 Vincent F. M. Segers
Vincent F. M. Segers Dirk L. Brutsaert
Dirk L. Brutsaert Gilles W. De Keulenaer
Gilles W. De Keulenaer- 1Laboratory of Physiopharmacology, University of Antwerp, Antwerp, Belgium
- 2Department of Cardiology, University Hospital Antwerp, Edegem, Belgium
- 3Department of Cardiology, Middelheim Hospital, Antwerp, Belgium
The heart is a highly structured organ consisting of different cell types, including myocytes, endothelial cells, fibroblasts, stem cells, and inflammatory cells. This pluricellularity provides the opportunity of intercellular communication within the organ, with subsequent optimization of its function. Intercellular cross-talk is indispensable during cardiac development, but also plays a substantial modulatory role in the normal and failing heart of adults. More specifically, factors secreted by cardiac microvascular endothelial cells modulate cardiac performance and either positively or negatively affect cardiac remodeling. The role of endothelium-derived small molecules and peptides—for instance NO or endothelin-1—has been extensively studied and is relatively well defined. However, endothelial cells also secrete numerous larger proteins. Information on the role of these proteins in the heart is scattered throughout the literature. In this review, we will link specific proteins that modulate cardiac contractility or cardiac remodeling to their expression by cardiac microvascular endothelial cells. The following proteins will be discussed: IL-6, periostin, tenascin-C, thrombospondin, follistatin-like 1, frizzled-related protein 3, IGF-1, CTGF, dickkopf-3, BMP-2 and−4, apelin, IL-1β, placental growth factor, LIF, WISP-1, midkine, and adrenomedullin. In the future, it is likely that some of these proteins can serve as markers of cardiac remodeling and that the concept of endothelial function and dysfunction might have to be redefined as we learn more about other factors secreted by ECs besides NO.
Introduction
The heart is a muscular pump consisting of myocytes, endothelial cells (ECs), fibroblasts, stem cells, and inflammatory cells (Segers and Lee, 2008; Kamo et al., 2015). Cardiac tissue is a highly organized structure of cells and extracellular matrix with an intricate multidirectional communication between cells. All cells present in the myocardium secrete autocrine, juxtacrine, and paracrine factors that modulate function of neighboring cells (Figure 1). Intercellular communication plays crucial roles in cardiac development and normal cardiac function in the adult organism, but also in the pathophysiology of cardiac remodeling and heart failure development. In particular, factors secreted by cardiac microvascular ECs play a crucial role in normal cardiac function and during cardiac remodeling.
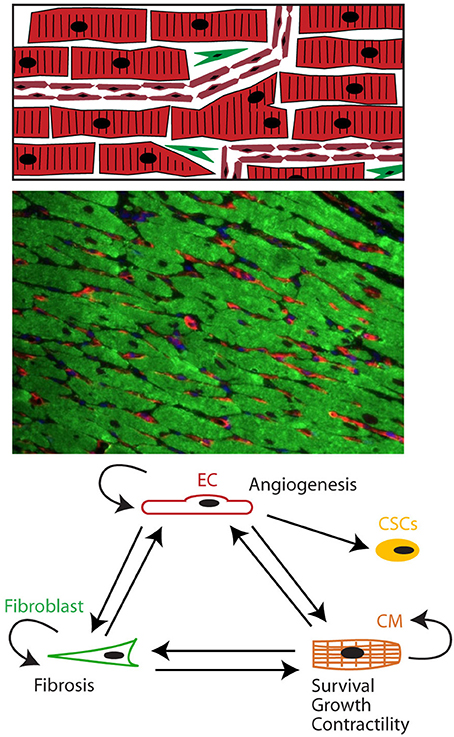
Figure 1. The heart as a pluricellular organ. (Upper) The heart is a highly organized pluricellular tissue consisting of myocytes (red, striated), capillary ECs (red, smaller elongated cells), and to a lesser extent fibroblasts (green spindle shaped) and stem cells. (Middle) Fluorescent staining of myocardial tissue with myocytes depicted in green and endothelial cells in red. Myocytes and endothelial cells are in close contact with each other. (Lower) Cells communicate through autocrine, juxtacrine and paracrine signals.
The role of endothelium-derived small molecules and peptides has been extensively studied and is relatively well defined. For instance, nitric oxide (NO) affects cardiac contractility by inducing an earlier onset of relaxation resulting in a longer diastole and favoring diastolic filling (Brutsaert, 2003; Balligand et al., 2009). Another example is endothelin-1, which has positive inotropic effects (Moravec et al., 1989) and induces a hypertrophic response in cardiomyocytes (Drawnel et al., 2013). However, ECs do not only secrete small molecules and peptides but also numerous proteins. Information on the role of these proteins in normal cardiac biology and cardiac remodeling is limited and scattered throughout the literature.
Another issue is that the cardiotrophic effects of certain secreted proteins are not always linked to the source of the proteins, which is in a number of instances the cardiac microvascular endothelium. In recent years, a number of excellent papers have been published describing cardioprotective effects of specific endogenous proteins (Oshima et al., 2008; Shimano et al., 2011; Frangogiannis, 2012; Zhang et al., 2014), without discussing their source. Moreover, signaling proteins in the heart are sometimes referred to as “matricellular proteins” (Frangogiannis, 2012), a term that ignores the origin of these proteins and suggests that they are a static part of the extracellular matrix. Cardiac microvascular ECs are the most abundant cell type—not in total volume but in total number—in adult myocardium (Pinto et al., 2015), are in direct contact with adjacent cardiomyocytes and fibroblasts, and actively secrete many proteins.
In this review, we will link specific proteins that modulate cardiac contractility or cardiac remodeling to their expression by cardiac microvascular ECs using publicly available expression libraries. In physiology, there are numerous feed-back and feed-forward mechanisms that are part of an intricate multidirectional communication network. Similar feed-back and feed-forward mechanisms are present in the communication between ECs, cardiomyocytes, and fibroblasts in the heart. For example, when ECs send a signal to cardiomyocytes, these will respond with a signal that enhances or attenuates the original signal. To limit the degree of complexity in this review, we will focus on signals secreted by microvascular ECs present in the myocardium and ignore signals from other cells. We will narrow the focus of this review further by discussing endothelial-derived proteins; many excellent reviews can be found on small molecules and peptides secreted by cardiac ECs (Brutsaert, 2003; Chatzizisis et al., 2007; Duncker and Bache, 2008; Kamo et al., 2015; Lim et al., 2015). The overall aim of the present review is to provide new insights in the role of microvascular endothelial cells in pathophysiology of cardiac remodeling beyond secretion of NO. Furthermore, we want to summarize evidence about either the protective or the adverse effect of endothelium-derived proteins, regarding to cardiac contractility, cardiac remodeling, and different cardiac diseases.
Methodology
Inclusion of endothelial-derived proteins in this review was based on publicly available micro-array datasets in Geo Datasets (Table 1). Micro-array data were extracted from GSE45820 which contained mRNA expression levels of CD31 positive cardiac ECs isolated with flow cytometry cell sorting. These cardiac ECs were derived from mice with or without thoracic aorta constriction (TAC) (Moore-Morris et al., 2014). Relative gene expression between cardiac ECs from TAC mice was compared to wild type mice. We selected all genes encoding secreted proteins with at least a two-fold upregulation in mRNA expression and with a known function in adult cardiac physiology. The advantage of this strategy is that proteins were selected in a non-biased way. However, many secreted endothelial-derived proteins with important functions in cardiac biology will be missed using this strategy. For instance, neuregulin-1 upregulation in this micro-array database was less than two-fold, but its important roles in endothelial-cardiomyocyte communication have been well-established (Lemmens et al., 2006). Furthermore, one has to bear in mind that none of these proteins is produced exclusively by ECs. Proteins secreted by one specific cell type are rare in mammalian biology. Nevertheless, an endothelial source is an important physiological characteristic for a secreted protein, allowing specific regulatory features of synthesis and secretion and providing anatomical advantages for action. However, the endothelium is a fragile organ, easily disturbed during aging and environmental stimuli (Table 2). Therefore, an endothelial origin of a pathway may reveal an enhanced vulnerability of this pathway to certain (patho-)physiological conditions.
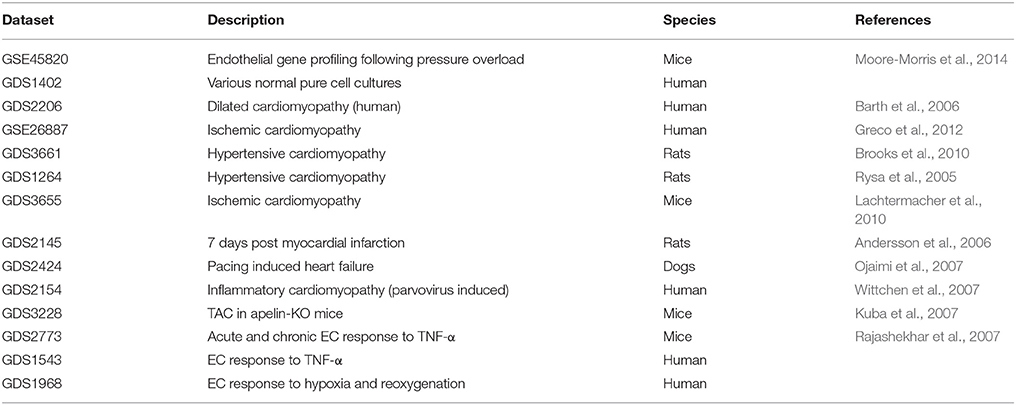
Table 1. Data sets used in this manuscript.
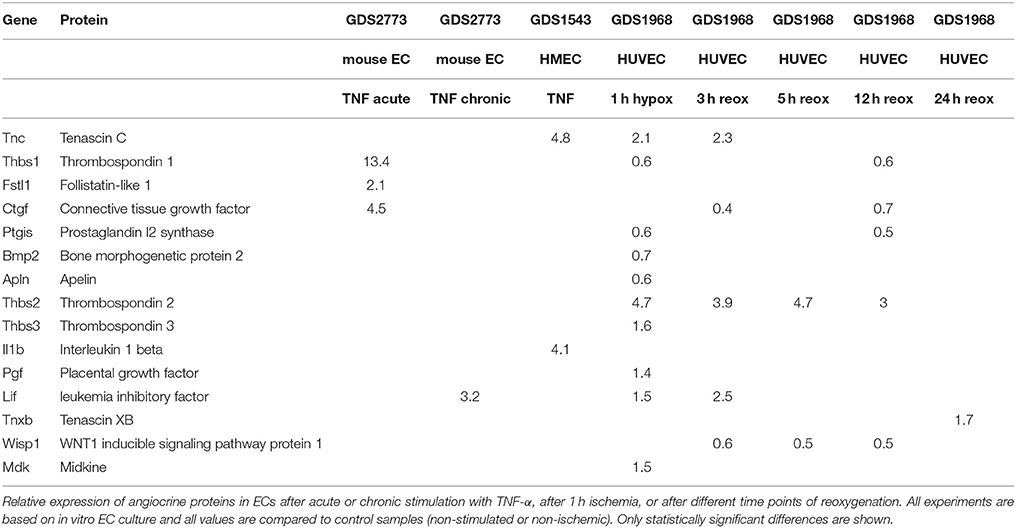
Table 2. Relative expression of angiocrine proteins upon TNF-α or hypoxia in cell culture.
This review is based on comprehensive search of peer-reviewed literature on PubMed. Inclusion of references was based on relevance to the topic, quality of the manuscript and consistency with the literature. Search terms included the following: heart, cardiac, ECs, endothelium, cardiomyocytes, fibroblasts, stem cell, hypertrophy, cardiac fibrosis, heart failure, cardiomyopathy, lactate, oxygen, vasodilation, mechanotransduction, adenosine, and vasopressin.
Furthermore, we checked expression of these proteins in cardiac ECs and other ECs using other publically available expression data sets. We confirmed the expression of all proteins mentioned in this manuscript using publicly available datasets of various normal pure human cells including cardiac microvascular ECs and cardiac fibroblasts (GDS1402, Table 3). We also checked expression of these genes in samples from human myocardial biopsies of patients with dilated cardiomyopathy (GDS2206), samples from hypertrophic hearts induced by exercise in rats (GDS654), and samples from hypertrophic hearts from various induction models (GDS598).
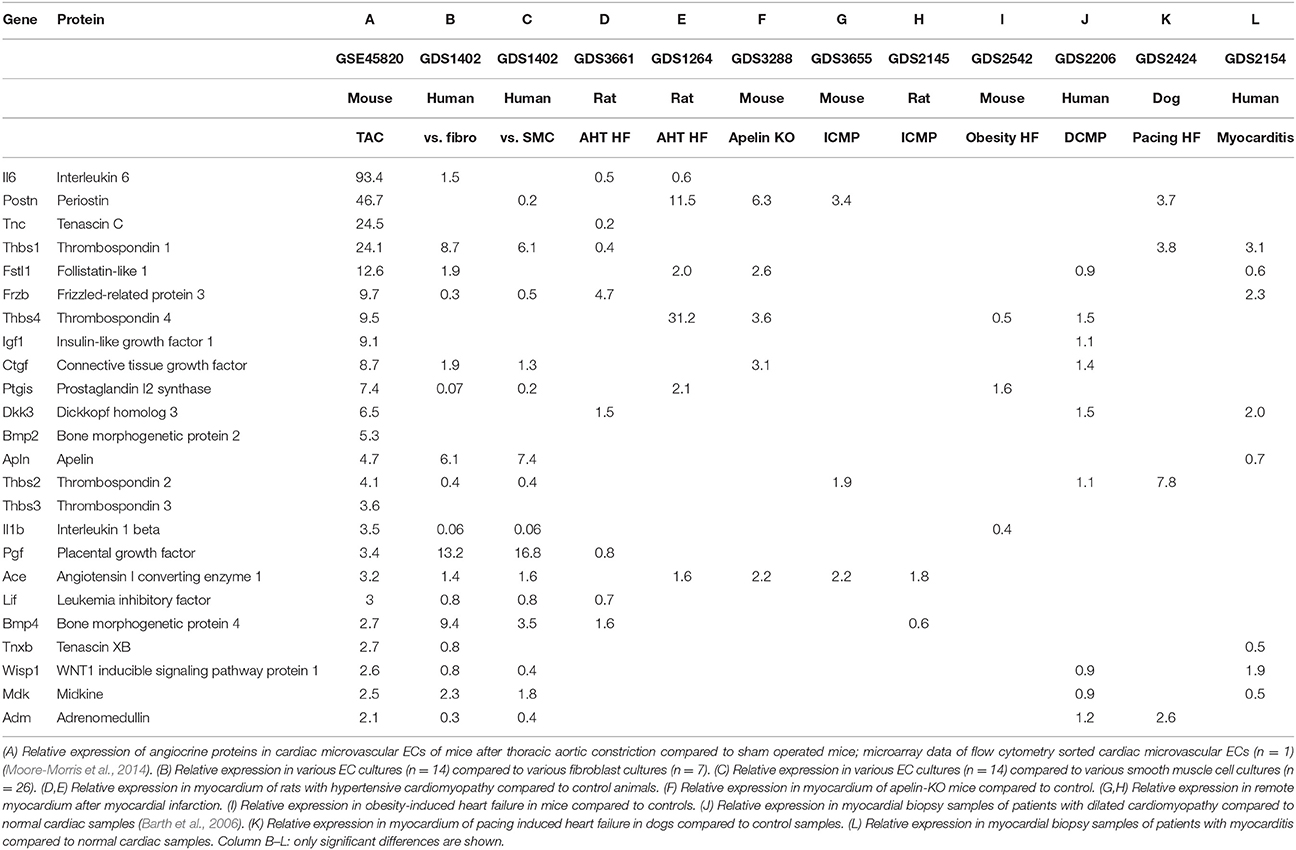
Table 3. Relative expression of angiocrine proteins in models of cardiac overload or compared to other cell types.
Cardiac Microvascular Endothelial Cells
Cardiac muscle is a tissue with high metabolic needs and therefore receives blood supply from a dense vascular and capillary network. Capillary density in the myocardium is around 3,000–4,000/mm2, which is substantially higher than in skeletal muscle where it is around 500–2,000/mm2 (Duncker and Bache, 2008). Microvascular ECs lining these capillaries not only serve as a barrier between blood and the myocardial tissue, but also communicate with adjacent cardiomyocytes by exchanging small molecules, peptides, proteins, microvesicles, and microRNAs (Figure 2) (Brutsaert, 2003). These secreted angiocrine substances constitute the endothelial effector function of the myocardium. Conceptually, one could discriminate the effector functions based on the target cell type, but alternatively one could also discriminate based on target processes, e.g., hypertrophy or fibrosis. The effector function of ECs has been first described almost 30 years ago, when it was shown that vascular ECs produce NO which induces relaxation of underlying smooth muscle cells (Palmer et al., 1987). Subsequently it has been shown that NO produced by endocardial endothelium also modulates contractility of cardiomyocytes (Brutsaert, 2003). Later, it has been shown that ECs communicate with cardiomyocytes by other signal molecules including prostaglandins and short peptides like endothelin (Brutsaert, 2003; Kamo et al., 2015). In recent years it has also been shown that proteins can modulate cardiac contractility (Lemmens et al., 2004) and have protective effects on cardiac remodeling (Liu et al., 2006): the best characterized example is neuregulin-1 (Vermeulen et al., 2016, 2017).
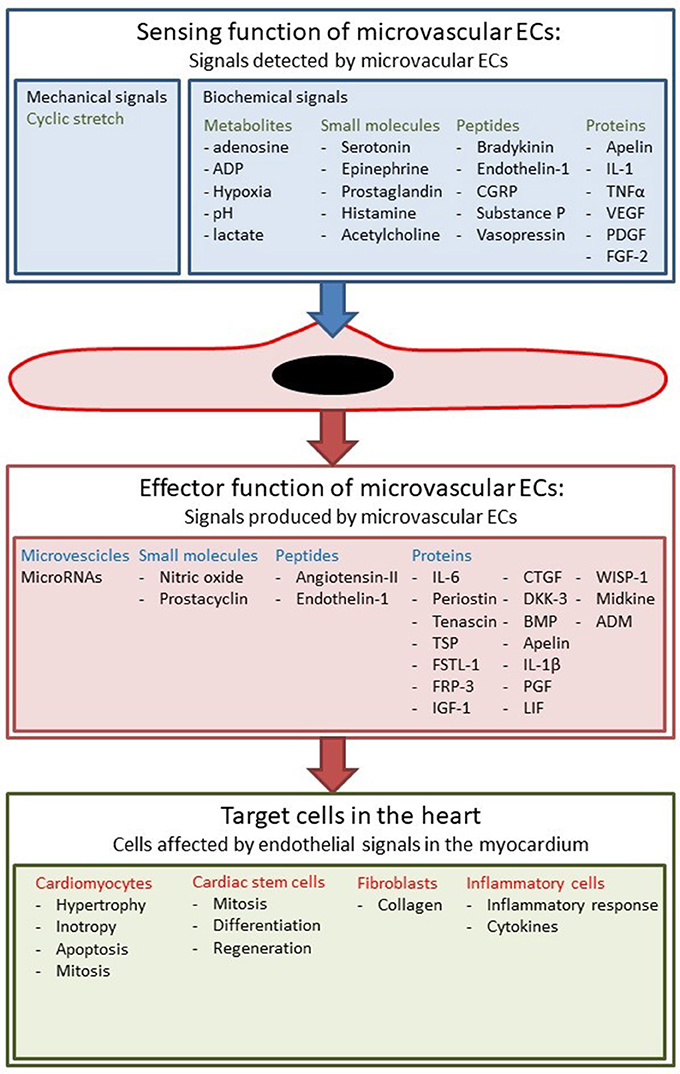
Figure 2. Sensing and effector function of cardiac ECs. ECs sense different biochemical and mechanical stimuli and communicate with other cell types in the myocardium.
Besides this effector function, ECs also have a sensing function to detect changes in hemodynamic, chemical, neurohormonal, and mechanical stimuli (Figure 2). The best known example of this sensing function is the secretion of vasodilatory substances such as NO in response to changes in shear stress (Chatzizisis et al., 2007; Duncker and Bache, 2008; Davies, 2009). However, shear stress is only important in arteries and larger arterioles, because flow rates in the microcirculation are much lower (Boulpaep, 2009). Nevertheless, ECs in specific microcirculations such as the heart or skeletal muscle are subjected to mechanical stress such as cyclical stretching and compression, and load-dependent strain. Furthermore, all ECs have receptors for metabolites, neurohormonal factors, cytokines, and growth factors; they harbor these receptors not only to regulate their own cellular physiology, but also to transduce signals to neighboring cells, for instance underlying cardiomyocytes. An interesting example is the responsiveness of ECs to estrogens by secreting more NO, a phenomenon that could explain some of the gender differences in many cardiovascular diseases (Gavin et al., 2009).
Cardiac Endothelial Cells Secrete Small Molecules That Modulate Cardiac Contractility and Cardiac Remodeling
ECs have many effector functions that occur in different organs such as regulation of coagulation or inflammatory cell infiltration, but they have also effector functions that are specific to certain tissues. ECs located in epicardial coronary arteries are a small minority of all ECs in the heart, but their role in modulating vascular smooth muscle function is extensively studied (Duncker and Bache, 2008). In this review we focus on the effector function of the microvascular ECs in the heart.
The vast majority of ECs in the heart are located in the microcirculation. These ECs produce paracrine factors, which can modulate cardiomyocyte contractility, growth and survival (Figure 3). Similar to ECs in coronary arteries, these paracrine factors include NO, prostacyclin, Ang-II, and ET-1 (Brutsaert, 2003). Detailed discussion of the paracrine effects of NO is outside the scope of this review (Brutsaert, 2003; Balligand et al., 2009). In small concentrations, NO has positive inotropic effects, whereas in higher concentrations it has negative inotropic effects (Mohan et al., 1996; Brutsaert, 2003; Balligand et al., 2009). The most reproducible effect of NO on cardiac contractility, however, is an earlier onset of relaxation (positive lusitropy) resulting in a longer diastole and favoring diastolic filling and coronary perfusion (Brutsaert, 2003; Balligand et al., 2009). In the long run, production of NO by endothelial NOS has antihypertrophic effects in models of cardiac hypertrophy (Palmer et al., 1987; Massion and Balligand, 2007). Paulus et al. recently proposed a novel paradigm for pathophysiology of heart failure with preserved ejection fraction (HFpEF). In this paradigm, a co-morbidity-induced dysfunction of cardiac microvascular endothelium plays a central role in development of cardiomyocyte hypertrophy and stiffness (Paulus and Tschope, 2013). Microvascular endothelial dysfunction leads to decreased NO production, decreased cGMP content and protein kinase G (PKG) activity in adjacent cardiomyocytes which results in development of hypertrophy and increased cardiomyocyte stiffness (Paulus and Tschope, 2013).
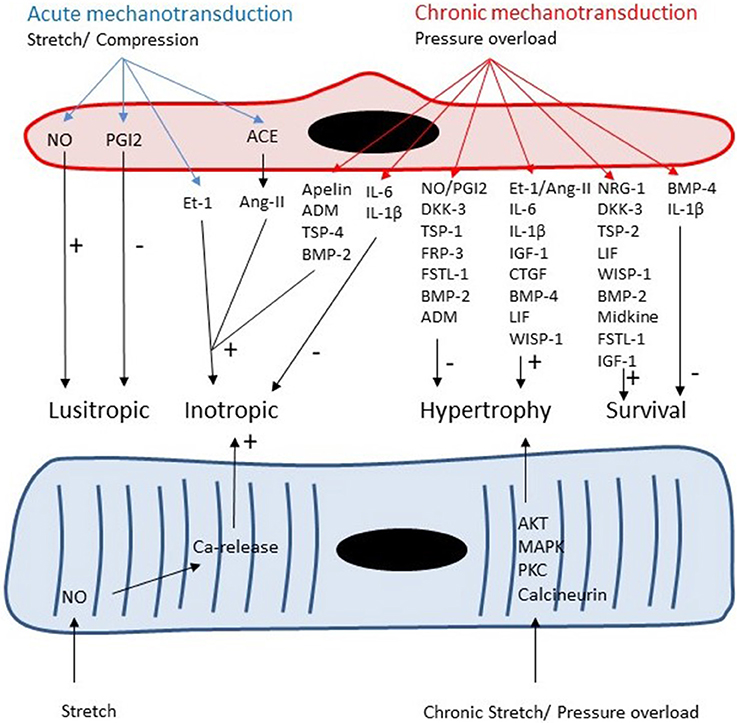
Figure 3. Both cardiomyocytes and microvascular ECs are responsive to acute and chronic changes in loading conditions. Autocrine and paracrine signaling leads to acute changes in lusitropy and inotropy of cardiomyocytes and to chronic changes in cardiomyocyte growth and survival.
The effects of prostacyclin on cardiac contractility range from a positive to a negative inotropic effect (Brutsaert, 2003). The main effect of prostacyclin on contractility is a delayed onset of relaxation and this effect opposes the action of NO (Brutsaert, 2003). The role of prostacyclin in cardiac remodeling is less well defined, but there is evidence that prostacyclin has anti-hypertrophic effects (Ritchie et al., 2004) and that the hypertrophic response is exaggerated in prostacyclin-receptor knockout mice (Hara et al., 2005; Harding and Murray, 2011). For the effects of other prostaglandins on cardiac remodeling, we refer the reader to ref (Harding and Murray, 2011). In the dataset used in this manuscript to select endothelium-derived proteins, prostaglandin I2 synthase mRNA is upregulated 7.4-fold in ECs derived from left ventricle of mice after aortic banding (Table 3).
Locally produced Ang-II is important in normal cardiac function with the most consistent effect being positive inotropy (Freer et al., 1976; Baker and Singer, 1988) and delayed relaxation (Meulemans et al., 1990; Brutsaert, 2003). The role of the renin-angiotensin-aldosterone system in cardiac hypertrophy is well characterized and led to the successful implementation of ACE inhibitors and angiotensin receptors blockers in daily clinical practice of heart failure (Weber and Brilla, 1991; Sadoshima and Izumo, 1993; Paul et al., 2006). In the dataset used in this manuscript to select proteins, Angiotensin converting enzyme (ACE) mRNA is upregulated 3.2-fold in ECs after aortic banding (Table 3).
The effects of Et-1 on cardiac contractility are diverse, but the most reproducible response is a positive inotropic effect (Moravec et al., 1989). Long term activation of the Et-1 pathway induces a hypertrophic response in cardiomyocytes and has been implicated in heart failure soon after its discovery (Drawnel et al., 2013); circulating and tissue levels of Et-1 are increased in patients with heart failure (Lerman et al., 1992; Loffler et al., 1993). However, studies with Et-1 receptor blockers in patients with heart failure have been disappointing (O'connor et al., 2003; Anand et al., 2004). This could be partly explained by the essential role of Et-1 for maintenance of normal cardiac contractility and for the adaptive stress response of cardiac tissue (Hathaway et al., 2015). Furthermore, Et-1 has been shown to have anti-apoptotic properties on cardiomyocytes (Kakita et al., 2001; Ogata et al., 2003; Drawnel et al., 2013). Interestingly, endothelium-specific Et-1 knockout mice show an exaggerated hypertrophic response to aortic banding (Heiden et al., 2014).
Microvascular Endothelial Cells Secrete Proteins That Modulate Cardiomyocyte Function and Cardiac Remodeling
ECs could be viewed as a single continuous organ of considerable size throughout the organism, instead of an additional cell type present in separate organs. They form an active secretory organ that not only has a major influence on the proteome in blood plasma but also on the proteome in the interstitial space of the capillaries. The secretome of ECs plays an essential role in development and normal physiology of all organs. In the heart, ECs are essential for normal development of the heart through various pathways including Notch and Wnt signaling pathways. The endothelial secretome is crucial in adult myocardium for maintenance of normal myocardial function and adequate response to various hemodynamic stimuli (e.g., pressure overload).
Data from a recently performed micro-array by Moore-Morris et al. show a marked upregulation of various secreted proteins by cardiac microvascular ECs upon chronic pressure overload in mice (Moore-Morris et al., 2014). Upregulated angiocrine proteins with a known cardiac function are shown in Table 3. This list cannot be assumed to be complete, because the micro-array is based on a single sample of sham and TAC operated mice precluding statistical analysis. However, this list is remarkably similar to a list that can be construed by comprehensive review of the literature on the effects of various secreted proteins on cardiac function, hypertrophy and remodeling. The fact that cells were freshly isolated from intact hearts with flow cytometry has two important advantages: the cells that were isolated are pure ECs and gene expression is analyzed on cells in a condition that matches their in vivo condition as closely as possible. There are also some drawbacks in using this list of proteins. For example, proteins that regulate cardiac contractility but not cardiac remodeling are perhaps not represented, neither proteins that are regulated by posttranslational modifications instead of transcription. We will use this list of proteins with differential expression between pressure-overloaded and normal hearts as an index list of proteins for further review, but one should keep in mind the advantages and drawbacks discussed.
We confirmed expression of these genes in cardiac microvascular ECs based on publicly available microarray data on various pure cell cultures (GEO Dataset: GDS1402). This microarray experiment contains 16 primary EC cultures, 7 fibroblast cell cultures, and 26 vascular smooth muscle cell cultures. We compared expression of different genes between ECs and fibroblasts and between ECs and vascular smooth muscle cells (Table 3). Table 3 only shows values for expression levels that are significantly different; for a number of proteins—e.g., TSP-4, IGF-1, or BMP-2—expression levels in ECs are comparable to expression in fibroblasts and vascular smooth muscle cells. Some proteins—e.g., IL-1β or prostacyclin synthase—have a higher expression in vascular smooth muscle cells and fibroblasts compared to ECs. Expression of TSP-1, BMP-4, and PGF is markedly higher in ECs compared to fibroblasts or vascular smooth muscle cells. These micro-arrays have been performed on cultured cells and one has to be careful with extrapolating these data to the in vivo situation because expression levels may be altered by the artificial cell culture environment and growth factors used in cell culture medium.
We evaluated expression of a panel of angiocrine proteins in micro-array experiments of different heart failure models, including biopsy samples of different forms of cardiomyopathy in humans and different animal models of heart failure (Table 3). Different forms of heart failure are included in this experiment: hypertensive cardiomyopathy, ischemic cardiomyopathy, dilated cardiomyopathy, myocarditis, and obesity induced cardiomyopathy. Most of the angiocrine proteins are up- or down-regulated in one or more of these heart failure models (Table 3), but none of the proteins is significantly altered in all of them. The large variety in underlying pathophysiology of these heart failure models is the main reason for variability in expression levels of angiocrine proteins. We included different models of heart failure, because heart failure is a heterogeneous disease, not only because of different causal factors, but also because of differences in genetic susceptibility, comorbidities, and even differences in a single patient when disease progresses over time. Furthermore, in contrast to the experiment performed by Moore-Morris et al. (first column of Table 3), all these expression data are based on biopsies or tissue samples and therefore are a mixture of different cell types. Although the number of cardiomyocytes and ECs can be expected to remain fairly constant, induction of heart failure will lead to changes in relative abundances of different cell types in the heart and therefore might affect expression levels. Relative changes in cell numbers will be different between different models of heart failure: e.g., fibroblast proliferation is more pronounced in certain models. Another caveat when interpreting Table 3 is the fact that not all genes are included in all micro-arrays, e.g., TSP-3 is only present in a minority of micro-array panels.
Abundance of angiocrine proteins is not only dependent on transcriptional activity, but also on translation, posttranslational modification and secretion. Therefore, we searched literature for mass-spectrometry data on the secretome of ECs. Specific data on cardiac microvascular ECs are not available, but mass spectrometry data have been published on the secretome of HUVECs (Tunica et al., 2009), endothelial progenitor cells (Hemmen et al., 2011), and EA.hy926 ECs (HUVEC hybridoma cell line) (Brioschi et al., 2013; Kwon et al., 2015).
A recent study investigated the in vitro response of endothelial responses to endotoxins (Kwon et al., 2015). Although the method used in this study simulates the pathophysiology of sepsis rather than cardiac remodeling, many inflammatory pathways are also activated in cardiac remodeling. Interestingly, endotoxins upregulate secretion of some of the proteins present in our index list; e.g., thrombospondin-1 secretion increases 1.2-fold, follistatin-related protein 1 secretion increases 1.2-fold, and connective tissue growth factor increases 1.8-fold (Kwon et al., 2015; Table 4). In a separate mass-spectrometry study in the same EC line it was shown that atorvastatin decreases protein secretion of thrombospondin-1, thrombospondin-2, and connective tissue growth factor (Brioschi et al., 2013). HMG-CoA reductase inhibitors have been said to have pleiotropic effects on other organ systems besides their cholesterol lowering effects (Mihos et al., 2014). Stimulation or inhibition of specific angiocrine proteins could be part of these pleiotropic effects of statins.
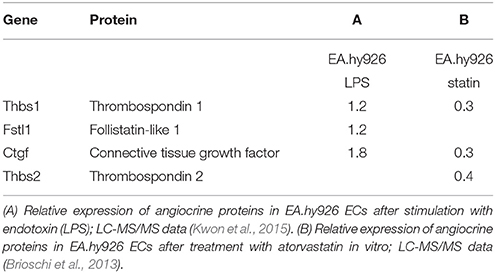
Table 4. Expression of angiocrine proteins as determined by mass-spectrometry.
Fibroblasts are generally considered to be the main source of extracellular matrix proteins, but ECs themselves are an important source of extracellular matrix proteins as well. ECs increase production of extracellular matrix proteins in response to pressure overload (Table 5) and therefore could significantly contribute to formation of extracellular matrix during fibrogenesis. Moreover, ECs also secrete numerous pro-fibrotic factors in response to hemodynamic stress.
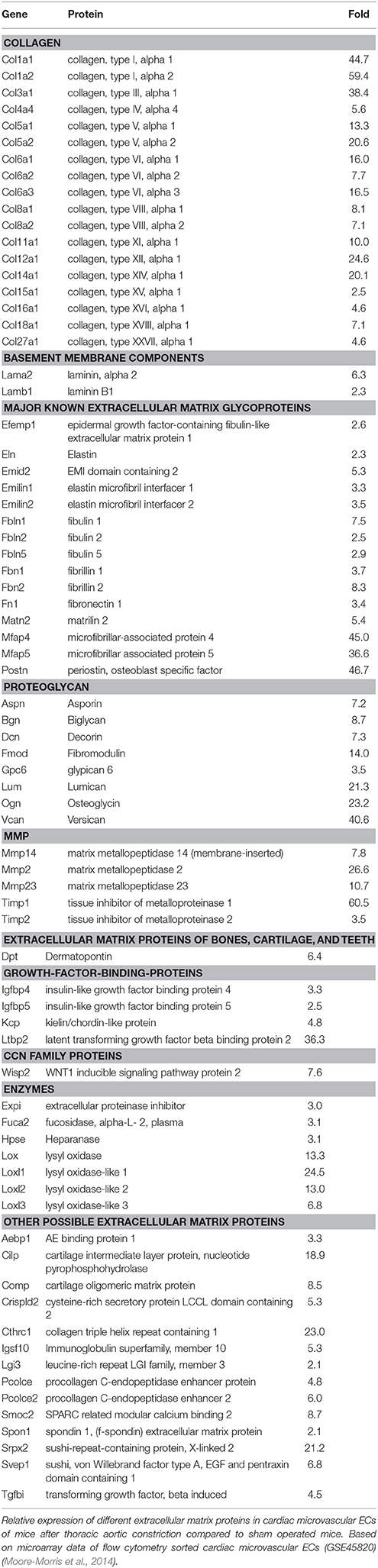
Table 5. Expression of extracellular matrix proteins by endothelial cells during cardiac overload.
Endothelium-Derived Proteins Modulating Cardiac Contractility and Cardiac Remodeling
In this section, we will discuss endothelium-derived proteins with known effects on cardiac function and/or remodeling. All proteins showed an increased expression in endothelial cells in response to pressure overload (Table 3), and they will be discussed in order of magnitude of this response.
Interleukin-6
It is well established that inflammatory cytokines including tumor necrosis factor-α, interleukin-1, and interleukin-6 (IL-6) play important roles in early and later stages of cardiac remodeling and heart failure (Paulus, 2000). In heart failure patients, inflammatory cytokines are elevated in the myocardium but also in plasma and have paracrine and endocrine functions (Paulus, 2000). Inflammatory cytokines can be produced by immune cells but also other cell types including ECs. Many excellent reviews cover the role of IL-6 signaling pathways in heart failure and cardiac remodeling (Fischer and Hilfiker-Kleiner, 2007; Fontes J. A. et al., 2015).
The effects of IL-6 on cardiomyocyte contractility have been well documented in vitro. IL-6 induces reversible negative inotropic effects on isolated hamster papillary muscles (Finkel et al., 1992), downregulation of SERCA2 in neonatal rat ventricular myocytes (Villegas et al., 2000), reduced expression of cardiac myosin heavy chain isoforms, and a loss of cardiac actin in rat cardiac myocytes (Patten et al., 2001). The negative inotropic effects of IL-6 in isolated cardiomyocytes can be partially explained by stimulation of iNOS expression and NO production (Yu et al., 2003). IL-6 increases NO production through activation of the JAK2/STAT3 pathway (Yu et al., 2003).
Besides negative inotropic effects, IL-6 also has clear effects on cardiac remodeling. Inhibition of IL-6 in a mouse model of transplant rejection decreases cardiomyocyte hypertrophy and cardiac fibrosis, indicating that IL-6 has pro-hypertrophic and pro-fibrotic properties (Diaz et al., 2009). The pro-hypertrophic effects of IL-6 have been recently confirmed in mice with a genetic deletion of IL-6, which showed attenuation of the hypertrophic response to pressure overload (Zhao et al., 2016) and attenuation of the fibrotic response to Ang-II infusion (González et al., 2015). Moreover, experiments with transgenic mice overexpressing α-adrenergic receptors suggest that part of the hypertrophic response of cardiomyocytes to catecholamines is mediated by endothelium-derived IL-6 (Papay et al., 2013; Figure 4). Furthermore, endothelium-derived IL-6 has also been implicated in the adaptive hypertrophic response to placental growth factor, an endothelial growth factor (Accornero et al., 2011). As discussed in a later section, placental growth factor stimulates EC growth and release of growth factors—including IL-6—from ECs (Accornero and Molkentin, 2011), and thus has indirect trophic effects on myocytes.
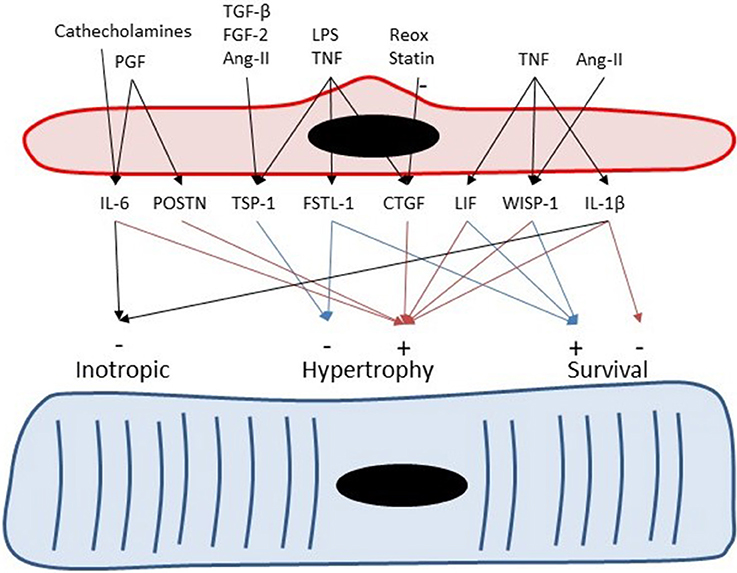
Figure 4. Endothelial cells can serve as paracrine intermediates. Placental growth factor (PGF) induces expression of periostin (Postn) and IL-6 in non-myocytes including cardiac microvascular ECs. Periostin and IL-6 have been implicated to play a role in adaptive hypertrophy induced by PGF (Accornero et al., 2011). Catecholamines induces cardiac hypertrophy partly by induction of endothelium-derived IL-6 (Papay et al., 2013). Transforming growth factor-β, fibroblast growth factor-2 (Frangogiannis et al., 2005), and Ang-II (Fischer et al., 2001) induce TSP-1 expression in CMVECs.
Subcutaneous injection of high doses of recombinant IL-6 in wild-type rats induces a dose-dependent dilatation of the left ventricle leading to heart failure within 7 days (Janssen et al., 2005), whereas continuous infusion of a lower doses of IL-6 in rats leads to ventricular hypertrophy and fibrosis (Meléndez et al., 2010). Consistently, administration of an antibody against the IL-6 receptor decreases cardiac remodeling after myocardial infarction in mice (Kobara et al., 2010). These experiments indicate that increased levels of IL-6 have detrimental effects on cardiac remodeling. However, other experiments indicate that IL-6 has protective effects on cardiac biology as well. For instance, IL-6 KO mice show cardiac dilatation, increased accumulation of interstitial collagen, and a decreased capillary density (Banerjee et al., 2009).
Periostin
Periostin is a secreted protein of 90 kDa, which contains 4 repetitive fasciclin domains and is involved in cell adhesions (Snider et al., 2009). Periostin is involved in normal fibrogenesis but also pathological fibrosis and interacts directly with other extracellular matrix proteins such as fibronectin, tenascin-C, and collagen I/V (Snider et al., 2009). Periostin can serve as a ligand for selected integrins, such as αvβ3, αvβ5, and α4β6, where it can affect the ability of cells to migrate or undergo a epithelial-mesenchymal transformation during pathological disease progression (Conway and Molkentin, 2008). In the adult heart periostin is induced following myocardial infarction, pressure overload, or generalized cardiomyopathy (Conway and Molkentin, 2008; Frangogiannis, 2012). The effects of periostin on cardiomyocyte contractility are unknown, but periostin does play a role in myocardial fibrosis and hypertrophy (Frangogiannis, 2012). It has been shown that periostin knockout mice have reduced fibrosis and hypertrophy following pressure overload, whereas periostin overexpressing transgenic mice spontaneously developed hypertrophy with aging (Oka et al., 2007).
It has been suggested that recombinant periostin had regenerative properties and can induce cardiomyocyte proliferation after myocardial infarction (Kuhn et al., 2007), but these results have been contested by other investigators (Conway and Molkentin, 2008). Therefore, more studies are needed to investigate that regenerative properties of periostin.
Tenascin-C
Tenascins (Tn) are a family of multimeric extracellular matrix glycoproteins characterized by a N-terminal globular domain and heptad repeats, which facilitate multimerization (Tucker and Chiquet-Ehrismann, 2009). Tenascins play important roles in cell adhesion and motility (Tucker and Chiquet-Ehrismann, 2009). Tn-C is the best characterized tenascin and is highly expressed in tendons and embryonic extracellular matrix (Tucker and Chiquet-Ehrismann, 2009). Tn-C has a wide range of effects on cell adhesion, motility, differentiation, growth control, and extracellular matrix organization via multiple cell surface receptors (Tucker and Chiquet-Ehrismann, 2009). Tn-C is expressed in various ECs including aortic ECs, pulmonary artery ECs, and HUVECs (Golledge et al., 2011; Table 6). Tn-C is secreted by ECs, but also has dynamic effects on ECs by inhibiting cardiac EC spreading and enhancing migration in response to angiogenic growth factors (Ballard et al., 2006). Tn-C has both pro- and antiangiogenic properties (Tucker and Chiquet-Ehrismann, 2009).

Table 6. Various effects of endothelial derived proteins.
Tn-C is almost absent in normal adult myocardium, but reappears during cardiac remodeling in response to pathologic insults, such as acute myocardial infarction, myocarditis, ischemia-reperfusion injury, and hypertensive cardiac fibrosis (Imanaka-Yoshida, 2012). Studies on direct effects of Tn-C on cardiomyocyte hypertrophy are missing, but Tn-C plays an important role during cardiac remodeling by loosening cell adhesion, upregulating MMPs, and enhancing inflammatory responses (Imanaka-Yoshida, 2012). These effects of Tn-C help cell rearrangement and allow myofibroblasts and capillary vessels to spread into the restoring tissue, but these might also cause tissue vulnerability, resulting in ventricular dilatation (Imanaka-Yoshida, 2012). Consistently, deletion of TN-C significantly reduces ventricular remodeling and improves cardiac function after coronary artery ligation in mice (Nishioka et al., 2010).
Thrombospodins
Expression of thrombospondin-4 (TSP-4) has been documented in human coronary artery ECs and smooth muscle cells (Stenina et al., 2003). Pressure overload leads to an increase in TSP-1 and TSP-4 expression in cardiac tissue (Mustonen et al., 2008; Xia et al., 2011), which confirms the micro-array dataset of Table 3. More specifically, immunostaining localized TSP-4 to ECs in hypertrophied hearts of spontaneously hypertensive rats (Mustonen et al., 2008). Upregulation of TSP-4 expression by increased loading conditions has also been shown in models of vasopressin infusion or myocardial infarction (Dawson et al., 2013). Therefore, it has been suggested that TSP-4 is an endothelial specific marker of cardiac overload (Mustonen et al., 2008). TSP-1 also shows load-dependent expression in ECs (Dawson et al., 2013), but compared to TSP-4 also shows a more widespread expression in different cell types (Frangogiannis et al., 2005). Expression of TSP-1 in ECs is also regulated by transforming growth factor-β, fibroblast growth factor-2 (Frangogiannis et al., 2005), and Ang-II (Chua et al., 1997; Fischer et al., 2001) (Figure 4).
TSP-4 serves as an endothelium-derived mechano-signaling molecule with a central role in adaptive contractile responses to acute stress, and appears to play a crucial role in the transition to chronic cardiac dilatation (Cingolani et al., 2011). Hearts of TSP-4 KO mice failed to acutely augment contractility or activate stretch-response pathways on exposure to acute pressure overload (Cingolani et al., 2011). In these complete KO mice, TSP-4 was deleted in all cells. Isolated cardiac trabeculae of TSP-4 KO mice failed to enhance contractility and cellular calcium increase after stretch, in contrast to wild-type trabeculae (Cingolani et al., 2011). However, the contractility response could be restored in TSP-4 KO cardiac trabeculae incubated with recombinant TSP-4 (Cingolani et al., 2011). Interestingly, when TSP-4 KO myocytes were isolated, they responded normally to stretch, indicating that TSP-4 secreted by other cells—e.g., microvascular ECs—is crucial for a normal response to stretch.
TSP-1 KO mice display an increased hypertrophic response to pressure overload (Xia et al., 2011). Similarly, TSP-4 KO mice also display an increased hypertrophic response (Frolova et al., 2012; Palao et al., 2016). Little is known on the role of TSP-2 in cardiomyocyte hypertrophy, but TSP-2 has been shown to be protective against doxorubicin induced cardiomyocyte toxicity (van Almen et al., 2011). Furthermore, TSP-2 also protects against age-related dilated cardiomyopathy in mice (Swinnen et al., 2009). In summary, TSP-1,−2,−3, and−4 are upregulated in ECs during pressure overload, but most data in the literature focus on TSP-1 and−4, that both seem to serve as endothelium-derived anti-hypertrophic factors.
Besides having anti-hypertrophic properties, TSP-1 is an important activator of transforming growth factor-β signaling. Latency-associated peptide is a peptide that forms a complex with transforming growth factor-β and in this state inactivates transforming growth factor-β. Binding of TSP-1 to latency-associated peptide leads to a conformational change leading to release and activation of transforming growth factor-β (Frangogiannis, 2012). As stated before, TSP-1 expression is increased by pressure overload and plays an important role in the fibrotic response of myocardial tissue. TSP-1 null mice have a low collagen content in the heart with increased infiltration of dysfunctional fibroblasts (Xia et al., 2011). Hearts of TSP-1 null mice are prone to chamber dilatation in response to pressure overload because of loss of extracellular matrix integrity. In contrast to TSP-1 KO mice, TSP-4 KO mice display an increased deposition of extracellular matrix in response to pressure overload (Frolova et al., 2012), indicating that TSP-4 serves as an endothelium-derived suppressor of exaggerated fibrosis. It has also been shown that TSP-2 has anti-fibrotic effects in age-related cardiac dilatation (Swinnen et al., 2009).
Follistatin-Like 1
Follistatin-like 1 (FSTL-1) is an extracellular glycoprotein with limited homology to the follistatin family of proteins (Ouchi et al., 2008). It has been reported that FSTL-1 is secreted by skeletal myocytes (Ouchi et al., 2008), myocardial tissue after myocardial infarction (Oshima et al., 2008), and cardiomyocytes and nonmyocytes (Shimano et al., 2011). Expression of FSTL-1 in ECs has been confirmed by immunohistochemistry in synovial ECs (Li et al., 2011) and cardiac microvascular ECs (Shimano et al., 2011). Numerous studies indicate that FSTL-1 has cardioprotective properties during cardiac remodeling. FSTL-1 has anti-apoptotic effects on cardiomyocytes by stimulation of the AKT pathway (Oshima et al., 2008). Furthermore, FSTL-1 shows anti-hypertrophic properties in murine animal models (Shimano et al., 2011; Tanaka et al., 2016). Data from small and large animal models also indicate that FSTL-1 has protective effects on ischemia/reperfusion injury (Ogura et al., 2012). Moreover, data indicate that FSTL-1 is upregulated after myocardial infarction and prevents cardiac rupture by activating cardiac fibroblasts (Maruyama et al., 2016). Consistently, FSTL-1 also has pro-fibrotic effects in models of lung fibrosis (Dong et al., 2015).
Frizzled-Related Protein 3
Frizzled-related protein 3 (FRP-3) is a member of the Wnt signaling pathway and plays important roles during cardiac embryogenesis. Expression of FRP-3 in ECs has also been confirmed by immunohistological staining in reproductive tissues (Partl et al., 2014). Currently, there are no known effects of FRP-3 on cardiac contractility, but it has been shown that FRP-3 expression is increased in failing human myocardium, with a decline following LV assist device therapy (Askevold et al., 2014), indicating that FRP-3 is related to heart failure progression, and its secretion dependent on ventricular wall strain. It has also been shown that FRP-3 mRNA levels are increased in left ventricles of mice post-myocardial infarction (Askevold et al., 2014). Like other members of the Wnt signaling pathway, evidence indicates that FRP-3 plays a role in cardiac remodeling, but direct effects on cardiomyocytes or fibroblasts have not been examined yet.
Insulin-Like Growth Factor-1
Insulin-like growth factor-1 (IGF-1) is a protein with growth promoting actions on multiple tissues including the myocardium. IGF-1 is part of the growth hormone /IGF-1 pathway and promotes cell survival via the phosphatidylinositol 3 kinase (PI3K)/Akt signaling pathway (Figure 4; Michele et al., 2013). Expression of IGF-1 has also been confirmed in brain microvascular ECs (Wang et al., 2013), endothelial progenitor cells (Urbich et al., 2005), and adipose tissue microvascular ECs (Kern et al., 1989).
It has been well-established that IGF-1 plays important roles in physiological and pathological cardiac remodeling and heart failure (Ellison et al., 2012). IGF-1 promotes cardiac growth and improves cardiac contractility and ejection fraction (Ren et al., 1999). IGF-1 also improves cardiac function after myocardial infarction by promoting tissue remodeling (Ren et al., 1999). We refer the reader to many excellent reviews on the role of IGF-1 in cardiac remodeling (Ren et al., 1999; Opgaard and Wang, 2005; Michele et al., 2013).
Connective Tissue Growth Factor
Connective tissue growth factor (CTGF) is a protein that plays an essential role in skeletal development and extracellular matrix production (Accornero et al., 2015). CTGF plays crucial roles in fibrotic responses, for instance in models of bleomycin-induced skin fibrosis (Liu et al., 2010). CTGF binds with various proteoglycans and integrins, but a specific cell surface receptor for CTGF has not been identified, so it is currently unclear how CTGF modifies cellular responses to injury. Although CTGF expression is strongly induced during cardiac remodeling, its role in cardiac remodeling remains controversial. Recent studies using transgenic mice suggest that CTGF is not involved in cardiac remodeling, hypertrophy, or fibrosis at baseline, nor with aging, after pressure overload, or with neuroendocrine agonist stimulation (Accornero et al., 2015; Fontes M. S. et al., 2015). However, in vitro data indicate that CTGF induces hypertrophy in cardiomyocytes (Hayata et al., 2008).
Dickkopf-3 (DKK-3)
Dickkopf (DKK) proteins are secreted regulators of the Wnt signaling pathway which include DDK-1 to DKK-4 (Krupnik et al., 1999; Niehrs, 2006). DKK-3 plays a role in embryological development of various organs including the heart, bone, the neural epithelium, and limb buds (Niehrs, 2006). DKK-3 is also expressed in various adult tissues and acts as a tumor suppressor of different malignancies (Veeck and Dahl, 2012). Expression of DKK-3 in tumor tissues is mostly located in microvascular ECs (Untergasser et al., 2008; Fong et al., 2009). DKK-3 is expressed in the adult heart (Krupnik et al., 1999), more specifically in microvascular ECs (Fong et al., 2009; Tunica et al., 2009; Zhang et al., 2014).
DKK-3 has been identified as a protein playing a role in cardiac remodeling. Genomic studies have shown that DKK3 is negatively correlated with myocardial mass in rat models of hypertrophy (Cerutti et al., 2006). Furthermore, it has been shown that overexpression of DKK-3 protects against aortic banding induced hypertrophy whereas knockout of DKK-3 aggravates the hypertrophic response (Zhang et al., 2014). It has also been shown that DKK-3 has protective effects in models of dilated cardiomyopathy (Lu et al., 2016) and myocardial infarction (Bao et al., 2015).
Bone Morphogenetic Protein-2 and -4
Bone morphogenetic protein-2 (BMP-2) and bone morphogenetic protein-4 (BMP-4) play important roles during heart development in vertebrates and are members of the transforming growth factor-β superfamily (Balligand et al., 2009). The role of BMP-2 in regeneration of the adult heart is incompletely defined, but BMP-2 is frequently used to stimulate differentiation of stem cells for cardiac regeneration therapies (DeGeorge et al., 2008; Kim et al., 2011; Wan et al., 2011). Similarly, BMP-4 has been shown to induce differentiation of embryonic stem cells (Hosseinkhani et al., 2007) and induced pluripotent stem cells into cardiomyocytes (Ren et al., 2011). However, BMP-4 has also been reported to have inhibiting properties on cardiomyocyte differentiation (Taha et al., 2007). It has been shown that cyclic stretch of ECs in vitro induces expression of BMP-2 (Balligand et al., 2009) and that BMP-2 has positive inotropic effects on isolated adult cardiomyocytes by activation of PI3K (Ghosh-Choudhury et al., 2003).
Recent evidence indicates that BMP-4 as well as BMP-2 also play key roles in cardiac remodeling. Whereas BMP-2 has anti-hypertrophic effects on adult cardiomyocytes, BMP-4 has pro-hypertrophic and pro-apoptotic effects on cardiomyocytes (Sun et al., 2013; Lu et al., 2014). Moreover, BMP-4 also has pro-fibrotic effects (Sun et al., 2013) whereas BMP-2 has anti-fibrotic effects (Wang et al., 2012).
Moreover, BMP-2 promotes angiogenesis in various tumors (Langenfeld and Langenfeld, 2004) and during osteogenesis (Carano and Filvaroff, 2003). BMP-4 has also potent pro-angiogenic properties and induces capillary sprouting (Zhou et al., 2007).
Apelin
Apelin is the ligand for the previously orphaned G protein–coupled receptor APJ (Japp et al., 2010). Apelin is expressed throughout the organism with particularly high levels in vascular endothelium (Chandrasekaran et al., 2008). APJ receptors are present on many different cell types including ECs, cardiomyocytes, and vascular smooth muscle cells (Japp et al., 2010). Apelin exerts major effects on both vascular tone and cardiac contractility. Both in isolated rat hearts and in vivo, apelin is a positive inotropic substance (Szokodi et al., 2002; Ashley et al., 2005). Together with Et-1 and adrenomedullin, apelin is among the most potent endogenous inotropic substances on a molar base (Szokodi et al., 2002). Apelin exerts its inotropic action by increasing the availability of intracellular calcium rather than enhancing myofilament calcium sensitivity (Chandrasekaran et al., 2008). In the failing heart, this increase in contractility is even more pronounced (Chandrasekaran et al., 2008). Apelin not only increases inotropy, but also decreases left ventricular pre- and afterload by its pronounced vasodilatory effects (Ashley et al., 2005). Therefore, apelin seems to be a beneficial endothelium-derived protein that increases inotropy and decreases cardiac loading when the myocardium is confronted with pressure overload. Interestingly, unlike most other inotropic agents, apelin does not induce cardiomyocyte hypertrophy (Chandrasekaran et al., 2008).
It has recently been shown that apelin also has anti-fibrotic effects in models of pressure overload (Pchejetski et al., 2012; Zhong et al., 2016) and myocardial infarction (Zhang et al., 2016) with direct inhibitory effects on fibroblasts (Pchejetski et al., 2012; Zhong et al., 2016).
Interleukin-1β
Interleukin-1β (IL-1β) is an inflammatory cytokine which is expressed in multiple tissues and by multiple cell types including ECs. In experimental models of pressure overload and cardiac hypertrophy, IL-1β expression is upregulated in the hypertrophied heart, predominantly localized in ECs and interstitial macrophages (Bujak and Frangogiannis, 2009). Similar to IL-6, IL-1β also has a negative inotropic effect on cardiomyocytes (Bujak and Frangogiannis, 2009). This negative inotropic effect is mediated through NO-dependent and NO-independent pathways (Bujak and Frangogiannis, 2009). Furthermore, IL-1β inhibits the β-adrenergic agonist-mediated increase in cAMP and cardiomyocyte contractility and IL-1β is an essential mediator in sepsis-induced contractile dysfunction (Bujak and Frangogiannis, 2009).
Extensive evidence suggests that IL-1 has pro-hypertrophic and pro-apoptotic effects on cardiomyocytes (Bujak and Frangogiannis, 2009). IL-1β induces cardiomyocyte apoptosis by activation of Bak and Bcl-xL through pathways involving NO (Bujak and Frangogiannis, 2009). Furthermore, IL-1β induces cardiomyocyte hypertrophy, upregulates atrial natriuretic factor (ANF) and suppresses expression of calcium regulatory genes (Bujak and Frangogiannis, 2009). Furthermore, IL-1β has well known pro-inflammatory properties. In IL-1-receptor KO hearts, collagen deposition was markedly decreased, in both the healing scar and the peri-infarct area (Bujak and Frangogiannis, 2009). IL-1β directly enhances fibrosis by upregulating expression of Ang-II receptors on cardiac fibroblasts and by stimulating fibroblast migration (Bujak and Frangogiannis, 2009). Beyond its pro-inflammatory and fibrogenic properties, IL-1 also promotes extracellular matrix remodeling by enhancing matrix metalloproteinase expression (Bujak and Frangogiannis, 2009).
Placental Growth Factor
Placental growth factor (PGF) is secreted by cardiomyocytes (Accornero and Molkentin, 2011) but also by cardiac microvascular ECs in which expression is upregulated by chronic pressure overload (Table 3) (Accornero and Molkentin, 2011; Moore-Morris et al., 2014). PGF is part of the vascular endothelium growth factor superfamily and binds to the VEGF-1 receptor which is expressed by ECs (Accornero et al., 2011). PGF has a limited role in normal cardiac homeostasis, but has been shown to be crucial in adaptive angiogenic responses (Accornero and Molkentin, 2011). Because CMVECs are at the same time both secretor and receptor cells for PGF, PGF might be part of an autocrine endothelial signaling system.
Deletion or overexpression of PGF does not alter cardiac function or morphology at baseline, but PGF is an essential component of the hypertrophic response to pathological stimuli such as pressure overload (Accornero et al., 2011). In contrast to wild-type mice, PGF KO mice do not form additional capillaries in response to aortic banding and rapidly develop heart failure (Accornero et al., 2011), whereas mice overexpressing PGF show an increased angiogenic response. PGF expression increases in response to hypertrophic stimuli (Accornero and Molkentin, 2011) and stimulates EC growth but also secretion of growth factors from ECs and fibroblasts, including IL-6 and periostin (Accornero and Molkentin, 2011). These growth factors stimulate cardiomyocyte cell growth. Based on these data, it has been suggested that PGF is a stress-response factor that suppresses disease in the heart by maintaining capillary/vessel density as well as providing protective trophic effects to cardiomyocytes (Accornero and Molkentin, 2011).
Leukemia Inhibitory Factor
Leukemia inhibitory factor (LIF) is a member of the IL-6 family of cytokines and induces hypertrophy in cardiomyocytes by activation of the LIF receptor and gp130, which functions as a co-receptor (Kodama et al., 1997). The hypertrophic effects of LIF are mediated by the JAK-STAT signaling pathway (Kodama et al., 1997). Most of these pro-hypertrophic effects have been demonstrated in isolated cardiomyocytes, but little is known on the role of LIF in pathophysiology of cardiac remodeling or heart failure (Zouein et al., 2013). However, a large body of evidence exists on the protective effects of the JAK-STAT pathway during acute cardiac stress (Zouein et al., 2013) and it has been shown that LIF protects against ischemia-reperfusion injury (Zouein et al., 2013).
LIF may play a role in cardiac regeneration as it has been shown to contribute to homing of bone marrow-derived progenitors, as well as differentiation of resident cardiac stem cells into ECs (Zouein et al., 2013). LIF not only protected against cardiomyocyte death in a mouse myocardial infarction model, but enhanced neovascularization, and induced homing of bone marrow cells to the heart and their differentiation into cardiac myocytes (Zouein et al., 2013).
Wnt1-Induced Secreted Protein-1 (WISP-1)
Wnt1-induced secreted protein-1 (WISP-1) is a member of the cysteine-rich 61, CTGF, and nephroblastoma overexpressed (CCN) family of growth factors (Colston et al., 2007). WISP-1 expression is upregulated during cardiac remodeling and expression is stimulated by tumor necrosis factor (Venkatachalam et al., 2009). It has been shown that WISP-1 induces cardiomyocyte hypertrophy in vitro by activation of Akt (Colston et al., 2007) and protects against doxorubicin-induced cardiomyocyte death (Venkatesan et al., 2010). It has also been shown that WISP-1 expression is induced by Ang-II stimulation and that WISP-1 is an important mediator of Ang-II induced cardiomyocyte hypertrophy (Shanmugam et al., 2011). WISP-1 also has pro-fibrotic effects by inducing fibroblast proliferation (Colston et al., 2007). Data on the role of WISP-1 are mostly limited to in vitro experiments and the role of WISP-1 in cardiac remodeling in vivo is largely unexplored.
Midkine
Midkine is an heparin-binding growth factor that binds to different receptors forming a multireceptor complex (Yamazaki et al., 1998). Midkine protects the heart from ischemia/reperfusion injury and infarction via its anti-apoptotic effects (Kadomatsu et al., 2014). Furthermore, midkine promotes EC proliferation, leading to angiogenesis and it also enhances inflammatory cell infiltration into lesions (Kadomatsu et al., 2014). The pro-angiogenic effects of midkine have been implicated to be the major reason why midkine protects against cardiac remodeling after myocardial infarction (Takenaka et al., 2009). The downside of midkine being a strong pro-angiogenic protein is that it has growth stimulating effects on different tumors. Although midkine has protective effects after myocardial infarction, it has also been shown to increase cardiac hypertrophy during pressure overload (Netsu et al., 2014).
Adrenomedullin
Adrenomedullin (ADM) is a 52-amino acid protein which belongs to the calcitonin gene-related peptide family. ADM is mainly produced by ECs and vascular smooth muscle cells in different organs (Krzeminski, 2016). ADM is a potent vasodilator that reduces systemic and pulmonary vascular resistance, induces renal vasodilation, and increases glomerular blood flow and filtration rate (Krzeminski, 2016). Research indicates that ADM has positive inotropic effects which involve the activation of adenylyl cyclase and cyclic AMP production in cardiomyocytes (Szokodi et al., 1998; Ihara et al., 2000). It has also been shown that ADM enhances cardiac contractility via mechanisms involving facilitation of Ca2+ release (Krzeminski, 2016). However, the effect of ADM on myocardial contractility is controversial because some authors claim it to have a negative inotropic effect mediated by the NO-cGMP pathway or to have no effect on myocardial contractility (Ikenouchi et al., 1997). Another report shows that ADM has negative inotropic effects on human isolated ventricular myocytes (Mukherjee et al., 2002). These discrepancies could partly be explained by interspecies variability in contractile responses.
ADM also has anti-hypertrophic effects and anti-fibrotic effects, thus protects the heart during cardiac remodeling (Kato and Kitamura, 2015). Moreover, ADM also has pro-angiogenic effects in different tissues (Kato and Kitamura, 2015). Taken together, current evidence indicates that ADM is beneficial in a number of cardiovascular diseases because it has protective effects on cardiac remodeling.
Angiocrine Proteins as Biomarkers for Cardiac Disease
ECs are the only cells in the myocardium that are in direct contact with circulating blood. Therefore, proteins secreted by cardiac ECs are more likely to reach the circulation—and will do so in higher concentrations—than proteins from other cell types in the heart. Therefore, specific proteins secreted by ECs could serve as biomarkers of heart failure or cardiac remodeling.
All the proteins discussed in the current paper have been shown to be upregulated in an animal model of pressure overload (Moore-Morris et al., 2014). Some of the proteins discussed in this paper also have been shown to have increased circulation plasma levels in patients with heart failure. For instance, a large body of evidence indicates that circulating levels of IL-6 are increased in patients with heart failure and provide important prognostic information (Wollert and Drexler, 2001). Current evidence on circulating proteins in different forms of heart failure is presented in Table 7. Endothelium-derived proteins can be up- or down-regulated in different forms of heart failure. For instance circulating periostin levels are decreased after myocardial infarction (Cheng et al., 2012), but are increased in patients with dilated cardiomyopathy (Norum et al., 2017).
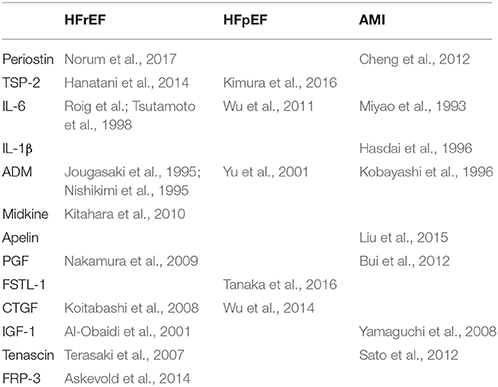
Table 7. Circulating endothelial-derived proteins as biomarkers for cardiac disease.
Conclusions
By listing currently known secreted endothelial-derived proteins and summarizing their effects on cardiac function or remodeling, an extended view on the (cardiac) endothelium as an (intrinsic) modulatory component of cardiac function emerges. It illustrates the diversity of paracrine pathways through which the endothelium affects the multiple functions and adaptive responses of the heart, which obviously is more complicated than secretion of nitric oxide. Accordingly, there is little doubt that a state of “endothelial activation” or “endothelial dysfunction” has a larger impact on cardiac function and heart failure progression than currently anticipated (and may diverge from the traditional NO-derived views, too often related to the pathophysiology of atherosclerosis; Figure 5).
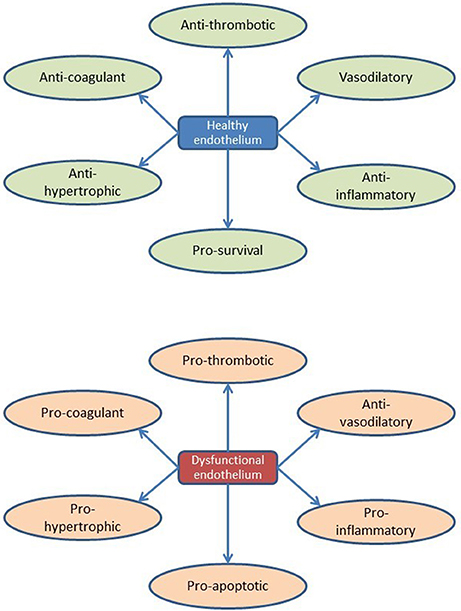
Figure 5. Overview of endothelial function and dysfunction during cardiac remodeling.
Given the complexity of the cross-talk between ECs and cardiomyocytes, one may wonder what is missing in our current understanding: (1) For many proteins, stimuli of synthesis and secretion from ECs are incompletely defined. Figure 2 gives a non-exhaustive summary of known stimuli, but these might differ between different proteins. (2) Also, the target cells of endothelium-derived proteins are incompletely characterized. In the current review, we focused on cardiomyocytes, but most proteins have an effect on multiple cell types. (3) We described the actions of different secreted proteins separately, but in reality actions of different proteins are not isolated from one another but enhance or oppose each other. Classically, cardiovascular experiments study the effect of one actor (e.g., a secreted protein) on one target response in a particular cell type (e.g., cardiomyocyte hypertrophy) at one level of complexity (e.g., cellular level). In these “one-dimensional” experiments, however, a lot of information is lost because only a single response is analyzed at a single level of complexity and—at the same time—data on interactions between different pathways and at different levels of complexity are not recorded. A more integrated approach will be necessary to study interdependency and synergy of different pathways. Ultimately, unraveling of paracrine signaling networks will be necessary to fully understand cardiac biology. (4) Drugability of the different paracrine pathways is still largely unexplored, with some notable exceptions such as NO, inflammatory factors, or neuregulin-1. (Segers and Lee, 2010, 2011; De Keulenaer et al., 2017). (5) In the future, endothelial function and dysfunction might have to be redefined as we learn more about other factors secreted by ECs. Currently, definition and evaluation of endothelial function is mostly based on secretion of NO and vasodilatory responses.
Author Contributions
VS: designed and wrote the manuscript; DB and GD: critically revised the manuscript.
Funding
This work was supported by an IOF-SBO grant of the University of Antwerp and by a grant of the Fund for scientific research Flanders (FWO), 1.5.011.18N.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Accornero, F., and Molkentin, J. D. (2011). Placental growth factor as a protective paracrine effector in the heart. Trends Cardiovasc. Med. 21, 220–224. doi: 10.1016/j.tcm.2012.05.014
Accornero, F., Van Berlo, J. H., Benard, M. J., Lorenz, J. N., Carmeliet, P., and Molkentin, J. D. (2011). Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ. Res. 109, 272–280. doi: 10.1161/CIRCRESAHA.111.240820
Accornero, F., Van Berlo, J. H., Correll, R. N., Elrod, J. W., Sargent, M. A., York, A., et al. (2015). Genetic analysis of connective tissue growth factor as an effector of transforming growth factor β signaling and cardiac remodeling. Mol. Cell. Biol. 35, 2154–2164. doi: 10.1128/MCB.00199-15
Al-Obaidi, M. K., Hon, J. K. F., Stubbs, P. J., Barnes, J., Amersey, R. A., Dahdal, M., et al. (2001). Plasma insulin-like growth factor-1 elevated in mild-to-moderate but not severe heart failure. Am. Heart J. 142, 11A–15A. doi: 10.1067/mhj.2001.118116
Anand, I., McMurray, J., Cohn, J. N., Konstam, M. A., Notter, T., Quitzau, K., et al. (2004). Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet 364, 347–354. doi: 10.1016/S0140-6736(04)16723-8
Andersson, K. B., Florholmen, G., Winer, L. H., Tønnessen, T., and Christensen, G. (2006). Regulation of neuronal type genes in congestive heart failure rats. Acta Physiol. 186, 17–27. doi: 10.1111/j.1748-1716.2005.01503.x
Ashley, E. A., Powers, J., Chen, M., Kundu, R., Finsterbach, T., Caffarelli, A., et al. (2005). The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc. Res. 65, 73–82. doi: 10.1016/j.cardiores.2004.08.018
Askevold, E. T., Aukrust, P., Nymo, S. H., Lunde, I. G., Kaasbøll, O. J., Aakhus, S., et al. (2014). The cardiokine secreted Frizzled-related protein 3, a modulator of Wnt signalling, in clinical and experimental heart failure. J. Intern. Med. 275, 621–630. doi: 10.1111/joim.12175
Baker, K. M., and Singer, H. A. (1988). Identification and characterization of guinea pig angiotensin II ventricular and atrial receptors: coupling to inositol phosphate production. Circ. Res. 62, 896–904. doi: 10.1161/01.RES.62.5.896
Ballard, V. L., Sharma, A., Duignan, I., Holm, J. M., Chin, A., Choi, R., et al. (2006). Vascular tenascin-C regulates cardiac endothelial phenotype and neovascularization. FASEB J. 20, 717–719. doi: 10.1096/fj.05-5131fje
Balligand, J. L., Feron, O., and Dessy, C. (2009). eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol. Rev. 89, 481–534. doi: 10.1152/physrev.00042.2007
Banerjee, I., Fuseler, J. W., Intwala, A. R., and Baudino, T. A. (2009). IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am. J. Physiol. Heart Circ. Physiol. 296, H1694–H1704. doi: 10.1152/ajpheart.00908.2008
Bao, M.-W., Cai, Z., Zhang, X.-J., Li, L., Liu, X., Wan, N., et al. (2015). Dickkopf-3 protects against cardiac dysfunction and ventricular remodelling following myocardial infarction. Basic Res. Cardiol. 110, 1–17. doi: 10.1007/s00395-015-0481-x
Barth, A. S., Kuner, R., Buness, A., Ruschhaupt, M., Merk, S., Zwermann, L., et al. (2006). Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. J. Am. Coll. Cardiol. 48, 1610–1617. doi: 10.1016/j.jacc.2006.07.026
Boulpaep, E. L. (2009). “The microcirculation,” in Medical Physiology, eds W. F. Boron and E. L. Boulpaep (Cleveland, OH: Elsevier), 461–482.
Brioschi, M., Lento, S., Tremoli, E., and Banfi, C. (2013). Proteomic analysis of endothelial cell secretome: a means of studying the pleiotropic effects of Hmg-CoA reductase inhibitors. J. Proteomics 78, 346–361. doi: 10.1016/j.jprot.2012.10.003
Brooks, W. W., Shen, S. S., Conrad, C. H., Goldstein, R. H., and Bing, O. H. (2010). Transition from compensated hypertrophy to systolic heart failure in the spontaneously hypertensive rat: structure, function, and transcript analysis. Genomics 95, 84–92. doi: 10.1016/j.ygeno.2009.12.002
Brutsaert, D. L. (2003). Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 83, 59–115. doi: 10.1152/physrev.00017.2002
Bui, A. H., Bonaca, M. P., Sabatine, M. S., Ray, K. K., Rifai, N., Cannon, C. P., et al. (2012). Elevated concentration of placental growth factor (PlGF) and long term risk in patients with acute coronary syndrome in the PROVE IT-TIMI 22 trial. J. Thromb. Thrombolysis 34, 222–228. doi: 10.1007/s11239-012-0704-z
Bujak, M., and Frangogiannis, N. G. (2009). The role of Interleukin-1 in the pathogenesis of heart disease. Arch. Immunol. Ther. Exp. 57, 165–176. doi: 10.1007/s00005-009-0024-y
Carano, R. A. D., and Filvaroff, E. H. (2003). Angiogenesis and bone repair. Drug Discov. Today 8, 980–989. doi: 10.1016/S1359-6446(03)02866-6
Cerutti, C., Kurdi, M., Bricca, G., Hodroj, W., Paultre, C., Randon, J., et al. (2006). Transcriptional alterations in the left ventricle of three hypertensive rat models. Physiol. Genomics 27, 295–308. doi: 10.1152/physiolgenomics.00318.2005
Chandrasekaran, B., Dar, O., and McDonagh, T. (2008). The role of apelin in cardiovascular function and heart failure. Eur. J. Heart Fail. 10, 725–732. doi: 10.1016/j.ejheart.2008.06.002
Chatzizisis, Y. S., Coskun, A. U., Jonas, M., Edelman, E. R., Feldman, C. L., and Stone, P. H. (2007). Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodelingmolecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 49, 2379–2393. doi: 10.1016/j.jacc.2007.02.059
Cheng, C.-W., Wang, C.-H., Lee, J.-F., Kuo, L.-T., and Cherng, W.-J. (2012). Levels of blood periostin decrease after acute myocardial infarction and are negatively associated with ventricular function after 3 months. J. Invest. Med. 60, 523–528. doi: 10.2310/JIM.0b013e3182408549
Chua, C. C., Hamdy, R. C., and Chua, B. H. L. (1997). Regulation of thrombospondin-1 production by angiotensin II in rat heart endothelial cells. Biochim. Biophys. Acta 1357, 209–214. doi: 10.1016/S0167-4889(97)00029-3
Cingolani, O. H., Kirk, J. A., Seo, K., Koitabashi, N., Lee, D.-I., Ramirez-Correa, G., et al. (2011). Thrombospondin-4 is required for stretch-mediated contractility augmentation in cardiac muscle. Circ. Res. 109, 1410–1414. doi: 10.1161/CIRCRESAHA.111.256743
Colston, J. T., De La Rosa, S. D., Koehler, M., Gonzales, K., Mestril, R., Freeman, G. L., et al. (2007). Wnt-induced secreted protein-1 is a prohypertrophic and profibrotic growth factor. Am. J. Physiol. Heart Circ. Physiol. 293, H1839–H1846. doi: 10.1152/ajpheart.00428.2007
Conway, S. J., and Molkentin, J. D. (2008). Periostin as a heterofunctional regulator of cardiac development and disease. Curr. Genomics 9, 548–555. doi: 10.2174/138920208786847917
Davies, P. F. (2009). Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 6, 16–26. doi: 10.1038/ncpcardio1397
Dawson, K., Aflaki, M., and Nattel, S. (2013). Role of the Wnt-Frizzled system in cardiac pathophysiology: a rapidly developing, poorly understood area with enormous potential. J. Physiol. 591, 1409–1432. doi: 10.1113/jphysiol.2012.235382
DeGeorge, B. R., Rosenberg, M., Eckstein, V., Gao, E., Herzog, N., Katus, H. A., et al. (2008). BMP-2 and FGF-2 synergistically facilitate adoption of a cardiac phenotype in somatic bone marrow c-kit(+)/Sca-1(+) stem cells. Clin. Transl. Sci. 1, 116–125. doi: 10.1111/j.1752-8062.2008.00034.x
De Keulenaer, G. W., Segers, V. F. M., Zannad, F., and Brutsaert, D. L. (2017). The future of pleiotropic therapy in heart failure. Lessons from the benefits of exercise training on endothelial function. Eur. J. Heart Fail. 19, 603–614. doi: 10.1002/ejhf.735
Diaz, J. A., Booth, A. J., Lu, G., Wood, S. C., Pinsky, D. J., and Bishop, D. K. (2009). Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am. J. Transpl. 9, 1773–1783. doi: 10.1111/j.1600-6143.2009.02706.x
Dong, Y., Geng, Y., Li, L., Li, X., Yan, X., Fang, Y., et al. (2015). Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J. Exp. Med. 212, 235–252. doi: 10.1084/jem.20121878
Drawnel, F. M., Archer, C. R., and Roderick, H. L. (2013). The role of the paracrine/autocrine mediator endothelin-1 in regulation of cardiac contractility and growth. Br. J. Pharmacol. 168, 296–317. doi: 10.1111/j.1476-5381.2012.02195.x
Duncker, D. J., and Bache, R. J. (2008). Regulation of coronary blood flow during exercise. Physiol. Rev. 88, 1009–1086. doi: 10.1152/physrev.00045.2006
Ellison, G. M., Waring, C. D., Vicinanza, C., and Torella, D. (2012). Physiological cardiac remodelling in response to endurance exercise training: cellular and molecular mechanisms. Heart 98, 5–10. doi: 10.1136/heartjnl-2011-300639
Finkel, M. S., Oddis, C. V., Jacob, T. D., Watkins, S. C., Hattler, B. G., and Simmons, R. L. (1992). Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 257, 387–389. doi: 10.1126/science.1631560
Fischer, J. W., Stoll, M., Hahn, A. W. A., and Unger, T. (2001). Differential regulation of thrombospondin-1 and fibronectin by angiotensin II receptor subtypes in cultured endothelial cells. Cardiovasc. Res. 51, 784–791. doi: 10.1016/S0008-6363(01)00345-5
Fischer, P., and Hilfiker-Kleiner, D. (2007). Survival pathways in hypertrophy and heart failure: the gp130-STAT3 axis. Basic Res. Cardiol. 102, 279–297. doi: 10.1007/s00395-007-0658-z
Fong, D., Hermann, M., Untergasser, G., Pirkebner, D., Draxl, A., Heitz, M., et al. (2009). Dkk-3 expression in the tumor endothelium: a novel prognostic marker of pancreatic adenocarcinomas. Cancer Sci. 100, 1414–1420. doi: 10.1111/j.1349-7006.2009.01208.x
Fontes, J. A., Rose, N. R., and Ciháková, D. (2015). The varying faces of IL-6: from cardiac protection to cardiac failure. Cytokine 74, 62–68. doi: 10.1016/j.cyto.2014.12.024
Fontes, M. S., Kessler, E. L., Van Stuijvenberg, L., Brans, M. A., Falke, L. L., Kok, B., et al. (2015). CTGF knockout does not affect cardiac hypertrophy and fibrosis formation upon chronic pressure overload. J. Mol. Cell. Cardiol. 88, 82–90. doi: 10.1016/j.yjmcc.2015.09.015
Frangogiannis, N. G. (2012). Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 92, 635–688. doi: 10.1152/physrev.00008.2011
Frangogiannis, N. G., Ren, G., Dewald, O., Zymek, P., Haudek, S., Koerting, A., et al. (2005). Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 111, 2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354
Freer, R. J., Pappano, A. J., Peach, M. J., Bing, K. T., McLean, M. J., Vogel, S., et al. (1976). Mechanism for the postive inotropic effect of angiotensin II on isolated cardiac muscle. Circ. Res. 39, 178–183. doi: 10.1161/01.RES.39.2.178
Frolova, E. G., Sopko, N., Blech, L., Popović, Z. B., Li, J., Vasanji, A., et al. (2012). Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 26, 2363–2373. doi: 10.1096/fj.11-190728
Gavin, K. M., Seals, D. R., Silver, A. E., and Moreau, K. L. (2009). Vascular endothelial estrogen receptor α is modulated by estrogen status and related to endothelial function and endothelial nitric oxide synthase in healthy women. J. Clin. Endocrinol. Metab. 94, 3513–3520. doi: 10.1210/jc.2009-0278
Ghosh-Choudhury, N., Abboud, S. L., Chandrasekar, B., and Ghosh Choudhury, G. (2003). BMP-2 regulates cardiomyocyte contractility in a phosphatidylinositol 3 kinase-dependent manner. FEBS Lett. 544, 181–184. doi: 10.1016/S0014-5793(03)00507-6
Golledge, J., Clancy, P., Maguire, J., Lincz, L., and Koblar, S. (2011). The role of tenascin C in cardiovascular disease. Cardiovasc. Res. 92, 19–28. doi: 10.1093/cvr/cvr183
González, G. E., Rhaleb, N.-E., D'ambrosio, M. A., Nakagawa, P., Liu, Y., Leung, P., et al. (2015). Deletion of interleukin-6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II-high salt-induced hypertension. J. Hypertens. 33, 144–152. doi: 10.1097/HJH.0000000000000358
Greco, S., Fasanaro, P., Castelvecchio, S., D'alessandra, Y., Arcelli, D., Di Donato, M., et al. (2012). MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes 61, 1633–1641. doi: 10.2337/db11-0952
Hanatani, S., Izumiya, Y., Takashio, S., Kimura, Y., Araki, S., Rokutanda, T., et al. (2014). Circulating thrombospondin-2 reflects disease severity and predicts outcome of heart failure with reduced ejection fraction. Circ. J. 78, 903–910. doi: 10.1253/circj.CJ-13-1221
Hara, A., Yuhki, K., Fujino, T., Yamada, T., Takayama, K., Kuriyama, S., et al. (2005). Augmented cardiac hypertrophy in response to pressure overload in mice lacking the prostaglandin I2 receptor. Circulation 112, 84–92. doi: 10.1161/CIRCULATIONAHA.104.527077
Harding, P., and Murray, D. B. (2011). The contribution of prostaglandins versus prostacyclin in ventricular remodeling during heart failure. Life Sci. 89, 671–676. doi: 10.1016/j.lfs.2011.07.025
Hasdai, D., Scheinowitz, M., Leibovitz, E., Sclarovsky, S., Eldar, M., and Barak, V. (1996). Increased serum concentrations of interleukin-1 beta in patients with coronary artery disease. Heart 76, 24–28. doi: 10.1136/hrt.76.1.24
Hathaway, C. K., Grant, R., Hagaman, J. R., Hiller, S., Li, F., Xu, L., et al. (2015). Endothelin-1 critically influences cardiac function via superoxide-MMP9 cascade. Proc. Natl. Acad. Sci. U.S.A. 112, 5141–5146. doi: 10.1073/pnas.1504557112
Hayata, N., Fujio, Y., Yamamoto, Y., Iwakura, T., Obana, M., Takai, M., et al. (2008). Connective tissue growth factor induces cardiac hypertrophy through Akt signaling. Biochem. Biophys. Res. Commun. 370, 274–278. doi: 10.1016/j.bbrc.2008.03.100
Heiden, S., Vignon-Zellweger, N., Masuda, S., Yagi, K., Nakayama, K., Yanagisawa, M., et al. (2014). Vascular endothelium derived endothelin-1 is required for normal heart function after chronic pressure overload in mice. PLoS ONE 9:e88730. doi: 10.1371/journal.pone.0088730
Hemmen, K., Reinl, T., Buttler, K., Behler, F., Dieken, H., Jansch, L., et al. (2011). High-resolution mass spectrometric analysis of the secretome from mouse lung endothelial progenitor cells. Angiogenesis 14, 163–172. doi: 10.1007/s10456-011-9200-x
Hosseinkhani, M., Hosseinkhani, H., Khademhosseini, A., Bolland, F., Kobayashi, H., and Gonzalez, S. P. (2007). Bone morphogenetic protein-4 enhances cardiomyocyte differentiation of cynomolgus monkey ESCs in knockout serum replacement medium. Stem Cells 25, 571–580. doi: 10.1634/stemcells.2006-0225
Ihara, T., Ikeda, U., Tate, Y., Ishibashi, S., and Shimada, K. (2000). Positive inotropic effects of adrenomedullin on rat papillary muscle. Eur. J. Pharmacol. 390, 167–172. doi: 10.1016/S0014-2999(00)00011-X
Ikenouchi, H., Kangawa, K., Matsuo, H., and Hirata, Y. (1997). Negative inotropic effect of adrenomedullin in isolated adult rabbit cardiac ventricular myocytes. Circulation 95, 2318–2324. doi: 10.1161/01.CIR.95.9.2318
Imanaka-Yoshida, K. (2012). Tenascin-C in cardiovascular tissue remodeling: from development to inflammation and repair. Circ. J. 76, 2513–2520. doi: 10.1253/circj.CJ-12-1033
Janssen, S. P., Gayan-Ramirez, G., Van den Bergh, A., Herijgers, P., Maes, K., Verbeken, E., et al. (2005). Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation 111, 996–1005. doi: 10.1161/01.CIR.0000156469.96135.0D
Japp, A. G., Cruden, N. L., Barnes, G., Van Gemeren, N., Mathews, J., Adamson, J., et al. (2010). Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation 121, 1818–1827. doi: 10.1161/CIRCULATIONAHA.109.911339
Jougasaki, M., Wei, C.-M., McKinley, L. J., and Burnett, J. C. (1995). Elevation of circulating and ventricular adrenomedullin in human congestive heart failure. Circulation 92, 286–289. doi: 10.1161/01.CIR.92.3.286
Kadomatsu, K., Bencsik, P., Görbe, A., Csonka, C., Sakamoto, K., Kishida, S., et al. (2014). Therapeutic potential of midkine in cardiovascular disease. Br. J. Pharmacol. 171, 936–944. doi: 10.1111/bph.12537
Kakita, T., Hasegawa, K., Iwai-Kanai, E., Adachi, S., Morimoto, T., Wada, H., et al. (2001). Calcineurin pathway is required for endothelin-1-mediated protection against oxidant stress-induced apoptosis in cardiac myocytes. Circ. Res. 88, 1239–1246. doi: 10.1161/hh1201.091794
Kamo, T., Akazawa, H., and Komuro, I. (2015). Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ. Res. 117, 89–98. doi: 10.1161/CIRCRESAHA.117.305349
Kato, J., and Kitamura, K. (2015). Bench-to-bedside pharmacology of adrenomedullin. Eur. J. Pharmacol. 764, 140–148. doi: 10.1016/j.ejphar.2015.06.061
Kern, P. A., Svoboda, M. E., Eckel, R. H., and Van Wyk, J. J. (1989). Insulinlike growth factor action and production in adipocytes and endothelial cells from human adipose tissue. Diabetes 38, 710–717. doi: 10.2337/diab.38.6.710
Kim, Y. Y., Ku, S.-Y., Liu, H.-C., Cho, H.-J., Oh, S. K., Moon, S. Y., et al. (2011). Cryopreservation of human embryonic stem cells derived-cardiomyocytes induced by BMP2 in serum-free condition. Reprod. Sci. 18, 252–260. doi: 10.1177/1933719110385130
Kimura, Y., Izumiya, Y., Hanatani, S., Yamamoto, E., Kusaka, H., Tokitsu, T., et al. (2016). High serum levels of thrombospondin-2 correlate with poor prognosis of patients with heart failure with preserved ejection fraction. Heart Vessels 31, 52–59. doi: 10.1007/s00380-014-0571-y
Kitahara, T., Shishido, T., Suzuki, S., Katoh, S., Sasaki, T., Ishino, M., et al. (2010). Serum midkine as a predictor of cardiac events in patients with chronic heart failure. J. Card. Fail. 16, 308–313. doi: 10.1016/j.cardfail.2009.12.014
Kobara, M., Noda, K., Kitamura, M., Okamoto, A., Shiraishi, T., Toba, H., et al. (2010). Antibody against interleukin-6 receptor attenuates left ventricular remodelling after myocardial infarction in mice. Cardiovasc. Res. 87, 424–430. doi: 10.1093/cvr/cvq078
Kobayashi, K., Kitamura, K., Hirayama, N., Date, H., Kashiwagi, T., Ikushima, I., et al. (1996). Increased plasma adrenomedullin in acute myocardial infarction. Am. Heart J. 131, 676–680. doi: 10.1016/S0002-8703(96)90270-7
Kodama, H., Fukuda, K., Pan, J., Makino, S., Baba, A., Hori, S., et al. (1997). Leukemia inhibitory factor, a potent cardiac hypertrophic cytokine, activates the JAK/STAT pathway in rat cardiomyocytes. Circ. Res. 81, 656–663. doi: 10.1161/01.RES.81.5.656
Koitabashi, N., Arai, M., Niwano, K., Watanabe, A., Endoh, M., Suguta, M., et al. (2008). Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur. J. Heart Fail. 10, 373–379. doi: 10.1016/j.ejheart.2008.02.011
Krupnik, V. E., Sharp, J. D., Jiang, C., Robison, K., Chickering, T. W., Amaravadi, L., et al. (1999). Functional and structural diversity of the human Dickkopf gene family. Gene 238, 301–313. doi: 10.1016/S0378-1119(99)00365-0
Krzeminski, K. (2016). The role of adrenomedullin in cardiovascular response to exercise – a review. J. Hum. Kinet. 53, 127–142. doi: 10.1515/hukin-2016-0017
Kuba, K., Zhang, L., Imai, Y., Arab, S., Chen, M., Maekawa, Y., et al. (2007). Impaired heart contractility in apelin gene–deficient mice associated with aging and pressure overload. Circ. Res. 101, e32–e42. doi: 10.1161/CIRCRESAHA.107.158659
Kuhn, B., Del Monte, F., Hajjar, R. J., Chang, Y. S., Lebeche, D., Arab, S., et al. (2007). Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat. Med. 13, 962–969. doi: 10.1038/nm1619
Kwon, O. K., Lee, W., Kim, S. J., Lee, Y.-M., Lee, J. Y., Kim, J. Y., et al. (2015). In-depth proteomics approach of secretome to identify novel biomarker for sepsis in LPS-stimulated endothelial cells. Electrophoresis 36, 2851–2858. doi: 10.1002/elps.201500198
Lachtermacher, S., Esporcatte, B. L., Montalvao, F., Costa, P. C., Rodrigues, D. C., Belem, L., et al. (2010). Cardiac gene expression and systemic cytokine profile are complementary in a murine model of post-ischemic heart failure. Braz. J. Med. Biol. Res. 43, 377–389. doi: 10.1590/S0100-879X2010007500014
Langenfeld, E. M., and Langenfeld, J. (2004). Bone morphogenetic protein-2 stimulates angiogenesis in developing tumors. Mol. Cancer Res. 2, 141–149.
Lemmens, K., Fransen, P., Sys, S. U., Brutsaert, D. L., and De Keulenaer, G. W. (2004). Neuregulin-1 induces a negative inotropic effect in cardiac muscle: role of nitric oxide synthase. Circulation 109, 324–326. doi: 10.1161/01.CIR.0000114521.88547.5E
Lemmens, K., Segers, V. F., Demolder, M., and De Keulenaer, G. W. (2006). Role of neuregulin-1/ErbB2 signaling in endothelium-cardiomyocyte cross-talk. J. Biol. Chem. 281, 19469–19477. doi: 10.1074/jbc.M600399200
Lerman, A., Kubo, S. H., Tschumperlin, L. K., and Burnett, J. C. Jr. (1992). Plasma endothelin concentrations in humans with end-stage heart failure and after heart transplantation. J. Am. Coll. Cardiol. 20, 849–853. doi: 10.1016/0735-1097(92)90183-N
Li, D., Wang, Y., Xu, N., Wei, Q., Wu, M., Li, X., et al. (2011). Follistatin-like protein 1 is elevated in systemic autoimmune diseases and correlated with disease activity in patients with rheumatoid arthritis. Arthritis Res. Ther. 13, 1–12. doi: 10.1186/ar3241
Lim, S. L., Lam, C. S., Segers, V. F., Brutsaert, D. L., and De Keulenaer, G. W. (2015). Cardiac endothelium-myocyte interaction: clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur. Heart J. 36, 2050–2060. doi: 10.1093/eurheartj/ehv132
Liu, H.-T., Chen, M., Yu, J., Li, W.-J., Tao, L., Li, Y., et al. (2015). Serum apelin level predicts the major adverse cardiac events in patients with ST elevation myocardial infarction receiving percutaneous coronary intervention. Medicine 94:e449. doi: 10.1097/MD.0000000000000449
Liu, S., Taghavi, R., and Leask, A. (2010). Connective tissue growth factor is induced in bleomycin-induced skin scleroderma. J. Cell Commun. Signal. 4, 25–30. doi: 10.1007/s12079-009-0081-3
Liu, X., Gu, X., Li, Z., Li, X., Li, H., Chang, J., et al. (2006). Neuregulin-1/erbB-activation improves cardiac function and survival in models of ischemic, dilated, and viral cardiomyopathy. J. Am. Coll. Cardiol. 48, 1438–1447. doi: 10.1016/j.jacc.2006.05.057
Loffler, B. M., Roux, S., Kalina, B., Clozel, M., and Clozel, J. P. (1993). Influence of congestive heart failure on endothelin levels and receptors in rabbits. J. Mol. Cell. Cardiol. 25, 407–416. doi: 10.1006/jmcc.1993.1047
Lu, D., Bao, D., Dong, W., Liu, N., Zhang, X., Gao, S., et al. (2016). Dkk3 prevents familial dilated cardiomyopathy development through Wnt pathway. Lab. Invest. 96, 239–248. doi: 10.1038/labinvest.2015.145
Lu, J., Sun, B., Huo, R., Wang, Y.-C., Yang, D., Xing, Y., et al. (2014). Bone morphogenetic protein-2 antagonizes bone morphogenetic protein-4 induced cardiomyocyte hypertrophy and apoptosis. J. Cell. Physiol. 229, 1503–1510. doi: 10.1002/jcp.24592
Maruyama, S., Nakamura, K., Papanicolaou, K. N., Sano, S., Shimizu, I., Asaumi, Y., et al. (2016). Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol. Med. 8, 949–966. doi: 10.15252/emmm.201506151
Massion, P. B., and Balligand, J. L. (2007). Relevance of nitric oxide for myocardial remodeling. Curr. Heart Fail. Rep. 4, 18–25. doi: 10.1007/s11897-007-0021-6
Meléndez, G. C., McLarty, J. L., Levick, S. P., Du, Y., Janicki, J. S., and Brower, G. L. (2010). Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 56, 225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635
Meulemans, A. L., Andries, L. J., and Brutsaert, D. L. (1990). Does endocardial endothelium mediate positive inotropic response to angiotensin I and angiotensin II? Circ. Res. 66, 1591–1601.
Michele, A., Emanuele, B., Eduardo, B., Pasquale, P.-F., Raffaele, N., Luigi, S., et al. (2013). The GH/IGF-1 axis in chronic heart failure. Endocr. Metab. Immune Disord. Drug Targets 13, 76–91. doi: 10.2174/1871530311313010010
Mihos, C. G., Pineda, A. M., and Santana, O. (2014). Cardiovascular effects of statins, beyond lipid-lowering properties. Pharmacol. Res. 88, 12–19. doi: 10.1016/j.phrs.2014.02.009
Miyao, Y., Yasue, H., Ogawa, H., Misumi, I., Masuda, T., Sakamoto, T., et al. (1993). Elevated plasma interleukin-6 levels in patients with acute myocardial infarction. Am. Heart J. 126, 1299–1304. doi: 10.1016/0002-8703(93)90526-F
Mohan, P., Brutsaert, D. L., Paulus, W. J., and Sys, S. U. (1996). Myocardial contractile response to nitric oxide and cGMP. Circulation 93, 1223–1229. doi: 10.1161/01.CIR.93.6.1223
Moore-Morris, T., Guimaraes-Camboa, N., Banerjee, I., Zambon, A. C., Kisseleva, T., Velayoudon, A., et al. (2014). Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Invest. 124, 2921–2934. doi: 10.1172/JCI74783
Moravec, C. S., Reynolds, E. E., Stewart, R. W., and Bond, M. (1989). Endothelin is a positive inotropic agent in human and rat heart in vitro. Biochem. Biophys. Res. Commun. 159, 14–18. doi: 10.1016/0006-291X(89)92397-8
Mukherjee, R., Multani, M. M., Sample, J. A., Dowdy, K. B., Zellner, J. L., Hoover, D. B., et al. (2002). Effects of adrenomedullin on human myocyte contractile function and β-adrenergic response. J. Cardiovasc. Pharmacol. Ther. 7, 235–240. doi: 10.1177/107424840200700406
Mustonen, E., Aro, J., Puhakka, J., Ilves, M., Soini, Y., Leskinen, H., et al. (2008). Thrombospondin-4 expression is rapidly upregulated by cardiac overload. Biochem. Biophys. Res. Commun. 373, 186–191. doi: 10.1016/j.bbrc.2008.05.164
Nakamura, T., Funayama, H., Kubo, N., Yasu, T., Kawakami, M., Momomura, S.-I., et al. (2009). Elevation of plasma placental growth factor in the patients with ischemic cardiomyopathy. Int. J. Cardiol. 131, 186–191. doi: 10.1016/j.ijcard.2007.10.050
Netsu, S., Shishido, T., Kitahara, T., Honda, Y., Funayama, A., Narumi, T., et al. (2014). Midkine exacerbates pressure overload-induced cardiac remodeling. Biochem. Biophys. Res. Commun. 443, 205–210. doi: 10.1016/j.bbrc.2013.11.083
Niehrs, C. (2006). Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 25, 7469–7481. doi: 10.1038/sj.onc.1210054
Nishikimi, T., Saito, Y., Kitamura, K., Ishimitsu, T., Eto, T., Kangawa, K., et al. (1995). Increased plasma levels of adrenomedullin in patients with heart failure. J. Am. Coll. Cardiol. 26, 1424–1431. doi: 10.1016/0735-1097(95)00338-X
Nishioka, T., Onishi, K., Shimojo, N., Nagano, Y., Matsusaka, H., Ikeuchi, M., et al. (2010). Tenascin-C may aggravate left ventricular remodeling and function after myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 298, H1072–H1078. doi: 10.1152/ajpheart.00255.2009
Norum, H. M., Broch, K., Michelsen, A. E., Lunde, I. G., Lekva, T., Abraityte, A., et al. (2017). The notch ligands DLL1 and periostin are associated with symptom severity and diastolic function in dilated cardiomyopathy. J. Cardiovasc. Transl. Res. 10, 401–410. doi: 10.1007/s12265-017-9748-y
O'connor, C. M., Gattis, W. A., Adams, K. F. Jr., Hasselblad, V., Chandler, B., Frey, A., et al. (2003). Tezosentan in patients with acute heart failure and acute coronary syndromes: results of the Randomized Intravenous TeZosentan Study (RITZ-4). J. Am. Coll. Cardiol. 41, 1452–1457. doi: 10.1016/S0735-1097(03)00194-3
Ogata, Y., Takahashi, M., Ueno, S., Takeuchi, K., Okada, T., Mano, H., et al. (2003). Antiapoptotic effect of endothelin-1 in rat cardiomyocytes in vitro. Hypertension 41, 1156–1163. doi: 10.1161/01.HYP.0000064342.30653.24
Ogura, Y., Ouchi, N., Ohashi, K., Shibata, R., Kataoka, Y., Kambara, T., et al. (2012). Therapeutic impact of follistatin-like 1 on myocardial ischemic injury in preclinical models. Circulation 126, 1728–1738. doi: 10.1161/CIRCULATIONAHA.112.115089
Ojaimi, C., Qanud, K., Hintze, T. H., and Recchia, F. A. (2007). Altered expression of a limited number of genes contributes to cardiac decompensation during chronic ventricular tachypacing in dogs. Physiol. Genomics 29, 76–83. doi: 10.1152/physiolgenomics.00159.2006
Oka, T., Xu, J., Kaiser, R. A., Melendez, J., Hambleton, M., Sargent, M. A., et al. (2007). Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 101, 313–321. doi: 10.1161/CIRCRESAHA.107.149047
Opgaard, O. S., and Wang, P. H. (2005). IGF-I is a matter of heart. Growth Hormone IGF Res. 15, 89–94. doi: 10.1016/j.ghir.2005.02.002
Oshima, Y., Ouchi, N., Sato, K., Izumiya, Y., Pimentel, D. R., and Walsh, K. (2008). Follistatin-like 1 is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation 117, 3099–3108. doi: 10.1161/CIRCULATIONAHA.108.767673
Ouchi, N., Oshima, Y., Ohashi, K., Higuchi, A., Ikegami, C., Izumiya, Y., et al. (2008). Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. J. Biol. Chem. 283, 32802–32811. doi: 10.1074/jbc.M803440200
Palao, T., Rippe, C., Van Veen, H., Vanbavel, E., Swärd, K., and Bakker, E. N. T. P. (2016). Thrombospondin-4 knockout in hypertension protects small-artery endothelial function but induces aortic aneurysms. Am. J. Physiol. Heart Circ. Physiol. 310, H1486–H1493. doi: 10.1152/ajpheart.00046.2016
Palmer, R. M., Ferrige, A. G., and Moncada, S. (1987). Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327, 524–526. doi: 10.1038/327524a0
Papay, R. S., Shi, T., Piascik, M. T., Naga Prasad, S. V., and Perez, D. M. (2013). α1A-adrenergic receptors regulate cardiac hypertrophy in vivo through interleukin-6 secretion. Mol. Pharmacol. 83, 939–948. doi: 10.1124/mol.112.084483
Partl, J. Z., Fabijanovic, D., Skrtic, A., Vranic, S., Martic, T. N., and Serman, L. (2014). Immunohistochemical expression of SFRP1 and SFRP3 proteins in normal and malignant reproductive tissues of rats and humans. Appl. Immunohistochem. Mol. Morphol. 22, 681–687. doi: 10.1097/PAI.0000000000000019
Patten, M., Kramer, E., Bunemann, J., Wenck, C., Thoenes, M., Wieland, T., et al. (2001). Endotoxin and cytokines alter contractile protein expression in cardiac myocytes in vivo. Pflugers Arch. 442, 920–927.
Paul, M., Mehr, A. P., and Kreutz, R. (2006). Physiology of local renin-angiotensin systems. Physiol. Rev. 86, 747–803. doi: 10.1152/physrev.00036.2005
Paulus, W. J., and Tschope, C. (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 62, 263–271. doi: 10.1016/j.jacc.2013.02.092
Pchejetski, D., Foussal, C., Alfarano, C., Lairez, O., Calise, D., Guilbeau-Frugier, C., et al. (2012). Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur. Heart J. 33, 2360–2369. doi: 10.1093/eurheartj/ehr389
Pinto, A. R., Ilinykh, A., Ivey, M. J., Kuwabara, J. T., D'antoni, M., Debuque, R. J., et al. (2015). Revisiting cardiac cellular composition. Circ. Res. 118, 400–409. doi: 10.1161/CIRCRESAHA.115.307778
Rajashekhar, G., Grow, M., Willuweit, A., Patterson, C. E., and Clauss, M. (2007). Divergent and convergent effects on gene expression and function in acute versus chronic endothelial activation. Physiol. Genomics 31, 104–113. doi: 10.1152/physiolgenomics.00157.2006
Ren, J., Samson, W. K., and Sowers, J. R. (1999). Insulin-like growth factor i as a cardiac hormone: physiological and pathophysiological implications in heart disease. J. Mol. Cell. Cardiol. 31, 2049–2061. doi: 10.1006/jmcc.1999.1036
Ren, Y., Lee, M. Y., Schliffke, S., Paavola, J., Amos, P. J., Ge, X., et al. (2011). Small molecule Wnt inhibitors enhance the efficiency of BMP-4-directed cardiac differentiation of human pluripotent stem cells. J. Mol. Cell. Cardiol. 51, 280–287. doi: 10.1016/j.yjmcc.2011.04.012
Ritchie, R. H., Rosenkranz, A. C., Huynh, L. P., Stephenson, T., Kaye, D. M., and Dusting, G. J. (2004). Activation of IP prostanoid receptors prevents cardiomyocyte hypertrophy via cAMP-dependent signaling. Am. J. Physiol. Heart Circ. Physiol. 287, H1179–1185. doi: 10.1152/ajpheart.00725.2003
Roig, E., Orús, J., Paré, C., Azqueta, M., Filella, X., Perez-Villa, F., et al. (1998). Serum interleukin-6 in congestive heart failure secondary to idiopathic dilated cardiomyopathy. Am. J. Cardiol. 82, 688–690. doi: 10.1016/S0002-9149(98)00388-9
Rysa, J., Leskinen, H., Ilves, M., and Ruskoaho, H. (2005). Distinct upregulation of extracellular matrix genes in transition from hypertrophy to hypertensive heart failure. Hypertension 45, 927–933. doi: 10.1161/01.HYP.0000161873.27088.4c
Sadoshima, J., and Izumo, S. (1993). Molecular characterization of angiotensin-II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts - critical role of the at(1) receptor subtype. Circ. Res. 73, 413–423. doi: 10.1161/01.RES.73.3.413
Sato, A., Hiroe, M., Akiyama, D., Hikita, H., Nozato, T., Hoshi, T., et al. (2012). Prognostic value of serum tenascin-C levels on long-term outcome after acute myocardial infarction. J. Card. Fail. 18, 480–486. doi: 10.1016/j.cardfail.2012.02.009
Segers, V. F., and Lee, R. T. (2008). Stem-cell therapy for cardiac disease. Nature 451, 937–942. doi: 10.1038/nature06800
Segers, V. F., and Lee, R. T. (2010). Protein therapeutics for cardiac regeneration after myocardial infarction. J. Cardiovasc. Transl. Res. 3, 469–477. doi: 10.1007/s12265-010-9207-5
Segers, V. F., and Lee, R. T. (2011). Biomaterials to enhance stem cell function in the heart. Circ. Res. 109, 910–922. doi: 10.1161/CIRCRESAHA.111.249052
Shanmugam, P., Valente, A. J., Prabhu, S. D., Venkatesan, B., Yoshida, T., Delafontaine, P., et al. (2011). Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 50, 928–938. doi: 10.1016/j.yjmcc.2011.02.012
Shimano, M., Ouchi, N., Nakamura, K., Van Wijk, B., Ohashi, K., Asaumi, Y., et al. (2011). Cardiac myocyte follistatin-like 1 functions to attenuate hypertrophy following pressure overload. Proc. Natl. Acad. Sci. U.S.A. 108, E899–E906. doi: 10.1073/pnas.1108559108
Snider, P., Standley, K. N., Wang, J., Azhar, M., Doetschman, T., and Conway, S. J. (2009). Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 105, 934–947. doi: 10.1161/CIRCRESAHA.109.201400
Stenina, O. I., Desai, S. Y., Krukovets, I., Kight, K., Janigro, D., Topol, E. J., et al. (2003). Thrombospondin-4 and its variants: expression and differential effects on endothelial cells. Circulation 108, 1514–1519. doi: 10.1161/01.CIR.0000089085.76320.4E
Suleiman, M. S., Singh, R. J. R., and Stewart, C. E. H. (2007). Apoptosis and the cardiac action of insulin-like growth factor I. Pharmacol. Ther. 114, 278–294. doi: 10.1016/j.pharmthera.2007.03.001
Sun, B., Huo, R., Sheng, Y., Li, Y., Xie, X., Chen, C., et al. (2013). Bone morphogenetic protein-4 mediates cardiac hypertrophy, apoptosis, and fibrosis in experimentally pathological cardiac hypertrophy. Hypertension 61, 352–360. doi: 10.1161/HYPERTENSIONAHA.111.00562
Swinnen, M., Vanhoutte, D., Van Almen, G. C., Hamdani, N., Schellings, M. W. M., D'hooge, J., et al. (2009). Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation 120, 1585–1597. doi: 10.1161/CIRCULATIONAHA.109.863266
Szokodi, I., Kinnunen, P., Tavi, P., Weckström, M., Tóth, M., and Ruskoaho, H. (1998). Evidence for cAMP-independent mechanisms mediating the effects of adrenomedullin, a new inotropic peptide. Circulation 97, 1062–1070. doi: 10.1161/01.CIR.97.11.1062
Szokodi, I., Tavi, P., Földes, G., Voutilainen-Myllylä, S., Ilves, M., Tokola, H., et al. (2002). Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ. Res. 91, 434–440. doi: 10.1161/01.RES.0000033522.37861.69
Taha, M. F., Valojerdi, M. R., and Mowla, S. J. (2007). Effect of bone morphogenetic protein-4 (BMP-4) on cardiomyocyte differentiation from mouse embryonic stem cell. Int. J. Cardiol. 120, 92–101. doi: 10.1016/j.ijcard.2006.08.118
Takenaka, H., Horiba, M., Ishiguro, H., Sumida, A., Hojo, M., Usui, A., et al. (2009). Midkine prevents ventricular remodeling and improves long-term survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 296, H462–H469. doi: 10.1152/ajpheart.00733.2008
Tanaka, K., Valero-Muñoz, M., Wilson, R. M., Essick, E. E., Fowler, C. T., Nakamura, K., et al. (2016). Follistatin like 1 regulates hypertrophy in heart failure with preserved ejection fraction. JACC Basic Transl. Sci. 1, 207–221. doi: 10.1016/j.jacbts.2016.04.002
Terasaki, F., Okamoto, H., Onishi, K., Sato, A., Shimomura, H., Tsukada, B., et al. (2007). Higher serum tenascin-C levels reflect the severity of heart failure, left ventricular dysfunction and remodeling in patients with dilated cardiomyopathy. Circ. J. 71, 327–330. doi: 10.1253/circj.71.327
Tsutamoto, T., Hisanaga, T., Wada, A., Maeda, K., Ohnishi, M., Fukai, D., et al. (1998). Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J. Am. Coll. Cardiol. 31, 391–398. doi: 10.1016/S0735-1097(97)00494-4
Tucker, R. P., and Chiquet-Ehrismann, R. (2009). The regulation of tenascin expression by tissue microenvironments. Biochim. Biophys. Acta 1793, 888–892. doi: 10.1016/j.bbamcr.2008.12.012
Tunica, D. G., Yin, X., Sidibe, A., Stegemann, C., Nissum, M., Zeng, L., et al. (2009). Proteomic analysis of the secretome of human umbilical vein endothelial cells using a combination of free-flow electrophoresis and nanoflow LC-MS/MS. Proteomics 9, 4991–4996. doi: 10.1002/pmic.200900065
Untergasser, G., Steurer, M., Zimmermann, M., Hermann, M., Kern, J., Amberger, A., et al. (2008). The Dickkopf-homolog 3 is expressed in tumor endothelial cells and supports capillary formation. Int. J. Cancer 122, 1539–1547. doi: 10.1002/ijc.23255
Urbich, C., Aicher, A., Heeschen, C., Dernbach, E., Hofmann, W. K., Zeiher, A. M., et al. (2005). Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J. Mol. Cell. Cardiol. 39, 733–742. doi: 10.1016/j.yjmcc.2005.07.003
van Almen, G. C., Swinnen, M., Carai, P., Verhesen, W., Cleutjens, J. P. M., D'hooge, J., et al. (2011). Absence of thrombospondin-2 increases cardiomyocyte damage and matrix disruption in doxorubicin-induced cardiomyopathy. J. Mol. Cell. Cardiol. 51, 318–328. doi: 10.1016/j.yjmcc.2011.05.010
Veeck, J., and Dahl, E. (2012). Targeting the Wnt pathway in cancer: the emerging role of Dickkopf-3. Biochim. Biophys. Acta 1825, 18–28. doi: 10.1016/j.bbcan.2011.09.003
Venkatachalam, K., Venkatesan, B., Valente, A. J., Melby, P. C., Nandish, S., Reusch, J. E. B., et al. (2009). WISP1, a pro-mitogenic, pro-survival factor, mediates Tumor Necrosis Factor-α (TNF-α)-stimulated cardiac fibroblast proliferation but inhibits TNF-α-induced cardiomyocyte death. J. Biol. Chem. 284, 14414–14427. doi: 10.1074/jbc.M809757200
Venkatesan, B., Prabhu, S. D., Venkatachalam, K., Mummidi, S., Valente, A. J., Clark, R. A., et al. (2010). WNT1-inducible signaling pathway protein-1 activates diverse cell survival pathways and blocks doxorubicin-induced cardiomyocyte death. Cell. Signal. 22, 809–820. doi: 10.1016/j.cellsig.2010.01.005
Vermeulen, Z., Hervent, A. S., Dugaucquier, L., Vandekerckhove, L., Rombouts, M., Beyens, M., et al. (2017). Inhibitory actions of the NRG-1/ErbB4 pathway in macrophages during tissue fibrosis in heart, skin and lung. Am. J. Physiol. Heart. Circ. Physiol. 313, H934–H945. doi: 10.1152/ajpheart.00206.2017
Vermeulen, Z., Segers, V. F., and De Keulenaer, G. W. (2016). ErbB2 signaling at the crossing between heart failure and cancer. Basic Res. Cardiol. 111:60. doi: 10.1007/s00395-016-0576-z
Villegas, S., Villarreal, F. J., and Dillmann, W. H. (2000). Leukemia Inhibitory Factor and Interleukin-6 downregulate sarcoplasmic reticulum Ca2+ ATPase (SERCA2) in cardiac myocytes. Basic Res. Cardiol. 95, 47–54. doi: 10.1007/s003950050007
Wan, C.-R., Chung, S., and Kamm, R. D. (2011). Differentiation of embryonic stem cells into cardiomyocytes in a compliant microfluidic system. Ann. Biomed. Eng. 39, 1840–1847. doi: 10.1007/s10439-011-0275-8
Wang, J., Tang, Y., Zhang, W., Zhao, H., Wang, R., Yan, Y., et al. (2013). Insulin-like growth factor-1 secreted by brain microvascular endothelial cells attenuates neuron injury upon ischemia. FEBS J. 280, 3658–3668. doi: 10.1111/febs.12359
Wang, S., Sun, A., Li, L., Zhao, G., Jia, J., Wang, K., et al. (2012). Up-regulation of BMP-2 antagonizes TGF-β1/ROCK-enhanced cardiac fibrotic signalling through activation of Smurf1/Smad6 complex. J. Cell. Mol. Med. 16, 2301–2310. doi: 10.1111/j.1582-4934.2012.01538.x
Weber, K. T., and Brilla, C. G. (1991). Pathological hypertrophy and cardiac interstitium - fibrosis and renin-angiotensin-aldosterone system. Circulation 83, 1849–1865. doi: 10.1161/01.CIR.83.6.1849
Wei, K., Serpooshan, V., Hurtado, C., Diez-Cuñado, M., Zhao, M., Maruyama, S., et al. (2015). Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 525, 479–485. doi: 10.1038/nature15372
Wittchen, F., Suckau, L., Witt, H., Skurk, C., Lassner, D., Fechner, H., et al. (2007). Genomic expression profiling of human inflammatory cardiomyopathy (DCMi) suggests novel therapeutic targets. J. Mol. Med. 85, 257–271. doi: 10.1007/s00109-006-0122-9
Wollert, K. C., and Drexler, H. (2001). The role of interleukin-6 in the failing heart. Heart Fail. Rev. 6, 95–103. doi: 10.1023/A:1011401825680
Wu, C. K., Lee, J. K., Chiang, F. T., Yang, C. H., Huang, S. W., Hwang, J. J., et al. (2011). Plasma levels of tumor necrosis factor-alpha and interleukin-6 are associated with diastolic heart failure through downregulation of sarcoplasmic reticulum Ca2+ ATPase. Crit. Care Med. 39, 984–992. doi: 10.1097/CCM.0b013e31820a91b9
Wu, C.-K., Wang, Y.-C., Lee, J.-K., Chang, S.-N., Su, M.-Y., Yeh, H.-M., et al. (2014). Connective tissue growth factor and cardiac diastolic dysfunction: human data from the Taiwan Diastolic Heart Failure Registry and molecular basis by cellular and animal models. Eur. J. Heart Fail. 16, 163–172. doi: 10.1002/ejhf.33
Xia, Y., Dobaczewski, M., Gonzalez-Quesada, C., Chen, W., Biernacka, A., Li, N., et al. (2011). Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension 58, 902–911. doi: 10.1161/HYPERTENSIONAHA.111.175323
Yamaguchi, H., Komamura, K., Choraku, M., Hirono, A., Takamori, N., Tamura, K., et al. (2008). Impact of serum insulin-like growth factor-1 on early prognosis in acute myocardial infarction. Int. Med. 47, 819–825. doi: 10.2169/internalmedicine.47.0736
Yamazaki, T., Komuro, I., and Yazaki, Y. (1998). Signalling pathways for cardiac hypertrophy. Cell. Signal. 10, 693–698. doi: 10.1016/S0898-6568(98)00036-9
Yu, C., Cheung, B., Leung, R., Wang, Q., Lai, W., and Lau, C. (2001). Increase in plasma adrenomedullin in patients with heart failure characterised by diastolic dysfunction. Heart 86, 155–160. doi: 10.1136/heart.86.2.155
Yu, X., Kennedy, R. H., and Liu, S. J. (2003). JAK2/STAT3, Not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J. Biol. Chem. 278, 16304–16309. doi: 10.1074/jbc.M212321200
Zhang, X., Hu, W., Feng, F., Xu, J., and Wu, F. (2016). Apelin-13 protects against myocardial infarction-induced myocardial fibrosis. Mol. Med. Rep. 13, 5262–5268. doi: 10.3892/mmr.2016.5163
Zhang, Y., Liu, Y., Zhu, X. H., Zhang, X. D., Jiang, D. S., Bian, Z. Y., et al. (2014). Dickkopf-3 attenuates pressure overload-induced cardiac remodelling. Cardiovasc. Res. 102, 35–45. doi: 10.1093/cvr/cvu004
Zhao, L., Cheng, G., Jin, R., Afzal, M. R., Samanta, A., Xuan, Y.-T., et al. (2016). Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ. Res. 118, 1918–1929. doi: 10.1161/CIRCRESAHA.116.308688
Zhong, J. C., Zhang, Z. Z., Xu, R., Chen, L. J., Guo, S. J., Xu, Y. L., et al. (2016). Os 36-07 Treatment with apelin-13 prevents pressure overload-induced aortic adventitial remodeling and fibrosis in hypertensive rats with tac. J. Hypertens. 34(Suppl. 1), e403–e404. doi: 10.1097/01.hjh.0000501036.54612.96
Zhou, Q., Heinke, J., Vargas, A., Winnik, S., Krauss, T., Bode, C., et al. (2007). ERK signaling is a central regulator for BMP-4 dependent capillary sprouting. Cardiovasc. Res. 76, 390–399. doi: 10.1016/j.cardiores.2007.08.003
Keywords: cardiac remodeling, endothelium, intercellular communication, proteins, heart failure
Citation: Segers VFM, Brutsaert DL and De Keulenaer GW (2018) Cardiac Remodeling: Endothelial Cells Have More to Say Than Just NO. Front. Physiol. 9:382. doi: 10.3389/fphys.2018.00382
Received: 29 January 2018; Accepted: 28 March 2018;
Published: 11 April 2018.
Edited by:
Gabriele Giacomo Schiattarella, University of Naples Federico II, ItalyReviewed by:
Luciana Venturini Rossoni, Universidade de São Paulo, BrazilFouad Zouein, American University of Beirut, Lebanon
Copyright © 2018 Segers, Brutsaert and De Keulenaer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent F. M. Segers, dmluY2VudC5zZWdlcnNAdWFudHdlcnBlbi5iZQ==