
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 15 October 2018
Sec. Craniofacial Biology and Dental Research
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01426
 Natalina Quarto1,2*
Natalina Quarto1,2* Siny Shailendra1
Siny Shailendra1 Nathaniel P. Meyer1
Nathaniel P. Meyer1 Siddharth Menon1Andrea Renda2Michael T. Longaker1*
Siddharth Menon1Andrea Renda2Michael T. Longaker1*Craniofacial development is a program exquisitely orchestrated by tissue contributions and regulation of genes expression. The basic helix–loop–helix (bHLH) transcription factor Twist1 expressed in the skeletal mesenchyme is a key regulator of craniofacial development playing an important role during osteoskeletogenesis. This study investigates the postnatal impact of Twist1 haploinsufficiency on the osteoskeletal ability and regeneration on two calvarial bones arising from tissues of different embryonic origin: the neural crest-derived frontal and the mesoderm-derived parietal bones. We show that Twist1 haplonsufficiency as well Twist1-sh-mediated silencing selectively enhanced osteogenic and tissue regeneration ability of mesoderm-derived bones. Transcriptomic profiling, gain-and loss-of-function experiments revealed that Twist1 haplonsufficiency triggers its selective activity on mesoderm-derived bone through a sharp downregulation of the bone-derived hormone Fgf23 that is upregulated exclusively in wild-type parietal bone.
The mammalian skull vault is a structure formed by five intramembranous flat bones: the paired frontal and parietal bones and the unpaired interparietal bone (Morriss-Kay, 2001). The adjacent osteogenic fronts of these intramembranous bones form the sutures, which accommodate the growth of skull vault and underlying brain. The morphogenesis of the skull vault is accomplished in two phases: the first is defined by the genesis, migration and early specification of skeletogenic precursor cells. The second the differentiation of the skeletogenic mesenchymal precursor cells and subsequent appositional growth of the bones. Calvarial bones arise from two embryonic tissues, namely, the neural crest and the mesoderm. The mixed developmental origin of the mammalian skull vault has been clearly defined by employing a Wnt1-Cre/conditional LacZ reporter R26R double transgene mouse tracing exclusively neural crest cells, and showing that the frontal and squamosal bones are neural crest-derived, while the parietal bone is of mesoderm-derived (Jiang et al., 2002). Later, Yoshida et al. (2008) elegantly validated the same dual embryonic origin of the frontal and parietal bones, by performing the reciprocal study using the Mesp1-Cre transgene combined with R26R, which uniquely identified mesodermal cells.
Recently, important progress has been accomplished in our understanding of how enhanced activation of specific signaling pathways contributes to differences in the osteogenic capacity and bone repair observed between neural crest-derived and mesoderm-derived calvarial bones. Our previous investigations unveiled that disparate embryonic tissue origin translates into regional differences in osteogenic and regenerative ability of parietal and frontal bones. Furthermore, our findings revealed that skeletal differences observed between the two calvarial bones are controlled by the integration of multiple and converging osteogenic signaling pathways differentially activated between frontal and parietal bones (Senarath-Yapa et al., 2013). We identified an enhanced activation of endogenous FGF, BMP and canonical Wnt (cWnt) signaling exclusively in the neural crest-derived frontal bone (Quarto et al., 2009a,b; Li et al., 2015), whereas enhanced activation of TGF-β signaling is found in the mesoderm-derived parietal bone (Li et al., 2013; Senarath-Yapa et al., 2016).
During the development of skeleton and bone tissue ossification, multiple morphogenetic growth factor and hormone signaling pathways impact transcriptional regulators to trigger the osteogenic phenotype (Karsenty, 2008; Karsenty et al., 2009; Karsenty and Wagner, 2002). Twist1 is an evolutionarily highly conserved transcription factor (Murre et al., 1989; Miraoui and Marie, 2010) originally identified in Drosophila (Thisse et al., 1987; Leptin, 1991). This transcription factor plays extensive roles in skeletogenesis, directing embryonic skeletal patterning, fetal skeletal development, and bone remodeling (Howard et al., 1997; Rice et al., 2000; Yousfi et al., 2001; O’Rourke and Tam, 2002; Bialek et al., 2004; Marie et al., 2008; Wu et al., 2008; Bildsoe et al., 2009). Studies in Drosophila development showed that Twist is expressed in mesodermal and cranial neural crest (CNC) cells (Thisse et al., 1988) and plays a key-role in mesoderm-specification and myogenesis (Thisse et al., 1987; Leptin, 1991; Baylies and Bate, 1996; Cripps and Olson, 1998). Moreover, the expression pattern of Twist1 in mouse suggests that this transcription factor regulates genes controlling the specification and the differentiation of cranial mesenchyme (Bildsoe et al., 2009). Twist1 is also found expressed in vivo in mesoderm-derived osteoblast progenitors as well as in vitro in calvaria murine osteoprogenitors (Murray et al., 1992) suggesting that Twist1 may play a key-regulator role during osteoblast differentiation. In humans heterozygous loss of TWIST1 function causes the Saethre–Chotzen syndrome. Individuals affected by this genetic condition have premature fusion of coronal suture, known as craniosynostosis. (Howard et al., 1997; Marie et al., 2008). Like humans, Twist1 mutant mice also are affected by coronal suture craniosynostosis (el Ghouzzi et al., 1997; Carver et al., 2002).
The current study focuses on the role of transcription factor Twist1 and its interplay with osteoskeletal phenotype of calvarial bones of different embryonic tissue origin. Our results indicate that Twist1 haploinsufficiency and/or Twist1 shRNA silencing enhanced the osteoskeletal phenotype of mesoderm-derived parietal bone and osteoblasts. This selective osteoskeletal induction triggered by Twist1 haploinsufficiency is mediated by a significant downregulation of Fgf23, a crucial player in balancing homeostasis of bone mineralization (Shimada et al., 2001; Larsson et al., 2004; Shimada et al., 2004). Our findings highlight new functional aspects of Twist1 haploinsufficiency in the context of calvarial bone skeletogenesis/regeneration and point to Fgf23 as putative target of Twist1 activity.
Experiments using animals were carried out following Stanford University Animal Care and Use Committee guidelines. All research has been approved by Stanford APLAC, with a number of protocol #9999, following approved guidelines by the Stanford University’s Institutional Review Board. Twist1+/− mice were purchased from Jackson Laboratory (Bar Harbor, ME, United States). Genotyping was performed as previously described (Chen and Behringer, 1995; Behr et al., 2011a) CD-1 mice were from (Charles Rivers Laboratories, Inc., Wilmington, MA, United States).
Frontal and parietal calvarial bones were harvested from postnatal day 21 (pN21) Twist1 haploinsufficient and wild-type mice (from the same litter) using a dissecting microscope. After meticulous removal of periosteum and dura mater from the calvaria skull, frontal and parietal bones were dissected free from surrounding sutures. Bones were then mechanically minced and digested using 0.2% dispase II and 0.1% collagenase A (Roche Diagnostics, Indianapolis, IN, United States) in serum-free medium in a shaker water bath at 37°C. Digestion was carried out six times (10 min each) as previously described (Quarto et al., 2009b; Li et al., 2013). The first two digestions were discarded. The last four digestions were pooled, centrifuged, and resuspended in non-inductive growth media α-minimal essential medium (α-MEM)-GlutaMax supplemented with 10% FBS and 100 IU/ml Penicillin/Streptomycin (GIBCO, Invitrogen, Carlsbad, CA, United States) as previously reported (Quarto et al., 2009b).
For the osteogenic assay osteoblasts were seeded in six-well plates at 5 × 105 per well. After 24 h, cells were cultured in inductive osteogenic medium (day 0) comprising of α-MEM-GlutaMax supplemented with 10% FBS, 100 IU/ml penicillin, 100 IU/ml streptomycin, 10 mMβ-glycerophosphate, and 100 μg/ml ascorbic acid (Sigma-Aldrich, St. Louis, MO, United States). Experiments were carried out using only first and second passage cells. Osteoblasts were collected at different time points (d0, d3, d10, and d28) and RNA isolated for the analysis of osteogenic markers. Extracellular matrix mineralization and bone nodules formation were analyzed by Alizarin red and/or von Kossa staining procedures as described previously (Quarto et al., 2009b, 2015).
Total RNA was isolated either from osteoblasts or frontal and parietal bones using TRIzol method according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA, United States). Upon DNAse I (Ambion; Austin, TX, United States) treatment to clear genomic DNA, RNA was reverse transcribed using a SuperScript III First-Strand kit (Invitrogen, Carlsbad, CA, United States) as previously described (Quarto et al., 2008, 2009b). Osteogenic gene expression profile was examined by RT-qPCR. The relative mRNA level in each sample was normalized to its Gapdh content. Values are provided as relative to Gapdh expression. The results are presented as means ± SD of triplicate.
Primers sequence are as follows: mTwist1-Forward: 5′-TTCAGACCCTCAAACTGGCG-3′; mTwist1-Reverse: 5′-CTGGGAATCTCTGTCCACGG-3′; mFgf23Forward: 5′-CGAGGAAAGAGTTCACGCCT-3′; mFgf23-Reverse: GCGTCTTTCTTCGACTTGCC; mOsx-Forward: 5′-AGCGACCACTTGAGCAAACAT-3′; mOsx-Reverse; 5′-GCGGCTGATTGGCTTCTTCT-3′. Annealing temperature at 58°C. Primers sequence for Bglap (osteocalcin) and Gapdh and PCR conditions were reported previously (Quarto et al., 2009b). The results are presented as means ± SD of triplicate.
Animal experiments were performed in accordance with Stanford University Animal Care and Use Committee guidelines. Briefly, after anesthesia with an intraperitoneal injection of ketamine 100 mg/kg + xylazine 20 mg/kg + acepromazine 3 mg/kg and disinfection of the surgical site of the mice, an incision was made laterally to the sagittal midline to expose the frontal and parietal bones. The pericranium was removed using a sterile cotton swab. Using diamond coated trephine bits and saline irrigation, bilateral full-thickness calvarial defects (2-mm in diameter) were created in the non-suture associated right frontal and left parietal bones of pN21 mice (wild-type = 5 mice, Twist1+/− n = 5 mice). Meticulous care was taken in order to protect the underlying dura mater or neighboring cranial sutures (Quarto et al., 2009b; Behr et al., 2011b, 2012).
The rate of healing μCT-scanning was performed as previously described (Behr et al., 2011b, 2012). Briefly, mice were scanned with a high-resolution MicroCAT II scanner (ImTek Inc., Knoxville, TN, United States) with an X-ray voltage of 80 kVP and an anode current of 450 μA. X-ray data reconstruction was performed with Cobra EXXIM (EXXIM Computing Corp., Livermore, CA, United States), and Micro View Software (GE Healthcare, Chicago, IL, United States). Each mouse was scanned with a CT-phantom, which is used to calibrate each scan. The precise threshold for regenerating calvarial bone was determined equivalent to 510 Houndsfield Units. The rest-defect areas were then determined with the Magic Wand Tool in Photoshop (Adobe Systems, San Jose, CA, United States). The area of the calvarial defects was evaluated by quantifying pixels in the defect. Percentage healing was then determined by dividing the rest defect area by the mean of defect size 1 day postoperatively. Mice were scanned 24 h post-surgery (time 0) and at week 1 2, 3, 4, 5, 6, and 7. For statistical analysis was used the Mann–Whitney test. A ∗p-value < 0.05 was considered statistically significant. For determining bone mineral density (BMD), standardized regions in the uninjured frontal and parietal bones were chosen and analyzed with the BMD tool in MicroView. The threshold range was set between 900 and 3500 voxels. The software automatically performed data analysis and calculations. Measurements were performed on four mice for each strain.
Osteoblasts (pN21) were plated onto 12-well plates to 50% confluency 1 day before transduction. Cells were transduced with either Fgf23shRNA lentiviral particles (sc-39487-V) or Twist1shRNA lentiviral particles (sc-38605-V) Santa Cruz Biotechnology, Santa Cruz, CA, United States) and shRNA (scramble) lentiviral particles-A (sc-108080) was used as control (Santa Cruz Biotechnology, Santa Cruz, CA, United States). Transductions were performed according to the manufacturer’s protocol and as previously reported (Quarto et al., 2015). The efficiency of transduction was monitored 48 h after co-transduction using copGFP Control Lentiviral Particles (sc-108084), Santa Cruz Biotechnology, Santa Cruz, CA, United States) and found to be 85–87%. Two days later, the transduced cells underwent to puromycin selection (5 μg/mL) for 5 days. Efficiency of Fgf23 and/or Twist1 knockdown was assessed either at gene expression level by RT-PCR analysis or at protein level by immunoblotting analysis. Transduced cell were cultured in presence of puromicin thoroughly all the time of experiments.
Immunoblotting analysis was performed using the following primary rabbit antibodies: rabbit anti-Twist1 (H-8: sc-15393) (dilution 1:200, Santa Cruz Biotechnology, Santa Cruz, CA, United States), rat monoclonal anti-FGF23 (MAB2629) (dilution 1:200, R&D System, Minneapolis, MN, United States) and anti-β-Actin (ab8227) (dilution 1:5,000; Abcam, Cambridge, MA, United States). Cell lysate proteins (40–80 μg) were resolved by NuPAGE 4–12% bis-Tris-HCl sodium dodecyl sulfate-polyacrylamide gel (Novex, Life Technologies, Carlsbad, CA, United States). Proteins were transferred to a polyvinylidene fluoride membrane (Bio-Rad, Inc., Hercules, CA, United States) and probed with specific antibody. Horseradish peroxidase-conjugated secondary anti-rabbit (7074S) and anti-rat antibodies (7077S) (dilution 1:2000; Cell Signaling, Danvers, MA, United States) were used. Immunoblotted proteins were visualized by enhanced chemiluminescence (Amersham Biosciences, Buckinghamshire, United Kingdom).
Cell-media were collected from subconfluent cells cultured for 48 h in growth medium supplemented with 2% FCS. Media were then concentrated 50-fold using Centricon filters (3000 NMWL, Millipore Corporation, Billerica, MA, United States). Collection and concentration of the media were carried out at 4°C. The volume of each conditioned medium was normalized by cell numbers. Protein concentration was determined by BCA protein assay (Pierce Biotechnology, Rockford, IL, United States). Media were analyzed for FGF23 concentrations by enzyme-linked immunosorbent assay (ELISA) using Quantakine mouse FGF23 kit (R&D Systems, Minneapolis, MN, United States), according to the manufacturer’s instructions. Photometric detection was done with an ELISA reader at 370-nm wavelength. Each sample was run in triplicate. The results are the mean ± SD of three independent experiments.
RNA-seq was performed by the Stanford Functional Genomics Facility (SFGF) core at Stanford University1. Total RNAs isolated from wild-type and Twist1+/− frontal and parietal bones (mice = 12/each strain) were purified using a Qiagen MiRNeasy Kit (Cat # 217004, Qiagen, Inc, Valencia, CA, United States), according to the manufacture’s instructions. Libraries were constructed using KAPA Stranded mRNA-Seq Kit (Cat # KK8420, Kapa Biosystem, Wilmington, MA, United States) following the manufacture’s instructions. Briefly, magnetic oligo-dt beads were used for poly (A) capture, followed by first strand synthesis using random primers, second strand synthesis, converting cDNA:RNA hybrid to double-stranded cDNA and incorporating dUTP into the second cDNA strand. Subsequently, A-tailing to add dAMP to the 3′-ends of the dscDNA library fragments was performed. An adapter ligation was carried out, where dsDNA adapters with 3′dTMP overhangs were ligated to the A-tailed library insert fragments. Library amplification was then performed to amplify library fragments carrying appropriate adapter sequences at both ends using high-fidelity, low-bias PCR. Amplified libraries were run on bio-analyzer to determine the library fragment size distribution and to detect the presence of excessive adapter dimer molecules using the High Sensitivity DNA Assay (Cat # 5067-4626 Agilent Technologies, Santa Clara, CA, United States). The quantified libraries were then pooled at equal nanomoles of individual libraries. After final QC the pooled libraries were sequenced on Illumina Next-Seq (Illumina, San Diego, CA, United States).
FastQC 0.11.22 from the Babraham Institute was used to perform quality control checks on raw sequence data. The mouse reference genome (mm9, NCBI Build 37) was obtained from the UCSC Genome Browser download site. The corresponding reference annotations (GTF) were obtained from the iGenomes site hosted by Illumina. The splice-aware aligner STAR [version 2.4.2a, (doi: 10.1093/bioinformatics/bts635)] was used to align reads to the mouse reference genome. Cuffdiff (Cufflinks version 2.2.1) was used for gene expression quantification (in FPKM) and for detection of differentially expressed genes based on the BAM alignments from STAR. The cummeRbund R package (version 2.12.1 with R version 3.2.2) was used for visualization and sample clustering. Venn (version 0.8.4) was used to generate dynamic Venn diagrams3 for differential gene expression based on the outputs from Cuffdiff. Hierarchical clustering of the gene expression data was done on log2 (FPKM + 1) values using the Cluster (version 3.0) software and the output (CDT, GTR, and ATR files) was visualized as heatmaps using Java TreeView (version 1.1.6r4)4. GSEA (version 2.2.0, DOI: 10.1073/pnas.0506580102) was used to perform gene set enrichment analysis to identify statistically significant gene sets showing concordant differences between two biological states. Each replicate corresponding to wild-type and Twist+/− frontal and parietal bones had, on an average, 63 million single-end reads of length 75 bp. Mapping of reads to the mm9 reference genome using STAR aligner resulted in, on an average, 74% uniquely mapped reads and 15% multi-mapped reads resulting in 89% mapped reads overall. Transcript abundances in terms of FPKMs were determined using Cuffdiff and the large amount of data produced from the Cuffdiff RNA-Seq differential expression analysis were then visualized using the cummeRbund R package as follows. Density plots were examined to assess the distributions of FPKM scores across samples
To analyze the formation/mineralization rate of wild-type and Twist1+/− frontal and parietal bones, we harvested skulls from pN2 mice and cultured for 2 days in complete a-MEM medium supplemented with calcein (1 μg/ml) (Millipore-Sigma, St. Louis, MO, United States). Where requested mouse recombinant FGF23 protein (100 ng/ml, R&D System, Minneapolis, MN, United States) was added to the cultures. Specimens were fixed in 10% neutral buffered formalin overnight at 4°C, embedded in OCT and cut into 8-μm section. Images of frozen sections were captured under a fluorescence microscope (Leica DMI 4000B) using the same exposure time, and the rate of fluorescence measured using J image densitometer. Sixty images selected randomly from 3 bones (for each group) were analyzed and data presented as average fluorescence intensity. Experiments were performed twice. Of note, technical aspects arising from this type of experiment dictated the choice of pN2 mice. Specifically, in order to preserve the calcein incorporation into the bony tissue, the specimens were not decalcified by EDTA, because this treatment would dissolve the calcein incorporated into the bone. Therefore, to be able to cut the bone into sections the tissue has to be soft and skull of pN2 meets this need. Of note, at pN2 Twist1 gene is already differentially expressed between frontal and parietal bones (see Figure 1).
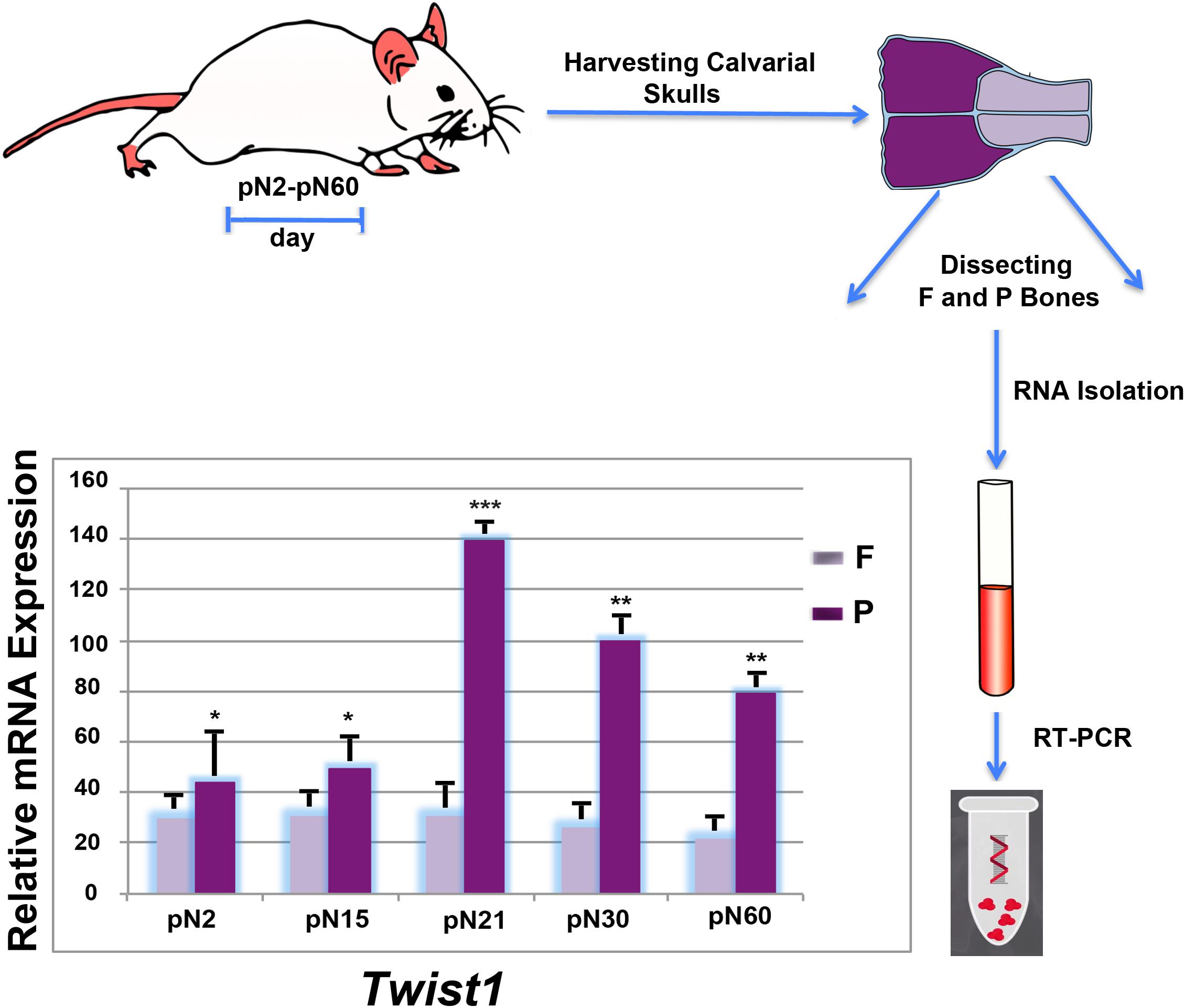
FIGURE 1. Differential postnatal expression of Twist1 in frontal and parietal bones. Time-course RT-qPCR analysis performed on frontal and parietal bone tissues (n = 10) revealed a differential expression of Twist1 with higher levels in parietal bone. The relative mRNA level in each sample is normalized to housekeeping gene Gapdh. Values are presented as relative to Gapdh expression. Values: ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001.
Data are presented as mean-SD of three independent samples. Statistical comparisons between groups were done using a two-tailed Student’s t test, ∗p ≤ 0.05; ∗∗p ≤ 0.01; and ∗∗∗p ≤ 0.001 were considered significant.
While the spatio-temporal expression profile of Twist1 during mouse embryonic development has been widely investigated (Chen and Behringer, 1995; Fuchtbauer, 1995; Stoetzel et al., 1995; Gitelman, 1997; Bildsoe et al., 2009), its postnatal expression pattern in bones of different embryonic tissue origin such as, the neural crest-derived frontal bone and paraxial mesoderm-derived parietal bone has not been described. Therefore, we began our study by profiling the postnatal Twist1 expression in these two bones at different time points. The analysis obtained by RT-PCR revealed a distinct expression pattern between the frontal and parietal bones, which was characterized, starting from day pN2 by a significant increase of Twist1 expression in the mesodermal-derived parietal bone which reach a peak at day pN21, followed by downregulation at later time points. Conversely, in the neural crest-derived frontal bone Twist1 expression was markedly lower and remained steady (Figure 1). This finding opens the question: does Twist1 haploinsufficiency differently impact frontal and parietal bones? In order to address this question, we compared both, in vitro and in vivo, the osteoskeletal ability of frontal and parietal bones derived from haploinsufficient Twist1 and wild-type mice.
Our previous studies demonstrated that neural crest-derived frontal osteoblasts (FOb) and mesoderm-derived parietal osteoblasts (POb) display a different osteogenic potential, with FOb differentiating more robustly than POb (Quarto et al., 2009b).
In the current study, we investigated whether a similar osteogenic profile would be observed in FOb and POb derived from haploinsufficient Twist1 calvarial bones. Confluent wild-type and Twist1+/− osteoblast cultures were incubated in osteogenic differentiation medium for 28 days followed by Alizarin red and von Kossa staining to assess for deposition of calcium into the extracellular matrix. As expected and previously described (Quarto et al., 2009b; Li et al., 2013) both staining methods revealed a robust mineralization of extracellular matrix and bone nodules formation in wild-type FOb compared to wild type POb (Figures 2A–C). Surprisingly, Twist1+/− POb displayed greater osteogenic potential than wild-type POb, whereas Twist1+/− FOb had less osteogenic potential than the wild-type counterpart (Figures 2A–C). This unique profile was also confirmed at molecular level by the expression of osteogenic markers specifically, the early marker osterix (Osx) and the late marker osteocalcin (Bglap) (Figure 2D). Thus, Twist1 haploinsufficiency positively impacts the osteogenic ability of mesoderm-derived osteoblasts whilst impairing that of neural crest-derived osteoblasts.
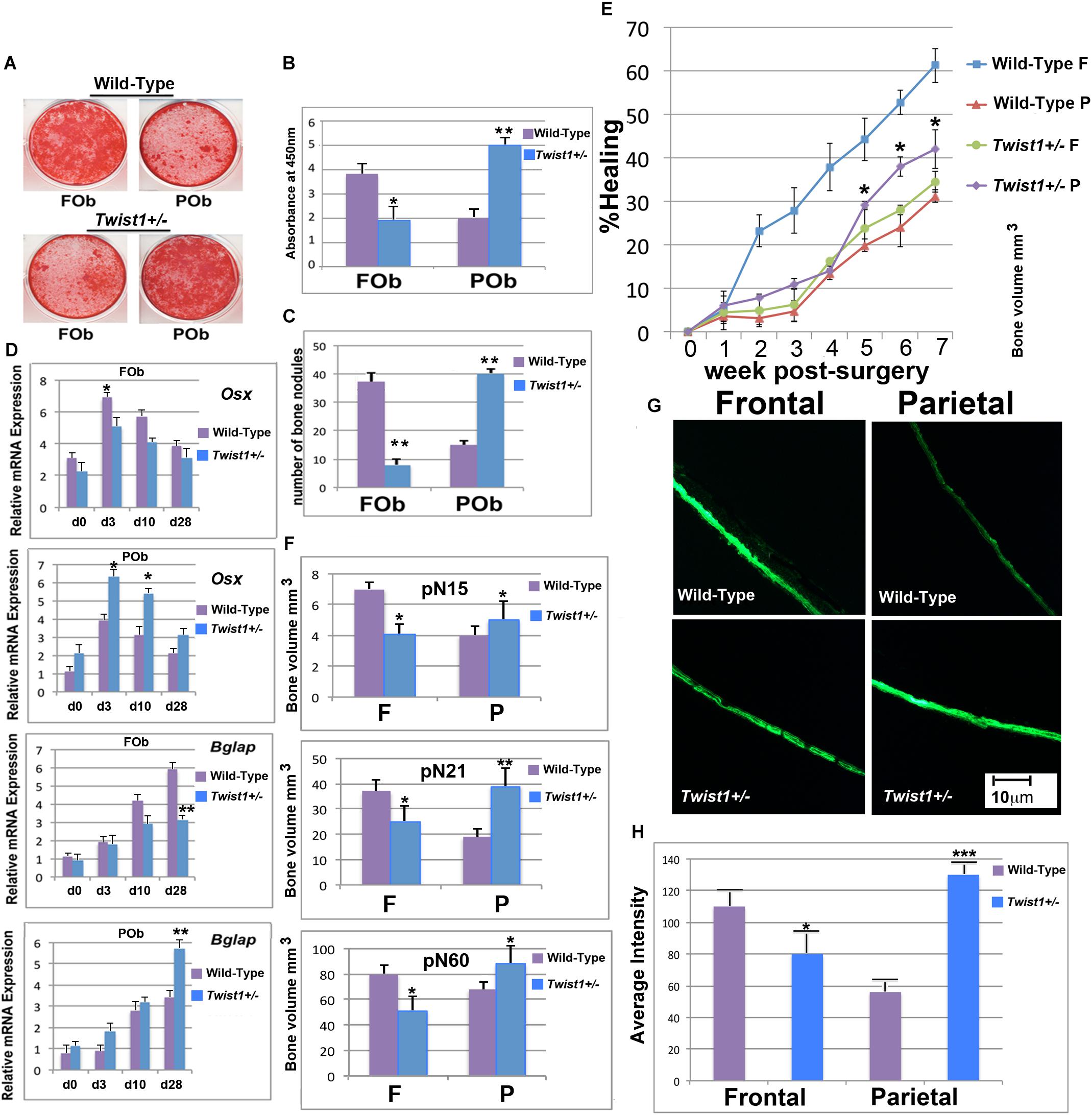
FIGURE 2. Twist1 haploinsufficiency enhances the osteoskeletal potential of mesoderm-derived parietal bone. (A) FOb and POb cultured under osteogenic conditions for 28 days and stained with Alizarin Red. (B) Quantification of Alizarin Red staining revealing a more intense mineralization of the extracellular matrix in Twist1+/− POb as compared to wild-type POb. (C) Number of large bone nodules formation identified by von-Kossa staining at osteogenic differentiation day 28. (D) Relative mRNA expression of early (Osx) and late (Bglap) osteogenic markers in wild-type and Twist1+/− osteoblasts cultures. Results are representative of three experiments performed. (E) Time course of frontal and parietal bone healing defects, quantification of defect repair (n? = ?5/each bone) based on μCT results are analyzed using magic wand tool in Photoshop after standardization of threshold for regenerating calvarial bone. Results are representative of two independent experiments performed. Statistical analysis was performed using the Mann–Whitney Test. ∗P ≤ 0.05. (F) Quantification of bone thickness in uninjured wild type and Twist1+/− Frontal and Parietal bones ∗P ≤ 0.05. (G) Bone formation in wild-type and Twist1+/− frontal and parietal bones assessed by culturing skulls for 48 h in presence of calcein (1 μg/ml). (H) Quantification of calcein incorporation. Histomorphometric analysis of calcein accumulation was obtained using Image J64 densitometry analysis. Scale bar, 10 μm. Results are representative of three independent experiments performed. Values: ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001.
Having observed an enhanced in vitro osteogenic potential of Twist1 haploinsufficient POb as compared to wild-type POb, we sought to investigate whether a similar osteoskeletal phenotype would be observed in vivo. To this aim, we evaluated the calvarial healing ability of frontal and parietal bones in wild-type and Twist1+/− mice. To explore in vivo healing, we introduced calvarial defects of 2-mm in diameter in the right frontal and left parietal bones of postnatal day 21 mice (n = 5). The healing rate of each defect was monitored for 7 weeks by microcomputed tomography (μCT). As previously reported (Quarto et al., 2009b; Li et al., 2015), μCT scanning of defects showed significantly less bone regeneration in wild-type parietal bone as compared to wild-type frontal bone (Figure 2E). Conversely, Twist1 haploinsufficient parietal bone healed significantly better than wild type parietal bone to an extent similar to that of the wild type frontal bone (Figure 2E). Moreover, μCT analysis performed at different time points on uninjured frontal and parietal bones revealed differences in bone density, with Twist1+/− parietal bone showing increased bone thickness as compared to wild-type parietal bone, and Twist1+/− frontal bone having decreased bone density (Figure 2F). The latter observation found further support from calcein labeling experiments performed on ex-vivo skull explants derived from wild-type and Twist1+/− mice. As shown in Figures 2G,H, Twist1+/− parietal bones incorporated significant higher amount of calcein as indicated by the intensity of fluorescence and its quantification. This outcome demonstrates the ability of Twist1+/− parietal bone to mineralize more robustly than wild-type, thus reflecting bone density properties. Taken together, results garnered from in vivo analysis mirrored those obtained from in vitro, supporting that Twist1 haploinsufficiency leads to a significant enhancement of the osteogenic capacity of mesoderm-derived parietal bone while negatively impacting the neural crest-derived frontal bone.
Next, we sought to investigate the molecular mechanism(s) through which Twist1 haploinsuffiency could enhance the osteoskeletal ability of the parietal bone and derived osteoblasts. Our initial approach was to analyze the genomic profile of Twist1+/− and wild-type frontal and parietal bones by bulk RNA-seq technique. Out of 24,015 mouse mm9 genes analyzed, this analysis revealed differential expression of 365 between wild-type parietal and Twist1+/− statistically different (p-value < = 0.0001) with a fold change greater than three. Between wild-type and Twist+/− frontal bones 217 genes were differentially expressed (p-value < = 0.0001) with a fold change greater than three. A greater differential gene expression was observed between wild-type frontal and parietal bones. Venn diagram and scatter plot analysis illustrate up or down regulated top genes with a fold change greater than three between the different bones (Figures 3A,B). Among the 365 genes differentially expressed between wild-type and Twist1+/− parietal bones 310 were upregulated and 55 downregulated, while 233 were upregulated, and 4 downregulated between wild-type and Twist1 frontal bones. The wild-type frontal and Twist1+/− bones were the most closely correlated with a Pearson correlation coefficient (r = 98), than wild-type parietal and Twist1+/− bones (r = 96) (Figure 3B). Among the genes differentially expressed between Twist1+/− and wild-type parietal bones, we found potentially of interest the Fgf23 gene, which encodes a peptide hormone regulating bone mineralization and levels of serum phosphate (Shimada et al., 2001, 2004; Yamazaki et al., 2002; Riminucci et al., 2003; Larsson et al., 2004; Wang et al., 2008). We identified Fgf23 as part of an unique signature of genes exclusively upregulated in wild-type parietal bones and significantly down-regulated in Twist1+/− parietal bone with a fold change greater than six thus, representing one of the top genes significantly down-regulated. Therefore, we thought that it could be a candidate gene to investigate. In addition to Fgf23, in this signature were also identified clusters of microRNAs (Figure 3C).
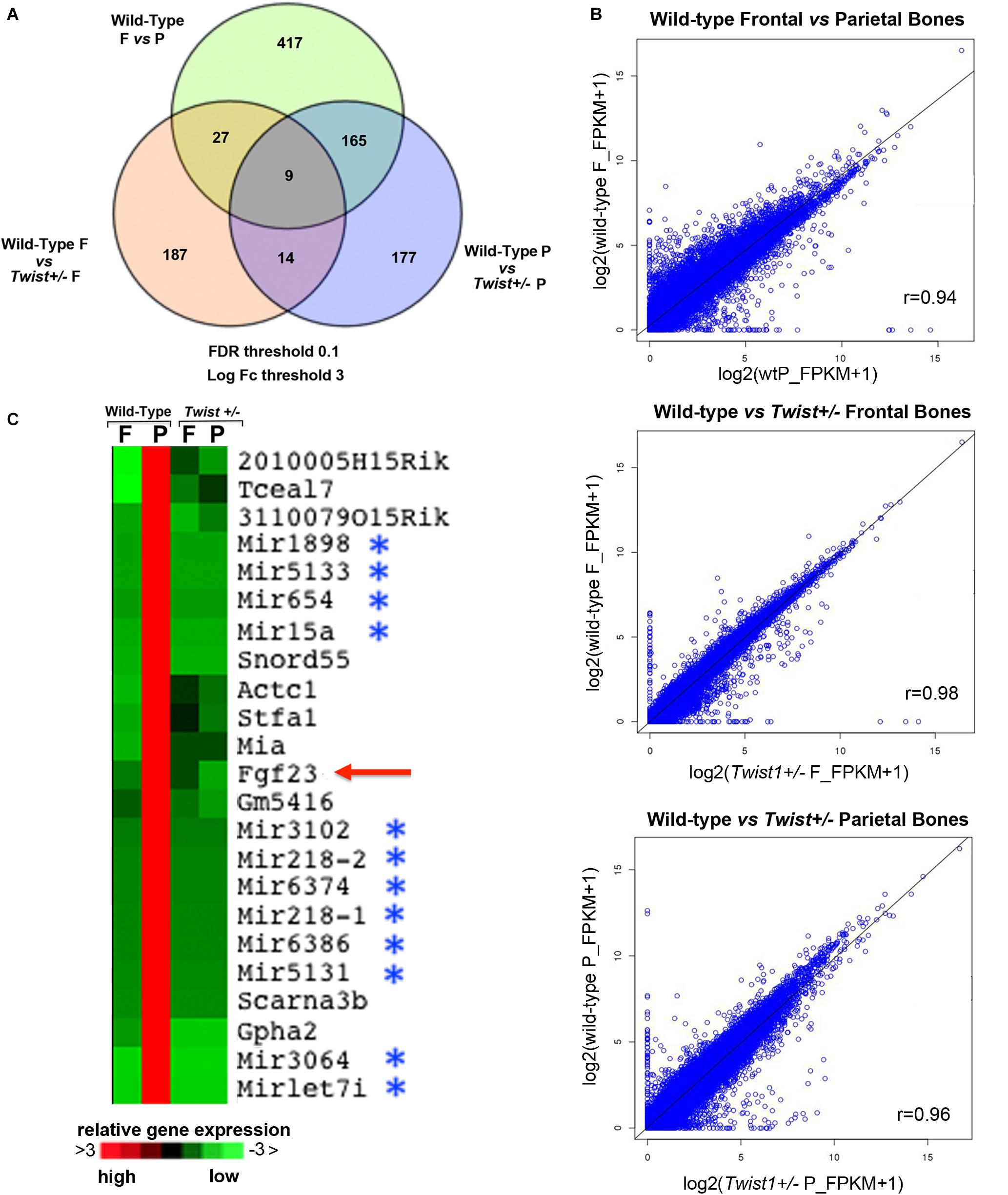
FIGURE 3. Transcriptomic profiles of wild-type and Twist+/− frontal and parietal bones. RNA-Seq analysis to assess gene expression changes caused by Twist1 haploinsufficiency in calvarial bone of pN21 mice revealed significant differences. (A) Venn diagram showing the comparison of top genes found differentially expressed (up and down regulated) in wild-type and Twist+/− Frontal (F) and Parietal (P) bones for FDR < 0.1 and log2 (fold change) >3, with fold change as ratios of gene-level FPKM values. Their gene expression levels are affected ≥3 fold. (B) Scatterplot representation showing the fold change Scatter plots of gene expression of genes [based on log2 (FPKM + 1) expression values with each dot representing a gene]. In the bottom of each graph the pair-wise Pearson’s correlation (r) for all genes is shown. (C) Heatmap showing Fgf23 as a component of a unique signature of genes that are exclusively upregulated in wild-type parietal bones. (upregulation in Red; downregulation in Green).
As a first step, prior to investigating the potential involvement of Fgf23 downregulation in enhancing POb osteogenic differentiation, we validated its differential expression by PCR analysis in bone tissues on which bulk RNA-seq analysis was performed. This analysis confirmed a significant downregulation of Fgf23 in Twist1+/− parietal bone as compared to wild-type (Figure 4A), thus validating the data obtained from RNA-seq analysis.
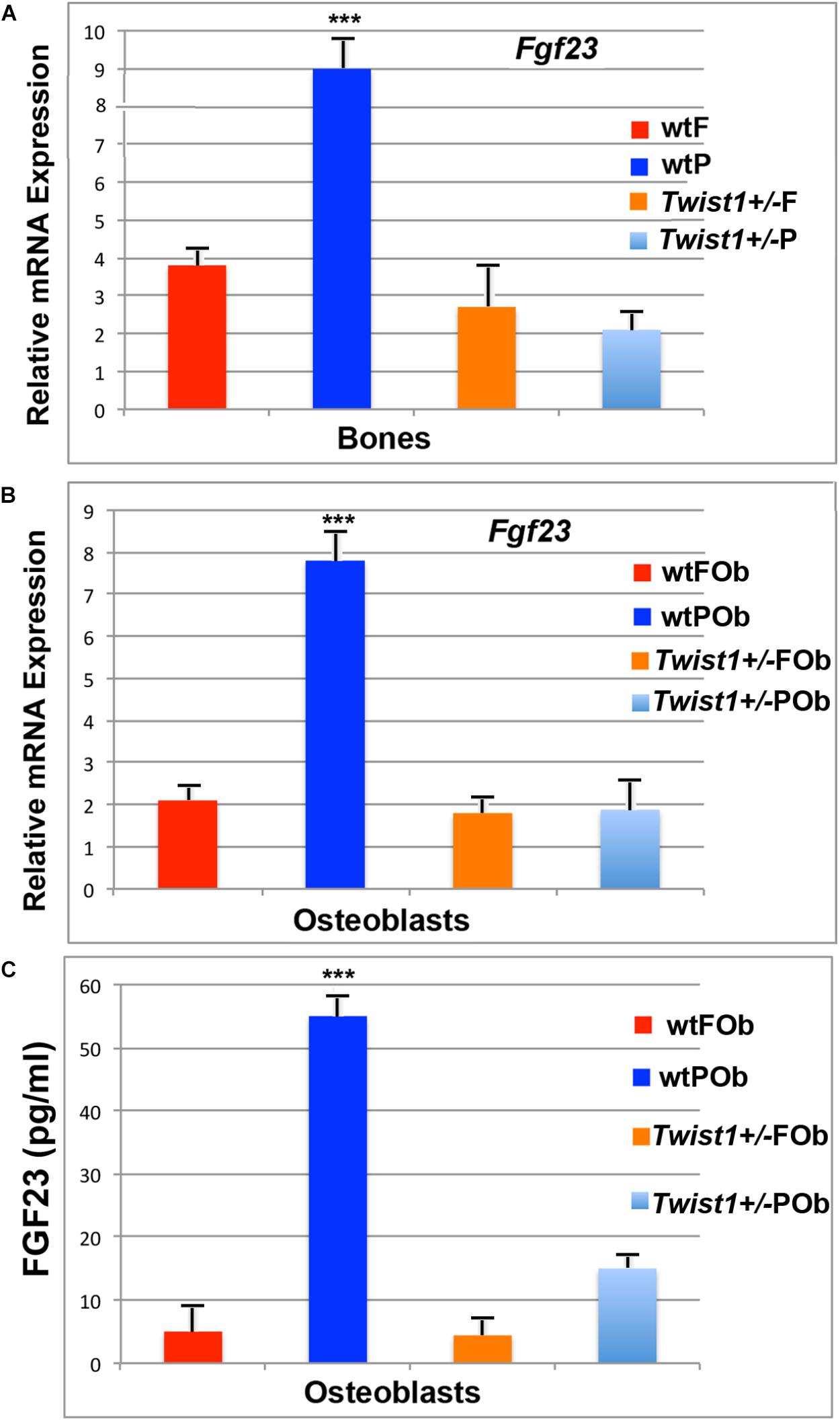
FIGURE 4. Validation of Fgf23 in calvarial bones and derived osteoblasts. (A) Validation of Fgf23 downregulation obtained by performing PCR analysis on bone tissues. (B) RT-qPCR analysis showing Fgf23 down-regulation in osteoblast primary cultures (depleted of pericranium and dura mater- derived cells). (C) ELISA assay performed on media collected from osteoblast cells and concentrated by 20-fold. Elevated level of secreted FGF23 protein is detected in the medium of wild-type POb, whereas in Twist+/− POb protein levels are sharply decreased to values similar to that of wild-type and Twist+/− FOb. Value: ∗∗∗p ≤ 0.001.
Previous studied have reported that Fgf23 is primarily expressed in osteoblasts and osteocytes (Riminucci et al., 2003; Mirams et al., 2004) and systemically circulates (Yamazaki et al., 2002). Additional PCR analysis performed on osteoblast cultures also revealed the same Fgf23 expression pattern as previously identified by RNA-seq analysis in bone tissues. Similarly, Fgf23 was found to be downregulated approximately seven-fold in Twist1+/− POb as compared to wild-type POb (Figure 4B). This finding is of interest and indicates that the expression pattern of Fgf23 and the downregulation initially observed in parietal bones comprising of pericranium, bone plate and dura-mater, is intrinsic of osteoblast cells which were derived from calvarial bones depleted of pericranium and dura-mater tissues. Consistent with this finding, ELISA detected high level of secreted FGF23 protein exclusively in wild-type POb, whereas, it was dramatically reduced in Twist1+/− POb (Figure 4C). Indeed, the validation of our RNA-seq results by RT-PCR builds support to further investigate the potential functional role that Fgf23 down-regulation may play in enhancing the osteogenic potential of Twist1+/− POb.
Having confirmed the differential expression by PCR and protein analysis we sought to investigate the potential role Fgf23 might play in the osteogenic phenotype observed between wild type and Twist1+/− POb. For this purpose, we performed a set of loss- and gain-of-function experiments. We began by treating osteoblasts with exogenously added FGF23 protein to assess whether this treatment would abrogate the increased osteogenic differentiation observed in Twist1+/− POb. Outcomes from an osteogenic differentiation assay performed with or without FGF23 protein revealed a dramatic effect on Twist1+/− POb. As shown in Figures 5A,B, Alizarin red staining detected a significant reduction of the extracellular matrix mineralization in FGF23 treated Twist1+/− POb in comparison to untreated POb. These biochemical differences were further supported by the expression profile of late osteogenic marker Bglap (Figure 5C). Conversely, FGF23 treatment had little effect on Twist1+/− FOb osteogenic profile which was already impaired by Twist1 haploinsufficiency (Supplementary Figures S1A–C). Moreover, FGF23 treatment significantly impacted wild-type FOb, but had little effect on wild-type POb (Supplementary Figures S1D–F). The differential effect elicited by FGF23 on wild-type FOb and POb may reflect differences in the threshold level of endogenous FGF23 protein between FOb and POb, and therefore, their responsiveness. Taken together, these data strongly suggest that Fgf23 downregulation may play a role in eliciting greater osteogenic capacity of Twist1+/− POb.
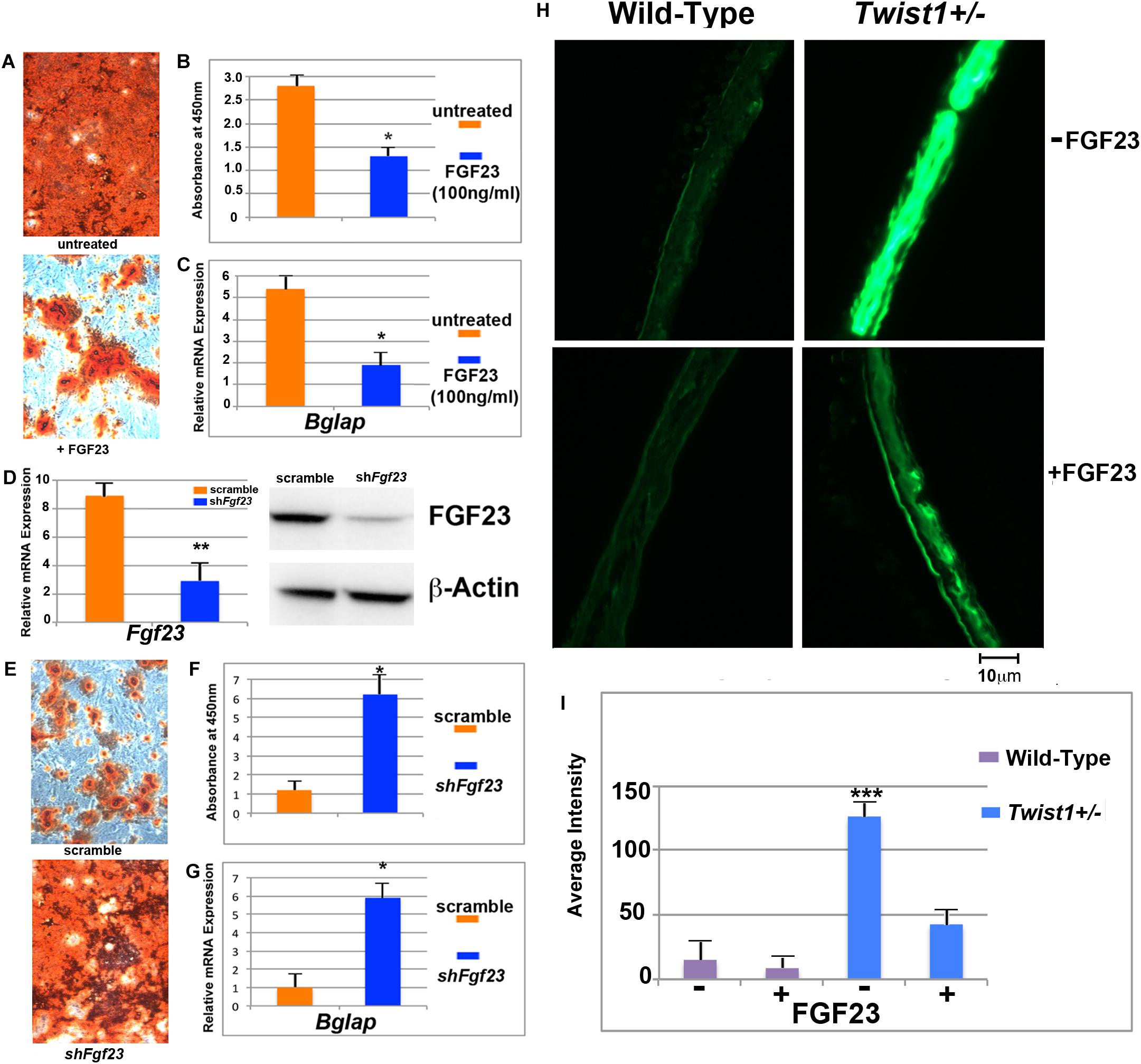
FIGURE 5. Inhibition of osteogenesis and bone mineralization by exogenous FGF23 protein. (A) Treatment with FGF23 protein (100 ng/ml) suppresses markedly osteogenic differentiation of Twist+/− POb as revealed by Alizarin red staining performed at differentiation day 28. (B) Quantification of Alizarin red staining for the osteogenic assay shown in panel A (C) RT-qPCR analysis of the osteogenic marker osteocalcin (Bglap) confirms that FGF23 treatment abrogates the enhanced osteogenic of Twist1+/− POb. (D) Validation of effective Fgf23 silencing monitored by RT-qPCR (left panel) and immunoblotting analysis (right panel). (E) Fgf23 silencing enhances the osteogenic capacity of wild-type POb mirroring that of Twist1+/− POb as revealed by Alizarin red staining. (F) Quantification of Alizarin red staining for the osteogenic assay shown in panel E. (G) osteocalcin (Bglap) expression by RT-qPCR analysis confirms increased terminal differentiation in shFgf23 POb compared to scramble POb. (H) Exogenous added FGF23 also decreases dramatically bone mineralization in parietal bone organ culture. Calcein was added to the cultures for 48 h. Immunofluorescence showing calcein accumulation into the bone. (I) Histomorphometric analysis of calcein accumulation. Scale bar, 10 μm. Results are representative of three independent experiments performed. Values: ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001.
Next, we performed a counter part experiment seeking to investigate whether silencing of Fgf23 in wild-type POb would potentiate their in vitro osteogenic ability mimicking that of Twist1+/− POb. Fgf23 silencing was successfully accomplished by transducing wild-type POb with shFgf23 lentiviral particles. Upon puromicin selection, expression of Fgf23 decreased approximately by 73% in shFgf23 cells as compared to scramble transduced cells (Figure 5D, left panel). Immunoblotting analysis confirmed decrease of FGF protein (Figure 5D, right panel). Then, we performed an osteogenic differentiation assay to evaluate the osteogenic ability of shFgf23POb in comparison to scramble POb. The analysis provided clear evidence on the key role that downregulation of Fgf23 plays in enhancing the osteogenic capacity observed in Twist1+/− POb. This was indicated by a robust extracellular matrix mineralization as assessed by Alizarin red staining (Figure 5E) and its quantification (Figure 5F) as well as elevated expression level of Bglap (Figure 5G) in Twist1+/− POb relative to scramble POb.
Fgf23 is a key player in regulating calcium homeostasis during bone cell differentiation and mineralization, and is abundantly expressed and secreted from immature and mature osteocytes within the bone matrix (Riminucci et al., 2003; Liao, 2013; Michigami, 2014; Guo and Yuan, 2015). Our results suggested that the osteoskeletal phenotype observed in Twist1+/− parietal bone and POb is at least in part mediated by Fgf23 downregulation. To further strengthen this observation we tested the ability of wild-type and Twist1+/− parietal bone tissue explants in culture to incorporate calcein either in presence or absence of FGF23 protein. After 48 h in culture, incorporation of calcein into the bone explants was monitored by immunofluorescence. We observed that treatment with FGF23 dramatically decreased calcein labeling in Twist1+/− parietal bone (Figure 5H, bottom panel) compared to untreated Twist1+/− control bone (Figure 5H, top panel). The histomorphometric quantification analysis of calcein incorporation in untreated Twist1+/− parietal bone showed a more intense immunofluorescence staining than FGF23 treated bone (Figure 5I). No significant differences were found between untreated and FGF23 treated wild-type parietal bones (Figure 5H, left panel, Figure 5I), this would reflect a lack of responsiveness by the cells due to high endogenous level of FGF23 and possible downregulation of its receptors. Taken together, these findings demonstrated that FGF23 treatment significantly inhibited bone mineralization on ex vivo Twist1+/− parietal bone explants and further support its role in controlling the osteoskeletal properties between wild-type and Twist1+/− parietal bones.
To gain further support to our findings, we next asked two questions: First, does silencing of Twist1 in wild-type POb phenocopy Twist1 haploinsufficient POb? Second, does it lead to downregulation of Fgf23 as observed in Twist1 haploinsufficient POb? To address these questions POb were transduced with shTwist1 or scramble lentiviral particles followed by puromicin selection. Silencing of Twist1 was assessed by PCR and immunoblotting analyses (Supplementary Figures S2A–C). Stable transduced cells after puromicin selection were first analyzed for their osteogenic ability. As shown in Figures 6A–C, shTwist1 POb robustly mineralized their extracellular matrix and expressed significant higher levels of Bglap than scramble POb. Importantly, in addition to the enhanced osteogenic differentiation we observed a concomitant downregulation of Fgf23 in shTwist1 POb. This was revealed either at the gene expression level by PCR analysis or at protein level by immunoblotting analysis performed on cultured shTwist1 POb (Figures 6D,E). Evidence of the functional effectiveness of Twist1 silencing was provided by the expression analysis in shTwist1 POb of three known Twist1 target genes: Zeb, Snail, and Cdh1 (Leptin, 1991; Rose and Malcolm, 1997; Kang and Massague, 2004; Peinado et al., 2007; Vesuna et al., 2008) and previously identified by our bulk RNA-seq as being differentially expressed between wild-type and Twist1 haploinsufficient parietal bones (Figure 6F). Remarkably, PCR analysis performed on scramble and shTwist1 POb (Figure 6G) mirrored the expression pattern of Zeb, Snail, Cdh1 previously observed in wild-type and Twist1 haploinsufficient parietal bones. The above findings strongly suggest a direct involvement of Twist1 haploinsufficiency in mediating the downregulation of Fgf23, which in turn confers a greater osteogenic ability to POb.
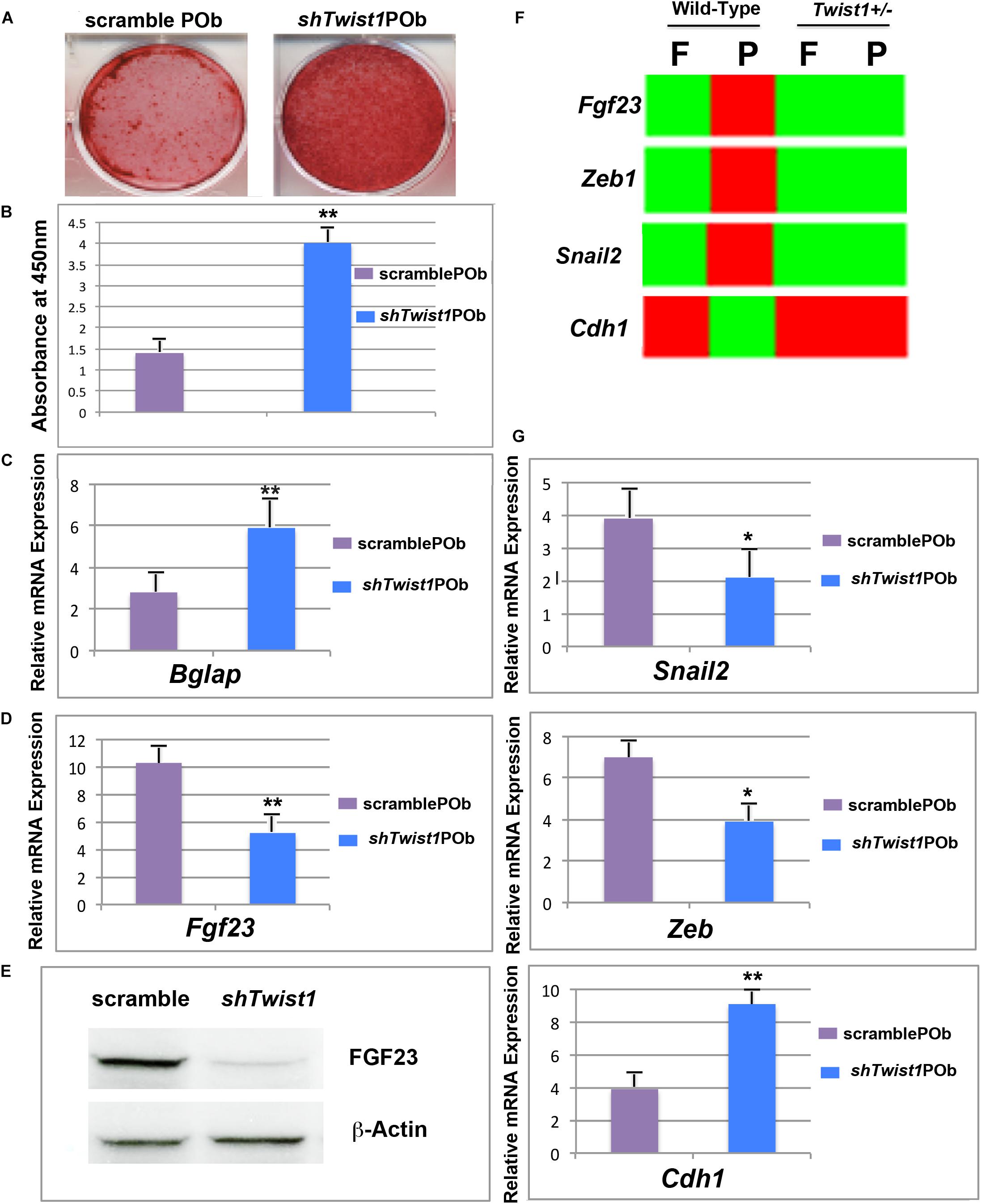
FIGURE 6. Twist1 silencing phenocopies the osteoskeletal profile of Twist1 haploinsufficiency in POb by downregulating Fgf23. (A) Osteogenic differentiation assay performed on shTwist1 POb revealed a robust extracellular matrix mineralization. (B) Quantification of Alizarin red staining for the osteogenic assay shown in panel A. (C) RT-qPCR analysis showing significant upregulation of Bglap as compared to scramble POb. (D) RT-qPCR analysis performed on stable transduced cells upon puromicin selection reveals that sh-mediated Twist1 silencing triggers a significant downregulation of Fgf23 expression. (E) Immunoblotting analysis using anti-FGF23 antibody confirms decreased FGF23 at protein level in shTwist1 POb compared to scramble POb. (F) Bulk RNA-seq analysis showing the effect of Twist1 haploinsufficiency on some of its target genes such Zeb, Snail1, and Cdh1 and comparison in wild-type versus Twist1 haploinsufficiency parietal bones. (G) As assessed by RT-qPCR analysis a similar pattern is observed in stable transduced shTwist1 POb and scramble POb upon puromicin selection and culturing them. Values: ∗p ≤ 0.05, ∗∗p ≤ 0.01.
Previous studies have described Twist1 as a key-regulator of craniofacial development, however, the role of Twist1 haploinsufficiency in skeletal development and regeneration of postnatal calvarial bones has not yet been fully delineated. Herein, we describe the impact of Twist1 haploinsufficiency on two calvarial bones of different embryonic tissue: the neural crest-derived frontal bone and mesoderm-derived parietal bone. We provide evidence that Twist1 haploinsufficiency enhances the osteoskeletal ability of the parietal bone compared to the corresponding wild-type bone by reaching a level similar to that of wild-type frontal bone. By performing bulk RNA-seq analysis we unveiled a distinct Fgf23 upregulation in wild-type parietal bone, which is significantly suppressed by Twist1 haploinsufficiency. Of note, the expression pattern of Fgf23 observed in mesoderm-derived parietal bone is sustained from early to late postnatal stages (Supplementary Figure S3).
Several lines of evidence support the role of Fgf23 downregulation in promoting a greater osteoskeletal capacity of Twist1 haploinsufficient parietal bone. First, gain-of-function experiment showed that treatment with FGF23 protein significantly decreased osteogenic differentiation of Twist1+/− POb as indicated by poor extracellular matrix mineralization and expression of specific osteogenic markers. Conversely, loss-of function by silencing Fgf23 in wild-type POb enhanced their osteogenic differentiation mirroring the profile observed in Twist1+/− POb. Second, bone density analysis and calcein labeling both detected a significant increased mineralization in Twist1 haploinsufficient parietal bone where Fgf23 is dramatically downregulated in comparison to wild-type. This finding was further corroborated by a complementary experiment showing that culturing ex vivo Twist1 haploinsufficient parietal bones in presence of exogenous FGF23 protein led to decreased calcein-labeling, and therefore, mineralization to levels comparable to that of wild-type parietal bone. The latter result highlights a direct role of Fgf23 downregulation in matrix mineralization of mesoderm-derived bones.
Impaired skeletal development is one major feature found in genetic disorders attributed to FGF23 overexpression (Liao, 2013). Mutation of this gene in humans causes autosomal dominant hypophosphatemic rickets (ADHR) (Shimada et al., 2002). Furthermore, high serum levels of FGF23 have been detected in patients affected by fibrous dysplasia (FD) of bone, which is a skeletal disorder with bone-forming cells failing to mature and aberrantly produce fibrous, or connective, tissue (Riminucci et al., 2003). Increased serum FGF23 has also been reported in X-linked hypophosphatemia (XLH) (Jonsson et al., 2003). Indeed, results garnered from our study further support FGF23 as a growth factor playing a central role in calcium homeostasis and acting on bone cell differentiation and mineralization.
Our findings unravel a novel role of Twist1 haploinsuffiency in the context of calvarial bone differentiation and regeneration. Haploinsuffiency of this bHLH transcription factor triggers a sharp downregulation of Fgf23 expression, which in turn enhances, selectively, the osteoskeletal ability of mesoderm-derived parietal bone, both in vitro and in vivo (Figure 7). Interestingly, Twist1 haploinsuffiency negatively impacts the osteoskeletal property of the neural crest-derived frontal bone. The latter observation is not fully unexpected and recapitulates embryonic effects of loss of Twist1 on CNC-derived frontal bone (Bildsoe et al., 2009). Our data indeed, reinforce the crucial role for Twist1 in skeletogenic differentiation of the CNC and point to a differential effect of loss of Twist1 in mesoderm-derived parietal bones. However, this observation deserves further investigation aimed to identify potential players in controlling the negative effect elicited postnatally by Twist1 haploinsufficiency in frontal bone.
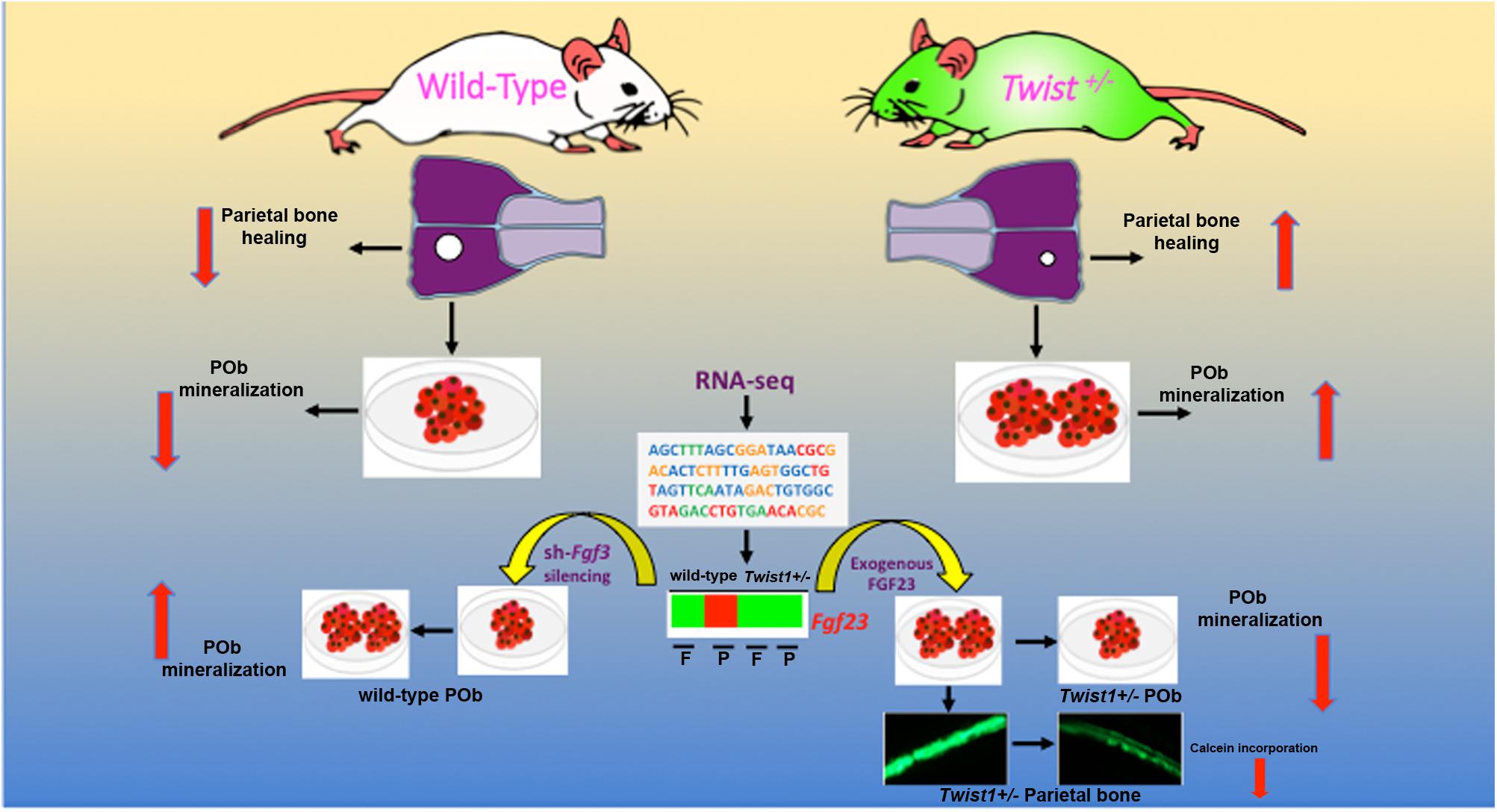
FIGURE 7. Twist1-haploinsufficiency-Fgf23-downregulation axis. Schematic representation of the molecular mechanism mediating the effect of Twist1 haploinsufficiency in enhancing the osteoskeletal ability of mesoderm-derived parietal bone via downregulation of Fgf23.
Taken together, our data point to a postnatal impact of Twist1 haploinsufficiency in conferring a higher osteogenic ability to the mesoderm-derived parietal bone relative to the neural crest-derived frontal bone. Since, the coronal suture is comprised mostly of mesoderm-derived tissue (Jiang et al., 2002; Yoshida et al., 2008), our finding triggers interest to investigating whether a same unique Fgf23 expression pattern, as that observed in wild-type and Twist1+/− parietal bones, is also present between Twist1+/− coronal synostotic and wild-type coronal patent sutures. A potential downregulation of Fgf23 within the mesoderm tissue of Twist+/− coronal suture could indeed induce synostosis.
Fgf23 gene transcription in bone remains largely undefined, therefore the finding that either Twist1 haploinsufficiency or its sh-mediated silencing suppressed the expression of Fgf23 in mesoderm-derived parietal bone and osteoblasts is interesting and raises the question whether this outcome is a direct or indirect effect elicited by Twist1. Is Twist1 a transcriptional activator of Fgf23 gene in parietal bones? Are there tissue-specific co-activators involved in the process? These are important and challenging questions that deserve to be addressed. Twist1 binds conserved cis-regulatory elements known as E-boxes present on gene promoters, triggering either transcriptional activation or repression of genes (Hamamori et al., 1997; Murre et al., 1989; Massari and Murre, 2000). Initial analysis of murine Fgf23 gene have identified within its core promoter region E-Box motifs (CANNTG), thus suggesting that Fgf23 could be a direct target of Twist1 (data not shown). This hypothesis finds support from our shTwist1+/− silencing experiments on POb showing that Twist1 silencing recapitulated the expression pattern of Fgf23 as well as that of specific Twist1 markers initially identified by RNA-Seq analysis in Twist1+/− parietal bones.
In conclusion, from our study emerged two novel findings: first, Twist1 haploinsufficiency selectively promotes a greater osteoskeletal ability and regeneration of mesoderm-derived calvarial bone, whilst negatively impacting neural crest-derived calvarial bone. A previous study has described that TWIST-1 haploinsufficiency enhanced osteogenesis of osteoblast cells derived from parietal bones of patients Saethre–Chotzen syndrome, however, the study is missing the analysis of osteoblasts derived from a frontal bone (Camp et al., 2018). Thus, by focusing our analysis on two calvarial bones of different tissue origin, we have highlighted a dual role of Twist1 haploinsufficiency. A second finding is the identification of a molecular mechanism though which Twist1 haploinsufficiency triggers preferentially osteoskeletal induction. Herein, we provide substantial evidence that this occurs via downregulation of Fgf23, which is uniquely expressed at high threshold levels in wild-type parietal bone. The innate mechanism presented in this paper, through which Twist1 haploinsufficiency preferentially triggers osteogenic induction of parietal bones may be beneficial to optimize treatments for skeletal regeneration, reconstruction and repair of mesoderm-derived bone, as well as to alleviate skeletal abnormalities caused by Twist1 haploinsufficiency.
NQ designed and performed the experiments, analyzed the data, and wrote the manuscript. SS, NM, and SM performed the experiments. AR and ML edited the manuscript.
This work was supported by the Gunn/Olivier fund, the Hagey Laboratory for Pediatric Regenerative Medicine, and the National Institutes of Health NIH grants R01 DE19434, R56DE025597 to ML.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Chris Duldulao for his technical assistance. A special thank for the RNA-seq data curation and formal analysis to Dr. Ramesh V. Nair, Bioinformatics Director, Bioinformatics-as-a-Service (BaaS) under the Genetics Bioinformatics Service Center (GBSC), a Stanford School of Medicine service center operated by the Department of Genetics. This work is dedicated to the memory of Mrs. Irma Giustino Weiss.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.01426/full#supplementary-material
Baylies, M. K., and Bate, M. (1996). twist: a myogenic switch in Drosophila. Science 272, 1481–1484. doi: 10.1126/science.272.5267.1481
Behr, B., Longaker, M. T., and Quarto, N. (2011a). Craniosynostosis of coronal suture in twist1 mice occurs through endochondral ossification recapitulating the physiological closure of posterior frontal suture. Front. Physiol. 2:37. doi: 10.3389/fphys.2011.00037
Behr, B., Sorkin, M., Lehnhardt, M., Renda, A., Longaker, M. T., and Quarto, N. (2012). A comparative analysis of the osteogenic effects of BMP-2, FGF-2, and VEGFA in a calvarial defect model. Tissue Eng. Part A 18, 1079–1086. doi: 10.1089/ten.TEA.2011.0537
Behr, B., Tang, C., Germann, G., Longaker, M. T., and Quarto, N. (2011b). Locally applied vascular endothelial growth factor A increases the osteogenic healing capacity of human adipose-derived stem cells by promoting osteogenic and endothelial differentiation. Stem Cells 29, 286–296. doi: 10.1002/stem.581
Bialek, P., Kern, B., Yang, X., Schrock, M., Sosic, D., Hong, N., et al. (2004). A twist code determines the onset of osteoblast differentiation. Dev. Cell 6, 423–435. doi: 10.1016/S1534-5807(04)00058-9
Bildsoe, H., Loebel, D. A., Jones, V. J., Chen, Y. T., Behringer, R. R., and Tam, P. P. (2009). Requirement for Twist1 in frontonasal and skull vault development in the mouse embryo. Dev. Biol. 331, 176–188. doi: 10.1016/j.ydbio.2009.04.034
Camp, E., Anderson, P. J., Zannettino, A. C. W., Glackin, C. A., and Gronthos, S. (2018). Tyrosine kinase receptor c-ros-oncogene 1 inhibition alleviates aberrant bone formation of TWIST-1 haploinsufficient calvarial cells from Saethre-Chotzen syndrome patients. J. Cell. Physiol. 233, 7320–7332. doi: 10.1002/jcp.26563
Carver, E. A., Oram, K. F., and Gridley, T. (2002). Craniosynostosis in Twist heterozygous mice: a model for Saethre-Chotzen syndrome. Anat. Rec. 268, 90–92. doi: 10.1002/ar.10124
Chen, Z. F., and Behringer, R. R. (1995). twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 9, 686–699. doi: 10.1101/gad.9.6.686
Cripps, R. M., and Olson, E. N. (1998). Twist is required for muscle template splitting during adult Drosophila myogenesis. Dev. Biol. 203, 106–115. doi: 10.1006/dbio.1998.9040
el Ghouzzi, V., Le Merrer, M., Perrin-Schmitt, F., Lajeunie, E., Benit, P., Renier, D., et al. (1997). Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat. Genet. 15, 42–46. doi: 10.1038/ng0197-42
Fuchtbauer, E. M. (1995). Expression of M-twist during postimplantation development of the mouse. Dev. Dyn. 204, 316–322. doi: 10.1002/aja.1002040309
Gitelman, I. (1997). Twist protein in mouse embryogenesis. Dev. Biol. 189, 205–214. doi: 10.1006/dbio.1997.8614
Guo, Y. C., and Yuan, Q. (2015). Fibroblast growth factor 23 and bone mineralisation. Int. J. Oral Sci. 7, 8–13. doi: 10.1038/ijos.2015.1
Hamamori, Y., Wu, H. Y., Sartorelli, V., and Kedes, L. (1997). The basic domain of myogenic basic helix-loop-helix (bHLH) proteins is the novel target for direct inhibition by another bHLH protein. Twist Mol. Cell. Biol. 17, 6563–6573. doi: 10.1128/MCB.17.11.6563
Howard, T. D., Paznekas, W. A., Green, E. D., Chiang, L. C., Ma, N., Ortiz de Luna, R. I., et al. (1997). Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 15, 36–41. doi: 10.1038/ng0197-36
Jiang, X., Iseki, S., Maxson, R. E., Sucov, H. M., and Morriss-Kay, G. M. (2002). Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 241, 106–116. doi: 10.1006/dbio.2001.0487
Jonsson, K. B., Zahradnik, R., Larsson, T., White, K. E., Sugimoto, T., Imanishi, Y., et al. (2003). Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N. Engl. J. Med. 348, 1656–1663. doi: 10.1056/NEJMoa020881
Kang, Y., and Massague, J. (2004). Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 118, 277–279. doi: 10.1016/j.cell.2004.07.011
Karsenty, G. (2008). Transcriptional control of skeletogenesis. Annu. Rev. Genomics Hum. Genet. 9, 183–196. doi: 10.1146/annurev.genom.9.081307.164437
Karsenty, G., Kronenberg, H. M., and Settembre, C. (2009). Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 25, 629–648. doi: 10.1146/annurev.cellbio.042308.113308
Karsenty, G., and Wagner, E. F. (2002). Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2, 389–406. doi: 10.1016/S1534-5807(02)00157-0
Larsson, T., Marsell, R., Schipani, E., Ohlsson, C., Ljunggren, O., Tenenhouse, H. S., et al. (2004). Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 145, 3087–3094. doi: 10.1210/en.2003-1768
Leptin, M. (1991). Twist and snail as positive and negative regulators during Drosophila mesoderm development. Genes Dev. 5, 1568–1576. doi: 10.1101/gad.5.9.1568
Li, S., Meyer, N. P., Quarto, N., and Longaker, M. T. (2013). Integration of multiple signaling regulates through apoptosis the differential osteogenic potential of neural crest-derived and mesoderm-derived Osteoblasts. PLoS One 8:e58610. doi: 10.1371/journal.pone.0058610
Li, S., Quarto, N., Senarath-Yapa, K., Grey, N., Bai, X., and Longaker, M. T. (2015). Enhanced activation of canonical wnt signaling confers mesoderm-derived parietal bone with similar osteogenic and skeletal healing capacity to neural crest-derived frontal bone. PLoS One 10:e0138059. doi: 10.1371/journal.pone.0138059
Liao, E. (2013). FGF23 associated bone diseases. Front. Med. 7, 65–80. doi: 10.1007/s11684-013-0254-6
Marie, P. J., Kaabeche, K., and Guenou, H. (2008). Roles of FGFR2 and twist in human craniosynostosis: insights from genetic mutations in cranial osteoblasts. Front. Oral Biol. 12, 144–159. doi: 10.1159/0000115036
Massari, M. E., and Murre, C. (2000). Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 20, 429–440. doi: 10.1128/MCB.20.2.429-440.2000
Mirams, M., Robinson, B. G., Mason, R. S., and Nelson, A. E. (2004). Bone as a source of FGF23: regulation by phosphate? Bone 35, 1192–1199.
Miraoui, H., and Marie, P. J. (2010). Pivotal role of Twist in skeletal biology and pathology. Gene 468, 1–7. doi: 10.1016/j.gene.2010.07.013
Morriss-Kay, G. M. (2001). Derivation of the mammalian skull vault. J. Anat. 199, 143–151. doi: 10.1046/j.1469-7580.2001.19910143.x
Murray, S. S., Glackin, C. A., Winters, K. A., Gazit, D., Kahn, A. J., and Murray, E. J. (1992). Expression of helix-loop-helix regulatory genes during differentiation of mouse osteoblastic cells. J. Bone Miner. Res. 7, 1131–1138. doi: 10.1002/jbmr.5650071004
Murre, C., McCaw, P. S., Vaessin, H., Caudy, M., Jan, L. Y., Jan, Y. N., et al. (1989). Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 58, 537–544. doi: 10.1016/0092-8674(89)90434-0
O’Rourke, M. P., and Tam, P. P. (2002). Twist functions in mouse development. Int. J. Dev. Biol. 46, 401–413.
Peinado, H., Olmeda, D., and Cano, A. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428. doi: 10.1038/nrc2131
Quarto, N., Behr, B., Li, S., and Longaker, M. T. (2009a). Differential FGF ligands and FGF receptors expression pattern in frontal and parietal calvarial bones. Cells Tissues Organs 190, 158–169. doi: 10.1159/000202789
Quarto, N., Senarath-Yapa, K., Renda, A., and Longaker, M. T. (2015). TWIST1 silencing enhances in vitro and in vivo osteogenic differentiation of human adipose-derived stem cells by triggering activation of BMP-ERK/FGF signaling and TAZ upregulation. Stem Cells 33, 833–847. doi: 10.1002/stem.1907
Quarto, N., Wan, D. C., Kwan, M. D., Panetta, N. J., Li, S., and Longaker, M. T. (2009b). Origin matters: differences in embryonic tissue origin and Wnt signaling determine the osteogenic potential and healing capacity of frontal and parietal calvarial bones. J. Bone Miner. Res. 25, 1680–1694. doi: 10.1359/jbmr.091116
Quarto, N., Wan, D. C., and Longaker, M. T. (2008). Molecular mechanisms of FGF-2 inhibitory activity in the osteogenic context of mouse adipose-derived stem cells (mASCs). Bone 42, 1040–1052. doi: 10.1016/j.bone.2008.01.026
Rice, D. P., Aberg, T., Chan, Y., Tang, Z., Kettunen, P. J., Pakarinen, L., et al. (2000). Integration of FGF and TWIST in calvarial bone and suture development. Development 127, 1845–1855.
Riminucci, M., Collins, M. T., Fedarko, N. S., Cherman, N., Corsi, A., White, K. E., et al. (2003). FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J. Clin. Invest. 112, 683–692. doi: 10.1172/JCI18399
Rose, C. S., and Malcolm, S. (1997). A TWIST in development. Trends Genet. 13, 384–387. doi: 10.1016/S0168-9525(97)01296-1
Senarath-Yapa, K., Li, S., Meyer, N. P., Longaker, M. T., and Quarto, N. (2013). Integration of multiple signaling pathways determines differences in the osteogenic potential and tissue regeneration of neural crest-derived and mesoderm-derived calvarial bones. Int. J. Mol. Sci. 14, 5978–5997. doi: 10.3390/ijms14035978
Senarath-Yapa, K., Li, S., Walmsley, G. G., Zielins, E., Paik, K., Britto, J. A., et al. (2016). Small molecule inhibition of transforming growth factor beta signaling enables the endogenous regenerative potential of the mammalian calvarium. Tissue Eng. Part A 22, 707–720. doi: 10.1089/ten.TEA.2015.0527
Shimada, T., Kakitani, M., Yamazaki, Y., Hasegawa, H., Takeuchi, Y., Fujita, T., et al. (2004). Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest. 113, 561–568. doi: 10.1172/JCI200419081
Shimada, T., Mizutani, S., Muto, T., Yoneya, T., Hino, R., Takeda, S., et al. (2001). Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. U.S.A. 98, 6500–6505. doi: 10.1073/pnas.101545198
Shimada, T., Muto, T., Urakawa, I., Yoneya, T., Yamazaki, Y., Okawa, K., et al. (2002). Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 143, 3179–3182. doi: 10.1210/endo.143.8.8795
Stoetzel, C., Weber, B., Bourgeois, P., Bolcato-Bellemin, A. L., and Perrin-Schmitt, F. (1995). Dorso-ventral and rostro-caudal sequential expression of M-twist in the postimplantation murine embryo. Mech. Dev. 51, 251–263. doi: 10.1016/0925-4773(95)00369-X
Thisse, B., el Messal, M., and Perrin-Schmitt, F. (1987). The twist gene: isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res. 15, 3439–3453. doi: 10.1093/nar/15.8.3439
Thisse, B., Stoetzel, C., Gorostiza-Thisse, C., and Perrin-Schmitt, F. (1988). Sequence of the twist gene and nuclear localization of its protein in endomesodermal cells of early Drosophila embryos. EMBO J. 7, 2175–2183. doi: 10.1002/j.1460-2075.1988.tb03056.x
Vesuna, F., van Diest, P., Chen, J. H., and Raman, V. (2008). Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem. Biophys. Res. Commun. 367, 235–241. doi: 10.1016/j.bbrc.2007.11.151
Wang, H., Yoshiko, Y., Yamamoto, R., Minamizaki, T., Kozai, K., Tanne, K., et al. (2008). Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J. Bone Miner. Res. 23, 939–948. doi: 10.1359/jbmr.080220
Wu, S. Y., Yang, Y. P., and McClay, D. R. (2008). Twist is an essential regulator of the skeletogenic gene regulatory network in the sea urchin embryo. Dev. Biol. 319, 406–415. doi: 10.1016/j.ydbio.2008.04.003
Yamazaki, Y., Okazaki, R., Shibata, M., Hasegawa, Y., Satoh, K., Tajima, T., et al. (2002). Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J. Clin. Endocrinol. Metab. 87, 4957–4960. doi: 10.1210/jc.2002-021105
Yoshida, T., Vivatbutsiri, P., Morriss-Kay, G., Saga, Y., and Iseki, S. (2008). Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev. 125, 797–808. doi: 10.1016/j.mod.2008.06.007
Keywords: Twist1, haploinsuffiency, Fgf23, downregulation, enhancement, osteoskeletogenesis
Citation: Quarto N, Shailendra S, Meyer NP, Menon S, Renda A and Longaker MT (2018) Twist1-Haploinsufficiency Selectively Enhances the Osteoskeletal Capacity of Mesoderm-Derived Parietal Bone Through Downregulation of Fgf23. Front. Physiol. 9:1426. doi: 10.3389/fphys.2018.01426
Received: 06 June 2018; Accepted: 19 September 2018;
Published: 15 October 2018.
Edited by:
Gianpaolo Papaccio, Università degli Studi della Campania “Luigi Vanvitelli” Naples, ItalyReviewed by:
Joan Therese Richtsmeier, Pennsylvania State University, United StatesCopyright © 2018 Quarto, Shailendra, Meyer, Menon, Renda and Longaker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalina Quarto, cXVhcnRvQHVuaW5hLml0 Michael T. Longaker, bG9uZ2FrZXJAc3RhbmZvcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.