 Cristina Carvalho
Cristina Carvalho Paula I. Moreira
Paula I. Moreira
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 03 July 2018
Sec. Vascular Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00806
This article is part of the Research Topic Oxidative Stress Revisited - Major Role in Vascular Diseases View all 13 articles
The brain is one of the most exquisite organs in the body with high metabolic demands, and requires a tight regulation of the surrounding environment. This tight control is exerted by the neurovascular unit (NVU) comprising different cell types, where endothelial cells play the commander-in-chief role. Thus, it is assumable that even slight perturbations in NVU might affect, in some cases irreversibly, brain homeostasis and health. In this line, recent findings support the two-hit vascular hypothesis for neurodegenerative conditions, where vascular dysfunction underlies the development of neurodegenerative diseases, such as Alzheimer’s disease (AD). Knowing that endothelial cells are rich in mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, two major reactive oxygen species (ROS) sources, this review aims to gather information on how oxidative stress is in the front line of vascular alterations observed in brain aging and neurodegenerative conditions, particularly AD. Also, a brief discussion about the therapeutic strategies aimed to protect against cerebrovascular diseases is included.
The brain integrates and regulates several central and peripheral signals to maintain body homeostasis (Ronnett et al., 2009). So, it is not surprising that the brain is an organ with a high energy demand, although it represents only 2% of the body weight (Mergenthaler et al., 2013). In fact, it is widely known that proper neuronal activity entails high amounts of energy. However, the capability of the brain to store energy is very reduced, requiring a constant supply of energy substrates, namely glucose, through blood flow to fulfill its energy needs (Ohta et al., 1992). For that reason the brain receives about 15% of cardiac output and accounts for 20% of total body oxygen consumption (Moreira et al., 2009; Nunomura et al., 2009; Ronnett et al., 2009). At this point, it is worth mentioning the neurovascular coupling, where neurons, glial cells and blood vessels communicate to each other to regulate cerebral blood flow (CBF) and vessels permeability depending on location and neuronal activity in order to efficiently maintain energy substrates supply to satisfy the metabolic needs (Gordon et al., 2007). Cerebral blood vessels comprise unique properties forming the blood–brain barrier (BBB), a physical barrier that permits the passage of water, some gases and lipophilic molecules by passive diffusion and the selective transport of certain molecules (e.g., glucose) and protects against external toxins and pathogens. Thus, even slight alterations in BBB properties can be responsible for the onset/progression of neurological diseases. From all the BBB constituents (endothelial, mural and glia cells, astrocytes and macrophages), endothelial cells that form blood vessels, play a major role in BBB proper functioning (Daneman and Prat, 2015). Blood vessels control the influx and efflux transport allowing, for example, the entry of glucose and amino acids from the blood into the central nervous system (CNS) and the removal of specific waste products from the CNS into the blood (Daneman and Prat, 2015). More recently, Kubíková et al. (2017), performed a mapping of brain microvessels obtained from two healthy human adult brain samples and found that in certain brain areas microvessels density is higher than others, which can reflect the different susceptibilities to vascular damage. Even slight alterations in brain vasculature can underlie different neurodegenerative events. Indeed, it is widely described that conditions interfering with brain vasculature, such as stiffening of cerebral arteries or increased vessel tortuosity, caused by diabetes and hypertension among other conditions, can induce BBB breakdown underlying the development of neurodegenerative conditions such as Alzheimer’s disease (AD) (Carvalho et al., 2010, 2013, 2014; Steinman et al., 2017). It is also known that in many cases BBB integrity is deeply affected by oxidative stress. In fact, increased reactive oxygen species (ROS) production contribute to endothelium dysfunction and increased permeability of BBB (Enciu et al., 2013). These alterations are mainly attributed to the redistribution and/or altered expression of critical tight junction proteins such as claudin-5 and occludin (Schreibelt et al., 2007; Lochhead et al., 2010).
Although it is widely accepted that vascular changes play a crucial role in neurodegenerative diseases, none of the available therapies are effective when translated to clinical trials revealing some gaps in the mechanisms behind the vascular and tissue brain changes under pathological conditions. Thus, studies aimed to develop new non-invasive techniques to better understand why and when changes occur bring a new hope for the treatment of conditions characterized by cerebrovascular alterations (Vaas et al., 2017). In the next section we will discuss the role of oxidative stress in brain vascular alterations putting the focus on mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox).
It is widely known that cells homeostasis depends on the regulated levels of ROS. Low/moderate levels of ROS can act as signaling molecules, which are crucial to maintain normal cells function, while uncontrolled generation of ROS causes oxidative damage contributing to cells dysfunction and damage (Chen et al., 2017).
Physiological ROS levels can play important roles in cerebral vasculature (De Silva and Miller, 2016). Studies performed in animal models show that ROS can contribute to the regulation of brain perfusion through their action in vascular tone control (Miller et al., 2009; Grochowski et al., 2018). Indeed, physiological levels of ROS play a major role as cerebral vasodilators (Figure 1). For example, it has been shown that the addition of NADPH, the substrate for Nox, in cerebral vessels in vitro and in vivo cause hydrogen peroxide (H2O2)-dependent vasodilatation (Didion and Faraci, 2002; Park et al., 2004; Miller et al., 2005; De Silva and Miller, 2016).
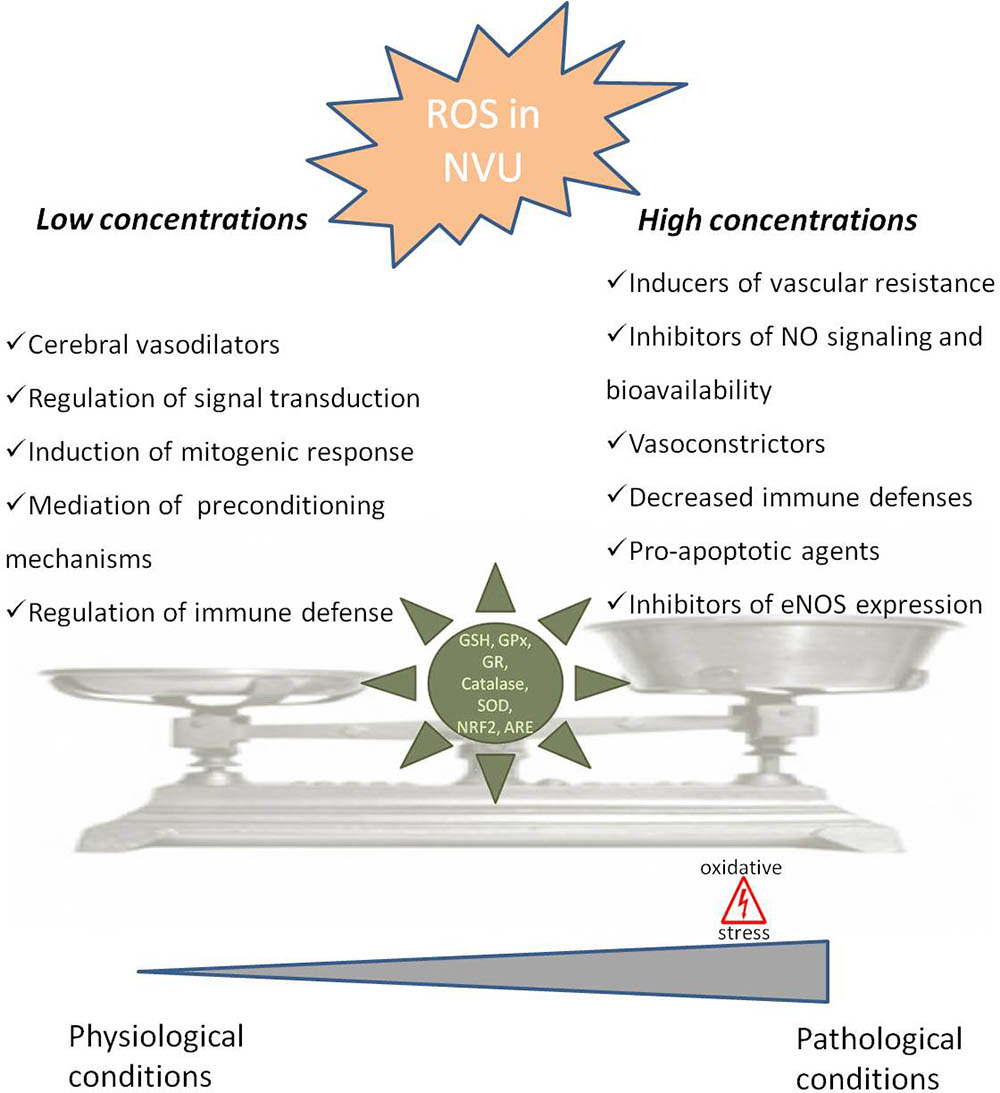
FIGURE 1. Dual role of reactive oxygen species in neurovascular unit. In neurovascular unit (NVU) reactive oxygen species (ROS) play a controversial role with low/moderate levels of ROS acting as signaling molecules, which are crucial to maintain normal cells function, while uncontrolled generation of ROS causes oxidative damage contributing to cellular dysfunction and cells damage. In physiological situations ROS can act as cerebral vasodilators, regulators of signal transduction, inducers of mitogenic response and immune defense mechanisms, regulators of blood flow and effectors of preconditioning mechanisms. However, when ROS concentrations pass a certain threshold, they lead to an increase in blood flow resistance, decreased nitric oxide (•NO) bioavailability, decreased vasodilatation and immune response, increased apoptosis and decreased endothelial nitric oxide synthase (eNOS) expression, leading to pathological conditions. The delicate balance of ROS levels is regulated by cellular antioxidant systems such as glutathione (GSH), glutathione peroxidase (GPx), glutathione reductase (GR), superoxide dismutase (SOD), catalase, nuclear factor erythroid 2-related factor 2 (NRF2) and antioxidant response element (ARE).
The neurovascular unit (NVU), containing neurons, astrocytes, pericytes, microglia and endothelial cells, is equipped with a powerful antioxidant defense systems that include glutathione (GSH), glutathione peroxidase, glutathione reductase, superoxide dismutase and catalase (Tayarani et al., 1987; Halliwell, 2001). GSH in particular has been shown to play an important role in the maintenance of BBB integrity (Figure 1) (Agarwal and Shukla, 1999). Also, the nuclear factor erythroid 2-related factor 2 (NRF2) seems to play a major defense role by modulating microglial dynamics (Rojo et al., 2010), by protecting astrocytes and neurons from toxic insults (Lee et al., 2003; Vargas and Johnson, 2009) and by regulating the expression of antioxidant enzymes (Shah et al., 2007; Yan et al., 2008). Additionally, NRF2 induces secondary defense proteins via interaction with the antioxidant response element (ARE) in the promoter region of target genes (Figure 1). Interestingly, astrocytes have a higher capability of efficiently increase GSH and ARE-linked gene expression, which allows astrocytes to be more protected than neurons against moderate levels of oxidative stress (Vargas and Johnson, 2009).
A failure in the ability of NVU cells to maintain the proper balance between ROS production and their neutralization causes the disruption of NUV and brain homeostasis predisposing to neurodegenerative conditions (Carvalho et al., 2013; Wevers and de Vries, 2016). It has been shown that following an injury microglia and astrocytes produce high levels of ROS via Nox, which seem to have harmful effects in the expression of important molecules involved in BBB integrity (e.g., ZO-1, claudin-5 and occluding). Pericytes, which are in close proximity to endothelial cells, play a vital role in the integrity of BBB (Armulik et al., 2010). Under pathological conditions, pericytes are highly susceptible to oxidative stress (Shah et al., 2013) resulting from overproduction of mitochondrial ROS (Shah et al., 2013). Moreover, it has been demonstrated that ROS production by activated microglia causes pericytes apoptosis (Ding et al., 2017). However, there is a lack of information regarding the role of ROS in pericytes under physiological conditions. Endothelial cells signaling seems to be crucial in the regulation of NVU proper functioning with increased ROS formation playing a major role in the alteration of NVU function and coupling (Girouard and Iadecola, 2006; Faraco et al., 2016).
It was also shown that ROS-induced changes in microcirculation can have profound implications in brain vascular pathophysiology due to alterations in blood flow resistance and, consequently, in regulation of blood pressure (Figure 1) (Staiculescu et al., 2014). Both superoxide (O2•-) and H2O2 are able to cause both relaxation as well as contraction of cerebral blood vessels depending on the concentration and presence of other species (Faraci and Sobey, 1998; Allen and Bayraktutan, 2009; Freeman and Keller, 2012). In fact, oxidative stress can impair cerebral vascular function via the disruption of endothelium-dependent nitric oxide (•NO) signaling (Figure 1) (Girouard et al., 2007; Mayhan et al., 2008; Miller et al., 2010b). It is widely described that the reaction of O2•- with •NO leads to a decrease in •NO bioavailability and, consequently, a decrease in its vasodilator, anti-proliferative, and anti-inflammatory properties (De Silva and Miller, 2016). Furthermore, increased levels of H2O2, a common vasodilator in cerebral circulation, can act as a pro-apoptotic agent in cerebral vascular cells (Figure 1) (Li et al., 2003). Increased levels of ROS can also lead to an increase in Rho kinase signaling (Aghajanian et al., 2009) interfering with endothelial nitric oxide synthase (eNOS) expression and activity, affecting •NO production (Figure 1) (Faraco et al., 2013).
Considering vascular cells, there are several ROS sources such as mitochondrial electron-transport chain, cyclooxygenases (COXs), lipoxygenases, cytochrome P450 reductases, xanthine oxidase, nitric oxide synthase (NOS) and Nox (Miller et al., 2010a). However, in this review, we only discuss mitochondria and Nox, both of them widely described as main producers of ROS either in physiological and pathological conditions in endothelial cells.
Reporting to the history of biology evolution, mitochondria are described as organelles derived from aerobic bacteria that in ancient times invaded proto-eukaryotic cells as parasites (Dromparis and Michelakis, 2012). From there, a symbiotic relationship evolved with mutual benefits and mitochondria became intracellular organelles (Dromparis and Michelakis, 2012). Although mitochondria are widely described as energy producers through the highly conserved oxidative phosphorylation (OXPHOS) process (Cadenas and Davies, 2000; Chan, 2006), nowadays the role of mitochondria reached substantially higher importance in cell homeostasis due to their involvement in several vital processes such as cell growth and differentiation, cell cycle control and death (Osellame et al., 2012; Carvalho et al., 2015), intermediary metabolism, calcium (Ca2+) homeostasis and signaling, and apoptosis (Chan, 2006).
However, mitochondrial energy production can be a double-edge sword. Indeed, OXPHOS is not 100% efficient and during this process an electron leak occurs between mitochondrial complexes leading to the production of ROS such as O2•-, •NO, hydroxyl radical (HO•), peroxynitrite (ONOO-), and H2O2 (Richter and Kass, 1991; Ricquier and Bouillaud, 2000; Goetz and Luch, 2008). As previously mentioned, low/moderate ROS levels exert a beneficial role by activating protective mechanisms (Valko et al., 2007; Correia et al., 2010; Alfadda and Sallam, 2012). However, when ROS levels reach critical values they lead to oxidative stress and activate anomalous signaling mechanisms that can lead to cells degeneration and death (Brown and Borutaite, 2001; Sheu et al., 2006).
Brain endothelial cells seem to possess a number of mitochondria higher than that observed in peripheral endothelial cells (Oldendorf et al., 1976; Alyautdin et al., 2014). However, in comparison with other cell types with higher energy requirements, mitochondria content in endothelial cells is modest. In rodent models, mitochondria compose 2–6% of the cell volume as opposed to 28% in hepatocytes and 32% in cardiac myocytes (Dromparis and Michelakis, 2012; Kluge et al., 2013). These observations support the idea that mitochondria is not a major source of energy in brain endothelial cells. In fact, several studies support the idea that brain endothelial cells obtain a large proportion of their energy from anaerobic glycolytic metabolism of glucose (Spahr et al., 1989; Mertens et al., 1990; Culic et al., 1997). Actually, mitochondria are more likely to serve primarily as essential signaling organelles in the vascular endothelium (Quintero et al., 2006; Tang et al., 2014; Busija et al., 2016).
The activation of mitochondria by physiological stimuli or pharmacological agents leads to the liberation of vasoactive factors by the endothelium, which exert a major role in the modulation and maintenance of BBB integrity and brain homeostasis (Busija and Katakam, 2014). Moreover, it is far known that aging leads to a decrease in mitochondria number in cerebral endothelial cells associated with the loss of BBB integrity (Mooradian, 1988). The BBB high demand of mitochondrial activation comes with a price, the prospect for an increased ROS production that in physiological conditions is regulated by antioxidant enzymes such as glutathione reductase, manganese superoxide dismutase, catalase, among others (Freeman and Keller, 2012). Additionally, it has been demonstrated that vascular endothelial growth factor (VEGF) exerts its functions in endothelial cells migration through mitochondrial ROS (Wang et al., 2011), a process involved in several physiological processes such as wound healing and vascular repair (Freeman and Keller, 2012). Moreover, studies in human coronary resistance arteries showed that mitochondrial ROS are involved in endothelium regulation of vascular homeostasis (Liu et al., 2003). In the same study, the authors reported that mitochondrial ROS have an influence in vascular regulation and health. Indeed, the authors tested mitochondrial complexes I and III and Nox inhibitors and observed that only the mitochondrial inhibitors were able to exert effects in the regulation of flow-induced dilation (Liu et al., 2003). Besides the above evidence more studies are needed to explore the role of mitochondrial ROS in cerebral vasculature during disease progression, in contrast to the extensive literature concerning systemic vessels.
The Nox family is also broadly studied due to its main catalytic function of ROS production by transference of an electron to molecular oxygen (Takac et al., 2012). The Nox family is composed by different isoforms: Nox1 to 5, Duox1 and Duox2 with a broad expression in different organs and tissues and with different cellular locations at vascular walls (de Almeida et al., 2017). Except for Nox-5 and DUOX-1 and -2, Nox are phagocytic oxidases, whose main task is to generate ROS to kill foreign pathogens at homeostasis. Of note, the major source of endothelial cells ROS comes from NOX-1, -2, -4, and -5 (Pendyala et al., 2009; Drummond and Sobey, 2014). Different Nox isoforms seem to produce different forms of ROS; Nox1 and 2 mainly produce O2•-; Nox4 mainly produces H2O2 and Nox5 seems to produce both O2•- and H2O2 (Dikalov et al., 2008; Helmcke et al., 2009). Under physiological conditions, Nox-derived ROS seem to exert an important role in the regulation of vasodilatation (Togliatto et al., 2017). Although it is considered that under physiological conditions Nox activity is constitutively low, when its function increases with the consequent increase in ROS production, it can trigger ROS production by other sources (Landmesser et al., 2003). Indeed, it has been described that Nox-derived ROS play a major role in coordinating some physiological processes such as innate immunity, modulation of redox-dependent signaling cascades, and can act as cofactors in the production of hormones (Drummond et al., 2011). Furthermore, it is described that H2O2 produced by Nox family can exert an endothelium-derived hyperpolarizing role causing vasodilatation and reducing blood pressure in mice (Ray et al., 2011). The Nox activity depends on the stimuli, such as cytokines (De Keulenaer et al., 1998), growth factors (Brandes et al., 2001), hyperlipidemia, and high glucose (Jansen et al., 2013). Additionally, Nox family is known for its role as oxygen sensors, modulating the different responses to hypoxia through hypoxia-inducible factor 1 alpha (HIF-1α) mRNA induction and HIF-1α stabilization (Gorlach et al., 2001), a process that is responsible for alterations in gene expression due to the oxidation of target proteins.
However, the overproduction of ROS by Nox family plays a major role in disruption of vascular homeostasis, which can underlie the development of neurodegenerative diseases. Of notice, so far only Nox2 and Nox4 were associated with endothelium dysfunction probably due to their major role as vascular ROS producers, in comparison to the other Nox isoforms (Matsuno et al., 2005; Takac et al., 2012).
Compelling evidence shows that Nox-derived oxidative stress causes many of the deleterious effects of angiotensin II on the cerebral vasculature. It was observed that angiotensin II acutely and chronically, increases O2•- production by Nox in rodent cerebral vessels (Girouard et al., 2006, 2007; De Silva and Faraci, 2016). Also, functional alterations on rodents’ cerebral arterioles following angiotensin II treatment are prevented by co-treatment with the ROS scavenger MnTBAP and the Nox2 peptide inhibitor gp91ds-tat (Girouard et al., 2006). It was also reported that angiotensin II can also activate and increase O2•- production in cerebral vessels by activating Nox1 (Jackman et al., 2009).
Interestingly, although not extensively studied, the Nox family activation and expression seems to be seasonal, a fact that could be correlated with seasonal alterations in oxidative stress and endothelium dysfunction observed in humans and rats unveiling a new strategic pathway to future studies of clinical relevance (Hopkins et al., 2011; Konior et al., 2014).
The aging process causes several alterations in brain blood vessels such as decreased elasticity, increased cerebrovascular remodeling and calcification, gradual cerebrovascular wall stiffness, low-grade and widespread inflammation and oxidative stress (Camici et al., 2015), factors that increase the risk for cerebrovascular diseases. So far, there is no agreement of how aging process occurs although several theories can be found in the literature. One of the most accepted theory for aging was first proposed by Harman (1956) and is known as “free radical theory of aging,” which postulates that ROS levels increase with age, being responsible for deoxyribonucleic acid (DNA), proteins and lipids oxidative damage (Harman, 1956).
Recent findings demonstrated the existence of BBB permeability alterations during normal aging in human hippocampus using advanced dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) (Montagne et al., 2015). It is also known that aging increases BBB susceptibility to different challenges and its permeability is less controlled allowing the entrance of neurotoxic substances, which affect brain homeostasis (Zlokovic, 2011; Caplan et al., 2017). In fact, BBB dysfunction is more pronounced under pathological conditions, including in mild cognitive impairment (MCI) individuals, supporting the idea that vascular alterations are early events in cognitive deficits (Figure 2) (Montagne et al., 2015). Using a multiphoton microscope, Han et al. (2015) observed increased levels of O2•- in the cerebral vessels of aged PS1/APP transgenic mice, a model of AD. Also, electron and confocal microscopy studies revealed that in F344 rats aortas, the expression of the mitochondrial biogenesis factors, such as mitochondrial transcription factor A and peroxisome proliferator-activated receptor-γ coactivator-1-alpha (PGC-1α), decreases with aging (Ungvari et al., 2008). Moreover, an increase in mitochondrial H2O2 production was observed in aged arterial rat vessels leading to an activation of nuclear factor-κB (NF-κB) and induction of inflammatory phenotypic changes in aged vasculature (Ungvari et al., 2007). Furthermore, an age-dependent decline in mitochondrial complexes I, III, and IV subunits expression was observed in rat aortas, although the authors did not specify if all the subunits of the three complexes were evaluated or not (Ungvari et al., 2008). Also, Wenzel et al. (2008) were able to show an age-dependent increase in mitochondrial ROS production and mitochondrial DNA lesions causing aorta vascular dysfunction in two different mice models, the ALDH-2–/– and MnSOD–/– models, which are deficient in aldehyde dehydrogenase-2 (ALDH-2) and manganese superoxide dismutase (MnSOD), respectively. Furthermore, the in vitro use of mitochondrial complex I inhibitor rotenone, after angiotensin II-induced O2•- production, showed significant improvements in endothelial dysfunction and tolerance in human aortic endothelial cells (Dikalov et al., 2014; Mikhed et al., 2015). Furthermore, the use of preconditioning strategies as a therapeutic intervention is emerging and under extensive scrutiny. Indeed, studies from our laboratory were able to show that treating rat brain endothelial cells with non-lethal cyanide concentrations, which lead to increased mitochondrial ROS production, was able to confer protection against a posterior high glucose-mediated damage, preventing apoptotic cell death (Correia et al., 2012). Importantly, Correia et al. (2012) were able to prove that in the absence of mitochondrial DNA, using mitochondrial-depleted DNA human teratocarcinoma NT2 cells, this effect was abrogated, emphasizing mitochondrial role in endothelium protection by preconditioning (Correia et al., 2012). Moreover, the use of mitochondrial-targeted antioxidants such as MitoTempo, showed promising results in counteracting age-related and microgravity-induced and hyperglycemia-induced endothelial dysfunction (Carvalho et al., 2014; Zhang et al., 2014). Indeed, Mitotempo was able to protect endothelial primary cultures of diabetic mouse model (db/db) and rat cerebral arteries against amyloid β (Aβ) toxicity (Carvalho et al., 2014) and lead to improvements in spatial working memory and motor skill learning in young 5xFAD mouse models of AD (Lu et al., 2015). Moreover, it was also able to recover mitochondrial dysfunction observed in rat cerebral arteries of animals exposed to microgravity conditions through reduction of mitochondrial ROS levels, increased mitochondrial potential and improvement in mitochondrial respiratory chain function (Zhang et al., 2014).
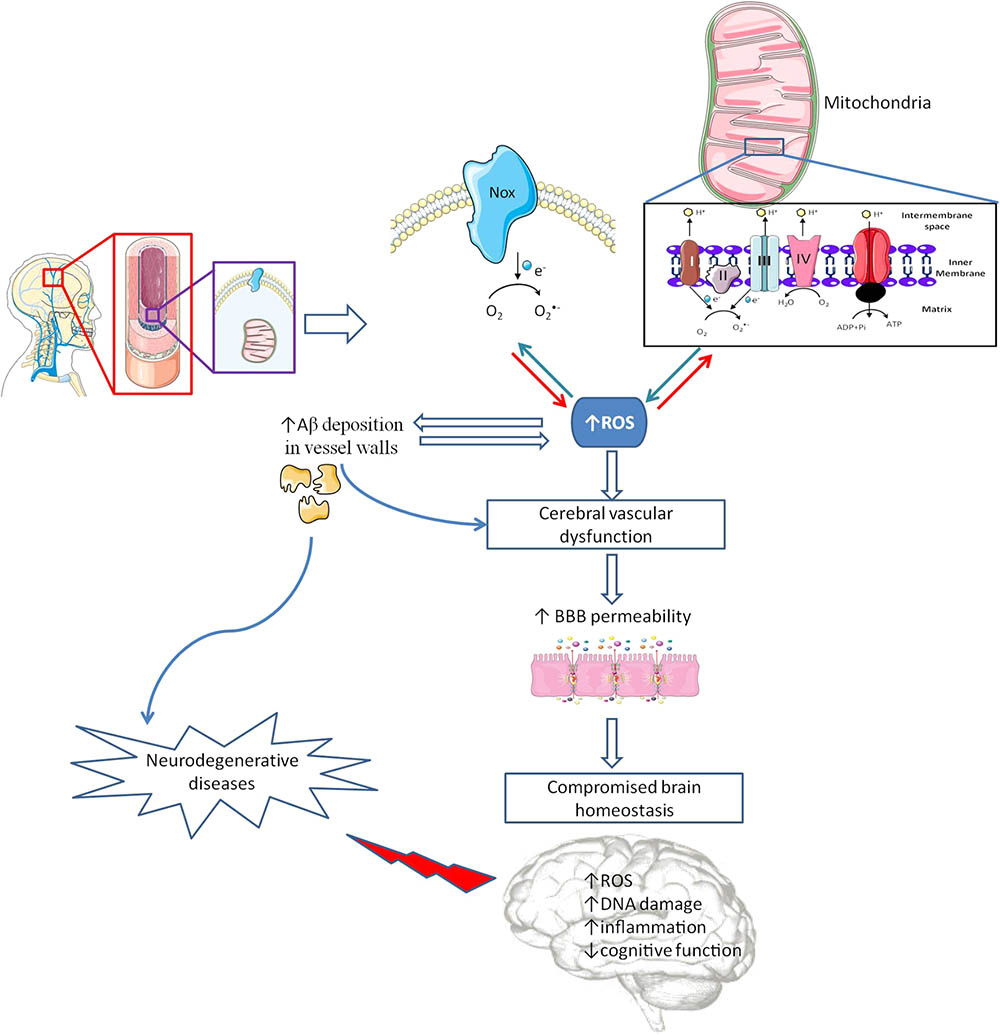
FIGURE 2. Cerebrovascular alterations and brain damage. The brain is a very delicate organ that relies on an extremely controlled environment and on a constant energy supply due to its high metabolic demand, which depend on blood–brain barrier (BBB) proper functioning. Besides other cell types (astrocytes, pericytes, microglia and a basement membrane made from structural proteins) endothelial cells (EC) assume the major role in controlling BBB integrity. Inside EC, mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox) act as main sources of reactive oxygen species (ROS). In pathological conditions, an increased ROS levels cause cerebrovascular dysfunction, which promotes an increase in BBB leakage interfering with brain energy supply and homeostasis, and increasing amyloid β (Aβ) peptide deposition in vascular walls. Alterations in BBB features are associated with oxidative stress and damage and inflammatory processes, among other deleterious alterations, contributing to cognitive defects and, eventually, the development of neurodegenerative diseases.
Mitochondrial-derived oxidative stress can also activate vascular inflammation in aged carotid arteries and vessels (Csiszar et al., 2007; Ungvari et al., 2007) leading to atherosclerosis through activation of endothelial NF-κB, which is responsible for the upregulation of adhesion molecules and increased monocyte adhesiveness in aged aortic arteries (Ungvari et al., 2007). Moreover, changes in Nox activity and/or expression is widely described in aged brain vessels (Park et al., 2007; Mayhan et al., 2008). Nevertheless, the mechanistic role of Nox in the aging process remains obscure (Sahoo et al., 2016).
Data from the literature also show that in cultured aged cerebromicrovascular endothelial cells (CMVECs) the mRNA expression levels of Nox2 and 4 subunits were upregulated compared with young CMVECs, a phenomenon similar to that observed in the myocardium, leading to increased levels of oxidative stress (Toth et al., 2014). Although further studies must be performed in order to corroborate this hypothesis, it seems that, similarly to the myocardium, in CMVECs the increased expression of Nox 2 and 4 causes oxidative stress, which activates the renin–angiotensin–aldosterone system (RAAS) (Wang et al., 2010). This activation can occur through a ROS-induced increase in angiotensinogen (AGT), a 60-kDa α2-globulin glycoprotein that constitutes the precursor of RAAS; stimulation of renin, the enzyme responsible for the initiation of the RAAS pathway; and release or regulation of angiotensin converting enzyme (ACE) activity, crucial for the formation of angiotensin II, the major effector of RAAS (Morato et al., 2017). Furthermore, since Nox 4 is usually localized in mitochondria, its increased expression can also contribute to increased levels of mitochondrial ROS potentiating the aging process as well as age-related diseases (Sahoo et al., 2016). The use of Nox inhibitors showed promising results in reducing oxidative stress levels and in improving endothelial function in several disease models (Girouard et al., 2006; Kahles et al., 2007; Matsumoto et al., 2007; Miller et al., 2010a). Also the use of resveratrol seems to be effective in restoring cerebromicrovascular endothelial function through a downregulation of Nox-derived ROS production in aged mice (Toth et al., 2014).
Despite the extensive literature stating that age-associated oxidative stress can lead to cerebrovascular dysfunction and cognitive decline, there is a significant gap in our knowledge regarding the exact mechanisms underlying these defects (Tarantini et al., 2017a).
Although the previous alterations seem to be common features in physiological aging of vessels, somewhere over the way those defects can become more pronounced triggering neurodegenerative events (Kling et al., 2013). Indeed, recent reports suggest a major role for the NVU in the initiation of neurodegeneration (Nelson et al., 2016). In fact, NVU dysfunction leads to an increased BBB permeability with the subsequent entry of neurotoxic molecules into the brain disturbing its homeostasis. A decrease in the removal of neurotoxic substances from the brain, and a deficient nutrient delivery system eventually culminate in neuronal loss and synaptic dysfunction (Zlokovic, 2008). Thus, a decline in cerebrovascular function in age-associated diseases, such as AD, is easily understandable (Girouard and Iadecola, 2006). Indeed, it is estimated that about 60–90% of AD patients also present cerebrovascular alterations including cerebral amyloid angiopathy (CAA), microinfarcts and ischemic lesions and microvascular degeneration (Jellinger and Mitter-Ferstl, 2003; Bell and Zlokovic, 2009).
It has been recently reported the existence of BBB and vascular alterations in several diseases such as Parkinson’s disease (PD), Huntington’s disease and cerebral small vessel disease (Lin et al., 2013, 2015; Lee and Pienaar, 2014; St-Amour et al., 2015; Al-Bachari et al., 2017) although there is a gap concerning the mechanisms underlying those alterations and the role of vascular oxidative stress in these pathologies.
Concerning AD, the most common form of dementia in the elderly, more information exists about the vascular alterations that occur in this disease (Gallart-Palau et al., 2016). Several clinical and basic evidence points to the existence of a major contribution of both large artery and small vessel disease in the pathogenesis of AD (Iadecola, 2013; De Strooper and Karran, 2016; Faraci, 2017). Having this into account, the two-hit vascular hypothesis of AD emerged, postulating that cerebrovascular damage is an initial insult that is self-sufficient to initiate neuronal injury and neurodegeneration, but can also promote accumulation of Aβ peptide, a neurotoxic peptide, in the brain (Nelson et al., 2016). Thus, events that compromise vascular health can initiate a cascade of deleterious events that culminate in AD development. In fact, previous studies from our laboratory demonstrated that some risk factors for AD, such as hyperglycemia, increase the susceptibility of brain endothelial cells to Aβ peptide, a phenomenon related to ROS overproduction (Carvalho et al., 2014). Moreover, several genetic [apolipoprotein E4 (APOE4), phosphatidylinositol-binding clathrin assembly protein (PICALM), clusterin, presenilin 1, amyloid precursor protein (APP), mesenchyme homeobox gene 2 (MEOX2)] (Nelson et al., 2016) and non-genetic (hypertension, metabolic syndrome, hypercholesterolemia, atherosclerosis, alcohol and substance abuse, among others) (Kivipelto et al., 2002; Skoog and Gustafson, 2006; Nelson et al., 2016; Campos-Pena et al., 2017) risk factors for AD seem to be linked with alterations in vascular function supporting the idea that vascular alterations play a major role in AD pathology.
Recent studies in wild type mice showed that, even under normal conditions, there is a different regional susceptibility of the cerebrovasculature to oxidative stress (Austin et al., 2015). Vessels from cortex and hippocampus, the two main areas compromised in AD, contain significantly higher levels of intracellular O2•- and increased protein levels of both Nox 2 and 4 (Austin et al., 2015). Other studies revealed significant changes in blood vessels morphology, decreased vascular density and increased vessels tortuosity in AD brains (Hunter et al., 2012). Moreover, the use of recent technologies such as arterial spin labeling magnetic resonance imaging (MRI), functional blood-oxygen-level-dependent (BOLD)-MRI, fluorodeoxyglucose-positron emission tomography (FDG-PET), and single-photon emission computerized tomography (SPECT), showed decreased levels of CBF in early phases of human AD progression (Daneman and Prat, 2015). Recently, Lourenço et al. (2017) showed that reduced CBF changes found in triple transgenic for AD (3xTg-AD) mice preceded memory dysfunction thus suggesting that cerebrovascular dysfunction could be the primary cause of neurovascular uncoupling in AD. Those observations are supported by MRI, transcranial doppler and SPECT studies revealing different patterns of decreased CBF coincident with the brain regions where neuropathological alterations are more relevant in AD (Hu et al., 2010; Pimentel-Coelho and Rivest, 2012). Those alterations seem to be closely related with the levels of oxidative stress in the cerebrovasculature. Studies from our laboratory showed that 3xTg-AD mice present an increased BBB permeability, in cortex and hippocampus (Carvalho et al., 2013), these alterations being correlated with decreased aconitase activity, an enzyme whose activity is inhibited by O2•- (Carvalho et al., 2013). The increased levels of ROS appeared to be the result of impaired activity of mitochondrial enzymatic complexes I–III (Figure 2). Moreover, and similarly to our observations in mitochondria isolated from whole brain of 3xTg-AD mice (Carvalho et al., 2012), Golgi silver impregnation studies using multiple samples from the hippocampus and cortices of AD patients showed that brain endothelial cells present enlarged mitochondria, disruption of the mitochondrial cristae and reduced abundance (Baloyannis and Baloyannis, 2012).
An interesting fact is that even though mitochondrial morphology is very similar in male and female rodent cerebral arteries, major differences can be observed concerning mitochondrial protein mass, respiration, and function (Rutkai et al., 2015), which is in accordance with sex/gender differences observed in several brain-related studies (Giordano et al., 2013; Christov-Moore et al., 2014; Ingalhalikar et al., 2014; Sun et al., 2015) including the susceptibility to develop AD (Candeias et al., 2017).
Mitochondrial ROS overproduction also increases Aβ deposition in vessels walls, which is a common feature in many cases of AD (Figure 2). Indeed, 34–35% of human AD cases present CAA, predominantly in the occipital lobe (Love et al., 2014). Aβ deposition in vessels walls seems to initiate a vicious cycle, Aβ increases ROS levels, which in turn potentiate Aβ deposition (Figure 2) (Park et al., 2011). There is also evidence showing that Aβ can induce Nox activation and ROS production, which are responsible for the activation of multiple mechanisms involved in vascular dysfunction and decreased levels of tight junction proteins mRNA (Carrano et al., 2011). However, Nox driven ROS production does not seem to affect Aβ production in Tg2576 mice, a mouse model of AD, lacking the key Nox 2 subunit (Park et al., 2008, 2011).
Endothelial ROS overproduction upregulates the endothelial production and release of endothelin-1, a well know vasoconstrictor that appears to be increased in AD (Palmer et al., 2013). Likewise, ROS overproduction seems to be responsible for reduced •NO bioavailability mainly by its fast reaction with O2•- generating ONOO- (Park et al., 2005; Tong et al., 2005) and for a decreased activity of potassium channels (Erdos et al., 2004), two major factors required for endothelial-mediated dilations in brain vasculature (Hamel et al., 2016). Moreover, Aβ-induced nitrosative stress in endothelial cells is also responsible for DNA damage, resulting in poly (ADP-ribose) polymerase (PARP) activation, ultimately leading to a large increase in intracellular Ca2+ through transient receptor potential melastatin-2 channels activation (Figure 2) (Park et al., 2014).
Reactive oxygen species overproduction also interferes with HIF-1α reducing its expression and activity which will restrain the stimulus to promote angiogenesis and new vessels formation (Ogunshola and Antoniou, 2009) leading to a vicious cycle of impaired capillary perfusion, hypoxia and oxidative stress (Mamelak, 2017).
Recognizing vascular ROS production as one of the main causes of BBB dysfunction in aging and dementia, particularly in AD, raises the prospect that pharmacotherapy targeting the major ROS producers, namely mitochondria and/or Nox, might open a new venue to stop or, at least, slow dementia progression (Sweeney et al., 2015). Indeed, relying on the overwhelming evidence that mitochondrial dysfunction could be in the genesis of neurovascular dysfunction, several molecules with potential therapeutic effects were designed to target ROS, boost mitochondrial function and decrease free radical production and oxidative damage (Tong et al., 2005; Hamel et al., 2008; Moon et al., 2014) in an attempt to improve neurovascular health. Some of those molecules ameliorated the cognitive performance of mouse models of cerebrovascular disease (Tong et al., 2005). It was observed that the steroid hormone 17β-estradiol (estrogen) modulates the expression of several transcriptional regulators causing a decrease in PGC-1α, and an increase in PGC-1β, PGC-1-related coactivator (PRC), nuclear respiratory factor 1 (NRF-1) and mitochondrial transcription factor A (TFAM), which protect cerebral blood vessels (Table 1) (Kemper et al., 2014). These protective effects were due to increased mitochondrial biogenesis and antioxidant enzymes (Kemper et al., 2014). However, the use of antioxidant therapy in cerebrovascular disease still needs validation. Indeed, several promising approaches failed in clinical trials (Tarantini et al., 2017b), opening an intense debate about the reasons for the failure of those molecules in humans. Merely as an example, the use of tempol showed promising results in improving vasodilator responses of cerebral arterioles in aged APP transgenic mice with CAA (Han et al., 2015) but so far there is no evidence of its efficacy in clinical trials. Furthermore, two major concerns arise from the use of antioxidants as therapeutic approaches: (1) physiological ROS concentrations are important in normal cell functioning and the use of antioxidants may interfere with ROS signaling and (2) the concentrations of antioxidants revealed to be suboptimal doses in certain situations. Indeed, it is known now that supra-physiological concentrations are required to compete with the constant reaction that usually occurs between O2•- and •NO (Drummond et al., 2011). Large part of the studies failed to prove that antioxidants reach the vasculature at appropriate/therapeutic concentrations (De Silva and Miller, 2016).
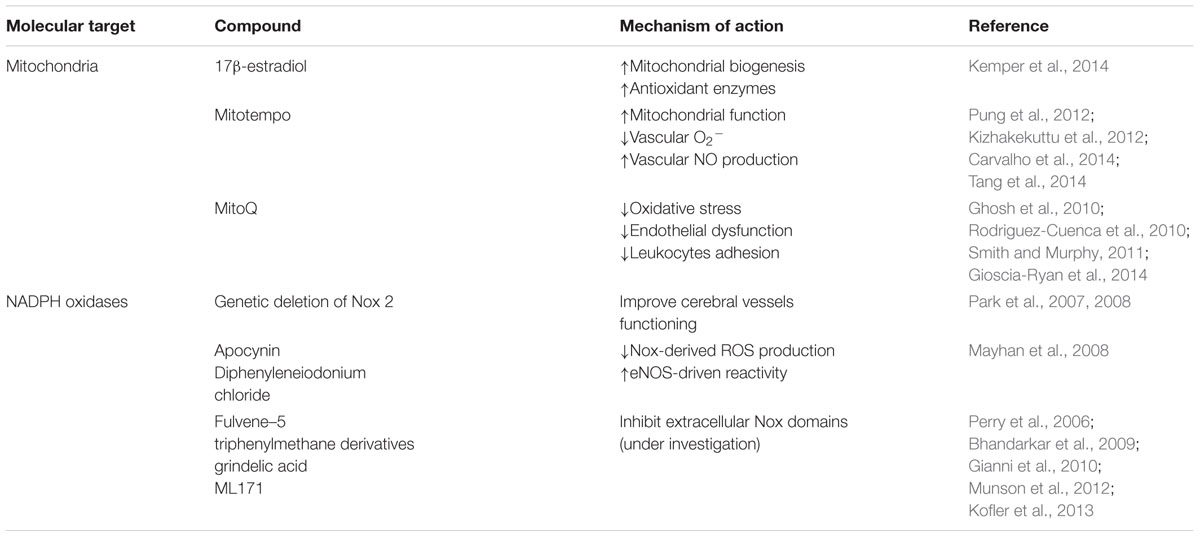
TABLE 1. Antioxidant based therapies.
To overcome the failure of the traditional antioxidants, researchers are developing new specific-targeted antioxidants. However, evidence about the specific-target antioxidants effects in endothelial cells is scarce. Moreover, mitochondria-targeted antioxidants decrease the dose required and limit toxic side effects rendering them a promising therapeutic approach (Smith and Murphy, 2011). It was observed that MitoTempo, a mitochondria-targeted antioxidant, was able to counteract Aβ-induced damage under hyperglycemic conditions in rat and mouse primary cultures of endothelial cells (Carvalho et al., 2014). Additionally, it was shown that MitoTempo was able to improve mitochondrial function and coronary collateral growth after ischemia/reperfusion in Zucker fatty rats (Table 1) (Pung et al., 2012). Recent studies also showed that MitoTempo was able to decrease vascular O2•- and increase vascular •NO production improving endothelial-dependent relaxation in angiotensin II-induced hypertensive C57Bl/6 mice (Tang et al., 2014). It was also observed that MitoTempo improves endothelial function and reduces mitochondrial O2•- levels in subcutaneous arterioles isolated from type 2 diabetic patients (Table 1) (Kizhakekuttu et al., 2012). Moreover, MitoQ, another mitochondria-targeted antioxidant was able to reduce oxidative stress, without causing adverse effects, on wild-type mice (Rodriguez-Cuenca et al., 2010; Smith et al., 2011) and animal models of neurodegenerative diseases such as AD and PD (Ghosh et al., 2010; Manczak et al., 2010). MitoQ was able to improve age-related arterial endothelial dysfunction in C57BL/6 mice (Table 1) (Gioscia-Ryan et al., 2014). Furthermore, MitoQ has been shown to prevent cocaine-induced cardiac dysfunction in Wistar rats (Vergeade et al., 2010). However, the use of mitochondrial-directed drugs should take in account two major limitations: first, the lack of organ-specificity, leading to a greater accumulation in mitochondria-rich tissues; and second the typically used chemicals tend to accumulate in the matrix and the matrix-facing surface of the inner mitochondrial membrane, over other important mitochondrial compartments (Leitao-Rocha et al., 2015). The majority of mitochondria-targeted compounds were already able to identify and get ahead of these limiting factors in human clinical trials, defining the amount of the compound that can be administered safely. For example, MitoQ, was already developed as a pharmaceutical by Antipodean Pharmaceuticals Inc. (Smith et al., 2008) and clinical trials are running, namely in type 2 diabetic patients, where MitoQ treatment was able to decrease ROS levels and significantly reduced the adhesion of leukocytes to endothelial cells in type 2 diabetic individuals (Escribano-Lopez et al., 2016).
Likewise, approaches designed to target Nox-derived ROS overproduction are also under intense scrutiny. Indeed, the genetic deletion or inhibition of Nox2 seems effective in improving cerebral vessels functioning in aged APP mice (Park et al., 2007, 2008). Apocynin and diphenyleniodonium are widely used to ameliorate Nox-related cerebrovascular dysfunction, however, both compounds present non-specific effects and cannot be used in the clinic (Table 1) (De Silva and Miller, 2016). In this line, several attempts are being made to synthesize new Nox inhibitors including fulvene-5, triphenylmethane derivatives, grindelic acid, and ML171 (Table 1) (De Silva and Miller, 2016). However, results are scarce and further studies are needed to demonstrate that those compounds are potential therapeutics for the treatment of neurodegenerative conditions characterized by oxidative stress-associated vascular alterations.
In sum, despite the existing information about the role of cerebrovascular oxidative stress in neurodegenerative conditions, a long way is still ahead to clarify the mechanisms of age-associated vascular damage and subsequent neurodegenerative conditions. New information is crucial for the design of more effective therapeutic strategies.
CC performed literature search and wrote the paper. PM provided a critical revision of the paper.
This work was funded by European funds from FEDER, through the Programa Operacional Factores de Competitividade – COMPETE 2020 (HealthyAging2020: CENTRO-01-0145-FEDER-000012) and Fundação para a Ciência e a Tecnologia (PEst-C/SAU/LA0001/2013-2014 and fellowship SFRH/BPD/107741/2015 to CC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with the authors.
Agarwal, R., and Shukla, G. S. (1999). Potential role of cerebral glutathione in the maintenance of blood-brain barrier integrity in rat. Neurochem. Res. 24, 1507–1514. doi: 10.1023/A:1021191729865
Aghajanian, A., Wittchen, E. S., Campbell, S. L., and Burridge, K. (2009). Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One 4:e8045. doi: 10.1371/journal.pone.0008045
Al-Bachari, S., Vidyasagar, R., Emsley, H. C., and Parkes, L. M. (2017). Structural and physiological neurovascular changes in idiopathic Parkinson’s disease and its clinical phenotypes. J. Cereb. Blood Flow Metab. 37, 3409–3421. doi: 10.1177/0271678X16688919
Alfadda, A. A., and Sallam, R. M. (2012). Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012:936486. doi: 10.1155/2012/936486
Allen, C. L., and Bayraktutan, U. (2009). Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 4, 461–470. doi: 10.1111/j.1747-4949.2009.00387.x
Alyautdin, R., Khalin, I., Nafeeza, M. I., Haron, M. H., and Kuznetsov, D. (2014). Nanoscale drug delivery systems and the blood-brain barrier. Int. J. Nanomedicine 9, 795–811. doi: 10.2147/IJN.S52236
Armulik, A., Genove, G., Mae, M., Nisancioglu, M. H., Wallgard, E., Niaudet, C., et al. (2010). Pericytes regulate the blood-brain barrier. Nature 468, 557–561. doi: 10.1038/nature09522
Austin, S. A., Santhanam, A. V., d’Uscio, L. V., and Katusic, Z. S. (2015). Regional heterogeneity of cerebral microvessels and brain susceptibility to oxidative stress. PLoS One 10:e0144062. doi: 10.1371/journal.pone.0144062
Baloyannis, S. J., and Baloyannis, I. S. (2012). The vascular factor in Alzheimer’s disease: a study in Golgi technique and electron microscopy. J. Neurol. Sci. 322, 117–121. doi: 10.1016/j.jns.2012.07.010
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 118, 103–113. doi: 10.1007/s00401-009-0522-3
Bhandarkar, S. S., Jaconi, M., Fried, L. E., Bonner, M. Y., Lefkove, B., Govindarajan, B., et al. (2009). Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J. Clin. Invest. 119, 2359–2365. doi: 10.1172/JCI33877
Brandes, R. P., Viedt, C., Nguyen, K., Beer, S., Kreuzer, J., Busse, R., et al. (2001). Thrombin-induced MCP-1 expression involves activation of the p22phox-containing NADPH oxidase in human vascular smooth muscle cells. Thromb. Haemost. 85, 1104–1110. doi: 10.1055/s-0037-1615970
Brown, G. C., and Borutaite, V. (2001). Nitric oxide, mitochondria, and cell death. IUBMB Life 52, 189–195. doi: 10.1080/15216540152845993
Busija, D. W., and Katakam, P. V. (2014). Mitochondrial mechanisms in cerebral vascular control: shared signaling pathways with preconditioning. J. Vasc. Res. 51, 175–189. doi: 10.1159/000360765
Busija, D. W., Rutkai, I., Dutta, S., and Katakam, P. V. (2016). Role of mitochondria in cerebral vascular function: energy production, cellular protection, and regulation of vascular tone. Compr. Physiol. 6, 1529–1548. doi: 10.1002/cphy.c150051
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. doi: 10.1016/S0891-5849(00)00317-8
Camici, G. G., Savarese, G., Akhmedov, A., and Luscher, T. F. (2015). Molecular mechanism of endothelial and vascular aging: implications for cardiovascular disease. Eur. Heart J. 36, 3392–3403. doi: 10.1093/eurheartj/ehv587
Campos-Pena, V., Toral-Rios, D., Becerril-Perez, F., Sanchez-Torres, C., Delgado-Namorado, Y., Torres-Ossorio, E., et al. (2017). Metabolic syndrome as a risk factor for Alzheimer’s disease: is abeta a crucial factor in both pathologies? Antioxid. Redox Signal. 26, 542–560. doi: 10.1089/ars.2016.6768
Candeias, E., Duarte, A. I., Sebastiao, I., Fernandes, M. A., Placido, A. I., Carvalho, C., et al. (2017). Middle-aged diabetic females and males present distinct susceptibility to Alzheimer disease-like pathology. Mol. Neurobiol. 54, 6471–6489. doi: 10.1007/s12035-016-0155-1
Caplan, L. R., Biller, J., Leary, M. C., Lo, E. H., Thomas, A. J., Yenari, M., et al. (2017). Primer on Cerebrovascular Diseases. Amsterdam: Elsevier Science.
Carrano, A., Hoozemans, J. J., van der Vies, S. M., Rozemuller, A. J., van Horssen, J., and de Vries, H. E. (2011). Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 15, 1167–1178. doi: 10.1089/ars.2011.3895
Carvalho, C., Cardoso, S., Correia, S. C., Santos, R. X., Santos, M. S., Baldeiras, I., et al. (2012). Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes 61, 1234–1242. doi: 10.2337/db11-1186
Carvalho, C., Correia, S., Santos, R., Cardoso, S., Santos, M., and Moreira, P. (2010). “Diabetes, mitochondria and brain endothelium dysfunction: a dangerous triad for neurodegeneration?,” in Mitochondria: Structure, Functions and Dysfunctions, ed. O. Svensson (Hauppauge, NY: Nova Science Publishers, Inc.), 561–577.
Carvalho, C., Correia, S. C., Cardoso, S., Placido, A. I., Candeias, E., Duarte, A. I., et al. (2015). The role of mitochondrial disturbances in Alzheimer, Parkinson and Huntington diseases. Expert Rev. Neurother. 15, 867–884. doi: 10.1586/14737175.2015.1058160
Carvalho, C., Katz, P. S., Dutta, S., Katakam, P. V., Moreira, P. I., and Busija, D. W. (2014). Increased susceptibility to amyloid-beta toxicity in rat brain microvascular endothelial cells under hyperglycemic conditions. J. Alzheimers Dis. 38, 75–83. doi: 10.3233/JAD-130464
Carvalho, C., Machado, N., Mota, P., Correia, S., Cardoso, S., Santos, R., et al. (2013). Type 2 diabetic and Alzheimer’s disease mice present similar behavioral, cognitive and vascular anomalies. J. Alzheimer Dis. 35, 623–635. doi: 10.3233/JAD-130005
Chan, D. C. (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252. doi: 10.1016/j.cell.2006.06.010
Chen, Q., Wang, Q., Zhu, J., Xiao, Q., and Zhang, L. (2017). Reactive oxygen species: key regulators in vascular health and diseases. Br. J. Pharmacol. 175, 1279–1292. doi: 10.1111/bph.13828
Christov-Moore, L., Simpson, E. A., Coude, G., Grigaityte, K., Iacoboni, M., and Ferrari, P. F. (2014). Empathy: gender effects in brain and behavior. Neurosci. Biobehav. Rev. 46(Pt 4), 604–627. doi: 10.1016/j.neubiorev.2014.09.001
Correia, S. C., Carvalho, C., Cardoso, S., Santos, R. X., Santos, M. S., Oliveira, C. R., et al. (2010). Mitochondrial preconditioning: a potential neuroprotective strategy. Front. Aging Neurosci. 2:138. doi: 10.3389/fnagi.2010.00138
Correia, S. C., Santos, R. X., Cardoso, S. M., Santos, M. S., Oliveira, C. R., and Moreira, P. I. (2012). Cyanide preconditioning protects brain endothelial and NT2 neuron-like cells against glucotoxicity: role of mitochondrial reactive oxygen species and HIF-1alpha. Neurobiol. Dis. 45, 206–218. doi: 10.1016/j.nbd.2011.08.005
Csiszar, A., Labinskyy, N., Orosz, Z., Xiangmin, Z., Buffenstein, R., and Ungvari, Z. (2007). Vascular aging in the longest-living rodent, the naked mole rat. Am. J. Physiol. Heart Circ. Physiol. 293, H919–H927. doi: 10.1152/ajpheart.01287.2006
Culic, O., Gruwel, M. L., and Schrader, J. (1997). Energy turnover of vascular endothelial cells. Am. J. Physiol. 273(1 Pt 1), C205–C213. doi: 10.1152/ajpcell.1997.273.1.C205
Daneman, R., and Prat, A. (2015). The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 7:a020412. doi: 10.1101/cshperspect.a020412
de Almeida, A., Ribeiro, T. P., and de Medeiros, I. A. (2017). Aging: molecular pathways and implications on the cardiovascular system. Oxid. Med. Cell. Longev. 2017:7941563. doi: 10.1155/2017/7941563
De Keulenaer, G. W., Alexander, R. W., Ushio-Fukai, M., Ishizaka, N., and Griendling, K. K. (1998). Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem. J. 329(Pt 3), 653–657. doi: 10.1042/bj3290653
De Silva, T. M., and Faraci, F. M. (2016). Microvascular dysfunction and cognitive impairment. Cell. Mol. Neurobiol. 36, 241–258. doi: 10.1007/s10571-015-0308-1
De Silva, T. M., and Miller, A. A. (2016). Cerebral small vessel disease: targeting oxidative stress as a novel therapeutic strategy? Front. Pharmacol. 7:61. doi: 10.3389/fphar.2016.00061
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Didion, S. P., and Faraci, F. M. (2002). Effects of NADH and NADPH on superoxide levels and cerebral vascular tone. Am. J. Physiol. Heart Circ. Physiol. 282, H688–H695. doi: 10.1152/ajpheart.00576.2001
Dikalov, S. I., Nazarewicz, R. R., Bikineyeva, A., Hilenski, L., Lassegue, B., Griendling, K. K., et al. (2014). Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 20, 281–294. doi: 10.1089/ars.2012.4918
Dikalov, S. I., Dikalova, A. E., Bikineyeva, A. T., Schmidt, H. H., Harrison, D. G., and Griendling, K. K. (2008). Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 45, 1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013
Ding, X., Zhang, M., Gu, R., Xu, G., and Wu, H. (2017). Activated microglia induce the production of reactive oxygen species and promote apoptosis of co-cultured retinal microvascular pericytes. Graefes Arch. Clin. Exp. Ophthalmol. 255, 777–788. doi: 10.1007/s00417-016-3578-5
Dromparis, P., and Michelakis, E. D. (2012). Mitochondria in vascular health and disease. Annu. Rev. Physiol. 75, 95–126. doi: 10.1146/annurev-physiol-030212-183804
Drummond, G. R., Selemidis, S., Griendling, K. K., and Sobey, C. G. (2011). Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 10, 453–471. doi: 10.1038/nrd3403
Drummond, G. R., and Sobey, C. G. (2014). Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol. Metab. 25, 452–463. doi: 10.1016/j.tem.2014.06.012
Enciu, A. M., Gherghiceanu, M., and Popescu, B. O. (2013). Triggers and effectors of oxidative stress at blood-brain barrier level: relevance for brain ageing and neurodegeneration. Oxid. Med. Cell. Longev. 2013:297512. doi: 10.1155/2013/297512
Erdos, B., Snipes, J. A., Miller, A. W., and Busija, D. W. (2004). Cerebrovascular dysfunction in Zucker obese rats is mediated by oxidative stress and protein kinase C. Diabetes 53, 1352–1359. doi: 10.2337/diabetes.53.5.1352
Escribano-Lopez, I., Diaz-Morales, N., Rovira-Llopis, S., de Maranon, A. M., Orden, S., Alvarez, A., et al. (2016). The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 10, 200–205. doi: 10.1016/j.redox.2016.10.017
Faraci, F. M. (2017). Disease highlights the cellular diversity of neurovascular units: sign in stranger. Circ. Res. 121, 203–205. doi: 10.1161/CIRCRESAHA.117.311386
Faraci, F. M., and Sobey, C. G. (1998). Role of potassium channels in regulation of cerebral vascular tone. J. Cereb. Blood Flow Metab. 18, 1047–1063. doi: 10.1097/00004647-199810000-00001
Faraco, G., Moraga, A., Moore, J., Anrather, J., Pickel, V. M., and Iadecola, C. (2013). Circulating endothelin-1 alters critical mechanisms regulating cerebral microcirculation. Hypertension 62, 759–766. doi: 10.1161/HYPERTENSIONAHA.113.01761
Faraco, G., Sugiyama, Y., Lane, D., Garcia-Bonilla, L., Chang, H., Santisteban, M. M., et al. (2016). Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J. Clin. Invest. 126, 4674–4689. doi: 10.1172/JCI86950
Freeman, L. R., and Keller, J. N. (2012). Oxidative stress and cerebral endothelial cells: regulation of the blood-brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta 1822, 822–829. doi: 10.1016/j.bbadis.2011.12.009
Gallart-Palau, X., Lee, B. S., Adav, S. S., Qian, J., Serra, A., Park, J. E., et al. (2016). Gender differences in white matter pathology and mitochondrial dysfunction in Alzheimer’s disease with cerebrovascular disease. Mol. Brain 9:27. doi: 10.1186/s13041-016-0205-7
Ghosh, A., Chandran, K., Kalivendi, S. V., Joseph, J., Antholine, W. E., Hillard, C. J., et al. (2010). Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 49, 1674–1684. doi: 10.1016/j.freeradbiomed.2010.08.028
Gianni, D., Taulet, N., Zhang, H., DerMardirossian, C., Kister, J., Martinez, L., et al. (2010). A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem. Biol. 5, 981–993. doi: 10.1021/cb100219n
Giordano, G., Tait, L., Furlong, C. E., Cole, T. B., Kavanagh, T. J., and Costa, L. G. (2013). Gender differences in brain susceptibility to oxidative stress are mediated by levels of paraoxonase-2 expression. Free Radic. Biol. Med. 58, 98–108. doi: 10.1016/j.freeradbiomed.2013.01.019
Gioscia-Ryan, R. A., LaRocca, T. J., Sindler, A. L., Zigler, M. C., Murphy, M. P., and Seals, D. R. (2014). Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 592, 2549–2561. doi: 10.1113/jphysiol.2013.268680
Girouard, H., and Iadecola, C. (2006). Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol. 100, 328–335. doi: 10.1152/japplphysiol.00966.2005
Girouard, H., Park, L., Anrather, J., Zhou, P., and Iadecola, C. (2006). Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler. Thromb. Vasc. Biol. 26, 826–832. doi: 10.1161/01.ATV.0000205849.22807.6e
Girouard, H., Park, L., Anrather, J., Zhou, P., and Iadecola, C. (2007). Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 27, 303–309. doi: 10.1161/01.ATV.0000253885.41509.25
Goetz, M. E., and Luch, A. (2008). Reactive species: a cell damaging rout assisting to chemical carcinogens. Cancer Lett. 266, 73–83. doi: 10.1016/j.canlet.2008.02.035
Gordon, G. R., Mulligan, S. J., and MacVicar, B. A. (2007). Astrocyte control of the cerebrovasculature. Glia 55, 1214–1221. doi: 10.1002/glia.20543
Gorlach, A., Diebold, I., Schini-Kerth, V. B., Berchner-Pfannschmidt, U., Roth, U., Brandes, R. P., et al. (2001). Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22(phox)-containing NADPH oxidase. Circ. Res. 89, 47–54. doi: 10.1161/hh1301.092678
Grochowski, C., Litak, J., Kamieniak, P., and Maciejewski, R. (2018). Oxidative stress in cerebral small vessel disease. Role of reactive species. Free Radic. Res. 52, 1–13. doi: 10.1080/10715762.2017.1402304
Halliwell, B. (2001). Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 18, 685–716. doi: 10.2165/00002512-200118090-00004
Hamel, E., Nicolakakis, N., Aboulkassim, T., Ongali, B., and Tong, X. K. (2008). Oxidative stress and cerebrovascular dysfunction in mouse models of Alzheimer’s disease. Exp. Physiol. 93, 116–120. doi: 10.1113/expphysiol.2007.038729
Hamel, E., Royea, J., Ongali, B., and Tong, X. K. (2016). Neurovascular and cognitive failure in Alzheimer’s disease: benefits of cardiovascular therapy. Cell. Mol. Neurobiol. 36, 219–232. doi: 10.1007/s10571-015-0285-4
Han, B. H., Zhou, M. L., Johnson, A. W., Singh, I., Liao, F., Vellimana, A. K., et al. (2015). Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. U.S.A. 112, E881–E890. doi: 10.1073/pnas.1414930112
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. doi: 10.1093/geronj/11.3.298
Helmcke, I., Heumuller, S., Tikkanen, R., Schroder, K., and Brandes, R. P. (2009). Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid. Redox Signal. 11, 1279–1287. doi: 10.1089/ARS.2008.2383
Hopkins, N. D., Stratton, G., Tinken, T. M., Ridgers, N. D., Graves, L. E., McWhannell, N., et al. (2011). Seasonal reduction in physical activity and flow-mediated dilation in children. Med. Sci. Sports Exerc. 43, 232–238. doi: 10.1249/MSS.0b013e3181ebe90e
Hu, W. T., Wang, Z., Lee, V. M., Trojanowski, J. Q., Detre, J. A., and Grossman, M. (2010). Distinct cerebral perfusion patterns in FTLD and AD. Neurology 75, 881–888. doi: 10.1212/WNL.0b013e3181f11e35
Hunter, J. M., Kwan, J., Malek-Ahmadi, M., Maarouf, C. L., Kokjohn, T. A., Belden, C., et al. (2012). Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer’s disease. PLoS One 7:e36893. doi: 10.1371/journal.pone.0036893
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Ingalhalikar, M., Smith, A., Parker, D., Satterthwaite, T. D., Elliott, M. A., Ruparel, K., et al. (2014). Sex differences in the structural connectome of the human brain. Proc. Natl. Acad. Sci. U.S.A. 111, 823–828. doi: 10.1073/pnas.1316909110
Jackman, K. A., Miller, A. A., Drummond, G. R., and Sobey, C. G. (2009). Importance of NOX1 for angiotensin II-induced cerebrovascular superoxide production and cortical infarct volume following ischemic stroke. Brain Res. 1286, 215–220. doi: 10.1016/j.brainres.2009.06.056
Jansen, F., Yang, X., Franklin, B. S., Hoelscher, M., Schmitz, T., Bedorf, J., et al. (2013). High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovasc. Res. 98, 94–106. doi: 10.1093/cvr/cvt013
Jellinger, K. A., and Mitter-Ferstl, E. (2003). The impact of cerebrovascular lesions in Alzheimer disease–a comparative autopsy study. J. Neurol. 250, 1050–1055. doi: 10.1007/s00415-003-0142-0
Kahles, T., Luedike, P., Endres, M., Galla, H. J., Steinmetz, H., Busse, R., et al. (2007). NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke 38, 3000–3006. doi: 10.1161/STROKEAHA.107.489765
Kemper, M. F., Stirone, C., Krause, D. N., Duckles, S. P., and Procaccio, V. (2014). Genomic and non-genomic regulation of PGC1 isoforms by estrogen to increase cerebral vascular mitochondrial biogenesis and reactive oxygen species protection. Eur. J. Pharmacol. 723, 322–329. doi: 10.1016/j.ejphar.2013.11.009
Kivipelto, M., Laakso, M. P., Tuomilehto, J., Nissinen, A., and Soininen, H. (2002). Hypertension and hypercholesterolaemia as risk factors for Alzheimer’s disease: potential for pharmacological intervention. CNS Drugs 16, 435–444. doi: 10.2165/00023210-200216070-00001
Kizhakekuttu, T. J., Wang, J., Dharmashankar, K., Ying, R., Gutterman, D. D., Vita, J. A., et al. (2012). Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 32, 2531–2539. doi: 10.1161/ATVBAHA.112.256024
Kling, M. A., Trojanowski, J. Q., Wolk, D. A., Lee, V. M., and Arnold, S. E. (2013). Vascular disease and dementias: paradigm shifts to drive research in new directions. Alzheimers Dement. 9, 76–92. doi: 10.1016/j.jalz.2012.02.007
Kluge, M. A., Fetterman, J. L., and Vita, J. A. (2013). Mitochondria and endothelial function. Circ. Res. 112, 1171–1188. doi: 10.1161/CIRCRESAHA.111.300233
Konior, A., Schramm, A., Czesnikiewicz-Guzik, M., and Guzik, T. J. (2014). NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 20, 2794–2814. doi: 10.1089/ars.2013.5607
Kubíková, T., Kochova, P., Tomasek, P., Witter, K., and Tonar, Z. (2017). Numerical and length densities of microvessels in the human brain: correlation with preferential orientation of microvessels in the cerebral cortex, subcortical grey matter and white matter, pons and cerebellum. J. Chem. Neuroanat. 88, 22–32. doi: 10.1016/j.jchemneu.2017.11.005
Landmesser, U., Dikalov, S., Price, S. R., McCann, L., Fukai, T., Holland, S. M., et al. (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111, 1201–1209. doi: 10.1172/JCI14172
Lee, H., and Pienaar, I. S. (2014). Disruption of the blood-brain barrier in Parkinson’s disease: curse or route to a cure? Front. Biosci. 19, 272–280. doi: 10.2741/4206
Lee, J. M., Shih, A. Y., Murphy, T. H., and Johnson, J. A. (2003). NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J. Biol. Chem. 278, 37948–37956. doi: 10.1074/jbc.M305204200
Leitao-Rocha, A., Guedes-Dias, P., Pinho, B. R., and Oliveira, J. M. (2015). Trends in mitochondrial therapeutics for neurological disease. Curr. Med. Chem. 22, 2458–2467. doi: 10.2174/0929867322666150209160317
Li, J., Li, W., Su, J., Liu, W., Altura, B. T., and Altura, B. M. (2003). Hydrogen peroxide induces apoptosis in cerebral vascular smooth muscle cells: possible relation to neurodegenerative diseases and strokes. Brain Res. Bull. 62, 101–106. doi: 10.1016/j.brainresbull.2003.08.011
Lin, C. Y., Hsu, Y. H., Lin, M. H., Yang, T. H., Chen, H. M., Chen, Y. C., et al. (2013). Neurovascular abnormalities in humans and mice with Huntington’s disease. Exp. Neurol. 250, 20–30. doi: 10.1016/j.expneurol.2013.08.019
Lin, S. L., Liao, A. Y., Yeh, S. J., and Lin, J. Y. (2015). The analysis of cardio-respiratory signals and cerebral autoregulation based on CO2 reactivity with healthy subjects and Parkinson’s patients. Technol. Health Care 24(Suppl. 1), S195–S203. doi: 10.3233/THC-151069
Liu, Y., Zhao, H., Li, H., Kalyanaraman, B., Nicolosi, A. C., and Gutterman, D. D. (2003). Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ. Res. 93, 573–580. doi: 10.1161/01.RES.0000091261.19387.AE
Lochhead, J. J., McCaffrey, G., Quigley, C. E., Finch, J., DeMarco, K. M., Nametz, N., et al. (2010). Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J. Cereb. Blood Flow Metab. 30, 1625–1636. doi: 10.1038/jcbfm.2010.29
Lourenço, C. F., Ledo, A., Barbosa, R. M., and Laranjinha, J. (2017). Neurovascular uncoupling in the triple transgenic model of Alzheimer’s disease: impaired cerebral blood flow response to neuronal-derived nitric oxide signaling. Exp. Neurol. 291, 36–43. doi: 10.1016/j.expneurol.2017.01.013
Love, S., Chalmers, K., Ince, P., Esiri, M., Attems, J., Jellinger, K., et al. (2014). Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am. J. Neurodegener. Dis. 3, 19–32.
Lu, L., Guo, L., Gauba, E., Tian, J., Wang, L., Tandon, N., et al. (2015). Transient cerebral ischemia promotes brain mitochondrial dysfunction and exacerbates cognitive impairments in young 5xFAD mice. PLoS One 10:e0144068. doi: 10.1371/journal.pone.0144068
Mamelak, M. (2017). Energy and the Alzheimer brain. Neurosci. Biobehav. Rev. 75, 297–313. doi: 10.1016/j.neubiorev.2017.02.001
Manczak, M., Mao, P., Calkins, M. J., Cornea, A., Reddy, A. P., Murphy, M. P., et al. (2010). Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 20(Suppl. 2), S609–S631. doi: 10.3233/JAD-2010-100564
Matsumoto, T., Kobayashi, T., Wachi, H., Seyama, Y., and Kamata, K. (2007). Vascular NAD(P)H oxidase mediates endothelial dysfunction in basilar arteries from Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Atherosclerosis 192, 15–24. doi: 10.1016/j.atherosclerosis.2006.06.005
Matsuno, K., Yamada, H., Iwata, K., Jin, D., Katsuyama, M., Matsuki, M., et al. (2005). Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112, 2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709
Mayhan, W. G., Arrick, D. M., Sharpe, G. M., and Sun, H. (2008). Age-related alterations in reactivity of cerebral arterioles: role of oxidative stress. Microcirculation 15, 225–236. doi: 10.1080/10739680701641421
Mergenthaler, P., Lindauer, U., Dienel, G. A., and Meisel, A. (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. doi: 10.1016/j.tins.2013.07.001
Mertens, S., Noll, T., Spahr, R., Krutzfeldt, A., and Piper, H. M. (1990). Energetic response of coronary endothelial cells to hypoxia. Am. J. Physiol. 258(3 Pt 2), H689–H694. doi: 10.1152/ajpheart.1990.258.3.H689
Mikhed, Y., Daiber, A., and Steven, S. (2015). Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int. J. Mol. Sci. 16, 15918–15953. doi: 10.3390/ijms160715918
Miller, A. A., Budzyn, K., and Sobey, C. G. (2010a). Vascular dysfunction in cerebrovascular disease: mechanisms and therapeutic intervention. Clin. Sci. 119, 1–17. doi: 10.1042/CS20090649
Miller, A. A., De Silva, T. M., Judkins, C. P., Diep, H., Drummond, G. R., and Sobey, C. G. (2010b). Augmented superoxide production by Nox2-containing NADPH oxidase causes cerebral artery dysfunction during hypercholesterolemia. Stroke 41, 784–789. doi: 10.1161/STROKEAHA.109.575365
Miller, A. A., Drummond, G. R., De Silva, T. M., Mast, A. E., Hickey, H., Williams, J. P., et al. (2009). NADPH oxidase activity is higher in cerebral versus systemic arteries of four animal species: role of Nox2. Am. J. Physiol. Heart Circ. Physiol. 296, H220–H225. doi: 10.1152/ajpheart.00987.2008
Miller, A. A., Drummond, G. R., Schmidt, H. H., and Sobey, C. G. (2005). NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ. Res. 97, 1055–1062. doi: 10.1161/01.RES.0000189301.10217.87
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
Moon, G. J., Kim, S. J., Cho, Y. H., Ryoo, S., and Bang, O. Y. (2014). Antioxidant effects of statins in patients with atherosclerotic cerebrovascular disease. J. Clin. Neurol. 10, 140–147. doi: 10.3988/jcn.2014.10.2.140
Mooradian, A. D. (1988). Effect of aging on the blood-brain barrier. Neurobiol. Aging 9, 31–39. doi: 10.1016/S0197-4580(88)80013-7
Morato, M., Reina-Couto, M., Pinho, D., Albino-Teixeira, A., and Sousa, T. (2017). “Regulation of the renin-angiotensin-aldosterone system by reactive oxygen species,” in Renin-Angiotensin System - Past, Present and Future, ed. A. Tolekova (Rijeka: InTech).
Moreira, P. I., Duarte, A. I., Santos, M. S., Rego, A. C., and Oliveira, C. R. (2009). An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J. Alzheimers Dis. 16, 741–761. doi: 10.3233/JAD-2009-0972
Munson, J. M., Fried, L., Rowson, S. A., Bonner, M. Y., Karumbaiah, L., Diaz, B., et al. (2012). Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci. Transl. Med. 4:127ra136. doi: 10.1126/scitranslmed.3003016
Nelson, A. R., Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2016). Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 1862, 887–900. doi: 10.1016/j.bbadis.2015.12.016
Nunomura, A., Hofer, T., Moreira, P. I., Castellani, R. J., Smith, M. A., and Perry, G. (2009). RNA oxidation in Alzheimer disease and related neurodegenerative disorders. Acta Neuropathol. 118, 151–166. doi: 10.1007/s00401-009-0508-1
Ogunshola, O. O., and Antoniou, X. (2009). Contribution of hypoxia to Alzheimer’s disease: is HIF-1alpha a mediator of neurodegeneration? Cell. Mol. Life Sci. 66, 3555–3563. doi: 10.1007/s00018-009-0141-0
Ohta, S., Meyer, E., Thompson, C. J., and Gjedde, A. (1992). Oxygen consumption of the living human brain measured after a single inhalation of positron emitting oxygen. J. Cereb. Blood Flow Metab. 12, 179–192. doi: 10.1038/jcbfm.1992.28
Oldendorf, W. H., Cornford, M. E., and Brown, W. J. (1976). The large apparent metabolic work capacity of the blood-brain barrier. Trans. Am. Neurol. Assoc. 101, 157–160.
Osellame, L. D., Blacker, T. S., and Duchen, M. R. (2012). Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 26, 711–723. doi: 10.1016/j.beem.2012.05.003
Palmer, J. C., Tayler, H. M., and Love, S. (2013). Endothelin-converting enzyme-1 activity, endothelin-1 production, and free radical-dependent vasoconstriction in Alzheimer’s disease. J. Alzheimers Dis. 36, 577–587. doi: 10.3233/JAD-130383
Park, L., Anrather, J., Girouard, H., Zhou, P., and Iadecola, C. (2007). Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J. Cereb. Blood Flow Metab. 27, 1908–1918. doi: 10.1038/sj.jcbfm.9600491
Park, L., Anrather, J., Zhou, P., Frys, K., Pitstick, R., Younkin, S., et al. (2005). NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. 25, 1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005
Park, L., Anrather, J., Zhou, P., Frys, K., Wang, G., and Iadecola, C. (2004). Exogenous NADPH increases cerebral blood flow through NADPH oxidase-dependent and -independent mechanisms. Arterioscler. Thromb. Vasc. Biol. 24, 1860–1865. doi: 10.1161/01.ATV.0000142446.75898.44
Park, L., Wang, G., Moore, J., Girouard, H., Zhou, P., Anrather, J., et al. (2014). The key role of transient receptor potential melastatin-2 channels in amyloid-beta-induced neurovascular dysfunction. Nat. Commun. 5:5318. doi: 10.1038/ncomms6318
Park, L., Wang, G., Zhou, P., Zhou, J., Pitstick, R., Previti, M. L., et al. (2011). Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc. Natl. Acad. Sci. U.S.A. 108, 5063–5068. doi: 10.1073/pnas.1015413108
Park, L., Zhou, P., Pitstick, R., Capone, C., Anrather, J., Norris, E. H., et al. (2008). Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 105, 1347–1352. doi: 10.1073/pnas.0711568105
Pendyala, S., Usatyuk, P. V., Gorshkova, I. A., Garcia, J. G., and Natarajan, V. (2009). Regulation of NADPH oxidase in vascular endothelium: the role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid. Redox Signal. 11, 841–860. doi: 10.1089/ARS.2008.2231
Perry, B. N., Govindarajan, B., Bhandarkar, S. S., Knaus, U. G., Valo, M., Sturk, C., et al. (2006). Pharmacologic blockade of angiopoietin-2 is efficacious against model hemangiomas in mice. J. Invest. Dermatol. 126, 2316–2322. doi: 10.1038/sj.jid.5700413
Pimentel-Coelho, P. M., and Rivest, S. (2012). The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer’s disease. Eur. J. Neurosci. 35, 1917–1937. doi: 10.1111/j.1460-9568.2012.08126.x
Pung, Y. F., Rocic, P., Murphy, M. P., Smith, R. A., Hafemeister, J., Ohanyan, V., et al. (2012). Resolution of mitochondrial oxidative stress rescues coronary collateral growth in Zucker obese fatty rats. Arterioscler. Thromb. Vasc. Biol. 32, 325–334. doi: 10.1161/ATVBAHA.111.241802
Quintero, M., Colombo, S. L., Godfrey, A., and Moncada, S. (2006). Mitochondria as signaling organelles in the vascular endothelium. Proc. Natl. Acad. Sci. U.S.A. 103, 5379–5384. doi: 10.1073/pnas.0601026103
Ray, R., Murdoch, C. E., Wang, M., Santos, C. X., Zhang, M., Alom-Ruiz, S., et al. (2011). Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 31, 1368–1376. doi: 10.1161/ATVBAHA.110.219238
Richter, C., and Kass, G. E. (1991). Oxidative stress in mitochondria: its relationship to cellular Ca2+ homeostasis, cell death, proliferation, and differentiation. Chem. Biol. Interact. 77, 1–23. doi: 10.1016/0009-2797(91)90002-O
Ricquier, D., and Bouillaud, F. (2000). Mitochondrial uncoupling proteins: from mitochondria to the regulation of energy balance. J. Physiol. 529(Pt 1), 3–10. doi: 10.1111/j.1469-7793.2000.00003.x
Rodriguez-Cuenca, S., Cocheme, H.M., Logan, A., Abakumova, I., Prime, T.A., Rose, C., et al. (2010). Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic. Biol. Med. 48, 161–172. doi: 10.1016/j.freeradbiomed.2009.10.039
Rojo, A. I., Innamorato, N. G., Martin-Moreno, A. M., De Ceballos, M. L., Yamamoto, M., and Cuadrado, A. (2010). Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 58, 588–598. doi: 10.1002/glia.20947
Ronnett, G. V., Ramamurthy, S., Kleman, A. M., Landree, L. E., and Aja, S. (2009). AMPK in the brain: its roles in energy balance and neuroprotection. J. Neurochem. 109(Suppl. 1), 17–23. doi: 10.1111/j.1471-4159.2009.05916.x
Rutkai, I., Dutta, S., Katakam, P. V., and Busija, D. W. (2015). Dynamics of enhanced mitochondrial respiration in female compared with male rat cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 309, H1490–H1500. doi: 10.1152/ajpheart.00231.2015
Sahoo, S., Meijles, D. N., and Pagano, P. J. (2016). NADPH oxidases: key modulators in aging and age-related cardiovascular diseases? Clin. Sci. 130, 317–335. doi: 10.1042/CS20150087
Schreibelt, G., Kooij, G., Reijerkerk, A., van Doorn, R., Gringhuis, S. I., van der Pol, S., et al. (2007). Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 21, 3666–3676. doi: 10.1096/fj.07-8329com
Shah, G. N., Morofuji, Y., Banks, W. A., and Price, T. O. (2013). High glucose-induced mitochondrial respiration and reactive oxygen species in mouse cerebral pericytes is reversed by pharmacological inhibition of mitochondrial carbonic anhydrases: implications for cerebral microvascular disease in diabetes. Biochem. Biophys. Res. Commun. 440, 354–358. doi: 10.1016/j.bbrc.2013.09.086
Shah, Z. A., Li, R. C., Thimmulappa, R. K., Kensler, T. W., Yamamoto, M., Biswal, S., et al. (2007). Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience 147, 53–59. doi: 10.1016/j.neuroscience.2007.02.066
Sheu, S. S., Nauduri, D., and Anders, M. W. (2006). Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim. Biophys. Acta 1762, 256–265. doi: 10.1016/j.bbadis.2005.10.007
Skoog, I., and Gustafson, D. (2006). Update on hypertension and Alzheimer’s disease. Neurol. Res. 28, 605–611. doi: 10.1179/016164106X130506
Smith, R. A., Adlam, V. J., Blaikie, F. H., Manas, A. R., Porteous, C. M., James, A. M., et al. (2008). Mitochondria-targeted antioxidants in the treatment of disease. Ann. N. Y. Acad. Sci. 1147, 105–111. doi: 10.1196/annals.1427.003
Smith, R. A., Hartley, R. C., and Murphy, M. P. (2011). Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Signal. 15, 3021–3038. doi: 10.1089/ars.2011.3969
Smith, R. A., and Murphy, M. P. (2011). Mitochondria-targeted antioxidants as therapies. Discov. Med. 11, 106–114.
Spahr, R., Krutzfeldt, A., Mertens, S., Siegmund, B., and Piper, H. M. (1989). Fatty acids are not an important fuel for coronary microvascular endothelial cells. Mol. Cell. Biochem. 88, 59–64. doi: 10.1007/BF00223424
Staiculescu, M. C., Foote, C., Meininger, G. A., and Martinez-Lemus, L. A. (2014). The role of reactive oxygen species in microvascular remodeling. Int. J. Mol. Sci. 15, 23792–23835. doi: 10.3390/ijms151223792
St-Amour, I., Aube, B., Rieux, M., and Cicchetti, F. (2015). Targeting cerebrovascular impairments in Huntington’s disease: a novel treatment perspective. Neurodegener. Dis. Manage. 5, 389–393. doi: 10.2217/nmt.15.41
Steinman, J., Koletar, M. M., Stefanovic, B., and Sled, J. G. (2017). 3D morphological analysis of the mouse cerebral vasculature: comparison of in vivo and ex vivo methods. PLoS One 12:e0186676. doi: 10.1371/journal.pone.0186676
Sun, Y., Lee, R., Chen, Y., Collinson, S., Thakor, N., Bezerianos, A., et al. (2015). Progressive gender differences of structural brain networks in healthy adults: a longitudinal, diffusion tensor imaging study. PLoS One 10:e0118857. doi: 10.1371/journal.pone.0118857
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2015). Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer’s disease. J. Cereb. Blood Flow Metab. 35, 1055–1068. doi: 10.1038/jcbfm.2015.76
Takac, I., Schroder, K., and Brandes, R. P. (2012). The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr. Hypertens. Rep. 14, 70–78. doi: 10.1007/s11906-011-0238-3
Tang, X., Luo, Y. X., Chen, H. Z., and Liu, D. P. (2014). Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 5:175. doi: 10.3389/fphys.2014.00175
Tarantini, S., Fulop, G. A., Kiss, T., Farkas, E., Zolei-Szenasi, D., Galvan, V., et al. (2017a). Demonstration of impaired neurovascular coupling responses in TG2576 mouse model of Alzheimer’s disease using functional laser speckle contrast imaging. Geroscience doi: 10.1007/s11357-017-9980-z [Epub ahead of print].
Tarantini, S., Tran, C. H. T., Gordon, G. R., Ungvari, Z., and Csiszar, A. (2017b). Impaired neurovascular coupling in aging and Alzheimer’s disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. 94, 52–58. doi: 10.1016/j.exger.2016.11.004
Tayarani, I., Chaudiere, J., Lefauconnier, J. M., and Bourre, J. M. (1987). Enzymatic protection against peroxidative damage in isolated brain capillaries. J. Neurochem. 48, 1399–1402. doi: 10.1111/j.1471-4159.1987.tb05677.x
Togliatto, G., Lombardo, G., and Brizzi, M. F. (2017). The future challenge of reactive oxygen species (ROS) in hypertension: from bench to bed side. Int. J. Mol. Sci. 18:E1988. doi: 10.3390/ijms18091988
Tong, X. K., Nicolakakis, N., Kocharyan, A., and Hamel, E. (2005). Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J. Neurosci. 25, 11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005
Toth, P., Tarantini, S., Tucsek, Z., Ashpole, N. M., Sosnowska, D., Gautam, T., et al. (2014). Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 306, H299–H308. doi: 10.1152/ajpheart.00744.2013
Ungvari, Z., Labinskyy, N., Gupte, S., Chander, P. N., Edwards, J. G., and Csiszar, A. (2008). Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am. J. Physiol. Heart Circ. Physiol. 294, H2121–H2128. doi: 10.1152/ajpheart.00012.2008
Ungvari, Z., Orosz, Z., Labinskyy, N., Rivera, A., Xiangmin, Z., Smith, K., et al. (2007). Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 293, H37–H47. doi: 10.1152/ajpheart.01346.2006
Vaas, M., Deistung, A., Reichenbach, J. R., Keller, A., Kipar, A., and Klohs, J. (2017). Vascular and tissue changes of magnetic susceptibility in the mouse brain after transient cerebral ischemia. Transl. Stroke Res. doi: 10.1007/s12975-017-0591-x [Epub ahead of print].
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84. doi: 10.1016/j.biocel.2006.07.001
Vargas, M. R., and Johnson, J. A. (2009). The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 11:e17. doi: 10.1017/S1462399409001094
Vergeade, A., Mulder, P., Vendeville-Dehaudt, C., Estour, F., Fortin, D., Ventura-Clapier, R., et al. (2010). Mitochondrial impairment contributes to cocaine-induced cardiac dysfunction: prevention by the targeted antioxidant MitoQ. Free Radic. Biol. Med. 49, 748–756. doi: 10.1016/j.freeradbiomed.2010.05.024
Wang, M., Zhang, J., Walker, S. J., Dworakowski, R., Lakatta, E. G., and Shah, A. M. (2010). Involvement of NADPH oxidase in age-associated cardiac remodeling. J. Mol. Cell. Cardiol. 48, 765–772. doi: 10.1016/j.yjmcc.2010.01.006
Wang, Y., Zang, Q. S., Liu, Z., Wu, Q., Maass, D., Dulan, G., et al. (2011). Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. Am. J. Physiol. Cell Physiol. 301, C695–C704. doi: 10.1152/ajpcell.00322.2010
Wenzel, P., Schuhmacher, S., Kienhofer, J., Muller, J., Hortmann, M., Oelze, M., et al. (2008). Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc. Res. 80, 280–289. doi: 10.1093/cvr/cvn182
Wevers, N. R., and de Vries, H. E. (2016). Morphogens and blood-brain barrier function in health and disease. Tissue Barriers 4:e1090524. doi: 10.1080/21688370.2015.1090524
Yan, W., Wang, H. D., Hu, Z. G., Wang, Q. F., and Yin, H. X. (2008). Activation of Nrf2-ARE pathway in brain after traumatic brain injury. Neurosci. Lett. 431, 150–154. doi: 10.1016/j.neulet.2007.11.060
Zhang, R., Ran, H. H., Cai, L. L., Zhu, L., Sun, J. F., Peng, L., et al. (2014). Simulated microgravity-induced mitochondrial dysfunction in rat cerebral arteries. FASEB J. 28, 2715–2724. doi: 10.1096/fj.13-245654
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. doi: 10.1016/j.neuron.2008.01.003
Keywords: neurovascular unit, endothelial cells, oxidative stress, mitochondria, NADPH oxidases, Alzheimer’s disease
Citation: Carvalho C and Moreira PI (2018) Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 9:806. doi: 10.3389/fphys.2018.00806
Received: 19 February 2018; Accepted: 08 June 2018;
Published: 03 July 2018.
Edited by:
Cristina M. Sena, University of Coimbra, PortugalReviewed by:
Jingyan Han, Boston University, United StatesCopyright © 2018 Carvalho and Moreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Carvalho, Y3Jpc3RpbmEuaW0uY2FydmFsaG9AZ21haWwuY29t Paula I. Moreira, dmVudGFAY2kudWMucHQvcGltb3JlaXJhQGZtZWQudWMucHQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.