 Jennifer A. Courtney
Jennifer A. Courtney James F. Cnota
James F. Cnota Helen N. Jones
Helen N. Jones
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Physiol. , 07 August 2018
Sec. Integrative Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01045
This article is part of the Research Topic Recent Advances in Placenta-Heart Interactions View all 6 articles
Congenital heart disease (CHD) is the most common birth defect, affecting ~1% of all live births (van der Linde et al., 2011). Despite improvements in clinical care, it is the leading cause of infant mortality related to birth defects (Yang et al., 2006) and burdens survivors with significant morbidity (Gilboa et al., 2016). Furthermore, CHD accounts for the largest proportion (26.7%) of birth defect-associated hospitalization costs—up to $6.1 billion in 2013 (Arth et al., 2017). Yet after decades of research with a primary focus on genetic etiology, the underlying cause of these defects remains unknown in the majority of cases (Zaidi and Brueckner, 2017). Unexplained CHD may be secondary to undiscovered roles of noncoding genetic, epigenetic, and environmental factors, among others (Russell et al., 2018). Population studies have recently demonstrated that pregnancies complicated by CHD also carry a higher risk of developing pathologies associated with an abnormal placenta including growth disturbances (Puri et al., 2017), preeclampsia (Auger et al., 2015; Brodwall et al., 2016), preterm birth (Laas et al., 2012), and stillbirth (Jorgensen et al., 2014). Both the heart and placenta are vascular organs and develop concurrently; therefore, shared pathways almost certainly direct the development of both. The involvement of placental abnormalities in congenital heart disease, whether causal, commensurate or reactive, is under investigated and given the common developmental window and shared developmental pathways of the heart and placenta and concurrent vasculature development, we propose that further investigation combining clinical data, in vitro, in vivo, and computer modeling is fundamental to our understanding and the potential to develop therapeutics.
The heart and placenta develop concurrently, with heart tube specification occurring at days 16–21 and a rudimentary villous tree forming by day 21 of gestation (Schleich, 2002; Kaufmann et al., 2004; Khong, 2004; Linask, 2013; Tyser et al., 2016).
Approximately 12 days post conception, the placenta comprises a layer of extra-embryonic mesoderm and two trophoblast populations, a core of cytotrophoblast cells covered by a layer of syncytiotrophoblast. Over the next 3–8 days the mesenchymal cell cores of the primary villi transform into first hemangiogenic precursor cells (Huppertz and Peeters, 2005). The villous trophoblast at this early stage of development is paramount in regulating the development of the placental vasculature with several components of key signaling pathways only expressed in the trophoblast, Figure 1A (Rossant and Cross, 2001). At the same time the heart forms, first as bilateral cardiogenic plates of mesoderm, then as a primitive heart tube as the two plates fuse during gastrulation (Gittenberger-de Groot et al., 2005; Lindsey et al., 2014). By day 20, the heart tube beats steadily, and includes a single ventricle and outflow tract (Schleich, 2002) Figure 1B (Gilbert and Barresi, 2016).
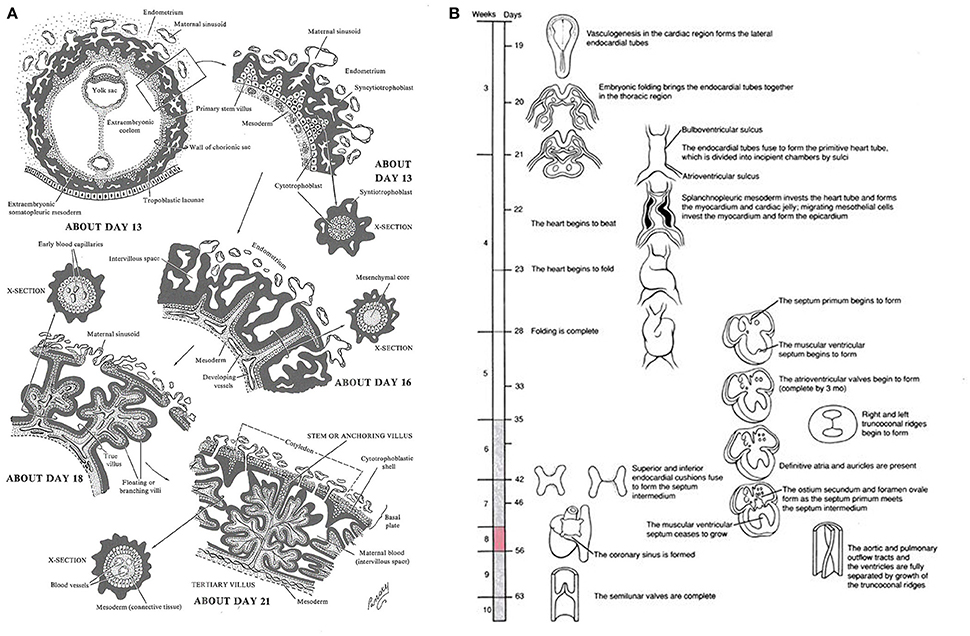
Figure 1. (A) Timeline of human early placental development: Adapted from Lifemap Discovery (Edgar et al., 2013), “At the beginning of week 3 primary stem villi, consisting of a cytotrophoblast core covered by a syncytial layer, appear. Extraembryonic mesodermal cells or cytotrophoblast penetrate the core of the primary villi and grow in the direction of the decidua to form secondary stem villi and by the end of week 3 the mesodermal cells differentiate into blood cells and small blood vessels, forming the villous capillary system, and creating tertiary villi. By week 4, capillaries in the tertiary villi contact capillaries developing in the mesoderm of the chorionic plate and in the connecting stalk, eventually contact the intraembryonic circulatory system, and connect the placenta and the embryo. Thus, in week 4, when the heart begins to beat, the placental villous system is able to supply the embryo with oxygen and nutrients.” (B) Timeline of human embryonic heart development: Adapted from Lifemap Discovery (Edgar et al., 2013). “The human heart develops on day 18 or 19 following fertilization. In response to induction signals from the underlying endoderm, the mesoderm in the cardiogenic area forms the cardiogenic cords. A hollow center forms within the cords, giving rise to the endocardial tubes. With lateral folding of the embryo, the paired endocardial tubes approach each other and fuse into a single tube called the primitive heart tube. The primitive heart tube develops into five distinct unpaired regions and begins to pump blood”.
Vascularization of the human placenta occurs by local de-novo vasculogenesis within the mesenchymal core of the secondary villi. This occurs prior to infiltration of fetal vessels or blood into the placenta with the progenitor cells derived directly from placental mesenchymal cells (Demir et al., 1989). Shortly after this, Hofbauer cells (macrophages of placental origin) develop inside the villous core in the vicinity of the vasculogenic precursor cells suggesting a putative paracrine role for these cells during the early stages of placental vasculogenesis (Demir and Erbengi, 1984; Cervar et al., 1999). During very early placental development, Vascular endothelial growth factor (VEGF) is highly expressed in cytotrophoblast cells and also in Hofbauer cells. On the other hand, the respective receptors, Flt-1 and Flk-1 are expressed on the vasculogenic and angiogenic precursor cells (Charnock-Jones et al., 2004). With this constellation an increase in the expression of VEGF and its receptors may orchestrate the temporal and spacial regulation of the differentiation and maturation of villous vascularization (Castellucci et al., 2000; Kingdom et al., 2000). Concurrently, heart looping occurs during week 4 and is controlled by gene regulatory networks for left-right patterning and local gradients (Lenhart et al., 2013; Sylva et al., 2014). Septa and valves form via tissue folding and cardiac cushions that undergo epithelial-mesenchyme transition during week 5 (Anderson et al., 2003). Cardiac vessel morphogenesis occurs concurrently via inductive signaling of epicardium from underlying myocardium (Lin et al., 2012). Fetal vascular development and circulation is established between 25 and 60 days and despite an extensive capillary network within early villi, there is little evidence of fetal-placental circulation until late first trimester (Thornburg and Louey, 2013). Once established, signaling between the placenta and fetal organs via the fetal-placental circulation may impact growth and remodeling of both the fetal heart and the placenta, which, in the placenta occurs throughout gestation until term (Figure 2).
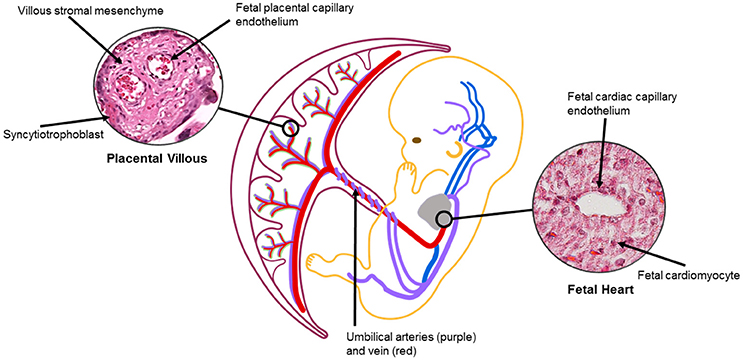
Figure 2. The placental and heart are connected via fetal vasculature. Signaling between trophoblast/endothelial and/or cardiomyocyte/endothelial cells may impact fetal vasculature, leading to changes in placental and heart structures (Jia et al., 2018). Oxygenation, flow, cell crosstalk can also affect organ development and remodeling via vascular changes throughout gestation. Placental villous image from Boyd Collection Centre for Trophoblast Research at the University of Cambridge. https://www.trophoblast.cam.ac.uk/Resources/ndp-index1.
Mechanisms of heart and placental development share common regulatory pathways, for example, cardiomyocyte specification and extra villous trophoblast invasion are both regulated by Notch and Wnt (De Falco et al, 2007; de la Pompa and Epstein, 2012). The endocardial cushion develops from neural crest cells to form heart valves and septa and requires similar genes as the placenta for proper formation and remodeling, including VegF and Connexin 43 (Maschhoff and Baldwin, 2000; Dor et al., 2001; Dunk et al., 2012).
Studies in both human and mouse demonstrate the effects of alterations in these genes and their associations with failure to develop appropriately. Connexin 43 mediates cell-cell interactions in cardiomyocytes, and aids in trophoblast fusion and intercellular placental communication (Maschhoff and Baldwin, 2000; Dunk et al., 2012). Babies with fetal growth restriction have lower levels of VCAM1 than those with normal growth trajectories (Rajashekhar et al., 2003). Targeted disruption of VCAM1 in mice leads to impaired placentation and fetal death, and severe abnormalities in the developing heart (Gurtner et al., 1995; Kwee et al., 1995), and deletion of the upstream gene FOXO1 attenuates VCAM1 expression and leads to similar outcomes (Ferdous et al, 2011). The loss of PPAR gamma in mice results in abnormal placental and cardiac development (Barak et al., 1999) with embryonic lethality mid-gestation. Using tetraploid embryos, PPAR gamma restoration to the inner cell mass did not overcome the placental or cardiac defects, however, importantly, when PPAR gamma was restored to the trophectoderm alone fetuses survive and cardiac malformations were absent.
Common molecular pathways also direct vasculogenesis and angiogenesis in both the heart and placenta. Loss of Cited2 in mice causes significant heart and fetal growth defects, disruption of left-right patterning, and fetal death (Bamforth et al., 2001, 2004; Lopes Floro et al., 2011). Placenta-specific mouse knockouts of Cited2 reveal disorganized labyrinthine placental vasculature by day E14.5 due to an impaired HIF-signaling response (Withington et al., 2006). Interestingly, targeted deletion of Cited2 in trophoblasts but not endothelial cells disrupted capillary patterning in the placenta (Moreau et al., 2014). Heart-specific knockout of Cited2 revealed similar myocardial and coronary vascular abnormalities (MacDonald et al., 2012), revealing that signaling by both trophoblasts and cardiomyocytes impacts local capillary growth and development. In contrast, a recent study by Rhee et al. (2018) identifies the necessity of endothelial cell signaling for regulation of cardiac development. The authors demonstrated left ventricular non-compaction when the chromatin remodeler Ino80 was knocked out in coronary vessel progenitor cells or in all endothelium, with a more severe heart phenotype in the endothelial-specific knockout. Unfortunately, this study did not include assessment of the placenta to identify if the endothelial-specific knockout also impacted placental development. Ino80 is a ubiquitous cell proliferation promoter, and disruption of endothelial cell proliferation pathways may underlie loss of conceptus or congenital defects with impacts on both heart and placenta.
Importantly, many murine studies published on genes necessary for heart development demonstrate a common phenotype, usually reported as embryonic lethal, that upon further analysis is due to malformation of a placental vascular tree, for example in the Hand1 and NKx2.5 null mice, driving the need to understand the roles of these genes in both placental and vascular development (Olson and Srivastava, 1996; Firulli et al., 2010). Recently, the Deciphering the Mechanisms of Developmental Disorders (DMDD) consortium demonstrated that 68% of the 103 mouse lines they investigated had placental phenotypes (Perez-Garcia et al., 2018). There was a strong correlation between heart and vascular abnormalities and placental defects; additionally, common genes affected heart and vascular morphology in the study.
We have previously published impaired placental vascular tree development in one subtype of CHD in humans (Jones et al., 2015) and more recently demonstrated it in another (TGA). Furthermore, to investigate mechanisms of this disruption we performed RNA-sequencing on human placentas from fetuses with CHDs and compared them to gestational-age matched controls. Term placentas from TGA, HLHS, and control births were collected under institutional IRB approval. RNA was isolated from FFPE samples using RecoverAll™ Total Nucleic Acid Isolation Kit for FFPE. RNA-Sequencing using Ullumina HiSeq was performed by CCHMC and UC Core facilities. RNA-sequencing analysis was done using AltAnalyze version 2.0 (Kallisto for alignment) to identify differentially expressed genes and clustering performed using GoElite. Methods described in Supplementary Materials. Clustering analysis identified pathways involved in heart looping, left/right patterning and symmetry, muscle development, and ATP synthesis were significantly downregulated in the placentas of the Hypoplastic Left Heart Syndrome (HLHS) fetuses compared to controls (Figure 3 and Supplemental Table 1), indicating disruption to pathways associated with both heart and placental development.
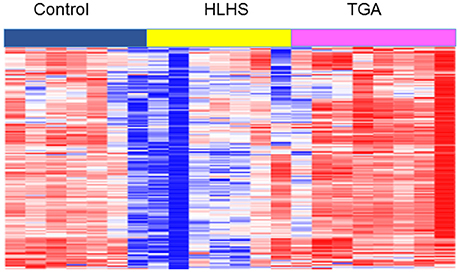
Figure 3. Heat map of RNA-sequencing profiles of term placentas from TGA, HLHS, and gestational-age matched controls from Cluster 1 of GoElite cluster analysis. Pathways significantly changed (p < 0.05) include determination of left/right symmetry, left/right patterning, heart looping, heart development, MHC protein complex, and ATP synthesis. A complete list of genes and pathways is included in Supplementary Material.
Based on studies of blood flow and cardiac remodeling during development, it is possible that placental dysfunction may significantly contribute to the incidence of CHD. Whether the placenta contributes to the development of CHD or whether disruption of common developmental pathways contribute to placental dysfunction and CHDs is not known.
Developmental abnormalities which impair normal blood flow through the embryonic heart, including improper placentation and villous vascular tree development with resulting impairment of flow, may contribute to the occurrence of CHD in humans. Alteration in placental hemodynamics could impact both inflow via the umbilical vein and outflow via the umbilical artery. Midgett et al. (2017) disrupted blood flow to inflow and outflow tracts of chick embryos at HH18 (looped tubular beating heart), causing malformations of developing heart structures. Defects were shown to be flow dependent, with more restriction to the outflow tract causing more significant heart defects.
However, the study of feto-placental circulation by Doppler ultrasound in humans with CHD has resulted in conflicting results. Evidence of lower cerebral resistance, i.e., brain sparing, flow patterns have been demonstrated in multiple studies but likely varies by type of cardiac defect (Hahn et al., 2016; Ruiz et al., 2017). However, the majority of umbilical flow patterns fall within the normal range. As the antenatal diagnosis of CHD most commonly occurs in the second trimester of pregnancy, these clinically measurable tools are being applied long after the development of the cardiac defect and may not reflect early hemodynamic disturbances that could contribute to the etiology of CHD. Impaired blood flow may occur much earlier in pregnancy with extensive cardiac remodeling occurring to compensate for the disrupted flow.
Cardiac remodeling also occurs in response to hypoxia, which increases VegFR-3 (Flt4) expression and subsequently VegFC. This signaling pathway improves vasculogenesis, angiogenic sprouting, and fetal cardiac development (Helske et al., 2001). Gadd45G has been shown to be sensitive to hypoxia in the heart during ischemia, leading to apoptosis of cardiomyocytes, fibrosis, and left ventricular dysfunction (Lucas et al., 2015). Isoform Gadd45A in placenta is also induced by hypoxia and leads to poor migration of trophoblasts (Liu et al., 2014). Lower levels of both isoforms lead to better outcomes. Impaired placentation, fibrin deposition, and reduced placental vasculature impact the gas exchange capabilities of the placenta and therefore oxygen levels available to the fetus via the umbilical vein may be reduced which could also contribute to hypoxia-induced cardiac remodeling. Diminished umbilical venous oxygen levels on fetal MRI further support these hypotheses (Sun et al., 2015).
During implantation, maternal signals direct embryonic cell invasion into the decidua, and maternal immunity determines depth of trophoblast invasion (Su and Fazleabas, 2015). Disruptions in uterine signaling or an overactive maternal immune response may limit invasion, leading to placental insufficiency and concurrent cardiac anomalies (Thornburg et al., 2010). Once the maternal-placental and fetal-placental circulations are established, the impact of the maternal or external environment, via the maternal circulation, may impact both placental and heart development directly (factors able to cross the placenta, e.g., flame retardant chemicals; Gorini et al., 2014) or may indirectly impact cardiac growth and development through abnormal signaling from the dysfunctional placenta. While existing understanding of CHD causation has largely focused on genetic etiologies (Zaidi and Brueckner, 2017), epidemiologic and experimental studies also support environmental contributions. Linask (2013) proposed that the maternal environment can also influence heart development. Multiple observational studies have linked multivitamins and folic acid supplementation to CHD (Botto et al., 2004). Proposed mechanisms are related to both DNA synthesis and methylation with numerous basic and translational studies addressing the question. Studies have associated maternal hyperlipidemia to an increased risk of CHD (Smedts et al., 2012; Wong et al., 2018), and maternal diabetes correlates strongly to certain CHDs in mouse studies (Hachisuga et al., 2015). Mouse studies indicate that folate deficiencies coupled with other environmental exposures (alcohol, lithium, Wnt3A deficiency) led to cardiac anomalies, while administering high folate levels in mice and humans before pregnancy prevented cardiac birth defects (Han et al., 2009; Serrano et al., 2010; Czeizel et al., 2015). Other maternal influences, such as hyperlipidemia and/or hyperglycemia, maternal obesity and maternal smoking have been associated with an increased risk of CHD (Alverson et al., 2011; Smedts et al., 2012).
Current treatment for many complex CHDs is palliative surgery shortly after birth. Babies born small for gestational age (SGA) who also have a CHD exhibit higher mortality after the first surgical correction than those babies with normal birth weights (Best et al., 2017). We have identified significant abnormalities in placentas from fetuses with either Hypoplastic Left Heart Syndrome (HLHS) or Transposition of the Great Arteries (TGA). Placental transport capabilities appear diminished in HLHS placentas, while microvascular changes are seen in the placentas of fetuses with both TGA and HLHS (Jones et al., 2015; Jones, unpublished data) and appear to be independent of fetal weight, as TGA babies are frequently significantly larger at birth than HLHS babies (Puri et al., 2017; Alsaied et al., 2018).
Previously, we (Jones et al., 2015) noted disruptions in the vascular trees in placentas of babies born with HLHS. Recent histopathological analyses of placentas from babies born with TGA show similar abnormalities, yet the babies generally weighed more at birth than the HLHS cohort (Jones, unpublished data). In addition, Doppler ultrasound abnormalities were absent despite the growth restriction and placental dysregulation (Gembruch et al., 2003; Parra-Saavedra et al., 2013). This suggests that the formation of the vascular tree in the placenta of CHD fetuses is abnormal despite subtype and while the potential for flow disruption and feedback may exist, it may be more likely that this disruption reflects abnormal vascular formation during early pregnancy and leads to the question if this is seen in other vascular beds of the CHD fetus (Sun et al., 2015).
To investigate the involvement of the placenta in congenital heart disease further, the combination of clinical data, human samples, murine models, and in vitro multi-cell models will be necessary. Furthermore, moving beyond examining the placenta only at the time of delivery, and utilizing newer technologies for studying the placenta during pregnancy in humans will be vital. Collection of human placental samples as well as the use of samples of maternal blood throughout pregnancy from the multiple subtypes of congenital heart disease will enable us to establish if the common placental perturbations we have already identified are indeed apparent across the spectrum of CHDs, and if those changes can be identified by assessing and characterizing extracellular vesicles within the maternal circulation during pregnancy.
The use of “heart-specific” animal models will enable us to study the progression of placental disruption in association with cardiac malformations at time points in gestation that we cannot access in humans, such as during the early first trimester. Furthermore, using capabilities designed in our lab we will investigate if correcting the identified placental defects during early pregnancy can impact fetal, cardiac and vascular bed development and growth. We hypothesize that very early signaling events impact fetal vasculature, affecting both placenta and heart, and that correcting the signaling pathways via the placenta may offer avenues for potential therapies.
Early signaling mechanisms between the fetus and the placenta remain largely under-investigated but may represent a mechanism that also links heart and placenta development and function. Investigations using co-cultured human cells in vitro will again enable us to identify if disturbances in cardiac cells or trophoblast cells can impact each other via the fetal vasculature and endothelium.
At this current time it is impossible to say if, in humans, placental changes may contribute to the cardiac anomalies in congenital heart disease or if the placental changes recently identified are due to a shared disrupted etiology which impact fetal development and growth concurrently. Clearly studies from murine models and chick embryo studies would suggest that placental structural and vascular changes contribute to congenital heart defects as described above. We aim to determine if this is true in humans and if this may provide a path for the potential treatment of CHD in utero.
This protocol was approved by the Institutional Review Boards of Cincinnati Children's Hospital Medical Center and Good Samaritan Hospital (Cincinnati, Ohio) under study number 2010–2610. Patient data were identified prior to analysis. Animal studies were approved by Cincinnati Children's Hospital Medical Center under IACUC protocol 2015-0078.
JAC, JC, and HJ contributed to the writing of the manuscript, data generation.
Funding is provided by Cincinnati Children's Hospital Medical Center Fetal Research Gift Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AM and handling editor declared their shared affiliation at time of review.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.01045/full#supplementary-material
1. ^Centre for Trophoblast Research. Boyd Collection. Available online at: https://www.trophoblast.cam.ac.uk/Resources/ndp-index (Accessed May 2, 2018).
Alsaied, T., Tseng, S., King, E., Hahn, E., Divanovic, A., Habli, M., et al. (2018). The effect of fetal hemodynamics on fetal growth in single ventricle and transposition of the great arteries. Ultrasound Obstet. Gynecol. doi: 10.1002/uog.18936. [Epub ahead of print].
Alverson, C. J., Strickland, M. J., Gilboa, S. M., and Correa, A. (2011). Maternal smoking and congenital heart defects in the Baltimore-Washington infant study. Pediatrics 127, e647–e653. doi: 10.1542/peds.2010-1399
Anderson, R. H., Webb, S., Brown, N. A., Lamers, W., and Moorman, A. (2003). Development of the heart. 2. Septation of the atriums and ventricles. Heart 89, 949–958. doi: 10.1136/heart.89.8.949
Arth, A. C., Tinker, S. C., Simeone, R. M., Ailes, E. C., Cragan, J. D., and Grosse, S. D. (2017). Inpatient hospitalization costs associated with birth defects among persons of all ages-United States, 2013. MMWR Morb. Mortal Wkly. Rep. 66, 41–46. doi: 10.15585/mmwr.mm6602a1
Auger, N., Fraser, W. D., Healy-Profitos, J., and Arbour, L. (2015). Association between preeclampsia and congenital heart defects. JAMA 314, 1588–1598. doi: 10.1001/jama.2015.12505
Bamforth, S. D., Bragança, J., Eloranta, J. J., Murdoch, J. N., Marques, F. I., Kranc, K. R., et al. (2001). Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat. Genet. 29, 469–474. doi: 10.1038/ng76
Bamforth, S. D., Bragança, J., Farthing, C. R., Schneider, J. E., Broadbent, C., Michell, A. C., et al. (2004). Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat. Genet. 36, 1189–1196. doi: 10.1038/ng1446
Barak, Y., Nelson, M. C., Ong, E. S., Jones, Y. Z., Ruiz-Lozano, P., Chien, K. R., et al. (1999). PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 4, 585–595. doi: 10.1016/S1097-2765(00)80209-9
Best, K. E., Tennant, P. W. G., and Rankin, J. (2017). Survival, by birth weight and gestational age, in individuals with congenital heart disease: a population-based study. JAHA 6:e005213. doi: 10.1161/JAHA.116.005213
Botto, L. D., Olney, R. S., and Erickson, J. D. (2004). Vitamin supplements and the risk for congenital anomalies other than neural tube defects. Am. J. Med. Genet. 125C, 12–21. doi: 10.1002/ajmg.c.30004
Brodwall, K., Leirgul, E., Greve, G., Vollset, S. E., Holmstrom, H., Tell, G. S., et al. (2016). Possible common aetiology behind maternal preeclampsia and congenital heart defects in the child: a cardiovascular diseases in Norway project study. Paediatr. Perinat. Epidemiol. 30, 76–85. doi: 10.1111/ppe.12252
Castellucci, M., Kosanke, G., Verdenelli, F., Huppertz, B., and Kaufmann, P. (2000). Villous sprouting: fundamental mechanisms of human placental development. Hum. Reprod. Update 6, 485–494.
Cervar, M., Blaschitz, G., Dohr, G., and Desoye, G. (1999). Paracrine regulation of distinct trophoblast functions in vitro bys placental macrophages. Cell Tissue Res. 295, 297–305. doi: 10.1007/s004410051236
Charnock-Jones, D. S., Kaufmann, P., and Mayhew, T. M. (2004). Aspects of human fetoplacental vasculogenesis and angiogenesis. I. Molecular recognition. Placenta 25, 103–113. doi: 10.1016/j.placenta.2003.10.004
Czeizel, A. E., Vereczkey, A., and Szabó, I. (2015). Folic acid in pregnant women associated with reduced prevalence of severe congenital heart defects in their children: a national population-based case-control study. Eur. J. Obstet. Gynecol. Reprod. Biol. 193, 34–39. doi: 10.1016/j.ejogrb.2015.06.024
De Falco, M., Cobellis, L., Giraldi, D., Mastrogiacomo, A., Perna, A., Colacurci, N., et al. (2007). Expression and distribution of notch protein members in human placenta throughout pregnancy. Placenta 28, 118–126. doi: 10.1016/j.placenta.2006.03.010
de la Pompa, J. L., and Epstein, J. A. (2012). Coordinating tissue interactions: notch signaling in cardiac development and disease. Dev. Cell 22, 244–254. doi: 10.1016/j.devcel.2012.01.014
Demir, R., and Erbengi, T. (1984). Some new findings about Hofbauer cells in the chorionic villi of the human placenta. Acta Anat. 119, 18–26.
Demir, R., Kaufmann, P., Castellucci, M., Erbengi, T., and Katowski, A. (1989). Fetal vasculogenesis and angiogenesis in human placental villi. Acta Anat. 136, 190–203.
Dor, Y., Camenisch, T. D., Itin, A., Fishman, G. I., McDonald, J. A., Carmeliet, P., et al. (2001). A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 128, 1531–1538. Available online at: http://dev.biologists.org/content/develop/128/9/1531.full.pdf
Dunk, C. E., Gelhaus, A., Drewlo, S., Baczyk, D., Pötgens, A. J. G., Winterhager, E., et al. (2012). The molecular role of Connexin 43 in human trophoblast cell fusion. Biol. Reprod. 86:115. doi: 10.1095/biolreprod.111.096925
Edgar, R., Mazor, Y., Rinon, A., Blumenthal, J., Golan, Y., Buzhor, E., et al. (2013). LifeMap discovery: the embryonic development, stem cells, and regenerative medicine research portal. PLoS ONE 8:e66629. doi: 10.1371/journal.pone.0066629
Ferdous, A., Morris, J., Abedin, M. J., Collins, S., Richardson, J. A., and Hill, J. A. (2011). Forkhead factor FOXO1 is essential for placental morphogenesis in the developing embryo. Proc. Natl. Acad. Sci. U.S.A. 108, 16307–16312. doi: 10.1073/pnas.1107341108
Firulli, B. A., McConville, D. P., Byers, J. S., Vincentz, J. W., Barnes, R. M., and Firulli, A. B. (2010). Analysis of a HAND1 hypomorphic allele reveals a critical threshold for embryonic viability. Dev. Dyn. 239, 2748–2760. doi: 10.1002/dvdy.22402
Gembruch, U., Meise, C., Germer, U., Berg, C., and Geipel, A. (2003). Venous Doppler ultrasound in 146 fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 22, 345–350. doi: 10.1002/uog.242
Gilbert, S. F., and Barresi, M. J. F. (2016). Developmental Biology, 11 edn. Sinauer Associates. doi: 10.1002/ajmg.a.38166
Gilboa, S. M., Devine, O. J., Kucik, J. E., Oster, M. E., Riehle-Colarusso, T., Nembhard, W. N., et al. (2016). Congenital heart defects in the United States: estimating the magnitude of the affected population in 2010. Circulation 134, 101–109. doi: 10.1161/CIRCULATIONAHA.115.019307
Gittenberger-de Groot, A. C., Bartelings, M. M., Deruiter, M. C., and Poelmann, R. E. (2005). Basics of cardiac development for the understanding of congenital heart malformations. Pediatr. Res. 57, 169–176. doi: 10.1203/01.PDR.0000148710.69159.61
Gorini, F., Chiappa, E., Gargani, L., and Picano, E. (2014). Potential effects of environmental chemical contamination in congenital heart disease. Pediatr. Cardiol. 35, 559–568. doi: 10.1007/s00246-014-0870-1
Gurtner, G. C., Davis, V., Li, H., McCoy, M. J., Sharpe, A., and Cybulsky, M. I. (1995). Targeted disruption of the murine VCAM1 gene: essential role of VCAM-1 in chorioallantoic fusion and placentation. Genes Dev. 9, 1–14.
Hachisuga, M., Oki, S., Kitajima, K., Ikuta, S., Sumi, T., Kato, K., et al. (2015). Hyperglycemia impairs left–right axis formation and thereby disturbs heart morphogenesis in mouse embryos. Proc. Natl. Acad. Sci. U.S.A. 112, E5300–E5307. doi: 10.1073/pnas.1504529112
Hahn, E., Szwast, A., Cnota, J., Levine, J. C., Fifer, C. G., Jaeggi, E., et al. (2016). Association between fetal growth, cerebral blood flow and neurodevelopmental outcome in univentricular fetuses. Ultrasound Obstet. Gynecol. 47, 460–465. doi: 10.1002/uog.14881
Han, M., Serrano, M. C., Lastra-Vicente, R., Brinez, P., Acharya, G., Huhta, J. C., et al. (2009). Folate rescues lithium-, homocysteine- and Wnt3A-induced vertebrate cardiac anomalies. Dis. Model. Mech. 2, 467–478. doi: 10.1242/dmm.001438
Helske, S., Vuorela, P., Carpén, O., Hornig, C., Weich, H., and Halmesmäki, E. (2001). Expression of vascular endothelial growth factor receptors 1, 2 and 3 in placentas from normal and complicated pregnancies. Mol. Hum. Reprod. 7, 205–210. doi: 10.1093/molehr/7.2.205
Huppertz, B., and Peeters, L. L. (2005). Vascular biology in implantation and placentation. Angiogenesis 8, 157–167. doi: 10.1007/s10456-005-9007-8
Jia, L., Zhou, X., Huang, X., Xu, X., Jia, Y., Wu, Y., et al. (2018). Maternal and umbilical cord serum-derived exosomes enhance endothelial cell proliferation and migration. FASEB J. 32, 1–10. doi: 10.1096/fj.201701337RR
Jones, H. N., Olbrych, S. K., Smith, K. L., Cnota, J. F., Habli, M., Ramos-Gonzales, O., et al. (2015). Hypoplastic left heart syndrome is associated with structural and vascular placental abnormalities and leptin dysregulation. Placenta 36, 1078–1086. doi: 10.1016/j.placenta.2015.08.003
Jorgensen, M., McPherson, E., Zaleski, C., Shivaram, P., and Cold, C. (2014). Stillbirth: the heart of the matter. Am. J. Med. Genet. A 164A, 691–699. doi: 10.1002/ajmg.a.36366.
Kaufmann, P., Mayhew, T. M., and Charnock-Jones, D. S. (2004). Aspects of human fetoplacental vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 25, 114–126. doi: 10.1016/j.placenta.2003.10.009
Khong, T. Y. (2004). Placental vascular development and neonatal outcome. Semin. Neonatol. 9, 255–263. doi: 10.1016/j.siny.2003.11.010
Kingdom, J., Huppertz, B., Seaward, G., and Kaufmann, P. (2000). Development of the placental villous tree and its consequences for fetal growth. Eur. J. Obstet. Gynecol. Reprod. Biol. 92, 35–43. doi: 10.1016/S0301-2115(00)00423-1
Kwee, L., Baldwin, H. S., Shen, H. M., Stewart, C. L., Buck, C., Buck, C. A., et al. (1995). Defective development of the embryonic and extraembryonic circulatory systems in vascular cell adhesion molecule (VCAM-1) deficient mice. Development 121, 489–503.
Laas, E., Lelong, N., Thieulin, A. C., Houyel, L., Bonnet, D., Ancel, P. Y., et al. (2012). Preterm birth and congenital heart defects: a population-based study. Pediatrics 130, e829–e837.
Lenhart, K. F., Holtzman, N. G., Williams, J. R., and Burdine, R. D. (2013). Integration of nodal and BMP signals in the heart requires FoxH1 to create left-right differences in cell migration rates that direct cardiac asymmetry. PLoS Genet. 9:e1003109. doi: 10.1371/journal.pgen.1003109.
Lin, F. J., You, L. R., Yu, C. T., Hsu, W. H., Tsai, M. J., and Tsai, S. Y. (2012). Endocardial cushion morphogenesis and coronary vessel development require Chicken ovalbumin upstream promoter-transcription factor II. Arterioscler. Thromb. Vasc. Biol. 32, e135–e146. doi: 10.1161/ATVBAHA.112.300255
Linask, K. K. (2013). The heart-placenta axis in the first month of pregnancy: induction and prevention of cardiovascular birth defects. J. Preg. 2013:320413. doi: 10.1155/2013/320413
Lindsey, S. E., Butcher, J. T., and Yalcin, H. C. (2014). Mechanical regulation of cardiac development. Front. Physiol. 5:318. doi: 10.3389/fphys.2014.00318
Liu, X., Mu, H., Luo, X., Xiao, X., Ding, Y., Yin, N., et al. (2014). Expression of Gadd45α in human early placenta and its role in trophoblast invasion. Placenta 35, 370–377. doi: 10.1016/j.placenta.2014.03.020
Lopes Floro, K., Artap, S. T., Preis, J. I., Fatkin, D., Chapman, G., Furtado, M. B., et al. (2011). Loss of Cited2 causes congenital heart disease by perturbing left-right patterning of the body axis. Hum. Mol. Genet. 20, 1097–1110. doi: 10.1093/hmg/ddq554
Lucas, A., Mialet-Perez, J., Daviaud, D., Parini, A., Marber, M. S., and Sicard, P. (2015). Gadd45γ regulates cardiomyocyte death and post-myocardial infarction left ventricular remodelling. Cardiovasc. Res. 108, 254–267. doi: 10.1093/cvr/cvv219
MacDonald, S. T., Bamforth, S. D., Braganca, J., Chen, C. M., Broadbent, C., Schneider, J. E., et al. (2012). A cell-autonomous role of Cited2 in controlling myocardial and coronary vascular development. Eur. Heart J. 34,2557–2565. doi: 10.1093/eurheartj/ehs056
Maschhoff, K. L., and Baldwin, H. S. (2000). Molecular determinants of neural crest migration. Am. J. Med. Genet. 97, 280–288. doi: 10.1002/1096-8628(200024)97:4<280::AID-AJMG1278>3.0.CO;2-N
Midgett, M., Thornberg, K., and Rugonyi, S. (2017). Blood flow patterns underlie developmental heart defects. Am. J. Physiol. Heart Circ. Physiol. 312, H632–H642. doi: 10.1152/ajpheart.00641.2016
Moreau, J. L., Artap, S. T., Shi, H., Chapman, G., Leone, G., Sparrow, D. B., et al. (2014). Cited2 is required in trophoblasts for correct placental capillary patterning. Dev. Biol. 392, 62–79. doi: 10.1016/j.ydbio.2014.04.023
Olson, E. N., and Srivastava, D. (1996). Molecular pathways controlling heart development. Science 272, 671–676.
Parra-Saavedra, M., Crovetto, F., Triunfo, S., Savchev, S., Peguero, A., Nadal, A., et al. (2013). Placental findings in late-onset SGA births without Doppler signs of placental insufficiency. Placenta 34, 1136–1141. doi: 10.1016/j.placenta.2013.09.018
Perez-Garcia, V., Fineberg, E., Wilson, R., Murray, A., Icoresi Mazzeo, C., Tudor, C., et al. (2018). Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 555, 463–468. doi: 10.1038/nature26002
Puri, K., Warshak, C. R., Habli, M. A., Yuan, A., Sahay, R. D., King, E. C., et al. (2017). Fetal somatic growth trajectory differs by type of congenital heart disease. Pediatr. Res. 83, 669–676. doi: 10.1038/pr.2017.275
Rajashekhar, G., Loganath, A., Roy, A. C., and Wong, Y. C. (2003). Expression and secretion of the vascular cell adhesion molecule-1 in human placenta and its decrease in fetal growth restriction. J. Soc. Gynecol. Investig. 10, 352–360. doi: 10.1016/S1071-5576(03)00121-7
Rhee, S., Chung, J. I., King, D. A., D'amato, G., Paik, D. T., Duan, A., et al. (2018). Endothelial deletion of Ino80 disrupts coronary angiogenesis and causes congenital heart disease. Nat. Commun. 9:368. doi: 10.1038/s41467-017-02796-3
Rossant, J., and Cross, J. C. (2001). Placental development: lessons from mouse mutants. Nat. Rev. Genet. 2, 538–548. doi: 10.1038/35080570
Ruiz, A., Cruz-Lemini, M., Masoller, N., Sanz-Cortés, M., Ferrer, Q., Ribera, I., et al. (2017). Longitudinal changes in fetal biometry and cerebroplacental hemodynamics in fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 49, 379–386. doi: 10.1002/uog.15970.
Russell, M. W., Chung, W. K., Kaltman, J. R., and Miller, T. A. (2018). Advances in the understanding of the genetic determinants of congenital heart disease and their impact on clinical outcomes. J. Am. Heart Assoc. 2:7. doi: 10.1161/JAHA.117.006906
Schleich, J. M. (2002). Development of the human heart: days 15–21. Heart 87:487. doi: 10.1136/heart.87.5.487
Serrano, M., Han, M., Brinez, P., and Linask, K. K. (2010). Fetal alcohol syndrome: cardiac birth defects in mice and prevention with folate. Am. J. Obstet. Gynecol. 203, 75.e77–75.e15. doi: 10.1016/j.ajog.2010.03.017
Smedts, H. P., van Uitert, E. M., Valkenburg, O., Laven, J. S., Eijkemans, M. J., Lindemans, J., et al. (2012). A derangement of the maternal lipid profile is associated with an elevated risk of congenital heart disease in the offspring. Nutr. Metab. Cardiovasc. Dis. 22, 477–485. doi: 10.1016/j.numecd.2010.07.016
Su, R. W., and Fazleabas, A. T. (2015). Implantation and Establishment of pregnancy in human and nonhuman primates. Adv. Anat. Embryol. Cell Biol. 216, 189–213. doi: 10.1007/978-3-319-15856-3_10
Sun, L. Q., MacGowan, C. K., Sled, J. G., Yoo, S. J., Manlhiot, C., Porayette, P., et al. (2015). Reduced fetal cerebral oxygen consumption is associated with smaller brain size in fetuses with congenital heart disease. Circulation 131, 1313–1323. doi: 10.1161/CIRCULATIONAHA.114.013051
Sylva, M., van den Hoff, M. J., and Moorman, A. F. (2014). Development of the human heart. Am. J. Med. Genet. A 164A, 1347–1371. doi: 10.1002/ajmg.a.35896.
Thornburg, K., O'Tierney, P., and Louey, S. (2010). The placenta is a programming agent for cardiovascular disease. Placenta 31, S54–S59. doi: 10.1016/j.placenta.2010.01.002
Thornburg, K. L., and Louey, S. (2013). Uteroplacental circulation and fetal vascular function and development. Curr. Vasc. Pharmacol. 11, 748–757. doi: 10.2174/1570161111311050012
Tyser, R. C., Miranda, A. M., Chen, C., Davidson, S. M., Srinivas, S., and Riley, P. R. (2016). Calcium handling precedes cardiac differentiation to initiate the first heartbeat. Elife 5:e17113. doi: 10.7554/eLife.17113
van der Linde, D., Konings, E. E., Slager, M. A., Witsenburg, M., Helbing, W. A., Takkenberg, J. J., et al. (2011). Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 58, 2241–2247. doi: 10.1016/j.jacc.2011.08.025
Withington, S. L., Scott, A. N., Saunders, D. N., Lopes Floro, K., Preis, J. I., Michalicek, J., et al. (2006). Loss of Cited2 affects trophoblast formation and vascularization of the mouse placenta. Dev. Biol. 294, 67–82. doi: 10.1016/j.ydbio.2006.02.025
Wong, P., Denburg, A., Dave, M., Levin, L., Morinis, J. O, Suleman, S., et al. (2018). Early life environment and social determinants of cardiac health in children with congenital heart disease. Paediatr. Child Health 23, 92–95. doi: 10.1093/pch/pxx146
Yang, Q., Chen, H., Correa, A., Devine, O., Mathews, T. J., and Honein, M. A. (2006). Racial differences in infant mortality attributable to birth defects in the United States, 1989-2002. Birth Defects Res. Part A Clin. Mol. Teratol. 76, 706–713. doi: 10.1002/bdra.20308
Keywords: placentation, congential heart defects, hemodynamics, heart development, molecular mechanisms
Citation: Courtney JA, Cnota JF and Jones HN (2018) The Role of Abnormal Placentation in Congenital Heart Disease; Cause, Correlate, or Consequence? Front. Physiol. 9:1045. doi: 10.3389/fphys.2018.01045
Received: 13 February 2018; Accepted: 13 July 2018;
Published: 07 August 2018.
Edited by:
Sandra Rugonyi, Oregon Health & Science University, United StatesReviewed by:
Alina Maloyan, Oregon Health & Science University, United StatesCopyright © 2018 Courtney, Cnota and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helen N. Jones, aGVsZW4uam9uZXNAY2NobWMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.