 Mathew J. Platt
Mathew J. Platt Jason S. Huber
Jason S. Huber Nadya Romanova
Nadya Romanova Keith R. Brunt
Keith R. Brunt Jeremy A. Simpson
Jeremy A. Simpson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 15 May 2018
Sec. Integrative Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.00472
A growing proportion of heart failure (HF) patients present with impairments in both ventricles. Experimental pressure-overload (i.e., transverse aortic constriction, TAC) induces left ventricle (LV) hypertrophy and failure, as well as right ventricle (RV) dysfunction. However, little is known about the coordinated progression of biventricular dysfunction that occurs in TAC. Here we investigated the time course of systolic and diastolic function in both the LV and RV concurrently to improve our understanding of the chronology of events in TAC. Hemodynamic, histological, and morphometric assessments were obtained from the LV and RV at 2, 4, 9, and 18 weeks post-surgery.
Results: Systolic pressures peaked in both ventricles at 4 weeks, thereafter steadily declining in the LV, while remaining elevated in the RV. The LV and RV followed different structural and functional timelines, suggesting the patterns in one ventricle are independent from the opposing ventricle. RV hypertrophy/fibrosis and pulmonary arterial remodeling confirmed a progressive right-sided pathology. We further identified both compensation and decompensation in the LV with persistent concentric hypertrophy in both phases. Finally, diastolic impairments in both ventricles manifested as an intricate progression of multiple parameters that were not in agreement until overt systolic failure was evident.
Conclusion: We establish pulmonary hypertension was secondary to LV dysfunction, confirming TAC is a model of type II pulmonary hypertension. This study also challenges some common assumptions in experimental HF (e.g., the relationship between fibrosis and filling pressure) while addressing a knowledge gap with respect to temporality of RV remodeling in pressure-overload.
Heart failure (HF), a chronic condition characterized by the inability of the heart to meet the metabolic demands of the body, is an increasing epidemic and a leading cause of mortality in the developed world (Benjamin et al., 2017). As the general population continues to age, the prevalence of HF will increase (Heidenreich et al., 2011). HF can develop acutely (e.g., infarction, broken heart syndrome, drug overdose) or over years as a result of chronic stress (e.g., hypertension, atherosclerosis, diabetes, cardiomyopathy). While all HF is progressive, each cause has defining characteristics that manifest in their own time-frame for reasons that are not always expected or explainable. Unfortunately, the majority of clinical knowledge on this progression is either retrospective or derived from late-stage patients only after years of remodeling precipitate symptoms. Alternatively, animal models are used to investigate the chronology of cardiac events to a particular stress over time. Temporal studies in animal models provide insight into the time course of remodeling patterns (Mirsky et al., 1983; Inoko et al., 1994; Klotz et al., 2006), and are valuable in the search for disease mechanisms and treatment targets (Foster et al., 2017).
Transverse aortic constriction (TAC) was developed to investigate the left ventricle (LV) response to pressure-overload (Rockman et al., 1991) and is suggested as a model of pulmonary hypertension (Chen et al., 2012; Mohammed et al., 2012; van Nierop et al., 2013). From a clinical standpoint, there is a growing understanding that poor right ventricle (RV) function is an independent predictor of HF patient outcomes (Ghio et al., 2001; Karaye et al., 2010; Meyer et al., 2010). At the same time, an increasing proportion of patients with LV dysfunction are also being diagnosed with concurrent RV dysfunction (Zakeri and Mohammed, 2015; Rosenkranz et al., 2016). Experimental models like TAC recapitulate this ‘biventricular phenotype’ and are therefore of growing importance to pre-clinical investigations. In this study, we address a knowledge gap in the TAC model with respect to the chronology of hemodynamic, morphological and kinetic changes in the RV as compared to the LV. We hypothesize that pulmonary hypertension and the associated RV pathology will occur secondary to LV dysfunction (i.e., type II pulmonary hypertension), and that the patterns of remodeling will be distinct from those observed in the LV.
Diastolic dysfunction is growing concern requiring more research, given that it is more prevalent than systolic dysfunction in HF patients (Hogg et al., 2004), the general population (Fischer et al., 2003; Wan et al., 2014), and particularly in women (Ferreira et al., 2015). There are numerous parameters used to assess cardiac diastole, however, no studies have compared these different indices for their relative capacity to grade diastolic dysfunction in a model of HF over time. This makes comparisons between experimental models using different parameters difficult to interpret. In the present study, we asked the question: do invasive indices of diastolic function (i.e., dP/dtmin, filling pressures, Tau) agree over time in TAC? We hypothesize that different indices of diastolic function will progress independently given the complexity of cardiac diastole and, likely, the different aspects captured by each parameter.
The salient findings in this study are: (1) Pulmonary hypertension was secondary to the increase in LV filling pressure and dysfunction, and RV and pulmonary vasculature remodeling was temporally distinct from the LV. (2) Determining the severity of diastolic dysfunction in either ventricle over time was complex, given that diastolic parameters were not in agreement until overt systolic dysfunction was apparent. And (3) the time course of structural and kinetic changes in both ventricles was interesting: interstitial fibrosis did not correlate with filling pressures in either ventricle, LV pressures did not predict RV pressures, and hypertrophy in the LV and RV had both early and late stage phenotypes that were not predictable from prevailing hemodynamics. Together, this study improves our understanding of the biventricular response to pressure-overload and firmly establishes TAC as a model of type II pulmonary hypertension.
Briefly, 9-week-old male (32–40 g) CD-1 mice (Charles River Laboratory International Inc.), were anesthetized with an isoflurane/oxygen mix (2%:100%), intubated and connected to a ventilator (Harvard Apparatus). Mice were ventilated at 200 breaths per minute at 300 μL per breath. TAC was done as previously described (Allwood et al., 2014; Foster et al., 2017). Briefly, the 2nd and 3rd ribs were separated from their cartilaginous connections with the sternum to expose the aortic arch. The transverse aorta was isolated and constricted to 26-gauge blunted needle with 7-0 silk thread. Sham surgery was similar to TAC, absent only the placement of the 7-0 thread. All mice were housed on a 12-h light/dark cycle and with food and water ad libitum. This study was approved by the Animal Care Committee at the University of Guelph and all experiments were carried out in accordance with the guidelines from the Canadian Council on Animal Care.
Mice were anesthetized with an isoflurane/oxygen mix (2%/100%). Echocardiography was performed using the Vevo2100 system (VisualSonics Inc., Toronto, ON, Canada) with the 40 MHz MS550D ultrasound transducer. Mice were kept at 37°C throughout data collection using a TH-5 rectal probe thermometer (Physiotemp Instruments LLC, Clifton, NJ, United States). Acquired M-mode images were analyzed with the LV-trace function from the cardiac package (VisualSonics Inc., Toronto, ON, Canada), and data was obtained over at least 5 heart beats as previously described (Platt et al., 2017). All measurements were made between noon and 5 pm.
Mice were sacrificed at 2, 4, 9, or 18 weeks after TAC or sham surgery. Again, an isoflurane/oxygen mix (2%:100%) was used, and again mice were kept at 37°C while a 1.2F catheter (FTS-1211B-0018; Scisense Inc., London, ON, Canada) was inserted into the RV via the right jugular and into the LV via the right carotid. Hemodynamic signals were digitized at a sampling rate of 2 kHz and recorded using iWorx® analytic software (Labscribe2, Dover, NH, United States). Data sets were analyzed with the removal of respiration artifacts. The presence of pulmonary hypertension was determined using the 99th percentile upper reference limit (i.e., greater than three standard deviations above the mean right ventricular systolic pressure in sham mice). Thus, 32.5 mmHg was deemed the physiological threshold for pulmonary hypertension in our model. Animals designated for morphometric analysis had organs removed, cleaned and weighed. Animals designated for histological analysis were exsanguinated and 10 mL of 1x PBS, 10 mL of 0.5 mol L-1 KCl, and 10 mL of 10% buffered formalin (VWR, Mississauga, ON, Canada) were perfused through the right carotid artery. Tissues were harvested, stored in 10% buffered formalin for 24 h, and then transferred to 70% ethanol until tissue processing.
Cross sectional slices (∼5 μm) were obtained from the mid papillary region of the heart. Paraffin embedded sections were stained with either Picro-Sirius Red (PSR; 500 ml of saturated picric acid solution and 0.5 g of Direct Red 80 from Sigma Aldrich), to visualize interstitial fibrosis (staining cytoplasm yellow and collagen red), or Wheat Germ (Sigma Aldrich) to assess cardiomyocyte cross-sectional area (CSA) (staining cell borders green). Images were acquired using an Olympus FSX 100 light microscope and analyzed using Cell Sense software (Olympus, Tokyo, Japan). CSA was quantified only from cardiomyocytes with centralized nuclei. For analysis of lung tissue, fresh Verhoff Van Geison stain was prepared with reagents obtained from VWR (Mississauga, ON, Canada). This stains elastic fibers and nuclei purple/black, collagen fibers red and cytoplasm yellow. The medial thickness of muscularized arteries was expressed as a percentage of external medial diameter. Arteries with internal diameters < 35 μm were selected for quantifying the ratio of muscularized to non-muscularized arteries (Sheikh et al., 2014).
All results are expressed as means ± SE unless otherwise indicated. Statistical analysis was performed using Prism (Graphpad Software Inc.). Statistical significance between sham and TAC mice at individual time-points was determined using the two-tailed Students t-test. A one-way ANOVA (Tukeys post hoc) was used to determine differences within one group over 18 weeks as all measures were terminal. Differences between sequential time-points were subsequently determined using a two-tailed Students t-test. Regression analyses were done with Prism 6.0. The threshold for significance was p < 0.05 in all cases unless otherwise specified.
To account for changes in function as the mice aged, TAC’s were compared to age-matched surgical shams at all time-points. Representative tracings and a summary of invasive hemodynamic function over 18 weeks of TAC are presented in Figure 1 and Table 1 (for sham data, see Table 2).
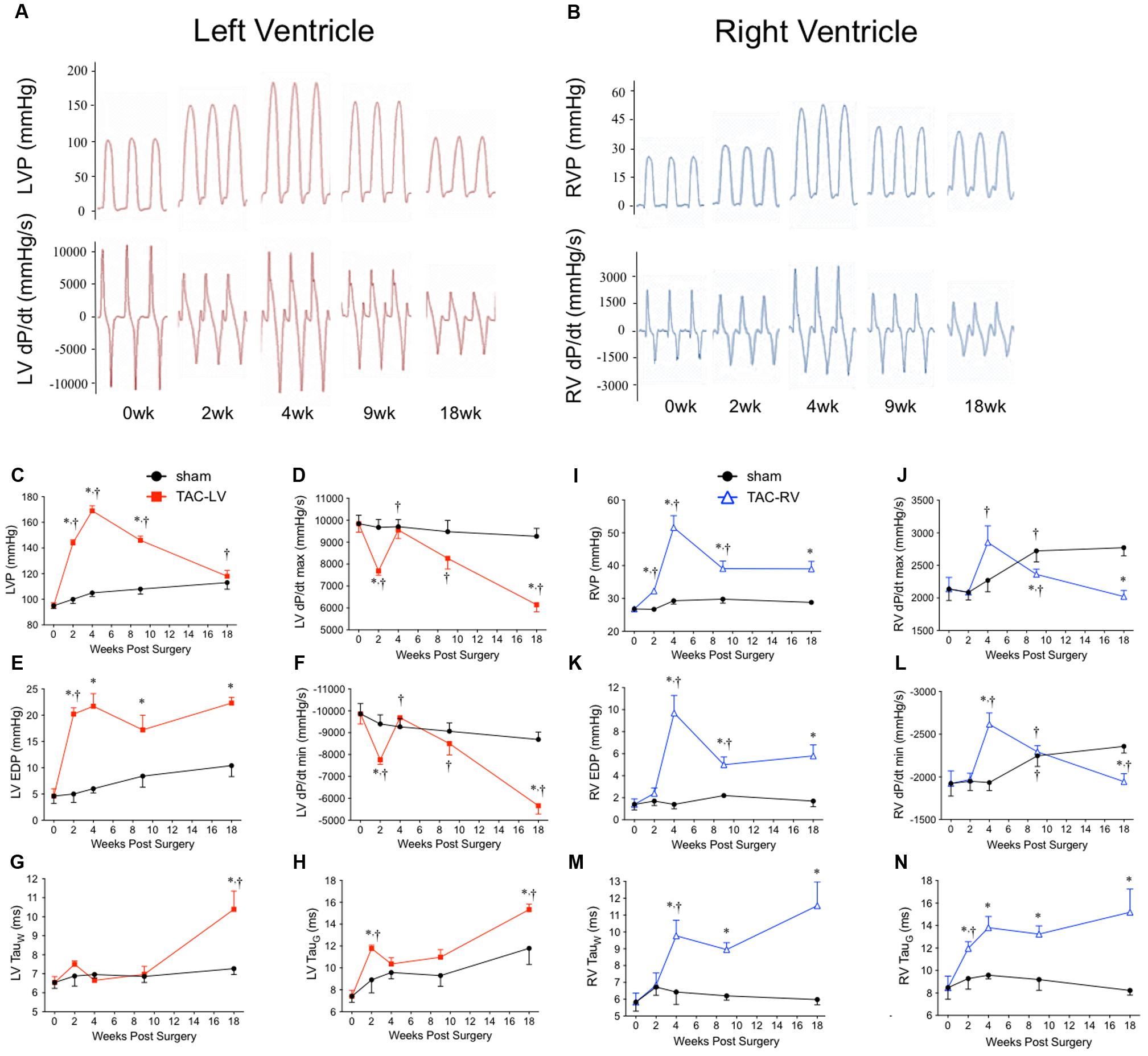
FIGURE 1. Hemodynamic characterization of the transverse aortic constriction (TAC) model in male CD1 mice over 18 weeks. Left panel (red) = left ventricular (LV) function. Right panel (blue) = right ventricular (RV) function. Representative hemodynamic tracings over 18 weeks from (A) the LV and (B) the RV. (C) Peak LV pressure (LVP), (D) LV dP/dtmax, (E) left ventricular end diastolic pressure (LV EDP), (F) LV dP/dtmin, (G) Tau (Weiss), and (H) Tau (Glantz) profile the temporal changes in LV hemodynamics over 18 weeks of TAC. (I) Peak RV pressure (RVP), (J) RV dP/dtmax, (K) right ventricular end diastolic pressure (RV EDP), (L) RV dP/dtmin, (M) Tau (Weiss), and (N) Tau (Glantz) profile the temporal changes in RV hemodynamics over 18 weeks of TAC. ∗p < 0.05 vs. age-matched sham. †p < 0.05 vs. previous time point.
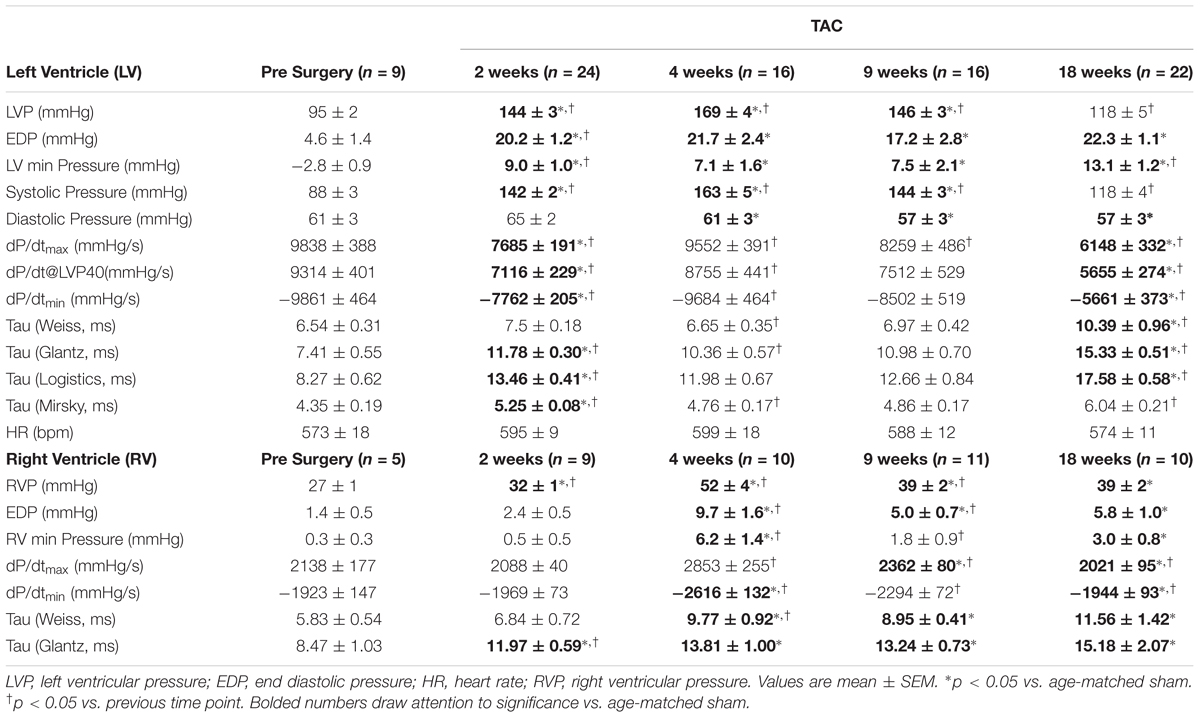
TABLE 1. Invasive hemodynamics from the left and right ventricles in TAC.
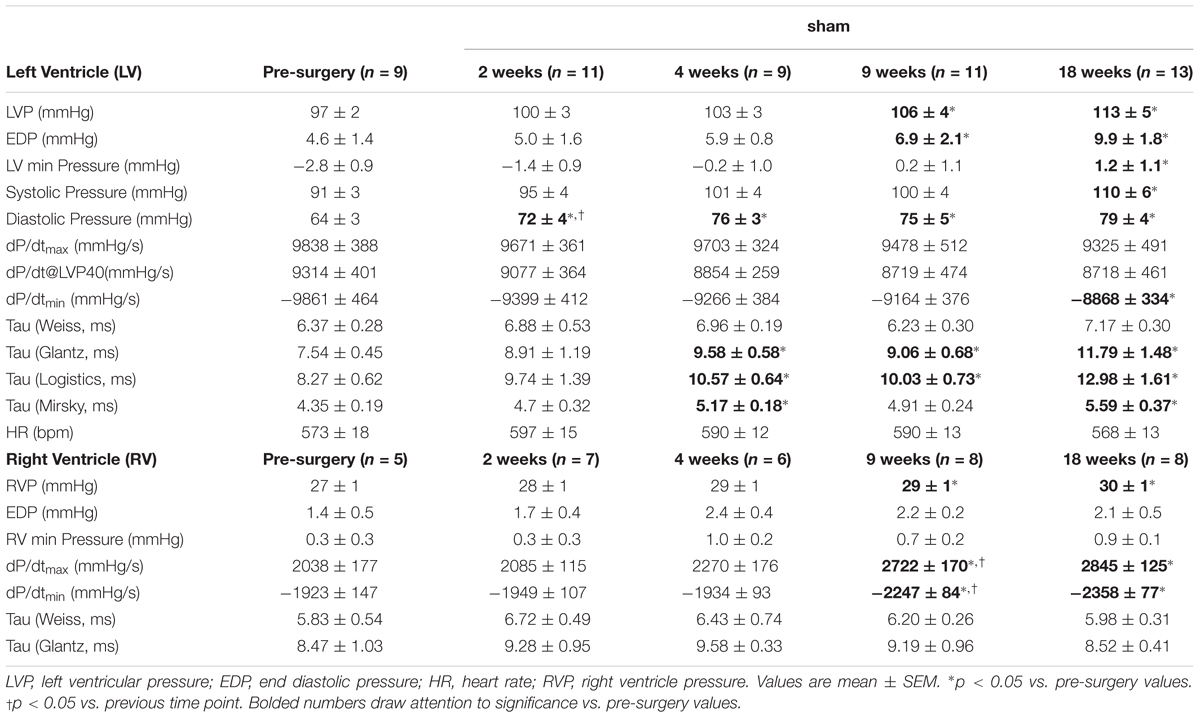
TABLE 2. Invasive hemodynamics from the left and right ventricles in sham.
LV pressure (LVP; Figure 1C) peaked at 4 weeks before progressively declining toward sham values by 18 weeks. RV pressure (RVP; Figure 1I) peaked at 4 weeks, but unlike the LVP, plateaued from 9 to 18 weeks. Of importance for the chronology of this model, pulmonary hypertension did not develop until 4 weeks. Only 11% (1/9) of TAC mice at 2 weeks had pulmonary hypertension. By 4 weeks, incidence of pulmonary hypertension rose to 90% (9/10) and was 75% thereafter (16/21).
To evaluate systolic function, the first positive derivative of ventricular pressure (dP/dtmax) was used as an index of contractility (Figures 1D,J). LV dP/dtmax initially decreased at 2 weeks before rebounding by 4 weeks and progressively declining thereafter. In the RV, dP/dtmax was unchanged until increasing at 4 weeks before also declining thereafter.
To assess diastolic dysfunction, we used three different indices: the first negative derivative of ventricular pressure (dP/dtmin), end diastolic pressure (EDP) and two methods to calculate the relaxation time constant (i.e., Tau Weiss; TauW and Tau Glantz; TauG) (Figures 1E–H,K–N and Table 1). LV EDP increased ∼400% by 2 weeks and plateaued thereafter (Figure 1E). LV dP/dtmin followed the same trend as LV dP/dtmax, initially reduced at 2 weeks, normalizing by 4 weeks and then progressively declining out to 18 weeks (Figure 1F). LV TauW increased only by 18 weeks (Figure 1G) while TauG was elevated both at 2 and 18 weeks (Figure 1H). In the RV, EDP initially increased by 4 weeks before partially returning toward sham levels at an elevated plateau between 9 and 18 weeks (Figure 1K). RV dP/dtmin followed the same pattern as RV dP/dtmax, increasing at 4 weeks before declining out to 18 weeks (Figure 1L). RV TauG increased at 2 weeks (Figure 1N) while TauW increased at 4 weeks (Figure 1M); both stayed elevated from sham thereafter.
In sham mice, LV and RV pressure, as well as systolic and diastolic function also changed over 18 weeks. LVP increased by ∼20%, while RVP increased by ∼10% (Figures 1C,I). Interestingly, while LV dP/dtmax was stable (changing by <10% over 18 weeks), RV dP/dtmax increased ∼30% between 2 and 9 weeks (Figures 1D,J). In the LV, most measures of diastolic function declined by 18 weeks (EDP and TauG increased by ∼60%, while dP/dtmin declined by 10%), yet TauW remained unchanged (Table 2). Alternatively, in the RV, measures of diastolic function either improved (dP/dtmin increased by ∼40%) or were unchanged (EDP, TauG and TauW were stable; Table 2).
Elevations in EDP are sometimes reconciled as the product of fibrotic remodeling. To investigate this relationship over time in our model, histological sections of the ventricles were stained with PSR (Figure 2A). In the LV, interstitial fibrosis progressively increased over 18 weeks, initially increasing ∼8 fold by 2 weeks with a further 50% increase out to 18 weeks (Figure 2B). In the RV, interstitial fibrosis increased linearly ∼10 fold by 4 weeks, stabilizing thereafter (Figure 2B).
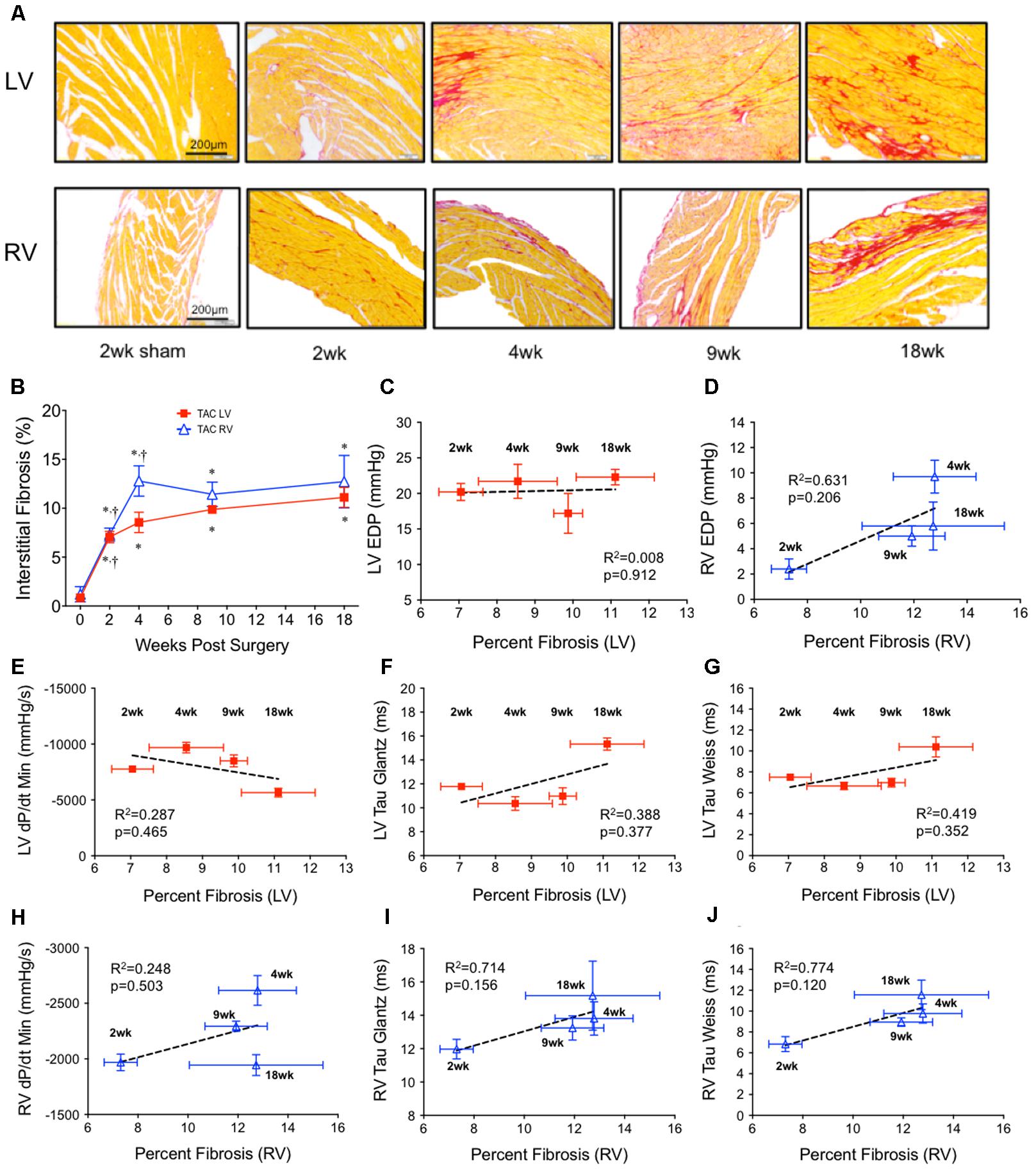
FIGURE 2. Ventricular interstitial fibrosis in both ventricles over 18 weeks of TAC. (A) Representative images stained with Picrosirius Red (top panel; left ventricle; LV, bottom panel; right ventricle; RV). (B) Average interstitial fibrosis over time in both ventricles (n = 6 for each point). Sham was unchanged over 18 weeks and was represented by the point at x = 0. ∗p < 0.05 vs. age-matched sham. †p < 0.05 vs. previous time point. Linear regression was run on correlations between average fibrosis and all invasive indicies of diastolic function: (C) LV end diastolic pressure (LV EDP), (D) RV end diastolic pressure (RV EDP), (E) LV dP/dtmin, (F) LV Tau Glantz, (G) LV Tau Weiss, (H) RV dP/dtmin, (I) RV Tau Glantz, (J) RV Tau Weiss.
Regression did not find a significant non-zero relationship between EDP and fibrosis over time in either the LV (Figure 2C) or RV (Figure 2D). Of an interesting note was that interstitial fibrosis increased ∼500% in the RV by 2 weeks without a concurrent change in RV EDP.
To investigate if fibrosis correlated with any other parameter of diastolic function, we compared the relationship between fibrosis and dP/dtmin, TauG and TauW in both the LV (Figures 2E–G) and RV (Figures 2H–J). No parameter consistently correlated over time. At best, in the RV only, there was a dichotomy between early and late fibrosis and diastolic dysfunction.
To investigate the hypertrophic response following LV pressure-overload, whole hearts (ventricles, septum and atria) were weighed and normalized to tibial length (TL). Weights were normalized to TL as increasing BW skewed the HW/BW ratio in sham mice, while HW/TL in the same mice was stable across normal growth (Figure 3). Representative H&E (top panel) and PSR (bottom panels) cross-sectional images of the heart are presented in Figure 4A. Whole heart hypertrophy increased steadily (Figure 4B), culminating in significantly larger hearts by 18 weeks of TAC (Figure 4C). To evaluate chamber-specific hypertrophy, atrial, LV, RV and septal weights were normalized to TL. The atrial/TL and LV/TL ratio increased steadily across all time points(Figures 4D,E). RV/TL did not increase until 4 weeks of TAC (Figure 4F) while the septum/TL ratio increased to 4 weeks where it plateaued and increased proportional to sham out to 18 weeks (Figure 4G). Expanded morphometrics are presented in Table 3 (TAC) and Table 4 (sham).
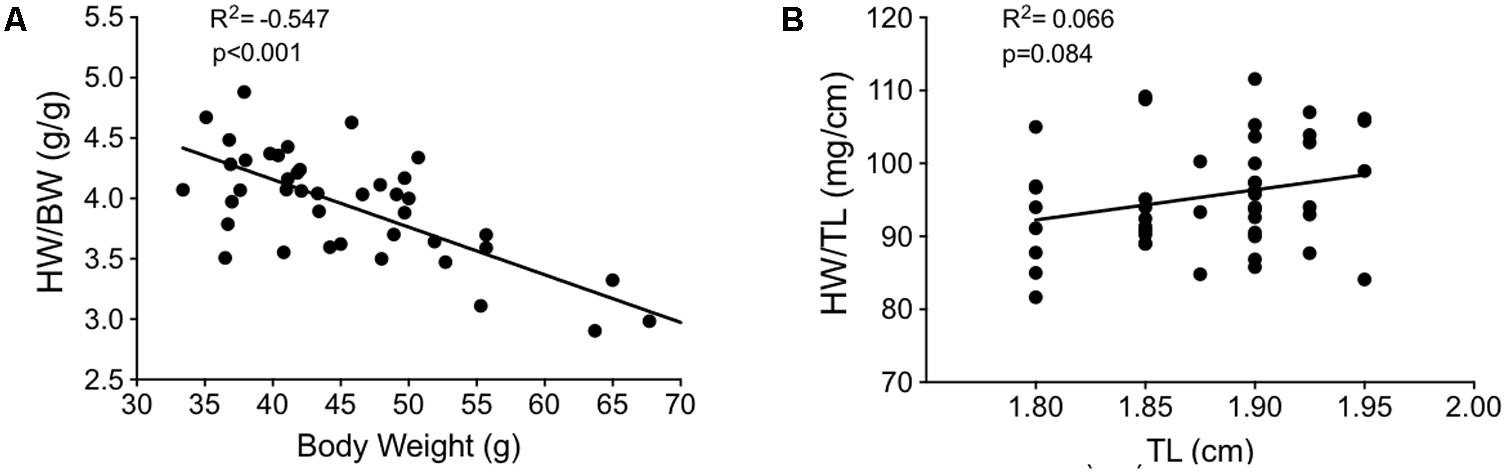
FIGURE 3. Correlation plots between measures of body growth and cardiac growth in sham mice between 9 and 27 weeks of age (n = 40). (A) Heart weight/body weight (HW/BW) plotted against body weight. (B) Heart weight/tibial length (HW/TL) plotted against tibial length. The lack of slope in (B) indicates HW/TL is less impacted by growth (i.e., changes in the denominator) than HW/BW. This suggests that normalizing cardiac weight to TL is a better method for determining cardiac hypertrophy between groups of varying weight/age.
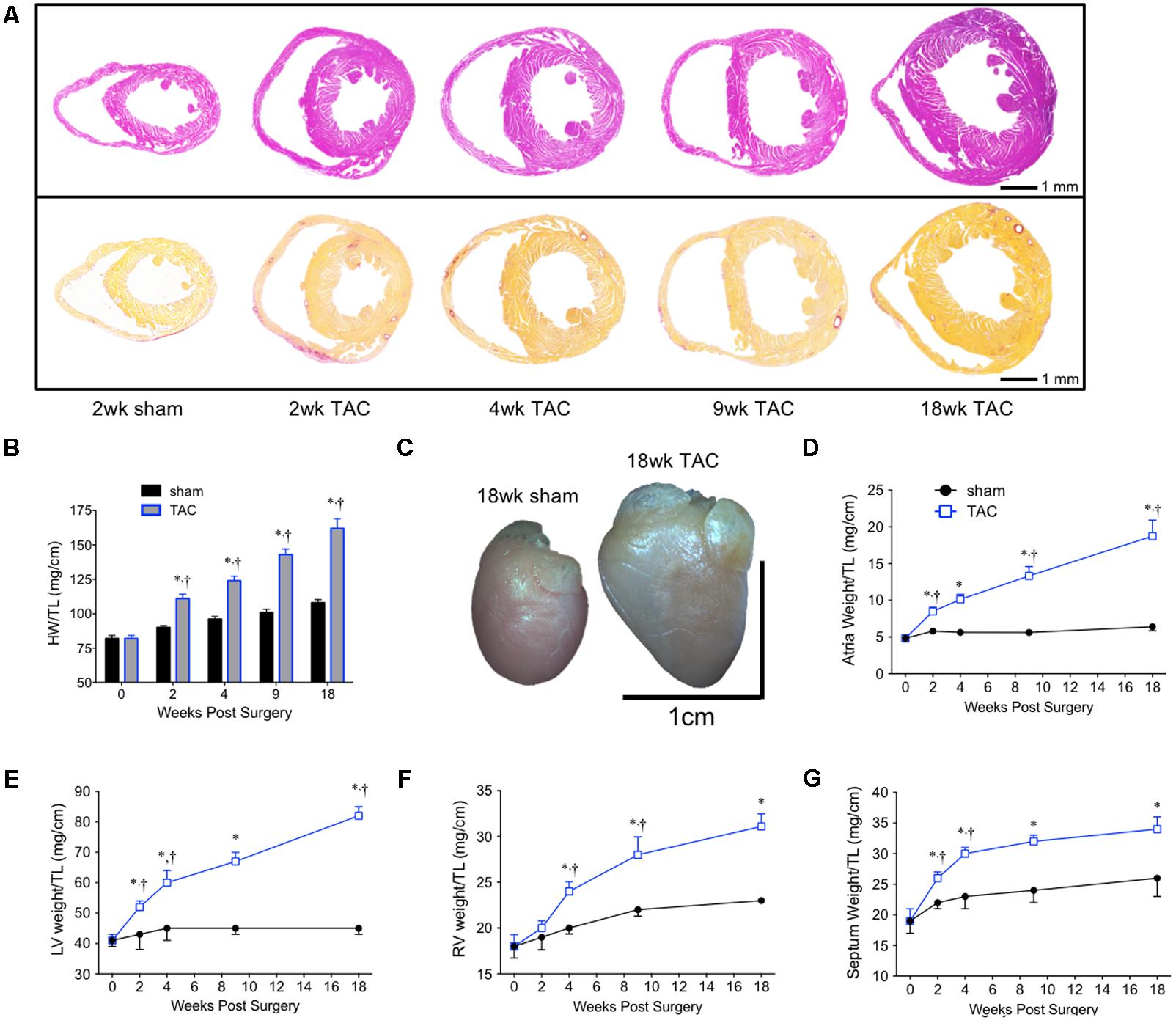
FIGURE 4. Summary of cardiac morphometrics following 18 weeks of TAC. (A) Representative cross-sectional images of the heart stained with H&E (top row) and PSR (bottom row). (B) Whole heart weight normalized to tibial length (HW/TL). (C) An 18 week sham (left) and TAC (right) heart. Normalized chamber-specific growth curves from the (D) atria, (E) left ventricle (LV), (F) right ventricle (RV), and (G) septum are presented. ∗p < 0.05 vs. age-matched sham. †p < 0.05 vs. previous time point.
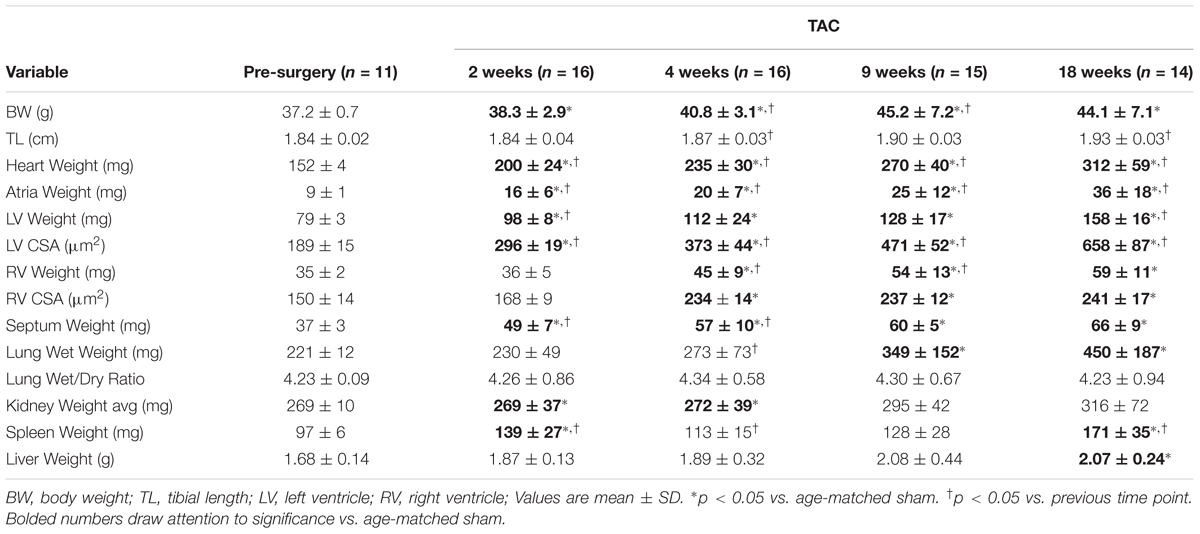
TABLE 3. Morphometric characteristics of TAC mice.
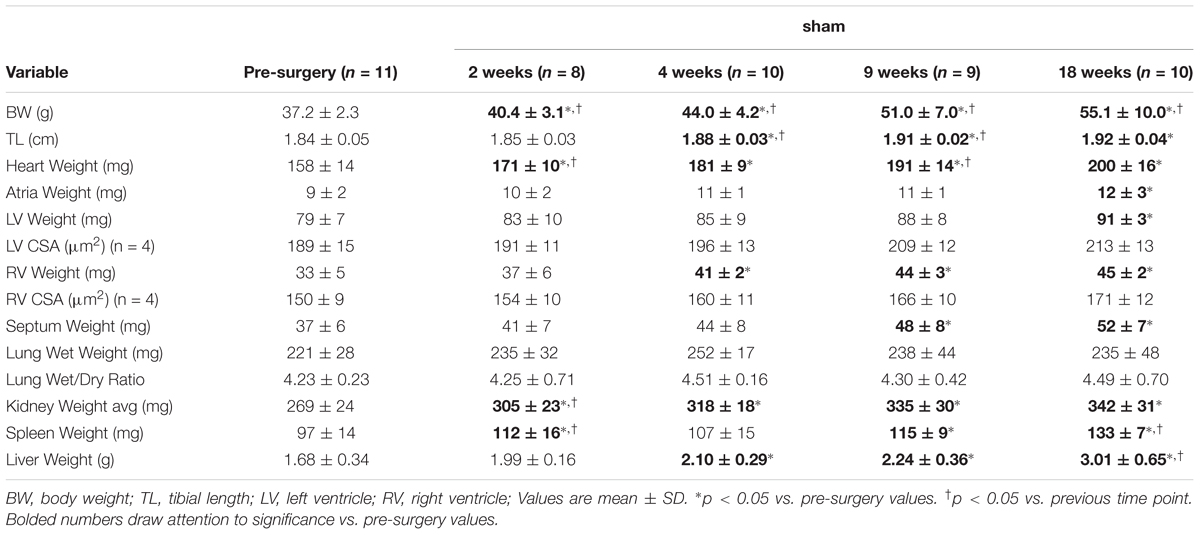
TABLE 4. Morphometric characteristics of sham mice.
To quantify concentric hypertrophy, cardiomyocyte CSA was assessed in both the LV and RV (Figure 5). LV CSA increased progressively over 18 weeks, while RV CSA only increased between 2 and 4 weeks, remaining stable thereafter (Figure 5B). The linear increase in LV weight and LV CSA over 18 weeks suggested the rate of hypertrophy was constant and likely (predominantly) concentric (Figure 5C). In contrast, the pattern in the RV had two parts. Initially (0–4 weeks), RV CSA and weight increased linearly (Figure 5D). Between 4 and 18 weeks, however, RV weight continued to increase while RV CSA plateaued. While this increased weight could be attributed to extracellular proteins or cardiomyocyte hyperplasia, RV fibrosis was static between 4 and 18 weeks (Figure 2B) and, adult cardiomyocyte hyperplasia is accepted to be low to negligible. This suggested the weight was coming from an increase in cardiomyocyte cell length (i.e., eccentric hypertrophy).
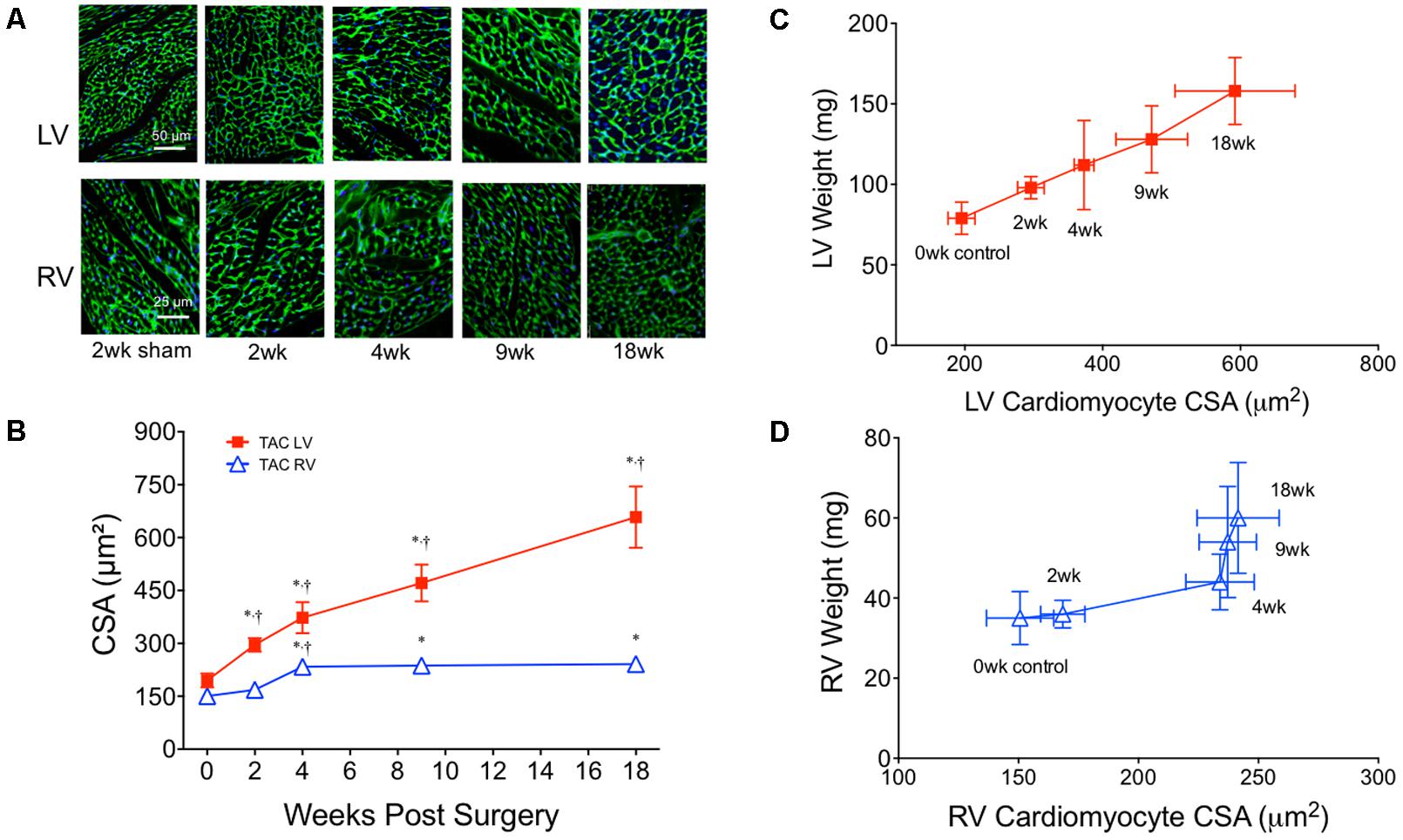
FIGURE 5. Cardiomyocyte cross-sectional area (CSA) from both the left (LV) and right (RV) ventricles over 18 weeks of TAC. (A) Representative images from the LV (top row) and RV (bottom row) stained with wheat germ agglutinin-3. (B) Averaged cardiomyocyte CSA over 18 weeks of TAC (n = 6 for each point). Complete sham CSA data is presented in Table 4. Correlation plots between ventricle weight and CSA in the (C) LV and (D) RV. ∗p < 0.01 vs. age-matched sham. †p < 0.01 vs. previous time point.
Representative M-mode images from the LV of sham and TAC mice are profiled in Figure 6A. LV dilation (increased LV end diastolic dimensions) began at 4 weeks and progressed to 18 weeks (Figure 6B). Cardiac output decreased by 2 weeks and remained stable until 9 weeks before decreasing further by 18 weeks (Figure 6C). Ejection fraction, a common variable used to clinically grade LV function, followed the same pattern as cardiac output with the secondary decline occurring between 9 and 18 weeks (Figure 6D). Complete echocardiographic data from sham and TAC mice is summarized in Table 5.
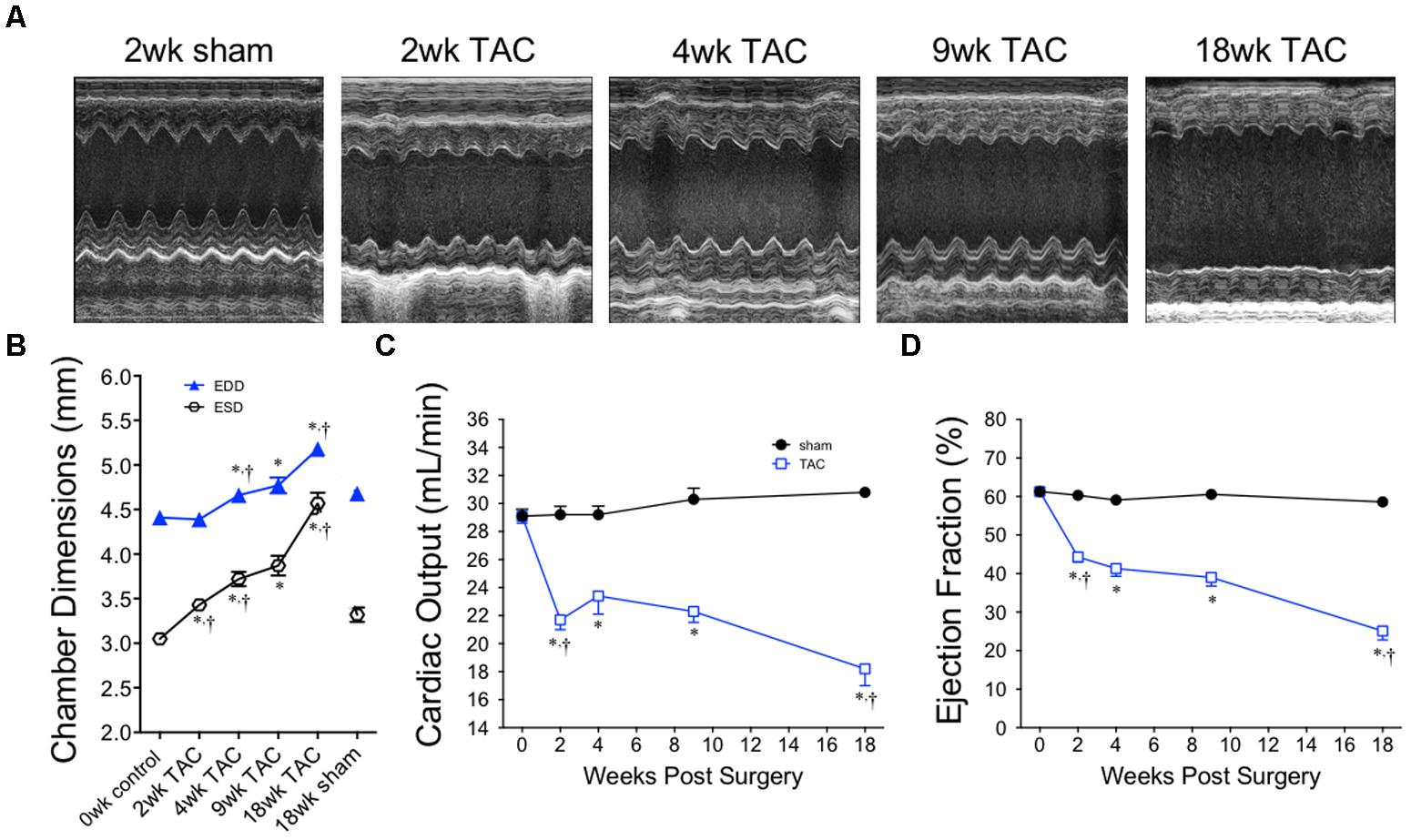
FIGURE 6. Chamber size and echocardiographic function over 18 weeks of TAC. (A) Representative M-mode images acquired from the mid-papillary region of the LV. (B) Left ventricular end diastolic (EDD) and end systolic (ESD) dimensions, (C) cardiac output and (D) ejection fraction demonstrate dilation and a decline in cardiac function, especially between the 9 and 18 week time-points. Heart rate was statistically unchanged between all time points. ∗p < 0.01 vs. age-matched sham. †p < 0.01 vs. previous time point.
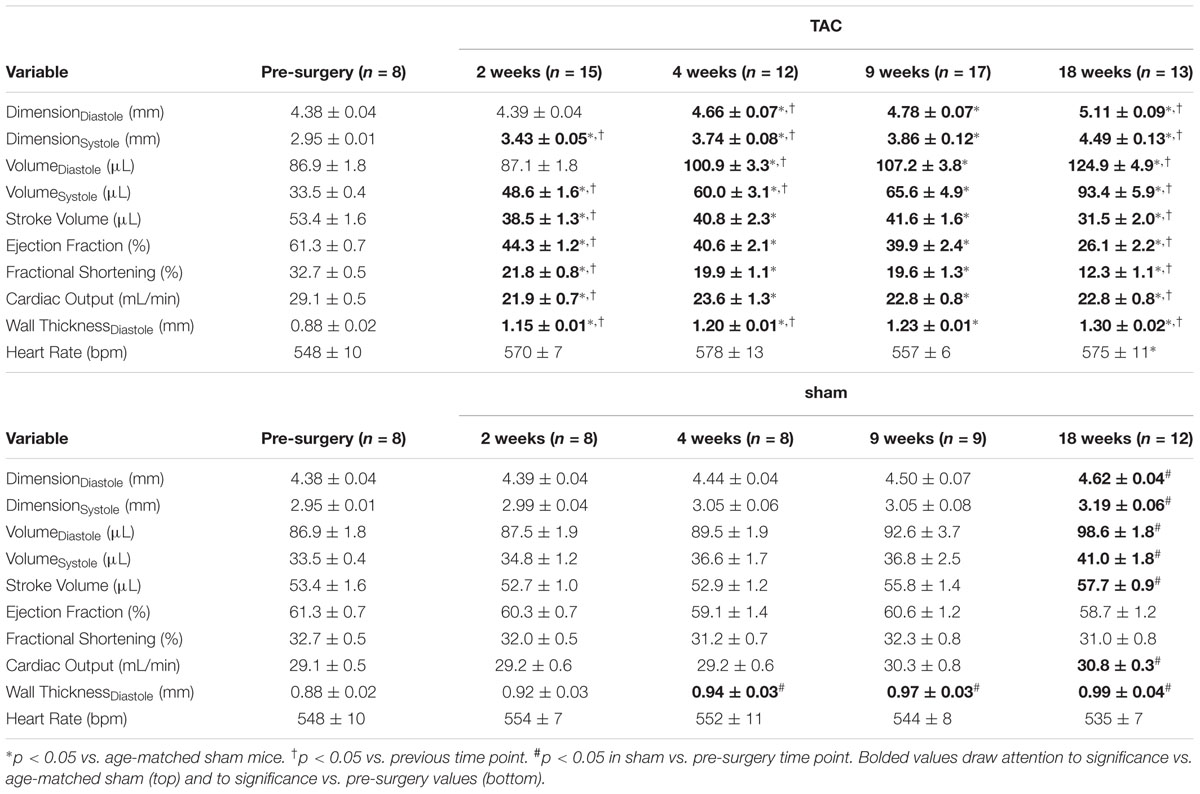
TABLE 5. Expanded echocardiographic function.
To confirm the increase in RVP was pathological, we looked for evidence of pulmonary vascular remodeling within the pulmonary circulation by histology (Figure 7A). We found increased large-vessel wall thickness by 2 weeks with a neomuscularization of the smaller vessels by 4 weeks; both continuously increased thereafter (Figures 7B,C). By 9 weeks, this was associated with an increased total lung weight without evidence of pulmonary edema (i.e., static lung wet/dry weight ratio; Table 2). Pulmonary vascular resistance, calculated using the formula PVR = (RVP–LV EDP)/cardiac output), was reduced at 2 weeks, before peaking at 4 weeks and remaining elevated thereafter (Figure 7D).
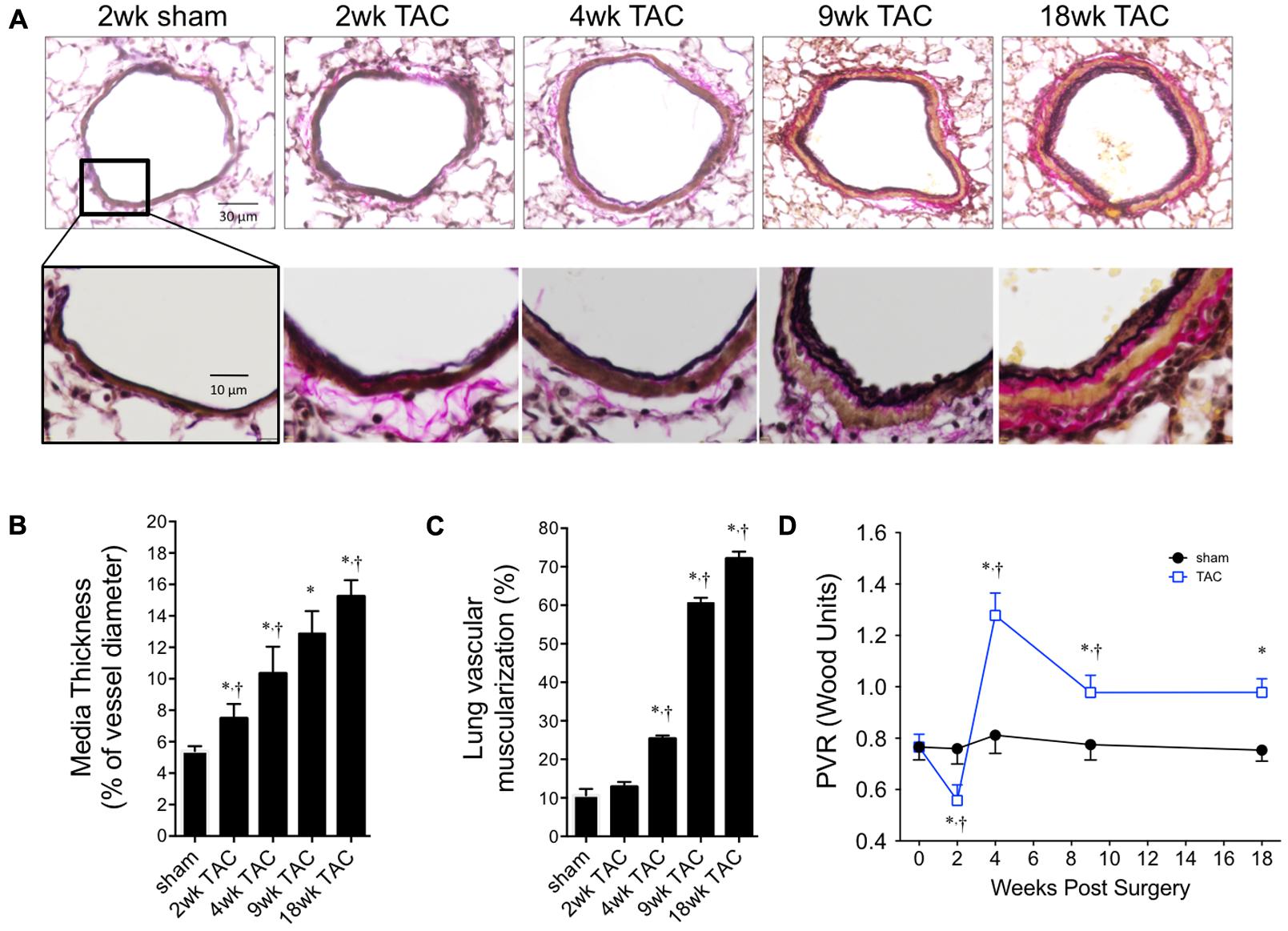
FIGURE 7. Pulmonary vascular remodeling over 18 weeks of TAC. (A) Representative cross-sectional images of whole pulmonary arteries (top panel) and vessel walls (bottom panel). (B) Thickness of vascular media was calculated as a percent of external diameter to normalize for varying vessel sizes. (C) Percent of muscularized small arterioles (<35 μm) in the lung. Sham values did not change over the 18 weeks study period and are represented as ‘sham’. (D) Pulmonary vascular resistance (PVR) was calculated from first principles; Resistance = ΔPressure/Flow. ΔPressure was the gradient between right ventricle systolic pressure and left ventricle end diastolic pressure. Flow was cardiac output. ∗p < 0.01 vs. age-matched sham. †p < 0.05 vs. previous time point.
This study is the first to report a longitudinal, biventricular evaluation of cardiac function and morphology in the murine pressure-overload model. Here we mapped the pathophysiological development of type II pulmonary hypertension, pulmonary vascular remodeling and RV dysfunction in TAC for the first time. There were two phases of function, compensatory and decompensatory. In the compensatory phase, both the LV and RV remodeled concentrically in line with increased pressure. During decompensation, the LV continued to hypertrophy concentrically despite declining pressure, while in the RV the data suggested a shift to eccentric hypertrophy. We also demonstrated that no one parameter was sufficient to explain diastolic function with each index (e.g., fibrosis, EDP, tau and dP/dtmin) following a different pattern over time. And finally, fibrosis did not correlate with diastolic pressure in either ventricle over time, calling into question how fibrosis per se is related to elevations in EDP.
TAC is commonly used to investigate LV hypertrophy as pressure drives proportional concentric cardiac growth (Grossman et al., 1975). The relationship between pressure and hypertrophy over time however, is more complex. In this model, we show early LV hypertrophy was concentric and proportional to pressure (0–4 weeks), but during decompensation (4–18 weeks), concentric hypertrophy persisted despite declining systolic pressures. While the former is well recognized in the literature, the latter (i.e., hypertrophy with declining pressure) is typically only reported at very late time points or in more severe models (Norton et al., 2002; Rothermel et al., 2005; Chen et al., 2012; Mohammed et al., 2012). Thus, pressure-independent hypertrophy is relative and a hallmark of decompensation. Importantly, we confirm the type of hypertrophy during decompensation as concentric. Cardiac hypertrophy is an independent risk factor for cardiovascular morbidity and mortality (Levy et al., 1990), and a primary target of emerging HF therapies (Frey et al., 2004). To our knowledge however, the molecular mechanism(s) by which cardiomyocytes undergo concentric hypertrophy through these two stages have not been directly compared and contrasted. The existence of two phases of concentric hypertrophy is relevant because anti-hypertrophic therapies are often initiated early in a model (e.g., Takimoto et al., 2005). This leaves the impact of therapies in the decompensated stage of cardiac remodeling largely unknown; therapies initiated in this later phase may not have the same outcome as those initiated early on. This is particularly important given the aged clinical population that largely presents after years of hypertrophic remodeling, only as their failing heart begins to precipitate symptoms. As these patients are approaching–or already experiencing–decompensation, comparatively assessing therapies between the compensatory and decompensatory phases is an important future focus of pressure-overload studies.
Both LV hypertrophy and eventual decompensation are ubiquitous following pressure-overload. The variability is in the degree of hypertrophy and the time-to-decompensation. The differences reported between models of pressure-overload are largely contingent on three main features. First, the degree of constriction (gauge of needle) impacts both the amount of hypertrophy and rate of dilation. Smaller gauges [i.e., 24-gauge (Pradhan et al., 2016), 25-gauge (van Nierop et al., 2013)] lead to mild concentric hypertrophy, no early dilation and mild dysfunction. Intermediate gauges [i.e., 27-gauge (Nakamura et al., 2001; Liao et al., 2002; van Nierop et al., 2013)] lead to more substantial short-term hypertrophy, a period of stable or recovered cardiac function, and then dilatation and decompensation within 4 to 9 weeks. Severe constrictions (28-gauge, Rothermel et al., 2005) develop concentric hypertrophy in tandem with rapid dilatation and mortality. Second, animal selection significantly affects the response to pressure-overload. Strain and gender differences contribute to the genetic variation (Barrick et al., 2007, 2009), while different ages and/or weights impact the relative severity of the same-sized constriction (e.g., 27-gauge induces a 20% increase in LV weight by 3 weeks in 6–8 weeks old mice (Rothermel et al., 2005) versus a 50% increase in 14–16 weeks old mice (Kapur et al., 2012)]. Third, and finally, the relative proximity of the band to the heart (i.e., ascending vs. transverse vs. abdominal aortic constriction) also impacts the magnitude of hypertrophy and rate of decompensation (Foster et al., 2017). These data indicate the need for direct intra-study comparisons of hypertrophy and its molecular mechanisms, and that inter-study comparisons be made with these factors in mind. Here, we report early concentric hypertrophy and recovered function (0–4 weeks) before dilatation and decompensation (18 weeks). These data suggest our model was moderate, and thus valuable to investigate the transitional phase between compensatory remodeling and decompensated HF.
Interstitial fibrosis correlates with a stiffening of the LV wall, which impairs ventricular relaxation and contributes (among other things) to increased passive filling pressures (EDP) (Jalil et al., 1989; Weber et al., 1993; Conrad et al., 1995; Yamamoto et al., 2002; Kass et al., 2004; Matsusaka et al., 2006). Increased EDP is part of the diagnosis of diastolic dysfunction (Mandinov et al., 2000; Paulus et al., 2007), thus any cause of increased EDP is a clinically important event. Here we show that over time, an increase in EDP was neither dependent on a change in interstitial fibrosis, nor guaranteed by an increase in interstitial fibrosis. In the LV, fibrosis increased from 7 to 11% between 2 and 18 weeks without commensurate increases in EDP. In the RV, fibrosis increased 5-fold by 2 weeks while EDP remained unchanged. Interestingly, other reports have described disagreement between the onset and severity of fibrosis and the relative diastolic pressures. Wu et al. (2011) reported a threefold increase in LV fibrosis in their model without any change in EDP. Hamdani et al. (2014) reported increased left ventricular interstitial fibrosis in a diabetic model in the absence of changes to EDP. A study by Perrino (2006), reported variable LV interstitial fibrosis with static LVEDP in three different TAC groups. And finally, treating with sildenafil to reduce fibrosis twofold in a TAC model did not alter EDP (Pradhan et al., 2016). These data suggest gross fibrosis does not explain changes in diastolic pressure in TAC. For example, collagen cross-linking is more predictive of LV stiffness and diminished diastolic function than total collagen content, both in experimental models (Badenhorst, 2003; López et al., 2012) and in humans (Ravassa et al., 2017). What remains to be established is a definitive approach to capturing ECM remodeling related to diastolic dysfunction, which will be critical in developing future therapies.
In shams, we report appreciable changes in cardiac structure and function over the 18 week study period. This was especially true in the RV, where between 2 and 9 weeks invasive measures of contractility and relaxation increased by ∼33%. In the same time frame, the change in LV function was <10%. This suggests the healthy RV may reach functional maturity in a different timeframe than the LV. Importantly, evidence of contractile dysfunction in the TAC RV was only significant by 18 weeks because of the increase in RV function in aged-matched shams. This demonstrates the importance of mapping structure and function in shams/controls, particularly in longitudinal studies where early time points may misrepresent the mature phenotype.
Type-II pulmonary hypertension is the most prevalent form of pulmonary hypertension (Rosenkranz et al., 2016). Reported in over 50% of HF patients (Hoeper et al., 2005, 2009; Lam et al., 2009), its presence increases 3-year mortality by 20–50% (positively correlated with severity of pulmonary hypertension) (Bogaard et al., 2009). While numerous models exist for investi gating the pathophysiology and treatment options for type-I pulmonary hypertension, the literature on experimental models of type-II pulmonary hypertension is comparatively sparse (Stenmark et al., 2009). In type-II pulmonary hypertension, the primary condition is LV dysfunction; increased LV filling pressures (EDP) precede the increase in pulmonary artery pressures (Adusumalli and Mazurek, 2017). This differs etiologi cally from type I pulmonary hypertension, which originates as a primary pathology of the pulmonary vasculature (Simonneau et al., 2009). Here, we corroborate previous findings demonstrating pulmonary hypertension and RV dysfunction are prominent features of the LV pressure-overload model (Chen et al., 2012). We further verified that the onset of pulmonary hypertension was secondary to the increase in EDP, and that pathological remodeling in the pulmonary vasculature was secondary to the spike in RVP. This confirms TAC is a model of type II pulmonary hypertension with the instigating pathology in the LV, which migrates to the RV and associated vasculature. As current therapies for type I pulmonary hypertension (i.e., pulmonary vasodilators) are either ineffective (Naeije and Huez, 2007) or detrimental (Guazzi and Arena, 2010) in patients with HF (type II), there is an urgent need for novel therapies that address this population. TAC presents an ideal model to investigate both the pathophysiology of type II pulmonary hypertension, as well as novel treatments for this prevalent condition.
Remodeling patterns in the LV cannot be applied to the RV; the RV is developmentally (Zaffran, 2004), physiologically (Littlejohns et al., 2014), and molecularly (Fields et al., 1978) distinct from the LV and pumps into a different vascular bed against differing hemodynamic loads. Here we report early concentric hypertrophy between 2 and 4 weeks that occurred in tandem with increased RVP, followed by a secondary phase where RV weight increased without any further increase in cardiomyocyte CSA–suggestive of a shift to eccentric hypertrophy. The lack of concentric hypertrophy was unexpected given sustained pulmonary hypertension. This contrasted the LV where cardiomyocyte CSA increased out to 18 weeks despite declining pressures and suggests the RV does not respond similarly to the stimuli driving continued concentric growth in the late-stage LV. Emerging studies suggest RV hypertrophy may be determinant to mortality; a defining feature of decompensation is a disproportionate increase in RV mass compared to LV mass (Bing et al., 1995; Norton et al., 2002; Brooks et al., 2010; Chen et al., 2012). Generally, cardiac hypertrophy studies are LV-centric (Weeks and McMullen, 2011; Maillet et al., 2013) and more attention should be provided to the RV.
The RV is, at least in part, hemodynamically dependent on the LV (Santamore and Dell’Italia, 1998). One example of this is in the flow across the lung, which is a function of the pressure gradient between the RV (RVP) and the LV (LVEDP). An increase in LVEDP is thought to cause a reflexive increase in RVP for the maintenance of flow through the pulmonary system (Chen et al., 2012). Our analysis does not show this to be entirely true as a curvilinear relationship is a better fit (Figure 8). This may be due to extensive recruitment and distension of pulmonary capillaries to reduce pulmonary vascular resistance in early situations of elevated EDP.
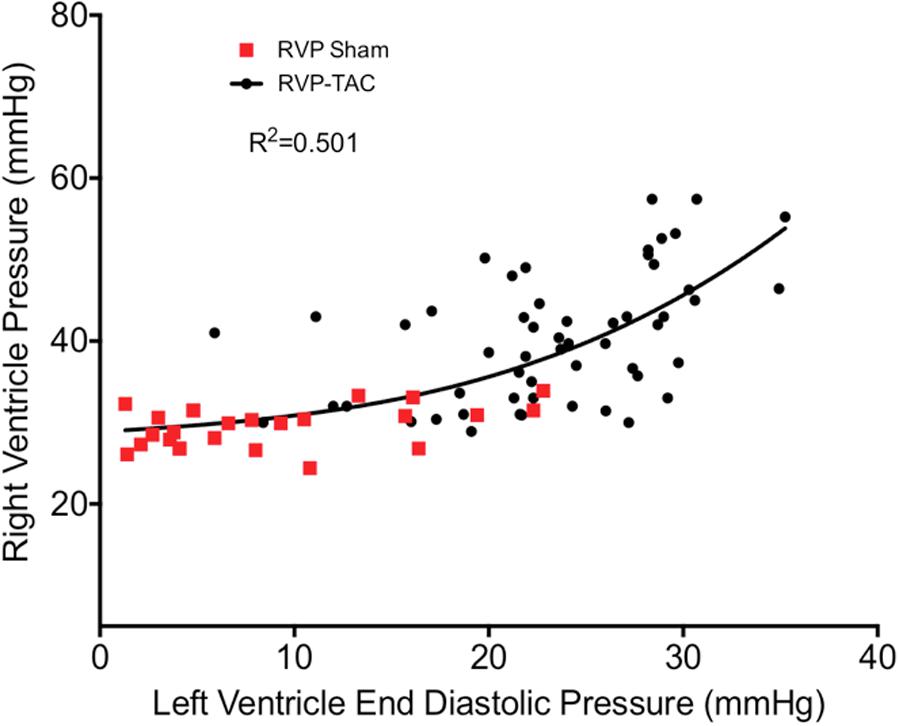
FIGURE 8. Correlation plot between right ventricle systolic pressure and left ventricular end diastolic pressure in both sham (n = 23) and TAC (n = 55). The curve of best fit was drawn using a non-linear third order polynomial (cubic) regression.
Finally, in light of the varying patterns between the LV and RV with regards to pressure and hypertrophy, we introduce an updated depiction of hypertrophy progression in pressure-overload (Figure 9).
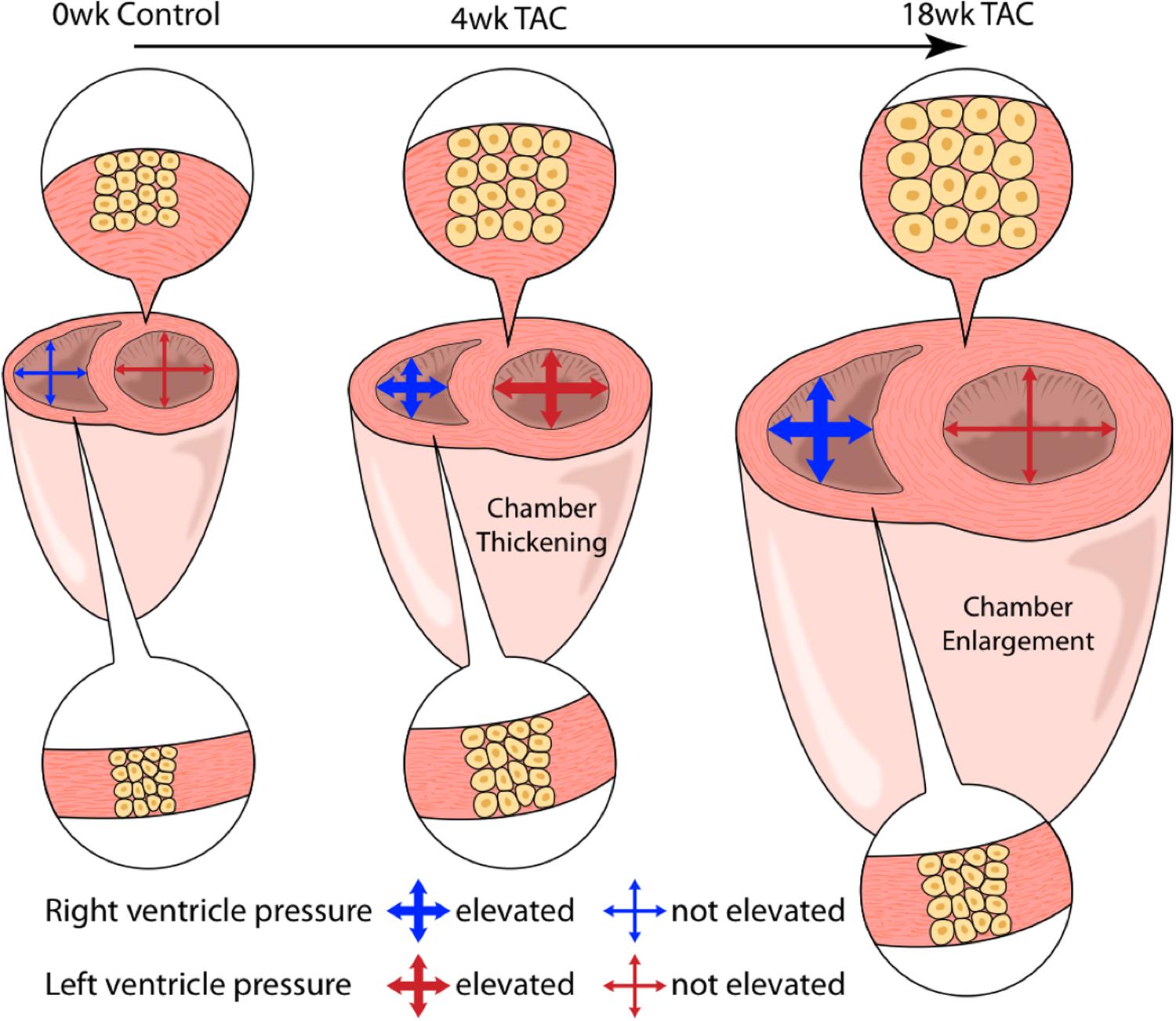
FIGURE 9. An artistic depiction of the hypertrophic response in both ventricles over 18 weeks of TAC. At 4 weeks, both the right (RV) and left (LV) ventricles present with concentric hypertrophy (thickening of ventricle walls with an increase in cardiomyocyte cross-sectional area (CSA). LV concentric hypertrophy continued out to 18 weeks despite declining systolic pressures. Alternatively, RV cardiomyocyte CSA was static despite sustained systolic pressure-overload. Here, total weight increased in the absence of changes in CSA, suggestive of cardiomyocyte lengthening (eccentric hypertrophy).
The concept of “diastolic dysfunction,” which grew out of clinical observation, is cumbersome to diagnose and a validated gold standard is missing. Indeed, what we define as dysfunction is only in terms of referencing ‘normal’ function, which is dynamic as the cardiovascular system develops and ages. In experimental models, the standard is to measure diastolic function invasively (e.g., EDP, dP/dtmin, and Tau). This differs from the clinical reliance on indirect surrogate measures (e.g., symptoms, E/A ratio and serum BNP levels). Of concern is whether the surrogate measurements remain in agreement with the fundamental pathophysiology. We found over time, diastolic function was not easily reconciled by any one parameter, suggesting diastolic dysfunction is more a spectrum of impairments than a singular entity.
As components of diastolic function, we interpret EDP as a measure of pressure-stress to relaxation, Tau as the relative time required for relaxation, and dP/dtmin as the instantaneous maximum rate-of-relaxation. We show that diastolic parameters were incongruent with each other over time, a finding reported in other pressure-overload studies (Takimoto et al., 2005; Perrino, 2006; Chen et al., 2012). Indeed, each parameter likely has subtle and unique inferential value. If both EDP and Tau are increased, is this the same disease state as increased EDP when Tau is unchanged? As most studies rely on a single parameter to assess diastolic function, this question remains difficult to answer. Caution should be taken when evaluating development of diastolic dysfunction in different etiologies (e.g., channelopathy vs. interstitial fibrosis) as they may not conform to a single diastolic measure. Rather, each measure likely varies in sensitivity to identify dysfunction, or captures a particular physiological nuance of diastolic function.
One discrepancy to highlight is the difference in sensitivity between Tau methods to assess diastolic function. Particularly, Tau Glantz (TauG) detected diastolic impairments by 2 weeks while Tau Weiss (TauW) was unchanged. The discrepancy between Tau measures may exist because TauW (approximated in isolated hearts) assumes an independence of Tau from volume loading and changes in EDP (Weiss et al., 1976). TauG, however, is an updated method derived from observations in intact dog hearts, and accounts for the impact of variable loading conditions (i.e., changing diastolic pressures) (Raff and Glantz, 1981). Given the chronically elevated EDP in our (and most) pressure overload model(s), TauG may be the more sensitive parameter. Yet, it remains uncertain at this time which is the superior measure that avoids type-I/II statistical errors and, thus, until more physiological studies are performed, both measures should be used with a clear knowledge as to their underlying assumptions.
Additionally, it was unexpected that in sham mice, while systolic function remained stable, the thresholds for diastolic dysfunction were met (EDP, dP/dtmin, and TauG decreased from pre-surgical values). This is consistent with human data where diastolic function is more fragile with aging than systolic function (Fischer et al., 2003). These steady declines suggest the “thresholds” used to identify diastolic dysfunction may need to be adjusted in each study based on reference values relative to aging.
Finally, parameters for grading diastolic function in the LV were also impaired in the RV following TAC. However, RV diastolic dysfunction remains largely undefined, both clinically and in basic research, and gold standard indicies of RV diastolic function remain to be determined in a patient population (Axell et al., 2015).
HF, either clinically or experimentally, is not defined by the presence or absence of pulmonary edema. Indeed, congestion is neither always expected in HF nor always due to pulmonary edema. In fact, pulmonary edema is rare in patients with HF (Mahdyoon et al., 1989; Tarvasmäki et al., 2014; Melenovsky et al., 2015), particularly when compared to the reference values in age-matched individuals without HF (Kataoka and Matsuno, 2008). This is sometimes despite the presence of hemodynamic congestion (this was part of the motivation to shift terminology from congestive HF to HF with preserved or reduced ejection fraction). Consistent with HF patients, a feature of TAC is congestion without pulmonary edema (Chen et al., 2012; Foster et al., 2017). This is not to say that pulmonary edema never occurs in experimental models of HF. Both in our study and in others, pulmonary edema is observed along with rapid decompensation instead of reaching pre-determined time-points (Inoko et al., 1994; Kihara and Sasayama, 1997; Siri et al., 1997; Chung et al., 1998; van Nierop et al., 2013). Thus, when present, pulmonary edema in rodent models may be more indicative of impending mortality than part of the pathophysiology of morbidity.
While we characterize cardiac function and morphology across multiple time points, we do so in young healthy male mice when the clinical HF population is variably aged and both sexes. Indeed, the necessary next step is to apply this ‘mapping’ approach using aged male and female subjects. Future pre-clinical models might also benefit from an assessment of frequently used clinical variables such as blood biomarkers (i.e., BNP; Doust et al., 2005), Doppler echo, or frailty indexes (Singh et al., 2014) to further improve the alignment between experimental models and the clinical population. As well, isolated cardiomyocyte measurements of length and width would provide insight into eccentric hypertrophy.
The goal of HF research is to translate findings to the clinical population to improve survival and quality of life. As too few drugs make the transition from bench to bedside (Mak et al., 2014; Fernández-Avilés et al., 2018), there is a need to improve the success of translational research. Several reports have emphasized that the current designs of preclinical rodent studies are not fundamentally rigorous enough (Kloner, 2013; Ovize et al., 2013; Vander Heide and Steenbergen, 2013). This demands an improved understanding of the basic pathophysiology of the models used to investigate preclinical therapies (Houser et al., 2012). A bi-ventricular temporal assessment incorporating multiple indices of diastolic function addresses this in part, adding to our understanding of pressure-overload HF and specifically the time course of pathology in the RV.
MP, JH, KB, and JS conceptualized the study, designed the experiments, drafted the manuscript, critically revised the manuscript for intellectual content, and gave the final approval for the article. MP, NR, and JH conducted the experiments. All authors analyzed and/or interpreted the experimental results and read and gave permission to the final draft of the manuscript.
This work was supported by grants from National Science Research Engineering Council (KB and JS), a Canadian Institutes of Health Research grant (MOP 111159 to JS), and a Heart and Stroke Foundation of Canada grant (JS and KB). JS is also a New Investigator of the Heart and Stroke Foundation of Canada. We further acknowledge the philanthropic support for cardiovascular research from Betty and Jack Southen, of London ON and from Matthew O’Hara, of Guelph ON to the laboratory of JS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We sincerely thank Ian Smith for his generation of Figure 9.
BW, body weight; CSA, cross-sectional area; EDP, End Diastolic Pressure; HF, heart failure; HW, heart weight; LV, left ventricle; LVP, left ventricular pressure; RV, right ventricle; RVP, right ventricular pressure; TAC, transverse aortic constriction; TL, tibia length.
Adusumalli, S., and Mazurek, J. A. (2017). Pulmonary hypertension due to left ventricular cardiomyopathy: is it the result or cause of disease progression? Curr. Heart Fail. Rep. 14, 507–513. doi: 10.1007/s11897-017-03682
Allwood, M. A., Kinobe, R. T., Ballantyne, L., Romanova, N., Melo, L. G., Ward, C. A., et al. (2014). Heme oxygenase-1 overexpression exacerbates heart failure with aging and pressure overload but is protective against isoproterenol-induced cardiomyopathy in mice. Cardiovasc. Pathol. 23, 231–237. doi: 10.1016/j.carpath.2014.03.007
Axell, R. G., Hoole, S. P., Hampton-Till, J., and White, P. A. (2015). RV diastolic dysfunction: time to re-evaluate its importance in heart failure. Heart Fail. Rev. 20, 363–373. doi: 10.1007/s10741-015-9472-0
Badenhorst, D. (2003). Cross-linking influences the impact of quantitative changes in myocardial collagen on cardiac stiffness and remodelling in hypertension in rats. Cardiovasc. Res. 57, 632–641. doi: 10.1016/S0008-6363(02)00733-2
Barrick, C. J., Dong, A., Waikel, R., Corn, D., Yang, F., Threadgill, D. W., et al. (2009). Parent-of-origin effects on cardiac response to pressure overload in mice. Am. J. Physiol. Heart Circ. Physiol. 297, H1003–H1009. doi: 10.1152/ajpheart.00896.2008
Barrick, C. J., Rojas, M., Schoonhoven, R., Smyth, S. S., and Threadgill, D. W. (2007). Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal- and background-dependent development of concentric left ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 292, H2119–H2130. doi: 10.1152/ajpheart.00816.2006
Benjamin, E. J., Blaha, M. J., Chiuve, S. E., Cushman, M., Das, S. R., Deo, R., et al. (2017). Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation 135:e646. doi: 10.1161/CIR.0000000000000485
Bing, O. H., Brooks, W. W., Robinson, K. G., Slawsky, M. T., Hayes, J. A., Litwin, S. E., et al. (1995). The spontaneously hypertensive rat as a model of the transition from compensated left ventricular hypertrophy to failure. J. Mol. Cell. Cardiol. 27, 383–396. doi: 10.1016/S0022-2828(08)80035-1
Bogaard, H. J., Abe, K., Vonk Noordegraaf, A., and Voelkel, N. F. (2009). The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 135, 794–804. doi: 10.1378/chest.08-0492
Brooks, W. W., Shen, S. S., Conrad, C. H., Goldstein, R. H., and Bing, O. H. L. (2010). Transition from compensated hypertrophy to systolic heart failure in the spontaneously hypertensive rat: structure, function, and transcript analysis. Genomics 95, 84–92. doi: 10.1016/j.ygeno.2009.12.002
Chen, Y., Guo, H., Xu, D., Xu, X., Wang, H., Hu, X., et al. (2012). Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: heart failure causes severe lung disease. Hypertension 59, 1170–1178. doi: 10.1161/HYPERTENSIONAHA.111.186072
Chung, E. S., Perlini, S., Aurigemma, G. P., Fenton, R. A., Dobson, J. G., and Meyer, T. E. (1998). Effects of chronic adenosine uptake blockade on adrenergic responsiveness and left ventricular chamber function in pressure overload hypertrophy in the rat. J. Hypertens. 16, 1813–1822. doi: 10.1097/00004872-199816120-00015
Conrad, C. H., Brooks, W. W., Hayes, J. A., Sen, S., Robinson, K. G., and Bing, O. H. (1995). Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation 91, 161–170. doi: 10.1161/01.CIR.91.1.161
Doust, J. A., Pietrzak, E., Dobson, A., and Glasziou, P. (2005). How well does B-type natriuretic peptide predict death and cardiac events in patients with heart failure: systematic review. BMJ 330:625. doi: 10.1136/bmj.330.7492.625
Fernández-Avilés, F., Sanz-Ruiz, R., Climent, A. M., Badimon, L., Bolli, R., Charron, D., et al. (2018). Global overview of the transnational alliance for regenerative therapies in cardiovascular syndromes (TACTICS) recommendations: a comprehensive series of challenges and priorities of cardiovascular regenerative medicine. Circ. Res. 122, 199–201. doi: 10.1161/CIRCRESAHA.117.312099
Ferreira, R. G., Worthington, A., Huang, C.-C., Aranki, S. F., and Muehlschlegel, J. D. (2015). Sex differences in the prevalence of diastolic dysfunction in cardiac surgical patients. J. Card. Surg. 30, 238–245. doi: 10.1111/jocs.12506
Fields, J. Z., Roeske, W. R., Morkin, E., and Yamamura, H. I. (1978). Cardiac muscarinic cholinergic receptors. Biochemical identification and characterization. J. Biol. Chem. 253, 3251–3258.
Fischer, M., Baessler, A., Hense, H. W., Hengstenberg, C., Muscholl, M., Holmer, S., et al. (2003). Prevalence of left ventricular diastolic dysfunction in the community. Results from a Doppler echocardiographic-based survey of a population sample. Eur. Heart J. 24, 320–328. doi: 10.1016/S0195-668X(02)00428-1
Foster, A. J., Platt, M. J., Huber, J. S., Eadie, A. L., Arkell, A. M., Romanova, N., et al. (2017). Central-acting therapeutics alleviate respiratory weakness caused by heart failure-induced ventilatory overdrive. Sci. Transl. Med. 9:eaag1303. doi: 10.1126/scitranslmed.aag1303
Frey, N., Katus, H. A., Olson, E. N., and Hill, J. A. (2004). Hypertrophy of the heart: a new therapeutic target? Circulation 109, 1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB
Ghio, S., Gavazzi, A., Campana, C., Inserra, C., Klersy, C., Sebastiani, R., et al. (2001). Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J. Am. Coll. Cardiol. 37, 183–188. doi: 10.1016/S0735-1097(00)01102-5
Grossman, W., Jones, D., and McLaurin, L. P. (1975). Wall stress and patterns of hypertrophy in the human left ventricle. J. Clin. Invest. 56, 56–64. doi: 10.1172/JCI108079
Guazzi, M., and Arena, R. (2010). Pulmonary hypertension with left-sided heart disease. Nat. Rev. Cardiol. 7, 648–659. doi: 10.1038/nrcardio.2010.144
Hamdani, N., Hervent, A.-S., Vandekerckhove, L., Matheeussen, V., Demolder, M., Baerts, L., et al. (2014). Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovasc. Res. 104, 423–431. doi: 10.1093/cvr/cvu223
Heidenreich, P. A., Trogdon, J. G., Khavjou, O. A., Butler, J., Dracup, K., Ezekowitz, M. D., et al. (2011). Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Associatio. Circulation 123, 933–944. doi: 10.1161/CIR.0b013e31820a55f5
Hoeper, M. M., Barberà, J. A., Channick, R. N., Hassoun, P. M., Lang, I. M., Manes, A., et al. (2009). Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J. Am. Coll. Cardiol. 54, S85–S96. doi: 10.1016/j.jacc.2009.04.008
Hoeper, M. M., Markevych, I., Spiekerkoetter, E., Welte, T., and Niedermeyer, J. (2005). Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur. Respir. J. 26, 858–863.
Hogg, K., Swedberg, K., and McMurray, J. (2004). Heart failure with preserved left ventricular systolic function; epidemiology, clinical characteristics, and prognosis. J. Am. Coll. Cardiol. 43, 317–327. doi: 10.1016/j.jacc.2003.07.046
Houser, S. R., Margulies, K. B., Murphy, A. M., Spinale, F. G., Francis, G. S., Prabhu, S. D., et al. (2012). Animal models of heart failure: a scientific statement from the American Heart Association. Circ. Res. 111, 131–150. doi: 10.1161/RES.0b013e3182582523
Inoko, M., Kihara, Y., Morii, I., Fujiwara, H., and Sasayama, S. (1994). Transition from compensatory hypertrophy to dilated, failing left ventricles in Dahl salt-sensitive rats. Am. J. Physiol. 267, H2471–H2482. doi: 10.1152/ajpheart.1994.267.6.H2471
Jalil, J. E., Doering, C. W., Janicki, J. S., Pick, R., Shroff, S. G., and Weber, K. T. (1989). Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ. Res. 64, 1041–1050. doi: 10.1161/01.RES.64.6.1041
Kapur, N. K., Wilson, S., Yunis, A. A., Qiao, X., Mackey, E., Paruchuri, V., et al. (2012). Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation 125, 2728–2738. doi: 10.1161/CIRCULATIONAHA.111.080002
Karaye, K. M., Habib, A. G., Mohammed, S., Rabiu, M., and Shehu, M. N. (2010). Assessment of right ventricular systolic function using tricuspid annular-plane systolic excursion in Nigerians with systemic hypertension. Cardiovasc. J. Afr. 21, 186–190. doi: 10.5830/CVJA-2010-031
Kass, D. A., Bronzwaer, J. G. F., and Paulus, W. J. (2004). What mechanisms underlie diastolic dysfunction in heart failure? Circ. Res. 94, 1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6
Kataoka, H., and Matsuno, O. (2008). Age-related pulmonary crackles (rales) in asymptomatic cardiovascular patients. Ann. Fam. Med. 6, 239–245. doi: 10.1370/afm.834
Kihara, Y., and Sasayama, S. (1997). Transition from compensatory hypertrophy to dilated failing left ventricle in Dahl-Iwai salt-sensitive rats. Am. J. Hypertens. 10, 78S–82S. doi: 10.1016/S0895-7061(97)00080-0
Kloner, R. A. (2013). Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ. Res. 113, 451–463. doi: 10.1161/CIRCRESAHA.112.300627
Klotz, S., Hay, I., Zhang, G., Maurer, M., Wang, J., and Burkhoff, D. (2006). Development of heart failure in chronic hypertensive Dahl rats: focus on heart failure with preserved ejection fraction. Hypertension 47, 901–911. doi: 10.1161/01.HYP.0000215579.81408.8e
Lam, C. S. P., Roger, V. L., Rodeheffer, R. J., Borlaug, B. A., Enders, F. T., and Redfield, M. M. (2009). Pulmonary hypertension in heart failure with preserved ejection fraction. J. Am. Coll. Cardiol. 53, 1119–1126. doi: 10.1016/j.jacc.2008.11.051
Levy, D., Garrison, R. J., Savage, D. D., Kannel, W. B., and Castelli, W. P. (1990). Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 322, 1561–1566. doi: 10.1056/NEJM199005313222203
Liao, Y., Ishikura, F., Beppu, S., Asakura, M., Takashima, S., Asanuma, H., et al. (2002). Echocardiographic assessment of LV hypertrophy and function in aortic-banded mice: necropsy validation. Am. J. Physiol. Heart Circ. Physiol. 282, H1703–H1708. doi: 10.1152/ajpheart.00238.2001
Littlejohns, B., Heesom, K., Angelini, G. D., and Suleiman, M.-S. (2014). The effect of disease on human cardiac protein expression profiles in paired samples from right and left ventricles. Clin. Proteomics. 11, 1–14. doi: 10.1186/1559-0275-11-34
López, B., Querejeta, R., González, A., Larman, M., and Díez, J. (2012). Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension 60, 677–683. doi: 10.1161/HYPERTENSIONAHA.112.196113
Mahdyoon, H., Klein, R., Eyler, W., Lakier, J. B., Chakko, S. C., and Gheorghiade, M. (1989). Radiographic pulmonary congestion in end-stage congestive heart failure. Am. J. Cardiol. 63, 625–627. doi: 10.1016/0002-9149(89)90912-0
Maillet, M., van Berlo, J. H., and Molkentin, J. D. (2013). Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 14, 38–48. doi: 10.1038/nrm3495
Mak, I. W., Evaniew, N., and Ghert, M. (2014). Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 6, 114–118.
Mandinov, L., Eberli, F. R., Seiler, C., and Hess, O. M. (2000). Diastolic heart failure. Cardiovasc. Res. 45, 813–825. doi: 10.1016/S0008-6363(99)00399-5
Matsusaka, H., Ide, T., Matsushima, S., Ikeuchi, M., Kubota, T., Sunagawa, K., et al. (2006). Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension 47, 711–717. doi: 10.1161/01.HYP.0000208840.30778.00
Melenovsky, V., Andersen, M. J., Andress, K., Reddy, Y. N., and Borlaug, B. A. (2015). Lung congestion in chronic heart failure: haemodynamic, clinical, and prognostic implications. Eur. J. Heart Fail. 17, 1161–1171. doi: 10.1002/ejhf.417
Meyer, P., Filippatos, G. S., Ahmed, M. I., Iskandrian, A. E., Bittner, V., Perry, G. J., et al. (2010). Effects of right ventricular ejection fraction on outcomes in chronic systolic heart failure. Circulation 121, 252–258. doi: 10.1161/CIRCULATIONAHA.109.887570
Mirsky, I., Pfeffer, J. M., Pfeffer, M. A., and Braunwald, E. (1983). The contractile state as the major determinant in the evolution of left ventricular dysfunction in the spontaneously hypertensive rat. Circ. Res. 53, 767–778. doi: 10.1161/01.RES.53.6.767
Mohammed, S. F., Storlie, J. R., Oehler, E. A., Bowen, L. A., Korinek, J., Lam, C. S. P., et al. (2012). Variable phenotype in murine transverse aortic constriction. Cardiovasc. Pathol. 21, 188–198. doi: 10.1016/j.carpath.2011.05.002
Naeije, R., and Huez, S. (2007). Expert opinion on available options treating pulmonary arterial hypertension. Expert Opin. Pharmacother. 8, 2247–2265. doi: 10.1517/14656566.8.14.2247
Nakamura, A., Rokosh, D. G., Paccanaro, M., Yee, R. R., Simpson, P. C., Grossman, W., et al. (2001). LV systolic performance improves with development of hypertrophy after transverse aortic constriction in mice. Am. J. Physiol. Heart Circ. Physiol. 281, H1104–H1112. doi: 10.1152/ajpheart.2001.281.3.H1104
Norton, G. R., Woodiwiss, A. J., Gaasch, W. H., Mela, T., Chung, E. S., Aurigemma, G. P., et al. (2002). Heart failure in pressure overload hypertrophy: the relative roles of ventricular remodeling and myocardial dysfunction. J. Am. Coll. Cardiol. 39, 664–671. doi: 10.1016/S0735-1097(01)01792-2
Ovize, M., Thibault, H., and Przyklenk, K. (2013). Myocardial conditioning: opportunities for clinical translation. Circ. Res. 113, 439–450. doi: 10.1161/CIRCRESAHA.113.300764
Paulus, W. J., Tschöpe, C., Sanderson, J. E., Rusconi, C., Flachskampf, F. A., Rademakers, F. E., et al. (2007). How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur. Heart J. 28, 2539–2550. doi: 10.1093/eurheartj/ehm037
Perrino, C. (2006). Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J. Clin. Invest. 116, 1547–1560. doi: 10.1172/JCI25397
Platt, M. J., Huber, J. S., Brunt, K. R., and Simpson, J. A. (2017). Pulmonary flow as an improved method for determining cardiac output in mice after myocardial infarction. J. Am. Soc. Echocardiogr. 30, 612.e–623.e. doi: 10.1016/j.echo.2017.02.008
Pradhan, K., Sydykov, A., Tian, X., Mamazhakypov, A., Neupane, B., Luitel, H., et al. (2016). Soluble guanylate cyclase stimulator riociguat and phosphodiesterase 5 inhibitor sildenafil ameliorate pulmonary hypertension due to left heart disease in mice. Int. J. Cardiol. 216, 85–91. doi: 10.1016/j.ijcard.2016.04.098
Raff, G. L., and Glantz, S. A. (1981). Volume loading slows left ventricular isovolumic relaxation rate. Evidence of load-dependent relaxation in the intact dog heart. Circ. Res. 48, 813–824. doi: 10.1161/01.RES.48.6.813
Ravassa, S., López, B., Querejeta, R., Echegaray, K., San José, G., Moreno, M. U., et al. (2017). Phenotyping of myocardial fibrosis in hypertensive patients with heart failure. Influence on clinical outcome. J. Hypertens. 35, 853–861. doi: 10.1097/HJH.0000000000001258
Rockman, H. A., Ross, R. S., Harris, A. N., Knowlton, K. U., Steinhelper, M. E., Field, L. J., et al. (1991). Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 88, 8277–8281. doi: 10.1073/pnas.88.18.8277
Rosenkranz, S., Gibbs, J. S. R., Wachter, R., De Marco, T., Vonk Noordegraaf, A., and Vachiéry, J.-L. (2016). Left ventricular heart failure and pulmonary hypertension. Eur. Heart J. 37, 942–954. doi: 10.1093/eurheartj/ehv512
Rothermel, B. A., Berenji, K., Tannous, P., Kutschke, W., Dey, A., Nolan, B., et al. (2005). Differential activation of stress-response signaling in load-induced cardiac hypertrophy and failure. Physiol. Genomics 23, 18–27. doi: 10.1152/physiolgenomics.00061.2005
Santamore, W. P., and Dell’Italia, L. J. (1998). Ventricular interdependence: significant left ventricular contributions to right ventricular systolic function. Prog. Cardiovasc. Dis. 40, 289–308. doi: 10.1016/S0033-0620(98)80049-2
Sheikh, A. Q., Lighthouse, J. K., and Greif, D. M. (2014). Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 6, 809–817. doi: 10.1016/j.celrep.2014.01.042
Simonneau, G., Robbins, I. M., Beghetti, M., Channick, R. N., Delcroix, M., Denton, C. P., et al. (2009). Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 54, S43–S54. doi: 10.1016/j.jacc.2009.04.012
Singh, M., Stewart, R., and White, H. (2014). Importance of frailty in patients with cardiovascular disease. Eur. Heart J. 35, 1726–1731. doi: 10.1093/eurheartj/ehu197
Siri, F. M., Jelicks, L. A., Leinwand, L. A., and Gardin, J. M. (1997). Gated magnetic resonance imaging of normal and hypertrophied murine hearts. Am. J. Physiol. 272, H2394–H2402. doi: 10.1152/ajpheart.1997.272.5.H2394
Stenmark, K. R., Meyrick, B., Galiè, N., Mooi, W. J., and McMurtry, I. F. (2009). Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L1013–L1032. doi: 10.1152/ajplung.00217.2009
Takimoto, E., Champion, H. C., Li, M., Belardi, D., and Ren, S. (2005). Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 11, 214–222. doi: 10.1038/nm1175
Tarvasmäki, T., Harjola, V.-P., Nieminen, M. S., Siirilä-Waris, K., Tolonen, J., Tolppanen, H., et al. (2014). Acute heart failure with and without concomitant acute coronary syndromes: patient characteristics, management, and survival. J. Card. Fail. 20, 723–730. doi: 10.1016/j.cardfail.2014.07.008
van Nierop, B. J., van Assen, H. C., van Deel, E. D., Niesen, L. B. P., Duncker, D. J., Strijkers, G. J., et al. (2013). Phenotyping of left and right ventricular function in mouse models of compensated hypertrophy and heart failure with cardiac MRI. PLoS One 8:e55424. doi: 10.1371/journal.pone.0055424
Vander Heide, R. S., and Steenbergen, C. (2013). Cardioprotection and myocardial reperfusion: pitfalls to clinical application. Circ. Res. 113, 464–477. doi: 10.1161/CIRCRESAHA.113.300765
Wan, S.-H., Vogel, M. W., and Chen, H. H. (2014). Pre-clinical diastolic dysfunction. J. Am. Coll. Cardiol. 63, 407–416. doi: 10.1016/j.jacc.2013.10.063
Weber, K. T., Brilla, C. G., and Janicki, J. S. (1993). Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc. Res. 27, 341–348. doi: 10.1093/cvr/27.3.341
Weeks, K. L., and McMullen, J. R. (2011). The athlete’s heart vs. the failing heart: can signaling explain the two distinct outcomes? Physiology (Bethesda) 26, 97–105. doi: 10.1152/physiol.00043.2010
Weiss, J. L., Frederiksen, J. W., and Weisfeldt, M. L. (1976). Hemodynamic determinants of the time-course of fall in canine left ventricular pressure. J. Clin. Invest. 58, 751–760. doi: 10.1172/JCI108522
Wu, X., Simpson, J., Hong, J. H., Kim, K.-H., Thavarajah, N. K., Backx, P. H., et al. (2011). MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1L613V mutation. J. Clin. Invest. 121, 1009–1025. doi: 10.1172/JCI44929
Yamamoto, K., Masuyama, T., Sakata, Y., Nishikawa, N., Mano, T., Yoshida, J., et al. (2002). Myocardial stiffness is determined by ventricular fibrosis, but not by compensatory or excessive hypertrophy in hypertensive heart. Cardiovasc. Res. 55, 76–82. doi: 10.1016/S0008-6363(02)00341-3
Zaffran, S. (2004). Right ventricular myocardium derives from the anterior heart field. Circ. Res. 95, 261–268. doi: 10.1161/01.RES.0000136815.73623.BE
Keywords: diastolic dysfunction, pulmonary hypertension, left heart disease, concentric remodeling, mouse models
Citation: Platt MJ, Huber JS, Romanova N, Brunt KR and Simpson JA (2018) Pathophysiological Mapping of Experimental Heart Failure: Left and Right Ventricular Remodeling in Transverse Aortic Constriction Is Temporally, Kinetically and Structurally Distinct. Front. Physiol. 9:472. doi: 10.3389/fphys.2018.00472
Received: 27 November 2017; Accepted: 16 April 2018;
Published: 15 May 2018.
Edited by:
Joaquin Garcia-Estañ, Universidad de Murcia, SpainReviewed by:
Sean P. Didion, University of Mississippi Medical Center, United StatesCopyright © 2018 Platt, Huber, Romanova, Brunt and Simpson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy A. Simpson, amVyZW15c0B1b2d1ZWxwaC5jYQ==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.