 Seyeon Park
Seyeon Park Seunghyun Ahn1
Seunghyun Ahn1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 19 June 2018
Sec. Redox Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00762
This article is part of the Research Topic Vitamin C in Cancer and Infectious Diseases: Physiological, Biochemical and Therapeutic Interventions View all 8 articles
There is an ongoing interest in cellular antioxidants and oxidants as well as cellular mechanisms underlying their effects. Several reports suggest that vitamin C (L-ascorbic acid) functions as a pro-oxidant with selective toxicity against specific types of tumor cells. In addition, reduced glutathione plays an emerging role in reducing oxidative stress due to xenobiotic toxins such as metals and oxidants associated with diseases such as cancer, cardiovascular disease, and stroke. High-dose intravenous vitamin C and intravenous glutathione have been used as complementary, alternative, and adjuvant medicines. Here, we review the molecular mechanisms underlying the regulation of oxidation/reduction systems, focusing on the altered metabolomics profile in cancer cells following treatment with pharmacological vitamin C. This review focuses on the role of vitamin C in energy metabolism in terms of adenosine triphosphate, cysteine, and reduced glutathione levels, affecting cancer cell death.
From a systems biology perspective, the integrated use of proteomics, genomics, and transcriptomics is extremely important for translational metabolomics-based research (Shin et al., 2016). Microarray analysis and qPCR have been performed to investigate the effect of vitamin C on gene expression. A recent study has reported that a series of genes in embryonic stem cells are differentially regulated by vitamin C treatment (Shin et al., 2004). Most of these upregulated genes belong to gene families that regulate neurogenesis, neuronal maturation, and neurotransmission (Shin et al., 2004; Belin et al., 2010). Based on the observation that vitamin C treatment suppresses the expression of PMP22, a myelin gene that is overexpressed in one of the hereditary motor and sensory neuropathies, it has been suggested that vitamin C induces dose-dependent suppression of PMP22 expression by inhibiting the production of cAMP, a regulator of CREB-binding promoter located in PMP22 (Hai et al., 2001; Kaya et al., 2007; Belin et al., 2010). Vitamin C acts as a competitive inhibitor of adenylate cyclase, and represses the expression of a variety of genes under the control of cAMP-dependent pathway (Belin et al., 2010). Microarray data suggested that 5 days of vitamin C supplementation under normal physiological condition, but not under cancer condition, induce an upregulation of calnexin isoform (Canali et al., 2014). On the other hand, microarray analysis using human colon carcinoma HT29 cells has shown that vitamin C downregulated the expression of translational initiation factor subunits, tRNA synthetases, and genes crucial for cell cycle progression accompanied by S-phase arrest of proliferative cells induced by vitamin C (Belin et al., 2009). In addition, microarray analysis using mouse models grafted with HT29 cells has consistently shown a decreased expression of translational initiation factor and tRNA synthetases in tumors following vitamin C treatment (Belin et al., 2009).
Proteomics research also elucidates the protein expression in terms of post-translational modifications triggered by a specific stimulus independent of protein neo-synthesis. Post-translational modifications such as phosphorylation of tyrosine or serine/threonine, sulfur oxidation of cysteine, and glutathionylation represent key mechanisms of cell stimulation related to oxidative stress. Our group conducted proteomics analyses of the effect of vitamin C on cancer at the cellular level and in mouse models grafted with tumor cells (Park et al., 2006, 2009). When human leukemia cell line NB4 was treated with relatively high concentration (0.5 mM) of vitamin C, approximately 200 differentially expressed spots were detected by two-dimensional electrophoresis. This proteomics analysis suggested that the domain polymerization state of quaternary structure protein composed of four domains via disulfide bond was altered in response to vitamin C treatment. One of these proteins included protein disulfide isomerase (PDI) belonging to the thiol/disulfide exchange catalyst superfamily. It acts as a protein-thiol-oxidoreductase enzyme. It also shares sequence homology with thioredoxin (Park, 2013). Another protein was immunoglobulin heavy chain binding protein (BiP), a multi-domain chaperone identical to chaperone Hsp70. BiP binds via a disulfide bond to the α-subunit of prolyl 4-hydroxylase (P4-H), a partner of PDI (John and Bulleid, 1996). P4-H is a multimeric protein composed of α-subunit and β-subunit. Its α-subunit is catalytically more important than its β-subunit. In addition, its β-subunit is identical to the multifunctional PDI enzyme (John and Bulleid, 1996). These results suggest that vitamin C oxidizes intracellular levels of reduced glutathione and the valence change of glutathione and reduced glutathione results in disulfide bond rearrangement in the quaternary structure of proteins such as PDI and BiP. Our previous study also demonstrated that changes in intracellular valence of glutathione between reduced glutathione occur shortly after exposure to vitamin C (Park et al., 2004).
Regional changes in oxidation state induced by vitamin C lead to a variety of alterations involving sulfur oxidation in the cellular milieu and result in transitions in the protein quaternary structure. The oxidation state of cysteine sulfur is important for the determination of the tertiary structure of proteins (Park, 2013). An important example of protein influenced by regional changes in oxidative state associated with vitamin C is glyceraldehyde 3-phosphate dehydrogenase (GAPDH) involved in glycolysis metabolism. It has been reported that GAPDH activity is reduced by reactive oxygen species (ROS) or vitamin C treatment (Hwang et al., 2009; Yun et al., 2015). High concentration of vitamin C generating ROS suppresses GAPDH via Cys glutathionylation (Hwang et al., 2009; Yun et al., 2015). The role of GAPDH in vitamin C-dependent alterations suggests that vitamin C influences glucose metabolism via altered oxidation/reduction status. It also suggests an interface between proteomics analysis and metabolomics approach to determine the effect of vitamin C.
Metabolomics is appropriate for the study of biological processes induced by endogenous developmental changes or drugs and other xenobiotics via endogenous metabolome (Oskouie and Taheri, 2015). Approximately 38000 chemical compounds in metabolites are generally detected in the human body according to a recent report (Oskouie and Taheri, 2015). Metabolome is typically composed of carbohydrates, amino acids, lipids, nucleotides, and other organic compounds. Metabolites exhibit varying levels of volatility and polarity, and therefore, a variety of analytical technologies are employed in metabolomics studies (Oskouie and Taheri, 2015). The most common methodologies used for the identification of metabolites include nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (Meister and Anderson, 1983).
Metabolic profiling indicates altered metabolomics in the cancer cells during cancer progression. Therefore, metabolite-based investigations of various cancers represent a useful approach to identify diagnostic, therapeutic, and prognostic biomarkers in cancer. It has been suggested that glucose metabolism in tumor cells varies from that of normal cells. Glucose metabolism is known to be associated with sustainable proliferation. Otto Warburg suggested that tumor cells metabolize approximately ten-fold more glucose to lactate under aerobic conditions than normal tissues in a given time (Koppenol et al., 2011). Moreover, higher conversion of glucose to lactic acid under aerobic condition in cancer cells is accompanied by retaining mitochondrial respiration (Koppenol et al., 2011).
Specific metabolic proteins have been identified as potential oncoproteins (Table 1). For example, pyruvate kinase type M2 is an oncoprotein expressed in squamous cell carcinoma (Wong et al., 2008). Specific oncoproteins alter cancer cell metabolism by directly regulating key metabolic enzymes and pathways (Nagarajan et al., 2016). For example, oncogenic transcription factor MYC activates the transcription of glycolytic enzyme genes and glucose transporters that enhance aerobic glycolysis (Shim et al., 1997; Osthus et al., 2000; Ahuja et al., 2010). In addition, oncogenic kinase Akt activates hexokinase 2, phosphofructokinase 1 (PFK1), and phosphofructokinase 2 (PFK2). It also induces localization of glucose transporters to the cell surface, resulting in enhanced glycolysis (Deprez et al., 1997; Robey and Hay, 2009). It is well known that mitochondrial metabolism is regulated by oncoprotein Bcl-2 (Krishna et al., 2011). Ha-Ras and β-catenin oncoproteins reprogram metabolic flows in mouse liver tumors (Unterberger et al., 2014). Hepatitis B X-interacting protein, an oncoprotein, also enhanced glucose metabolism by suppressing the synthesis of cytochrome c oxidase 2 and pyruvate dehydrogenase alpha 1 in breast cancer (Liu et al., 2015).
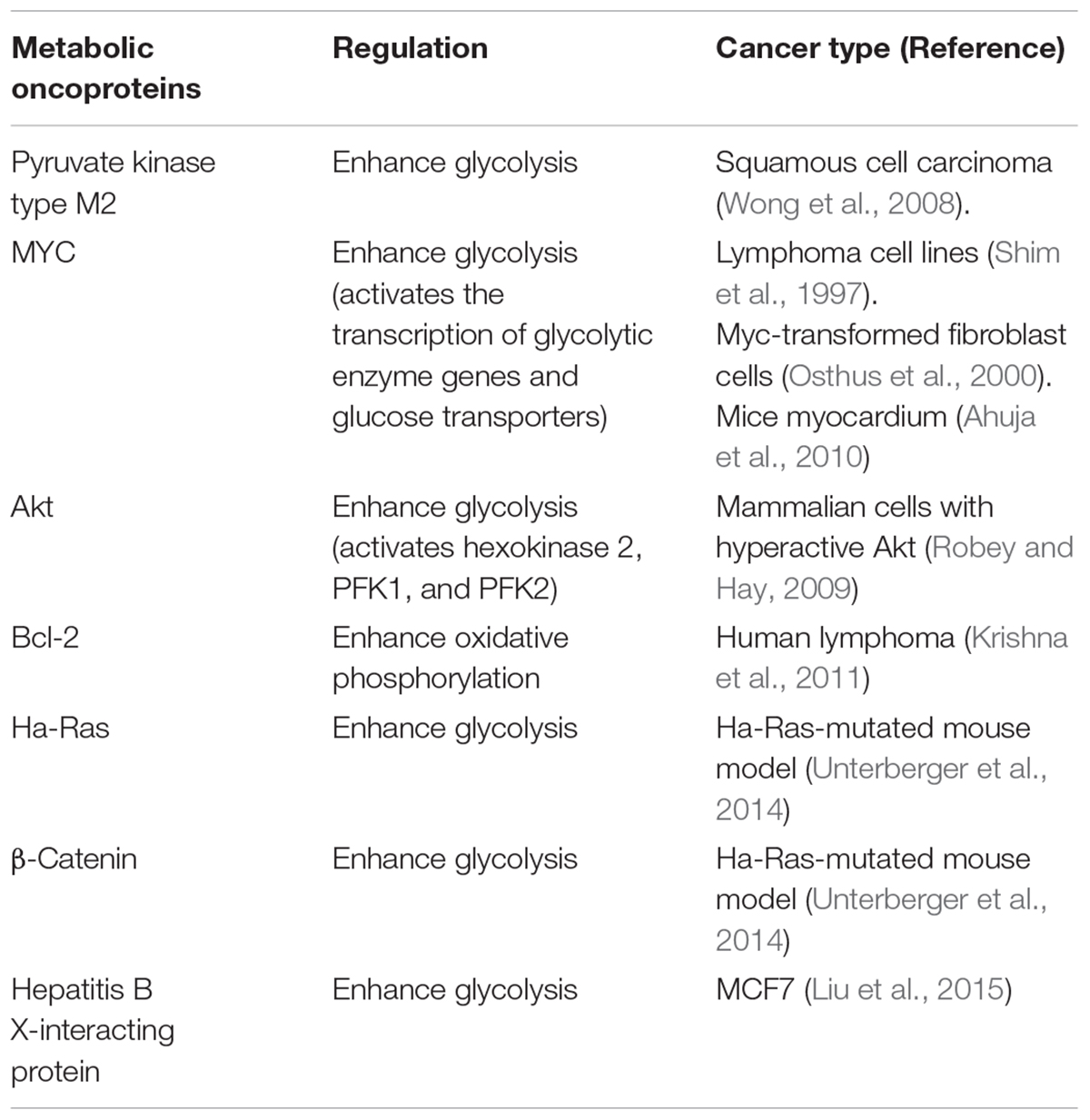
TABLE 1. Metabolic proteins identified as oncoproteins.
Parkin (PARK2), a Parkinson disease-associated gene, is a glucose metabolism-related tumor suppressor whose expression is diminished in tumors (Zhang et al., 2011). Parkin deficiency activates glycolysis and reduces mitochondrial respiration, leading to the Warburg effect (Zhang et al., 2011). In Parkinson deficient cells, mitochondrial dysfunction and enhanced oxidative stress are observed (Zhang et al., 2011).
Other evidence implicating oncogenes in aerobic glycolysis include phosphorylation of a variety of glycolytic enzymes by oncogenic Src kinase and enhanced glucose uptake by oncogenic Ras activation in fibroblasts (Cooper et al., 1984; Flier et al., 1987). Ras oncogene links metabolomic alterations by vitamin C with tumor suppression by vitamin C. A recent report found that high-dose vitamin C is selectively toxic to human colorectal cancer cells carrying either K-Ras or B-Raf mutations (Yun et al., 2015). Mutant K-Ras or B-Raf activate downstream mitogen-activated protein kinase (MAPK) pathway, leading to the up-regulated expression of GLUT1, a glucose transporter that imports dehydroascorbate (DHA, an oxidized form of vitamin C) into cells (Yun et al., 2009, 2015). Imported DHA is then reduced back to vitamin C by oxidizing glutathione, resulting in depletion of glutathione, and high levels of intracellular ROS (Vera et al., 1993, 1995; Yun et al., 2015). It has been suggested that such oxidative stress in highly glycolytic K-Ras- or B-Raf-mutant cells triggers inactivation of GAPDH via Cys oxidation, leading to abnormal glycolysis that is rarely seen in K-Ras or B-Raf wild-type cells (Yun et al., 2015).
In addition to glucose metabolism, vitamin C induces specific changes in other cellular metabolic pathways in cancer cells. Oxidative stress is an important mechanism of vitamin C in cancer cells. Glutathione-related metabolism also affects cancer progression by vitamin C because glutathione is a major cellular antioxidant.
An underlying hypothesis is that ROS production is an inevitable consequence of electron transport combined with oxidative phosphorylation under physiological conditions. High levels of ROS induce cellular senescence or death. However, oxidation-evading mechanisms of tumor cells differ from that of normal cells (Andrisic et al., 2018). As discussed above, a distinct feature of many cancer cells is their metabolic dependence on anaerobic glycolysis in spite of functional glucose metabolism at the expense of oxygen (Koppenol et al., 2011). Although energetically less efficient, glycolysis produces ATP at a much faster rate by avoiding mitochondrial oxidative phosphorylation (Andrisic et al., 2018). Therefore, cancer cells are protected from deleterious ROS generation that normally should be expected during enhanced proliferation (Andrisic et al., 2018). In addition, enhanced glycolysis is likely to act as a pentose phosphate pathway shunt to provide NADPH and substrates for nucleotide synthesis. NADPH also acts as a reducing agent for oxidized GSH and provides intracellular redox balance (Andrisic et al., 2018). Nonetheless, production of ROS is stimulated in cancer cells compared with that of normal cells (Trachootham et al., 2009). Therefore, cancer cells generally up-regulate multiple antioxidant systems including GSH and thioredoxin, buffering ROS levels to allow tumor cell progression (Harris et al., 2015). Although thioredoxin is not so abundant as GSH in cells, it reduces ROS and is regenerated in a GSH-independent manner by thioredoxin reductase (Holmgren and Lu, 2010). Because GSH and thioredoxin pathways synergistically contribute to cancer cell survival, it has been suggested that blocking both GSH and thioredoxin pathways inhibits cancer promotion (Harris et al., 2015). Cancer cells show metabolic alterations to manage oxidative stress, and therefore, a recent study has suggested that glutathione synthetic pathway is a promising therapeutic target (Beatty, 2015). Mass spectrometry was used to conduct metabolomics profiling of triple-negative breast cancer (TNBC) compared with control cells (Beatty, 2015). TNBC does not represent oncogenic HER2 amplification. It does not express estrogen receptor or progesterone receptor. TNBC is an aggressive and genetically heterogeneous subset of breast cancer, which is refractory to usual targeted therapies (Beatty, 2015). A distinct feature of TNBC metabolic profiling is that levels of glutathione, a cellular redox buffer, are lower in TNBC cell lines compared with the controls (Beatty, 2015). Glutathione biosynthesis is required to suppress ROS in TNBC cells. Thus, inhibition of glutathione biosynthesis leads to reduced tumor cell growth both in vitro and in vivo (Beatty, 2015), illustrating the role of GSH metabolic alterations in cancer. Metabolomics contributes to a better understanding of cancer therapeutically. Likewise, malignant mesothelioma is a fatal cancer with no effective cure. Recently, disabling mitochondrial peroxide metabolism or reducing Akt signaling suppressed mesothelioma malignancy (Tomasetti et al., 2014; Cunniff et al., 2015). This finding may be linked to the examination showing that ROS induced by high dose of ascorbate in mesothelioma inhibited cell death (Takemura et al., 2010).
Vitamin C (L-ascorbic acid) is a well-known reducing agent that is easily oxidized to dehydroascorbate (DHA) in solution. Physiologically, vitamin C is transported into cells as ascorbate in specific cell types by sodium-dependent ascorbic acid transporters. It can also be administered into cells in oxidized DHA form facilitated by glucose transporters (GLUTs) (Nishikimi and Yagi, 1991; Vera et al., 1993; Tsukaguchi et al., 1999; Rumsey et al., 2000; Liang et al., 2001). Following the transportation of DHA into cells via glucose transporters, it is reduced to ascorbate using GSH, and is trapped inside the cells where it accumulates as ascorbic acid (Vera et al., 1993, 1995). Therefore, vitamin C is considered as a pro-oxidant that produces oxidative stress (Halliwell and Foyer, 1976; Heikkila and Cabbat, 1983). Accordingly, vitamin C enhances arsenic trioxide (As2O3)-induced cytotoxicity in multiple myeloma cells by decreasing intracellular GSH levels (Grad et al., 2001). A clinical study has reported such results in patients with multiple myeloma treated with a combination of vitamin C and As2O3 (Bahlis et al., 2001). In vitro, vitamin C suppresses the growth of mouse myeloma cells. In vivo, vitamin C inhibited the growth of leukemic progenitor cells isolated from a patient with acute myeloid leukemia (AML) in our previous study (Park et al., 1971, 1992; Park, 1985). In a few clinical studies, manipulation of vitamin C levels in AML patients has produced clinical benefit (Park et al., 2001, 2002). Based on such result, complementary and alternative medicine practitioners have used high concentrations of vitamin C to treat their patients (Meister and Anderson, 1983; Park et al., 2001, 2002; Park, 2013). The physiological concentration of vitamin C is <0.1 mM in plasma. Plasma vitamin C concentrations (1–10 mM, depending on cell lines) that are toxic to cancer cells in vitro can be attained clinically by i.v., and not via oral administration of a high dose of vitamin C (Park, 2013).
Recent studies have found that serum concentrations of GSH are associated with various disease conditions (Droge and Breitkreutz, 2000; Prousky, 2008; Forman et al., 2009; Smeyne and Smeyne, 2013). For example, decreased serum concentration of GSH has been linked to cancer and neurodegenerative disease susceptibility (Smeyne and Smeyne, 2013). Because GSH is so poorly absorbed in the gastrointestinal system, i.v. GSH (rather than most oral GSH supplements) represents another complementary and alternative medicine therapy (Chen et al., 2011).
We have previously reported that in vitro treatment with 0.25–2.0 mM vitamin C induces apoptosis of leukemia cells (Park et al., 2004). Vitamin C-stimulated oxidation of GSH to dimerized oxidized form (GSSG) leads to accumulation of hydrogen peroxide (H2O2), resulting in the induction of apoptosis. A number of previous reports also suggested that high-dose vitamin C kills cancer cells by acting as a pro-drug that generates H2O2 (Chen et al., 2008; Takemura et al., 2010; Du et al., 2012; Uetaki et al., 2015). The direct role of H2O2 in the induction of apoptosis in acute myeloid leukemia (AML) cells has been confirmed using catalase to completely abrogate vitamin C-induced apoptosis (Park et al., 2004).
A recent metabolomics study has suggested an important relationship between vitamin C and GSH in terms of glucose metabolism, including glycolysis, citric acid cycle (tricarboxylic acid; TCA cycle), and pentose phosphate pathway (Uetaki et al., 2015). A list of metabolites associated with metabolic perturbations related to glucose metabolism is provided in Table 2, which is in line with a previous report showing that vitamin C influenced glucose metabolism (Hwang et al., 2009; Yun et al., 2015). Levels of metabolites associated with upstream glycolysis, partial TCA cycle (such as citrate and cis-aconitate), and pentose phosphate pathway (PPP) are increased in response to high-dose vitamin C while levels of metabolites downstream of glycolysis are decreased with the exception of citrate and cis-aconitate (Uetaki et al., 2015). This finding may be attributed to GAPDH inactivation by vitamin C-induced oxidative stress. Upstream metabolites of GAPDH accumulate whereas downstream metabolites of GAPDH were depleted. ATP concentrations are decreased in response to high-dose vitamin C, indicating that high-dose vitamin C interfered with glycolytic and citric acid cycle energy flux, consequently diminishing ATP production (Uetaki et al., 2015). The authors have suggested that vitamin C-mediated oxidative stress in turn induces the depletion of NADH, which inhibits glycolytic flux. The decreased ATP level due to inhibition of energy metabolism causes cell death (Uetaki et al., 2015).
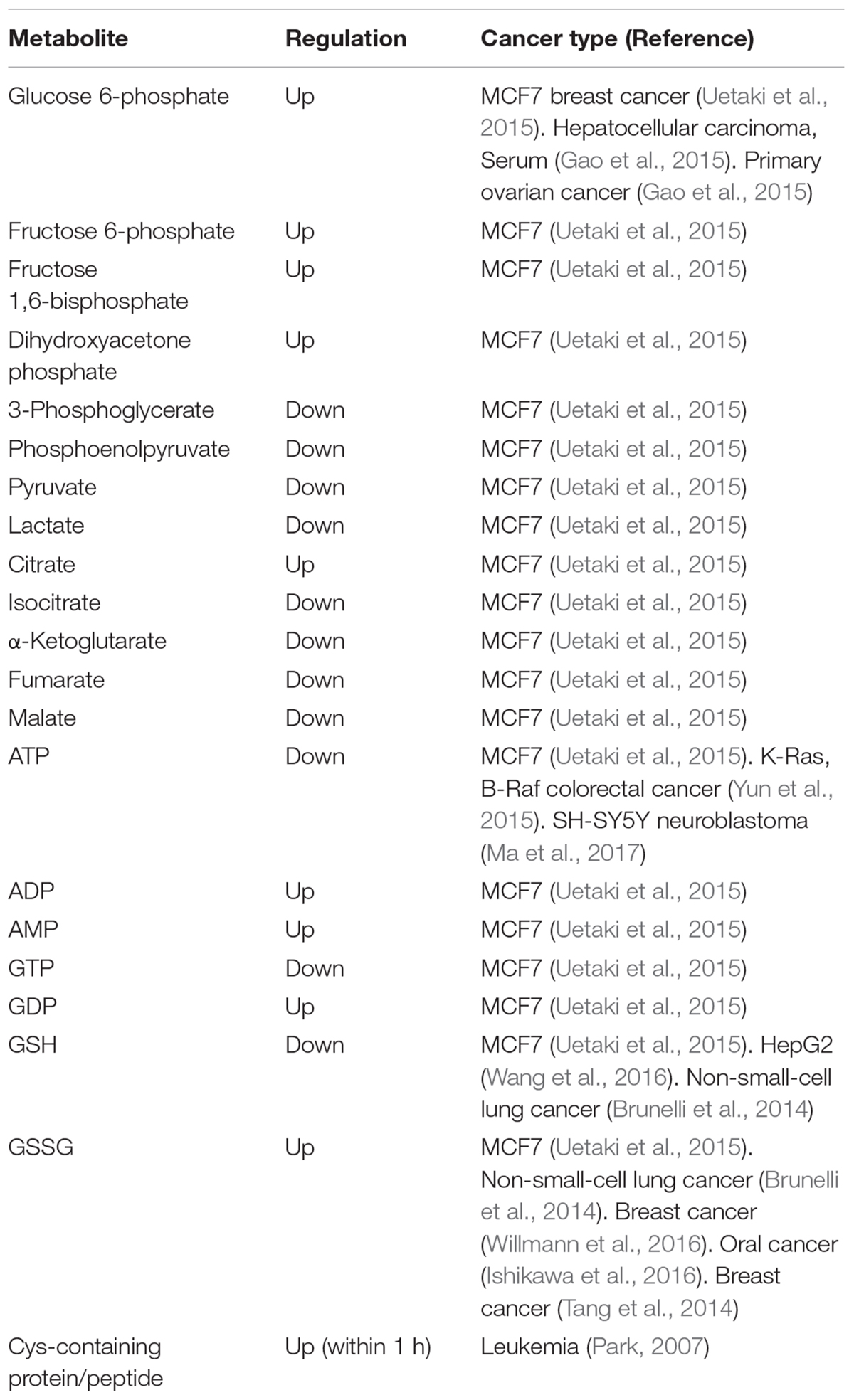
TABLE 2. Metabolites of glucose and GSH metabolism altered by vitamin C (Metabolite alteration by high concentration (>1 mM) of vitamin C in glucose and GSH metabolism).
GSH plays a significant role in cellular defense against oxidative stress by reducing free radicals and ROS. It acts in various cysteine-mediated intracellular processes, including the metabolism of cysteine amino acids and biosynthesis of leukotrienes and DNA (Larsson et al., 1983; Meister and Anderson, 1983). GSH is synthesized via sequential steps of two enzyme reactions containing γ-glutamylcysteine synthetase (γ-GCS) and GSH synthase. γ-GCS catalyzes the rate-limiting step of GSH synthesis (Meister and Anderson, 1983). GSTs are a major group of detoxification enzymes that conjugate GSH to reactive metabolites. Multiple forms of GST isozymes have been identified (Shepherd et al., 2000). To date, eight distinct classes (α, κ, μ, φ, π, θ, σ, and ζ) encoding soluble cytosolic GSTs have been identified in mammals on the basis of their degree of sequence identity (Hayes and McLellan, 1999). GST-P1 is a gene that encodes a GST belonging to the π class. GST-A1, A2, A3, and A4 genesc encode human GST subunits belonging to the α class. GST-M1, M2, M3, M4, and M5 genes encode GST subunits belonging to the μ class (Sheehan et al., 2001). Substantial evidence suggests that ROS play an important role in cellular signaling linked to transcriptional machinery or act as a second messenger (Griffith and Meister, 1979; Palmer and Paulson, 1997; Kunsch and Medford, 1999; Hensley et al., 2000; Sheehan et al., 2001; Carcamo et al., 2002a,b). Furthermore, evidence indicates that phase II detoxification enzymes such as GSH S-transferase, NAD(P)H:quinone oxidoreductase1, UDP-glucuronosyltransferase, and epoxide hydrolase can be induced by various compounds, including food phytochemicals (Wattenberg, 1981; Nakamura et al., 2000). Our previous data established the regulation of GSH levels via transcriptional regulation of glutathione synthase and GST synthesis by vitamin C (Park, 2007). The role of vitamin C-induced changes in GSH/GSSG ratio was first established in this report. We have investigated the relationship of vitamin C with GSH in leukemia cell lines. We found that vitamin C-induced decrease in intracellular GSH/GSSG ratio and H2O2 accumulation led to transcriptional induction of intracellular protein and protection against oxidative stress, such as γ-GCS in HL-60 and NB-4 cells. Although the effect of H2O2 accumulation induced by vitamin C was eliminated by catalase, vitamin C-mediated transcriptional induction of these enzymes has been observed, indicating that the altered GSH/GSSG ratio was more important than H2O2 accumulation in inducing the activity of enzymes that protect against oxidative stress (Park, 2007).
A redox cycle requires adequate support via GSH reductase and GSH peroxidase for defense against redox stress. In addition, relatively high concentrations of GSH via synthesis and active transport of GSSG or GSH S-conjugates are needed. Stimulation of γ-GCS transcription increases GSH concentration (Goto et al., 1995). Our observations suggested that vitamin C stimulated the expression of γ-GCS, resulting in an increase in the level of GSH via de novo synthesis at the expense of cysteine (Park, 2007). Concentrations of GSH in three types of myeloid leukemia cells were elevated within 3 h after treatment with vitamin C and gradually returned to their baseline levels by 12 h (Park, 2007). Such increase in the concentration of GSH was associated with enhanced expression of γ-GCS. GSH synthesis and GST activation in response to vitamin C occurred rapidly (in 1 h) (Park, 2007). The elevated expression of γ-GCS in response to vitamin C is accompanied by corresponding increase in the concentration of GSH, representing an important function of vitamin C in cellular GSH homeostasis (Park, 2007).
Cysteine is known to be a rate-limiting precursor for GSH synthesis (Watanabe and Bannai, 1987). Therefore, we investigated cysteine uptake in AML cells after treatment with vitamin C (Park, 2007). Intracellular L-Cys incorporation was measured in intact HL-60, NB4, and KG1 cells exposed to vitamin C using 35S-labeled-L-Cys containing media (Park, 2007). The rate of uptake in the absence of vitamin C was very low (at most 119% of baseline by 16 h) (Park, 2007). However, it peaked after 1 h and 3 h (Park, 2007). An inhibitor of gamma-glutamylcysteine synthetase, buthionine sulfoximine potently inhibited the second peak, suggesting glutathione synthesis following the incorporation of cysteine. These results indicate that vitamin C induced GSH synthesis in parallel with intracellular cysteine uptake. Interestingly, intracellular GSH levels in these AML cells incubated with vitamin C peaked around 3 h and declined thereafter, while the increase in [35S]-L-Cys incorporation occurred at 3 h and continued (Park, 2007). This result demonstrated that transporation of [35S]-L-Cys into cells through cysteine uptake is followed by incorporation and intracellular transfer. Thus, the sulfhydryl transfer system might be affected by vitamin C.
In view of the signaling effects of vitamin C, the association between vitamin C and glutathione in myeloid cells may partly explain the potential effect of vitamin C on cellular signal transduction. It appears that vitamin C has a positive effect on sulfhydryl (-SH) uptake. Considering that intracellular concentration of glutathione determines cellular thiol-disulfide redox potential to a large extent, it might regulate a variety of cellular processes via disulfide bridge formation and protein glutathionylation.
Recently, biological and pre-clinical studies suggest that high dose intravenous vitamin C combined with conventional chemotherapy agent synergistically increase the effectiveness of cancer therapy. (Espey et al., 2011; Hoffer et al., 2015). A phase I study states that high dose intravenous vitamin C in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer did not reveal increased toxicity (Monti et al., 2012). In view of the metabolic effect, we conclude that vitamin C plays a key role in the challenges associated with glucose and GSH metabolism (Figure 1 and Table 2). Vitamin C induces high ROS level and oxidation of GSH. Accompanied by direct glutathionylation of GAPDH in glycolysis, glucose metabolism was altered by vitamin C treatment. Furthermore, changes in reduced glutathione ratio triggered by vitamin C resulted in altered GSH metabolism via de novo synthesis. From the information available, it seems clear that vitamin C is involved in a variety of oxidative mechanisms. Therefore, vitamin C may be an adjuvant medicine combined with conventional chemotherapy drug to induce cancer cell death. In the future, another issue pertaining to vitamin C is whether its use as an adjuvant medicine is valid in all populations or only in some populations depending on the range of intakes. Therefore, further studies are required to identify the molecular targets of vitamin C sensitivity such as transporter.
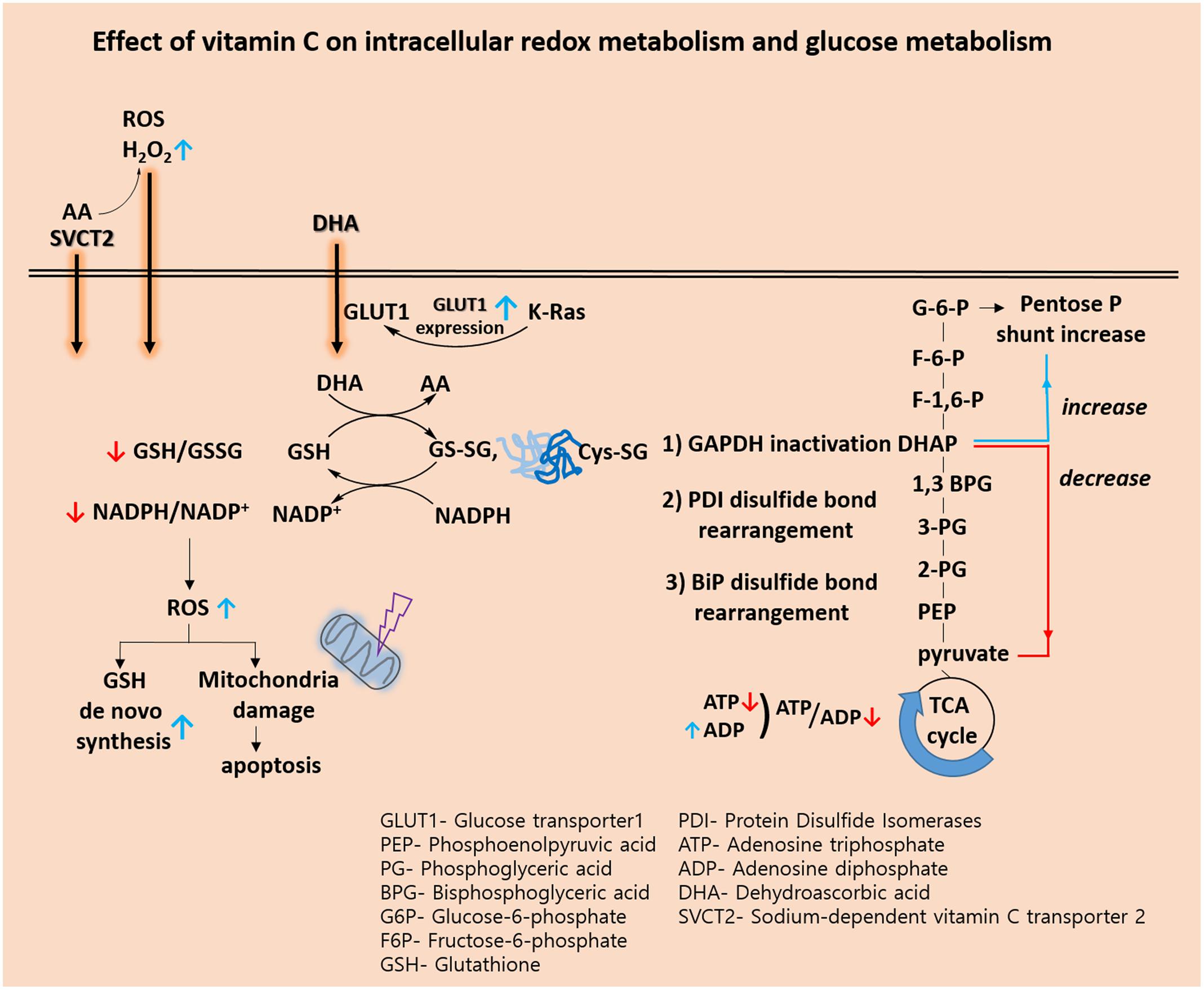
FIGURE 1. Effect of vitamin C on intracellular redox metabolism and glucose metabolism.
SP conceived the idea and wrote the original draft. SA and YS drafted the manuscript. YY and CY reviewed and supervised the manuscript writing process.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahuja, P., Zhao, P., Angelis, E., Ruan, H., Korge, P., Olson, A., et al. (2010). Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J. Clin. Invest. 120, 1494–1505. doi: 10.1172/JCI38331
Andrisic, L., Dudzik, D., Barbas, C., Milkovic, L., Grune, T., and Zarkovic, N. (2018). Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biol. 14, 47–58. doi: 10.1016/j.redox.2017.08.009
Bahlis, N., Jordan-Mcmurry, I., Grad, J., Reis, I., Neel, J., Kharfan-Dabaja, M., et al. (2001). Phase I results from a phase I/II study of arsenic trioxide (As2O3) and ascorbic acid (AA) in relapsed and chemorefractory multiple myeloma. Blood 98:375a.
Beatty, A. (2015). Metabolite profiling reveals the glutathione biosynthetic pathway as a therapeutic target in triple negative breast cancers. Cancer Res. 75:3552. doi: 10.1158/1538-7445.AM2015-3552
Belin, S., Kaya, F., Burtey, S., and Fontes, M. (2010). Ascorbic Acid and gene expression: another example of regulation of gene expression by small molecules? Curr. Genomics 11, 52–57. doi: 10.2174/138920210790217936
Belin, S., Kaya, F., Duisit, G., Giacometti, S., Ciccolini, J., and Fontes, M. (2009). Antiproliferative effect of ascorbic acid is associated with the inhibition of genes necessary to cell cycle progression. PLoS One 4:e4409. doi: 10.1371/journal.pone.0004409
Brunelli, L., Caiola, E., Marabese, M., Broggini, M., and Pastorelli, R. (2014). Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget 5, 4722–4731. doi: 10.18632/oncotarget.1958
Canali, R., Natarelli, L., Leoni, G., Azzini, E., Comitato, R., Sancak, O., et al. (2014). Vitamin C supplementation modulates gene expression in peripheral blood mononuclear cells specifically upon an inflammatory stimulus: a pilot study in healthy subjects. Genes Nutr. 9:390. doi: 10.1007/s12263-014-0390-x
Carcamo, J. M., Borquez-Ojeda, O., and Golde, D. W. (2002a). Vitamin C inhibits granulocyte macrophage-colony-stimulating factor-induced signaling pathways. Blood 99, 3205–3212.
Carcamo, J. M., Pedraza, A., Borquez-Ojeda, O., and Golde, D. W. (2002b). Vitamin C suppresses TNF alpha-induced NF kappa B activation by inhibiting I kappa B alpha phosphorylation. Biochemistry 41, 12995–13002.
Chen, P., Stone, J., Sullivan, G., Drisko, J. A., and Chen, Q. (2011). Anti-cancer effect of pharmacologic ascorbate and its interaction with supplementary parenteral glutathione in preclinical cancer models. Free Radic. Biol. Med. 51, 681–687. doi: 10.1016/j.freeradbiomed.2011.05.031
Chen, Q., Espey, M. G., Sun, A. Y., Pooput, C., Kirk, K. L., Krishna, M. C., et al. (2008). Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 11105–11109. doi: 10.1073/pnas.0804226105
Cooper, J. A., Esch, F. S., Taylor, S. S., and Hunter, T. (1984). Phosphorylation sites in enolase and lactate dehydrogenase utilized by tyrosine protein kinases in vivo and in vitro. J. Biol. Chem. 259, 7835–7841.
Cunniff, B., Newick, K., Nelson, K. J., Wozniak, A. N., Beuschel, S., Leavitt, B., et al. (2015). Disabling mitochondrial peroxide metabolism via combinatorial targeting of peroxiredoxin 3 as an effective therapeutic approach for malignant mesothelioma. PLoS One 10:e0127310. doi: 10.1371/journal.pone.0127310
Deprez, J., Vertommen, D., Alessi, D. R., Hue, L., and Rider, M. H. (1997). Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J. Biol. Chem. 272, 17269–17275. doi: 10.1074/jbc.272.28.17269
Droge, W., and Breitkreutz, R. (2000). Glutathione and immune function. Proc. Nutr. Soc. 59, 595–600. doi: 10.1017/S0029665100000847
Du, J., Cullen, J. J., and Buettner, G. R. (2012). Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 1826, 443–457. doi: 10.1016/j.bbcan.2012.06.003
Espey, M. G., Chen, P., Chalmers, B., Drisko, J., Sun, A. Y., Levine, M., et al. (2011). Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic. Biol. Med. 50, 1610–1619. doi: 10.1016/j.freeradbiomed.2011.03.007
Flier, J. S., Mueckler, M. M., Usher, P., and Lodish, H. F. (1987). Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 235, 1492–1495. doi: 10.1126/science.3103217
Forman, H. J., Zhang, H., and Rinna, A. (2009). Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 30, 1–12. doi: 10.1016/j.mam.2008.08.006
Gao, R., Cheng, J., Fan, C., Shi, X., Cao, Y., Sun, B., et al. (2015). Serum metabolomics to identify the liver disease-specific biomarkers for the progression of hepatitis to hepatocellular carcinoma. Sci. Rep. 5:18175. doi: 10.1038/srep18175
Goto, S., Yoshida, K., Morikawa, T., Urata, Y., Suzuki, K., and Kondo, T. (1995). Augmentation of transport for cisplatin-glutathione adduct in cisplatin-resistant cancer cells. Cancer Res. 55, 4297–4301.
Grad, J. M., Bahlis, N. J., Reis, I., Oshiro, M. M., Dalton, W. S., and Boise, L. H. (2001). Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells. Blood 98, 805–813. doi: 10.1182/blood.V98.3.805
Griffith, O. W., and Meister, A. (1979). Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 254, 7558–7560.
Hai, M., Bidichandani, S. I., and Patel, P. I. (2001). Identification of a positive regulatory element in the myelin-specific promoter of the PMP22 gene. J. Neurosci. Res. 65, 508–519. doi: 10.1002/jnr.1181
Halliwell, B., and Foyer, C. H. (1976). Ascorbic acid, metal ions and the superoxide radical. Biochem. J. 155, 697–700. doi: 10.1042/bj1550697
Harris, I. S., Treloar, A. E., Inoue, S., Sasaki, M., Gorrini, C., Lee, K. C., et al. (2015). Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222. doi: 10.1016/j.ccell.2014.11.019
Hayes, J. D., and McLellan, L. I. (1999). Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 31, 273–300. doi: 10.1080/10715769900300851
Heikkila, R. E., and Cabbat, F. S. (1983). Ascorbate-induced lipid peroxidation and inhibition of [3H]spiroperidol binding in neostriatal membrane preparations. J. Neurochem. 41, 1384–1392. doi: 10.1111/j.1471-4159.1983.tb00836.x
Hensley, K., Robinson, K. A., Gabbita, S. P., Salsman, S., and Floyd, R. A. (2000). Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 28, 1456–1462. doi: 10.1016/S0891-5849(00)00252-5
Hoffer, L. J., Robitaille, L., Zakarian, R., Melnychuk, D., Kavan, P., Agulnik, J., et al. (2015). High-dose intravenous vitamin C combined with cytotoxic chemotherapy in patients with advanced cancer: a phase I-II clinical trial. PLoS One 10:e0120228. doi: 10.1371/journal.pone.0120228
Holmgren, A., and Lu, J. (2010). Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem. Biophys. Res. Commun. 396, 120–124. doi: 10.1016/j.bbrc.2010.03.083
Hwang, N. R., Yim, S. H., Kim, Y. M., Jeong, J., Song, E. J., Lee, Y., et al. (2009). Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 423, 253–264. doi: 10.1042/BJ20090854
Ishikawa, S., Sugimoto, M., Kitabatake, K., Sugano, A., Nakamura, M., Kaneko, M., et al. (2016). Identification of salivary metabolomic biomarkers for oral cancer screening. Sci. Rep. 6:31520. doi: 10.1038/srep31520
John, D. C., and Bulleid, N. J. (1996). Intracellular dissociation and reassembly of prolyl 4-hydroxylase:the alpha-subunits associated with the immunoglobulin-heavy-chain binding protein (BiP) allowing reassembly with the beta-subunit. Biochem. J. 317, 659–665. doi: 10.1042/bj3170659
Kaya, F., Belin, S., Bourgeois, P., Micaleff, J., Blin, O., and Fontes, M. (2007). Ascorbic acid inhibits PMP22 expression by reducing cAMP levels. Neuromuscul. Disord. 17, 248–253. doi: 10.1016/j.nmd.2006.12.008
Koppenol, W. H., Bounds, P. L., and Dang, C. V. (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337. doi: 10.1038/nrc3038
Krishna, S., Low, I. C., and Pervaiz, S. (2011). Regulation of mitochondrial metabolism: yet another facet in the biology of the oncoprotein Bcl-2. Biochem. J. 435, 545–551. doi: 10.1042/BJ20101996
Kunsch, C., and Medford, R. M. (1999). Oxidative stress as a regulator of gene expression in the vasculature. Circ. Res. 85, 753–766. doi: 10.1161/01.RES.85.8.753
Larsson, A., Orrenius, S., Holmgren, A., and Mannervik, B. (1983). Functions of Glutathione: Biochemical, Physiological, Toxicological, and Clinical Aspects. New York, NY: Raven Press.
Liang, W. J., Johnson, D., and Jarvis, S. M. (2001). Vitamin C transport systems of mammalian cells. Mol. Membr. Biol. 18, 87–95. doi: 10.1080/09687680110033774
Liu, F., Zhang, W., You, X., Liu, Y., Li, Y., Wang, Z., et al. (2015). The oncoprotein HBXIP promotes glucose metabolism reprogramming via downregulating SCO2 and PDHA1 in breast cancer. Oncotarget 6, 27199–27213. doi: 10.18632/oncotarget.4508
Ma, E., Chen, P., Wilkins, H. M., Wang, T., Swerdlow, R. H., and Chen, Q. (2017). Pharmacologic ascorbate induces neuroblastoma cell death by hydrogen peroxide mediated DNA damage and reduction in cancer cell glycolysis. Free Radic. Biol. Med. 113, 36–47. doi: 10.1016/j.freeradbiomed.2017.09.008
Meister, A., and Anderson, M. E. (1983). Glutathione. Annu. Rev. Biochem. 52, 711–760. doi: 10.1146/annurev.bi.52.070183.003431
Monti, D. A., Mitchell, E., Bazzan, A. J., Littman, S., Zabrecky, G., Yeo, C. J., et al. (2012). Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One 7:e29794. doi: 10.1371/journal.pone.0029794
Nagarajan, A., Malvi, P., and Wajapeyee, N. (2016). Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2, 365–377. doi: 10.1016/j.trecan.2016.06.002
Nakamura, Y., Ohigashi, H., Masuda, S., Murakami, A., Morimitsu, Y., Kawamoto, Y., et al. (2000). Redox regulation of glutathione S-transferase induction by benzyl isothiocyanate: correlation of enzyme induction with the formation of reactive oxygen intermediates. Cancer Res. 60, 219–225.
Nishikimi, M., and Yagi, K. (1991). Molecular basis for the deficiency in humans of gulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am. J. Clin. Nutr. 54, 1203S–1208S. doi: 10.1093/ajcn/54.6.1203s
Oskouie, A. A., and Taheri, S. (2015). Recent developments and application of metabolomics in cancer diseases. J. Paramed. Sci. 6, 116–135.
Osthus, R. C., Shim, H., Kim, S., Li, Q., Reddy, R., Mukherjee, M., et al. (2000). Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 275, 21797–21800. doi: 10.1074/jbc.C000023200
Palmer, H. J., and Paulson, K. E. (1997). Reactive oxygen species and antioxidants in signal transduction and gene expression. Nutr. Rev. 55, 353–361. doi: 10.1111/j.1753-4887.1997.tb01561.x
Park, C., Kim, W., Kim, K., Lee, M., Jung, C., Han, S., et al. (2001). Clinical response achieved in AML by manipulation of L-ascorbic acid (LAA) levels. Blood 98, 218B–219B.
Park, C. H. (1985). Biological nature of the effect of ascorbic acids on the growth of human leukemic cells. Cancer Res. 45, 3969–3973.
Park, C. H., Bergsagel, D. E., and Mcculloch, E. A. (1971). Ascorbic acid: a culture requirement for colony formation by mouse plasmacytoma cells. Science 174, 720–722. doi: 10.1126/science.174.4010.720
Park, C. H., Kimler, B. F., Bodensteiner, D., Lynch, S. R., and Hassanein, R. S. (1992). In vitro growth modulation by L-ascorbic acid of colony-forming cells from bone marrow of patients with myelodysplastic syndromes. Cancer Res. 52, 4458–4466.
Park, S. (2007). “Apoptosis of leukemia cells induced by L-ascorbic acid and arsenic triocide: the effect of exidative stress and glutathione homeostasis,” in Cell Apoptosis and Cancer, ed. A. W. Talyor (Hauppauge, NY: Nova Science Publishers), 87–111.
Park, S. (2013). The effects of high concentrations of vitamin C on cancer cells. Nutrients 5, 3496–3505. doi: 10.3390/nu5093496
Park, S., Ahn, E. S., Lee, S., Jung, M., Park, J. H., Yi, S. Y., et al. (2009). Proteomic analysis reveals upregulation of RKIP in S-180 implanted BALB/C mouse after treatment with ascorbic acid. J. Cell. Biochem. 106, 1136–1145. doi: 10.1002/jcb.22097
Park, S., Han, S. S., Park, C. H., Hahm, E. R., Lee, S. J., Park, H. K., et al. (2004). L-Ascorbic acid induces apoptosis in acute myeloid leukemia cells via hydrogen peroxide-mediated mechanisms. Int. J. Biochem. Cell Biol. 36, 2180–2195. doi: 10.1016/j.biocel.2004.04.005
Park, S., Kim, K., Jung, C., Lee, M., Park, K., Kim, J., et al. (2002). Apoptotic signal transduction as a molecular mechanism for the clinical response achieved by L-ascorbic acid (LAA) in acute myeloid leukemia (AML). Blood 100:562.
Park, S., Lee, J., and Yeom, C. H. (2006). A proteomic approach to the identification of early molecular targets changed by L-ascorbic acid in NB4 human leukemia cells. J. Cell. Biochem. 99, 1628–1641. doi: 10.1002/jcb.20971
Prousky, J. (2008). The treatment of pulmonary diseases and respiratory-related conditions with inhaled (nebulized or aerosolized) glutathione. Evid. Based Complement. Alternat. Med. 5, 27–35. doi: 10.1093/ecam/nem040
Robey, R. B., and Hay, N. (2009). Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 19, 25–31. doi: 10.1016/j.semcancer.2008.11.010
Rumsey, S. C., Daruwala, R., Al-Hasani, H., Zarnowski, M. J., Simpson, I. A., and Levine, M. (2000). Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J. Biol. Chem. 275, 28246–28253. doi: 10.1074/jbc.M000988200
Sheehan, D., Meade, G., Foley, V. M., and Dowd, C. A. (2001). Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 360, 1–16. doi: 10.1042/bj3600001
Shepherd, A. G., Manson, M. M., Ball, H. W., and Mclellan, L. I. (2000). Regulation of rat glutamate-cysteine ligase (gamma-glutamylcysteine synthetase) subunits by chemopreventive agents and in aflatoxin B(1)-induced preneoplasia. Carcinogenesis 21, 1827–1834. doi: 10.1093/carcin/21.10.1827
Shim, H., Dolde, C., Lewis, B. C., Wu, C. S., Dang, G., Jungmann, R. A., et al. (1997). c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. U.S.A. 94, 6658–6663. doi: 10.1073/pnas.94.13.6658
Shin, D. M., Ahn, J. I., Lee, K. H., Lee, Y. S., and Lee, Y. S. (2004). Ascorbic acid responsive genes during neuronal differentiation of embryonic stem cells. Neuroreport 15, 1959–1963. doi: 10.1097/00001756-200408260-00025
Shin, J. M., Kamarajan, P., Fenno, J. C., Rickard, A. H., and Kapila, Y. L. (2016). Metabolomics of head and neck cancer: a mini-review. Front. Physiol. 7:526. doi: 10.3389/fphys.2016.00526
Smeyne, M., and Smeyne, R. J. (2013). Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 62, 13–25. doi: 10.1016/j.freeradbiomed.2013.05.001
Takemura, Y., Satoh, M., Satoh, K., Hamada, H., Sekido, Y., and Kubota, S. (2010). High dose of ascorbic acid induces cell death in mesothelioma cells. Biochem. Biophys. Res. Commun. 394, 249–253. doi: 10.1016/j.bbrc.2010.02.012
Tang, X., Lin, C. C., Spasojevic, I., Iversen, E. S., Chi, J. T., and Marks, J. R. (2014). A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 16:415. doi: 10.1186/s13058-014-0415-9
Tomasetti, M., Nocchi, L., Staffolani, S., Manzella, N., Amati, M., Goodwin, J., et al. (2014). MicroRNA-126 suppresses mesothelioma malignancy by targeting IRS1 and interfering with the mitochondrial function. Antioxid. Redox Signal. 21, 2109–2125. doi: 10.1089/ars.2013.5215
Trachootham, D., Alexandre, J., and Huang, P. (2009). Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591. doi: 10.1038/nrd2803
Tsukaguchi, H., Tokui, T., Mackenzie, B., Berger, U. V., Chen, X. Z., Wang, Y., et al. (1999). A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 399, 70–75.
Uetaki, M., Tabata, S., Nakasuka, F., Soga, T., and Tomita, M. (2015). Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci. Rep. 5:13896. doi: 10.1038/srep13896
Unterberger, E. B., Eichner, J., Wrzodek, C., Lempiainen, H., Luisier, R., Terranova, R., et al. (2014). Ha-ras and beta-catenin oncoproteins orchestrate metabolic programs in mouse liver tumors. Int. J. Cancer 135, 1574–1585. doi: 10.1002/ijc.28798
Vera, J. C., Rivas, C. I., Fischbarg, J., and Golde, D. W. (1993). Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 364, 79–82. doi: 10.1038/364079a0
Vera, J. C., Rivas, C. I., Velasquez, F. V., Zhang, R. H., Concha, I. I., and Golde, D. W. (1995). Resolution of the facilitated transport of dehydroascorbic acid from its intracellular accumulation as ascorbic acid. J. Biol. Chem. 270, 23706–23712. doi: 10.1074/jbc.270.40.23706
Wang, N., Wei, J., Liu, Y., Pei, D., Hu, Q., Wang, Y., et al. (2016). Discovery of biomarkers for oxidative stress based on cellular metabolomics. Biomarkers 21, 449–457. doi: 10.3109/1354750X.2016.1153720
Watanabe, H., and Bannai, S. (1987). Induction of cystine transport activity in mouse peritoneal macrophages. J. Exp. Med. 165, 628–640. doi: 10.1084/jem.165.3.628
Wattenberg, L. W. (1981). Inhibition of carcinogen-induced neoplasia by sodium cyanate, tert-butyl isocyanate, and benzyl isothiocyanate administered subsequent to carcinogen exposure. Cancer Res. 41, 2991–2994.
Willmann, L., Schlimpert, M., Hirschfeld, M., Erbes, T., Neubauer, H., Stickeler, E., et al. (2016). Alterations of the exo- and endometabolite profiles in breast cancer cell lines: a mass spectrometry-based metabolomics approach. Anal. Chim. Acta 925, 34–42. doi: 10.1016/j.aca.2016.04.047
Wong, T. S., Liu, X. B., Chung-Wai Ho, A., Po-Wing Yuen, A., Wai-Man, Ng, R., et al. (2008). Identification of pyruvate kinase type M2 as potential oncoprotein in squamous cell carcinoma of tongue through microRNA profiling. Int. J. Cancer 123, 251–257. doi: 10.1002/ijc.23583
Yun, J., Mullarky, E., Lu, C., Bosch, K. N., Kavalier, A., Rivera, K., et al. (2015). Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350, 1391–1396. doi: 10.1126/science.aaa5004
Yun, J., Rago, C., Cheong, I., Pagliarini, R., Angenendt, P., Rajagopalan, H., et al. (2009). Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559. doi: 10.1126/science.1174229
Keywords: vitamin C, cancer, metabolomics, glutathione metabolism, glucose metabolism
Citation: Park S, Ahn S, Shin Y, Yang Y, and Yeom CH (2018) Vitamin C in Cancer: A Metabolomics Perspective. Front. Physiol. 9:762. doi: 10.3389/fphys.2018.00762
Received: 28 March 2018; Accepted: 30 May 2018;
Published: 19 June 2018.
Edited by:
Jin Wang, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Simona Martinotti, Università degli Studi del Piemonte Orientale, ItalyCopyright © 2018 Park, Ahn, Shin, Yang and Yeom. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seyeon Park, c3lwYXJrMjFAZG9uZ2R1ay5hYy5rcg== Chang H. Yeom, bHltcGh5Y2hAaGFubWFpbC5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.