 Klaus-Dieter Schlüter
Klaus-Dieter Schlüter Hanna Sarah Kutsche
Hanna Sarah Kutsche Christine Hirschhäuser
Christine Hirschhäuser Rolf Schreckenberg
Rolf Schreckenberg Rainer Schulz
Rainer Schulz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Physiol., 17 December 2018
Sec. Redox Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01799
This article is part of the Research TopicOxidative Stress in Myocardial and Neural RemodelingView all 10 articles
Reactive oxygen species (ROS) exert signaling character (redox signaling), or damaging character (oxidative stress) on cardiac tissue depending on their concentration and/or reactivity. The steady state of ROS concentration is determined by the interplay between its production (mitochondrial, cytosolic, and sarcolemmal enzymes) and ROS defense enzymes (mitochondria, cytosol). Recent studies suggest that ROS regulation is different in the left and right ventricle of the heart, specifically by a different activity of superoxide dismutase (SOD). Mitochondrial ROS defense seems to be lower in right ventricular tissue compared to left ventricular tissue. In this review we summarize the current evidence for heart chamber specific differences in ROS regulation that may play a major role in an observed inability of the right ventricle to compensate for cardiac stress such as pulmonary hypertension. Based on the current knowledge regimes to increase ROS defense in right ventricular tissue should be in the focus for the development of future therapies concerning right heart failure.
Oxidative stress is defined as a condition by which an imbalance occurs between the production of reactive oxygen species (ROS) and the antioxidant defense system. The term ROS includes molecules that have one or more unpaired electrons (i.e., superoxide and hydroxyl) and non-radicals that are able to generate free radicals (i.e., hydrogen peroxide). Intracellular sources of ROS are the electron transport chain of the mitochondria, monoamine oxidase (MAO), p66shc (for review, see Di Lisa et al., 2017), xanthine oxidase (XO), uncoupling proteins (UCP, depending on the mitochondrial membrane potential; for review see Cadenas, 2018), uncoupled nitric oxide (NO) synthase (NOS), sodium-potassium ATPase (NKA), and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) (for review, see Egea et al., 2017). The defense system contains enzymes such as superoxide dismutase (SOD), catalase, glutathione peroxidase, and coupled NOS. Figures 1, 2 give an overview about ROS sources and ROS defense systems in the heart. Whereas subtle changes in ROS act as intracellular signaling pathways (redox signaling) high levels of ROS can cause cell damage and dysfunction (oxidative stress) (for review, see Egea et al., 2017).
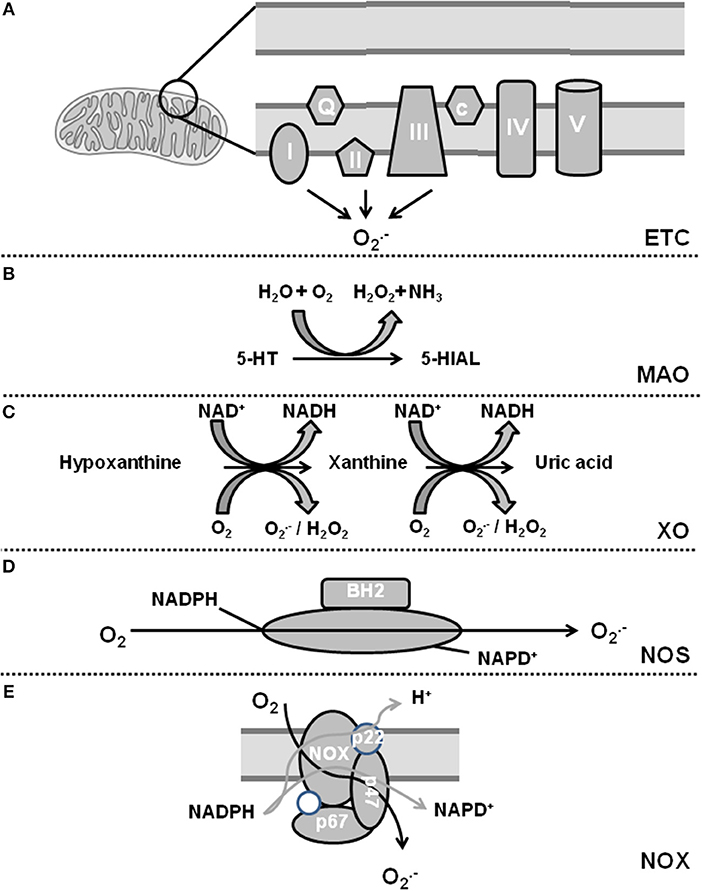
Figure 1. Sources of reactive oxygen species in cardiomyocytes. (A) Complex I and III of the electron transport chain (ETC) constitutively release and complex II can be activated by NOX-dependent ROS. (B) Monoamine oxidase (MAO) generates H2O2. (C) Xanthine oxidase (XO) catalyzes a two-step reaction leading to additional release of H2O2. (D) Uncoupling of nitric oxide synthase (NOS) leads to generation of . (E) NADPH oxidase (NOX) generates also upon activation.
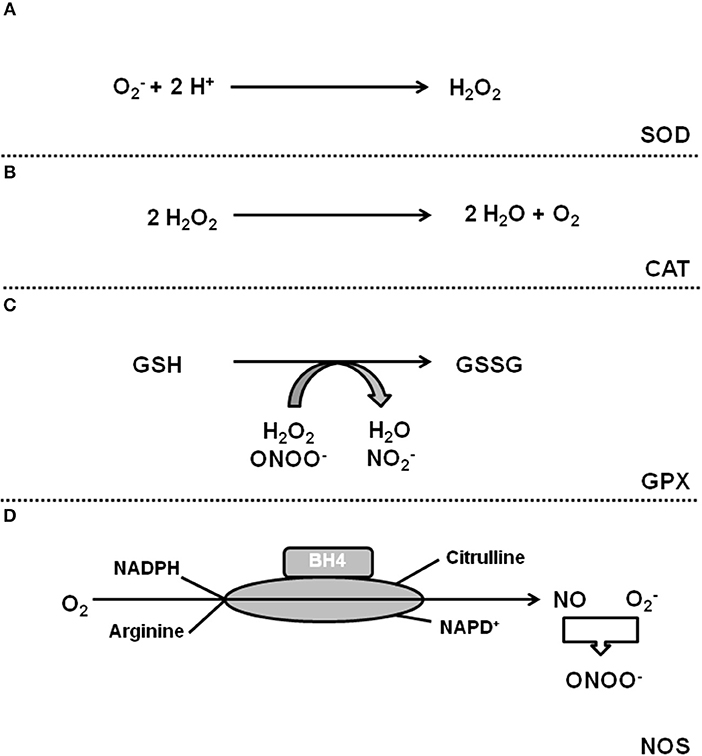
Figure 2. ROS defense systems in cardiomyocytes. (A) Superoxide dismutases catalyze the formation of H2O2 that can be detoxificated by catalase (B). (C) Gluatathione peroxidase (GPX) reduces H2O2. (D) Coupled NOS generates NO that neutralizes .
The following review article now summarizes our current understanding about similarities and differences in ROS handling between LV and RV. We searched the current literature (PubMed, MedLine data bank until July, 2018) using the terms “right heart and ROS,” “pulmonary hypertension and ROS,” “RV failure and ROS,” “LV failure and ROS,” “RV hypertrophy and ROS” and “LV hypertrophy and ROS.” Most studies dealing with ROS and RV hypertrophy used models of pulmonary hypertension induced by banding, monocrotaline injection or chronic hypoxia. In these models ROS contribute to pulmonary hypertension and RV remodeling. In many studies, effects of ROS reduction on RV hypertrophy were secondary to reduced pulmonary pressure (reviewed by Wong et al., 2013). In the current review we therefore focus on studies directly assessing ROS and ROS-dependent effects in RV tissue and compare these results to established concepts generated from the LV.
Oxidative stress in cardiomyocytes occurs during chronic pressure or volume overload of the heart (Gladden et al., 2013; Hansen et al., 2016), cardiac ischemia/ reperfusion (Riba et al., 2017), cardiomyopathy (Ishikawa et al., 2005), diabetes (Guido et al., 2017), chemotherapy-induced heart failure in the left (Mouli et al., 2015; Li et al., 2018), and right ventricle (Anghel et al., 2018), poison such as cigarette smoke (Talukder et al., 2011), chronic kidney disease (Duni et al., 2017), or aging (Chang et al., 2017), or as a response to congenital heart failure (Iacobazzi et al., 2016). Within the heart other sources of ROS are cells adjacent to cardiomyocytes such as inflammatory cells (Xu et al., 2011; Hernandez-Resendiz et al., 2018), endothelial cells (Burger et al., 2011), stem cells (Mandraffino et al., 2017), and cardiac fibroblasts (Ciulla et al., 2011). Redox signaling contributes to cardiac hypertrophy and even more important oxidative stress contributes to the transition of adaptive to maladaptive cardiac hypertrophy, named maladaptive remodeling. Oxidative stress can damage cells by growth factor-independent activation of cardiac growth regulation (Calamaras et al., 2015), can inactivate NO leading to loss of myocyte-specific NO function (Rassaf et al., 2006; Lüneburg et al., 2016), can directly reduce cardiomyocyte function by oxidative modification of sarcomere proteins such as tropomyosin (Heusch et al., 2010b; Canton et al., 2011) or sarcoplasmatic reticulum proteins (i.e., SERCA; Qin et al., 2017), can induce a calcium desensitization of myofibrils (Wang et al., 2008), can activate the Na-K-ATPase (Wang et al., 2017a), can damage mitochondrial function (Ide et al., 2001; Sverdlov et al., 2016), or can induce cell death (apoptosis, necrosis; Redza-Dutordoir and Averill-Bates, 2016). Therefore, ROS defense strategies of the cells are necessary for cell survival and functional stabilization in both ventricles.
The different chambers of the heart are derived from different embryonic origin, namely the first (left ventricle, LV) and second heart field (right ventricle, RV). In rodent hearts, cardiomyocytes isolated from the LV or RV differ in size, number of mono-nucleated cells, cellular adaptation to culture conditions, and cell shortening (Schlüter, 2016; summarized in Figure 3). This gives raise to speculations that the LV and RV may differentially respond to cardiac stresses. Pressure overload is associated with adaptations performed on the transcriptional level. Many of them are similar between the RV and LV. However, some genes are up-regulated in the pressure-overloaded RV only, including genes involved in Wnt signaling (Dickkopf 3, Sfrp2, and Wif1), annexin A7, clusterin/apolipoprotein J, neuroblastoma suppression of tumorigenicity 1 (Nbl1), formin-binding protein (Fnbp4), and the lectin-like oxidized low-density lipoprotein (oxLDL) receptor (LOX; Reddy and Bernstein, 2015). Differences occur also on the level of miRNA (Reddy and Bernstein, 2015). Considering the high impact of ROS for cardiac adaptation to pressure overload, it is also important to understand such differences with respect to ROS formation, ROS defense, and ROS-dependent cellular responses. Indeed, mitochondria isolated from the LV or RV of rat hearts generates different amounts of ROS (Schreckenberg et al., 2015; summarized in Figure 3). Treatment of isolated perfused rat hearts with serotonin, a substrate for MAO, results in the promotion of protein carbonylation as evidenced by increased ROS formation, specifically in the RV but not the LV. Interestingly, no differences between RV and LV antioxidant enzymes and serotonin receptors/transporter are detected (Liu et al., 2008).
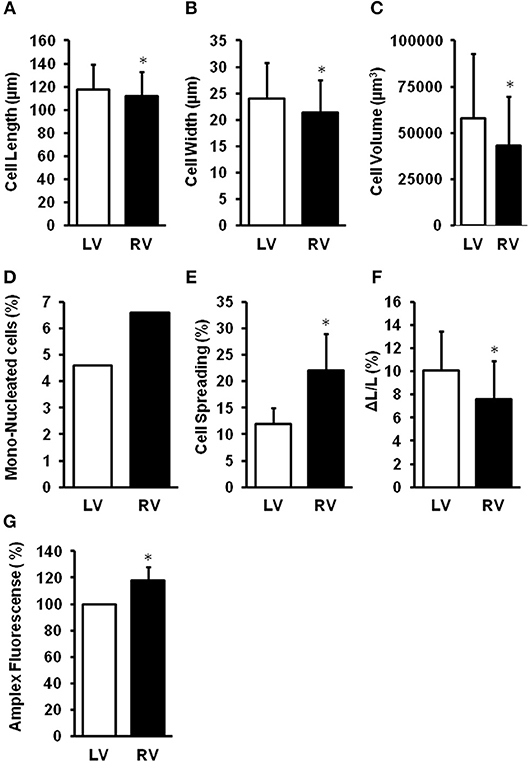
Figure 3. Differences between right (RV) and left (LV) ventricular cardiomyocytes. LV cardiomyocytes are longer (A), wider (B), larger (C), less mono-nucleated (D), have a reduced cell spreading (E) as adaptation to culture conditions, and a stronger load-free cell shortening (F). Furthermore, mitochondria from RV generate more ROS (G). Data depicted from Schreckenberg et al. (2015) and Schlüter (2016). *p < 0.05 vs. LV.
Pressure overload induces the NOX subunit gp91 (Li et al., 2002; Byrne et al., 2003; Tanaka et al., 2005; Grieve et al., 2006; Liu et al., 2006, 2010; Guggilam et al., 2007; DeMarco et al., 2008; Nisbet et al., 2009; Chemaly et al., 2011; Xu et al., 2011; Ogura et al., 2013; Frazziano et al., 2014; Matsuda et al., 2015; Ma et al., 2016; Sirker et al., 2016; Zhu et al., 2017). In human right and left heart failure the p47phox subunit of NOX also translocates to the sarcrolemma (Nediani et al., 2007). Increased NOX expression is associated with increased formation of superoxide anions (Nediani et al., 2007; Ogura et al., 2013; Dos Santos Lacerda et al., 2017; Türck et al., 2018). Furthermore, hypoxia, leading to pulmonary hypertension, and RV hypertrophy, increases RV expression of NOX2/4 (Liu et al., 2014; Ye et al., 2016; Zhu et al., 2017). In the monocrotaline-induced pulmonary hypertension rat model, Nox4 expression is induced in cardiomyocytes but also in the intercellular area (mainly co-localizing with fibroblasts) (He et al., 2017). In the RV, NOX4 is also regulated by the α1A-receptor (Cowley et al., 2017); stimulation of this receptor decreases NOX4 expression during pulmonary hypertension. NOX-dependent ROS modifies mitochondrial function by increased release of ROS from complex II of the mitochondria during the transition from RV hypertrophy to failure (Redout et al., 2007) (ROS induced ROS release). There are differences between the RV and LV ventricles in the primary source of ROS production. In the RV, NOX, and mitochondrial complex II activity both increase during the transition to heart failure, whereas, in the LV, NOX appears to be the primary source of ROS generation (Redout et al., 2010).
Xanthine oxidoreductase (XO) activity remains unaltered in the monocrotaline-induced RV hypertrophy model but its activity increases with the transition to RV failure. XO is mainly localized in CD68+ inflammatory cells based on studies with an affinity-purified polyclonal antibody (de Jong et al., 2000). In another rat model of pulmonary hypertension-induced RV failure, metabolomics analysis revealed an increase in xanthine, and uric acid in the hypertrophied RV, suggesting the production of ROS by XO. Furthermore, the RV level of α-tocopherol nicotinate declined, consistent with oxidative stress decreasing antioxidants (Wang et al., 2017). XO also contributes to ROS formation in LV (Moris et al., 2017).
Uncoupled NOS is another protein involved in the generation of oxidative stress in the LV secondary to pressure overload (Takimoto et al., 2005). In the RV, uncoupled NOS contributes to ROS generation, too. In caveolin-1 (Cav-1) knockout mice subjected to chronic hypoxia the transition from RV hypertrophy to failure is accelerated compared to wild-type mice and caused by uncoupling of RV endothelial NOS and increased protein tyrosine nitration; all changes are prevented by re-expressing an endothelial-specific Cav-1 transgene (avoiding NOS uncoupling) or by NOS inhibition without modifying the extent of pulmonary hypertension (Cruz et al., 2012). Also, in a pharmacological model of hypertension chronic administration of L-NAME leads to uncoupling of NOS in RV (Schreckenberg et al., 2015).
Uncoupling of NOS can be caused by reduced substrate (arginine) for NOS. Reasons for substrate limitation can be an induction of arginase that leads to substrate limitation (Heusch et al., 2010; Schreckenberg et al., 2015), increased plasma concentrations of asymmetric dimethyl arginine (ADMA), a natural circulating inhibitor of NOS (Lüneburg et al., 2016), or depletion of NOS with tetrahydrobiopterin (BH4, Shimizu et al., 2013).
A proteomic analysis of the normal rabbit and porcine RV and LV free walls shows equivalent cellular aerobic capacity, volume of mitochondria, mitochondrial enzyme content (cytochrome c oxidase, respiratory complexes 1 and 3–5, aconitase, SOD), and mitochondrial enzyme activities (Phillips et al., 2011). Interestingly, mitochondrial membrane potential, a surrogate of overall mitochondrial function, is lower in the resting RV compared to the LV (Nagendran et al., 2008), while—at least in rats—ROS formation in mitochondria isolated from the RV is slightly higher than in LV mitochondria (Schreckenberg et al., 2015). At last in part, the latter might be the consequence of a reduced ROS defense capacity (Borchi et al., 2010; Manni et al., 2016). Comparing mitochondria from hypertrophic RV with those of non-hypertrophic LV revealed differences in electrone transport chain activity and ATP generating enzyme expression Gupto et al., 2016.
While the mitochondrial protein profiles of the RV and LV are quite similar at rest, they diverge when subjected to an increased afterload (Phillips et al., 2011), and mitochondrial membrane potential increases with RV hypertrophy (Nagendran et al., 2008). This hyperpolarization of mitochondria, indicating reduced oxidative phosphorylation, is related to an activation of the nuclear factor of activated T cells (NFAT) pathway and is reversed by dichloroacetate, an inhibitor of pyruvate dehydrogenase kinases (PDK) (Nagendran et al., 2008). Thus, an increase in PDK activity in RV hypertrophy contributes to the decreased oxidation of pyruvate in mitochondria and an increased conversion of pyruvate to lactate in the cytosol. An increase in glycolytic hexokinase and lactate dehydrogenase activities occurs following monocrotaline-induced pulmonary hypertension at the stage of compensated RV hypertrophy (Balestra et al., 2015), further supporting the concept of a metabolic switch from mitochondrial oxidative phosphorylation to glycolysis in the compensated phase of RV hypertrophy (Paulin and Michelakis, 2014; Sutendra and Michelakis, 2014). Indeed, a decreased mitochondrial oxygen usage and an increased anaerobic glycolysis has been described in patients with pulmonary hypertension (Wong et al., 2011) (for a detailed review, see Freund-Michel et al., 2014), and the decrease in mitochondrial oxidative phosphorylation during the development of RV hypertrophy has been suggested to decrease mitochondrial ROS formation (for review, see Paulin and Michelakis, 2014).
The increase in glucose uptake and the mitochondrial hyperpolarization are lost with the progression of RV hypertrophy to failure (Nagendran et al., 2008). For the LV, ROS sensors revealed increased mitochondrial ROS in resting and contracting cardiomyocytes during the progression to heart failure. Pathway analysis of mitochondrial ROS-sensitive networks indicated that increased mitochondrial ROS in failing cardiomyocytes disrupts the normal coupling between cytosolic signals and nuclear gene programs driving mitochondrial function, calcium handling, action potential repolarization, and antioxidant enzymes (Dey et al., 2018). Indeed, in the RV, during the transition from RV hypertrophy to RV failure, mitochondrial ROS defense system (SOD-2) is down-regulated (Redout et al., 2007).
Another key regulator that is decreased during RV failure is the peroxisome proliferator-activated receptor gamma coactivator (PGC) 1α, leading to impaired fatty acid oxidation, decreased mitochondrial mass and number, and reduced oxidative capacity, potentially contributing to increased ROS production (Karamanlidis et al., 2011; Gomez-Arroyo et al., 2013). In an animal model of pulmonary hypertension-induced RV failure, fatty acid oxidation decreases secondary to the failure of palmitoylcarnitine to stimulate oxygen consumption. In humans with pulmonary hypertension, RV long-chain fatty acids and triglyceride contents are increased and ceramide, a mediator of lipotoxicity, accumulates (Brittain et al., 2016).
In rats treated with monocrotaline to increase pulmonary artery pressure without inducing RV hypertrophy, RV hydrogen peroxide increases but SOD, catalase, and glutathione peroxidase activities are also enhanced (Siqueira et al., 2018).
During pressure overload–induced LV hypertrophy, antioxidant enzymes are activated in the compensated stage but their activity decreases during the onset of LV failure. In contrast, only the antioxidant enzyme catalase becomes activated in some (Ecarnot-Laubriet et al., 2003) but not all studies (Pichardo et al., 1999) while SOD and glutathione peroxidase are not activated at all in the compensated stage of RV hypertrophy secondary to pulmonary hypertension, predisposing the RV to ROS induced damage at an earlier stage than in the LV (Pichardo et al., 1999; Ecarnot-Laubriet et al., 2003; Schreckenberg et al., 2015). With a progression of from RV hypertrophy to failure, down-regulation of antioxidant enzymes, and increased ROS production occurs in a mice model of pulmonary hypertension (Aziz et al., 2015; Reddy and Bernstein, 2015), although in one model of monocrotaline-induced RV failure, glutathione peroxidase increases while catalase, and SOD activities are similar to sham animals (Türck et al., 2018).
Despite some controversial results the general view, however, remains that increased ROS formation and decreased ROS defense leads to increased oxidative stress driving the progression from RV hypertrophy to RV failure.
(Patho)physiological conditions known to activate p38 mitogen activated protein (MAP) kinase are often associated with increased ROS formation (Wenzel et al., 2001, 2006, 2007). Indeed, p38 MAP kinase is activated by oxidative stress (Redout et al., 2007). An activation of p38 MAP kinase pathways is linked to cardiac hypertrophy and dysfunction and in RV and LV of end-stage failing human hearts, p38 MAP kinase and extracellular-signal regulated kinase (ERK), but not c-Jun N-terminal kinases (JNK), are activated; a significant correlation between protein kinase activities is observed between LV and RV from the same heart (Nediani et al., 2007).
Increased ROS subsequently modifies tropomyosin, induces matrix metalloproteases (MMPs 2, 9, and 13), sensitizes β-adrenoceptors (via induction of protein kinase C-ε), and causes endothelial dysfunction in the right ventricle (Cheng et al., 2009; Lu et al., 2011; Cau et al., 2013; Schreckenberg et al., 2015).
In LV tissue, ROS is associated with an induction of p90rsk and the sodium-proton-exchanger (NHE) and furthermore, via ROS-dependent formation of lipid peroxidation-derived aldehydes (Cingolani et al., 2011). Furthermore, ROS activates the mammalian target of rapamycin (mTOR)-p70s6k pathway (Calamaras et al., 2015). Both pathways (NHE and mTOR-p70s6k) are also involved in growth factor-dependent acceleration of protein synthesis (Simm et al., 1998; Schäfer et al., 2002). Commonly ROS and growth factors activate also the ERK pathway but the latter is not directly linked to the regulation of protein synthesis (Pönicke et al., 2001; Calamaras et al., 2015).
Apart from NOX, activation of the renin-angiotensin-system is apparent in the RV during pressure overload (for review, see Ameri et al., 2016). Compared to normal hearts, however, angiotensin II binding is diminished in the failing RV of pulmonary artery hypertension patients due to angiotensin II type 1 receptor down-regulation, despite RV myocardial angiotensin converting enzyme (ACE) activity being increased (Zisman et al., 1998). Interestingly, the ACE DD genotype, associated with an increased myocardial ACE activity, is more frequent in patients with pulmonary hypertension than in healthy individuals, but it is also associated with preserved RV function in pulmonary hypertension patients (Abraham et al., 2003).
A specific role for LOX-1 in RV hypertrophy and failure has been suggested. First, oxLDL receptors cross react with NOX (Ogura et al., 2013). Furthermore, ventricular expression of oxLDL receptors is induced under hypoxia leading to pulmonary hypertension and RV hypertrophy (Zhu et al., 2017). Cross-reactivity of oxLDL receptors with angiotensin II receptors type 1 has also been reported. In all these cases, NOX is subsequently activated favoring oxidative stress. It seems that this mechanism plays an important role in right heart failure.
In general, attenuation of mitochondrial-derived oxidative stress is a reasonable therapeutic concept to attenuate RV hypertrophy and transition to RV failure (for review, see Maarman et al., 2017).
As expected from the findings that ROS is increased in RV hypertrophy and transition to RV failure, trapping molecules targeting mitochondrial ROS (mitoTEMPO), folic acid, EUK-134, a synthetic antioxidant mimicking the activity of SOD, attenuate RV hypertrophy (Redout et al., 2010; Qipshidze et al., 2012; Datta et al., 2015).
Regulation of SOD, in particular SOD-1 (located in the cytosol), and SOD-2 (located in mitochondria), has been proven to attenuate hypertrophy and even more important transition to heart failure. In a pharmacological rat model of hypertension (L-NAME induced hypertension) SOD-2 was induced in the LV but not in the RV (Schreckenberg et al., 2015). Up-regulation of SOD-2 in the LV was associated with less oxidative stress and preserved function in the presence of hypertrophy. Similarly, induction of SOD-2 activity has repeatedly reported to improve cardiac function. Interestingly, at least for the LV multiple strategies to improve SOD activity work, such as the natural product Sheng-Mai-San (Chai et al., 2016), inhibition of the renin-angiotensin-system (Tanaka et al., 2005), calcium antagonists (Umemoto et al., 2004), or resveratrol (Danz et al., 2009). Whether any of these mechanisms is sufficient to increase SOD activity in RV remains unclear. As mentioned above, SOD is induced during hypertrophy in LV tissue (Date et al., 2002; Lu et al., 2010; Qiao et al., 2014; Aziz et al., 2015; Schreckenberg et al., 2015). Failure to increase SOD as an adaptive mechanism to rescue mitochondrial and cytosolic ROS is associated with heart failure (Redout et al., 2007; Koga et al., 2013). In a model of bronchopulmonary dysplasia, SOD-2 expression but not activity is induced leaving ROS formation unchanged. This underlines the importance of SOD-2 activity for protection against ROS-derived damage. Failure of the RV to up-regulate SOD-2 expression and activity might be a key step for right heart failure.
Other treatment using secoisolariciresinol diglucoside (Puukila et al., 2017), dehydroepiandrosterone (Alzoubi et al., 2013; Rawat et al., 2014), trimethoxystilbene (Liu et al., 2014), pterostilbene (Dos Santos Lacerda et al., 2017), trapidil (Türck et al., 2018), and α1A-adrenoceptor stimulation with A61603 (Cowley et al., 2017) and finally fenofibrate (Galhotra et al., 2018) attenuate both RV hypertrophy and dysfunction and decreases RV ROS levels at the same time; however, a causality between changes in ROS and preservation of RV morphology and/or function could not be proven.
In contrast to the pharmacological approaches, neither the genetic deletion of sirtuin 3 (Waypa et al., 2013) nor the up-regulation of thioredoxin 2 (Adesina et al., 2017) affected RV hypertrophy during pulmonary hypertension. Sirtuin-3 is a nicotinamide adenine dinucleotide–dependent deacetylase that activates forkhead box O3a (FOXO3)-dependent up-regulation of SOD-2 (Sundaresan et al., 2009). Thioredoxin 2 is a mitochondrial located protein involved in ROS defense of the organelle (Dunn et al., 2010).
β-Blockers may also considered as a therapeutic option in right heart failure. β-Adrencoeptor signaling is sensitized by ROS. At least in the left ventricle carvedilol, a β-blocker with antioxidative properties, was able to attenuate the hypertrophic response to anthracylines (Arozal et al., 2011). In rats with monocrotaline-induced pulmonary hypertension bucindolol treatment decreases RV necrosis, fibrosis, and infiltration of inflammatory cells and improves RV systolic function. In addition, bucindolol promotes a decrease in the cardiac sympathovagal balance by reducing sympathetic drive and increasing parasympathetic drive (Lima-Seolin et al., 2017). Changes in ROS were not measured. In a model of hypertension (two-kidney one-clip), β-blockers attenuated ROS and MMP2, a ROS-dependent regulated MMP, independent of its antioxidative property suggesting that direct stimulation of β-adrenoceptors increases ROS in ventricular tissue (Rizzi et al., 2014).
There are multiple reports that treatment regimes affecting the angiotensin-NOX-ROS axis attenuate hypertrophy and heart failure, but also few examples showing no effects (Table 1).
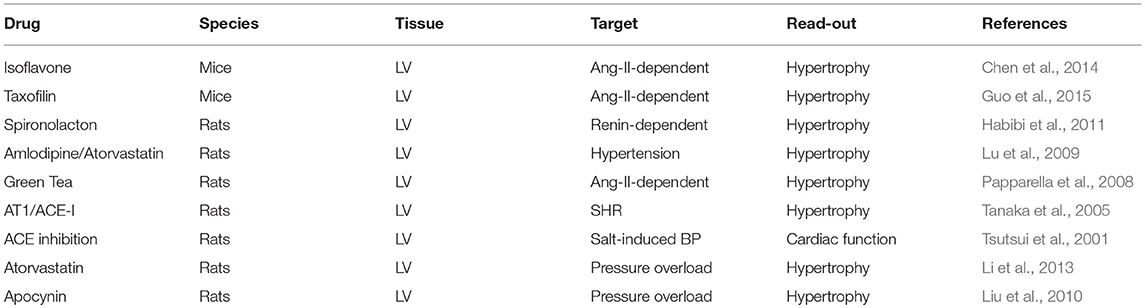
Table 1. Treatment of the angiotensin-NOX-ROS axis and effects on hypertrophy.
A coupling between ROS, cardiac hypertrophy and heart failure has been established for the LV. Concerning the RV only few data are available that directly analyzes right heart hypertrophy in the context of ROS signaling. As it stands there is consensus that RV tissue has a reduced oxidative defense capacity thereby favoring oxidative stress especially during the transition from RV hypertrophy to failure. Whether ROS targets in the RV include those proteins that are directly linked to cardiac growth is unclear and questionable. In contrast, oxidative modification of proteins leading to failure seems to be similar between both ventricles. Table 2 highlights the findings on ROS formation, defense, and targets in RV in comparison to LV.
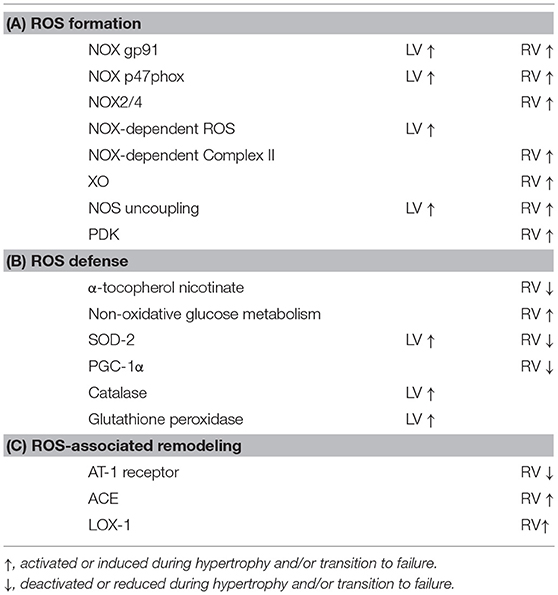
Table 2. Differences between LV and RV in ROS handling leading to hypertrophy and failure.
K-DS and RaS wrote the manuscript, performed literature search, and work on the conception. HK and CH provided data to Figure 3 and read and improved the manuscript. RoS read and added conceptional ideas and data to the chapter defense system.
K-DS and RaS received funding from the German Research Foundation (SFB/CRC 1213 B05).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Claudia Lorenz (Alcedis GmbH) for proofreading of our manuscript.
Abraham, W. T., Raynolds, M. V., Badesch, D. B., Wynne, K. M., Groves, B. M., Roden, R. L., et al. (2003). Angiotensin-converting enzyme DD genotype in patients with primary pulmonary hypertension: increased frequency and association with preserved haemodynamics. J. Renin. Angiot. Aldoster. Syst. 4, 27–30. doi: 10.3317/jraas.2003.003
Adesina, S. E., Wade, B. E., Bijli, K. M., Kang, B. Y., Williams, C. R., Ma, J., et al. (2017). Hypoxia inhibits expression and function of mitochondrial thioredoxin 2 to promote pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L599–L608. doi: 10.1152/ajplung.00258.2016
Alzoubi, A., Toba, M., Abe, K., O'Neill, K. D., Rocic, P., Fagan, K. A., et al. (2013). Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 304, H1708–H1718. doi: 10.1152/ajpheart.00746.2012
Ameri, P., Bertero, E., Meliota, G., Cheli, M., Canepa, M., Brunelli, C., et al. (2016). Neurohormonal activation and pharmacological inhibition in pulmonary arterial hypertension and related right ventricular failure. Heart Fail. Rev. 21, 539–547. doi: 10.1007/s10741-016-9566-3
Anghel, N., Herman, H., Balta, C., Rosu, M., Stan, M. S., Nita, D., et al. (2018). Acute cardiotoxicity induced by doxorubicin in right ventricle is associated with increase of oxidative stress and apoptosis in rats. Histol. Histopathol. 33, 365–378. doi: 10.14670/HH-11-932
Arozal, W., Sari, F. R., Watanabe, K., Arumugam, S., Veeraveedu, P. T., Ma, M., et al. (2011). Carvedilol-afforded protection against daunorubicin-induced cardiomyopathic rats in vivo: effects on cardiac fibrosis and hypertrophy. ISRN Pharmacol. 8:430549. doi: 10.5402/2011/430549
Aziz, A., Lee, A. M., Ufere, N. N., Damiano, R. J., Townsend, R. R., and Moon, M. R. (2015). Proteomic profiling of early chronic pulmonary hypertension: evidence for both adaptive and maladaptive pathology. J. Pulm. Respir. Med. 5:1000241. doi: 10.4172/2161-105X.1000241
Balestra, G. M., Mik, E. G., Eerbeek, O., Specht, P. A., van der Laarse, W. J., and Zuurbier, C. J. (2015). Increased in vivo mitochondrial oxygenation with right ventricular failure induced by pulmonary arterial hypertension: mitochondrial inhibition as driver of cardiac failure? Respir. Res. 16:6. doi: 10.1186/s12931-015-0178-6
Borchi, E., Bargelli, V., Stillitano, F., Giordano, C., Sebastiani, M., Nassi, P. A., et al. (2010). Enhanced ROS production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochim. Biophys. Acta 1802, 331–338. doi: 10.1016/j.bbadis.2009.10.014
Brittain, E. L., Talati, M., Fessel, J. P., Zhu, H., Penner, N., Calcutt, M. W., et al. (2016). Fatty acid metabolic defects and right ventricular lipotoxicity in human pulmonary arterial hypertension. Circulation 133, 1936–1944. doi: 10.1161/CIRCULATIONAHA.115.019351
Burger, D., Montezano, A. C., Nishgaki, N., He, Y., Carter, A., and Touyz, R. M. (2011). Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/Rho kinase pathways targeted to lipid rafts. Arterioscler. Thromb. Vasc. Biol. 31, 1898–1907. doi: 10.1161/ATVBAHA.110.222703
Byrne, J. A., Grieve, D. J., Bendall, J. K., Li, J. M., Gove, C., Lambeth, J. D., et al. (2003). Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II–induced cardiac hypertrophy. Circ. Res. 93, 802–804. doi: 10.1161/01.RES.0000099504.30207.F5
Cadenas, S. (2018). Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta 1859, 940–950. doi: 10.1016/j.bbabio.2018.05.019
Calamaras, T. D., Lee, C., Lan, F., Ido, Y., Siwik, D. A., and Colucci, W. S. (2015). Lipid peroxidation product 4-hydroxy-trans-2-nonenal (HNE) causes protein synthesis in cardiac myocytes via activated mTORC1-P70S6K-RPS6 signaling. Free Radic. Biol. Med. 82, 137–146. doi: 10.1016/j.freeradbiomed.2015.01.007
Canton, M., Menazza, S., Sheeran, F. L., Polverino de Laureto, P., Di Lisa, F., Pepe, S., et al. (2011). Oxidation of myofibrillar proteins in human heart failure. J. Am. Coll. Cardiol. 57, 300–309. doi: 10.1016/j.jacc.2010.06.058
Cau, S. B., Barato, R. C., Celes, M. R., Muniz, J. J., Rossi, M. A., and Tanus-Santos, J. E. (2013). Doxycycline prevents acute pulmonary embolism-induced mortality and right ventricular deformation in rats. Cardiovasc. Drugs Ther. 27, 259–267. doi: 10.1007/s10557-013-6458-9
Chai, C. Z., Mo, W. L., Zhuang, X. F., Kou, J. P., Yan, Y. Q., and Yu, B. Y. (2016). Protective effects of Sheng-Mai-San on right ventricular dysfunction during chronic intermittent hypoxia in mice. Evid. Based Complement. Alternat. Med. 2016:4682786. doi: 10.1155/2016/4682786
Chang, Y. M., Chang, H. H., Lin, H. J., Tsai, C. C., Tsai, C. T., Chang, H. N., et al. (2017). Inhibition of cardiac hypertrophy effects in D-galactose-induced senescent hearts by alpinate oxyphyllae fructus treatment. Evid. Based Complement. Alternat. Med. 2017:2624384. doi: 10.1155/2017/2624384
Chemaly, E. R., Hadri, L., Zhang, S., Kim, M., Kohlbrenner, E., Sheng, J., et al. (2011). Long-term in vivo resistin overexpression induces myocardial dysfunction and remodeling in rats. J. Mol. Cell. Cardiol. 51, 144–155. doi: 10.1016/j.yjmcc.2011.04.006
Chen, G., Pam, S. Q., Chen, C., Pan, S. F., Zhang, X. M., and He, Q. Y. (2014). Puerarin inhibits angiotensin II-induced cardiac hypertrophy via the redox-sensitive ERK1/2, p38 and NF-κB pathways. Acta Pharmacol. Sin. 35, 463–475. doi: 10.1038/aps.2013.185
Cheng, Y. S., Dai, D. Z., and Dai, Y. (2009). Isoproterenol disperses distribution of NADPH oxidase, MMP-9, and pPKCε in the heart, which are mitigated by endothelin receptor antagonist CPU0213. Acta Pharmacol. Sin. 30, 1099–1106. doi: 10.1038/aps.2009.104
Cingolani, O. H., Pérez, N. G., Ennis, I. L., Alvarez, M. C., Mosca, S. M., Schinella, G. R., et al. (2011). In vivo key role of reactive oxygen species and NHE-1 activation in determining excessive cardiac hypertrophy. Pflugers Arch. Eur. J. Physiol. 462, 733–743. doi: 10.1007/s00424-011-1020-8
Ciulla, M. M., Paliotti, R., Carini, M., Magrini, F., and Aldini, G. (2011). Fibrosis, enzymatic and non-enzymatic cross-links in hypertensive heart disease. Cardiovasc. Hematol. Disord. Drug Targets 11, 61–73. doi: 10.2174/187152911798347025
Cowley, P. M., Wang, G., Joshi, S., Swigart, P. M., Lovett, D. H., Simpson, P. C., et al. (2017). α1A-Subtype adrenergic agonist therapy for the failing right ventricle. Am. J. Physiol. Heart Circ. Physiol. 313, H1109–H1118. doi: 10.1152/ajpheart.00153.2017
Cruz, J. A., Bauer, E. M., Rodriguez, A. I., Gangopadhyay, A., Zeineh, N. S., Wang, Y., et al. (2012). Chronic hypoxia induces right heart failure in caveolin-1-/- mice. Am. J. Physiol. Heart Circ. Physiol. 302, H2518–H2527. doi: 10.1152/ajpheart.01140.2011
Danz, E. D., Skramsted, J., Henry, N., Bennett, J. A., and Keller, R. S. (2009). Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic. Biol. Med. 46, 1589–1597. doi: 10.1016/j.freeradbiomed.2009.03.011
Date, M. O., Morita, T., Yamashita, N., Nishida, K., Yamaguchi, O., Higuchi, Y., et al. (2002). The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model. J. Am. Coll. Cardiol. 39, 907–912. doi: 10.1016/S0735-1097(01)01826-5
Datta, A., Kim, G. A., Taylor, J. M., Gugino, S. F., Farrow, K. N., Schumacker, P. T., et al. (2015). Mouse lung development and NOX1 induction during hyperoxia are developmentally regulated and mitochondrial ROS dependent. Am. J. Physiol. Lung. Cell Mol. Physiol. 309, L369–L377. doi: 10.1152/ajplung.00176.2014
de Jong, J. W., Schoemaker, R. G., de Jonge, R., Bernocchi, P., Keijzer, E., Harrison, R., et al. (2000). Enhanced expression and activity of xanthine oxidoreductase in the failing heart. J. Mol. Cell. Cardiol. 32, 2083–2089. doi: 10.1006/jmcc.2000.1240
DeMarco, V. G., Habibi, J., Whaley-Connell, A. T., Schneider, R. I., Heller, R. L., Bosanquet, J. P., et al. (2008). Oxidative stress contributes to pulmonary hypertension in the transgenic (mRen2)27 rat. Am. J. Physiol. Heart Circ. Physiol. 294, H2659–H2668. doi: 10.1152/ajpheart.00953.2007
Dey, S., DeMazumder, D., Sidor, A., Foster, D. B., and O'Rourke, B. (2018). Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ. Res. 123, 356–371. doi: 10.1161/CIRCRESAHA.118.312708
Di Lisa, F., Giorgio, M., Ferdinandy, P., and Schulz, R. (2017). New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. Br. J. Pharmacol. 174, 1690–1703. doi: 10.1111/bph.13478
Dos Santos Lacerda, D., Türck, P., Gazzi de Lima-Seolin, B., Colombo, R., Duarte Ortiz, V., Poletto Bonetto, J. H., et al. (2017). Pterostilbene reduces oxidative stress, prevents hypertrophy and preserves systolic function of right ventricle in cor pulmonale model. Br. J. Pharmacol. 174, 3302–3314. doi: 10.1111/bph.13948
Duni, A., Liakopoulos, V., Rapsomanikis, K. P., and Dounousi, E. (2017). Chronic kidney disease and disproportionally increased cardiovascular damage: does oxidative stress explain the burden? Oxidat. Med. Cell. Longevity. 2017:9036450. doi: 10.1155/2017/9036450
Dunn, L. L., Buckle, A. M., Cooke, J. P., and Ng, M. K. (2010). The emerging role of the thioredoxin system in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 30, 2089–2098. doi: 10.1161/ATVBAHA.110.209643
Ecarnot-Laubriet, A., Rochette, L., Vergely, C., Sicard, P., and Teyssier, J. R. (2003). The activation pattern of the antioxidant enzymes in the right ventricle of rat in response to pressure overload is of heart failure type. Heart Dis. 5, 308–312. doi: 10.1097/01.hdx.0000089836.03515.a9
Egea, J., Fabregat, I., Frapart, Y. M., Ghezzi, P., Görlach, A., Kietzmann, T., et al. (2017). European contribution to the study of ROS: a summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 13, 94–162. doi: 10.1016/j.redox.2017.05.007
Frazziano, G., Al Ghouleh, I., Baust, J., Shiva, S., Champion, H. C., and Pagano, P. J. (2014). Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension: indications for a seminal role for mitochondrial Nox4. Am. J. Physiol. Heart Circ. Physiol. 306, H197–H205. doi: 10.1152/ajpheart.00977.2012
Freund-Michel, V., Khoyrattee, N., Savineau, J. P., Muller, B., and Guibert, C. (2014). Mitochondria: roles in pulmonary hypertension. Int. J. Biochem. Cell Biol. 55, 93–97. doi: 10.1016/j.biocel.2014.08.012
Galhotra, P., Prabhakar, P., Meghwani, H., Mohammed, S. A., Banerjee, S. K., Seth, S., et al. (2018). Beneficial effects of fenofibrate in pulmonary hypertension in rats. Mol. Cell. Biochem. 449, 185–194. doi: 10.1007/s11010-018-3355-3
Gladden, J. D., Zelickson, B. R., Guichard, J. L., Ahmed, M. I., Yancey, D. M., Ballinger, S., et al. (2013). Xanthine oxidase inhibition preserves left ventricular systolic but not diastolic function in cardiac volume overload. Am. J. Physiol. Heart Circ. Physiol. 305, H1440–H1450. doi: 10.1152/ajpheart.00007.2013
Gomez-Arroyo, J., Mizuno, S., Szczepanek, K., Van Tassell, B., Natarajan, R., dos Remedios, C. G., et al. (2013). Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ. Heart Fail. 6, 136–144. doi: 10.1161/CIRCHEARTFAILURE.111.966127
Grieve, D. J., Byrne, J. A., Siva, A., Layland, J., Johar, S., Cave, A. C., et al. (2006). Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J. Am. Coll. Cardiol. 47, 817–826. doi: 10.1016/j.jacc.2005.09.051
Guggilam, A., Haque, M., Kerut, E. K., McIlwain, E., Lucchesi, P., Seghal, I., et al. (2007). TNF-α blockade decreases oxidative stress in the paraventricular nucleus and attenuates sympathoexcitation in heart failure rats. Am. J. Physiol. Heart Circ. Physiol. 293, H599–H609. doi: 10.1152/ajpheart.00286.2007
Guido, M. C., Marques, A. F., Tavares, E. R., Tavares de Melo, M. D., Salemi, V. M. C., and Maranhão, R. C. (2017). The effects of diabetes induction on the rat heart: differences in oxidative stress, inflammatory cells, and fibrosis between subendocardial and interstitial myocardial areas. Oxidat. Med. Cell. Longevity 2017:5343972. doi: 10.1155/2017/534397
Guo, H., Zhang, X., Cui, Y., Zhou, H., Xu, D., Shan, T., et al. (2015). Taxifolin protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Toxicol. Appl. Pharmacol. 287, 168–177. doi: 10.1016/j.taap.2015.06.002
Gupto, A. A., Cordero-Reyes, A. M., Youker, K. A., Matsunami, R. K., Engler, D. A., Li, S., et al. (2016). Differential mitochondrial function in remodeled right and nonremodeled left ventricles in pulmonary hypertension. J. Card. Fail. 22, 73–81. doi: 10.1016/j.cardfail.2015.09.001
Habibi, J., DeMarco, V. G., Ma, L., Pulakat, L., Rainey, W. E., Whaley-Connell, A. T., et al. (2011). Mineralocorticoid receptor blockade improves diastolic function independent of blood pressure reduction in a transgenic model of RAAS overexpression. Am. J. Physiol. Heart Circ. Physiol. 300, H1484–H1491. doi: 10.1210/endo-meetings.2011.PART4.P2.P3-440
Hansen, T., Galougahi, K. K., Celermajer, D., Rasko, N., Tang, O., Bubb, K. J., et al. (2016). Oxidative and nitrosative signalling in pulmonary arterial hypertension — implications for development of novel therapies. Pharmacol. Therap. 165, 50–62. doi: 10.1016/j.pharmthera.2016.05.005
He, J., Li, X., Luo, H., Li, T., Zhao, L., Qi, Q., et al. (2017). Galectin-3 mediates the pulmonary arterial hypertension-induced right ventricular remodeling through interacting with NADPH oxidase 4. J. Am. Soc. Hypertens. 11, 275–289.e2. doi: 10.1016/j.jash.2017.03.008
Hernandez-Resendiz, S., Chinda, K., Ong, S. B., Cabrera-Fuentes, H., Zazueta, C., and Hausenloy, D. J. (2018). The role of redox dysregulation in the inflammatory response to acute myocardial ischaemia-reperfusion injury - adding fuel to the fire. Curr. Med. Chem. 25, 1275–1293. doi: 10.2174/0929867324666170329100619
Heusch, P., Aker, S., Boengler, K., Deindl, E., van de Sand, A., Klein, K., et al. (2010). Increased inducible nitric oxide synthase and arginase II expression in heart failure: no net nitrite/nitrate production and protein S-nitrosylation. Am. J. Physiol. Heart Circ. Physiol. 299, H446–H453. doi: 10.1152/ajpheart.01034.2009
Heusch, P., Canton, M., Aker, S., van de Sand, A., Konietzka, I., Rassaf, T., et al. (2010b). The contribution of reactive oxygen species and p38 mitogen-activated protein kinase to myofilament oxidation and progression of heart failure in rabbits. Br. J. Pharmacol. 160, 1408–1416. doi: 10.1111/j.1476-5381.2010.00793.x
Iacobazzi, D., Suleiman, M. S., Ghorbel, M., George, S. J., Caputo, M., and Tulloh, R. M. (2016). Cellular and molecular basis of RV hypertrophy in congential heart disease. Heart 102, 12–17. doi: 10.1136/heartjnl-2015-308348
Ide, T., Tsutsui, H., Hayashidani, S., Kang, D., Suematsu, N., Nakamura, K., et al. (2001). Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial Infarction. Circ. Res. 88, 529–535. doi: 10.1161/01.RES.88.5.529
Ishikawa, K., Kimura, S., Kobayashi, A., Sato, T., Matsumoto, H., Ujiie, Y., et al. (2005). Increased reactive oxygen species and anti-oxidative response in mitochondrial cardiomyopathy. Circ. J. 69, 617–620. doi: 10.1253/circj.69.617
Karamanlidis, G., Bautista-Hernandez, V., Fynn-Thompson, F., Del Nido, P., and Tian, R. (2011). Impaired mitochondrial biogenesis precedes heart failure in right ventricular hypertrophy in congenital heart disease. Circ. Heart Fail. 4, 707–713. doi: 10.1161/CIRCHEARTFAILURE.111.961474
Koga, K., Kenessey, A., and Ojamaa, K. (2013). Macrophage migration inhibitory factor antagonizes pressure overload-induced cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 304, H282–H293. doi: 10.1152/ajpheart.00595.2012
Li, J. M., Gall, N. P., Grieve, D. J., Chen, M., and Shah, A. M. (2002). Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40, 477–484. doi: 10.1161/01.HYP.0000032031.30374.32
Li, M., Sala, V., De Santis, M. C., Cimino, J., Cappello, P., Pianca, N., et al. (2018). Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation 138, 696–711. doi: 10.1161/CIRCULATIONAHA.117.030352
Li, R., Fang, W., Cao, S., Li, Y., Wang, J., Xi, S., et al. (2013). Differential expression of NAD(P)H oxidase isoforms and the effect of atorvastatin on cardiac remodeling in two-kidney two-clip hypertensive rats. Pharmazie 68, 261–269. doi: 10.1691/ph.2013.2782
Lima-Seolin, B. G., Colombo, R., Bonetto, J. H. P., Teixeira, R. B., Donatti, L. M., Casali, K. R., et al. (2017). Bucindolol improves right ventricle function in rats with pulmonary arterial hypertension through the reversal of autonomic imbalance. Eur. J. Pharmacol. 798, 57–65. doi: 10.1016/j.ejphar.2016.12.028
Liu, B., Luo, X. J., Yang, Z. B., Zhang, J. J., Li, T. B., Zhang, X. J., et al. (2014). Inhibition of NOX/VPO1 pathway and inflammatory reaction by trimethoxystilbene in prevention of cardiovascular remodeling in hypoxia-induced pulmonary hypertensive rats. J. Cardiovasc. Pharmacol. 63, 567–576. doi: 10.1097/FJC.0000000000000082
Liu, J., Zhou, J., An, W., Lin, Y., Yang, Y., and Zang, W. (2010). Apocynin attenuates pressure overload-induced cardiac hypertrophy in rats by reducing levels of reactive oxygen species. Can. J. Physiol.Pharmacol. 88, 745–752. doi: 10.1139/Y10-063
Liu, J. Q., Zelko, I. N., Erbynn, E. M., Sham, J. S., and Folz, R. J. (2006). Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L2–L10. doi: 10.1152/ajplung.00135.2005
Liu, L., Marcocci, L., Wong, C. M., Park, A. M., and Suzuki, Y. J (2008). Serotonin-mediated protein carbonylation in the right heart. Free Radic. Biol. Med. 45, 847–854. doi: 10.1016/j.freeradbiomed.2008.06.008
Lu, J. C., Cui, W., Zhang, H. L., Liu, F., Han, M., Liu, D. M., et al. (2009). Additive beneficial effects of amilodipine and atorvastatin in reversing advanced cardiac hypertophy in elderly spontaneously hypertensive rats. Clin. Exptl. Pharmacol. Physiol. 36, 1110–1119. doi: 10.1111/j.1440-1681.2009.05198.x
Lu, X., Dang, C. Q., Guo, X., Molloi, S., Wassall, C. D., Kemple, M. D., et al. (2011). Elevated oxidative stress and endothelial dysfunction in right coronary artery of right ventricular hypertrophy. J. Appl. Physiol. 110, 1674–1681. doi: 10.1152/japplphysiol.00744.2009
Lu, Z., Xu, X., Hu, X., Fassett, J., Zhu, G., Tao, Y., et al. (2010). PGC-1a regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antiox. Redox. Signal. 13, 1011–1022. doi: 10.1089/ars.2009.2940
Lüneburg, N., Siques, P., Brito, J., Arriaza, K., Pena, E., Klose, H., et al. (2016). Long-term chronic intermittent hypobaric hypoxia in rats causes an imbalance in the asymmetric dimethylarginine/nitric oxide pathway and ROS activity: a possible synergistic for altitude pulmonary hypertension? Pulm. Med. 2016:6578578. doi: 10.1155/2016/6578578
Ma, L., Ambalavanan, N., Liu, H., Sun, Y., Jhala, N., and Bradley, W. E. (2016). TLR4 regulates pulmonary vascular homeostasis and remodeling via redox signaling. Front. Biosci. 21, 397–409. doi: 10.2741/4396
Maarman, G. J., Schulz, R., Sliwa, K., Schermuly, R. T., and Lecour, S. (2017). Novel putative pharmacological therapies to protect the right ventricle in pulmonary hypertension: a review of current literature. Br. J. Pharmacol. 174, 497–511. doi: 10.1111/bph.13721
Mandraffino, G., Aragona, C. O., Cairo, V., Scuruchi, M., Lo Gullo, A., D'ascola, A., et al. (2017). Circulating progenitor cells in hypertensive subjects: effectiveness of a treatment with olmesartan in improving cell number and miR profile in addition to expected pharmacological effects. PLoS ONE 12:e0173030. doi: 10.1371/journal.pone.0173030
Manni, M. E., Rigacci, S., Borchi, E., Bargelli, V., Miceli, C., Giordano, C., et al. (2016). Monoamine oxidase is overactivated in left and right ventricles from ischemic hearts: an intriguing therapeutic target. Oxidat. Med. Cell. Longevity 2016:4375418. doi: 10.1155/2016/4375418
Matsuda, S., Umemoto, S., Yoshimura, K., Itoh, S., Murata, T., and Fukai, T. (2015). Angiotensin II activates MCP-1 and induces cardiac hypertrophy and dysfunction via Toll-like receptor 4. Atheroscler. Thromb. 26, 833–844. doi: 10.5551/jat.27292
Moris, D., Spartalis, M., Tzatzaki, E., Spartalis, E., Karachaliou, G. S., Triantafyllis, A. S., et al. (2017). The role of reactive oxygen species in myocardial redox signaling and regulation. Ann. Transl. Med. 5:324. doi: 10.21037/atm.2017.06.17
Mouli, S., Nanayakkara, G., AlAlasmari, A., Eldoumani, H., Fu, X., Berlin, A., et al. (2015). The role of frataxin in doxorubicin-mediated cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 309, H844–H859. doi: 10.1152/ajpheart.00182.2015
Nagendran, J., Gurtu, V., Fu, D. Z., Dyck, J. R., Haromy, A., Ross, D. B., et al. (2008). A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J. Thorac. Cardiovasc. Surg. 136, 168–178. doi: 10.1016/j.jtcvs.2008.01.040
Nediani, C., Borchi, E., Giordano, C., Baruzzo, S., Ponziani, V., Sebastiani, M., et al. (2007). NADPH oxidase-dependent redox signaling in human heart failure: relationship between the left and right ventricle. J. Mol. Cell. Cardiol. 42, 826–834. doi: 10.1016/j.yjmcc.2007.01.009
Nisbet, R. E., Graves, A. S., Kleinhenz, D. J., Rupnow, H. L., Reed, A. L., Fan, T. H., et al. (2009). The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am. J. Respir. Cell Mol. Biol. 40, 601–609. doi: 10.1165/2008-0145OC
Ogura, S., Shimosawa, T., Mu, S., Sonobe, T., Kawakami-Mori, F., Wang, H., et al. (2013). Oxidative stress augments pulmonary hypertension in chronically hypoxic mice overexpressing the oxidized LDL receptor. Am. J. Physiol. Heart Circ. Physiol. 305, H155–H162. doi: 10.1152/ajpheart.00169.2012
Papparella, I., Ceolotto, G., Montemurro, D., Antonello, M., Garbisa, S., Rossi, G. P., et al. (2008). Green tea attenuates angiotensin II-induced cardiac hypertrophy in rats by modulating reactive oxygen species production and the Src/Epidermal growth factor receptor/Act signalling pathway. J. Nutr. 138, 1596–1601. doi: 10.1093/jn/138.9.1596
Paulin, R., and Michelakis, E. D. (2014). The metabolic theory of pulmonary arterial hypertension. Circ Res. 115, 148–164. doi: 10.1161/CIRCRESAHA.115.301130
Phillips, D., Aponte, A. M., Covian, R., Neufeld, E., Yu, Z. X., and Balaban, R. S. (2011). Homogenous protein programming in the mammalian left and right ventricle free walls. Physiol. Genom. 43, 1198–1206. doi: 10.1152/physiolgenomics.00121.2011
Pichardo, J., Palace, V., Farahmand, F., and Singal, P. K. (1999). Myocardial oxidative stress changes during compensated right heart failure in rats. Mol. Cell. Biochem. 196, 51–57. doi: 10.1023/a:1006914111957
Pönicke, K., Schlüter, K. D., Heinroth-Hoffmann, I., Seyfarth, T., Goldberg, M., Piper, H. M., et al. (2001). Naunyn Schmiedebergs. Arch. Pharmacol. 364, 444–453. doi: 10.1007/s002100100469
Puukila, S., Fernandes, R. O., Türck, P., Carraro, C. C., Bonetto, J. H. P., de Lima-Seolin, B. G., et al. (2017). Secoisolariciresinol diglucoside attenuates cardiac hypertrophy and oxidative stress in monocrotaline-induced right heart dysfunction. Mol. Cell. Biochem. 432, 33–39. doi: 10.1007/s11010-017-2995-z
Qiao, W., Zhang, W., Gai, Y., Zhao, L., and Fan, J. (2014). The histone acetyltransferase MOF overexpression blunts cardiac hypertrophy by targeting ROS in mice. Biochem. Biophys. Res. Commun. 448, 379–384. doi: 10.1016/j.bbrc.2014.04.112
Qin, F., Siwik, D. A., Pimentel, D. R., Morgan, R. J., Biolo, A., Tu, V. H., et al. (2017). Cytosolic H2O2 mediates hypertrophy, apoptosis, and decreased SERCA activity in mice with chronic hemodynamic overload. Am. J. Physiol. Heart Circ. Physiol. 306, H1453–H1463. doi: 10.1152/ajpheart.00084.2014
Qipshidze, N., Tyagi, N., Metreveli, N., Lominadze, D., and Tyagi, S. C. (2012). Autophagy mechanism of right ventricular remodeling in murine model of pulmonary artery constriction. Am. J. Physiol. Heart Circ. Physiol. 302, H688–H696. doi: 10.1152/ajpheart.00777.2011
Rassaf, T., Poll, L. W., Brouzos, P., Lauer, T., Totzeck, M., Kleinbongard, P., et al. (2006). Positive effects of nitric oxide on left ventricular function in humans. Eur. Heart J. 27, 1699–1705. doi: 10.1093/eurheartj/ehl096
Rawat, D. K., Alzoubi, A., Gupte, R., Chettimada, S., Watanabe, M., Kahn, A. G., et al. (2014). Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 64, 1266–1274. doi: 10.1161/HYPERTENSIONAHA.114.03261
Reddy, S., and Bernstein, D. (2015). Molecular mechanisms of right ventricular failure. Circulation 132, 1734–1742. doi: 10.1161/CIRCULATIONAHA.114.012975
Redout, E. M., van der Toorn, A., Zuidwijk, M. J., van de Kolk, C. W. A., van Echteld, C. J. A., Musters, R. J. P., et al. (2010). Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 298, H1038–H1047. doi: 10.1152/ajpheart.00097.2009
Redout, E. M., Wagner, M. J., Zuidwijk, M. J., Boer, C., Musters, R. J. P., van Hardeveld, C., et al. (2007). Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res. 75, 770–781. doi: 10.1016/j.cardiores.2007.05.012
Redza-Dutordoir, M., and Averill-Bates, D. A. (2016). Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 1863, 2977–2992. doi: 10.1016/j.bbamcr.2016.09.012
Riba, A., Deres, L., Sumegi, B., Toth, K., Szabados, E., and Halmosi, R. (2017). Cardioprotective effect of resveratrol in a postinfarction heart failure model. Oxidative Med. Cell. Longevity 2017:6819281. doi: 10.1155/2017/6819281
Rizzi, E., Guimaraes, D. A., Ceron, C. S., Prado, C. M., Pinheiro, L. C., Martins-Oliveira, A., et al. (2014). β1-Adrenergic blockers exert antioxidant effects, reduce matrix metalloproteinase activity, and improve renovascular hypertension-induced cardiac hypertrophy. Free Radical. Biol. Med. 73, 308–317. doi: 10.1016/j.freeradbiomed.2014.05.024
Schäfer, M., Schäfer, C., Piper, H. M., and Schlüter, K. D. (2002). Hypertrophic responsiveness of cardiomyocytes to α- and β-adrenoceptor stimulation requires sodium-proton-exchanger-1 (NHE-1) activation but not cellular alkalization. Eur. J. Heart Fail. 4, 249–254. doi: 10.1016/S1388-9842(02)00016-8
Schlüter, K. D. (ed.). (2016). “Ways to study the biology of cardiomyocytes,” in Cardiomyocytes – Active Players in Cardiac Disease (Cham: Springer International Publishing), 3–23. doi: 10.1007/978-3-319-31251-4_1
Schreckenberg, R., Rebelo, M., Deten, A., Weber, M., Rohrbach, S., Pipicz, M., et al. (2015). Specific mechanisms underlying right heart failure: the missing upregulation of superoxide dismutase-2 and its decisive role in antioxidative defense. Antioxdi. Redox Signal. 23, 1220–1232. doi: 10.1089/ars.2014.6139
Shimizu, S., Ishibashi, M., Kumagai, S., Wajima, T., Hiroi, T., Kurihara, T., et al. (2013). Decreased cardiac mitochondrial tetrahydrobiopterin in a rat model of pressure overload. Int. J. Mol. Med. 31, 589–596. doi: 10.3892/ijmm.2013.1236
Simm, A., Schlüter, K. D., Diez, C., Piper, H. M., and Hoppe, J. (1998). Activation of p70S6 kinase by β-adrenoceptor agonists on adult cardiomyocytes. J. Mol. Cell. Cardiol. 30, 2059–2067. doi: 10.1006/jmcc.1998.0768
Siqueira, R., Colombo, R., Conzatti, A., de Castro, A. L., Carraro, C. C., Tavares, A. M. V., et al. (2018). Effects of ovariectomy on antioxidant defence systems in the right ventricle of female rats with pulmonary arterial hypertension induced by monocrotaline. Can. J. Physiol. Pharmacol. 96, 295–303. doi: 10.1139/cjpp-2016-0445
Sirker, A., Murdoch, C. E., Protti, A., Sawyer, G. J., Santos, C. X. C., Martin, D., et al. (2016). Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J. Mol. Cell. Cardiol. 98, 11–17. doi: 10.1016/j.yjmcc.2016.07.003
Sundaresan, N. R., Gupta, M., Kim, G., Rajamohan, S. B., Isbatan, A., and Gupta, M. P. (2009). Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2771. doi: 10.1172/JCI39162
Sutendra, G., and Michelakis, E. D. (2014). The metabolic basis of pulmonary arterial hypertension. Cell Metab. 19, 558–573. doi: 10.1016/j.cmet.2014.01.004
Sverdlov, A. L., Elezaby, A., Qin, F., Behring, J. B., Luptak, I., Calamaras, T. D., et al. (2016). Mitochondrial reactive oxygen species mediate cardiac structural, functional, and mitochondrial consequences of diet-induced metabolic heart disease. J. Am. Heart. Assoc. 5:e002555. doi: 10.1161/JAHA.115.002555
Takimoto, E., Champion, H. C., Li, M., Ren, S., Rodriguez, E. R., Tavazzi, B., et al. (2005). Oxidant stress from nitric oxide synthase−3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Invest. 115, 1221–1231. doi: 10.1172/JCI21968
Talukder, M. A., Johnson, W. M., Varadharaj, S., Lian, J., Kearns, P. N., El-Mahdy, M. A., et al. (2011). Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. Am. J. Physiol. Heart Circ. Physiol. 300, H388–H396. doi: 10.1152/ajpheart.00868.2010
Tanaka, M., Umemoto, S., Kawahara, S., Kubo, M., Itoh, S., Umeji, K., et al. (2005). Angiotensin II type 1 receptor antagonist and angiotensin-converting enzyme inhibitor altered the activation of Cu/Zn-containing superoxide dismutase in the heart of stroke-prone spontaneously hypertensive rats. Hypertens. Res. 28, 67–77. doi: 10.1291/hypres.28.67
Tsutsui, H., Ide, T., Hayashidami, S., Kinugawa, S., Suematsu, N., Utsumi, H., et al. (2001). Effects of ACE inhibition on left ventricular failure and oxidative stress in Dahl salt-sensitive rats. J. Cardiovasc. Pharmacol. 37, 725–733. doi: 10.1097/00005344-200106000-00010
Türck, P., Lacerda, D. S., Carraro, C. C., de Lima-Seolin, B. G., Teixeira, R. B., Poletto Bonetto, J. H., et al. (2018). Trapidil improves hemodynamic, echocardiographic and redox state parameters of right ventricle in monocrotaline-induced pulmonary arterial hypertension model. Biomed. Pharmacother. 103, 182–190. doi: 10.1016/j.biopha.2018.04.001
Umemoto, S., Tanaka, M., Kawahara, S., Kubo, M., Umeji, K., Hashimoto, R., et al. (2004). Calcium antagonist reduces oxidative stress by upregulating Cu/Zn superoxide dismutase in stroke-prone spontaneously hypertensive rats. Hypertens. Res. 27, 877–885. doi: 10.1291/hypres.27.877
Wang, L., Lopaschuk, G. D., and Clanachan, A. S. (2008). H2O2-induced left ventricular dysfunction in isolated working rat hearts is independent of calcium accumulation. J. Mol. Cell. Cardiol. 45, 787–795. doi: 10.1016/j.yjmcc.2008.08.010
Wang, X., Liu, J., Drummond, C. A., and Shapiro, J. I. (2017a). Sodium potassium adenosine triphosphatase (Na/K-ATPase) as a therapeutic target for uremic cardiomyopathy. Exp. Opin. Ther. Targets 21, 531–541. doi: 10.1080/14728222.2017.1311864
Wang, X., Shults, N. V., and Suzuki, Y. J. (2017). Oxidative profiling of the failing right heart in rats with pulmonary hypertension. PLoS ONE 12:e0176887. doi: 10.1371/journal.pone.0176887
Waypa, G. B., Osborne, S. W., Marks, J. D., Berkelhamer, S. K., Kondapalli, J., and Schumacker, P. T. (2013). Sirtuin 3 deficiency does not augment hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 49, 885–891. doi: 10.1165/rcmb.2013-0191OC
Wenzel, S., Abdallah, Y., Helmig, S., Schäfer, C., Piper, H. M., and Schlüter, K.-D. (2006). Contribution of PI 3-kinase isoforms to angiotzensin II- and α-adrenoceptor-mediated signaling pathways in cardiomyocytes. Cardiovasc. Res. 71, 352–362. doi: 10.1016/j.cardiores.2006.02.004
Wenzel, S., Rohde, C., Wingerning, S., Roth, J., Kojda, G., and Schlüter, K. D. (2007). Lack of endothelial nitric oxide synthase-derived nitric oxide formation favors hypertrophy in adult ventricular cardiomyocytes. Hypertension 49, 193–200. doi: 10.1161/01.HYP.0000250468.02084.ce
Wenzel, S., Taimor, G., Piper, H. M., and Schlüter, K. D. (2001). Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-β expression in adult ventricular cardiomyocytes. FASEB J. 15, 2291–2293. doi: 10.1096/fj.00-0827fje
Wong, C. M., Bansal, G., Pavlickova, L., Marocci, L., and Suzuki, Y. J. (2013). Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid. Redox Signal. 18, 1789–1796. doi: 10.1089/ars.2012.4568
Wong, Y. Y., Ruiter, G., Lubberink, M., Raijmakers, P. G., Knaapen, P., Marcus, J. T., et al. (2011). Right ventricular failure in idiopathic pulmonary arterial hypertension is associated with inefficient myocardial oxygen utilization. Circ. Heart Fail. 4, 700–706. doi: 10.1161/CIRCHEARTFAILURE.111.962381
Xu, Q., Dalic, A., Fang, L., Kiriazis, H., Ritchie, R. H., Sim, K., et al. (2011). Myocardial oxidative stress contributes to transgenic β2-adrenoceptor activation induced cardiomyopathy and heart failure. Br. J. Pharmacol. 162, 1012–1028. doi: 10.1111/j.1476-5381.2010.01043.x
Ye, J. X., Wang, S. S., Ge, M., and Wang, D. J. (2016). Suppression of endothelial PGC-1α is associated with hypoxia-induced endothelial dysfunction and provides a new therapeutic target in pulmonary arterial hypertension. Am. J. Physiol. Lung. Cell Mol. Physiol. 310, L1233–L1242. doi: 10.1152/ajplung.00356.2015
Zhu, T. T., Zhang, W. F., Luo, P., Qian, Z. X., Li, F., Zhang, Z., et al. (2017). LOX-1 promotes right ventricular hypertrophy in hypoxia-exposed rats. Life Sci. 174, 35–42. doi: 10.1016/j.lfs.2017.02.016
Keywords: cardiac remodeling, heart failure, oxidative stress, pulmonary hypertension, uncoupling protein, MAO
Citation: Schlüter K-D, Kutsche HS, Hirschhäuser C, Schreckenberg R and Schulz R (2018) Review on Chamber-Specific Differences in Right and Left Heart Reactive Oxygen Species Handling. Front. Physiol. 9:1799. doi: 10.3389/fphys.2018.01799
Received: 06 September 2018; Accepted: 29 November 2018;
Published: 17 December 2018.
Edited by:
Thao P. Nguyen, UCLA David Geffen School of Medicine, United StatesReviewed by:
Marcos Lopez, University of Chicago, United StatesCopyright © 2018 Schlüter, Kutsche, Hirschhäuser, Schreckenberg and Schulz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaus-Dieter Schlüter, a2xhdXMtZGlldGVyLnNjaGx1ZXRlckBwaHlzaW9sb2dpZS5tZWQudW5pLWdpZXNzZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.