 Natasha Whitehead
Natasha Whitehead Jonathan F. Gill
Jonathan F. Gill Marijke Brink
Marijke Brink Christoph Handschin
Christoph Handschin- 1Biozentrum, University of Basel, Basel, Switzerland
- 2Department of Biomedicine, University of Basel and University Hospital Basel, Basel, Switzerland
Aging is associated with a decline in cardiac function due to a decreased myocardial reserve. This adverse cardiac remodeling comprises of a variety of changes, including a reduction in mitochondrial function and a decline in the expression of the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a central regulator of mitochondrial biogenesis and metabolic adaptation in the myocardium. To study the etiological involvement of PGC-1α in cardiac aging, we used mouse models mimicking the modest down- and upregulation of this coactivator in the old and the exercised heart, respectively. Young mice with reduced cardiac expression of PGC-1α recapitulated part of the age-related impairment in mitochondrial gene expression, but otherwise did not aggravate the aging process. Inversely however, moderate overexpression of PGC-1α counteracts numerous key age-related remodeling changes, e.g., by improving blood pressure, age-associated apoptosis, and collagen accumulation, as well as in the expression of many, but not all cardiac genes involved in mitochondrial biogenesis, dynamics, metabolism, calcium handling and contractility. Thus, while the reduction of PGC-1α in the heart is insufficient to cause an aging phenotype, moderate overexpression reduces pathological remodeling of older hearts and could thereby contribute to the beneficial effects of exercise on cardiac function in aging.
Introduction
Aging causes a decline in cardiac function as a result of decreased myocardial reserve and adverse remodeling (Dai et al., 2016). Key molecular phenotypes of cardiac aging include alterations in stress response pathways (Lakatta, 1993), mitochondrial function (Judge et al., 2005; Dai et al., 2012a; Tocchi et al., 2015), cardiac energy metabolism (Lee et al., 2002; Lopaschuk et al., 2010), calcium signaling (Koban et al., 1998; Hobai and O'Rourke, 2001; Bers, 2006), contractility (Zile and Brutsaert, 2002; Strait and Lakatta, 2012), cardiomyocyte death (Kwak, 2013b) and extracellular matrix (ECM) remodeling (Nadal-Ginard et al., 2003; Kwak, 2013a; Horn and Trafford, 2016). These changes are mediated by a complex interconnected network of transcriptional and posttranslational processes that initially may be beneficial, aimed at the maintenance of cardiac function, but in the long term often are detrimental to the heart (Volkova et al., 2005), resulting in age-related cardiac remodeling with impaired cardiac reserve and thus increasing the risk for heart failure and other cardiovascular events.
Intriguingly, various heart pathologies have been associated with mitochondrial dysfunction and altered cardiac metabolism (Dillon et al., 2012). Thus, not surprisingly, mitochondrial gene expression is prominently altered in the aging myocardium (Anderson and Prolla, 2009). As a consequence, aged hearts often reveal a mitochondrial impairment (Ventura-Clapier et al., 2008; Barton et al., 2016) while adequate mitochondrial function and integrity is vital for cellular homeostasis and cardiac performance (Chaudhary et al., 2011). Unfortunately, the underlying molecular mechanisms of these and other age-related changes are largely unknown. Notably however, the aged heart shows reduced levels of the peroxisome proliferator-activated receptor y coactivator 1α (PGC-1α) as well as downstream transcription factor binding partners including the estrogen-related receptor α (ERRα), the peroxisome proliferator-activated receptor α (PPARα) and the mitochondrial transcription factor A (TFAM) (Finck and Kelly, 2006; Scarpulla, 2008; Vina et al., 2009; Dillon et al., 2012; Vega and Kelly, 2017).
The transcriptional network that is controlled by PGC-1α and its transcription factor partners is intrinsically linked to the control of mitochondrial biogenesis and metabolic adaptation in various tissues, including skeletal, and cardiac muscle (Dillon et al., 2012; Kupr and Handschin, 2015; Schnyder et al., 2017; Vega and Kelly, 2017). Loss-of-function animal models for heart PGC-1α exhibit an inability to meet the energy demands precipitated by increased cardiac work load, primarily due to reduced mitochondrial fatty acid oxidation and ATP synthesis efficiency (Arany et al., 2005; Leone et al., 2005; Lai et al., 2008; Lehman et al., 2008; Martin et al., 2014). An analogous reduction in PGC-1α expression has been reported in different cardiac pathologies, linked to a switch from oxidative metabolism to glycolysis (Finck and Kelly, 2006; Vega and Kelly, 2017). Conversely, high-level cardiac-specific overexpression also leads to impaired heart function, in this case accompanied by uncontrolled mitochondrial biogenesis, loss of sarcomere structure and a dilated cardiomyopathy (Lehman et al., 2000). Thus, deregulation of PGC-1α in either direction evokes adverse effects in the heart, demonstrating the critical role of this transcriptional coactivator in the maintenance of cardiac health (Anderson and Prolla, 2009). Importantly, a moderate increase in PGC-1α expression, e.g., as observed in some endurance exercise studies (O'Neill et al., 2007; Kim et al., 2008; Riehle et al., 2014; Vettor et al., 2014; Tam et al., 2015), or even the prevention of PGC-1α deterioration, could elicit beneficial effects in the heart. For example, low level overexpression of PGC-1α in the myocardium promotes an excitation-contraction (E-C) coupling phenotype that is prototypic for physiological hypertrophy of the heart, in addition to altering the expression of genes involved in the regulation of the circadian clock, heat shock, excitability, calcium signaling and contraction (Mutikainen et al., 2016).
In this study, we used both gain- and loss-of-function models for moderate overexpression and reduction of PGC-1α in the heart, respectively, to study the involvement of this coactivator in controlling cardiac aging. While a modest downregulation of PGC-1α in young animals was linked to a mitochondrial gene expression signature similar to that observed in an old heart, this reduction was insufficient to elicit a premature aging phenotype in regards to other aspects of age-related cardiac remodeling. Inversely however, a moderate elevation of PGC-1α blunted or prevented a broad range of age-associated changes in the heart of old mice.
Materials and Methods
Experimental Animals
Male, 3 month old and 24 month old PGC-1α muscle-specific knockout (MKO) and transgenic (MTg) mice of the same C57BL/6 background as well as their respective littermate controls (WT) were obtained from in-house breeding of previously described lines. The reduction of PGC-1α gene expression in the MKO animals resulted from human skeletal actin (HSA) promoter-driven cre expression in floxed PGC-1α mice (Perez-Schindler et al., 2013). The overexpression of PGC-1α in the MTg mice resulted from the muscle creatine kinase (MCK) promoter to drive the expression of a PGC-1α transgene (Lin et al., 2002). Both animal models depict their primary change in skeletal muscle, but due to the low expression of the HSA and MCK promoters in cardiomyocytes, a more modest reduction and elevation, respectively, of PGC-1α in the heart is achieved. The MKO mice were homozygous for the floxed PGC-1α and heterozygous for the HSA-cre allele. The MTg animals were heterozygous for the MCK-PGC-1α transgene. All mice were kept in a conventional facility under a 12/12 h light/dark cycle and had free access to food and water. This study was carried out in accordance with the principles of the Basel Declaration and with Federal and Cantonal Laws regulating the care and use of experimental animals in Switzerland, as well as institutional guidelines of the Biozentrum and the University of Basel. The protocol with all methods described here was approved by the “Kantonales Veterinäramt” of the Kanton Basel-Stadt, under consideration of the well-being of the animals and the 3R principle.
Blood Pressure Measurements
Arterial blood pressure (BP) was measured with a non-invasive tail cuff system (BP-2000 blood pressure analysis system, Visitech Systems). Blood pressure was measured over 5 consecutive days with 5 pre-measurements and 15 measurements per day. Mice were rotated in terms of their position and measurements were performed at the same time each day.
Tissue Collection
Animals were sacrificed by carbon dioxide inhalation and mouse length was measured from the tail base to the nose with mice lying straight on the dissection plate. Immediately after death, hearts were dissected, washed in Dulbecco's phosphate buffered saline (PBS, Sigma-Aldrich) and cut transversely in half. The upper half was flash frozen in liquid nitrogen for RNA and protein extraction. The lower part was frozen in cooled isopentane and embedded in 7% Tragacanth for preparation of cryosections. Transverse heart area and collagen quantification were assessed by consecutive cutting of the sections starting at the tip until the left ventricle was reached. Transverse heart area was determined on 5–6 sections per heart in at least 4 mice per group by using a threshold in which the entire tissue could be visualized after conversion of the color into primary images and using the functions “Analyze” and “Measure” to determine area and perimeter in Fiji.
RNA Isolation and Gene Expression Analysis
Total cardiac mRNA was extracted from frozen, crushed samples with the help of Lysing matrix tubes (MP Biomedicals), 1 ml Tri reagent (Sigma-Aldrich) and FastPrep FP120. The procedure was carried out according to the manufacturer's instructions. RNA concentration and purity was measured with a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific). The RNA purity was determined with the ratio of 260/280 nm and 260/230 nm. A purification step was carried out on samples with purities beneath 1.7.
RNA quality was assessed using a 2100 Bioanalyzer (G2938B, Agilent Technologies). The lowest RIN detected was 8.2. 400 ng of total RNA was treated with DNase I (Invitrogen) and then reverse transcribed into cDNA using SuperScript II Reverse Transcriptase (Invitrogen) and random hexanucleotide mix (Roche).
The relative expression levels of each gene of interest were quantified with the ΔΔCT method using FastStart Essential DNA Green Master (Roche) and the LightCycler 480 system (Roche). The average of the values obtained for the housekeeping genes TATA-binding protein (TBP), hypoxanthine guanine phosphoribosyl transferase (HPRT) and 18S rRNA was used for normalization Primer sequences are listed in Table 1.

Table 1. Primer pairs for real-time RT-qPCR analysis.
Protein Isolation and Western Blotting
Cardiac proteins were extracted from 30 mg of crushed samples using 300 μl ice-cold tissue lysis buffer [50 mM Tris-HCl (pH 7.5), 1 mM EDTA, 0.5 mM EGTA, 1% NP-40 substitute, 150 mM NaCl, 0.2% Na-deoxycholate, 1 mM dithiothreitol (DTT), complete mini protease inhibitor (Roche), PhosStop easy Pack phosphatase inhibitor (Roche), 10 mM nicotinamide], Pellet Pestel Motor (Kontes) and MISONIX ultrasonic liquid processor. Then, the samples were shaken for 30 min at 1,300 rpm at 4°C and centrifuged at 13,000 g for 10 min at 4°C. The protein concentration of the supernatant was determined by the albumin standard method (Thermo Scientific), samples were diluted in lysis buffer to a final concentration of 3 μg/μl containing 1x Laemmli Sample Buffer (Bio-Rad) and 20% β-mercaptoethanol (Bio-Rad) and subsequently boiled for 5 min.
Thirty microgram of proteins were loaded and separated on Mini-PROTEAN TGX Stain-free Precast gels (4–20%, Bio-Rad). The transfer was carried out for 1 h at 100 V onto nitrocellulose membranes. Membranes were blocked for 1 h in either 5% bovine albumin serum (BSA) or in 5% BSA with Tris buffered saline and Tween 20 (TBST) for 60 min and incubated overnight at 4°C with primary antibodies (see Table 2 for details): Myosin (skeletal, slow) (200 kDa; Sigma-Aldrich) (diluted 1:5,000) and Caspase 3 (17,19,35 kDa; Cell Signaling) (diluted 1: 1,000), both in 3% BSA. For protein detection, membranes were incubated for 1 h with the secondary antibody: Anti-Mouse (Dako) diluted 1:10,000 in 3% BSA, respectively 3% milk. Antibody detection was carried out using an appropriate chemiluminescence horseradish peroxidase (HRP) substrate detection kit (Thermo Scientific). The imaging and quantification of Western blots was done with the Fusion software (Fusion). Total protein was used as a loading control. Imaging of total protein was carried out on gel and membrane using a stain-free enabled imaging system (Fusion). Representative Western blots of two animals per group are shown, quantification was performed on n = 6 per group.
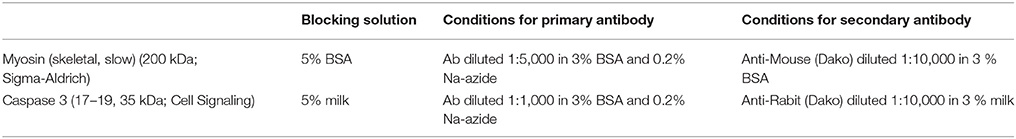
Table 2. Antibody conditions.
Picrosirius Red Staining
Eight micro meters-thick heart cryo-sections were thawed and stained for 1 h in Picrosirius Red (0.5 g Sirius Red under the name Direct Red 80, Sigma-Aldrich, in 500 ml 1.3% saturated aqueous solution of picric acid, Sigma-Aldrich). Sections were washed in acidified water (5 ml glacial acetic acid in 1 l distilled water), dehydrated by 3 incubations with 100% ethanol, cleared in xylene and mounted with Histomount (Invitrogen). Quantification of the collagen amount from the Picrosirius Red staining was carried out with the software Ilastik 1.1 and Fiji. Briefly, 12 images representing the true staining (collagen = red; background = yellow) as well as artifacts such as dirt, wrinkles, blood, image noise, blood vessels, unevenness of staining were cropped to train the program illastik to recognize these problem zones and create a masked image allowing an equal quantification of collagen of all used images (5–6 whole sections per group, sections obtained from different animals). The area covered by collagen in these masked images was measured using the software Fiji.
Hydroxyproline Assay
Ten microgram of frozen heart tissue was diluted in 100 μl nuclease free water and homogenized using the MISONIX ultrasonic liquid processor and transferred to a pressure-safe pyrex glass tube. The same amount of 37% HCl was added to the samples, which were then heated at 120°C for 3 h. For each assay, 10 μl of sample was transferred to a 96-well plate and evaporated at 60°C to dryness. Chloramine T (55 mM chloramine T), 10% 2-propanol in acetate citrate buffer (0.8M sodium acetate trihydrate, 240 mM citric acid, 1.2% glacial acetic acid, 850 mM NaOH in 1 l ddH2O) and Ehrlich's reagent [10 mM p-dimethylaminobenzaldehyde in 2-propanol/perchloric acid (2:1 v/v)] were added and the absorbance at 560 nm was measured with Infinite M1000 (Tecan). The amount of hydroxyproline was calculated with the help of a hydroxyproline standard (Sigma-Aldrich).
Statistical Analysis
All statistical calculations were done with GraphPad PRISM version 6.07. Statistical significance was calculated using 2-Way ANOVA and all values were displayed as means ± standard errors of the means (SEM). */#: p < 0.05; **/##: p < 0.01; ***/###: p < 0.001; ****/####: p < 0.0001 indicates age/genotype related significant differences. In the case of p values close, but slightly above to the significance cut-off of p < 0.05, actual p-values are indicated.
Results
Moderate Overexpression of PGC-1α Prevents Age-Related Increases in Diastolic Blood Pressure
First, general cardiovascular traits were analyzed in our models of moderate PGC-1α deficiency and overexpression in comparison to WT mice. All three mouse lines exhibited an age-linked increase in heart weight (Figure 1A) associated with an elevated cardiac area (Figure 1B). Intriguingly, the cardiac area was already significantly larger in the young PGC-1α MTg mice while the old MKO animals showed a reduced cardiac area compared with the respective age-matched WT mice (Figure 1B). Despite the parallel trend for heart weight and area, the age-related decrease in gene expression of the growth- and hypertrophy-regulating kinase mammalian target of rapamycin (mTOR) in the WT group was diametrically opposite in the MTg mice (Figure 1C). Furthermore, a strong increase in vascular endothelial growth factor (VEGF) gene expression was observed in both young and old MTg mice (Figure 1D), which might be indicative of improved tissue vascularization in this model. Even though some studies reported depressed heart rates with increased age (Chaudhary et al., 2011; Moghtadaei et al., 2016), we observed an increase in the WT as well as MKO animals (Figure 1E). The MTg mice exhibited an increased heart rate already at young age (Figure 1D). Overall, blood pressures were lower in MKO and MTg mice than in WT mice (Figures 1F,G). Systolic blood pressures did not change with age, whereas as published, diastolic pressures were significantly higher in the old than in the young mice of (Figure 1F). Notably, there is an age-dependent increase of diastolic blood pressure in the WT and MKO lines, which is prevented by the moderate PGC-1α overexpression in the MTg mice (see Gill et al., 2018, replicated in Figure 1G).
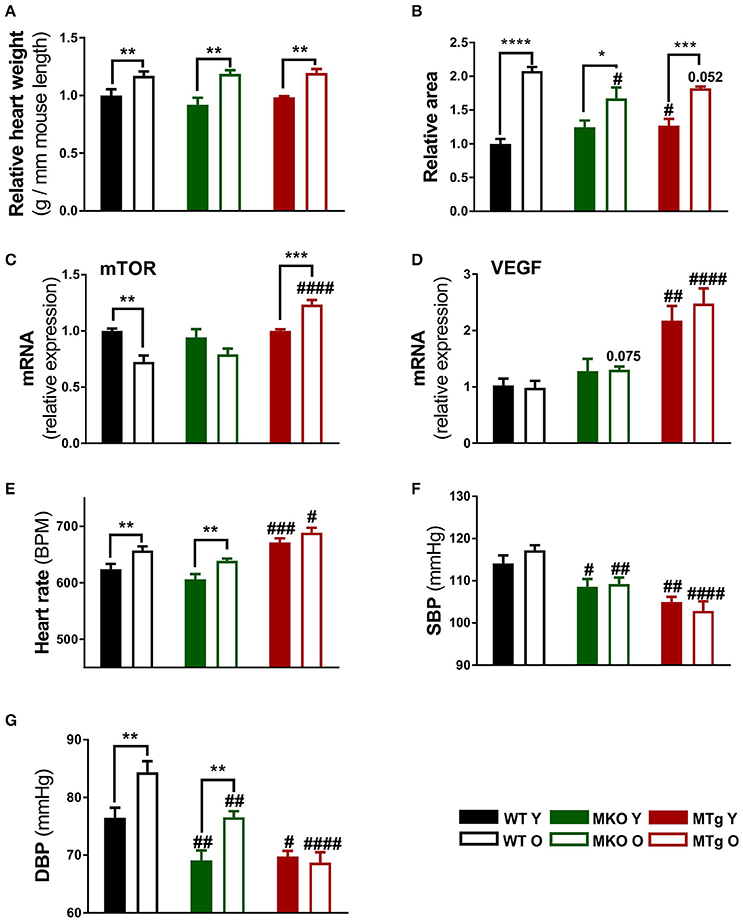
Figure 1. Moderate PGC-1α modulation reduces blood pressure. (A) Heart weight to mouse length ratios were assessed after sacrifice at 3 and 24 months of age for the young (filled bars) and old (open bars) mice, respectively (n = 6/group). (B) Transverse heart area was measured on microscopy sections using Fiji software (n = 4/group). Relative area refers to normalization to the area measured in WT Y animals. (C,D) Gene expression of mTOR and VEGF (n = 6/group). (E–G) Heart rate, systolic and diastolic blood pressure were measured with BP-2000 Blood Pressure Analysis System (Visitech Systems) (n = 10–12/group). Data are means ± SEM. Significant differences (p < 0.05) associated with age and phenotype are indicated by asterisks (*) and by hashtags (#, compared to WT), respectively (*/#: p < 0.05; **/##: p < 0.01; ***/###: p < 0.001; ****/####: p < 0.0001 indicates age/genotype related significant differences). DBP, diastolic blood pressure; mTOR, mammalian target of rapamycin; O, old; SBP, systolic blood pressure; Y, young; WT, wildtype; MKO, muscle knockout; MTg, muscle transgenic.
Moderate Overexpression of PGC-1α Counteracts Age-Associated ECM Remodeling
In older hearts, programmed cell death and necrosis induce not only loss of contractile tissue, but also a reactive compensatory hypertrophy of remaining cardiomyocytes, accumulation of collagen and fibrosis (Jugdutt, 2003). In all three mouse lines, mRNA expression of collagen I and III was reduced with age (Figure 2A). Strikingly however, picrosirius red staining revealed that an accumulation of collagen protein in aged WT mice was completely abrogated in the MTg animals (Figures 2B–D). This discrepancy between transcript and protein levels could stem from an adaptive, repressive process to reduce collagen expression, which however is dominated by the altered balance between ECM synthesis and degradation due to aging-induced modulation of matrix metalloprotease (MMP) and tissue inhibitors of metalloproteinases (TIMP) activities (Kwak, 2013a). Apoptosis may be an additional potential initiator of ECM remodeling. In line with the effect on collagen accumulation, moderate overexpression of PGC-1α in the MTg model prevented the age-linked increase in cleaved caspase 3, a known marker of apoptosis (Figures 2E–G). Finally, moderate elevation of PGC-1α also counteracted the age-associated diminished gene expression of the telomeric repeat-binding factor 1 and 2 (TERF1 and TERF2) (Figures 2H,I), two proteins that protect mammalian telomers (Palm and de Lange, 2008; Bernardes de Jesus and Blasco, 2012; Moslehi et al., 2012). The young MKO mice already exhibit levels of TERF1 corresponding to old WT animals (Figure 2H).
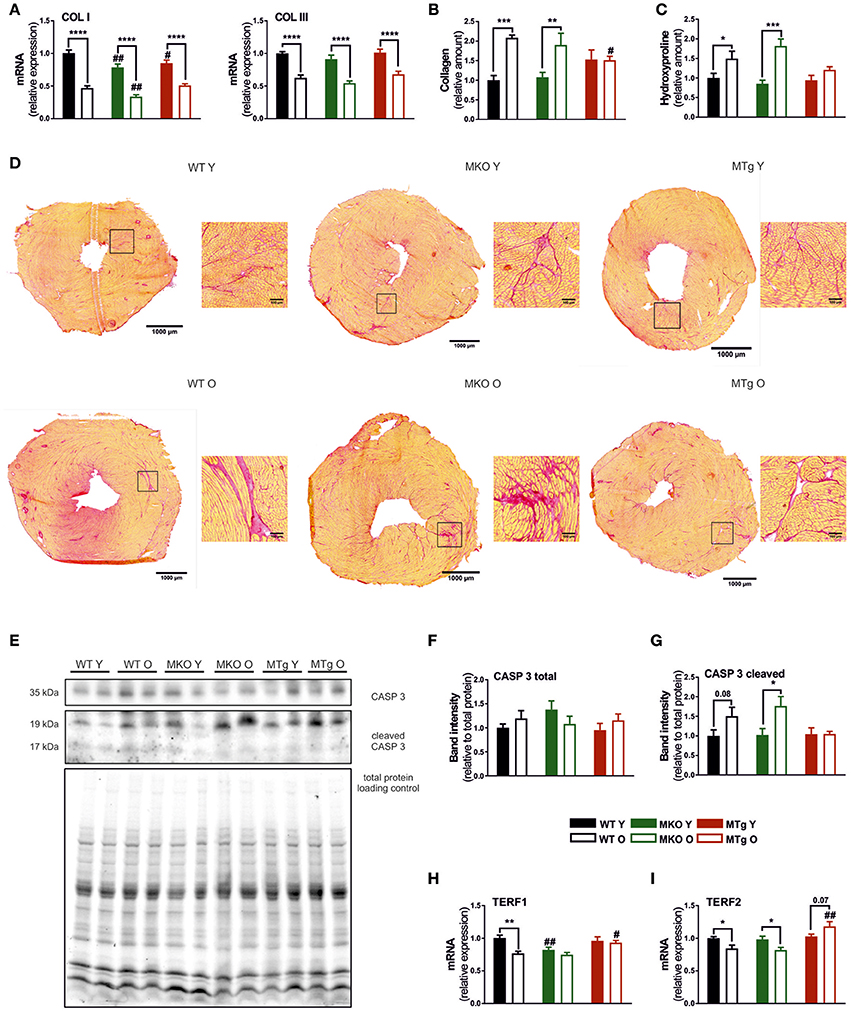
Figure 2. PGC-1α overexpression reduces age-dependent collagen accumulation and the age-induced expression of telomere-modulating genes. (A) Gene expression of collagen type I and III (n = 6/group). (B) Relative collagen content measured with Picrosirius Red staining (n = 5–6/group). (C) Relative content of hydroxyproline measured with hydroxyproline assay (n = 6/group). (D) Picrosirius Red staining done on 8 μm thick cryosections, red indicates collagen. (E) Western blot of caspase 3 and cl. caspase 3, normalized to total protein loading control. Western blot quantification (n = 6/group) of (F) caspase 3 and (G) cleaved caspase 3 (n = 6/group). (H,I) Expression of TERF1 and TERF2 mRNA (n = 6/group). Data are means ± SEM of values from each group. Significant age/genotype-associated differences (p < 0.05) are indicated by asterisks (*) and by hashtags (#, compared to WT) respectively (*/#: p < 0.05; **/##: p < 0.01; ***p < 0.001; ****p < 0.0001 indicates age/genotype related significant differences). CASP, caspase; COL, collagen; O, old; TERF, telomeric repeat-binding factor; Y, young.
Moderate PGC-1α Overexpression Counteracts the Age-Associated Downregulation of Mitochondrial Gene Expression
Similar to other heart pathologies, cardiac aging leads to the decreased expression of genes involved in mitochondrial biogenesis and function (Dai et al., 2012b). In line, the level of several mitochondrial transcripts was reduced in old compared to young WT mice (Figure 3), notably also the levels of PGC-1α and the estrogen-related receptor α (ERRα), one of the major transcription factor binding partners of PGC-1α in the regulation of metabolic gene expression (Huss et al., 2002). This reduction was also observed for some of these genes in the MKO mice (Figure 3) while importantly, the transcript levels of several mitochondrial genes were already reduced in young MKO animals comparable to old WT expression (Figure 3). Moreover, mitochondrial gene expression was further reduced in MKO animals compared to old WT mice. Strikingly, moderate overexpression of PGC-1α not only almost completely blunted the age-associated reduction in gene expression, but was also sufficient to raise the transcript levels of most genes in young and old MTg hearts above those of young WT mice (Figure 3). These patterns were not only observed in genes encoding proteins important for mitochondrial biogenesis, Krebs cycle and oxidative phosphorylation (OXPHOS) (Figures 3A–I), but also in mitochondrial fission and fusion gene expression (Figures 3J–O). WT mice showed an age-dependent decrease in the fatty acid β-oxidation (FAO) transcripts (Figures 3P–R) indicating a possible decreased reliance of cardiomyocytes on lipid metabolism in line with the reduction in OXPHOS gene expression. Interestingly, the FAO gene expression did not follow the WT pattern in the MKO mice. More consistently, we observed a pattern of increased transcript levels of the FAO genes, and an abrogation of the age effect in the MTg mice compared to the age-matched WT animals (Figures 3P–R).
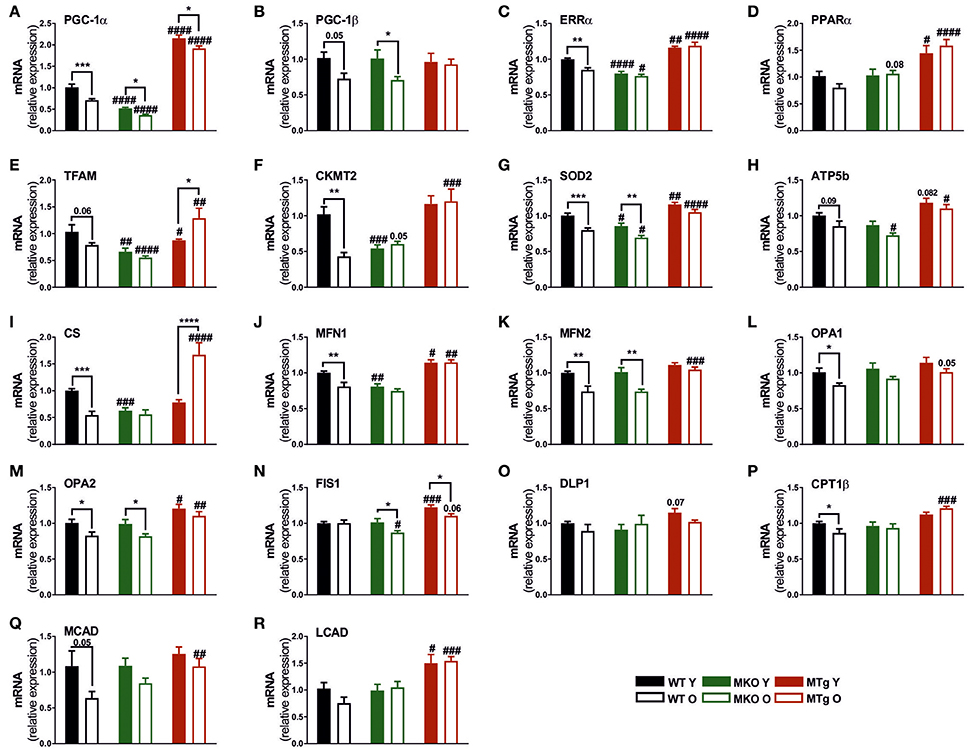
Figure 3. PGC-1α overexpression protects against the age-related reduction of genes involved in mitochondrial biogenesis, dynamics, oxidative phosphorylation and fatty acid oxidation. (A–F) Gene expression profile of genes involved in mitochondrial biogenesis and function (n = 6/group). (G–I) Gene expression levels of mitochondrial superoxide dismutase (SOD2), OXPHOS and TCA cycle genes (n = 6/group). (J–M) Mitochondrial fusion gene expression profile (n = 6/group). (N,O) Mitochondrial fission gene expression profile (n = 6/group). (P–R) Fatty acid β-oxidation (FAO) gene expression profile. Data are means ± SEM of values from each group. Significant age/genotype-associated differences (p < 0.05) are indicated by asterisks (*) and by hashtags (#, compared to WT) respectively (*/#: p < 0.05; **/##: p < 0.01; ***/###: p < 0.001; ****/####: p < 0.0001 indicates age/genotype related significant differences). ATP5B ATP synthase, H+ transporting, mitochondrial F1 complex, beta polypeptide; CKMT2, creatine kinase, mitochondrial 2; CPT1β, carnitine palmitoyltransferase 1β; CS, citrate synthase; DLP1, dynamin-like protein 1; FIS1, fission, mitochondrial 1; LCAD, long chain acyl-CoA dehydrogenase; MCAD, medium chain acyl-CoA dehydrogenase; MFN, mitofusin; O, old; Opa, mitochondrial dynamin like GTPase; SOD2, mitochondrial superoxide dismutase; TCA, tricaboxylic acid; Y, young.
Moderate PGC-1α Overexpression Induces a Gene Expression Profile Favoring Calcium Handling and Contractile Function
In addition to mitochondrial gene expression, age-related cardiac changes also include a decline in transcription of genes encoding proteins for calcium handling and contractility (Koban et al., 1998; Hobai and O'Rourke, 2001; Zile and Brutsaert, 2002; Bers, 2006; Strait and Lakatta, 2012). The age-related decrease in sarcoplasmic/endoplasmic reticulum Ca2+ transporting ATPase 2 (SERCA2) gene expression was observed in WT and MKO mice, but was completely prevented by the moderate overexpression of PGC-1α in the MTg mice (Figure 4A). Interestingly, the MTg animals also exhibited elevated transcript levels of phospholamban (PLB) and sodium/calcium exchanger protein 1 (NCX1), two additional genes involved in the regulation of sarcoplasmic reticulum (SR) calcium handling (Figures 4B,C), which could indicate overall improved calcium clearance. A blunting of the age effect in the MTg mice was also observed for α-myosin heavy chain (α-MHC) expression (Figure 4D). Curiously, an increase in β-MHC transcript levels was found in both old MKO and MTg, but not WT animals (Figure 4E). In contrast however, aging increased slow myosin protein levels only in WT and MKO mice, while the overexpression of PGC-1α in the MTg animals prevented this switch (Figures 4F,G). Finally cardiac α-actinin (ACTC1) expression was elevated in young and old MTg mice (Figure 4H), while cardiac troponin I3 (TNNI3) transcripts were significantly increased only in old MTg animals compared to the WT and MKO counterparts (Figure 4I).
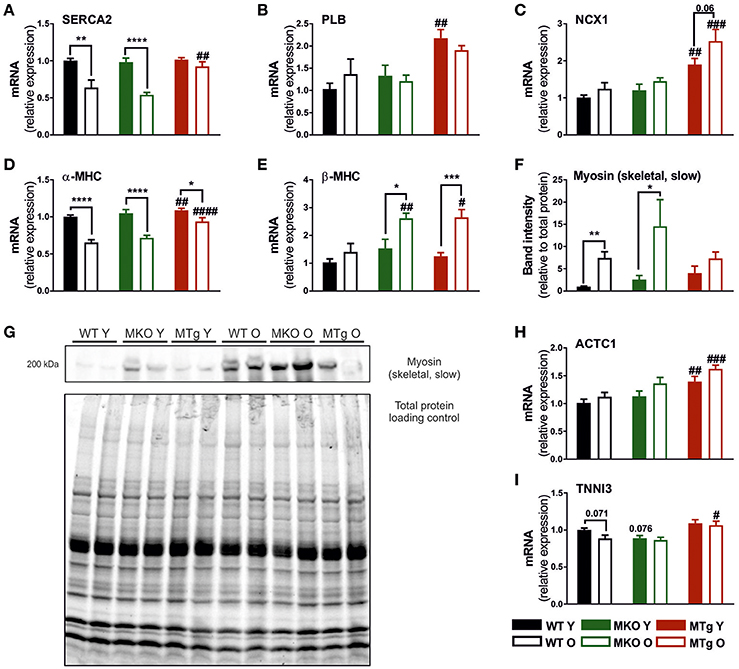
Figure 4. Increased PGC-1α blunts the age-associated regulation of calcium homeostasis and contractile genes. Gene expression levels of (A) SERCA2, (B) PLN, (C) NCX1, (D) α-MHC, (E) β-MHC, (H) ACTC1, and (I) TNNI3 (n = 6/group). (F,G) Western blot and quantification (n = 6/group) of myosin (skeletal, slow), which represents β-MHC in the heart, normalized to total protein loading control. Data are means ± SEM of values from each group. Significant age/genotype-associated differences (p < 0.05) are indicated by asterisks (*) and by hashtags (#, compared to WT) respectively (*/#: p < 0.05; **/##: p < 0.01; ***/###: p < 0.001; ****/####: p < 0.0001 indicates age/genotype related significant differences). ACTC1, actin, alpha, cardiac muscle 1; PLB, phospholamban; MHC, myosin heavy chain; NCX1, sodium/calcium exchanger protein 1; O, old; SERCA2, sarcoplasmic/endoplasmic reticulum Ca2+ transporting ATPase 2; TNNI3, troponin I3, cardiac type; Y, young.
Discussion
Aging affects the function of every organ in our body. In the heart, age-related remodeling is closely linked to an increased risk for cardiac disease and heart failure. Similar to other pathological contexts, a shift in metabolism from oxidative substrate usage toward glycolysis, accompanied by a reduction in mitochondrial number and function, has been described in older hearts. This metabolic switch is often observed together with a decrease in the expression of PGC-1α (Dorn et al., 2015). Indeed, we have also observed an age-linked decline in PGC-1α expression (Figure 3A). While the mechanistic underpinnings of this regulation are unclear, it is conceivable that analogous to skeletal muscle, cardiac PGC-1α gene expression is based on the aging-associated reduction in contractile function, but maybe also due to changes in the neuroendocrine milieu, or metabolic properties. Inversely, exercise, which exerts beneficial effects on cardiac function, elevates PGC-1α gene expression in cardiac and skeletal muscle (Vega et al., 2017). We have now used transgenic models of moderate reduction and overexpression, respectively, to study the involvement of PGC-1α in the cardiac aging process. Notably, relatively small changes in PGC-1α gene expression resulted in striking changes in the transcriptional program in the young and old heart (summarized in Table 3). Determination of mitochondrial function would be of interest in future studies, since several aspects of mitochondrial respiration are altered in old skeletal muscle of gain- and loss-of-function models for PGC-1α (Gill et al., 2018). Strikingly, moderate overexpression of PGC-1α in the heart in the range of 1.5- to 2-fold quantitatively mimicking the increase in PGC-1α gene expression after exercise (O'Neill et al., 2007; Kim et al., 2008; Riehle et al., 2014; Vettor et al., 2014; Tam et al., 2015) blunted or prevented various aspects of age-associated transcriptional cardiac remodeling. As a key regulatory factor in the control of mitochondrial biogenesis and oxidative metabolism, PGC-1α overexpression resulted in a marked elevation of a number of mitochondrial and other metabolic genes. The modulation of several mitochondrial fission and fusion genes could indicate that PGC-1α is linked to higher mitochondrial dynamics and ultimately healthier mitochondria in cardiac myocytes (Chan, 2006; Chaudhary et al., 2011). Moreover, PGC-1α could reduce the levels of reactive oxygen species (ROS) (St-Pierre et al., 2006), which increase in the aged heart, cause oxidative damage to proteins, lipids and DNA, leading to the dysregulation of redox-sensitive signaling pathways (Judge et al., 2005). Such a PGC-1α-dependent effect has for example been described in diabetic nephropathy (Guo et al., 2015). A moderate elevation of PGC-1α in line with that observed in the trained heart thus seems sufficient to mitigate the aging-associated deterioration in mitochondrial function and cellular metabolism. Of note, superphysiological overexpression of cardiac PGC-1α has been linked to exacerbated mitochondrial biogenesis replacing myofibrillar structure, and ultimately leading to cardiomyopathy (Lehman et al., 2000; Russell et al., 2004).
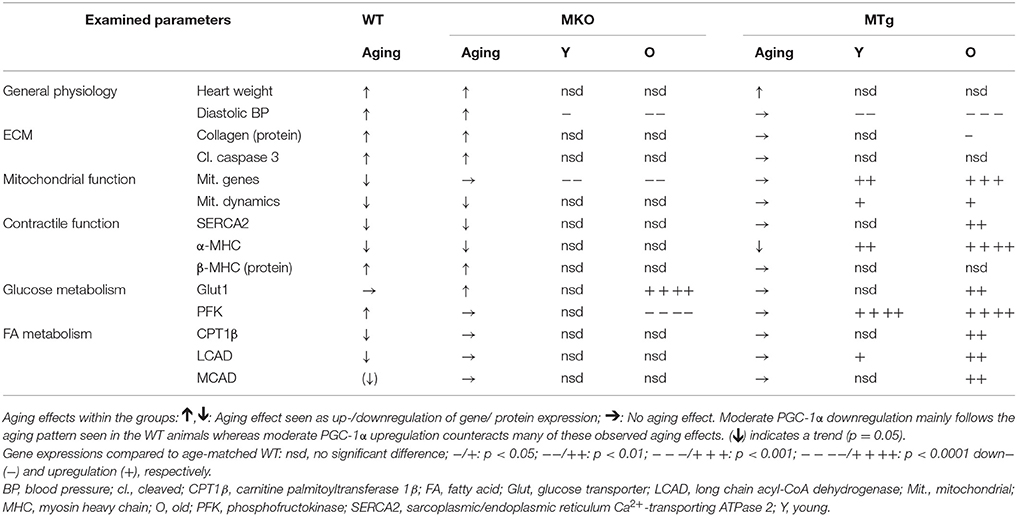
Table 3. Overview of age-related changes in WT, MKO, and MTg groups and of the MKO and MTg compared to WT animals.
Reduced mitochondrial function in aging has been associated with enhanced apoptosis, ECM remodeling and fibrosis (Chaudhary et al., 2011; Martin-Fernandez and Gredilla, 2016). The mitigation of age-associated cleavage of caspase 3, accumulation of collagens and expression of TERF2 in the MTg animals implies a broad effect of cardiac PGC-1α to reduce different aspects of this adverse remodeling. A similar outcome has been reported in a mouse model for pressure overload-induced cardiac hypertrophy, in which PGC-1α reduced apoptosis and fibrosis in severely stressed hearts (Pereira et al., 2014). Inversely, adequate mitochondrial function is essential to provide the primary source of energy that fuels the contractile apparatus according to the cellular demand (Chan, 2006). Moreover, tightly controlled calcium handling, and appropriate expression of contractile proteins are crucial for normal heart function. In the MTg mice, PGC-1α prevented the drop in SERCA2 expression, which is downregulated with age and other pathological cardiac conditions (Lompre et al., 1991). The concomitant increase in PLB and NCX1 transcript levels suggest a broader PGC-1α-dependent remodeling of cardiac calcium handling, analogous to the regulation of SR calcium homeostasis in skeletal muscle (Summermatter et al., 2012). This hypothesis is in line with previous findings describing that increased PGC-1α levels improve contractile and diastolic performance of cardiomyocytes by increasing calcium reuptake into the SR due to elevated SERCA2 activity, thereby preventing cellular calcium overload in the heart (Chen et al., 2010). In another study using the MTg animals as a model for modest cardiac elevation of PGC-1α, a strengthening of E-C coupling inducing favorable changes in excitability, calcium signaling and contraction has been reported (Mutikainen et al., 2016). Thus, together with the prevention of the aging-linked shift in contractile protein expression, e.g., the elevation of β-MHC protein, PGC-1α overexpression regulates different programs that are involved in contractile function of the heart.
Finally, several of our findings support that the age-associated cardiac remodeling differs between the MTg and the other two lines. In contrast to the old WT and MKO mice, which both show the same features typical of pathological cardiac hypertrophy, possibly associated with their higher diastolic blood pressures, the results obtained for the old PGC-1α-overexpressing mice suggest a more physiological form of hypertrophy (Ellison et al., 2012). First of all, physiological hypertrophy is characterized by improvements in oxidative phosphorylation, calcium handling as well as reduced fibrosis and apoptosis (Mann and Rosenzweig, 2012). Several of these traits were indeed found in our MTg line. Second, increased mTOR expression has been linked to physiological eccentric hypertrophy (Gielen et al., 2010; Ikeda et al., 2015) and was also observed in our MTg mice. Furthermore, the increase in VEGF expression in the MTg mice could be indicative of a higher tissue vascularization, another feature of physiological cardiac hypertrophy. Consistent with our findings, restoration of cardiac levels of PGC-1α resulted in an increase in VEGF and a preservation of capillary density in mice with a transverse aortic constriction (Pereira et al., 2014). Finally, the changes in calcium handling proteins observed in the MTg animals suggest improved E-C coupling, in line with observations done in exercise-induced physiological hypertrophy of the heart (Mutikainen et al., 2016).
Collectively, the results obtained in the MTg mice suggest that modest overexpression of PGC-1α in the heart is sufficient to prevent many adverse changes evoked by aging in the heart while others remain unaffected, or even show an unexpected phenotype (e.g., heart rate). Of note, some of the potentially beneficial effects are already observed in young MTg animals, and are preserved into old age. Inversely however, the mild reduction in PGC-1α transcript levels in the MKO mice elicited a more restricted response, even though this reduction of ~40–50% quantitatively recapitulates the decrease in PGC-1α gene expression in different contexts and models of cardiac pathologies (Garnier et al., 2009; Schilling et al., 2011). The requirement for adequate PGC-1α gene expression was most notable in the regulation of several mitochondrial genes. Most of the other parameters that were studied here were unaffected by the reduction in PGC-1α in the MKO model, with the exception of TERF1. This surprisingly mild phenotype could be caused by limitations of the animal model, which harbors a life-long reduction in PGC-1α that could trigger adaptive responses, or in which the reduction might simply be insufficient to induce a more severe aging phenotype in the heart. Unfortunately, cardiac-specific knockout mice for PGC-1α have not been reported yet, and therefore, the phenotype of mice with a complete ablation of PGC-1α in the heart is unknown. Second, the cardiac phenotype of the MKO and the MTg animals might be confounded by the more marked knockout and overexpression in skeletal muscle, e.g., by modulation of the secretion of myokines or differences in locomotion and endurance. Furthermore, it is conceivable that the numerous compensatory, redundant, adaptive, maladaptive and reparative mechanisms, which are engaged to provide cardio-protection in a pathological context (Strait and Lakatta, 2012), circumvent an absolute requirement for PGC-1α to ensure proper regulation of different biological programs. In line with this hypothesis, whole body ablation of PGC-1α results only in a mild phenotype, which is dramatically exacerbated in a PGC-1α/PGC-1β double-knockout context (Lai et al., 2008). Then, aging is a progressive phenomenon, in which most aging-linked changes are initiated and escalated at the age of 1–24 months in C57Bl6/J mice (Flurkey et al., 2007). Based on the time of sacrifice, we obviously can only draw direct conclusions from our data on hearts of mice at the age of 24 months. It is entirely possible that these effects might differ in quantitative and qualitative outcome at earlier or later time points. Finally, it is obviously conceivable that cardiac PGC-1α is largely dispensable for the aging phenotype.
Conclusion
PGC-1α is a pleiotropic transcriptional coregulator that controls complex networks and biological programs in various tissues (Kupr and Handschin, 2015). Using genetic mouse models to mimic the reduction in cardiac PGC-1α expression in pathological contexts and the increase in PGC-1α transcript levels in exercise, respectively, we have now delineated the contribution of this coactivator to the aging response of the heart. The MKO mice exhibited a precocious aging phenotype in regard to mitochondrial gene expression, but as a single hit, a reduction in cardiac PGC-1α is insufficient to trigger or exacerbate the adverse remodeling of older hearts. Strikingly however, the MTg animals were largely protected against the pathological plasticity induced by aging, indicating that elevation of PGC-1α, e.g., as observed after endurance training, could be a potential intervention to mitigate the decline in heart function and reduce aging-associated cardiac pathologies. Thus, since our analyses were largely focused on the transcriptional program, future studies should aim at a more functional assessment of contractile function, potentially combined with interventional approaches to boost cardiac PGC-1α levels.
Author Contributions
JG, NW, MB, and CH designed the study. NW performed the experiments. NW, JG, MB, and CH analyzed and interpreted the data. NW and CH wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was funded by the Swiss National Science Foundation, the European Research Council (ERC) Consolidator grant 616830-MUSCLE_NET, Swiss Cancer Research grant KFS-3733-08-2015, the Swiss Society for Research on Muscle Diseases (SSEM), SystemsX.ch, the Novartis Stiftung für Medizinisch-Biologische Forschung and the University of Basel. JG was supported by a Fellowship for Excellence of the Biozentrum Basel International PhD Program.
Abbreviations
BP, blood pressure; E-C coupling, excitation-contraction coupling; ECM, extracellular matrix; FA, fatty acid; FAO, fatty acid oxidation; MTg, muscle-specific PGC-1α transgenic; MKO, muscle-specific PGC-1α knockout; OXPHOS, oxidative phosphorylation; PGC-1α, peroxisome proliferator-activated receptor γ coactivator 1α; SR, sarcoplasmic reticulum; VEGF, vascular endothelial growth factor; WT, Wild type.
References
Anderson, R., and Prolla, T. (2009). PGC-1α in aging and anti-aging interventions. Biochim. Biophys. Acta 1790, 1059–1066. doi: 10.1016/j.bbagen.2009.04.005
Arany, Z., He, H., Lin, J., Hoyer, K., Handschin, C., Toka, O., et al. (2005). Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 1, 259–271. doi: 10.1016/j.cmet.2005.03.002
Barton, G., Sepe, J., Mckiernan, S., Aiken, J., and Diffee, G. (2016). Mitochondrial and metabolic gene expression in the aged rat heart. Front. Physiol. 7:352. doi: 10.3389/fphys.2016.00352
Bernardes de Jesus, B., and Blasco, M. (2012). Assessing cell and organ senescence biomarkers. Circ. Res. 111, 97–109. doi: 10.1161/CIRCRESAHA.111.247866
Bers, D. (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiology 21, 380–387. doi: 10.1152/physiol.00019.2006
Chan, D. (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252. doi: 10.1016/j.cell.2006.06.010
Chaudhary, K., El-Sikhry, H., and Seubert, J. (2011). Mitochondria and the aging heart. J. Geriatr. Cardiol. 8, 159–167. doi: 10.3724/SP.J.1263.2011.00159
Chen, M., Wang, Y., and Qu, A. (2010). PGC-1 alpha accelerates cytosolic Ca2+ clearance without disturbing Ca2+ homeostasis in cardiac myocytes. Biochem. Biophys. Res. Commun. 396, 894–900. doi: 10.1016/j.bbrc.2010.05.018
Dai, D. F., Chen, T., Johnson, S. C., Szeto, H., and Rabinovitch, P. (2012a). Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 16, 1492–1526. doi: 10.1089/ars.2011.4179
Dai, D. F., Rabinovitch, P. S., and Ungvari, Z. (2012b). Mitochondria and Cardiovascular aging. Circ. Res. 110, 1109–1124. doi: 10.1161/CIRCRESAHA.111.246140
Dai, D., Chiao, Y., Wessells, R., Bodmer, R., Szeto, H., and Rabinovitch, P. (2016). “Cardiac Aging,” in Handbook of the Biology of Aging, 8 Edn. eds M. Kaeberlein and G. Martin (San Diego, CA: Academic Press), 459–494.
Dillon, L. M., Rebelo, A. P., and Moraes, C. T. (2012). The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 64, 231–241. doi: 10.1002/iub.608
Dorn, G. W. II., Vega, R. B., and Kelly, D. P. (2015). Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 29, 1981–1991. doi: 10.1101/gad.269894.115
Ellison, G. M., Waring, C. D., Vicinanza, C., and Torella, D. (2012). Physiological cardiac remodelling in response to endurance exercise training: cellular and molecular mechanisms. Heart 98, 5–10. doi: 10.1136/heartjnl-2011-300639
Finck, B. N., and Kelly, D. P. (2006). PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 116, 615–622. doi: 10.1172/JCI27794
Flurkey, K., Currer, J., and Harrison, D. (2007). “The mouse in aging research,” in The Mouse in Biomedical Research, eds J. Fox, S. Barthold, M. Davisson, C. Newcomer, F. Quimby and A. Smith. [Burlington, MA: American College Laboratory Animal Medicine (Elsevier)], 637–672.
Garnier, A., Zoll, J., Fortin, D., N'guessan, B., Lefebvre, F., Geny, B., et al. (2009). Control by circulating factors of mitochondrial function and transcription cascade in heart failure: a role for endothelin-1 and angiotensin II. Circ. Heart Fail. 2, 342–350. doi: 10.1161/CIRCHEARTFAILURE.108.812099
Gielen, S., Schuler, G., and Adams, V. (2010). Cardiovascular effects of exercise training: molecular mechanisms. Circulation 122, 1221–1238. doi: 10.1161/CIRCULATIONAHA.110.939959
Gill, J. F., Santos, G., Schnyder, S., and Handschin, C. (2018). PGC-1alpha affects aging-related changes in muscle and motor function by modulating specific exercise-mediated changes in old mice. Aging Cell 17:e12697. doi: 10.1111/acel.12697
Guo, K., Lu, J., Huang, Y., Wu, M., Zhang, L., Yu, H., et al. (2015). Protective role of PGC-1alpha in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS ONE 10:e0125176. doi: 10.1371/journal.pone.0125176
Hobai, I., and O'Rourke, B. (2001). Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation 103, 1577–1584. doi: 10.1161/01.CIR.103.11.1577
Horn, M., and Trafford, A. (2016). Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 93, 175–185. doi: 10.1016/j.yjmcc.2015.11.005
Huss, J. M., Kopp, R., and Kelly, D. (2002). Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J. Biol. Chem. 277, 40265–40274. doi: 10.1074/jbc.M206324200
Ikeda, M., Ide, T., Fujino, T., Matsuo, Y., Arai, S., Saku, K., et al. (2015). The Akt-mTOR axis is a pivotal regulator of eccentric hypertrophy during volume overload. Sci Rep 5:15881. doi: 10.1038/srep15881
Judge, S., Jang, Y., Smith, A., Hagen, T., and Leeuwenburgh, C. (2005). Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 19, 419–421. doi: 10.1096/fj.04-2622fje
Jugdutt, B. (2003). Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr. Drug Targets Cardiovasc. Haematol. Disord. 3, 1–30. doi: 10.2174/1568006033337276
Kim, J., Wende, A. R., Sena, S., Theobald, H. A., Soto, J., Sloan, C., et al. (2008). Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 22, 2531–2543. doi: 10.1210/me.2008-0265
Koban, M. U., Moorman, A. F., Holtz, J., Yacoub, M. H., and Boheler, K. (1998). Expressional analysis of the cardiac Na–Ca exchanger in rat development and senescence. Cardiovasc. Res. 37, 405–423. doi: 10.1016/S0008-6363(97)00276-9
Kupr, B., and Handschin, C. (2015). complex coordination of cell plasticity by a PGC-1alpha-controlled transcriptional network in skeletal muscle. Front. Physiol. 6:325. doi: 10.3389/fphys.2015.00325
Kwak, H. (2013a). Aging, exercise, and extracellular matrix in the heart. J. Exerc. Rehabil. 9, 338–347. doi: 10.12965/jer.130049
Kwak, H. (2013b). Effects of aging and exercise training on apoptosis in the heart. J. Exerc. Rehabil. 9, 212–219. doi: 10.12965/jer.130002
Lai, L., Leone, T. C., Zechner, C., Schaeffer, P. J., Kelly, S., Flanagan, D., et al. (2008). Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 22, 1948–1961. doi: 10.1101/gad.1661708
Lakatta, E. (1993). Cardiovascular regulatory mechanisms in advanced age. Physiol. Rev. 73, 413–467. doi: 10.1152/physrev.1993.73.2.413
Lee, C. K., Allison, D. B., Brand, J., Weindruch, R., and Prolla, T. (2002). Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc. Natl. Acad. Sci. U.S.A. 99, 14988–14993. doi: 10.1073/pnas.232308999
Lehman, J. J., Barger, P. M., Kovacs, A., Saffitz, J. E., Medeiros, D. M., and Kelly, D. P. (2000). Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106, 847–856. doi: 10.1172/JCI10268
Lehman, J. J., Boudina, S., Banke, N., Sambandam, N., Han, X., Young, D., et al. (2008). The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am. J. Physiol. Heart Circ. Physiol. 295, H185–H196. doi: 10.1152/ajpheart.00081.2008
Leone, T. C., Lehman, J. J., Finck, B. N., Schaeffer, P. J., Wende, A., Boudina, S., et al. (2005). PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3:e101. doi: 10.1371/journal.pbio.0030101
Lin, J., Wu, H., Tarr, P., Zhang, C., Wu, Z., Boss, O., et al. (2002). Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 418, 797–801. doi: 10.1038/nature00904
Lompré, A. M., Lambert, F., Lakatta, E. G., and Schwartz, K. (1991). Expression of sarcoplasmic reticulum Ca(2+)-ATPase and calsequestrin genes in rat heart during ontogenic development and aging. Circ. Res. 69, 1380-1388. doi: 10.1161/01.RES.69.5.1380
Lopaschuk, G. D., Ussher, J. R., Folmes, C. D., Jaswal, J. S., and Stanley, W. C. (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258. doi: 10.1152/physrev.00015.2009
Mann, N., and Rosenzweig, A. (2012). Can exercise teach us how to treat heart disease? Circulation 126, 2625–2635. doi: 10.1161/CIRCULATIONAHA.111.060376
Martin, O. J., Lai, L., Soundarapandian, M. M., Leone, T., Zorzano, A., Keller, M. P., et al. (2014). A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 114, 626–636. doi: 10.1161/CIRCRESAHA.114.302562
Martín-Fernández, B., and Gredilla, R. (2016). Mitochondria and oxidative stress in heart aging. Age 38, 225–238. doi: 10.1007/s11357-016-9933-y
Moghtadaei, M., Jansen, H., Mackasey, M., Rafferty, S., Bogachev, O., Sapp, J., et al. (2016). The impacts of age and frailty on heart rate and sinoatrial node function. J. Physiol. 594, 7105–7126. doi: 10.1113/JP272979
Moslehi, J., Depinho, R., and Sahin, E. (2012). Telomeres and mitochondria in the aging heart. Circ. Res. 110, 1226–1237. doi: 10.1161/CIRCRESAHA.111.246868
Mutikainen, M., Tuomainen, T., Naumenko, N., Huusko, J., Smirin, B., Laidinen, S., et al. (2016). Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha1 induces a cardiac excitation-contraction coupling phenotype without metabolic remodelling. J. Physiol. 594, 7049–7071. doi: 10.1113/JP272847
Nadal-Ginard, B., Kajstura, J., Leri, A., and Anversa, P. (2003). Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ. Res. 92, 139–150. doi: 10.1161/01.RES.0000053618.86362.DF
O'Neill, B. T., Kim, J., Wende, A. R., Theobald, H. A., Tuinei, J., Buchanan, J., et al. (2007). A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 6, 294–306. doi: 10.1016/j.cmet.2007.09.001
Palm, W., and de Lange, T. (2008). How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334. doi: 10.1146/annurev.genet.41.110306.130350
Pereira, R., Wende, A., Crum, A., Hunter, D., Olsen, C., Rawlings, T., et al. (2014). Maintaining PGC-1alpha expression following pressure overload-induced cardiac hypertrophy preserves angiogenesis but not contractile or mitochondrial function. FASEB J. 28, 3691–3702. doi: 10.1096/fj.14-253823
Pérez-Schindler, J., Summermatter, S., Santos, G., Zorzato, F., and Handschin, C. (2013). The transcriptional coactivator PGC-1alpha is dispensable for chronic overload-induced skeletal muscle hypertrophy and metabolic remodeling. Proc. Natl. Acad. Sci. U.S.A. 110, 20314–20319. doi: 10.1073/pnas.1312039110
Riehle, C., Wende, A. R., Zhu, Y., Oliveira, K. J., Pereira, R. O., Jaishy, B. P., et al. (2014). Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol. Cell. Biol. 34, 3450-3460. doi: 10.1128/MCB.00426-14
Russell, L. K., Mansfield, C. M., Lehman, J. J., Kovacs, A., Courtois, M., Saffitz, J. E., et al. (2004). Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 94, 525–533. doi: 10.1161/01.RES.0000117088.36577.EB
Scarpulla, R. (2008). Transcriptional paradigms in mammalian Mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638. doi: 10.1152/physrev.00025.2007
Schilling, J., Lai, L., Sambandam, N., Dey, C. E., Leone, T. C., and Kelly, D. P. (2011). Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor gamma coactivator-1 signaling. Circ. Heart Fail. 4, 474–482. doi: 10.1161/CIRCHEARTFAILURE.110.959833
Schnyder, S., Kupr, B., and Handschin, C. (2017). Coregulator-mediated control of skeletal muscle plasticity - A mini-review. Biochimie 136, 49–54. doi: 10.1016/j.biochi.2016.12.011
St-Pierre, J., Drori, S., Uldry, M., Silvaggi, J. M., Rhee, J., Jäger, S., et al. (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408. doi: 10.1016/j.cell.2006.09.024
Strait, J. B., and Lakatta, E. (2012). Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin. 8, 143–164. doi: 10.1016/j.hfc.2011.08.011
Summermatter, S., Thurnheer, R., Santos, G., Mosca, B., Baum, O., Treves, S., et al. (2012). Remodeling of calcium handling in skeletal muscle through PGC-1alpha: impact on force, fatigability, and fiber type. Am. J. Physiol. Cell Physiol. 302, C88–C99. doi: 10.1152/ajpcell.00190.2011
Tam, B. T., Pei, X. M., Yung, B. Y., Yip, S. P., Chan, L., Wong, C. S., et al. (2015). Autophagic adaptations to long-term habitual exercise in cardiac muscle. Int. J. Sports Med. 36, 526–534. doi: 10.1055/s-0034-1398494
Tocchi, A., Quarles, E. K., Basisty, N., Gitari, L., and Rabinovitch, P. (2015). Mitochondrial dysfunction in cardiac aging. Biochim. Biophys. Acta 1847, 1424–1433. doi: 10.1016/j.bbabio.2015.07.009
Vega, R., and Kelly, D. (2017). Cardiac nuclear receptors: architects of mitochondrial structure and function. J. Clin. Invest. 127, 1155–1164. doi: 10.1172/JCI88888
Vega, R. B., Konhilas, J. P., Kelly, D. P., and Leinwand, L. A. (2017). Molecular mechanisms underlying cardiac adaptation to exercise. Cell Metab. 25, 1012–1026. doi: 10.1016/j.cmet.2017.04.025
Ventura-Clapier, R., Garnier, A., and Veksler, V. (2008). Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc. Res. 79, 208–217. doi: 10.1093/cvr/cvn098
Vettor, R., Valerio, A., Ragni, M., Trevellin, E., Granzotto, M., Olivieri, M., et al. (2014). Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: role in adaptation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 306, E519–E528. doi: 10.1152/ajpendo.00617.2013
Viña, J., Gomez-Cabrera, M., Borras, C., Froio, T., Sanchis-Gomar, F., Martinez-Bello, V., et al. (2009). Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 61, 1369–1374. doi: 10.1016/j.addr.2009.06.006
Volkova, M., Garg, R., Dick, S., and Boheler, K. (2005). Aging-associated changes in cardiac gene expression. Cardiovasc. Res. 66, 194–204. doi: 10.1016/j.cardiores.2004.11.016
Keywords: PGC-1α, aging, myocardium, remodeling, transcriptional regulation, heart
Citation: Whitehead N, Gill JF, Brink M and Handschin C (2018) Moderate Modulation of Cardiac PGC-1α Expression Partially Affects Age-Associated Transcriptional Remodeling of the Heart. Front. Physiol. 9:242. doi: 10.3389/fphys.2018.00242
Received: 05 December 2017; Accepted: 06 March 2018;
Published: 21 March 2018.
Edited by:
Kenneth S. Campbell, University of Kentucky, United StatesReviewed by:
Bradford G. Hill, University of Louisville, United StatesKatia aquilano, Università degli Studi di Roma Tor Vergata, Italy
Copyright © 2018 Whitehead, Gill, Brink and Handschin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Handschin, Y2hyaXN0b3BoLmhhbmRzY2hpbkB1bmliYXMuY2g=