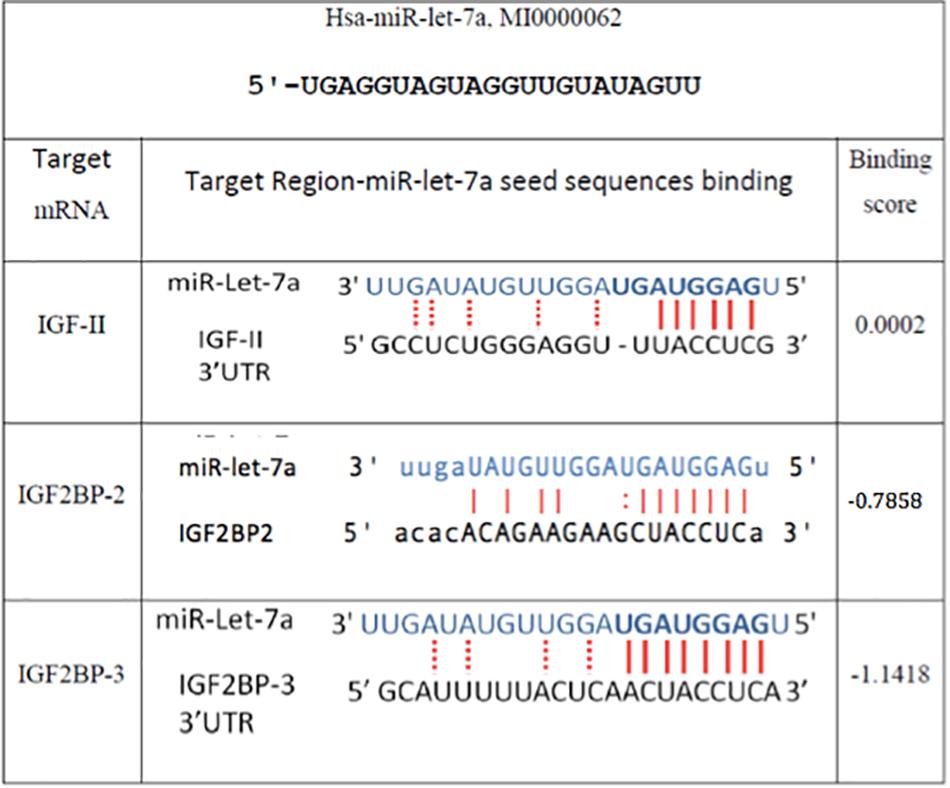
Figure 2. In silico prediction of miR-let-7a target genes. Miranda (see text footnote 1), RNA hybrid (see text footnote 3), and Target Scan (www.targetscan.org/) online miR target prediction software suggested that IGF-II and IGF2BP-2 and 3 are putative targets for miR-let-7a. The binding scores illustrated are according to Miranda software.
To experimentally validate that miR-let-7a could regulate the abundance of these potential target, the expression of miR-let-7a was increased by a respective mimic in Huh7 cells. Transfection with miR-let-7a mimics led to a significant suppression of IGF-II and IGF2BP family transcripts (Figure 3). In detail, miR-let-7a/mimic levels were elevated around 150 fold, which led to a significant downregulation of IGF-II mRNA in Huh7 cells compared to mock controls (p = 0.0429) (Figure 3A). Transfection of miR-let-7a mimics also resulted in a significant down regulation of IGF2BP-2 and -3 on the mRNA level (p = 0.0042 and p = 0.003, respectively) (Figures 3B,C). In addition, western immunoblotting analyses confirmed a negative effect of miR-let-7a on IGF2BP family members (Figures 3D,E) (p = 0.0012).
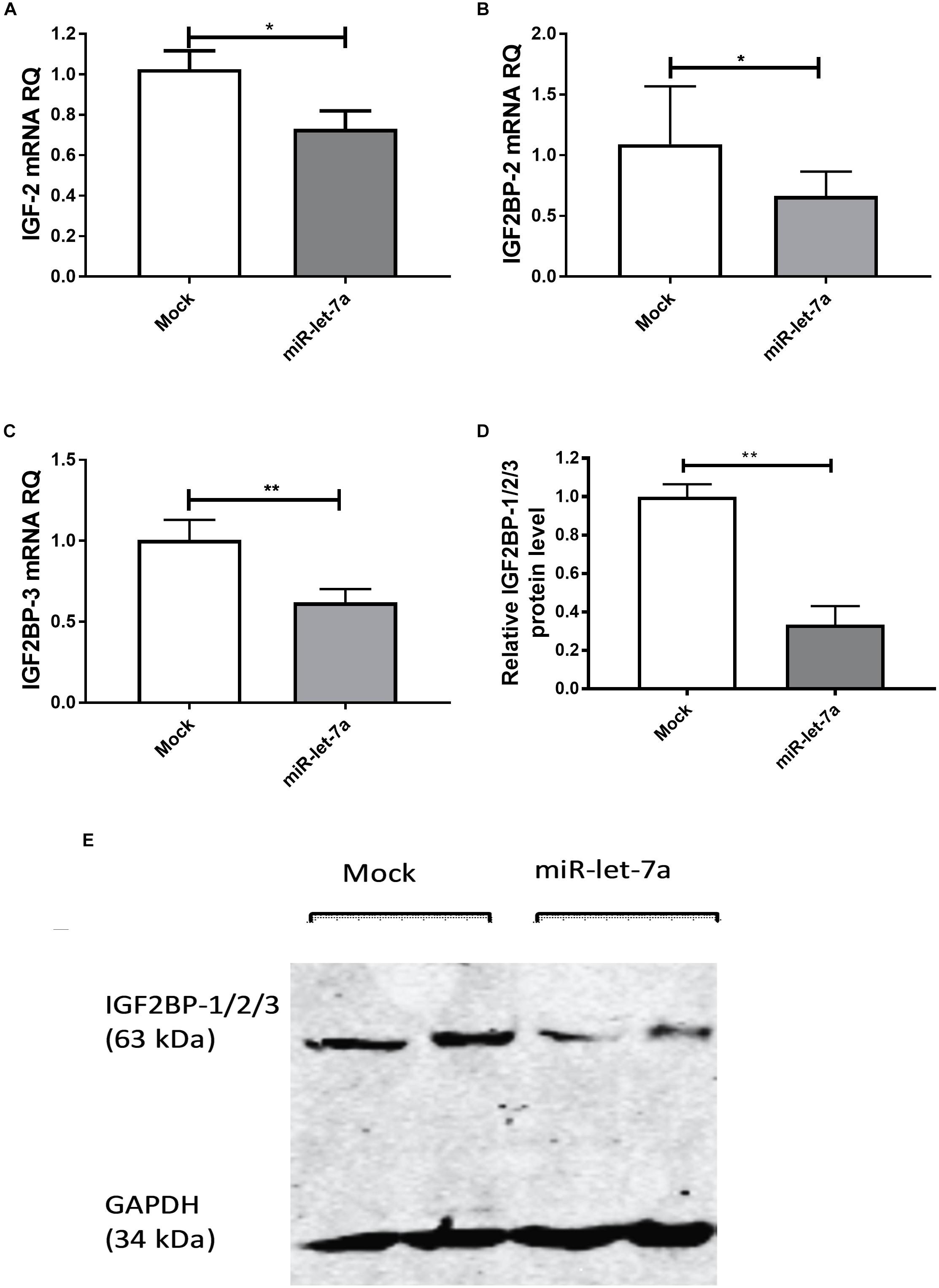
Figure 3. miR-let-7a suppresses IGF-II, IGF2BP-2 and -3. Upon mimicking of miR-let-7a in Huh7 cells, IGF-II mRNA expression was significantly downregulated compared to mock controls (P < 0.05) (A). Mimicking of miR-let-7a in Huh7 cells resulted in a significant downregulation of IGF2BP-2 and IGF2BP-3 mRNA (P < 0.05 and P < 0.01, respectively) (B,C) and protein (P < 0.01) (D,E) levels. RQ, relative quantitation; ∗∗P < 0.01; ∗P < 0.05.
The link between epigenetically regulated miR-let-7a and the identified IGF-signaling pathway constituents is further supported by decitabine treatment experiments. In accordance with our initial hypothesis (elevated expression of miR-let-7a after inhibition of methyltransferase activity; Figure 1B), this drug is leading to a drastic reduction of IGF-II and IGF2BP-2/3 in HCC cells (Figure 4).
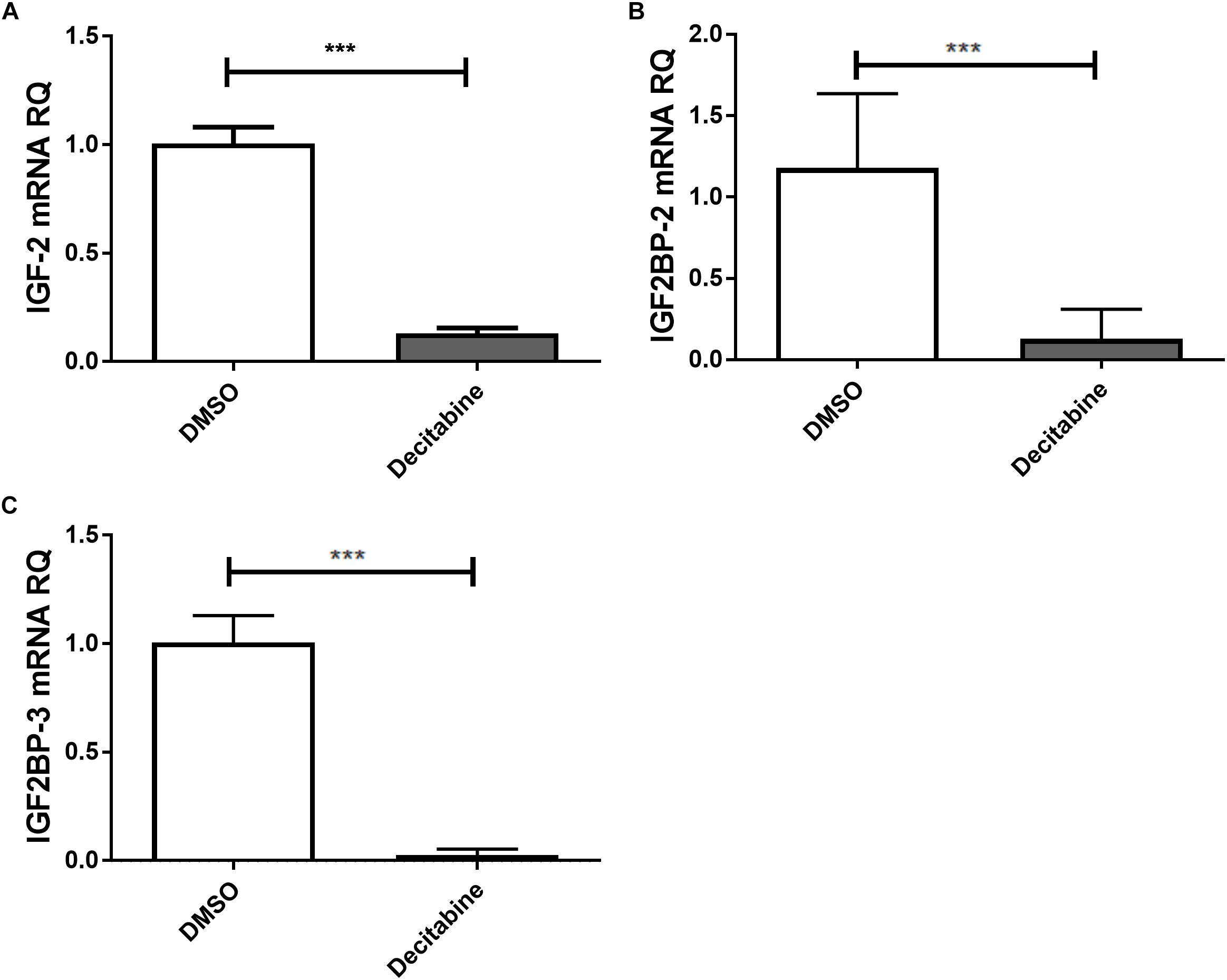
Figure 4. Decitabine represses IGF-II and IGF2BP-2 and 3. Huh7 cells were treated with 5 μM decitabine, a DNA methyltransferase inhibitor, for 5 days. This led to a marked suppression of IGF-II (A), IGF2BP2 (B), and IGF2BP3 (C) mRNA expression compared to untreated controls (P < 0.001). RQ, relative quantitation; ∗∗∗P < 0.001.
To confirm that miR-let-7a is directly interacting with the identified target mRNA for IGF-II, IGF2BP-2 and IGF2BP-3, a luciferase reporter assay was performed. For that purpose, Huh7 cells were transfected with pmiRGLO vector harboring the in silico predicted miR-let-7a binding site on IGF-II, IGF2BP2 or IGF2BP3 3’UTR alone or co-transfected with miR-let-7a mimics. In accordance with our previous data, transfection of miR-let-7a mimics decreased the luciferase activity of IGFII by 52.5%. In addition, both analyzed IGF2BP family members were significantly reduced by 47.3% (IGF2BP2) and 44.0% (IGF2BP-3) (Figure 5). Together, these results show that miR-let-7a can directly repress IGF-II and in a direct manner, but can also repress it indirectly through targeting its binding proteins, IGF2BP-2 and -3.
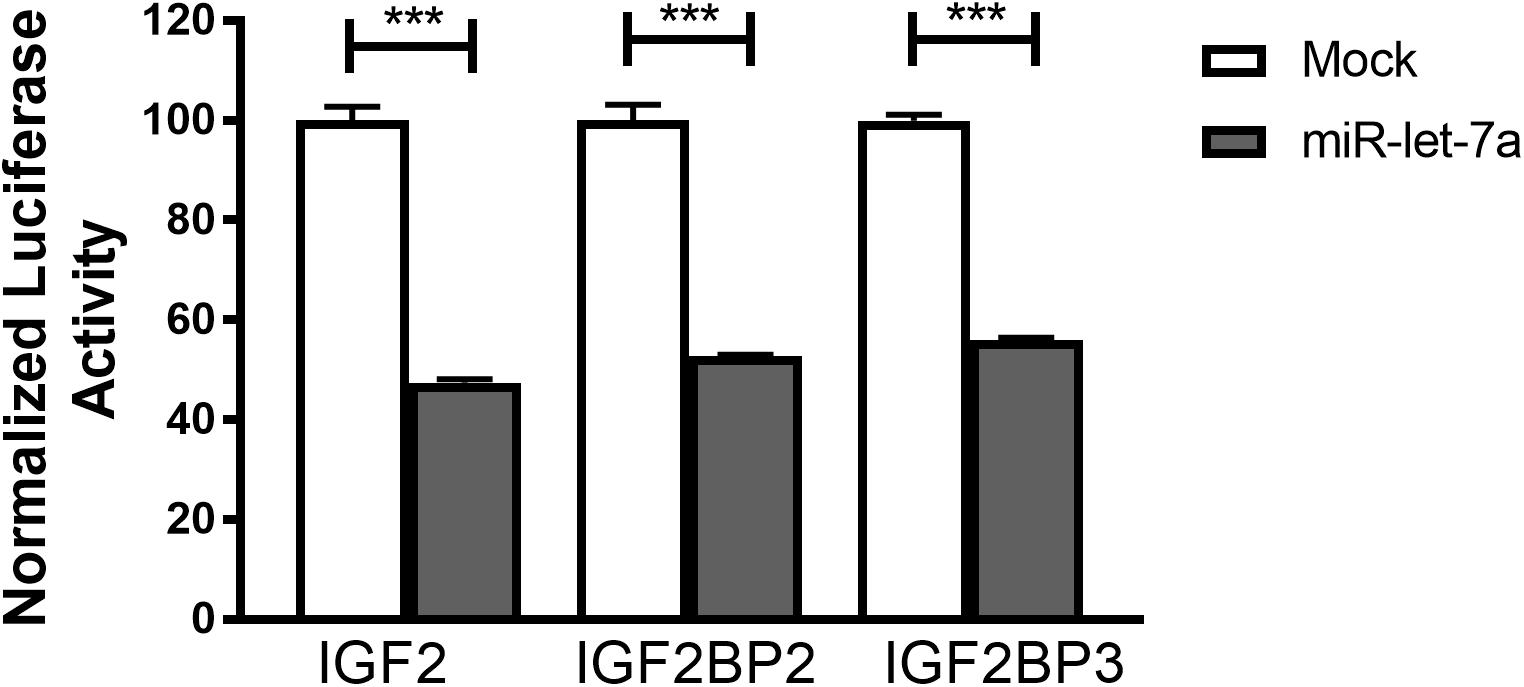
Figure 5. IGF-II, IGF2BP2, and IGF2BP3 are direct targets of miR-let-7a. Luciferase reporter assay shows that miR-let-7a directly targets IGF-II, IGF2BP2, and IGF2BP3, where Huh7 cells were transfected with pmiRGLO vector harboring the in silico predicted miR-let-7a binding site on IGF2, IGF2BP2, and IGF2BP3 3’UTR alone or co-transfected with miR-let-7a mimics. Mimics decreased the luciferase activity of IGF2 (pmirGLO_IGF2) by 52.5% (P < 0.0001) and IGF2BP-2 (pmirGLO_IGF2BP2) and IGF2BP-3 (pmirGLO_IGF2BP3) by a mean of 47.28% (P < 0.0001) and 44.03% (P < 0.0001), respectively; ∗∗∗P < 0.001.
Hepatocellular carcinoma is the fifth most common type of cancer and due to high rate of recurrence and limited chemotherapy and radiotherapy efficacy it is the second most deadly type of cancer (Ferlay et al., 2015; Mazzanti et al., 2016). HCC is characterized by dysregulation of many oncogenic signaling pathways including the insulin-like growth factor (IGF) axis. In human hepatocarcinogenesis, overexpression of the secreted ligand IGF-II is of special importance for tumor formation and activation of the relevant receptor IGF-1R correlates with poor patient prognosis (Breuhahn et al., 2006). Elevated IGF-II defines HCC patients with specific clinical features (Breuhahn et al., 2004) and supports tumor cell proliferation and migration (Nussbaum et al., 2005, 2008). In this study we now demonstrate that this oncogenic signaling axis is affected by miR-let-7a in a multi-modal manner: IGF-II as well as IGF-II-stabilizing proteins of the IGF2BP family are negatively regulated by miR-let-7a.
Up to our knowledge, our results show for the first time that the expression of miR-let-7a is downregulated in HCV-induced HCC tissues. That was further confirmed by TCGA data, where miR-let-7a shows an 0.8 fold decrease in tumor tissues compared to normal tissues (Li et al., 2014). Our finding is also in line with other findings where miR-let-7a was found to be downregulated in breast (Lu et al., 2011), lung (Brueckner et al., 2007), and pancreatic (Torrisani et al., 2009) cancers. However, a recent study has shown that miR-let-7a is downregulated in early stage HCC tissues compared to adjacent non-tumor as well as cirrhotic and chronic hepatitis tissues, but its expression in HCC was comparable to that of normal liver (Shi et al., 2017). This raises questions about potential underlying mechanisms responsible for such restricted expression in HCC. A posttranscriptional mode of miR-let-7a regulation has already been described in HCC, where the overexpressed protein Lin28B binds to the precursor miR-let-7a (pre-miR-let-7a), blocking its dicer processing and inducing its degradation (Heo et al., 2008). In this study, we tackled the question of miR-let-7a transcriptional regulation in HCC, possibly through DNA methylation. We were able to show that the identified CpG island in the MIRLET7A3 gene is hypermethylated in HCC compared to healthy liver. The same CpG island was found to be hypermethylated in other cancer types such as epithelial ovarian cancer (Lu et al., 2007) and breast cancer (Vrba et al., 2013). However, a substantial impact of such hypermethylation on the expression of miR-let-7a was only validated in leukemia (Ko et al.). This hypermethylation was found to correlate with advanced tumor stages and poor prognostic outcomes, which sheds light on the potential significance of miRNAs coded in this region (Lu et al., 2007, 2011). Interestingly, the same CpG island was found to be hypomethylated in some lung adenocarcinoma tissues and this correlated with overexpression of miR-let-7a (Brueckner et al., 2007).
IGF2BPs constitute a family of three “oncofetal” proteins whose expression is abolished in adult tissue, yet highly re-expressed in many cancers to promote tumor invasiveness (Bell et al., 2013). Among them, IGF2BP-1 has been reported to inhibit IGF-II mRNA translation during development (Nielsen et al., 1999) while on the other hand IGF2BP-2 and -3 were previously shown to promote IGF-II mRNA translation in rhabdomyosarcoma and leukemia respectively (Liao et al., 2005; Dai et al., 2011). IGF-II signaling can be tuned by IGF2BPs, which are reported to influence the fate of IGF-II as well as IGF1 receptor (IGF1R) mRNA (Liao et al., 2011; Fawzy et al., 2016). For further investigation in HCC, we knocked down IGF2BP-2/-3, which led to a marked decrease in IGF-II expression (data not shown). This verifies the importance of IGF2BP-2 and 3 for the stability of IGF-II mRNA making it more available for the translation machinery. This finding contradicts other studies where knockdown of IGF2BP-2 and -3 was found to have no effect on IGF-II mRNA levels but rather led to decreased translation of the protein in rhabdomyosarcoma and glioblastoma cell, respectively (Dai et al., 2011; Suvasini et al., 2011). However, an IGF2BP-induced stabilization of mRNAs has been described for IGF2BP-1 which was reported to protect BTRC, C-MYC, and CD44 against miRs and the degradation machinery (Noubissi et al., 2006; Vikesaa et al., 2006; Elcheva et al., 2009). Due to the different effects of IGF2BPs on IGF-II in varying cell types, the shielding effect of IGF2BP-2 and -3 needs further validation in other cancer types.
Our findings that miR-let-7a targets both IGF2BP-2 and 3, and since IGF2BP-2 and 3 are essential for IGF-II stability, we can say that miR-let-7a can target IGF-II in HCC via two mechanisms; directly through binding to its mRNA and indirectly through targeting its stabilizers; IGF2BP-2/3. Similarly, we have previously shown that another miR-let-7 family member, namely miR-let-7i directly suppresses IGF-1R, as well as indirectly through targeting IGF2BPs in HCC (Fawzy et al., 2016), thus highlighting the crucial role played by miR-let-7 family in regulating IGF-axis members in HCC.
The epigenetic regulation of miR-let-7a and its role in regulating the identified IGF-axis members is further supported by decitabine treatment experiments. Where inhibition of DNA methyltransferases (DNMT) using decitabine induces miR-let-7a and drastically reduces IGF-II and IGF2BP-2/3 in HCC cells.
In conclusion, we provided a link between DNA methylation, miR expression and oncogenic signaling. We showed that the epigenetically repressed miR-let-7a has multilayered implications on the IGF signaling in HCC. Thus, this work enhances our understanding of the molecular mechanisms by which miR-let-7a exerts its tumor suppressor activity in HCC. This study presents miR-let-7a as a promising candidate for clinical trials with potential use in cancer therapy.
AA conceived and supervised the study. AA and KB designed the experiments. AW, NE-E, RA, and MA performed the experiments. HET co-supervised the work process. KH and GE provided liver tissues and patients clinical data. KB provided new tools and reagents. AW, NE-E, KB, and AA analyzed the data. AW wrote the manuscript. NE-E, KB, and AA made the manuscript revisions.
This work was supported by the Science and Technology Development Fund (STDF) under grant number 4242 as well as the Alexander von Humboldt-Georg Forster Research Fellowship for Experienced Researchers number EGY 1160930 GF-E. Moreover, it was supported by two Deutsche Forschungsgemeinschaft (DFG) grants BR 2960/3-1 to KB and AA and SCHI 273/6-1 that supported the stay of AA in Germany for this research work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Mohamed Hassan and Prof. Dr. Sherif El-Khamisy, Center for Genomics, Zewail City of Science and Technology for providing the mimics for miR-let-7a from their grant program who helped in the completion of this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.01918/full#supplementary-material
Assal, R. A., El Tayebi, H. M., Hosny, K. A., Esmat, G., and Abdelaziz, A. I. (2015). A pleiotropic effect of the single clustered hepatic metastamiRs miR-96-5p and miR-182-5p on insulin-like growth factor II, insulin-like growth factor-1 receptor and insulin-like growth factor-binding protein-3 in hepatocellular carcinoma. Mol. Med. Rep. 12, 645–650. doi: 10.3892/mmr.2015.3382
Bae, M. H., Lee, M. J., Bae, S. K., Lee, O. H., Lee, Y. M., Park, B. C., et al. (1998). Insulin-like growth factor II (IGF-II) secreted from HepG2 human hepatocellular carcinoma cells shows angiogenic activity. Cancer Lett. 128, 41–46.
Bell, J. L., Wachter, K., Muhleck, B., Pazaitis, N., Kohn, M., Lederer, M., et al. (2013). Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell. Mol. Life Sci. 70, 2657–2675. doi: 10.1007/s00018-012-1186-z
Breuhahn, K., Longerich, T., and Schirmacher, P. (2006). Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 25, 3787–3800.
Breuhahn, K., Vreden, S., Haddad, R., Beckebaum, S., Stippel, D., Flemming, P., et al. (2004). Molecular profiling of human hepatocellular carcinoma defines mutually exclusive interferon regulation and insulin-like growth factor II overexpression. Cancer Res. 64, 6058–6064.
Brueckner, B., Stresemann, C., Kuner, R., Mund, C., Musch, T., Meister, M., et al. (2007). The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 67, 1419–1423.
Dahiya, N., Sherman-Baust, C. A., Wang, T. L., Davidson, B., Shih, I. M., Zhang, Y., et al. (2008). MicroRNA expression and identification of putative miRNA targets in ovarian cancer. PLoS One 3:e2436. doi: 10.1371/journal.pone.0002436
Dai, N., Rapley, J., Angel, M., Yanik, M. F., Blower, M. D., and Avruch, J. (2011). mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 25, 1159–1172. doi: 10.1101/gad.2042311
El Tayebi, H. M., Salah, W., El Sayed, I. H., Salam, E. M., Zekri, A. R., Zayed, N., et al. (2011). Expression of insulin-like growth factor-II, matrix metalloproteinases, and their tissue inhibitors as predictive markers in the peripheral blood of HCC patients. Biomarkers 16, 346–354. doi: 10.3109/1354750X.2011.573095
El Tayebi, H. M., Waly, A. A., Assal, R. A., Hosny, K. A., Esmat, G., and Abdelaziz, A. I. (2015). Transcriptional activation of the IGF-II/IGF-1R axis and inhibition of IGFBP-3 by miR-155 in hepatocellular carcinoma. Oncol. Lett. 10, 3206–3212.
Elcheva, I., Goswami, S., Noubissi, F. K., and Spiegelman, V. S. (2009). CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol. Cell 35, 240–246. doi: 10.1016/j.molcel.2009.06.007
Fawzy, I. O., Hamza, M. T., Hosny, K. A., Esmat, G., and Abdelaziz, A. I. (2016). Abrogating the interplay between IGF2BP1, 2 and 3 and IGF1R by let-7i arrests hepatocellular carcinoma growth. Growth Factors 34, 42–50. doi: 10.3109/08977194.2016.1169532
Ferlay, J., Soerjomataram, I., Dikshit, R., Eser, S., Mathers, C., Rebelo, M., et al. (2015). Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386. doi: 10.1002/ijc.29210
Gardiner-Garden, M., and M. Frommer (1987). CpG islands in vertebrate genomes. J. Mol. Biol. 196, 261–282. doi: 10.1016/0022-2836(87)90689-9
Habashy, D. A., El Tayebi, H. M., Fawzy, I. O., Hosny, K. A., Esmat, G., and Abdelaziz, A. I. (2016). Interplay between microRNA-17-5p, insulin-like growth factor-II through binding protein-3 in hepatocellular carcinoma. World J. Hepatol. 8, 976–984. doi: 10.4254/wjh.v8.i23.976
Heo, I., Joo, C., Cho, J., Ha, M., Han, J., and Kim, V. N. (2008). Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol. Cell 32, 276–284. doi: 10.1016/j.molcel.2008.09.014
Jeng, Y. M., Chang, C. C., Hu, F. C., Chou, H. Y., Kao, H. L., Wang, T. H., et al. (2008). RNA-binding protein insulin-like growth factor II mRNA-binding protein 3 expression promotes tumor invasion and predicts early recurrence and poor prognosis in hepatocellular carcinoma. Hepatology 48, 1118–1127. doi: 10.1002/hep.22459
Kessler, S. M., Pokorny, J., Zimmer, V., Laggai, S., Lammert, F., Bohle, R. M., et al. (2013). IGF2 mRNA binding protein p62/IMP2-2 in hepatocellular carcinoma: antiapoptotic action is independent of IGF2/PI3K signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G328–G336. doi: 10.1152/ajpgi.00005.2012
Ko, Y.-C., Fang, W.-H., Lin, T.-C., Hou, H.-A., Chen, C.-Y., Tien, H.-F., et al. (2014). MicroRNA let-7a-3 gene methylation is associated with karyotyping, CEBPA promoter methylation, and survival in acute myeloid leukemia. Leuk. Res. 38, 625–631. doi: 10.1016/j.leukres.2014.03.008
Lahm, H., Gittner, K., Krebs, O., Sprague, L., Deml, E., Oesterle, D., et al. (2002). Diethylnitrosamine induces long-lasting re-expression of insulin-like growth factor II during early stages of liver carcinogenesis in mice. Growth Horm. IGF Res. 12, 69–79.
Li, J.-H., Liu, S., Zhou, H., Qu, L.-H., and Yang, J.-H. (2014). starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–D97. doi: 10.1093/nar/gkt1248
Liao, B., Hu, Y., and Brewer, G. (2011). RNA-binding protein insulin-like growth factor mRNA-binding protein 3 (IMP-3) promotes cell survival via insulin-like growth factor II signaling after ionizing radiation. J. Biol. Chem. 286, 31145–31152. doi: 10.1074/jbc.M111.263913
Liao, B., Hu, Y., Herrick, D. J., and Brewer, G. (2005). The RNA-binding protein IMP-3 is a translational activator of insulin-like growth factor II leader-3 mRNA during proliferation of human K562 leukemia cells. J. Biol. Chem. 280, 18517–18524.
Liu, Y., Xia, Y., Dai, W., Han, H., Dong, Y., Cai, J., et al. (2014). Cholesterol-conjugated let-7a mimics: antitumor efficacy on hepatocellular carcinoma in vitro and in a preclinical orthotopic xenograft model of systemic therapy. BMC Cancer 14:889. doi: 10.1186/1471-2407-14-889
Lu, L., Katsaros, D., de la Longrais, I. A., Sochirca, O., and Yu, H. (2007). Hypermethylation of let-7a-3 in epithelial ovarian cancer is associated with low insulin-like growth factor-II expression and favorable prognosis. Cancer Res. 67, 10117–10122.
Lu, L., Katsaros, D., Zhu, Y., Hoffman, A., Luca, S., Marion, C. E., et al. (2011). Let-7a regulation of insulin-like growth factors in breast cancer. Breast Cancer Res. Treat. 126, 687–694. doi: 10.1007/s10549-010-1168-5
Mazzanti, R., Arena, U., and Tassi, R. (2016). Hepatocellular carcinoma: where are we? World J. Exp. Med. 6, 21–36.
Nielsen, J., Christiansen, J., Lykke-Andersen, J., Johnsen, A. H., Wewer, U. M., and Nielsen, F. C. (1999). A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol. Cell. Biol. 19, 1262–1270.
Noubissi, F. K., Elcheva, I., Bhatia, N., Shakoori, A., Ougolkov, A., Liu, J., et al. (2006). CRD-BP mediates stabilization of [beta]TrCP1 and c-myc mRNA in response to [beta]-catenin signalling. Nature 441, 898–901.
Nussbaum, T., Samarin, J., Ehemann, V., Bissinger, M., Ryschich, E., Khamidjanov, A., et al. (2008). Autocrine insulin-like growth factor-II stimulation of tumor cell migration is a progression step in human hepatocarcinogenesis. Hepatology 48, 146–156. doi: 10.1002/hep.22297
Nussbaum, T., Vreden, S., Farsad, M., Schirmacher, P., and Breuhahn, K. (2005). Insulin-like growth factor (IGF)-II in human hepatocarcinogenesis–a potential therapeutic target? Verh. Dtsch. Ges. Pathol. 89, 254–260.
O’Hara, A. J., Wang, L., Dezube, B. J., Harrington, W. J. Jr., Damania, B., and Dittmer, D. P. (2009). Tumor suppressor microRNAs are underrepresented in primary effusion lymphoma and Kaposi sarcoma. Blood 113, 5938–5941. doi: 10.1182/blood-2008-09-179168
Park, S.-M., Shell, S., Radjabi, A. R., Schickel, R., Feig, C., Boyerinas, B., et al. (2007). Let-7 prevents early cancer progression by suppressing expression of the embryonic gene HMGA2. Cell Cycle 6, 2585–2590.
Poirier, K., Chalas, C., Tissier, F., Couvert, P., Mallet, V., Carrie, A., et al. (2003). Loss of parental-specific methylation at the IGF2 locus in human hepatocellular carcinoma. J. Pathol. 201, 473–479.
Rahmoon, M. A., Youness, R. A., Gomaa, A. I., Hamza, M. T., Waked, I., El Tayebi, H. M., et al. (2017). MiR-615-5p depresses natural killer cells cytotoxicity through repressing IGF-1R in hepatocellular carcinoma patients. Growth Factors 35, 76–87. doi: 10.1080/08977194.2017.1354859
Reinhart, B. J., Slack, F. J., Basson, M., Pasquinelli, A. E., Bettinger, J. C., Rougvie, A. E., et al. (2000). The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906.
Rougvie, A. E., and Ambros, V. (1995). The heterochronic gene lin-29 encodes a zinc finger protein that controls a terminal differentiation event in Caenorhabditis elegans. Development 121, 2491–2500.
Schulman, B. R. M., Esquela-Kerscher, A., and Slack, F. J. (2005). Reciprocal expression of lin-41 and the microRNAs let-7 and mir-125 during mouse embryogenesis. Dev. Dyn. 234, 1046–1054.
Sempere, L., Freemantle, S., Pitha-Rowe, I., Moss, E., Dmitrovsky, E., and Ambros, V. (2004). Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 5:R13.
Shi, W., Zhang, Z., Yang, B., Guo, H., Jing, L., Liu, T., et al. (2017). Overexpression of microRNA let-7 correlates with disease progression and poor prognosis in hepatocellular carcinoma. Medicine 96:e7764. doi: 10.1097/MD.0000000000007764
Slack, F. J., Basson, M., Liu, Z., Ambros, V., Horvitz, H. R., and Ruvkun, G. (2000). The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol. Cell 5, 659–669.
Suvasini, R., Shruti, B., Thota, B., Shinde, S. V., Friedmann-Morvinski, D., Nawaz, Z., et al. (2011). Insulin growth factor-2 binding protein 3 (IGF2BP3) is a glioblastoma-specific marker that activates phosphatidylinositol 3-kinase/mitogen-activated protein kinase (PI3K/MAPK) pathways by modulating IGF-2. J. Biol. Chem. 286, 25882–25890. doi: 10.1074/jbc.M110.178012
Takamizawa, J., Konishi, H., Yanagisawa, K., Tomida, S., Osada, H., Endoh, H., et al. (2004). Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 64, 3753–3756.
Torrisani, J., Bournet, B., du Rieu, M. C., Bouisson, M., Souque, A., Escourrou, J., et al. (2009). let-7 MicroRNA transfer in pancreatic cancer-derived cells inhibits in vitro cell proliferation but fails to alter tumor progression. Hum. Gene Ther. 20, 831–844. doi: 10.1089/hum.2008.134
Vikesaa, J., Hansen, T. V., Jonson, L., Borup, R., Wewer, U. M., Christiansen, J., et al. (2006). RNA-binding IMPs promote cell adhesion and invadopodia formation. EMBO J. 25, 1456–1468.
Vrba, L., Munoz-Rodriguez, J. L., Stampfer, M. R., and Futscher, B. W. (2013). miRNA gene promoters are frequent targets of aberrant DNA methylation in human breast cancer. PLoS One 8:e54398. doi: 10.1371/journal.pone.0054398
Wachter, D. L., Kristiansen, G., Soll, C., Hellerbrand, C., Breuhahn, K., Fritzsche, F., et al. (2012). Insulin-like growth factor II mRNA-binding protein 3 (IMP3) expression in hepatocellular carcinoma. A clinicopathological analysis with emphasis on diagnostic value. Histopathology 60, 278–286. doi: 10.1111/j.1365-2559.2011.04091.x
Wu, J., and Zhu, A. (2011). Targeting insulin-like growth factor axis in hepatocellular carcinoma. J. Hematol. Oncol. 4:30. doi: 10.1186/1756-8722-4-30
Keywords: miR-let-7a, methylation, IGF-II, IGF2BP, HCC
Citation: Waly AA, El-Ekiaby N, Assal RA, Abdelrahman MM, Hosny KA, El Tayebi HM, Esmat G, Breuhahn K and Abdelaziz AI (2019) Methylation in MIRLET7A3 Gene Induces the Expression of IGF-II and Its mRNA Binding Proteins IGF2BP-2 and 3 in Hepatocellular Carcinoma. Front. Physiol. 9:1918. doi: 10.3389/fphys.2018.01918
Received: 10 September 2018; Accepted: 20 December 2018;
Published: 24 January 2019.
Edited by:
Richard T. Waldron, Cedars-Sinai Medical Center, United StatesReviewed by:
Deepak Gurbani, University of Texas Southwestern Medical Center, United StatesCopyright © 2019 Waly, El-Ekiaby, Assal, Abdelrahman, Hosny, El Tayebi, Esmat, Breuhahn and Abdelaziz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed I. Abdelaziz, YWhtZWQuaWhhYi5hYmRlbGF6aXpAZ21haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.