 Andrew P. Holmes
Andrew P. Holmes Clare J. Ray
Clare J. Ray Andrew M. Coney
Andrew M. Coney Prem Kumar
Prem Kumar
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 16 May 2018
Sec. Integrative Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00562
This article is part of the Research Topic Physiological and Pathological Responses to Hypoxia and High Altitude View all 36 articles
The mammalian carotid body (CB) is the primary arterial chemoreceptor that responds to acute hypoxia, initiating systemic protective reflex responses that act to maintain O2 delivery to the brain and vital organs. The CB is unique in that it is stimulated at O2 levels above those that begin to impact on the metabolism of most other cell types. Whilst a large proportion of the CB chemotransduction cascade is well defined, the identity of the O2 sensor remains highly controversial. This short review evaluates whether the mitochondria can adequately function as acute O2 sensors in the CB. We consider the similarities between mitochondrial poisons and hypoxic stimuli in their ability to activate the CB chemotransduction cascade and initiate rapid cardiorespiratory reflexes. We evaluate whether the mitochondria are required for the CB to respond to hypoxia. We also discuss if the CB mitochondria are different to those located in other non-O2 sensitive cells, and what might cause them to have an unusually low O2 binding affinity. In particular we look at the potential roles of competitive inhibitors of mitochondrial complex IV such as nitric oxide in establishing mitochondrial and CB O2-sensitivity. Finally, we discuss novel signaling mechanisms proposed to take place within and downstream of mitochondria that link mitochondrial metabolism with cellular depolarization.
The mammalian carotid body (CB) is a highly specialized sensory organ derived from the neural crest. The sensory units of the CB are the ‘glomus’ or ‘type I’ cells that respond to a variety of stimuli including hypoxia, hypercapnia, acidosis and hormones thereby allowing the CB to function as a polymodal receptor (Kumar and Bin-Jaliah, 2007; Ribeiro et al., 2013; Thompson et al., 2016). Type I cell activation leads to stimulation of adjacent chemoafferent fibers that relay sensory information into the central nervous system. The physiological consequence of CB stimulation is therefore the initiation of series of systemic protective reflexes characterized by increased ventilation, tachycardia, systemic vasoconstriction and adrenaline release from the adrenal medulla (Kumar, 2009).
There is now a general consensus that a series of key processes contribute to the CB hypoxic chemotransduction cascade. These include attenuation of outward K+ current, type I cell depolarization, Ca2+ influx through L-type Ca2+ channels, neurosecretion and chemoafferent excitation (Kumar and Prabhakar, 2012; Lopez-Barneo et al., 2016). Single type I cells are exquisitely sensitive to O2 and rapid (within ms) activation of the hypoxic chemotransduction cascade initiates at PO2s of between 20–40 mmHg (Biscoe and Duchen, 1990; Buckler and Vaughan-Jones, 1994; Buckler and Turner, 2013); a level considerably greater than that at which cell metabolism is affected in O2-insensitive cells.
What remains highly controversial is the molecular identity of the specific O2 sensor within the type I cell. We would argue that the physiological O2 sensor should exhibit certain key features: (1) expression in the type I cell permitting intrinsic O2 sensitivity; (2) the ability to bind O2; (3) its binding of O2 occurs over the physiological range at which the type I cell is stimulated; (4) it is required for the CB to be stimulated by hypoxia; and (5) it is able to activate the CB transduction cascade within milliseconds. Many proposed sensors fit one or two of these criteria but few have been shown to adequately comply with all five.
In mammalian cells, O2 is the terminal electron acceptor in the mitochondrial respiratory chain. Continuous binding and reduction of O2 in the CuB/haem a3 (cytochrome a3) binuclear center of complex IV drives mitochondrial electron transport and promotes activation of the mitochondrial ATP synthase. The long-established, mitochondrial hypothesis for chemoreception proposes that CB excitation induced by hypoxia is initiated by a reduction in O2 dependent mitochondrial energy respiration. This review will briefly critique the current evidence as to whether the mitochondria can be considered the functionally relevant O2 sensors within the CB.
All mitochondrial poisons induce chemoafferent excitation (Krylov and Anichkov, 1968; Mulligan et al., 1981; Obeso et al., 1989; Donnelly et al., 2014; Holmes et al., 2016, 2017) leading to rapid increases in ventilation (Owen and Gesell, 1931), heart rate and arterial blood glucose (Alvarez-Buylla and de Alvarez-Buylla, 1988). Chemoafferent responses are rapid, dose dependent and reversible (Donnelly et al., 2014; Holmes et al., 2016) and the magnitude of the rise in chemoafferent frequency caused by saturating concentrations is consistent with those evoked by severe hypoxia or anoxia (Krylov and Anichkov, 1968; Mulligan et al., 1981; Obeso et al., 1989). Furthermore, mitochondrial inhibitors and uncouplers augment neurotransmitter secretion, confirming an action through the type I cell rather than the afferent nerve endings (Obeso et al., 1989; Rocher et al., 1991). Despite the strong consistency between all of the different types of mitochondrial poisons (both in the older and more recent studies), it should be noted that some of the pharmacological agents used may not have acted selectively on the mitochondria and so the conclusions should be viewed with a certain degree of caution.
Mitochondrial poisons cause fast (within ms) type I cell depolarization and increases in [Ca2+]i. The size and timing of the [Ca2+]i rise observed using many different mitochondrial inhibitors or uncouplers closely resembles that seen in hypoxia (Biscoe et al., 1989; Biscoe and Duchen, 1990; Wyatt and Buckler, 2004; Buckler, 2011). As with hypoxia (Buckler and Vaughan-Jones, 1994; Urena et al., 1994), the increases in [Ca2+]i are dependent on cellular depolarization and extracellular Ca2+ influx through voltage gated Ca2+ channels (Wyatt and Buckler, 2004). TASK1/3, TREK-1, BKCa, Kv4.1, and Kv4.3, have all been shown to be expressed in the CB and inhibited by hypoxia (Buckler et al., 2000; Sanchez et al., 2002; Williams et al., 2004; Yamamoto and Taniguchi, 2006; Kim et al., 2009; Kreneisz et al., 2009; Turner and Buckler, 2013; Wang et al., 2017b). Of these, TASK1/3 and TASK-like K+ currents are diminished by a multitude of mitochondrial inhibitors leading to membrane depolarization (Barbe et al., 2002; Wyatt and Buckler, 2004; Buckler, 2011; Turner and Buckler, 2013; Kim D. et al., 2015).
Recent evidence has revealed the presence of TRP and other non-selective Ca2+-activated cation currents in type I cells that are activated by hypoxia (Kumar et al., 2006; Kang et al., 2014; Kim I. et al., 2015; Wang et al., 2017a). Although intriguing, the full functional relevance of these currents in type I cell O2-sensing remains to be further characterized, and in particular whether these currents can be upregulated to preserve O2 sensing in the absence of TASK channels (Turner and Buckler, 2013). Current evidence suggests that mitochondrial inhibitors are also capable of increasing these inward depolarizing currents (Kim I. et al., 2015; Wang et al., 2017a).
The presence of functional mitochondria does appear necessary for the CB to respond fully to hypoxia. For instance, cyanide, rotenone and FCCP all attenuate TASK channel currents in such a way that prevents any further reduction by hypoxia (Wyatt and Buckler, 2004). In intact CB preparations oligomycin, cyanide and azide all reduce or abolish subsequent chemoafferent responses to hypoxia (Mulligan et al., 1981; Donnelly et al., 2014). Some of the attenuation observed in these experiments may have been due to impairment of oxidative phosphorylation in the chemoafferent fibers, limiting their excitability. Any such impairment is not apparent in response to physiological levels of hypoxia, since the PO2 that activates the type I cells occurs at a much higher level than those which would decrease mitochondrial function in the chemoafferent fibers. As such, chemoafferent responses to sustained hypoxia are better maintained than those in response to sustained high doses of mitochondrial inhibitors (Mulligan et al., 1981).
In a recent study the importance of mitochondrial complex I was tested by developing mice deficient in Ndufs2 (a gene coding for NADH dehydrogenase [ubiquinone] iron-sulfur protein 2- a component of complex I that participates in ubiquinone binding) in tyrosine hydroxylase positive cells (Fernandez-Aguera et al., 2015). Type I cells isolated from these mice were insensitive to hypoxia; they lacked any hypoxia-induced K+ current attenuation, [Ca2+]i elevation or neurotransmitter release. Furthermore, these mice failed to increase respiratory frequency when breathing 10% O2. This work supported a previous study in which type I cell hypoxic chemosensitivity was abolished in the presence of rotenone (Ortega-Saenz et al., 2003). The authors propose a mechanism whereby exposure to hypoxia promotes reverse electron transport and ROS/NADH generation via complex I which is driven by a high rate of succinate oxidation at complex II. Accordingly, they have recently shown that genetic and pharmacological deactivation of complex II completely blocks type I cell hypoxic sensitivity (Gao et al., 2017).
This intriguing and elegant hypothesis does, however, show some discrepancies with evidence from earlier reports. For instance, similar experiments performed on CBs with heterozygous Sdhd knock out displayed an augmented, rather than depressed basal activity and had a completely preserved hypoxic response (Piruat et al., 2004). Furthermore, when rat type I cells were exposed to tetramethyl-p-phenylenediamine (TMPD) and ascorbate in the presence of rotenone, there was still a robust elevation in Ca2+ upon hypoxic stimulation (Wyatt and Buckler, 2004). This would suggest that feeding electrons into cytochrome c is sufficient to sustain type I cell hypoxic sensitivity even when complex I activity (and ROS generation) is inhibited. Complex IV activity rather than complex I and II may therefore be necessary for hypoxic chemotransduction. The same report also showed that application of H2O2 was unable to excite the type I cell directly. This observation is consistent with the lack of effect of multiple anti-oxidants used in other CB preparations and animal species (Sanz-Alfayate et al., 2001; Agapito et al., 2009; Gomez-Nino et al., 2009). Interestingly, using novel ex vivo CB culture techniques combined with FRET based ROS sensors, Bernardini et al. (2015) deduced that type I cell ROS actually decreases in hypoxia due to reduction in NADPH oxidase activity (an alternative ROS source). Clearly there is a need for reconciliation between these findings.
The evidence that mitochondria are required for CB O2 chemotransduction and that mitochondrial inhibitors can cause chemoexcitation, is not enough to define them as the O2-sensors in the CB. Clearly, mitochondria are able to bind O2. However, the Km of the cytochrome a3 for O2 is reported to be <1 mmHg in isolated mitochondria and between 1–5 mmHg in dissociated cells and tissue preparations, with little variation existing between different cell types (Wilson et al., 1988; Tamura et al., 1989). This is far lower than the PO2 at which the CB type I cells begin to be activated and, for this reason, is a common argument against the mitochondrial hypothesis.
However, there is now a substantial body of evidence indicating that the CB type I cell mitochondria are unique. Experiments performed by Mills and Jobsis (1970, 1972), were the first to identify an unusually low affinity cytochrome a3 within the CB. Using absorbance spectra, they estimated that 43–67.5% of total cytochrome a3 within the intact CB preparation had a remarkably low O2 affinity. This fraction was reported to be almost 100% reduced at PO2s between 7–9 mmHg and 50% reduced at a PO2 as high as 90 mmHg. In contrast, the remaining fraction was only 50% reduced at a PO2 of approximately 0.8 mmHg, comparable to cytochrome a3 found in other tissues (Gnaiger, 2001). Thus, the CB appeared to express both low and high affinity subtypes of cytochrome a3. At that time, the specific cellular location(s) of each was unclear. Later experiments utilized the photolabile binding of CO, to deduce that saturation of cytochrome a3 with CO prevented any additional chemoafferent excitation during hypoxia, implying that not only was the cytochrome a3 in the CB unusual, it was also required for O2-sensing (Wilson et al., 1994; Lahiri et al., 1999). It should be pointed out that the concentrations of CO used in these studies could have directly modified the activity of the BKCa channel (Williams et al., 2004, 2008) and the generation of H2S (Yuan et al., 2015) and as such some of the observations could be related to mechanisms independent of the mitochondria.
In dissociated rabbit type I cell clusters, mitochondrial electron transport begins to be inhibited at a high PO2 value of approximately 40 mmHg (Duchen and Biscoe, 1992a). PO2-NADH response curves demonstrate a significant ‘right shift’ in type I cells compared to sensory neurons, indicative of a heightened and distinctive O2 sensitivity. In addition, mitochondrial depolarization occurs at higher PO2s compared to O2-insensitive cells (Duchen and Biscoe, 1992b). More recent work has verified the “right shifts” in both PO2-NADH and PO2-rh-123 response curves in rat type I cells, confirming that the unusually low mitochondrial O2 affinity is conserved in multiple species (Turner and Buckler, 2013). By isolating complex IV activity with a cocktail of mitochondrial inhibitors plus TMPD and ascorbate, the authors were able to reveal that complex IV activity is a component of the mitochondria with the exceptionally low O2 affinity. Importantly, type I cell hypoxic response curves for electron transport inhibition, mitochondrial depolarization and complex IV run-down display considerable overlap with the rise in Ca2+, indicating that these processes are intimately linked. Therefore, it does appear that type I cell mitochondria have a highly specialized low affinity for O2 due to an altered function/subtype of cytochrome a3 in complex IV that predisposes CB energy metabolism to being impaired at high O2 tensions. It is likely that the high affinity cytochrome a3 in the CB described by Mills & Jöbsis is located in the non-O2 sensing tissue such as the, nerve endings, blood vessels and type II cells.
Understanding the mechanism linking a fall in mitochondrial O2 consumption with K+ channel inhibition (or cation channel activation) is contentious. As previously mentioned, there could be a role for elevated mitochondrial ROS generation but this is still to be validated (Fernandez-Aguera et al., 2015). Another possibility is an alteration in cytosolic nucleotides. Switching from a cell attached to inside-out patch configuration diminishes background K+ channel activity, suggesting that a basal level of an intracellular factor(s) activates TASK channels in normoxia (Varas et al., 2007). Addition of 5 mM MgATP in the inside-out configuration can restore about 50% of this background K+ channel activity. Both mitochondrial inhibition and hypoxia also significantly elevate free Mg2+, consistent with a decrease in MgATP. Thus, the fall in MgATP during hypoxia is likely to attenuate a significant proportion of TASK-current leading to depolarization. However, the remaining modulators that account for the other 50% of TASK current are still to be identified.
Another proposed mediator of TASK channel activity that is sensitive to changes in cytosolic nucleotide concentrations is AMPK (Wyatt et al., 2007). However, initial favorable studies based on pharmacological evidence have since been challenged by the finding that the AMPK-α1α2 deficient CB retains complete O2-sensitivity (Mahmoud et al., 2016). Other groups have also shown that pharmacological targeting of AMPK does not impact on the hypoxia-induced K+ channel inhibition (Kim et al., 2014). Discrepancies may arise from the non-selectivity of the drugs used to evaluate AMPK function and potential redundancy mechanisms known to develop in genetic animal models (Nowak et al., 1997). A final hypothesis is that a build-up in lactate upon mitochondrial inhibition in hypoxia activates the olfactory receptor Olfr78 (Chang et al., 2015). However, the concentration of lactate necessary to elevate Ca2+ in an intact CB preparation appears to be quite high (30 mM) and whether local levels reach this threshold during hypoxia is uncertain. We await a mechanism demonstrating how activation of Olfr78 (a G-protein-coupled receptor) modulates TASK or cation channel activity. A summary of the proposed O2-sensitive mitochondrial signaling pathways in the CB is presented in Figure 1.
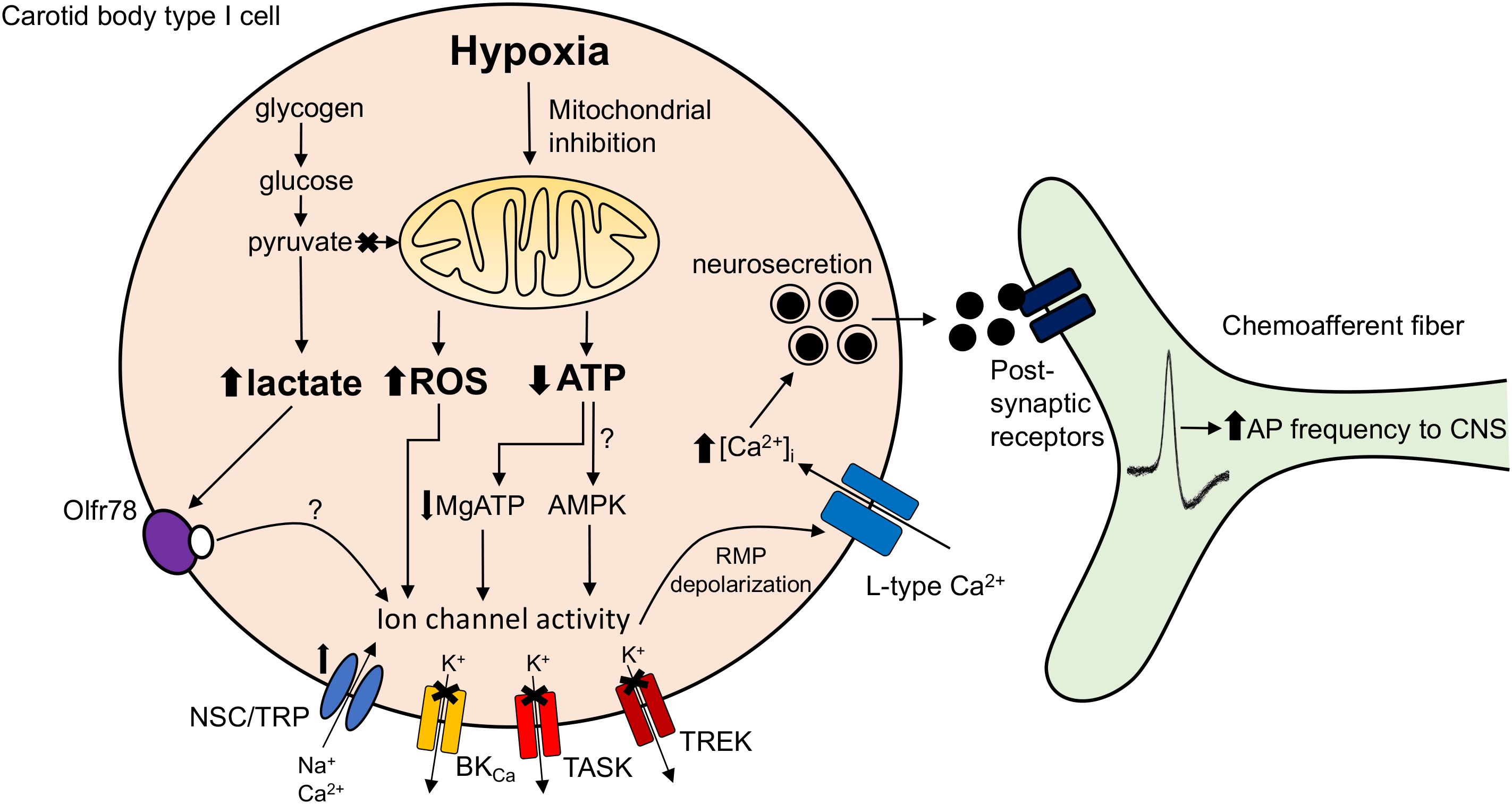
FIGURE 1. Carotid body mitochondrial signaling mechanisms activated during hypoxia. Hypoxia-induced mitochondrial inhibition is proposed to increase lactate generation (Chang et al., 2015), augment mitochondrial complex I reactive oxygen species (ROS) production (Fernandez-Aguera et al., 2015) or reduce mitochondrial ATP synthesis (Buckler and Turner, 2013). These changes are proposed to directly or indirectly (e.g., via Olf78 receptor activation, reduced MgATP concentration or stimulation of AMP-activated protein kinase; AMPK) modify ion channel function leading to resting membrane potential (RMP) depolarization. This causes opening of L-type Ca2+ channels, neurosecretion and an increase in discharge frequency of the adjacent chemoafferent fibers. TRP, transient receptor potential channel; NSC, non-selective cation channel; BKCa, large conductance Ca2+-activated K+ channel; TASK, TWIK-related acid-sensitive K+ channel; TREK, TWIK-related K+ channel; AP, action potential.
We propose that there are 2 potential means to account for the extraordinary O2 sensitivity of type I cell mitochondria. First, there could be a high level of production of a cytosolic factor that is able to freely diffuse into the mitochondria and then compete with O2 binding in complex IV (Figure 2). We predict that this competition would render mitochondrial electron transport more susceptible to subsequent falls in O2. We recently tested this by applying exogenous nitrite to CBs and subsequently measuring hypoxic sensitivity (Holmes et al., 2016). Nitrite is reduced within the mitochondria to generate local NO, a recognized competitive inhibitor of complex IV (Brown and Cooper, 1994; Cleeter et al., 1994; Castello et al., 2006, 2008; Basu et al., 2008). Moderate basal inhibition of the CB mitochondria by nitrite exaggerated the subsequent chemoafferent excitation during hypoxia signifying an increase in CB O2 sensitivity. Therefore, we validated the idea that CB hypoxic sensitivity could be adjusted by a factor capable of competing with O2 in the mitochondria and suggested a physiological role for endogenous NO in establishing type I cell mitochondrial O2-sensitivity. Measurable amounts of NO have been detected in mitochondrial of type I cells (Yamamoto et al., 2006). A possible source is nitric oxide synthase 3 (NOS-3) given its location within the type I cell (Yamamoto et al., 2006). Interestingly, mice with reduced NOS-3 have a dampened hypoxic ventilatory response and a depressed CB function (Kline et al., 2000). One explanation for this is an adaptation to chronic hypoxia brought about by reduced CB blood flow. However, this is unlikely as there is no significant type I cell hyperplasia/hypertrophy (McGregor et al., 1984; Tatsumi et al., 1991). Instead, the blunted CB activity could be due to the lack of NO acting on the CB mitochondria. Consideration of the precise compartmentalization of NO should also be taken into account. Whilst NO in the mitochondria induces chemostimulation, its action in other regions is likely to have opposing effects via modulation of soluble guanylate cyclase and L-type Ca2+/BKCa channels (Summers et al., 1999; Iturriaga et al., 2000; Silva and Lewis, 2002; Valdes et al., 2003). In addition, other diffusible cytosolic factors have been implicated in CB O2 sensing including H2S and CO (Peng et al., 2010; Yuan et al., 2015). Both of these gasses are capable of inhibiting type I cell mitochondria (Wilson et al., 1994; Lahiri et al., 1999; Buckler, 2011). Future experiments are required to evaluate if these substances act by setting type I cell mitochondrial O2 sensitivity.
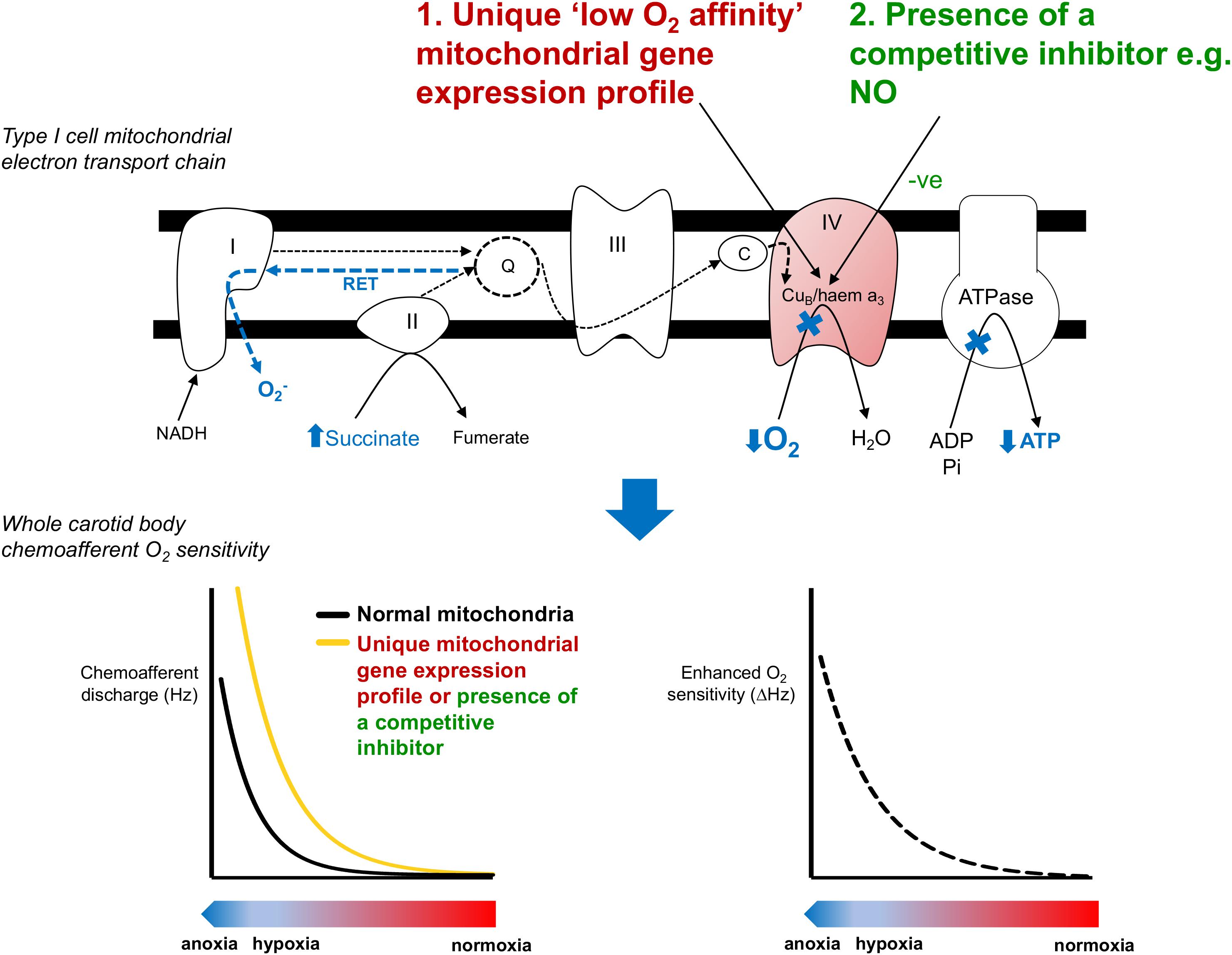
FIGURE 2. Potential mechanisms underlying the unique low O2 affinity mitochondria in the carotid body. Inhibition of mitochondrial function in the carotid body type I cell occurs at PO2 values well above those that inhibit metabolism in other O2 insensitive cells. This activates a number of proposed signaling pathways (shown in blue). The unique low O2 affinity of the carotid body mitochondria could be due to (1) a unique mitochondrial gene expression profile or (2) the presence of a competitive inhibitor such as NO (Holmes et al., 2016). The impact of lower mitochondrial O2 affinity is to cause a right-shift in the PO2-chemoafferent response curve, thereby enhancing the functional O2 sensitivity and allowing the carotid body to respond over a more physiological range of arterial PO2s. RET, reverse electron transport.
A second explanation for the low O2-affinity of the type I cell mitochondria is that it has a unique gene expression profile (Figure 2). Exploring gene expression in the CB is challenging due to its relatively small size and heterogeneity. However, advances in molecular biology techniques now make it possible to perform whole genome analysis using just micrograms of tissue or even single cells. RNA-sequencing analysis has now revealed a number of mitochondrial related genes that have a particularly high expression in the type I cell (Zhou et al., 2016). Of these Ndufa4l2 and Cox4i2 have been shown to be more highly expressed in the CB compared with tissue from the superior cervical ganglion (Gao et al., 2017). Whether these two genes contribute to the low mitochondrial O2 affinity remains to be determined but these findings do support the idea that CB mitochondria have a unique genetic signature encoding their mitochondrial complexes. We would expect many more genetic studies to probe this further until the type I cell mitochondria can be accurately modeled to pinpoint the structural conformation underlying its low O2 affinity. An interesting comparator may be the mitochondria isolated from guinea pig CB which does not appear to have any inherent O2 sensitivity (Gonzalez-Obeso et al., 2017).
On current evidence, it is very hard to disprove the mitochondrial hypothesis of CB O2 sensing. The mitochondria seem to fulfill all five criteria that we have proposed for adequate O2-sensors. What is less clear is a mechanistic understanding of how the low O2 sensitivity of the CB mitochondria is achieved and if mitochondria are involved in establishing pathological changes in CB function.
AH, CR, AC, and PK all contributed to the writing and editing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agapito, M. T., Sanz-Alfayate, G., Gomez-Nino, A., Gonzalez, C., and Obeso, A. (2009). General redox environment and carotid body chemoreceptor function. Am. J. Physiol. Cell Physiol. 296, C620–C631. doi: 10.1152/ajpcell.00542.2008
Alvarez-Buylla, R., and de Alvarez-Buylla, E. R. (1988). Carotid sinus receptors participate in glucose homeostasis. Respir. Physiol. 72, 347–359. doi: 10.1016/0034-5687(88)90093-X
Barbe, C., Al-Hashem, F., Conway, A. F., Dubuis, E., Vandier, C., and Kumar, P. (2002). A possible dual site of action for carbon monoxide-mediated chemoexcitation in the rat carotid body. J. Physiol. 543(Pt 3), 933–945. doi: 10.1113/jphysiol.2001.015750
Basu, S., Azarova, N. A., Font, M. D., King, S. B., Hogg, N., Gladwin, M. T., et al. (2008). Nitrite reductase activity of cytochrome c. J. Biol. Chem. 283, 32590–32597. doi: 10.1074/jbc.M806934200
Bernardini, A., Brockmeier, U., Metzen, E., Berchner-Pfannschmidt, U., Harde, E., Acker-Palmer, A., et al. (2015). Type I cell ROS kinetics under hypoxia in the intact mouse carotid body ex vivo: a FRET-based study. Am. J. Physiol. Cell Physiol. 308, C61–C67. doi: 10.1152/ajpcell.00370.2013
Biscoe, T. J., and Duchen, M. R. (1990). Responses of type I cells dissociated from the rabbit carotid body to hypoxia. J. Physiol. 428, 39–59. doi: 10.1113/jphysiol.1990.sp018199
Biscoe, T. J., Duchen, M. R., Eisner, D. A., O’Neill, S. C., and Valdeolmillos, M. (1989). Measurements of intracellular Ca2+ in dissociated type I cells of the rabbit carotid body. J. Physiol. 416, 421–434. doi: 10.1113/jphysiol.1989.sp017769
Brown, G. C., and Cooper, C. E. (1994). Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 356, 295–298. doi: 10.1016/0014-5793(94)01290-3
Buckler, K. J. (2011). Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 463, 743–754. doi: 10.1007/s00424-012-1089-8
Buckler, K. J., and Turner, P. J. (2013). Oxygen sensitivity of mitochondrial function in rat arterial chemoreceptor cells. J. Physiol. 591(Pt 14), 3549–3563. doi: 10.1113/jphysiol.2013.257741
Buckler, K. J., and Vaughan-Jones, R. D. (1994). Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J. Physiol. 476, 423–428. doi: 10.1113/jphysiol.1994.sp020143
Buckler, K. J., Williams, B. A., and Honore, E. (2000). An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J. Physiol. 525(Pt 1), 135–142. doi: 10.1111/j.1469-7793.2000.00135.x
Castello, P. R., David, P. S., McClure, T., Crook, Z., and Poyton, R. O. (2006). Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 3, 277–287. doi: 10.1016/j.cmet.2006.02.011
Castello, P. R., Woo, D. K., Ball, K., Wojcik, J., Liu, L., and Poyton, R. O. (2008). Oxygen-regulated isoforms of cytochrome c oxidase have differential effects on its nitric oxide production and on hypoxic signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 8203–8208. doi: 10.1073/pnas.0709461105
Chang, A. J., Ortega, F. E., Riegler, J., Adison, D. V. M., and Krasnow, M. A. (2015). Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 527, 240–244. doi: 10.1038/nature15721
Cleeter, M. W., Cooper, J. M., Darley-Usmar, V. M., Moncada, S., and Schapira, A. H. (1994). Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 345, 50–54. doi: 10.1016/0014-5793(94)00424-2
Donnelly, D. F., Kim, I., Mulligan, E. M., and Carroll, J. L. (2014). Non-additive interactions between mitochondrial complex IV blockers and hypoxia in rat carotid body responses. Respir. Physiol. Neurobiol. 190, 62–69. doi: 10.1016/j.resp.2013.09.009
Duchen, M. R., and Biscoe, T. J. (1992a). Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J. Physiol. 450, 13–31.
Duchen, M. R., and Biscoe, T. J. (1992b). Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J. Physiol. 450, 33–61.
Fernandez-Aguera, M. C., Gao, L., Gonzalez-Rodriguez, P., Pintado, C. O., Arias-Mayenco, I., Garcia-Flores, P., et al. (2015). Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I Signaling. Cell Metab. 22, 825–837. doi: 10.1016/j.cmet.2015.09.004
Gao, L., Bonilla-Henao, V., Garcia-Flores, P., Arias-Mayenco, I., Ortega-Saenz, P., and Lopez-Barneo, J. (2017). Gene expression analyses reveal metabolic specifications in acute O-2-sensing chemoreceptor cells. J. Physiol. 595, 6091–6120. doi: 10.1113/jp274684
Gnaiger, E. (2001). Bioenergetics at low oxygen: dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir. Physiol. 128, 277–297. doi: 10.1016/s0034-5687(01)00307-3
Gomez-Nino, A., Agapito, M. T., Obeso, A., and Gonzalez, C. (2009). Effects of mitochondrial poisons on glutathione redox potential and carotid body chemoreceptor activity. Respir. Physiol. Neurobiol. 165, 104–111. doi: 10.1016/j.resp.2008.10.020
Gonzalez-Obeso, E., Docio, I., Olea, E., Cogolludo, A., Obeso, A., Rocher, A., et al. (2017). Guinea Pig oxygen-sensing and carotid body functional properties. Front. Physiol. 8:12. doi: 10.3389/fphys.2017.00285
Holmes, A. P., Ray, C. J., Pearson, S. A., Coney, A. M., and Kumar, P. (2017). Ecto-5′-nucleotidase (CD73) regulates peripheral chemoreceptor activity and cardiorespiratory responses to hypoxia. J. Physiol. doi: 10.1113/JP274498 [Epub ahead of print].
Holmes, A. P., Turner, P. J., Buckler, K. J., and Kumar, P. (2016). Moderate inhibition of mitochondrial function augments carotid body hypoxic sensitivity. Pflugers Arch. 468, 143–155. doi: 10.1007/s00424-015-1745-x
Iturriaga, R., Villanueva, S., and Mosqueira, M. (2000). Dual effects of nitric oxide on cat carotid body chemoreception. J. Appl. Physiol. 89, 1005–1012. doi: 10.1152/jappl.2000.89.3.1005
Kang, D., Wang, J., Hogan, J. O., Vennekens, R., Freichel, M., White, C., et al. (2014). Increase in cytosolic Ca2+ produced by hypoxia and other depolarizing stimuli activates a non-selective cation channel in chemoreceptor cells of rat carotid body. J. Physiol. 592(Pt 9), 1975–1992. doi: 10.1113/jphysiol.2013.266957
Kim, D., Cavanaugh, E. J., Kim, I., and Carroll, J. L. (2009). Heteromeric TASK-1/TASK-3 is the major oxygen-sensitive background K+ channel in rat carotid body glomus cells. J. Physiol. 587(Pt 12), 2963–2975. doi: 10.1113/jphysiol.2009.171181
Kim, D., Kang, D., Martin, E. A., Kim, I., and Carroll, J. L. (2014). Effects of modulators of AMP-activated protein kinase on TASK-1/3 and intracellular Ca(2+) concentration in rat carotid body glomus cells. Respir. Physiol. Neurobiol. 195, 19–26. doi: 10.1016/j.resp.2014.01.020
Kim, D., Kim, I., Wang, J., White, C., and Carroll, J. L. (2015). Hydrogen sulfide and hypoxia-induced changes in TASK (K2P3/9) activity and intracellular Ca(2+) concentration in rat carotid body glomus cells. Respir. Physiol. Neurobiol. 215, 30–38. doi: 10.1016/j.resp.2015.04.012
Kim, I., Fite, L., Donnelly, D. F., Kim, J. H., and Carroll, J. L. (2015). “Possible role of TRP channels in rat glomus cells,” in Arterial Chemoreceptors in Physiology and Pathophysiology, eds C. Peers, P. Kumar, C. N. Wyatt, E. Gauda, C. A. Nurse, and N. Prabhakar (Berlin: Springer-Verlag), 227–232. doi: 10.1007/978-3-319-18440-1_25
Kline, D. D., Yang, T., Premkumar, D. R., Thomas, A. J., and Prabhakar, N. R. (2000). Blunted respiratory responses to hypoxia in mutant mice deficient in nitric oxide synthase-3. J. Appl. Physiol. 88, 1496–1508. doi: 10.1152/jappl.2000.88.4.1496
Kreneisz, O., Benoit, J. P., Bayliss, D. A., and Mulkey, D. K. (2009). AMP-activated protein kinase inhibits TREK channels. J. Physiol. 587(Pt 24), 5819–5830. doi: 10.1113/jphysiol.2009.180372
Krylov, S. S., and Anichkov, S. V. (1968). “The effect of metabolic inhibitors on carotid chemoreceptors,” in Arterial Chemoreceptors, ed. R. W. Torrance (Oxford: Blackwell), 103–109.
Kumar, P. (2009). Systemic effects resulting from carotid body stimulation-invited article. Adv. Exp. Med. Biol. 648, 223–233. doi: 10.1007/978-90-481-2259-2_26
Kumar, P., and Bin-Jaliah, I. (2007). Adequate stimuli of the carotid body: more than an oxygen sensor? Respir. Physiol. Neurobiol. 157, 12–21. doi: 10.1016/j.resp.2007.01.007
Kumar, P., Pearson, S., and Gu, Y. C. (2006). A role for TRP channels in carotid body chemotransduction? FASEB J. 20, A1229–A1229. doi: 10.1007/978-3-319-18440-1_25
Kumar, P., and Prabhakar, N. R. (2012). Peripheral chemoreceptors: function and plasticity of the carotid body. Compr. Physiol. 2, 141–219. doi: 10.1002/cphy.c100069
Lahiri, S., Ehleben, W., and Acker, H. (1999). Chemoreceptor discharges and cytochrome redox changes of the rat carotid body: role of heme ligands. Proc. Natl. Acad. Sci. U.S.A. 96, 9427–9432. doi: 10.1073/pnas.96.16.9427
Lopez-Barneo, J., Gonzalez-Rodriguez, P., Gao, L., Fernandez-Aguera, M. C., Pardal, R., and Ortega-Saenz, P. (2016). Oxygen sensing by the carotid body: mechanisms and role in adaptation to hypoxia. Am. J. Physiol. Cell Physiol. 310, C629–C642. doi: 10.1152/ajpcell.00265.2015
Mahmoud, A. D., Lewis, S., Juricic, L., Udoh, U. A., Hartmann, S., Jansen, M. A., et al. (2016). AMP-activated protein kinase deficiency blocks the hypoxic ventilatory response and thus precipitates hypoventilation and Apnea. Am. J. Respir. Crit. Care Med. 193, 1032–1043. doi: 10.1164/rccm.201508-1667OC
McGregor, K. H., Gil, J., and Lahiri, S. (1984). A morphometric study of the carotid body in chronically hypoxic rats. J. Appl. Physiol. 57, 1430–1438. doi: 10.1152/jappl.1984.57.5.1430
Mills, E., and Jobsis, F. F. (1970). Simultaneous measurement of cytochrome a3 reduction and chemoreceptor afferent activity in the carotid body. Nature 225, 1147–1149. doi: 10.1038/2251147a0
Mills, E., and Jobsis, F. F. (1972). Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. J. Neurophysiol. 35, 405–428. doi: 10.1152/jn.1972.35.4.405
Mulligan, E., Lahiri, S., and Storey, B. T. (1981). Carotid body O2 chemoreception and mitochondrial oxidative phosphorylation. J. Appl. Physiol. 51, 438–446. doi: 10.1152/jappl.1981.51.2.438
Nowak, M. A., Boerlijst, M. C., Cooke, J., and Smith, J. M. (1997). Evolution of genetic redundancy. Nature 388, 167–171. doi: 10.1038/40618
Obeso, A., Almaraz, L., and Gonzalez, C. (1989). Effects of cyanide and uncouplers on chemoreceptor activity and ATP content of the cat carotid body. Brain Res. 481, 250–257. doi: 10.1016/0006-8993(89)90801-9
Ortega-Saenz, P., Pardal, R., Garcia-Fernandez, M., and Lopez-Barneo, J. (2003). Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J. Physiol. 548(Pt 3), 789–800. doi: 10.1113/jphysiol.2003.039693
Owen, H., and Gesell, R. (1931). Peripheral and central chemical control of pulmonary ventilation. Proc. Soc. Exp. Biol. Med. 28, 0765–0766. doi: 10.3181/00379727-28-5523
Peng, Y. J., Nanduri, J., Raghuraman, G., Souvannakitti, D., Gadalla, M. M., Kumar, G. K., et al. (2010). H2S mediates O2 sensing in the carotid body. Proc. Natl. Acad. Sci. U.S.A. 107, 10719–10724. doi: 10.1073/pnas.1005866107
Piruat, J. I., Pintado, C. O., Ortega-Saenz, P., Roche, M., and Lopez-Barneo, J. (2004). The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol. Cell. Biol. 24, 10933–10940. doi: 10.1128/MCB.24.24.10933-10940.2004
Ribeiro, M. J., Sacramento, J. F., Gonzalez, C., Guarino, M. P., Monteiro, E. C., and Conde, S. V. (2013). Carotid body denervation prevents the development of insulin resistance and hypertension induced by hypercaloric diets. Diabetes Metab. Res. Rev. 62, 2905–2916. doi: 10.2337/db12-1463
Rocher, A., Obeso, A., Gonzalez, C., and Herreros, B. (1991). Ionic mechanisms for the transduction of acidic stimuli in rabbit carotid body glomus cells. J. Physiol. 433, 533–548. doi: 10.1113/jphysiol.1991.sp018442
Sanchez, D., Lopez-Lopez, J. R., Perez-Garcia, M. T., Sanz-Alfayate, G., Obeso, A., Ganfornina, M. D., et al. (2002). Molecular identification of Kvalpha subunits that contribute to the oxygen-sensitive K+ current of chemoreceptor cells of the rabbit carotid body. J. Physiol. 542(Pt 2), 369–382.
Sanz-Alfayate, G., Obeso, A., Agapito, M. T., and Gonzalez, C. (2001). Reduced to oxidized glutathione ratios and oxygen sensing in calf and rabbit carotid body chemoreceptor cells. J. Physiol. 537(Pt 1), 209–220. doi: 10.1111/j.1469-7793.2001.0209k.x
Silva, J. M., and Lewis, D. L. (2002). Nitric oxide enhances Ca(2+)-dependent K(+) channel activity in rat carotid body cells. Pflugers Arch. 443, 671–675. doi: 10.1007/s00424-001-0745-1
Summers, B. A., Overholt, J. L., and Prabhakar, N. R. (1999). Nitric oxide inhibits L-type Ca2+ current in glomus cells of the rabbit carotid body via a cGMP-independent mechanism. J. Neurophysiol. 81, 1449–1457. doi: 10.1152/jn.1999.81.4.1449
Tamura, M., Hazeki, O., Nioka, S., and Chance, B. (1989). In vivo study of tissue oxygen metabolism using optical and nuclear magnetic resonance spectroscopies. Annu. Rev. Physiol. 51, 813–834. doi: 10.1146/annurev.ph.51.030189.004121
Tatsumi, K., Pickett, C. K., and Weil, J. V. (1991). Attenuated carotid body hypoxic sensitivity after prolonged hypoxic exposure. J. Appl. Physiol. 70, 748–755. doi: 10.1152/jappl.1991.70.2.748
Thompson, E. L., Ray, C. J., Holmes, A. P., Pye, R. L., Wyatt, C. N., Coney, A. M., et al. (2016). Adrenaline release evokes hyperpnoea and an increase in ventilatory CO2 sensitivity during hypoglycaemia: a role for the carotid body. J. Physiol. 594, 4439–4452. doi: 10.1113/jp272191
Turner, P. J., and Buckler, K. J. (2013). Oxygen and mitochondrial inhibitors modulate both monomeric and heteromeric TASK-1 and TASK-3 channels in mouse carotid body type-1 cells. J. Physiol. 591(Pt 23), 5977–5998. doi: 10.1113/jphysiol.2013.262022
Urena, J., Fernandez-Chacon, R., Benot, A. R., Alvarez, de Toledo, G. A., and Lopez-Barneo, J. (1994). Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc. Natl. Acad. Sci. U.S.A. 91, 10208–10211. doi: 10.1073/pnas.91.21.10208
Valdes, V., Mosqueira, M., Rey, S., Del Rio, R., and Iturriaga, R. (2003). Inhibitory effects of NO on carotid body: contribution of neural and endothelial nitric oxide synthase isoforms. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L57–L68. doi: 10.1152/ajplung.00494.2001
Varas, R., Wyatt, C. N., and Buckler, K. J. (2007). Modulation of TASK-like background potassium channels in rat arterial chemoreceptor cells by intracellular ATP and other nucleotides. J. Physiol. 583(Pt 2), 521–536. doi: 10.1113/jphysiol.2007.135657
Wang, J. J., Hogan, J. O., and Kim, D. (2017a). Voltage- and receptor-mediated activation of a non-selective cation channel in rat carotid body glomus cells. Respir. Physiol. Neurobiol. 237, 13–21. doi: 10.1016/j.resp.2016.12.005
Wang, J. J., Hogan, J. O., Wang, R., White, C., and Kim, D. (2017b). Role of cystathionine-gamma-lyase in hypoxia-induced changes in TASK activity, intracellular Ca2+ and ventilation in mice. Respir. Physiol. Neurobiol. 246, 98–106. doi: 10.1016/j.resp.2017.08.009
Williams, S. E., Brazier, S. P., Baban, N., Telezhkin, V., Muller, C. T., Riccardi, D., et al. (2008). A structural motif in the C-terminal tail of slo1 confers carbon monoxide sensitivity to human BK(Ca) channels. Pflugers Arch. Eur. J. Physiol. 456, 561–572. doi: 10.1007/s00424-007-0439-4
Williams, S. E., Wootton, P., Mason, H. S., Bould, J., Iles, D. E., Riccardi, D., et al. (2004). Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science 306, 2093–2097. doi: 10.1126/science.1105010
Wilson, D. F., Mokashi, A., Chugh, D., Vinogradov, S., Osanai, S., and Lahiri, S. (1994). The primary oxygen sensor of the cat carotid body is cytochrome a3 of the mitochondrial respiratory chain. FEBS Lett. 351, 370–374. doi: 10.1016/0014-5793(94)00887-6
Wilson, D. F., Rumsey, W. L., Green, T. J., and Vanderkooi, J. M. (1988). The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J. Biol. Chem. 263, 2712–2718.
Wyatt, C. N., and Buckler, K. J. (2004). The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type I cells. J. Physiol. 556(Pt 1), 175–191. doi: 10.1113/jphysiol.2003.058131
Wyatt, C. N., Mustard, K. J., Pearson, S. A., Dallas, M. L., Atkinson, L., Kumar, P., et al. (2007). AMP-activated protein kinase mediates carotid body excitation by hypoxia. J. Biol. Chem. 282, 8092–8098. doi: 10.1074/jbc.M608742200
Yamamoto, Y., Konig, P., Henrich, M., Dedio, J., and Kummer, W. (2006). Hypoxia induces production of nitric oxide and reactive oxygen species in glomus cells of rat carotid body. Cell Tissue Res. 325, 3–11. doi: 10.1007/s00441-006-0178-4
Yamamoto, Y., and Taniguchi, K. (2006). Expression of tandem P domain K+ channel, TREK-1, in the rat carotid body. J. Histochem. Cytochem. 54, 467–472. doi: 10.1369/jhc.5A6755.2005
Yuan, G., Vasavda, C., Peng, Y. J., Makarenko, V. V., Raghuraman, G., Nanduri, J., et al. (2015). Protein kinase G-regulated production of H2S governs oxygen sensing. Sci. Signal. 8:ra37. doi: 10.1126/scisignal.2005846
Keywords: carotid body, hypoxia, mitochondria, nitric oxide, arterial chemoreceptor, O2 sensor, metabolism, mitochondrial inhibitors
Citation: Holmes AP, Ray CJ, Coney AM and Kumar P (2018) Is Carotid Body Physiological O2 Sensitivity Determined by a Unique Mitochondrial Phenotype? Front. Physiol. 9:562. doi: 10.3389/fphys.2018.00562
Received: 30 January 2018; Accepted: 30 April 2018;
Published: 16 May 2018.
Edited by:
Rodrigo Iturriaga,Pontificia Universidad Católica de Chile, ChileReviewed by:
Gavin Clive Higgins, Monash University, AustraliaCopyright © 2018 Holmes, Ray, Coney and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prem Kumar, cC5rdW1hckBiaGFtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.