 Margreet C. M. Vissers
Margreet C. M. Vissers Andrew B. Das
Andrew B. Das- Centre for Free Radical Research, Department of Pathology and Biomedical Science, University of Otago, Christchurch, Christchurch, New Zealand
Whether vitamin C (ascorbate) has a role to play as an anti-cancer agent has been debated for decades. Ascorbate has been used by cancer patients in an unregulated environment, either as a dietary supplement or in pharmacological doses administered by infusion, with numerous reports of clinical benefit, but in the absence of rigorous clinical trial data. The design of appropriate clinical trials has been hindered by a lack of understanding of the mechanism(s) of action that would inform the choice of effective dose, timing of administration and likely responsive cancer models. More recently, expanded understanding of the biological activities of ascorbate has led to a number of plausible hypotheses for mechanisms of anti-cancer activity. Prominent among these are the generation of significant quantities of hydrogen peroxide by the autoxidation of supra-physiological concentrations of ascorbate and stimulation of the 2-oxoglutarate-dependent dioxygenase family of enzymes (2-OGDDs) that have a cofactor requirement for ascorbate. Hydrogen peroxide generation is postulated to generate oxidative stress that preferentially targets cancer cells. The 2-OGDDs include the hydroxylases that regulate the hypoxic response, a major driver of tumor survival, angiogenesis, stem cell phenotype and metastasis, and the epigenetic histone and DNA demethylases. The latter are of particular interest, with recent studies suggesting a promising role for ascorbate in the regulation of the ten-eleven translocase (TET) DNA demethylases in hematological cancers. Support for these proposed mechanisms has come from many in vitro studies, and xenograft animal models have consistently shown an anti-cancer effect of ascorbate administration. However, decisive evidence for any particular mechanism(s) of action is not yet available from an in vivo setting. With a number of early phase clinical trials currently underway, evidence for potential mechanism(s) of action is required to inform the most appropriate study design and choice of cancer model. Hopefully such information will result in sound clinical data that will avert adding any further controversy to this already contentious debate.
Introduction
Whether vitamin C has a role to play in the development and regulation of cancer growth has been a topic of investigation and discussion for decades. There is a substantial body of literature that documents potential anti-tumor effects of ascorbate in in vitro and in vivo settings, with many reporting cytotoxicity toward cancer cells and a slowing of tumor growth in animal models (for reviews, see Du et al., 2012; Park, 2013; Fritz et al., 2014; Wilson et al., 2014; Mata et al., 2016; Cimmino et al., 2018; Klimant et al., 2018). Human clinical studies, however, have been infrequent, with most recent phase I/II studies aiming to determine the tolerability of pharmacological doses of ascorbate for patients with advanced cancer (Hoffer et al., 2008, 2015; Stephenson et al., 2013). Some of these studies have suggested that high dose ascorbate treatment may have a clinical benefit for patients with pancreatic cancer (Monti et al., 2012; Cieslak and Cullen, 2015) and other advanced cancers (Hoffer et al., 2015) but too few patients have been analyzed in these studies to date. In addition, the use of dietary vitamin C supplements has been linked to improved patient outcome in breast cancer (Harris et al., 2013, 2014).
Interest in the use of ascorbate for cancer was initiated in the 1970’s when Pauling and Cameron advocated for its use following their observations of apparently extended survival in patients with advanced cancer treated with intravenous infusions of ∼10 g ascorbate daily (Cameron and Pauling, 1976, 1978). At the time, these claims were met with incredulity by many, mostly because the available information on the biological activity of ascorbate could not be reconciled with a plausible anti-cancer mechanism. There were proposals that ascorbate deficiency might interfere with the synthesis of connective tissues, particularly collagen (McCormick, 1959; Cameron and Pauling, 1973), but there was no explanation that could explain the need for intravenous administration, as deficiency could be easily overcome by dietary supplementation. The Mayo Clinic attempted to repeat Pauling and Cameron’s results in a randomized placebo-controlled study in patients with advanced cancer, but they failed to detect any clinical benefit after daily oral administration of 10 g of vitamin C (Moertel et al., 1985). The fundamental difference between the two data sets is now believed to be the oral versus intravenous route of ascorbate, which is known to result in significantly altered pharmacokinetics of the vitamin (Padayatty and Levine, 2001; Padayatty et al., 2004). When viewed alongside a more sophisticated understanding of tumor biology, this information of the pharmacokinetics and functions of vitamin C has led to the development of new hypotheses for potential mechanisms of anti-cancer activity. This crucial information was unavailable in the 1970’s, but has now engendered renewed consideration of the potential role for ascorbate in cancer.
High dose ascorbate infusions are an easily accessed therapy used by both medical practitioners and complementary and alternative medicine providers, but without robust evidence of clinical efficacy (Padayatty et al., 2010). There is an acknowledged need for good clinical studies to determine the anticancer efficacy of this practice and to establish appropriate clinical guidelines. Case reports suggest that there may be circumstances under which vitamin C can provide a clinical benefit (these are usually high intravenous doses, given as a course over months) (Drisko et al., 2003; Padayatty et al., 2006; Mikirova et al., 2016; Raymond et al., 2016) and recent phase I trials have indicated that high-dose vitamin C is a useful adjunct to chemotherapy (Welsh et al., 2013; Hoffer et al., 2015). However, given the prevalence of ascorbate use by cancer patients, clinical advantage is not commonly seen, suggesting that only a subset of patients may benefit. This highlights the problem: how to identify the potentially responsive patients and determine the best treatment regimen, dose and frequency of intervention? To address these questions, it is essential that we understand how ascorbate might act in cancer. Currently a number of hypotheses are under investigation and in this review we will consider the evidence to date in support of these, evaluated against what we know about ascorbate chemistry and biology.
Uptake and Metabolism of Ascorbate
Ascorbate is a small, highly water-soluble molecule derived from glucose and present in all plants and animals (Hornig, 1981; Smirnoff and Wheeler, 2000; Padayatty and Levine, 2016). Most animals synthesize it in the liver or kidneys but it has become a vitamin for humans, other primates, guinea pigs and fruit bats, which have mutations in gulonolactone oxidase, the terminal enzyme in the biosynthetic pathway (Chatterjee et al., 1975; Banhegyi et al., 1997; Linster and Van Schaftingen, 2007; Du et al., 2012). In all animals, ascorbate is transported to the tissues via the circulation and most cells concentrate the vitamin to levels many-fold above plasma levels (average ∼50 μM) through active uptake by the sodium-dependent vitamin C transporters SVCT1 and SVCT2 (Tsao, 1997; May and Qu, 2005; Savini et al., 2008; Harrison and May, 2009; Mandl et al., 2009; Nualart et al., 2014). An exception to this is the red blood cell, which does not express SVCT2 and accumulates ascorbate by uptake of dehydroascorbate (DHA) via the GLUTs (Tu et al., 2017). Tissue levels vary considerably, with some organs, notably brain, adrenals, liver, and white cells containing concentrations up to 20 mM (Tsao, 1997). High intracellular levels are thought to reflect a demand for ascorbate as an essential enzyme cofactor (Tsao, 1997; May and Qu, 2005).
The high water-solubility and active transport of ascorbate in the body means that this compound is readily acquired and distributed, but not stored, and turnover is constant. The reliance on dietary intake in humans can therefore lead to deficiency if adequate intake is not maintained and this is pertinent to the effects of ascorbate supplementation in cancer. Turnover appears to be accelerated during periods of illness (Bonham et al., 1999; Fain et al., 2003; Gan et al., 2008; Evans-Olders et al., 2010) and, although information is sparse, many cancer patients are found to be ascorbate deficient when measurements have been made (Anthony and Schorah, 1982; Ray and Husain, 2001; Mayland et al., 2005; Shah et al., 2009; Badid et al., 2010; Nagamma et al., 2014). These low plasma levels are likely to correlate with lower tissue levels; this has been demonstrated in white blood cells (Levine et al., 2001) and in a mouse model of ascorbate dependency low plasma levels resulted in almost complete tissue deficiency (Vissers et al., 2011). In contrast, animals that synthesize ascorbate are known to maintain plasma saturation levels regardless of health challenges, dramatically increasing production to compensate for increased turnover when ill (Chatterjee et al., 1975; Michels and Frei, 2013; Campbell et al., 2015). This is an important point for consideration in experiments using animal tumor xenograft models. Unless the study is carried out with guinea pigs or a mutant animal such as the Gulo-/- knock-out mouse, the plasma and tissue ascorbate levels can be maintained at a high baseline level by endogenous synthesis and supplementation may have little impact. Wild-type animals, and knock-out animals bred onto a wild-type background such as the Gulo-/- mouse, also demonstrate a limited capacity for ascorbate uptake through the gut (Michels and Frei, 2013). Hence, if these animals are used in an experimental setting, dietary supplementation may have limited effect, and plasma and tissue levels should be monitored throughout to confirm ascorbate status and the applicability of the model for the human condition. It should be noted that such measurements are rarely carried out, and this can complicate the interpretation of published experimental data.
The Chemistry and Functions of Ascorbate
The chemistry of ascorbate dictates its biological activity. It is possibly best known for its capacity to act as an antioxidant: this property reflects its ability to readily undergo one- or two-electron oxidation, generating the relatively stable ascorbyl radical or DHA, respectively. DHA is unstable at neutral pH, and unless it is reduced in vivo by glutathione or thioredoxin to regenerate reduced ascorbate, it will rapidly decompose to diketogulonic, oxalic and threonic acids (Washburn and Wells, 1999; Smirnoff, 2000), which are eliminated from the body via the kidneys (Washko et al., 1992, 1993; Washburn and Wells, 1999; Linster and Van Schaftingen, 2007). Consequently, DHA represents only a very small fraction of the ascorbate present in vivo and is not likely to be present at concentrations above 1–2 μM (Dhariwal et al., 1991; Michels and Frei, 2013; Pullar et al., 2018). The amount present in vivo may often be overestimated when blood or tissues are analyzed, as it seems that most of the DHA detected is likely to reflect oxidation in air during sample processing (Dhariwal et al., 1991; Levine et al., 1998; Michels and Frei, 2013; Pullar et al., 2018). The oxidation of ascorbate and breakdown of DHA is likely to account for daily turnover, and this may be exacerbated during periods of illness (Chatterjee et al., 1975; Bonham et al., 1999; Fain et al., 2003; Gan et al., 2008; Evans-Olders et al., 2010).
Ascorbate is able to chelate and reduce transition metal ions, particularly Fe3+ and Cu2+ (Du et al., 2012; Padayatty and Levine, 2016), and this property contributes to its ability to promote iron uptake from the diet (Lane and Richardson, 2014). It also enables a pro-oxidant activity through the recycling of Fe2+ (Eq. 1), and in the presence of oxygen leads to the generation of superoxide, H2O2 and highly reactive oxidants such as the hydroxyl radical by promoting the Fenton reaction (Eq. 2) and Haber–Weiss chemistry (Winterbourn, 1981; Buettner, 1987; Smirnoff, 2000; Koppenol, 2001; Levine et al., 2011; Michels and Frei, 2013) (Figure 1).
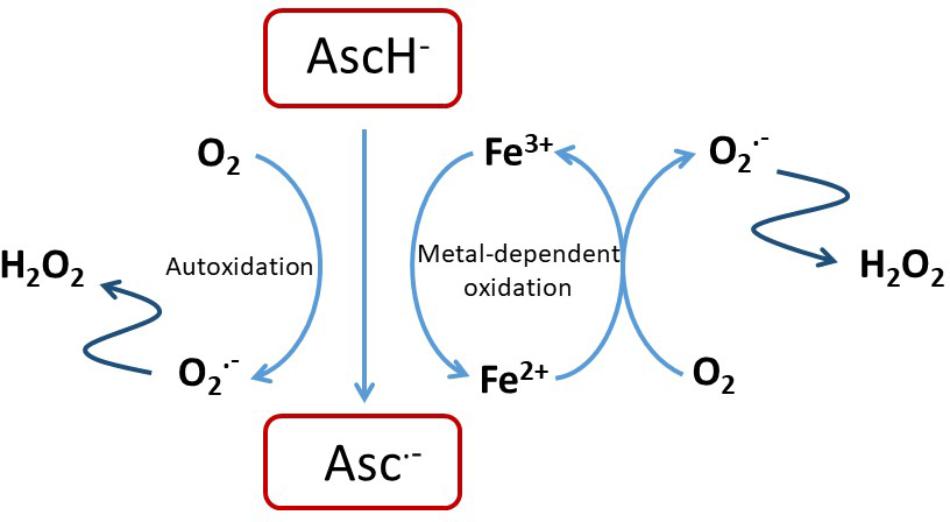
FIGURE 1. Pro-oxidant activity of ascorbate. Reactions of ascorbate with oxygen or free transition metal ions such as Fe2+/3+ or Cu+/2+ can result in the generation of H2O2 that is itself cytotoxic of that can undergo further reaction to exacerbate oxidative stress (Shah et al., 2009; Vissers et al., 2011; Nualart et al., 2014).
The ability to reduce transition metals is likely to be fundamental to the activity of ascorbate as an essential co-factor for many Fe- and Cu-containing enzymes; in this capacity it ensures that enzyme activity is optimized by the maintenance of the active-site metals in the reduced state (Kivirikko et al., 1989; Rebouche, 1991; Ozer and Bruick, 2007; Mandl et al., 2009; Grosso et al., 2013; Vissers et al., 2014).
Anti-Cancer Activities of Ascorbate
All the known properties of ascorbate are being considered for the investigation of its potential anti-cancer activity. There are numerous studies that have demonstrated a cytotoxic effect of ascorbate on tumor cells in vitro, either alone or in combination with chemotherapeutics (Wells et al., 1995; Reddy et al., 2001; Pathak et al., 2002; Wozniak and Anuszewska, 2002; Guerriero et al., 2006; Kassouf et al., 2006; Chen et al., 2007, 2012; Martinotti et al., 2011; Verrax et al., 2011; Ma et al., 2014; Cieslak et al., 2015; Xia et al., 2017) or radiation (Herst et al., 2012; Castro et al., 2014). The cytotoxicity in many of these studies reflects the oxidative stress resulting from the H2O2 generated in cell culture medium when ascorbate is present at concentrations of 1 mM or above, and manifests as increased cell cycle arrest, p53 upregulation, decreased ATP levels, compromised mitochondrial function, suppression of antioxidant gene expression NrF-2 and/or cell death by apoptosis (Tarumoto et al., 2004; Fromberg et al., 2011; Rouleau et al., 2016; Yang et al., 2017). Anti-cancer effects have also been demonstrated with ascorbate levels well below 1 mM: levels as low as 100 μM or even 1 μM in the culture medium enhanced the susceptibility of cancer cells to etoposide, cisplatin, or doxorubicin (Kurbacher et al., 1996; Reddy et al., 2001; Tarumoto et al., 2004; An et al., 2011). The mechanism of action at these low concentrations remains unclear, but potentially involves modification of cell survival pathways involving p53 (Kurbacher et al., 1996; Reddy et al., 2001; Tarumoto et al., 2004; An et al., 2011). The highly variable experimental designs and differences in outcome in these in vitro studies highlight the complex nature of investigations involving the addition of ascorbate to cell culture systems, with both artifacts and actual cell functional changes being possible. The interactions of ascorbate with chemotherapeutics are likely to reflect the redox properties of extracellular ascorbate and the many intracellular co-factor activities. There is also a clinical concern that ascorbate could impede the action of chemotherapy drugs (Ludke et al., 2009, 2010; Perrone et al., 2009), but this area remains poorly understood. There is significant consensus that ascorbate can act synergistically with gemcitabine (Kassouf et al., 2006; Espey et al., 2011; Martinotti et al., 2011; Volta et al., 2013; Cieslak et al., 2015), and treatment with a combination of the two agents did not result in any adverse events in recent phase I/II trials (Monti et al., 2012; Welsh et al., 2013).
Consideration of all the potential interactions between ascorbate and chemotherapeutics is beyond the scope of this review, but it should be noted that a clear understanding of this area will be important when considering introducing ascorbate into a clinical cancer setting. Instead, we aim to consider the evidence for the most current hypotheses that suggest that the anti-cancer activity of ascorbate reflects either its redox, pro-oxidant or enzyme co-factor activity (Figure 2). A significant body of literature is emerging that tests these ideas and we will consider the extent to which the current evidence supports these mechanisms.
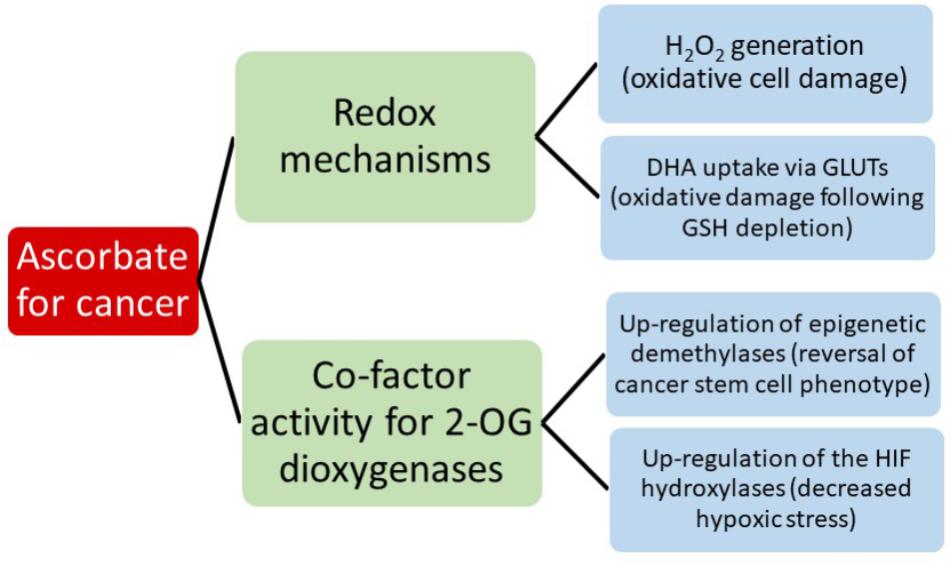
FIGURE 2. A summary of the current hypotheses that may contribute to anti-cancer activity by ascorbate.
Mouse Xenograft Models
The use of mouse tumor xenograft models is common to many investigations, and there is a high degree of consistency in the reported results: administration of supplemental ascorbate, either through the diet or via high dose peritoneal administration, results in a decreased growth rate with a range of different tumors (Gao et al., 2007; Chen et al., 2008; Cha et al., 2011, 2013; Espey et al., 2011; Ma et al., 2014; Campbell et al., 2015, 2016b; Yun et al., 2015). The similarity of the data despite differences in tumor models and dosage regimes for ascorbate has reinforced support for ascorbate having an anti-cancer function, but in most cases the mechanism of action is inferred from data obtained in vitro. Very few studies have reported changes in the tumor biology that allow insight into the mechanism of action and conclusions beyond a simple correlation between tumor growth and ascorbate availability. Without this information, the in vivo data from animal studies cannot directly test a working hypothesis and this will be considered below.
Ascorbate as a Pro-oxidant
The realization that intravenous ascorbate infusion results in plasma concentrations in the mM range, albeit transiently (half-life ∼2 h) (Padayatty et al., 2004), led to the suggestion that autoxidation of ascorbate and/or the stimulation of metal-catalyzed production of reactive oxygen species could generate significant quantities of H2O2 that are selectively toxic to cancer cells (Carosio et al., 2007; Belin et al., 2009). In tissue culture media, ascorbate concentrations above 1 mM are known to be cytotoxic (Buettner, 1988; Miller et al., 1990; Halliwell et al., 2000; Chen et al., 2005, 2007, 2008). The rate of ascorbate autoxidation is slowed when iron chelators are added to the medium and cytotoxicity can be inhibited by catalase, supporting the dependency of this phenomenon on the generation of H2O2 (Halliwell et al., 2000; Clement et al., 2001; Wee et al., 2003; Chen et al., 2007). Cancer cells have been shown to be more susceptible to H2O2 and oxidative stress than primary cell lines which is related to the metabolism of H2O2 and antioxidant capacity (Doskey et al., 2010; Du et al., 2010; Erudaitius et al., 2010; Olney et al., 2013; Schoenfeld et al., 2017). In contrast to tissue culture media, autoxidation of ascorbate is slow in plasma and at neutral pH (Buettner, 1988), but the ascorbyl radical has been detected in biological fluids that can contain traces of catalytic Fe (Buettner and Chamulitrat, 1990; Chen et al., 2007).
As indicated above, ascorbate-mediated cytotoxicity toward cancer cells in vitro has been carried out with concentrations in excess of the 1 mM threshold that induces significant H2O2 (Chen et al., 2011; Espey et al., 2011; Fromberg et al., 2011; Rouleau et al., 2016; Kim et al., 2018). These observations have fuelled the pilot clinical studies and phase I clinical trials involving the intravenous administration of pharmacological doses of ascorbate that aim to achieve mM levels in the circulation and extracellular fluid of the tumor (Hoffer et al., 2008, 2015; Monti et al., 2012; Stephenson et al., 2013; Welsh et al., 2013). While there are suggestions of a clinical benefit from these vitamin C infusions (Hoffer et al., 2008, 2015; Stephenson et al., 2013; Welsh et al., 2013), it remains a challenge to determine whether this is due to an ascorbate-mediated pro-oxidant effect. It is unclear whether H2O2 is generated in sufficient concentrations in a tumor in vivo to damage the cancer cells. Given that the autoxidation of ascorbate is highly dependent on the availability of oxygen, there is a question around the impact of the hypoxic tumor environment on the generation of H2O2. To our knowledge, definitive evidence for H2O2-mediated cytotoxicity initiated by supra-physiological ascorbate concentrations in vivo, such as detection of oxidative damage in tumors exposed to high dose ascorbate in situ, is not yet available.
Cancer Cell Uptake of DHA and Associated Oxidative Stress
Dehydroascorbate is structurally similar to glucose and can be taken up into cells via the GLUTs and this may contribute to the intracellular pool in red blood cells, in neutrophils at sites of infection and in other regions of the body (Rumsey et al., 1997, 2000; Corpe et al., 2005; Wilson, 2005; Michels and Frei, 2013). Once inside the cell, DHA is reduced by GSH, NADH, and NADPH-dependent enzymes, thereby potentially depleting the cell of these crucial molecules (May et al., 1997, 1999; May, 2002, 2011). A connection between this phenomenon and the upregulation of GLUT1 in KRAS and BRAF mutant cells was recently proposed to account for the anti-tumor activity of ascorbate in colorectal cancer (Yun et al., 2015). KRAS and BRAF mutations are common in colorectal cancer and are associated with upregulation of GLUT1 and a glycolytic phenotype. Cultured cancer cells harboring mutations in KRAS and BRAF were shown to take up DHA via GLUT1 when incubated with mM concentrations of ascorbate, and this was associated with a loss of cell viability. The results were extrapolated into an in vivo mouse model with implanted mutant KRAS and BRAF cancer cells in which tumor growth was slowed when the animals were administered high doses of vitamin C by daily intraperitoneal injection of 4 g/kg. The data in this comprehensive study clearly show that the upregulated expression of GLUT1 can result in rapid uptake of DHA by these cancer cell lines, but there is much less certainty about the causal relationship between this and the observed cytotoxicity. At the 1–2 mM concentrations of ascorbate used, the generation of H2O2 in the medium via autoxidation, as discussed above, could equally result in depletion of GSH and cytotoxicity. This possibility could be eliminated by including a H2O2 scavenger such as catalase in the culture medium, and without this information, it remains uncertain whether cell death resulted from the uptake of DHA and subsequent intracellular redox stress, as was proposed, or whether it was due to H2O2 generated in the medium. Whether KRAS-mutant tumor growth inhibition in the vitamin C-treated mice was related to markers of oxidative stress in the tumor was not tested. Higher levels of ascorbate, and fewer intestinal polyps, were reported in KrasG12D mice treated with tamoxifen than in mice without this mutation, but whether these polyps showed higher levels of oxidative stress was not determined. Metabolic markers of glycolysis were decreased in KRAS and BRAF mutant cells in culture, and although this may be consistent with the hypothesis under investigation, it could equally be effected by downregulation of a hypoxic phenotype or by H2O2-mediated toxicity.
As previously discussed, DHA is unstable at neutral pH and is generally not detected in plasma or tissue samples (Dhariwal et al., 1991; Michels and Frei, 2013; Pullar et al., 2018). Therefore, when considering the likelihood of DHA uptake via the GLUTs contributing to the anti-tumor activity of ascorbate, it will be important to demonstrate that DHA exists in vivo in sufficiently high concentrations to be able to compete with physiological concentrations of glucose. Interestingly, ascorbate was recently reported to enhance the cytotoxicity of cetuximab in KRAS mutant colon cancer cell lines and co-incidentally reverse the glycolytic Warburg phenotype (Aguilera et al., 2016). Also, in another study, this activity was associated with the cellular accumulation of ascorbate in an SVCT2-dependent manner (Jung et al., 2016). Together, these studies paint a complex picture of the interaction of ascorbate with colon cancer cells.
Downregulation of the Hypoxic Phenotype
Rapid cell division and poor blood vessel formation in a developing tumor result in local areas of oxygen and nutrient deprivation, leading to activation of the HIFs. These transcription factors determine the successful tumor adaptation to a stressful microenvironment, driving the transcription of numerous genes involved in glycolysis and glucose transport, angiogenesis, metastasis and resistance to chemo- and radio-therapy (Semenza, 2010, 2016; Ratcliffe, 2013). High HIF activity has been shown to promote the expression of a stem cell phenotype in breast cancer (De Francesco et al., 2015; Semenza, 2015, 2016) and is associated with a poor prognosis in a number of cancers (Dachs and Tozer, 2000; Vleugel et al., 2005; Cao et al., 2009; Semenza, 2010, 2015, 2016; Volinia et al., 2012; Wang et al., 2012, 2014; Zhang et al., 2012; Deb et al., 2014; Liu et al., 2014; Li et al., 2016; Schoning et al., 2017). Consequently the HIFs are now considered an important target for cancer therapy.
Activation of the HIFs is controlled by proline and asparaginyl hydroxylases (the HIF hydroxylases) that modify the regulatory HIF-α subunit (Bishop and Ratcliffe, 2014; Pugh and Ratcliffe, 2017), targeting the protein for proteasomal degradation and preventing the formation of an active transcriptional complex. The HIF hydroxylases belong to the family of iron-containing dioxygenases that utilize oxygen and 2-oxoglutarate as substrates (Ozer and Bruick, 2007), and for which ascorbate functions as a co-factor, possibly by maintaining the active site Fe in the reduced state (Koivunen et al., 2004). The co-factor role is specific to ascorbate (Myllyla et al., 1978), which is structurally specific for the hydroxylase active site (Kaczmarek et al., 2009). When cells are deficient in ascorbate, HIF hydroxylase activity is compromised and HIF transcription activity is increased, particularly in response to conditions of mild or moderate hypoxia (Knowles et al., 2003; Vissers et al., 2007; Kuiper et al., 2014a; Campbell et al., 2016a). These observations have led to the hypothesis that increasing ascorbate supply to cancer cells could stimulate the activity of the HIF hydroxylases and decrease the activation of the HIFs, thereby slowing tumor growth rates.
There is a growing body of evidence consistent with this hypothesis. Investigations with the vitamin C-dependent Gulo-/- mouse have indicated a strong association between tumor ascorbate content, HIF activation and tumor growth in vivo following variable dietary intake or intra-peritoneal injection of pharmacological ascorbate (Campbell et al., 2015, 2016a). The daily administration of high dose vitamin C to tumor-bearing Gulo-/- mice indicated that HIF-1 transcriptional activity was depressed in association with ascorbate uptake. Elevation of tumor ascorbate content was transient following intraperitoneal infusion and levels were maintained with daily, but not alternate day, administration. Decreased HIF-1 expression and slowed tumor growth also required daily administration and ascorbate levels were associated with reduced tumor microvessel density and decreased regions of hypoxia, suggesting more effective oxygen delivery throughout the tumor (Campbell et al., 2016b).
Retrospective analysis of human tumor tissue has identified an inverse association between the amount of ascorbate present in the tumor and markers of HIF activation in endometrial, colorectal and thyroid cancer tissue (Kuiper et al., 2010, 2014b; Jozwiak et al., 2015). Higher tumor ascorbate was associated with increased disease-free survival for colorectal cancer patients (Kuiper et al., 2014b). Taken together, these data provide strong evidence for an ascorbate-mediated downregulation of HIF transcriptional activity that would be consistent with slower tumor growth.
Epigenetic Mechanisms of Action
Genetic and epigenetic changes are inextricably linked with the development of cancer (Baylin and Jones, 2011, 2016). One of the most striking epigenetic changes in cancer is global DNA hypomethylation, which can result in genomic instability and increased chromosomal fragility (Berman et al., 2011; Ehrlich and Lacey, 2013). Furthermore, hypomethylation can activate the transcription of transposable elements and oncogenes, providing multiple mechanisms by which it can contribute to oncogenesis. In addition to global hypomethylation, hypermethylation localized to the promotors of tumor suppressor genes has also been established (Baylin and Jones, 2011). In contrast, most gene promotors that contain CpG islands are not methylated in adult stem cells, and this is crucial for these genes to remain active or ready to be activated (Shen and Laird, 2013). These findings are encouraging for the development of cancer therapies because epigenetic changes are reversible, whereas the cancer-driving mutations themselves are much more difficult to target and revert.
The role of ascorbate in epigenetics has received increasing attention in multiple contexts, from the normal functioning of cells to the treatment of cancer (Lorsbach et al., 2003; Monfort and Wutz, 2013; Young et al., 2015; Camarena and Wang, 2016; Gillberg et al., 2017; Cimmino et al., 2018; Mastrangelo et al., 2018). Of particular relevance to this discussion is the direct involvement of the 2-OGDDs in epigenetic regulation, specifically the ten-eleven translocase (TET) and JMJC families. The TET family was named after the initial discovery of TET1 as a translocation partner of MLL in AML with t(10;11)(q22;q23) (Ono et al., 2002; Lorsbach et al., 2003). Subsequent homology searches in mammalian genomes detected two other members of this family, TET2 and TET3, which along with TET1 catalyze the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) (Tahiliani et al., 2009). Further oxidation of 5-hmC to 5-formylcytosine (5fC) and then 5-carboxylcytosine (5caC) is also carried out by the TET proteins. Collectively, these oxidative steps lead to both active and passive demethylation of DNA (He et al., 2011; Kohli and Zhang, 2013). The JMJC family contains more than 20 proteins that collectively are able to demethylate mono-, di-, and tri- methylated histone lysine residues (Tsukada et al., 2006; Monfort and Wutz, 2013). Each demethylation step involves the oxidation of a methyl group, followed by the spontaneous removal of formaldehyde. These discoveries have established the TET and JMJC dioxygenases as integral components for active demethylation of DNA and histones, respectively.
Altered expression or mutations affecting these dioxygenases have been detected in solid tumors from a wide range of tissues as well as in hematological malignancies (Gillberg et al., 2017). Given that ascorbate is required for optimal activity of both families (Klose et al., 2006; Tsukada et al., 2006; Minor et al., 2013) and that that the majority of mutations in these cancers affect one copy of the gene, it is possible that ascorbate could compensate by increasing the remaining enzyme activity. The majority of evidence supporting this mechanism comes from models of AML. Next generation sequencing of large cohorts of patients with AML have shown that approximately 10% of patients carry a mutation in TET2 that mostly affects only one allele (Ley et al., 2013; Papaemmanuil et al., 2016). JMJC mutations are much less frequent with changes to KDM5A and KDM6A each detected in ∼1% of patients. Interestingly, mutations in IDH are mutually exclusive with TET2, with IDH1 or IDH2 affected in approximately 20% of cases. The reason for this exclusivity demonstrates the strong link between metabolism and epigenetics. IDH converts isocitrate to 2-OG (a required cofactor for TET and JmjC enzymes). In contrast, mutant IDH (mutIDH) generates 2-hydroxyglutarate (2-HG) instead, which has been shown to impair hematopoiesis by inhibiting TET2 (Figueroa et al., 2010; Losman et al., 2013).
Building on this information, three recent studies have explored the potential of ascorbate to restore normal cell function in mouse and cellular models of leukemia involving either mutant TET2 or IDH (Agathocleous et al., 2017; Cimmino et al., 2017; Shenoy et al., 2017). Cimmino et al. (2017) used a transgenic mouse model whereby TET2 knockdown was inducible and reversible using RNAi. They showed that TET2 knockdown resulted in aberrant self-renewal, loss of myeloid lineage markers, significant hypermethylation, and decreased transcription of genes that are hypermethylated in AML patients. Both TET2 restoration and ascorbate (250 μM) reversed these changes. It is interesting to note that the addition of catalase (1 unit/mL of medium) had no effect on ascorbate’s ability to reverse aberrant self-renewal in re-plating assays. Furthermore, ascorbate at this concentration did not increase intracellular dichlorofluorescein fluorescence, an indicator of intracellular reactive oxygen species, which could be detected with as little as 50 μM H2O2 (Wang et al., 2014).
Agathocleous et al. (2017) used a number of different mouse models including various combinations of Tet2 deficiency, Gulo-/-, and Flt3ITD (an activating mutation that cooperates with Tet2 loss to induce AML). Firstly, they found that human and mouse HSCs had unusually high ascorbate levels that correlated with expression levels of the ascorbate transporter Slc23a2. Secondly, they showed that Gulo-/- mice had increased number HSC numbers that was in part due to decreased TET2 activity. In an elegant series of experiments involving transplantation of Flt3ITD donor bone marrow cells into either wild-type or Gulo-/- recipients, they showed that ascorbate deficiency was able to cooperate with Flt3ITD to promote leukemogenesis in a manner similar to Tet2 loss. Furthermore, the development of myeloid leukemia was accelerated when Tet2Δ/+;Flt3ITD donor bone marrow cells were transplanted into Gulo-/- recipients with respect to wild-type recipients. Finally, supplementation of a 1% ascorbate diet (Harlan) to Tet2Δ/+;Flt3ITD;Gulo-/-mice at 7 weeks, significantly prolonged survival compared to those that were not supplemented. Related experiments using Tet2Δ/Δ;Flt3ITD donor bone marrow cells showed that these effects were mediate via Tet2 dependent as well as Tet2 independent mechanisms. It is possible that Tet2 independent effects are mediated via Tet1 or Tet3, but this will need to be explored in future experiments.
The third study utilized HOXA9-immortalized mouse bone marrow cells expressing IDH1R132H in order to explore the effects of ascorbate treatment (Mingay et al., 2018). Ascorbate treatment (345 μM) promoted DNA demethylation at enhancers that are implicated in myeloid differentiation. Ascorbate treatment also resulted in the promotor demethylation and increased expression of several key hematopoietic genes. It is important to note that 2-phosphate ascorbic acid was used for cell culture experiments. This was done in order to explicitly avoid the extracellular production of H2O2.
Cumulatively, these data suggest that ascorbate is able to mitigate the effect of Tet2 loss in models of leukemia at least in part by upregulating TET2 activity. Given the low concentrations of ascorbate used, it is unlikely that the effects seen are mediated via the production of H2O2. These findings warrant the investigation of using ascorbate to improve TET activity in patients with heterozygous TET or IDH mutations. This is particularly relevant for AML, where TET2 and IDH mutations are early drivers (Papaemmanuil et al., 2016). TET2 mutations occur in the white blood cells of otherwise healthy adults with clonal hematopoiesis (Busque et al., 2012), and it is conceivable that ascorbate supplementation could prevent progression to myelodysplastic syndrome or overt AML. One recent clinical trial in AML patients looked at ascorbate supplementation as an adjunct to DNA methyltransferase inhibition (DNMTi) therapy in elderly patients aged 60–87 (Zhao et al., 2018). They compared ascorbate plus low-dose decitabine prior to aclarubicin and cytarabine (A-DCAG, n = 39) vs. DCAG alone (n = 34). Clinical remission was significantly higher after one round of treatment in the ascorbate group, which was associated with a higher overall survival. Unfortunately, no data was provided with regards to TET2 mutation status in the patient group. Future clinical trials involving ascorbate will need to stratify response by mutation status if epigenetic mechanisms of action are to be validated in patient cohorts.
Pharmacokinetic Considerations for Ascorbate in Cancer
The hypotheses discussed above all require adequate access for ascorbate to the tumor cells via effective distribution throughout the tumor environment. An understanding of the pharmacokinetics of ascorbate delivery into the extracellular tumor space and uptake into the cells is essential to make sense of the impact of ascorbate administration and to design effective treatment protocols for clinical intervention studies. As noted above, there is a fundamental difference between the plasma pharmacokinetics after oral or intravenous administration (Padayatty et al., 2004). Modeling the profile of plasma levels following intravenous infusion predicted that a peak concentration in the mM range would be achievable, and this has subsequently been measured in a number of studies (Hoffer et al., 2008; Robitaille et al., 2009; Stephenson et al., 2013). In tumor-bearing Gulo-/- mice, elevated ascorbate levels were seen to persist in the tumor for more than 24 h post administration of high dose intraperitoneal injection, and this was attributed to increased stability in the hypoxic tumor environment (Campbell et al., 2016b).
From all current evidence, including animal studies and the information available from the available human clinical studies, there does seem to be an advantage for intravenous infusion for cancer, but optimal protocols for dosage and frequency of administration have not been developed. A dose of ∼1 g/kg is often administered to cancer patients (Padayatty et al., 2010) and is apparently well tolerated with minimal side-effects (Stephenson et al., 2013). That the frequency of ascorbate administration may also be important is suggested by the finding that the anti-tumor activity of ascorbate was more effective following daily administration to Gulo-/- mice than when the infusions were given on alternate days (Campbell et al., 2016b).
Tumor cells, as all tissue cells, are dependent on the plasma supply for nutrient delivery. The poor vasculature in tumors prevents effective oxygen delivery and drives the upregulation of the hypoxic response (Ratcliffe, 2013; Semenza, 2016). Oxygen has a diffusion distance of around 100 μm in tissues and this coincides with the inter-vessel distance in well-perfused normal tissues (Ratcliffe, 2013; Semenza, 2016). It is possible, therefore, that ascorbate delivery is similarly compromised when the vasculature is dysfunctional and disorganized. When the diffusion of ascorbate through tissue layers was modeled in an in vitro setting, it was found to diffuse through a multi-cell layer in a peri-cellular manner (Kuiper et al., 2014c). Based on the diffusion parameters and on the measured kinetics of uptake into the cells, the likely distribution of ascorbate in a tumor, at variable plasma concentrations, was modeled. These data indicated that, when plasma concentrations are very low, there is virtually no ascorbate in the tissues and only those cells in the immediate vicinity of the blood vessel are able to accumulate any ascorbate (Figure 3). This scenario, modeled for 10 μM plasma levels, represents a state approaching scurvy. When plasma concentrations are 50 μM, a level considered in the healthy range but below plasma saturation (Carr and Frei, 1999; Levine et al., 2001), low levels of ascorbate penetrate up to 100 μm from the blood vessel, but cellular accumulation to physiological low mM concentrations is only seen in the cells immediately adjacent to the blood vessel (Figure 3). Optimal intracellular levels could be achieved when plasma is saturated (100 μM), but the diffusion zone does not extend beyond 100 μm. The inference is that 100 μM plasma ascorbate, which is the maximum achievable by oral intake, would not be sufficient to ensure effective distribution to the cells throughout a poorly perfused tumor. When plasma levels were increased to the mM range, the model indicated optimal uptake of cells even at 200 μm from the blood vessel. This information supports the rationale for a number of the hypotheses discussed in this review that indicate an advantage of intravenous ascorbate infusions over oral intake.
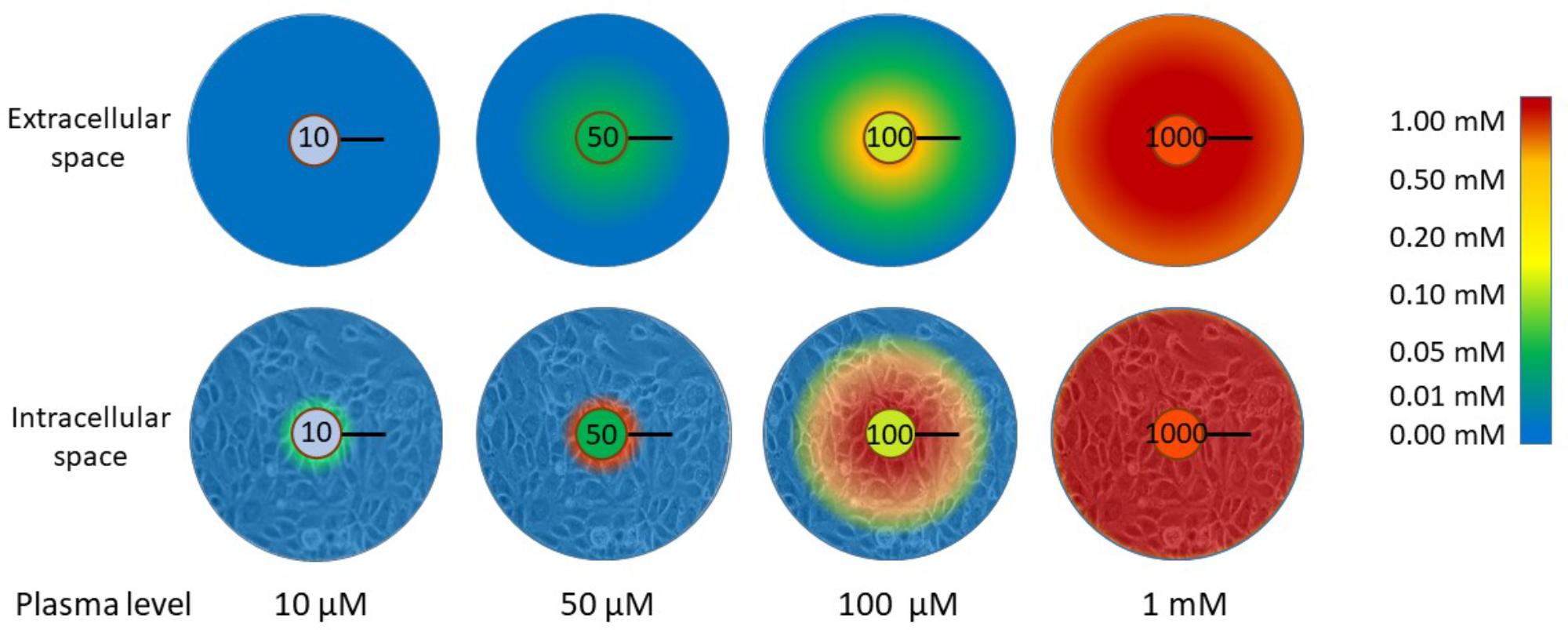
FIGURE 3. The distribution of ascorbate through extracellular tissue and into tissue cells in relation to plasma concentration. When plasma levels are low (10 μM), virtually no ascorbate is able to reach the tissues. This represents a state close to scurvy (complete deficiency). There is a significant difference in the capacity for ascorbate to accumulate in tissue cells when plasma levels are in the ‘healthy but not saturated’ range (50 μM) or saturated (100 μM). Below saturation, only cells adjacent to the blood vessel wall accumulate physiological intracellular concentrations (low mM). Diffusion of ascorbate does not extend beyond 100 μm with plasma ascorbate ≤100 μM and cells beyond this limited distance cannot accumulate ascorbate. To reach beyond this diffusion limit, supra-physiological levels are required. The diffusion distance for oxygen in tissues is 100 μm and this is the average distance between blood vessels in well-perfused tissues (Ludke et al., 2009, 2010; Volta et al., 2013). The data represented in this figure are from Kuiper et al. (2014c). The black bars represent 100 μm.
Summary
Recent discoveries have expanded our knowledge of the biological functions of ascorbate, and have highlighted a number of interesting hypotheses that suggest there is a good rationale to investigate the feasibility of using ascorbate as an adjunct treatment for cancer. We believe that the research progress described in this review will soon lead to the development of effective protocols for the use of ascorbate in the cancer clinical setting. Clinical studies underway will help identify which patients might benefit from ascorbate treatment. Given its lack of toxicity, ready availability and low cost, it is our hope that good information on the mechanism(s) of action will aid translation of the new information into clinical practice.
Author Contributions
MV and AD reviewed the manuscript. MV contributed two-thirds of the writing and formulated the proposal. AD was largely responsible for the sections on epigenetics and other co-factor activities of ascorbate.
Funding
The writing of this review was funded by the employing institute, The University of Otago, Christchurch, New Zealand. AD has been supported by a grant from the Bone Marrow Cancer Research Trust.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
2-OGDD, 2-oxoglutarate-dependent dioxygenase; AML, acute myeloid leukemia; DHA, dehydroascorbate; GLUT, glucose transporter; HIF, hypoxia-inducible factor; HSC, hematopoietic stem cell; IDH, isocitrate dehydrogenase; JMJC, Jumonji-C-domain-containing; SVCT, sodium dependent vitamin C transporter.
References
Agathocleous, M., Meacham, C. E., Burgess, R. J., Piskounova, E., Zhao, Z., Crane, G. M., et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. doi: 10.1038/nature23876
Aguilera, O., Munoz-Sagastibelza, M., Torrejon, B., Borrero-Palacios, A., Del Puerto-Nevado, L., Martinez-Useros, J., et al. (2016). Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 7, 47954–47965. doi: 10.18632/oncotarget.10087
An, S. H., Kang, J. H., Kim, D. H., and Lee, M. S. (2011). Vitamin C increases the apoptosis via up-regulation p53 during cisplatin treatment in human colon cancer cells. BMB Rep. 44, 211–216. doi: 10.5483/BMBRep.2011.44.3.211
Anthony, H. M., and Schorah, C. J. (1982). Severe hypovitaminosis C in lung-cancer patients: the utilization of vitamin C in surgical repair and lymphocyte-related host resistance. Br. J. Cancer 46, 354–367. doi: 10.1038/bjc.1982.211
Badid, N., Ahmed, F. Z., Merzouk, H., Belbraouet, S., Mokhtari, N., Merzouk, S. A., et al. (2010). Oxidant/antioxidant status, lipids and hormonal profile in overweight women with breast cancer. Pathol. Oncol. Res. 16, 159–167. doi: 10.1007/s12253-009-9199-0
Banhegyi, G., Braun, L., Csala, M., Puskas, F., and Mandl, J. (1997). Ascorbate metabolism and its regulation in animals. Free Radic. Biol. Med. 23, 793–803. doi: 10.1016/S0891-5849(97)00062-2
Baylin, S. B., and Jones, P. A. (2011). A decade of exploring the cancer epigenome - biological and translational implications. Nat. Rev. Cancer 11, 726–734. doi: 10.1038/nrc3130
Baylin, S. B., and Jones, P. A. (2016). Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 8:a019505. doi: 10.1101/cshperspect.a019505
Belin, S., Kaya, F., Duisit, G., Giacometti, S., Ciccolini, J., and Fontes, M. (2009). Antiproliferative effect of ascorbic acid is associated with the inhibition of genes necessary to cell cycle progression. PLoS One 4:e4409. doi: 10.1371/journal.pone.0004409
Berman, B. P., Weisenberger, D. J., Aman, J. F., Hinoue, T., Ramjan, Z., Liu, Y., et al. (2011). Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 44, 40–46. doi: 10.1038/ng.969
Bishop, T., and Ratcliffe, P. J. (2014). Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a historical overview and future perspectives. Hypoxia 2, 197–213.
Bonham, M. J. D., Abu-Zidan, F. M., Simovic, M. O., Sluis, K. B., Wilkinson, A., Winterbourn, C. C., et al. (1999). Early ascorbic acid depletion is related to the severity of acute pancreatitis. Br. J. Surg. 86, 1296–1301. doi: 10.1046/j.1365-2168.1999.01182.x
Buettner, G. R. (1987). Activation of oxygen by metal complexes and its relevance to autoxidative processes in living systems. Bioelectrochem. Bioenerg. 18, 29–36. doi: 10.1016/0302-4598(87)85005-5
Buettner, G. R. (1988). In the absence of catalytic metals ascorbate does not autoxidize at pH 7: ascorbate as a test for catalytic metals. J. Biochem. Biophys. Methods 16, 27–40. doi: 10.1016/0165-022X(88)90100-5
Buettner, G. R., and Chamulitrat, W. (1990). The catalytic activity of iron in synovial fluid as monitored by the ascorbate free radical. Free Radic. Biol. Med. 8, 55–56. doi: 10.1016/0891-5849(90)90144-8
Busque, L., Patel, J. P., Figueroa, M. E., Vasanthakumar, A., Provost, S., Hamilou, Z., et al. (2012). Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 44, 1179–1181. doi: 10.1038/ng.2413
Camarena, V., and Wang, G. (2016). The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. 73, 1645–1658. doi: 10.1007/s00018-016-2145-x
Cameron, E., and Pauling, L. (1973). Ascorbic acid and the glycosaminoglycans. An orthomolecular approach to cancer and other diseases. Oncology 27, 181–192. doi: 10.1159/000224733
Cameron, E., and Pauling, L. (1976). Supplemental ascorbate in the supportive treatment of cancer: prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. U.S.A. 73, 3685–3689. doi: 10.1073/pnas.73.10.3685
Cameron, E., and Pauling, L. (1978). Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. U.S.A. 75, 4538–4542. doi: 10.1073/pnas.75.9.4538
Campbell, E. J., Vissers, M. C., Bozonet, S., Dyer, A., Robinson, B. A., and Dachs, G. U. (2015). Restoring physiological levels of ascorbate slows tumor growth and moderates HIF-1 pathway activity in Gulo(-/-) mice. Cancer Med. 4, 303–314. doi: 10.1002/cam4.349
Campbell, E. J., Vissers, M. C., and Dachs, G. U. (2016a). Ascorbate availability affects tumor implantation-take rate and increases tumor rejection in Gulo-/- mice. Hypoxia 4, 41–52.
Campbell, E. J., Vissers, M. C., Wohlrab, C., Hicks, K. O., Strother, R. M., Bozonet, S. M., et al. (2016b). Pharmacokinetic and anti-cancer properties of high dose ascorbate in solid tumours of ascorbate-dependent mice. Free Radic. Biol. Med. 99, 451–462. doi: 10.1016/j.freeradbiomed.2016.08.027
Cao, D., Hou, M., Guan, Y. S., Jiang, M., Yang, Y., and Gou, H. F. (2009). Expression of HIF-1alpha and VEGF in colorectal cancer: association with clinical outcomes and prognostic implications. BMC Cancer 9:432. doi: 10.1186/1471-2407-9-432
Carosio, R., Zuccari, G., Orienti, I., Mangraviti, S., and Montaldo, P. G. (2007). Sodium ascorbate induces apoptosis in neuroblastoma cell lines by interfering with iron uptake. Mol. Cancer 6:55. doi: 10.1186/1476-4598-6-55
Carr, A. C., and Frei, B. (1999). Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am. J. Clin. Nutr. 69, 1086–1107. doi: 10.1093/ajcn/69.6.1086
Castro, M. L., McConnell, M. J., and Herst, P. M. (2014). Radiosensitisation by pharmacological ascorbate in glioblastoma multiforme cells, human glial cells, and HUVECs depends on their antioxidant and DNA repair capabilities and is not cancer specific. Free Radic. Biol. Med. 74, 200–209. doi: 10.1016/j.freeradbiomed.2014.06.022
Cha, J., Roomi, M. W., Ivanov, V., Kalinovsky, T., Niedzwiecki, A., and Rath, M. (2011). Ascorbate depletion increases growth and metastasis of melanoma cells in vitamin C deficient mice. Exp. Oncol. 33, 226–230.
Cha, J., Roomi, M. W., Ivanov, V., Kalinovsky, T., Niedzwiecki, A., and Rath, M. (2013). Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int. J. Oncol. 42, 55–64. doi: 10.3892/ijo.2012.1712
Chatterjee, I. B., Majumder, A. K., Nandi, B. K., and Subramanian, N. (1975). Synthesis and some major functions of vitamin C in animals. Ann. N. Y. Acad. Sci. 258, 24–47. doi: 10.1111/j.1749-6632.1975.tb29266.x
Chen, P., Stone, J., Sullivan, G., Drisko, J. A., and Chen, Q. (2011). Anti-cancer effect of pharmacologic ascorbate and its interaction with supplementary parenteral glutathione in preclinical cancer models. Free Radic. Biol. Med. 51, 681–687. doi: 10.1016/j.freeradbiomed.2011.05.031
Chen, P., Yu, J., Chalmers, B., Drisko, J., Yang, J., Li, B., et al. (2012). Pharmacological ascorbate induces cytotoxicity in prostate cancer cells through ATP depletion and induction of autophagy. Anti Cancer Drugs 23, 437–444. doi: 10.1097/CAD.0b013e32834fd01f
Chen, Q., Espey, M. G., Krishna, M. C., Mitchell, J. B., Corpe, C. P., Buettner, G. R., et al. (2005). Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. U.S.A. 102, 13604–13609. doi: 10.1073/pnas.0506390102
Chen, Q., Espey, M. G., Sun, A. Y., Lee, J. H., Krishna, M. C., Shacter, E., et al. (2007). Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 8749–8754. doi: 10.1073/pnas.0702854104
Chen, Q., Espey, M. G., Sun, A. Y., Pooput, C., Kirk, K. L., Krishna, M. C., et al. (2008). Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 11105–11109. doi: 10.1073/pnas.0804226105
Cieslak, J. A., and Cullen, J. J. (2015). Treatment of pancreatic cancer with pharmacological ascorbate. Curr. Pharm. Biotechnol. 16, 759–770. doi: 10.2174/138920101609150715135921
Cieslak, J. A., Strother, R. K., Rawal, M., Du, J., Doskey, C. M., Schroeder, S. R., et al. (2015). Manganoporphyrins and ascorbate enhance gemcitabine cytotoxicity in pancreatic cancer. Free Radic. Biol. Med. 83, 227–237. doi: 10.1016/j.freeradbiomed.2015.02.018
Cimmino, L., Dolgalev, I., Wang, Y., Yoshimi, A., Martin, G. H., Wang, J., et al. (2017). Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079.e20–1095.e20. doi: 10.1016/j.cell.2017.07.032
Cimmino, L., Neel, B. G., and Aifantis, I. (2018). Vitamin C in stem cell reprogramming and cancer. Trends Cell Biol. doi: 10.1016/j.tcb.2018.04.001 [Epub ahead of print].
Clement, M. V., Ramalingam, J., Long, L. H., and Halliwell, B. (2001). The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxid. Redox Signal. 3, 157–163. doi: 10.1089/152308601750100687
Corpe, C. P., Eck, P., Wang, J., Al-Hasani, H., and Levine, M. (2005). Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J. Biol. Chem. 288, 9092–9101. doi: 10.1074/jbc.M112.436790
Dachs, G. U., and Tozer, G. M. (2000). Hypoxia modulated gene expression: angiogenesis, metastasis and therapeutic exploitation. Eur. J. Cancer 36, 1649–1660. doi: 10.1016/S0959-8049(00)00159-3
De Francesco, E. M., Bonuccelli, G., Maggiolini, M., Sotgia, F., and Lisanti, M. P. (2015). Vitamin C and Doxycycline: a synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget 8, 67269–67286. doi: 10.18632/oncotarget.18428
Deb, S., Johansson, I., Byrne, D., Nilsson, C., Investigators, K., Constable, L., et al. (2014). Nuclear HIF1A expression is strongly prognostic in sporadic but not familial male breast cancer. Mod. Pathol. 27, 1223–1230. doi: 10.1038/modpathol.2013.231
Dhariwal, K. R., Hartzell, W. O., and Levine, M. (1991). Ascorbic acid and dehydroascorbic acid measurements in human plasma and serum. Am. J. Clin. Nutr. 54, 712–716. doi: 10.1093/ajcn/54.4.712
Doskey, C. M., Buranasudja, V., Wagner, B. A., Wilkes, J. G., Du, J., Cullen, J. J., et al. (2010). Tumor cells have decreased ability to metabolize H2O2: implications for pharmacological ascorbate in cancer therapy. Redox Biol. 10, 274–284. doi: 10.1016/j.redox.2016.10.010
Drisko, J. A., Chapman, J., and Hunter, V. J. (2003). The use of antioxidants with first-line chemotherapy in two cases of ovarian cancer. J. Am. Coll. Nutr. 22, 118–123. doi: 10.1080/07315724.2003.10719284
Du, J., Cullen, J. J., and Buettner, G. R. (2012). Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 1826, 443–457. doi: 10.1016/j.bbcan.2012.06.003
Du, J., Martin, S. M., Levine, M., Wagner, B. A., Buettner, G. R., Wang, S. H., et al. (2010). Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. 16, 509–520. doi: 10.1158/1078-0432.CCR-09-1713
Ehrlich, M., and Lacey, M. (2013). DNA hypomethylation and hemimethylation in cancer. Adv. Exp. Med. Biol. 754, 31–56. doi: 10.1007/978-1-4419-9967-2_2
Erudaitius, D., Huang, A., Kazmi, S., Buettner, G. R., and Rodgers, V. G. (2010). Peroxiporin expression is an important factor for cancer cell susceptibility to therapeutic H2O2: implications for pharmacological ascorbate therapy. PLoS One 12:e0170442. doi: 10.1371/journal.pone.0170442
Espey, M. G., Chen, P., Chalmers, B., Drisko, J., Sun, A. Y., Levine, M., et al. (2011). Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic. Biol. Med. 50, 1610–1619. doi: 10.1016/j.freeradbiomed.2011.03.007
Evans-Olders, R., Eintracht, S., and Hoffer, L. J. (2010). Metabolic origin of hypovitaminosis C in acutely hospitalized patients. Nutrition 26, 1070–1074. doi: 10.1016/j.nut.2009.08.015
Fain, O., Paries, J., Jacquart, B., Le Moel, G., Kettaneh, A., Stirnemann, J., et al. (2003). Hypovitaminosis C in hospitalized patients. Eur. J. Intern. Med. 14, 419–425. doi: 10.1016/j.ejim.2003.08.006
Figueroa, M. E., Abdel-Wahab, O., Lu, C., Ward, P. S., Patel, J., Shih, A., et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567. doi: 10.1016/j.ccr.2010.11.015
Fritz, H., Flower, G., Weeks, L., Cooley, K., Callachan, M., McGowan, J., et al. (2014). Intravenous vitamin C and cancer: a systematic review. Integr. Cancer Ther. 13, 280–300. doi: 10.1177/1534735414534463
Fromberg, A., Gutsch, D., Schulze, D., Vollbracht, C., Weiss, G., Czubayko, F., et al. (2011). Ascorbate exerts anti-proliferative effects through cell cycle inhibition and sensitizes tumor cells towards cytostatic drugs. Cancer Chemother. Pharmacol. 67, 1157–1166. doi: 10.1007/s00280-010-1418-6
Gan, R., Eintracht, S., and Hoffer, L. J. (2008). Vitamin C deficiency in a university teaching hospital. J. Am. Coll. Nutr. 27, 428–433. doi: 10.1080/07315724.2008.10719721
Gao, P., Zhang, H., Dinavahi, R., Li, F., Xiang, Y., Raman, V., et al. (2007). HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12, 230–238. doi: 10.1016/j.ccr.2007.08.004
Gillberg, L., Orskov, A. D., Liu, M., Harslof, L. B. S., Jones, P. A., and Gronbaek, K. (2017). Vitamin C - A new player in regulation of the cancer epigenome. Semin. Cancer Biol. doi: 10.1016/j.semcancer.2017.11.001 [Epub ahead of print].
Grosso, G., Bei, R., Mistretta, A., Marventano, S., Calabrese, G., Masuelli, L., et al. (2013). Effects of vitamin C on health: a review of evidence. Front. Biosci. 18, 1017–1029. doi: 10.2741/4160
Guerriero, E., Sorice, A., Capone, F., Napolitano, V., Colonna, G., Storti, G., et al. (2006). Vitamin C effect on mitoxantrone-induced cytotoxicity in human breast cancer cell lines. PLoS One 9:e115287. doi: 10.1371/journal.pone.0115287
Halliwell, B., Clement, M. V., Ramalingam, J., and Long, L. H. (2000). Hydrogen peroxide. Ubiquitous in cell culture and in vivo? IUBMB Life 50, 251–257. doi: 10.1080/15216540051080930
Harris, H. R., Bergkvist, L., and Wolk, A. (2013). Vitamin C intake and breast cancer mortality in a cohort of Swedish women. Br. J. Cancer 109, 257–264. doi: 10.1038/bjc.2013.269
Harris, H. R., Orsini, N., and Wolk, A. (2014). Vitamin C and survival among women with breast cancer: a meta-analysis. Eur. J. Cancer 50, 1223–1231. doi: 10.1016/j.ejca.2014.02.013
Harrison, F. E., and May, J. M. (2009). Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 46, 719–730. doi: 10.1016/j.freeradbiomed.2008.12.018
He, Y. F., Li, B. Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. doi: 10.1126/science.1210944
Herst, P. M., Broadley, K. W., Harper, J. L., and McConnell, M. J. (2012). Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radic. Biol. Med. 52, 1486–1493. doi: 10.1016/j.freeradbiomed.2012.01.021
Hoffer, L. J., Levine, M., Assouline, S., Melnychuk, D., Padayatty, S. J., Rosadiuk, K., et al. (2008). Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann. Oncol. 19, 1969–1974. doi: 10.1093/annonc/mdn377
Hoffer, L. J., Robitaille, L., Zakarian, R., Melnychuk, D., Kavan, P., Agulnik, J., et al. (2015). High-dose intravenous vitamin C combined with cytotoxic chemotherapy in patients with advanced cancer: a phase I-II clinical trial. PLoS One 10:e0120228. doi: 10.1371/journal.pone.0120228
Hornig, D. (1981). Metabolism and requirements of ascorbic acid in man. S. Afr. Med. J. 60, 818–823.
Jozwiak, P., Krzeslak, A., Wieczorek, M., and Lipinska, A. (2015). Effect of glucose on GLUT1-dependent intracellular ascorbate accumulation and viability of thyroid cancer cells. Nutr. Cancer 67, 1333–1341. doi: 10.1080/01635581.2015.1078823
Jung, S. A., Lee, D. H., Moon, J. H., Hong, S. W., Shin, J. S., Hwang, I. Y., et al. (2016). L-Ascorbic acid can abrogate SVCT-2-dependent cetuximab resistance mediated by mutant KRAS in human colon cancer cells. Free Radic. Biol. Med. 95, 200–208. doi: 10.1016/j.freeradbiomed.2016.03.009
Kaczmarek, M., Cachau, R. E., Topol, I. A., Kasprzak, K. S., Ghio, A., and Salnikow, K. (2009). Metal ions-stimulated iron oxidation in hydroxylases facilitates stabilization of HIF-1 alpha protein. Toxicol. Sci. 107, 394–403. doi: 10.1093/toxsci/kfn251
Kassouf, W., Highshaw, R., Nelkin, G. M., Dinney, C. P., and Kamat, A. M. (2006). Vitamins C and K3 sensitize human urothelial tumors to gemcitabine. J. Urol. 176(4 Pt 1), 1642–1647. doi: 10.1016/j.juro.2006.06.042
Kim, T. J., Byun, J. S., Kwon, H. S., and Kim, D. Y. (2018). Cellular toxicity driven by high-dose vitamin C on normal and cancer stem cells. Biochem. Biophys. Res. Commun. 497, 347–353. doi: 10.1016/j.bbrc.2018.02.083
Kivirikko, K. I., Myllyla, R., and Pihlajaniemi, T. (1989). Protein hydroxylation: prolyl 4-hydroxylase, an enzyme with four cosubstrates and a multifunctional subunit. FASEB J. 3, 1609–1617. doi: 10.1096/fasebj.3.5.2537773
Klimant, E., Wright, H., Rubin, D., Seely, D., and Markman, M. (2018). Intravenous vitamin C in the supportive care of cancer patients: a review and rational approach. Curr. Oncol. 25, 139–148. doi: 10.3747/co.25.3790
Klose, R. J., Yamane, K., Bae, Y., Zhang, D., Erdjument-Bromage, H., Tempst, P., et al. (2006). The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 442, 312–316. doi: 10.1038/nature04853
Knowles, H. J., Raval, R. R., Harris, A. L., and Ratcliffe, P. J. (2003). Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 63, 1764–1768.
Kohli, R. M., and Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. doi: 10.1038/nature12750
Koivunen, P., Hirsila, M., Gunzler, V., Kivirikko, K. I., and Myllyharju, J. (2004). Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 279, 9899–9904. doi: 10.1074/jbc.M312254200
Koppenol, W. H. (2001). The Haber-Weiss cycle–70 years later. Redox Rep. 6, 229–234. doi: 10.1179/135100001101536373
Kuiper, C., Dachs, G. U., Currie, M. J., and Vissers, M. C. (2014a). Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Radic. Biol. Med. 69, 308–317. doi: 10.1016/j.freeradbiomed.2014.01.033
Kuiper, C., Dachs, G. U., Munn, D., Currie, M. J., Robinson, B. A., Pearson, J. F., et al. (2014b). Increased tumor ascorbate is associated with extended disease-free survival and decreased hypoxia-inducible factor-1 activation in human colorectal cancer. Front. Oncol. 4:10. doi: 10.3389/fonc.2014.00010
Kuiper, C., Molenaar, I. G., Dachs, G. U., Currie, M. J., Sykes, P. H., and Vissers, M. C. (2010). Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 70, 5749–5758. doi: 10.1158/0008-5472.CAN-10-0263
Kuiper, C., Vissers, M. C., and Hicks, K. O. (2014c). Pharmacokinetic modeling of ascorbate diffusion through normal and tumor tissue. Free Radic. Biol. Med. 77, 340–352. doi: 10.1016/j.freeradbiomed.2014.09.023
Kurbacher, C. M., Wagner, U., Kolster, B., Andreotti, P. E., Krebs, D., and Bruckner, H. W. (1996). Ascorbic acid (vitamin C) improves the antineoplastic activity of doxorubicin, cisplatin, and paclitaxel in human breast carcinoma cells in vitro. Cancer Lett. 103, 183–189. doi: 10.1016/0304-3835(96)04212-7
Lane, D. J., and Richardson, D. R. (2014). The active role of vitamin C in mammalian iron metabolism: much more than just enhanced iron absorption! Free Radic. Biol. Med. 75, 69–83. doi: 10.1016/j.freeradbiomed.2014.07.007
Levine, M., Daruwala, R. C., Park, J. B., Rumsey, S. C., and Wang, Y. (1998). Does vitamin C have a pro-oxidant effect? Nature 395, 231; author reply 232. doi: 10.1038/26137
Levine, M., Padayatty, S. J., and Espey, M. G. (2011). Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv. Nutr. 2, 78–88. doi: 10.3945/an.110.000109
Levine, M., Wang, Y., Padayatty, S. J., and Morrow, J. (2001). A new recommended dietary allowance of vitamin C for healthy young women. Proc. Natl. Acad. Sci. U.S.A. 98, 9842–9846. doi: 10.1073/pnas.171318198
Ley, T. J., Miller, C., Ding, L., Raphael, B. J., Mungall, A. J., Robertson, A., et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074. doi: 10.1056/NEJMoa1301689
Li, M., Xiao, D., Zhang, J., Qu, H., Yang, Y., Yan, Y., et al. (2016). Expression of LPA2 is associated with poor prognosis in human breast cancer and regulates HIF-1alpha expression and breast cancer cell growth. Oncol. Rep. 36, 3479–3487. doi: 10.3892/or.2016.5206
Linster, C. L., and Van Schaftingen, E. (2007). Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 274, 1–22. doi: 10.1111/j.1742-4658.2006.05607.x
Liu, L., Ning, X., Sun, L., Zhang, H., Shi, Y., Guo, C., et al. (2014). Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 99, 121–128.
Lorsbach, R. B., Moore, J., Mathew, S., Raimondi, S. C., Mukatira, S. T., and Downing, J. R. (2003). TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 17, 637–641. doi: 10.1038/sj.leu.2402834
Losman, J. A., Looper, R. E., Koivunen, P., Lee, S., Schneider, R. K., McMahon, C., et al. (2013). (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625. doi: 10.1126/science.1231677
Ludke, A., Akolkar, G., Ayyappan, P., Sharma, A. K., and Singal, P. K. (2009). Time course of changes in oxidative stress and stress-induced proteins in cardiomyocytes exposed to doxorubicin and prevention by vitamin C. PLoS One 12:e0179452. doi: 10.1371/journal.pone.0179452
Ludke, A., Sharma, A. K., Bagchi, A., and Singal, P. K. (2010). Vitamin C modulates doxorubicin-induced cardiotoxicity by reducing P53 and map kinase activation. Free Radic. Biol. Med. 49(Suppl.):S26. doi: 10.1016/j.freeradbiomed.2010.10.040
Ma, Y., Chapman, J., Levine, M., Polireddy, K., Drisko, J., and Chen, Q. (2014). High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 6:222ra18. doi: 10.1126/scitranslmed.3007154
Mandl, J., Szarka, A., and Banhegyi, G. (2009). Vitamin C: update on physiology and pharmacology. Br. J. Pharmacol. 157, 1097–1110. doi: 10.1111/j.1476-5381.2009.00282.x
Martinotti, S., Ranzato, E., and Burlando, B. (2011). In vitro screening of synergistic ascorbate-drug combinations for the treatment of malignant mesothelioma. Toxicol. In Vitro 25, 1568–1574. doi: 10.1016/j.tiv.2011.05.023
Mastrangelo, D., Pelosi, E., Castelli, G., Lo-Coco, F., and Testa, U. (2018). Mechanisms of anti-cancer effects of ascorbate: cytotoxic activity and epigenetic modulation. Blood Cells Mol. Dis. 69, 57–64. doi: 10.1016/j.bcmd.2017.09.005
Mata, A. M., Carvalho, R. M., Alencar, M. V., Cavalcante, A. A., and Silva, B. B. (2016). Ascorbic acid in the prevention and treatment of cancer. Rev. Assoc. Med. Bras. 62, 680–686. doi: 10.1590/1806-9282.62.07.680
May, J. M. (2002). Recycling of vitamin C by mammalian thioredoxin reductase. Methods Enzymol. 347, 327–332. doi: 10.1016/S0076-6879(02)47032-2
May, J. M. (2011). The SLC23 family of ascorbate transporters: ensuring that you get and keep your daily dose of vitamin C. Br. J. Pharmacol. 164, 1793–1801. doi: 10.1111/j.1476-5381.2011.01350.x
May, J. M., Mendiratta, S., Hill, K. E., and Burk, R. F. (1997). Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J. Biol. Chem. 272, 22607–22610. doi: 10.1074/jbc.272.36.22607
May, J. M., Mendiratta, S., Qu, Z. C., and Loggins, E. (1999). Vitamin C recycling and function in human monocytic U-937 cells. Free Radic. Biol. Med. 26, 1513–1523. doi: 10.1016/S0891-5849(99)00017-9
May, J. M., and Qu, Z. C. (2005). Transport and intracellular accumulation of vitamin C in endothelial cells: relevance to collagen synthesis. Arch. Biochem. Biophys. 434, 178–186. doi: 10.1016/j.abb.2004.10.023
Mayland, C. R., Bennett, M. I., and Allan, K. (2005). Vitamin C deficiency in cancer patients. Palliat. Med. 19, 17–20. doi: 10.1191/0269216305pm970oa
McCormick, W. J. (1959). Cancer: a collagen disease, secondary to a nutritional deficiency. Arch. Pediatr. 76, 166–171.
Michels, A. J., and Frei, B. (2013). Myths, artifacts, and fatal flaws: identifying limitations and opportunities in vitamin C research. Nutrients 5, 5161–5192. doi: 10.3390/nu5125161
Mikirova, N., Hunnunghake, R., Scimeca, R. C., Chinshaw, C., Ali, F., Brannon, C., et al. (2016). High-dose intravenous vitamin C treatment of a child with neurofibromatosis type 1 and optic pathway glioma: a case report. Am. J. Case Rep. 17, 774–781. doi: 10.12659/AJCR.899754
Miller, D. M., Buettner, G. R., and Aust, S. D. (1990). Transition metals as catalysts of “autoxidation” reactions. Free Radic. Biol. Med. 8, 95–108. doi: 10.1016/0891-5849(90)90148-C
Mingay, M., Chaturvedi, A., Bilenky, M., Cao, Q., Jackson, L., Hui, T., et al. (2018). Vitamin C-induced epigenomic remodelling in IDH1 mutant acute myeloid leukaemia. Leukemia 32, 11–20. doi: 10.1038/leu.2017.171
Minor, E. A., Court, B. L., Young, J. I., and Wang, G. (2013). Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 288, 13669–13674. doi: 10.1074/jbc.C113.464800
Moertel, C. G., Fleming, T. R., Creagan, E. T., Rubin, J., O’Connell, M. J., and Ames, M. M. (1985). High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 312, 137–141. doi: 10.1056/NEJM198501173120301
Monfort, A., and Wutz, A. (2013). Breathing-in epigenetic change with vitamin C. EMBO Rep. 14, 337–346. doi: 10.1038/embor.2013.29
Monti, D. A., Mitchell, E., Bazzan, A. J., Littman, S., Zabrecky, G., Yeo, C. J., et al. (2012). Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One 7:e29794. doi: 10.1371/journal.pone.0029794
Myllyla, R., Kuutti-Savolainen, E. R., and Kivirikko, K. I. (1978). The role of ascorbate in the prolyl hydroxylase reaction. Biochem. Biophys. Res. Commun. 83, 441–448. doi: 10.1016/0006-291X(78)91010-0
Nagamma, T., Baxi, J., and Singh, P. P. (2014). Status of oxidative stress and antioxidant levels in smokers with breast cancer from western Nepal. Asian Pac. J. Cancer Prev. 15, 9467–9470. doi: 10.7314/APJCP.2014.15.21.9467
Nualart, F., Mack, L., Garcia, A., Cisternas, P., Bongarzone, E. R., Heitzer, M., et al. (2014). Vitamin C transporters, recycling and the bystander effect in the nervous system: SVCT2 versus gluts. J. Stem Cell Res. Ther. 4:209. doi: 10.4172/2157-7633.1000209
Olney, K. E., Du, J., van ’t Erve, T. J., Witmer, J. R., Sibenaller, Z. A., Wagner, B. A., et al. (2013). Inhibitors of hydroperoxide metabolism enhance ascorbate-induced cytotoxicity. Free Radic. Res. 47, 154–163. doi: 10.3109/10715762.2012.755263
Ono, R., Taki, T., Taketani, T., Taniwaki, M., Kobayashi, H., and Hayashi, Y. (2002). LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 62, 4075–4080.
Ozer, A., and Bruick, R. K. (2007). Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat. Chem. Biol. 3, 144–153. doi: 10.1038/nchembio863
Padayatty, S. J., and Levine, M. (2001). New insights into the physiology and pharmacology of vitamin C. Can. Med. Assoc. J. 164, 353–355.
Padayatty, S. J., and Levine, M. (2016). Vitamin C: the known and the unknown and Goldilocks. Oral Dis. 22, 463–493. doi: 10.1111/odi.12446
Padayatty, S. J., Riordan, H. D., Hewitt, S. M., Katz, A., Hoffer, L. J., and Levine, M. (2006). Intravenously administered vitamin C as cancer therapy: three cases. Can. Med. Assoc. J. 174, 937–942. doi: 10.1503/cmaj.050346
Padayatty, S. J., Sun, A. Y., Chen, Q., Espey, M. G., Drisko, J., and Levine, M. (2010). Vitamin C: intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS One 5:e11414. doi: 10.1371/journal.pone.0011414
Padayatty, S. J., Sun, H., Wang, Y., Riordan, H. D., Hewitt, S. M., Katz, A., et al. (2004). Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann. Intern. Med. 140, 533–537. doi: 10.7326/0003-4819-140-7-200404060-00010
Papaemmanuil, E., Gerstung, M., Bullinger, L., Gaidzik, V. I., Paschka, P., Roberts, N. D., et al. (2016). Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221. doi: 10.1056/NEJMoa1516192
Park, S. (2013). The effects of high concentrations of vitamin C on cancer cells. Nutrients 5, 3496–3505. doi: 10.3390/nu5093496
Pathak, A. K., Singh, N., Khanna, N., Reddy, V. G., Prasad, K. N., and Kochupillai, V. (2002). Potentiation of the effect of paclitaxel and carboplatin by antioxidant mixture on human lung cancer h520 cells. J. Am. Coll. Nutr. 21, 416–421. doi: 10.1080/07315724.2002.10719244
Perrone, G., Hideshima, T., Ikeda, H., Okawa, Y., Calabrese, E., Gorgun, G., et al. (2009). Ascorbic acid inhibits antitumor activity of bortezomib in vivo. Leukemia 23, 1679–1686. doi: 10.1038/leu.2009.83
Pugh, C. W., and Ratcliffe, P. J. (2017). New horizons in hypoxia signaling pathways. Exp. Cell Res. 356, 116–121. doi: 10.1016/j.yexcr.2017.03.008
Pullar, J. M., Bayer, S., and Carr, A. C. (2018). Appropriate handling, processing and analysis of blood samples is essential to avoid oxidation of vitamin C to dehydroascorbic acid. Antioxidants 7:29. doi: 10.3390/antiox7020029
Ratcliffe, P. J. (2013). Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J. Physiol. 591(Pt 8), 2027–2042. doi: 10.1113/jphysiol.2013.251470
Ray, G., and Husain, S. A. (2001). Role of lipids, lipoproteins and vitamins in women with breast cancer. Clin. Biochem. 34, 71–76. doi: 10.1016/S0009-9120(00)00200-9
Raymond, Y. C., Glenda, C. S., and Meng, L. K. (2016). Effects of high doses of vitamin c on cancer patients in Singapore: nine cases. Integr. Cancer Ther. 15, 197–204. doi: 10.1177/1534735415622010
Rebouche, C. J. (1991). Ascorbic acid and carnitine biosynthesis. Am. J. Clin. Nutr. 54(Suppl. 6), 1147S–1152S. doi: 10.1093/ajcn/54.6.1147s
Reddy, V. G., Khanna, N., and Singh, N. (2001). Vitamin C augments chemotherapeutic response of cervical carcinoma HeLa cells by stabilizing P53. Biochem. Biophys. Res. Commun. 282, 409–415. doi: 10.1006/bbrc.2001.4593
Robitaille, L., Mamer, O. A., Miller, W. H. Jr., Levine, M., Assouline, S., Melnychuk, D., et al. (2009). Oxalic acid excretion after intravenous ascorbic acid administration. Metabolism 58, 263–269. doi: 10.1016/j.metabol.2008.09.023
Rouleau, L., Antony, A. N., Bisetto, S., Newberg, A., Doria, C., Levine, M., et al. (2016). Synergistic effects of ascorbate and sorafenib in hepatocellular carcinoma: new insights into ascorbate cytotoxicity. Free Radic. Biol. Med. 95, 308–322. doi: 10.1016/j.freeradbiomed.2016.03.031
Rumsey, S. C., Daruwala, R., Al-Hasani, H., Zarnowski, M. J., Simpson, I. A., and Levine, M. (2000). Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J. Biol. Chem. 275, 28246–28253. doi: 10.1074/jbc.M000988200
Rumsey, S. C., Kwon, O., Xu, G. W., Burant, C. F., Simpson, I., and Levine, M. (1997). Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 272, 18982–18989. doi: 10.1074/jbc.272.30.18982
Savini, I., Rossi, A., Pierro, C., Avigliano, L., and Catani, M. V. (2008). SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids 34, 347–355. doi: 10.1007/s00726-007-0555-7
Schoenfeld, J. D., Sibenaller, Z. A., Mapuskar, K. A., Wagner, B. A., Cramer-Morales, K. L., Furqan, M., et al. (2017). O2- and H2O2-mediated disruption of fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 31, 487.e8–500.e8. doi: 10.1016/j.ccell.2017.02.018
Schoning, J. P., Monteiro, M., and Gu, W. (2017). Drug resistance and cancer stem cells: the shared but distinct roles of hypoxia-inducible factors HIF1alpha and HIF2alpha. Clin. Exp. Pharmacol. Physiol. 44, 153–161. doi: 10.1111/1440-1681.12693
Semenza, G. L. (2010). HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56. doi: 10.1016/j.gde.2009.10.009
Semenza, G. L. (2015). Regulation of the breast cancer stem cell phenotype by hypoxia-inducible factors. Clin. Sci. 129, 1037–1045. doi: 10.1042/CS20150451
Semenza, G. L. (2016). The hypoxic tumor microenvironment: a driving force for breast cancer progression. Biochim. Biophys. Acta 1863, 382–391. doi: 10.1016/j.bbamcr.2015.05.036
Shah, F. D., Patel, J. B., Shukla, S. N., Shah, P. M., and Patel, P. S. (2009). Evaluation of plasma non-enzymatic antioxidants in breast cancer etiology. Asian Pac. J. Cancer Prev. 10, 91–96.
Shen, H., and Laird, P. W. (2013). Interplay between the cancer genome and epigenome. Cell 153, 38–55. doi: 10.1016/j.cell.2013.03.008
Shenoy, N., Bhagat, T., Nieves, E., Stenson, M., Lawson, J., Choudhary, G. S., et al. (2017). Upregulation of TET activity with ascorbic acid induces epigenetic modulation of lymphoma cells. Blood Cancer J. 7:e587. doi: 10.1038/bcj.2017.65
Smirnoff, N. (2000). Ascorbic acid: metabolism and functions of a multi-facetted molecule. Curr. Opin. Plant Biol. 3, 229–235. doi: 10.1016/S1369-5266(00)00069-8
Smirnoff, N., and Wheeler, G. L. (2000). Ascorbic acid in plants: biosynthesis and function. Crit. Rev. Biochem. Mol. Biol. 35, 291–314. doi: 10.1080/10409230008984166
Stephenson, C. M., Levin, R. D., Spector, T., and Lis, C. G. (2013). Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother. Pharmacol. 72, 139–146. doi: 10.1007/s00280-013-2179-9
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Tarumoto, T., Nagai, T., Ohmine, K., Miyoshi, T., Nakamura, M., Kondo, T., et al. (2004). Ascorbic acid restores sensitivity to imatinib via suppression of Nrf2-dependent gene expression in the imatinib-resistant cell line. Exp. Hematol. 32, 375–381. doi: 10.1016/j.exphem.2004.01.007
Tsao, C. S. (1997). “An overview of ascorbic acid chemistry and biochemistry,” in Vitamin C in Health and Disease. Antioxidants in Health and Disease, 1st Edn, eds L. Packer and J. Fuchs (New York, NY: Marcel Dekker), 25–58.
Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., et al. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. doi: 10.1038/nature04433
Tu, H., Wang, Y., Li, H., Brinster, L. R., and Levine, M. (2017). Chemical transport knockout for oxidized vitamin C, dehydroascorbic acid, reveals its functions in vivo. EBioMedicine 23, 125–135. doi: 10.1016/j.ebiom.2017.08.017
Verrax, J., Dejeans, N., Sid, B., Glorieux, C., and Calderon, P. B. (2011). Intracellular ATP levels determine cell death fate of cancer cells exposed to both standard and redox chemotherapeutic agents. Biochem. Pharmacol. 82, 1540–1548. doi: 10.1016/j.bcp.2011.07.102
Vissers, M. C., Bozonet, S. M., Pearson, J. F., and Braithwaite, L. J. (2011). Dietary ascorbate intake affects steady state tissue concentrations in vitamin C-deficient mice: tissue deficiency after suboptimal intake and superior bioavailability from a food source (kiwifruit). Am. J. Clin. Nutr. 93, 292–301. doi: 10.3945/ajcn.110.004853
Vissers, M. C., Gunningham, S. P., Morrison, M. J., Dachs, G. U., and Currie, M. J. (2007). Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radic. Biol. Med. 42, 765–772. doi: 10.1016/j.freeradbiomed.2006.11.023
Vissers, M. C., Kuiper, C., and Dachs, G. U. (2014). Regulation of the 2-oxoglutarate-dependent dioxygenases and implications for cancer. Biochem. Soc. Trans. 42, 945–951. doi: 10.1042/BST20140118
Vleugel, M. M., Greijer, A. E., Shvarts, A., van der Groep, P., van Berkel, M., Aarbodem, Y., et al. (2005). Differential prognostic impact of hypoxia induced and diffuse HIF-1alpha expression in invasive breast cancer. J. Clin. Pathol. 58, 172–177. doi: 10.1136/jcp.2004.019885
Volinia, S., Galasso, M., Sana, M. E., Wise, T. F., Palatini, J., Huebner, K., et al. (2012). Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc. Natl. Acad. Sci. U.S.A. 109, 3024–3029. doi: 10.1073/pnas.1200010109
Volta, V., Ranzato, E., Martinotti, S., Gallo, S., Russo, M. V., Mutti, L., et al. (2013). Preclinical demonstration of synergistic active nutrients/drug (AND) combination as a potential treatment for malignant pleural mesothelioma. PLoS One 8:e58051. doi: 10.1371/journal.pone.0058051
Wang, T., Gilkes, D. M., Takano, N., Xiang, L., Luo, W., Bishop, C. J., et al. (2012). Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. U.S.A. 111, E3234–E3242. doi: 10.1073/pnas.1410041111
Wang, W., He, Y. F., Sun, Q. K., Wang, Y., Han, X. H., Peng, D. F., et al. (2014). Hypoxia-inducible factor 1alpha in breast cancer prognosis. Clin. Chim. Acta 428, 32–37. doi: 10.1016/j.cca.2013.10.018
Washburn, M. P., and Wells, W. W. (1999). The catalytic mechanism of the glutathione-dependent dehydroascorbate reductase activity of thioltransferase (glutaredoxin). Biochemistry 38, 268–274. doi: 10.1021/bi980480v
Washko, P. W., Wang, Y., and Levine, M. (1993). Ascorbic acid recycling in human neutrophils. J. Biol. Chem. 268, 15531–15535.
Washko, P. W., Welch, R. W., Dhariwal, K. R., Wang, Y., and Levine, M. (1992). Ascorbic acid and dehydroascorbic acid analyses in biological samples. Anal. Biochem. 204, 1–14. doi: 10.1016/0003-2697(92)90131-P
Wee, L. M., Long, L. H., Whiteman, M., and Halliwell, B. (2003). Factors affecting the ascorbate- and phenolic-dependent generation of hydrogen peroxide in Dulbecco’s Modified Eagles Medium. Free Radic. Res. 37, 1123–1130. doi: 10.1080/10715760310001607041
Wells, W. W., Rocque, P. A., Xu, D. P., Meyer, E. B., Charamella, L. J., and Dimitrov, N. V. (1995). Ascorbic acid and cell survival of adriamycin resistant and sensitive MCF-7 breast tumor cells. Free Radic. Biol. Med. 18, 699–708. doi: 10.1016/0891-5849(94)00188-P
Welsh, J. L., Wagner, B. A., van’t Erve, T. J., Zehr, P. S., Berg, D. J., Halfdanarson, T. R., et al. (2013). Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother. Pharmacol. 71, 765–775. doi: 10.1007/s00280-013-2070-8
Wilson, J. X. (2005). Regulation of vitamin C transport. Annu. Rev. Nutr. 25, 105–125. doi: 10.1146/annurev.nutr.25.050304.092647
Wilson, M. K., Baguley, B. C., Wall, C., Jameson, M. B., and Findlay, M. P. (2014). Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac. J. Clin. Oncol. 10, 22–37. doi: 10.1111/ajco.12173
Winterbourn, C. C. (1981). Hydroxyl radical production in body fluids: roles of metal ions, ascorbate and superoxide. Biochem. J. 198, 125–131. doi: 10.1042/bj1980125
Wozniak, G., and Anuszewska, E. L. (2002). Influence of vitamins C and E on cytotoxic activity of adriamycin in chosen cell cultures. Acta Pol. Pharm. 59, 31–35.
Xia, J., Xu, H., Zhang, X., Allamargot, C., Coleman, K. L., Nessler, R., et al. (2017). Multiple myeloma tumor cells are selectively killed by pharmacologically-dosed ascorbic acid. EBioMedicine 18, 41–49. doi: 10.1016/j.ebiom.2017.02.011
Yang, Y., Lu, X., Liu, Q., Dai, Y., Zhu, X., Wen, Y., et al. (2017). Palmitoyl ascorbate and doxorubicin co-encapsulated liposome for synergistic anticancer therapy. Eur. J. Pharm. Sci. 105, 219–229. doi: 10.1016/j.ejps.2017.05.038
Young, J. I., Zuchner, S., and Wang, G. (2015). Regulation of the epigenome by vitamin C. Annu. Rev. Nutr. 35, 545–564. doi: 10.1146/annurev-nutr-071714-034228
Yun, J., Mullarky, E., Lu, C., Bosch, K. N., Kavalier, A., Rivera, K., et al. (2015). Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350, 1391–1396. doi: 10.1126/science.aaa5004
Zhang, H., Wong, C. C., Wei, H., Gilkes, D. M., Korangath, P., Chaturvedi, P., et al. (2012). HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 31, 1757–1770. doi: 10.1038/onc.2011.365
Keywords: ascorbate, hypoxia-inducible factor, hydrogen peroxide, iron-mediated autoxidation, antioxidant, epigenetic demethylases, ten-eleven translocases, HIF hydroxylases
Citation: Vissers MCM and Das AB (2018) Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence. Front. Physiol. 9:809. doi: 10.3389/fphys.2018.00809
Received: 09 April 2018; Accepted: 08 June 2018;
Published: 03 July 2018.
Edited by:
Jin Wang, Fudan University, ChinaReviewed by:
Simona Martinotti, Università degli Studi del Piemonte Orientale, ItalyVenkat R. Pannala, Biotechnology HPC Software Applications Institute (BHSAI), United States
Copyright © 2018 Vissers and Das. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margreet C. M. Vissers, bWFyZ3JlZXQudmlzc2Vyc0BvdGFnby5hYy5ueg==